pdf, 12.1 MB - QUANTUM - Johannes Gutenberg
Werbung

Johannes Gutenberg-Universität Mainz
Fachbereich 08 (Physik, Mathematik und Informatik)
VORLESUNG:
MASSENSPEKTROMETRIE UND TEILCHENFALLEN
Dr. habil. Klaus Blaum
Mainz, Wintersemester 2007/08
Dieses Skript wurde in Zusammenarbeit mit Prof. Dr. Klaus Wendt (Institut für Physik,
Johannes Gutenberg-Universität Mainz) erstellt, dem an dieser Stelle herzlichst gedankt sei.
Inhalt
1 Einleitende Betrachtungen
1.1 Messprinzip und Dispersion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2 Historische Entwicklung in der Atommassenspektrometrie . . . . . . . . . . . . .
1.3 Kenngrößen der Spektrometrie . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1
2
6
11
I
13
GRUNDLAGEN
2 Ionenerzeugung
2.1 Ionisationsprozesse . . . . . . . . . . . . . . .
2.2 Elektronenstoßionisation . . . . . . . . . . . .
2.3 Oberflächenionisation . . . . . . . . . . . . .
2.4 Plasmaionisation . . . . . . . . . . . . . . . .
2.5 Nichtresonante und resonante Laserionisation
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
14
14
15
19
22
23
3 Ionennachweis
3.1 Fluoreszenzschirm und Photographische Platte . . . . .
3.2 Faraday-Detektor . . . . . . . . . . . . . . . . . . . . . .
3.3 Mikrokanalplatten-Detektor und Daly-Detektor . . . . .
3.4 Sekundärelektronenvervielfacher und Channeltron . . . .
3.5 Nichtdestruktive Nachweismethoden . . . . . . . . . . .
3.5.1 Nichtdestruktiver Ionennachweis mittels FT-ICR
3.5.2 Optischer nichtdestruktiver Ionennachweis . . . .
3.6 Untergrund und Totzeitkorrektur . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
25
25
25
27
29
31
31
33
33
.
.
.
.
.
36
36
39
42
44
45
.
.
.
.
.
.
.
.
.
.
4 Ionenoptik
4.1 Extraktionsoptiken und Emittanz . . . . . . . . .
4.2 Raumladungskompensation und Perveanz . . . .
4.3 Child-Langmuir Limit der Stromdichte im Strahl
4.4 Pierce-Geometrie für parallele Ionenstrahlen . . .
4.5 Transfermatrizen . . . . . . . . . . . . . . . . . .
II
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
METHODEN DER MASSENSPEKTROMETRIE
5 Flugzeitmassenspektrometrie
5.1 Quellen mit konstanter Energie bzw. konstantem Impuls . . . . . . . . . . . . . .
5.2 Ein- und zweistufige lineare Flugzeitmassenspektrometer . . . . . . . . . . . . . .
5.3 Reflektron - Flugzeitmassenspektrometer . . . . . . . . . . . . . . . . . . . . . . .
i
49
50
51
52
56
ii
INHALT
6 Radiofrequenz-Massenspektrometer
59
7 Quadrupolmassenspektrometrie
7.1 Entwicklungsgeschichte . . . . . . . . . . . . . . . . . . . . . . . . .
7.2 Die ideale lineare Paulfalle . . . . . . . . . . . . . . . . . . . . . . .
7.2.1 Hyperbolisches Quadrupolpotential . . . . . . . . . . . . . .
7.2.2 Bewegungsgleichungen in der idealen linearen Paulfalle . . .
7.2.3 Stabilitätsdiagramme und Massenauflösungsvermögen . . .
7.2.4 Frequenzspektrum der Lösungen und Ionenflugbahnen . . .
7.3 Die reale lineare Paulfalle . . . . . . . . . . . . . . . . . . . . . . .
7.3.1 Potentialbeschreibung und Bewegungsgleichungen . . . . .
7.3.2 Einschussbedingungen und maximales Auflösungsvermögen
7.3.3 Transmissionspeakformen des Quadrupol-Massenfilters . .
7.4 Nachbarmassenunterdrückung . . . . . . . . . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
62
62
63
64
65
66
70
72
73
75
78
79
8 Hochselektive Verfahren der Massenspektrometrie
8.1 Vorkommen und Bestimmung von Ultraspurenisotopen . . . . . . . . . . . . . . .
82
83
9 Beschleunigermassenspektrometrie
86
10 Resonanzionisations-Massenspektrometrie
10.1 Anregungspfade der RIMS, Selektivität und Effizienz . . . . . . . . . . . . . . . .
96
98
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
11 Massenspektrometrie in Speicherringen
104
11.1 Entwicklungsgeschichte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
11.2 Funktionsweise eines Speicherrings . . . . . . . . . . . . . . . . . . . . . . . . . . 104
11.3 Massenspektrometrie im Speicherring . . . . . . . . . . . . . . . . . . . . . . . . . 107
12 Ionenfallen und Präzisionsmassenspektrometer
12.1 Die Paulfalle . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
12.1.1 Die ideale dreidimensionale Paulfalle . . . . . . . . . . . .
12.1.2 Die reale dreidimensionale Paulfalle . . . . . . . . . . . .
12.2 Die Penningfalle . . . . . . . . . . . . . . . . . . . . . . . . . . .
12.2.1 Kurze Historie . . . . . . . . . . . . . . . . . . . . . . . .
12.2.2 Die ideale Penningfalle . . . . . . . . . . . . . . . . . . . .
12.2.3 Die reale Penningfalle . . . . . . . . . . . . . . . . . . . .
12.2.3.1 Elektrische Feldfehler . . . . . . . . . . . . . . .
12.2.3.2 Magnetische Feldfehler . . . . . . . . . . . . . .
12.2.3.3 Asymmetrie der Fallengeometrie und Fehljustage
12.2.3.4 Einfluss von gespeicherten Ionen anderer Massen
12.2.4 Anregung der Ionenbewegung . . . . . . . . . . . . . . . .
12.2.4.1 Dipolanregung . . . . . . . . . . . . . . . . . . .
12.2.4.2 Quadrupolanregung . . . . . . . . . . . . . . . .
12.2.5 Massenbestimmung und Kalibrierung . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
111
111
111
112
116
116
116
118
119
120
120
121
122
122
123
124
Tabellen
2.1
Austrittsarbeit φs , Ionisationspotential φi und Elektronenaffinität Ae für verschiedene Materialien oder Elemente . . . . . . . . . . . . . . . . . . . . . . . . .
21
3.1
Typische Spezifikationen eines Mikrokanalplatten-Detektors. . . . . . . . . . . . .
28
7.1
Gemessene Nachbarmassenunterdrückung für drei Massenbereiche . . . . . . . . .
81
8.1
Atomare und molekulare Isobare bei 14 u . . . . . . . . . . . . . . . . . . . . . .
82
9.1
Anwendungen von Ultraspurenisotopen
95
. . . . . . . . . . . . . . . . . . . . . . .
12.1 Eigenfrequenzen in einer hyperbolischen Penningfalle . . . . . . . . . . . . . . . . 121
iii
Abbildungen
1.1
1.2
1.3
1.4
1.5
1.6
1.7
1.8
1.9
1.10
1.11
1.12
Begriffsbestimmung zu Spektrum, Spektroskopie und Spektrometrie . . . . . . .
Einzelschritte und Messprinzip in der Spektrometrie . . . . . . . . . . . . . . . .
Dispersion in der Spektrometrie . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Spezifische Einzelprozesse der Massenspektrometrie . . . . . . . . . . . . . . . . .
Notwendige Prozesse in der Probeneinbringung . . . . . . . . . . . . . . . . . . .
Prinzipskizze des Thompson’schen Parabelspektrographen . . . . . . . . . . . . .
Photo des Originalaufbaus des Thompson’schen Parabelspektrographen . . . . .
Massenparabeln für Hg, Co und Ne . . . . . . . . . . . . . . . . . . . . . . . . . .
Erste wirkliche Massenspektren von Aston . . . . . . . . . . . . . . . . . . . . . .
Schnittbild des Aston’schen Massenspektrometers . . . . . . . . . . . . . . . . . .
Photo des Aston’schen Massenspektrometers . . . . . . . . . . . . . . . . . . . .
Schnittbild des Dempster’schen Massenspektrometers und Spektrum der Magnesiumisotope . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.13 Zusammenstellung der Kenngrößen der Spektrometrie allgemein . . . . . . . . . .
1.14 Kenngrößen der Empfindlichkeit . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.15 Kenngrößen der Selektivität . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
10
11
11
12
2.1
2.2
2.3
2.4
2.5
2.6
2.7
2.8
2.9
2.10
Prinzip der Elektronenstoßionisation . . . . . . . . . . . . . . . . .
Ionisierungsenergien der chemischen Elemente . . . . . . . . . . . .
Ionisationswirkungsquerschnitte . . . . . . . . . . . . . . . . . . . .
Vergleich der Ionisationswirkungsquerschnitte zwischen Experiment
Ionisationspotentiale als Funktion des Ladungszustandes . . . . . .
Cross-Beam Elektronenstoßionenquelle . . . . . . . . . . . . . . . .
Ionisationseffizienz als Funktion des Ionisationspotentials . . . . . .
Oberflächenionenquellen . . . . . . . . . . . . . . . . . . . . . . . .
Prinzip einer Plasmaionenquelle . . . . . . . . . . . . . . . . . . . .
Plasmaionenquellen . . . . . . . . . . . . . . . . . . . . . . . . . . .
. . . . . . . .
. . . . . . . .
. . . . . . . .
und Theorie
. . . . . . . .
. . . . . . . .
. . . . . . . .
. . . . . . . .
. . . . . . . .
. . . . . . . .
15
16
17
18
18
19
20
22
23
24
3.1
3.2
3.3
3.4
3.5
3.6
3.7
3.8
3.9
3.10
Nachweis der Neonisotope durch Thompson 1912 . . . . . .
Photo und Aufbau eines MCP Detektors . . . . . . . . . . .
Elektronischer Schaltplan und Signal eines MCP Detektors
Nachweiseffizienz eines Vielkanalplatten-Detektors . . . . .
Funktionsweise eines Daly-Detektors . . . . . . . . . . . . .
Funktionsweise eines Sekundärelektronenvervielfacher . . .
Funktionsweise eines Channeltron-Detektors . . . . . . . . .
Channeltron-Detektor in off-axis Geometrie . . . . . . . . .
Prinzip eines Off-Axis Channeltron-Detektor . . . . . . . .
Prinzipieller Aufbau eines nichtdestruktiven Nachweises . .
.
.
.
.
.
.
.
.
.
.
26
27
27
28
29
30
30
31
32
32
iv
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
1
2
3
4
5
7
7
8
8
9
9
v
ABBILDUNGEN
3.11 Optischer Nachweis eines Einzelions . . . . . . . . . . . . . . . . . . . . . . . . .
3.12 Bestimmung der Totzeit eines Channeltron-Detektors . . . . . . . . . . . . . . . .
33
35
4.1
4.2
4.3
37
38
4.4
4.5
4.6
4.7
4.8
Einfluss der Plasmadichte auf die Strahleigenschaften . . . . . . . . . . . . . . . .
Emittanz, dargestellt als einhüllende Ellipse . . . . . . . . . . . . . . . . . . . . .
(a) Darstellung der Emittanzellipse für verschiedene Strahlformen.
(b)
Erläuterung der Transportparameter . . . . . . . . . . . . . . . . . . . . . . . . .
Funktion R(z), die die Entwicklung eines bei z = 0 achsenparallelen Strahls unter
Berücksichtigung der Raumladung beschreibt . . . . . . . . . . . . . . . . . . . .
Bedingungen K = K0 (Raumladungsgrenze) und K = 10−4 · K0 für Protonen
und Elektronen dargestellt . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Child-Langmuir Limit der Stromdichte: (a) Skizze der Geometrie (b) Potenzialverlauf . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Pierce-Geometrie für parallele Ionenstrahlen . . . . . . . . . . . . . . . . . . . . .
(a) Berechnung einer Extraktionsoptik (b) Detailausschnitt der Ionisatoroberfläche und des Verlaufs des Plasmapotenzials . . . . . . . . . . . . . . . . . . . .
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
41
41
43
45
46
5.1
5.2
5.3
5.4
Beschleunigung im gesamten Feld - konstante Energie . . . . . . . . .
Beschleunigung mit einem kurzen Impulsübertrag - konstanter Impuls
Prinzipaufbau eines zweistufigen linearen Flugzeitmassenspektrometers
Prinzipaufbau eines Reflektron - Flugzeitmassenspektrometers . . . . .
.
.
.
.
51
52
53
57
6.1
6.2
Aufbau des MISTRAL Massenspektrometers mit einer idealen Ionentrajektorie .
Transmissionssignal von 30 Na mit einer Halbwertszeit von 48ms . . . . . . . . . .
60
61
7.1
7.2
7.3
7.4
7.5
7.6
7.7
7.8
7.9
7.10
7.11
7.12
Nobelpreisverleihung 1989 an Wolfgang Paul mit Urkunde . . . . . . . . . . .
Ideale lineare Paulfalle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
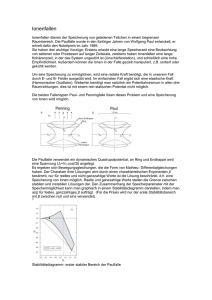
Stabilitätsdiagramm der Mathieu’schen Differentialgleichung . . . . . . . . . .
Stabilitätsdiagramm für die lineare Paulfalle . . . . . . . . . . . . . . . . . . .
Stabilitätsdiagramme 1. Ordnung im (U, V )-Raum . . . . . . . . . . . . . . .
Stabile und instabile Ionenflugbahnen . . . . . . . . . . . . . . . . . . . . . .
Reale lineare Paulfalle mit runder Stabgeometrie . . . . . . . . . . . . . . . .
Elektrodenkonfigurationen für Radiofrequenzionenfallen . . . . . . . . . . . .
Nichtlineare Resonanzlinien im Stabilitätsdiagramm des Massenfilters . . . .
Phasenraum-Akzeptanzellipsen für verschiedene Arbeitspunkte . . . . . . . .
Entwicklung der Massenpeakformen . . . . . . . . . . . . . . . . . . . . . . .
Massenspektren mit großem dynamischem Bereich für verschiedene Elemente
.
.
.
.
.
.
.
.
.
.
.
.
63
64
67
68
69
72
73
73
76
77
78
80
8.1
8.2
8.3
Auflösung des molekularen Massendubletts bei 119 u . . . . . . . . . . . . . . . .
Relative Häufigkeiten seltener Isotope in der Erdkruste . . . . . . . . . . . . . . .
Lebensdauern und Zerfallsarten langlebiger Radioisotope . . . . . . . . . . . . . .
83
84
85
9.1
9.2
9.3
9.4
Prinzipielle Selektionsschritte in einer mittelgroßen AMS Anlage . . . . . . . . .
Prinzip einer Cäsiumsputterquelle für hohe Ströme negativer Ionen . . . . . . . .
Unterdrückung von molekularen Isobareninterferenzen im Gasstripper . . . . . .
Erzeugung verschiedener Ladungszustände in einer kleinen und einer konventionellen AMS Anlage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Startdetektor für eine Flugzeitmessung in der AMS . . . . . . . . . . . . . . . . .
Besetzung der Ladungszustände in Abhängigkeit des Gasdrucks im gasgefüllten
Magneten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
87
88
89
9.5
9.6
.
.
.
.
38
.
.
.
.
.
.
.
.
.
.
.
.
89
90
91
vi
ABBILDUNGEN
9.7
(a) Ionisationskammer mit einseitig segmentierter Elektrode (b) Vergleich
spezifischen Energieverlustes für die Isobaren 41 K, 41 Ca . . . . . . . . . . .
9.8 Zweidimensionaler Scatterplot mit mehreren Ladungszuständen . . . . . . .
9.9 Grundplan der Züricher AMS-Anlage . . . . . . . . . . . . . . . . . . . . . .
9.10 Standardisotope der AMS mit typischen Anwendungsgebieten . . . . . . . .
des
. . .
. . .
. . .
. . .
10.1
10.2
10.3
10.4
10.5
10.6
.
.
.
.
.
.
Größe der Isotopieverschiebung als Funktion der Neutronenzahl . . . . . . .
Prinzipdarstellung der Resonanzionisations-Massenspektrometrie . . . . . .
Anregungs- und Ionisationspfade in der RIMS . . . . . . . . . . . . . . . . .
Ein- bis dreistufig resonante Anregungs- und Ionisationsleitern für Calcium
Entwicklung von der ein- bis zur dreistufig resonanten RIMS . . . . . . . .
Prinzip einer Laserionenquelle . . . . . . . . . . . . . . . . . . . . . . . . . .
.
.
.
.
.
.
92
93
94
94
. 97
. 98
. 99
. 100
. 101
. 102
11.1
11.2
11.3
11.4
11.5
Schematische Darstellung des ESR Speicherrings an der GSI Darmstadt . . . . .
Photo des ESR Speicherrings an der GSI Darmstadt . . . . . . . . . . . . . . . .
Betatron-Oszillation entlang der idealen Ionenbahn . . . . . . . . . . . . . . . . .
Schematische Darstellung des Prinzips der Massenmessung im ESR . . . . . . . .
Schottky-Spektrum von simultan aufgenommenen und gekühlten exotischen Nukliden im Experimentierspeicherring . . . . . . . . . . . . . . . . . . . . . . . . . .
11.6 Spektrum der Umlaufdauer mit dem ESR im isochronen Massenmodus . . . . . .
109
110
12.1 Elektrodenkonfiguration zur Erzeugung eines Quadrupolpotentials . . . . . .
12.2 Pseudopotential in einer Paulfalle . . . . . . . . . . . . . . . . . . . . . . . . .
12.3 Trajektorie eines geladenen Teilchens in einer Paulfalle . . . . . . . . . . . . .
12.4 Theoretische nichtlineare Resonanzen in einer Paulfalle . . . . . . . . . . . . .
12.5 Experimentelle nichtlineare Resonanzen in einer Paulfalle . . . . . . . . . . .
12.6 Zylindrische und hyperbolische Penningfalle . . . . . . . . . . . . . . . . . . .
12.7 Eigenbewegungen eines Ions in der Penningfalle . . . . . . . . . . . . . . . . .
12.8 Radiale Ionentrajektorien in der Penningfalle . . . . . . . . . . . . . . . . . .
12.9 Dreidimensionale Ionentrajektorien in der Penningfalle . . . . . . . . . . . . .
12.10Energieniveauschema eines Ions in der Penningfalle . . . . . . . . . . . . . . .
12.11Radiale Segmentierung der Ringelektrode einer Penningfalle . . . . . . . . . .
12.12Konversion einer reinen Magnetronbewegung in eine reine Zyklotronbewegung
12.13Nuklidkarte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
112
112
113
114
115
117
118
119
120
122
123
124
126
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
. .
105
106
106
108
Kapitel 1
Einleitende Betrachtungen
Die Begriffe des Spektrums, der Spektroskopie und Spektrometrie sind prinzipiell sehr
allgemein definiert und betreffen Strahlung wie auch Teilchen. Eine halbwegs konsistente Definition ist etwa gegeben durch:
Abb. 1.1: Begriffsbestimmung zu Spektrum, Spektroskopie und Spektrometrie
Speziell massen-spektrometrische Untersuchungen können sehr unterschiedliche Zielsetzungen haben. Bei analytischen Fragestellungen steht in der Regel die Bestimmung einer unbekannten Stoffzusammensetzung im Vordergrund, wobei die Massenpeaks im Spektrum zugeordnet
und quantifiziert werden müssen. Primäre Herausforderung ist dabei die Relativ- bzw. Absolutbestimmung von Fläche oder Höhe der Massenpeaks. Diese werden üblicherweise durch Ionenstrom oder Zählratenintegration bestimmt. Bei grundlagen-orientierten Aufgaben zur Massenbestimmung hingegen wird die Lage eines Massenpeaks, also der Massenwert selbst, mit
hoher Präzision vermessen. Hierzu werden auch häufig indirekte Methoden z.B. über Zerfallsenergiemessung eingesetzt.
1
2
KAPITEL 1. EINLEITENDE BETRACHTUNGEN
Leider gibt es aus Sicht des Physikers kein empfehlenswertes generelles und umfassendes
Lehrbuch zum Thema Massenspektrometrie. Während die Historie sehr gut in verschiedenen
Werken dargelegt ist [Duck86, Hoff96, Klie68, Ogat70, Whit1986], sei daher gerade bzgl. der
Kenngrößen der Massenspektrometrie als analytischem Verfahren auch auf die Literatur aus der
analytischen Chemie verwiesen [Frit76, Koch74, Skoo85, Vand93].
1.1
Messprinzip und Dispersion
Allgemeine Prinzipien der Spektrometrie
Zur Strukturierung lässt sich jedes massenspektrometrische Messverfahren prinzipiell in
eine Reihe von Einzelkomponenten zerlegen. Diese müssen nicht in allen Fällen individuell
durchlaufen werden, stellen aber nur in ihrer Kombination den dispersiven Charakter der
Massenspektrometrie:
Abb. 1.2: Einzelschritte und Messprinzip in der Spektrometrie
1.1. MESSPRINZIP UND DISPERSION
3
Es ist lehrreich die Dispersion ganz allgemein in der Spektrometrie einzuführen und daraus
den einfachen Spezialfall der Massenspektrometrie zu extrahieren:
Abb. 1.3: Dispersion in der Spektrometrie
4
KAPITEL 1. EINLEITENDE BETRACHTUNGEN
Spezifische Komponenten der Massenspektrometrie
Die Unterteilung der Massenspektrometrie in Einzelkomponenten kann ganz detailliert
erfolgen. Hierbei können wir bereits die einzelnen möglichen Verfahren und Schritte benennen,
gerade auch, um die in der Chemie übliche Verwendung von Abkürzungen einzuführen und
diese an einer Stelle zusammenzustellen:
Abb. 1.4: Spezifische Einzelprozesse der Massenspektrometrie mit Abkürzungen und ihre
Einordnung
Massenspektrometrie ist also ganz deutlich als physikalisches Messverfahren zu sehen, das
eine breite experimentelle Unterstützung von Seiten der Chemie und Elektronik voraussetzt
und ein nahezu grenzenloses interdisziplinäres Anwendungsspektrum aufweist.
1.1. MESSPRINZIP UND DISPERSION
5
Die Vorlesung behandelt nur den mittleren der Physik zugeordneten Bereich, während
die anderen Komponenten nur sporadisch und ganz am Rande gestreift werden.
Ein Beispiel physikalisch-chemischer Forschung und Entwicklung in der Massenspektrometrie
betrifft etwa die Optimierung der Probeneinbringung: Die zu untersuchende Probe kann in
allen möglichen Ausgangsphasen vorliegen. Zur massenspektrometrischen Bestimmung wird
in nahezu ausnahmslos die Auflösung in Molekül- oder Atomionen im Hochvakuum benötigt.
(Ausnahme ist etwa die Ionenmobilitätsspektrometrie, die bei Atmosphärendruck abläuft).
Üblicherweise wird aber vor Einbringung ins Vakuum die Überführung in die gasförmige Phase
vorgeschaltet bzw. vorbereitet. Zur Erzeugung von Atomionen ist dabei in fast allen Fällen die
Reduktion der typischerweise als Oxid vorliegenden Komponente notwendig. Dabei kommen
verschiedene Verfahren in der Probenvorbereitung zum Einsatz:
Abb. 1.5: Notwendige Prozesse in der Probeneinbringung als Vorstufe zur Atomisation und
Ionisation
6
KAPITEL 1. EINLEITENDE BETRACHTUNGEN
1.2
1886
Historische Entwicklung in der Atommassenspektrometrie
Eugen Goldstein entdeckt die Kanalstrahlen – erste Erzeugung eines Ionenstrahls
Zu dieser Zeit ist weder die Atomhypothese akzeptiert, noch sind Isotope bekannt
(das Neutron wird 1920 postuliert und erst 1932 entdeckt werden!)
Seine Apparatur ist eine Hochspannungs-Gasentladungsröhre mit einer
durchlöcherten Kathode und einem Austritt ins Hochvakuum über eine differentielle Pumpstrecke. Es verbrennt etwa 1 - 10 cm3 /h ( 1021 Moleküle/h =
3x1017 Moleküle/sec) und es wird etwa 4x10−5 A nachgewiesen, das entspricht
immerhin einer Ausbeute von etwa 10−4 .
Fast zur gleichen Zeit werden in wenigen Labors weltweit die Eigenschaften dieser Strahlen
untersucht:
1897
J.J. Thompson (Cambridge Nobelpreis 1906 für die Entdeckung des Elektrons)
entwickelt dazu den Parabelspektrographen.
~ und B
~ stehen parallel zueinander,
Ein elektrisches und magnetisches Feld der Feldstärken E
oBdA in y-Richtung bei z = Flugrichtung, v = Geschwindigkeit und U = Energie. Es gilt dann
für die jeweilige transversale Ablenkung durch das jeweilige Feld.
Bei Annäherung durch Kreisbögen mittels tan αe,m ≈ αe,m ≈ l/re,m gilt
r
lE
e
e E
e B
αe ≈
und αm ≈ lB
=l
=l
2
2U
mv
2mU
mv
Die Flugstrecke kann man bei kleinen Winkeln multiplikativ berücksichtigen. Daraus folgt dann
für die Gesamtablenkung in x und y:
y ≈ lL
e B
e E
, x ≈ lL
,
m v2
mv
woraus über Quadrieren eine Parabelform folgt
y≈
1 m E 2
x .
lL e B 2
1.2. HISTORISCHE ENTWICKLUNG IN DER ATOMMASSENSPEKTROMETRIE
7
Abb. 1.6: Prinzipskizze des Thompson’schen Parabelspektrographen
Pro Masse erhält man also eine Parabel mit niederenergetischeren Teilchen nahe dem Scheitelpunkt und hochenergetischeren Teilchen bei höheren Werten der Parabel.
Abb. 1.7: Photo des Originalaufbaus des Thompson’schen Parabelspektrographen
Bei der Untersuchung von Neon fand Thompson mit seinem Parabelspektrograph nahe zusammen liegende aber deutlich getrennte Parabeln. Darüber wurde im Jahre 1910 erstmals die
Existenz von unterschiedlichen Isotopen gefunden, also die Isotopie entdeckt:
20 N e
( − 90, 51%)
( − 0, 27%) - erst 1919 von Aston gefunden!
22 N e ( − 9, 22%)
21 N e
8
KAPITEL 1. EINLEITENDE BETRACHTUNGEN
Abb. 1.8: Massenparabeln für Hg, Co und Ne
Die Strommessung über eine Photoplatte ist dabei nur qualitativ ohne Intensitätsbestimmung. Ein konkurrierendes Labor leitet
1898
Wilhelm Wien (Berlin, Nobelpreis 1911). Er entwickelt den nach ihm benannten Wienfilter, ein Geschwindigkeitsfilter, in welchem sich senkrecht zueinan~ und B
~ Felder bei konstanter Energie der
der und zur Flugrichtung stehende E
Teilchen immer gerade für einen Massenwert kompensieren.
1919
F. Aston (Nobelpreis 1922) arbeitet im Labor von Thompson und baut dort den
ersten Spektrographen mit Geschwindigkeitsfokussierung einer Masse auf einen
Punkt, d.h. er gibt die Energiebestimmung auf, erhält aber dafür das erste
Massenspektrum mit genauer Massenbestimmung.
Abb. 1.9: Erste wirkliche Massenspektren von Aston
1.2. HISTORISCHE ENTWICKLUNG IN DER ATOMMASSENSPEKTROMETRIE
9
~ und B
~ -Feld, sowie Blenden
Das Spektrometer setzt sich aus einer Gasentladungsröhre, E
und einer Photoplatte zusammen. Die hohe Energieunschärfe der verwendeten Plasmaröhre
macht dabei Probleme.
Abb. 1.10: Schnittbild des Aston’schen Massenspektrometers
Aston detektiert mit dieser trotzdem viel leistungsfähigeren Apparatur erstmals auch das
Spurenisotop 21 N e mit einer natürlichen Häufigkeit von nur 0,2 %. Außerdem wird der
Massendefekt, also die bindungsbedingte Abnahme der Masse der Einzelkonstituenten erstmals
experimentell zugänglich.
Dies ist die erste experimentelle Bestätigung der von Einstein
postulierten Äquivalenz von Masse und Energie.
Abb. 1.11: Photo des Aston’schen Massenspektrometers
10
1918
KAPITEL 1. EINLEITENDE BETRACHTUNGEN
A.J. Dempster (in Chicago) realisiert die Richtungsfokussierung in einem 180
Magneten und verwendet erstmals eine energiescharfe Ionenquelle, die statt
Gasentladung mittels Elektronenstoß funktioniert.
Abb. 1.12: links: Schnittbild des Dempster’schen Massenspektrometers mit 180 Magnet, energiescharfer Ionenquelle und Elektrometerdetektor, rechts: Spektrum der Magnesiumisotope
Durch Strommessung über ein Elektrometer kann er genaue Isotopenverhältnisse bestimmen,
gibt aber die genaue Ermittlung der Peaklagen zur Massenbestimmung auf. Im Jahre 1929
schlägt Dempster die Doppelfokussierung vor.
1.3. KENNGRÖSSEN DER SPEKTROMETRIE
1.3
11
Kenngrößen der Spektrometrie
Auflösung und Nachbarmassenunterdrückung
Eine Zusammenstellung der in der Spektrometrie wichtigen Kenngrößen zur Charakterisierung
und Quantifizierung der Leistungsfähigkeit eines Verfahrens gibt:
Abb. 1.13: Zusammenstellung der Kenngrößen der Spektrometrie allgemein
Hierbei sind die Kenngrößen der Empfindlichkeit miteinander korreliert, wobei eine quantitative Definition aus der Untergrundzählrate und über den gewählten Intervallfaktor mit statistisch
belegten Wahrscheinlichkeiten erfolgt.
Abb. 1.14: Kenngrößen der Empfindlichkeit
12
KAPITEL 1. EINLEITENDE BETRACHTUNGEN
Die Kenngrößen der Selektivität eines massenspektrometrischen Verfahrens, das
Auflösungsvermögen und die Nachbarmassenunterdrückung, müssen in ihrer Definition
sauber getrennt werden. Sie sind korreliert aber es kann kein quantitativer Zusammenhang
formuliert werden, da dieser stark von der jeweiligen Form des erhaltenen Massenpeaks und
seinen Ausläufern abhängt. Zusätzlich muss klar angegeben werden, wie Peakbreiten gemessen
werden; typisch sind auf halber Höhe (FWHM), bei 10 % oder bei 5 %.
Abb. 1.15: Kenngrößen der Selektivität
Teil I
GRUNDLAGEN
13
Kapitel 2
Ionenerzeugung
2.1
Ionisationsprozesse
Ionenquellen (engl.: ion sources) sind von großer Bedeutung in der Massenspektrometrie. Nur
durch effiziente Produktion und Ionisation der in den Massenspektrometer einzubringenden
Teilchen lassen sich Präzisionsexperimente mit kleinsten Mengen von Ionen durchführen.
Die Ionisationsmethoden sind vielfältig und zahlreiche Ionenquellen sind zumeist nur für eine
bestimmte Anwendung entwickelt und optimiert. Heute in der Massenspektrometrie eingesetzte
Ionisationsverfahren sind:
1. Elektronenstoßionisation (Electron impact ionization EII)
2. Oberflächenionisation (Surface ionization SI)
3. Laserionisation (Laser ionization LI)
4. Laserdesorption (Laser desorption LD)
5. Matrix unterstützte Laserdesorption/Ionisation
tion/ionisation MALDI)
(Matrix
assisted
laser
desorp-
6. Sekundärionen-Massenspektrometrie (Secondary ion mass spectrometry SIMS)
7. Schnelle Atomstöße (Fast-atom bombardment FAB)
8. Chemische Ionisation (Chemical Ionization CI)
9. Elektrospray Ionisation (Electrospray ionization ESI)
10. Feld Ionisation (Field ionization FI)
11. Thermospray Desorption/Ionisation (Thermo-spray desorption/ionization TSDI)
In diesem Kapitel sollen nur die wichtigsten und häufigst eingesetzten Ionisationsmechanismen für effiziente Ionisation der Neutralatome vorgestellt werden. Als Literaturquelle dienen
u.a. die Lehrbücher von L. Vályi, Atom and Ion Sources [Vály1977], I.G. Brown, The Physics
and Technology of Ion Sources [Brow1989], and B. Wolf, Handbook of Ion Sources [Wolf1995].
14
2.2. ELEKTRONENSTOSSIONISATION
2.2
15
Elektronenstoßionisation
Ionisation von Neutralteilchen durch Kollision bzw. inelastische Stöße mit Elektronen in einem
Gas ist der fundamentalste Ionisationsmechanismus. Diese Methode, die in Abb. 2.1 schematisch
dargestellt ist, wird als Elektronenstoßionisation bezeichnet [Maer1985]. Bei diesem Ionisationsprozess wird ein freies Elektron im Gas durch ein angelegtes elektrisches Feld auf eine Energie
beschleunigt die ausreichend ist, um bei einer Kollision mit dem Neutralteilchen eine Ionisation
hervorzurufen. Die Reaktion lautet: e− + A → 2e− + I. Eine bestimmte Elektronenmindestenergie wird für die Ionisation benötigt, d.h. die Elektronenenergie muss hoch genug sein, um
das äußerste gebundene Elektron vom Neutralatom zu entfernen (Ee > eφi ). Dies ist das so
genannte erste Ionisationspotential bzw. die Ionisierungsenergie φi .
Abb. 2.1: Prinzip der Elektronenstoßionisation. Gezeigt ist die Elektronenstoßionenquelle mit
Filament, Repeller und Linsensystem.
In Abb. 2.2 ist die Ionisierungsenergie der Elemente als Funktion ihrer Ordnungszahl aufgetragen. Bei der Ionisierungsenergie handelt es sich um die zur Herauslösung des am leichtesten gebundenen Elektrons aus einem neutralen Atom notwendigen Energie. Die auffallenden Ähnlichkeiten in den chemischen und physikalischen Eigenschaften der Elemente in jeder
senkrechten Spalte des Periodensystems sind ein deutlicher Hinweis darauf, dass der Aufbau der
Atome einer Systematik folgt.
Die Ionisationswahrscheinlichkeit ist abhängig von der Elektronenenergie: für Energien unterhalb eφi ist sie Null, für Energien drei- bis viermal eφi ist sie maximal und oberhalb nimmt
sie wieder ab. Dieses Verhalten ist in Abb. 2.3(a) dargestellt.
16
KAPITEL 2. IONENERZEUGUNG
Abb. 2.2: Eine Kurve der Ionisierungsenergien der chemischen Elemente als Funktion ihrer
Ordnungszahl. Deutlich erkennbar ist die regelmäßige Wiederholung gewisser Eigenschaften
über die sechs vollständigen Perioden des Periodensystems. Die Anzahl der Elemente in jeder
dieser Perioden ist ebenfalls angegeben [Hall2003].
Nahe der Ionisationsschwelle kann der Ionisationsquerschnitt abgeschätzt werden über:
σi = C(Ee − E0i )n
(2.1)
wobei C eine Konstante ist. Für die Ionisation von Atomen gilt n = 1.1269 und für die Ionisation von Ionen beträgt n = 1.056 (bei unterschiedlicher Konstante C). Abb. 2.3(b) zeigt die
Änderung des Ionisationswirkungsquerschnitts für ausgewählte Edelgase bei Elektronenstoßionisation nahe der Ionisationsgrenze. Der Wirkungsquerschnitt ist dabei in Einheiten von a0 ,
d.h. dem Bohr’schen Radius mit a0 = (4πε0 ~2 )/(e2 me ) = 0.529 Å und 1Å= 10−10 m angegeben.
Im klassischen Ansatz kann der inelastische Stoß, der zur Ionisation, führt als Zweiteilchenstoß betrachtet werden, bei dem nur die beiden beteiligten Elektronen (Stoßelektron und Atomelektron) berücksichtigt werden. Wird das Atomelektron als in Ruhe während des Stoßes
angenommen, so ist der Energieübertrag E vom freien Stoßelektron auf das gebundene Elektron
gegeben durch [Land1938]:
E=
4m1 m2
(m1 + m2 )2
1+
Ee2
ρ2 ve42
(e1 e2 )2
m1 m2
m1 +m2
2
(2.2)
wobei m1 , m2 ; e1 , e2 ; v1 , v2 ; Ee1 , Ee2 die Massen, Ladungen, Geschwindigkeiten und Energien
des freien und gebundenen Elektrons bezeichnen. Für Elektron-Elektron-Stöße hat man e1 =
e2 = e und m1 = m2 = m und es resultiert die Thomson-Gleichung der Form
E=
Ee2
1+
ρ2 2
e4 Ee2
.
(2.3)
17
2.2. ELEKTRONENSTOSSIONISATION
(a)
(b)
Abb. 2.3: (a) Verhalten des Ionisationswirkungsquerschnitts als Funktion der Elektronenstoßenergie für H1 , H2 , He, Ne, N2 und Ar. (b) Variation des Ionisationswirkungsquerschnitts von
Argon, Xenon und Neon Atomen als Funktion der Elektronenstoßenergie nahe der Ionisationsschwelle [Vály1977].
Der Kollisions- bzw. Streuparameter ρ ist gegeben durch
"
#
E 2
E
e4
e4
E
2
=
−
f
.
ρ =
(E i )2 Ee2
Ee2
(E i )2
Ee2
(2.4)
Hier bezeichnet E i die Ionisationsenergie. Unter der Annahme, dass jeder Energietransfer E >
E i zu einer Ionisation führt, kann der Ionisations-Stoßwirkungsquerschnitt σi als Funktion der
Anzahl N der Elektronen in der äußeren Schale angegeben werden mit
e4
E
2
σi = N πρ = N π i 2 f
.
(2.5)
(E )
Ee2
Gleichung (2.5) beschreibt qualitativ sehr gut die Energieabhängigkeit des Ionisationswirkungsquerschnitts, sagt aber einen falschen Wert (im Vergleich zum Experiment) für das Maximum
voraus. Abbildung 2.4 zeigt den Vergleich zwischen experimentell gewonnener Wirkungsquerschnittskurve und verschiedenen theoretischen Modellen, u.a. dem hier vorgestellten klassischen
Näherungsmodell nach Thomson [Thom1912].
Die o.a. klassische Beschreibung kann noch verfeinert werden indem die Bewegung des im
Atom gebundenen Elektrons mitberücksichtigt wird [Gryz1959]. Die theoretische Vorhersage
über die Lage des Maximums für den Ionisationswirkungsquerschnitt als Funktion der Elektronenenergie stimmt dabei schon recht gut mit dem experimentellen Wert überein (siehe Abb.
2.4). Es fällt jedoch auf, dass die Abweichungen zwischen Theorie und Experiment bei hohen
Stoßenergien sehr stark sind. Dies liegt daran, dass die Wechselwirkung während der Kollision
zwischen dem Elektron und dem verbleibenden Atom in den Modellen nicht mit berücksichtigt
wird. Eine bessere Übereinstimmung zwischen Theorie und Experiment kann nur erreicht werden, wenn eine vollständig quantenmechanische Rechnung durchgeführt wird (siehe z.B. quantenmechanische Näherung durch Bethe [Beth1930]). An dieser Stelle soll jedoch auf eine tiefergehende Beschreibung verzichtet werden.
18
KAPITEL 2. IONENERZEUGUNG
Abb. 2.4: Vergleich der Ionisationswirkungsquerschnitte zwischen Experiment und verschiedenen
theoretischen Modellen [Vály1977].
Neben der einfachen Ionisation durch Elektronenstoß können auch höher geladene bzw.
hochgeladene Ionen produziert werden, sofern die Elektronenstoßenergie ausreichend ist, um
die weiteren Elektronen aus der Atomhülle zu entfernen. Allerdings werden dazu immer höhere
Energien benötigt, da das Ionisationspotential mit der Anzahl der aus der Atomhülle entfernten Elektronen ansteigt. Abbildung 2.5 zeigt berechnete Ionisationspotentiale [Carl1970] als
Funktion des Ladungszustandes.
Abb. 2.5: Berechnete Ionisationspotential für alle Ladungszustände aller Elemente [Wolf1995].
Die Zahlenwerte sind entnommen aus [Carl1970].
19
2.3. OBERFLÄCHENIONISATION
Wechselwirkungsregion
5.34 cm
WolframFilament
Ionisationsraum
Extraktionslinse
Linse 3
Linse 2
Linse 1
6.06 cm
Abb. 2.6: Die Cross-Beam Elektronenstoßionenquelle.
Eine kommerzielle Cross-Beam-Elektronenstoßionenquelle mit Linsensystem und zwei
Wolfram-Filamenten ist in Abb. 2.6 zu sehen. Sie wird gemeinsam mit einem QuadrupolMassenspektrometer der Firma ABB Extrel vertrieben. Die von der Wolframkathode emittierten
Elektronen werden je nach Einstellung mit 3-200 V Spannung in den Ionisationsraum beschleunigt, so dass das Gas in der Wechselwirkungsregion abhängig von der Elektronenenergie
teilweise stoßionisiert wird. Die entstandenen Ionen werden über die Extraktionselektrode
abgezogen und durch die Ionenlinsen in den Quadrupol-Massenfilter fokussiert.
2.3
Oberflächenionisation
Atome können bei Kontakt mit einer heißen Metalloberfläche ionisiert werden. Dies bezeichnet
man als Kontaktionisation oder Oberflächenionisation.
Oberflächenionisation kann ein sehr effizienter Weg sein, um Elemente mit niedrigem Ionisationspotential, wie z.B. Alkalis (≤ 5 eV) zu ionisieren. Das gilt auch für Elemente mit hoher
Elektronenaffinität zur Bildung von negativen Ionen, wie beispielsweise die Halogene (≥ 1.8 eV).
Oberflächenionenquellen für positive Ionenerzeugung bestehen aus einem HochtemperaturIonisator welcher aus einem Material mit hoher Austrittsarbeit, wie z.B. Wolfram, Rhenium,
Iridium oder Zeolite, hergestellt ist. Zur Generierung von negativen Ionen verwendet man Materialien mit niedriger Austrittsarbeit, so z.B. Wolfram beschichtet mit einer Monolage Cäsium
oder Platin beschichtet mit Kohlenstoff.
Solange die Verweildauer der Teilchen auf der Oberfläche lange genug ist, um mit der heißen
Oberfläche in ein thermisches Gleichgewicht zu kommen (typischerweise 10−5 bis 10−3 s), so
ist die Ionisationswahrscheinlichkeit durch eine Form der Langmuir-Saha Gleichung [Lang1925]
gegeben:
−1
g0
ni
.
(2.6)
= 1 + ee(φi −φs )/kT
Pi =
n0 + ni
gi
Hier bezeichnen ni und n0 die Anzahl an Ionen oder Atomen die von der Oberfläche verdampft
20
KAPITEL 2. IONENERZEUGUNG
werden, gi und g0 die statistischen Gewichte der Ion- und Atomzustände (für Alkalimetalle gilt
gi /g0 = 1/2), φi ist das Ionisationspotential des Atoms, φs die Austrittsarbeit des Metalls und T
die Temperatur der heißen Metalloberfläche. Der Bruchteil an Ionisation, der für die meisten φi −
φs Kombinationen erzeugt werden kann ist, in der Regel sehr gering. Aber für die Alkalimetalle
und Erdalkalis (Li, Na, K, Rb, Cs, Ca, Sr, Ba, usw.) mit sehr niedriger Austrittsarbeit erreicht
man auf heißen refraktiven Metallplatten (Ta, W, Re, Ir, Pt, usw.) mittels Oberflächenionisation
hohe bis sehr hohe Ionisationswahrscheinlichkeiten. Einige Beispiele: Pi (K auf Pt bei 1500 K)=
1.0, Pi (Cs auf W bei 1500 K)= 0.99 und Pi (Ba auf Re bei 2200 K)= 0.12. Abbildung 2.7 zeigt
an weiteren Beispielen die Ionisationseffizienz von verschiedenen Elementen auf einer Oberfläche
mit Austrittsarbeit φs = 5.25. Beträgt die Differenz φs − φi ≥ 0.5, so ist die Ionisationseffizienz
größer 90% und für φs = φi beträgt die Effizienz 33% beim gewählten Beispiel. Abhängig von
den statistischen Gewichten g, die durch 2J + 1 gegeben sind - wobei J der Gesamtdrehimpuls
des Ions bzw. Atoms ist - kann die Ionisationseffizienz auch deutlich geringer sein.
Abb. 2.7: Die Ionisationseffizienz als Funktion des Ionisationspotentials für eine Oberfläche mit
Austrittsarbeit φs = 5.25.
Für die Erzeugung von negativen Ionen gilt eine ähnliche Formel wie (2.6), jedoch mit der
Differenz zwischen Austrittsarbeit der Elektronen aus der Oberfläche φs und der Elektronenaffinität des Atoms oder Moleküls Ae im Exponenten (φs − Ae ) [Alto1986].
In Tabelle 2.1 sind Werte von Austrittsarbeiten für verschiedene Materialien sowie Ionisationspotentiale und Elektronenaffinitäten für verschiedene Elemente und Moleküle aufgelistet
die geeignet sind zur Oberflächenionisation [Wolf1995].
Obwohl die Ionisationseffizienz mit steigender Temperatur abnimmt, muss der Ionisator
doch heiß genug sein, um das entsprechende Element zu verdampfen. Zudem muss die Diffusion oder die Oberflächenbedeckung niedrig genug sein (≤ 10% einer Monolage), um die
Ionisationsbedingungen (φs ) des Ionisationsmaterials zu erhalten. Daher ist die Kontrolle der
Oberflächenbedeckung die Hauptaufgabe bei der Entwicklung von Oberflächenionenquellen. Die
Lebensdauer der Ionenquellen kann bei kontinuierlichem Betrieb mehr als 2000 h betragen, wobei
dies natürlich sehr stark vom Ionenquellenaufbau und dem Reservoir für die zu ionisieren-
21
2.3. OBERFLÄCHENIONISATION
Tabelle 2.1: Austrittsarbeit φs , Ionisationspotential φi und Elektronenaffinität Ae für verschiedene Materialien oder Elemente [Smit1967,
McDa1972, Alto1986, Alto1993].
Material
φs (eV)
φi (eV)
Ni
Mo
Ta
W
W+O
Ir
Pt
Re
Li
Na
Al
K
Ca
Ga
Rb
Sr
In
Cs
Ba
La
Rear earth metals
Th
4.61
4.15
4.12
4.54
6
5.40
5.32
4.85
2.46
2.28
4.2
2.25
3.2
4.16
2.13
2.74
–
1.81
2.11
3.3
∼ 3.5
3.38
Ba auf W
Cs auf W
Th auf W
1.56
1.36
2.63
7.6
7.2
7.8
8.0
–
9.0
9.0
7.9
5.4
5.1
6.0
4.3
6.1
6.0
4.2
5.7
5.8
3.9
5.2
5.6
5.6-6.9
∼4
BaO
SrO
BaO + SrO
Cs-Oxide
1.5
2.0
0.95
0.75
LaB6
ThO2
TaC
ZrO2
MgO
BeO
Al2 O3
SiO2
CuO
W-Oxide
Ni-Oxide
Pt-Oxide
2.70
2.54
3.14
4.2
4.4
4.7
4.7
5.0
5.34
6.24
6.34
6.55
Ae (eV)
1.1
1.3
0.6
0.6
–
1.9
2.5
0.2
0.6
0.55
0.45
0.5
-1.5
0.3
0.49
-1
0.3
0.4
-0.5
0.5
0.5-0.5
–
2.8
22
KAPITEL 2. IONENERZEUGUNG
den Atome abhängt. Zwei Beispiele von Oberflächenionenquellen sind in Abb. 2.8 gezeigt.
Oberflächenionenquellen haben den Vorteil, dass sie über einen langen Zeitraum hinweg von
bis zu mehreren tausend Stunden stabile Ionenstrahlbedingungen liefern. Zudem sind sie ideal
geeignet für schwierige Elemente wie z.B. Halogene und Alkalis und zeichnen sich durch eine sehr
gute Elementselektivität aus. Nachteile dieser Ionisationsmethode sind die hohe Wärmeabgabe
an die Umgebung und der nur bedingte Einsatz für Elemente mit niedriger Austrittsarbeit und
niedrigem Ionisationspotential bzw. hoher Elektronenaffinität.
Abb. 2.8: Oberflächenionenquellen nach Daley [Dale1971] (links) und Souzis [Souz1990] (rechts).
Eine Thermoionisations-Ionenquelle basiert auf dem gleichen Prinzip wie eine
Oberflächenionenquelle, allerdings wird sie in Verbindung mit einer heißen Zelle als Ionisator eingesetzt. Dadurch kann die Ionisationseffizienz erhöht werden, denn die Teilchen sind in
der Kavität eingefangen und machen zuerst zahlreiche Stöße mit den Wänden ehe sie die Quelle
verlassen. Elemente mit Ionisationspotentialen bis zu 8 eV können in diesen Ionenquellen
ionisiert werden wobei Temperaturen bis zu 2700 K vorliegen [Kirc1990].
2.4
Plasmaionisation
Die Plasmaionenquelle wird benutzt um Elemente zu ionisieren, die mittels Oberflächenionisation
nicht zugänglich sind. Das Plasma wird dabei durch eine Gasmischung (typischerweise Ar und
Xe, aber auch SF6 ) generiert, welches durch Elektronenbeschuss ionisiert wurde. Dabei werden
die Elektronen von einem Filament (zumeist Wolfram) emittiert, beschleunigt und in einem
Magnetfeld eingefangen, um das Plasma zu generieren und die Ionisationseffizienz zu erhöhen.
Abbildung 2.9 zeigt das Prinzip der Plasmaionisation von Elementen mit hohem
Schmelzpunkt (die also schwer verdampfbar sind) am Beispiel einer SF6 Plasmaionenquelle [Sait1998]. Die Metalle mit hohem Schmelzpunkt wie z.B. B, Mo und Si reagieren
mit aktiven Fluorionen im Plasma um Metallfluoride mit genügend hohem Dampfdruck zu
erzeugen. Diese Metallfluoride werden dann verdampft und anschließend durch Beschuss mit
Elektronen und Ionen, die im SF6 Plasma generiert wurden, in ihre Komponenten (Metallionen
und Flurionen) zerlegt. Das Prinzip ist also sehr ähnlich einer Elektronenstoßionenquelle.
Weitere Beispiele von Plasmaionenquellen, wie sie bei der Ionisation von Radionukliden
am on-line Isotopenseparator ISOLDE am CERN in Genf (Schweiz) eingesetzt werden, sind in
Abb. 2.10 gezeigt. Die beiden Plasmaionenquellen unterscheiden sich lediglich in der Temperatur des Transferröhrchens für die Atome. Für die Produktion von Edelgasisotopen wird das
2.5. NICHTRESONANTE UND RESONANTE LASERIONISATION
23
Abb. 2.9: Prinzip einer SF6 Plasmaionenquelle zur Ionisation von Elementen mit hohem
Schmelzpunkt.
Transferröhrchen zwischen Target und Gasplasma durch einen ständigen Wasserfluss gekühlt.
Dadurch wird der Transport von schwer flüchtigen Elementen unterdrückt und folglich die Isobarenkontamination der Ionenstrahlen reduziert.
2.5
Nichtresonante und resonante Laserionisation
Im Gegensatz zu nichtresonanten Laserionenquellen, bei denen die Ionen in einem Plasma hoher Leistungsdichte (hervorgerufen durch die starke Fokussierung eines Laserstrahls) produziert
werden, nutzen resonante Laserionenquellen die selektive Anregung und den selektiven Photoionisationsübergang eines bestimmten Elementes aus. Durchstimmbare Hochleistungslasersysteme sind notwendig um eine hohe Ionisationseffizienz zu erreichen. Aufgrund der hohen
Isobaren- und Isotopenselektivität, die man mit resonanter Laserionisation erreicht, ist die
Laserionenquelle die zur Zeit häufigst eingesetzte Ionenquelle zur Ionisation von kurzlebigen
Radionukliden für on-line Präzisionsmassenexperimente [Köst2003]. Aufgrund der Vielfalt an
Anwendungen wird die resonante und nichtresonante Laserionisation in einem eigenen Kapitel
zu einem späteren Zeitpunkt behandelt.
24
KAPITEL 2. IONENERZEUGUNG
Abb. 2.10: Plasmaionenquellen mit heißem (oben) und kaltem (unten) Transferröhrchen (”line”)
wie sie beim on-line Isotopenseparator ISOLDE am CERN in Genf zur Ionisation von Radionukliden eingesetzt werden.
Kapitel 3
Ionennachweis
3.1
Fluoreszenzschirm und Photographische Platte
Der Fluoreszenzschirm ist die älteste Technik zur Sichtbarmachung von geladenen Teilchen
und wurde bereits 1886 eingesetzt. Dazu trifft das Ion auf eine beschichtete Oberfläche aus
üblicherweise Glas oder Zinkblende (Zinksulfid) und ruft Fluoreszenz hervor. Diese Methode ist
sehr unempfindlich und es werden ca. 108 Ionen/Sekunde für den Nachweis benötigt.
Die später im Jahre 1910 entwickelte photographische Platte aus Silberhalogenen hatte bereits eine deutlich höhere Empfindlichkeit. So konnten 104 Ionen auf eine Fläche von 0.1 mm2
nachgewiesen werden. Zudem weist diese Nachweismethode einen hohen dynamischen Bereich
von bis zu neun Größenordnungen (104 − 1013 Ionen/s) und ein hohes Auflösungsvermögen von
> 105 auf. Die photographische Platte wurde in der Massenspektrometrie u.a. 1912 von Thompson zum erstmaligen Nachweis von Elementisotopen am Beispiel von Neon eingesetzt (siehe Abb.
3.1). Die Nachteile dieser Nachweismethode sind zahlreich:
1. massenabhängige Nachweisempfindlichkeit
∼
Z
M
1/2
(3.1)
mit Z/M : Ladung zu Massenverhältnis.
2. keine genaue Isotopenhäufigkeitsbestimmung möglich
3. die Signalintensität nimmt nicht linear mit der Anzahl der auftreffenden Teilchen zu
4. Aufladung der Plattenoberfläche.
3.2
Faraday-Detektor
Ein Faraday-Auffänger oder Faraday-Detektor (FC, von engl. Faraday Cup ) ist ein Detektor zur Messung von Ionen- oder Elektronenströmen. Faraday-Auffänger werden beispielsweise
in Massenspektrometern als Alternative oder zusätzlich zum Sekundärelektronen-Vervielfacher
(SEV, siehe Kap. 3.4) verwendet. Vorteil des Faraday-Auffängers ist seine Zuverlässigkeit und
Robustheit und die Möglichkeit den Ionenstrom oder Elektronenstrom absolut zu messen. Zudem ist die Empfindlichkeit zeitlich konstant und nicht massenabhängig. Nachteil ist die geringe
Nachweisempfindlichkeit (Imin ≈ 10−16 A bzw. ≈ 103 Ionen) und die geringe Bandbreite (d.h.
lange Reaktionszeit).
25
26
KAPITEL 3. IONENNACHWEIS
Abb. 3.1: Einsatz einer photographischen Platte 1912 durch Thompson für den erstmaligen
Nachweis von Elementisotopen am Beispiel von Neon [Asto1929].
Der prinzipielle Aufbau ist recht einfach. Ein Faraday-Detektor besteht aus einem Metallbecher (Faradaybecher), der in den zu messenden Ionenstrahl (Elektronenstrahl) gebracht wird.
Da der Faradaybecher auf konstantem Potenzial gehalten wird, müssen die aufgefangenen Ionen
(Elektronen) durch Elektronen, welche über einen angeschlossenen hochohmigen Widerstand
(typisch 109 − 1011 Ω) in den Faradaybecher zufließen bzw. abfließen können, neutralisiert werden. Am Widerstand fällt deswegen eine Spannung ab welche ein Maß für den Ionenstrom
(Elektronenstrom) ist und z.B. mit einem Verstärker/Elektrometer gemessen werden kann.
Wird verhindert, dass reflektierte Ionen/Elektronen oder aus der Detektoroberfläche herausgeschlagene Sekundärelektronen den Faradaybecher verlassen, kann mit einem FaradayAuffänger direkt die Anzahl der aufgefangenen Ladungsträger pro Zeiteinheit bestimmt werden.
Das kann durch die geometrische Form des Faradaybechers und durch auf negativem Potenzial liegende Suppressor-Elektroden erreicht werden, welche die Sekundärelektronen wieder zum
Detektor zurückzwingen.
Bei Neutralgas-Faraday-Auffängern wird die Suppressor-Elektrode positiv vorgespannt, so
dass die durch den Impakt von neutralen Atomen erzeugten Sekundärelektronen vom FaradayAuffänger weggeleitet werden. Zum Ladungausgleich müssen deswegen Elektronen über den
hochohmigen Widerstand nachfließen, womit ein Signal detektiert werden kann.
3.3. MIKROKANALPLATTEN-DETEKTOR UND DALY-DETEKTOR
3.3
27
Mikrokanalplatten-Detektor und Daly-Detektor
Ein Mikrokanalplatten-Detektor (MCP - micro channel plate) besteht aus einer großen Anzahl
parallel angeordneter, sehr dünner Kanäle (Durchmesser zwischen 10 µm und 100 µm, Länge
zu Durchmesser Verhältnis zwischen 40 und 100), die aus einem Material (z.B. Bleiglas) bestehen, das einen hohen elektrischen Widerstand von etwa 100 MΩ zwischen den Kanälen aufweist
[Wiza1979]. Die Kanalachsen sind typischerweise leicht geneigt (∼ 8◦ ) gegen die MCP Eintrittsfläche. Abbildung 3.2 zeigt einzelne MCP Platten und ihr Zusammenbau. Zwischen den
(a)
(b)
(c)
Abb. 3.2: Einzelne MCP Platten (a) und ihr Zusammenbau (b,c). Die Zeichnung in (b) zeigt
zwei aufeinander abgestimmte MCP Platten (grün) in Chevron Konfiguration. Die Metallanode
ist in blau und die Keramikisolatoren sind in rot dargestellt.
beiden Enden der Kanäle wird eine elektrische Spannung von etwa 1000 V angelegt. Dazu kann
die in Abb. 3.3 gezeigte Spannungsteilerschaltung benutzt werden. Das resultierende Pulssignal
ist ebenfalls in Abb. 3.3 gezeigt. Die Spezifikationen eines Mikrokanalplatten-Detektors sind in
Abb. 3.3: Elektronischer Schaltplan und typisches Pulssignal für einen MikrokanalplattenDetektor (mit zusätzlichem Phosphorbildschirm).
Tab. 3.1 aufgelistet.
Ein Ion, das auf das MCP trifft, erzeugt ein primäres Elektron, das im Kanal beschleunigt
wird und eine Lawine von sehr vielen (103 − 104 ) auslöst. Mittels einer zweiten Platte (siehe
Abb. 3.2b/c, Chevron Konfiguration) erhält man einen ausreichend starken Strompuls, der von
einem Vielkanalzähler zeitgerecht abgespeichert wird.
Typische Nachweiseffizienzen eines Mikrokanalplatten-Detektors für verschiedene Teilchen
sowie als Funktion der Teilchenenergie zeigt Abb. 3.4. Für Ionen mit einer kinetischen Energie
von etwa 2 keV beträgt die Nachweiseffizienz ca. 25-30%. Für den Nachweis von Ionen ist bei
28
KAPITEL 3. IONENNACHWEIS
Tabelle 3.1:
Detektors.
Typische Spezifikationen eines Mikrokanalplatten-
Eigenschaft
Verstärkungsfaktor
Dunkelzählrate
Maximale Zählrate
Widerstand
Maximale Arbeitsspannung
Bereich Arbeitstemperatur
Bereich Arbeitsdruck (max.)
Pulsanstiegsdauer
Pulsbreite
Wert
107 bei 2200 V
5 Ereignisse/Sekunde/cm2
108 /Sekunde
66-400 M Ω
2200 V
-50◦ bis 100◦
1.0 · 10−6 mbar
≈ 0.5 ns
≈ 1 ns
niedriger Zählrate (< 1 MHz) ein Channeltron-Detektor (siehe Kap. 3.4) eindeutig einem MCP
Detektor vorzuziehen.
e- for Channeltron
(2)
ions for MCP
(1)
e-for MCP
Abb. 3.4: Nachweiseffizienz eines Mikrokanalplatten-Detektors für verschiedene Teilchen (links)
und als Funktion der Teilchenenergie (rechts) [BURL2004].
Ein Daly-Detektor, benannt nach N. R. Daly [Daly1960], ist ein Ionendetektor bestehend
aus einem Metallknopf, einem Szintillator (Phosphorschirm) und einem Photomultiplier bzw.
Sekundärelektronenvervielfacher. Ionen die auf den bei -20 kV betriebenen polierten Metallknopf treffen lösen einen Schauer an Sekundärelektronen aus. Zwischen dem Metallknopf und
dem Szintillator ist ein Hochspannung angelegt, so dass die Elektronen auf den Szintillator
(Phosphorschirm) hin beschleunigt werden und dort Photonen auslösen. Diese Photonen werden verstärkt und durch den Photomultiplier außerhalb der Vakuumkammer detektiert. Im
Analogmodus kann dieses System eine Verstärkung haben die einen Faktor 100 größer ist als
die bei einem Faradaycup. Die Funktionsweise des Daly-Detektors ist in Abb. 3.5 dargestellt
[Daly1960].
Daly-Detektoren werden häufig in Massenspektrometern verwendet. Vorteil des DalyDetektors ist, dass der Photomultiplier durch ein Glasfenster, das die Photonen durchlässt,
vom Inneren des Massenspektrometers separiert werden kann, was eine sonst mögliche Kontamination verhindert und die Lebensdauer verlängert. Daly-Detektoren sind vor allem für Ionenstrahlen mit niedriger Intensität (ca. 10−13 A) geeignet, bei denen das elektrische Rauschen
3.4. SEKUNDÄRELEKTRONENVERVIELFACHER UND CHANNELTRON
29
Daly conversion
electrode
Abb. 3.5: Funktionsweise eines Daly-Detektors bestehend aus einer Konversionselektrode, Phosphorschirm und Photomultiplier.
von Faraday-Verstärkern signifikant wird. Nachteil ist, dass der Daly-Detektor nur mit einem
positiven Ionenstrahl benutzt werden kann. Allerdings erlauben Channeltron-Detektoren (siehe
Kap. 3.4) den Einsatz sowohl bei positiven als auch negativen Ionenstrahlen. Vorteile des DalyKollektorsystems sind:
1. hoher Verstärkungsfaktor
2. ausgezeichnetes Signal zu Rausch Verhältnis
3. Nachweiseffizienz von typischerweise höher 95%
4. gute Langzeitstabilität .
3.4
Sekundärelektronenvervielfacher und Channeltron
Der Sekundärelektronenvervielfacher (siehe Abb. 3.6) besteht aus einer lichtempfindlichen
Photokathode, einem elektronenoptischen Eingangssystem, einem Vervielfachersystem und
einer Anode. Die im Szintillator erzeugten Photonen gelangen durch das Eintrittsfenster auf die Photokathode des Sekundärelektronenvervielfacher und lösen dort über den
Photoeffekt Photoelektronen aus. Bei den meisten für Szintillationszählern verwendeten
Sekundärelektronenvervielfachern ist die Photokathode als dünne Metallschicht aufgedampft
(halbdurchlässige Photokathode). Als Kathodenmaterial eignen sich Kombinationen verschiedener Alkali- und Erdalkalimetalle, die den Empfindlichkeitsbereich für die nachzuweisende
Photonenstrahlung bestimmen. Beim Ionen- bzw. Elektronennachweis spielt das eine untergeordnetere Rolle.
Durch das elektronenoptische Eingangssystem werden die Photoelektronen bzw. Elektronen/Ionen beschleunigt und auf die erste Elektrode (erste Dynode) des Vervielfachersystems fokussiert. In dem aus mehreren Dynoden bestehenden Vervielfachungssystem wird
durch Sekundärelektronenemission der Kathodenstrom verstärkt und gelangt schlussendlich zur
Anode.
30
KAPITEL 3. IONENNACHWEIS
n
Io
n/
tro
k
Ele
Abb. 3.6: Funktionsweise eines Sekundärelektronenvervielfacher mit Szintillator, Lichtleiter,
Photokathode, Dynoden, Anode, Reflektor, lichtdichtes Gehäuse und Spannungsteiler.
Der Anodenstrom erzeugt durch den Spannungsabfall an einem Widerstand das Ausgangssignal. Die Höhe des Spannungsimpulses ist proportional der im Szintillator entstehenden und auf
die Photokathode auftreffenden Lichtmenge. Die Lichtmenge ihrerseits entspricht der im Detektor absorbierten Strahlungsenergie.
In modernen Massenspektrometern wie z.B. bei Quadrupolmassenfiltern und Penningfallenmassenspektrometern ist höchste Nachweiseffizienz für den Einteilchennachweis unabdingbar.
Daher erfolgt heute der Nachweis bei diesen beiden Methoden zumeist mit einem ChanneltronDetektor, dessen Funktionsprinzip in Abb. 3.7 erläutert ist.
Abb. 3.7: Funktionsweise eines Channeltron-Detektors.
Eine mögliche Geometrie eines Channeltrons ist in Abb. 3.8 dargestellt. Der Detektor ist
in off-axis-Anordnung, d.h. außerhalb der Zentralachse angebracht. Dadurch wird verhindert,
dass Neutralteilchen, die beispielsweise den Massenfilter unselektiert durchlaufen, oder gestreute
31
3.5. NICHTDESTRUKTIVE NACHWEISMETHODEN
UV-Photonen die Zählrate verfälschen. Aufgrund der gekrümmten und trichterähnlichen Form
sowie des Spannungsabfalls von bis zu 3 kV entlang des zentralen Kanals1 Spannung arbeitet
das aus Bleiglas gefertigte und mit einem Halbleitermaterial beschichtete Channeltron als
Sekundärelektronenvervielfacher. Da die Sekundärelektronenausbeute von der Geschwindigkeit
der auftreffenden Ionen abhängt, d.h. bei gegebener Energie von deren Masse, nimmt die Nachweisempfindlichkeit mit zunehmender Masse ab. Um diese Form der Massendiskriminierung zu
vermeiden, erfolgt der Ionennachweis indirekt: Die transmittierten Ionen werden von einer dem
Channeltron gegenüber angebrachten und auf -4 kV Hochspannung liegenden Konversionsdynode abgezogen. Anschließend folgt der Nachweis der herausgeschlagenen Elektronen im Channeltron, das auf deutlich kleinerem Potential (-2 kV) liegt. Dieser Vorgang ist zur Verdeutlichung
in Abb. 3.9 dargestellt.
+
e-
Abb. 3.8: Channeltron-Detektor in off-axis Geometrie (Model 402A-H) der Firma DeTech
(Palmer, MA, USA). Die Gesamthöhe beträgt ca. 8 cm.
3.5
3.5.1
Nichtdestruktive Nachweismethoden
Nichtdestruktiver Ionennachweis mittels FT-ICR
Das Grundprinzip des nichtdestruktiven Fouriertransformation–Ionenzyklotronresonanz (FT–
ICR) Nachweises über induzierte Spiegelströme ist in Abb. 3.10 am Beispiel einer Ionenfalle
schematisch dargestellt. Ein gespeichertes Ion der Ladung q bzw. eine Ionenwolke mit N Einzelladungen (Q = N · q) wird zu einer kohärenten Bewegung mit einem bestimmten Radius angeregt. Dies geschieht durch ein kurzes, resonantes Radiofrequenz (RF)–Signal, das dipolartig
an zwei gegenüberliegenden Fallenelektroden angelegt wird. In Abb. 3.10 sind dies die geteilten
Hälften der Ringelektrode, was der Beobachtung der reduzierten Zyklotronbewegung dient. Die
sich bewegende Ladung Q influenziert in den Metallelektroden der Penningfalle eine zeitlich
veränderliche Ladungsverteilung. Sind die Elektroden mit einem Widerstand R verbunden, fällt
dort ein Wechselspannungssignal ab, das die Frequenzanteile aller kohärent angeregten Ionen
1
Die Höhe des Spannungsabfalls ist abhängig vom gewählten Detektormodell. Typischerweise werden Spannungen von nicht mehr als 2.1-2.3 kV angelegt.
32
KAPITEL 3. IONENNACHWEIS
(a)
(b)
Abb. 3.9: Prinzip eines Off-Axis Channeltron-Detektor ohne (a) und mit (b) Einsatz einer
Konversionsdynode.
enthält. Das Spannungssignal, auch als Transient bezeichnet, wird nach seiner Verstärkung
aufgenommen, indem man es in diskreten Zeitschritten mit der Abtastfrequenz (engl.: sampling frequency) νs digitalisiert. Eine Fouriertransformation in den Frequenzbereich liefert das
Frequenz- oder Massenspektrum, das die relativen Anteile der einzelnen Ionensorten wiedergibt.
MAGNETIC
FIELD
z
PENNING
TRAP
I
FFT
SPECTRUM
ANALYSER
LOW NOISE
AMPLIFIER
EXCITED ION AT
CYCLOTRON ORBIT
SEGMENTED
RING ELECTRODE
MASS SPECTRUM
INDUCED AC-CURRENT
I
dP/df
FFT
time
frequency
Abb. 3.10: Prinzipieller Aufbau für eine nichtdestruktive Bestimmung von Eigenfrequenzen.
Gespeicherte Ionen werden durch ein Radiofrequenzsignal zu einer kohärenten Bewegung angeregt. Nach der Verstärkung der in den Elektroden influenzierten Spiegelströme erhält man
die für einzelne Ionensorten charakteristische Frequenzinformation durch eine Fourieranalyse.
3.6. UNTERGRUND UND TOTZEITKORREKTUR
3.5.2
33
Optischer nichtdestruktiver Ionennachweis
Eine sehr effiziente Methode, um die Anwesenheit von Atomen bzw. Ionen z.B. in einer Teilchenfalle nachzuweisen, ist ihr laserinduziertes Fluoreszenzlicht zu detektieren. Dieses Verfahren ist
allerdings limitiert auf Teilchen die ein Energieniveauschema besitzen, das die Anregung mit
verfügbaren Lasern erlaubt. Dieser optische nichtdestruktive Nachweis basiert auf der Tatsache, dass die Lebensdauer eines angeregten atomaren bzw. ionischen Energieniveaus in der
Größenordnung von 10−7 liegt, wenn es über elektrische Dipolstrahlung zerfällt. Wiederholte
Anregung des gleichen Ions mit einem Laser bei Sättigungsintensität führt dann zu einer Fluoreszenzzählrate von 107 Photonen pro Sekunde. Unter der Annahme, dass ein Photonendetektionssystem verwendet wird mit einem Akzeptanzöffnungswinkel von 10◦ , einer Photomultipliernachweiseffizienz von 10% sowie Filter- und Transmissionsverluste von 90%, so kann ein Bruchteil
von etwa 10−3 der Photonen detektiert werden. Dies führt zu einem leicht nachweisbaren Signal
und man erreicht höchste Nachweisempfindlichkeit, wie die in Abb. 3.11 gezeigte optische
Detektion eines einzelnen Bariumions in einer Paulfalle eindrucksvoll verdeutlicht. Die optische
Abb. 3.11: Optischer Nachweis eines einzelnen Bariumions in einer Paulfalle.
Nachweismethode setzt jedoch voraus, dass das Ion sich permanent im Laserstrahl bewegt. Dies
erfordert eine Kühlung der Ionenbewegung in einem Maße, dass die Bewegungsamplitude kleiner
als der Durchmesser des Laserstrahls ist.
3.6
Untergrund und Totzeitkorrektur
Die Totzeitverluste in der Nachweiselektronik basieren auf zwei Effekten: (1) Der Totzeit der
Ausleseelektronik und (2) der Totzeit des Detektors. Zur Minimierung der Ausleseelektroniktotzeit werden häufig zwei Counter in einer Art Ping-Pong-Konfiguration eingesetzt, d.h. während
der eine Counter zählt, wird der andere Counter ausgelesen, auf Null gesetzt und für den nächsten
Zählbetrieb vorbereitet. Dadurch können die Totzeitverluste in der Ausleseelektronik, die im
34
KAPITEL 3. IONENNACHWEIS
Bereich von ≈ 1 ns liegen, vernachlässigt werden. Nicht zu vernachlässigen sind jedoch die Verluste aufgrund des limitierten Auflösungsvermögens von Pulspaaren. In allen Detektorsystemen
ist eine gewisse Zeit zwischen zwei aufeinanderfolgenden Ereignissen nötig, um diese als zwei getrennte Pulse zu registrieren. Unter Totzeit des Detektors versteht man daher im Allgemeinen
die Zeit, innerhalb der die Nachweiselektronik mit der Erkennung eines Ereignisses beschäftigt
ist und keine weiteren Ereignisse registrieren kann.
Bei der Totzeitkorrektur muss zwischen einem retriggerbaren und einem nicht-retriggerbaren
Detektor unterschieden werden [Leo1994]. In einem nicht-retriggerbaren Detektor wird nach
jedem registrierten Signal der Detektor für eine fest vorgegebene Zeit τ für neue Ereignisse
blockiert. Solange die Zählrate unterhalb der Sättigung bleibt, kann die Korrektur gemäß
m
(3.2)
n=
1−m·τ
erfolgen. Dabei stellt m die gemessene, n die wahre Zählrate und τ die Totzeit des Detektors dar.
Bei einem retriggerbaren System, wie z.B. im Falle des Channeltrons, verlängert ein innerhalb der
Totzeit eintreffendes zweites Signal den Verarbeitungsprozess des ersten Signals. Mit steigender
Ereignisrate kann die Verarbeitung eines Signals nicht beendet werden und die Zählrate geht
gegen Null. In diesem Fall ist der Zusammenhang zwischen beobachteter und tatsächlicher
Zählrate durch
m = n · e−n·τ
(3.3)
gegeben. Diese Gleichung lässt sich bei bekanntem m und τ und gesuchtem n nur numerisch
lösen. Für Zählraten bis 0.1 · τ −1 beträgt die Abweichung des Verhältnisses der aus Gl. (3.2)
und (3.3) bestimmten Zählraten allerdings weniger als 0.5% und Gl. (3.2) kann demzufolge als
gute Näherung verwendet werden. Dies liegt darin begründet, dass eine Taylorentwicklung der
beiden Gleichungen um τ = 0 in erster Ordnung das gleiche Ergebnis liefert.
Zur Bestimmung der Totzeit τ eines retriggerbaren Detektors wird gewöhnlich die
Umkehrpunkt-Methode gewählt, bei der ab einer bestimmten Zählrate trotz weiterer Erhöhung
des Ionenstromes die registrierte Ereigniszahl abnimmt. Die maximale Zählrate mmax wird
genau dann beobachtet, wenn die wahre Zählrate gerade der inversen Totzeit entspricht, also
nmax = τ −1 . Durch Einsetzen dieses Wertes in Gl. (3.3) folgt für die Totzeit des Detektors der
Ausdruck τ = (e · mmax )−1 . Diese Methode kann jedoch zur Schädigung des Detektors führen,
da häufig sehr hohe Zählraten zum Erreichen des Umkehrpunktes notwendig sind. Aus diesem
Grund ist die Zwei-Isotopenmethode - sofern realisierbar - zu bevorzugen [Whit1986, Fahe1998],
die eine sehr präzise Bestimmung von τ auch bei niedrigen Zählraten garantiert. Dazu trägt
man für ein Element mit zwei (A, B) oder mehr Isotopen die bei unterschiedlichen Zählraten
nA gemessenen Isotopenverhältnisse R = A/B gegen nA auf. Aus der Regressionsgeraden
ergibt sich die Totzeit zu
τ=
Steigung
(R − R0 )/nA
R − R0
=
=
.
nA · (1 − R0 )
1 − R0
1 − Achsenschnittpunkt
(3.4)
R0 bezeichnet hierbei das wahre Isotopenverhältnis ohne Totzeitverluste. Abb. 3.12 zeigt
hierzu ein Beispiel [Blau2000]. Mittels Elektronenstoßionisation (siehe Kap. 2.1) wurden die
Isotopenverhältnisse 40 Ca/42 Ca und 40 Ca/44 Ca für verschiedene Zählraten des Isotops 40 Ca
bestimmt und gemäß Abb. 3.12 aufgetragen. Die eingezeichneten Fehler resultieren aus den statistischen Fehlern der gemessenen Zählraten. Die fehlergewichteten Regressionsgeraden liefern
nach Gl. (3.4) eine gemittelte Totzeit von τ = (25.9 ± 1.6) ns. Dies steht in hervorragender Übereinstimmung mit dem vom Hersteller angegebenen Wert von τ ≈ 25 ns für diesen
Channeltron-Detektortyp. Mit Alterung des Channeltrons nimmt dieser Wert jedoch leicht zu,
so dass die Messung etwa halbjährlich wiederholt werden sollte.
35
3.6. UNTERGRUND UND TOTZEITKORREKTUR
156
Isotopenverhältnis
152
τ = (27.2 ± 1.9) ns
148
144
40
Ca / 42Ca
50
48
τ = (24.6 ± 1.3) ns
46
40
44
44
Ca / Ca
4.0x105
8.0x105
1.2x106
1.6x106
2.0x106
40
Ca Zählrate / s
Abb. 3.12: Bestimmung der Totzeit eines Channeltron-Detektors aus der Änderung des Isotopenverhältnisses 40 Ca/42 Ca und 40 Ca/44 Ca als Funktion der gemessenen Zählrate des 40 Ca-Isotops.
Kapitel 4
Ionenoptik
4.1
Extraktionsoptiken und Emittanz
Aufgabe der Ionenoptik in einem Massenspektrometer ist die Formung und der möglichst
verlustfreie Transport eines Ionenensembles, hierbei kommt der Erzeugung eines ideal parallelen
Ionenstrahls besondere Bedeutung zu. Im Gegensatz zum Licht hat man es aber in der
Teilchenoptik mit einem Strahl aus geladenen Teilchen zu tun, die Kräfte aufeinander ausüben.
Das heißt, die Teilchen bewegen sich nicht nur unter dem Einfluss von außen auf sie einwirkenden
elektrischen und magnetischen Felder, sondern sie werden in ihren Bahnen durch die Ladungen
aller Teilchen beeinflusst. Dieser Effekt wir in der so genannten Raumladung zusammengefasst,
der bei der Erzeugung und Handhabung eines Ionenstrahls eine ganz entscheidende Rolle
spielt. Darüber hinaus sind auftretende Plasma- und Oberflächenpotenziale, Abbildungsfehler
der ionenoptischen und der dispersiven Elemente sowie mitreisende Elektronen im Strahl
problematisch für die Erzeugung und den Erhalt eines ideal parallelen als auch eines hinreichend kollimierten Strahls. Gute Gesamtzusammenstellungen der notwendigen theoretischen
Hintergründe und der in diesem Kapitel auftretenden Größen finden sich in [Groß83], [Hint97],
[Hump90].
Weiter unten wird sich zeigen, dass die Geometrie der Ionenquelle und des Beschleunigungssystems die bestimmenden Größen für die Qualität des Strahls im Massenspektrometer
und damit für dessen Auflösung und Transmission sind.
Als Beispiel hierzu zeigt Abbildung 4.1 die Eigenschaften eines Strahls, der aus einer
Plasmaoberfläche extrahiert wird in Abhängigkeit von der Dichte des Plasmas. Da auch bei
Oberflächenionisation aufgrund der notwendigen hohen Temperatur zur effizienten Ionisation
eine dünne Plasmaschicht, das so genannte thermische Plasma, entsteht, ist dieser Einfluss
typisch und dominant für nahezu alle Ionenquellentypen. Für eine gegebene Geometrie und
gegebene Plasmabedingungen muss er berücksichtigt werden, es gibt i.A. nur ein Potenzial, das
einen Strahl gewünschter Qualität erzeugt.
Ein Maß für die Strahlqualität des Ionenstrahls ist die Emittanz. Sie ist definiert als das
Volumen des Phasenraums, das die Teilchen des Strahls ausfüllen. Legt man ein kartesisches
Koordinatensystem mit den Ortskoordinaten qi und den Impulskoordinaten pi zugrunde, so ist
das Volumenelement des durch die diese Koordinaten aufgespannten sechsdimensionalen Phasenraums gegeben durch dV = dp1 · dp2 · dp3 · dq1 · dq2 · dq3 . Nach dem Liouvilleschen Theorem ist
das Phasenraumvolumen bei Bewegung in konservativen elektrischen und magnetischen Feldern
36
4.1. EXTRAKTIONSOPTIKEN UND EMITTANZ
37
Abb. 4.1: Einfluss der Plasmadichte auf die Strahleigenschaften, P: Plasma, O: Quellenöffnung,
E: Extraktionselektrode: (a) divergenter Strahl für ein überdichtes Plasma, (b) paralleler Strahl
bei optimaler mittlerer Plasmadichte, (c) ”unterdichtes” Plasma und resultierender konvergenter
Strahl
erhalten. Es wird für ein Ionenensemble daher durch die Form und Größe der Ionenquelle
und der primären Beschleunigung festgelegt. Als Emittanz definiert man dann dieses effektive
Phasenraumvolumen zu
Z Z Z Z Z Z
ε=
dp1 · dp2 · dp3 · dq1 · dq2 · dq3
(4.1)
Üblicherweise sind die beiden transversalen (x,y) sowie die longitudinale Raumrichtung (z) entkoppelt, so dass man die transversalen Emittanzen jeweils einzeln betrachten kann. Diese können
dann durch die Position (qi ) und den Winkel (φi ) der Bewegungsrichtung der Teilchen senkrecht
zur Strahlachse dargestellt werden, wobei sich der Winkel ergibt aus
pi
mit i = x, y
(4.2)
φi = arctan
pz
Eine grobe obere Abschätzung für die transversale Emittanz gibt dann die Größe εmax = qi · φi
für einen Punkt nahe eines Fokusses an, einen genaueren Wert hingegen die Randemittanz, die
durch eine, das Strahlvolumen im Phasenraum einhüllende, Ellipse geformt wird:
εRand = γ · x2 + 2α · x · φx + β · φ2x
(4.3)
Aus der Lage der Ellipse kann man auf die räumlichen Eigenschaften des Strahls, aus der
Größe auf die Qualität des Ionenstrahls, schließen. In Abbildung 4.3(a) sind entsprechend die
Emittanzellipsen für parallele, fokussierte, konvergente und divergente Strahlen dargestellt.
Wie in Abbildung 4.3(b) erläutert, beschreiben die Parameter α, β und γ die Halbachsen der
Ellipse sowie ihre räumliche Orientierung. Sie werden als Transportparameter bezeichnet.
Um einen möglichst optimalen Strahl zu erzeugen und zu erhalten, entwickelt man eine
der Emittanz angepasste Ionenoptik, ein System aus Linsen, die auf unterschiedlichen Potenzialen liegen. Eine exakte Berechnung einer Ionenoptik und ihrer Einwirkung auf den Ionenstrahl, unter Berücksichtigung von Raumladungseffketen erfordert eine Computersimulation,
38
KAPITEL 4. IONENOPTIK
Abb. 4.2: Emittanz, dargestellt als einhüllende Ellipse (gestrichelt) zur wirklichen Phasenraumfläche eines Ionenstrahls (grau)
Abb. 4.3: (a) Darstellung der Emittanzellipse für verschiedene Strahlformen: (1) paralleler, (2)
fokussierter, (3) divergenter, (4) konvergenter Strahl. (b) Erläuterung der Transportparameter
4.2. RAUMLADUNGSKOMPENSATION UND PERVEANZ
39
wobei die Laplacegleichung für eine vorgegebene Elektrodengeometrie auf räumlichen Gitterpunkten berechnet und dann die Bewegungsgleichugen für festgelegte Teilchensorte, -energie
und -dichte schrittweise numerisch integriert wird. Beispiele solcher Programme sind SIMION
[Dahl00a, Dahl00b], PBguns [Boer99], Axcel-GSI [Späd91], MAFIA, KOBRAS [Späd00] und andere. Früher wurden sie auch mit Hilfe von Simulationen an Modellen in einem ”elektrolytischen
Trog” ermittelt (Bemerkung: vergleiche den Versuch im Anfängerpraktikum).
4.2
Raumladungskompensation und Perveanz
Für die Einflüsse durch Raumladungen ist die Dichte im Strahl entscheidend. Bei einem
schwachen Strom, sind die Teilchen so weit voneinander entfernt, dass Raumladungseffekte
keine nennenswerte Rolle spielen. Bei größeren Strömen jedoch wirken die mit den Teilchen
verknüpften Ladungen und Ströme aufeinander und müssen berücksichtigt werden. Dazu macht
man zwei vereinfachende Annahmen:
(a) Die Ladung ist im Strahl kontinuierlich verteilt
(b) Der Strahl ist laminar
Im Folgenden soll die Entwicklung eines zunächst parallelen Strahls in der Zeit betrachtet werden. Das elektrische und das magnetische Feld sind durch Strom- bzw. Ladungsdichten gegeben.
1
Er =
Rε0
ZR
µ0
Bϕ =
R
ZR
rρ(r)dr
(4.4)
rj(r)dr
(4.5)
0
0
Darüber hinaus kann aufgrund von Annahme (a) die Ladungsdichte ersetzt werden durch
ρ(r) =
j(r)
,
v
(4.6)
wobei v für die Teilchengeschwindigkeit steht.
Die Differentiation nach der Zeit kann ersetzt werden durch die Differentiation nach der Koordinate z (Strahlrichtung):
d
d
=v .
(4.7)
dt
dz
Setzt man in den folgenden Ansatz
F = ṗ = m · b = m ·
d2 r
dt2
(4.8)
~ + ~v × B)
~
F~ = q(E
die Gleichungen 4.4 bis 4.7 ein und berücksichtigt bei der Bildung des Kreuzprodukts, dass
das B-Feld in azimutaler Richtung und die Teilchengeschwindigkeit in z-Richtung weisen, dann
erhält man mit c2 = 1/(µ0 · ε0 )
q
d2 R
m 2 =
dz
Rv 3 ε0
ZR
0
rj(r)dr(1 −
v2
)
c2
(4.9)
40
KAPITEL 4. IONENOPTIK
Die eins in der Klammer resultiert aus dem elektrischen Feld, während der Term v 2 /c2 den
Beitrag des magnetischen Feldes darstellt. Man sieht daran, dass im nicht relativistischen Fall,
dieser Beitrag vernachlässigbar klein ist und die Formel vereinfacht werden kann. Setzt man
darüber hinaus die Stromdichte als konstant mit j(r) = j0 für r ≤ R und j(r) = 0 für r > R
dann erhält man folgende Gleichung:
q
d2 R
= 3
· j0 (z) · R.
2
dz
2v mε0
(4.10)
(Bemerkung: Die Stromdichte und der Radius ändern sich längs des Strahls, der Gesamtstrom
I = πR2 j0 bleibt aber konstant.)
Für eine weitere Interpretation der Gleichung ersetzt man die Geschwindigkeit durch die
Beschleunigungsspannung U
mv 2
, und
U=
2q
die Stromdichte durch I = πR2 j0 . Darüber hinaus führt man die so genannte ”Perveanz” eines
Ionenstrahls ein. Sie ist definiert als
K=
I
U 3/2
=
πR2 j0
.
U 3/2
(4.11)
Als weitere ”Abkürzung” definiert man eine für jede Teilchensorte charakteristische Konstante
r
2q
K0 = 2πε0 ·
.
(4.12)
m
−6 A für Protonen.
Sie beträgt K0 = 3.4 × 10−5 V A
3/2 für Elektronen und K0 = 0.77 × 10
V 3/2
Mit K und K0 lässt sich Gleichung (4.10) wie folgt umschreiben
K 1
d2 R
=
· .
2
dz
K0 R
(4.13)
q
ln R
Diese Gleichung lässt sich durch numerische Integration (die Transformation ξ =
R0
ermöglicht die Rückführung auf das Dawson Integral) lösen. Man erhält dann zumindest in
impliziter Form die Abhängigkeit des Strahlradius von der Laufstrecke und kann so den Radius
R(z) zeichnerisch darstellen. (Lösung durch Rückführung auf das Dawson-Integral).
In Abbildung 4.4 ist
des Strahlradius in Einheiten von R0 und die Laufstrecke
p die Entwicklung
1
in Einheiten von K/K0 R0 dargestellt. R0 ist so gewählt das, bei diesem Radius die Bahn
achsenparallel ist, also dR
dz = 0 bei R = R0 . Damit ist R0 der kleinste mögliche Radius und für
die weiteren Rechnungen wurde angenommen das, das für den Fall z = 0 gilt.
4.2. RAUMLADUNGSKOMPENSATION UND PERVEANZ
41
Abb. 4.4: Dargestellt ist die Funktion R(z), die die Entwicklung eines bei z = 0 achsenparallelen
Strahls unter Berücksichtigung der Raumladung beschreibt
Der Abbildung kann man entnehmen, dass ein Strahl mit einer Perveanz K = K0 schon nach
einer Laufstrecke von 2R0 seinen Radius verdoppelt hat. Bei einem Strahl mit einer größeren
Perveanz geht es sogar noch schneller. Soll ein Strahl seinen Radius erst nach einer Laufstrecke
von etwa 100 Radien verdoppelt haben, ist eine Perveanz von K < 10−4 · K0 notwendig. In
diesem Fall wäre dann die Raumladung vernachlässigbar.
Man bezeichnet die Bedingung K = K0 auch als Raumladungsgrenze. Damit ist der maximal
mögliche Strom bei gegebener Spannung für jede Teilchensorte vorgegeben. In Abbildung 4.5
sind die Bedingungen K = K0 und K = 10−4 · K0 für Elektronen und Protonen graphisch
dargestellt. In den schraffierten Bereichen sind Raumladungseffekte nicht vernachlässigbar und
der Bereich oberhalb der Raumladungsgrenze ist mit unbeschleunigten Strahlen praktisch nicht
zugänglich.
Abb. 4.5: Die Bedingungen K = K0 (Raumladungsgrenze) und K = 10−4 · K0 sind für Protonen und Elektronen dargestellt. In den schraffierten Bereichen zwischen den Grenzen ist die
Raumladung nicht vernachlässigbar.
42
KAPITEL 4. IONENOPTIK
Zur Erinnerung: Die bisher gemachten Überlegungen gelten für einen laminaren Strahl, wobei
die Stromdichte über den gesamten Strahlquerschnitt kontinuierlich verteilt ist und an den
Strahlgrenzen plötzlich auf Null abfällt.
4.3
Child-Langmuir Limit der Stromdichte im Strahl
In diesem Kapitel sollen einige Effekte, die bei der Beschleunigung eines Strahls auftreten, etwas
genauer betrachtet werden. Dazu geht man zunächst von einem Teilchenstrahl aus, der an der
Stelle z = 0 in der x, y-Ebene mit der kinetischen Energie E = 0 erzeugt wird. Seine Ausdehnung
in der x, y-Ebene wird als unendlich angesehen und es wirkt in dieser Ebene keine Kraft auf die
Teilchen. Eine Skizze der Geometrie ist in Abbildung 4.6(a) gegeben. Der Potenzialverlauf φ(z)
lässt sich dann einfach aus den Gleichungen
ρv = j = const
mv 2
+ qφ = E = const
2
ρ
∆φ = −
ε0
zu
j
∆φ = −
ε0
2(E − qφ)
m
−1/2
(4.14)
(4.15)
berechnen. Das Potenzial folgt dann unter der Annahme j → 0 einem linearen Verlauf, wie er
in Abbildung 4.6(b) dargestellt ist.
Bei wachsender Stromdichte kommt nun zu der äußeren beschleunigenden Kraft ein Beitrag
resultierend aus der Raumladung hinzu. Sie reduziert die Kraft auf die Teilchen, wenn diese
bei z = 0 starten. Man kann die Stromdichte so lange steigern, bis die Kraft bei z = 0 gegen
Null geht. Damit ist die maximale Stromdichte erreicht, die Teilchen würden sonst in die Quelle
zurückbeschleunigt. In diesem Grenzfall folgt das Potenzial einem Gesetz in der Form
φ = φ0 + const · z 4/3
(4.16)
Sein Verlauf ist in Abbildung 4.6(b) ebenfalls dargestellt.
Die maximal mögliche Stromdichte ist durch das Child-Langmuir-Limit
jmax =
2
U 3/2
K0 · 2
9π
d
(4.17)
gegeben, wobei U für die Potenzialdifferenz zwischen Anfang und Ende der Beschleunigungsstrecke steht und L die Länge der Strecke ist.
Ein Beispiel für einen typischen Hochspannungs-Massenseparator (der Mainzer RISIKO
Massenseparator) mit einer Beschleunigungsspannung bei 30 keV und einer Masse von M=100+
liefert:
mA
jmax = 7 cm
2 ⇒ Imax = 0.55µA für 0.1mm Öffnung
Imax = 3.4µA für 0.25mm Öffnung
Imax = 340µA für 2.5mm Öffnung
4.3. CHILD-LANGMUIR LIMIT DER STROMDICHTE IM STRAHL
43
Abb. 4.6: (a) Die auf der linken Elektrode ohne kinetische Energie entstehenden Ladungsträger
werden durch eine rechts anliegende Spannung beschleunigt. Unter Annahme einer in x- und yRichtung unendlichen Ausdehnung fliegen die Ladungsträger genau in die z-Richtung. (b) Bei
geringem Strom verläuft das Potential von links nach rechts linear. Bei einem größtmöglichen
(raumladungsbegrenzten) Strom folgt es einem z4/3 -Gesetz.
Für typische Massenspektrometer mit einer niedrigeren Beschleunigungsspannung von 3 keV
ergibt sich für die gleiche Masse:
µA
jmax = 200 cm
Öffnung
2 ⇒ Imax = 20nA für 0.1mm
Imax = 125nA für 0.25mm Öffnung
Imax = 12.5µA für 2.5mm Öffnung
Eine geringe Raumladung wirkt in diesen Fällen ähnlich wie eine schwache Zerstreuungslinse,
deren Wirkung durch entsprechende optische Komponenten kompensiert werden kann. Das
heißt, der Strahl wird durch die Raumladung divergent und durch eine Linse wieder konvergent.
Bei zunehmender Perveanz, also bei einem intensiven Strahl, der in einem homogenen Feld
beschleunigt wird, kann dieser Verbreiterung mit Hilfe einer bestimmten Feldkonfiguration
der Pierce-Anordnung entgegengewirkt werden. Sie soll im nächsten Kapitel etwas genauer
vorgestellt werden.
Betrachtet man zunächst aber einen in x- und y- Richtung begrenzten Strahl, und Elektroden mit einem kreisförmigen Querschnitt zum Strahltransport, so führen Felddurchgriffe
an den Elektroden je nach Strahl (positive oder negative Ladung) zu einer fokussierenden
bzw. defokussierenden Wirkung. Um diese Wirkung auszugleichen, sind entsprechende
Linsensysteme oder die so genannte Pierce-Geometrie [Pier54] notwendig.
44
KAPITEL 4. IONENOPTIK
4.4
Pierce-Geometrie für parallele Ionenstrahlen
Die Pierce-Geometrie ist eine optimierte Elektrodengeometrie für die Ionenquelle und
Beschleunigungsregion zur Erzeugung eines idealen parallelen Ionenstrahls für einen raumladungsbegrenzten Strahl.
In dieser Geometrie bilden die Elektroden zwischen denen das beschleunigende Potenzial
angelegt wird einen festgelegten Winkel mit der Strahlachse, wie in Abbildung 4.7 dargestellt.
Der Wert beträgt etwa 67,5◦ , die für die Elektrode auf der Quellenseite bis zur Strahlradius
beibehalten wird, während er auf der Extraktionsseite bis auf knapp 90◦ anwächst - der
Abstand der Elektroden ist in etwa auf 8 Strahlradien festgelegt. Aus den eingezeichneten
Äquipotenziallinien entnimmt man dann eine fokussierende Kraft auf den Ionenstrahl, die ohne
weitere Einwirkung zu einem exakt parallelen Strahlverlauf führen sollte.
Die Pierce-Geometrie ist allerdings nur eine Näherung, da
1) die Plasmaoberfläche nicht linear ist bzw. ein falsches Potenzial in der Extraktionsöffnung
vorliegt (vgl. Aus- bzw. Einbeulungen an der Quellenöffnung von Abbildung 4.1),
2) die Elektroden eine endliche Ausdehnung haben,
3) die Ionenemission und damit die Ladungsdichte im Strahl nicht konstant über R ist,
4) die Extraktionsöffnung in der zweiten Elektrode das Feld verzerrt und
5) der Strahl nicht monoenergetisch ist.
Besonders aus dem letzten Punkt resultiert eine starke Abhängigkeit der Fokussierungseigenschaften einer Ionenquellen von der Energieverteilung, die nicht über eine entsprechende
Geometrie kompensiert werden kann. Eine Nachfokussierung zur Parallelisierung des Ionenstrahls in einer Rohrlinse (typischerweise eine so genannten Einzellinse mit drei Rohrstücken
auf Masse, Linsenpotenzial, Masse) ist daher in allen Fällen notwendig.
Daraus ergeben sich folgende experimentelle Auswirkungen:
1) Man konstruiert einen möglichst konvergenten, im Allgemeinen aber leicht divergenten
Strahl an der Extraktion.
2) Das Child-Langmuir-Limit reduziert sich etwa um einen Faktor 2.
3) Die Emittanz hängt stark von der Energieverteilung ab und wird weit unter dem theoretisch erwarteten Werten limitiert von (ohne Herleitung)
#
r
1/2 "
eV
kT
(4.18)
mm ·
ε[π mm mrad] = 0.0653 · r ·
A
amu
Beispiel: RISIKO, M = 100+ , r = 0.25mm, kT = 1eV : εmin = 1.6 × 10−3 π mm mrad,
experimentell ergibt sich die Emittanz zu εexp = 1.5 × 10−1 π mm mrad, sie ist also knapp einen
Faktor 100 schlechter. Dies kann auf die schon diskutierten Einflüsse des Plasmapotentials in
der Quelle, bzw. an den Oberflächen der Quelleneinzelteile zurückgeführt werden, die in den
Abschätzungen nicht berücksichtigt werden.
45
4.5. TRANSFERMATRIZEN
Abb. 4.7: Mit Elektroden der schraffiert dargestellten Form kann im Innenbereich ein seitlich
begrenzter Strahl existieren, der sich ebenso verhält wie der entsprechende Ausschnitt aus dem
unendlich ausgedehnten Strahl. Das heißt, dass Einflüsse auf den Strahl durch Raumladungen
und z.B. Elektrodenöffnungen ausgeglichen werden können.
Ein entsprechendes reales Strahlprofil zeigt Abbildung 4.8, das mit dem Simulationscode
PBgun aus dem Oak Ridge National Laboratory für einen dortigen 20 keV Massenseparator
angefertigt wurde und trotz optimaler Einstellung der Ionenoptikparameter einen deutlich divergenten Strahl aus der Extraktionsoptik zeigt.
4.5
Transfermatrizen
Zu einer ersten, näherungsweisen Berechnung von ionenoptischen Systemen macht man sich
die Analogie zur geometrischen Strahlenoptik zu Nutze. Unter Vernachlässigt von Abbildungsfehlern, die zu höheren Ordnungen in den Bewegungsgleichungen führen würden, kann man in
erster Ordnung lineare Gleichungen ansetzen, die in Form eines einfach zu handhabenden und
klar zu formulierenden Matrixformalismus dargestellt werden können [Woll87, Woll99]. Transfer
eines Strahles durch ein ionenoptisches System wird dann durch konsekutive Anwendung der
Matrizen jeder einzelnen Komponente auf den Strahlvektor über Matrizenmultiplikation erzielt:
(Strahlvektornachher ) = (Mn ) · · · (M3 ) · (M2 ) · (M1 ) · (Strahlvektorvorher )
(4.19)
x
α
Der Strahlvektor charakterisiert dabei den Ionenstrahl. Er ist gegeben durch:
δk
δM
46
KAPITEL 4. IONENOPTIK
Abb. 4.8: Berechnung einer Extraktionsoptik unter Berücksichtigung der Beiträge des thermischen Plasmas einer Oberflächenionisationsquelle und des damit verbundenen Potenzials. Die
Geometrie ist Pierce-nahe, wird aber durch eine zusätzliche Repellerelektrode beeinflusst. Es
wurde ein Strom von 1µA positiv geladener Cs Ionen angenommen. Abbildung (b) zeigt den
Detailausschnitt der Ionisatoroberfläche und des Verlaufs des Plasmapotenzials
Mit
x = Strahlausdehnung senkrecht zur opt. Achse
α = vx /vz = W inkel zur optischen Achse
δk =
k−k0
k0
δM =
=
∆k
k
M/q−M0 /q0
M0 /q0
)
beides analog zur geometr. Optik
= Abweichung von der Sollenergie mit k0 = 1/2 · M0 · v 2
=
∆M
M
= relative Abweichung von der Sollmasse M0 /q0 .
Aufgrund der Erhaltung des Phasenraums (Liouville’scher Satz) sowie der Energie und
der Masse gilt:
det T ≡ 1
(4.20)
47
4.5. TRANSFERMATRIZEN
mit
x1
α
1
=T
δk1
δM 1
x0
x/x
x/α
x/δk
x/δM
x0
α α/x
α/α
α/δk
α/δM
α0
0
·
·
=
δk0 δk /x δk /α δk /δk δk /δM δk0
δM 0
δM /x δM /α δM /δk δM /δM
δM 0
Bei nichtdispersiven Elementen kann eine Reduktion auf eine 2×2 Strahlmatrix
(4.21)
x/x x/α
α/x α/α
!
mit den Elementen x für Größe und α für Winkel analog zur Optik durchgeführt werden. Das Element x/x stellt dabei die Vergrößerung dar, das Element x/α ≡ 0 für abbildende
Systeme.
Beispiele für entsprechende Strahlmatrizen sind:
!
1 d
Driftstrecke der Länge d:
0 1
Beschleunigung auf das Potenzial U = U0 + Uplus auf der Länge d, ausgehend vom
1
Startpotenzial U0 :
0
Einzellinse:
1
0
− f1
1
2·d
U/U0 +1
√ 1
U/U0 +1
√
!
cos φ
− sin φ
Elektrisches Sektorfeld: re
0
0
cos φ
− sin φ
Magnetisches Sektorfeld: rm
0
0
re · sin φ re · (1 − cos φ) 0
cos φ
sin φ
0
1
0
0
0
0
1
rm · sin φ 1/2 · rm · (1 − cos φ) 1/2 · rm · (1 − cos φ)
cos φ
1/2 · sin φ
1/2 · sin φ
0
1
0
0
0
1
48
KAPITEL 4. IONENOPTIK
In den beiden letztgenannten, nicht rotationssymmetrischen Fällen wird nur die Einwirkung
auf die Ablenkrichtung betrachtet, während keine Einwirkung auf die dazu orthogonale
transversale Richtung angenommen wird. Die idealen Radien für beide Sektorfelder sind dabei
durch die elektrische bzw. magnetische Feldstärke E und B gegeben:
re =
2 · k0
M0 · v02
=
q0 · E
q0 · E
bzw.
rm =
M0 · v0
q0 · B
Um auch Elemente zu berechnen, die gleichzeitig auf beide transversale Komponenten einwirken,
also etwa Quadrupollinsen, Solenoide oder entsprechend geformte magnetische bzw. elektrische
Felder, kann ein aufwändigerer Ansatz verwendet werden, in welchem der vollständige Strahlvektor (x, x’, y, y’, z, z’) betrachtet wird.
Teil II
METHODEN DER
MASSENSPEKTROMETRIE
49
Kapitel 5
Flugzeitmassenspektrometrie
Die Dispersion findet bei Flugzeitmassenspektrometern (TOF-MS von Time-of-flight) im
Zeitraum und nicht im Ortsraum statt. Dies ist der wesentliche Unterschied zu statischen Spektrometern, der sowohl Vor- als auch Nachteile mit sich bringt. Da die Selektion auf der simplen
Untersuchung der Kinematik basiert und die Massenabhängigkeit der Fluggeschwindigkeit ausnutzt, sind Flugzeitmassenspektrometer eigentlich vom Prinzip her die einfachsten dispersiven
Systeme, stellen aber hohe Anforderungen an die experimentelle Realisation. Dies betrifft
besonders die gepulste Erzeugung der Ionen, die Präzision der Elektrodengeometrie sowie die
Zeitauflösung des Nachweissystems. Für die meisten Anwendungen ist eine klassische, nichtrelativistische Berechnung der Flugzeit bei weitem ausreichend; der über die Vernachlässigung
relativistischer Beiträge eingebrachte Fehler liegt i.A. um mehrere Größenordnungen unterhalb
der experimentellen Ungenauigkeiten.
Die Messung der Flugzeit wird durch einen gepulsten Start der Ionen begonnen. Typische
Beschleunigungsenergien liegen im Bereich von 1 bis 10 kV. Nichtrelativistisch gerechnet erhalten einfach geladene Ionen der Masse m = 100 u dabei für U = 1 keV gemäß e · U = 1/2 m · v 2
eine Geschwindigkeit von v ≈ 10−4 ·c = 3·104 m/s - sie sind also hinreichend nicht-relativistisch.
Bei einer vernünftig zu realisierenden Flugstrecken im Bereich von typisch 3 Metern beträgt
damit die Gesamtflugzeit etwa 100 µs, der zeitliche Abstand zweier benachbarter Massen
am Detektor liegt hingegen nur im Bereich von 1 µs. Um akzeptable Massenauflösungen zu
erhalten und zumindest 10 bis 100 Messpunkte zur Identifikation eines Massenpeak zu haben,
sind Zeitauflösungen im Detektor und der nachfolgenden Elektronik von etwa 10 ns notwendig.
Entscheidender Vorteil der Flugzeitmassenspektrometer ist aber die Möglichkeit pro
Ionenpuls ein gesamtes Massenspektrum in ein und demselben Detektor aufzunehmen. Dies
ist mit statischen Spektrometern - wenn überhaupt - dann nur über simultane Detektion in
mehreren bzw. ausgedehnten, ortsauflösenden Detektoren, über Aufnahme und Entwicklung
von photografischen Platten oder über einen meistens langsamen und daher nicht als simultan
zu bezeichnenden Massenscan möglich. Es folgt daraus, dass trotz des gepulsten Betriebs, der ja
zwangsläufig ein Puls-Pauseverhältnis (einen Dutycycle) von signifikant kleiner 100% bedingt,
durch die Parallelmessung hohe Effizienz erreicht werden kann. Dazu muss die Datennahme in
hinreichend hoher Abfolge im kHz Bereich erfolgen. Während die schnelle Datennahme schon
seit mehr als 50 Jahren möglich ist, sprengten die dann anfallenden hohen Datenströme bis
vor nicht allzu langer Zeit die Aufnahme- und Speicherkapazität der Auswerteelektronik und
Speichermedien. Empfindliche Flugzeitmassenspektrometer mit guter Auflösung sind daher
z.B. für analytische Anwendungen erst seit wenigen Jahren konkurrenzfähig und kommerziell
50
5.1. QUELLEN MIT KONSTANTER ENERGIE BZW. KONSTANTEM IMPULS
51
erhältlich, ihre Qualität hängt sehr stark von dem eingebauten Datenaufnahme- und Rechnersystem ab.
Zur Pulsung der Ionen sind prinzipiell zwei Ansätze möglich:
1. Gepulste Erzeugung der Ionen,
z.B. durch Pulslaserdesorption, Pulsung eines Primärionenstrahls, der Sekundärionen
aus dem Material heraussputtert, gepulstes Düseneinlasssystem, oder Ähnliches. Die
notwendige kurze Pulsdauer stellt hier oftmals ein experimentelles Problem dar.
2. Gepulste Beschleunigung der Ionen,
dies ist der experimentell weitaus einfachere Ansatz, da die Beschleunigungsspannung
mit vergleichsweise einfachen elektronischen Mitteln in wenigen ns von feldfrei auf den
gewünschten Wert geschaltet werden kann. Die Erzeugung der Ionen kann dann sowohl
kontinuierlich als auch gepulst mit hinreichender Synchronisation erfolgen, wobei nun die
Zeitauflösung in der Ionisation unkritisch ist.
5.1
Quellen mit konstanter Energie bzw. konstantem Impuls
Man unterscheidet bei TOF-Massenspektrometern zwischen Quellen mit konstanter Energie
und konstantem Impuls. Beide können bei gepulster Erzeugung sowie Beschleunigung der
Ionen eingesetzt werden. Wir betrachten den einfachen Fall eines anfänglich ruhenden Ions,
also verschwindender Startenergie und Startimpuls.
Konstante Energie:
Das zu analysierende Ion der Ladung q befindet sich in einem Plattenkondensator. Die
Spannung U wird schlagartig aufgebaut und liegt an, bis das Teilchen die Strecke s durchlaufen
hat und das Feld durch die als Netz oder Blende ausgebildete negative Platte verlassen hat, wie
in Abb. 5.1 dargestellt:
Abb. 5.1: Beschleunigung im gesamten Feld - konstante Energie
Die Energie beträgt dann E = F · s = q · U ·
der Masse m.
s
s1
= 1/2 m · v 2 = const. , sie ist unabhängig von
52
KAPITEL 5. FLUGZEITMASSENSPEKTROMETRIE
Konstanter Impuls:
Das zu analysierende Ion der Ladung q befindet sich wieder in einem Plattenkondensator.
Die Spannung U wird schlagartig aufgebaut, bleibt aber nur für eine Zeit anliegen, die kurz
gegenüber der Flugzeit bis zur negativen Platte ist, danach driftet das Ion im feldfreien Raum
weiter (Abb. 5.2).
Abb. 5.2: Beschleunigung mit einem kurzen Impulsübertrag - konstanter Impuls
Der Impulsübertrag entspricht der einwirkenden Kraft F = dp
dt , daraus berechnet sich der Impuls
durch Integration zu p = q ·U · st0 = m·v = const. , er ist wiederum unabhängig von der Masse m.
m
Die Auflösung < = ∆m
ist in beiden Fällen durch die Zeitauflösung bestimmt.
man durch Ableiten leicht zeigen kann, gilt für den Fall
konstanter Energie:
<=
m
∆m
=
t
2·∆t
konstanten Impulses:
<=
m
∆m
=
t
∆t
Wie
,
;
diese ist also um einen Faktor 2 besser. Beiträge von einer Orts- und Energieunschärfe
∆W
k·T
sind jedoch deutlich unterschiedlich. In beiden Fällen gilt ∆t
t = WGesamt ≈ e·U , wenn man eine
thermische Energieunschärfe voraussetzt. Da man im 2. Fall des konstanten Impulses aber nur
einen kleinen Teil - typisch etwa 1% - der angelegten Beschleunigungsspannung durchlaufen
kann, ist die erreichbare Auflösung trotzdem deutlich geringer. Üblich und hier diskutiert sind
daher nur TOF’s vom Typ der konstanten Energie.
5.2
Ein- und zweistufige lineare Flugzeitmassenspektrometer
Betrachtet werde ein zweistufiges TOF entsprechend der Abbildung 5.3 mit den dort angegebenen Größen.
Die resultierende Gesamtflugzeit bis zum Detektor ist die bestimmende Größe, aus der
man die Massenauflösung ermitteln kann. Dazu werden die entsprechenden experimentellen
Größen, i.e. Länge und Spannung der 1. Beschleunigungsregion, Länge und Spannung der 2.
Beschleunigungsregion geeignet optimiert, so dass an der Detektorposition ein Zeitfokus für alle
Massen entsteht, der möglichst unabhängig von Unschärfebeiträgen ist.
5.2. EIN- UND ZWEISTUFIGE LINEARE FLUGZEITMASSENSPEKTROMETER
53
Abb. 5.3: Prinzipaufbau eines zweistufigen linearen Flugzeitmassenspektrometers
Die Flugzeit wird bestimmt als Funktion der Größen
anfängliche Startenergie
W0 = 1/2 m · v02 ≈ k · T ,
Energiegewinn in der 1. Beschleunigungsstrecke
W1 = q · U1 ,
Energiegewinn in der 2. Beschleunigungsstrecke
W2 = q · U2 ,
relative Ortsunschärfe
relative Energieunschärfe
∆=
s0
s1
und
ε=
W0
W1
≈
k·T
q·U1 .
Sehr hilfreich zur Darstellung sind die abgeleiteten Größen
Gesamtenergie
Wtot = W1 + W2 ,
Verhältnis Gesamtenergie zu derjenigen der ersten Stufe
und die kompensierte Unschärfe
k0 =
Wtot
W1
=
W1 +W2
W1
δ = ε − ∆.
Auf der Basis dieser Größen kann die Flugzeit in jeder Stufe berechnet werden. Die
Rechnung selbst ist trivial aber aufwendig und wird hier für die erste Stufe schrittweise
durchgeführt:
Die Beschleunigung entlang des Feldes ist gegeben durch
W1
d2 s
=
2
dt
s1 · m
(5.1)
Durch Integration erhält man die Geschwindigkeit
W1
ds
=
·t+
dt
s1 · m
r
2 · W0
m
(5.2)
2 · W0
· t + s0 =: s1
m
(5.3)
und den Weg
W1
s=
· t2 +
2s1 · m
r
54
KAPITEL 5. FLUGZEITMASSENSPEKTROMETRIE
Die Integrationskonstanten werden durch die Anfangsbedingungen festgelegt. Durch Gleichsetzen des Wegs mit der Fluglänge s1 in der ersten Beschleunigungsstrecke erhält man die Flugzeit
t über eine quadratische Gleichung. Hierbei ist nur die positive Lösung physikalisch sinnvoll.
s
√
2 · m · W0
2 · m · W0 2 2 · m · (s0 − s1 )
· s1 −
· s1 +
· s1
t1 = −
W1
W1
W12
( r
)
r
r
2·m
W0
W0 s 0
(5.4)
· −
+
−
+1
= s1 ·
W1
W1
W1 s 1
r
o
√
2·m n √
· − ε+ 1+δ
= s1 ·
W1
Analog kann die Flugzeit t2 in der 2. Beschleunigungsstrecke
)
(r
r
√
2 · m W1
W1 + W2
t2 = s 2 ·
·
·
+δ− 1+δ
W1 W2
W1
sowie diejenige in der Driftstrecke, td , berechnet werden, in der eine konstante Geschwindigkeit
v2 vorliegt.
(r
)−1
r
1
2·m
W1 + W2
1
td = d · ·
·
+δ
(5.5)
=d·
2
W1
W1
v2
Die Gesamtflugzeit als Funktion von s1 ist dann gegeben durch
T (s1 ) = t1 + t2 + td
(
)
√
r
√
√
W1 d
2·m
W1 + W2
W1
=
·
+
+ δ · s2 ·
+
1 + δ · s1 − s2 ·
− ε · s1
W1
W2
W1
W2
2
(5.6)
Den Zeitfokus, also den Punkt an dem die Beiträge der Orts- und Energieunschärfe ∆ und ε, ausgedrückt hier in der Größe der kompensierten Unschärfe δ = ∆−ε, minimal werden, erhalten wir
als Extremwert dieser Funktion in Bezug auf den Parameter δ. Dazu nutzen wir eine Taylor’sche
Reihenentwicklung und betrachten das Verschwinden der einzelnen Entwicklungsordnungen, die
sehr schnell konvergieren:
∆T = T (δ) − T (0) =
∞
X
T (n) (δ = 0) n
·δ
n!
n=1
Für den Fall δ = ε = 0 erhalten wir dann unter Verwendung der
Wtot = W1 + W2 und k0
r
√
p
2·m
2 · k0
T (δ = ε ≡ 0) =
·
· s2 + k0 ·
k0 · s1 + √
Wtot
k0 + 1
(5.7)
abgeleiteten Größen
d
2
(5.8)
Damit ergibt sich die Position des Zeitfokus erster Ordnung ( dT
dδ ≡ 0) bei einer Driftstrecke von
1
s2
3/2
√ ·
d = 2 · s1 · k0 · 1 −
(5.9)
k0 + k0 s1
2
d T
Für den Zeitfokus zweiter Ordnung ( dT
dδ ≡ 0 und dδ2 ≡ 0) ergibt sich die Zusatzbedingung
3
s2
d
= 1−
(5.10)
·
s1
k0
2 · s1
5.2. EIN- UND ZWEISTUFIGE LINEARE FLUGZEITMASSENSPEKTROMETER
55
Aussage und Auswirkung dieser Beziehungen soll im Folgenden detailliert unter verschiedenen
Aspekten diskutiert werden:
1. Einstufiges Flugzeitmassenspektrometer
Betrachten wir die erhaltenen Beziehungen zuerst an einem TOF-MS mit einer einstufigen Beschleunigungsstrecke. Dafür gilt: s2 = 0 und k0 = 1, aus (5.9) folgt dann d = 2 · s1 ,
der Zeitfokus erster Ordnung ist also bei der doppelten Länge der Beschleunigungsstrecke
festgelegt und kann durch Wahl der Geräteparameter nicht beeinflusst werden. Die Bedingung
(5.10) führt darüber hinaus zu einer mathematisch unerfüllbaren Gleichung, was bedeutet, dass
bereits der Beitrag der 2. Ordnung prinzipiell nicht kompensiert werden kann.
Insgesamt ist dies sehr unbefriedigend und führt, auch aufgrund der zwangsläufig kurzen
Driftstrecke von d = 2 · s1 zu schlechter Auflösung. Einstufige Beschleunigung ist daher generell
nicht akzeptabel, wird nicht realisiert und nicht weiter betrachtet.
2. Massenabhängigkeit
Alle erhaltenen Beziehungen sind massenunabhängig, der Zeitfokus liegt also für alle
Massen am selben Punkt und kann daher auch wirklich als Detektorposition genutzt werden.
Eine räumliche Verschiebung wie etwa bei den gebogenen Fokalebenen magnetischer Massenspektrometer liegt, zumindest bis zur 2. Ordnung, nicht vor und muss nicht kompensiert werden.
3. Anfangsenergieunschärfe
Der Einfluss einer Anfangsenergieunschärfe ist nicht vollständig kompensiert, der entsprechende
√
Term ε · s1 in Gleichung (5.6) wurde bisher vernachlässigt.
4. Massenauflösung
Bei Vernachlässigung der Energieunschärfe lässt sich die Auflösung bestimmen aus
∆T ≈
T (3) (δ = 0) 3
· δ + höhere Ordnungen
6
(5.11)
Dieser Beitrag muss dann gerade der Flugzeitdifferenz für benachbarte Massen entsprechen:
n
o
∆T (m + 1, m) = T (m + 1) − T (m) = const. · (m + 1)1/2 − m1/2
(5.12)
T (m)
1 1/2
= T (m) · (1 + ) − 1 ≈
m
2·m
Die Massenauflösung erhält man nun durch Einsetzen von (5.8) in (5.12) und Wahl der Parameter
gemäß (5.9) und (5.10). Über eine etwas aufwändige Umformung kann man (5.11) für die
Näherungen k0 >> 1 und k0 >> s2 /s0 folgt [Wile55]:
<=
m
k0
≈ 16 · 2
∆m
∆
(5.13)
56
KAPITEL 5. FLUGZEITMASSENSPEKTROMETRIE
5. Beispiel: Beiträge der Orts- und Energieunschärfe
Als aufschlussreiches numerisches Beispiel sei ein kleines TOF-MS diskutiert, wie es von
Wiley bereits 1955 aufgebaut wurde [Wile55]. Die Daten sind die folgenden:
Komponente
Größe
Feldstärke
Energie
Flugzeit
[cm]
[V/cm]
[eV]
[µs]
1. Beschleunigung
s1 = 0.2
320
W1 = 64
0.035· m1/2
2. Beschleunigung
s2 = 1.2
1280
W2 = 1536
0.035· m1/2
Driftstrecke
d = 40.0
0
Wtot = 1600
0.69· m1/2
Ortsunschärfe
∆1 = 0.2
ε1 = 0
∆2 = 0.02
Energieunschärfe
W0 = 0.1
ε2 = 0.0016
±0.035· m1/2
Die Spannungen ergeben einen Wert von k0 ≈ 25. Bei der angenommenen Ortsunschärfe
∆1 in Größe der Länge der gesamten 1. Beschleunigungsstrecke ergibt sich damit immerhin
noch eine Massenauflösung gemäß (5.13) von < ≈ 400. Bei Reduktion auf eine Ortsunschärfe
∆2 von nur 1/10 der Länge der 1. Beschleunigungsstrecke sogar theoretisch ein Betrag von
< ≈ 40000. Beides ist relativ bzw. extrem hoch.
Berücksichtigen wir nun aber zusätzlich eine nur geringfügige Energieunschärfe von z.B.
ε2 = 0.0016, also 1.6 Promille. Gemäß (5.6) trägt diese zur Zeitunschärfe bei mit
r
√
2·m
· s1 · ε
(5.14)
∆T =
W1
Damit folgt [Wile55]
m
T
1
√ ·
<=
=
=
∆m
2 · ∆T
4· ε
(
k0 + 1
1/2
k0
1/2
−
k0
−1
1/2
k0 + k0
k0
√
≈
4· ε
s2
·
s1
)
(5.15)
Es ergibt sich bereits damit also durch diesen geringen Beitrag von nur ca. 1.6 Promille, eine Reduktion um einen Faktor 3 auf einen Wert von < ≈ 150 bzw. eine völlige Zerstörung des Wertes
für vernachlässigbare Ortsunschärfe. Eine auch nur geringe Energieunschärfe hat also einen
entscheidenden Einfluss auf die Auflösung. Dies wird häufig über den Aufbau eines Reflektrons
kompensiert.
5.3
Reflektron - Flugzeitmassenspektrometer
Durch Einbau eines Ionenspiegels in ein Flugzeitmassenspektrometer entsteht ein Reflektron
[Mamy73]. Abbildung. 5.4 zeigt der Übersichtlichkeit halber eine vereinfachte Version mit
einstufiger Beschleunigungsregion und einstufigem Ionenspiegel, wobei die Entfernung des
räumlichen Zeitfokus eingezeichnet ist. Ein jeweils zweistufiges System ist einfach vorstellbar.
57
5.3. REFLEKTRON - FLUGZEITMASSENSPEKTROMETER
Abb. 5.4: Prinzipaufbau eines Reflektron - Flugzeitmassenspektrometers
Über die energieabhängige, unterschiedliche Eindringtiefe kann eine Kompensation der
Anfangsenergieunschärfe erfolgen. Idealerweise ist dabei wiederum die primäre Beschleunigungsstrecke zweistufig ausgelegt, damit die Ortsunschärfe am Zeitfokus bis zur 2. Ordnung
kompensiert werden kann. Der Ionenspiegel wird dann günstigerweise auch zweistufig aufgebaut, indem durch verschiedene Gitter (oder Gitterlos durch Ionenlinsen) ein schwächer
abbremsendes Feld mit einem stärkeren voll abbremsenden und die Flugrichtung umkehrenden
Feld kombiniert wird. Da Quelle und Detektor zwangsläufig nicht auf einer Achse installiert
sein können, muss eine geringe seitliche Verkippung der ionenoptischen Komponenten und eine
kurvenförmige Flugbahn der Ionen im Reflektron akzeptiert werden.
2
d T
Bei geeigneter Auslegung kann das zweistufige Reflektron die Größen dT
dε ≡ 0 und dε2 ≡ 0
kompensieren. Die Rechnung verläuft dabei analog zur zweistufigen Beschleunigung im linearen
TOF, ist aber deutlich aufwändiger. Sie liefert Resultate für die Größen
s3
=
Länge der 1. Abbremsregion des Ionenspiegels
W3
=
Energie der 1. Abbremsregion des Ionenspiegels
s4
=
Eindringtiefe
(W4
=
Wtot - W3 = vorgegeben)
Der Rechenweg, der zu diesen Größen führt, verwendet wiederum die Flugzeiten, wobei die 1.
Abbremszeit sich ergibt zu
(
)
r
p
√
s3
W3
·
t3 = 2 · m · Wtot ·
1−ε− 1+ε−
(5.16)
W3
Wtot
und die Umkehrzeit (im 2. Abbremsfeld) zu
s4
2 · m · Wtot ·
·
Wtot
r
W3
Wtot
(5.17)
TRef lektron = t1 + t2 + td1 + 2 · t3 + t4 + td2
(5.18)
t4 = 2 ·
p
1+ε−
Die Gesamtflugzeit ergibt sich dann zu
58
KAPITEL 5. FLUGZEITMASSENSPEKTROMETRIE
Sie muss nun wieder Taylor-entwickelt werden, diesmal nach ε und dann die für den Zeitfokus
optimalen Werte der oben genannten Parameter ermittelt werden. Für die weitere Entwicklung
sowie Ergebnisse sei auf [Cott97] verwiesen. Probleme bereiten der Winkel in der Flugbahn
und der daraus resultierende seitliche Versatz, der zu einer Abhängigkeit von der transversalen
Anfangsgeschwindigkeit führt. Der Erfinder des Reflektrons konnte eine Auflösung von > 3000
demonstrieren, später konnte dies auf Werte > 10 000 gesteigert werden. Die endgültige Grenze
der Auflösung ist durch Feldinhomogenitäten, Aufladungseffekte und die zeitliche Startunschärfe
der Ionen gegeben.
Kapitel 6
Radiofrequenz-Massenspektrometer
Wir gehen nun von den statischen Massenspektrometern über zu den Radiofrequenz (RF)Massenspektrometern. Damit konnten nach ca. 1960 relative Massengenauigkeiten von 10−7
und mit moderneren Systemen sogar noch höhere Genauigkeiten erreicht werden. Die Massenmessungen mit RF-Spektrometern basieren fast alle auf der Messung von Frequenzen, in erster
Linie der Zyklotronfrequenz νc . Das ist die Umlauffrequenz eines freien, geladenen Teilchens
mit der Ladung q und der Masse m in einem homogenen Magnetfeld der Stärke B. Bewegt sich
das Teilchen senkrecht zum Magnetfeld, so wird die Beschleunigung durch die Lorentz-Kraft
bestimmt die gerade gleich der Zentrifugalkraft ist, um das Teilchen auf einer Kreisbahn zu
halten. Daraus resultiert für die Zyklotronfrequenz
νc =
1 qB
·
,
2π m
(6.1)
sie ist also interessanterweise unabhängig vom Bahnradius.
Allgemein kann man sagen, dass die höchsten Präzisionen bei Messungen unterschiedlicher
Größen immer dann erreicht werden, wenn die Messung auf einer Frequenzbestimmung
basiert. Beispiele hierfür sind Präzisionsmassenmessungen in Ionenfallen durch Bestimmung der Zyklotronfrequenz des gespeicherten Ions, optische Frequenzmessungen z.B. der
2S − 1S-Übergang im Wasserstoff und Zeitmessungen mit Atomuhren. Im Folgenden soll der
Radiofrequenz-Transmissions-Massenspektrometer MISTRAL beispielhaft vorgestellt werden.
Eine ausführliche Beschreibung der Apparatur findet sich in [Simo1995, Lunn1996]. Die
Ionenfallen und Speicherringe in denen ebenfalls Frequenzmessungen an gespeicherten Ionen
erfolgen, werden zu einem späteren Zeitpunkt diskutiert.
MISTRAL (Mass measurements at ISolde with a Transmission RAdiofrequency spectrometer onLine) [Lunn2000] ist am on-line Isotopenseparator ISOLDE [Kugl2000] am CERN [CERN2005]
in Genf/Schweiz aufgebaut. Es dient der präzisen Bestimmung von Massen extrem kurzlebiger Radionukliden mit Halbwertszeiten im Bereich von 10 ms. Die Anwendungsfelder von
Präzisionsmassenmessungen können in [Boll2001, Klug2003, Lunn2003, Boll2004] nachgelesen
werden.
Eine schematische Darstellung des MISTRAL Spektrometers und eine typischen Ionentrajektorie ist in Abb. 6.1 dargestellt. Eingeschossene Ionen bei voller ISOLDE Strahlenergie von
60 keV machen zwei Umläufe im homogenen Magnetfeld. Die transmittierten Ionen werden mit
einem Sekundärelektronenvervielfacher (siehe Kap. 3 der Vorlesung) nachgewiesen. Mit einer
Einschussschlitzbreite von 0,4 mm und einem Flugradius von 0,5 m wird eine Auflösung von 2500
ohne Einsatz der Modulationsradiofrequenz erreicht. Um eine Messung durchzuführen, erfolgt
eine Modulation der longitudinalen kinetischen Energie. Das rechte Teilbild in Abb. 6.1 zeigt
den Aufbau des Modulators. Er kommt nach einer halben und nach drei halben Runden zum
59
60
KAPITEL 6. RADIOFREQUENZ-MASSENSPEKTROMETER
Abb. 6.1: Aufbau des MISTRAL Massenspektrometers mit einer idealen Ionentrajektorie. Ionen
werden vom ISOLDE Strahlrohr mit einer Energie von 60 keV eingeschossen. Referenzionen zur
Kalibration des Magnetfeldes liefert die off-line Ionenquelle. Im Teilbild rechts ist die Elektrodenstruktur des Modulators zu erkennen. Das untere Teilbild zeigt eine Ionenbahn im Magneten
mit dem 0,4 mm Einschussschlitz gefolgt von dem ersten Modulator nach einer halben Runde,
dann der phasendefinierte Schlitz mit einer Breite von bis zu 5 mm, anschließend der zweite
Modulator nach drei halben Runden und schlussendlich der Austrittsschlitz. Das linke Teilbild
zeigt das Ionensignal von 39 K als Funktion der Radiofrequenz. Es sind drei Harmonische (um
3400) zu sehen. Die Massenauflösung ist größer als 100 000 [Lunn2000].
Einsatz, so dass die Ionen dazwischen eine volle Zyklotronbahn im Magnetfeld beschreiben. Eine
Radiofrequenzspannung wird an die zentrale Modulatorelektrode angelegt. Abhängig von der
Phase der RF-Spannung zum Zeitpunkt des Durchflugs der Ionen durch den Modulator, bewirkt
die resultierende longitudinale Beschleunigung einen größeren oder kleineren Zyklotronradius im
Vergleich zur ”normalen” Flugbahn (alle Flugbahnen sind isochron). Die Ionen passieren einen
0,4 mm Austrittsschlitz wenn der Nettoeffekt der beiden Modulationen gerade Null beträgt. Das
passiert, wenn die Radiofrequenzspannung ein ”ganzzahliges-plus- 21 ”-Vielfaches der Zyklotronfrequenz ist:
1
νRF = n +
νc .
(6.2)
2
D.h. die Ionen spüren während der zweiten Modulation genau das Gegenteil dessen, was sie
während der ersten Modulation gespürt haben. Für harmonische Zahlen (z.B. größer als 1000)
und einer Radiofrequenzspannung von etwa 200 V zeigt das Ionensignal als Funktion der Radiofrequenz schmale Transmissionspeaks in gleichmäßigen Abständen der Zyklotronfrequenz.
61
Abb. 6.2: Gemessener Transmissionspeak von 30 Na (T1/2 = 48 ms). Die Messung besteht aus
64 Frequenzschritten mit einem Zeitfenster von jeweils 320 ms. Insgesamt wurden 25 Scans
aufgenommen. Die Zentrumsfrequenz resultiert aus einer Dreiecksanpassung und entspricht der
2421. Harmonischen der Zyklotronfrequenz im 0,38679 T starken Magnetfeld bei einer Ionenenergie von 60 keV. Die Massenauflösung beträgt etwa 50 000 und wurde zur Verbesserung der
Transmission so gering gewählt [Lunn2000].
Die Auflösung kann mehr als 100 000 betragen, wie im linken Teilbild der Abb. 6.1 zu sehen ist.
Für eine richtige Massenmessung wird immer alternierend das Ion mit unbekannter Masse und
ein Ion mit gut bekannter Masse ohne Änderung des Magnetfeldes eingeschossen. Dadurch
kann das Magnetfeld kalibriert werden. Durch den schnellen Ionenwechsel können zeitliche
Veränderungen der Magnetfeldstärke korrigiert und somit systematische Unsicherheiten
minimiert werden. Die Massenauflösung R = m/∆m eines Radiofrequenz-TransmissionsMassenspektrometers hängt von der harmonischen Zahl n, der Austrittsschlitzweite w und der
Amplitudenmodulation des Flugbahndurchmessers Dm ab. Sie ist gegeben durch [Coc1988]
R = 2πn
Dm
.
w
(6.3)
Aufgrund der hohen Transportenergie und des schnellen Messverfahrens können sehr kurz-lebige
Nuklide untersucht werden. Als Beispiel ist in Abb. 6.2 das Transmissionssignal des Radionuklids
30 Na mit einer Halbwertszeit von nur 48 ms dargestellt. Die Massenauflösung ist ca. 50 000 und
die erreichte relative Massengenauigkeit beträgt typischerweise wenige 10−7 . Alle Massenwerte
von Nukliden werden in einer Datenbank gesammelt, der sogenannten Atomic-Mass Evaluation.
Die jüngste Ausgabe ist die AME2003 [Audi2003].
Kapitel 7
Quadrupolmassenspektrometrie
7.1
Entwicklungsgeschichte
Anfang der 50er Jahre entwickelte Wolfgang Paul die Idee, dass elektrische und magnetische Multipolfelder Teilchen mit einem magnetischen oder elektrischen Dipolmoment in zwei Dimensionen
fokussieren können. Linsen für Atom- und Molekülstrahlen wurden verwirklicht [Frie1951]. Die
Frage: ,,Was geschieht, wenn man geladene Teilchen - Ionen oder Elektronen - in solche Multipolfelder injiziert?”1 führte 1953 zur Entwicklung des Quadrupol-Massenspektrometers (QMS)
[Paul1953]. Es benutzt zur Massentrennung nicht nur die fokussierenden und defokussierenden Kräfte eines hochfrequenten elektrischen Quadrupolfeldes, sondern macht auch von den
Stabilitätseigenschaften der Bewegung Gebrauch. Die theoretischen und experimentellen Entwicklungen des linearen QMS wurden von der Gruppe um Paul bestimmt [Paul1955]. Die klassische Veröffentlichung ,,Das elektrische Massenfilter als Massenspektrometer und Isotopentrenner ” von Paul, Reinhard und von Zahn [Paul1958] bildete den vorläufigen Höhepunkt dieser
Forschungsarbeiten. Durch Erweiterung der Methoden der zweidimensionalen Fokussierung auf
drei Dimensionen konnten Ionenfallen, die der Speicherung von Ionen in einem räumlich begrenzten Bereich dienen, entwickelt werden [Fisc1958, Fisc1959, Ghos1995, Majo2004]. Wie Abb. 7.1
zeigt, führten die vorgenannten Arbeiten zur Verleihung des Nobelpreises 1989 an Wolfgang Paul
(1913-1993), Universität Bonn, ,,für die Entwicklung der Ionenkäfigtechnik ”.
Die Anwendungsmöglichkeiten und Eigenschaften der linearen Paulfalle bzw. des Quadrupols
wurden nach 1958 in einer Reihe von theoretischen und experimentellen Arbeiten detailliert untersucht und publiziert, wie z.B. [Busc1961, Brub1961, Brub1964, Lee1971, Aust1992, Blau1997,
Tito1998a, Tito1998b]. Besonders hervorzuheben sind die Arbeiten von Dawson, deren Resultate großteils in dem grundlegenden Werk und Lehrbuch: ,,Quadrupole Mass Spectrometry
and its Applications” [Daws1995] zusammengefasst sind. In jüngster Zeit führten dann vor
allem Computersimulationen zu einem tieferen Einblick über die Ionenbewegung in der linearen Paulfalle [Munt1995, Reub1996, Blau1998b, Blau2000a]. Da die dreidimensionale Paulfalle
lediglich eine Erweiterung der linearen Paulfalle darstellt und im Idealfall die Ionenbewegung in
radialer und axialer Richtung als unabhängig voneinander betrachtet werden kann, werden in
der Vorlesung die Grundlagen der Ionenbewegungen für den zwei- und dreidimensionalen Fall
getrennt behandelt.
Allgemein erhält man eine elastische Bindung an eine Achse oder an einen Punkt im Raum,
wenn eine zur Fallenachse rücktreibende Kraft auf die Teilchen einwirkt:
−
→ −
F ∼→
r n.
1
Entnommen aus dem Nobel-Vortrag von Wolfgang Paul am 12. Oktober 1989 in Stockholm.
62
(7.1)
63
7.2. DIE IDEALE LINEARE PAULFALLE
Abb. 7.1: Verleihung des Nobelpreises durch König Carl XVI. Gustav von Schweden und
zugehörige Urkunde an Wolfgang Paul am 12. Oktober 1989. Aufnahme: Foto Klein, Bonn.
Mit der Festlegung auf n = 1, d.h. eine linear mit dem Abstand r ansteigende Kraft:
−
→
−
→
→
F = −e · ∇Φ = −c−
r mit c = const.,
(7.2)
folgt ein Potential Φ(x, y, z, t), das eine rein quadratische Abhängigkeit von den kartesischen
Koordinaten x, y, z besitzt. Das Potential ist demnach von der Form
→
Φ(−
r , t) = Φ0 (t) · (ax2 + by 2 + cz 2 )
.
(7.3)
Bei Vernachlässigung von Raumladungen in der Elektrodenanordnung liefert die LaplaceGleichung ∆Φ = 0 die Bedingung a + b + c = 0. Die beiden einfachsten Möglichkeiten, diese zu
erfüllen, sind:
1. a = −b und c = 0; es resultiert das zweidimensionale Feld des Quadrupolmassenfilters
bzw. der linearen Ionenfalle,
2. a = b und c = −2a; es resultiert die dreidimensionale Konfiguration der Ionenfalle.
Die Ionenfalle wird zu einem späteren Zeitpunk in der Vorlesung behandelt.
7.2
Die ideale lineare Paulfalle
In der linearen Paulfalle bewegen sich geladene Teilchen in einem hochfrequenten elektrischen
Vierpolfeld, wobei die für die Stabilität der Ionenbahnen entscheidende Eigenschaft direkt die
spezifische Ladung Ze/m ist. Der Betrag der aus dem quasistationären Wechselfeld des linearen
Paulfalle resultierenden Feldstärke ist proportional dem Abstand r von der vorgegebenen zAchse eines Koordinatensystems. Die Realisation eines idealen Vierpolfeldes erfolgt durch die
Verwendung hyperbelförmiger Elektroden.
64
KAPITEL 7. QUADRUPOLMASSENSPEKTROMETRIE
-Φ0
+Φ0
2r0
+Φ0
y
z
-Φ0
x
Abb. 7.2: Elektrodenanordnung der idealen, endlich großen linearen Paulfalle und Verlauf der
Äquipotentiallinien, die eine vierzählige Symmetrie in Bezug auf die z-Achse aufweisen.
7.2.1
Hyperbolisches Quadrupolpotential
Bei Verwendung von vier hyperbolisch geformten Elektroden, die sich entlang der z-Achse erstrecken und mit der Wahl a = −b = 1/r02 für das Potential der linearen Paulfalle folgt aus
Gl. (7.3):
(x2 − y 2 )
→
Φ(−
r , t) = Φ0 (t) ·
.
(7.4)
r02
Dabei beträgt der Abstand zwischen zwei gegenüberliegenden felderzeugenden Elektroden 2r0 .
Legt man die Spannung 2Φ0 an das Elektrodenpaar an, so ist das Potential auf den Elektroden ±Φ0 . Eine Darstellung der Elektrodenkonfiguration mit resultierenden Äquipotentiallinien
(x2 − y 2 ) = const. zeigt Abb. 7.2. Aus Gl. (7.4) ergeben sich die Feldstärkekomponenten zu:
Ex = − Φ0 · x/r02 ,
Ey = Φ0 · y/r02 , Ez = 0 .
(7.5)
Schießt man die positiven Ionen in z-Richtung bei konstanter Spannung +Φ0 ein, so führen sie
harmonische Schwingungen in der (x, z)-Ebene aus. Aufgrund des umgekehrten Vorzeichens für
das Feld Ey wird ihre Amplitude in der y-Richtung exponentiell ansteigen, d.h. die resultierende
Kraft ist in der (y, z)-Ebene defokussierend; die Teilchen stoßen gegen die Elektroden und gehen
verloren. Durch Anlegen einer zusätzlichen periodischen Wechselspannung kann dieses Verhalten
vermieden werden. Aufgrund des periodischen Wechsels des Vorzeichens der elektrischen Kraft
erhält man in beiden Raumrichtungen alternierend Fokussierung und Defokussierung. Dies führt
bei geeigneter Frequenz über das Prinzip der starken Fokussierung zu einer stabilen Ionenbahn.
Ist die an die jeweils gegenüberliegenden Elektroden der Paulfalle angelegte Spannung Φ0 durch
eine Gleichspannung U und eine Wechselspannung V mit der Frequenz Ω
Φ0 (t) = U + V · cos Ωt
(7.6)
65
7.2. DIE IDEALE LINEARE PAULFALLE
gegeben, so lautet das ideale Quadrupolpotential:
(x2 − y 2 )
→
Φ(−
r , t) = (U + V · cos Ωt) ·
.
r02
7.2.2
(7.7)
Bewegungsgleichungen in der idealen linearen Paulfalle
Betrachten wir im Folgenden ein einfach positiv geladenes Ion der Masse m im Quadrupolpotential (7.7). Aus der Differentialgleichung
··
−
→ →
→
r +e ∇Φ(−
r , t) = 0
m−
(7.8)
resultieren die Bewegungsgleichungen in x-, y-, und z-Richtung:
ẍ +
2e
· (U + V · cos Ωt) · x = 0
mr02
(7.9)
ÿ −
2e
· (U + V · cos Ωt) · y = 0
mr02
(7.10)
z̈ = 0 .
(7.11)
Auf den ersten Blick würde man erwarten, dass der zeitabhängige Teil der Kraft im zeitlichen
Mittel verschwindet, was in einem homogenen Feld auch der Fall ist. In einem inhomogenen,
periodischen Feld wie dem Quadrupolfeld bleibt jedoch eine kleine mittlere Kraft übrig, die
immer in Richtung des abnehmenden Feldes wirkt, also hier in Richtung der Mittelachse. Aus
diesem Grund bleiben die Ionen unter geeigneten Bedingungen in radialer Richtung gespeichert, ohne die Elektroden zu berühren. Die Ionen führen nach Gl. (7.9) und (7.10) periodische Schwingungsbewegungen um die z-Achse aus, die durch eine periodisch sich verändernde
·
Rückstellkraft hervorgerufen werden. Integration von Gl. (7.11) liefert z = const., d.h. die in
z-Richtung eingeschossenen Ionen bewegen sich mit konstanter Geschwindigkeit in z-Richtung
durch das Feld. Durch Einführung der dimensionslosen Transformationsparameter
ax = −ay =
4eV
8eU
, qx = −qy =
,
2
2
mr0 Ω
mr02 Ω2
Ωt = 2ξ
(7.12)
erhält man aus Gl. (7.9) und (7.10) die Mathieu’schen Differentialgleichungen:
d2 x
+ (ax + 2qx · cos 2ξ) · x = 0 ,
dξ 2
(7.13)
d2 y
− (ay + 2qy · cos 2ξ) · y = 0
dξ 2
(7.14)
.
Dabei gibt a/4 das Verhältnis von potentieller Energie eU im Gleichspannungsfeld zu kinetischer
Energie mv 2 /2 = mr02 Ω2 /2 der Schwingung mit Amplitude r0 und q/2 das Verhältnis von Epot
im Wechselspannungsfeld zu Ekin an.
Das Verhalten der Ionen ergibt sich aus den Eigenschaften der Lösungen der Schwingungsgleichungen (7.13) und (7.14) mit periodischen Koeffizienten. Beide Bewegungskomponenten
gehorchen der gleichen Differentialgleichung, so dass es genügt, die Mathieu’sche Differentialgleichung in der Normalform
d2 u
+ (au − 2qu · cos 2ξ) · u = 0
dξ 2
mit
u = x, y
(7.15)
66
KAPITEL 7. QUADRUPOLMASSENSPEKTROMETRIE
zu betrachten [McLa1947, Meix1954]. Alle Lösungen lassen sich als Linearkombination zweier
linear unabhängiger Lösungen in einer Fourierreihe der Form
u(ξ) = αI · eµu ξ
∞
X
s=−∞
c2s,u e2isξ + αII · e−µu ξ
∞
X
c2s,u e−2isξ
(7.16)
s=−∞
darstellen. Mit Hilfe der Euler’schen Gleichung e±ix = cos x ± i sin x und dem charakteristischen Exponenten µu = iβu kann die allgemeine Lösung der Mathieuschen Differentialgleichung (7.16) zu
u(ξ) = A ·
∞
P
s=−∞
c2s,u cos [(2s + βu )ξ] + B ·
∞
P
s=−∞
c2s,u sin [(2s + βu )ξ]
(7.17)
umgeschrieben werden, wobei die Koeffizienten A := αI + αII und B := i(αI − αII ) durch die
Anfangsbedingungen [u(ξ = 0), u̇(ξ = 0)] bestimmt sind. Die Konstante βu wird durch die
Transformationsparameter au und qu in einer Kettenbruchgleichung festgelegt:
βu2 = au +
2
qu
(2+βu )2 −au −
(4+βu )2 −au −
2
qu
+
(2−βu )2 −au −
(4−βu )2 −au −
2
qu
2
qu
(6+βu )2 −au −...
2
qu
.
(7.18)
2
qu
(6−βu )2 −au −...
Die Koeffizienten c2s,u der Reihenentwicklung (7.16) und (7.17) hängen ebenfalls nur vom Arbeitspunkt (au , qu ) und nicht von den Anfangsbedingungen ab und können über Rekursionsformeln berechnet werden [Marc1989]. Wegen
1
lim |c2s,u | |s| = 0
s→±∞
(7.19)
konvergieren sie für große |s| schnell gegen Null.
7.2.3
Stabilitätsdiagramme und Massenauflösungsvermögen
Die Mathieu-Gleichung (7.15) hat zwei Klassen von Lösungen:
• Stabile Bewegung: die Ionen schwingen mit begrenzter Amplitude in x- und y-Richtung
und durchqueren das Quadrupolfeld in z-Richtung, ohne an die Elektroden zu stoßen; u(ξ)
bleibt für ξ → ∞ beschränkt;
• Instabile Bewegung: die Schwingungsamplituden in x- oder in y-Richtung wachsen exponentiell an, so dass die Teilchen von den Elektroden weggefangen werden ehe sie das Ende
des Quadrupols erreichen; u(ξ) wächst für ξ → ∞ über alle Grenzen.
Der Lösungstyp ist nach Gl. (7.16) bzw. (7.17) eindeutig durch µu bzw. βu bestimmt, d.h. ob
Stabilität besteht, hängt nur von den Parametern au und qu ab, dagegen nicht von den Anfangsbedingungen der Ionenbewegung, z.B. ihrer Geschwindigkeit und ihrem Ort. Für komplexe
µu ist die Lösung instabil, wenn αI 6= 0 ist. Falls µu reell und µu 6= 0 ist, wächst einer der beiden
Terme eµu ξ oder e−µu ξ uneingeschränkt; die Lösungen sind ebenfalls instabil. Es zeigt sich also,
dass die Lösungen (7.17) nur dann periodisch und stabil sind sowie die Amplituden der Ionenbewegung beschränkt, wenn βu reell und nicht ganzzahlig ist. Die Lösungen mit ganzzahligem
67
7.2. DIE IDEALE LINEARE PAULFALLE
a )
1 4
1 2
s ta b le
1 2
0 .2
b )
1 0
8
1 0
0 .0
6
8
-0 .1
4
6
2
-0 .2
z
0
-0 .3
a
a
a
z
4
c )
0 .1
-2
2
-6
-2
-0 .5
-8
z -s ta b le
r-s ta b le
-1 0
-4
-6
-0 .4
-4
0
-0 .6
-1 2
-1 6
-1 2
-8
-4
q
0
4
8
1 2
1 6
z a n d r -s ta b le
-0 .7
0
2
4
6
8
q
1 0
1 2
z
1 4
1 6
1 8
1
0 .0
0 .2
0 .4
0 .6
0 .8
q
1 .0
1 .2
1 .4
z
Abb. 7.3: (a) Bereiche stabiler Lösungen der Mathieu’schen Differentialgleichung. (b) Bereiche
stabiler Lösungen für die axiale und radiale Bewegung der Ionen in der Paulfalle. (c) Vergrößerter
Ausschnitt aus Abb. (b). Der Punkt kennzeichnet einen möglichen Arbeitspunkt. Die Linie
stellt eine Arbeitsgerade dar für welche die Paulfalle als Massenfilter betrieben wird. Weitere
Erläuterungen im Text.
βu heißen Mathieu’sche Funktionen und bilden die Grenze zwischen Stabilität und Instabilität. Trägt man in einem Koordinatensystem mit den Achsen au und qu die Bereiche stabiler
und instabiler Lösungen ein, so erhält man das in Abb. 7.3 dargestellte Stabilitätsdiagramm
der Mathieu’schen Differentialgleichung. Die schattierten Bereiche repräsentieren dabei stabile
Lösungen; sie müssen aufgrund der Unabhängigkeit der Lösungen von Gl. (7.15) vom Vorzeichen des qu -Parameters symmetrisch zur au -Achse sein. Da die Ionenbewegung nur dann stabil
ist, wenn sich für beide Bewegungskomponenten (x, y) stabile Bahnen ergeben, müssen beide
Arbeitspunkte (ax , qx ) und (ay , qy ) im stabilen Bereich liegen. Nach Gl. (7.12) gilt ax = −ay
und qx = −qy . Daher fallen bei Spiegelung der (au , qu )-Ebene an der au - und qu -Achse die
beiden Arbeitspunkte a = ax = −ay und q = qx = −qy zusammen. Infolgedessen erhält man die
überlappenden Gebiete der (x, y)-Stabilität. Die ersten drei stabile Bereiche für eine lineare
Paulfalle bzw. einen Quadrupolmassenfilter sind in Abb. 7.4(A) mit I, II und III beschriftet.2
Die Grenzen der jeweiligen Stabilitätsbereiche sind durch die Mathieu’schen Funktionen der
ganzzahligen charakteristischen Exponenten βu gegeben, die auch charakteristische Kurven
genannt werden. Sie können durch Potenzreihenentwicklung von Gl. (7.18) um qu2 = 0 berechnet
werden [Blau1997]. Für den in der Praxis relevanten ersten Stabilitätsbereich einer linearen
Paulfalle gilt 0 ≤ βu ≤ 1. Er ist in Abb. 7.4(B) vergrößert dargestellt. Ein Ion bleibt nur dann
auf einer stabile Bahn, wenn sein zugehöriger Arbeitspunkt (au , qu ) innerhalb des dreiseitigen
Gebietes liegt, dessen Spitze durch
(a0 , q0 ) = (0.237, 0.706)
(7.20)
gegeben ist. Da die höheren Stabilitätsbereiche II und III mit Mathieu-Parametern (au , qu )II ≈
(0.02, 7.57) und (au , qu )III ≈ (3, 3) in der Praxis nur eine untergeordnete Rolle spielen, soll hier
nicht weiter auf sie eingegangen werden; näheres dazu findet man in [Daws1995] und [Du1999].
Die mathematischen Grenzen βy = 0 und βx = 1 des 1. Stabilitätsbereiches sind als rein theoretisch anzusehen. Für eine lineare Paulfalle bzw. realen Quadrupolmassenfilter endlicher Größe
2
Die Reihenfolge der Stabilitätsbereiche entspricht der Notation von [Daws1995]. Andere Autoren verwenden
teilweise eine abweichende Notation.
68
KAPITEL 7. QUADRUPOLMASSENSPEKTROMETRIE
Abb. 7.4: (A) Die Stabilitätsbereiche (schattiert) der linearen Paulfalle bzw. des Quadrupolmassenfilters. (B) Ausschnitt I aus (A): Der erste Bereich gleichzeitiger Stabilität in x- und
y-Richtung. Weitere Erläuterungen im Text.
müssen die Grenzen des realen Stabilitätsbereiches zwingend innerhalb dieses Gebietes liegen,
da die Ionenbahnen noch zusätzlich durch weitere Bedingungen, wie etwa den endlichen Abstand
der Elektroden, eingeschränkt werden (siehe Kap. 7.3.2).
Bei einer vorgegebenen Wahl der Spannungen U , V sowie festen Werten für r0 und Ω liegen
gemäß Gl. (7.12) alle Massen m auf einer Arbeitsgeraden au /qu = 2U/V = const. (siehe Abb.
7.4(B)), die nicht von der Masse und Ladung des eingeschossenen Ions, sondern nur vom
Verhältnis der Gleichspannungs- und Wechselspannungsamplitude U/V abhängt. Legt man die
Arbeitsgerade mit konstantem au /qu einer linearen Paulfalle durch den Nullpunkt des (au , qu )Diagramms, so definieren die Schnittpunkte der Arbeitsgeraden mit den Begrenzungslinien
βy = 0 und βx = 1 des Stabilitätsdreiecks ein Intervall [q1 , q2 ], welchem mit der Transformationsgleichung (7.12) eindeutig ein Massenintervall ∆m zugeordnet werden kann. Nur die darin enthaltenen Ionenmassen gelangen auf stabilen Bahnen durch das Spektrometer (in dem gewählten
Beispiel die Masse m2 ). Die anderen Ionenmassen werden gegen die Quadrupolstäbe beschleunigt oder lateral aus dem Feld geworfen. Zu schwere Ionen (m1 ) sind x-stabil und y-instabil,
während zu leichte Ionen (m3 ) x-instabil und y-stabil sind. Eine Vergrößerung der Steigung der
Arbeitsgeraden bewirkt eine Annäherung an die Spitze des Stabilitätsdreiecks; der Massenbereich ∆m wird schmäler und nähert sich ∆m = 0. Im Extremfall U/V = a0 /(2q0 ) ≈ 0.168 ist der
Quadrupol-Massenfilter nur noch für Ionen einer exakt bestimmten Masse durchlässig bzw. die
lineare Paulfalle speichert nur noch eine bestimme Masse.
In der Nähe der Spitze des Stabilitätsdreiecks lässt sich das theoretische Massenauflösungsvermögen < = m/∆m aus den beiden Schnittpunkten der Arbeitsgeraden mit den Randkurven
des stabilen Bereiches als Funktion des Quotienten U/V berechnen:
<≈
0.126
0.126
=
0.16784 − U/V
(0.16784 − γ) + δ/V
.
(7.21)
Die Auflösung kann über den Quotienten U/V , d.h. über die Steigung der Arbeitsgeraden
gemessen in Bruchteilen von 0.16784 = a0 /2q0 , variiert werden. Experimentell wird dies
gewöhnlich durch Anpassung der Beziehung U = γV − δ erreicht. Dabei bestimmt der Arbeitsparameter γ die Auflösung. Der Parameter δ ist ein zusätzlicher Gleichspannungsoffset.
7.2. DIE IDEALE LINEARE PAULFALLE
69
Für δ = 0 wird eine konstante Auflösung < des Massenfilters garantiert. Mit der Wahl
γ = 0.16784 und δ 6= 0 ist die Auflösung < proportional zur Wechselspannung V und somit auch
proportional zur Ionenmasse m. Dies ist die Bedingung für einen konstanten ∆m Modus, bei
dem die Breite des transmittierten Massenbereichs konstant und unabhängig von der Masse
ist. Die Beziehung (7.21) wurde unter der Voraussetzung hergeleitet, dass das Quadrupolfeld
in z-Richtung unendlich lang ist, denn nur für t → ∞ streben die instabilen Ionenbahnen
gegen ∞. In der Praxis genügt es zu verlangen, dass die Ionen eine bestimmte Anzahl n
von Hochfrequenzperioden im Massenfilter bzw. in der Paulfalle der Länge L ausführen, die
groß genug ist, um die Amplituden instabiler Bahnen so weit aufzuschaukeln, dass die Ionen
schließlich auf die Elektroden treffen und weggefangen werden. Die Frequenz des Wechselfeldes
bzw. die Länge der lineare Paulfalle muss also gewährleisten, dass die Verweilzeit der Ionen im
Feld sehr viel größer ist als die Dauer einer Hochfrequenzperiode.
Durch Umformung der Transformationsgleichungen (7.12) zu
U=
ay mr02 Ω2
qy mr02 Ω2
qx mr02 Ω2
ax m2 r02 Ω2
=−
, V =
=−
8e
8e
4e
4e
(7.22)
wird die Wirkungsweise des Quadrupols als Massenspektrometer deutlich. Bei konstantem r0 und Ω erhält man durch Einsetzen der einzelnen Ionenmassen aus dem (au , qu )Stabilitätsdiagramm die Stabilitätsdiagramme für die einzelnen Massen im (U, V )-Raum.
Abb. 7.5: Die in den (U, V )-Raum transformierten Stabilitätsdiagramme 1. Ordnung für die
Massen m1 < m2 < m3 .
Wie Abb. 7.5 verdeutlicht, entsprechen die Stabilitätsdiagramme für die Massen m1 < m2 < m3
der Form nach demjenigen im (au , qu )-Raum, sind aber nach Gl. (7.22) mit massengewichteten
Faktoren gestreckt. Ändert man die an den Elektroden liegenden Spannungen U und V gleichzeitig und proportional, d.h. au /qu bleibt konstant, so bewegt man sich entlang der Arbeitsgeraden
und bringt Ionen immer größer werdender Masse nacheinander in den stabilen Bereich. Es
resultiert ein massenaufgelöstes Spektrum.
Auf der V -Achse, d.h. für U = 0, wird der Massenfilter bzw. die lineare Paulfalle im ,,rf-only”
Modus betrieben [Daws1985]. Es liegt Stabilität für 0 < qu < qmax = 0.92 vor (siehe Abb.
7.4(B)). Daraus folgt, dass sich alle Ionen mit Masse q 4eV
2 2 < m < ∞ auf stabilen Bahnen
u r0 Ω
bewegen. In diesem Falle wirkt das Quadrupolfeld wie ein Hochpass-Massenfilter. Man erhält
70
KAPITEL 7. QUADRUPOLMASSENSPEKTROMETRIE
ein integrales Spektrum, da jeweils an den Schnittpunkten der Stabilitätsdreiecke mit der
Abszisse die Beträge der leichteren, aber stetig ansteigenden Massen weggeschnitten werden.
7.2.4
Frequenzspektrum der Lösungen und Ionenflugbahnen
Aus der allgemeinen Lösung der Mathieu’schen Differentialgleichung (7.17) folgt, dass die Bahnen aller Ionen gleicher Masse sich nur in den Konstanten A und B entsprechend den verschiedenen Anfangsbedingungen u0 , u̇0 und Ωt0 unterscheiden. Daraus ergibt sich das Frequenzspektrum der Ionenbewegung für einen festen Arbeitspunkt (au , qu ) unter Verwendung von Ωt = 2ξ
zu:
Ω
ωs,u = |2s + βu |
(7.23)
s = 0, ±1, ±2, ...
u = x, y .
2
Wegen der Konvergenz der Koeffizienten c2s,u nach Gl. (7.19) für große |s| nehmen die Anteile
der höheren Frequenzen rasch ab. Es ist zu betonen, dass Ionen unterschiedlicher Masse verschiedene Arbeitspunkte im Stabilitätsdiagramm und folglich verschiedene Koeffizienten c2s,u
und βu haben. Die Frequenzspektren ihrer Bewegung, insbesondere die Grundfrequenz
ω0,u = βu
Ω
2
für
s=0 ,
(7.24)
sind verschieden.
Zur Beschreibung der Ionenbewegung im Quadrupolmassenfilter bzw. in der linearen Paulfalle
ist es am einfachsten, die beiden Spezialfälle mit den Arbeitspunkten nahe des Koordinatenursprungs und nahe der Stabilitätsspitze näher zu betrachten. Für den ersten Fall βu2 1 kann
die Kettenbruchgleichung (7.18) zu
βu2 = au +
qu2
2
für
|au | qu 1
(7.25)
genähert werden. Diese Näherung wird häufig bei Ionenfallen angewendet und adiabatische
Näherung genannt. Es genügt, die Koeffizienten c2s,u mit s = 0, ±1 zu betrachten, für die in
der Näherung (7.25) eine einfache Formel angegeben werden kann:
c2,u = c−2,u = −
qu
c0,u
4
.
(7.26)
Die allgemeine Lösung der Mathieu’schen Differentialgleichung lautet demnach:
u(t) = c0,u
p
A2
mit
+
B2
i
qu
cos Ωt cos
· 1−
2
h
ϕ = arctan
A2 − B 2
A2 + B 2
βu
Ωt − ϕ
2
(7.27)
.
(7.28)
Sie setzt sich aus zwei überlagerten Bewegungen zusammen:
1. der Makrobewegung, einer langsamen Schwingung mit der Frequenz ωu =
βu
2 Ω,
und
2. der Mikrobewegung,
einer schnellen Schwingung mit der Führungsfeldfrequenz Ω, deren
Amplitude 12 qu jedoch klein ist gegen die der ersten Bewegung.
7.2. DIE IDEALE LINEARE PAULFALLE
71
Aus der Harmonizität der Schwingung folgt der Ansatz, die Bewegung der Ionen über ein effektives statisches Pseudopotential darzustellen, wobei man ausgehend von den Mathieuschen
Differentialgleichungen (7.15) die Bewegung in einen Anteil niedrigerer und einen höherer
(getrieben durch das Führungsfeld) Frequenz aufteilt. Dadurch lassen sich die Gleichungen
lösen und man erhält die Bewegung eines harmonischen Oszillators mit den Säkularfrequenzen
r
r
Ω qu2
Ω qu2
− au und ωy =
+ au
(7.29)
ωx =
2
2
2
2
in einem Pseudopotential der Tiefe
1
eU
e2 V 2
Dx = mr0 ωx2 =
+
2
2
4mr02 Ω2
(7.30)
bzw. analog
Dy =
1
e2 V 2
eU
mr0 ωy2 =
.
−
2
2
2
2
4mr0 Ω
(7.31)
Der zweite Spezialfall nahe der Spitze des Stabilitätsdreiecks bei βx ≈ 1 und βy ≈ 0 (siehe Abb.
7.4(B)) ist vor allem für den Quadrupol-Massenfilter mit hohem Auflösungsvermögen m/∆m
relevant. Die stabilen und instabilen Ionenbewegungen, die für den hyperbolischen QuadrupolMassenfilter endlicher Ausdehnung resultieren, sollen anhand von Flugbahnsimulationen diskutiert werden. Abb. 7.6 zeigt typische aus diesem Spezialfall resultierende Flugbahnen eines
,,stabilen”3 Ions der Masse m2 (A), eines ,,instabilen”, zu leichten Ions der Masse m1 < m2
(B) sowie eines ,,instabilen”, zu schweren Ions der Masse m3 > m2 (C) in der (x, z)-Ebene
(oben) und (y, z)-Ebene (unten). Der Ioneneintritt in den Massenfilter erfolgte jeweils parallel
zur z-Achse. Man beachte die unterschiedlichen Achsenskalierungen für die einzelnen Projektionen. Die Frequenzen der jeweiligen Bewegung können Gl. (7.23) entnommen werden. Aus
der Gleichung für das Potential in der Nähe der z-Achse folgt, dass die Komponenten der elektrischen Feldstärke (7.5) in der x- und y-Richtung jeweils der Koordinate x bzw. y proportional
und unabhängig von der anderen Koordinate sind. Für positive Ionen wirkt das reine Gleichspannungsfeld nach Gl. (7.9) in der x-Richtung fokussierend, sie werden anfänglich zur Achse
beschleunigt (Abb. 7.6(A), oben). Die Ionen führen ohne ein Wechselfeld stabile harmonische
Schwingungen aus. Durch das Hinzufügen einer Wechselspannung V wird die Ionenbewegung
ähnlich der eines Schwingungssystems, das ganz in der Nähe seiner Resonanzfrequenz angeregt
wird [Brub1961]. Die Amplitude erreicht nach wenigen Schwingungen den bis zu 15fachen Anfangswert und klingt dann wieder ab; der Verlauf ist periodisch. Die Schwingungsfrequenz in der
x-Richtung: ω0,x = β2x Ω ≈ 12 Ω ist etwa halb so groß wie die angelegte Wechselspannungsfrequenz
Ω.
In der y-Richtung wirkt gemäß Gl. (7.10) das reine Gleichspannungsfeld auf positive Ionen
defokussierend (Abb. 7.6(A), unten). Für achsennahe Eintrittsorte erfahren die Ionen zu Beginn nach außen gerichtete, defokussierende Kräfte, die jedoch mit wachsender Auslenkung für
stabile Ionenflugbahnen durch die zunehmende Wirkung des Wechselfeldes rückläufig gemacht
werden. Das Ion erreicht wieder die z-Achse. Durch die Differenz der entgegengesetzt wirkenden Beschleunigung des Gleich- und Wechselfeldes wird die mittlere zeitliche Bahn des Ions
bestimmt. Die resultierende Beschleunigung ist proportional zur Auslenkung des Ions, d.h. die
Bewegung ist harmonisch. Wie für die x-Ionenbewegung beträgt auch hier die Maximalamplitude etwa das 15fache des Anfangswertes. Wegen βy ≈ 0 setzt sich die y-Ionenbewegung
3
Die Begriffe ,,stabile” bzw. ,,instabile” Ionen werden hier als vereinfachte Bezeichnungen für Ionen mit stabilen
(beschränkte Maximalamplitude) bzw. instabilen (unbeschränkte Maximalamplitude) Flugbahnen verwendet.
72
KAPITEL 7. QUADRUPOLMASSENSPEKTROMETRIE
x-Auslenkung [mm]
(A)
(B)
(C)
8
8
0.12
6
6
0.09
4
4
0.06
2
2
0.03
0
0
0.00
-2
-2
-0.03
-4
-4
-0.06
-6
-6
-8
-8
8
0.09
y-Auslenkung [mm]
6
z
-0.09
-0.12
8
6
0.06
4
4
2
0.03
0
0.00
0
-0.03
-2
2
-2
z
-4
-4
-0.06
-6
-6
-8
-0.09
m2
x, y stabil
-8
m1 < m2
x instabil, y stabil
m 3 > m2
x stabil, y instabil
Abb. 7.6: Flugbahn eines stabilen Ions der Masse m2 (A), eines instabilen, zu leichten Ions
der Masse m1 < m2 (B) sowie eines instabilen, zu schweren Ions der Masse m3 > m2 (C).
Darstellung jeweils in der (x, z)- und (y, z)-Ebene.
im Wechselfeld nach Gl. (7.23) und (7.27) aus einer Makroschwingung, mit der langsamen
β
Frequenz ω0,y = 2y Ω, und einer schnelleren etwa synchron mit der Frequenz
Ω der Wechβ
selspannung erfolgenden Mikroschwingung, mit der Frequenz ω1,y = 1 + 2y Ω, zusammen.
Zusätzlich treten die Frequenzen der höheren Entwicklungsterme mit s > 1 auf, jedoch mit
deutlich kleineren Amplituden; sie sind in Abb. 7.6 nicht sichtbar.
Zu leichte Ionen mit m1 < m2 führen aufgrund zu starker defokussierender Kräfte in der xRichtung eine instabile Bewegung aus (Abb. 7.6(B), oben), d.h. ihre Schwingungsamplitude
schaukelt sich in der (x, z)-Ebene exponentiell auf, bis sie von den Elektroden weggefangen
werden. In der (y, z)-Ebene verläuft die Schwingungsbewegung stabil (Abb. 7.6(B), unten).
Für zu schwere Ionen mit m3 > m2 ist der Effekt gerade umgekehrt (Abb. 7.6(C)). Entlang
einer Arbeitsgeraden sind Ionen mit (au , qu )-Parametern außerhalb des Stabilitätsdreiecks also
immer nur in einer Koordinate instabil, während die Ionenbewegung für die andere Koordinate
stabil verläuft.
7.3
Die reale lineare Paulfalle
Gegenüber der im vorangegangenen Abschnitt diskutierten idealisierten Paulfalle besitzt die
reale lineare Paulfalle, wie sie in Abb. 7.7 dargestellt ist, eine Reihe von Unvollkommenheiten,
die Abweichungen von der idealen Potentialverteilung hervorrufen und dadurch die Ionenbewegung beeinflussen. Darin eingeschlossen sind die endliche Dimensionierung, die Näherung
73
7.3. DIE REALE LINEARE PAULFALLE
Austrittsblende
L
Detektor
Massen separierter
Ionenstrahl
y
Linsen
z
Quadrupolfilterstäbe
−Φ0
Ionenquelle
R
x
r0
+Φ0
Abb. 7.7: Schematische Darstellung eines realen Quadrupol-Massenfilters bzw. linearen Paulfalle
mit runder Stabgeometrie. Die Einlage kennzeichnet die Polarität des Potentials Φ0 in Bezug
zu den kartesischen Koordinaten x und y, den freien Feldradius r0 und den Stabradius R.
hyperbolischer Äquipotentialflächen durch kreisrunde Stäbe als Elektroden sowie Herstellungsund Justagefehler. Es ist daher zwingend notwendig, auch bzw. gerade die reale lineare Paulfalle
bzw. den realen Massenfilter zu betrachten. Einige mögliche Elektrodenkonfigurationen für Radiofrequenzionenfallen können Abb. 7.8 entnommen werden.
Abb. 7.8: Mögliche Elektrodenkonfigurationen für Radiofrequenzionenfallen.
Im Folgenden werden zunächst die Abweichungen vom idealen Quadrupolpotential durch die Verwendung runder Stäbe endlicher Länge und deren Auswirkungen auf die Bewegungsgleichungen
und die maximal erreichbare Auflösung diskutiert. Anschließend wird auf die Beschränkung
der maximalen Schwingungsamplitude durch den endlichen Abstand der Elektroden eingegangen. Weitere Abweichungen wie z.B. Justagefehler der Stabelektroden, Potentialschwankungen,
Raumladungseffekte und Einwirkungen äußerer elektrischer und magnetischer Felder seien hier
zwar erwähnt, werden aber nicht im Detail betrachtet.
7.3.1
Potentialbeschreibung und Bewegungsgleichungen bei runder Stabgeometrie
Elektroden mit hyperbolischem Querschnitt zur Erzeugung eines echten Quadrupolpotentials,
wie es der Mathieu’schen Differentialgleichung (7.15) zugrunde liegt, werden erst seit wenigen
Jahren in Quadrupol-Massenfiltern eingesetzt. Auch die linearen Paulfallen sind zumeist aus
runden Stäben aufgebaut. Die resultierenden Abweichungen vom idealen Feldverlauf machen
74
KAPITEL 7. QUADRUPOLMASSENSPEKTROMETRIE
sich vor allem dann bemerkbar, wenn Resonanzeffekte auftreten, bei denen die Einwirkungen
kleiner Störungen sich zeitlich akkumulieren. Bei bestimmten (au , qu )-Werten innerhalb des Stabilitätsdreiecks kommt es zu instabilen Ionenbewegungen, die zu Transmissionseinbußen führen.
Diesen Effekt bezeichnet man als nichtlineare Resonanzen [Busc1961, Daws1969, Daws1995],
wobei die nichtlinearen Terme zu resonanter Anregung von Schwingungen mit großen bzw.
unbeschränkten Amplituden führen. Eine Vielzahl von Studien wurden dazu in der dreidimensionalen Ionenfalle durchgeführt, da die nichtlinearen Resonanzen zu einem rapiden Verlust gespeicherter Ionen führen können [Wang1994, Fran1995, Wert1996]. Die allgemeinen
Resonanzbedingungen für nichtlineare Terme in der linearen Paulfalle werden im Folgenden
hergeleitet.
Die Potentialverteilung in der (x, z)-Ebene eines Quadrupols bzw. einer linearen Paulfalle mit
runden Stäben kann als eine unendliche Multipolentwicklung ausgedrückt werden [Busc1961],
die ebenfalls die Laplace-Gleichung erfüllt. In Zylinderkoordinaten gilt:
Φ(r, t) = [U + V cos(ωt)] ·
∞
X
m=0
Cm
r
r0
m
cos(mθ)
(7.32)
mit θ = arctan(y/x). Terme mit r −m treten nicht auf, da die Bedingung Φ(r, t) = 0 auf der Achse
erfüllt sein muss. Die Entwicklungskoeffizienten Cm können als Funktion des Elektrodenradius
nummerisch ermittelt werden [Deni1971]. Im Falle einer perfekten vierfach-Symmetrie, bei der
alle vier Stäbe den identischen Radius R haben und präzise im Abstand r0 von der z-Achse
angebracht sind, dreht sich das Vorzeichen des Potentials um, wenn sich θ um ±π/2 ändert.
Demzufolge muss die Bedingung
π
= −1
(7.33)
cos m
2
erfüllt werden. Dies impliziert, dass der Ausdruck für das Potential (7.32) nur die Terme mit
m = 2, 6, 10, 14, ..., 2(2n + 1), ... annehmen kann. In allgemeiner Form folgt:
Φ(r, t) = [U + V cos(ωt)] ·
∞
P
Cn
n=0
r
r0
2(2n+1)
× cos [2 (2n + 1) θ] .
(7.34)
Der erste Term (n = 0) in dieser Multipolentwicklung ist der Quadrupolterm, der zweite (n = 1)
wird als Duodekapolterm, der dritte (n = 2) als 20-Pol-Term, usw. bezeichnet. Für den Spezialfall hyperbolischer Stäbe bleibt nur der erste Term übrig. Es resultiert die Mathieu’sche Differentialgleichung in der Normalform (7.15). Aufgrund der (r/r0 )-Abhängigkeit werden die
Terme höherer Ordnung erst in der Nähe der Quadrupolstäbe, d.h. im äußeren Feldbereich,
relevant und können zu einer messbaren Abweichung vom idealen Quadrupolpotential führen.
Für die Herleitung der Bewegungsgleichungen muss die Beziehung (7.34) differenziert werden.
In rechtwinkligen kartesischen Koordinaten lauten die Lösungen [Blau1998b]:
d2 x
dξ 2
= − [a + 2q cos(2ξ)] ·
∞
X
Cn (2n + 1) ×
∞
X
Cn (2n + 1) ×
n=0
x2 + y 2
r02
2n
× {x cos [2 (2n + 1) θ] + y sin [2 (2n + 1) θ]}
d2 y
dξ 2
= − [a + 2q cos(2ξ)] ·
n=0
x2 + y 2
r02
× {y cos [2 (2n + 1) θ] − x sin [2 (2n + 1) θ]}
(7.35)
2n
(7.36)
75
7.3. DIE REALE LINEARE PAULFALLE
mit ξ, a = ax = −ay und q = qx = −qy gemäß Gl. (7.12). Wichtig ist, dass der Quadrupolterm
(n = 0) beider Bewegungsgleichungen nur von einer einzigen Koordinate x oder y abhängt. Im
Gegensatz dazu sind die Bewegungsgleichungen in der x- und y-Richtung für die höheren Multipolterme der Feldentwicklung gekoppelt. Die beste Approximation an ein reines Quadrupolpotential erhält man durch Wahl des Elektrodenradius R derart, dass C1 = 0 ist und damit der
Beitrag des 12-Pol-Terms, der die stärkste Abweichung des realen vom idealen Quadrupolpotential beinhaltet, verschwindet. Dayton und Mitarbeiter [Dayt1954] bestimmten als erste
empirisch, dass sich das reale Quadrupolfeld bei einem Stab- zu freiem Feldradiusverhältnis
von R/r0 = 1.148 dem Hyperbelfeld am besten annähert. Diese Anpassung liefert das größte
Gebiet um die Achse, in dem der Feldstärkegradient konstant ist. In der Praxis werden die
Quadrupolstäbe in ein geerdetes Gehäuse eingebaut, welches nur geringfügig größer ist als der
Elektrodenaufbau selbst und somit zur Potentialverteilung beiträgt. Der Einfluss des Gehäuses
auf den optimalen Stabradius ist aufgrund der Radialsymmetrie gering, es vergrößert aber stark
den elektrischen Feldgradienten in der Nähe der 45◦ Ebenen zwischen den Stäben [Deni1971].
In einer späteren Arbeit [Lee1971] wird anhand von semianalytischen Rechnungen zur Potentialverteilung das ideale Verhältnis von Stab- zu Feldradius mit
R
= 1.14511
r0
=⇒
C1 = 0
(7.37)
angegeben. Mit diesem Wert, der bei einer Fertigungspräzision der Stäbe von etwa 10 − 50 µm
nur bis zur dritten Nachkommastelle relevant ist, resultiert als erster nicht verschwindender
Störterm in der Multipolentwicklung (7.34) der 20-Pol-Term mit n = 2. Dieser Beitrag sowie der
von Termen höherer Ordnung ist in der Nähe der z-Achse der linearen Paulfalle vernachlässigbar,
wird aber in der nahen Umgebung der Stäbe signifikant.
Eine Folge der Störterme höherer Ordnung ist das Auftreten von nichtlinearen Resonanzen,
im Englischen auch häufig ,,stop-bands” genannt. Resonanzen zwischen der Ionenbewegung und einem Multipolmoment gerader Ordnung N des Feldes findet man bei den x-,
y-Bewegungsfrequenzen, die der Beziehung
Ω0 = Ω = n
βy
βx
Ω + (N − n) Ω
2
2
für
n = 0, 2, 4, 6, ..., N
(7.38)
genügen. Wird die bei der Herleitung von Gl. (7.34) geforderte Spiegelsymmetrie gegenüber der
z = 0 Ebene gebrochen, so können grundsätzlich Multipolglieder beliebiger Ordnung auftreten.
Ungerade Ordnungen beschreiben dabei asymmetrische, gerade Ordnungen hingegen spiegelsymmetrische Abweichungen vom idealen Quadrupolfeld. Neben den Fehljustagen stellen Frequenzfehler der Wechselspannung eine weitere Quelle für Störterme dar, weil sie Oberwellen im
zeitlichen Feldverlauf hervorrufen. Diese Störglieder sind aber nur von untergeordneter Bedeutung. Die dominierenden Resonanzlinien niedrigster gerader und ungerader Multipolordnung
sind in Abb. 7.9 mit Angabe der zugehörigen Resonanzbedingung eingezeichnet. Fährt der Arbeitspunkt bei einem Massenscan entlang der Arbeitsgeraden über die entsprechenden Stellen
im Stabilitätsbereich hinweg, so sollte die Transmission durch den Massenfilter bzw. die lineare
Paulfalle an den markierten Stellen ein lokales Minimum aufweisen. Eine quantitative Aussage
über den Ionenverlust beim Durchlaufen einer nichtlinearen Resonanz kann an dieser Stelle nicht
gegeben werden.
7.3.2
Einschussbedingungen und maximales Auflösungsvermögen
Wie bereits erwähnt, entscheidet bei einem vorgegebenen Feld allein die spezifische Ladung
Ze/m eines Ions bzw. der Arbeitspunkt (au , qu ), ob die Teilchenbahn stabil oder instabil ist.
76
KAPITEL 7. QUADRUPOLMASSENSPEKTROMETRIE
aa/q
2U/V
u/qu==2U/V
Abb. 7.9: Die nichtlinearen Resonanzlinien 3. (strichpunktiert), 4. (punktiert), 6. (durchgezogen) und 10. (gestrichelt) Ordnung im Stabilitätsdiagramm des Massenfilters.
Anfangsort sowie Richtung und Größe der Anfangsgeschwindigkeit haben keinen Einfluss auf
die Stabilität. Die zusätzliche Einschränkung der Amplitude stabiler Bahnen durch die endliche
Ausdehnung des Elektrodensystems führt allerdings zu einer oberen Grenze für die Transversalgeschwindigkeit der Ionen in x- und y-Richtung sowie zu einer Begrenzung des Einschussbereiches der Ionen. So dürfen die maximalen Schwingungsamplituden xmax und ymax , die selbst
bei parallelem Einschuss zur z-Achse leicht eine Größenordnung über den Startwerten liegen,
nicht den freien Feldradius r0 (siehe Abb. 7.7) überschreiten. Die Maximalamplitude einer Ionenflugbahn ergibt sich aus der Bewegungsgleichung (7.17) zu:
|umax | =
p
A2
+
B2
·
∞
X
s=−∞
|c2s,u |
mit
u = x, y
.
(7.39)
Die Koeffizienten A (ξ0 , u0 , u̇0 ) und B (ξ0 , u0 , u̇0 ) sind durch die Anfangsbedingungen vorgegeben
und können aus dem Fundamentalsystem
u (ξ0 ) = A · uI (ξ0 ) + B · uII (ξ0 )
(7.40)
u̇ (ξ0 ) = A · u̇I (ξ0 ) + B · u̇II (ξ0 )
berechnet werden. Für den Betrag der maximalen Schwingungsamplitude folgt:
|umax | =
∞
X
1
·
|c2s,u | ·
− u̇I · uII s=−∞
uI · u̇II
q
[u0 · u̇II (ξ0 ) − u̇0 · uII (ξ0 )]2 + [u̇0 · uI (ξ0 ) − u0 · u̇I (ξ0 )]2
.
(7.41)
7.3. DIE REALE LINEARE PAULFALLE
77
Trägt man die zu einer festen Phase ξ0 gehörigen Orts/Geschwindigkeits-(u0 , u̇0 )-Paare in
ein Phasenraum-Diagramm ein, so erhält man eine Kollektion von Akzeptanzellipsen
[Daws1995, Mars1998]. In Abb. 7.10 sind einige ausgewählte Akzeptanzellipsen für au = 0
und βu = 0.2, 0.5, 0.8 dargestellt. Normiert auf den freien Feldradius umax = const. = r0 des
Abb. 7.10: Phasenraum-Akzeptanzellipsen für verschiedene Punkte βu im Stabilitätsdreieck.
Die Eintrittsphasen sind: (a) ξ0 = 0 (b) ξ0 = π/4 (c) ξ0 = π/2 (d) ξ0 = 3π/4.
Quadrupols beschreiben diese den kompletten Satz an Anfangsparametern, die zu einer Transmission des Ions bei der entsprechenden Phase des Wechselfeldes führen. Im Falle der ,,rf-only”
betriebenen linearen Paulfalle (U = 0) sind die x- und y-Ionenbewegungen und demzufolge auch
die resultierenden Akzeptanzellipsen identisch. Dies gilt nicht für au 6= 0 bzw. U 6= 0. Maximale
Transmission der erwünschten Masse wird nur dann erreicht, wenn das Phasenraumvolumen
des (im Ionisationsprozess) erzeugten Ionenensembles innerhalb der dunkelgrau schraffierten
Flächen liegt. Denn nur in diesem Bereich ist gewährleistet, dass die (umax , u̇max )-Paare für alle
Einschussphasen kleiner als die gegebenen Randbedingungen bleiben.
Eine weitere Limitierung realer Quadrupole bzw. linearer Paulfallen gegenüber dem idealisierten
Fall ist die endliche Länge der Stäbe. Um eine vollständige Massenselektion zu erreichen,
müssen alle Ionen falscher Masse eine genügend starke transversale Beschleunigung erfahren
und zudem lange genug im Feld verweilen. Wolfgang Paul veröffentlichte in seiner Arbeit von
1958 [Paul1958] eine einfache Beziehung zwischen dem maximalen Auflösungsvermögen
<max und der Anzahl an Hochfrequenzperioden N , die das Ion während des Durchquerens des
Massenfilters ausführt:
<max = m/∆m ≈ N 2 /12.25 .
(7.42)
∆m gibt die Halbwertsbreite (FWHM) des Massenpeaks m an. Diese Abschätzung ist gültig
für vernünftige Transmissionswerte und wurde in einer Reihe von weiteren Arbeiten bestätigt
(siehe z.B. [Daws1995, Blau1998b]). Sie basiert auf der Tatsache, dass bei hoher Auflösung die
Arbeitsgerade nahe der Spitze des Stabilitätsdreiecks verläuft. In diesem Fall erfolgt gemäß
Kap. 7.2.4 die Mikroschwingung des Ions in der
p y-Richtung nahezu synchron zur Hochfrequenz
ν des Wechselfeldes und es gilt: N = νt ≈ νL m/2E. Die Anzahl an RF-Zyklen kann demnach
durch Verringerung der axialen Geschwindigkeit des Ions, durch Erhöhung von ν und/oder
durch Vergrößerung der Stablänge des Massenfilters L erhöht werden. Bei einer vorgegebenen
Auflösung <max liefert dies eine obere Grenze für die maximal erlaubte axiale Ionenenergie Emax :
2
<max ≈ 0.0426L2cm νMHz
mamu /Emax,eV .
(7.43)
Für ein System mit L = 21 cm und ν = 2.9 MHz wäre beispielsweise zum Erreichen einer
Auflösung <max > 1000 bei einer Massenzahl m = 40 amu eine Ionenenergie Emax < 6 eV
78
KAPITEL 7. QUADRUPOLMASSENSPEKTROMETRIE
Voraussetzung. Wird der Massenfilter im Bereich niedriger Auflösung eingesetzt, so bestimmen
die experimentellen Parameter U und V die Auflösung und man kann näherungsweise Gl. (7.21)
des idealen unendlich langen Quadrupol-Massenfilters verwenden.
7.3.3
Transmissionspeakformen des Quadrupol-Massenfilters
In diesem Abschnitt sollen die Auswirkungen der vorgenannten Abweichungen auf die allgemeine
Form eines Massenpeaks beschrieben werden, wie es in Abb. 7.11 skizziert ist [Blau2000a]. Der
durch die Steigung der Arbeitsgeraden vorgegebene Bereich ∆qu , der zur stabilen Transmission führt, ist in Abb. 7.11(A) dargestellt. Der ideale Massenfilter sollte einen rechteckförmigen
Massenpeak liefern (Abb. 7.11(B)). Wie diskutiert, führt die endliche transversale Ausdehnung
des realen Quadrupols zu einem Verlust von Ionen auf an sich stabilen Bahnen, wenn deren
Startparameter nicht in der Akzeptanzellipse des Massenfilters liegen. Das Akzeptanzvolumen nimmt nahe der Grenzen des Stabilitätsdreiecks stark ab, und man erhält einen Massen-
(A)
Arbe
r
itsge
a de
(B)
(C)
(D)
Abb. 7.11: Entwicklung der realen Massenpeakformen. Gezeigt sind die Spitze des ersten Stabilitätsbereiches (A), die erwarteten Peakformen für ein ideales unendliches Feld (B), für ein
transversal limitiertes Feld (C) sowie für ein endliches Feld limitiert in transversaler und longitudinaler Dimension (D).
peak mit Rundungen. Dies rührt daher, dass Ionen mit (au , qu )-Werten nahe den Stabilitätsgrenzen deutlich größere Maximalamplituden besitzen als in der Mitte des stabilen Be-
7.4. NACHBARMASSENUNTERDRÜCKUNG
79
reiches und demzufolge ein Teil der Ionen trotz stabiler Flugbahnen aufgrund des limitierten
freien Feldradius r0 von den Stäben weggefangen werden. Wegen der unterschiedlichen x- und
y-Schwingungsbewegung (siehe Abb. 7.6) ist dieser Effekt zur größeren Massenseite hin ausgeprägter. Folglich wird der flache Transmissionspeak asymmetrisch rund mit einer leichten
Verschiebung des Peakmaximums zur kleineren Massenseite hin, wie in Abb. 7.11(C) verdeutlicht. Aufgrund seiner endlichen Länge besitzt ein realer Quadrupol ein zwar kleines, aber
endliches Akzeptanzvolumen für Ionen falscher Masse. Dies führt dazu, dass Ionen auf instabilen Flugbahnen den Quadrupol passieren können, wenn ihre Startparameter innerhalb dieser
Akzeptanzellipse liegen. Daraus resultieren gemäß Abb. 7.11(D) Ausläufer an den Massenpeaks,
die zur kleineren Massenseite hin stärker sind (wiederum aufgrund der unterschiedlichen x-yBewegungsklassen). Die Peakform wird darüberhinaus durch Randfelder am Massenfiltereintritt
und -austritt [Brub1968, Daws1971, Hunt1989, McIn1989] sowie durch die in Kap. 7.3.1 hergeleiteten nichtlinearen Resonanzen verändert. Letztere bewirken zusätzliche schmale Strukturen
auf den Massenpeaks. Die exakten Lagen der möglichen nichtlinearen Resonanzen können durch
die Multipolentwicklung der Quadrupol-Feldverteilung (7.34) bzw. durch Abb. 7.9 vorhergesagt
werden. Für die Massenanalyse sind insbesondere die Resonanzlinien, die durch die Spitze des
Stabilitätsdreiecks hindurchgehen, von größerer Bedeutung. Denn nur diese werden bei hohem
Massenauflösungsvermögen von der Arbeitsgeraden au /qu = 2U/V geschnitten. Sie führen zu
Unsicherheiten in der Transmission und somit zu Ionenverlusten.
7.4
Nachbarmassenunterdrückung und Isotopenverhältnismessungen
Zur Bestimmung der Nachbarmassenunterdrückung eines Quadrupolmassenspektrometers und
für Isotopenverhältnismessung wird häufig eine Elektronenstoßionenquelle eingesetzt, obwohl
deren Selektivität im Ionisationsprozess beschränkt ist. Bei den folgenden Messungen wurden
die zu ionisierenden Teilchen gezielt mit Hilfe einer Atomstrahlquelle erzeugt. Dazu gehörten u.a.
Lithium, Calcium und Gadolinium. Die Messungen zeigten, dass eine nahezu selektive Ionisation
bei Vermeidung der meisten molekularen Interferenzen erreicht werden kann, wenn die Elektronenstoßenergie zwischen 8 und 12 eV gewählt wird. Aufgrund der niedrigen Ionisationspotentiale
der genannten Elemente Li, Ca und Gd (IPLi = 5.39 eV, IPCa = 6.11 eV, IPGd = 6.15 eV) findet
trotz der niedrigen Elektronenenergie noch eine effiziente Ionisation statt, während die Ionisation
der molekularen Spezies um mehrere Größenordungen unterdrückt wird.
Unter diesen optimierten Bedingungen wurden Massenspektren mit hohem dynamischem Bereich gemessen und die beobachtete Linienform sowie die ermittelten Selektivitätswerte mit den
Berechnungen über Akzeptanzellipsenflächen verglichen. Die aus mehreren Einzelscans resultierenden Summenspektren für Ionenmassen im Bereich von Lithium (A), Calcium (B) und
Gadolinium (C) sind in normierter logarithmischer Darstellung in Abb. 7.12 gezeigt. Die experimentellen Punkte wurden mit konventioneller Elektronenstoßionisation (EI) aufgenommen
und mit den simulierten Peakformen unter Berücksichtigung des Stoßuntergrundes verglichen.
Das Massenspektrometer wurde im Modus konstanter Peakbreite ∆m10% betrieben. Um in
allen Fällen einen guten Kompromiss zwischen Transmission und Nachbarmassenunterdrückung
zu erreichen, wurden für die verschiedenen Elemente unterschiedliche Peakbreiten gewählt. Für
Li und Gd betrug ∆m10% ∼ 0.65 amu und für Ca ∆m10% ∼ 0.4 amu. Die Auflösungswerte
sind demzufolge < ≈ 11, 100 und 250 bei A = 7, 40 und 158. Im Falle des Calciums kann
die Ionisation von Kalium aufgrund des geringen Ionisationspotentials (IPK = 4.34 eV) nicht unterdrückt werden. Die Peaks bei den Massenzahlen 39 und 41 werden daher auf die Isotope
39,41 K zurückgeführt. Die Frequenz des Wechselfeldes betrug in (A) und (B) 2.9 MHz. Zum Er-
80
KAPITEL 7. QUADRUPOLMASSENSPEKTROMETRIE
0
10
(A)
(B)
(C)
-1
Norm.
[AU]
Norm. Intensity
Intensität [AU]
10
-2
10
-3
10
-4
10
-5
10
-6
10
-7
10
-8
10
6
7
39
40
41
157
158
Mass[amu]
[u]
Masse
Abb. 7.12: Massenspektren mit großem dynamischem Bereich für Lithium (A), Calcium (B)
und Gadolinium (C). Die experimentellen Ergebnisse (Punkte), welche durch selektive Elektronenstoßionisation aufgenommen wurden, werden mit den über die Akzeptanzflächenmethode
simulierten Peakfomen (Linie) verglichen. Nähere Erläuterungen dazu im Text.
reichen des Massenbereiches von Gd musste die Hochfrequenz auf ν = 1.2 MHz reduziert werden
(C). Dies führt u.a. dazu, dass die Ausläufer des Gd-Massenpeaks nicht so schnell abfallen wie
es für Li und Ca der Fall ist.
Bei den zugehörigen Simulationen der Akzeptanzellipsenflächen wurde die Ionenenergie mit 9 eV
für Li und Ca und mit 6 eV für Gd bei einer Verteilung von ∆E = 1.3 eV angenommen. Das
Verhältnis U/V wurde so lange variiert, bis die simulierte Peakbreite ∆m10% mit der experimentell beobachteten übereinstimmte. Die resultierenden Transmissionskurven für A = 7, 40
und 158 sind in Abb. 7.12 als durchgezogene Linien dargestellt. Die Übereinstimmung mit den
experimentell beobachteten Peaks ist über acht Größenordnungen hinweg ausgezeichnet. Vor
allem die korrekte Beschreibung der Peakflanken ist für den Einsatz des QMS in der Spurenanalyse wichtig, denn sie verdeutlicht, dass die Simulationsrechnungen zuverlässige a priori
Abschätzungen für die Nachbarmassenunterdrückung geben können. Sowohl Experiment als
auch Theorie bestätigen die Vorhersage, dass die Peakform asymmetrisch mit einer leichten
Verschiebung des Maximums zur kleineren Massenseite hin ist. Bei geringeren Peakbreiten,
wie im Falle von Ca, wird dieser Effekt noch stärker betont. Die Bestimmung der Peakintensitätsverhältnisse für verschiedene Isotope eines Elements erlaubt die sehr präzise Bestimmung
von Isotopenverhältnissen und damit den Einsatz eines Quadrupolmassenfilters u.a. in der
analytischen Chemie und der Spurenanalyse.
Zur unabhängigen Bestimmung des Untergrundes bzw. des Überlapps zweier Massenpeaks wurde
das QMS jeweils einige hundert Sekunden lang auf die Massenzahl A − 1/2, A und A + 1/2 stabil
eingestellt. Die daraus resultierenden Abschätzungen für die Nachbarmassenunterdrückung,
d.h. die mittlere Selektivität des QMS für ein Isotop mit der Massenzahl A gegenüber den
81
7.4. NACHBARMASSENUNTERDRÜCKUNG
Tabelle 7.1: Gemessene Nachbarmassenunterdrückung S auf der halben Massenposition zur
kleineren und größeren Massenseite hin für drei unterschiedliche Massenbereiche. Die mit ?
markierten Werte sind durch Untergrund limitiert.
Element
Masse [amu]
Lithium
6-7
Calcium
40 - 48
Gadolinium
152 - 160
S = Nstabil,A /NA+1/2
S = Nstabil,A /NA−1/2
1.6 (4) · 108 ?
5.2 (8) · 107
2.1 (4) · 105
5.3 (7) · 104
1.1 (3) · 108 ?
3.8 (5) · 107
Nachbarisotopen, sind in Tab. 7.1 angegeben. Die bei einem Restgasdruck von p = 5 · 10−7 mbar
erreichten Nachbarmassenunterdrückungen liegen im Bereich von 108 für Li und Ca sowie im
Bereich von 105 für Gd. Der vorhergesagte steilere Abfall der Peakflanken zu höheren Massen
hin konnte experimentell bestätigt werden.
Kapitel 8
Hochselektive Verfahren der
Massenspektrometrie
Ihrem Namen entsprechend führt Massenspektrometrie generell eine Trennung nach unterschiedlichen Atom- bzw. Molekülmassen, präziser ausgedrückt, nach Ionenmassen durch. Eine
Trennung von atomaren bzw. molekularen Isobaren über die Dispersion ist mittels konventioneller massenspektrometrischer Techniken nur dann möglich, wenn eine hinreichende Massendifferenz zwischen diesen vorliegt. Dabei liegen für die meisten Isotope mit geraden Massen im
Bereich von 36 ≤ A ≤ 200 atomare Isobare vor, während im gesamten Massenbereich speziell
bei analytischen Anwendungen auch molekulare Isobare zu berücksichtigen sind. Betrachten wir
eine vergleichsweise einfache aber typische Massenregion, diejenige von A = 14, gemäß Tabelle
8.1:
Isotope Moleküle
Masse (MeV/c2 )
∆ Masse (MeV/c2 )
14 C
m/∆ m
13044.0422
14 N
12 CH
2
13043.8861
0.1561
83562
13055.6004
11.5582
1129
13 CH
13051.4364
7.3942
1766
Tabelle 8.1: Atomare und molekulare Isobare bei 14 u
Wie in allen Fällen entlang der Nuklidkarte ist die Massendifferenz zwischen den atomaren Isobaren 14 C und 14 N minimal. Man würde eine Massenauflösung von < > 100000 benötigt, um
diese aufzulösen. Molekulare Isobare haben üblicherweise eine deutlich höhere Masse, sind also
im Prinzip auch konventionell abtrennbar. Dabei ist aber außer der Auflösung auch die Nachbarmassenunterdrückung (siehe Kapitel 1) von Bedeutung, da natürlich die Flanke des benachbarten
Peaks ausreichend weit unterdrückt sein muss, um eine präzise Aussage über eine Häufigkeit oder
ein Isotopenverhältnis zu geben. Dies ist besonders dann der Fall, wenn extrem seltene Isotope
untersucht werden sollen (siehe unten). Bei in der Größenordnung ähnlichen Häufigkeiten kann
hingegen eine sehr hohe Selektion über höchste Auflösung sowohl durch magnetische SektorfeldMassenspektrometer, als auch in Ionenfallen erzielt werden. Ein entsprechendes Beispiel zeigt
Abb. 8.1. Dort wurde mit dem großen Sektorfeld-Massenspektrometer von Mattauch und Herzog im MPI für Chemie in Mainz eine Auflösung von < = 196000 demonstriert und damit
molekulare Isobare auf der Masse 119 aufgelöst.
82
8.1. VORKOMMEN UND BESTIMMUNG VON ULTRASPURENISOTOPEN
83
Abb. 8.1: Auflösung des molekularen Massendubletts bei 119 u.
Die Massenzahlen der Moleküle kann man dabei in recht guter Näherung durch Summation
der einzelnen beteiligten Isotopenmassen erhalten. Abweichungen aufgrund der molekularen
Bindung liegen etwa im Bereich von 10−6 und darunter. Die Massen von Abb. 8.1 liegen bei
119.08305 u bzw. 119.08767 u und damit weit oberhalb derjenigen des einzigen stabilen Isotops
auf der Masse, 119 Sn, mit 118.9034 u.
8.1
Vorkommen und Bestimmung von Ultraspurenisotopen
In der Natur gibt es eine ganze Reihe von sehr seltenen Isotopen. Dies sind im Wesentlichen
langlebige Radioisotope, die durch verschiedene Entstehungsmechanismen gebildet werden und
dann in einer Gleichgewichtshäufigkeit zwischen Erzeugung und Zerfall vorliegen. Typische
Häufigkeiten der Bedeutsamsten dieser Isotope in der Erdkruste (die Werte variieren stark im
inhomogenen Erdinneren) sind in Abbildung 8.2 dargestellt, wobei ihre relative Häufigkeit zu
dem häufigsten stabilen Isotop des gleichen Elements angegeben ist. Lebensdauern und Zerfallsarten langlebiger Isotope sind in Abbildung 8.3 angegeben.
Zusätzlich zu diesen natürlichen Radioisotopen treten noch eine ganze Reihe von anthropogenen
Isotopen auf, die vornehmlich durch friedlichen bzw. militärischen Einsatz der Kernenergie
freigesetzt wurden. Das Interesse an der Ultraspurenbestimmung seltenster Isotope natürlichen
und künstlichen Ursprungs ist sehr vielfältig. Viele dieser Spezies stellen empfindliche
Proben für geochemische und umweltrelevante Untersuchungen der Atmosphäre, Hydrosphäre,
Lithosphäre bis hin zur Technosphäre dar [Kuts00]. Eine Unmenge an Informationen steckt
in ihrer Produktionsmechanismen, den Transportvorgängen und ihren Zerfallskanälen, die
zusammen für ihre Seltenheit auf der Erde verantwortlich sind. Weiterführende Aspekte betreffen kosmochemische und astrophysikalische Untersuchungen an extraterrestrischem Material,
sowie Studien aus den Bereichen der Kern- und Teilchenphysik. Mit künstlich angereicherten
Proben können darüber hinaus vielfältige Untersuchungen im ernährungswissenschaftlichen
und bio-medizinischen Bereich sowie in den Materialwissenschaften durchgeführt werden.
Aufgrund der extremen Seltenheit sind neben der möglichst vollständigen Unterdrückung
84
KAPITEL 8. HOCHSELEKTIVE VERFAHREN DER MASSENSPEKTROMETRIE
molekularer Isobarenkontaminationen auf der gesuchten Masse zusätzlich höchste Spezifikationen bezüglich der Isotopenselektion, also der Nachbarmassenunterdrückung, und
auch gute Nachweiseffizienz erforderlich.
In diesem Bereich haben sich aktuell zwei
massenspektrometrische Verfahren etabliert, die im Folgenden vorgestellt werden sollen:
Beschleuniger-Massenspektrometrie (AMS = Accelerator-MS) [Tuni98] und hochauflösende
Laser-Resonanzionisations-Massenspektrometrie (RIMS) [Leto87, Hurs88], speziell in der
Ausprägung als spektral hochauflösendes Verfahren (HR-RIMS) [Payn94, Wend02, Wend05].
Komplementär wird gerade ein sehr konkurrenzfähiges quantenoptisches Verfahren entwickelt,
die selektive Atomzählung in einer MOT (ATTA = Atom Trap Trace Analysis) [Chen99, Lu02].
Einen detaillierten Vergleich dieser drei Verfahren gibt [Luwe03].
Abb. 8.2: Relative Häufigkeiten seltener Isotope in der Erdkruste
8.1. VORKOMMEN UND BESTIMMUNG VON ULTRASPURENISOTOPEN
85
Abb. 8.3: Lebensdauern und Zerfallsarten langlebiger Radioisotope, • = β − -Zerfall, ◦ = β + Zerfall, = α-Zerfall, die Marken a, b und c geben das Alter des Universums, des Sonnensystems
und der Erde an.
Kapitel 9
Beschleunigermassenspektrometrie
Die Beschleuniger-Massenspektrometrie kombiniert gleich eine ganze Reihe von selektiven
Prozessen und kann damit aktuell die höchste Unterdrückung der Beiträge von Isobarenkontaminationen und Nachbarmassen erreichen. Die einzelnen Prozessschritte setzen sich dabei
jeweils aus ganz konventionellen Techniken zusammen, die teilweise bereits behandelt wurden.
Diese sind
1. Selektive Ionisation für ausgewählte Spezies durch Wahl von negativer Ionisation in einer
Sputterionenquelle,
2. Erste konventionelle Massenfilterung in einem magnetischen Sektorfeld (Niederenergieseite),
3. Beschleunigung in einem Tandembeschleuniger auf Energien im Bereich von einigen hundert kVolt bis wenige 10 MVolt,
4. Umladung der negativen Ionen im Beschleunigerterminal durch Elektronenstripping in
einem Gas- oder Folientarget in einen Ladungszustand von 1+ bis etwa 7+, je nach Energie,
5. Energiefilterung und zweite konventionelle Massenfilterung in einem magnetischen Sektorfeld (Hochenergieseite),
6. ggf. Flugzeitmessung zur weiteren Massenselektion und Untergrundunterdrückung
7. ggf. Verwendung eines gasgefüllten Sektorfeldmagneten zur Kompression mehrerer
Ladungszustände zur weiteren Selektivitätssteigerung,
8. Element- und massenselektiver Teilchennachweis über ein kernphysikalisches ∆ E/E- Detektorteleskop.
Die wichtigsten Prozessschritte sind in Abb.9.1 entlang einer typischen mittelgroßen
Beschleuniger-Massenspektrometrieanlage aufgezeigt. Das eingebundene Photo zeigt den
Einbau einer Charge von Sputtertargets in die Quelle, die dann automatisiert nacheinander
durchgemessen werden. Zur Kalibrierung der Isotopenverhältnismessung und zur Bestimmung des Untergrundslevels und möglicher Kontaminationen werden dabei regelmäßig auch
entsprechende Kalibrationsproben und Blanks zwischen gemessen.
86
87
Der Plot an der Stelle des Detektors verdeutlicht die typische zweidimensionale Detektorauslesung, in der über die Auftragung von Energieverlust zu Gesamtenergie eine Zuordnung von
Ladungszahl und Masse für jedes Ereignis möglich wird.
Abb. 9.1: Prinzipielle Selektionsschritte in einer mittelgroßen AMS Anlage
Betrachten wir nun die einzelnen Prozessschritte:
1. Selektive negative Ionisation in einer Sputterionenquelle
Das Prinzip der Sputterionisation beruht auf der Beschleunigung positiv geladener Ionen
von einer heißen Oberfläche (oder einer eigenständigen Ionenquelle für den Primärionenstrahl)
auf das sogenannte Sputtertarget, das die zu untersuchende Probe enthält. Man verwendet
häufig Cäsium, da es eine sehr gute Oberflächenionisierbarkeit mit Effizienzen von nahe 100%
hat und dank seiner recht hohen Masse gute Sputterausbeuten liefert. Beschleunigungsspannungen der Primärionen liegen dabei typisch bei einigen kVolt. Ein Cs-Ion schlägt dann
aufgrund seiner hohen Energie gleich einige und sehr unterschiedliche Targetspezies heraus. Es
werden sowohl neutrale, einfach sowie mehrfach, positiv als auch negativ geladene Fragmente in
Form von Atomen, Ionen, Molekülen und Clustern erzeugt. Nur die einfach negativ geladenen
Atome oder Moleküle sind für die AMS von Interesse. Da dieser Bruchteil nicht sonderlich
hoch ist, müssen starke Cs+ -Ionenstrahlen zum Einsatz kommen. Daher werden in der AMS
üblicherweise großflächige Ionisatoren für das Cs eingesetzt und nicht gerichtete, gut fokussierte
Primärionenstrahlen wie bei ortsauflösender Sekundärionen-Massenspektrometrie (siehe später).
Der große Vorteil für die AMS liegt in der Tatsache begründet, dass viele Spezies keine negativen
Ionen bilden. So hat die Abwesenheit eines negativen Ions des Stickstoffs als Isobar zu C14
zur Etablierung der Radiokarbondatierung per AMS geführt. Dafür hat Libby den Nobelpreis
gewonnen. Andererseits wird die Anzahl der mit AMS zugänglichen Elemente dadurch stark
88
KAPITEL 9. BESCHLEUNIGERMASSENSPEKTROMETRIE
eingeschränkt. Routinemäßig sind dies heute weltweit nur etwa 8 bis 10 Isotope, wobei diese
in vielen Fällen als negative Molekülionen gebildet werden, z.B. CaF oder CaH. Dies reduziert
die Ionisationseffizienz und setzt eine aufwändige und für Kontaminationen anfällige chemische
Probenpräparation voraus Ein Diagramm einer typischen Quelle zeigt Abbildung 9.2
Abb. 9.2: Prinzip einer Cäsiumsputterquelle für hohe Ströme negativer Ionen
2. Konventionelles Sektorfeldmassenspektrometer auf der Niederenergieseite
Hier werden alle primär gebildeten unerwünschten Massen unterdrückt. Bei einer Beschleunigungsspannung im Bereich von üblicherweise 30 bis 60 kV werden für das magnetische
Sektorfeld typisch Werte von 5 bis 6 Größenordnungen Unterdrückung erreicht. Begrenzt
wird dies bei großen Massendifferenzen zwischen gesuchtem Isotop und Kontamination durch
den Restgasdruck über gaskinetische Stöße, bei Nachbarmassen auch durch die verwendete
Schlitzbreite. Als Besonderheit wird bei AMS-Anlagen oftmals eine schnelle Potentialänderung
beim Magnetdurchgang verwendet: zur Kalibrierung der Resultate muss schnell und häufig zur
Isotopenverhältnismessung zwischen unterschiedlichen Massen hin- und hergeschaltet werden.
Die Umschaltung des Primärmagneten von Masse A des gesuchten Isotops zu Masse B des
Referenzisotops wäre langsam und mit einer hohen Hysterese des Magnetfeldes verbunden.
Eine Änderung der Beschleunigungsspannung über eine Nachbeschleunigung vor Eintritt
und entsprechende Abbremsung nach dem Austritt ist viel günstiger. Es wird dazu nur die
Magnetkammer auf das notwendige Zusatzpotential im Bereich von kV gelegt und Ein- und
Austritt geeignet ionenoptisch ausgelegt. Das Umschalten der Isotope kann dadurch nahezu
beliebig schnell erfolgen und die Ionenoptik muss nicht mit verändert werden.
3. Beschleunigung auf hohe Energie
Dieser Schritt hat mehrere Vorteile: durch die hohe Energie der Ionen können im 2.
Teil der Anlage kernphysikalische Detektionsverfahren zum Einsatz kommen, die eine genaue
Zuordnung von Ladungszahl Z und Massenzahl A zulassen und daher eine vollständige
Identifikation jedes Ereignisses erlauben. Zusätzlich erfolgt in der Mitte des üblicherweise
verwendeten Tandembeschleunigers, im Terminal, eine Umladung, damit das Potential doppelt
ausgenutzt werden kann.
89
4. Umladung
Auf dem hohen Potential des Beschleunigerterminals ist eine Gasstrecke oder eine dünne
Folie angebracht, die der Ionenstrahl passieren muss. Es werden dabei mehrere Außenelektronen abgestreift und die negativen Ionen in positive verwandelt. Dabei wird auch ein vollständiger
Molekülaufbruch erzielt und alle molekularen Isobareninterferenzen um viele Größenordnungen
unterdrückt. Danach durchläuft der nun (mehrfach) positiv geladene Ionenstrahl das gleiche
Potential diesmal umgekehrt und wird entsprechend seiner Ladungszahl um ein Vielfaches der
Terminalspannung beschleunigt. Ein 5 MV Tandem erreicht so Strahlenergien von 25 MeV,
falls der Ladungszustand 4+ extrahiert wird.
Eine Prinzipskizze der Wirkungsweise des Gasstrippers für das Beispiel des
wobei alle isobaren Kohlenwasserstoffe gecrackt werden.
14 C
gibt Abb. 9.3,
Abb. 9.3: Unterdrückung von molekularen Isobareninterferenzen im Gasstripper
Beim Strippingprozess wird je nach der Energie des Beschleunigers ein entsprechendes Band
von positiven Ladungszuständen erzeugt. Dies zeigt Abbildung 9.4 für unterschiedliche
Terminalenergien des Beschleunigers im typischen Arbeitsbereich von 400 keV bis 7 MeV.
Abb. 9.4: Erzeugung verschiedener Ladungszustände in einer kleinen ( 400 kV) und einer konventionellen AMS Anlage (Terminalspannung 3 - 7 MV)
Der jeweils mit höchster Effizienz erzeugte Zustand kann durch ein einfaches Kriterium, das
Bohr bereits 1948 aufstellte, abgeschätzt werden. Bohr sagte voraus, dass Elektronen, deren
Orbitalgeschwindigkeit kleiner als die Ionengeschwindigkeit relativ zu den ruhenden Target-
90
KAPITEL 9. BESCHLEUNIGERMASSENSPEKTROMETRIE
spezies ist, durch Kollision verloren gehen. Dieses einfache sogenannte Bohr’sche Kriterium
führt zu einem mittleren Ladungszustand von
r
p
E/2
·
m
E·Z
1
v
ion
1/3
1/3
∼
·Z
≈
·Z
≈
·
(9.1)
q̄ =
v0
v0
v0
2·m
2
e
die Bohr’sche Geschwindigkeit des im Atom umlaufenden Elektrons
wobei v0 = α · c = 4π·ε
0~
ist. Die Hochzahl wurde später empirisch zu 0.45 korrigiert. Der Ladungszustand ist also
in einfacher nichtrelativistischer Näherung proportional der Wurzel der Energie sowie der
Kernladung des Projektils und umgekehrt proportional zu derjenigen seiner Masse. Es ist also
verständlich, dass bei höherer Beschleunigerenergie in höheren Ladungszuständen gearbeitet
werden kann, wodurch die Selektivität des Verfahrens wächst, da dort die Unterdrückung
jeglichen Isobarenuntergrunds besser ist.
5. Elektrische Energie- und mag. Massenselektion auf der Hochenergieseite
Durch die gaskinetischen Stöße im Strippingprozess leidet natürlich die Strahlqualität.
Deren Wirkungsquerschnitt ist zwar um viele Größenordnungen kleiner als derjenige des
Stripping, aber trotzdem werden einige Ionen von falscher Energie und Winkel erzeugt. Erstere
werden über eine Energieselektion in einem elektrischen Sektorfeld herausgefiltert, die zweite
Massenselektion unterdrückt Molekülfragmente falscher Masse, die zufällig gerade auf gleicher
Energie erzeugt wurden.
6. Flugzeitmassenmessung (optional)
Besonders im Bereich hoher Massen installiert man häufig eine zusätzliche Flugzeitstrecke
von einigen Metern Länge zur weiteren Erhöhung der Selektivität. Aufgrund der hohen
Projektilenergie kann man als Start- und Stoppdetektoren einfach dünne Aluminiumfolien in
den Strahlengang stellen, wie in Abb. 9.5 dargestellt, ohne die Strahlqualität nennenswert
zu verringern.
Aus diesen Folien werden durch jedes transmittierende Ion Elektronen
herausgeschlagen, die man mit einem Channeltrondetektor mit hoher Zeitauflösung im
Nanosekunden-Bereich quantitativ nachweisen kann.
Abb. 9.5: Startdetektor für eine Flugzeitmessung in der AMS, das Stoppsignal generiert der
weiter unten diskutierte kernphysikalische Detektor
91
7. Kompression der Ladungszustände im gasgefüllten Magneten (optional)
Die vormals unerwünschten gaskinetischen Stöße im Restgas kann man zur weiteren Unterdrückung von Isobaren nutzen. In einem Sektorfeldmagneten, der typisch mit wenigen Torr
Gasdruck geflutet ist, kommt es zu einer Vielzahl von Stößen zwischen den schnellen Ionen und
den Gasatomen. Dabei nehmen hohe (positive) Ladungszustände vornehmlich Elektronen auf,
niedrigere geben welche ab. Insgesamt stellt sich wieder ein mittlerer Ladungszustand ein, der
durch die empirische Beziehung
ν
· Z 0.45
(9.2)
q̄ ∼
=
ν0
gegeben ist. Dieser ist aufgrund höherer Dichte des Gases als im Stripper viel schmaler als
dort. Für Projektile unterschiedlicher Ladungszahl ergibt sich demnach eine unterschiedliche
Ablenkung, die über die magnetische Steifigkeit der jeweiligen Teilchensorte beschrieben wird
m·ν
ν0
B·ρ ∼
∼ 0.45
=
q̄ · e
Z
(9.3)
Es wird damit eine Kompression der Ladungszustände in einen einzelnen q̄ erzielt und dadurch
im Magneten eine weitere Selektion erreicht. Aufgrund hoher Transmissionsverluste von bis
zu einem Faktor 10 bleibt aber ein erwarteter Effizienzgewinn aus. Die Abhängigkeit der
Ausbildung des mittleren Ladungszustands vom Gasdruck zeigt Abb. 13.6.
Abb. 9.6: Besetzung der Ladungszustände in Abhängigkeit des Gasdrucks im gasgefüllten Magneten
8. Element- und massenselektiver Ionennachweis
Die Verwendung eines Detektorteleskops, das gleichzeitig den spezifischen Energieverlust
pro Strecke als auch die Gesamtenergie misst, ermöglicht eine zweidimensionale Auftragung
K, 41 Ca jedes einzelnen Zählereignisses. Nach der Bethe-Bloch-Formel ist der spezifische
92
KAPITEL 9. BESCHLEUNIGERMASSENSPEKTROMETRIE
Energieverlust gegeben durch
dE
k · Z2
≈
(9.4)
dx
v2
wobei der Vorfaktor alle Eigenschaften des abbremsenden Mediums im Detektor enthält. Daraus
lässt sich ableiten, dass die Flugstrecke im Detektor bis zum Stillstand nach dem Reichweitegesetz von Geiger näherungsweise gegeben ist durch
r≈k·
W02
Z2 · M
(9.5)
Alternativ zum Energieverlust kann daher auch die Restenergie nach Abbremsung in einem
Großteil des Detektors, die sogenannte ”Residual Energy”, zur Bestimmung der Ordnungszahl
Z des Projektils verwendet werden. Die gesamte deponierte Energie hingegen ist proportional
der Masse M des Teilchens.
Ein typischer Detektor für die AMS ist eine Ionisationskammer in der eine der Elektroden, etwa die positiv geladene, längs des Flugwegs segmentiert ist, wie in Abb. 9.7(a)
dargestellt. An dieser können über die akkumulierten Elektronen längs des Abbremsweges
sowohl Signale für den spezifischen Energieverlust als auch für die Restenergie abgegriffen
werden. Die zweite durchgehende, hier negative Elektrode misst die beim Abbremsen erzeugten
positiven Ionen und darüber die Gesamtenergie. Alternativ kommen heute auch Halbleiterzähler
mit hoher Energieauflösung sowie auch im Testbetrieb bolometrische Kryodetektoren zum
Einsatz.
Abb. 9.7: (a) Ionisationskammer mit einseitig segmentierter Elektrode zur gleichzeitigen ∆E,
ERes und E Messung ; (b) Vergleich des spezifischen Energieverlustes für die Isobaren 41 K, 41 Ca
Aus den Messgrößen kann man immer einen zweidimensionalen Plot erzeugen, der eine Auftrennung nach Z und M zulässt. Erschwert wird dies aber durch das Auftreten der unterschiedlichen
Ladungszustände im Detektor. Dies ist in dem Messergebnis der Abb. 9.8, die einen typischen
zweidimensionalen Scatterplot für schwere Element zeigt, deutlich.
93
Abb. 9.8: Zweidimensionaler Scatterplot mit mehreren Ladungszuständen, vertikal Gesamtenergie, horizontal spezifischer Energieverlust
Aufgrund der umfangreichen Experimentiertechnik, zu der zusätzlich eine sehr aufwändige
chemische Probenvorbereitung kommt, die wir hier nicht besprochen haben, gibt es nur etwa
50 AMS-Anlagen weltweit. Führende Anlagen in Europa stehen an der ETH Zürich sowie im
VERA (Vienna Environmental Research Accelerator) in Wien [Kuts00]. Ein Grundplan des
Züricher Labors ist in Abb. 9.9 gegeben [Sute04]. Neben dem großen 4 MeV Tandem, der über
mehrere Beamlines von einigen 10 m Länge verfügt, sind hier auch die richtungsweisenden und
in Zürich entwickelten Kleinbeschleunigeranlagen Tandy und Micadas eingezeichnet, mit denen
die AMS sich in Richtung table-top Apparatur zu entwickeln sucht [Sute98, Sute04]. Beide
haben bereits erfolgreiche Demonstrationen bzgl. Radiokarbondatierung mit nur unwesentlich
eingeschränkten Spezifikationen in Vergleich zur großen Maschine gezeigt.
Insgesamt ist die mit der AMS nachweisbare Anzahl von Ultraspurenisotopen recht begrenzt. Die sieben Standardisotope, die an den meisten AMS Anlagen weltweit routinemäßig
untersucht werden, sind am Ende des Kapitels in Abb. 9.10 mit einer Nennung der typischen
Anwendungsgebiete und ihrer Erzeugungsmechanismen aus der Höhenstrahlung angegeben. In
den meisten Fällen können mit typischen mittelgroßen AMS Maschinen (∼ 5 MV) Isotopenverhältnisse von 10−13 bis maximal hinunter zu 10−15 erreicht.
Eine detaillierte Aufstellung der Anwendungsgebiete der AMS und der dazu verwendeten
Radioisotope gibt Tabelle 9.1 am Ende des Kapitels [Kuts00].
94
KAPITEL 9. BESCHLEUNIGERMASSENSPEKTROMETRIE
Abb. 9.9: Grundplan der Züricher AMS-Anlage mit der großen Beschleunigeranlage (4 MV)
sowie den Kleinbeschleunigern Tandy (400 kV Terminalspannung) und Micadas (100 kV)
Abb. 9.10: Standardisotope der AMS mit typischen Anwendungsgebieten vor einem Diagramm
ihrer Entstehung durch Höhenstrahlung
95
Tabelle 9.1: Anwendungen von Ultraspurenisotopen
Kapitel 10
ResonanzionisationsMassenspektrometrie
Als alternativer Ansatz zur Beschleunigermassenspektrometrie für die Bestimmung von Ultraspurenisotopen verwendet die Resonanzionisations-Massenspektrometrie (RIMS) die Selektivität
der resonanten optischen Anregung zur Unterdrückung von Isobaren und zur weiteren Reduktion
der Beiträge von Nachbarisotopen [Herr94]. Dabei kommt keine Hintereinander- und Ineinanderschachtelung verschiedener selektiver Prozesse zum Einsatz, sondern es wird ausschließlich
die Ionisation an einem ansonsten konventionellen Massenspektrometer durch Lasereinsatz optimiert und auf zumindest ein Element, häufig auch auf nur ein Isotop, eingeschränkt. Die Elementselektivität (Isobarenunterdrückung) wird durch die spektral weit auseinander liegenden
Resonanzlinien aller Elemente bzw. möglicher molekularer Isobare erreicht. Die Isotopenselektion (Nachbarmassenunterdrückung) erfordert die spektrale Auflösung der Isotopieverschiebung
benachbarter Isotope eines Elements. Dieser Effekt ist auf zwei Beiträge zurückzuführen:
• die unterschiedliche Masse des Kerns führt zu einer unterschiedlichen Mitbewegung im
Schwerpunktssystem Masseneffekt). Diese ist wiederum in einen Zweikörperanteil zwischen
Kern und Valenzelektron (den normalen Masseneffekt) und einen Vielkörperanteil zwischen
allen Elektronen und dem Kern (den spezifischen Masseneffekt) aufzuteilen.
• die unterschiedliche endliche Kerngröße verschiedener Isotope führt zu einer Variation des
Feldes (Feld- oder Volumeneffekt).
Beide Effekte sind sowohl vorzeichenmäßig als auch in ihrer Größenverteilung gegenläufig. Der
Masseneffekt ist bei leichten Kernen stark ausgeprägt, der Volumeneffekt bei schweren. In der
Mitte der Nuklidkarte heben sie sich gerade auf, so dass sehr kleine Isotopieverschiebungen
um Masse 100 resultieren. Dies ist in Abb. 10.1 gezeigt. Zusätzlich muss bei der RIMS
auch die Hyperfeinstrukturaufspaltung berücksichtigt werden, die bei Isotopen mit ungerader
Massenzahl und daher nicht verschwindendem Kernspin I 6= 0 zu einer Aufspaltung der
Resonanzlinien führt. Dies hat eine die Selektivität beeinflussende Verschiebung der einzelnen
HFS Komponenten sowie eine Änderung der Anregungswahrscheinlichkeit zur Folge. Die
Hintergründe und Details zu beiden Effekten gehen über die Zielsetzung dieser Vorlesung
hinaus, es wird dazu auf jedes gute Atomphysiklehrbuch verwiesen, z.B. [Bran95, Demt93].
Die verwendete Laserstrahlung muss also je nach Anforderung spektral ausreichend schmalbandig sein, um entweder ein Element auszuwählen, oder sogar ein Isotop zu selektieren.
Gepulste abstimmbare Laser haben dabei aufgrund des auf der Heisenberg’schen Unschärferelation basierenden Fourierlimits eine minimale spektrale Breite des Inversen ihrer
96
97
Abb. 10.1: Größe der Isotopieverschiebung als Funktion der Neutronenzahl
Pulslänge, die aber üblicherweise durch gerätespezifische Unzulänglichkeiten nicht realisiert
wird:
∆ω · τ ≥ 1
(10.1)
Typische Breiten liegen im Bereich von mehreren GHZ, so dass eine Auflösung der Isotopieverschiebung nicht möglich ist und ausschließlich elementselektive Anwendungen bzw.
Unterdrückung von molekularen Verunreinigungen in Frage kommen. Kontinuierliche Laser
können extrem schmalbandig betrieben werden und bei typischen spektralen Breiten um
1 MHz die Auflösung der Isotopieverschiebung und Hyperfeinstruktur gewährleisten. Eine
Unterdrückung von vielen Größenordnungen wird aber durch die nicht zu vernachlässigende
Anregung in der Flanke jeder optischen Resonanz limitiert. Hierzu müssen experimentellen
Gegebenheiten (keine experimentellen Verbreiterungseffekte) und Auswahl der Übergänge
optimiert werden.
Der prinzipielle Prozess der Resonanzionisations-Massenspektrometrie ist vergleichsweise
einfach [Leto87, Hurs88, Payn94]. Er ist in Abb. 10.2 dargestellt. Die zu untersuchende Probe
wird zuerst in einen Atomdampf überführt und dabei der Strahlung von einem oder mehreren
Lasern ausgesetzt. Die durch den Anregungs- und Ionisationsprozess gebildeten Resonanzionen
werden durch ein elektrisches Feld beschleunigt, durchlaufen eine konventionelle Massenselektion
und werden mit einem Teilchendetektor quantitativ durch Zählung nachgewiesen.
98
KAPITEL 10. RESONANZIONISATIONS-MASSENSPEKTROMETRIE
Abb. 10.2: Prinzipdarstellung der Resonanzionisations-Massenspektrometrie
Der Einsatz der stufenweisen resonanten optischen Anregung und nachfolgenden Ionisation mittels schmalbandigen kontinuierlichen Lasern in der so genannten hochauflösenden (HR-) RIMS
führt dabei zu ähnlichen Spezifikationen wie diejenigen der AMS, sofern in jedem Schritt die
Isotopieverschiebung aufgelöst werden kann:
• im Prinzip vollständige Isobarenunterdrückung (> 108 ),
• bis zu extrem hohen Werten steigerbare Isotopenselektivität (> 1012 ),
• gute Effizienz, damit niedrige Nachweisgrenzen bzw. hohe Empfindlichkeit (∼ 10−5 ),
• geringer Untergrund (∼ 20 mHz Zählrate entspricht ∼ 40 Untergrundereignisse/ Stunde
Messzeit) und
• geringe zeitliche Messdauer (typisch ∼ 15 min).
Zum Einsatz des Verfahrens ist auf der einen Seite also die Verwendung von mehreren spektral präzise abgestimmten und kontrollierten Lasern notwendig, auf der anderen Seite ist das
Verständnis und die Beschreibung der mehrstufig resonanten Anregungs- und Ionisationsprozesse
gefragt. Hier nimmt die RIMS deutliche Anleihen bei Atomphysik und Quantenoptik.
10.1
Anregungspfade der RIMS, Selektivität und Effizienz
Typische Anregungs- und Ionisationspfade der resonanten Ionisation sind in Abbildung 10.3
dargestellt. Aufgrund der energetischen Lage der Ionisationsgrenze relativ zum atomaren
Grundzustand um 5 - 9 eV werden aus mehreren Gründen üblicherweise 3 Laser im nahen
ultravioletten, sichtbaren bzw. bis hin zum nahen infraroten Spektralbereich verwendet. Erste
10.1. ANREGUNGSPFADE DER RIMS, SELEKTIVITÄT UND EFFIZIENZ
99
Anregungsschritte aus dem Grundzustand liegen für fast alle Elemente im blauen bis ultravioletten Spektralbereich, aufgrund der 1/n2 Abhängigkeit der Balmerformel sind höhere Übergänge
dagegen deutlich niederenergetischer im roten bis infraroten.
Abb. 10.3: Anregungs- und Ionisationspfade in der RIMS
Die Zahlen auf den Übergangspfeilen der Abb. 10.3 geben typische Wirkungsquerschnitte für
optische Anregungen an. Alle Übergänge zwischen tiefliegenden Zuständen im Atom haben in
Resonanz hohe Wirkungsquerschnitte von
σ=
λ2
2π
(10.2)
Diese liegen im Bereich von etwa 10−10 cm2 . Bei Einstrahlung von gut abgestimmtem, leistungsstarkem Laserlicht (Photonenzahl nγ groß) und ausreichender Wechselwirkungszeit tInt
kann gemäß
< = σopt · tInt · nγ
(10.3)
eine hohe Übergangsrate erzeugt werden. Diese ist nur durch die Sättigung beschränkt,
die in einem geschlossenen Zweiniveausystem durch induzierte Emissionsprozesse zu einer
Gleichbesetzung von Grund- und angeregtem Zustand führen würde. Aufgrund des ”Lecks”
der Weiteranregung im angeregten Zustand kann aber in der RIMS prinzipiell ein vollständiger
Besetzungstransfer über die Zwischenzustände bis zur Ionisation erreicht werden. Hierbei ist die Berücksichtigung von Verlusten aus der Anregungsleiter heraus in sogenannte
Fallenzustände sowie in vielen Fällen auch die Berücksichtigung von kohärenten Prozessen
(Rabi-Oszillationen, Outler-Townes Aufspaltung, STIRAP - Stimulated Rapid Adiabatic
Passage u.ä.) notwendig [Shor90]. Zur vollständigen Beschreibung wird der Dichtematrixformalismus herangezogen, mit dem eine sehr elegante und vollständige Darstellung aller
auftretenden resonanten Prozesse möglich ist. Es ist dazu eine numerische Integration der
resultierenden Gleichungen über die experimentellen Bedingungen notwendig, die Aussagen
über Frequenz- und Leistungsabhängigkeiten jedes einzelnen optischen Anregungsschrittes
und damit die Form und Intensität jeder Resonanz zulässt. Die nachfolgende Ionisation kann
100
KAPITEL 10. RESONANZIONISATIONS-MASSENSPEKTROMETRIE
über eine Ratengleichung miteinbezogen werden, so dass der vollständige mehrstufige Resonanzionisationsprozess komplett beschrieben wird. Bei bekannten Eigenschaften der beteiligten
atomaren Zustände, (Lebensdauern, Verzweigungsverhältnissen und Aufspaltungen) erreicht
man damit Vorhersagen und Analysen der erreichbaren Isotopenselektivität und Effizienz
des Gesamtprozesses in Abhängigkeit von den Parametern aller Laserfelder, wie etwa Intensitäten, Verstimmungen und Polarisationen [Bush99, Nört00]. Eine Beschreibung geht über
den Rahmen dieser Vorlesung hinaus und kann etwa in einer Quantenoptikvorlesung stattfinden.
Limitierender Schritt des Gesamtprozesses ist meistens die Ionisation selbst. Sie führt
entweder direkt in das Kontinuum, ist damit nicht resonant und besitzt daher einen sehr
kleinen Wirkungsquerschnitt von nur etwa 10−17 cm2 . Der Wert liegt damit um mehr als 6
Größenordnungen unter dem typischen Wirkungsquerschnitt für starke optische Übergänge.
In Fällen komplexer atomarer Spektren kann die resultierende Effizienzeinbuße durch die
Abstimmung des Ionisationsschrittes auf autoionisierende Zustände, doppelt angeregte atomare
Zustände oberhalb des ersten Ionisationspotentials, vermieden werden [Bush03]. Diese haben
aufgrund ihres resonanten Charakters einen vergleichsweise großen Wirkungsquerschnitt von bis
zu 10−11 cm2 und zerfallen instantan mit hoher Wahrscheinlichkeit in ein freies Elektron und
ein positiv geladenes Ion. Alternativ kann auch eine effiziente Ionisation aus hochangeregten
gebundenen Rydbergzuständen durch ein starkes elektrisches Feld (Feldionisation) oder den
Einsatz eines Far Infrared (FIR)-Lasers (z.B. eines CO2 -Lasers im Frequenzbereich um 10 µm)
erfolgen, der eine extrem hohe Photonendichte zur Verfügung stellen kann.
Die in Abb. 10.3 gezeigte Vielfalt der Anregungsprozesse kann aus einer anfangs ein- bis
endgültig dreistufigen resonanten Anregung mit jeweils dem nötigen nachfolgenden Ionisationsschritt abgeleitet werden. Entsprechende Anregungs- und Ionisationsleitern sind für das
Beispiel des Calciumatoms (IP = 6,1132 eV) in Abbildung 10.4 dargestellt. Anhand dieser
Entwicklung wird die Notwendigkeit der mehrstufig resonanten Anregung deutlich.
Abb. 10.4: Ein- bis dreistufig resonante Anregungs- und Ionisationsleitern für Calcium
Die verschiedenen Anregungsleitern geben sehr unterschiedliche Spezifikationen für die Selektivität als auch die Anregungseffizienz. Dies kann anhand der experimentellen Daten der RIMS
10.1. ANREGUNGSPFADE DER RIMS, SELEKTIVITÄT UND EFFIZIENZ
101
an einem thermischen Atomstrahl (transversale Anordnung der Laserstrahlen), wie in Abb.
10.5 dargestellt, diskutiert werden:
Abb. 10.5: Entwicklung von der ein- bis zur dreistufig resonanten RIMS
Gezeigt sind die Anregungen mit spektral schmalbandigem Licht. Bei der einstufig resonanten
Anregung mit direkter nichtresonanter 1 + 10 Ionisation, kann die Dopplerverbreiterung
der sich bewegenden Atome nicht kompensiert werden. Trotz transversaler Anregung zur
Atomstrahlrichtung werden Linienbreiten um 100 MHz FHWM realisiert und eine lange starke
Dopplerflanke über viele GHz Ausdehnung resultiert. Im gesamten Spektralbereich aller
stabilen Isotope des Calciums (40 ≤ A ≤ 48) werden nur Werte von maximal 102 für die
optische Isotopenselektivität erreicht. Zusätzlich ist eine auf dem Bild nicht ersichtliche sehr
niedrige Anregungseffizienz im Bereich von nur 10−8 zu verzeichnen, da der Übergang ins
Kontinuum nur sehr schwach ist und nur geringe Laserleistung um 1 W im uv zur Verfügung
gestellt werden kann. Die 2stufige Anregung 2 + 10 ist bereits deutlich besser. Eine weitgehende
Dopplerkompensation durch antikollineare Einstrahlung des roten gegen das blaue Laserlicht
ist möglich und resultiert in deutlich schmäleren Linien und einer Erhöhung der Isotopenselektivität auf Werte > 106 . Auch die Effizienz gewinnt um etwa 2 Größenordnungen, da die
nichtresonante Ionisation vom höher liegenden Zwischenzustand aus einen deutlich höheren
Wirkungsquerschnitt hat und im grünen Spektralbereich bis zu 10 Watt Laserleistung zur
Verfügung gestellt werden kann. Optimale Resultate liefert aber erst die dreistufig resonante
3 + 10 Anregung: vollständige Dopplerkompensation und ein weiterer Selektionsschritt führen
zu Selektivitätswerten um 1010 und Effizienzen von bis zu 10−3 durch Verwendung eines 30
Watt Infrarot Lasers. Die im Bild eingezeichneten Kurven sind theoretische Berechnungen
mittels des Dichtematrixformalismus, wobei nur die experimentellen Daten der Laserleistung
gefittet wurden. Für die dreistufige Anregung ist keine Abstimmung aller drei Laser über die
Isotopieverschiebungen der einzelnen Isotope möglich. Daher wurde nur in einem Sprung die
Selektivität an der Position des gesuchten Isotops 41 Ca bestimmt.
102
KAPITEL 10. RESONANZIONISATIONS-MASSENSPEKTROMETRIE
Die RIMS gibt es in einer ganzen Reihe von experimentellen Ausprägungen. Dabei wird jeweils
eine Kombination mit einem geeigneten Massenspektrometer, je nach den Parametern des
Laserionisationsprozesses gewählt.
1. Pulslaser RIMS mit Flugzeitmassenspektrometern
Offensichtlich ist die gepulste Laserionisation aufgrund der kurzen Pulsdauer von typisch
10 - 50 ns eine ideale Quelle für ein Flugzeitmassenspektrometer. Hierbei werden elementselektive Anwendungen bearbeitet, aber nur eine begrenzte Isotopenselektivität im Bereich um 103
aufgrund der spektralen Breite der Laser und der geringen Nachbarmassenunterdrückung der
TOF-MS erreicht. Zur Realisation hoher Empfindlichkeit gibt es konkurrierende Ansätze:
a. Es wird ein kontinuierlicher Atomisationsprozess mit hochrepetierenden Lasern mit Pulsraten im Bereich von 10 kHz kombiniert. Der Laser- Dutycycle (das Puls- zu Pauseverhältnis)
beträgt dann etwa 10−4 . Er wird aber durch die vergleichsweise lange Aufenthaltszeit (∼
5 µs) der thermischen Atome im ausgedehnten Laserstrahldurchmesser von typisch 3 bis
5 mm Durchmesser weitgehend kompensiert, so dass Ionisationseffizienzen von > 10−4
möglich sind.
b. Die Ionisation findet in einer weitgehend geschlossenen Kammer, einer sogenannten Laserionenquelle statt. Die Atome haben dann nach jedem Wandstoß erneut die Möglichkeit
ionisiert zu werden und nur eine geringe Wahrscheinlichkeit durch das kleine Extraktionsloch die Quelle zu verlassen [Alkh89, Ames90]. Dieser Ansatz wird heutzutage zu effizienten und selektiven Ionisation kurzlebiger Radionuklide an den verschiedenen on-line
Massenseparatoren (ISOLDE im CERN, ISAC am TRIUMF in Vancouver, und ähnliche)
als dominante Ionisationstechnik eingesetzt, wobei die erreichbare Effizienz linear mit der
Laserpulsrate ist. Es kommen daher wiederum nur hochrepetierende Laser zum Einsatz, es
werden aber Ionisationseffizienzen von bis zu 30% und hohe Isobarenselektivitäten erzielt,
die nur durch die Oberflächenionisation an den heißen Kammerwänden begrenzt sind. Eine
Prinzipskizze zeigt Abb. 10.6.
Abb. 10.6: Prinzip einer Laserionenquelle
c. Alternative wird eine gepulste Laserablation oder der Einsatz einer synchronisierten gepulsten Sputteratomisation zusammen mit niedriger Pulsrate im 1 bis 10 Hz Bereich verwendet werden. In beiden Fällen ist zusätzlich eine Ortsauflösung durch Fokussierung
der desorbierenden Strahlung möglich. Man erreicht bei Laserdesorption üblicherweise
den Bereich von etwa 10 - 100 µm Auflösung, bei Sputtern kann bei gut fokussiertem
Primärionenstrahl bis in den 1 µm Bereich und darunter (Nano-SIMS) vorgedrungen werden. Die Verfahren sind noch in der Entwicklung.
10.1. ANREGUNGSPFADE DER RIMS, SELEKTIVITÄT UND EFFIZIENZ
103
2. Hochauflösende RIMS am thermischen Atomstrahl
Die HR-RIMS verwendet kontinuierliche Laserstrahlung in transversaler Geometrie an
einem gut kollimierten Atomstrahl. Durch Einstrahlung der Laser in antikollinearer Geometrie
können alle experimentellen Linienverbreiterungseffekte (z.B. die Dopplerverbreiterung) stark
reduziert werden und spektrale Auflösung im Bereich weniger MHz erreicht werden. Dies
bildet eine ideale Ionenquelle für ein Quadrupolmassenspektrometer, da hier eine präzise
Lokalisation in einem nahezu feldfreien Raum erfolgt und nur eine sehr geringe Beschleunigung
der Laserionen nötig ist. Blaum konnte zeigen, dass durch Einsatz der Laserionisation die
Nachbarmassenunterdrückung im QMS deutlich auf Werte bis > 108 gesteigert werden kann,
dies ist auf die verringerte Energiebreite bei der Erzeugung der Laserionen im Vergleich zu
Elektronenstoßionisation zurückzuführen [Blau98]. Mit dreistufiger resonanter Laseranregung
und ionisation konnte die aktuell weltweit höchste Isotopenselektivität mit laserbasierten
Methoden (ohne Voranreicherung) von 3 · 1012 im Ultraspurennachweis von 41 Ca gezeigt werden
[Bush01]. Damit erreicht die HR-RIMS ähnliche Spezifikationen wie die weitaus aufwändigere
AMS.
3. Hochauflösende RIMS in kollinearer Geometrie am schnellen Atomstrahl
Für den Fall verschwindender Isotopieverschiebung im Bereich um A ∼ 100 kann die
Dopplerverschiebung optischer Resonanzlinien zur Selektion in der optischen Anregung
herangezogen werden. Das Verfahren basiert auf einem schnellen Atomstrahl, der durch
Neutralisation eines 10 bis 50 kVolt Ionenstrahls erzeugt wird, und parallel mit dem Strahl
eines oder mehrerer Laser angeregt und ionisiert wird. Es ist vergleichsweise aufwändig und hat
sich daher trotz guter Resultate für den 89,90 Sr Ultraspurennachweis nicht etablieren können.
Es sei auf dazu nur auf die Literatur verwiesen [Wend02, Wend05].
Kapitel 11
Massenspektrometrie in
Speicherringen
11.1
Entwicklungsgeschichte
Seit dem Nachweis der Elektronenkühlung von Budker und Kollegen 1976 [Budk1976,
Budk1978], sind Speicherringe zu einem universellen Werkzeug entwickelt worden.
Es
werden Fragestellungen aus dem Bereich der Atomphysik, Molekülphysik, Kernphysik und
Hochenergiephysik behandelt.
Die ersten Speicherringe bzw. Kreisbeschleuniger wurden zunächst für Hochenergie- und Kernphysikexperimente entwickelt und arbeiteten vorwiegend mit leichten geladenen Teilchen wie
Protonen. Um die Anzahl der im Ring akkumulierten Teilchen erhöhen zu können, schlug Budker
1966 vor, dem umlaufenden Ionenstrahl einen energiescharfen, ,,kalten” Elektronenstrahl gleicher mittlerer Geschwindigkeit zu überlagern [Budk1966]. Diese sogenannte Elektronenkühlung
wurde 1974 im eigens dafür gebauten Protonenspeicherring NAP-M in Novosibirsk erfolgreich
demonstriert [Budk1976].
Seitdem wurden eine Vielzahl von Speicherringen in Betrieb genommen, an denen atomphysikalische Experimente durchgeführt werden. Eine Übersicht geben beispielsweise [Müll1994,
Lars1995, Mokl1996, Müll1997]. Zu diesen Speicherringen gehören der TSR in Heidelberg
[Jaes1990], ASTRID in Aarhus [Sten1988], CRYRING in Stockholm [Dana1993] und ESR in
Darmstadt [Fran1987]. An den Niederenergie-Speicherringen können geladene Teilchen bis
zu einer Maximalenergie von etwa 30 MeV/u (β ≈ 0.25) gespeichert werden. Die höchsten
Ladungszustände, die an diesen Ringen erreicht werden, sind meist durch die Maximalenergie
des vorgelagerten Beschleunigers bestimmt und reichen bis q ≈ 50. Anders beim ESR in Darmstadt (siehe Abb. 11.1): Durch die Beschleunigung im Schwerionensynchrotron (SIS) werden
Energien erreicht, bei denen selbst Uran vollständig ionisiert werden kann.
11.2
Funktionsweise eines Speicherrings
Die Funktionsweise eines Speicherrings sei am Beispiel des Experimentierspeicherrings (ESR)
an der Gesellschaft für Schwerionenforschung (GSI) in Darmstadt erläutert [Stöh1998]. Er ist
der einzige Ring, in dem alle Ionen einschließlich nackter Uranionen akkumuliert werden können
[Fran1987]. Mit einer Bahnlänge von 108.36 m besitzt er genau den halben Umfang des SIS, so
dass von den 4 Ionenpaketen, die im SIS umlaufen, jeweils 2 gleichzeitig in den ESR eingeschossen
werden können. Die magnetische Steifigkeit beträgt 10 Tm, wodurch es möglich ist, nackte
Uranionen bei einer maximalen Energie von 560 MeV/u zu speichern. Üblicherweise erfolgen die
104
105
11.2. FUNKTIONSWEISE EINES SPEICHERRINGS
Experimente mit schweren Ionen bei einer Energie von ca. 300 MeV/u, was einer Geschwindigkeit
von β ' 0.65 entspricht und eine Umlauffrequenz von etwa 2 · 106 s−1 zur Folge hat.
Abbildung 11.1 zeigt den Speicherring mit seinen Diagnose- und Experimentiereinrichtungen
schematisch. Ein Photo des ESR Speicherrings ist in Abb. 11.2 gezeigt.
Deutlich sind die
Injektion
vom SIS/FRS
Reinjektion SIS
Umladedetektor
" Nord "
Quadrupol
Fokussierungsmagnete
6+
C to U92+
Ionenarten
Ionenenergien 50 - 370 MeV/u
Umfang
108,36 m
Schottkypickup
Elektronen-
GasjetTarget
Kühler
HV Terminal
DC-Ionenstrahltransformator
Ablenkdipol
0
10 m
RF-Kavität
Umladedetektor
" Süd "
Abb. 11.1: Schematische Darstellung des Experimentierspeicherrings ESR an der Gesellschaft für
Schwerionenforschung in Darmstadt mitsamt seinen Diagnose- und Experimentiereinrichtungen.
großen Ablenk- und Fokussierungsmagnete zu erkennen (orange: Dipolmagnet, rot: Quadrupolmagnete). Als ionenoptische Elemente besitzt der Ring sechs 60◦ Dipolmagnete und sechs
Quadrupol-Triplets bzw. Dublets, die benötigt werden, um die Ionen nahe der idealen Bahn
zu halten. Diese Bahn kann als ein geschlossenes Orbital betrachtet werden, auf dem sich die
Teilchen mit der Energie EP und dem Impuls pP bewegen, die durch die Magnetfelder und den
Krümmungsradius der Magnete bestimmt sind. Tatsächlich kommt es jedoch zu einer Art sinusförmiger Schwingung der Ionen um diese sogenannte Sollbahn. So fokussieren die Quadrupolmagnete jeweils in einer Ebene und wirken senkrecht zu dieser Ebene defokussierend. Da der
Ionenstrahl eine Vielzahl von Quadrupolmagneten durchläuft, kommt es zu Oszillationen um die
ideale Bahn. Der Verlauf ist in Abb. 11.3 schematisch dargestellt. Die Amplitude A(s) dieser
Schwingung, die auch Betatronoszillation genannt wird, setzt sich zusammen aus der Amplitudenfunktion βy (s), die durch die magnetischen Führungsfelder festgelegt ist, und der Emittanz
des Strahls y . y repräsentiert eine transversale Koordinate, während s die longitudinale Koor-
106
KAPITEL 11. MASSENSPEKTROMETRIE IN SPEICHERRINGEN
Abb. 11.2: Photo des Experimentierspeicherrings ESR. Die Dipolmagnete sind in der Farbe
orange, die Quadrupolmagnete in rot zu sehen.
dinate ist. Die Anzahl der Schwingungen pro Umlauf ergibt sich aus βy (s) durch
I
1
1
Qy =
ds .
2π
β(s)
(11.1)
c
Es ist zu beachten, dass die Umlauffrequenz weder ganzzahlig noch 1/2, 1/3, etc. gewählt werden
darf (vgl. Abb. 11.3). In diesem Fall würden die von Feldfehlern hervorgerufenen Bahnstörungen
resonant verstärkt werden, da die Teilchen bei jedem Umlauf eine phasengleiche Auslenkung
erfahren.
Wichtig ist auch die Beziehung zwischen dem Impuls der umlaufenden Teilchen pP und ihrer
Abb. 11.3: Betatron-Oszillation entlang der idealen Ionenbahn [Stöh1998].
107
11.3. MASSENSPEKTROMETRIE IM SPEICHERRING
Umlauffrequenz ν0 . Abweichungen ∆pP vom Sollimpuls führen zu einer Änderung der Teilchenbahn, was wiederum eine veränderte Umlauffrequenz zur Folge hat. Zwischen der relativen Impulsänderung ∆pP /pP und der relativen Frequenzänderung ∆ν0 /ν0 besteht die lineare Beziehung
1
1
∆pP
∆ν0
=
−
.
(11.2)
·
ν0
γ 2 γt2
pP
Der Proportionalitätsfaktor berücksichtigt zum einen die relativistische
p Beziehung zwischen Im2
puls und Geschwindigkeit eines Teilchens (der Term 1/γ mit γ = 1/ 1 − β 2 ) und zum anderen
die Beziehung zwischen Impuls- und Bahnänderung (1/γt2 ) [Schl1997]. γt wird auch als transition-γ bezeichnet, das definiert ist durch df /dEP |γ=γt = 0, d.h. für γ = γt hat eine Energie- bzw.
Impulsänderung keine Frequenzänderung zur Folge. Hingegen ist für γ < γt mit zunehmendem
Impuls immer eine Frequenzerhöhung verbunden.
11.3
Massenspektrometrie im Speicherring
Ein Speicherring kann als hochauflösender Massenanalysator eingesetzt werden, indem die
Massen durch die präzise Messung ihrer Umlauffrequenzen bestimmt werden. Die Beziehung
zwischen Umlauffrequenz ν, Masse-Ladungs-Verhältnis m/q und Geschwindigkeit v unterschiedlicher im Ring zirkulierender Ionen ist gegeben durch:
∆ν
1 ∆(m/q) ∆v
=− 2
+
ν
v
γt m/q
γ2
1− 2
γt
.
(11.3)
Die Größe γ ist der Lorentz-Faktor der Ionen und γt ist ein ionenoptischer Parameter, der den
Übergangspunkt des Speicherrings beschreibt. Um eine eindeutige Beziehung zwischen Frequenz
und Masse zu erhalten, muss der zweite (geschwindigkeitsabhängige) Term auf der rechten Seite
von Gl. (11.3) zum Verschwinden gebracht werden. Das kann auf zwei unterschiedliche Arten
geschehen und somit stehen zwei komplementäre Massenspektrometriemethoden zur Verfügung,
wie in Abb. 11.4 für den Experimentierspeicherring an der GSI gezeigt [Klug2004].
Bei der Schottky-Massenspektrometrie (SMS) [Fran1995, Rado1997, Rado2000] wird der Speicherring im Standardmodus (γt = 2.4) betrieben und Elektronenkühlen eingesetzt, so dass
∆v/v → 0 und die Umlauffrequenz wird aus der Analyse des Schottky-Rauschens bestimmt.
SMS ist ein vielseitiges Werkzeug für die Spektroskopie von instabilen, hochgeladenen Nukliden mit hoher Effizienz und Auflösungsvermögen, Einzelionenempfindlichkeit und in-situKalibrierung. Es gestattet die Messung von Masse und Lebensdauer der gespeicherten Ionen.
Die Schottky-Spektroskopie [Bore1974] wird verbreitet zur nichtdestruktiven Strahldiagnose in
Kreisbeschleunigern und Speicherringen eingesetzt. Die gespeicherten Ionen zirkulieren im Speicherring mit einer Umlauffrequenz von ca. 2 MHz. Bei jedem Umlauf induzieren sie Spiegelladungen in zwei elektrostatischen Abnahmeelektroden, die in der Ringöffnung angebracht sind
[Beck1990]. Weil alle elektronengekühlten Ionen dieselbe exakt definierte Geschwindigkeit besitzen, zeigen ihre Umlauffrequenzen eine eineindeutige Beziehung zu ihren Masse-LadungsVerhältnissen m/q. Die Umlauffrequenz wird durch die Geschwindigkeit der Ionen und die Länge
ihrer geschlossenen Umlaufbahn bestimmt. Die Signale von beiden Aufnahmeplatten werden
mittels rauscharmen Verstärkern verstärkt und anschließend aufsummiert. Schließlich offenbart
das Schottky-Rauschen, nach Fouriertransformation in den Frequenzraum, die hochaufgelösten
Massen von unterschiedlichen gleichzeitig gespeicherten und gekühlten Ionensorten [Schl1997].
Ein typisches Frequenzspektrum hochgeladener Ionen im Bereich der Seltenen Erden zeigt
Abb. 11.5. Wie im Inset von Abb. 11.5 gezeigt, ermöglicht SMS Einzelionenselektivität mit
108
KAPITEL 11. MASSENSPEKTROMETRIE IN SPEICHERRINGEN
Abb. 11.4: Schematische Darstellung des Prinzips der Massenmessung im Experimentierspeicherring an der GSI. Die Bewegung von bis zu vier verschiedenen Sorten, die durch ihr
Masse-Ladungs-Verhältnis (m/q)1...4 charakterisiert sind, ist zu erkennen. Bei der SchottkyMassenspektrometrie (links) werden Ionen durch Elektronenkühlen gekühlt und besitzen die
gleiche mittlere Geschwindigkeit v während die Ionen bei der isochronen Massenspektrometrie
(rechts) “heiß” sind und unterschiedliche Geschwindigkeiten haben.
hohem Auflösungsvermögen und hoher Effizienz. Während eines Experimentes können mehr als
100 neue Massen gemessen werden.
Die grau markierten Massen in Abb. 11.5 waren zuvor unbekannt. Die gleichzeitige Speicherung
von bekannten und unbekannten (schwarz markiert) Massen erlaubt eine in-situ-Kalibrierung.
Für die Frequenzen zweier unterschiedlicher Ionen k und l kann man in erster Näherung schreiben
[Rado2000]:
(m/q)k − (m/q)l
fk − fl
= −αp
,
(11.4)
fk
(m/q)k
wo αp der sogenannte “momentum compaction factor” ist, der definiert ist als
αp =
dC/C
,
d(Bρ)/(Bρ)
(11.5)
und der das Verhältnis von relativer Änderung der Umlaufbahnlänge (C) zu relativer Änderung
der magnetischen Steifigkeit (Bρ) beschreibt. αp hat typischerweise einen Wert von ungefähr
0.15. Die Breite der Frequenzverteilung jeder Ionensorte k ist bestimmt durch [Rado2000]:
1
1
δpk
δvk
δfk
=
=
,
(11.6)
− αp ·
− αp · γk2 ·
fk
pk
vk
γk2
γk2
wo δpk /pk und δvi /vi die relativen Breiten des Impulses beziehungsweise die Geschwindigkeitsverteilungen für die k-te Ionensorte bedeuten. Die Gleichungen (11.4) und (11.6) bilden die Ba-
11.3. MASSENSPEKTROMETRIE IM SPEICHERRING
109
Abb. 11.5: Schottky-Spektrum von simultan aufgenommenen und gekühlten exotischen Nukliden im Experimentierspeicherring. Die Frequenzachse zeigt die Abweichung der 32. Harmonischen der korrespondierenden Umlauffrequenzen der Ionen (v/c ≈ 0.67) von der Frequenz eines
stabilisierten lokalen Oszillators bei etwa 59.33 MHz. Das Inset zeigt Grundzustand und isomeren Zustand von komplett gestripptem 143 Sm. Mittels Schottky-Massenspektrometrie wird
Einzelionensensitivität erreicht [Klug2004].
sis für die Schottky Massenspektrometrie, das heißt zur Massenmessung von gespeicherten und
identifizierten Ionen genügt die Messung ihrer Umlauffrequenzen. Die Genauigkeit der Methode
kann erhöht werden, indem innerhalb des akzeptierten m/q Bereichs gleichzeitig verschiedene
Ladungszustände des gleichen interessierenden Ions gespeichert und untersucht werden.
Für die Isochrone Massenspektrometrie (IMS) [Haus2000], eine Technik, die es ermöglicht,
Lebensdauern unterhalb einer Millisekunde zu erforschen, wird der ESR im isochronen Modus
bei einem reduzierten Wert γt = 1.4 betrieben. Bei dieser spezifischen ionenoptischen Einstellung wird die Umlauffrequenz von Ionen mit dem gleichen Masse-Ladungs-Verhältnis unabhängig
von ihrer Geschwindigkeit. Bei dieser sogenannten Übergangsenergie, bei der die injizierten Ionen einen Lorentz-Faktor von γ = γt besitzen, wird ihre Umlauffrequenz aus der Flugzeit für
jeden Umlauf bestimmt [Troe1992]. Dies erlaubt die präzise Massenbestimmung von heißen,
ungekühlten Kernen [Stad2004]. Die Nuklide mit bekannten Massen, die gleichzeitig mit den
zu untersuchenden Ionen gespeichert sind, werden zur Kalibrierung des Spektrums eingesetzt,
wodurch die bislang unbekannten Massen gewonnen werden können. Zum Nachweis der umlaufenden Ionen ist eine Folie in der Ringöffnung montiert. Bei jedem Durchgang eines Ions
durch diese Folie werden reichlich Delta-Elektronen produziert. Die Elektronen werden mittels
zweier Mikrokanalplatten-Detektoren nachgewiesen und die Signale dienen als Zeitstempel für
jeden Umlauf eines jeden gespeicherten Ions [Haus2001]. Die injizierten Ionen zirkulieren typischerweise einige hundert mal im Speicherring mit einer Umlaufdauer von etwa 500 ns. Die
110
KAPITEL 11. MASSENSPEKTROMETRIE IN SPEICHERRINGEN
maximale beobachtete Anzahl an Umläufen war annähernd 3500 und es können die Massen von
kurzlebigen Nukliden mit Halbwertszeiten im Bereich von ein paar Millisekunden oder sogar
Mikrosekunden (siehe Abb. 11.6) gemessen werden [Klug2004, Stad2004].
/
Abb. 11.6: Ein Spektrum der Umlaufdauer wurde während eines Experiments mit dem Experimentierspeicherring im isochronen Massenmodus aufgenommen. Die Leistungsfähigkeit von IMS
zur Untersuchung von Kernen mit Halbwertszeiten bis hinunter in den Mikrosekundenbereich
ist dargestellt.
Im isochronen Modus des Experimentierspeicherrings an der GSI in Darmstadt wurde ein Massenauflösungsvermögen von R = 110000 mit einer mittleren Massengenauigkeit von etwa 100 keV
demonstriert [Bosh2002, Stad2004]. Im Schottky-Massenspektrometrie-Modus wurde sogar ein
Auflösungsvermögen von bis zu R = 2 · 106 und eine relative Massengenauigkeit von δm/m =
2 · 10−7 erreicht [Litv2004, Litv2005]. Die erreichbare Halbwertszeit liegt für den isochronen
Modus des Experimentierspeicherrings bei einigen Mikrosekunden [Klug2004, Stad2004] und im
Schottky-Modus bei ungefähr einer Sekunde. Letztere ist durch die lange Kühldauer heißer
Fragmente limitiert, was es unmöglich macht, die interessantesten Regionen kurzlebiger Kerne
auf beiden Seiten des Tals der Stabilität zu erreichen.
Kapitel 12
Ionenfallen und
Präzisionsmassenspektrometer
12.1
Die Paulfalle
12.1.1
Die ideale dreidimensionale Paulfalle
Eine dreidimensionale Speicherung der Ionen macht neben der in Kapitel 7 beschriebenen radialen Speicherung auch die axiale Speicherung erforderlich. Da die Form und Art der Endelektroden bei den verschiedenen bisher in Experimenten verwendeten linearen Fallen nicht einheitlich
ist, müsste man genau genommen jeden speziellen Fall einzeln untersuchen. Ziel ist es aber
immer, ein möglichst harmonisches statisches Potential zu erzeugen. Die unterschiedlichen Realisierungen wie z.B. hyperbolische Endkappen, gelochte Endbleche, Ringe um die Quadrupolstangen, Überlagerung von End- und Hauptsegmenten, Endstifte oder Endsegmente orientieren
sich meist an der Zugänglichkeit der Ionen für Anregung und Nachweis und man findet eher
selten Hinweise auf Gründe, die das Potential betreffen.
Grundsätzlich tritt allerdings durch ein elektrostatisches Potential in axialer Richtung natürlich
ein defokussierender Effekt in radialer Richtung ein. Die Schwächung des Quadrupolpotentials
wird in [Ghos1995] folgendermaßen beschrieben: Das statische Potential in axialer Richtung
kann als harmonisches Potential angenähert werden
1 2
κU
2
2
(12.1)
x +y
Φs = 2 z −
2
z0
wobei z die halbe Länge des Endelektroden-Abstands und κ ein von der Fallengeometrie
abhängender Faktor ist. U ist die an den Endelektroden anliegende Spannung. Die Elektrodenkonfiguration für die Erzeugung eines axialsymmetrischen Quadrupolpotentials zeigt
Abb. 12.1.
s
2κqU
ωz =
(12.2)
mz02
ist die axiale Schwingungsfrequenz der Ionen im parabolischen Pseudopotential (siehe Abb. 12.2
links) der Falle. Das Pseudopotential wird geschwächt durch die Hinzunahme des statischen
Potentials (Abb. 12.2 rechts)
m
(ωr0 )2 x2 + y 2
Φr =
(12.3)
2q
wodurch die effektive Kreisfrequenz der Makrobewegung ωr in folgender Weise reduziert wird:
r
1
(12.4)
ωr0 = ωr2 − ωz2 .
2
111
112
KAPITEL 12. IONENFALLEN UND PRÄZISIONSMASSENSPEKTROMETER
Abb. 12.1: Grundlegende Elektrodenkonfiguration zur Erzeugung eines axialsymmetrischen
Quadrupolpotentials. Die inneren Flächen der Elektroden sind Hyperboloide und folgen den
Äquipotentialflächen des elektrischen Feldes.
R F + D C
z
e le c tr ic p o te n tia l V
e le c tr ic p o te n tia l V
R F o n ly
r
z
r
Abb. 12.2: Pseudopotential in einer Paulfalle. Links: nur ein rf Potential wird an die Falle
angelegt. Rechts: ein zusätzlich angelegtes DC Potential.
Eine Lissajous-ähnliche Trajektorie eines geladenen Mikroteilchens in einer Paulfalle zeigt
Abb. 12.3 [Wuer1959].
12.1.2
Die reale dreidimensionale Paulfalle
In der experimentellen Wirklichkeit treten einige Abweichungen vom idealen Verhalten von
Paulfallen auf. Es gibt eine Reihe von Effekten, die dazu führen, dass die Potentiale nicht mehr
die exakte Form haben, was teilweise erhebliche Auswirkungen auf Speicherdauer, Laserkühlen
etc. hat. Eine recht detaillierte Untersuchung (sowohl theoretisch als auch experimentell) findet
sich für die klassische Paulfalle bei [Ghos1995, Majo2004]. Die genannten Effekte wurden bereits
für die lineare Paulfalle behandelt und sollen daher hier nur qualitativ diskutiert werden.
Störungen des idealen Quadrupolpotentials werden u.a. verursacht durch:
- Justageungenauigkeiten beim Zusammenbau der Falle sowie die technische Unmöglichkeit,
unendlich ausgedehnte hyperbolische Elektroden zu bauen. In der Praxis verwendet man runde
Stangen, da diese exakter gefertigt werden können und das Potential in der Nähe der Fallenachse
12.1. DIE PAULFALLE
113
Abb. 12.3: Beobachtete Lissajous-ähnliche Trajektorie eines geladenen Mikroteilchens in einer
Paulfalle [Wuer1959].
dennoch gut als Quadrupolpotential angenommen werden kann.
- Kontaktpotentiale durch Ablagerungen auf den Kupferelektroden, verursacht vom Ofen beim
Erzeugen der Ionen. Die dadurch entstehenden statischen Dipolfelder führen zu einer verschobenen Gleichgewichtslage der Ionen außerhalb der Fallenmitte. Somit sind die Ionen dem
Führungsfeld ausgesetzt, was in einer erhöhten Mikrobewegung resultiert. Der Dopplereffekt 1.
und 2. Ordnung wird so größer und die Aufheizung durch Restgasstöße nimmt zu.
- die Raumladung größerer Ionenwolken: die Ionen beeinflussen durch ihre Ladungsverteilung
das Pseudopotential. Der sich daraus ergebende Abschirmeffekt führt zu einer Verschiebung der
realen Speicherparameter gegenüber den theoretischen.
Aufgrund von Abweichungen vom idealen Quadrupolfeld treten auch innerhalb des stabilen
Bereichs Instabilitäten auf. Diese werden dadurch ausgelöst, dass die aufgeführten Störungen
zu höheren Multipolkomponenten des Potentials führen und bei bestimmten Verhältnissen der
Säkularfrequenzen ein Energietransfer von der einen in die andere Bewegungsrichtung stattfinden kann. Dies führt zu einer Aufweitung der Teilchenbahn und dadurch zum Verlust der
Ionen. Diese sogenannten nichtlinearen Resonanzen treten entlang bestimmter Linien im
Stabilitätsdiagramm auf. Weitere Folgen sind, dass die Säkularfrequenzen ortsabhängig werden (d.h. eine Bahnamplitudenänderung entspricht einer effektiven Änderung der Speicherparameter) und die Entkopplung der Bewegungen in axialer und radialer Richtung nicht mehr
streng gilt. Die theoretischen Instabilitätslinien bzw. nichtlinearen Resonanzen im ersten
Stabilitätsbereich der Paulfalle sind für die Störungen der Ordnung n = 3 bis n = 8 in
Abb. 12.4 dargestellt. Die Ergebnisse der entsprechenden experimentellen Untersuchungen zeigt
Abb. 12.5 [Alhe1996b, Alhe1997b].
Eine sorgfältige Justage beim Bau der Falle, sowie ein Ausgleichen von Kontaktpotentialen durch
zusätzlich an einzelne Elektroden angelegte Korrekturspannungen und die Wahl eines stabilen
Arbeitspunkts sind die dem Experimentator zur Verfügung stehenden Maßnahmen. In einzelnen
Fällen, wie z.B. zur Isotopentrennung [Alhe1996], kann eine Anregung der Ionen durch zusätzlich
angelegte Multipolfelder oder durch gezieltes Ausnutzen der Instabilitäten durchaus erwünscht
sein.
114
KAPITEL 12. IONENFALLEN UND PRÄZISIONSMASSENSPEKTROMETER
Abb. 12.4: Theoretische Instabilitätslinien bzw. nichtlinearen Resonanzen im ersten Stabilitätsbereich der Paulfalle für Störungen der Ordnung n = 3 bis n = 8.
12.1. DIE PAULFALLE
115
Abb. 12.5: Experimentelle Instabilitätslinien bzw. nichtlinearen Resonanzen im ersten Stabilitätsbereich einer realen Paulfalle mit gespeicherten H+
2 Ionen. Die Instabilitätslinien sind
gemäß Abb. 12.4 zugeordnet, wobei Ω/2π auf 1 normiert wurde. Die Intensität der Grauschattierung ist proportional zur Anzahl der gespeicherten Ionen.
116
12.2
KAPITEL 12. IONENFALLEN UND PRÄZISIONSMASSENSPEKTROMETER
Die Penningfalle
Dieser Abschnitt behandelt die theoretischen Grundlagen zur Speicherung eines geladenen
Teilchens in einer Penningfalle. Abweichungen von den idealen Speicherbedingungen durch
die Speicherfelder selbst, sowie durch die Anwesenheit von mehreren Ionensorten werden betrachtet. Die notwendigen Schritte zur geeigneten Präparation und Anregung der gespeicherten
Ionen werden aufgezeigt. Eine ausführliche Beschreibung des Systems eines geladenen Teilchens
in einer Penningfalle befindet sich in dem Übersichtsartikel von L.S. Brown und G. Gabrielse
[Brow1986] sowie in [Boll1990] im Hinblick auf Massenbestimmungen. In diesem Kapitel sind
die wesentlichen Merkmale kurz aufgezeigt.
12.2.1
Kurze Historie
Aus dem Gauß’schen Gesetz folgt, dass ein elektrostatisches Feld allein ein geladenes Teilchen
nicht gleichzeitig in allen drei Raumrichtungen einschließen kann. Durch Überlagerung eines
elektrischen 3D-Multipolfeldes mit einem magnetischen Dipolfeld ist es allerdings möglich. Dabei
verhindert das in z-Richtung anliegende Magnetfeld das Ausbrechen des Ions in radialer Richtung, während das elektrische Feld den axialen Einschluss erledigt. Die erste experimentelle Verwirklichung dieser Idee gelang Frans Michel Penning in den 1930er Jahren. Hans Dehmelt realisierte 1959 ein elektrisches Quadrupolfeld durch das Anlegen einer einfachen Gleichspannung an
Elektroden, deren Form sich den hyperbolischen Äquipotentialflächen des 3D-Quadropolfeldes
annähert. Diese Geometrie wird bis heute für Präzisions-Penningfallen verwendet.
12.2.2
Die ideale Penningfalle
Die dreidimensionale Speicherung eines geladenen Teilchens in einer Penningfalle beruht auf
seiner Bewegung im elektromagnetischen Feld. Befindet sich ein Teilchen der Ladung q und der
Masse m in einem starken, homogenen Magnetfeld B, das in z–Richtung zeigt, so bewegt es sich
mit der Zyklotronfrequenz
q
(12.5)
ωc = B
m
auf Kreisbahnen um die magnetischen Feldlinien und ist somit in radialer Richtung gebunden.
Eine Speicherung in allen Raumrichtungen erhält man durch die zusätzliche Überlagerung eines
schwachen, elektrostatischen Quadrupolpotentials der Form
1 2
V0
2
(12.6)
V (z, ρ) = 2 z − ρ ,
2d
2
gegeben in zylindrischen Koordinaten.
Ein solches Potential kann durch eine Elektrodengeometrie von drei Rotationshyperboloiden erreicht werden, deren Oberflächen den Äquipotentialflächen identisch sind. Abbildung 12.6 (a)
zeigt die geometrische Anordnung einer hyperbolischen Penningfalle, bestehend aus einer
geschlossenen Ringelektrode und den beiden Endkappen. Mit anderen Käfigformen, wie z.B.
der zylindrischen Falle in Abb. 12.6 (b) mit offenen Zylinderelementen als Elektroden, lässt sich
ebenso ein speicherndes Potential in axialer Richtung erzeugen. V0 ist die angelegte Fallenspannung entsprechender Polarität zwischen der Ringelektrode und den beiden Endelektroden mit
dem jeweiligen Minimalabstand ρ0 bzw. z0 zum Fallenzentrum. Die Größe
ρ20
1
2
2
z +
(12.7)
d =
2 0
2
ist ein Maß für die Dimension der Falle.
117
12.2. DIE PENNINGFALLE
(a)
(b)
B
z
B
z
z0
V0
r
0
r
V0
r
Abb. 12.6: Penningfalle mit hyperbolischen (a) und zylindrischen (b) Elektroden. Das Magnetfeld ist jeweils entlang der Fallenachse gerichtet. Zur Speicherung wird zwischen der Ringelektrode und den Endkappen die Fallenspannung V0 entsprechender Polarität angelegt.
Löst man die Bewegungsgleichungen [Brow1986] für alle Raumkoordinaten,
~ ,
mz̈ = qEz
und
mρ
~¨ = q E~ρ + ρ~˙ × B
(12.8)
mit den elektrischen Feldstärken
V0
Ez = − 2 z
d
und
E~ρ =
V0
2d2
ρ
~,
(12.9)
so erhält man als resultierende Bewegung die Überlagerung dreier entkoppelter Schwingungen
(siehe Abb. 12.7) mit den charakteristischen Eigenfrequenzen
r
r
ωc
qV0
ωc2 ωz2
und
ω
=
±
−
.
(12.10)
ωz =
±
md2
2
4
2
Die harmonische, axiale Schwingung mit der Frequenz ωz ist von der angelegten Fallenspannung
V0 und den Geometrieparameter d abhängig. Die Bewegung in der Radialebene besteht aus den
beiden überlagerten Kreisbewegungen mit einer schnellen reduzierten Zyklotronfrequenz ω+ und
einer langsamen Magnetronfrequenz ω− . Eine Reihenentwicklung der radialen Eigenfrequenzen
liefert die folgende Beziehungen:
V0
ω− ≈ 2
(12.11)
2d B
und
V0
ω+ ≈ ωc − 2 ,
(12.12)
2d B
für die radialen Eigenfrequenzen. Dies zeigt, dass die Magnetronfrequenz in erster Näherung
unabhängig von der Masse des gespeicherten Teilchens ist. Zwischen allen Eigenfrequenzen
118
KAPITEL 12. IONENFALLEN UND PRÄZISIONSMASSENSPEKTROMETER
b
a
Endkappe
B
c
Magnetronbewegung (ν-)
z
r
Ringelektrode
rr+
V=
Modifizierte Zyklotronbewegung (ν+)
Axiale Bewegung (νz)
Abb. 12.7: Schematische Darstellung der drei idealerweise unabhängigen Eigenbewegungen eines
gespeicherten Teilchens in einer Penningfalle (a): Eine harmonische Schwingung im speichernden
elektrischen Potential in axialer Richtung (ωz ), sowie die Überlagerung einer schnellen Kreisbewegung mit der reduzierten Zyklotronfrequenz (ω+ ) und der langsamen Magnetronbewegung
(ω− ) in der Radialebene (b). Die Amplituden der Gesamtionenbewegung (c) liegt zur Vermeidung von Feldfehlern idealerweise unter einem Millimeter.
gelten die Relationen:
ωc = ω+ + ω− ,
ωc2
=
2
ω+
+
2
ω−
+
ωz2 ,
(12.13)
(12.14)
ω− < ωz < ω+ ,
(12.15)
2ω+ ω− = ωz2 .
(12.16)
Einige Beispiele zur Ionenbewegung in der Penningfalle sind in den Abbildungen 12.8 und 12.9
zusammengefasst. Radiale Trajektorienprojektionen sind in Abb. 12.8 dargestellt, dreidimensionale Trajektorien zeigt Abb. 12.9.
Zur Bestimmung der Masse eines gespeicherten Teilchens ergibt sich zum einen die Möglichkeit
die freie Zyklotronfrequenz (12.5) direkt über einen Flugzeitnachweis zu messen. Zum anderen
kann die Masse über die individuelle Beobachtung der Eigenschwingungen (12.14) durch Detektion des influenzierten Signals in den Fallenelektroden ermittelt werden.
Als Beispiel sind in Tabelle 12.1 typische Werte der Eigenfrequenzen für eine hyperbolische
Penningfalle aufgelistet. Die Fallendimensionen sind durch den Radius ρ0 = 6,38 mm und
den einfachen Abstand zur Endkappe z0 = 5,5 mm gegeben. Die Speicherparameter betragen
B = 7 T für das Magnetfeld und V0 = 10 V als typische Fallenspannung. Betrachtet werden
Ionen der Massen A = 1 u, A = 133 u, A = 197 u und A = 250 u.
12.2.3
Die reale Penningfalle
Abweichungen von der zuvor beschriebenen idealen Penningfalle mit harmonischem Speicherpotential, wie z.B. mögliche Inhomogenitäten und eine Dejustage der Achse des Magnetfeldes,
ergeben für die gespeicherten Teilchen eine Frequenzabhängigkeit von den Bewegungsamplituden
und eine mögliche Kopplung der einzelnen Eigenbewegungen. Dies schränkt die Präzision einer
Messung der Schwingungsfrequenzen ein. Zudem modifiziert sich mit zunehmender Anzahl von
geladenen Teilchen in der Falle die Tiefe des Speicherpotentials und einzelne Ionen tasten mit
ihren unterschiedlichen Bewegungsamplituden verschiedene Speicherfelder ab, was insgesamt zu
einer Verbreiterung und Verschiebung des Resonanzschwerpunkts einer Frequenzmessung führen
kann. Die genannten Aspekte werden im Folgenden diskutiert.
119
1
1
0.5
0.5
y / x0
y / x0
12.2. DIE PENNINGFALLE
0
-0.5
0
-0.5
-1
-1 -0.50 0.5 1
-1 -0.50 0.5 1
x / x0
(b)
x / x0
1
1
0.5
0.5
y / x0
y / x0
(a)
-1
0
-0.5
-0.5
-1
-1 -0.50 0.5 1
(c)
0
-1
-1 -0.50 0.5 1
(d)
x / x0
x / x0
Abb. 12.8: Einige radiale Projektionen von Ionenbewegungen mit r− = 2, 5r+ . Periodische
Bahnen für√(a) ω+ /ω− = 2; (b) ω+ /ω− = 8; (c) ω+ /ω− = 9/2; (d) quasiperiodische Bahn für
ω+ /ω− = 2 17.
12.2.3.1
Elektrische Feldfehler
Die Harmonizität des elektrischen Potentials ist gestört durch die nur endliche Ausdehnung der
Elektrodenoberflächen, die Bohrungen in den Endkappen der Falle zum Einfang und Ausschuss
der Ionen, die Segmentierung der Ring- und der Endkappenelektroden zum Einkoppeln des
Anregungssignals und zur Detektion des Ionenstromes, sowie durch mechanische Fertigungstoleranzen und Ungenauigkeiten beim Zusammenbau der Fallenelektroden. Das reale Speicherpotential lässt sich für Bewegungsamplituden ρ in der Nähe der Fallenmitte (ρ ρ0 ) durch eine
Entwicklung nach Legendrepolynomen angeben
∞
ρ k
1 X
0
Pk (cos θ) .
(12.17)
V = Videal + V0
Ck
2
d
k=0
Wegen der Symmetrie unter der Transformation von z → −z, verschwinden die ungeraden
Ordnungen in k. Der Term mit k = 2 verändert die Stärke des Quadrupolpotentials und führt
zu einer Modifikation von ωz zu
qV0
(1 + C2 ),
(12.18)
ωz2 =
md2
und damit auch zur Verschiebung von ω+ und ω− mit jeweils entgegengesetztem Vorzeichen.
Die Summenfrequenz ωc bleibt unverändert. Terme höherer Ordnung (k ≥ 4) führen jedoch zu
unerwünschten Frequenzverschiebungen, da diese von den Bewegungsamplituden der gespeicherten Teilchen abhängen. Zusätzliche Elektroden zwischen der Ringelektrode und den Endkappen,
120
KAPITEL 12. IONENFALLEN UND PRÄZISIONSMASSENSPEKTROMETER
1
z/ x0 0
-1
-5
(a)
5
0
0
x/ x0
0
5 -5
y/ x0
1
z/ x0 0
-1
-5
(b)
5
0 y/ x
0
0
x/ x0 0
5 -5
Abb. 12.9: Trajektorien in drei Dimensionen mit den Startbedingungen r− = 50r
√ + , rz = 0, 2r+ .
(a) Periodische Bahn für ω+ /ωz = 6; (b) quasiperiodische Bahn für ω+ /ωz = 35.
bzw. an die Endkappe anschließend, ermöglichen das Anlegen einer Korrekturspannung, die anharmonische Anteile des Potentialverlaufs kompensieren soll.
12.2.3.2
Magnetische Feldfehler
Neben Einflüssen aufgrund von Inhomogenitäten des supraleitenden Magneten führt das starke,
homogene Magnetfeld B0 zu einer Magnetisierung der in den Magneten eingebrachten Materialien der Ionenfallenapparatur und damit je nach Größe und Betrag ihrer magnetischen Suszeptibilität zu lokalen Veränderungen in der Feldstärke und in den Eigenfrequenzen. Die linearen Beiträge zur Magnetfeldänderung werden durch die Ionenbewegung weggemittelt. Terme
der nächsthöheren Ordnung, durch einen quadratischen Offset ∆B(β2 ) zum magnetischen Feld
beschrieben:
∆B
(12.19)
= β2 z 2 − ρ2 /2 · z − z · ρ ,
B0
ergeben analog zum Fall der elektrischen Feldfehler eine Frequenzverschiebung, die von der
Größe der Bewegungsamplituden abhängt:
ρ2+
ρ2−
∆ωc
ωc
ωc
2
= β2 (z −
1−
−
1+
.
(12.20)
ωc
4
ω+ − ω−
4
ω+ − ω−
12.2.3.3
Asymmetrie der Fallengeometrie und Fehljustage
Eine Verkippung der Magnetfeldachse relativ zur Symmetrieachse der Fallenelektroden und
somit zur Achse des elektrischen Potentials, sowie die Abweichung des Quadrupolpotentials
von einer perfekten Zylindersymmetrie können allgemein durch
1
1
1 2
2
2
2
2
2
(12.21)
V = mωz z − (x + y ) − ε (x − y )
2
2
2
beschrieben werden, wobei zusätzlich zum Quadrupolpotential (12.6) die quadratischen
Änderungen durch den Asymmetrieparameter ε modelliert werden. Die Komponenten des
magnetischen Feldes, das um die Winkel θ und φ verkippt ist, schreiben sich zu
Bx = B sinθ cosφ,
(12.22)
121
12.2. DIE PENNINGFALLE
Tabelle 12.1: Eigenfrequenzen νi = ωi /2π von einfach geladenen Ionen
in einer hyperbolischen Penningfalle mit den Betriebsparametern ρ0 =
6,38 mm, z0 = 5,5 mm, V0 = 10 V und B = 7 T für Ionensorten
verschiedener Masse.
A/u
1
133
197
250
ν+
107,6 MHz
804,7 kHz
541,8 kHz
426,0 kHz
νz
982,8 kHz
85,22 kHz
70,02 kHz
62,16 kHz
ν−
4,487 kHz
4,512 kHz
4,525 kHz
4,535 kHz
By = B sinθ sinφ,
(12.23)
Bz = B cosθ.
(12.24)
Als wichtigstes Resultat des so gewonnenen Gleichungssystems folgt eine Invarianzbeziehung
zwischen den Eigenfrequenzen ωi einer nicht vollkommenen Penningfalle analog zu Gl. (12.14)
ω 2c = ω 2+ + ω 2− + ω 2z ,
(12.25)
die dazu dienen kann, die Justage des Magnetfeldes zu überprüfen.
12.2.3.4
Einfluss von gespeicherten Ionen anderer Massen
Werden im Verlauf einer Frequenzmessung mehrere Ionen unterschiedlicher Masse gleichzeitig in
der Penningfalle gespeichert, so können die kontaminierenden Ionen einen Einfluss auf die Lage
der Linienmitte der zu untersuchenden Ionensorte haben. Diese Verunreinigungen sind durch
den Produktionsprozess der Ionen vorgegeben. Beispielsweise handelt es sich an ISOLTRAP
im Wesentlichen um weitere Isobare oder isomere Zustände eines Nuklids. Die Ergebnisse systematischer Untersuchungen mit 10 bis 30 gespeicherten Ionen, sowie einer Simulation zweier
Ionen verschiedener Masse unter Einfluss der Coulombwechselwirkung sind in [Köni1991] und
[Boll1992] diskutiert. Haben die Ionen die gleiche Masse, so werden beide gleichermaßen mit
einem Radiofrequenzfeld angeregt und damit keine Frequenzverschiebung beobachtet. Bei unterschiedlichen Massen hängt das Vorzeichen der Frequenzverschiebung vom Unterschied beider Resonanzfrequenzen relativ zu deren Linienbreiten (FWHM) ab. Sind die Frequenzen beider Ionensorten innerhalb ihrer Linienbreiten nicht zu trennen, so wird eine Resonanzkurve
beobachtet, die schmäler ist als die Überlagerung beider Einzelresonanzen und beide Frequenzen verschieben sich in Richtung des gemeinsamen Schwerpunktes. Ist der Unterschied beider
Ionenmassen so groß, dass deren Resonanzkurven einzeln aufzulösen sind, so beobachtet man eine
Verschiebung beider Resonanzen zu niedrigeren Frequenzen hin. Der Betrag der Verschiebung
ist in beiden Fällen von der Anzahl der gespeicherten Ionen abhängig.
122
12.2.4
KAPITEL 12. IONENFALLEN UND PRÄZISIONSMASSENSPEKTROMETER
Anregung der Ionenbewegung
Jede der drei idealerweise voneinander entkoppelten Eigenbewegungen eines gespeicherten Ions
stellt für sich gesehen einen frequenzscharfen harmonischen Oszillator dar, dem durch die
resonante Einstrahlung eines elektrischen Wechselfeldes von außen Energie zugeführt werden
kann. Abbildung 12.10 zeigt die Energieniveaus der einzelnen Eigenschwingungen, die sich zur
Abb. 12.10: Energiediagramm von harmonischen Oszillatorniveaus eines spinlosen Teilchens
gespeichert in einer idealen Penningfalle (siehe Abb. 12.7). Dabei bezeichnet ω+ die reduzierte
Zyklotronfrequenz, ωz die axiale Frequenz und ω− die Magnetronfrequenz.
Gesamtenergie des gespeicherten Teilchens addieren. Die Besonderheit der Magnetronbewegung
ist, dass sie hauptsächlich durch ihren negativen Anteil an potentieller Energie bestimmt wird.
Ein Erhöhen der Quantenzahl n− bzw. ein Vergrößern des Magnetronradius bedeutet in diesem
Fall einen Verlust an potentieller Energie. Die resonante Dipolanregung kann allgemein dazu
dienen, einzelne Eigenfrequenzen genau zu bestimmen. Eine Quadrupolanregung bei einer Summenfrequenz νi +νj führt zu einer Kopplung der beiden Bewegungen i und j mit einem Übergang
zwischen den einzelnen Niveauschemata. Da die Eigenfrequenzen zum Teil massenabhängig sind
(Gl. (12.5), Gl. (12.10)), lassen sich somit gespeicherte Ionenwolken manipulieren bzw. einzelne
Ionenspezies nach ihrer Masse selektieren. Eine quantenmechanische Betrachtung der Bewegung
eines gespeicherten Ions in der Penningfalle erfolgt in den Übungen.
12.2.4.1
Dipolanregung
Eine Dipolanregung bei einer bestimmten Eigenfrequenz wird im Experiment angewendet, wenn
die einzelne Eigenbewegung einer Ionensorte manipuliert werden soll. Dies kann dazu dienen, die
Bewegungsamplitude bewusst zu vergrößern, oder um schließlich unerwünschte, kontaminierende
Ionensorten aus der Falle zu entfernen. Zuvor muss die Eigenfrequenz über die resonante Anregung bestimmt werden. Sie kann beispielsweise durch eine Minimierung der Ionenzählrate nach
dem Ausschuss aus der Penningfalle erkannt werden. Das Einstrahlen eines Dipolfeldes Ex für
123
12.2. DIE PENNINGFALLE
(a)
(b)
−Uq
r
+Ud
r0
−Ud
r
r0
+Uq
+Uq
−Uq
Abb. 12.11: Radiale Segmentierung der Ringelektrode zur Einstrahlung eines elektromagnetischen Wechselfeldes. (a) Das Einstrahlen einer Radiofrequenz an zwei gegenüberliegenden
Ringsegmenten führt zu einem Dipolfeld. (b) Ein Quadrupolfeld lässt sich durch eine Anregung
zwischen den jeweils gegenüberliegenden Segmenten eines viergeteilten Ringes erzeugen.
die radiale x–Komponente
Ud
· cos (ωrf t − φrf ) · x̂ ,
E~x =
a
(12.26)
geschieht über eine Wechselspannung der Amplitude Ud beim Radius a und der Frequenz ωrf .
Sie wird an zwei gegenüberliegenden Ringsegmenten der Falle (siehe Abb. 12.11 (a)) angelegt.
Wird die Wechselspannung über die Endkappen eingestrahlt, so wirkt sie auf die axiale
Bewegung. Bei geeigneter Phasenbeziehung zwischen Ionenbewegung φion und anregendem Feld
φrf erhält man ein Anwachsen der Bewegungsamplitude, die der Dauer der Anregung Trf und
der Anregungsamplitude Ed proportional ist.
12.2.4.2
Quadrupolanregung
Die Quadrupolanregung bei der Summe von einzelnen Eigenfrequenzen kann zur Kopplung von
Eigenbewegungen sowie zur Frequenzbestimmung benutzt werden. Möchte man z.B. zusammengesetzte Frequenzen messen, wie die zur Massenbestimmung verwendete reine Zyklotronfrequenz ωc = ω+ + ω− (12.13), so erfolgt die Anregung über ein azimutales Quadrupolfeld
mit der Frequenz ωrf , das an den jeweils gegenüberliegenden Segmenten der vierfach geteilten
Ringelektrode eingestrahlt wird (siehe Abb. 12.11 (b)):
2Uq
E~x = 2 · cos (ωrf t − φrf ) · y x̂ ,
a
(12.27)
2Uq
E~y = 2 · cos (ωrft − φrf ) · xŷ .
a
(12.28)
Dies bewirkt eine Kopplung der beiden Radialbewegungen, die im Resonanzfall ωrf = ωc zu einer
vollständigen periodischen Konversion zwischen den beiden Bewegungsradien ρ+ und ρ− führt
[Köni1995a, Köni1995b]. Abbildung 12.12 zeigt die berechnete Entwicklung der Bewegungsradien einer vollständigen Konversion einer anfangs reinen Magnetronbewegung in eine reine reduzierte Zyklotronbewegung. Die radiale kinetische Energie (ohne Dämpfung) Erad ändert sich
124
KAPITEL 12. IONENFALLEN UND PRÄZISIONSMASSENSPEKTROMETER
(a)
(b)
Abb. 12.12: Konversion einer reinen Magnetronbewegung in eine reine Zyklotronbewegung aufgrund der Anregung durch ein azimutales Quadrupolfeld der Zyklotronfrequenz ωc = ω+ + ω− .
Teil (a) und (b) zeigen die erste und die zweite Hälfte der Konversion. Die durchgezogene
Kreislinie in (a) deutet den Startradius der Magnetronbewegung an (aus [Boll1990]).
wegen Erad,i ∼ ρi 2 · ωi 2 ebenfalls periodisch mit T = 2 Tconv , wobei
Tconv = π ·
m a2
a2
·
(ω+ − ω− ) ≈ π
B.
q 2Uq
2Uq
(12.29)
Mit ω+ ω− und somit (ω+ − ω− ) ≈ ωc ist die notwendige Anregungsdauer Trf in erster
Näherung nur durch das Magnetfeld B und die eingestrahlte Amplitude Uq bestimmt.
12.2.5
Massenbestimmung und Kalibrierung
Die Massenbestimmung in einer Penningfalle basiert auf der Bestimmung der Zyklotronfrequenz
νc = qB/(2πm) von Ionen der Ladung q, die in einem homogenen und stabilen Magnetfeld der
Stärke B gespeichert sind. Genauer gesagt, die bestimmbare Größe ist das Ladungs-MasseVerhältnis, wobei normalerweise der Ladungszustand des Ions bekannt ist.
In hochpräzisen Massenmessungen sind die wichtigen Beziehungen zwischen den Eigenfrequenzen der Ionenbewegung und der reinen Zyklotronfrequenz bereits durch die Gleichungen (12.13)
und (12.14) gegeben. Die unabhängige Bestimmung der Eigenfrequenzen und die Benutzung
dieser Gleichungen erlauben die Massenbestimmung der gespeicherten Ionen unter der Voraussetzung, dass die Magnetfeldstärke B bekannt ist. Es gibt zwei Vorgehensweisen: Entweder
werden die Eigenfrequenzen bestimmt, indem die korrespondierenden Bewegungen durch ein RFDipolfeld getrieben werden [Brow1986] oder man bestimmt direkt die Summenfrequenz durch
Quadrupolanregung [Boll1990, Köni1995a].
Damit die Atommasse m aus der gemessenen Frequenz gewonnen werden kann, muss das Magnetfeld kalibriert werden. Dies wird durch Messung der Zyklotronfrequenz eines Referenzions
mit wohlbekannter Masse mref , beispielsweise 23 Na, 85 Rb und 133 Cs, realisiert. Sie wurden in
einem Penningfallenexperiment mit einer relativen Unsicherheit kleiner 2 · 10−10 [Brad1999] bestimmt. Nimmt man an, dass sämtliche Ionensorten einfach geladen sind q = +e, kann die
Atommasse m des interessierenden Ions wie folgt berechnet werden:
m=
νc,ref
(mref − me ) + me ,
νc
(12.30)
125
12.2. DIE PENNINGFALLE
wo νc,ref die Zyklotronfrequenz des Referenzions, νc die Zyklotronfrequenz des interessierenden
Ions, mref die Masse des Referenzions und me die Elektronenmasse bezeichnen. Allerdings trägt
die Unsicherheit der Atommasse des Referenzions auf diese Weise auch zur Unsicherheit des
Massenergebnisses bei.
Deshalb werden die mittels Penningfallen-Massenspektrometrie gewonnenen Massenresultate
meistens in Form des Verhältnisses r der Zyklotronfrequenzen angegeben:
r=
νc,ref
mion
= ion .
νc
mref
(12.31)
Auf diese Weise lässt sich die Masse des interessierenden Ions jederzeit mit dem aktuellsten
bekannten Massewert des Referenzions bestimmen. Das ursprüngliche Ergebnis der Massenspektrometrie ist völlig unabhängig von Änderungen des bekannten Werts der Referenzmasse.
Während bei der Speicherring-Massenspektrometrie die Zyklotronresonanzen der beiden Ionensorten gleichzeitig aufgenommen werden können, da beide gemeinsam gespeichert sind, wird bei
der Penningfallen-Massenspektrometrie die Referenzmessung einmal vor und einmal nach der
Messung des zu untersuchenden Ions durchgeführt. Die gemessenen Zyklotronfrequenzen werden
später auf den Zeitpunkt der Messung des interessierenden Ions interpoliert. Somit besteht eine
komplette Penningfallen-Massenmessung aus drei separaten Zyklotronfrequenzbestimmungen:
der des interessierenden Ions, der vorausgehenden und der nachfolgenden des Referenzions.
Vor kurzem wurde die Möglichkeit aufgezeigt, Kohlenstoffcluster als Massenreferenzen
für hochpräzise on-line Massenmessungen an einfach geladenen Radionukliden einzusetzen
[Blau2002]. Derzeit wird am Penningfallen-Massenspektrometer ISOLTRAP eine neue Methode
zur Magnetfeldkalibrierung mittels einfach geladener Kohlenstoffcluster C+
n (n bis zu 20)
eingesetzt [Kell2003]. Die Vorteile dieser jüngsten Entwicklung sind offensichtlich [Blau2003]:
Es ist eine Vielzahl von Referenzmassen verfügbar, die höchstens sechs Atommasseneinheiten
vom interessierenden Ionen entfernt liegen (siehe Abb. 12.13). Folglich sind systematische
masseabhängige Unsicherheiten, die mit zunehmendem Abstand zwischen der gemessenen Masse
und der Referenzmasse ansteigen, minimiert. Jede Unsicherheit der Referenzmasse ist per
Definition eliminiert und man kann nicht nur direkte sondern auch absolute Massenmessungen
durchführen, da die Masse eines interessierenden Ions direkt auf den mikroskopischen Massenstandard bezogen ist, denn die vereinheitlichte Atommasseneinheit u ist definiert als 1/12 der
Masse von 12 C [Quin1998]. Ein weiterer Vorteil ist, dass Cluster im Allgemeinen mittels CrossReferenzmessungen interne Kontrollen gegen systematische Fehler und die Überprüfung des
gegenwärtigen Genauigkeitslimits für Massenmessungen ermöglichen [Kell2003b, Blau2003b].
Teilweise werden Kohlenstoff-Wasserstoff-Verbindungen als Massenreferenz verwendet, um über
ein ideales q/A-Dublett zu verfügen [Brad1999].
Bei der Massenspektrometrie an hochgeladenen Ionen wie zum Beispiel an SMILETRAP oder
in Speicherringen erhält man die Atommasse durch Korrektur für die fehlenden q Elektronen
mit Masse me und ihre Bindungsenergie EB . Die gesamten Bindungsenergien der Elektronen
werden durch Aufsummierung der relevanten experimentellen Ionisierungsenergien berechnet,
die beispielsweise in [Kell1987] tabelliert sind. Für Ionen mit Z < 20 ist die relative Massenunsicherheit in EB < 10−11 . Für schwerere Atome berechnete Elektronen-Bindungsenergien
haben Massenunsicherheiten von etwa 100 eV [Scof2001]. Für abgeschlossene Elektronenschalen
und Schalen mit einem zusätzlichen oder einem fehlenden Elektron können relativistische Rechnungen mit einer Unsicherheit von der Größenordnung 20 eV [Rodr2001] angestellt werden.
Wie die Massenmessungen an einfach geladenen Ionen, werden bei hochgeladenen Ionen die
idealen Massenmessungen mit verschiedenen Ladungszuständen von 12 C als Massenreferenz
durchgeführt. Die Ladung von 12 C wird so gewählt, dass die Messung an SMILETRAP möglichst
mit q/A Dublett-Ionen durchgeführt wird [Berg2002].
126
KAPITEL 12. IONENFALLEN UND PRÄZISIONSMASSENSPEKTROMETER
Abb. 12.13: Die Nuklidkarte zeigt alle Nuklide mit Halbwertszeiten über 100 ms (offene
Quadrate). Stabile Nuklide sind mit vollen Quadraten und Schalenabschlüsse mit gestrichelten
Linien gekennzeichnet. Der Bereich bekannter Nuklide wird durch die äußeren Linien markiert.
Die durchgezogenen diagonalen Linien zeigen die Isobaren von Kohlenstoffclustern 12 C+
n.
Literatur
[Alhe1996] R. Alheit, K. Enders, and G. Werth: Isotope separation by nonlinear resonances in
a Paul trap, Appl. Phys. B 62, 511 (1996).
[Alhe1996b] R. Alheit, S. Kleinadam, F. Vedel, M. Vedel, and G. Werth: Higher order non-linear
resonances in a Paul trap, Int. J. Mass Spectrom. Ion Processes 154, 155 (1996).
[Alhe1997b] R. Alheit: Speicherung und laserinduzierte Photodissoziation von H+
2 in einer
Paulfalle, Dissertation, Institut für Physik, Universität Mainz (1997).
[Alkh89] Alkhazov, G.D., Berlovich, E.Y., and Panteleyev, V.N., A new highly efficient selective
laser ion source, Nucl. Instr. Meth. Phys. Res. A 280, 141 - 143 (1989).
[Alto1986] G.D. Alton, Surf. Sci. 175 (1986) 226.
[Alto1993] G.D. Alton, Nucl. Instr. and Methods B 73 (1993) 221.
[Ames90] Ames, F., Brumm, T., Jäger, K., Kluge, H.-J., Suri, B.M., Rimke, H., Trautmann, N.,
and Kirchner, R., A high-temperature laser ion source for trace analysis and other applications,
Appl. Phys. B 51, 200 - 206 (1990).
[Asto1929] F.W. Aston, Isotope, Hirzel, Leipzig (1929).
[Audi2003] G. Audi, A.H. Wapstra, C. Thibault, Nucl. Phys. A 729 (2003) 337.
[Aust1992] W.E. Austin, J.H. Leck, and J.H. Batey: Study of the performance of a group of
quadrupole mass spectrometers, J. Vac. Sci. Technol. A 10, 3563 (1992).
[Beck1990] K. Beckert, S. Cocher, B. Franzke, U. Schaaf (Darmstadt, GSI), in: P. Marin, P.
Mandrillon (Eds.), EPAC 90 - Proc. 2nd Europ. Part. Accel. Conf., Nice, 1990., Éditions
Frontières, Gif-sur-Yvette, 1990.
[Berg2002] I. Bergström, C. Carlberg, T. Fritioff, G. Douysset, J. Schönfelder, R. Schuch, Nucl.
Instr. and Meth. A 487 (2002) 618.
[Beth1930] H.A. Bethe, Ann. der Phys. 5 (1930) 325.
[Blau1997] K. Blaum: Optimierung eines Quadrupol-Massenspektrometers zur isotopenselektiven Ultraspurenbestimmung, Diplomarbeit, Institut für Physik, Universität Mainz (1997).
[Blau98] K. Blaum, Ch. Geppert, P. Müller, W. Nörtershäuser, E.W. Otten, A. Schmitt, N.
Trautmann, K. Wendt, and B.A. Bushaw, Properties and performances of a quadrupole mass
filter used for resonance ionization mass spectrometry, Int. Journal Mass. Spec. Ion Proc.,
181, 67-87 (1998).
127
128
LITERATUR
[Blau1998b] K. Blaum, C. Geppert, P. Müller, W. Nörtershäuser, E.W. Otten, A. Schmitt,
N. Trautmann, K. Wendt, and B.A. Bushaw: Properties and performance of a quadrupole
mass filter used for resonance ionization mass spectrometry, Int. J. Mass Spectr. Ion Processes
181, 67 (1998).
[Blau2000] K. Blaum, Resonante Laserionisations-Massenspektrometrie an Gadolinium zur Isotopenhäufigkeitsanalyse mit geringsten Mengen, Dissertation, Johannes Gutenberg-Universität
Mainz (2000).
[Blau2000a] K. Blaum, B.A. Bushaw, Ch. Geppert, P. Müller, W. Nörtershäuser, and K. Wendt:
Peak shape for a quadrupole mass spectrometer: comparison of computer simulation and experiment, Int. J. Mass Spectrom. 202, 81 (2000).
[Blau2002] K. Blaum, G. Bollen, F. Herfurth, A. Kellerbauer, H.-J. Kluge, M. Kuckein, E.
Sauvan, C. Scheidenberger, L. Schweikhard, Eur. Phys. J. A 15 (2002) 245.
[Blau2003] K. Blaum, G. Huber, H.-J. Kluge, L. Schweikhard, Eur. Phys. J. D 24 (2003) 145.
[Blau2003b] K. Blaum, A. Herlert, G. Huber, H.-J. Kluge, J. Maul, L. Schweikhard, Anal.
Bioanal. Chem. 377 (2003) 1133.
[Boer99] J.E. Boers, PBgun - an interactive PC programme for the simulation of electron and
ion beams and guns, Thunderbird Simulations, 1999.
[Boll1990] G. Bollen, R.B. Moore, G. Savard, and H. Stolzenberg: The Accuracy of HeavyIon Mass Measurements using Time of Flight-Ion Cyclotron Resonance in a Penning Trap,
J. Appl. Phys. 68, 4355 (1990).
[Boll1992] G. Bollen, H.-J. Kluge, M. König, T. Otto, G. Savard, H. Stolzenberg, R.B. Moore,
G. Rouleau, G. Audi, and the ISOLDE Collaboration: Resolution of Nuclear Ground and
Isomeric States by a Penning Trap Mass Spectrometer, Phys. Rev. C 46, R2140 (1992).
[Boll2001] G. Bollen, Nucl. Phys. A 693 (2001) 3.
[Boll2004] G. Bollen, Lect. Notes Phys. 651 (2004) 169.
[Bore1974] J. Borer et al., in: Proc. IXth Conf. on High Energy Accelerators, Stanford, 1974,
p. 53.
[Bosh2002] F. Bosch, Nucl. Phys. A 701 (2002) 509c.
[Brad1999] M.P. Bradley, J.V. Porto, S. Rainville, J.K. Thompson, D.E. Pritchard, Phys. Rev.
Lett. 83 (1999) 4510.
[Bran95] B.H. Brandsden and C.J. Joachan, Physics of Atoms and Molecules, Longman publications, Wiley and Sons, New York 1995.
[Brow1986] L.S. Brown and G. Gabrielse: Geonium Theory: Physics of a Single Electron or Ion
in a Penning Trap, Rev. Mod. Phys. 58, 233 (1986).
[Brow1989] I.G. Brown, The Physics and Technology of Ion Sources, Wiley, New York, 1989.
[Brub1961] W.M. Brubaker: The quadrupole mass filter, Proc. Int. Conf. Instrum. Meas. 1, 305
(1961).
129
LITERATUR
[Brub1964] W.M. Brubaker, J. Tuul:
Rev. Sci. Instrum. 35, 1007 (1964).
Performance studies of a quadrupole mass filter,
[Brub1968] W.M. Brubaker: An improved quadrupole mass analyser, Advan. Mass Spectrom.
4, 293 (1968).
[Budk1966] G.I. Budker: An effective method of damping particle oscillations in proton and
antiproton storage rings, Sov. J. Atom. Energy 22, 438 (1967).
[Budk1976] G.I. Budker, N.S. Dikansky, V.I. Kudelainen et al.: Experimental studies of electron
cooling, Part. Acc. 7, 197 (1976).
[Budk1978] G.I. Budker and A.N. Skrinskii: Electron cooling and new possibilities in elementary
particle physics, Sov. Phys. Uspeki 21, 277 (1978).
[BURL2004] BURLE Web Seite: www.burle.com.
[Busc1961] F.v. Busch and W. Paul Über nichtlineare Resonanzen im elektrischen Massenfilter
als Folge von Feldfehlern, Z. Phys. 164, 588 (1961).
[Bush99] B.A. Bushaw, W. Nörtershäuser, and K. Wendt, Lineshapes and optical selectivity
in high-resolution double-resonance ionization mass spectrometry, Spectrochim. Acta B 54,
321-332 (1999).
[Bush01] B.A. Bushaw, W. Nörtershäuser, P. Müller and K. Wendt, Diode-laser-based resonance
ionization mass spectrometry of the long-lived radionuclide 41 Ca with abundance sensitivity <
10 −12 , J. Radioanal. Nucl. Chem. 247, 351-356 (2001).
[Bush03] B.A. Bushaw, W. Nörtershäuser, K. Blaum, K. Wendt, Studies of narrow autoionizing
resonances in gadolinium, Spectrochim. Acta B58, 1083-1095 (2003)
[Carl1970] T.A. Carlson, C.W. Nestor, Jr., N. Wasserman, and J.D. McDowell, At. Data. 2
(1970) 63.
[CERN2005] http://www.cern.ch
[Chen99] C.Y. Chen, Y.M. Li, K. Bailey, T.P. O’Connor, L. Young, Z.-T. Lu, Ultrasensitive
isotope trace analysis with a magneto optical trap, Science 286, 1139 (1999).
[Coc1988] A. Coc, R. Le Gac, M. de Saint Simon, C. Thibault, and F. Touchard, Nucl. Instrum.
Methods Phys. Res. A 271 (1988) 512.
[Cott97] R. J. Cotter, Time of Flight Mass Spectrometry, Americal Chemistry Society prof. Ref.
books 1997.
[Dahl00a] D.A. Dahl, SIMION 3D, Ion Source Software, Idaho National Engineering Laboratory
2000.
[Dahl00b] D.A. Dahl, SIMION for the personal computer in reflection, Int. J. Mass Spec 200,
3-25 (2000).
[Dale1971] H.L. Daley and J. Perel, Rev. Sci. Instrum. 42 (1971) 1324.
[Daly1960] N.R.Daly, Scintillation Type Mass Spectrometer Ion Detector, Rev. Sci. Instr. 3/31
(1960) 264-267.
130
LITERATUR
[Dana1993] H. Danared: Electron cooling at CRYRING, Phys. Scripta 48, 405 (1993).
[Daws1969] P.H. Dawson and N.R. Whetten: Non-linear resonances in quadrupole mass spectrometers due to imperfect fields. II. The quadrupole mass filter and the monopole mass spectrometer, Int. J. Mass Spectrom. Ion Phys. 3, 1 (1969).
[Daws1971] P.H. Dawson: Fringing fields in the quadrupole mass filter, Int. J. Mass Spectrom. Ion Phys. 6, 33 (1971).
[Daws1985] P.H. Dawson: Performance characteristics of an r.f.-only quadrupole, Int. J. Mass
Spectrom. Ion Processes 67, 267 (1985).
[Daws1995] P.H. Dawson (Ed.): Quadrupole Mass Spectrometry and its Applications, AIP, New
York (1995); originally published by Elsevier, Amsterdam (1976)
[Dayt1954] I.E. Dayton, F.C. Shoemaker and R.F. Mozley: The measurement of twodimensional fields. Part II: Study of a quadrupole magnet, Rev. Sci. Instrum. 25, 485 (1954).
[Demt93] Demtröder, W., Laserspektroskopie, Springer, Berlin (1993).
[Deni1971] D.R. Denison: Operating parameters of a quadrupole in a grounded cylindrical housing, J. Vac. Sci. Technol. 8, 266 (1971).
[Du1999] Z. Du, D.J. Douglas, and N. Konenkov: Elemental analysis with quadrupole mass
filters operated in higher stability regions, J. Anal. At. Spectrom. 14, 1111 (1999).
[Duck86] H.E. Duckworth et al, Mass Spectrometry, Cambridge University Press, 1986.
[Fahe1998] A.J. Fahey, Measurements of dead time and characterization of ion counting systems
for mass spectrometry, Rev. Sci. Instrum. 69 (1998) 1282-1288.
[Fisc1958] E. Fischer, O. Osberghaus, and W. Paul:
Ein
Ber. d. Wirtsch. Ministeriums Nordrhein-Westfalen, Nr. 415 (1958).
Ionenkäfig,
Forsch.-
[Fisc1959] E. Fischer: Die dreidimensionale Stabilisierung von Ladungsträgern in einem Vierpolfeld, Z. Phys. 156, 1 (1959).
[Fran1987] B. Franzke: The heavy ion storage and cooler ring project at GSI, Nucl. Inst. Meth.
B24/25, 18 (1987).
[Fran1995] J. Franzen, R.-H. Gabling, M. Schubert, and Y. Wang in: R.E. March, and
J.F.J. Todd (Eds.), Practical aspects of ion trap mass spectrometry. Fundamentals of ion
trap mass spectrometry 1, 49 (1995).
[Fran1995b] B. Franzke, K. Beckert, H. Eickhoff, F. Nolden, H. Reich, U. Schaaf, B. Schlitt, A.
Schwinn, M. Steck, Th. Winkler, Phys. Scr. T 59 (1995) 176.
[Frie1951] H. Friedberg, and W. Paul: Optische Abbildung mit neutralen Atomen, Naturwissenschaften 38, 159 (1951).
[Frit76] J.S. Fritz and G.H. Schenk, Quantitative Analytical Chemistry, Allyn and Bacon Inc.
1976.
[Ghos1995] P.K. Ghosh: Ion Traps, International Series of Monographs on Physics 90, Clarendon Press, Oxford (1995).
LITERATUR
131
[Groß83] J. Großer, Einführung in die Teilchenoptik, Teubner Studienbücher, Stuttgart 1983.
[Gryz1959] M. Gryzinski, Phys. Rev. 115 (1959) 374.
[Hall2003] D. Halliday, R. Resnick, and J. Walker, Physik, WILEY-VCH, Weinheim (2003).
[Haus2000] M. Hausmann, F. Attallah, K. Beckert, F. Bosch, A. Dolinskiy, H. Eickhoff, M.
Falch, B. Franczak, B. Franzke, H. Geissel, Th. Kerscher, O. Klepper, H.-J. Kluge, C.
Kozhuharov, K.E.G. Löbner, G. Münzenberg, F. Nolden, Yu.N. Novikov, T. Radon, H. Schatz,
C. Scheidenberger, J. Stadlmann, M. Steck, T. Winkler, H. Wollnik, Nucl. Instr. and Meth.
A 446 (2000) 569.
[Haus2001] M. Hausmann, J. Stadlmann, F. Attallah, K. Beckert, P. Beller, F. Bosch, H. Eickhoff, M. Falch, B. Franczak, B. Franzke, H. Geissel, T. Kerscher, O. Klepper, H.-J. Kluge,
C. Kozhuharov, Y.A. Litvinov, K.E.G. Löbner, G. Münzenberg, N. Nankov, F. Nolden, Y.N.
Novikov, T. Ohtsubo, T. Radon, H. Schatz, C. Scheidenberger, M. Steck, Z. Sun, H. Weick,
H. Wollnik, Hyperfine Interactions 132 (2001) 289.
[Herr94] Herrmann, G., Kluge, H.-J., Passler, G., Trautmann, N., and Wendt, K.,
Resonanzionisations-Spektroskopie mit Lasern, Phys. Bl. 50 (10) , 929 - 933 (1994).
[Hint97] F. Hinterberger, Physik der Teilchenbeschleuniger, Springer Verlag, Berlin 1997.
[Hoff96] E. de Hoffmann et al, Mass Spectrometry Principles and Applications, Wiley, 1996.
[Hump90] S. Humphries, Charged Particle Beams, John Wiley Sons, Inc., New York 1990.
[Hunt1989] K.L. Hunter and B.J. McIntosh: An improved model of the fringing fields of a
quadrupole mass filter, Int. J. Mass Spectrom. Ion Processes 87, 157 (1989).
[Hurs88] G.S. Hurst and M.G. Payne, Principles and Applications of Resonance Ionisation Spectroscopy, Adam Hilger, Bristol, 1988.
[Jaes1990] E. Jaeschke et al.: First electron cooling of heavy ions at the new Heidelberg storage
ring TSR, Part. Acc. 32, 97 (1990).
[Kell1987] R.L. Kelly, J. Phys. Chem. Ref. Data 16, Supplement no. 1 (1987) 1.
[Kell2003] A. Kellerbauer, K. Blaum, G. Bollen, F. Herfurth, H.-J. Kluge, M. Kuckein, E.
Sauvan, C. Scheidenberger, L. Schweikhard, Eur. Phys. J. D 22 (2003) 53.
[Kell2003b] A. Kellerbauer, Int. J. Mass Spectrom. 229 (2003) 107.
[Kirc1990] R. Kirchner, Nucl. Instr. and Methods A 292 (1990) 203.
[Klie68] H. Klienitz, Hrg., Massenspektrometrie, Verlag Chemie, 1968.
[Klug2003] H.-J. Kluge, K. Blaum, F. Herfurth, W. Quint, Phys. Scr. T 104 (2003) 167.
[Klug2004] H.-J. Kluge, K. Blaum, C. Scheidenberger, Nucl. Instr. and Meth. A 532 (2004) 48.
[Koch74] O.G. Koch und G.A. Koch-Dedic, Handbuch der Spurenanalyse, Springer, Heidelberg,
1974.
[Köni1991] M. König: Massenspektrometrische Auflösung von Isomer und Grundzustand in
einer Penningfalle und Untersuchungen zur Coulombwechselwirkung gespeicherter Ionen,
Diplomarbeit, Institut für Physik, Universität Mainz (1991).
132
LITERATUR
[Köni1995a] M. König, G. Bollen, H.-J. Kluge, T. Otto, and J. Szerypo: Quadrupole Excitation
of Stored Ion Motion at the True Cyclotron Frequency, Int. J. Mass. Spectrom. Ion Process.
142, 95 (1995).
[Köni1995b] M. König: Präzisionsmassenbestimmung instabiler Cäsium- und Bariumisotope in
einer Penningfalle und Untersuchungen der Ionenbewegung bei azimutaler Quadrupolanregung, Dissertation, Institut für Physik, Universität Mainz (1995).
[Köst2003] U. Köster et al., Spectrochim. Acta 58B (2003) 1047.
[Kugl2000] E. Kugler, Hyperfine Interactions 129 (2000) 23.
[Kuts00] W. Kutschera, Das Sortieren von Atomen ”one by one”, Physik in unserer Zeit, 31,
203 (2000).
[Lang1925] I. Landmuir and K.H. Kingdon, Proc. R. Soc. London A 107 (1925) 61.
[Land1938] L.D. Landau and E. Lifsitz, Mechanics, Moscow, Nauka, 1938.
[Lars1995] M. Larsson: Atomic and molecular physics with ion storage rings, Rep. Progr. Phys.
58, 1267 (1995).
[Lee1971] G.E. Lee-Whiting and L. Yamazaki: Semi-analytical calculations for circular
quadrupoles, Nucl. Instrum. Meth. 94, 319 (1971).
[Leo1994] W.R. Leo, Techniques for Nuclear and Particle Physics Experiments. A How-to Approach, Springer, Berlin, New York (1994).
[Leto87] V.S. Letokhov, Laser Photoionization Spectroscopy, Academic Press, Orlando, 1987.
[Litv2004] Yu.A. Litvinov, H. Geissel, Yu.N. Novikov, Z. Patyk, T. Radon, C. Scheidenberger,
F. Attallah, K. Beckert, F. Bosch, M. Falch, B. Franzke, M. Hausmann, Th. Kerscher, O.
Klepper, H.-J. Kluge, C. Kozhuharov, K.E.G. Löbner, G. Münzenberg, F. Nolden, M. Steck,
H. Wollnik, Nucl. Phys. A 734 (2004) 473.
[Litv2005] Yu.A. Litvinov, H. Geissel, T. Radon, F. Attallah, G. Audi, K. Beckert, F. Bosch,
M. Falch, B. Franzke, M. Hausmann, M. Hellström, Th. Kerscher, O. Klepper, H.-J. Kluge,
C. Kozhuharov, K.E.G. Löbner, G. Münzenberg, F. Nolden, Yu.N. Novikov, W. Quint, Z.
Patyk, H. Reich, C. Scheidenberger, B. Schlitt, M. Steck, K. Sümmerer, L. Vermeeren, M.
Winkler, Th. Winkler, H. Wollnik, Nucl. Phys. A 756 (2005) 3.
[Lu02] Z.T. Lu, Atom Trap Trace Analysis, McGraw Hill 2002 Yearbook of Science and Technologies, 2001.
[Lunn1996] D. Lunney et al., Hyp. Interact. 99 (1996) 105.
[Lunn2000] D. Lunney, G. Bollen, Hyperfine Interactions 129 (2000) 249.
[Lunn2003] D. Lunney, J.M. Pearson, C. Thibault, Rev. Mod. Phys. 75 (2003) 1021.
[Luwe03] Z.-T. Lu and K. D. A. Wendt, Laser-Based Methods for Ultrasensitive Trace-Isotope
Analyses, Rev. Sci. Instrum. 74, 1169 (2003).
[Maer1985] T.D. Maerk and G.H. Dunn, Eds., Electron Impact Ionization, Springer, New York,
1985. See, in particular, T.D. Maerk, Partial ionization cross sections, chap. 5 therein, and
G.H. Dunn, Electron-ion ionization, chap. 8 therein.
LITERATUR
133
[Majo2004] F.G. Major, V.N. Gheorghe, and G. Werth: Charged particle traps: The physics
and techniques of charged particle field confinement, Springer (2004).
[Mamy73] B.A. Mamyrin, V.I. Karataev, D.V. Shmikk, V.A. Zagulin, The Mass-Reflectron, a
New Nonmagnetic Time-of-Flight Mass Spectrometer with High Resolution, Sov. Phys.-JETP
Vol.37 No.1, 45 (1973).
[Marc1989] R.E. March and R.J. Hughes: Quadrupole Storage Mass Spectrometry, Chemical
Analysis Series Vol. 102, Wiley Interscience, New York (1989).
[Mars1998] A.G. Marshall, C.L. Hendrickson, and G.S. Jackson: Fourier Transform Ion Cyclotron Resonance Mass Spectrometry: A Primer, Mass. Spectr. Revs. 17, 1 (1998).
[McDa1972] E.W. McDaniel and M.R.C. McDowell, Case Studies in Atomic Collision Physics
II, North Holland, Amsterdam, 1972.
[McIn1989] B.J. McIntosh and K.L. Hunter: Influence of realistic fringing fields on the acceptance of a quadrupole mass filter, Int. J. Mass Spectrom. Ion Processes 87, 165 (1989).
[McLa1947] N.W. McLachlan: Theory and Application of Mathieu-Functions, Clarendon-Press,
Oxford (1947).
[Meix1954] J. Meixner and F.W. Schäfke: Mathieusche Funktionen und Sphäroidfunktionen,
Springer-Verlag, Berlin (1954).
[Mokl1996] P.H. Mokler und T. Stöhlker: The physics of highly-charged heavy ions revealed by
storage/cooler rings, Adv. Atom. Nucl. Opt. Phys. 37, 297 (1996).
[Müll1994] A. Müller: Atomic physics experiments at ion storage rings, Nucl. Inst. Meth. B 87,
34 (1994).
[Müll1997] A. Müller and A. Wolf, Heavy ion storage rings, in: Accelerator-based atomic physics
techniques and applications, (Ed. J. C. Austin and S. M. Shafroth), 147, AIP Press, Woodbury
(1997).
[Munt1995] F. Muntean: Transmission study for r.f.-only quadrupoles by computer simulation,
Int. J. Mass. Spectrom. Ion Processes 151, 197 (1995).
[Nört00] W. Nörtershäuser, B.A. Bushaw, P. Müller, and K. Wendt, Lineshapes in TripleResonance Ionization Spectroscopy, Appl. Optics 39, 5590-5600 (2000).
[Ogat70] K. Ogata and T. Hayakawa, Recent Development in Mass Spectroscopy, University
Park Press, 1970.
[Paul1953] W. Paul and H. Steinwedel:
Z. Naturforschung 8a, 448 (1953).
Ein neues Massenspektrometer ohne Magnetfeld,
[Paul1955] W. Paul and M. Raether: Das elektrische Massenfilter, Z. Phys. 140, 262 (1955).
[Paul1958] W. Paul, H.P. Reinhard, and U.v. Zahn: Das elektrische Massenfilter als Massenspektrometer und Isotopentrenner, Z. Phys. 152, 143 (1958).
[Payn94] Payne, M.G., Deng, L., and Thonnard, N., Applications of Resonance Ionization Mass
Spectrometry, Rev. Sci. Instrum. 65 (8) , 2433 - 2458 (1994).
134
LITERATUR
[Pier54] J.R. Pierce, Theory and Design of Electron Beams, D.V.- Nostrand Comp, Toronto
1954.
[Quin1998] T.J. Quinn, I.M. Mills, in: The International System of Units (SI), The SI Brochure,
7th Edition, Bureau International des Poids et Mesures, Sèvres, France, 1998, p. 152.
[Rado1997] T. Radon, Th. Kerscher, B. Schlitt, K. Beckert, T. Beha, F. Bosch, H. Eickhoff,
B. Franzke, Y. Fujita, H. Geissel, M. Hausmann, H. Irnich, H.C. Jung, O. Klepper, H.-J.
Kluge, C. Kozhuharov, G. Kraus, K.E.G. Löbner, G. Münzenberg, Yu. Novikov, F. Nickel,
F. Nolden, Z. Patyk, H. Reich, C. Scheidenberger, W. Schwab, M. Steck, K. Sümmerer, H.
Wollnik, Phys. Rev. Lett. 78 (1997) 4701.
[Rado2000] T. Radon, H. Geissel, G. Münzenberg, B. Franzke, Th. Kerscher, F. Nolden,
Yu.N. Novikov, Z. Patyk, C. Scheidenberger, F. Attallah, K. Beckert, T. Beha, F. Bosch,
H. Eickhoff, M. Falch, Y. Fujita, M. Hausmann, F. Herfurth, H. Irnich, H.C. Jung, O. Klepper,
C. Kozhuharov, Yu.A. Litvinov, K.E.G. Löbner, F. Nickel, H. Reich, W. Schwab, B. Schlitt,
M. Steck, K. Sümmerer, T. Winkler, H. Wollnik, Nucl. Phys. A 677 (2000) 75.
[Reub1996] A.J. Reuben, G.B. Smith, P. Moses, A.V. Vagov, M.D. Woods, D.B. Gordon, and
R.W. Munn: Ion trajectories in exactly determined quadrupole fields, Int. J. Mass Spectrom. Ion Processes 154, 43 (1996).
[Rodr2001] G.C. Rodrigues, M.A. Ourdane, J. Bieron, P. Indelicato, E. Lindroth, Phys. Rev.
A. 63 (2001) 012510.
[Sait1998] Y. Saitoh, Y. Ohkoshi, W. Yokota, Rev. Sci. Instrum. 69(2) (1998) 703.
[Schl1997] B. Schlitt, K. Beckert, F. Bosch, H. Eickhoff, B. Franzke, Y. Fujita, H. Geissel, M.
Hausmann, H. Irnich, O. Klepper, H.-J. Kluge, C. Kozhuharov, G. Kraus, G. Münzenberg, F.
Nickel, F. Nolden, Z. Patyk, T. Radon, H. Reich, C. Scheidenberger, W. Schwab, M. Steck,
K. Sümmerer, Th. Winkler, T. Beha, M. Falch, Th. Kerscher, K.E.G. Löbner, H.C. Jung, H.
Wollnik, Yu. Novikov, Nucl. Phys. A 626 (1997) 315c.
[Scof2001] J. Scofield, in: Ionization Energies, LLNL Internal report, Livermore, 94550 California, USA, 2001.
[Shor90] B.W. Shore, The Theory of Coherent Atomic Excitation, John Wiley and Sons, New
York, 1990.
[Simo1995] M. de Saint Simon et al., Phys. Scripta T 59 (1995) 406.
[Skoo85] D. A. Skoog, Principles of Instrumental Analysis, Saunders College Publ. Philadelphia,
1985.
[Smit1967] C.J. Smithells, Metals Reference Book, Vol. III, Butterworths, London, 1967.
[Souz1990] A.E. Souzis et al., Rev. Sci. Instrum. 61 (1990) 788.
[Späd91] P. Spädtke, Rev. Sci. Sinst. 63, 2647 (1991).
[Späd00] P. Spädtke, C. Mühle, Rev. Sci. Inst. 71, 820 (2000).
LITERATUR
135
[Stad2004] J. Stadlmann, M. Hausmann, F. Attallah, K. Beckert, P. Beller, F. Bosch, H. Eickhoff, M. Falch, B. Franczak, B. Franzke, H. Geissel, T. Kerscher, O. Klepper, H.-J. Kluge,
C. Kozhuharov, Yu.A. Litvinov, K.E.G. Löbner, M. Matoš, G. Münzenberg, N. Nankov, F.
Nolden, Yu.N. Novikov, T. Ohtsubo, T. Radon, H. Schatz, C. Scheidenberger, M. Steck, H.
Weick, H. Wollnik, Phys. Lett. B 586 (2004) 27.
[Sten1988] R. Stensgaard: ASTRID - The Aarhus storage ring, Phys. Scripta T22, 315 (1988).
[Stöh1998] T. Stöhlker: Atomphysik sehr starker Zentralfelder: Die Röntgenstrahlung der
schwersten Ein- und Zweielektronensysteme, Habilitationsschrift, Johann Wolfgang GoetheUniversität Frankfurt (1998).
[Sute98] M. Suter, A new generation of small facilities for accelerator mass spectrometry, Nucl.
Instr. Meth. Phys. Res. B 139, 150 (1998).
[Sute04] M. Suter, 25 years of AMS - a review of recent developments, Nucl. Instr. Meth. Phys.
Res. B 223, 139 (2004).
[Thom1912] J.J. Thomson, Phil. Mag. 23 (1912) 419.
[Tito1998a] V.V. Titov: Detailed study of the quadrupole mass analyzer operating within
the first, second, and third (intermediate) stability regions. I. Analytical approach,
J. Am. Soc. Mass Spectrom. 9, 50 (1998).
[Tito1998b] V.V. Titov: Detailed study of the quadrupole mass analyzer operating within the
first, second, and third (intermediate) stability regions. II. Transmission and resolution,
J. Am. Soc. Mass Spectrom. 9, 70 (1998).
[Troe1992] J. Trötscher, K. Balog, H. Eickhoff, B. Franczak, B. Franzke, Y. Fujita, H. Geissel,
Ch. Klein, J. Knollmann, A. Kraft, K.E.G. Löbner, A. Magel, G. Münzenberg, A. Przewloka,
D. Rosenauer, H. Schäfer, M. Sendor, D.J. Vieira, B. Vogel, Th. Winkelmann, H. Wollnik,
Nucl. Instr. and Meth. B 70 (1992) 455.
[Tuni98] C. Tuniz, J.R. Bird, D. Fink, G.F. Herzog, Accelerator Mass Spectrometry, CRC Press,
New York, 1998.
[Vály1977] L. Vályi, Atom and Ion Sources, Wiley, London, 1977.
[Vand93] C. Vandecasteele und C.B. Block, Modern Methods for Trace Element Determination,
Wiley Sons, NY, 1993.
[Wang1994] Y. Wang and J. Franzen: The non-linear ion trap. Part 3.: Multipole components
in three types of practical ion traps, Int. J. Mass Spectrom. Ion Processes 132, 155 (1994).
[Wend02] K. Wendt, The New Generation of Resonant Ionization Mass Spectrometers - Becoming Competitive in Selective Atomic Ultra Trace Determination?, Europ. Mass Spec. 8, 273
(2002).
[Wend05] K. Wendt, N. Trautmann, Recent developments in isotope ratio measurements by
resonance ionization mass spectrometry, Int. J. Mass Spectrom. 242, 161 (2005).
[Wert1996] G. Werth, R. Alheit, T. Gudjons, and S. Kleineidam: Some observations on higherorder non-linear resonances in a Paul trap, Rapid. Commun. Mass Spectrom. 10, 583 (1996).
136
LITERATUR
[Whit1986] F.A. White and G.M. Wood, Mass Spectrometry. Applications in Science and engineering, John Wiley & Sons, New York (1986).
[Wile55] W.C: Wiley, J. H. McLaren, Time of Flight Mass Spectrometer with Improved Resolution, Rev. Sci. Instrum. 26, 1150 (1955).
[Wiza1979] J.L. Wiza, Nucl. Instrum. and Meth. 162 (1979) 587-601.
[Wolf1995] B. Wolf, Ion Sources, CRC Press, New York, 1995.
[Woll87] H. Wollnik, Optics of Charged Particles, Academic Press, 1987.
[Woll99] H. Wollnik, Tutorial: Ion Optics in Mass Spectrometers, J. Mass Spec. 34, 991-1006
(1999).
[Wuer1959] R.F. Wuerker, H. Shelton, and R.V. Langmuir: J. Appl. Phys. 30, 342 (1959).