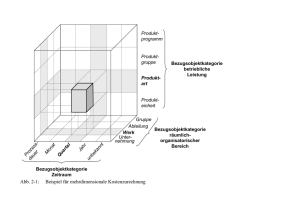
Christian Pasule - des Instituts für Physiologie und Biotechnologie
Werbung

Universität Hohenheim Institut für Physiologie und Biotechnologie der Pflanzen Prof. Dr. Andreas Schaller “Loss of function” Analyse der Matrix Metalloproteinase 1 (Le-MMP 1) in Tomate Diplomarbeit vorgelegt von Christian Pasule Hohenheim, Januar 2006 Betreut von Prof. Dr. Andreas Schaller Inhaltsverzeichnis Zusammenfassung.................................................................................................................... III 1 1.1 1.2 1.3 1.4 1.5 Einleitung ...................................................................................................................... 1 Matrix Metalloproteinasen ............................................................................................. 2 Primäre Zellwand ........................................................................................................... 3 Bakterielle Invasionsmechanismen ................................................................................ 5 Phenolische Verbindungen............................................................................................. 7 Ziele der Arbeit............................................................................................................... 9 2 2.1 2.1.1 2.1.2 2.2 2.3 2.4 2.4.1 2.4.2 2.4.3 2.4.4 2.4.5 2.4.6 2.4.7 Material und Methoden ............................................................................................. 10 Organismen................................................................................................................... 10 Pflanzliches Material .................................................................................................... 10 Bakterien Stämme ........................................................................................................ 10 Vektoren und Plasmide................................................................................................. 10 Oligonukleotide ............................................................................................................ 10 Puffer und Lösungen .................................................................................................... 11 Reverse Transkription .................................................................................................. 11 Gelelektrophorese......................................................................................................... 11 Einbettung von pflanzlichem Gewebe in Technovit 7100® ......................................... 12 Northern Blot ............................................................................................................... 12 Extraktion phenolischer Substanzen............................................................................. 14 Hochdruck-Flüssigkeitschromatographie (HPLC)....................................................... 15 Diskontinuierliche Polyacrylamidgelelektrophorese (DISK-PAGE) und Western Blot................................................................................................................. 15 Sonstige Puffer und Lösungen...................................................................................... 17 Medien.......................................................................................................................... 18 Methoden...................................................................................................................... 18 Reverse Transkription .................................................................................................. 18 Polymerase-Ketten-Reaktion (PCR) ............................................................................ 19 Auftrennung und Sichtbarmachung der DNA.............................................................. 19 Reinigung der PCR-Produkte aus Agarosegelen.......................................................... 20 Reinigung der PCR-Produkte ....................................................................................... 20 Einbettung von Gewebe von Tomate in Technovit 7100® ........................................... 20 Schneiden von Dünnschnitten am Rotationsmikrotom ................................................ 21 Färben mit Toluidin Blau ............................................................................................. 21 Färben mit Sudan Black ............................................................................................... 21 Untersuchung der Segmente mit dem Lichtmikroskop ............................................... 22 Northern Blot................................................................................................................ 22 Hybridisierung von Northern Blots mit radiomarkierten Sonden .............................. 24 Strippen der Blots ......................................................................................................... 25 Extraktion phenolischer Verbindungen (nach Goldwasser et al. 1999) ....................... 25 Bestimmung des Gehalts an löslichen und gebundenen Phenolen (Folin-Ciocalteu) (nach Goldwasser et al,. 1999) ..................................................................................... 26 Bestimmung des Gehalts an Lignin (nach Goldwasser et al., 1999)............................ 26 HPLC-Analyse phenolischer Verbindungen ............................................................... 26 Infektion durch Xanthomonas campestris pv. campestris............................................ 27 Isolierung von Blattproteinen ....................................................................................... 28 2.5 2.6 2.7 2.7.1 2.7.2 2.7.3 2.7.4 2.7.5 2.7.6 2.7.7 2.7.8 2.7.9 2.8 2.8.1 2.8.2 2.8.3 2.8.4 2.8.5 2.8.6 2.8.7 2.8.8 2.8.9 I 2.9 Bestimmung der Proteinkonzentration (Bradford Test) ............................................... 28 2.9.1 Diskontinuierliche Polyacrylamidgelelektrophorese (DISK-PAGE) und Western Blot................................................................................................................. 28 3 3.1 3.2 3.3 3.4 3.5 3.6 Ergebnisse ................................................................................................................... 30 “Silencing” von LeMMP-1........................................................................................... 30 Histologische Untersuchungen..................................................................................... 32 Northern Blot................................................................................................................ 36 Extraktion phenolischer Substanzen............................................................................. 37 HPLC-Analyse phenolischer Verbindungen ................................................................ 39 Infektion durch Xanthomonas campestris pv. campestris............................................ 41 4 Diskusion ..................................................................................................................... 44 5 Literatur ...................................................................................................................... 49 6 6.1 6.2 6.3 Anhang......................................................................................................................... 55 cDNA Sequenz der LeMMP-1 und LeMMP-2 ............................................................ 55 Abkürzungsverzeichnis ................................................................................................ 56 SI-Einheiten.................................................................................................................. 57 Eidesstattliche Erklärung.......................................................................................................... 58 Danksagung.............................................................................................................................. 59 II Zusammenfassung Zusammenfassung Im Vorfeld dieser Arbeit, wurden transgene Tomatenpflanzen von Elke Sieferer erzeugt, die ein Hairpin-Konstrukt zur Unterdrückung der LeMMP-1 Expression durch RNA-Interferenz tragen. Diese transgenen Tomatenpflanzen, dienten in dieser Arbeit als Werkzeug, zur funktionellen Charakterisierung der LeMMP-1. Hairpin-Pflanzen zeigen bereits drei bis vier Wochen nach der Keimung Veränderungen in der Oberflächenstruktur des Hypocotyls. Die Veränderungen, werden drei bis vier Wochen nach der Keimung als dunkle, nekrotisch erscheinende Flecken sichtbar. Mit fortschreitendem Alter der Pflanzen wird das gesamte Hypocotyl von den oberflächlichen Veränderungen erfasst. Mikroskopische Untersuchungen von Dünnschnitten der Hypocotyle von HairpinPflanzen, und deren Färbung mit Toluidin Blau zeigten, dass es zur Akkumulation phenolischer Substanzen in Cortex-Zellen unterhalb der Epidermis kommt. Färbungen mit Sudanfarbstoffen zeigten eine Imprägnierung der Zellwände mit Suberin und Cutin. Weiterhin zeigte sich ein verändertes Zellmuster im Cortex von Hairpin-Pflanzen. Die mikroskopischen Beobachtungen deuten auf eine erhöhte Akkumulation von phenolischen Substanzen in Hairpin-Pflanzen. Extraktion löslicher und gebundener phenolischer Substanzen und deren Konzentrationsbestimmung, bestätigte diese Annahme. Neben der quantitativen wurde auch eine qualitative Veränderung in der Zusammensetzung der phenolischen Substanzen in Hairpin-Pflanzen beobachtet. Die Akkumulation von Phenolen kann auf eine Induktion der Phenylalanin Ammoniumlyase zurückgeführt werden. Induktion der PAL und Akkumulation von Phenolen stellen typische Reaktionen der Pathogenabwehr dar. Daher wurde überprüft, ob es Unterschiede in der Resistenz gegenüber Phytopathogenen zwischen Hairpin und UC82B-Pflanzen gibt. Die Pflanzen wurden mit dem Phytopathogen Xanthomonas campestris pv. campestris Stamm 93-1 infiziert, und eine bakterielle Wachstumskurve wurde erstellt. Es zeigte sich ein vermindertes Bakterienwachstum in Hairpin-Pflanzen. SDS-PAGE und Western Blot Analysen von Proteinextrakten aus Blättern von infizierten UC82B-Pflanzen zeigten eine Induktion der LeMMP-1 infolge der Infektion mit Xanthomonas campestris. Die Befunde deuten auf eine Rolle der LeMMP-1 sowohl in der Pathogenese, als auch in der Entwicklung des Stängel-Cortex in Tomatenpflanzen. III Einleitung 1. Einleitung Die Tomate (Lycopersicon esculentum), hat ihren Ursprung in Mittel- und Südamerika. Sie wurde bereits 200 v. Chr. von den Azteken und Inkas kultiviert. Christoph Kolumbus führte die Tomate nach 1492 in Europa ein. Sie gehört deshalb in Europa zu den hemerochoren Pflanzen, und wegen ihrer Einführung nach 1492 zu den Neozoe. Erste Beschreibungen der Tomate stammen aus Italien aus dem Jahre 1522. In Deutschland begann erst ab 1925 der ``Siegeszug`` der Tomate, die davor eher als Zierpflanze Verwendung hatte. Tomaten sind einjährige, frostempfindliche Pflanzen, die 30-150 cm groß werden können. Die gesamte Pflanze ist mit Drüsenhaaren überzogen. Der Stamm ist nicht verholzt, und macht einen “krautigen” Eindruck. Die Blätter sind meist unregelmäßig fiederschnittig bis lappig gezähnt. Ihre Blütezeit haben sie von Juli bis Oktober, und die Blüten haben eine gelbe Farbe. Die Frucht der Tomate kann je nach Sorte sehr unterschiedlich sein in Größe und Farbe. Botanisch gesehen ist die Tomatenfrucht eine Beere. Heutzutage ist die Tomate eine wichtige und weit verbreitete Kulturpflanze, mit einer enormen wirtschaftlichen Bedeutung. Interessant als Forschungsobjekt ist die Tomate nicht nur aufgrund ihres relativ kleinen Genoms. Sie besitzt 12 Chromosomen bei einer Genomgröße von 950 Mb. Man schätzt, dass ungefähr 730 Mb des Genoms als Heterochromatin vorkommt, welches reich an repetitiven Sequenzen ist. Die restlichen 220 Mb des Genoms kommen als Euchromatin vor, wo man 90% der vorhandenen Gene vermutet (Mueller et al., 2005). Die Tomate ist ausserdem ein diploider Organismus, der leicht mit Agrobakterium tumefaciens zu transformieren ist. Die Fähigkeit zur Selbstbestäubung ermöglicht es, homozygote Inzuchtlinien zu erhalten, wodurch genetische Studien erleichtert werden. In 2003 wurde das “Tomato Sequencing Project” ins Leben gerufen, an dem zehn Länder beteiligt sind, mit dem Ziel das Genom der Tomate zu sequenzieren. Dies geschieht im Rahmen des “International Solanaceae Project”, mit dem Ziel eine Datenbank für den Informationsaustausch zu erstellen (Mueller et al., 2005). Die Tomate dient auch als Modellorganismus um Vorgänge wie Fruchtentwicklung (Tanksley, 2004), oder pflanzliche Abwehrmechanismus gegen phytopathogene Bakterien wie Pseudomonas syringae pv. tomato (Pedley und Martin, 2003) zu studieren. Die Wundantwort der Solanaceen ist in Tomate sehr gut untersucht und stellt ein Modell für systemische Reaktionen in Pflanzen dar (Ryan, 2000). Bei Untersuchungen zur Reaktionen von Tomatenpflanzen auf exogene Gabe des Pilztoxins Fusicoccin, fiel die Matrix-Metalloproteinase 1 (LeMMP-1) auf, welche Gegenstand der 1 Einleitung vorliegenden Arbeit ist. Microarray Analysen zeigten eine rasche und transiente Induktion der LeMMP-1 Expression nach Behandlung mit Fusicoccin, was auf eine mögliche Beteiligung des LeMMP-1 in der Pathogenese hindeutet (Frick und Schaller, 2002). 1.1 Matrix Metalloproteinasen Matrix Metalloproteinasen (MMPs) gehören zur Superfamilie der Metzinkine, der Zinkabhängigen Metallopeptidasen, die durch das Zink-bindende-Motiv HEXXHXXGXXH charakterisiert sind. MMPs werden als PrePro-Enzyme in einer zymogenen Form synthetisiert. Die aminoterminale Transmembrandomäne scheint Teil eines Signalpeptids zu sein, welches das Enzym für den cotranslationellen Transport in das endoplasmatisches Retikulum markiert. In der Propeptiddomäne befindet sich ein “Cystein-Switch”, welches das katalytische Zinkion bindet und das Zymogen dadurch inaktiviert. Durch enzymatische Spaltung der Propeptiddomäne wird das Enzym aktiviert (Bode et al., 1999). Die Regulation der MMPs kann auf transkriptioneller Ebene erfolgen, oder durch eine regulierte enzymatische Spaltung der Propeptiddomäne, was zur Aktivierung des Enzyms führt. Darüber hinaus ist eine Regulierung der Enzymaktivität durch endogene Inhibitoren (TIMPs = Tissue Inhibitors of Metalloproteinases) beschrieben (Gomez et al., 1997). In Vertebraten haben MMPs eine Funktion im Ab- und Umbau der extrazellulären Matrix, in der Entwicklung, Embryogenese und Organogenese. Aktuelle Forschung befasst sich auch mit der Rolle von MMPs bei der Entstehung von Krankheiten wie Krebs und Arthritis. Auch in höheren Pflanzen wurden einige MMPs identifiziert, deren Funktion allerdings noch weitgehend unbekannt ist. Bisherige Befunde deuten darauf hin, dass auch die MMPs in Pflanzen an Entwicklungsprozessen und Pathogenese beteiligt sein können. In Arabidopsis thaliana konnten fünf potentielle MMP-Gene identifiziert werden (Maidment et al., 1999). Für die At1-MMP konnte nach Expression in Escherichia coli die autokatalytische Aktivierung (Abspaltung des Propeptids) gezeigt werden (Maidment et al., 1999). Die At2-MMP scheint eine wichtige Funktion in Pflanzenwachstum, Morphogenese, Entwicklung, im Speziellen bei der Seneszenz und Blütenbildung zu haben (Golldack et al., 2002). In Gurke (Cucumis sativus) wird die Cs1-MMP im Endstadium der Seneszenz exprimiert (Delorme et al., 2000). Für die GmMMP-2 aus Sojabohne, wurde die Akkumulation des Transkripts nach Befall mit Phytophora sojae und Pseudomonas syringae pv. glycinea gezeigt (Liu et al., 2001). 2 Einleitung Die LeMMP-1 aus Tomate weist alle eben genannten typischen Merkmale und Strukturen von Matrix Metalloproteinasen auf. Von zwei potentiellen Transmembrandomänen ist eine Teil des N-terminalen Signalpeptides, die andere bildet einen Membrananker am CTerminus, in dessen Nähe eine ω-site zur Verknüpfung mit einem GPI-Anker prognostiziert wird. Es findet sich ebenso das Zink-Binde-Motiv HEXGHXXGXXH wie auch der konservierte „Met-turn“ im Motiv AIMYP. Im Propeptid befindet sich ein PRCG(V/N)(P/A)D-Motiv mit dem charakteristischen „Cystein-Switch“. Die cDNA Sequenz der LeMMP-1 (EST Klon cLEC15M21, Clemson University) umfasst 1181 Basenpaare mit einem offenen Leserahmen von 1104 Basenpaare und sie codiert für ein PrePro-Protein von 367 Aminosäuren bei einem errechneten Molekulargewicht von 40,2 kDa (Frick, 2002). Für die katalytische Domäne konnte eine caseinolytische und gelatinolytische Aktivität nachgewiesen werden. Chelatoren zweiwertiger Metallionen sowie in geringen Massen thiolmodifizierende Reagenzien hemmten die Aktivität der LeMMP-1. Maximale Aktivität wurde bei pH 6,3 in Gegenwart von 500 µM CaCl2 beobachtet (Frick und Schaller, 2002). Aufgrund der Charakteristika von pflanzlichen MMPs und der für die LeMMP-1 beobachteten Induzierbarkeit durch das Pilztoxin Fusicoccin wurde für die LeMMP-1 eine Funktion im Umbau der Zellwand (Apoplast) im Verbindung mit der Abwehr von mikrobiellen Pathogenen vorgeschlagen (Frick und Schaller, 2002). 1.2 Primäre Zellwand Die primären Wände pflanzlicher Zellen bestehen aus einer amorphen Matrix aus Hemicellulosen und Pektinen, sowie einem geringen Anteil an Strukturproteinen, in welche die Cellulosemikrofibrillen eingebettet sind. Hemicellulosen bilden eine heterogene Gruppe von Polysacchariden. Die am besten untersuchte Hemicellulose bei Dikotyledonen ist das Xyloglucan. Xyloglucan ist ein Polysaccharid mit β-1-4-verknüpften β-D-Glucosebausteinen, mit 1-6-verknüpften Seitenketten aus Xylose, Galaktose und oftmals, aber nicht immer, mit endständiger Fucose. Molekulare Modelle besagen, dass sich die terminale Fucose der Seitenkette zurückfaltet, und dabei mit der Glucankette in Wechselwirkung tritt. Die nächstgelegenen Glucosereste werden dabei in einer linearen Konfiguration fixiert. Die nun entstandene flache Struktur erleichtert die Anlagerung kurzer Bereiche des Xyloglucanrückgrats an die Oberfläche der Cellulosemikrofibrille (Levy et al., 1997). 3 Einleitung Pektine bilden wie Hemicellulosen eine heterogene Gruppe von Polysacchariden. Charakteristischerweise enthalten Pektine saure Zucker wie Galacturonsäure und neutrale Zucker wie Rhamnose, Galactose und Arabinose. Sie sind lösliche Zellwandpolysaccharide und können sehr einfach aufgebaut sein, wie Homogalacturonan (lineares Polymer aus 1-4 verknüpfter α-D-Galakturonsäure). Pektine können aber auch sehr komplex aufgebaut sein wie das Rhamnogalacturonan II, das aus mindestens zehn verschiedenen Zuckern besteht, mit einem komplizierten Verknüpfungsmuster. Pektine bilden molekulare Netzwerke, indem sich die Carboxylgruppen benachbarter Pektinmoleküle über Calcium-Ionen miteinander verbinden. Die Komplexität mancher Pektine deutet darauf hin, dass es sich hierbei nicht ausschließlich um Strukturkomponenten handelt! Eine mögliche Rolle als Elicitoren bei der Pathogenantwort wurde vorgeschlagen (Van Cutsem und Messiaen, 1993). Die Zellwand enthält auch mehrere Typen von Strukturproteinen (Showalter, 1993). Welche anhand ihrer Aminosäurezusammensetzung als hydroxyprolinreiche Glycoproteine, glycinreiche Proteine, prolinreiche Proteine usw. klassifiziert werden. Der Gehalt an Strukturproteinen kann in verschiedenen Zelltypen sehr unterschiedlich sein, und sich im Laufe der Entwicklung sowie in Reaktion auf Umweltstimuli verändern (Showalter, 1993). Die Synthese der Cellulosemikrofibrillen erfolgt an der Plasmamembran durch so genannte Rosettenkomplexe oder Terminalkomplexe (Brown et al., 1996). Die einzelnen Glucanketten der Cellulose bestehen aus 2000 bis 25000 Glucosemolekülen die β-1-4 verknüpft sind (Brown et al., 1996). Mehrere solcher Glucanketten bilden dicht gepackte Mikrofibrillen. Im Zuge der Differenzierung, kann der Primärwand eine Sekundärwand aufgelagert werden. Sekundärwände sind durch den Verlust der Verformbarkeit gekennzeichnet. Sie bestehen aus Cellulosemikrofibrillen, Hemicellulose und Lignin. Cellulosemikrofibrillen und Hemicellulose scheinen in der Sekundärwand strukturell besser organisiert zu sein als in der Primärwand. Die Sekundärwand lässt sich in drei Schichten (S1 bis S3) unterteilen. S1 besteht aus links- und rechts-gedrehten Mikrofibrillenhelices. S2 und S3, bestehen jeweils aus nur einer Art Mikrofibrillenhelices (entweder links- oder rechts-gedrehten). Die Orientierung der Helices in S2 und S3 unterscheiden sich jedoch. Während der Sekundärwandbildung, kommt es in der S1, S2, in der Primärwand und in der Mittellamelle seltener aber in S3 zur Lignifizierung (Goodwin et al., 1983). Sekundärwände sind häufig in Festigungsgewebe und Leitgewebe (Xylem) zufinden. Neben der Funktion in der Formgebung und als Festigungselemente, stellen pflanzliche Zellwände auch eine physikalische Barriere gegen das Eindringen phytophatogener Organismen dar. 4 Einleitung 1.3 Bakterielle Invasionsmechanismen Mikrobielle Pathogene nutzen unterschiedliche Strategien, um Pflanzen zu befallen. Die Invasion von Phytopathogenen kann aktiv oder passiv erfolgen. Viele Pilze durchdringen mit Hilfe von Druck und Zellwand-abbauenden Enzymen die Kutikula und Zellwand, um Pflanzen zu besiedeln. Wunden, sowie natürliche Öffnungen wie Stomata, Lentizellen, Nektarien und Hydathoden (Hugouvieux et al., 1998), um nur einige zu nennen, können ebenfalls Eintrittspforten für Pathogene darstellen. Fusicoccum amygdali, ein Pathogen auf Pfirsich und Mandelbäumen, sondert ein Toxin aus, das Fusicoccin, welches eine ProtonenATPase in der Plasmamembran aktiviert. Dies führt zur Hyperpolarisierung der Plasmamembran, gefolgt von Kalium Einstrom. Das Ergebnis ist eine unphysiologische Öffnung der Stomata, was dem Pathogen die Penetration erleichtert. Im Gegensatz zu Pilzen sind Bakterien nicht in der Lage, aktiv die Kutikula zu penetrieren. Die meisten phytopathogenen Bakterien sind fakultative Pathogene. Sie “warten” auf Gelegenheiten, um über natürliche Öffnungen oder Wunden in den Pflanzenorganismus einzudringen, was durch eine hohe Populationsdichte unterstützt wird. Um eine Infektion zu etablieren, benötigen die Phytopathogene verschiedene Virulenzfaktoren. Als Virulenzfaktoren können hydrolytische Enzyme, Toxine oder sonstige sekretierte Proteine (Effektorproteine) dienen. Xanthomonas campestris pv. campestris, z.B. besitzt eine Metalloprotease, welche in der Lage ist, ein prolinreiches Glycoprotein (gp120) abzubauen, welches mit der extrazellulären Matrix der Leitbündel assoziiert ist. Eine Aktivität wurde auch gegen hydroxyprolinreiche Glycoproteine wie Extensine in Kartoffel und Tomate gezeigt (Dow et al., 1998). Sekretionssysteme von Typ I-IV, wie sie bei vielen gramnegativen Phytopathogenen vorkommen, werden ebenfalls zu den Virulenzfaktoren gezählt. Das hrp-Gencluster (hrp, hypersensitive response and pathogenicity) von Xanthomonas campestris pv. campestris, einem Pathogen welches Paprika und Tomaten befällt, ist notwendig für die Pathogenität und die Induktion der Hypersensitivitätsreaktion (HR) (Bonas et al., 1991). Sequenzanalysen geben Hinweise auf die biochemische Funktion der hrp-Gene. Ähnlichkeiten zwischen hrp-Proteinen von Xanthomonas campestris pv. vesicatoria und Proteinen von Pathogenen von Säugetieren (wie z.B. Ysc und Spa/Mix von Yersinia spp und Shigella flexneri) weisen darauf hin, dass Hrp-Proteine an der Proteinsekretion durch (Fenselau et al., 1992; Van Gijsegem et al., 2000). Typ III Sekretionssysteme (TTSS), sind bei pathogenen Bakterien weit verbreitet. Elektronenmikroskopische Untersuchungen des TTSS von Shigella (Tamano et al., 2000, Blocker et al., 2001) zeigten die Existenz eines 5 Einleitung makromolekularen Komplexes, welcher die bakterielle Membranen durchspannt. Der Komplex besteht aus einem Basalkörper mit zwei oberen und zwei unteren Ringen, welche durch eine sich nach außen fortsetzende nadelähnliche Struktur miteinander verbunden sind („Nadelkomplex“). TTSS sind essentiell für die Pathogenität und Persistenz der Bakterien in den Wirtspflanzen (Minsavage et al., 1990). Xanthomonas campestris pv. vesicatoria Stämme mit einem Defekt im TTSS sind nicht mehr pathogen, und auch nicht mehr zur Vermehrung im Apoplasten fähig, da Sie auf Ressourcen wie Nährstoffe und Wasser aus den Wirtszellen angewiesen sind. Das TTSS wird für die Translokation von Effektorproteinen in die Wirtszelle benötigt, um lebensnotwendige Ressourcen verfügbar zu machen. Zahlreiche potentielle Effektorproteine wurden in Xanthomonas campestris pv. vesicatoria identifiziert, welche mit der TTSS-Pathogenitätsinsel co-reguliert werden (Noel et al., 2001). Ein Effektorprotein, das XopD, welches TTSS-abhängig sekretiert wird (Noel et al., 2002), besitzt eine proteolytische Aktivität gegen SUMO-gebundene Proteine in Wirtszellen. SUMO (small ubiquitin-like modifier) gehören zu einer Familie Ubiquitin-ähnlicher Proteine, welche durch ein System das ähnlich arbeitet wie das Ubiquitinkonjugatiossystem, kovalent mit eukaryontischen Proteinen verknüpft werden können (Melchoir, 2000; Kurepa et al., 2003). SUMO und SUMO Proteasen regulieren zahlreiche zelluläre Prozesse wie Signaltransduktionen, Progression des Zellzyklus, und die Stressantwort bei Pflanzen (Melchoir et al., 2000). Durch de-SUMO-ylierung von Proteinen der Wirtszellen, greift das Pathogen in den pflanzlichen Zellregulierungsmechanismus ein, um sein Fortbestehen in der infizierten Pflanze zu sichern. TTSS sind essentiell für die Pathogenität vieler phytopathogener Bakterien, da sie dadurch die Möglichkeit besitzen, die pflanzliche Zellwand zu überbrücken, und durch Injektion von Effektorproteinen in das Cytoplasma den pflanzlichen Stoffwechsel zu modifizieren (Alfano et al., 1997). Werden aber die Effektorproteine (oder deren Wirkung) von korrespondierenden Resistenzproteinen der Wirtspflanze erkannt, so wirken sie als „avirulence (Avr)“-Faktoren und lösen eine Abwehrreaktion aus, durch die das Wachstum und die Ausbreitung des Phytopathogens limitiert wird (Hotson et al., 2003). Die Verstärkung der pflanzlichen Zellwände durch Lignin oder weitere polymere phenolische Substanzen ist Bestandteil der pflanzlichen Abwehrreaktion (Review, Nicholson und Hammerschmidt, 1992). Durch die Lignifizierung der Zellwände bildet die Pflanze eine mechanische Barriere gegen eingedrungene Phytopathogene. Gleichzeitig erhöht die Lignifizierung die Widerstandsfähigkeit der Zellwände gegenüber zellwandabbauenden Enzymen. Die Diffusion von Toxinen in das Cytoplasma der Wirtszelle und der Austritt von Wasser und Nährstoffen 6 Einleitung aus der Wirtszelle in den Apoplasten werden erschwert. Es wurde gezeigt, dass Infektionen mit Cladosporium cucumerpum in Gurke die Ligninsynthese induziert (Robertson, 1987). Gurken, die resistent gegenüber Cladosporium cucumerpum sind, zeigen eine schnellere Akkumulation von Lignin als nicht-resistente Gurkenpflanzen (Hijwegen, 1963). Werden Kartoffelknollen mit Phytophora infestans infiziert, kommt es ebenfalls zur Einlagerung von polymeren phenolischen Verbindungen in der Knolle (Friend et al., 1973). Möglicherweise handelt es sich dabei um Chlorogensäurepolymere, da nach einer Infektion die Konzentration löslicher Chlorogensäure in der Kartoffelknolle stark absinkt (Friend, 1981). Neben der Rolle von Lignin in der mechanischen Verstärkung der Zellwände während der Pathogenantwort, können Ligninvorstufen und deren freie Radikale möglicherweise auch als toxische Substanzen in der direkten Pathogenabwehr fungieren (Ride, 1978). 1.4 Phenolische Verbindungen Pflanzen sind in der Lage zahlreiche phenolische Substanzen zu synthetisieren, mit vielfältigen Aufgaben. Sie dienen der Pathogenabwehr (Phytoalexine), als Frassschutz (Tannine), zur mechanischen Stabilität (Lignin), als Farbpigmente (Anthocyane) und der Allelopathie (Kaffeesäure). Durch ihre Vielfalt und unterschiedliche Funktionen, stellen phenolische Substanzen eine große heterogene Gruppe sekundärer Inhaltsstoffe in Pflanzen dar, die durch den Phenolring als gemeinsames Strukturmerkmal gekennzeichnet sind. Einige phenolische Verbindungen sind nur in organischen Lösungsmitteln löslich, einige sind wasserlösliche Carbonsäuren, und andere bilden große hydrophobe Makromoleküle. Phenolische Verbindungen werden in Pflanzen über unterschiedliche Stoffwechselwege synthetisiert. Die wichtigsten Stoffwechselwege sind der Shikimisäureweg, und der AcetatMalonat-Weg. Im Shikimisäureweg werden einfache Kohlenhydrat-Vorstufen aus der Glykolyse und dem Pentosephosphatweg in aromatische Aminosäuren umgewandelt. Dadurch sind Pflanzen in der Lage, die drei aromatischen Aminosäuren Phenylalanin, Tyrosin und Tryptophan herzustellen. Aus Phenylalanin entsteht unter Eliminierung von Amoniak die Zimtsäure. Zimtsäure ist ein Vorläufer vieler phenolischer Substanzen. Die Umwandlung von Phenylalanin zu Zimtsäure wird durch das Enzym Phenylalanin-Ammoniak-Lyase (PAL) katalysiert. Die PAL befindet sich an einem Scheideweg zwischen Primär- und Sekundärstoffwechsel, und ist damit ein wichtiger Regulator bei der Bildung vieler phenolischer Verbindungen. Die trans-Zimtsäure wird durch Addition von Hydroxylgruppen 7 Einleitung und weitere Substituenten an den aromatischen Ring in weitere phenolische Verbindungen umgewandelt. Die trans-Zimtsäure und ihre Derivate werden auch Phenylpropane genannt, weil sie einen Benzolring und eine Seitenkette mit drei Kohlenstoffatomen enthalten. Einfache phenolische Verbindungen wie Hydroxyzimtsäuren (Kaffeesäure und Ferulasäure) werden von Pflanzen an die Umgebung abgegeben, und entfalten dort ihre allelopathische Wirkung. In Konkurrenz um Wasser, Licht und Nährstoffe wird so das Wachstum benachbarter Pflanzen beeinträchtigt. Neben einfachen phenolischen Verbindungen, gibt es auch sehr komplex aufgebaute wie z.B. Lignin. Lignin ist ein Polymer aus stark verzweigten Phenylpropaneinheiten. Die genaue Struktur ist jedoch nicht bekannt. Im Allgemeinen besteht Lignin aus drei verschiedenen Phenylpropanalkoholen (Coniferyl-, Cumaryl-, und Sinapylalkohol), die durch Enzyme, welche freie Radikale als Zwischenprodukte erzeugen, zu einem Polymer verknüpft werden. Lignin befindet sich in den Zellwänden von Stützelementen und Leitgeweben. Es verleiht den Pflanzen mechanische Stabilität, und verstärkt das Gewebe des Stamms und der Leitgefäße, und erlaubt so einen aufrechten Wuchs der Pflanzen, und eine Weiterleitung von Wasser und Mineralstoffen durch das Xylem. Die Fähigkeit zur Bildung von Lignin, ist wahrscheinlich eine der wichtigsten Anpassungen der Pflanzen an das Landleben (Taiz und Zeiger, 2000). Suberin besteht wie Lignin aus den drei zuvor genannten Phenylpropanalkoholen (Monolignolen). Die Penylpropane in Suberin sind jedoch mit langkettigen Fettsäuren verestert. Dadurch ist Suberin sehr hydrophob. Aufgrund dieser Eigenschaft wird Suberin zur Bildung wasserundurchlässigen Schichten in den pflanzlichen Organismen verwendet (z.B. Caspary-Streifen der Endodermis, Kork). Flavonoide stellen die umfangreichste Gruppe der phenolischen Verbindungen dar. Sie bestehen aus zwei aromatischen Ringen, die über eine C3 Brücke miteinander verbunden sind. Nur einer der beiden aromatischen Ringe leitet sich wie die zuvor genannten phenolischen Verbindungen von der Zimtsäure ab (B-Ring). Der zweite aromatische Ring (ARing) stammt vom Acetat-Malanat-Weg ab. Flavonoide können als Antioxidantien fungieren (z.B. Quercetin). Sie reagieren mit freien Radikalen, und inaktivieren sie. Sie dienen auch als Frass-Schutz (z.B. Naringenin-7-Rhamnoglucosid in Citrusfrüchten), oder zum Schutz gegen UV-Strahlung. Auch eine wichtige Gruppe pflanzlicher Pigmente, die Anthocyane, gehören zu den Flavonoiden. Viele phenolische Substanzen haben antimikrobielle Wirkung und wirken als Phytoalexine. Sie werden als Reaktion auf Infektionen durch Bakterien oder Pilze von den Pflanzen synthetisiert, und helfen eine Ausbreitung der eingedrungenen Pathogene zu vermindern. Die 8 Einleitung Art der gebildeten Phytoalexine ist charakteristisch für die jeweilige Pflanzenfamilie. Hülsenfrüchtler z.B. bilden üblicherweise Isoflavonoide als Phytoalexine, während in Solanaceaen verschiedene Sesquiterpene anzutreffen sind. Phytoalexine lassen sich normalerweise nicht vor einer Infektion in Pflanzen nachweisen. Wie die Bildung komplexer polymerer Phenole stellt also auch die Bildung von Phytoalexine eine Abwehrreaktion auf Pathogenbefall dar. 1.5 Ziele der Arbeit In dieser Arbeit sollten LeMMP-1 “Loss of function” Tomatenpflanzen charakterisiert werden, um dadurch nähere Erkenntnisse über die Funktion des Enzyms zu bekommen. Die Rolle der LeMMP-1 in der Entwicklung von Tomatenpflanzen sollte histologisch untersucht werden. Die Bildung löslicher und polymerer Phenole in den „loss of function“-Mutanten sollte qualitativ und quantitativ erfasst werden. In wieweit sich die Akkumulation phenolischer Verbindungen auf die Resistenz gegenüber mikrobiellen Pathogenen auswirkt, sollte im Pathosystem Tomate/Xanthomonas campestris pv. Tomate untersucht werden. 9 Material und Methoden 2. Material und Methoden 2.1 Organismen 2.1.1 Pflanzliches Material Lycopersicon esculentum cv. UC82B ROYAL SLUIS (Holland) 2.1.2 Bakterien Stämme Xanthomonas campestris pv. campestris Geschenk von Harry J. Klee Stamm 93-1 Department of Horticultural Sciences of Florida, Gainesville, Florida 32611 2.2 Vektoren und Plasmide PAL (171) 580 bp Fragment von LePAL5 (Görlach et al., 1995) Ubq. (61) 1050 bp Fragment in pSK(-) (Frick und Schaller, 2002) 2.3 Oligonukleotide Primer Sequenz (5’ -----> 3’-Orientierung) MMP 15 CCTTTATTCATCGCCATACT MMP 13 TCTTGTTCGCCGGCCACCGT MMP 25b TGCCATTGCTTATGTTGTAATA MMP 23 GTCTTACCCTCGGGCCACTT ToMV F TCATCTGTATGGGCTGACCCTA ToMV R CAACCGTAGCGTCGTCTACCCT 10 Material und Methoden Aktin F TGTGGGAGATGAAGCTCAATCG Aktin R TCAAACTATCAGTGAGGTCACG PAL F M13 rev PAL R M13-21 Ubq. F T3 Ubq. R T7 2.4 Puffer und Lösungen 2.4.1 Reverse Transkription DNase I Fermentas, St.Leon-Rot M-MuLV Reverse Transkriptase Fermentas, St.Leon-Rot Taq-DNA-Polymerase Peqlab, Erlangen 2.4.2 Gelelektrophorese 50 x TAE-Puffer 242 g Tris/HCl 57,1 ml Essigsäure 100 ml 0,5 M EDTA pH 8,0 mit H2O auf 1 l aufgefüllt Zugabe von 14 µg/l EtBr (Ethidiumbromid) nach Verdünnen 10 x Ladepuffer 50% Glycerin 1 mM EDTA 11 Material und Methoden 2.4.3 Einbettung von pflanzlichem Gewebe in Technovit 7100® Fixierungslösung 5 % v/v Formaldehyd (37 %) 5 % v/v Essigsäure (99 %) 90 % Ethanol (70 %) Dehydrierung 75% Ethanol (v/v) 90% Ethanol (v/v) 100% Ethanol Präinfiltration EtOH/Technovit Basislösung 2:1 EtOH/Technovit Basislösung 1:1 EtOH/Technovit Basislösung 1:2 Technovit Basislösung unverdünnt Infiltrationsmedium Technovit 7100 (GMA mit Co-Katalysator XCL) Polyethylenglycol 400 (100 ml) Dibenzoylperoxid (Härter I) (1 g) Einbettungsflüssigkeit Infiltrationsmedium (30 ml) Technovit 7100 (Härter II) (1,5 ml) 2.4.4 Northern Blot 1 M KPP-Puffer 1 M K2HPO4 1 M KH2PO4 pH 6,5 durch mischen der beiden Lösungen einstellen TE-Puffer 10 mM Tris/HCl pH 8,0 1 mM EDTA 10 x RNA-Gelpuffer 0,4 M MOPS 12 Material und Methoden 100 mM Natriumacetat 10 mM EDTA pH 7,0 mit NaOH einstellen 10 x Ladepuffer 50 % Glycerol 1 mM EDTA Spatelspitze Bromphenolblau 20 x SSC 175,3 g NaCl 100,5 g Na3Citrate x 2 H2O pH 7,0 mit HCl einstellen Mit ddH2O auf 1 l auffüllen 20 % SDS 20 g Natriumdodecylsulfat auf 100 ml ddH2O Hybridisierungslösung 50 % Formamid 5 x SSC 50 mM KPP pH 7,0 Heringssperma DNA, partiell hydrolysiert, denaturiert 100 µg/ml 2 x Denhardt’s Reagenz 0,5 % SDS 50 x Denhardt’s Reagenz 5 g Ficoll 5 g Polyvinylpyrrolidon 5 g Rinderserumalbumin (BSA) Mit ddH2O auf 500 ml auffüllen, sterilfiltrieren Waschlösung 1 1 x SSC 0,5 % SDS 13 Material und Methoden Waschlösung 2 0,2 x SSC 0,5 % SDS Waschlösung zum Strippen 0,04 x SSC 0,04 % SDS 2.4.5 Extraktion phenolischer Substanzen 1 N HCL 8,3 ml HCl 37 % auf 100 ml mit ddH2O aufgefüllt 2 N HCL 16,6 ml HCl 37 % auf 100 ml mit ddH2O aufgefüllt 0,1 N HCL 0,83 ml HCl 37 % auf 100 ml mit ddH2O aufgefüllt 0,5 N NaOH 2 g NaOH auf 100 ml mit ddH2O aufgefüllt 37 % HCL Thioglycolsäure (98 %) Sigma-Aldrich Folin-Ciocalteu’s Phenolreagenz 2 N Sigma-Aldrich 20 % Na2CO3 20 g Na2CO3 auf 100 ml mit ddH2O aufgefüllt p-Cumarsäure Sigma-Aldrich 14 Material und Methoden 2.4.6 Hochdruck-Flüssigkeitschromatographie (HPLC) Phosphorsäure 85 % ROTISOLV® HPLC Gradient Grade Roth, Karlsruhe HPLC-Wasser Acetonitril Roth Zimtsäure (trans) Fluka, Buchs SG Ferulasäure Roth Sinapinsäure Roth Chlorogensäure Sigma-Aldrich Kaffeesäure Roth Methanol Roth 2.4.7 Diskontinuierliche Polyacrylamidgelelektrophorese (DISK-PAGE) und Western Blot Trenngelpuffer (4x) 1,5 M Tris/HCl pH 8,8 0,4 % (w/v) SDS Sammelgelpuffer (4x) 0,5 M Tris/HCl pH 6,8 0,4 % (w/v) SDS APS 10 % 10 % (w/v) Ammoniumperoxidisulfat in H2O 15 Material und Methoden Trenngel 12 %ig, für 2 Gele (1,5 mm) 5,4 ml Acrylamid-Stammlösung (rotiphorese® Gel40, Roth) 4,5 ml Trenngelpuffer 8,1 ml H2O 60 µl APS 10% 13µl Tetramethyläthylendiamin (TEMED) Komponenten mischen, ca. 2/3 hoch in Gelkassette gießen, mit H2O überschichten, polymerisieren lassen (ca. 30 min). Sammelgel 4,5 %ig für 2 Gele (1,5 mm) 0,75 ml Acrylamid-Stammlösung 1,7 ml Sammelgelpuffer 4,25 ml H2O 20 µl APS 10 % 12 µl TEMED Wasser vom festem Trenngel entfernen, Sammelgel auf Trenngel gießen, Taschenkamm einschieben, polymerisieren lassen (ca. 30 min) SDS PAGE-Puffer (Laufpuffer) 50 mM Tris/HCl pH 8,3 384 mM Glycin 0,1 % (w/v) SDS SDS Probenpuffer 200 mM Tris/HCl pH 6,8 400 mM Dithiothreit (DTT) 8 % (w/v) SDS 0,4 % (w/v) Bromphenolblau 40% (v/v) Glycerin Kathodenlösung 40 mM 6-Aminohexansäure 20 % (v/v) Methanol Anodenlösung 1 0,3 M Tris/HCl pH 10,4 20 % (v/v) Methanol 16 Material und Methoden Anodenlösung 2 25 mM Tris/HCl pH 10,4 20 % (v/v) Methanol TBS (Tris-Buffered Saline, 25 mM Tris) 8 g NaCl 0,2 g KCl 3 g Tris in 800 ml H2O lösen, pH auf 7,4 einstellen, mit H2O auf 1 l auffüllen (20x konzentriert hergestellt) TBS/Tween 0,1 % (v/v) Tween®20 in TBS Blockierungslösung 6 % (w/v) Magermilchpulver in TBS Tween sekundärer Antikörper Anti-Rabbit IgG, Heavy and Light Chain (Goat), Peroxidase Conjugate (Calbiochem, Darmstadt) “Enhanced Chemiluminescence” 2.5 Super Signal® West Dura Trial Kit (Pierce) Sonstige Puffer und Lösungen PCR-Puffer (5 x) 15 mM MgCl2 100 mM (NH4)2 SO4 0,08 % Triton X-100 20 % DMSO 250 mM KCl 50 mM Tris/HCl pH 8,3 Protein-Extraktionspuffer 500 mM NaCl 250 mM Tris/HCl pH 7,5 2,5 % (v/v) Triton X-100 50 mM β-Mercaptoethanol 17 Material und Methoden 10 µl/ml Protease-Inhibitor-Cocktail (SigmaAldrich, Steinheim), frisch zugesetzt Toluidin Blau O (Serva) 0,03 g Toluidin Blau O in 100 ml lösen Sudan Black (Serva) 0,1g Sudan Black in 50 ml 96 % Ethanol lösen 50 ml Glycerin hinzufügen 2.6 Medien Nutrient Broth 8 g/l Nutrient Broth No. 4 (Fluka) 15 g/l Agar Autoklaviert (20 min) 2.7 Methoden 2.7.1 Reverse Transkription Die reverse Transkriptionsreaktion wurde mit der RevertAID™ M-MuLV Reverse Transkriptase (MBI-Fermentas) durchgeführt. Für die reverse Transkriptionsreaktion wurde 4µg Gesamt RNA eingesetzt und mit DEPC behandeltem Wasser auf 16 µl aufgefüllt. Zu diesem Volumen wurde 4 µl DNase (RNase frei) und 2 µl 10 x Puffer zugegeben und für 30 min bei 37 °C inkubiert. Nach der Inkubationszeit wurden 2 µl EDTA hinzugefügt und zur Inaktiverung der DNase ein Hitzeschritt (15 min bei 70 °C) durchgeführt. 8 µl des Ansatzes wurden auf ein Kontrollgel aufgetragen. Für die reverse Transkription wurden zu 10 µl des Ansatzes 1 µl 10 mM oligo(dT) hinzugegeben und für 5 min bei 70 °C inkubiert, danach sofort auf Eis. Nun wurde mit 4 µl 5 x Reaktion-Puffer, 2 µl dNTP-Mix (10mM) und mit 2 µl Wasser auf 19 µl aufgefüllt, für 5 min bei 37 °C inkubiert, und wieder auf Eis gestellt. Zum Schluss gab man 200 U der Reversen Transkriptase hinzu und inkubierte 60 min bei 42 °C. 18 Material und Methoden Die Reaktion wurde durch einen Hitzeschritt (15 min bei 70 °C) gestoppt. Für die anschließende PCR wurde je 1,0 µl cDNA verwendet, der Rest wurde bei -20 °C eingefroren. 2.7.2 Polymerase-Ketten-Reaktion (PCR) Die Standard-PCR wurde in einem Volumen von 25/50 µl durchgeführt, die Ansätze enthielten dabei je nach Ausgangsmaterial (Plasmid-DNA, genomische DNA, cDNA) unterschiedliche Mengen an Templat-DNA, 5/10 µl 5x PCR-Puffer, 1/2 µl dNTPs (10 mM), je 1/2 µl “forward” und “reverse” Primer (10 µM) und 1 µl Taq-Polymerase (5 U/µl). Aufgefüllt wurde mit autoklaviertem ddH2O. Die verwendeten Programme bestanden aus folgenden Zyklen: PCR-Programm (1) PCR-Programm (2) PCR-Programm (3) 95 °C/2 min (1 x) 95 °C/5 min (1 x) 95 °C/5 min (1 x) 95 °C/30 sec 95 °C/30 sec 95 °C/30 sec 58 °C/40 sec 30 x 58 °C/30 sec 25-35 x 55 °C/30 sec 72 °C/60 sec 72 °C/45 sec 72 °C/45 sec 72 °C/3 min 72 °C/7 min 72 °C/7 min 35 x 2.7.3 Auftrennung und Sichtbarmachung der DNA Die Auftrennung von DNA erfolgte auf TAE-Agarosegelen (1 % Agarose, 1 µl 1% Ethidiumbromidlösung/ 120 ml Gelvolumen) in TAE-Puffer bei 80 bis 120 V. Vor dem Auftragen der Proben wurden diese mit 10 % (v/v) Ladepuffer versetzt. Als Größenstandard wurden 5 µl 1kb DNA-Ladder (GeneRulerTM 1kb DNA ladder, Fermentas) aufgetragen. Nach der Auftrennung wurde die DNA unter UV-Licht sichtbar gemacht und die Lage im Gel mit dem INFINITY-CAPT-SYSTEM, (Peqlab) dokumentiert. 19 Material und Methoden 2.7.4 Reinigung der PCR-Produkte aus Agarosegelen Das gewünschte DNA-Fragment wurde unter UV-Licht aus dem Gel ausgeschnitten mit Hilfe des QIAquick® Gel Extraction Kits (Qiagen, Hilden) nach Protokoll des Herstellers eluiert. 2.7.5 Reinigung der PCR-Produkte Die amplifizierten DNA-Fragmente wurden mit Hilfe des QIAquick® PCR Purification Kit (Qiagen, Hilden) nach Protokoll des Herstellers gereinigt. 2.7.6 Einbettung von Gewebe von Tomate in Technovit 7100® Hypocotyl, Blattstiel und Wurzel von Tomatenpflanzen (UC82B und “Hair pin”-Pflanzen) wurden unter fließendem Wasser von Verunreinigungen gereinigt, und mit einer Rasierklinge in 0,5 cm große Segmente geschnitten. Die Segmente, wurden in einem Gemisch aus Formaldehyd, Eisessig, und 70 % Ethanol fixiert (5 % Formaldehyd (37 %)), 5 % Eisessig (99 %), 90 % Ethanol (70 %)), welches unter Vakuum infiltriert wurde, bis keine Bläschenbildung mehr zu erkennen war. Die Fixierungslösung wurde 3-mal ausgetauscht. Die Segmente wurden für drei Tage in der Fixierungslösung belassen. Nach der Fixierung erfolgte ein Waschschritt, 30 min mit 75 % Ethanol. Anschließend wurde zweimal mit 75 % Ethanol 30 min infiltriert. Die Segmente wurden über Nacht in 75 % Ethanol belassen. Anschließend wurde das Gewebe dehydriert. Dazu wurde das Gewebe zunächst für sechs Stunden mit 90 % Ethanol, dann für 18 Stunden mit 95 % Ethanol infiltriert, bis keine Bläschen mehr zu erkennen waren. Anschließend wurde dreimal für drei Stunden mit absolutem Ethanol und abschließend für 18 Stunden mit absolutem Ethanol infiltriert. Es folgte die Infiltration eines Gemisches aus zwei Teilen Ethanol und einem Teil Technovit 7100 Basislösung, gefolgt von Ethanol/Technovit 1/1, Ethanol/Technovit 1/2, und reiner Technovit Basislösung. In der Basislösung wurden die Segmente für 18 Stunden belassen. Anschließend wurde Technovit Basislösung mit Härter I infiltriert (Vorbereitungsflüssigkeit). Der Vorbereitungsflüssigkeit wurde der Härter II zugegeben, die Segmente wurden in der 20 Material und Methoden Histoform S ausgerichtet, und bei Raumtemperatur (RT) zur Polymerisation belassen. Nach der Polymerisation, wurden die Technovit Blöcke mit Technovit 3040 auf die Histoblocks aufgeklebt. 2.7.7 Schneiden von Dünnschnitten am Rotationsmikrotom Der Histoblock wurde in den Objekthalter vom Rotationsmikrotom (1165/ROTOCUT, JUNG) fest eingespannt. Durch gleichmäßige Bewegung wurden Schnitte von 5 µm erstellt. Die Sektionen wurden mit einem Pinsel auf die Objektträger (Super Frost® Plus, MenzelGläser) übertragen, auf die vorher Wasser (100 µl) getropft wurde. Anschließend wurden die Schnitte bei 60 °C auf einer Heizplatte gestreckt. 2.7.8 Färben mit Toluidin Blau Die auf den Objektträgern haftenden Schnitte, wurden anschließend zur weiteren Untersuchung gefärbt. Dafür wurde 0,03 g Toluidin Blau O Salz in 100 ml Wasser gelöst. Anschließend gab man einen Tropfen (50 µl) der Färbelösung auf die Schnitte, und inkubierte diese 5 Minuten bei Raumtemperatur. Anschließend wurden sie unter fließendem Wasser gewaschen, um die überschüssige Farbe zu entfernen. 2.7.9 Färben mit Sudan Black 0,1 g Sudan Black Salz wurde in 50 ml 96 % Ethanol gelöst. Anschließend wurde die Suspension filtriert, und mit 50 ml Glycerin versetzt. Zum Färben wurden die Segmente mit je einem Tropfen (50 µl) der Sudan Black Färbelösung für 5 Minuten bei 60 °C inkubiert. Anschließend wurden die Objekte unter fließendem Wasser gewaschen. 21 Material und Methoden 2.8 Untersuchung der Segmente mit dem Lichtmikroskop Die gefärbten Sektionen wurden mit dem Lichtmikroskop (Axioskop 2 plus, Zeiss) im Hellfeld betrachtet. Hierzu wurden verschiedene Vergrößerungen verwendet (x 2,5, x 10, x 40). 2.8.1 Northern Blot Zur Kontrolle der Qualität der RNA und um das Expressionsmuster einzelner Gene zu überprüfen wandte man die Methode der Northern Blot Hybridisierung an. Dazu wurden 4 identische Gele hergestellt, ein Gel für die Ethidiumbromidfärbung, und die restlichen drei Gele zum Blotten. Als erster Schritt wurde ein denaturierendes Gel bestehend aus 3 g Agarose, 180 ml H2O, 25 ml 10 x RNA Gelpuffer und 45 ml Formaldehyd gegossen. Dann wurden 5,5µg RNA (max. 5,5µl) mit 1 µl 10 x RNA Gelpuffer, 3,5 µl Formaldehyde und 10 µl Formamide versehen (Endvolumen 20µl), 15min bei 65°C inkubiert, anschließend auf Eis abgeschreckt und dann anzentrifugiert. Nun wurden 2µl Ladepuffer zugefügt, die Gelkammer mit 1xGelpuffer gefüllt und 20µl der Probe geladen. Bei der Elektrophorese wurde eine Spannung von 120 V angelegt. Bei einer Laufstrecke des Bromphenolblau Farbstoffs von ca. 9 cm wurde die Elektrophorese gestoppt. Nach dem Lauf wurde das Gel zweimal 20 min in einem großen Volumen Wasser geschwenkt, anschließend der Teil für die Ethidiumbromidfärbung abgeschnitten, der anschließend 30min in Wasser mit 1µg/ml Ethidiumbromid inkubiert wurde. Die Gele die für das Blotten vorgesehen waren, wurden noch 15min in 10x SSC gewaschen. Anschließend wurde die Blottingapparatur in einer Glasschale aufgebaut, wobei 10x SSC als Transferpuffer diente. 22 Material und Methoden Aufbau der Blottingapparatur: 200g Glasplatte ca. 5cm Papierhandtücher 3 Lagen Gel-Blotting-Papier GB 002 Nitrocellulosemembran Gel 2 Lagen Gel-Blotting-Papier GB 002 Glasplatte Glasschale 10xSSC Die Gele wurden auf eine “Brücke” aus zwei Lagen Gel-Blotting-Papier GB 002 (Roth) gelegt. Rund um das Gel wurde mit Parafilm abgedichtet. Die Nitrocellulosemembran wurde zuerst mit sterilem ddH2O angefeuchtet, und anschließend in 10 x SSC getaucht. Die Nitrocellulosemembran wurde danach auf das Gel gelegt. Drei Stücke Gel-Blotting-Papier wurden in Größe des Gels geschnitten, mit 10 x SSC angefeuchtet, und einzeln auf die Nitrocellulosemembran aufgelegt. Auf das Gel-Blotting-Papier wurde nun ein Stapel Papierhandtücher aufgelegt, und mit einem Gewicht von ca. 200 g beschwert. Der Transfer erfolgte ÜN bei RT. Das Gel für die Ethidiumbromidfärbung wurde nach der Inkubation mit der Färbelösung über Nacht in einem großen Volumen Wasser gewaschen und am darauf folgenden Tag wurde das Ergebnis mit Hilfe einer Videodokumentationsapparatur festgehalten. Nach dem Transfer, wurde das Papier entfernt, und eine Ecke des Blots wurde zur Markierung abgeschnitten. Der Blot wurde kurz in 2 x SSC geschwenkt, anschließend auf ein Stück Gel-Blotting-Papier mit der Schichtseite nach oben angetrocknet. Danach wurde die RNA durch UV-Licht (Stratalinker, Stratagene) auf der Membran fixiert. Der Blot wurde bei RT vollständig getrocknet, und zwischen Filterpapier bei RT bis zur weiteren Verwendung aufbewahrt. 23 Material und Methoden 2.8.2 Hybridisierung von Northern Blots mit radiomarkierten Sonden Für die Hybridisierung wurden die Blot’s mit ddH2O befeuchtet, und anschließend in 5 x SSC überführt. Danach gab man die Blot’s aufgerollt und mit der RNA-Seite nach oben in die Hybridisierungsröhren, und man setzte 20 ml Prä-Hybridisierunslösung hinzu. Die PräHybridisierung erfolgte mindestens 2 Stunden bei 42 °C. Für die Markierung der Proben wurde 25 ng DNA in 21 µl ddH2O 5 min aufgekocht, danach auf Eis abgekühlt, und kurz anzentrifugiert. Auf Eis wurde hinzugefügt: 1 µl 500 µM dATP 1 µl 500 µM dTTP 1 µl 500 µM dGTP 20 µl 2,5 x Random Primer Lösung 5 µl (50 µCi) (α-32P)-dCTP mix 1 µl Klenow-Fragment Mischen, kurz anzentrifugieren, 10 Minuten bei 37 °C inkubieren. Danach 5 µl Stop-Puffer hinzufügen, gut mischen und kurz anzentrifugieren. Zur Reinigung der Sonde, wurde eine Bio-Spin-Column (BioRad) unten geöffnet, in ein 2 ml Eppendorfgefäß gestellt, und die Lösung innerhalb der Säule wurde entfernt. Es wurde 2 Minuten bei 3500 rpm (1000 x g) zentrifugiert, und der Durchlauf entfernt. Das Säulchen wurde in ein 1,5 ml SchraubdeckelEppendorfgefäß überführt. In der Mitte der Säule wurden 55 µl der radioaktiven Probe aufgetragen, und 4 Minuten bei 3500 rpm (1000 x g) zentrifugiert. Die benutzte Säule wurde entsorgt, und die Aktivität im Durchlauf wurde bestimmt. Die Probe wurde anschließend 5 Minuten gekocht, auf Eis abgeschreckt und kurz anzentrifugiert. Aus der Hybridisierungsröhre wurde die Prähybridisierungslösung entfernt und durch 10 ml Hybridisierungslösung ersetzt. Der Lösung wurde die denaturierte Sonde zugesetzt, und die Hybridisierung erfolgte bei 42 °C ÜN. Am darauffolgenden Tag wurden die Filter gewaschen. Die Hybridisierungslösung wurde ausgegossen und es wurde kurz mit Waschlösung 1 gespült. Dann wurde 5min in Lösung1 gewaschen, währenddessen auf 60°C geheizt wurde. Anschließend wurde 30min bei 60°C in Lösung 1 und 30min bei 60°C in Lösung2 gewaschen. Nach dem Waschen wurde der Filter in eine Schale mit Lösung2 überführt, zwischen Frischhaltefolie eingepackt damit diese nicht austrocknen und je nach Stärke der Aktivität zwischen 5-14 Tagen bei -80 °C auf einen Röntgenfilm exponiert. 24 Material und Methoden 2.8.3 Strippen der Blots Die Membranfilter wurden aus der Frischhaltefolie ausgepackt, in eine Glasschale mit etwas Waschlösung überführt. 400 ml der Waschlösung wurde vorher in einer Glasschale in der Mikrowelle bis zum Siedepunkt erhitzt, und die Membranfilter wurden damit übergossen. Anschließend wurde die Glasschale mit der Waschlösung und den Membranfilter nochmals für zwei Minuten in die Mikrowelle gestellt, und danach ließ man sie auf einen Schüttler abkühlen. Diese Waschung wurde noch zweimal wiederholt. Nach der Waschung wurden die Membranfilter auf Filterpapier getrocknet und bei RT gelagert. 2.8.4 Extraktion phenolischer Verbindungen (nach Goldwasser et al., 1999) Hypocotyle und Blattstiele von vier Wochen alten Tomaten-Pflanzen, wurden in flüssigem Stickstoff gemörsert. Anschließend wurde das Pflanzenpulver, in ein Eppendorfgefäß mit 1 ml 50 % Methanol übertragen, und 90 Minuten bei 80 °C inkubiert, um die löslichen Phenole zu extrahieren. Nach anschließender Zentrifugation bei 14 000 rpm für 15 Minuten in der Eppendorf Zentrifuge 5402, wurde der Überstand dekantiert und bis zur Bestimmung der löslichen Phenole im Kühlschrank gelagert. Zur Hydrolyse gebundener Phenole wurde das Sediment in 1 ml 0,5 N NaOH aufgenommen, und für 24 Stunden bei RT geschüttelt. Anschließend wurde mit 0,5 ml 1 N HCl neutralisiert. Nach Zentrifugation (15 min, 14 000 rpm in Eppendorf 5402) wurde der Überstand dekantiert und zur Bestimmung der gebundenen Phenole verwendet. Das Sediment, wurde zweimal mit je 1 ml ddH2O und einmal mit 1 ml Methanol gewaschen, und abzentrifugiert (15 min, 14 000 rpm in Eppendorf 5402). Nach der letzten Zentrifugation wurde der Überstand verworfen, und das Sediment für 48 Stunden in Alufolie bei 37 °C getrocknen. Anschließend wurde das Trockengewicht des Sediments bestimmt. Zur Bestimmung des Ligningehaltes wurde das trockene Sediment in 1 ml 2 N HCl mit 100 µl Thioglycolsäure aufgenommen, und für vier Stunden bei 95 °C im Heizblock (Eppendorf, Thermomixer Comfort) inkubiert. Nach vier Stunden wurde auf Eis abgekühlt, und 10 Min bei 14 000 rpm zentrifugiert. Der Überstand wurde verworfen, und das Sediment wurde zweimal in je 1 ml ddH2O gewaschen und zentrifugiert (15 min, 14 000 rpm, in Eppendorf 5402). Anschließend wurde das Sediment in 1 ml 0,5 N NaOH resuspendiert und ÜN bei 4 °C geschüttelt. Unlösliche Bestandteile wurden abzentrifugiert (10 min, 14 000 rpm), und der Überstand abgenommen. Das Sediment wurde in 0,3 ml ddH2O gewaschen und 25 Material und Methoden abzentrifugiert (10 min, 14 000 rpm), und der Überstand wurde mit dem Überstand vom vorherigen Schritt vereinigt. Die vereinigten Überstände wurden mit 0,2 ml konzentrierter HCl angesäuert, und bei 4 °C ÜN gefällt. Das Präzipitat wurde abzentrifugiert (15 min, 14 000 rpm), mit 1 ml 0,1 N HCl gewaschen, und erneut abzentrifugiert. Das Präzipitat wurde nun in 1 ml 0,5 N NaOH aufgenommen. 2.8.5 Bestimmung des Gehalts an löslichen und gebundenen Phenolen (Folin-Ciocalteu) (nach Goldwasser et al., 1999) Jeweils 100 µl der Methanol (lösliche Phenole) bzw. NaOH (gebundene Phenole) Extrakte, wurden mit 900 µl ddH2O auf 1 ml aufgefüllt, mit 0,5 ml 2 N Folin-Ciocalteu’s Phenol Reagenz versetzt, gut gemischt und für 3 Minuten bei RT inkubiert. Nach 3 Minuten wurde 2,5 ml 20 % Na2CO3 hinzugegeben, ordentlich vermischt, und für eine Stunde bei RT stehengelassen. Anschließend wurde die Konzentration der Phenole als p-CumarsäureÄquivalente, bei einer Wellenlänge von 725 nm im Photometer (Carry 100 BIO) ermittelt. 2.8.6 Bestimmung des Gehalts an Lignin (nach Goldwasser et al., 1999) Der Ligningehalt wurde Photometrisch bei 280 nm als Lignin-ähnliche Thioglycolsäurederivate bestimmt. Die Proben wurden dazu 10fach verdünnt, und die Absorption (OD280, Eppendorf, Bio Photometer) wurde ermittelt. Die Werte wurden angegeben als Extinktion/gTrockengewicht 2.8.7 HPLC-Analyse phenolischer Verbindungen Die Chromatographie gehört zu den Verfahren, bei denen Stoffgemische untersucht werden, indem man sie in ihre einzelnen Bestandteile auftrennt. Basis der Auftrennung ist die unterschiedliche Verteilung der Stoffe in zwei Phasen. Die mobile Phase (Flüssigkeit oder Gas) bewegt sich an der stationären Phase (Feststoff oder Flüssigkeit) vorbei und nimmt die Stoffe dabei unterschiedlich schnell mit. 26 Material und Methoden Bei der Säulen-Chromatographie befindet sich die stationäre Phase in einer Säule (NUCLEOSIL 5, C 18, 120A), durch die die mobile Phase fließt (Laufmittel A: 0,25 % H3PO4 in HPLC-Wasser: Laufmittel B: 0,25 % H3PO4 in Acetonitril). Die zu trennenden Proben werden auf der Säule festgehalten, und durch einen Gradienten, bestehend aus den Laufmitteln A und B von der Säule eluiert. Die Detektion erfolgte bei einer Wellenlänge von 254 nm. Injiziert wurde ein Volumen von 50 µl der methanolischen bzw. NaOH-Extrakte, bei einer Durchlaufgeshwindigkeit von 1 ml/min. Das verwendete HPLC-System war die La Chroma®, von Hitachi. Verwendete Gradienten: Zeit (min) Laufmittel A % Laufmittel B % Durchlauf (ml/min) 0 100 0 1 30 65 35 1 40 0 100 1 40,1 100 0 1 65 100 0 1 Zeit (min) Laufmittel A % Laufmittel B % Durchlauf (ml/min) 0 100 0 1 50 65 35 1 60 0 100 1 60,1 100 0 1 85 100 0 1 2.8.8 Infektion durch Xanthomonas campestris pv. campestris Sieben Wochen alte Tomatenpflanzen (5 x UC82B und 5 x HP 1.3 abc) wurden mit dem Pflanzenpathogen Xanthomonas campestris pv. campestris Stamm 93-1 infiziert. Die Bakterien wurden für drei Tage bei 30 °C auf Nutrient Broth-Medium (NB) (8 g NB mit 15 g Agar/l) kultiviert. Anschließend wurden über Nacht (ÜN) Kulturen von den Bakterien in NB Flüssigmedium angesetzt (8 g NB/l). Am folgenden Tag wurde die Bakteriensuspension auf einem OD600 Wert von 0,4 mit ddH2O verdünnt. Die Suspension wurde nochmals 1:100 27 Material und Methoden mit ddH2O verdünnt, zu einer endgültigen Konzentration von 106 colony-forming-units/ml (cfu/ml). Die Suspension (50 µl, 50 000 cfu) wurde anschließend mit einer Spritze (ERSTA® 1 ml) in die Blätter infiltriert. Infizierten Blätter von jeder Pflanze wurden nach 24 Stunden mit einem Skalpell entfernt, und in einem 2 ml Eppendorfgefäß in 1 ml Proteinextraktionspuffer mit 10 µl Protease-Inhibitor-Cocktail mit einem Minithorax (POLYTRON PT 1200, KINEMATICA AG) homogenisiert. Anschließend wurde eine Verdünnungsreihe in ddH2O erstellt (103, 104, 105, 106, 107-pro Probe/Pflanze), und je 100 µl (pro Probe/Pflanze) auf NB-Platten ausplattiert. Die Platten wurden für drei Tage bei 30 °C kultiviert, wonach die Anzahl der Kolonien auf den verschiedenen Platten ermittelt und gemittelt wurde. Anhand dieser Werte wurde eine Bakterienwachstumskurve mit Hilfe des Computerprogramms EXEL erstellt. Für das Experiment wurden fünf HP 1.3 abc und fünf UC82B benutzt. 2.8.9 Isolierung von Blattproteinen Die Extrakte von 2.8.7 wurden 5 Minuten bei 14 000 rpm und 4 °C in Eppendorf 5402 zentrifugiert. Der Überstand wurde in ein frisches Reaktionsgefäß überführt. Die Aufbewahrung des Proteinextraktes erfolgte bei -80 °C. 2.9 Bestimmung der Proteinkonzentration (Bradford Test) Zur Messung der Proteinkonzentration der Extrakte wurde von einer 1:250 –Verdünnung, 800 µl entnommen, und mit 200 µl Bradford Reagenz (Roti®-Quant, Roth) versetzt. Nach 10 Minuten bei RT wurde die Proteinkonzentration über die Absorptionsänderung bei 595 nm gegen BSA (Rinderserumalbumin Fraktion V, Roth, Karlsruhe) als Standard ermittelt. 2.9.1 Diskontinuierliche Polyacrylamidgelelektrophorese (DISK-PAGE) und Western Blot Proteinextrakte wurden 3:1 mit SDS Probenpuffer versetzt, für 10 min im Wasserbad gekocht und über Polyacrylamidgele (12 %) in SDS PAGE-Puffer bei 100 V aufgetrennt. Als 28 Material und Methoden Größenstandard wurden 3 µl „PageRulerTM Prestained Protein Ladder“ (Fermentas) aufgetragen. Anschließend wurden Proteine vom Gel auf Blotting Membranen (Westran® Clear Signal, zugeschnitten auf 9 x 5,5 cm, Schleicher & Schuell, Dassel) übertragen. Das ebenfalls auf 9 x 5,5 cm zugeschnittene Blotting-Papier (NOVABLOT Elektroden-Papier, Amersham) wurde 10 min in den entsprechenden Puffern gewässert (drei Papiere in Anodenlösung 1, sechs Papiere in Anodenlösung 2, sechs Papiere in Kathodenpuffer). Die Membran wurde kurz in Methanol getaucht und dann 10 min in Anodenlösung 1 gewässert. Die Graphitelektroden wurden etwa 30 min in H2O gewässert, abgetrocknet und die Blotting Apparatur wie aus der Abbildung ersichtlich luftblasenfrei aufgebaut. Kathode 6 Lagen Papier in Kathodenlösung Gel Membran 3 Lagen Papier in Anodenlösung 1 6 Lagen Papier in Anodenlösung 2 Anode Nach elektrophoretischem Transfer während 1,5 h bei 100 mA/Blot wurden die Membranen ÜN in Blockierungspuffer (6 % Magermilchpulver in TBS/Tween) unter Schütteln bei 4 °C abgesättigt. Die Membranen wurden anschließend mit dem primären Antikörper (antiLeMMP1, 1:400 in Blockierungspuffer) zwei Stunden bei RT auf einem Schüttler inkubiert. Die Membranen wurden anschließend dreimal während 5 Minuten mit TBS/Tween gewaschen. Die Inkubation mit dem sekundären Antikörper (Anti-Rabbit IgG, Heavy and Light Chain (Goat) Peroxidase Conjugate (Calbiochem, Darmstadt), 1:10.000 in Blockierungspuffer) erfolgte während 1 h bei Raumtemperatur. Anschließend wurde wie oben dreimal mit TBS/Tween gewaschen und der Blot mittels enhanced chemiluminescence (ECL) entwickelt. Dazu wurden 200 µl frisch angesetzte ECL-Lösung (Super Signal® West Dura Trial Kit) für 5 min hinzugegeben. Nach dem Abtropfen des Blots wurde dieser in Folie gelegt und in der Dunkelkammer auf Röntgenfilm (CL-XPosure™ Film, Pierce) exponiert. 29 Ergebnisse 3. Ergebnisse 3.1 “Silencing” von LeMMP-1 Zur funktionellen Charakterisierung der LeMMP-1 in Tomatenpflanzen sind im Vorfeld dieser Arbeit von Elke Sieferer transgene Pflanzen erzeugt worden, die ein Hairpin-Konstrukt zur Unterdrückung der LeMMP-1 Expression durch RNA-Interferenz (Wesley et al., 2001) tragen. Es stehen fünf transgene Linien zur Verfügung, die in der T0-Generation mit HP 1, HP 2, HP 3, HP 4 und HP 5 bezeichnet wurden. Individuen der T1 Generation wurden mit HP 1(2, 3, 4, 5) .x, die der T2 Generation mit HP 1 (2, 3, 4, 5) .x.y bezeichnet. Im Rahmen dieser Arbeit sollte zunächst überprüft werden, ob das Hairpin-Konstrukt tatsächlich zum “silencing” der LeMMP-1 Expression in den transgenen Linien geführt hat. Außerdem sollte überprüft werden, ob das “silencing” für LeMMP-1 spezifisch ist, oder ob eine zweite MMP der Tomate, die anhand von EST`s (expressed sequence tag) in Datenbanken nachgewiesen werden konnte, ebenfalls betroffen ist. Zum Nachweis des “silencing” auf Ebene der Transkripte wurde die Technik der Reversen Transkriptase (RT) – PCR eingesetzt. RNAPräparationen aus Hypocotylen und Sprossmaterial oberhalb der Kotyledonen von 24 Tage alten Keimlingen der Linien HP 1.6, HP 2.9, HP 5.4 und UC82B (Wildtyp Kontrolle) wurden von Andreas Schaller zur Verfügung gestellt. Die während der PCR eingesetzten Primerpaare sollten jeweils spezifisch cDNA-Fragmente von LeMMP-1 und LeMMP-2 amplifizieren (siehe Anhang). Zu Kontrollzwecken, und zur Abschätzung der Templatmenge wurde eine PCR auf Aktin (Abb. 3.1.1) durchgeführt. Es zeigte sich, dass die Konzentration des Aktintranskriptes in den acht zu untersuchenden Proben (Spuren 1-8 in Abb. 3.1.1) ähnlich hoch ist. Ein Vergleich mit der Kontrollreaktion mit genomischer DNA als Templat zeigte ausserdem, dass die RNAPräparation frei von DNA-Verunreinigungen waren (Spur 10 in Abb. 3.1.1). Die Abbildung 3.1.2 zeigt, dass die Transkripte von LeMMP-1 in Hypocotyle der transgenen Linien HP 1.6, HP 2.9, HP 5.4 im Vergleich zum Wildtyp deutlich vermindert sind. In Blättern zeigte sich jedoch keine Reduktion des LeMMP-1 Transkriptes. Das Transkript von LeMMP-2 liess sich nur in den Blättern von Kontrollpflanzen nachweisen (Abb. 3.1.2, Spur 1). Es wurde auch eine PCR mit Primern spezifisch für das Tomaten Mosaik Virus (TMV) durchgeführt, um zu überprüfen, ob die verwendeten Pflanzen mit dem TMV infiziert waren 30 Ergebnisse (Abb. 3.1.2). Dies wurde notwendig, da im Versuchszeitraum eine TMV Infektion in den Gewächshäusern des Instituts festgestellt wurde. Hier musste sichergestellt werden, dass die Versuchspflanzen nicht betroffen waren, da anderenfalls keine eindeutige Interpretationen der Ergebnisse zu erzielen sind. Als Positiv-Kontrolle wurde RNA aus Blattmaterial TMVinfizierter Pflanzen (zur Verfügung gestellt von J. Babo) eingesetzt (Abb. 3.1.2 Spur 9). Hier zeigte sich ein starkes Signal nach der RT-PCR. In den zu untersuchenden Proben (Spuren 18) liess sich die virale RNA nicht, oder nur in verschwindenden Mengen (Abb. 3.1.2, Spur 5) nachweisen. Abschliessend lässt sich sagen, dass in den transgenen HP Linien zumindest im Hypocotyl ein spezifisches “Silencing” der LeMMP-1 Expression erreicht worden war. Abb.3.1.1 Nachweis des Aktin Transkriptes durch RT-PCR nach 25, 30 und 35 Zyklen. Als Größenstandard wurde die 1 Kb DNA-Leiter von Fermentas verwendet. Die Länge der Fragmente ist in bp angegeben. In den Spuren 1-4 wurde RNA aus Blattmaterial, in den Spuren 5-8 wurde RNA aus Hypocotyle eingesetzt. 1 = UC82B, 2 = HP 1.6, 3 = HP 2.9, 4 = HP 5.4, 5 = UC82B, 6 = HP 1.6, 7 = HP 2.9, 8 = HP 5.4, 9 = RNA aus Blättern einer TMV infizierten Pflanze (UC82B), 10 = Genomische Tomaten-DNA, M=Größenstandard. 31 Ergebnisse Abb.3.1.2 3.2 Nachweis der Transkripte von LeMMP-1, LeMMP-2 und TMV durch RT-PCR nach 30 Zyklen. Als Größenstandard diente ebenfalls die 1 Kb DNA-Leiter von Fermentas. Die Länge der Fragmente ist in bp angegeben. In den Spuren 1-4 wurde RNA aus Blattmaterial, in den Spuren 58 wurde RNA aus Hypocotyle eingesetzt. 1 = UC82B, 2 = HP 1.6, 3 = HP 2.9, 4 = HP 5.4, 5 = UC82B, 6 = HP=1.6, 7 = HP=2.9, 8 = HP 5.4, 9 = RNA aus Blättern einer TMV infizierten Pflanze (UC82B), 10 = Genomische Tomaten-DNA, M=Größenstandard. Histologische Untersuchungen Die HP – Pflanzen zeigen im Vergleich zum Wildtyp (UC82B) einen ausgeprägten Phänotyp. Der Phänotyp ist im Hypocotyl am deutlichsten ausgeprägt. Dieser ist bereits drei bis vier Wochen nach der Keimung sichtbar. Es bilden sich dunkle, wahrscheinlich nekrotische Flecken, die sich mit fortschreitendem Alter der Pflanzen immer mehr ausdehnen, bis das gesamte Hypocotyl damit überzogen ist. Einhergehend kommt es zum Verlust der Trichome des Hypocotyls. Blattstiele zeigen einen ähnlichen Phänotyp, der beginnend an den ältesten Filderblätter zum Absterben und schliesslich zum Abwurf der betroffenen Blätter führt (Abb.3.2.1). Um die Veränderungen des Gewebes in den HP-Linien genauer zu untersuchen, wurden Hypocotyle, nach der in Material und Methoden beschriebenen Methode in Technovit 7100 eingebettet, am Rotationsmikrotom geschnitten, und anschliessend mikroskopisch untersucht. 32 Ergebnisse B A Abb.3.2.1 A) Hypocotyl HP 1.3 abc, B) Hypocotyl UC82B. Gut zu erkennen ist der dunkle, nekrotisch erscheinende Hypocotyl von HP 1.3 abc, der das ganze Hypocotyl vollständig überzieht. Ebenso erkennt man das Fehlen der Fliederblätter im Vergleich zu UC82B. Cortex Epidermis Trichom Xylem Leitbündel Phloem Vasculäres Kambium Mark Abb.3.2.2 Übersicht über die zelluläre Organisation im Hypocotyl einer vier Wochen alten UC82B Pflanze. Querschnitt wurde gefärbt mit Toluidin Blau. 33 Ergebnisse A B Abb.3.2.3 Hypocotyl-Querschnitte vier Wochen alter Tomatenpflanzen, gefärbt mit Toluidin Blau. A) HP 4.3, B) UC82B. Der Pfeil markiert den Bereich unter der Epidermis, wo verschiedenste phenolische Substanzen eingelagert werden. A B 2 1 Abb.3.2.4 A Abb.3.2.5 Hypocotyl-Querschnitte vier Wochen alter Tomatenpflanzen, gefärbt mit Toluidin Blau. A) HP 1.3 abc, B) UC82B. Der Pfeil (1) markiert die abgestorbene Epidermis. Pfeil (2) markiert das desorganisierte Gewebe des Cortex. B Hypocotyl-Querschnitte vier Wochen alter Tomatenpflanzen, gefärbt mit Toluidin Blau. A) HP 1.3 abc, B) UC82B. Der Pfeil markiert die beginnende Einlagerung phenolischer Substanzen. 34 Ergebnisse A Abb.3.2.6 A Abb.3.2.7 B Hypocotyl-Querschnitte vier Wochen alter Tomatenpflanzen, gefärbt mit Toluidin Blau. A) HP 1.3 abc, B) UC82B. B Hypocotyl-Querschnitte vier Wochen alter Tomatenpflanzen, gefärbt mit Sudan Black. A) HP 1.3 abc, B) UC82B. Pfeile markieren Zellen des Cortex, welche mit Sudan Black gefärbt sind. Abbildung 3.2.2. zeigt den Querschnitt durch das Hypocotyl einer Tomatenpflanze des Wildtyps (UC82B). Deutlich ist der geschlossene Zylinder sekundären Xylems zu sehen, der das Mark umschliesst. Die Abbildung 3.2.6. A, zeigt, dass der Zylinder sekundären Xylems in HP-Pflanzen noch nicht vollständig geschlossen ist. Im Mark von HP-Pflanzen sind keine Veränderungen zu erkennen. Das Cortex von HP-Pflanzen zeigt ebenfalls dramatische Unterschiede im Vergleich zum Wildtyp (UC82B) (Abb. 3.2.3-3.2.6). In HP-Pflanzen kommt es im Cortex, in den subepidermalen Zellschichten, zur Akkumulation von phenolischen Substanzen, die im Wildtyp-Cortex nicht vorkommen (Abb. 3.2.3). Ebenso kommt es in HPPflanzen zur Einlagerung von Cutin und Suberin (Abb. 3.2.7). Das Cortex erscheint in HPPflanzen zusätzlich äusserst desorganisiert (Abb. 3.2.4), und die Cortexzellen unterscheiden sich in ihrer Form vom Wildtyp-Cortexzellen. Zusätzlich scheint die Epidermis in HPPflanzen teilweise abgestorben zu sein (Abb. 3.2.4). Die Cortexzellen von HP-Pflanzen 35 Ergebnisse zeigen auch ein Zellmuster, dass an einem sekundären Abschlussgewebe erinnert (Abb. 3.2.6). Die Akkumulation der phenolischen Substanzen beginnt in den subepidermalen Zellschichten, was in der Abb. 3.2.5 zu erkennen ist. Zur Identifizierung der eingelagerten Substanzen wurde mit Toluidin Blau und Sudan Black gefärbt. Toluidin Blau dient zum Nachweis phenolischer Substanzen, wie z.B. Lignin. Es bietet jedoch keine Möglichkeit zur Bestimmung einzelner Substanzen. Toluidin Blau färbt Cellulose-Zellwände rot-violett und phenolische Bestandteile der Zellwand grün-blau. Die Sudanfarbstoffe erlauben es Substanzen wie Suberin und Cutin zu identifizieren, bietet jedoch nicht die Möglichkeit zwischen diesen zu unterscheiden. Generell kann gesagt werden, das HP-Pflanzen phenolische Substanzen im Cortex akkumulieren. Die Färbung mit Sudan Black zeigte, dass Suberin und Cutin eingelagert werden. Neben Suberin und Cutin, werden weitere phenolische Substanzen eingelagert, die jedoch nicht identifiziert sind. Die Tatsache, dass das Muster der Toluidin Blau-Färbung die Bereiche mit einbezieht, die von den Sudan-Farbstoffen gefärbt werden, und über diese hinausgeht, verstärkt die Vermutung, das es sich um weitere phenolische Substanzen handelt, die eingelagert werden. 3.3 Northern Blot Die mikroskopischen Untersuchungen zeigten, dass HP-Pflanzen phenolische Substanzen im Hypocotyl einlagern. Mit der Methode des Northern Blot, wurde die Expression der Phenylalanin-Ammoniak-Lyase (PAL), das Schlüsselenzyms in der Synthese von phenolischen Substanzen in Blättern von UC82B und HP 1.6/2.9/5.4 verglichen. Ubiquitin (UBQ) diente als positive Kontrolle, da man davon ausgeht, dass Ubiquitin in alle Geweben in gleichem Masse exprimiert wird. Ein Ethidiumbromid gefärbtes Gel, diente als Ladekontrolle (Abb. 3.3.1). In der Tat war für Ubiquitin in allen vier Spuren ein Signal gleicher Stärke zu beobachten. In zwei der drei HP-Linien, nicht aber im Wildtyp, liess sich das Transkript der PAL nachweisen. Die Synthese und Einlagerung von phenolische Verbindungen in den Hypocotylen der HP-Linien geht also einher mit einer gesteigerten Expression der PAL. 36 Ergebnisse Abb. 3.3.1 3.4 Nothern Blot Analyse. 5,5 µg totaler RNA aus Hypocotyle des Wildtyps und drei LeMMP-1 HPLinien wurden elektrophoretisch aufgetrennt, auf eine Nitrocellulosemembran übertragen und mit radiomarkierte cDNA Sonden für Phenylalanin Ammoniak Lyase (PAL) und Ubiquitin (UBQ) hybridisiert. Als Ladekontrolle diente ein zweites, mit Ethidiumbromid (EtBr) gefärbtes Gel. Extraktion phenolischer Substanzen HP-Pflanzen zeigten bei der Northern Blot Untersuchung (3.3) eine Induktion der PAL im Vergleich zum Wildtyp. Die PAL ist ein Schlüsselenzym in der Synthese von phenolischen Verbindungen, und katalysiert die Umwandlung von Phenylalanin zu Zimtsäure, dem wichtigsten Vorläufer aller phenolischen Verbindungen. Ausserdem wurde während der histologischen Untersuchungen (3.2) eine Einlagerung phenolischer Verbindungen im Hypocotyle der HP-Pflanzen beobachtet. Im folgendem sollte die Bildung löslicher und gebundener Phenole, sowie die Akkumulation von Lignin in den transgenen Linien im Vergleich zum Wildtyp quantifiziert werden. Untersucht wurden Hypocotyl und Blattstiele von vier Wochen alten UC82B und HP 1.3 abc Pflanzen. Es wurde die Konzentration in 37 Ergebnisse jeweils fünf unabhängigen Extrakte aus Organe von UC82b und fünf HP 1.3 abc Pflanzen bestimmt, und die Mittelwerte wurden graphisch dargestellt (Abb. 3.4.1-3.4.3). Zur Bestimmung der Konzentration an löslichen und gebundenen phenolischen Verbindungen wurde der “Folin-Ciocalteu”-Test (Goldwasser et al., 1999) durchgeführt, und die Konzentration der phenolischen Verbindungen wurde bei einer Wellenlänge von 725 nm als p-Cumarsäure-Äquivalente im Photometer bestimmt. B 600 600 500 500 400 400 Konz. µg/gFG Konz. µg/gFG A 300 200 100 200 100 0 0 UC82b Abb. 3.4.1 300 HP 1.3 abc UC82b HP 1.3 abc Lösliche phenolische Substanzen im Blattstiel (A) und im Hypocotyl (B). Gezeigt ist der Mittelwert von fünf unabhängigen Messungen. Die Fehlerbalken zeigen die Standardabweichung vom Mittelwert. Die Konzentration phenolischer Substanzen wurde bestimmt als p-CumarsäureÄquivalente bei einer Wellenlänge von 725 nm. B 350 700 300 600 250 500 Konz. µg/gFG Konz. µg/gFG A 200 150 100 50 300 200 100 0 0 UC82b Abb. 3.4.2 400 HP 1.3 abc UC82b HP 1.3 abc Gebundene phenolische Substanzen im Blattstiel (A) und im Hypocotyl (B). Gezeigt ist der Mittelwert von fünf unabhängigen Messungen. Die Fehlerbalken zeigen die Standardabweichung vom Mittelwert. Die Konzentration an phenolische Substanzen wurde bestimmt als p-CumarsäureÄquivalente bei einer Wellenlänge von 725 nm. 38 Ergebnisse Man beobachtete eine deutliche Erhöhung der Konzentration an löslichen und gebundenen phenolischen Verbindungen in Hypocotylen und Blattstielen von HP 1.3 abc. Die HP 1.3 abc Pflanzen zeigten eine nahezu um das Doppelte erhöhte Konzentration an löslichen und gebundenen phenolischen Verbindungen (Abb. 3.4.1 B und 3.4.2 B) im Hypocotyl. Die Konzentration an löslichen und gebundenen phenolischen Verbindungen im Blattstielen von HP 1.3 abc war ebenfalls deutlich erhöht (Abb. 3.4.1 A und 3.4.2 A). A B Extinction/gTG Extinction/gTG 500 400 300 200 100 0 UC82b Abb. 3.4.3 1200 1000 800 600 400 200 0 HP 1.3 abc UC82b HP 1.3 abc A) Ligningehalt des Blattstiels. B) Ligningehalt des Hypocotyls. Lignin wurde bestimmt als Lignin-ähnliche Thioglycolsäurederivate. Die Konzentration ist als E 280/gTG angegeben. Gezeigt ist der Mittelwert von fünf unabhängigen Experimenten. Die Fehlerbalken zeigen die Standardabweichung vom Mittelwert. Nach der Extraktion von löslichen und gebundenen phenolischen Verbindungen, wurde Lignin im Rückstand als Thioglycolsäurederivate bestimmt. Für Lignin (Abb.3.4.3) zeigte sich kein bedeutender Unterschied zwischen UC82B und HP 1.3 abc. 3.5 HPLC-Analyse phenolischer Verbindungen Mit Hilfe des HPLC-Systems wurden Extrakte löslicher und gebundener phenolischer Substanzen auf ihre Zusammensetzung untersucht. Als stationäre Phase wurde eine NUCLEOSIL 5, C 18, 120A Säule verwendet. Als Laufmittel diente 0,25 % H3PO4 in H2O und 0,25 % H3PO4 in Acetonitril. Detektiert wurde bei einer Wellenlänge von 254 nm. 39 0,25 100 0,2 80 0,15 60 0,1 40 0,05 20 0 Laufmittelkonzentration (%) E254 Ergebnisse Zimtsäure Sinapinsäure Ferulasäure Kaffeesäure Chlorogensäure Laufmittel A Laufmittel B 0 10 20 30 40 50 60 RT (min) Trennung der Substanzen, die als Standard benutzt wurden (siehe Legende). Für jede Substanz wurde 50 µl einer Lösung von 1mg/ml Methanol eingesetzt. 0,15 100 0,13 80 0,11 E254 0,09 60 0,07 40 0,05 0,03 20 0,01 -0,01 Laufmittelkonzentration (%) Abb. 3.5.1 UC82B HP 1.3 abc Laufmittel A Laufmittel B 0 10 20 30 40 50 60 RT (min) Trennung gebundener phenolischer Substanzen von UC82B und HP 1.3 abc aus dem Hypocotyl. Die aufgetragene Menge entspricht 23,8 µgFG. 0,15 100 0,13 80 0,11 E254 0,09 60 0,07 40 0,05 0,03 20 0,01 -0,01 Laufmittelkonzentration (%) Abb. 3.5.2 UC82B HP 1.3 abc Laufmittel A Laufmittel B 0 10 20 30 40 50 60 RT(min) Abb. 3.5.3 Trennung löslicher phenolischer Substanzen von UC82B und HP 1.3 abc aus dem Hypocotyl. Die aufgetragene Menge entspricht 15,7 µgFG. 40 Ergebnisse Die Trennung der gebundenen phenolischen Substanzen aus den Extrakten von Hypocotylen zeigte deutliche Unterschiede in Zusammensetzung und Konzentration phenolischer Substanzen zwischen HP 1.3 abc undUC82B (Abb. 3.5.2). Die in HP 1.3 abc akkumulierten Phenole liessen sich anhand der Retentionszeiten nicht eindeutig eines der Standardsubstanzen (Abb. 3.5.1) zuordnen und müssen daher unidentifiziert bleiben. Die Trennung der löslichen phenolischen Substanzen (Abb. 3.5.3) gibt keine Hinweise über Änderungen in Zusammensetzung oder Konzentration dieser Substanzen. Dieser Befund überrascht angesichts des Ergebnisses der Quantifizierung löslicher Phenole im Hypocotyl nach Folin-Ciocalteau (Abb. 3.4.1), oder aber ihre Konzentration lag unter der Nachweisgrenze des eingesetzten HPLC-Systems. 3.6 Infektion durch Xanthomonas campestris pv. campestris Microarray Analysen im Vorfeld dieser Arbeit zeigten eine Induktion der LeMMP-1 nach Behandlung mit dem Pilztoxin Fusicoccin. Dies deutete auf eine Funktion des Enzyms in der Pathogenabwehr. Um zu überprüfen, ob es Unterschiede in der Sensitivität gegenüber phytopathogenen Bakterien zwischen HP-Pflanzen und UC82B gibt, wurden vier Wochen alte HP 1.3 abc und UC82B Pflanzen mit dem phytopathogen Xanthomonas campestris pv. campestris Stamm 93-1 nach der in Material und Methoden beschriebenen Methode infiziert, und eine Bakterienwachstumskurve wurde erstellt. 1,00E+09 1,00E+08 cfu/Blatt 1,00E+07 1,00E+06 1,00E+05 UC82b 1,00E+04 HP 1.3 abc 1,00E+03 1,00E+02 1,00E+01 1,00E+00 0 1 2 3 4 5 6 7 8 Tage nach der Infektion Abb.3.6.1 Bakterielle Wachstumskurve erstellt aus den Mittelwerten der ermittelten Kolonienzahlen von HP 1.3 abc und UC82B an den angegebenen Zeitpunkten (siehe Material und Methoden). Die XAchse zeigt die Zeitpunkte der Probenentnahme nach der Infektion. Die Y-Achse zeigt die ermittelte Anzahl an colony-forming-units in infizierten Fliederblättern. 41 Ergebnisse Es zeigte sich ein vermindertes Wachstum von Xanthomonas campestris pv. campestris auf HP 1.3 abc Pflanzen im Vergleich zu UC82B. Während zum Zeitpunkt 0 (Zeitpunkt der Infiltration) und ein bis zwei Tage nach der Infiltration kein signifikanter Unterschied zu erkennen ist, werden die Unterschiede des bakteriellen Wachstums zwischen HP 1.3 abc und UC82B von dritten bis zum achten Tag nach der Infiltration immer größer (Abb. 3.6.1). A Abb. 3.6.2 A B Expression der LeMMP-1 in UC82B nach Infektion mit Xanthomonas campestris pv. campestris A) Western Blot, B) SDS-Page Gel, 0 bis 8 Tage nach Infiltration der Bakteriensuspension wurden Blattextrakte erstellt, und jeweils 15.6 µg Protein wurde analysiert. Der Pfeil markiert die Position des TMV-Hüllproteins. Als Grössenstandard diente die “Prestained Protein Ladder” (Fermentas; Grössenangaben in kDa). B M 0 1 2 3 4 5 6 7 8 TMV M 170 130 100 72 55 40 33 24 17 11 Abb. 3.6.3 Expression der LeMMP-1 in HP 1.3 abc nach Infektion mit Xanthomonas campestris pv. campestris A) Western Blot, B) SDS-Page Gel, 0 bis 8 Tage nach Infiltration der Bakteriensuspension wurden Blattextrakte erstellt, und jeweils 24,9 µg Protein wurde analysiert. Der Pfeil markiert die Position des TMV-Hüllproteins. Als Grössenstandard diente die “Prestained Protein Ladder” (Fermentas; Grössenangaben in kDa). Wie die Western Blot Analyse in Abb. 3.6.2 zeigt, induziert die Infiltration des Phytopathogen Xanthomonas campestris pv. campestris die Expression von LeMMP-1 im UC82B Wildtyp. Am zweiten Tag nach der Infektion ist das erste LeMMP-1 Signal zu 42 Ergebnisse erkennen, vier Tage nach der Infektion ist die höchste Konzentration erreicht, danach erfolgt eine Herunterregulierung des Enzyms, mit der niedrigsten Konzentration am achten Tag nach der Infektion (Abb. 3.6.2). In HP 1.3 abc war auch nach bakterieller Infektion kein Signal der Le-MMP1 zu erkennen (Abb. 3.6.3). Offenbar führt das Hairpin-Konstrukt zum effizienten “silencing” der LeMMP-1 in den transgenen Pflanzen. Wie bereits zuvor erwähnt, kam es im Versuchszeitraum zu einer TMV-Infektion in den Gewächshäusern des Instituts. In der Tat ist das Hüllprotein des TMV auf Coomassiegefärbten Gelen nachzuweisen (Pfeil in Abb. 3.6.2 B und Abb. 3.6.3 B). UC82B und HP 1.3 abc Pflanzen scheinen in Gleicherweise TMV-infiziert. Dabei ist es wahrscheinlich, dass die beobachteten Unterschiede in Resistenz gegenüber Xanthomonas campestris pv. campstris und Induktion der LeMMP-1-Expression nicht auf TMV, sondern auf den Genotyp der Versuchspflanzen zurückzuführen ist. Trotzdem sollten die Ergebnisse unter Vorbehalt betrachtet werden, und das Experiment sollte unbedingt wiederholt werden. 43 Diskussion 4. Diskussion Zur funktionellen Charakterisierung der LeMMP-1 in Tomaten wurden in dieser Arbeit transgene Pflanzen eingesetzt, in denen die Expression der LeMMP-1 durch RNAInterferenze unterdrückt sein sollte. Es galt zunächst zu überprüfen, ob das Hairpin-Konstrukt in den Tomatenpflanzen tatsächlich zur Unterdrückung der LeMMP-1 Expression auf Transkript- und Protein-Ebene geführt hatte. Mittels RT-PCR konnte gezeigt werden, dass die Menge des LeMMP-1-Transkriptes in Hypocotylen der Hairpin-Pflanzen vermindert war. Im Falle der Linie HP 1.6 war das Transkript nicht mehr nachweisbar (Abb.3.1.2). In Blättern der Hairpin-Pflanzen dagegen schien die Menge des LeMMP-1-Transkriptes nicht verändert zu sein (Abb. 3.1.2). Das Resultat ist äußerst überraschend, da auf Proteinebene die LeMMP-1 auch in Blättern nicht nachweisbar ist (Abb. 3.6.3 A). Daher sollten die Produkte der RT-PCR unbedingt durch Sequenzierung überprüft werden, ob es sich tatsächlich um die LeMMP-1 handelt. Es wäre möglich, dass es sich um cDNAs von Tomaten-Transkripten handelt, die eine gewisse Ähnlichkeit mit der LeMMP-1 aufweisen, und daher von den LeMMP-1Primerpaaren erkannt wurden. Da von Tomate noch nicht das gesamte Genom entschlüsselt wurde, muss man diese Möglichkeit in Betracht ziehen. Die Sequenzierung der PCR Produkte wird in dieser Frage Gewissheit geben. Das Transkript der LeMMP-2 liess sich mittels RT-PCR in Blättern, nicht aber in Hypocotylen von Tomatenpflanzen nachweisen (Abb. 3.1.2). Die Expression der LeMMP-2 war in Blättern der Hairpin-Pflanzen unterdrückt. Dieses Resultat ist zunächst überraschend, kann aber durch die grosse Sequenzähnlichkeit zwischen der LeMMP-1 und LeMMP-2 erklärt werden (siehe Anhang). Im Hinblick auf die Interpretationen der hier erzielten Ergebnisse ist wichtig, dass eine LeMMP-2 Expression in Hypocotylen des Wildtyps nicht nachweisbar war. Alle im Hypocotyl der transgenen Pflanzen beobachteten Veränderungen müssen also auf “silencing” von LeMMP-1 zurückzuführen sein. Mikroskopische Beobachtungen zeigten in Hypocotylen der Hairpin-Pflanzen einen Verlust der zellulären Organisation im Cortex (Abb. 3.2.4 A). Im Vergleich zu UC82B KontrollPflanzen waren die Cortexzellen der Hairpin-Pflanzen kleiner, zahlreicher und sehr unregelmässig geformt und angeordnet, was möglicherweise auf eine unregelmässige Zellteilung zurückzuführen ist. Gillmor et al. (2005) zeigten, dass Mutationen in einem Gen (PNT 1) in Arabidopsis thaliana, welches ein Enzym kodiert, das notwendig für die Synthese 44 Diskussion des GPI-Ankers ist, zur verzögerten Morphogenese (Übergang von Globulär- ins HerzStadium), Störung der Zellteilung (unregelmässige Zellteilungen im Proembryo und der Hypophyse) und zu Änderungen in den Bestandteilen der Zellwand führt (niedrigere Konzentration an Cellulose, erhöhte Konzentration von Pektinen, und unregelmässige und zufällige Ablagerung von Pektin, Xyloglucanen und Callose). All diese Veränderungen, welche zumindest teilweise an die in den LeMMP-1 Hairpin-Pflanzen beobachteten Veränderungen erinnern, waren auf die Unfähigkeit der Pflanzen zur Bildung des GPIAnkers, und dadurch zur GPI-Verankerung von Proteinen in der Plasmamembran zurückzuführen. Von nur sieben Proteinen aus Pflanzen ist eine GPI-Verankerung experimentell nachgewiesen, und die Sequenz bekannt (Eisenhaber et al., 2003). Vier davon sind Arabinogalactan Proteine (AGPs). Das sind extrazelluläre Glycoproteine, die einen Einfluss auf Wachstums und Entwicklungsprozesse haben (Schultz et al., 2000 und Zhao et al., 2002). So wird für das Protein LeAGP-1 wird eine Beteiligung am Dickenwachstum der Zellwände und bei der Lignifizierung angenommen(Gao und Showalter, 2000). Von Gillmor et al. (2005) werden die in der pnt1 Mutanten beobachteten Defekte in Embryonalentwicklung, Zellteilung und Zellwandsynthese in einen kausalen Zusammenhang mit dem Verlust des GPI-verankerten AGPs gestellt. Für die LeMMP-1 wird aber auch eine Verankerung in der Plasmamembran durch einen GPI-Anker prognostiziert (Zimmermann, 2004). Es ist also durchaus denkbar, dass ein Verlust der LeMMP-1 Funktion in der pnt1 Mutante zu den dort beobachteten Veränderungen beiträgt. Die in Hypocotylen der LeMMP-1 HP-Pflanzen beobachteten Störungen in der Zellteilung belegen eine Funktion der LeMMP-1 in der Entwicklung zumindest des Stängelcortex von Tomatenpflanzen. Durch Färbung mikroskopischer Schnitte, konnte hier auch eine veränderte Zusammensetzung der Zellwand gezeigt werden. Allerdings scheint es sich nicht wie in der pnt1 Mutante um eine Veränderung des Anteils an kristalliner Cellulose zu handeln. Durch Färbung mit Toluidin Blau konnte die Akkumulation von phenolischen Verbindungen in Zellwänden des Cortex nachgewiesen werden (Abb. 3.2). Der durch Toluidin Blau gefärbte Bereich war teilweise identisch mit der Färbung durch Sudan Black, das spezifisch hydrophobe Substanzen wie Cutin und Suberin färbt. Es ist also wahrscheinlich, dass die positive Phenolfärbung auf den phenolischen Anteil des Suberin zurückzuführen ist. Suberin und Cutin dienen als Matrix für Wachse, und bilden durch ihren hydrophoben Charakter wasserundurchlässige Schichten. Derart ausgerüstete Zellen finden häufig als innere (Caspary-Streifen) und äußere Häute (Wurzel) Verwendung, mit der Funktion den Wasserdurchtritt zu kontrollieren oder zu verhindern. Im Kork ist Suberin ebenfalls zu finden. Dieser dient dem nachträglichen 45 Diskussion Abschluss der Oberfläche des Pflanzenkörpers, wie etwa nach Verletzungen. Das in HairpinPflanzen beobachtete Absterben der Epidermis ist möglicherweise auf die Inkrustierung der Cortexzellen zurückzuführen, da dadurch die Versorgung der Epidermis mit Nährstoffen und Wasser vermindert ist. Die Extraktion löslicher und gebundener Phenole und deren Konzentrationsbestimmung, zeigte erhöhte Konzentrationen in Hairpin-Pflanzen (3.4). Diese Resultate stehen im Einklang mit der beobachteten Suberinisierung im Cortex von HP-Pflanzen. Es wurde gezeigt, dass die in verwundeten Kartoffelknollen eintretende Suberinisierung mit einer Steigerung der PALAktivität einhergeht (Bernards et al., 2000 und Oosterhaven et al., 1995). PAL katalysiert die Umwandlung von Phenylalanin zu Zimtsäure, welche im Phenylpropanweg zu weiteren phenolischen Substanzen umgewandelt wird. Auch in dieser Arbeit konnte mittels Northern Blot Analysen (3.3) die Akkumulation von PAL-Transkripten in Hypoctylen von HPPflanzen nachgewiesen werden. Ishizuka et al. (1991) zeigten ebenfalls, dass es zur rapiden Akkumulation von PAL-Transkripten nach Verwundung in Kartoffelknollen kommt. Eine chromatographische Trennung phenolischer Substanzen mittels der Methode der HPLC, bestätigte den zuvor beobachteten gesteigerten Gehalt an gebundenen (in der Zellwand veresterten) Phenolen, in LeMMP-1 Pflanzen. Auch liessen sich in Extrakten aus Hypocotylen von HP-Pflanzen Unterschiede in der Zusammensetzung der phenolischen Substanzen im Vergleich zu UC82B-Kontrollpflanzen feststellen. Eine Identifizierung der hier gebildeten Phenole durch Vergleich mit Referenzsubstanzen ist allerdings im Rahmen dieser Arbeit nicht mehr gelungen. Die Trennung löslicher phenolischer Substanzen führte zu keinem Ergebnis. Möglicherweise waren die zu trennenden Substanzen degradiert, oder ihre Konzentration lag unter der Nachweisgrenze. Die Zelluläre Organisation im Stängelcortex von LeMMP-1 HP-Pflanzen sowie die Suberinisierung der Zellwände erinnert stark an die Ausbildung eines sekundären Abschlussgewebes (Abb. 3.2). Ob es in diesem Zusammenhang auch zu einer Lignifizierung der Zellwände kommt, konnte noch nicht eindeutig geklärt werden. Die Tatsache, dass nur ein Teilbereich der mit Toluidin Blau als phenolhaltig identifizierten Zellwände mit Sudan Black gefärbt wurden, also suberinisiert waren, mag darauf hindeuten. Allerdings konnte eine signifikante Erhöhung des Ligningehaltes nicht nachgewiesen werden (Abb. 3.4.3). Hier sollte der Einsatz Lignin-spezifischer Färbetechniken in histologischen Untersuchungen Aufschluss geben. Weitere Hinweise könnte die Analyse von Markerenzymen liefern. Bernards et al. (2000) zeigten, dass es im Zusammenhang der Lignifizierung und Suberinisierung zur Induktion von Enzymen des Phenylpropanstoffwechsels kommt (PAL, 46 Diskussion Zimtalkohol Dehydrogenase, 4-Coumaryl-CoA Ligase). Die Cinnamoyl-CoA: NADP+ Oxidoreduktase aber wird charakteristischerweise nur während der Lignifizierung induziert. Dieses Enzym könnte also als molekularer Marker herangezogen werden, um zwischen Lignifizierung und Suberinisierung zu unterscheiden (Bernards et al., 2000). Mikroarray Analysen zeigten, dass die LeMMP-1 nach Behandlung mit dem Pilztoxin Fusicoccin induziert wird (Frick und Schaller, 2002). In dieser Arbeit wurde darüber hinaus eine Akkumulation der LeMMP-1 nach Infektion mit Xanthomonas campestris pv. campestris beobachtet (Abb. 3.6.1) Diese Befunde deuten auf eine mögliche Beteiligung des Enzyms bei der Pathogenantwort. Sollte die LeMMP-1 eine Rolle in der Pathogenabwehr spielen, würde man für HP-Pflanzen eine erhöhte Anfälligkeit gegenüber Phytopathogenen erwarten. Die erzielten Resultate zeigten jedoch ein im Vergleich zu UC82B-Kontrollpflanzen vermindertes bakterielles Wachstum auf HP-Pflanzen. Die Resistenz von Tomatenpflanzen gegenüber Xanthomonas campestris pv. campestris scheint also bei Verlust der LeMMP-1 erhöht zu sein.1 Dieser Widerspruch lässt sich möglicherweise durch die Tatsache erklären, dass phytopathogene Bakterien mit Hilfe von Effektorproteinen als Virulenzfaktoren in vielfältiger Weise in den Stoffwechsel der Wirtspflanze eingreifen. Es wäre denkbar, dass die Phytopathogene die Expression der LeMMP-1 induzieren um die Besiedlung der Wirtspflanze zu erleichtern. Die Induktion der LeMMP-1 könnte der Bildung phenolischer Substanzen und der Verstärkung der Zellwand entgegenwirken und so die Reaktionen der Pathogenabwehr vermindern. HP-Pflanzen haben die Fähigkeit zur LeMMP-1 Bildung verloren und damit ist den Bakterien möglicherweise ein wichtiger Faktor, zur Besiedlung der Pflanze entzogen. Vielleicht ist das verminderte Bakterien-Wachstum auf HP-Pflanzen aber auch ein sekundärer Effekt, der auf die im Hypocotylen beobachteten Veränderungen der Zellwand zurückzuführen sein könnte. Sollte es in den Blättern zu ähnlichen Veränderungen kommen, wäre es denkbar, dass die Einlagerung phenolischer Substanzen wie Suberin für die Bakterien, die Zugänglichkeit der Wirtszelle erschwert. Der hydrophobe Charakter von Suberin beeinträchtigt möglicherweise auch die Mobilität der Bakterien. Auch die Akkumulation löslicher phenolischer Substanzen, mag zu einer erhöhten Resistenz beitragen. Lösliche phenolische Substanzen wie Isoflavonoide sind in den letzten Jahren vor allem durch ihre Rolle als Phytoalexine bekannt geworden (Review, Nicholson und Hammerschmidt, 1992). Phytoalexine sind sekundäre Inhaltsstoffe mit antimikrobieller Wirkung, die als Reaktion auf eine Infektion gebildet werden, mit dem Ziel, die Ausbreitung der Pathogene in der Pflanze zu 1 Einschränkend soll hier nochmals erwähnt werden, dass es im Versuchszeitraum zu einer TMV-Infektion in den Gewächshäusern des Instituts kam, von der auch die Pflanzen des bakteriellen Infektionsexperiments betroffen waren. Die erzielten Ergebnisse sind deshalb mit Vorbehalt zu betrachten. 47 Diskussion verhindern. Ebenso wurde gezeigt, dass die PAL nach Pathogenbefall induziert wird, was die Pflanze zur Synthese phenolischer Substanzen veranlasst, um die Ausbreitung der Pathogene einzudämmen (Review, Nicholson und Hammerschmidt, 1992). Es ist weiterhin bekannt, dass die Ablagerung von Lignin und anderen polymeren, phenolischen Substanzen, einen Teil der pflanzlichen Reaktion auf Phytopathogene darstellt (Review, Nicholson und Hammerschmidt, 1992). Ob der Funktionsverlust der LeMMP-1 direkt oder indirekt für das verminderte bakterielle Wachstum in HP-Pflanzen verantwortlich ist, werden weitere Untersuchungen zeigen müssen. Infektionsexperimente mit Bakterien-Stämmen, die wegen Verlustes von Effektorproteinen in ihrer Pathogenität vermindert sind, könnten möglicherweise diese Frage beantworten. 48 Literatur 5. Literatur Alfano, J. R., Collmer, A. (1997): The type III (Hrp) secretion pathway of plant pathogenic bacteria: trafficking harpins, Avr proteins, and death. J. Bacteriol. 179, 5655-62. Bernards, M. A., Susag, L. M., Bedgar, D. L., Anterola, A. M., Lewis, N. G. (2000): Induced phenylpropanoid metabolism during suberization and lignification: a comparative analysis. J. Plant Physiol. 157, 601-7. Blocker, A., Jouihri, N., Larquet, E., Gounon, P., Ebel, F., Parsot, C., Sansonetti, P., Allaoui, A. (2001): Structure and composition of the Shigella flexneri, `needle complex', a part of its type III secreton. Mol. Microbiol 39, 652±663. Bode, W., Fernandez-Catalan, C., Tschesche, H., Grams, F., Nagase, H. and Maskos, K. (1999): Structural properties of matrix metalloproteinases. Cell. Mol. Life Sci. 55, 639-652. Bonas, U., Schulte, R., Fenselau, S., Minsavage, G. V., Staskawicz, B. J., Stall, R. E. (1991): Isolation of a gene cluster from Xanthomonas campestris pv. vesicatoria that determines pathogenicity and the hypersensitive response on pepper and tomato. Mol. Plant-Microbe Interact. 4, 81–88. Brown, R. M. Jr.; Saxena, I. M.; Kudlicka, K. (1996): Cellulose biosynthesis in in higher plants. Trends Plant Sci. 1, 149-155. Delorme, V. G. R., McCabe, P. F., Kim, D.-J. and Leaver, C. J. (2000): A Matrix Metalloproteinase Gene Is Expressed at the Boundary of Senescence and Programmed Cell Death in Cucumber. Plant Physiol. 123, 917-927. Dow, J. M., Davies, H. A. and Danials, M. J. (1998): A Metalloprotease from Xanthomonas campestris, That Specifically Degrades Proline/Hydroxyproline-Rich Glycoproteins of the Plant Extracellular Matrix. MPMI 11, 1085-1093. 49 Literatur Eisenhaber, B., Wildpaner, M., Schultz, C. J., Borner, G. H. H., Dupree, P., Eisenhaber, F. (2003): Glycosylphosphatidylinositol Lipid Anchoring of Plant Proteins. Sensitive Prediction from Sequence- and Genome-Wide Studies for Arabidopsis and Rice. Plant Physiol. 133, 1691-1701. Fenselau, S., Balbo, I., Bonas, U. (1992): Determinants of pathogenicity in Xanthomonas campestris pv. vesicatoria are related to proteins involved in secretion in bacterial pathogens of animals. Mol. Plant-Microbe Interact. 5, 390–396. Frick, U. B. (2002): Le1-MMP, eine Matrix Metalloproteinase von Lycopersicon esculentum: cDNA-Mikroarray Expressionsanalyse, molekulare und biochemische Charakterisierung. Diplomarbeit Universität Bern. Frick, U. B. and Schaller, A. (2002): cDNA microarray analysis of fusicoccin-induced changes in gene expression in tomato plants. Planta 216, 83-94. Friend, J. (1981): Plant phenolics, lignification and plant disease. Prog. Phytochem. 7, 197261. Friend, J., Reynolds, S. B., Aveyard, M. A. (1973): Phenylalanine ammonia lyase, chlorogenic acid and lignin in potato tuber inoculated with Phytophthora infestans. Physiol. Plant Pathol. 3, 495-507. Gao, M. and Showalter, A. M. (2000): Immunolocalization of LeAGP-1, a modular arabinogalactan-protein, reveals its developmentally regulated expression in tomato. Planta 210, 865-874. Gillmor, C. S., Lukowitz, W., Brininstool, G., Sedbrook, J. C., Hamann, T., Poindexter, P., Somerville, C. (2005): Glycosylphosphatidylinositol-Anchored Proteins Are Required for Cell Wall Synthesis and Morphogenesis in Arabidopsis. The Plant Cell 17, 1128-1140. 50 Literatur Görlach, J., Raesecke, H. R., Rentsch, D., Regenass, M., Roy, P., Zala, M., Keel, C., Boller, T., Amrhein, N., Schmid, J. (1995): Temporally distinct accumulation of transcripts encoding enzymes of the prechorismate pathway in elicitor-treated, cultured tomato cells. Proc Natl Acad Sci U S A 92, 3166-70. Goldwasser, Y., Hershenhorn, J., Plakhine, D., Kleifeld, Y., Rubin, B. (1999): Biochemical factors involved in vetch resistance to Orobanche aegyptiaca. Physiological and Molecular Plant Pathology 54, 87-96. Golldack, D., Popova, O. V. and Dietz, K.-J. (2002): Mutation of the Matrix Metalloproteinase At2-MMP Inhibits Growth and Causes Late Flowering and Early Senescence in Arabidopsis. J. Biol. Chem. 277, 5541-5547. Gomez, D. E., Alonso, D. F., Yoshiji, H., Thorgeirsson, U. P. (1997): Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur. J. Cell Biol. 74, 111122. Goodwin, T.W., and Mercer, E. I. (1983): The Plant Cell Wall. Introduction to Plant Biochemistry, Pergamon Press, New York, NY , pp. 55-91. Hotson, A., Chosed, R., Shu, H., Orth, K., Mudgett, M. B. (2003): Xanthomonas type III effector XopD targets SUMO-conjugated proteins in planta. Molecular Microbiology 50 (2), 377-389. Hijwegen, T. (1963): Lignification, a possible mechanism of active disease resistance against pathogens. Neth. J. Plant Pathol. 69, 314-17. Hugouvieux, V., Barber, C. E., Daniels, M. J. (1998) : Entry of Xanthomonas campestris pv. campestris into hydathodes of Arabidopsis thaliana leaves: a system for studying early infection events in bacterial pathogenesis. Mol. Plant Microbe Interact. 11, 537-43. Ishizuka, M., Yamada, F., Tanaka, Y., Takeuchi, Y., Imaseki, H. (1991): Sequential Induction of mRNAs for Phenylalanine Ammonia-lyase in Slices of Potato Tuber. Plant Cell Physiol. 32, 57-64. 51 Literatur Kurepa, J., Walker, J. M., Smalle, J., Gosink, M. M., Davis, S. J., Durham, T. L., Sung, D., Vierstra, R. D. (2003): The small ubiquitin-like modifier (SUMO) protein modification system in Arabidopsis. Accumulation of SUMO1 and -2 conjugates is increased by stress. J. Biol. Chem. 278, 6862–6872. Levy, S., Maclachlan, G., Stachelin, L. A. (1997): Xyloglucan sidechains modulate binding to cellulose during in vitro binding assays as predicted by conformational dynamics simulations. Plant Journal 11, 373-386. Liu, Y., Dammann, C., Bhattacharyya, M. K. (2001): The Matrix Metalloproteinase Gene GmMMP2 Is Activated in Response to Pathogenic Infections in Soybean. Plant Physiol. 127, 1788-1797. Maidment, J. M., Moore, D., Murphy, G. P., Murphy, G. and Clark, I. M. (1999): Matrix Metalloproteinase Homologues from Arabidopsis thaliana. J. Biol. Chem. 274, 34706-34710. Melchoir, F. (2000): SUMO-nonclassical ubiquitin. Annu Rev Cell Dev Biol 16, 591–626. Minsavage, G. V., Dahlbeck, D., Whalen, M. C., Kearny, B., Bonas, U., Staskawicz, B. J. and Stall, R. E. (1990): Gene-for-gene relationships specifying disease resistance in Xanthomonas campestris pv. vesicatoria–pepper interactions. Mol Plant–Microbe Interact 3, 41-47. Mueller, L. A., Tanksley, S. D., Giovannoni, J. J., van Eck, J., Stack, S., Choi, D., Kim, B. D., Chen, M., Cheng, Z., Li, C., Ling, H., Xue, Y., Seymour, G., Bishop, G., Bryan, G., Sharma, R., Khurana, J., Tyagi, A., Chattopadhyay, D., Singh, N. K., Stiekema, W., Lindhout, P., Jesse, T., Lankhorst, R. K., Bouzayen, M., Shibata, D., Tabata, S., Granell, A., Botella, M. A., Giuliano, G., Frusciante, L., Causse, M. and Zamir, D. (2005): The Tomato Sequencing Project, the first cornerstone of the International Solanaceae Project (SOL). Comp Funct Genom 6, 153–158. Nicholson, R. L. and Hammerschmidt, R. (1992): Phenolic compounds and their role in disease resistance. Annu. Rev. Phytopathol. 30, 369-389. 52 Literatur Noel, L., Thieme, F., Nennstiel, D., and Bonas, U. (2001): cDNA-AFLP analysis unravels a genome-wide hrpGregulon in the plant pathogen Xanthomonas campestris pv. vesicatoria. Mol. Microbiol 41, 1271–1281. Noel, L., Thieme, F., Nennstiel, D., and Bonas, U. (2002): Two novel type III-secreted proteins of Xanthomonas campestris pv. vesicatoria are encoded within the hrp pathogenicity island. J. Bacteriol 184, 1340–1348. Oosterhaven, K., Hartmans, K. J., Scheffer, J. J. C., van der Plas, L. H. W. (1995): S-Carvone Inhibits Phenylalanine Ammonia Lyase (PAL) Activity and Suberization During Wound Healing of Potato Tubers. J. Plant Physiol. 146, 288-294. Pedley K. F., Martin G. B. (2003): Molecular basis of Pto-mediated resistance to bacterial speck disease in tomato. Annu Rev Phytopathol 41, 215–243. Ride, J. P. (1978): The role of cell wall alterations in resistance to fungi. Ann. Appl. Biol. 89, 302-6. Robertson, B. (1987): Endo-polygalacturonase from Cladosporium cucumerinum elicits lignification in cucumber hypocotyls. Physiol. Mol. Plant Pathol. 31, 361-74. Ryan, C. A. (2000): The systemin signaling pathway: differential activation of plant defensive genes. Biochim Biophys Acta 1477, 112-21. Schultz, C. J., Johnson, K. L., Currie, G., Bacic, A. (2000): The Classical Arabinogalactan Protein Family of Arabidopsis. Plant Cell 12, 1751-1767. Showalter, A. M. (1993): Structure and function of plant cell wall proteins. Plant Cell 5, 9-23. Taiz, L. and Zeiger, E. (2000): Physiologie der Pflanzen. Spektrum Akademischer Verlag GmBH Heidelberg, Berlin. 53 Literatur Tamano, K., Aizawa, S., Katayama, E., Nonaka, T., Imajoh-Ohmi, S., Kuwae, A., Nagai, S., Sasakawa, C. (2000): Supramolecular structure of the Shigella type III secretion machinery: the needle part is changeable in length and essential for delivery of effectors. EMBO J. 19, 3876±3887. Tanksley S. D. (2004): The genetic, developmental, and molecular bases of fruit size and shape variation in tomato. Plant Cell. 16, 181–189. Van Cutsem, P. and Messiaen, J. (1993): Roles of Pectins in cell walls. Acta Hort 326, 93104. Van Gijsegem, F., Genin, S., Boucher, C. (1993): Conservation of secretion pathways for pathogenicity determinants of plant and animal bacteria. Trends Microbiol. 1, 175-80. Van Gijsegem, F., Vasse, J., Camus, J. C., Marenda, M., Boucher, C. (2000): Ralstonia solanacearum produces hrp dependent pili that are required for PopA secretion but not for attachment of bacteria to plant cells. Mol. Microbiol 36, 249±260. Wesley, S. V., Helliwell, C. A., Smith, N. A., Wang, M. B., Rouse, D. T., Liu, Q., Gooding, P. S., Singh, S. P., Abbott, D., Stoutjesdijk, P. A., Robinson, S. P., Gleave, A. P., Green, A. G., Waterhouse, P. M. (2001): Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J. 27, 581-90. Zhao, Z. D., Tan, L., Showalter, A. M., Lamport, D. T. A., Kieliszewski, M. J. (2002): Tomato LeAGP-1 arabinogalactan-protein purified from transgenic tobacco corroborates the Hyp contiguity hypothesis. Plant J. 31, 431-444. Zimmermann, D. (2005): Untersuchungen zur subzellulären Lokalisation der Metalloprotease LeMMP1 aus Tomate. Diplomarbeit Universität Hohenheim, Institut für Physiologie und Biotechnologie der Pflanzen. 54 Anhang 6. Anhang 6.1 cDNA Sequenz der LeMMP-1 und LeMMP-2 cDNA Sequenz der LeMMP-1 und LeMMP-2. Gezeigt sind die Ansatzstellen der verwendeten Primer für die LeMMP-1 (Rot) und LeMMP-2 (Grün) die zur Überprüfung des “Silencing” benutzt wurden. Sternchen (*) markieren homologe Bereiche zwischen der LeMMP-1 und LeMMP-2. LeMMP-1 LeMMP-2 ATATATATATAATAAAGAGCAGAGAGAAGTTGCAAGTAAGCAAATTAGAACAATTTAGCA -----------------------------------------------------------MMP 15 LeMMP-1 LeMMP-2 AGCAAGTTTCTGATGAGGATTCCTTTATTCATCGCCATACTTTTTGTTCTTAGTGTTCCA ---AA-----TGAGGAGGATTCATCTATACATTGCCATTGCTTATGTTGT-AATATTGCA ** *** ******** * *** *** ***** ** **** * * * ** ** MMP 25 b LeMMP-1 LeMMP-2 TTTCCATC-TTCAGCTCATTTCTTCCCAAATATTTCTTCAATTCCTCCTAATTTATTGAA AACTAGTTGTTCAGCTCATTTCTTTCCAAATATTTCATCAATCCCTTCTTCTTTACTCAA * *************** *********** ***** *** ** **** * ** LeMMP-1 LeMMP-2 ACCAAATGCCACTGCCTGGGATGCTTTTAACAAGTTATTAGGATGCCATTCCGGTCAGAC ACCTAATGCCACTGCTTGGACTTCTTTTCAAAAGTTGTTAGGATGTCAACCTGCTCAGAA *** *********** *** * ***** * ***** ******** ** * * ***** LeMMP-1 LeMMP-2 GGTCGACGGCTTAGCGAAAATCAAAAAATATTTTCACTACTTTGGATACATTAATAATTC AGTCGACGGCATTGCTAAAATCAAAAAATATTTTCAACATTTTGGTTACATTAATAATTT ********* * ** ******************** * ***** ************* LeMMP-1 LeMMP-2 TTCCA---CTAACTTCACTGATGATTTTGATGATACTCTTGAATCTGCTCTCAAGACCTA GAGTAGTTTTAATTTCACTGATGAATTTGATGATACTCTTGAATCTGCTCTCAAGACGTA * *** *********** ******************************** ** LeMMP-1 LeMMP-2 CCAGCTTAACTTCAACCTCAACACCACCGGTGTGCTCGACGCGAACACCATTCAGCATCT TCAGAGAAATTTCAACCTCAAAGCCACCGGTGTGCTCGATGCGCCCACCATTCAGCATCT *** ** *********** **************** *** *************** LeMMP-1 LeMMP-2 CATAAAACCCAGATGTGGAAACGCTGATGTAGTTAACGGTACTAGTACTATGAACTCCGG CATAAAACCCAGATGTGGAAATGCCGATCTAGTTAACGGTACTAGTACTATGAACGCTGG ********************* ** *** ************************** * ** LeMMP-1 LeMMP-2 TAAGCCACCGGCAGGTTCTCCGACGATGCACACCGTAGCTCACTACTCCTTCTTTCCGGG AAAGCC---------------------GCACACGGNGGCTCACTACTCCTTCTTTCCTGG ***** ****** * ******************** ** MMP 13 LeMMP-1 LeMMP-2 AAGTCCACGGTGGCCGGCGAACAAGAGAGATCTGACATATGCTTTTGCACCGCAGAATGG AAGACCAAAGTGGCCCGAGGGTAAGACTGATTTGACTTATGCCTTTCTACCGGCAAACAA *** *** ****** * * **** *** **** ***** *** **** ** MMP 23 55 Anhang LeMMP-1 LeMMP-2 ACTGACGGATGATATTAAGATTGTGTTCACACGAGCGTTTGATAGGTGGTCGGAGGTGAC TTTGACGGATGATATTAAGAGTGTCTTCTCACGTGCGTTTGATCGGTGGTCGGAGGTAAC ****************** *** *** **** ********* ************* ** LeMMP-1 LeMMP-2 TCCATTGACGTTTACTGAAATAGCATCGTACCAATCGGCTGATATTAAGATCGGGTTTTT CCCGTTGA-----------------------------GCT-------------------** **** *** LeMMP-1 LeMMP-2 LeMMP-1 CAGCGGAGATCACAACGATGGAGAGCCGTTTGATGGTCCTATGGGGACATTAGCACACGC ---------TCACGG--------------------------------------------**** GTTTTCCCCACCGGCGGGGCATTTTCACTTGGACGGCGAGGAGAACTGGGTGATCGACGG LeMMP-1 TGCGCCGATAGTTGATGGGAATTTCTTTTCTATATTGTCGGCGGTGGACCTTGAATCGGT LeMMP-1 TGCGGTTCATGAAATCGGGCATTTATTGGGTTTGGGTCATTCATCCGTAGAAGATGCTAT LeMMP-1 TATGTACCCGACTTTAGGAGCGGGTACCCGAAGAGTCGAGCTTAGAAATGATGATATATT LeMMP-1 GGGAGTCCAGGAGTTATACGGGTCTAACCCGAATTATACTGGGCCAAACCCAAATTTGAC LeMMP-1 TCCGAGCCAAGAGAGTGACACAAATGGAGCCCCGATATTTGAGTTATCATGGTTTCATGG LeMMP-1 GTTTCTTGGTTTATTCTTTGCTTTGTTCATTCAACTGTAGTTGATGGCATGATGTGATTA LeMMP-1 TTTTTTAATACTATATACTTGATTCATCTTTTTAAAAAAAAAAAAA 6.2 Abkürzungsverzeichnis Abb. Abbildung APS Ammoniumperoxidisulfat At Arabidopsis thaliana bp Basenpaare cDNA komplementäre DNA ddH2O doppelt destilliertes H2O DNA Desoxyribonukleinsäure dNTP Desoxynukleotidtriphosphat DISK-PAGE Diskontinuierliche Polyacrylamidgelelektrophorese ECL Enhanced Chemiluminescence ECM Extrazelluläre Matrix EDTA Ethylendiamintetraessigsäure EST „expressed sequence tag“ 56 Anhang GPI / GPI-Anker Glycosyl-Phosphatidylinositol (-Anker) HPLC High Pressure Liquid Chromatography kDa Kilo Dalton Le Lycopersicon esculentum NB Nutienth Broth MMP Matrix Metalloproteinase PAL Phenylalanin Amoniak Lyase PCR Polymerase Chain Reaction RNase Ribonuclease rpm rounds per minute RT (min) Retentionszeit (Minuten) RT Raumtemperatur RT-PCR Reverse Transcriptase Polymerase Chain Reaction SDS Natriumdodecylsulfat TBS Tris-Buffered Saline TEMED Tetramethyläthylendiamin TIMPs Tissue Inhibitors of Metalloproteinases TMV Tomaten Mosaik Virus Ubq. Ubiquitin ÜN Über Nacht z.B. zum Beispiel 6.3 SI-Einheiten °C Grad Celsius l Liter M Molar min Minuten m Milli- µ Mikro- 57 Eidesstattliche Erklärung Eidesstattliche Erklärung Ich versichere hiermit, dass ich die vorliegende Diplomarbeit selbständig angefertigt, keine anderen als die angegebenen Hilfsmittel benutzt und sowohl wörtliche, als auch sinngemäß entlehnte Stellen als solche kenntlich gemacht habe. Die Arbeit wurde in gleicher oder ähnlicher Form noch keiner anderen Prüfungsbehörde vorgelegt und auch nicht veröffentlicht. Hohenheim, 18. Januar 2006 58 Danksagung Danksagung Einen besonderen Dank geht an, • Herrn Professor Dr. Andreas Schaller, für die Vergabe des Diplomarbeitsthemas, Bereitstellung des Arbeitsplatzes und für die optimale Betreuung. • Herrn Professor Dr. Arthur Pfitzner, der freundlicherweise die Zweitkorrektur übernommen hat. • allen Mitarbeiter des Instituts, für die praktische und theoretische Unterstützung und für die angenehme Arbeitsatmosphäre. • meinen Eltern (Sponsoren), für die Materielle und Moralische Unterstützung während meines Studiums. • an meiner Freundin Anna Xianhua Chen, für ihre Unterstützung und ihre aufmunternden Worte, während des gesamten Studiums. 59