18. Juni 2015 Frühe Nutzenbewertung nach § 35a SGB V
Werbung

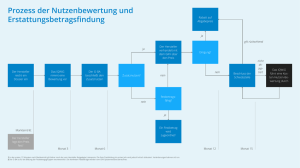
Dr. Christina Diessel Apothekerin TEL (030) 2700406-405 FAX (030) 2700406-9405 [email protected] www.bkk-dachverband.de 18. Juni 2015 Frühe Nutzenbewertung nach § 35a SGB V Im Januar 2011 startet in Deutschland das erste Mal ein Verfahren zur Nutzen-Bewertung neuer patentierter Arzneistoffe. Damit erfolgt ein Paradigmenwechsel in der Versorgung mit neuen Arzneimitteln im ambulanten Bereich. Konnte zuvor der pharmazeutische Unternehmer (pU) den Preis frei festsetzen, muss er sein neues Arzneimittel nun einer Bewertung unterziehen. Auf Basis dieser Bewertung wird ein angemessener Erstattungsbetrag verhandelt, der nach einem Jahr nach Marktzugang wirksam wird. Da das Verfahren im Rahmen des Gesetzes zur Neuordnung des Arzneimittelmarktes (AMNOG) im deutschen Gesundheitswesen etabliert wurde, erhielt die deutsche Zusatznutzenbewertung den weitaus griffigeren Namen „AMNOG-Bewertung“. Daneben wird auch häufig von der frühen Nutzenbewertung gesprochen, weil das Verfahren am Anfang des Lebenszyklus eines neuen Arzneimittels steht. Bei einer AMNOG-Bewertung wird der Zusatznutzen im Vergleich zum aktuellen Therapiestandard bewertet. Der aktuelle Therapiestandard wird nach den Kriterien der Evidenz-basierten Medizin vom G-BA ermittelt und als zweckmäßige Vergleichstherapie (zVT) bezeichnet. Im Gegensatz zu einigen anderen europäischen Ländern wie z. B. United Kingdom oder Frankreich sind neue Arzneimittel weiterhin direkt nach der Zulassung für den Patienten verfügbar und werden von der Gesetzlichen Krankenversicherung (GKV) erstattet. Im ersten Jahr ist die Preisbildung dem pU überlassen. Damit soll der schnelle Marktzugang innovativer Produkte gewährleistet bleiben, so dass eine zeitnahe Versorgung von Patienten in Deutschland sichergestellt ist. Eine AMNOG-Bewertung wird seit mehr als vier Jahren immer dann durchgeführt, wenn ein neues Arzneimittel auf den deutschen Markt kommt oder eine Zulassungserweiterung erhält. Zusätzlich kann eine frühe Nutzenbewertung durchgeführt werden, wenn neue Evidenz für ein zuvor bewertetes Arzneimittel vorliegt. Dahinter steht allerdings kein Automatismus. Der pU kann eine erneute Bewertung seines Arzneimittels beim Gemeinsamen Bundesausschuss (G-BA) beantragen, muss es aber nicht tun! Hat der G-BA einen Nutzenbeschluss befristet, schließt sich nach Ablauf der Frist eine neue Nutzenbewertung an. Eine Befristung wird vom G-BA stets begründet und wird meist in Fällen gewählt, wo zeitnah mit neuer medizinischer Evidenz zu rechnen ist. Die Beweislast für einen Zusatznutzen liegt auf der Seite des pU. Dazu reicht der pU ein Dossier beim G-BA ein. Reicht der pU kein oder ein unvollständiges Dossier ein, kann der G-BA den Zusatznutzen des betroffenen Präparates nicht beurteilen. In der Folge gilt der Zusatznutzen als nicht belegt. Die Bewertung des Dossiers wird innerhalb von drei Monaten nach Start des Verfahrens durchgeführt. In den meisten Fällen wird das Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen (IQWiG) vom G-BA mit der Bewertung betraut. Nach drei Monaten werden das Dossier und dessen Bewertung auf der Homepage des G-BA veröffentlicht und es beginnt ein Stellungnahmeverfahren. Im schriftlichen Teil des Stellungnahmeverfahrens können verschiedene Akteure des Gesundheitswesen Stellung nehmen (z. B. Sachverständige der medizinischen und pharmazeutischen Wissenschaft und Praxis, die Spitzenorganisationen der pharmazeutischen Industrie, betroffene pharmazeutische Unternehmer, die Berufsvertretung der Apotheker und die Dachverbände der Ärztegesellschaften der besonderen Therapierichtung). Neben dem betroffenen pU äußern sich oft der Verband Forschender Arzneimittelhersteller (VFA), die Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) und Fachgesellschaften aus der betroffenen Indikation zu den kritischen Punkten im Bewertungsverfahren. Die strittigen Aspekte werden in einer mündlichen Anhörung im Unterausschuss Arzneimittel (UA AM) des G-BA mit den Stellungnehmern diskutiert. Im Nachgang entscheidet der G-BA auf Basis aller im Verfahren gewonnenen Informationen über den Zusatznutzen des neuen Arzneimittels. Der Beschluss muss innerhalb von sechs Monaten nach Beginn des Verfahrens gefasst werden. Neben dem Ausmaß des Zusatznutzens und dessen Aussagegüte werden weitere Angaben zum Arzneimittel im Beschluss gemacht. Es werden die für die GKV zu erwartenden Jahrestherapiekosten je Patient für das neue Arzneimittel und seine zVT aufgeführt. Auch die maximale Mengenkomponente in Form von betroffenen Patientenzahlen findet Berücksichtigung. Neben dem Beschluss werden u. a. dessen Tragende Gründe und auch das Wortprotoll der mündlichen Anhörung veröffentlicht. Die Öffentlichkeit kann damit große Teile des Verfahrens nachvollziehen. Im folgenden Abschnitt wird auf die verschiedenen Ausprägungen im Nutzenbeschluss näher eingegangen. Die Inhalte bilden die Grundlage für die sich anschließenden Erstattungsbetragsverhandlungen nach § 130b SGB V. Aufteilung in Patientenpopulationen Eine getrennte Bewertung der Zielpopulation wird immer notwendig, wenn verschiedene Komparatoren ausgewählt werden. Die Zielpopulation wird mit der Arzneimittelzulassung festgelegt. Alle Patienten, die mit dem neuen Arzneimittel behandelt werden können, gehören zur Zielpopulation. Innerhalb einer Zielpopulation kann der G-BA verschiedene zVT in Abhängigkeit der verfügbaren medizinischen Evidenz wählen. Bspw. kann eine zVT, die für jüngere Patienten gut geeignet ist, bei älteren Patienten aufgrund von erhöhten Nebenwirkungsarten nicht mehr empfohlen werden. So wird die Indikationspopulation gemäß der Arzneimittelzulassung in verschiedene Anwendungsgebiete (AWG) unterteilt. Innerhalb eines AWG ist eine weitere Aufteilung möglich. Der G-BA und auch das IQWiG haben den gesetzlichen Auftrag den Zusatznutzen differenziert nach verschiedenen Patientenmerkmalen zu bewerten, wenn dieses Merkmal einen Einfluss auf den Effekt des Arzneimittels haben kann. Beispielsweise kann das Alter eines Patienten, eine erfolgte Vortherapie oder das Stadium der Erkrankung zu einer Aufteilung der betroffenen Patientenpopulation führen. Diese Auftrennung wird aus verschiedenen Perspektiven diskutiert. Meist werden die Gruppen post-hoc gebildet, d. h. eine isolierte Betrachtung der Gruppe war vor Beginn der Studie nicht vorgesehen. Mit diesem Vorgehen kauft man sich statistische Probleme ein, die sowohl die Qualität als auch die Quantität der gewon- nen Aussagen stark beeinflussen. Die Gruppenbildung macht in der sich anschließenden Verhandlung die Bildung eines Mischpreises über alle Teilpopulationen mit meist unterschiedlicher Bewertung notwendig. Die Bedeutung des Mischpreises wird hinsichtlich der Wirtschaftlichkeit unterschiedlich interpretiert. Zusatznutzen Der Zusatznutzen eines Arzneimittels kann folgende Ausprägungen annehmen: erheblich, beträchtlich, gering, nicht quantifizierbar und geringer (abfallend geordnet). Die Ausprägung geringerer Zusatznutzen ist verbal besser als „größerer Schaden“ zu beschreiben. Daneben wird die Kategorie „nicht belegt“ immer dann vergeben, wenn eine Bewertung nicht möglich ist (z. B. aufgrund fehlender Daten). Für die Einteilung in die Zusatznutzenkategorie werden die Ergebnisse für patientenrelevante Endpunkte berücksichtigt. Zwar sind die patientenrelevanten Endpunkte in der ArzneimittelNutzenbewertungsverordnung (AM-NutzenV) und der Verfahrensordnung des G-BA in verschiedene Gruppen (Mortalität, Morbidität, gesundheitsbezogene Lebensqualität, Nebenwirkungen) definiert, jedoch wird eine Zuteilung des einzelnen Endpunktes zur einzelnen Gruppe erst durch die Spruchpraxis des G-BA deutlich. Ähnlich wie in Frankreich wird die beste Zusatznutzenkategorie nur für seltene Ausnahmefälle genutzt. Am 19. Februar 2015 wurde der erste Beschluss zu dem Arzneimittel Hemangiol® (Propranolol) gefasst, in dem einem Arzneimittel ein erheblicher Zusatznutzen anerkannt wurde. Kommt der G-BA in seinem Beschluss zu dem Ergebnis, dass der Zusatznutzen nicht belegt ist, soll das neue Arzneimittel in eine Festbetragsgruppe eingeordnet werden. Ist das nicht möglich, schließt sich auch für dieses Arzneimittel mit nicht belegtem Zusatznutzen eine Bestattungsbetragsverhandlung an. Ergebnissicherheit Neben dem Ausmaß des Zusatznutzens wird auch die Aussagesicherheit vom G-BA bewertet. Ein nachgewiesener Zusatznutzen erhält einen Anhaltspunkt, einen Hinweis oder einen Beleg für seine Ergebnissicherheit. Dabei ist der Beleg die höchste Güte, die der G-BA verteilt, abgestuft danach folgen Hinweis und zuletzt Anhaltspunkt. Die Zuordnung im Einzelfall hängt maßgeblich von der Qualität der vorgelegte(n) Studie(n) ab und richtet sich nach international anerkannten Kriterien der Evidenz-basierten Medizin. Für die sich anschließende Erstattungsbetragsverhandlung ist die Nutzenkategorie in Kombination mit dem Grad der Ergebnissicherheit von Bedeutung. Damit ergeben sich bei einem Zusatznutzen 18 Kombinationsmöglichkeiten. (Eine Ausnahme bildet hier die Bewertung von Orphan Drugs, deren Zusatznutzen grundsätzlich mit der Zulassung als belegt gilt. Der G-BA bewertet in seinem Beschluss lediglich das Ausmaß des Zusatznutzens und nicht die Aussagesicherheit.) Epidemiologie Im G-BA-Beschluss wird die Zulassungspopulation des Arzneimittels quantifiziert und stellt die beteiligten Akteure oft vor Herausforderungen. Die Datenlage zu Inzidenz und Prävalenz in den einzelnen Teilpopulationen von Erkrankungen in Deutschland sind häufig nicht gut. Zu betonen ist hier, dass die Anzahl der Patienten im GKV-System in Deutschland dargestellt wird, die gemäß der Arzneimittelzulassung mit dem Arzneimittel therapiert werden könnten. Es handelt sich dabei um eine theoretische Obergrenze bei indikationsgemäßem Einsatz des Arzneimittels. Kosten (inklusive zusätzliche GKV-Kosten) Die Kosten werden als der GKV entstehende Kosten ermittelt, d. h. bei Arzneimitteln werden die gesetzlichen Rabatte abgezogen, um so einen realistischeren Betrag zu erhalten. Es erfolgt eine Gegenüberstellung der Kosten für das neue Arzneimittel und die seiner zVT. Es werden die Jahrestherapiekosten dargestellt. Neben den Arzneimittelkosten können zusätzlich der GKV entstehende Kosten abgebildet werden, wenn sie regulär bei einer Arzneimittelanwendung anfallen. Wenn die gleichen zusätzlichen GKV-Kosten bei einer Therapie mit dem neuen Arzneimittel und seiner zVT anfallen, werden sie im Beschluss nicht berücksichtigt. Beispielsweise werden die Kosten für Blutzuckerteststreifen bei den Jahrestherapiekosten der zVT berücksichtigt, wenn die zVT aus einem insulinhaltigen Therapieregime besteht und das neue Arzneimittel ausschließlich in oraler Form verabreicht wird und eine regelmäßige Blutzuckerkontrolle nicht nötig ist . Dagegen erfolgt keine Berücksichtigung, wenn beide Therapieregime insulinhaltig sind. Grafische Übersicht: frühe Nutzenbewertung nach § 35a SGB V