z - B CUBE Dresden
Werbung

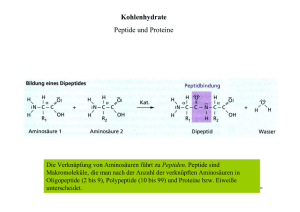
2.2. Peptide Peptide entstehen durch Kondensation der a-Carboxylgruppe einer Aminosäure mit der a-Aminogruppe einer anderen Aminosäure Peptid: bis zu ~30 linear über Peptidbindung verknüpfte Aminosäuren (AAs) Polypeptid(kette): >30 linear über Peptidbindung verknüpfte AAs Protein: gefaltete Polypeptidkette (enthält evtl. chemische Modifikationen) 51 Peptide (I) N-Terminus C-Terminus 5 Aminosäurereste: Pentapeptid Seryl-glycyl-tyrosyl-alanyl-leucin 52 Peptide (II) Ionisierung eines Peptids by pH 7.0 N-Terminus C-Terminus 53 2.3. Proteine Durchschnittliche molekulare Masse pro Aminosäure: 110 Da 54 Bestimmung der molekularen Masse von Proteinen (und anderer Moleküle) mittels Massenspektrometrie (MS): Messung der Wanderungsgeschwindigkeit geladener Moleküle im elektrischen Feld, kann das Masse-zu-Ladung-Verhältnis (m/z) jedes Moleküls bestimmt werden Prinzipieller Aufbau eines Massenspektrometers Elektrospray Ionisierung (ESI) 55 Proteinmoleküle werden in der Regel in einer sauren wäßrigen Lösung (z.B. 0.5% Ameisensäure / 49,75% H2O / 49,75% Methanol) in das Massenspektrometer injeziert. → sämtliche Proteinmoleküle sind dann vielfach positiv geladen → gegenüber dem ungeladenen Protein (a-Aminogruppe und a-Carboxylgruppe am N- bzw C-Terminus und Seitenketten der Aminosäuren sind ungeladen) erhöht sich die Ladung des Proteins mit jedem zusätzlich vorhandenen Proton um +1 und die Masse um +1 Da (Protonenemasse) → ein Proteinmolekül, dass z zusätzliche Protonen enthält besitzt die Ladung z(+1) und ist um die Masse z(1 Da) schwerer als das ungeladene Proteinmolekül; es gilt also: mungeladenes Protein + z mProtein mit zH+ = z z → mungeladenes Protein = z muss aus Massenspektrum berechnet werden ( Vereinfachung: das Vorzeichen der Ladung wird nicht angegeben und die Molekularmasse wird nur als Zahlenwert (ohne Da) angegeben mProtein mit zH+ -1 z ) kann direkt aus Massenspektrum abgelesen werden 56 Wie bestimmt man z aus dem Massenspektrum eines Proteins? Massenspektrum von Apomyoglobin z11 z12 z9 z8 z6 z4 z13 z3 z15 z2 z1 Die verschiedenen Signale stammen von Apomyoglobinmolekülen, die verschieden stark protoniert sind Benachbarte Signale differieren in der Protonenanzahl nur um 1 Proton Je kleiner der m/z Wert desto stärker ist das Proteinmolekül protoniert 57 Für die Signale gilt mProtein mit z H+ mProtein mit z H+ mProtein mit z H+ n 2 1 -1 - 1 =…= zn - 1 = z2 mungel. Protein = z1 z z z ( 1 ) ( ) 2 ( n ) Für die Ladungszahlen direkt benachbarter Signale gilt zn = zn+1 + 1 → Auflösen der obigen Gleichung nach zn ergibt: mProtein mit z zn = zn+1 mProtein mit z H+ n zn - H+ n+1 -1 mProtein mit z zn+1 H+ n+1 58 2.4. Sequenzierung von Proteinen Erstes komplett sequenziertes Protein: Insulin (Frederic Sanger 1955) Allgemeine Strategie der Proteinsequenzierung: a. Irreversible Spaltung der Disulfidbrücken (falls vorhanden) b. Irreversible Spaltung der Polypeptidkette in kleinere (5-20 AAs) überlappende Peptide (Proteolyse) c. Trennung der Peptide mittels Chromatographie d. Bestimmung der AA-Sequenz jedes Peptids e. Rekonstruktion der Proteinsequenz 59 Strategie der Proteinsequenzierung (I) 60 Strategie der Proteinsequenzierung (II) 61 a. Irreversible Spaltung der Disulfidbrücken Disulfidbrücke in Protein Mercaptoethanol Mercaptoethanoldisulfid Jodacetamid Jodwasserstoff carbamidomethylierte Proteine 62 b. Irreversible Spaltung der Polypeptidkette in kleinere (~5-25 AAs) überlappende Peptide (Proteolyse) → durch ortsspezifische Proteasen: 63 → durch Bromcyan (Cyanogenbromid): 64 c. Trennung der Peptide mittels Chromatographie Entscheidend für Auftrennung der Peptide: Unterschiede in Stärke ihrer Wechselwirkung mit der stationären Phase (Ausnahme: Gelfiltration) 65 → geeignete stationäre Phasen für Peptidtrennungen: Anionenaustauscher Kationenaustauscher Polymerkügelchen (Durchmesser: ~5-20 µm) Pore (Innendurchmesser: 2-200 nm) Umkehrphase SiO2-Kügelchen (Durchmesser: ~1-5 µm) Gelfiltration/Gelpermeation 66 d. Bestimmung der Peptidsequenz → durch Edman-Abbau: 67 → durch Edman-Abbau (Fortsetzung): (H+) Nach jedem Abspaltungszyklus: Analyse der erhaltenen PTH-AA mittels RP-Chromatographie 68 → durch Tandem-Massenspektrometrie (MS/MS oder MS2): Peptide (Mutterionen) Peptid Fragmente (Fragmentionen) 69 e. Rekonstruktion der Proteinsequenz → erfolgt Computer gestützt: - Identifizierung der Überlapp-Bereiche und Abfolge der Peptide - Identifizierung der Peptidsequenz aus Massen von Mutterionen und den zugehörigen Fragmentionen 70