3. Thermodynamik
Werbung

140
3. Thermodynamik
3. Thermodynamik
3.1. Grundlagen
3.1.1. Einführung
Die Thermody namik beschreibt die Zustände
und deren Änderung infolge der Wechselwirkung mit der Umgebung von kompliziert zusammengesetzten makroskopischen Systemen
durch eine geringe Anzahl makrosk op ischer
Variablen, wi e z. B. Druck oder Temperatur,
sowie durch thermodynam ische Potentiale.
Das System kann makroskop isch betrachtet
werden. Hierbei wird das gesamte System
durch makroskopisch meßbare Systemeigenschaften und deren Zusammenhänge beschrieben. Dies wird als phänomenologische Thermodynamik bezeichnet, die der älteste Zweig der
Thermodynamik ist.
Das System kann auch mikroskopisch betrachtet werden. Hierbei werden die makroskopischen Systemeigenschaften auf die Wechselwirkungen der Systembestandteile (Atome,
Moleküle) zurückgeführt. Die Beschreibung
erfolgt mit den statistischen Methoden der
klassischen Mechanik bzw. der Quantenmechanik. Beispielsweise erklärt die kinetische Gastheorie das Zustandekommen des
Gasdrucks und ermöglicht ein tieferes Verständnis des Temperaturbegriffs. Oder es können mit Hilfe der Statistik thermodynamische
Potentiale hergeleitet werden, aus denen sich
alle Zustandsgrößen und Materialeigenschaften (z. B. die spezifische Wärmekapazität) ergeben. In Bild 3- 1 sind diese Betrachtungsweisen gegenübergestellt.
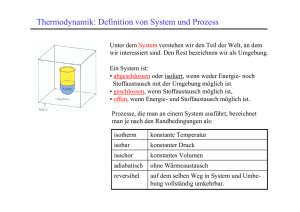
Ein thermodynamisches System kann mit
seiner Umgebung in Wechselwirkung stehen.
Fi ndet kein Austausch von Energie und Masse
über die Systemgrenzen statt, so ist das System
abgeschlossen. Wird nur die Arbeit W (z. B.
mechanische, elektrische, magnetische Arbeit)
ausgetauscht, liegt ein adiabates System vor.
Bei geschlossenen Systemen findet ein Austausch von Arbeit Wund Wärme Q und bei
offenen Systemen noch zusätzlich ein Masseaustausch statt.
Die wichtigsten Erkenntnisse in der Thermodynamik sind in vier Hauptsätzen formuliert.
Der erste Hauptsatz ist der Energieerhaltungssatz. Er besagt, daß die Änderung der inneren
Energie !1U durch Wärmezufuhr Q und (oder)
Arbeitsverrichtung W erfolgen kann. Der
zweite Hauptsatz sagt mit Hilfe des Entropiebegriffs etwas über die Richtung von Zustandsänderungen aus. Bei reversiblen Prozessen ist die Entropieänderung null ; bei irreversiblen Prozessen ist sie positiv, d. h., die Wärme
ist nicht vollständig in andere Energieformen
umwandelbar. Von der Thermodynamik irreversibler Prozesse sind die Transport- und
Ausgleichsvorgänge von besonderer praktischer
Bedeutung. Die E ntropie S läßt sich auch mikroskopisch als Wahrscheinlichkeitsfunktion
deuten (Logarithmus der Zustandswahrscheinlichkeit In P multipl iziert mit der BoltzmannKonstanten k) . Zustandsänderungen werden
in Richtung maximaler Wahrscheinlichkeit
(maximale Entropie) ablaufen. Der dritte
Hauptsatz (Satz von Nernst) zeigt, daß bei
Annäherung der Temperatur an den absoluten Nullpunkt (T -+ 0) die Entropie konstant
wird. Diese Konstante wird gleich null gesetzt. Aus dem dritten Hauptsatz folgt auch,
daß der absolute Nullpunkt (T= 0) nicht erreicht werden kann.
Ein thermodynamisches System - sei es gasförmig (ideale oder reale Gase), flüssig oder
fest - kann durch Zustandsgleichungen und
Zustandsfunktionen, die nur vom Anfangsund Endzustand abhängen, beschrieben werden. Zu den Zustandsfunktionen {thermodynamischen Potentialen) gehören die innere
Energie U, die Enthal pie H , die freie Energie
F, die freie Enthalpi e G und die Entropie S.
Mit den Zustandsgleichungen und Zustandsfunktionen ist die Beschreibung von Gleichgewichtszuständen und Gleichgewichtsbedingungen möglich.
3.1.2. Thermodynamische Grundbegriffe
Systeme
Ein räumlich abgrenzbarer Bereich, der herausgelöst von seiner Umgebung betrachtet
werden soll, wird als System bezeichnet. Nach
Art der Systemgrenzen werden verschiedenartige Systeme unterschieden, wie aus Tabelle
3-1 hervorgeht.
3.1.Grundlagen
I
I
THERMODYN A MIK
Umgebung
abgesch Iossen
adiabat
Systems
des
lw
1~1~1~1~1m
1
I
geschlossen
offen
LW = O+ W
Energiesatz (erster Hauptsatz)
makroskopische
Betrachtung
~/deale Gase
Beschreibung
pV = VRmT
des
.&:
.,0
gesamten
Systems
durch
makroskopische
Variable
(z. B.
p, V, T, p, C)
r-
"51
0
0
~
c::
Ql
reale Gase
(van der Waals)
1
E
0
c::
.&:
a.
I
Festkörper
V = f(p ,T)
"'
und Endzustand
abhängig (Zustandsfunktion)
Zustandsbeschreibung
des Systems
<z. B. p,
mikroskopische
Betrachtung
0
::T
nurvom Anfangs-
(fJ
Ia
~
"
Beschreibung
L...__
v. n
Gleichgewichtszustand
Gleichgewichtsbedingungen
~
des
innere
Systems
Energie U
iä
~ Enthalpie H
;;;·
H= U+p V
0
::T
~
Satz von Nernst
(111. Hauptsatz)
~
basierend
aufden
I--
statistischen
Methoden
~reieEnergieF
der
F=U-TS
V = f(p ,T)
~
"öl
"'"'c;;·
sehe Potentiale.
~ Rüssigkeiten
'111
,..--
thermodynami-
ZustandsGleichungen
141
,--
freie
Enthalpie G
G=H-TS
0
.,
c
:J
'"
Entropie
klassischen
bzw.
Quantenmechanik
:J
s
~
!!l.
~
lim S(p, T, V) =
T- 0
= konst=O
;<'
L...__
Richtung der Zustandsänderung (zweiter Hauptsatz)
-I
0, •• = l!.S
T
Entropie S
reversible Vorgänge
I ~S system + l1Sumgebu ng =
S = k · lnP
-
irreversible Vorgänge
0
I
ö.Ssystem + ll.Sumgebung > 0
S=f(Zeit)
Bild 3-1.
Strukturbild der Thermodynamik.
Zustand, Zustandsgrößen, Prozeßgrößen
In der Mechanik wird die Lage eines Punktes
im Raum durch drei Koordinaten festgelegt ;
in der Thermodynamik benutzt man Zustandsgrößen, um den Zustand eines Systems
zu beschreiben. Historisch bedingt wird zwischen den direkt meßbaren thermischen Zustandsgrößen
- Druckp,
Thermodynamik
irreversibler Prozeß
Transport- und
Ausgleichsvorgänge
(z. B. Wärmeleitung,
Diffusion)
-Volumen V,
- Temperatur T
und den davon abgeleiteten kalorischen Zustandsgrößen, wie z. B.
- innere Energie U,
- EnthalpieHund
- Entropie S
unterschieden.
142
3. Thermodynamik
Tabelle 3.1. Thermodynamische Systeme.
Bezeichnung
des Systems
Kennzeichen der Systemgrenzen
Beispiele
offen
durchlässig für Materie und Energie
Wärmeübertrager, Gasturbine
geschlossen
durchlässig für Energie, undurchlässig
für Materie
geschlossener Kühlschrank, Warmwasserheizung, Heißluftmotor
abgeschlossen
undurchlässig für Energie und Materie
verschlossenes Thermosgefäß
adiabat
undurchlässig für Materie und Wärme,
durchlässig für mechanische Arbeit
rasche Kompression in einem Gasmotor
Bleiben die Zustandsgrößen zeitlich konstant,
dann befindet sich das System in einem
Gleichgewichtszustand Der Zustand eines Systems kann auf verschiedene Weise verändert werden (z. B. durch Wärmezufuhr von
außen). Hat sich, ausgehend von dem Gleichgewichtszustand I, ein neuer Gleichgewichtszustand 2 eingestellt, dann haben alle Zustandsgrößen wieder wohldefinierte Werte
angenommen.
innere Energie U, Enthalpie H). Intensive
Größen sind davon unabhängig (zo B. Druck p,
Temperatur T). Wird eine extensive Größe
durch die Substanzmenge dividiert, ergibt sich
eine intensive Größe.
Eine spezifische Größe x ergibt sich nach
DIN 5490 aus einer gemessenen extensiven
Größe X, indem durch die Masse m des Systems dividiert wird:
I Die Ände-r-un_g_ !!..Z
- -e-in_e_r_Z
_u_s_t-an_d_s_g_ro_··ß- e- 2---,
hängt nicht von der Art der Prozeßführung ab, sondern nur vom Anfangs- und
Endzustand. Es gilt
!!..Z =
z2- z,.
(3-1)
Im Gegensatz zu den wegunabhängigen Zustandsgrößen sind Wärme und mechanische
Arbeit wegabhängige Prozeßgrößen. Die mit
dem System bei einer Zustandsänderung ausgetauschten Energiebeträge sind von dem
Verlauf des Prozesses abhängig.
Für jeden Gleichgewichtszustand sind die Zustandsgrößen durch eine Zustandsgleichung
miteinander verknüpft. So gilt z. B. für ideale
Gase ein einfacher Zusammenhang zwischen
Druck, Volumen und Temperatur (Abschn.
3.1.5). Bei realen Gasen ist der Zusammenhang komplizierter und muß empirisch und
mit Hilfe von Modellrechnungen ermittelt
werden (Abschn. 3.4).
Spezifische und molare Größen
Viele thermodynamische Größen sind extensiv, d.h., sie hängen von der Substanzmenge
(Masse m, Stoffmenge v) des Systems ab (z. B.
(3-2)
In der Maßeinheit einer spezifischen Größe
steht immer x = . kg - 1 Spezifische Größen
werden nach DIN 1345 mit kleinen Formelbuchstaben geschrieben.
0
•
0
Der Quotient aus einer gemessenen Größe X
und der Stoffmenge v ist die molare Größe X m,
die durch den Index m gekennzeichnet wird:
X
Xm= - 0
V
(3-3)
1
Die Maßeinheit einer molaren Größe enthält
stets xm = .. mol - 1 .
Jede spezifische Größe kann leicht in die entsprechende molare Größe umgerechnet werden. Aus GI. (3-2) und (3-3) folgt sofort
X= X m = X m V' oder
0
(H) I
3.1. Grundlagen
Darin ist M die Molmasse der betreffenden
Substanz (Einheit kg/mol).
143
p
Beispiel
3.1-1: Um m = 2 kg Wasser zu verdampfen, ist die
Verdampfungswärme Qd = 4,512 MJ erforderlich.
Wie groß sind die spezifische und die molare Verdampfungswärme von Wasser?
A
Lösung:
Für die spezifische Verdampfungswärme erhält man
qd = Qdlm = 2,256 MJ/kg. Die Molmasse von Wasser ist M = 18 g/mol. Somit beträgt die molare Verdampfungswärme
Qmd = 2,256 MJ/kg · 18 g/mol
= 40,6 kJ/mol.
3.1.3. Temperatur
Die Temperatur ist der menschlichen Empfindung direkt zugänglich und wird mit Begriffen wie "warm" und "kalt" umschrieben.
Körper, die sich auf verschiedener Temperatur befinden, können durch Befühlen unterschieden und entsprechend ihrer Temperatur
klassifiziert werden. Bringt man zwei Körper
verschiedener Temperatur in Kontakt, so stellt
man fest, daß der warme Körper kälter und der
kalte wärmer wird. Es findet ein Temperaturausgleich statt, der dann beendet ist, wenn das
System einen Gleichgewichtszustand erreicht
hat. Dieser Sachverhalt wird durch den nullten Hauptsatz der Thermodynamik ausgedrückt:
Im thermodynamischen Gleichgewicht haben alle Bestandteile eines Systems dieselbe Temperatur.
Der vorgenannte subjektive Temperaturbegriff muß natürlich durch eine Temperaturdefinition mit entsprechenden Meßvorschriften ersetzt werden. Die exakte Definition der
sog. thermodynamischen Temperatur geschieht
über den Wirkungsgrad einer idealen Wärmekraftmaschine und wird in Abschn. 3.3.5 behandelt.
Bereits im Jahr 1704 stellte G. AMONTONS
(I 663 bis 1705) fest, daß der Druck eines
Gases, dessen Volumen konstant gehalten
wird, von der Temperatur abhängt. Er schlug
vor, die Temperatur proportional zum Druck
des Gases zu setzen (T,....., p) und damit die
Bild 3-2. Prinzip eines Gasthermometers mit konstantem Gasvolumen. Durch Heben oder Senken des
Ausgleichsgefäßes A wird der Quecksilberspiegel im
linken Schenkel des U-Rohrs auf der Nullmarke gehalten.
p Druck
T absolute Temperatur.
Temperaturmessung auf eine Druckmessung
zurückzuführen. Man erreicht dies mit Hilfe
des in Bild 3-2 dargestellten Gasthermometers.
Es läßt sich zeigen, daß die Temperatur des
Gasthermometers für ideale Gase (Abschn.
3.1.4 und 3.1.5) identisch ist mit der oben erwähnten thermodynamischen Temperatur. Die
Abweichungen, die reale Gase zeigen, kann
man rechnerisch berücksichtigen.
Der im Gasthermometer bestimmte Gasdruck
p kann erst dann in eine Temperatur T umgerechnet werden, wenn die Proportionalitätskonstante zwischen Druck und Temperatur
festgelegt ist. Alle Experimente, besonders die
in Abschn. 3.1.4 geschilderten von Gay-Lussac,
zeigen, daß es einen absoluten Nullpunkt der
Temperatur gibt. Um eine Temperaturskala
festzulegen, ist daher nur noch die Temperatur eines weiteren Punktes zu definieren.
Dazu wurde der Tripelpunkt des Wassers zu
Tn = 273,16 K (Kelvin) festgelegt. Der Tripelpunkt ist der Zustand, bei dem in einem
Gefaß der feste, flüssige und gasförmige Aggregatzustand miteinander im Gleichgewicht
sind. Der Tripelpunkt des Wassers ist leicht
herzustellen und mit einer Toleranz von einigen Millikelvin reproduzierbar. Die 13. Gene-
144
3. Thermodynamik
ralkonferenz für Maße und Gewichte (GKMG)
legte 1967 als Einheit für die Temperatur fest:
Durch diese Definition wird erreicht, daß
Temperaturdifferenzen in beiden Einheiten
dieselbe Maßzahl haben.
I Kelvin ist der 273,16te Teil der thermodynamischen Temperatur des Tripelpunktes von Wasser.
Für den praktischen Gebrauch wurde die Internationale Temperaturskala von 1990 (ITS-90)
erarbeitet. Sie stützt sich auf 17 gut reproduzierbare thermodynamische Gleichgewichtszustände als definierende Fixpunkte (Tabelle 3-2)
und gilt als derzeit beste Darstellung thermodynamischer Temperaturen.
Die Einheit Kelvin (K) für die absolute Temperatur wurde zu Ehren von W. THOMSON
(1824 bis 1907), dem späteren Lord Kelvin
gewählt, auf den die Temperaturskala zurückgeht.
Die so definierte Kelvin-Skala hat dieselbe
Skalenteilung wie die bereits 1742 von A.
CELSIUS ( 1701 bis 1744) vorgeschlagene Skala,
bei der Schmelz- und Siedepunkte des Wassers unter Normdruck (0 oc bzw. 100 oq als
Fixpunkte dienen. Der Zusammenhang zwischen der absoluten Temperatur T in Kelvin
und der Temperatur 9 in Grad Celsius ergibt
sich aus
9
T
- = - - 27315.
oc K
'
(3-5)
Zur Interpolation zwischen den Fixpunkten wird
zwischen 0,65 K und 5 K die Temperatur aus dem
Dampfdruck von 3 He bzw. 4 He bestimmt; zwischen
3 K und 24,5561 K mit einem Gasthermometer.
Oberhalb 13,8033 K bis 1234,93 K werden Pt-Widerstandsthermometer und für noch höhere Temperaturen Spektralpyrometer eingesetzt.
Temperaturmessung
Jede physikalische Größe, die sich mit der
Temperatur ändert, kann zur Temperaturmessung herangezogen werden. Für die verschiedensten Meßaufgaben, Meßobjekte und Temperaturbereiche wurden unterschiedliche Meßverfahren entwickelt. Eine Zusammenstellung
gängiger Methoden enthält Tabelle 3-3. Die
Tabelle 3-2. Definierende Fixpunkte der ITS-90. Wenn nicht anders angegeben, beträgt der Druck
Pn = 101,325 kPa.
Gleichgewichtszustand
Siedepunkt von Helium bei verschiedenen
Dampfdrücken
Tripelpunkt des Gleichgewichtswasserstoffs
Siedepunkt von Wasserstoff beim Dampfdruck 32,9 kPa
und 102,2 kPa
Tripelpunkt des Neons
Tripelpunkt des Sauerstoffs
Tripelpunkt des Argons
Tripelpunkt des Quecksilbers
Tripelpunkt des Wassers
Schmelzpunkt der Galliums
Erstarrungspunkt des Indiums
Erstarrungspunkt des Zinns
Erstarrungspunkt des Zinks
Erstarrungspunkt des Aluminiums
Erstarrungspunkt des Silbers
Erstarrungspunkt des Goldes
Erstarrungspunkt des Kupfers
79 0
in K
3 bis 5
13,8033
17
20,3
24,5561
54,3584
83,8058
234,31 56
273,16
302,9146
429,7485
505,078
692,677
933,473
1234,93
1337,33
1357,77
{}90
in
oc
- 270,15 bis
- 268 ,15
- 259,3467
-256,15
-252,85
-248 ,5939
-218,791 6
-189,3442
-38,8344
0,01
29,7646
156,5985
231,928
419,527
660,323
961 ,78
1064,18
1084,62
3.1. Grundlagen
145
Tabelle 3-3. Temperaturmeßverfahren.
Thermometertyp
Meßbereich
in oc
Fehl ergrenzen
physikalisches Meßprinzip
- 200 bis
- 110 bis
- 90 bis
- 58 bis
- 38 bis
bis
Näherungsweise
in Größenordnung der Skalenteilung.
Details in
VDEIVDI 3511
Thermische Ausdehnung einer Flüssigkeit wird zur Temperaturmessung verwendet. Die Temperatur wird aus dem
Stand der Flüssigkeit in einer Glaskapillare ermittelt.
FlüssigkeitsGlasthermometer
....
0
.....
0
8
0
8....
0
..<::
.....
"'Oll
s::
Füllung:
Pentangemisch
Alkohol
Toluol
Hg-Tl
Quecksilber
Galliumlegierung
30
210
100
30
800
1000
FlüssigkeitsFederthermometer
- 35 bis 500
1 bis 2% des
Anzeigebereichs
Thermische Ausdehnung einer Flüssigkeit (z. B. Hg unter 100 bis 150 bar)
wird auf eine Rohr- oder Schneckenfeder übertragen.
DampfdruckFederthermometer
-50 bis 350
1 bis 2% des
Anzeigebereichs
Dampfdruck einer Flüssigkeit (Ethylether, Hexan, Toluol, Xylol) wird auf
eine Rohr- oder Schneckenfeder
übertragen .
0 bis 1000
1 bis 2% des AnZeigebereichs
Thermische Ausdehnung eines Metallstabs bewegt ein Meßwerk.
-50 bis 400
l bis 3% des Anzeigebereichs
Thermobimetall besteht aus zwei fest
miteinander verbundenen Schichten aus
Werkstoffen mit unterschiedlichen thermischen Ausdehnungskoeffizienten und
krümmt sich bei Temperaturänderung.
;:I
....
....
0
..<::
:;:I
il:l
0
..<::
u
"'
·a
ol
..<::
u
0
8
Stabausdehnungsthermometer
Bimetall thermometer
....
~
0
8
0
.5
-5
"'Oll
§
Thermoelemente
AuFe-NiCr
Cu-Konstantan
Fe-Konstantan
Ni Cr-Konstantan
Pt - PtRh
W-WMo
270 bis
200 bis
200 bis
200 bis
0 bis
0 bis
0
400
700
900
1600
3300
0,75% des Temperatur-Sollwerts, mindestens 3 K
Zwischen zwei Verbindungsstell en verschiedener Metalle entsteht eine Thermospannung, wenn di e Verbindungsstellen auf verschiedenen Temperaturen
sind (Seebeck-Effekt).
- 250 bis
- 60 bis
- 273 bis
40 bis
1000
180
400
270
0,3 bis 5 K
0,2 bis 2, 1 K
0,5 bis 1,5 K
Temperaturabhängigkeit des elektrisehen Widerstandes von Metallen und
Halbleitern dient zur Temperaturbestimmung.
-
....
..<::
'e
0
il:l
0
..<::
u
·s"'
~
0
0
Widerstandsthermometer
Platin
Nickel
Heißleiter
Kaltleiter
146
3. Thermodynamik
Tabelle 3-3. (Fortsetzung)
Thermometertyp
....
0)
....
0)
s0
8
Strahlungspyrometer
Spektralpyrom .
Bandstrahlungsp.
GesamtstrahIungspyrometer
Fehlergrenzen
physikalisches Meßprinzip
650 bis 5000
50 bis 2000
- 40 bis 3000
I bis 35 K
I bis 1,5%
des Bereichs
Temperatur eines Körpers wird aus der
Wärmestromdichte seiner elektromagnetischen Strahlung bestimmt.
Messung erfolgt entweder in engem
Spektralbereich, breitem Spektralband oder im gesamten Spektrum.
1150 bis 2000
200 bis 2200
10 bis 25 K
1 bis 1,5%
des Bereichs
Rote und grüne Strahlungsanteile von
Meßstelle und Referenzlampe werden
verglichen. Vergleich erfolgt subjektiv
durch Farbvergleich oder objektiv
durch Photoempfänger.
Meßbereich
in °C
0)
..<::
f-<
0)
"'
0
'<;]
bl)
1:1
::s
....
..<::
,;:s
....
0)
.0
1:1
....
0)
Verteilungspyrometer
Farbangleichpyr.
Verhältnispyrometer
Photothermometrie
250 bis 1000
±lK
Die Oberfläche eines heißen Körpers
wird mit infrarotempfindlichen Platten
photographisch aufgenommen. Zur Untersuchung von Temperaturfeldern geeignet.
Temperaturmeßfarben
40 bis 1350
±5K
Auf Meßkörper wird Farbe aufgebracht, die bei Erreichen einer bestimmten Temperatur den Farbton ändert.
Temperaturkennkörper
100 bis 1600
±7K
Zylindrische Körper aus Metallegierungenzeigen durch Schmelzen eine
bestimmte Temperatur an.
Segerkegel
600 bis 2000
..<::
~
....
0)
>
C!:l
0)
::E
0)
....
0)
"'
1:1
Mischung aus Ton und Feldspat wird
bei Erreichen einer bestimmten Temperatur weich, der Kegel neigt sich zur
Seite.
0
~
.0
akustisches
Thermometer
-271 bis- 253
Temperaturabhängigkeit der Schallgeschwindigkeit in Gasen ist ein Maß
für die Temperatur.
magnetisches
Thermometer
- 273 bis - 200
Magnetische Suszeptibilität paramagnetischer Salze hängt reziprok von der
absoluten Temperatur ab.
Glasfaserthermometer
50 bis
250 Auflösung
0,1 K
Die Fähigkeit einer Glasfaser, Lichtwellen zu führen, hängt vom temperaturempfindlichen Brechungsindex ab.
3.1. Grundlagen
VDEIVDI-Richtlinien 3511 geben eine ausführlichere Darstellung sowie eine Zusammenstellung der relevanten DIN-Normen.
Temperatur .9z gilt nach GI. (3-7) , wenn V1
das Volumen bei .9 1 ist
Vz = I~ = 11 [I + r:x ( .9z - .91) ]3 =
=VI [I + 3 r:x (.9z- .91) + 3 r:x Z(.9z- .91f
+ r:x3 (.9 z - .91)3].
3.1.4. Thermische Ausdehnung
Festkörper
Die meisten Festkörper dehnen sich bei Erwärmung aus. Die relative Verlängerung MI I
eines Stabes kann innerhalb bestimmter Grenzen proportional zur Temperaturänderung !!.T
gesetzt werden:
M
-=r:x!!.T
I
.
(3-6) •
(3-7)
Die beiden letzten Glieder der Klammer sind
gegenüber dem linearen Glied vemachlässigbar. Daher erhält man in guter Näherung
(3-8)
oder für die relative Volumenänderung
!!.V
- = y !!.T
Ist die Länge 11 bei der Temperatur .9 1 bekannt, so folgt für die Länge lz bei der Temperatur .9z
147
(3-9)
V
mit !!.T = Tz- T 1 = .9z - .91 und dem Raumausdehnungskoeffizienten
1
y = 3 r:x .
mit !!.T= Tz - T 1 = .9z - .9 1• Die Proportionalitätskonstante r:x ist der Längenausdehnungskoeffizient. Sie ist ein Materialparameter und
kann näherungsweise konstant gesetzt werden.
In der Wirklichkeit steigt der Längenausdehnungskoeffizient r:x mit der Temperatur leicht
an; Tabelle 3-4 enthält einige mit I06 multiplizierte Mittelwerte für die Temperaturbereiche
0 °C ~ .9 ~ 100 °C und 0 °C ~ .9 ~ 500 °C.
Mit der Längenausdehnung der Körper ist
zwangsläufig eine Volumenänderung verknüpft.
Für das Volumen Vz eines Würfels bei der
(3-10)
Beispiel
3.1-2: Eine Messingkugel (cx = 19 · 10- 6 K- 1) hat
bei der Temperatur 9 1 = 20 °C den Durchmesser
d 1 = 20,00 mm. Auf welche Temperatur ih muß sie
erwärmt werden, damit sie in einem Ring mit dem
Innendurchmesser d 2 = 20,03 mm stecken bleibt?
Wie hat sich das Kugelvolumen verändert?
Lösung:
Nach GI. (3-6) ist die Temperaturänderung
0,03 mm
79 K
20mm · 19 · 10- 6 K- 1
•
Also ist die erforderliche Temperatur 9 2 = 99 °C.
Die relative Volumenvergrößerung beträgt nach
GI. (3-9) und (3-10)
t:J.V
- = y!:J.T = 3 a !:J.T= 4,5 · 10- 3•
!:J.T = M
da
Tabelle 3-4. Mittlerer linearer Längenausdehnungskoeffizient r:x einiger Festkörper in verschiedenen Temperaturbereichen.
I 06 a
inK- 1
Temperaturbereich
Aluminium
Kupfer
Stahl C 60
rostfreier Stahl
Invarstahl
Quarzglas
gewöhnliches Glas
ooc ~ 9
~ 100 oc
23,8
16,4
11 ,1
16,4
0,9
0,51
9
106 (X
inK- 1
ooc~
~
V
e
9
500 °C
27,4
17,9
13,9
18,2
0,61
10,2
Die Dichte
eines Körpers ist umgekehrt
proportional zum Volumen. Für die Temperaturabhängigkeit gilt
e(.9)
m
Vo(l
+ y .9)
Ist eo= ml Vo die Dichte bei .9 0 = 0 o C, dann
ist die Dichte bei der Temperatur .9
Q ( .9)
Qo
= + Y .9
1
~
eo (I -
y .9) .
(3-11 )
148
3. Thermodynamik
I
Flüssigkeiten
Weil Flüssigkeiten keine Eigengestalt haben,
ist nur die Volumenänderung von Interesse.
Es gelten GI. (3-8) , (3-9) und (3-11); allerdings ist der Raumausdehnungskoeffizient y
größer als bei Festkörpern. Einige Zahlenwerte enthält Tabelle 3-5.
Tabelle 3-5. Raumausdehnungskoeffizient y
einiger Flüssigkeiten bei der Temperatur
.9= 20°C.
Stoff
10 3 yi nK-
Wasser
Quecksilber
Pentan
Ethylalkohol
Heizöl
0,208
0, 182
1,58
1,10
0,9 bis 1,0
1
y =0003661 K- 1 = - - '
273,15K
Ein Gas in diesem Grenzzustand wird als
ideales Gas bezeichnet.
Wie die graphische Darstellung des GayLussacschen Gesetzes in Bild 3-3 zeigt, wird
das Volumen bei .9 =- 273,15 oc gleich null.
Dies ist der absolute Nullpunkt der Temperatur. Natürlich gilt das Gay-Lussacsche Gesetz bei sehr tiefen Temperaturen nicht mehr.
Reale Gase kondensieren beim Abkühlen ;
selbst am absoluten Nullpunkt muß noch ein
bestimmtes Restvolumen, nämlich das Eigenvolumen der Atome, übrig bleiben. Die absolute Temperatur T erlaubt eine einfache Formulierung des Gay-Lussacschen Gesetzes:
V( T ) = V0 -
T
To
Bemerkenswert ist die Anomalie des Wassers.
Bei der Temperatur .9 = 4 °C hat die Dichte
ihr Maximum mit l2max = 0,999973 kg/dm 3.
Wenn im Winter ein See zufriert, sammelt
sich das Wasser von .9 = 4 °C und größter
Dichteam Grund; darüber liegen die kälteren
und leichteren Schichten. Weil die kalten
Schichten nicht absinken, erfolgt keine Wärmeübertragung durch Konvektion. Der Wärmetransport durch Wärmeleitung ist nicht
sehr effektiv (Abschn. 3.5), so daß tiefe Seen
nicht bis zum Grund durchgefrieren.
V
bzw. -T = konst. (3-12)
Hierbei ist T0 = 273, 15 K
-273,15 °C
Gase
Temperatur {}
Bei Gasen hängt das Volumen vom Druck
und der Temperatur ab. Messungen von
J. A. C. CHARLES (1746 bis 1823), die von
J. L. GAY-LUSSAC (1778 bis 1823) vertieft
wurden, ergaben, daß bei einem Gas unter
konstantem Druck das Volumen linear mit
der Temperatur gemäß GI. (3-9) variiert:
V(.9)
=
V0 (1
Bild 3-3. Zusammenhang zwischen dem Volumen V
und der Temperatur T eines idealen Gases bei konstantem Druck.
Wird das Volumen eines Gases konstant gehalten und die Tem peratur verändert, dann
variiert der Druck p gemäß
wenn V0 das Volumen bei .9 0 = 0 oc ist.
Experimente liefern für den Raumausdehnungskoeffizienten y im Gay-L ussacschen G esetz für fast alle Gase den gleichen Wert. Die
Unterschiede zwischen den einzelnen G asen
werden um so geringer, je ni edriger der Druck
p ist. Im Grenzfall p -+ 0 ergi bt sich für all e
Gase
+ Y .9)
(3-13)
p (T)= p0 I.._ bzw. PT = konst.
To
(3-14)
p(.9) = Po( l
+ y .9),
oder
Diese Gl eichung ist die Grundlage der Temperaturbestimmung nach Amontons mit Hilfe
des Gasthermometers.
3.1. Grundlagen
m
V.=n
3.1.5. Allgemeine Zustandsgleichung
idealer Gase
Qo
Das Volumen V und der Druck p einer abgeschlossenen Menge eines idealen Gases sind
bei konstanter Temperatur durch das Gesetz
von Boyle-Mariotte verknüpft:
p V= konst.
(3-15)
1
Der Zusammenhang wurde 1662 von R. BOYLE
(1627 bis 1691) und unabhängig von ihm 1679
von E. MARIOTTE (1620 bis 1684) experimentell gefunden.
Die Gesetze von Boyle-Mariotte, Gay-Lussac
und Charles, formuliert in GI. (3-15), (3-12)
sowie (3-14), lassen sich in einer Gleichung,
der Zustandsgleichung idealer Gase kombinieren:
pV
r=
konst.
(3-16)
T
Tn
(3-17)
zusammen. Somit wird aus GI. (3-17)
pV
Pn
-=--m.
T
ToQn
Die Werte für p 0 , Tn und Qo werden zusammengefaßt zu der individuellen (speziellen)
Gaskonstanten
(3-18)
1
Die Zustandsgleichung idealer Gase erhält
demnach die Form
(3-19)
1
Da die Gaskonstante R; von der Dichte Q0 des
Gases abhängt, ergibt sich für jede Gasart
eine eigene, individuelle Konstante.
1
Reale Gase befolgen GI. (3-16) um so besser,
je geringer der Druck und je höher die Temperatur ist. Die physikalischen Gründe hierfür sind in Abschn. 3.2.1 erläutert.
Die Zustandsgrößen Druck p, Volumen V und
Temperatur T einer konstanten Stoffmenge
eines idealen Gases gehorchen stets GI. (316). Durch Auflösung nach dem Druck ergibt
sich p = konst. · TI V
Werden das Gefäßvolumen und die Temperatur vorgegeben, dann hängt der Gasdruck und
damit die Konstante von der Gasmenge ab,
die sich im Gefäß befindet.
Zur Bestimmung der Konstante wird GI.
(3-16) in die Form
p V Po Vn
--
149
1
gebracht. Die Größen mit dem Index n beziehen sich auf den in DIN 1343 festgelegten
Normzustand mit der Normtemperatur Tn =
273,15 K (.9 0 = 0 °C) und dem Normdruck
Pn = 101 325 Pa.
Das Volumen Vn des Gases hängt mit der
Dichte Qn beim Normzustand und der Masse
·m gemäß
Beispiel
3.1-3: Wie groß ist die individuelle Gaskonstante
von Luft?
Lösung:
Die Dichte beim Normzustand beträgt Qn = I ,293
kg/m 3. Damit errechnet man für die Gaskonstante
R;
2
101 325 N m 273,15 K · 1,293 kg m- 3
=
286 •9 _J_.
kg K
Der Nachteil, für jedes Gas eine besondere
Gaskonstante in GI. (3-19) einsetzen zu müssen, entfallt, wenn in GI. (3-17) das Volumen Vn
durch die Stoffmenge v ausgedrückt wird.
Nach dem Satz von A. AvOGADRO (1776 bis
1856) benötigt eine bestimmte Teilchenmenge
eines idealen Gases bei bestimmten Werten des
Drucks und der Temperatur stets das gleiche
Volumen, und zwar unabhängig von der Gasart. Für die Stoffmenge v = 1 mol beträgt beim
Normzustand nach DIN 1443 das Molvolumen
Vmn = 22,414 drn3/mol. Somit ist das Volumen
V" der Teilchenmenge v
und GI. (3-17) erhält die Form
p- V
T
Pn Vmn
T"
= - - V.
!50
3. Thermodynamik
Die Konstanten der rechten Seite faßt man
zur universellen (molaren) Gaskonstante Rm
zusammen:
= Pn Vmn = 8 3145
R
T;.
m
'
Lösung:
J
molK·
Damit erhält man die Zustandsgleichung der
idealen Gase:
(3-2i ]
Diese Form hat den Vorteil, daß für alle Gase
dieselbe Gaskonstante verwendet werden kann.
Die individuelle Gaskonstante R; kann bei
Kenntnis der Molmasse M des Gases aus der
molaren Gaskonstante Rm berechnet werden.
Nach GI. (3-4), die den allgemeinen Zusammenhang zwischen spezifischen und molaren
Größen beschreibt, gilt
Rm
M
R; = - .
(3-21)
1
Die Anzahl der Teilchen in der Teilchenmenge
v = 1 mol wird durch die Avogadrosche Konstante angegeben:
NA = 6,0221 · 10 23 mol -
1
bar eingestellt hat. Wie groß sind die Teilchenanzahl N, die Teilchenmenge v und die Masse m des
Gases?
.
Mit der Avogadro-Konstante kann die rechte
Sei te von GI. (3-20) umgeformt werden:
Der Druck des Gases beträgt p = PL + Pü = 3,016 · 10 5
Pa. Die absolute Temperatur ist T= 295,15 K. Nach
GI. (3-22) folgt für die Teilchenanzahl
pV
3,016·10 5 Nm- 2 ·2·10- 3 m 3
N=-=
kT
1,381 · 10- 23 Nm K - 1 · 295,15 K
= 1,48. 10 23 .
Die Teilchenmenge ist
pV
N
v= - - = = 0,246mol.
TRm
NA
Helium hat die Molmasse M = 4,003 g/mol. Damit
ist die Masse des Gases m = v M = 0,985 g.
Der funktionale Zusammenhang der drei Zustandsgrößen Druck, Volumen und Temperatur in der Zustandsgleichung der idealen Gase
kann in einem dreidimensionalen Raum nach
Bild 3-4 anschaulich dargestellt werden. Alle
Gleichgewichtszustände liegen auf der gekrümmten Fläche. Schnitte durch die Fläche
bei konstanter Temperatur liefern die Hyperbeln des Boyle-Mariotteschen Gesetzes im
p, V-Diagramm. Schnitte bei konstantem
Druck erzeugen die Geraden des Gay-LussacT
Rm
pV=vNAN T.
A
HICnn ist V= v NA die Teilchenanzahl des
Systems. Der Quotient
k
Rm - UH065 · 10
.\~
23
J
K
wtrd al' Bolt:.mann-Konstante (L. BOLTZMANN,
1844 bis 1906) bezeichnet. Hiermit ergibt sich
eme wc1tere Form der Zustandsgleichung
1dcaler Gase
pJ=NkT.
(3-22)
Bei piel
0
3./-4: Ein Gefaß mit V= 2 I Inhalt wird bei der
Temperatur 9 = 22 oc evakuiert und anschließend
mit Helium gefüllt, bis sich gegenüber dem äußeren
Luftdruck PL = 1016 h Pa der Überdruck Po= 2,0
Bild 3-4. Zustandsfläche der Zustandsgleichung
idealer Gase.
p Druck, Vm molares Volumen, T absolute Temperatur
3.2. Kinetische Gastheorie
sehen Gesetzes im V, T-Diagramm, und
schließlich ergeben Schnitte bei konstantem
Volumen die Geraden des Charlesschen Gesetzes im p, T-Diagramm.
ZurÜbung
Ü 3.1-1: Ein Glasstab aus Pyrex-Glas und ein Maßstab aus Messing Ms 58 sind bei .9 1 = 20 oc genau
/ 1 = 1000 mm lang. Welche Länge liest man für den
Glasstab ab, wenn beide Körper auf .9 2 = 100 °C
erwärmt werden? (etalas ~ 3,2 . w- 6 K- 1; C<Ms =
19 · w-6 K - 1)
Ü 3.1-2: Eine kreisf6rmige Stahlplatte hat bei
.9 1 = 20 °C den Durchmesser d 1 = 1200 mm. Um
welchen Betrag nimmt ihre Fläche zu, wenn sie auf
.9 2 = 96 °C erwärmt wird?
Ü 3.1-3: Wie groß ist die Zugspannung in Eisenbahnschienen bei .9 1 = - 20 °C, wenn sie bei
.9 2 = + 20 °C spannungsfrei verschweißt wurden?
Der Elastizitätsmodul des Stahls beträgt E = 2 · 10 5
N/mm 2 (Abschn. 2.11).
Ü 3.1-4: Bei .9 1 = 20 oc beträgt die Dichte von
Quecksilber Q1 = 13,546 kg/dm 3• Bei welcher Temperatur .9 2 ist die Dichte (h = 13,5 kg/dm 3 ?
Ü 3.1-5: Wie groß ist die individuelle Gaskonstante
von Wasserdampf, wenn bei der Temperatur
.9 = 800 oc und dem Druck p = 9,807 bar das spezifische Volumen v = 0,5 m 3/kg beträgt?
Ü 3.1-6: In ein Gefaß mit dem Volumen V= 201
wird bei der Temperatur .9 = 22 °C Luft gepumpt,
bis sich der Überdruck Pü = 100 bar einstellt. Welche Masse hat das Gas, wenn der äußere Luftdruck
PL = I bar beträgt?
151
Ein ideales Gas zeichnet sich dadurch aus,
daß es die Zustandsgleichung idealer Gase
(GI. (3-15) und folgende in Abschn. 3.1-5))
befolgt. Ein reales Gas verhält sich dann
ideal, wenn die Teilchendichte gering und die
Temperatur wesentlich über der Siedetemperatur der Substanz liegt. In diesem Zustand
ist das Eigenvolumen der Moleküle sehr viel
kleiner als das Gefäßvolumen; außerdem sind
die zwischenmolekularen Kräfte vernachlässigbar, da diese eine sehr kurze Reichweite
haben.
Die Modellsubstanz des idealen Gases hat
folgende Eigenschaften:
- Das Gas besteht aus einer großen Anzahl
gleichartiger Teilchen, den Molekülen.
- Die räumliche Ausdehnung der Teilchen ist
so klein, daß ihr Eigenvolumen gegenüber
dem Gefäßvolumen vernachlässigbar ist
(Konzept des Massenpunktes).
- Zwischen den Teilchen existieren keine
Wechselwirkungskräfte, ausgenommen bei
einem Zusammenstoß.
- Die Zusammenstöße der Teilchen untereinander und mit den Gefäßwänden verlaufen
völlig elastisch innerhalb einer vernachlässigbaren Zeitspanne.
Der Druck, den ein Gas auf die Gefäßwand
ausübt, wurde bereits 1738 von Bernoulli so
erklärt, daß die Teilchen bei ihren Zusammenstößen mit der Wand an diese einen beY
Ü 3.1-7: In einem Gefaß mit V= I m 3 Inhalt befindet sich bei der Temperatur T = 250 K und dem
Druck p = 2,5 bar ein ideales Gas. Wie groß ist
dessen Teilchenmenge?
3.2. Kinetische Gastheorie
3.2.1. Gasdruck
Die bisher phänomenologisch eingeführten
Zustandsgrößen erhalten eine mechanische
Interpretation durch die kinetische Gastheorie.
Hierbei legt man die atomare Struktur der
Materie zugrunde und leitet die thermodynamischen Eigenschaften der Gase aus der
Bewegung der Gasmoleküle unter Anwendung der Gesetze der Mechanik ab.
X
8
z
Bild 3-5. Zur kinetischen Gastheorie: Wü1je/ mit einem Molekül der Geschwindigkeit v,.
x, y, z Koordinaten
a Kantenlänge
152
3.Thennodynanllk
stimmten Impuls übertragen und dadurch eine
Kraft ausüben. Zur Bestimmung des_ Drucks
sei zunächst nach Bild 3-5 ein Würfel der
Kantenlänge a als Gefäß betrachtet, in dem
sich lediglich ein Molekül der Masse mM befinden soll. Das Molekül bewege sich mit der
Geschwindigkeit vi und treffe auf die rechte
Wand des Würfels. Gemäß den Stoßgesetzen
von Abschn. 2.7 wird das Teilchen wie beim
optischen Reflexionsgesetz reflektiert und
gibt dabei den Impuls t'lpi = 2 mM vxi an die
Wand ab. Nach einer bestimmten Laufzeit !lt
wiederholt sich der Vorgang, so daß in regelmäßigen Abständen nach Bild 3-6 ein Kraftstoß auf die rechte Wand ausgeübt wird. Die
mittlere Kraft Fi auf die rechte Wand beträgt
!lpj
F = '
!lt
2mM Vx i
= _m_M_v
_i_i
mittleren Geschwindigkeitsquadrat
} N
v2 = v2·
X
N i=l XI
L:
vereinfachen zu
mM
a
2a lvxi
Bei den üblichen Teilchenanzahlen verschwindet das in Bild 3-6 angedeutete diskrete Auftreten der Stöße vollkommen. Tatsächlich
treffen beispielsweise bei einem mit Luft gefüllten Gefäß im Normzustand auf jeden
Quadratzentimeter der Wand je Sekunde
etwa 3 · 1023 Teilchen.
Die Geschwindigkeiten der einzelnen Moleküle messen zu wollen, ist ein hoffnungsloses
Unterfangen. Sinnvoll sind nur statistische
Aussagen, z. B. eine Berechnung des Mittelwerts. Der obige Ausdruck läßt sich mit dem
-2
P =-Nv
V
X.
Nun gilt für jedes Teilchen
v2 = vi + v; + vi.
Da bei vielen Teilchen alle Raumrichtungen
gleichmäßig vorkommen, gilt für die Mittelwerte der Geschwindigkeitsquadrate
Demnach erhält man für den Druck
(3-23)
Bild 3-6. Zur kinetischen Gastheorie: Kraftstöße auf
die Wand.
F; Kraft, t Zeit, a Kantenlänge, Vx; Geschwindigkeit
Damit ist der "Druck", von einem Molekül
herrührend,
_
Pi=
Fi
mMvii
mMvii
A =-a-3- =--V-
.
N un sollen sich N Teilchen mit verschiedenen
Geschwindigkeiten im Würfel befinden. Falls
sie untereinander nicht zusammenstoßen, ergibt sich der Druck auf die Wand durch
Summation über alleN Einzelbei träge:
( 2
2
2
2 )
p =mM
- Vx I + Vx 2 + Vx 3 + · · · + Vx N
Diese Grundgleichung der kinetischen Gastheorie ist auch gültig, wenn Zusammenstöße
zwischen den Teilchen stattfinden, sowie bei
beliebiger Gefäßform.
GI. (3 -23) läßt sich mit Hilfe der Dichte
e = m / V = N mM I V umschreiben:
(3-24)
Diese Beziehung kann benutzt werden, um
die mittleren Molekülgeschwindigkeiten in
Gasen zu berechnen. Als mittlere Geschwindigkeit vm wird die Wurzel aus dem mittleren Geschwindigkeitsquadrat v 2 definiert:
V
N
mM '\'
=-v LJ
i= l
2
vxi·
1
(3-25)
3.2. Kinetische Gastheorie
Beispiel
j-mMv 2 =
3.2-1: Beim Normznstand beträgt die Dichte von
Stickstoff l?n = 1,2505 kg/m 3• Wie groß ist die mittlere Geschwindigkeit?
Lösung:
V
m
=v
3·101325Nm- 2
1,2505 kg m- 3
=
3.2-2: Wie groß ist die mittlere Geschwindigkeit
und die Schallgeschwindigkeit c von Luft
bei 9 = 20°C?
Tabelle 3-6. Mittlere Geschwindigkeit vm und
Schallgeschwindigkeit c einiger Gase beim
Normzustand 9- 0 = 0 ac und Pn = 1,013 bar
(Q Dichte, x Isentropenexponent).
X
in
kg/m 3
0,1785
1,784
0,0899
1,4289
1,2505
1,2928
Beispiel
vm
x ist der in Abschn. 3.3.4 definierte Isentropenexponent, der im Bereich 1 < x ~ 5/3
liegt. Tabelle 3-6 enthält Werte der mittleren
Geschwindigkeit vm und der Schallgeschwindigkeit c für einige Gase.
Helium
Argon
Wasserstoff
Sauerstoff
Stickstoff
Luft
die zeigt, daß das mittlere Geschwindigkeitsquadrat proportional zur Temperatur ist.
Daraus folgt sofort für die Temperaturabhängigkeit der mittleren Geschwindigkeit:
(3-26)
c=ff.
Q
kT,
493 m/s.
Die mittlere Geschwindigkeit der Moleküle
ist in der Größenordnung der Schallgeschwindigkeit. Nach Gl. (5-186) gilt für die Schallgeschwindigkeit
Gas
153
1,67
1,67
1,41
1,40
1,40
1,40
Vm
c
in
m
mls
m/s
1305
413
1840
461
493
485
974
308
1260
315
337
331
Lösung:
Aus GI. (3-26) folgt
Vm20
~93
--
und Vm20 = I ,036 Vmo.
273
Mit vmo = 485 m/s (Tabelle 3-6) ergibt sich vm 2o =
502 m/s. Im gleichen Verhältnis nimmt die Schallgeschwindigkeit von c0 = 331 m/s auf c20 = 343 m/s
--=
Vmo
zu.
Eine sehr plastische Deutung des Temperaturbegriffs wird möglich durch Einführung
der mittleren kinetischen Energie Ekin eines
Teilchens der Masse mM:
-
Eldn
I
2
= 2mMv
Wird die Grundgleichung (3-23) der kinetischen Gastheorie in der Form
(3-27)
Aus Gl. (3-26) und (3-27) folgt
3
Ekin =2kT.
3.2.2. Thermische Energie und Temperatur
.
(3-28)
Dieser Ausdruck erlaubt eine anschauliche
Interpretation der phänomenologisch eingeführten Zustandsgrößen "Temperatur":
p V -IN
-3 mMv2
geschrieben, so ist eine Verwandtschaft mit
der allgemeinen Zustandsgleichung (3-22)
idealer Gase
p V=NkT
offensichtlich. Durch Gleichsetzen der rechten Seiten entsteht die Beziehung
Die Temperatur ist ein Maß für die mittlere kinetische Energie der Moleküle.
Durch die Verknüpfung von Temperatur und
kinetischer Energie wird auch wieder auf die
Existenz eines absoluten Temperatur-Nullpunkts hingewiesen, bei dem jede Teilchenbe-
154
3. Thermodynamik
wegung aufhört. (Die Quantentheorie lehrt,
daß bei T = 0 K noch eine Nullpunktsenergie
vorhanden ist.)
Der Exponent läßt sich leicht umformen:
Gleichverteilungssatz
Da die Teilchenanzahldichte n = N / V proportional zum Druck ist, gilt für das Verhältnis der
Teilchenanzahldichten in der Höhe h und am
Erdboden bei h = 0:
Die Modellsubstanz - die Grundlage der vorgenannten abgeleiteten Gleichungen - besteht aus punktförmigen Teilchen mit jeweils
f = 3 Freiheitsgraden. Da sich im zeitlichen
Mittel die Bewegung der Moleküle gleichmäßig auf alle drei Raumrichtungen verteilt,
kann man die kinetische Energie eines Moleküls in drei gleiche Teile aufspalten. Auf
jeden F reiheitsgrad entfällt somit die mittlere
thermische Energie pro Molekül
(3-29)
1
_ mMgh
Ph =Po e
kT
Der Zähler im Exponenten entspricht der Differenz der potentiellen Energie Mpo1 im
Schwerefeld zwischen den beiden betrachteten Zuständen, so daß auch gilt
nh
=
!J.Epo,
e - kT
no
D ieses Ergebnis kann verallgemeinert werden
auf Gase, deren Teilchen nicht punktförmig
sind (z. B. das hantelförmige N r Molekül) und
daher mehr als drei Freiheitsgrade haben:
Die thermische Energie eines Moleküls
verteilt sich gleichmäßig auf all e seine
Freiheitsgrade. Jeder Freiheitsgrad hat
die Energie Er= k T.
±
Dieser Gleichverteilungssatz (Äquipartionsprinzi p) liefert für die mittlere kinetische Energie
eines Moleküls mit f Freiheitsgraden
-
f
Ekjn = -
2
k T.
(3-30)
D er Gleichverteilungssatz verliert seine Gültigkeit bei tiefen Temperaturen, wo Quanteneffekte wirksam werden (Abschn. 3.3.3).
3.2.3. Geschwindigkeitsverteilung
der Gasmoleküle
Boltzmann-Faktor
Die barometrische Höhenformel gemäß G l.
(2-184) beschreibt die Druckabna hme in der
Atmosphäre mit zunehmender Höhe h:
_ eoTog h
PoT
Dieses Ergebnis lä ßt sich verallgemeinem auf
zwei beliebige Energiezustände E 1 und E 2 •
Werden auf diese beiden Energieniveaus N
Teilchen verteilt, dann gilt für die Besetzungszahlen bzw. Teilchenanzahldichten
(3-31)
Diese Exponentialfunktion ist als BoltzmannFaktor bekannt und spielt in den Gleichungen
der Gleichgewichtsstatistik eine große Rolle.
Der Boltzmann-Faktor gibt an, welcher
Bruchteil der Teilchen aufgrund ihrer thermischen Bewegung die Energieschwelle
E 2 - E 1 überschritten hat.
Er tritt auf in den G leichungen der Leitfähigkeit von Halbleitern, in der Diodenkennlinie,
beim Verdampfen von Flüssigkeiten und beim
Elektronena ustritt aus Glühkathoden, um einige Beispiele zu nennen.
Haben mehrere Zustände dieselbe Energie
(entartete Zustände) , dann kann dies durch
ein statistisches Gewicht g berücksichtigt werden. Aus Gl. (3-31 ) wird dann
(3-32)
3.2. Kinetische Gastheorie
Wenn ein System verschiedene Zustände mit
den Energien E 1 , E2 , ••• einnimmt, so ist die
Wahrscheinlichkeit dafür, daß der Zustand
mit der Energie Ei besetzt ist, gegeben durch
155
dann ergibt sich die Normierungskonstante C
aus der Forderung
00
Jf(v) dv =I.
0
(3-33)
Maxwellsehe Verteilungsfunktion
Bei einem Gas ändern sich infolge der Zusammenstöße zwischen den Gasmolekülen
ständig deren Geschwindigkeiten. Trotzdem
ist eine statistische Aussage darüber möglich,
mit welcher Wahrscheinlichkeit eine bestimmte Geschwindigkeit vorkommt. Nach GI.
(3-33) ist die Wahrscheinlichkeit für das Auftreten einer Geschwindigkeit zwischen v und
v + dv gegeben durch die Verteilungsfunktion
f( v)dv= Cg (v )e 2kTdv.
Darin berücksichtigt g (v) dv das statistische
Gewicht des Geschwindigkeitsintervalls.
Im dreidimensionalen Geschwindigkeitsraum
nach Bild 3-7 liegen die Spitzen aller Geschwindigkeitsvektoren mit den Beträgen zwischen v und v + dv in einer Kugelschale mit
dem Radius v und der Dicke dv. Die Anzahl
der möglichen Geschwindigkeitsvektoren ist
proportional zum Volumen dieser Kugelschale 4n v 2 dv. Setzt man
Dies ist der mathematische Ausdruck dafür,
daß ein Teilchen mit Sicherheit irgend eine
Geschwindigkeit zwischen null und unendlich
haben muß. Durch Bestimmung des Integrals
folgt
C=(~)312
2n kT
Die Maxwellsehe Geschwindigkeitsverteilung
lautet demnach
Sie wurde von J. C. MAXWELL im Jahr 1859
gefunden und 1876 von L. BOLTZMANN theoretisch begründet.
Bild 3-8 zeigt die Verteilungsfunktion für
Stickstoff-Moleküle bei den Temperaturen
T = 300 Kund T = 900 K.
1,5 ·10- 3
g(v)=4nv 2,
' ' , T = 900K
''
0,5·10- 3
500
0
''
''
' ........ ... __
1500 ~
1000
2000
Geschwindigkeit v
Bild 3-8. Maxwellsehe
}ur S tickstoffmolekü/e.
Geschwindigk eitsverteilung
Die wahrscheinlichste Geschwindigkeit Vw , also
diejenige, die am häufigsten auftritt, kann aus
GI. (3-34) durch Bestimmung des Maximums
ermittelt werden:
Bild 3-7. Zur Maxwellsehen Geschwindigkeitsverteilung: Geschwindigkeiten zwischen v und v+ dv.
=
V
w
lV~
fTkT = lf2
V3m·
V
(3-35)
156
3. Thermodynamik
Die durchschnittliche Geschwindigkeit v, also
der arithmetische Mittelwert der Geschwindigkeitsbeträge aller Teilchen, liegt zwischen
vw und vm:
Ist die Mindestgeschwindigkeit v0 sehr viel
größer als die mittlere Geschwindigkeit Vm,
dann gilt in guter Näherung für den Bruchteil x der reaktionsfähigen Teilchen
2 1 [E;
(3-36)
An vielen Prozessen sind nur jene Teilchen
beteiligt, deren Energie eine bestimmte
Schwelle überschreitet. Beispiele sind chemische Reaktionen, Glühemission von Elektronen aus Metallen, Stoßionisation in Gasen.
Mit Hilfe von GI. (3-34) läßt sich berechnen,
welcher Bruchteil der Teilchen die erforderliche Mindestenergie bzw. Mindestgeschwindigkeit besitzt.
Beispiel
3.2-3: Eine chemische Reaktion wird eingeleitet,
wenn die Gasatome eine Aktivierungsenergie von
EA = I eV = 1,6 . w- 19 J aufbringen. Welcher Bruchteil der Moleküle ist dazu in der Lage, wenn die
Masse der Moleküle mM = 4,65 · 10- 26 kg beträgt?
Die Temperatur sei T1 = 300 K bzw. T2 = 900 K.
Wie groß ist jeweils die mittlere Geschwindigkeit Vm?
Lösung:
Die Aktivierungsenergie
e~ntspricht
geschwindigkeit von v0 =
einer Mindest= 2625 m/s. Im
M
Vergleich hierzu sind die mittleren Geschwindigkeiten klein:
L'm 1
,
=
lD
kTI
- = 517 m/s und
mM
vm 2
,
= 895 m/s.
- EA
I
I IL..__x_=_v'i:._1t_V_ kTk_T_e_ k r_.- - - -(3--3-7)--'
·
ZurÜbung
Ü 3.2-1: Ein Gefäß mit V= II Inhalt ist mit Helium
gefüllt. Das Gas befindet sich im Normzustand.
a) Wie groß ist die mittlere Geschwindigkeit vm der
Atome? b) Wie groß ist die gesamte kinetische
Energie aller He-Atome, die sich in dem Gefäß
befinden?
Ü 3.2-2: Eine Orgelpfeife einer Kirchenorgel schwingt
bei .9 1 = 20 °C mit der Frequenz / 1 =440Hz. Die
Frequenz einer Pfeife ist proportional zur Schallgeschwindigkeit in der Luft. Welche Frequenz gibt
die Pfeife im Winter ab, wenn die Temperatur der
angesaugten Luft .9 2 = 5 °C beträgt? (Zur Temperaturabhängigkeit der Schallgeschwindigkeit siehe
Beispiel 3.2-2.Die Längenänderung der Pfeife ist
ein vernachlässigbarer Effekt.)
Ü 3.2-3: Wie groß ist die Wahrscheinlichkeit da-
für, daß Stickstoff-Moleküle bei Raumtemperatur (T = 300 K) Geschwindigkeiten im Intervall
1000 m/s ~ v ~ II 00 m/s haben? Wieviel Moleküle
erfüllen diese Bedingung, wenn das Gas beim
Normdruck das Volumen V= I I ausfüllt?
Ü 3.2-4: Bei der Glühemission von Wolfram müssen
die Elektronen die Austrittsarbeit WA = 4,5 eV
überwinden. Welcher Bruchteil der Elektronen ist
dazu bei Raumtemperatur bzw. bei T= 1500 Kin
der Lage? (Elektronengas wird näherungsweise wie
ein ideales Gas angesehen.)
Der Bruchteil x der Moleküle mit v ~ v0 beträgt
00
Jf(v) dv
x = :;
=
Jf(v) dv
00
Jf(v) d v.
''o
0
Eine numerische Integration mit einem programmierbaren Rechner liefert
für T 1 = 300 K: x 1 = 1,14 · I0- 16 und
für
T2 =
900 K: x 2 = 1,06 · I0- 5 .
Obwohl die Temperatur nur um den Faktor drei
variiert, verändert sich die Anzahl der reaktionsfähigen Teilchen um viele Größenordnungen.
3.3. Hauptsätze der
Thermodynamik
3.3.1. Wärme
Aus dem letzten Abschnitt geht hervor, daß
die Temperatur Iein Maß ist für die Energie,
die in der ungeordneten thermischen Bewe- ·
gung der Teilchen steckt. 1Bei Gasen und Flüssigkeiten ist dies die kinetische Energie der
Translation und Rotation der Moleküle sowie
die Schwingungsenergie der Molekülschwingungen. In Festkörpern schwingen die Atome
um ihre Ruhetagen; hierbei werden mit zu-
3.3. Hauptsätze der Thermodynamik
nehmender Temperatur die Schwingungsamplituden immer größer.
Bringt man zwei Körper, die sich auf verschiedenen Temperaturen befinden, in Kontakt, dann findet ein Temperaturausgleich
statt: Die Temperatur des kälteren Körpers
nimmt zu und die des wärmeren nimmt ab.
Dies bedeutet nach den vorgenannten Erläuterungen, daß vom warmen System an das
kalte System Energie übertragen wird. Diese
Energieübertragung belegt man mit dem Begriff Wärme:
ärme ist Energie, die aufgrund eines
Temperaturunterschieds zwischen zwei Systemen übertragen wird. Diese Energieübertragung hat eine eindeutige Richtung.
Die Wärme fließt stets in Richtung der
niedrigeren Temperatur. Der Wärmeübergang ist also ein irreversibler Prozeß.
Die SI-Maßeinheit der Wärme ist wie für
jede Energieform I J (Joule). Somit erhalten
die Wärmekapazitäten die Maßeinheiten C:
I J/(K) , c: I J/(kg K) , Cm: I J/(mol K).
Im älteren Schrifttum und im praktischen Gebrauch findet man häufig noch die früher
übliche Maßeinheit für die Wärme, die Kilokalorie. Für die Internationale Tafelkalorie
gilt der Umrechnungsfaktor I kcalrr=4,I868kJ.
(Molare Wärmekapazitäten einiger Gase enthält Tabelle 3-8, spezifische Wärmekapazitäten von einigen Festkörpern und Flüssigkeiten
Tabelle 3-12.)
Die Wärmekapazität kann nur in bestimmten
Grenzen als Konstante angesehen werden.
Tatsächlich hängt sie von der Temperatur ab.
Bei einer endlichen Temperaturänderung von
T 1 auf T2 beträgt die übertragene Wärme
T2
Q12=m
Wird einem Festkörper oder einer Flüssigkeit
Wärme zugeführt, dann ist dies immer mit
einer Temperaturerhöhung verknüpft, falls
kein Phasenübergang stattfindet (Abschn.
3.4.3). Um die Temperatur T eines Systems
um d T zu erhöhen, ist eine Wärmezufuhr
c5Q erforderlich, die proportional zu d T ist:
c5Q = CdT.
(3-38)
c
C-c.vn
(3-39)
T2
J c(T)dT= v J Cm(J)dT.
T,
T,
(3-41)
Ist das Temperaturintervall klein, kann die
Wärmekapazität näherungsweise als konstant
angenommen werden, und GI. (3-41) vereinfacht sich zu
QI 2 = m c(T2- TI) = v Cm(T2- TI).
(3-42)
1
Die Proportionalitätskonstante C ist die Wärmekapazität des Systems. Sie hängt von der
Art des Stoffs und von der Menge ab, sie ist
äiSo eine extensive Größe.
Je nachdem, ob die Wärmekapazität C auf
die Massemoder die Teilchenmenge v bezogen wird, ergibt sich die spezifische Wärmekapazität
c=m
157
Diese Gleichung gilt auch für einen größeren
Temperaturbereich, wenn anstatt der wahren
eine mittlere Wärmekapazität eingesetzt wird.
Beispiel
3.3-1: Wie groß ist die Wärme, die einem Bauteil
aus Eisen von der Masse m = 0,8 kg zugeführt werden muß, um es von .9 1 = 20 °C auf .9 2 = 400 °C zu
erwärmen?
Lösung:
oder die molare Wärmekapazität
c
(3-40)
Cm =-.
V
Nach GI. (3-4)
Cm =C M.
gilt
der
Zusammenhang
In diesem Temperaturintervall ist die spezifische
Wärmekapazität linear von der Temperatur abhängig c 1 = 465 J/(kg K) , c2 = 615 J/(kg K). Die
mittlere spezifische Wärmekapazität beträgt
c = 540 J/(kg K). Damit ist die erforderliche Wärme
Q l2
=
=
m c (.92- .91)
0,8 kg · 540 J/(kg K) · 380 K = 164 kJ.
158
3. Thermodynamik
Zur Veranschaulichung: Mit der gleichen Energie
könnte man das Bauteil von v 1 = 0 auf v2 = 640 m/s
beschleunigen.
Die spezifische bzw. molare Wärmekapazität
von Gasen hängt außer von der Gasart auch
ab von
- der Temperatur,
- dem Druck (nicht bei idealen Gasen) und
von
- der Prozeßführung.
Die umgesetzte Wärme kann deshalb i. a.
nicht nach GI. (3-4 1) berechnet werden, da je
nach Versuchsbedingungen eine ganz bestimmte Wärmekapazi tät einzusetzen wäre.
Für die Praxis sind besonders zwei Versuchsbedingungen von Bedeutung, für die die Wärmekapazitäten vieler Gase gemessen sind:
a) Temperaturänderung bei konstantem Volumen; die isochore Wärmekapazität wird
mit dem Index "v" gekennzeichnet: Cv,
Cy,
Cmv~
b) Temperaturänderung bei konstantem Druck;
Die isobare Wärmekapazität erhält den
Index "p": CP, cP, Cmp·
Kalorimetrie
Wärmekapazitäten werden in Kalorimetern
gemessen. Bild 3-9 zeigt das Prinzip eines Mischungskalorimeters, das geeignet ist, die
Wärmekapazität von Festkörpern und Flüssigkeiten zu messen. Im Ionern des gut isolierten Dewar-Gejäßes befindet sich eine Flüs-
sigkeit (meist Wasser) der Masse m 1 bei der
Temperatur T1 • Wird ein Körper der Masse
m 2 mit der Temperatur T2 in die Flüssigkeit
eingetaucht, so stellt sich nach einiger Zeit
die Mischungstemperatur Tm ein. Es muß
folgende _nergiebilanzgleichuni);rfüllt sein:
mi
C1
(Tm- TI)+ CK(Tm- TI)=
=
mz Cz(Tz- Tm).
CK ist die Wärmekapazität des Kalorimeters.
Daraus bestimmt sich die zu messende spezifische Wärmekapazität des Körpers 2:
(3-43)
Es ist einleuchtend, daß mit dieser Methode
die spezifische Wärmekapazität nur relativ zu
der des Wassers c 1 gemessen werden kann.
Aus diesem Grund hat man früher die spezifische Wärmekapazität des Wassers mit
c 1 = I kcal/ (kg K) festgelegt und darauf alle anderen Wärmekapazitäten bezogen.
Die Bestimmung der spezifischen Wärmekapazität cv von Gasen bei konstantem Volumen
ist verhältnismäßig schwierig. Das Gas wird
in ein Kalorimetergefaß eingeschlossen und z. B. mit einer elektrischen Heizung - aufgeheizt Da die Wärmekapazität des Gefäßes
sehr viel größer ist als die des Gases, ist das
Meßergebnis nicht sonderlich genau. Einfacher ist die Bestimmung der spezifischen
Wärmekapazität cP unter konstantem Druck:
=
r u-:
:
::
m2
=
c2
-----
Bild 3-9. Mischungska/orimeter.
m Masse,
c spezifische Wärmekapazität
1 Flüssigkeit, 2 Festkörper
Bild 3-10. Kalorimeter zur Bestimmung der isobaren
spezifischen Wärmekapazität cP von Gasen.
T Temperatur
3.3. Hauptsätze der Thermodynamik
Gemäß Bild 3-1 0 leitet man eine bestimmte
Menge erhitztes Gas in einer Rohrschlange
durch ein Wasserkalorimeter. Aus der Temperaturdifferenz T 1 - T2 , dem Massenstrom
und der Temperaturzunahme der Flüssigkeit
läßt sich die Wärmekapazität cp bestimmen.
c, kann aus cP berechnet werden (Abschn.
3.3.3).
Zur Übung
Ü 3.3-1: Die Wärmekapazität CK eines Kalorimeters soll bestimmt werden. Dazu wird ein Kupferblock der Masse m2 = 150 g und der Temperatur
3 2 = 35 °C in das Wasserbad der Masse m 1 = 250 g
und der Temperatur .9 1 = 15 °C getaucht Die Mischungstemperatur beträgt .9m = 15,9 °C.
Ü 3.3-2: In ein Kalorimeter, das mit Methylalkohol
der Masse m 1 = 0,3 kg gefüllt ist, wird eine Heizwicklung getaucht und mit elektrischem Strom geheizt. Die Heizleistung beträgt P = 100 W. Die
Temperaturzunahme der Flüssigkeit ist dT/dt =
0,119 Kls. Wie groß ist die spezifische Wärmekapazität von Methylalkohol, wenn die Wärmekapazität
des Kalorimeters CK = 95 J/K beträgt?
0 3.3-3: Um die isobare spezifische Wärmekapazität von Stickstoffmonoxid (NO) zu bestimmen,
wird das Gas gemäß Bild 3-10 durch ein Kalorimeter geleitet. Dieses ist mit m 1 = I kg Wasser gefüllt.
Die Wärmekapazität des Gefäßes ist vernachlässigbar. Die Temperaturdifferenz zwischen ein- und
ausströmendem Gas ist T1 - T2 = 5 K. Der Volumenstrom beträgt V= I 1/s. Die Dichte von NO ist
Q = 1,34 kg/m 3• Die Temperaturzunahme der Flüssigkeit ist d T 3 /dt = I ,6 · 10- 3 K/s. Wie groß ist die
isobare pezifische Wärmekapazität cP und die isobare molare Wärmekapazität C m, p?
Ü 3.3-4: Die spezifische Wärmekapazität der Festkörper entspricht bei tiefen Temperaturen dem
Debyesehen T3-Gesetz c = konst. T 3. Für Zink gilt
Cm = 1,76 J/mol K (T= 20 K). Welche Wärme muß
einem Bauteil der Masse m = 200 g entzogen werden, wenn es von T 2 = 20 K auf T1 = 4,2 K abgekühlt werden soll?
3.3.2. Erster Hauptsatz der
Thermodynamik
Aus der kinetischen Gastheorie folgt sehr einleuchtend, daß Wärme eine Energieform ist.
Diese Theorie wurde erst um die Mitte des
19. Jahrhunderts entwickelt. Bis zu Beginn des
19. Jahrhunderts war die Meinung vorherrschend, daß beim Wärmeübergang von einem
159
heißen auf eine kalten Körper ein Wärmestoff, das "Phlogiston", überwechselt. Von den
zahlreichen Experimenten, die im Lauf der
Zeit die Theorie des Wärmestoffs zu Fall
brachten, seien kurz zwei erwähnt:
Im Jahr 1797 beaufsichtigte Graf Rumford (B.
THOMPSON, 1753 bis 1814) das Kanonenbohren im Münchener Zeughaus. Mit Hilfe eines
von Pferden angetriebenen Bohrers wurde
eine Kanone aufgebohrt Die dabei entwikkelte Wärme wurde an Kühlwasser abgegeben. In 2,5 Stunden wurden 8,5 kg Wasser
zum Kochen gebracht. Rumford zog aus seinen Beobachtungen den Schluß, daß die
Temperaturerhöhung durch die mechanische
Arbeit der Pferde verrichtet wurde: "Mehr
Energie läßt sich erzeugen, indem man mehr
Pferdefutter verwendet." - 1799 brachte H.
DAVY ( 1778 bis 1829) zwei Eisstücke von
.9 = 0 °C durch Reiben zum Schmelzen. Auch
hierbei wurde die erforderliche Schmelzwärme durch mechanische Arbeit zugeführt.
Im Jahr 1842 erkannte der Arzt R. MAYER
( 1814 bis 1878) als erster die Existenz eines
allgemeinen Energieerhaltungssatzes, der außer
den bisher bekannten mechanischen Energieformen die Wärme mit einschließt. Er stellte
fest, daß der Energiesatz der Mechanik uneingeschränkt gilt, wenn die Wärme als weitere
Energieform berücksichtigt wird. Aus vorliegenden Daten der spezifischen Wärmekapazitäten cP und c. von Luft berechnete er als
erster da mechanische Wärmeäquivalent, also
den Umrechnungsfaktor der (damals) in Kalorien gemessenen Wärme in mechanische Energieeinheiten. Aufgrund ungenauer Meßdaten
erhielt Mayer einen Zahlenwert, der um 14%
vom korrekten Wert abwich.
Von 1843 bis 1850 bemühte sich J. P. JouLE
( 1818 bi 1889) in vielen verschiedenartigen
Experimenten um eine genaue Bestimmung
des mechani ·chen Wärmeäquivalents. Er erhielt einen Zahlenwert für das mechanische
Wärmeäquivalent, der lediglich um 1% von
dem heute anerkannten Wert 4,1868 kJ
(= I kcal) abweicht.
Unabhängig von Mayer entwickelte 1847
H. V. HELMHOLTZ (1821 bis 1894) den allgememen Energie atz, der außer mechanischer
und Wärmeenergie auch alle anderen Energieformen, wie z. B. elektrische, magnetische
160
3. Thermodynamik
und chemi ehe Energie, einschließt. Dieser
er. te Hauptsatz der Thermodynamik lautet:
In einem abgeschlos enen -Syst;~bleibt
der Gesamtbetrag der Energie konstant.
Innerhalb des Systems können die verschiedenen Energieformen ineinander umgewandelt werden.
l
Heimholt= kam zu seiner Schlußfolgerung aufgeund der Tatsache, daß es nicht gelingt, ein
Perpetuum mobile zu bauen, also eine Maschine, die tändig Arbeit abgibt, ohne gleichzeitig entsprechende Energie aufzunehmen.
Eine solche Maschine, die dem ersten Hauptsatz widersprechen würde, wäre ein Perpetuum
mobile erster Art.
halten ein positives Vorzeichen. Wenn das
System Energie nach außen abgibt, ist diese
negativ.
Die innere Energie ist eine Zustandsgröße
(Abschn. 3.1.2), d. h., sie hängt nur vom
augenblicklichen Zustand des Systems ab,
nicht aber davon, wie das System in diesen
Zustand gelangt ist. Wäre dies nicht so, dann
ließe sich ein perpetuum mobile konstruieren.
Speziell bei den idealen Gasen gilt nach
GI. (3-30) für die innere Energie
U= N
-
Etin =
f
N2 k T
=
f
v 2 Rm T.
(3-45)
Die innere Energie der idealen Gase hängt
außer von der Stoffmenge nur von der
Temperatur ab.
Es gibt kein Perpetuum mobile erster Art.
Dieser Erfahrungssatz ist schon recht alt. Bereits 1775 beschloß die französische Akademie der Wissenschaften, Vorschläge von Erfindern für ein Perpetuum mobile nicht mehr
zu prüfen.
Innere Energie
Die gesamte thermische Energie eines Systems, die in der ungeordneten Bewegung der
Teilchen steckt, wird nach Kelvin als innere
Energie U des Systems bezeichnet. Diese kann
nach den obigen Erläuterungen nur geändert
werden, wenn über die Systemgrenzen Energie mit der Umgebung ausgetauscht wird.
Die Energieübertragung umfaßt in d en folgenden Betrachtungen led iglich Wärme und
mechanische Arbeit, kann aber jederzeit auf
alle vorhandenen Energieformen ausgedehnt
werden. Für die Änderung dU der inneren
Energie gilt somit
r
Wird bei einer Zustandsänderung das Volumen konstant gehalten, dann kann am System
keine Volumenänderungsarbeit verrichtet werden. Nach GI. (3-44) gilt für eine solche isochore Zustandsänderung
dU=bQ
v-konst=
vCmvdT=mcvdT.
Da d1e innere Energie eine Zustandsgröße ist,
kann für eine beliebige Zustandsänderung,
d ie nicht isochor zu sein braucht, die Änderung d er inneren Energie nach der vorgenannten Beziehung berechnet werden:
dU= v CmvdT=mcv dT.
(3-46)
Für beliebige Zustandsänderungen idealer
G ase hängt die Änderung der inneren
Energie nur von der isochoren Wärmekapazität und der Temperaturänderung ab.
Bei einer endlichen Temperaturänderung ist
die gesamte Änderung der inneren Energie
T•
dU=bQ+bW.
Die Änderung der inneren Energie eines
geschlossenen Systems entspricht der Summe von übertragener Wärme und Arbeit.
!lU= U2- ul =V
T•
=m
J Cmv(T)dT
T,
JCv(T)dT
oder nach GI. (3-44)
!lU = U2- U1 = Ql2 +
Das Vorzeichen der umgesetzten Energiebeträge wird wie folgt festgelegt: Wärme und
Arbeit, die dem System zugeführt werden, er-
(3-47)
T,
w12.
(3-48)
Die umgesetzte Wärme Q 12 und die mechanische Arbeit W 12 sind Prozeßgrößen (Abschn.
3.3. Hauptsätze der Thermodynamik
3.1.2). Sie hängen von der Art der Prozeßführung ab, lassen sich also nicht nach der Art
der inneren Energie als Differenzzweier fester
Werte bechreiben.
Zur Berechnung der Volumenänderungsarbeit
bei einemgeschlossenen System sei die Kompression eines Gases gemäß Bild 3-11 betrachtet. In einem Zylinder mit verschiebbarem Kolben befindet sich ein Gas unter dem
Druck p. Zur Verschiebung des Kolbens mit
der Fläche A um die Strecke ds ist die Arbeit
ö W = F ds = pA ds erforderlich. Das Produkt
A ds = d V entspricht der Änderung des Gasvolumens. Das Differential der Arbeit ist also
- mit dem Minuszeichen nach der Vorzeichenvereinbarung ÖW=-pdV.
(3-49)
J
161
2
-------.......
b
~ .......
.......
v,
Volumen V
Bild 3-12. Volumenänderungsarbeit im p,V-Diagramm.
1, 2 Grenzpunkte, W12 Volumenänderungsarbeit, a,
b Wege
Enthalpie
Außer der inneren Energie U ist eine weitere
Zustandsgröße, die Enthalpie H, sehr nützlich:
l
Bild 3-11. Zur Bestimmung der Volumenänderungsarbeit.
A Kolbenfläche, F Kraft, p Druck, ds Wegelement
Wird das Volumen von V1 nach V2 geändert,
so ist die Gesamtarbeit
v.
W12=-
Jp(V)dV.
(3-50)
Vt
Bild 3-12 erlaubt eine anschauliche Interpretation:
H=U+pV.
(3-51)
1
Das totale Differential der Enthalpie ist
dH =dU+ p d V+ V dp. Für Zustandsänderungen, die unter konstantem Druck ablaufen,
vereinfacht es sich zu dH = dU+ p d V.
Mit der Volumenänderungsarbeit in geschlossenen Systemen ö W =- p d V ergibt sich
dH = dU - W. Diese Beziehung läßt sich
mit dem ersten Hauptsatz (GI. (3-44)) so
chreiben:
o
dH = ÖQ
p - konst
=V Cmp dT= m CpdT.
(3-52)
Die Volumenänderungsarbeit entspricht
der Fläche unter der Kurve der Zustandsänderung im p, V-Diagramm.
Bei einer isobaren Zustandsänderung ist
umgesetzte Wärmemenge gleich der Änderung der Enthalpie.
Es wird noch einmal deutlich, daß die Arbeit
als Prozeßgröße vom Weg im p, V-Diagramm
abhängt. Für dieselben Endpunkte 1 und 2 erfordert der Weg a eine geringere Arbeit als
derWeg b.
Die Einführung der Enthalpie vereinfacht
thermodynamische Berechnungen bei Zustandsänderungen, die bei konstantem Druck
ablaufen.
162
3. Thermodynamik
3.3.3. Berechnung der Wärmekapazitäten
In diesem Abschnitt soll gezeigt werden, daß
die isochore spezifische bzw. molare Wärmekapazität einfach gebauter Moleküle mit Hilfe der Ergebnisse der kinetischen Gastheorie
berechnet werden kann. Die isobaren Wärmekapazitäten cP und Cmp hängen mit den isochoren Wärmekapazitäten Cv und Cmv wie
folgt zusammen:
Die Temperatur eines idealen Gases der Teilchenmenge v soll isobar um d T erhöht werden. Die erforderliche Wärme ist
Die Temperaturabhängigkeit der inneren Energie wird durch GI. (3-45) beschrieben:
f
U(T)=2 v RmT .
Die Basis dieser Beziehung ist der Gleichverteilungssatz (Abschn. 3.2.2), nach dem die
thermische Energie eines Moleküls gleichmäßig auf seine verschiedenen Freiheitsgrade f
verteilt ist. Somit gilt für die isochore molare
Wärmekapazität
(3-56)
1
bQ ip=konst =V Cmp dT.
Die innere Energie ändert sich dabei nach
GI. (3-44) und (3-49) um
dU=bQ+ DW= vCmpdT-pdV.
Da die innere Energie eine Zustandsgröße ist,
läßt sich ihre Änderung für beliebige ZuStandsänderungen nach GI. (3-46) berechnen:
dU=
V
Cmv dT.
Durch Gleichsetzen dieser beiden Ausdrücke
erhält man
Die isobare molare Wärmekapazität folgt aus
GI. (3-53)
l
I
Cmp = (;
+ 1) R m.
(3-57)
und
(3-58)
.
_ _ _ _ _ _ _ _ _ _ _ ____J
Entsprechend sind die spezifischen Wärmekapazitäten
cV =1._
2 R-I
vCmvdT= vCmpdT - pdV
(3-59)
Aus der Zustandsgleichung idealer Gase ergibt sich d VldT= vRmiP und schließlich
(3-53)
Ebenso gilt mit der individuellen Gaskonstante Ri für die spezifischen Wärmekapazitäten
(3-54)
Die isochore molare Wärmekapazität kann
nun aus der inneren Energie des Systems
berechnet werden. Nach GI. (3-46) gilt
[
c•• ~
+~
(3-55)
Das Verhältnis von isobarer und isochorer
Wärmekapazi tä~ ist der Jsentropenexponent x,
der bei isentropen Zustandsänderungen eine
wichtige Rolle spielt (Abschn. 3.3.4). Mit GI.
(3-56) bis (3-59) folgt
Cmp Cp
2
x=--=-= 1 +-.
Cmv Cv
f
(3-60)
1
Zur Berechnung der Wärmekapazitäten von
Gasen nach GI. (3-56) bis (3-59) ist die
Kenntnis der Molekülform erforderlich, um
die möglichen Freiheitsgrade f des Moleküls
angeben zu können. Für verschiedene Molekültypen sind in Tabelle 3-7 die Freiheitsgrade und die daraus berechneten molaren
Wärmekapazitäten sowie der Isentropenexponent angegeben. Jedes Teilchen hat drei
Translationsfreiheitsgrade. Dazu kommen bei
3.3. Hauptsätze der Thermodynamik
163
Tabelle 3-7. Freiheitsgrade, molare Wärmekapazitäten Cm und Isentropenexponent x für verschiedene Molekülformen.
Molekülform
Symbol
Freiheitsgrade
Cmv
Translation
Rotation
Oszillation
gesamt
3
-
-
•
punktförmig
~mp
X
m
In
J
J
-molK
-molK
3
12,47
20,79
1,67
5
20,79
29, 10
1,40
starre Hantel
• •
3
2
schwingende Hantel
~
3
2
2
7
29,10
37,41
1,29
~
3
3
-
6
24,94
33,26
1,33
mehratomig, starr
mehratomigen Molekülen noch drei Freiheitsgrade der Rotation. Bei zweiatomigen Molekülen in Form einer gestreckten starren Hantel werden nur zwei Freiheitsgrade für die
Rotation angesetzt. Diese entfallen auf die
Rotation um Achsen, die senkrecht zur Hantelachse stehen. Die Rotation um die Hantelachse tritt nicht auf, da infolge des geringen
Massenträgheitsmoments dafür extrem hohe
Temperaturen nötig wären (Begründung a uf
S. 164). Für die Schwingung einer Hantel werden zwei Freiheitsgrade angesetzt, da bei eiTabelle 3-8. Gemessene molare Wärmekapazitäten Cm einiger Gase beim Normdruck
Pn = 1,013 bar und der Temperatur 9 = 20 oc.
Gas
Cmv
~mp
in
m
J
molK
--
X
J
mol K
Helium
Argon
He
Ar
12,47
12,47
20,80
20,80
1,67
1,67
Wasserstoff
Sauerstoff
Stickstoff
Luft
Chlor
H2
02
Nz
Cl 2
20,43
21 ,06
20,76
20,77
25,74
28,76
29,43
29,09
29, 10
34,70
1,41
1,40
1,40
1,40
1,35
Kohlendioxid
Schwefeldioxid
Methan
Ethan
Ammoniak
co2
S0 2
CH4
C2H6
NH3
28,46
31 ,40
26,19
43,12
27,84
36,96
40,39
34,59
51,70
36,84
1,30
1,29
1,32
1,20
1,31
nem schwingenden System im Mittel derselbe
Energiebetrag als kinetische und als potentielle Energie vorliegt (Abschn. 5.1 ).
Die theoretisch berechneten molaren Wärmekapazitäten in Tabelle 3-7 können nun mit
den gemessenen Werten in Tabelle 3-8 verglichen werden. Bei den Edelgasen stimmen die
Messungen hervorragend mit den theoretichen Berechnungen für punktförmige Teilchen überein. D1e zweiatomigen Gase zeigen
mit Ausnahme von Chlor eine gute Übereinstimmung mit den theoretischen Werten der
starren Hantel. Dies bedeuet: Die Moleküle
von H2 • 0 2 und N 2 verhalten sich bei Raumtemperatur wie starre Hanteln. Die Zahlenwerte \On Cl 2 hegen zwischen den erwarteten
für die starre und die schwingende Hantel.
Tatsächlich chwingt bei Raumtemperatur
etwa die Hälfte der ClrMoleküle, während
die andere Hälfte starr ist. Dieses auf den ersten Blick merkwürdige Verhalten wird verständlich, wenn die Temperaturabhängigkeit
der Wärmekapazität betrachtet wird.
Bild 3-13 zeigt den Verlauf der molaren Wärmekapazität Cmv von Wasserstoff in Abhängigkeit von der Temperatur. Offenbar verhält
ich H2 bei t1efen femperaturen wie ein einatomige Gas mit drei Freiheitsgraden. Mit
teigender Temperatur beginnen die Moleküle ab etwa T = 0 K zu rotieren, dies bewirkt emen Anstieg der Wärmekapazität. Bei
Raumtemperatur rotieren prakti eh alle Moleküle. Die Wärmekapazität mmmt erneut zu,
wenn ab etwa T = 800 K die Moleküle zu
schwingen beginnen. Die Schwelle, bei der
164
3.Thennodynanllk
....:;
"'0.Ci;
30
E
G)
-LIllol t<
'E
a:
"'
~
o
"5Cl
·~
20
:I
50
100
200
500
1000
2000
K
5000
10000
Temperatur T
Bild 3-J 3. Temperaturabhängigkeit der isochoren molaren Wärmekapazität Cmv von Wasserstoff. Wasserstoff
dissoziiert bei etwa T= 3200 K. Die fortgesetzte gestrichelte Linie gilt for ein stabiles zweiatomiges M o/ekül.
die Oszillation einsetzt, liegt für CI 2 tiefer als
für H 2 , so daß bei CI 2 unterhalb der Raumtemperatur bereits ein Großteil der Moleküle
schwingt.
Vom klassischen Gleichverteilungssatz her ist
das Ausfrieren von Freiheitsgraden mit abnehmender Temperatur nicht verständlich.
Nach den Gesetzen der Quantenmechanik
aber ist der Drehimpuls eines Moleküls gequantelt. Der minimale Drehimpuls Lmin beträgt f1 = h/2 1t mit der Planckschen Konstanten h. Damit ist die minimale Rotationsenergie eines Moleküls mit dem MassenträgheitsmomentJ
Ist die mittlere thermische Energie -} k T je
Freiheitsgrad kleiner als diese minimale Rotationsenergie, so wird das Molekül bei einem
Stoß i. a. nicht in Rotation versetzt werden
können. Nach den Regeln der Quantenmechanik ist auch die Schwingungsenergie gequantelt mit der Mindestenergie h f, f ist hierbei
die Schwingungsfrequenz. Diese Energie liegt
üblicherweise höher als die Schwellenenergie
fi.ir die Rotation.
Beispiel
3.3-2: Bei welcher Temperatur beginnen die Wasserstoff-Moleküle zu rotieren?
Lösung:
Die Grenze ist näherungsweise gegeben durch
-} k T ~-} f1 2/ J. Für das Massenträgheitsmoment gilt
J = 2mr 2. Mit m = 1,67. J0- 27 kg und r ~ 5. w-ll m
ergibt sich J ~ 8,35 · 10- 48 kg m 2. Die Temperaturschwelle ist dann etwa T~ f1 2!k J = 95 K.
Die letzte Gruppe der Gase in Tabelle 3-7
besteht aus mehratomigen Molekülen, die
jeweils mehrere Schwingungsformen haben
können. Bei Raumtemperatur sind die meisten Schwingungen noch nicht angeregt, so
daß keine Systematik in die gemessenen Wärmekapazitäten gebracht werden kann.
Bei kristallinen Festkörpern sitzen die einzelnen Atome bzw. Moleküle an festen Plätzen
eines Raumgitters. Punktförmige Atome können dabei Schwingungen in den drei Raumrichtungen ausführen. Da jede Schwingungsrichtung formal mit zwei F rei heitsgraden in
die Rechnung eingeht, haben die Atome jeweils sechs Freiheitsgrade für die Berechnung
der Wärmekapazität. Nach GI. (3-56) ist dann
die molare Wärmekapazität eines F estkörpers
3.3. Hauptsätze der Thermodynamik
J
Cmv = 3 Rm = 24,9 - - molK
Dieses Ergebnis ist als Dulong-Petitsches Gesetz (P. L. DULONG, 1785 bis 1838, und A. T.
PETIT, 1791 bis 1820) bekannt. Wie Bild 3-14
zeigt, wird das Dulong-Petitsche Gesetz bei
hohen Temperaturen gut befolgt, während
mit abnehmender Temperatur durch Ausfrieren der Freiheitsgrade die Wärmekapazität
gegen null geht.
kJ
kmol K
20
165
mit verschiebbarem Kolben eingeschlossen.
Die Prozeßführung sei so kontrolliert, daß zu
jeder Zeit Druck und Temperatur des Gases
mit Umgebungsdruck und -temperatur im
Gleichgewicht sind. Ferner erfolge die Bewegung des Kolbens reibungsfrei. Unter diesen
Voraussetzungen sind die beschriebenen Prozesse jederzeit umkehrbar (reversibel).
Für alle Prozesse wird anband einer Darstellung im p, V-Diagramm die umgesetzte Energie (mechanische Arbeit bzw. Wärme) berechnet. Alle Gleichungen werden mit molaren Größen geschrieben. Für Berechnungen
mit spezifischen Größen müssen lediglich folgende Vertauschungen durchgeführt werden:
vRm -+mRi,
vCmp-+ m Cp,
vCmv-+mcv.
cJ
~ 15
·;::;;
"'c.
"' 10
(Die wichtigsten Ergebnisse der folgenden Betrachtungen sind in Bild 3-22 am Ende von
Abschn. 3.3.4 tabellarisch zusammengefaßt.)
-"
Q)
§
:;:'"'
~ 5
3.3.4.1. Isotherme Zustandsänderung
0
E
0
100
200
300
400
500
K
Temperatur T
Bild 3-14. Temperaturabhängigkeit
Wärmekapazität einiger Festkörper.
der
molaren
Bei komplizierten Molekülkristallen (beispielsweise Eis) kommen außer den Schwingungen
auch Rotationen ganzer Molekülgruppen vor,
so daß die molare Wärmekapazität oberhalb
des Wertes liegt, den die Dulong-Petitsche
Regel angibt.
Die isotherme Zustandsänderung (T = konst)
kann nach Bild 3-15 so realisiert werden, daß
ein Zylinder mit guter Wärmeleitfähigkeit an
ein Wärmebad großer Wärmekapazität angekoppelt wird. Die Zustandsänderung soll sehr
langsam (quasistatisch) erfolgen. Die allgemeine Zustandsgleichung idealer Gase (GI.
(3-20)) nimmt im Fall konstanter Temperatur
die Form des Boyle-Mariotteschen Gesetzes
(GI. (3-15)) an:
p V= v Rm T= konst.
3.3.4. Spezielle Zustandsänderungen
idealer Gase
Zustandsänderungen, die in realen Systemen
ablaufen, sind meist recht komplex, lassen
sich aber durch verhältnismäßig einfach zu
behandelnde spezielle Zustandsänderungen
annähern.
Die Zustandsänderungen sollen mit einem
idealen Gas konstanter Teilchenmenge in einem geschlossenen System durchgeführt werden. Das Gas sei in einem dichten Zylinder
Bild 3-15.
derung.
Im p, V-Diagramm von Bild 3-16 ist die Isotherme eine Hyperbel. Das Gas wird vom Anfangszustand I auf den Endzustand 2 kompri-
166
3. Thermodynamik
dU= bQ+ öW=O
\
\
\
\
\
\
'\
'''
\T'
\
'' '
T"
.........
.........
........
'
........
oder
W12 = -
Ql2
an. Dies bedeutet, daß die gesamte bei einer
Kompression zugeführte Arbeit quantitativ
als Wärme an die Umgebung abgegeben werden muß. (Dieser Wärmeübergang findet nur
dann statt, wenn die Systemtemperatur höher
ist als die Umgebungstemperatur; damit der
Temperaturanstieg vernachlässigbar klein
bleibt, muß der Prozeß unendlich langsam
geführt werden.) Umgekehrt muß bei einer
isothermen Expansion die vom System nach
außen abgegebene Arbeit zunächst als Wärme
aus dem umgebenden Wärmebad dem System zufließen. Für die umgesetzte Wärme
gilt
v,
Volumen V
(3-62) ;
B ild 3-16. Isotherme Kompression vom Zustand I
zum Zustand 2.
Temperaturen der Isothermen: T < T' < T".
W12 Volumenänderungsarbeit
miert. Hierbei muß dem System eine Volumenänderungsarbeit zugeführt werden. Nach
GI. (3-50) ist diese Arbeit
v.
W 1z=- p(V)dV.
v,
J
Mit dem Boyle-Mariotteschen Gesetz p =
vRm Tl Vergibt sich hieraus
3.3.4.2. Isochore Zustandsänderung
Bei der isochoren Zustandsänderung wird
durch ein genügend steifes Gefäß das Volumen der eingeschlossenen Gasmenge konstant
gehalten. Die Zustandsgleichung idealer Gase
entspricht im Fall V= konst dem Gesetz von
Char/es und Gay-Lussac, GI. (3 -14) :
p
T
v Rm
V
- = - - = konst.
Im p, V-Diagramm nach Bild 3- 17· kann die
Isochore als vertikale Gerade dargestellt wer-
(3-61)
In Übereinstimmung mit der Vorzeichenkonvention von Abschn. 3.3.2 wird die zugeführte
Kompressionsarbeit positiv. Bei einer Expansion wi rd die abgegebene Arbeit negativ. Gemä ß der Bedeutung des Integrals kann die
Arbeit im p, V-Diagramm anschaulich sichtbar gemacht werden:
Die Volumenänderungsarb eit entspricht
der Fläche unter der Kurve im p, V-Di agramm.
Da bei einer isothermen Zustandsänderung
die innere Energie konstant bleibt (sie hängt
nur von Tab), nimmt der erste Hauptsatz die
Form
p, +-------~k
v, = Vz
Volumen
v
Bild 3-17. Isochore Erwärmung vom Zustand 1 zum
Zustand 2.
3.3. Hauptsätze der Thermodynamik
167
den. Bei der skizzierten isochoren Erwärmung
muß man dem System Wärme zuführen. Es
gilt 15Q = v Cmv d T und hieraus
(3-63)
1
Cmv ist in diesem Fall die mittlere molare
Wärmekapazität zwischen den Temperaturen
T 1 und T2 •
P1=P2
+----+----__...;:!..:
Da bei konstantem Volumen keine Volumenänderungsarbeit vorkommt, nimmt der erste
Hauptsatz die Form dU=bQ und U2 -U 1
= Q 12 an. Dies bedeutet, daß die zugeführte
Wärme ausschließlich der Erhöhung der inneren Energie dient.
v,
3.3.4.3. Isobare Zustandsänderung
Volumen V
Bild 3-19. Isobare Expansion vom Zustand 1 zum
Zustand 2.
W, 2 Volumenänderungsarbeit
Die isobare Zustandsänderung (p = konst)
kann nach Bild 3-18 verwirklicht werden.
Durch statische Belastung des Kolbens ist der
Druck im Innenraum konstant, unabhängig
von der Höhe des Kolbens. Die Zustandsgleichung idealer Gase nimmt die Form des
Gay-Lussacschen Gesetzes nach GI. (3-12) an:
(3-65)
Diese Arbeit ist bei einer Expansion negativ,
d. h., sie wird vom System nach außen abgegeben. Bei einer Kompression ist die Arbeit
positiv, da sie dem System zugeführt werden
muß.
Nach dem ersten Hauptsatz ist
V V Rm
-=--=konst.
T
p
b Q = dU- b W
oder
Q,2= U2- U, +p(V2- V,).
Bild 3-18. Realisierung der isobaren
Zustandsänderung.
Im p, V-Diagramm von Bild 3-19 ist die Isobare eine waagrechte Gerade. Die gezeigte
Expansion verläuft so, daß dem System von
Bild 3-18 durch eine geeignete Heizung die
Wärme Q 12 zugeführt wird, worauf sich der
Kolben nach oben schiebt. Für die erforderliche Wärme gilt bQ = v Cmp d T oder
(3-64)
1
Die Volumenänderungsarbeit entspricht der
Fläche unter der Isobare. Sie beträ_gt
Dies bedeutet, daß bei einer Erwärmung sowohl die Erhöhung der inneren Energie als
auch die abgegebene mechanische Arbeit
durch die zugeführte Wärme gedeckt werden
müssen. Zur Erinnerung: Bei der isochoren
Erwärmung wurde durch die zugeführte Wärme lediglich die innere Energie vergrößert.
Dies ist der anschauliche Grund, weshalb die
isobare Wärmekapazität stets größer ist als
die isochore: Cmp > Cmv·
3.3.4.4. Isentrope Zustandsänderung
Die isentrope Zustandsänderung kann in
einem adiabaten System realisiert werden, bei
dem jeglicher Wärmeübergang zur Umgebung unterbunden wird. Im Gegensatz zur
isothermen Zustandsänderung, bei der gemäß
168
3. Thermodynamik
Bild 3-15 ein guter Wärmekontakt zur Umgebung notwendig ist, muß der Zylinder jetzt
mit einer geeigneten Wärmeisolation versehen
werden. Die adiabate Zustandsänderung läßt
sich leicht verwirklichen, wenn der Prozeß
sehr schnell abläuft, so daß für eine Wärmeübertragung keine Zeit bleibt. Der Name
Isentrope rührt daher, daß die Zustandsgröße
Entropie, die in Abschn. 3.3.6 definiert ist, bei
einer reibungsfrei und quasistatisch verlaufenden Zustandsänderung konstant bleibt. Die
reversibel durchlaufende Adiabate ist mit der
Isentrope identisch (Einzelheiten hierzu in
Abschn. 3.3.6).
Bei einem adiabaten System ( l> Q = 0) nimmt
der erste Hauptsatz die Form d V= (> W oder
(!)
dU+pdV=O
an. Mit GI. (3-46) gilt
(2)
vCm v dT+pdV=O.
Die Änderung der Enthalpie ist nach GI. (3-51)
und (3-52)
vr'
T 1 Vt'- 1 = T2
T V"- 1 = konst.
oder
(3-67)
Schließlich läßt sich noch eine Beziehung
zwischen Druck und Temperatur herstellen:
PI-" Tl'= Pi->< n
oder
p ,_" T" = konst.
(3-68)
GI. (3-66) bis (3-68) werden als Poissonsche
Gleichungen bezeichnet. Sie wurden von D.
POISSON (1781 bis 1840) im Jahr 1822 gefunden.
Im p, V-Diagramm von Bild 3-20 ist eine isentrope Kompression dargestellt. Der Kurvenverlauf I nach 2 entspricht p = konst/V"
(GI. (3-66)) und ist stei ler als bei einer isothermen Zustandsänderung. Dies bedeutet,
daß die Temperatur des Systems während der
Kompression zunimmt. Umgekehrt kühlt sich
das Gas bei einer isentropen Entspannung ab.
d H = d U+pdV+ V dp= vCmp dT.
Mit GI. (I) ergibt sich hieraus
vCmpdT= Vdp.
(3)
Durch Elimination von d Taus GI. (2) und (3)
folgt
dp
p
Diese Gleichung läßt sich direkt integrieren.
Führt man noch zur Abkürzung den bereits in
GI. (3-60) definierten Jsentropenexponenten
(Adiabatenexponenten) x = Cmp/Cmv ein, so
ergibt sich
v2
p,
xln- = ln v,
P2
v,
Aus di eser Beziehung folgt sofort die lsentropengleichung (Adiabatengleichung)
l
p , Vl' = p 2 V2_' oder
p V "= konst.
(3-66)
J
Eine Verknüpfung zwischen Temperatur und
Volumen ergibt sich, wenn mit Hilfe der Zustandsgleichung idealer Gase der Druck eliminiert wird:
Volumen V
Bild 3-20. Jsentrope Kompression vom Zustand 1
zum Zustand 2.
Die Volumenänderungsarbeit läßt sich auch
hierbei als Fläche unter der Kurve ermitteln
bzw. durch Integration von GI. (3-66) berechnen:
v.
W,2=- Jp(V)dV,
v,
mitp(V)=p 1 V1"/V"ergibtsich
3.3. Hauptsätze der Thermodynamik
w,2 = ~[(~)"-'-1].
x-1
v2
(3-69)
Diese Beziehung ist mit Hilfe der Poissonschen Gleichungen und der Zustandsgleichung idealer Gase auf vielfältige Art und
Weise umformbar. Eine wesentlich einfachere
Berechnung der Arbeit hingegen ist durch den
ersten Hauptsatz möglich. Für ein adiabates
System (bQ = 0) nimmt dieser die Form
dU= «:5 W an. Dies besagt, daß die bei einer
isentropen Kompression zugeführte Volumenänderungsarbeit ausschließlich der Erhöhung
der inneren Energie dient. Diese beträgt aber
nach GI. (3-46) bW =dU= v Cmv dT bzw.
nach Integration
169
rung p V" = konst sind Extreme, die sich in
der Praxis kaum verwirklichen lassen. Bei der
Kompression bzw. Expansion eines Gases in
einem Verdichter oder Motor wird eher eine ·
poly trope Zustandsänderung der Form
p V"= konst.
(3-71)
1
ablaufen, wobei der Poly tropenexponent n im
allgemeinen zwischen I und x liegt : I < n < x
Im p, V-Diagramm von Bild 3-21 verläuft eine
solche pol ytrope Zustandsänderung innerhalb
des gekennzeichneten Gebiets. Für einen
realen Verdichtungsprozeß, wie er beispielswei se in einem ungekühlten Turboverdichter
stattfindet, ist n < x.
(3-70)
Beispiel
3.3-3: Eine Luftfeder besteht aus einem Zylinder
mit 250 mm Durchmesser und 500 mm Länge, der
durch einen verschiebbaren Kolben abgeschlossen
ist. Die Luft im Zylinder habe zunächst ebenso wie
die Umgebungsluft die Temperatur .9 1 = 20 °C und
den Druck p 1 = I bar. Welche kinetische Energie
hat ein auffahrendes Fahrzeug, wenn beim Aufprall
der Kolben 400 mm weit eindringt? Welche Temperatur und welcher Druck wird erreicht?
Lösung:
n =x
Der Enddruck ist nach GI. (3-66)
v, )"= I bar · (I5 )
--v;
P2 = p 1 (
1,4
= 9,52 bar.
Die Temperatur beträgt nach GI. (3-67)
T2 = T, (
~~
r-l (+r
= 293 K ·
= 558 K ;
.92 = 285 oc.
Die Teilchenmenge ist v = p 1 V1 I Rm T 1 = I ,01 mol.
Mit der molaren-Wärmekapazität Cm v=20,8J/mo!K
errechnet man die Kompressionsarbeit nach GI.
(3-70) zu W12 = 5567 J. Ein Teil dieser Arbeit, nämlich WL = ( V1 - V2) p 1 = 1963 J, wird von der Umgebungsluft geleistet, und nur die Differenz stammt
vom auffahrenden Fahrzeug. Demnach ist Ekin
= 3604 J.
3.3.4.5. Polytrope Zustandsänderung
Sowohl die isotherme Zustandsänderung p V 1
= konst als auch die isentrope Zustandsände-
Volumen V
Bild 3-21. Poly tropen.
n Polyrropenexponenr,
x l sentropenexponent;
hervorgehoben: Bereich 1 < n < x der Polytrope im
engeren Sinn
Die Polytropengleichung (3-71) beschreibt
aber a uch all e bisher beschriebenen Zustandsänd erungen. Da bei nimmt der Polytropenexponent fol gende Werte an:
-
Isotherme:
Isentrope:
Isobare:
Isochore:
n = I,
n = x,
n = 0,
n = oo .
Die Poissonschen Gleichungen (3-66) bis
(3 -68) gelten auch für polytrope Zustandsänderungen, wenn der Isentropenexponent x
durch den Polytropenexponenten n ersetzt
Zustandsänderung
Bedingung
p, V-Diagramm
thermische
Zustandsgrößen
erster
Hauptsatz
Wärme
Volumenänderungsarbeit
isotherm
dT=O
p
p V = konstant
c:SO + oW= 0
oo
oW = -p dV
T= konstant
=
- ow
Boyle-Mariotte
V
isochor
dV=O
p
~ = konstant
V= konstant
ow = O
dU=c:SO
0 12 =v Cmv(T2 - T,)
Charles
=
mc. (T2
-
W12 = 0
T1 )
V
i~bar
dp=O
V
p
p= konstant
T = konstant
dU= c:SO + oW
oO = v CmpdT
oW =pdV
Gay-Lussac
U2 - U 1 =
0 12 + W12
0 12 = vCmp(T2 - T,)
= mcp(T2 - T,)
W12 = p(V1
p VK
dU=oW
c:SO = 0
oW = vcm.dT
U2-U, = W,2
0,2 = 0
W1 2 = vCm.(T2
-
V2 )
V
isentrop
dS =0
00=0
S =konstant
p \
\
\
= konstant
T vK - 1 = konstant
p 1 - K TK = konstant
'
-
P2 v2- p,
...............
T,)
v1
X-1
V
vn
rvn -l
polytrop
p
p1 V
Bild 3-22.
Spezielle Zustandsänderungen idealer Gase.
= konstant
=konstant
nrn =
konstant
dU= c:SO+ oW
c:SO =dU- oW
oW = - pdV
0 12 =vRm(T2 -T1 )
W12 = - - (T2 - T1 )
n-1
(x~1- n~1)
vRm
3.3. Hauptsätze der Thermodynamik
wird. Ebenso gilt GI. (3-69) für die Berechnung der Volumenänderungsarbeit, wenn anstelle des Isentropenexponenten der Polytropenexponent eingesetzt wird.
Eine Zusammenstellung der wichtigsten Ergebnisse von Abschn. 3.3.4 zeigt Bild 3-22.
Zur Übung
Ü 3.3-5: Beim Dieselmotor wird im Kompressionstakt Luft so rasch verdichtet, daß keine Wärmeabgabe an die Umgebung erfolgt und die hohe Temperatur zur Entzündung des eingespritzten Kraftstoffs ausreicht. Gegeben sei ein Motor mit dem
Verdichtungsverhältnis V1I V2 = 20. Zu Beginn der
Kompression ist das Volumen V1 = 0,61 mit Luft
der Temperatur .9 1 = 27 °C und dem Druck p 1 =
950 mbar gefüllt. - a) Wie hoch ist die Endtemperatur .9 2 nach der Kompression? b) Welcher
Druck p 2 stellt sich ein? c) Welche Arbeit W12 muß
während der Kompression von außen am Kolben
verrichtet werden?
Ü 3.3-6: Ein Wetterballon hätte prall gefüllt das
Volumen Vmax =50m 3. Am Erdboden ist er nur
teilweise gefüllt worden: Beim Druck p 1 = I bar
und der Temperatur .9 1 = 7 °C nimmt das eingefüllte Wasserstoffgas nur das Volumen V1 = Vmax
ein.
t
a) Welche Gasmenge v und welche Masse m enthält
der Ballon?
b) Der Aufstieg geschieht so rasch, daß durch die
Ballonhülle praktisch keine Wärme übertragen
wird. In einer bestimmten Höhe ist der Innendruck gleich dem Außendruck p 2 = 0,2 bar. Welches Gasvolumen V2 enthält dann der Ballon?
c) Wie groß ist in diesem Fall die Temperatur T2
der Gasfüllung?
d) Sonneneinstrahlung heizt danach den Ballon auf.
Das Füllgas dehnt sich solange aus, bis der
Ballon prall gefüllt ist. Dabei bleibt der Druck
konstant (p 3 = p 2 ). Auf welchen Wert T 3 steigt
dabei die Gastemperatur?
e) Welche Wärme Q23 hat das Gas aufgenommen?
Ü 3.3-7: Eine abgeschlossene Menge eines idealen
Gases wird vom Ausgangszustand p 1 = I bar, V1 = 1 I
und .9 1 = 22 oc auf die Hälfte seines Volumens verdichtet. Während der Kompression wird Wärme
zugeführt, so daß rine Zustandsänderung gemäß
der Beziehung p V2 = konst durchlaufen wird.
a) Wie groß ist der erreichte Enddruck p 2 ? b) Welche Endtemperatur .9 2 stellt sich ein? c) Welche
Arbeit W12 wurde dem System bei der Kompression zugeführt? d) Wie groß ist die zugeführte
Wärme Q 12 , wenn das Gas aus Molekülen in Form
einer starren Hantel besteht, bei denen die Freiheitsgrade der Translation und Rotation angeregt
sind?
171
Ü 3.3-8: Wasserstoff mit der Teilchenmenge v wird
in einem Zylinder mit verschiebbarem Kolben einer
Zustandsänderung unterworfen. Der Ausgangszustand ist gekennzeichnet durch p 1 = I bar, V1 = 2 I
und .9 1 = 20 °C. Die Zustandsänderung erfolgt im
p, V-Diagramm längs einer Geraden vom Anfangszum Endzustand, der bestimmt ist durch den Druck
P2 = 2 bar und das Volumen V2 = 3 I. - a) Wie groß
ist die Teilchenmengetv des Gases? b) Wie groß ist
die Endtemperatur .9 2 ? c) Welche Arbeit W12 gibt
das Gas nach außen ab? d) Um welchen Betrag !lU
steigt die innere Energie des Gases? e) Welche
Wärmemenge Q12 wird bei der Zustandsänderung
zugeführt?
3.3.5. Kreisprozesse
Durchläuft ein System eine Folge von Zustandsänderungen, so daß der Endzustand
wieder mit dem Anfangszustand übereinstimmt, so handelt es sich um einen Kreisprozeß. Ein rechtsläufiger Kreisprozeß liegt
vor, wenn die Zustandsänderungen im p,
V-Diagramm im Uhrzeigersinn durchlaufen
werden. Beim Kreisprozeß in Bild 3-23 wird
während der Expansion von I nach 2 Volumenänderungsarbeit nach außen abgegeben,
die der Fläche unter der oberen Kurve entspricht. Bei der anschließenden Kompression
von 2 nach I wird Arbeit zugeführt, die der
Fläche unter der · unteren kurve entspricht.
Insgesamt> wird also bei einem· rechtsläufigen
Kreisprozeß mehr Arbeit abgegeben als zugeführt.
2
Volumen V
Bild 3-23. Rechtsläufiger Kreisprozeß.
1, 2 Zustandspunkte,
helle Graujläche: zugeführte Volumenänderungsarbeit, gesamte Graujläche: abgegebene Volumenänderungsarbeit, umfahrene Fläche: Nutzarb,eit
172
3. Thermodynamik
Die je Umlauf nach außen abgegebene
Nutzarbeit entspricht dem Flächeninhalt
der vom Kreisprozeß eingeschlossenen Figur im p, V-Diagramm. Sie kann als Kreisintegral geschrieben werden:
W=
f ö W = - f p d V.
(3-72)
Der erste Hauptsatz nimmt bei einem kompletten Umlauf die Form
fdU=O=f öQ+f bW
(3-73)
an. Das Kreisintegral über alle Änderungen
der inneren Energie ist null, da die innere
Energie als Zustandsgröße nach einem vollen
Umlauf wieder den Anfangswert annimmt.
Dies bedeutet, daß sich die je Zyklus abgegebene Nutzarbeit aus der Differenz der zuund abgeführten Wärmen ergibt.
Bei einem linksläufigen Kreisprozeß wird die
Figur im p, V-Diagramm im Gegenuhrzeigersinn durchlaufen. Da hierbei die abgegebene
Expansionsarbeit stets kleiner ist als die zugeführte Kompressionsarbeit, läuft der Prozeß nur, wenn mit Hilfe eines Motors periTabelle 3-9. Eigenschaften von Kreisprozessen.
Umlauf- rechtsläufig
sinn
linksläufig
Bezeich- Kraftmaschinennung
prozeß
Arbeitsmaschinenprozeß
Wärmefluß
Wärme wird bei
hoher Temperatur aufgenommen und bei tiefer Temperatur
abgegeben.
Wärme wird bei
tiefer Temperatur
aufgenommen und
bei hoher Temperatur abgegeben.
mechanische
Arbeit
Differenz von
zu- und abgeführter Wärme
wird als mechanische Nutzarbeit abgegeben.
Differenz von abund zugeführter
Wärme wird als
mechanische Arbeit zugeführt.
Beispiele
Verbrennungsmotor, Wärmekraftmaschine
Kältemaschine,
Wärmepumpe
--
odisch mechanische Arbei t zugeführt wird .
Tabelle 3-9 zeigt ei ne G egenüberstellung der
Eigenschaften von rechts- und linksläufigen
Kreisprozessen.
Die Kreisp rozesse, die im folgenden beschrieben werden, soll en reibungsfrei durchlaufen
werden. Ferner soll si ch das Gas stets im thermodynamischen G leichgewicht mit der Umgebung befinden. Unter diesen Voraussetzungen sind alle Kreis p rozesse reversibel führbar,
d. h., sie können sowohl rechts- als auch linksläufig sein.
3.3.5.1. Carnotscher Kreisprozeß
Rechtsläufiger Prozeß
Von S. CARNOT ( 1796 bis 1832) wurde ein
Kreisprozeß vorgeschlagen, mit dem Wärme
in einer periodisch arbeitenden Maschine in
mechanische Arbeit umgeform t werd en kann.
Nach Bild 3-24 verläuft der P rozeß im p, VDiagramm zwischen zwei Isothermen und
zwei Isentropen. Als Arbeitsmedium dient ein
ideales Gas der Teilchenmenge v. Folgend e
Einzelprozesse werden aneinandergerei ht:
->
2: Isotherme Kompression von VI auf v2
bei der tiefen Temperatur T 1:
zugeführte Arbeit
VI
In-'
v2
abgegebene Wärme
VI
Q 12 = - vRmT1 In-.
v2
2-> 3: Isentrope Kompression von V2 auf V3 ;
die Temperatur steigt von T 1 auf T 3 :
wl2
=
V Rm Tl
zugeführte Arbeit
W23 = vCmv(T3-TI)·
3 -> 4: Isotherme Expansion von V3 auf V4 bei
der hohen Temperatur T 3 :
zugeführte Wärme
abgegebene Arbeit
3.3. Hauptsätze der Thermodynamik
173
3
1-+2
2-+3
3-+4
isotherme
Kompression
isentrope
Kompression
isotherme
Expansion
B
li=~
~t9~
----
- - - - - -Wärmebad
r,
- -----
Wärmeisolation
Wärmebad
T3
4 ...... ,
isentrope
Expansion
'7:1
"""
I:J
Wärmeisolation
PT+---+------+-----+--V,
Volumen V
Bild 3-24. Garnotscher Kraftmaschinenprozeß.
Q Wärme, W Arbeit, rot umgrenzte Fläche: Nutzarbeit
4-> 1: Isentrope Expansion von V4 auf V1 ; die
Temperatur fällt von T3 auf T 1:
abgegebene Arbeit
w4I = - V
Cmv (T3- T,).
Die Nutzarbeit je Zyklus entspricht dem Inhalt der rot begrenzten Fläche im p, V-Diagramm von Bild 3-24. Sie beträgt
w = f {> w = w,2 + W23 + W34 + w4, .
Mit W 23 = - W 41 ergibt sich
W=
w,2 + W34
=-
v Rm (
T3 1n..!::i- T1 ln....!:i) .
v3
V2
Für die beiden Isentropen gilt nach GI. (3-67)
VJ'- I = T, v2x-l und
T3 vr' = T, V,"-'·
T3
Daraus folgt für die Volumina V4 /V3 = V1/V2
und schließlich für die Nutzarbeit
v4
v3
W=- vRm In--- (T3- T,).
Sie ist negativ, weil sie vom System nach
außen abgegeben wird.
Die Energieströme, die bei der Camot-Kraftmaschine (und im Prinzip bei jeder Wärmekraftmaschine) umgesetzt werden, sind in
Bild 3-25 anschaulich dargestellt. Von der zugeführten Wärme kann nur ein Teil (meist
der kleinere) als mechanische Arbeit abgegeben werden. Den anderen Teil muß das System als Abwärme an eine Wärmesenke tiefer Temperatur abführen. Aus dem ersten
Hauptsatz folgt die Bilanzgleichung
I Wl = Qzu- IQab I
174
3. Thermodynamik
hohen Wirkungsgrades die Temperatur der
Wärmequelle so hoch wie möglich sein.
Warmequelle
Temperatur T3
Qzu
r------!------;1-~--,
Wärmemenge, die von der
Wärmequelle dem
System zugeführt wird
'G:,\ \,_ _ _
I
I
I -. ..
I
I
--1
Wärmekraftmaschine
abgegebene
mechanische
Nutzarbeit
W = - j pdV
I
I
o
I
.-----''---..L----,
Wärmesenke
Temperatur T, < T3
Wärmemenge, die vom
System an die Wärmesenke abegegeben wird
Bild 3-25. Energiejlußdtagramm eines rechtsläufigen Carnot-Pro::esses.
oder, mit den Bezetchnungen des Carnot-Prozesses und richtigen Vorzeichen,
[- Ql2 + Q34 +
w= 0 .
(3-74)
Verschiedene Kreisprozesse lassen sich miteinander vergleichen durch Berechnung des
thermischen Wirkung5grades 'lth, der den
Nutzen (abgegebene Arbeit) zum Aufwand
(;ugeführte Wärme) ins Verhältnis setzt:
(3-75)
1/ ll
Bctm Carnot-Prozeß ist dte zugeführte Wärme
Qzu
= Q34
v4
3.3-4: Welcher thermische Wirkungsgrad ist mit
einem Camot-Prozeß erreichbar, der zwischen den
Temperaturen 93 = 500 °C und 9 1 =50 °C abläuft?
Lösung:
Nach GI. (3-76) ist
450K
1 C = - - = 0 58 = 58%.
"h
•
773 K
'
Der Carnot-Prozeß läßt sich praktisch nicht
realisieren, da zu viele widersprüchliche
Eigenschaften in einem System vereinigt sein
müßten. Seine große Bedeutung liegt in der
Abschätzung des maximalen Nutzeffekts einer
Wärmekraftmaschine, die zwischen zwei Temperaturgrenzen Wärme in Arbeit umwandeln
soll. Ein Vergleich verschiedener rechtsläufiger Kreisprozesse, die zwischen der Maximaltemperatur T 3 und der Minimaltemperatur T 1
ablaufen, zeigt, daß der höchstmögliche thermische Wirkungsgrad durch den Carnot-Prozeß erreicht wird.
Thermodynamische Temperatur
Da der thermische Wirkungsgrad des CarnotProzesses n ur von den Temperaturen der beteiligten Wärmebäder, aber nicht vom Arbeitsstoff ab hängt, ist eine TemperaturdefinitiOn möglich, die von speziellen Thermometereigenscha ften unabhängig ist. Nach GI.
(3-74) bis (3-76) gilt
= V Rrn T3 In-.
VJ
Damit ist d er thermische Wirkungsgrad
'lth,C
Beispiel
~-T--::
T, = I T3
Tl . T3
~
(J-'/ö) I
Der thermische Wirkungsgrad des CarnotProzesses ist nur von den Temperaturen
der beiden Wärmebäder abhängig.
Der thermische Wirkungsgrad des CarnotProzesses könnte dann 100% werden, wenn die
Temperatur der Wärmesenke T 1 = 0 K wäre.
Da in der Praxis die Wärmesenke z. B. das
Kühlwasser eines Flusses oder die Umgebungsluft ist, muß für die Erzielung eines
'lth
= Qzu- Qabl =I_ IQab l = I _ Tl.
Qzu
Qzu
T3
Hieraus folgt die Beziehung zwischen den
umgesetzten Wärmemengen und den Temperaturen der Wärmebäder:
I
I
' Qab
Qzu
= T,
T3
oder Q,2+ Q34=0.
Tl
T3
(3-77)
GI. (3-77) erlaubt nach W. Thomson (Lord
Kelvin) die Definition der thermodynamischen
Temperatur. Die Temperaturen zweier Wärmebäder lassen sich dadurch vergleichen, daß
man zwischen ihnen einen idealen CarnotProzeß ablaufen läßt und die übertragenen
Wärmen mißt. Wird die Temperatur eines
3.3. Hauptsätze der Thermodynamik
Wärmebads festgelegt, z. B. die Temperatur
von Wasser am Tripelpunkt mit TT,=273,16K,
dann kann die ganze Temperaturskala ausgemessen werden. Die so definierte thermodynamische Temperatur ist identisch mit der
Gastemperatur des Gasthermometers (Abschn.
3.1.3).
Linksläufiger Prozeß
175
temperatur T 3 , die Wärme Qzu entzogen und
dem System zugeführt. Als Wärmesenke dient
i.a. die Umgebung. Das Verhältnis von Nutzen
zu Aufwand wird bei linksläufigen Kreisprozessen als Leistungszahl bezeichnet. Bei einer
Kältemaschine ist der Nutzen die Wärme
Qzu, der Aufwand ist die Arbeit W des Antriebsmotors. Die Leistungszahl einer Kältemaschine wird deshalb definiert als
Beim linksläufigen Camot-Prozeß wird das
p, V-Diagramm von Bild 3-24 im Gegenuhr-
zeigersinn durchlaufen. Dabei wird bei der
tiefen Temperatur T 1 Wärme aus der Umgebung aufgenommen und bei der hohen Temperatur T 3 wieder abgegeben. Das Energieflußdiagramm des linksläufigen Prozesses ist
in Bild 3-26 dargestellt. Die Energiebilanz
sagt aus, daß die abgegebene Wärme betragsmäßig gleich ist der Summe aus zugeführter
Wärme und mechanischer Arbeit:
(3-78)
1
Wärmemenge, die vom
Oab
System an die Wärme.---L-__.:..._-L.._ _, senke abgegeben wird
I
:G:, \. __
1
Für den Carnot-Prozeß ergibt sich mit den
bereits berechneten Energiebeträgen
(3-80)
I
Die Leistungszahl ist um so günstiger, je
näher die Temperaturen von Wärmequelle
und Wärmesenke beeinander liegen.
Beispiel
Wärmesenke
Temperatur T3
t
(3-79)
3.3-5: Eine Kältemaschine nach Carnot soll eine
Kühlraumtemperatur von .9 1 = 5 °C bei einer Außentemperatur von .9 3 = 35 °C erreichen. Wie groß ist
die Leistungszahl eKc?
I
II
II ' , .... __
I
Arbeitsmaschine
I
I
I
zugeführte
mechanische
Arbeit
W=-fpdV
I
.---L---L---...,
Wärmemenge, die von der
Wärmequelle dem
System zugeführt wird
Wärmequelle
Temperatur r, < T3
Bild 3-26. Energieflußdiagramm eines linksläufigen
Carnot-Prozesses.
Der linksläufige K.reisprozeß kann auf zweierlei Arten genutzt werden:
a) Kältemaschine
Eine Kältemaschine hat die Aufgabe, einen
Raum zu kühlen, in dem z. B. Lebensmittel
gelagert werden. Der zu kühlende Raum dient
als Wärmequelle. Ihm wird bei der Temperatur T 1 , die niedriger ist als die Umgebungs-
Lösung:
Nach GI. (3-80) ist eK,c = 278 K/30 K = 9,27. Dies
bedeutet, daß die Leistung des Antriebsmotors nur
rund ein neuntel der Wärmeleistung sein muß, die
dem Kühlraum entzogen werden soll.
b) Wärmepumpe
Bei der Wärmepumpe ist die Wärmequelle
die Umgebung (z. B. Luft, Erdreich, Grundwasser), der die Wärme bei tiefer Temperatur
entzogen und dem System zugeführt wird.
Wärmesenke ist z. B. die Warmwasserheizung
eines Hauses. Der Nutzen bei der Wärmepumpe liegt also in der bei hoher Temperatur
abgegebenen Wärme Qab; der Aufwand ist
auch in diesem Fall die Arbeit des Antriebsmotors. Die Leistungszahl der Wärmepumpe
wird deshalb definiert als
(3-81)
176
3. Thermodynamik
Für den Carnot-Prozeß ergi bt sich
(3-82)
'7th,C
Die Leistungszahl der Wärm epumpe nach
Carnot ist immer größ er als eins, und zwar
um so größer, je kleiner der thermi sche
Wirkungsgrad eines rechtslä ufigen CarnotProzesses zwischen denselben Tempera turgrenzen ist, d. h. , je kleiner d ie Temperaturdifferenz T 3 - T 1 ist.
Beispiel
3. 3-6: Eine Wärmepumpe nimmt Wärme aus der
Umgebungsluft bei .9 1 = - 10 °C auf und gi bt Wärme an eine Warmwasserheizung mit der Vorlauftemperatur 9 3 = 40 oc ab. Wie groß ist die Leistungszahl nach Carnot ?
Lös ung:
Nach GI. (3-82) gilt ew.c = 313 K /50 K = 6,26.
In der Praxis werden Kältemasch inen und
Wärmepumpen meist mit Kältemitteln, wie
z. B. Frigen und Ammoniak, betrieben, die
während des Kreisprozesses Phasenänderungen (Abschn. 3.4.3) durchlaufen. Das Prinzip
des Kreislaufs zeigt Bild 3-27. In einem Verdampfer wtrd dem Oüssigen Kältemittel, das
gennl!cn Druck und niednge Temperatur hat,
die Wärme Q,u zugeführt, so daß es verdampft. Der Dampf wird in einem Kompressor verdichtet und somit erwärmt. Im Kondensator wird dem heißen Dampf die Wärmemenge Q.b entzogen, so daß das Kältemittel kondensiert. Die unter hohem Druck stehende Flüssigkeit wird durch ein DrosselKompressor
DrosselventiI
Biid j -27. Kreislauf einer Kompressor-Kältemaschine bzw. -Wärmepumpe.
venti l entspannt. Dabei kühlt sie sich ab und
wird dem Verdampfer für den nächsten Kreisla uf zugeleitet.
Di e Leistungszahlen realer Wärmepumpen
si nd niedriger a ls die Leistungszahl eines
Carnot-Prozesses. Für elektrisch betriebene
Luft/Wasser-Wä rmepumpen ist beispielsweise
cw ~ 3. Bei großen Anlagen, die mit einem
Di eselmotor angetrieben werden, sind die erreichbaren Leistungszahlen größer.
3.3.5.2. Technische Kreisprozesse
D ie Kreisprozesse, die in realen Maschinen
abl aufen, können durch idealisierte Vergleichspro::esse angenä hert werden. Bild 3-28 zeigt
ei ne Z usa mmenstellung von Vergleichsprozessen, d ie in techni schen Wärmekraftmaschinen
idealisiert a bl a ufen . Die Pfeile im p, V-Diagramm zeigen die Prozesse, bei denen Wärme
zu- bzw. abgeführt wird .
Obwohl Verbrennungsmo toren offene Systeme
sind, können sie näherungsweise als geschlossene Systeme angesehen werden. Beim Seiliger-Pro::eß (nach einem Vorschlag von M.
SEILIGER, 1922) wird Frischluft isentrop verdichtet. Nach Zündung des Luft-KraftstoffGemisches läuft eine Verbrennung ab, die
näherungsweise d urch eine isochore und isobare Wärmezufuhr beschrieben wird. Die Expansion des verbrannten Gemisches erfolgt
isentrop. Der nachfolgende Austa usch von
verbrannten Gasen d urch Frischluft wird als
isochore Wärmeabgabe angenähert. D er thermische Wirkungsgra d ist abhängig von den
Temperaturen der fünf Eckpunkte.
Ein Spezialfall des Seiliger-Prozesses mit
V2 = V3 = V4 ist der Otto-Prozeß (N. ÜTTO,
1832 bis 1892). Hierbei verbrennt das LuftKraftstoff-Gemisch nach der Zündung so
schnell, daß die Wärmezufuhr idealisierend
wie eine isochore Z ustandsänderung erfolgt.
Der thermische W irkungsgrad hängt ab vom
Kompressionsverhältnis G = VI I v2.
Ein weiterer Spezia lfall des Seiliger-Prozesses
mit P2 = PJ = P4 ist der Diesel-Prozeß (R.
D IESEL, 1858 bis 191 3). Der Kraftstoff wird so
in die komprimierte Luft eingespritzt, daß
di e Verbrennung näherungsweise isobar erfolgt. Bild 3-29 zeigt ein Original-p, V-Diagramm ein es Dieselmotors. Der thermische
3.3. Hauptsätze der Thermodynamik
Bezeichnung
SeiligerProzeß
Einzelprozesse
p, V-Diagramm
'I~
~
a.?
4
2
lwl
177
thermischer
Wirkungsgrad
2 lsentropen ,
2 Isochoren,
T]th = 1 -
1 Isobare
T5- Tl
T3- T2 +x(T4- T3)
:a..
c
V
Q)
0
0
E
cn
p
OttoProzeß
Cl
c
c
c
~
::l
c
Q)
.!:
~
E
c
2 Isochoren
T]th = 1- (
V
DieselProzeß
Q)
'\.
3
2
Wl
2 lsentropen,
1'lth=
1 Isochore,
1 Isobare
=1-
4
~
1 a.b
V
....
0
E
~~~
a"
::l
2
Wl
JouleProzeß
V
ID::l
<ll
Q)
cn
Cl
c
::l
E
'E
2 Isobaren
''Ii :
•
(!)
.!:
E
2 lsentropen,
2
Wl
:t:: ....
ocn
Q)
T1
Tlth = 1 --=11th c
T3 ' '
1
p
Ca>
a>c
<n·cn.C
21sochoren
EricssonProzeß
o'-
= 1- (
4
21sobaren
T1
TJth = 1 - T3 =1)th,C
O.u
~a.b
a.bl
Cl
4
V
Ci5
p
I
'1ilc
'-Ql
.:S:OJ
.....
<ll
c._
Ec
(lj<ll
0
ClausiusRankine-Prozeß
~
IWI
1
2 lsentropen,
2 Isobaren
4
Koexistenzgebiet
Technische Kreisprozesse.
h3- h4
11th= h3- h ,
h4
""1-h3
Ia••
V
Bild 3-28.
~:) - "
2 Isothermen,
r,
Q)(!)
x- 1
V
r,
IWI
-::l
.s= ....
ucn
cn<ll
T1
T]th = 1 - T2
1
GJ"-1
4a,.
r,
ra,.
.s=
u
cn
<ll
"C ~:-1)
r,
a:i
c
~: )" -
2 Isothermen,
3
·a;
::r:
Q)
(
p
StirlingProzeß
~
c
a> ·c-2
)"- 1
1 ••
.c
0
0
~:
1
4
.c
....
.s=
u
cn
<ll
Q
2 lsentropen,
178
3. Thermodynamik
Druck -
Kolbenweg -Diagramm
Indiz. Arbeitsmitteldruck
effektiver Arbeitsmitteldruck
mech. Wirkungsgrad
L
•
..0
pml- 5.09 bar
pme• 3 bar
ETA- 59.5 Y.
60
'u
.>/.
:J
L
c
10
HATZ E79
Bild 3-29.
20
30
n- 2773 l/mln M- 10.1 Nm
V-Nr. 5
15.11.85 k
40
50
P- 2.9 kW
70
60
Kolbenweg /mm
80
rachhochachule fOr Technik
Essllngen, Maschinenlabor 1
p, V-Diagramm eines Dieselmotors (Rechnerausdruck).
Wirkungsgrad des Diesel-Prozesses übertrifft
den des Otto-Prozesses, allerdings ist der mittlere Kolbendruck im Dieselmotor wesentlich
höher als im Ottomotor.
Das Arbeitsmedium beim Stir/ing-Prozeß
( R. STJRLING, 1790 bis 1878) ist ein Gas
(meistens Luft). Die Wärmezufuhr erfolgt bei
der isochoren Erwärmung und der isothermen
Expansion. Die während der isochoren Abkühlung abgegebene Wärme ist betragsmäßig
so groß wie die bei der isochoren Erwärmung
zugeführte: Q23 = - Q41 • Gelingt es, die abgegebene Wärme Q41 zwischenzuspeichern und
bei der isochoren Erwärmung wieder dem System zuzuführen, dann muß von außen her
nur noch die Wärme Q34 zugeführt werden,
und der thermische Wirkungsgrad erreicht
den Wert des Carnot-Prozesses.
Der Stirling-Prozeß kann nach Bild 3-30 näherungsweise so realisiert werden, daß ein Arbeitskolben und ein Verdrängerkolben, um
90 ° phasenverschoben, auf eine Kurbelwelle
arbeiten. Der Verdrängerkolben schiebt ·die
Luft im Zylinder hin und her und bringt sie
abwechselnd in Kontakt mit dem heißen bzw.
kalten Teil der Maschine. Der Regenerator
besteht aus Metallspänen, die beim Durch-
strömen der heißen Luft Wärme aufnehmen
und diese nachher wieder an die durchströmende kalte Luft abgeben.
Bild 3-31 zeigt ein Demonstrationsmodell eines
Heißluftmotors. Im Deckel ist eine Glühwendel eingebaut, die als elektrische Wärmequelle dient. Die Wärmesenke ist Kühlwasser,
das den unteren Teil des doppelwandigen
Zylinders durchfließt. Der Heißluftmotor
kann bezüglich des thermischen Wirkungsgrades bislang nicht mit den Verbrennungsmotoren konkurrieren, weil die interne Wärmeübertragung (Q 41 --+ Q23 ) nur unvollkommen
gelingt. Der linksläufige Stirling-Prozeß wurde z. B. bei der Philips-Gaskältemaschine verwirklicht, die mit dem Arbeitsmedium Wasserstoff oder Helium bei der Luftverflüssigung eingesetzt wird.
In der offenen Gasturbine, die hauptsächlich
bei Flugzeugen verwendet wird, läuft ein Prozeß ab, den man näherungsweise durch den
Joule-Prozeß beschreiben kann. Luft wird im
Verdichter isentrop komprimiert. In der Brennkammer wird eingespritzter Treibstoff (Kerosin) mit der heißen Luft verbrannt (isobare
Erwärmung) und anschließend in der Turbine
isentrop entspannt. Die verbrannten Gase
3.3. Hauptsätze der Thermodynamik
1-+2
2-+3
3-+4
4 -+1
Wärmebad der
tiefen Temperatur
isotherme
Kompression
Bild 3-30.
isochore
Erwärmung
179
isotherme
Expansion
r,
isochore
Abkühlung
Realisierung eines Stirlingschen Kreisprozesses.
werden beim realen Prozeß ausgestoßen. Der
idealisierte Kreisprozeß wird durch eine isobare Abkühlung geschlossen.
Ortsfeste Gasturbinen arbeiten nach dem geschlossenen Ericsson-Prozeß (J. ErucssoN, 1803
bis 1899), der von J. ACKERET und C. KELLER
näherungsweise verwirklicht wurde. Unter der
Voraussetzung, daß die bei den isobaren Zustandsänderungen umgesetzten Wärmemengen
intern übertragen werden können, erreicht der
Ericsson-Prozeß den thermischen Wirkungsgrad des Carnot-Prozesses.
In Dampfkraftanlagen läuft i. a. der ClausiusRankine-Prozeß (R. J. E. CLAUSIUS, 1822 bis
1885 ; W. J. M. RANKI E, 1802 bis 1872) ab.
Die Speisewasserpumpe erhöht von I nach 2
isentrop den Druck des Wassers. Durch isobare Wärmezufuhr wird das Wasser verdampft und der Heißdampf von 3 nach 4 in
der Turbine isentrop entspannt. Im Kondensator verflüssigt sich der entspannte Dampf
durch Wärmeabfuhr an das Kühlwasser, und
Kondensat wird wieder der Speisewasserpumpe zugeleitet. Der thermische Wirkungsgrad
ist im wesentlichen von der Enthalpie des
Dampfes vor und nach der Entspannung abhängig.
Zur Übung
Bild 3-31.
Demonstrationsmodell
eines Heißluftmotors.
Ü 3.3-9: Mit einem idealen Gas wird der rechtsläufige Kreisprozeß gemäß Bild 3-32 durchgeführt,
der sich aus Isobaren und Isochoren zusammensetzt.
Die Zustandsgrößen der Eckpunkte im p, V-Dia-
I 0
3. Thermodynamik
1400 min - 1 Zyklen durchlaufen werden? c) Wie
groß ist die erforderliche Leistung P des Antriebsmotors ? d) Welche Wärmeleistung IQ.b I wird an
die Umgebung abgegeben ?
33.6. Zweiter Hauptsatz
der Thermodynamik
Q.
.X
u
2
0
33.6;). Reversible und irreversible Prozesse
~ =
v2
Volumen V
Bild 3-32. Zu Aufgabe Ü 3.3-9: Kreisprozeß aus
2 isobaren und 21 ochoren.
gramm sind p 1 = 7,5 bar, P2 = 10 bar, V2 = I I, V3 =
1,51. Das Gas besteht aus zweiatomigen Molekülen,
die im betrachteten Temperaturbereich rotieren,
ohne zu schwingen. Die Teilchenmenge beträgt
v = 0,3 mol. - a) Wie groß sind die Temperaturen
T 1 , T2 und T3 ? b) Welche Nutzarbeit W wird je
Umlauf abgegeben? c) Welche Wärme Q," muß je
Zyklus zugeführt werden? d) Wie groß ist der thermische Wirkungsgrad '7th des Kreisprozesses? e)
Welchen Wirkungsgrad hätte eme Camot-Maschine. die zwischen denselben Maximal- und Minimaltemperaturen T1 und T 1 arbeitet?
L' 3.3-10: Eine
= 3 soll ein
Wärmepumpe mit der Leistungszahl
Haus heizen. Die erforderliche Heizleistung ist Q.b = 15 kW be1 fh = 45 °C. Die
Außentemperatur beträgt 9 1 = - 5 °C. - a) Welche
elektrische Leistung P nimmt der Motor auf? b)
Wie groß wäre d1e Leistung Pc des Antriebsmotors,
wenn ein Camot-Prozeß ablaufen würde?
cw
Ü 3.3-11: In einer mit Wasserstoff betriebenen Gaskältemaschine läuft ein linksläufiger Stirling-Prozeß
mit folgenden Einzelprozessen ab:
I
-+
2
-+
3
4
-+
-+
2: Isochore Erwärmung vom Anfangszustand
p 1 = 9 bar, V1 = 0,28 I und T 1 = 77 K auf
T2= 300 K;
3: Isotherme Kompression von V1 = V2 a uf
V3 = V4 = 0,14 I;
4: Isochore Abkühlung von T2 auf T 1;
1: Isotherme Expansion von V4 auf V1•
a) Wie groß ist die Leistungszahl cK des Prozesses
unter der Voraussetzung, daß die interne Wärmeübertragung - Q 34 = Q 12 ideal gelingt? b) Welche
Kältelei~tung
liefert die Maschine, wenn n =
Ozu
Wird vom elastischen Stoß zweier Billardkugeln eine Filmaufnahme gemacht und der
Film anschließend vorwärts- und rückwärtslaufend betrachtet, so kann ein Zuschauer,
der bei der Aufnahme nicht dabei war, nicht
sagen, welche Laufrichtung des Film das Experiment richtig wiedergibt. In beiden Richtungen könnte der Vorgang abgelaufen sein;
keine der beiden Varianten verletzt die Stoßgesetze. Solche umkehrbaren oder reversiblen
Vorgänge werden in der Mechanik beobachtet, wenn keine Wärmeentwicklung infolge
von Rei bung auftritt.
Ein Prozeß ist reversi bel, wenn bei seiner
Umkehr der Ausgangszustand wieder erreicht wird, ohne daß Änderungen in der
Umgebung zurückbleiben.
Reversible Zustandsänderungen von Gasen
si nd als idealisierte Grenzfälle denkbar, wenn
die Prozesse reibungsfrei und quasistatisch
verlaufen, so daß der Druck und die Temperatur des Gases zu jeder Zeit mit der Umgebung im Gleichgewicht sind.
Wird der Fall eines Apfels von einem Baum
gefilmt und der Film später rückwärtslaufend
betrachtet, so löst die Szene allgemeine Heiterkeit aus. Jedermann weiß aus Erfahrung,
daß dieser Vorgang irreversibel ist, also nicht
von all ein in umgekehrter Richtung abläuft.
Ein Vorgang ist irreversibel, wenn seine
Umkehr zum Ausgangszustand nur unter
äußerer Einwirkung möglich ist, wobei
eine Veränderung in der Umgebung zurückbleibt.
Beim unelasti schen Aufprall des Apfels auf
dem Boden wird seine kinetische Energie in
thermische Energie umgesetzt; die Temperatur des Apfels und der unmittelbaren Umge-
3.3. Hauptsätze der Thermodynamik
bung erhöht sich demnach geringfügig. Der
umgekehrte Vorgang, daß der Apfel sich abkühlt und dann nach oben hüpft, ist noch nie
beobachtet worden, obwohl er den ersten
Hauptsatz nicht verletzen würde.
Weitere Beispiele für irreversible Vorgänge
sind
- Diffusion: Stoffe breiten sich aufgrund eines
Konzentrationsgefälles so lange aus, bis die
Konzentration räumlich konstant ist. Konzentrationsunterschiede dagegen bauen sich
nicht von selbst auf;
- Wärmeübergang: Wärme geht von einem
warmen auf einen kalten Körper über, bis
die Temperatur ausgeglichen ist. Temperaturunterschiede jedoch entstehen nicht von
selbst;
- Chemische Reaktionen, die von selbst ablaufen: Wasserstoff verbindet sich mit
Sauerstoff zu Wasser. Für die Zersetzung
des Wassers in seine Bestandteile hingegen
muß Energie aufgewendet werden.
heuerer Betrag an innerer Energie steckt,
könnten durch geringfügiges Abkühlen des
Meerwassers nahezu unbegrenzte Energiereserven freigesetzt werden. Eine solche Maschine, die zwar den zweiten, nicht aber den
ersten Hauptsatz verletzt, wird als Perpetuum
mobile zweiter Art bezeichnet. Eine weitere
Formulierung des zweiten Hauptsatzes lautet
also:
I
Es gibt keine periodisch arbeitende Maschine, die Wärme aus einer Wärmequelle
entnimmt und vollständig in mechanische
Arbeit umwandelt.
Es gibt kein Perpetuum mobile zweiter Art.
Die linksläufigen Kreisprozesse zeigen, daß
Wärme unter Arbeitsaufwand einem kalten
Körper entzogen und einem warmen ' Körper
zugeführt werden kann (Bild 3-26). Clausius
formulierte 1850 den zweiten Hauptsatz so:
Wärme geht nicht von selbst von einem
kalten auf einen warmen Körper über.
Bei genauer Betrachtung sind alle natürlich
ablaufenden und technischen Prozesse irreversibel. Reversible Vorgänge sind lediglich
idealisierte Grenzfälle.
3.3.6.2. Formulierungen des zweiten
Hauptsatzes
Die Irreversibilität natürlicher und technischer Prozesse ist der Inhalt des zweiten
Hauptsatzes der Thermodynamik. Dieser legt
die Richtung der von selbst ablaufenden Vorgänge fest, die stets einem Gleichgewichtszustand zustreben. Eine klassische Formulierung des zweiten Hauptsatzes stammt von
Thomson (Lord Kelvin) aus dem Jahr 1851:
J
o.b
O.u
~·
I
I
I
I
_...,
/I
I
I
I
I
w
I
- -~
I' '~
I'
I
I
I
'l th,C =
~ o.b
0,5
Ew.c =
f
2
O.u
b)
t
O.u
'
IT0''~
I I
II
I I
I
'
w
-
-·
I
o.b
I
I
J
I
I
I
I
'$'
"
_...,.
./1
I
'l lh,s =
Die Erfahrung zeigt, daß eine Wärmekraftmaschine stets auch Wärme an eine Wärmesenke tiefer Temperaturen abgeben muß
(Bild 3-25). Ließe sich eine Maschine konstruieren, die ohne Wärmeabgabe bei tiefer
Temperatur auskäme, so wären die Energieprobleme der Menschheit für alle Zeiten gelöst. Da z. B. in den Weltmeeren ein unge-
181
0,75
Ew.c =
2
I
t
O.u
Bild 3-33. Es existiert keine Maschine, die einen höheren Nutzeffekt als die Carnot-Maschine lwt:
a) Kopplung von rechts- und linkslaufender CarnotMaschine, b) Kopplung einer rechtsläufigen " Super"Maschine mit einer linksläufigen Carnot-Maschine.
. Thermodynam •
band \On Bild -33 erkennt man, daß der
thermi he Wirkung grad de· amot-Proze e ni ht üb rtroffen werden kann. Zwi eben
den Temperaturgrenzen T3 = 600 K und T 1 =
K wirkt je ein rechts- und ein link läufiger Krei prozeß. Bild 3-33 a zeigt eine Carnot- \ 'ärmekraftma hine. die nach GI. (3-76)
den thermi. chen Wirkungsgrad 'llh.C = 0.5 hat
Ihre mechani he 'utzarbeit wird eingesetzt,
um eine Wärmepumpe zu betreiben, die nach
GI. ( - 2) die Lei tungszahl tw.c = 2 aufwei t.
Au den Daten geht klar hervor. daß im Endeffekt jedem Wärmebad die Wärmemenge,
die ihm eine M~ chine entnimmt, von der
anderen wieder zugeführt wird.
Bild 3-33 b zeigt eine hypotheti ehe .,Super"Wärmekraftma chine mit einem thermt chen
\Virkung grad, der den Camot:;chen übertrifft
(z. B. fJih, = 0,75).
'immt die e Ma chine
bei piel wei e die Wärmelei tung 4 kW vom
oberen Wärmebad auf. dann gibt ie Q = I kW
an da kalte Reservoir und P = 3 kW an die
Warmepumpe ab. Die amot- Wärmepumpe
mm mt a u dem unteren Wärmebad Q = 3 kW
an Wärmelei tung auf und gibt an da obere
Q = 6 kW ab. Die bedeutet hluß endl ich,
daß Wärme ohne äußere Arbeit zufuhr von
emem kalten auf emen warmen Körper übergeht, was gegen den zweiten Hauptsatz vertößt. Daraus folgt
bn höherer thermt eher Wirkung:;grad ab
der de Camot-Proze. e 1st mcht möglich.
33.6.3. Entropie
Die b1 herigen Formulierungen des zweiten
Hauptsatze können mathematisch ausgedrückt
werd en mit Htlfe der Zu tand größe Entropie,
d ie ge tattet, den Grad der Irreversibilität
ei ne Vorganges zu berechnen. Ausgangspunkt
der folgenden Betrachtungen i t der ideale
reversibel geftihrte Carnot-Prozeß (Bild 3-24).
Für die umgesetzten Wärmen und die Temperaturen der Wärmebäder gilt nach GI. (3-77)
Ql2+ Q34 = 0 .
T1
T3
Der Quotient von übertragener Wärme und
der ab oluten Temperatur, bei der sie übertragen wurde, wird als reduzierte Wärme be-
Volumen V
Bild 1·34. Er: atz eines beliebigen Kreisprozesses
durch ein System ~·on Carnot-Prozessen.
zeichnet. Offensichtlich i t die Summe der reduzierten Wärmen bei einem kompletten
Umlauf eine reversiblen Carnot-Proze ses
null. Wird ein belieb1ger Krei prozeß reveribel durchlaufen, dann kann er nach Bild 3-34
durch unendlich viele differentiell chmale
Camot-Prozes e er etzt werden. Auch hierbei
ist die umme aller reduzierten Wärmen null:
f c)~re• = 0.
(3-83)
I
Der Index rev soll daran erinnern, daß die
Prozeßführung revenbel ein muß.
Wenn d'te G ro" ße -c"> Qrev
·
komp Ietten
T- be1· emem
Umla uf keine Änderung erfahrt, erfüllt sie
d1 e Voraussetzungen, die an eine Zustandsgröße gestellt werden. Diese Zustandsgröße
bezeichnet man nach Clausius als Entropie S.
Ihr Differential ist definiert als
dS = f>Qrev
(3-84)
T .
Die Maßeinheit der Entropie ist J/K Der
Nullpunkt kann willkürlich gewählt werden.
Die Entropiedifferenz zwischen einem Ausgangszustand 1 und einem Endzustand 2 ist
---!lS =
s2- SI= J bQrev
l
T
0
(3-85)
I
3.3. Hauptsätze der Thermodynamik
183
Die Entropieänderung i t al Differenz zweier
Zustand größen ' egunabhängig. Zu ihrer Berechnung muß aber ein - wenig ten in Gedanken - reali ierbarer reversibler Weg bechritten werden. Bei reversibel geführten
adiabaten Zu land änderungen ist <>Qrev = 0.
omil gibt e keine Änderung der Entropie
(S 1 = S2); die Zustand änderungverläuft isenrrop.
Die Entropieänderung bei einer Zustandsänderung eine idealen Gases läßt sich aus
GI. (3-84) mit Hilfe de ersten Hauptsatzes
berechnen:
dS= bQrev = dU+pdV.
T
T
Mit GI. (3-46) für die Änderung der inneren
Energie ergibt sich daraus
dS= vC
mv
dT +Ldv.
T
T
Nach der Zu tandsgleichung idealer Gase ist
p / T= v Rm/Vund somit
dT
dV
dS= vCmvT+ vRmv ·
Wird die molare Wärmekapazität Cmv als
konstant vorausgesetzt, kann integriert werden:
Bild 3-35.
such.
Zu Beispiel 3.3-7: Gay-Lussacscher Ver-
hälter in einen zunächst evakuierten Rezipienten
trömen. Die Anordnung war nach außen wärmeisoliert (adiabates System). Gay-Lussac fand, daß
nach Erreichen des Gleichgewichtszustands die
Temperatur des Gases nicht verändert war und
schloß daraus, daß die innere Energie idealer Gase
nicht vom Volumen abhängt Wie groß ist die
Entropieänderung bei dem geschilderten Vorgang?
Lösung: Obwohl die Ausströmung ins Vakuum ein
hochgradig irreversibler Prozeß ist, läßt sich die
Entropieänderung mit Hilfe eines reversiblen Ersatzprozesses berechnen. Ein denkbarer Ersatzprozeß ist die isotherme Expansion mit jeweils dem
gleichen Anfangs- und Endzustand wie der tatsächliche Prozeß. Nach GI. (3-86) gilt dann mit T1 = T2
v2
AS = S2- S ,= vRmln-.
v,
Die Entropieänderung ist größer als null, weil
v2 > v, ist Ist z. B. V = 1 mol und V21V, = 2, dann
beträgt die Entropieänderung
1
(3-86)
Nach GI. (3-51) kann die innere Energie durch
die Enthalpie ausgedrückt ':\'erden:
dU= dH- p dV- Vdp.
Damit gilt
ds
dH- V dp =
T
V
c
mp
Hat der Carnotsche Kreisprozeß irreversible
Anteile (z. B. Reibungsarbeit oder Wärmeübertragung mit Temperaturgefälle zwischen
Wärmebad und Gas), so ist der thermische
Wirkungsgrad geringer als bei vollkommen
rever ibler Prozeßführung:
n
dT T
R
h
m
dp
p
und nach der Integration
1
AS = I mol · 8 314-- · ln 2= 5 76-.
'
molK
' K
• 1th,irr
--
Q, 2 + Q34
Q
<
n rev•tth,
34
T3- T,
TJ
Anstelle von GI. (3-77) gilt dann
Q, 2+ Q34 0
T
T < .
1
3
Für beliebige irreversible Kreisprozesse gilt
entsprechend (im Gegensatz zu Gl. (3-83), die
nur bei reversibler Prozeßführung gi"tig ist)
Beispiel
3.3-7: In einem berühmt gewordenen Versuch ließ
Gay-Lussac nach Bild 3-35 ein Gas aus einem Be-
(3-88)
184
3. Thermodynamik
I\
In einem adiabaten geschlossenen System
können von selbst nur Vorgänge ablaufen,
bei denen die Entropie ansteigt.
2
~#'..,....,.. -
>~e
\((e
/
;
Ein Beispiel für den Entropieanstieg ist die
Ausströmung eines Gases ins Vakuum (Beispiel 3.3-1 1). Ist ein System abgeschlossen,
dann ist die innere Energie des Systems konstant, und die Entropie des Systems strebt
einem Maximalwert zu, den sie im Gleichgewichtszustand erreicht hat.
/
/
/
/
/
I
I
Volumen V
Bild 3-36. Kreisprozeß mit reversiblem und irreversiblem Anteil.
Aus der Definitionsgleichung der Entropie
dS = bQreviT folgt, daß in einem T, S-Diagramm die reversibel umgesetzte Wärmemenge als Fläche unter der Kurve einer Zustandsänderung abgelesen werden kann:
liQrev = T dS
Nach Bild 3-36 sei jetzt ein Kreisprozeß betrachtet, der aus einem irreversiblen (I -+ 2)
und einem reversiblen (2-+ 1) Weg besteht.
Der Gesamtprozeß ist damit irreversibel, und
nach GI. (3-88) gilt
~
J
J
bQ = c'i Qirr + bQrev < O.
T
I
T
2
T
M1t GI. (3-85) kann man schreiben
2
.
bQirr
J-T- + S 1 -·~ 2< O.
C'
I
Betrachtet man msbesondere adiabate Systeme, bet denen keine Wärmeübertragung stattfindet ( liQirr = 0), dann gilt
S2 -
sl > o
2
Ql2,rev =
f T dS ·
(3-91)
I
Bild 3-37 zeigt das Wärmeschaubild des Carnot-Prozesses. Die zugeführte Wärme entspricht der F läche unterhalb der Geraden
3-4, die abgegebene Wärme ist sichtbar
als Fläche unterhalb der Geraden 1-2. Die
Nutzarbeit entspricht wie beim p, V-Diagramm dem Flächeninhalt der umfahrenen
Figur.
(3-89)
1-..
...
In einem adiabaten geschlossenen System
sind irreversibl e Prozesse stets mit einem
Anstieg der Entropie verknüpft. Bei reversi bler Prozeßführung bleibt die Entropie
konstant.
oder
3
4
T3
~
E
Q)
IWI
0.
E
~
e
::J
0
Ti
2
"'
m
.J:l
Mathematisch kann diese Aussage auch so
formuliert werden :
dS
!1;
0.
(3-90)
J
s2 = s3
Das Gleichheitszeichen gilt für reversible, das
Größer-als-Zeichen für irreversible Prozesse.
Da in der Natur von selbst nur irreversible
Prozesse ablaufen, gilt:
Entropie S
Bild 3-37. T,S-Diagramm des Carnot-Prozesses.
W Arbeit
1 bis 4 Zustandspunkte
3.3. Hauptsätze der Thermodynamik
185
3.3.6.4. Statistische Deutung der Entropie
Mit Hilfe statistisrher Betrachtungen soll gezeigt werden, daß die Entropie in engem Zusammenhang steht zu der Wahrscheinlichkeit,
mit der ein bestimmter Zustand realisiert
werden kann.
Es sei zunächst ein Gefäß mit dem Volumen
V betrachtet, in dem sich nur ein Gasmolekül
befindet. Die Wahrscheinlichkeit, das Molekül bei einer Kontrolle im Volumen V anzutreffen, ist PI = I, d. h., es ist - gemäß der
Voraussetzung - mit Sicherheit darin. Denkt
man sich jetzt das Gefäß halbiert, so ist die
Wahrscheinlichkeit dafür, das Molekül bei
einer Stichprobe im Teilvolumen V2 = ~ V1 zu
finden, Pl 12 = ~- Wird das Volumen in drei
gleiche Teile geteilt, dann ist die Wahrscheinlichkeit, daß das Volumen V3 = V besetzt
ist, pjl3 = t·
Dieses Gedankenspiel kann fortgesetzt werden bis zu einer n-fachen Unterteilung des
Raumes. Die Wahrscheinlichkeit, das Molekül in einer Zelle vom Volumen V0 =*V anzutreffen, ist dann Pl 1n =
Befinden sich zwei Moleküle im Volumen V,
dann ist die Wahrscheinlichkeit, beide gleichzeitig in derselben Zelle vom Volumen
V"=* V anzutreffen, gleich dem Produkt der
Einzelwahrscheinlichkeiten (Satz vom "sowohl
als auch"):
t
*·
Plln = pjln pjln = (
+)
2
Sind N Moleküle vorhanden, dann ist die
Wahrscheinlichkeit dafür, daß sich alle im
Volumen Vn = V aufhalten, gegeben durch
*
Nach Bild 3-38 werden nun zwei an sich sehr
unwahrscheinliche Zustände miteinander verglichen: N Moleküle sollen sich bei einer
Stichprobe im Teilvolumen V3 = V bzw.
V2 =~V aufhalten. Das thermodynamische
Wahrscheinlichkeitsverhältnis für die beiden
Zustände ist
~~~1~~~~
I
Zustand ll: Pu
= (
Jf
v2
Bild 3-38. Vergleich zwischen zwei thermodynamischen Zuständen.
w = 10 1•76 · 1022 • Um zu Zahlen vernünftiger
Größenordnungen zu gelangen, bestimmt man
den natürlichen Logarithmus des Wahrscheinlichkeitsverhältnisses :
I
lnw ~ Nln(~_)·_______<3--92)~i
Für das genannte Beispiel ergibt sich In w =
4,05 · 10 21• Diese Größe steht in einem direkten Zusammenhang mit der Entropiedifferenz zwi chen den Zuständen I und Il. Die
Änderung der Entropie vom Zustand I zum
Zustand li kann mit Hilfe eines reversiblen
Ersatzprozesses berechnet werden. Zu diesem
Zweck wird das Gas in einem Zylinder mit
verschiebbarem Kolben vom Ausgangsvolumen V3 isotherm auf das Endvolumen V2
entspannt. Die Entropieänderung ist nach
GI. (3-86)
~S =S 1 - S1=v Rm in ( ~:) =N k in ( ~:) .
Ein Vergleich mit GI. (3-92) zeigt den von
Bolt:::mann gefundenen Zusammenhang zwischen Entropieänderung und thermodynamischem Wahrscheinlichkeitsverhältnis:
~s =
k In w.
(3-93)
t
W=~=(~)N
P,
v3
Für ein Gas mit der Teilchenanzahl N = 10 22
ergibt sich die außerordentlich große Zahl
Für die Entropie selbst gilt
S = kln P.
(3-94)
Die Entropie eines Systems ist um so höher, je größer die Wahrscheinlichkeit ist,
mit welcher der Zustand des Systems realisiert werden kann.
186
3. Thermodynamik
Beispiel
3.3-8: Wie groß ist die Wahrscheinlichkeit dafür,
daß sich ein Stein mit der Masse m = I kg und der
Temperatur T = 300 K spontan abkühlt und dafür
um h = 10 cm in die Höhe springt?
Lösung:
Die Energiebilanz des Vorganges ist 6Epo1 = - !J.U
oder m g h = - m c !J.T. Daraus folgt für die Temperaturabnahme !J.T = - g h/ c. Mit der spezifischen
Wärmekapazität c ~ 0,8 kJ/kg K ergibt sich !J.T=
- 1,2 · I0- 3 K. Die Temperaturänderung ist so minimal, daß der Vorgang näherungweise als isotherm
betrachtet werden kann. Die Entropieänderung kann
durch folgenden Ersatzprozeß berechnet werden:
Dem Stein wird reversibel die Wärme Q, •• = - mgh
bei der Temperatur T entzogen. Dann ist die Entropieänderung
!J.S = Q, •• =- mgh =- 3,27 · 10 - 3 1/K .
T
T
Die negative Entropieänderung besagt nach der
klassischen Thermodynamik, daß der Vorgang nicht
beobachtet wird. Statistisch besteht jedoch eine wenn auch verschwi nd end kleine - Wahrscheinlichkeit dafür, daß der Vorgang eintritt. Das Wahrscheinlichkeitsverhältnis der beiden Zustände des
Steins ist
W
= .!..3._ = e11Si k =
e - 2,37 10
20
=
J0 - 10
20
•
Pt
Füllt man in einen Behälter weißen Sand und
schichtet darüber vorsichtig dunklen Sand,
dann werden sich die beiden Sandsorten beim
Schütteln des Gefäßes mischen. Dieser typisch irreversible Mischungsvorgang kann
vom Standpunkt der Wahrscheinlichkeitsrechnung so interpretiert werden, daß das System
vom unwahrscheinlichen Zustand hoher Ordnung in den wahrscheinlicheren Zustand großer Unordnung übergeht. Von selbst ablaufende Vorgänge gehen stets von geordneten
Zuständen in Richtung größerer Unordnung.
Da sie gleichzeitig mit einem Entropieanstieg
verknüpft sind, folgt :
Entropie ist ein Maß für
Unordnung eines Systems.
----
d~ Grad
l
--Das Prinzip des Entropieanstiegs gilt nur für
abgeschlossene Systeme, nicht aber für offene.
Ist ein offenes System weit entfernt vom thermischen Gleichgewicht, so bewirken einerseits die Energiezufuhr oder auch der Zu-
strom neuer Stoffe und andererseits die Umwandlung im System in andere Energie- und
Stofforrnen, daß sich im System ständig neue
Lagen der Systemteile zueinander, neuartige
Bewegungsabläufe oder neuartige Reaktionsabläufe bilden, an denen größere Bereiche des
Systems beteiligt sind. Unter den sich kurzzeitig bildenden, miteinander konkurrierenden Strukturen (Moden) kommt es ab einem
charakteristischen Schwellwert der Energieoder Stoffzufuhr plötzlich zu makroskopisch
wahrnehmbaren Ordnungszuständen. Durch
Selbstorganisation setzen sich jene neuartigen
Moden (Ordner) durch, die den anderen Systemteilen ihre Ordnung am erfolgreichsten
aufprägen (Versklavung) und die höchsten
Wachstumsraten haben. Aus der Unordnung
(Chaos) entstehen also in offenen Systemen
geordnete Strukturen. Welche Ordnungszustände sich unter gegebenen Randbedingungen bilden, ist Untersuchungsgegenstand der
von H. HAKEN (* 1927) begründeten Lehre
vom Zusammenwirken der Einzelteile offener
Systeme, der Synergetik.
Zur Übung
ü 3.3-12: Wie groß ist die Energie, die man mit
einem Perpetuum mobile zweiter Art aus dem
Meerwasser gewinnen könnte, wenn dieses um
63 = I oc abgekühlt würde? Die Masse des Meerwassers ist m ~ 1,4 · 10 21 kg. Wie lange würde dieser Energievorrat reichen bei einem mittleren Leistungsbedarf der Menschheit von ungefähr P =
13TW?
Ü 3.3-13: Stickstoff wird vom Normzustand Pn, Tn
und Vn = 1 1 a) isobar, b) isochor auf die Temperatur T 1 = 500 K erwärmt. Wie groß ist in beiden
Fällen die Entropieänderung ?
ü 3.3-14:
Welche Kurvenform hat eine Isochore im
T, S-Diagramm? Wie sieht demnach das T, S-Dia-
gramm des Stirling-Prozesses aus? Wie kann man
zeigen, daß der thermische Wirkungsgrad des
idealen Stirling-Prozesses mit funktionierender interner Wärmeübertragung mit dem des Carnot-Prozesses identisch ist?
Ü 3.3-15: Ein Teil aus Kupfer mit der Masse
m = I kg und der Temperatur 3 1 = 10 °C wird in
Kontakt gebracht mit einem gleich schweren Kupferteil mit 3 2 = 30 °C. - a) Um welchen Betrag
ändert sich die Entropie der beiden Körper beim
Temperaturausgleich, wenn kein Wärmetransport
zur Umgebung erfolgt?
b) Wie groß ist die Wahrscheinlichkeit, daß der
umgekehrte Vorgang von selbst abläuft?
3.3. Hauptsätze der Thermodynamik
3.3.7. Thermodynamische Potentiale
Der zweite Ha ur- .satz erlaubt Aussagen über
die Richtung von selbst ablaufender Prozesse
in adiabaten bzw. abgeschlossenen Systemen.
Viele Prozesse, besonders chemische Reaktionen, laufen bei konstanter Temperatur ab.
Hierbei kann die Richtung von selbst ablaufender Vorgänge mit der von Helmho/tz eingeführtenfreien Energie F bestimmt werden:
F= U- TS.
(3-95)
1
Das totale Differential dieser Zustandsgröße
ist dF =dU- T dS- S d T. Für isotherme Systeme gilt dT= 0 und dF= dU- TdS. Mit
dem ersten Hauptsatz dU= f> Qrev + f> Wrev
und der Definitionsgleichung (3-84) für die
Entropie T dS = f> Qrev folgt für reversible
Prozesse dF = f> Wrev oder- f> Wrev =- dF.
Die Arbeit, die ein isothermes System bei reversiblen Prozessen abgeben kann, entspricht
der Abnahme der freien Energie. Bei irreversibler Prozeßführung ist die abgegebene
Arbeit stets kleiner als bei reversibler Führung, also- f> Wirr< - (j Wrev· Damit gilt
-
()W~-
dF.
(3-96)
Das Gleichheitszeichen gilt im Fall reversibler, das Kleiner-als-Zeichen bei irreversibler Prozeßführung.
Der maximale Arbeitsbetrag, den ein isothermes System nach außen abgeben kann,
ist gleich der Abnahme der freien Energie.
Nimmt beispielsweise bei einer isothermen
chemischen Reaktion die innere Energie von
u! auf u2 ab, dann kann nicht die ganze
Differenz t:.U als Arbeit nach außen abgegeben werden, sondern nur der Anteil der freien
Energie M = t:.U- T t:.S. Der Teilbetrag T t:.S,
die gebundene Energie, wird in Wärme umgesetzt. Es wird ausdrücklich darauf hingewiesen, daß unter "Arbeit" in diesem Fall
nicht nur die Volumenänderungsarbeit f> Wv =
- p d V, sondern auch jede andere Form von
Arbeit f> W' (z. B. elektrische Arbeit bei elektrochemischen Reaktionen und Oberflächenarbeit) verstanden wird: [> W = [> Wv + [> W'.
187
Wird das Volumen eines Systems konstant
gehalten, dann ist f> Wv = 0, und aus GI. (3-96)
folgt- f>W' ~- dFoder dF- ()W' ~ 0.
Bei spontan ablaufenden Reaktionen wird
Nutzarbeit abgegeben, d. h. - [> W' ~ 0. Für
die freie Energie folgt daraus
dF~O.
(3-97)
In einem isotherm-isochoren System verlaufen reversible Vorgänge bei konstanter
freier Energie, irreversible Prozesse unter
Abnahme der freien Energie. Im Gleichgewicht hat die freie Energie ein Minimum.
Somit ist auch die Richtung chemischer Reaktionen aufgezeigt: In isotherm-isochoren
Systemen verlaufen chemische Reaktionen
spontan, wenn die freie Energie der Reaktionspartner nach der Reaktion geringer ist als
vorher.
Die freie Energie gehört zu den thermodynamischen Potentialen. Wie in der Mechanik die
Komponenten einer Kraft durch Differentiation des Potentials nach den Koordinaten ermittelt werden können, besteht in der Thermodynamik die Möglichkeit, alle Zustandsgrößen durch Differentiation aus thermodynamischen Potentialen zu gewinnen. Für die
freie Energie gilt
dF= dU- TdS- SdT.
Mit dem ersten und zweiten Hauptsatz
dU=bQ-pdV=TdS-pdV
folgt
dF=-pdV-SdT.
(1)
Das totale Differential der Funktion F (V, T)
kann geschrieben werden
dF=(oF) dV+(oF) dT .
0V T
oT v
(2)
Aus dem Vergleich der Beziehungen (l) und
(2) folgt
p= -(oF)
oV
und
(3-98)
T
S= -(oF) .
oT v
(3-99)
188
3. Thermodynamik
Ist ein thermodynamisches Potential als Funktion seiner natürlichen Variablen bekannt, so
kann man durch reine Differentiationsprozesse andere thermodynamische Potentiale
oder Zustandsgrößen gewinnen. Auf diese
Weise werden beispielsweise Dampftafeln berechnet.
Ein weiteres thermodynamisches Potential ist
die freie Enthalpie G oder Gibbssches Potential (J. W. GIBBS, 1839 bis 1903):
G=H- TS= U+p V- TS.
(3-1 ~
weißes und gra ues Zinn bei T = 0 dieselbe
Entropie: Sweiß (0) = Sgrau (0).
Bei Annäherung eines homogenen Systems an
den absoluten Nullpunkt ist im Gleichgewicht die molare Entropie unabhängig von
thermodynamischen Parametern (z. B. Druck,
Volumen, Kristallstruktur, Magnetfeld) und
nimmt einen konstanten Wert Smo an. Dieser
Nernstsche Wärmesatz wurde von P/anck erweitert, der die Entropie am absoluten Nullpunkt null setzte:
S0 = 0
Für das totale Differential der Zustandsgröße
G(p, T) gilt
für
T = 0.
(3-104)
Die Entropie reiner Stoffe ist am absoluten Temperaturnullpunkt null.
d G = dU+ p d V+ V dp - T d S- S d T.
Mit dU+pdV= DQ= TdS folgt dG= Vdp
- S dT. Durch Vergleich mit
dG=(oG) dp+(oG) dT
op
oT
T
p
ergeben sich
V=(oG)
op
und
(3-101)
T
S=-(oG) .
or
(3-102)
p
Dte freie Enthalpte hat eine ähnhche Bedeutung wie die freie Energie. In einem isothermisobaren System gilt
dG~O.
(3-103)
Das Gleichheitszeichen gilt für reversible, das
Kleiner-als-Zeichen für irreversible Vorgänge.
In isotherm-isobaren Systemen strebt die
freie Enthalpie ein Minimum an, das sie
im Gleichgewichtszustand erreicht hat.
3.3.8. Dritter Hauptsatz der
Thermodynamik
Durch experimentelle Untersuchungen fand
Nernst (W. NERNST, 1864 bis 1941) im Jahr
1906, daß die Entropie fester Körper am absoluten Temperaturnullpunkt nicht von der
Kristallmodifikation abhängt. So hat z. B.
Diese Festlegung der Entropie durch den
dritten Hauptsatz ist im Einklang mit der
statistischen Deutung der Entropie. Der
Gleichgewichtszustand am absoluten Nullpunkt zeichnet sich durch maximale Ordnung
aus. Die Unordnung und damit die Entropie
sind null.
Der dritte Hauptsatz ist nur gültig für reine
Stoffe. Mischkristalle haben bei T = 0 eine
endliche Entropie. Außerdem müssen die Systeme im thermodynamischen Gleichgewicht
sein. Dies ist z. B. bei Gläsern nicht der Fall.
Gläser haben auch bei T = 0 noch eine Unordnung, demnach ist S 0 > 0. Der Ü bergang
in eine geordnete kristalline Phase findet
nicht statt, weil bei tiefen Temperaturen die
Reaktionsgeschwindigkeiten vernachlässigbar
klein werden.
Die Entropie eines Systems kann nach dem
dritten Hauptsatz absolut berechnet werden
S (T) =
Jo DQrev
T
T c(T)
=mJ--dT
o T
.
(3-105)
Die Entropie blei bt nur dann endlich, wenn
die spezifische Wärmekapazität c (T) mit abnehmender Temperatur hinreichend schnell
gegen null geht. D ies ist in der Tat der Fall :
Bei vielen Festkörpern gilt bei tiefen Temperaturen das Debyesehe T 3 -Gesetz c(T) =
konst · T 3 (Abschn. 9.3.1.2).
3.4. Zustandsänderungen realer Gase
Aus dem dritten Hauptsatz folgt auch,
daß der thermische Ausdehnungskoeffizient
(o VloT)p und der Druckkoeffizient (oploT)v
bei Annäherung an den absoluten Nullpunkt
null werden.
In Abschn. 3.3.5 ist erwähnt, daß ein CamotProzeß, bei dem die tiefe Temperatur T 1 = 0
ist, einen thermischen Wirkungsgrad von
"Yfth = 1 hat. Bei einem reversiblen CamotProzeß (Bild 3-24) gilt nach dem zweiten
Hauptsatz für das Kreisintegral der Entropie
f ds = s,2 + S23 + S34 + s4, = o.
189
mit Ri als der spezifischen Gaskonstante. Für
reale Gase mit molekularen Wechselwirkungen wird die Zustandsgleichung mit dem
Realgasfaktor Z korrigiert:
(3-1 07)
Bild 3-39 zeigt den Verlauf der Realgasfaktoren Z von Luft in Abhängigkeit vom Druck p
(von 0 bis 300 bar).
Nun ist S 23 = S 41 = 0 wegen isentroper Prozeßführung. Nach dem dritten Hauptsatz ist
S 12 = 0 für T 1 = 0. Also gilt t S = S 34 = 0. Die
Entropieänderung während der isothermen
Expansion von 3 nach 4 ist aber nach
GI. (3-85)
s34 =
> o.
T3
Der Widerspruch löst sich nur, wenn die tiefe
Temperatur T 1 > 0 gesetzt wird. Daraus folgt:
Q34
Der absolute Temperaturnullpunkt läßt
sich nicht erreichen.
Der dritte Hauptsatz wird deshalb gelegentlich auch als Satz von der Unerreichbarkeil des
absoluten Nullpunkts bezeichnet.
3.4. Zustandsänderungen
realer Gase
Sind die Wechselwirkungen zwischen den
Gasmolekülen - beispielsweise in der Nähe
von Phasenumwandlungen - nicht mehr zu
vernachlässigen, so handelt es sich um reale
Gase. Mit der allgemeinen Zustandsgleichung
idealer Gase (Abschn. 3.1.5) läßt sich die
Dichte e aus der absoluten Temperatur T und
dem Druck p ableiten: p V= m Ri T ergibt
wegene= m/V
0
100
bar
300
Bild 3-39. Realgasfaktor Z von Luft.
Die Dichte von Gasgemischen ea errechnet
sich aus den jeweiligen Dichten e1 , Q2, ... en
und deren prozentualen Volumenanteilen:
~Qi Vi
(3-1 06)
200
Druckp
I I,__e_a_=__v_______c_3_-I_os_)__,
190
3. Thennodynarrllk
3.4.1. Van-der-Waalssche
Zustandsgleichung
80
bar
Die für Ideale Gase abgeleiteten Gesetze vernachlässigen zwei Einflußgrößen, die bei hohen Drücken und tiefen Temperaturen besonders deutlich in Erscheinung treten, nämlich
73
- die zwischen den Gasmolekülen stattfindenden Allziehungskräfte (Kohäsion) und
-das Eigenvolumen der Gase (Kovo/umen).
60
J. D. VAN DER WAALS (1837 bis 1923) hat den
Druck und das Volumen in der allgemeinen
Gasgleichung dementsprechend korrigiert.
Die van-der-Waalssche Zustandsgleichung lautet mit molaren Größen
(3-~
Darin sind Vm das Molvolumen und asowie b
gasspezifische Materialkonstanten.
Den Korrekturterm a/ V~ nennt man Binnendruck. Er berücksichtigt die Wirkung der zwischenmolekularen Anziehungskräfte ( Van-derWaa/s-Kräfie, Abschn. 9.1.1.1), die Kohäsionskräfte zwischen den Flüssigkeitsmolekülen,
die auch für die Oberflächenspannung verantwortlich sind (Abschn. 2.3.2.3. 1, Bild 2-98).
Im Ionern der Gasphase heben sich die zwi-,chenmolekularen Kräfte zwar auf, an den
Grenzflächen (z. 8. einer Gasoberfläche) aber
weisen sie eine resultierende Kraft in Richtung des Gasinneren auf. Drdurch erhöht sich
der Innendruck im Gas (Binnendruck); das
Korrekturglied ist deshalb positiv. Der Binnendruck Pbi ist proportional zur Dichte der
anziehenden Teilchen und zur Dichte der stoßenden Umgebungsteilchen. Insgesamt ist also der Binnendruck proportional zum Quadrat
der Dichte : Pbi "'Q 2, oder wegen Q"' 11 Vm ist
Pb;"' I/V~ .
Der Faktor b berücksichtigt das Wechselwirkungsvolumen der Molekülkräfte, das van-derWaa/ssche Ko1•olumen; es entspricht etwa dem
vierfachen Eigenvolumen des Moleküls. Die
van-der-Waalssche Zustandsgleichung stellt
für konstante Temperaturen (Isothermen) im
p, V-Diagramm eine Funktion dritten Grades
dar. Bild 3-40 zeigt den Verlauf der Isothermen für Kohlendioxid (C0 2 ). Unterhalb der
Isothermen für die kritische Temperatur Tk
Gebiet des
idealen Gases
70
Q.
-""
g
50
47
Ci
40
313 K
304 ((
293 K
30
273 K
0,1
0,2
0,3
0.4
Molvolumen Vm
0,5
0,6 m' 0,7
kmol
Bild 3-40. Verlauf der Isothermen for Kohlendioxid
im p, V-Diagramm.
(für C0 2 ist Tk = 304,2 K) weisen die Isothermen mit abnehmendem Molvolumen ein
Druckmaximum und ein Druckmimimum
auf. Dies widerspricht jedoch der experimentellen Erfahrung: Mit fallendem Volumen
durchläuft der Druck nicht die Kurve EDCBA
(Isotherme 273 K), sondern verläuft horizontal längs der Geraden ECA. Dies liegt daran,
daß bei realen Gasen ab dem Punkt E eine
Veiflüssigung eintritt, die am Punkt A abgeschlossen ist. Der bei weiterer Komprimierung erfolgende steile Druckanstieg rührt von
der im Vergleich zu Gasen sehr kleinen Kompressibilität von Flüssigkeiten her.
Der Druck PE in Bild 3-40, bei dem eine Verflüssigung einsetzt, ist der Dampfdruck. Nach
dem ersten Hauptsatz der Thermodynamik
(Abschn. 3.3.6) müssen die Flächeninhalte
über der Linie CDE und unter der Linie
ABC gleich sein. Werden für jede Isotherme
jeweils die Punkte E der beginnenden Verflüssigung des Gases und jeweils die Punkte A
des Endes der Verflüssigung miteinander verbunden, so ergibt sich ein Bereich, innerhalb
dessen eine Umwandlung von der gasförmigen Phase in die flüssige stattfindet (rot um-
3.4. Zustandsänderungen realer Gase
grenzte Zone in Bild 3-40). Links von diesem
Gebiet liegt nur die flüssige und rechts nur
die gasförmige Phase vor. Im Koexistenzgebiet sind beide Phasen vorhanden. Bei Wasser
heißen diese Gebiete: überhitzter Dampf (rein
gasförmiger Zustand), trocken gesättigter
Dampf (Grenzkurve) und Naßdampf (innerhalb des Verflüssigungsgebiets). Der höchste
Dampfdruckpunkt ist der kritische Punkt K.
Die zugehörige Temperatur ist die kritische
Temperatur Tk. Sie ist der Wendepunkt der
entsprechenden Isotherme. Die zugehörigen
Werte sind der kritische Druck Pk (für C0 2
ist Pk = 7,38 MPa) und das kritische Volumen
Vk (für C02 ist Vmk = 0,1275 m 3/kmol).
Oberhalb des Punktes K ist eine Verflüssigung durch alleinige Komprimierung (kleineres Volumen und höherer Druck) nicht
möglich. Tabelle 3-10 enthält die kritischen
Werte für Temperatur und Druck sowie die
van-der-Waalsschen Konstanten a und b für
einige ausgewählte Stoffe.
Da der kritische Punkt K einen Wendepunkt
mit waagrechter Tangente darstellt, können
die drei kritischen Werte von Gasen (Tb Pk
191
und Vk) durch folgende drei Bestimmungsgleichungen errechnet werden: p = f(V) (vander-Waalssche Zustandsgleichung für die
Isotherme T= Tk), (oplo V)r = 0 (waag(o 1p/o V 2 )r. = 0
rechte
Tangente)
und
(Wendepunkt). Aus (op/o V)r. = 0 und
(o 2p I oV 2 )r. = 0 folgen
Vmk = 3 b
und
8a
T, = - - k
27 b Rm.
(3-11 0)
(3-111)
Werden diese beiden Gleichungen in die van
der Waalssche Zustandsgleichung (3-109) eingesetzt, ergibt sich
a
(3-112)
Pk = 27 b2 .
1
Aus der Kombination aller drei Gleichungen
erhält man
~pkV._m_k_
=___
R____________(3--l13-)~
L_-
3
Tk
8
m·
Tabelle 3-10. Kritische Temperatur Tk, kritischer Druck Pk sowie van-der-Waalssche Konstanten
a und b verschiedener Stoffe.
Stoff
Tk
in
K
Pt
m
MPa
a
N 4
in10 5 ~2
kmol
b.
m 10
2
Elemente
Wasserstoff (H 2 )
Helium (He)
Stickstoff (N 2 )
Sauerstoff (0 2 )
33,240
5,2010
126,20
154,576
1,296
0,2275
3,400
5,043
0,2486
0,0347
1,366
1,382
2,666
2,376
3,858
3,186
Luft
132,507
3,766
1,360
3,657
417
647,30
405,6
304,2
7,70
22,120
11,30
7,3825
6,59
5,5242
4,246
3,656
5,63
3,041
3,730
4,282
190,56
370
425,18
4,5950
4,26
3,796
2,3047
9,37
13,89
4,310
9,03
11,64
anorganische Verbindungen
Chlor (CI 2 )
Wasser (H 2 0)
Ammoniak (NH 3 )
Kohlendioxid (C0 2 )
organische Verbindungen
Methan (CH 4 )
Propan (C 3 H 8 )
Butan (C 4 H 10 )
m3
kmol
192
3. Thermodynamik
Bei dem Vergleich mit dem Wert des Realgasfaktors Z (GI. (3-107)) ergibt sich für den
kritischen Punkt
(3-114)
1
Wenn die allgemeine Gasgleichung für ideale
Gase am kritischen Punkt gültig wäre, müßte
Zk = I sein. Der Realgasfaktor Z gibt also den
Grad der Abweichung von der allgemeinen
Gasgleichung an (Bild 3-39).
Sind zwei der kritischen Werte Pk, Vmk und ~
bekannt, dann können die van-der- Waalsschen
Konstanten a und b errechnet werden:
b
=
Vmk
= Rm ~
3
(3-115)
8 Pk '
2
a = 3Pk v;k = 27 b Pk .
(3-116)
men wird. Dieser Effekt wird Joule-ThomsonEffekt genannt (J. P. JouLE, 1818 bis 18.89,
und W. THOMSON, 1824 bis 1907). Die druckbezogenen Temperaturdifferenzen betragen
beispielsweise für Luft 11T/ 11p = 2,5 K/MPa
und für Kohlendioxid 11Tfl1p = 7,5 K/MPa.
Die Luftverflüssigung gelang erstmalig Linde
(C. V. LINDE, 1842 bis 1934) im Jahr 1876.
Genaue Rechnungen ergeben, daß der JouleThomson-Effekt auch zu einer Erwärmung
führen kann. Oberhalb der Inversionstemperatur Ti erwärmt sich ein Gas, und unterhalb
dieser kühlt es sich ab. Näherungsweise ist
2a
T~--.
'
Rmb
(3-117)
1
Da für die kritische Temperatur eines realen
Gases nach GI. (3 -111) Tk = 8 a/(27 bRm) gilt,
ist die Inversionstemperatur
(3-118)
1
Beispiel
3.4-1: Für Kohlendioxid (C0 2 ) gilt am kritischen
Punkt Tk = 304,2 K und Pk = 7,38 MPa. Es sollen
hieraus die van-der-Waalsschen Konstanten a und b
berechnet werden.
Lösung:
Nach GI. (3-115) gilt
b
Rm Tk
3
= - - = 0,0428 m /kmol,
8pk
nach GI. (3-116) gilt
a = 27 b 2 p
k
Nm 4
kmol
= 3 66 ·10 5 - - 2.
'
3.4.2. Gasverflüssigung
(Joule-Thomson-Effekt)
Bei einem realen Gas ist wegen der zwischenmolekularen Wechselwirkungen und des Eigenvolumens der Moleküle die innere Energie
U volumen- und druckabhängig. Wird ein
reales Gas adiabat (ohne Wärmeübertragung)
und ohne Arbeitsverrichtung (Drosselung)
entspannt, so kühlt es sich im Gegensatz zum
idealen Gas ab. Zur Überwindung der zwischenmolekularen Anziehungskräfte muß nämlich Energie aufgewendet werden, die aus
dem Vorrat der inneren Energie U entnom-
Weil für Luft, Stickstoff, Sauerstoff und Kohlendioxid die Inversionstemperatur Ti weit
oberhalb der Raumtemperatur liegt, kühlen
sich diese Gase nach dem Joule-ThomsonEffekt ab, während sich Wasserstoff bei
Raumtemperatur (Tk = 33,3 K) erwärmt. Deshalb wird Wasserstoff zwecks Verflüssigung
erst mit flüssigem Stickstoff vorgekühlt
In Bild 3-41 sind einige technisch bedeutsame
Temperaturen und die entsprechenden physikalischen Effekte zusammengestellt. Zwecks
Untersuchung von Werkstoffen bei tiefen
Temperaturen kühlt man die Proben mit flüssiger Luft (T=79 K) oder flüssigem Stickstoff
(T = 77 K) ab. Zur Untersuchung des supraleitenden Zustandes (Abschn. 9.2.3) kühlt
man meist mit flüssigem Helium (T=4,2K
bis 0,83 K). Um tiefere Temperaturen, die
durch den Joule-Thomson-Effekt nicht mehr
erreicht werden, zu erhalten, müssen paramagnetische Salze adiabat entmagnetisiert
werden. Infolge der während der Entmagnetisierung zunehmenden Unordnung der magnetischen Struktur wird - analog zum Verdampfungsprozeß - dem Stoff Wärme entzogen, so daß eine Abkühlung eintritt (z. B.
Cäsium-Titan-Alaun, T= 0,0034 K). Nach diesem magnetokalorischen Effekt werden Temperaturen bis T = 10- 2 K erzeugt. Noch tie-
3.4. Zustandsänderungen realer Gase
Adiabate
Entmagnetisierung
der Molekular- bzw.
Atomarmagnete
Adiabate
Entmagnetisierung
der Kernmagnete
Joule-Thomson-Effekt
KaliumChrom- CenumAiaun
tluorid
I
0,05
1Q-6
1Q-3
10- 2
I
0,13
4,2
1Q- 1
Schmelzpunkte
Wasser Gold
H2 N 2 Luft Eis
sieden
schmelzen
I \I
I/
/
He
0,84
193
20 77 79 273 373
1336
10 3
10
K
104
Temperatur T
Bild 3-41.
Physikalische Effekte und einige technisch bedeutsame Temperaturen.
fere Temperaturen (bis T = 10- 6 K) kann man
durch Entmagnetisierung von Atomkernen erreichen.
3.4.3. Phasenumwandlungen
Eine Phase ist ein räumlich abgegrenztes Gebiet eines Stoffes mit gleichen physikalischen
Eigenschaften. Der Begriff Phase kann sowohl
auf die drei Aggregatzustände der Materie
(fest, flüssig, gasförmig), als auch auf die verschiedenen Modifikationen desselben Stoffs
(z. B. r:t.- und y-Eisen) angewandt werden. Die
unterschiedlichen chemischen Bestandteile
werden Komponenten genannt und zweckmäßigerweise durch eine chemische Strukturformel angegeben.
~
Bild 3-42 zeigt die möglichen Phasenübergänge für die drei Aggregatzustände fest,
flüssig und gasförmig unter Berücksichtigung von Modifikationsänderungen innerhalb
des festen Zustands. Allen Phasenübergängen
ist gemeinsam, daß Wärme zu- bzw. abgeführt werden muß, ohne daß eine Temperaturänderung eintritt. Diese Wärme wird
deshalb als latente Wärme bezeichnet.
Wird beispielsweise der Phasenübergang von
fest nach flüssig betrachtet, dann dient
die zugeführte Wärme der Aufbrechung
des Festkörpergitters. Die bei konstantem
Druck und konstanter Temperatur zugeführte
Wärme erhöht die Enthalpie der Substanz:
Hnüssig = Hrest + L'lHs . L'lHs wird als Schmelz-
fest
flüssig
gasförmig
Modifikationsänderung
{Modifikationsenthalpie f:J.HM)
Schmelzen
Sublimieren
{Sch melzenthalpie
l:J.Hs)
{Sublimationsenthalpie
l:J.Hsub = l:J.Hs + f:J.Hv)
n
fest
flüssig
gasförmig
Bild 3-42.
Erstarren
Sieden
{Erstarrungsenthalpie - Mfs)
{Verdampfungsenthalpie f:J.Hv)
Desublimieren
{Desublimationsenthalpie
- flHsub = - l:J.Hs- flHv)
Kondensieren
{Kondensationsenthalpie - f:J.Hv)
Phasenübergänge und zugehörige Entha/pien (Einstoffsystem).
---
194
3. Thermodynamik
spezifische
Verdampfungsenthalpie
l:J.h
Siedepunkt
373
V
Druck p = 1013 h Pa
380
= 2257
kJ
kg
K
.....
....
....::I
340
Q.
320
E
Q)
E
,!
spezifische
Schmelzenthalpie
300
l:J.h = 334 kJ
s
kg
Schmelzpunkt 280
273
260
240
220
/
/
/
latente
Wärme
/
200
10
100
1000
837
419
85
kJ
kg
3094
spezifische Enthalpie h
Bild 3-43.
Temperaturverlauf der spezifischen Enthalpie von Wasser.
Tabelle 3-11. Schmelz- bzw. Verdampfungstemperatur 9 sowie spezifische Schmelzenthalpie llhs
und spezifische Verdampfungsenthalpie llhv verschiedener Stoffe beim Druck Pn = 1013 hPa.
Stoff
Verdampfen
Schmelzen
f)
in
oc
-
259,15
270,7
209,85
218,75
213
!!hs
in kJ/k:g
f)
!!hv
in °C
in kJ/ kg
Elemente
Wasserstoff (H 2)
Helium (He)
Stickstoff (N 2)
Sauerstoff (0 2)
Luft
58,6
3,52
25,75
13,82
-
252,75
268 ,94
195,75
182,95
192,3
461
20,9
201
214
197
anorganische Verbindungen
Chlor (CI 2)
Wasser (H 20)
Ammoniak (NH 3)
Kohlendioxid (C0 2)
- 100,95
0,00
-80
- 56,55
90,4
335
339
184
-34,45
100,00
-33,45
- 78,45
289
2257
1369
574
- 182,45
- 187,65
- 138,35
58,6
80,0
77,5
-161,45
-42,05
-0,65
510
426
386
organische Verbindungen
Methan (CH 4 )
Propan (C 3H 8 )
Butan (C4H 10 )
3.4. Zustandsänderungen realer Gase
enthalpie bezeichnet. Sie wird bei der Erstarrung wieder frei (- 11Hs) . Beim Übergang
vom festen in den gasförmigen Zustand muß
die Summe aus Schmelzenthalpie 11Hs und
Verdampfungsenthalpie 11Hv als Sublimationsenthalpie 11Hsub = 11Hs + !1Hv zugeführt
werden.
Bild 3-43 zeigt den Temperaturverlauf als
Funktion der zugeführten spezifischen Enthalpie für Wasser vom Aggregatzustand
fest (Eis) bis gasförmig (Wasserd ampf). In
Tabelle 3-11 sind die Schmelz- bzw. Siedepunkte sowie die spezifischen Sch melz- bzw.
Verdampfungsenthalpien
zusammengestellt
(die Siedepunkte und Verd ampfungsenthalpien beziehen sich auf den Normdruck
Pn "= 1,013 · 10 5 Pa).
gewichtszustand zurücktreibt, forttreibt oder
keinen Einfluß zeigt. In der Mechanik (Abschn.
2.9.3) liegt bei einem stabilen Gleichgewicht
ein Minimum der potentiellen Energie vor.
Unterschiede in der potentiellen Energie (Gradient des mechanischen Potentials) sind die
treibenden Kräfte, die im Minimum verschwinden. In der Wärmelehre können je nach
Systemzustand fünf Gleichgewichtsforderungen auftreten (Abschn. 3.3.7). Sie sind in Bild
3-44 zusammengestellt:
- Maximum der Entropie S
für ein abgeschlossenes System ohne Materie- und Energieaustausch ;
- Minimum der jre1en Enthalpie G
für ein isobar-isothermes System ;
- Minirnum der freien Energie F
für ein i ochor-isothermes System ;
3.4.3.1. Thermodynamisches Gleichgewicht
Ein physikalisches System befind et sich im
Gleichgewicht, wenn sein physikalischer Zustand gleich bleibt. Es gibt stabile, labile und
indifferente Gleichgewichte, je nachdem, ob
eine äußere Störung das System zum Gleichisobar
dp=O
- AJinimum der Enthalpie H
für ein isobar-adiabates System owie
- Minimum der inneren Energie U
für ein isochor-adiabates System.
isochor
dV =O
isotherm
dT=O
dU=O
Maximum der
Entropie
dS ;;;;; 0
Minimum der
freien Enthalpie
dG ~ 0
Minimum der
freien Energie
dF ~ 0
Minimum der
Enthalpie
dH ~ 0
Minimum der
inneren Energie
dU ~ 0
r Enthalpie H
I
freie Enthalpie
~G
I
=
u
I
I
Bild 3-44.
+
I
pV
195
I
-
freie Energ ie
TS
I
Fl
Gleichgewichtsbedingungenfor die verschiedenen thermodynamischen Zustände.
adiabat
00 = 0
196
3. Thermodynamik
Chemische Reaktionen, die isobar und isotherm spontan ablaufen, haben alle eine negative molare freie Enthalpie t...Gm. Dabei kann
entweder Wärme frei werden (t....H < 0), oder
der Endzustand der Reaktion weist eine sehr
viel höhere Entropie auf (t...S = (t....H-t...G)I
T< 0).
3.4.3.2. Gleichgewicht zwischen flüssiger
und gasförmiger Phase
Analog zur Maxwellsehen Geschwindigkeitsverteilung in Gasen (Abschn. 3.2.3) gibt es
auch in Flüssigkeiten eine temperaturabhängige Verteilungsfunktion. Es ist immer eine
bestimmte Anzahl von Teilchen vorhanden,
deren Geschwindigkeit und somit deren kinetische Energie groß genug ist, um gegen die
Kohäsionskräfte der Nachbarteilchen die
Flüssigkeitsoberfläche zu durchstoßen.
Betrachtet sei ein Gefäß, in dem sich eine
Flüssigkeit befindet. Wird der Gasraum evakuiert, so steigt der Dampfdruck so lange, bis
120
sich ein Gleichgewicht zwischen der Verdampfungs- und der Kondensationsrate einstellt. Dann liegt ein gesättigter Dampf vor,
und der zugehörige Dampfdruck heißt Sättigungsdampfdruck Ps. Er ist unabhängig vom
Volumen, da sich bei Vergrößerung bzw. bei
Verkleinerung des Volumens entsprechend
mehr Dampf bildet bzw. kondensiert. Auch
das Einbringen von Körpern oder anderen
Gasmolekülen beeinflußt also den Sättigungsdampfdruck nicht. Für die Dampfdrücke
eines Gasgemischs (Partialdrücke) gilt deshalb das Daltonsche Gesetz (J. DALTON, 1766
bis 1844):
Der gesamte Druck eines Gasgemisches
ist gleich der Summe der Partialdrücke:
n
Pges=
L Pi·
i=l
(3-119)
Der Sättigungsdampfdruck steigt mit zunehmender Temperatur, da zusätzlich Flüssigkeit
verdampft, und nimmt ab mit fallender Tem-
~---.----.---.---.----.---.---.--~----.---.---.----r---r-.----~
{}
-20
-10
0
5
10
15
20
25
30
35
40
50
102Pa
10-2p, 0,96 2,59 6,09
8,7
12,25 17,01 23,33 31,60 42,32 56,1 73,57 123
100
90
r::J:
80
-"'
tJ
-
70
1J
60
flüssiger
Bereich
:::J
-o
c.
E
"'
VI
Cl
c
:::J
Cl
B·
50
'"'
(/)
40
gasförmiger
Bereich
30
20
10
0
10
20
30
40
oc
Temperatur {)
Bild 3-45.
Verlauf des Sättigungsdampfdrucks Ps von Wasser in Abhängigkeit von der Temperatur.
50
3.4. Zustandsänderungen realer Gase
peratur, weil Dampf kondensiert. Bild 3-45
zeigt den Verlauf des Sättigungsdampfdruckes
p, von Wasser in Abhängigkeit von der Temperatur. Diese Dampfdruckkurve beschreibt
die für das Gleichgewicht zwischen flüssiger
und gasförmiger Phase maßgebenden Wertepaare von Sättigungsdampfdruck p, und Temperatur.
Die Dampfdruckkurve wird durch den Boltzmann-Faktor (GI. (3-31)) beschrieben:
6.E
Ps- e
(3-120)
kT
~
AE ist die Energie, die benötigt wird, um
vom flüssigen in den gasförmigen Zustand zu
gelangen.
Der Verlauf der Dampfdruckkurve kann genauer
berechnet werden. Hierbei geht man davon aus,
daß mit einem Mol verdampfender Flüssigkeit ein
Carnotscher Kreisprozeß (Abschn. 3.3.5) durchlaufen wird. Wie Bild 3-46 zeigt, wird die Flüssigkeit
auf dem Weg 3-4 bei der Temperatur T+ dT und
dem Sättigungsdruck p, + dp, durch Zufuhr der
molaren Verdampfungsenthalpie .iRmv verdampft.
Auf dem Weg 1-2 erfolgt bei der Temperatur T
und dem Dampfdruck p, eine Kondensation. Zunächst liegt das Volumen vg in gasf6rmigem Zustand vor, am Ende ist das Volumen V~ 1 flüssig.
(Die adiabaten Teilstücke 4- 1 und 2-3 sind infinitesimal klein und daher bedeutungslos.) Die in
diesem Diagramm verrichtete Arbeit ist - d W
=(Vg- V~ 1 )dp,. Nach GI. (3-75) und (3-76) läßt
sich der thermische Wirkungsgrad des Carnotschen
Kreisprozesses ermitteln aus
dT
17th= -T =
( vg - V~ 1 ) dp,
AH
mv
Ll
Ps
er
..C
"'c:Ol
4
dp,
-"'
0
::J
T+ dT
3
+ dp,
Ps
2
T
1
::J
Ol
i:
'"'
U)
VJL
V,R
Molvolumen Vm
Bild 3-46. Carnotscher Kreisprozeß für eine verdampfende und kondensierende Flüssigkeit.
197
D araus ergibt sich als Steigung der Dampfdruckkurve die C/ausius-Clapeyronsche-G/eichung (R. E. CLAUSIUS, 1822 bis 1888, und
B. P. E. CLAPEYRON, 1799 bis 1864):
dp 5
dT
AHmv
( vJ?- v~1 ) T ·
(3-121)
Da das Molvolumen des Dampfes vg stets größer
ist als das der Flüssigkeit V~1 , ist die Steigung positiv, d. h., der Sättigungsdampfdruck steigt - wie
erwartet - mit zunehmender Temperatur. Wird
das Molvolumen der Flüssigkeit V~ 1 vernachlässigt und der gesättigte Dampf als ideale Gas
betrachtet ( vg = Rm TI p,), dann gilt
dp,
dT
.iHmvPs
Rm T2
dp5
.iHmv dT
P. = ----;:;;-Ti
oder
0
Nach Integration erhält man
(3-122)
Dies entspricht dem Boltzmann-Faktor (GI. (3-120)) .
Die Dampfdruckkurve läßt sich unter Berücksichtigung der Temperaturabhängigkeit
der Verdampfungsenthalpie für viele Substanzen in folgender Form darstellen:
a
In ( - Ps) =---b
in T/T0 +c . (3-123)
Pso
T
a, b und c sind materialabhängige Konstanten. Die Dampfdruckkurve endet bei hohen
Temperaturen am kritischen Punkt.
Ist der Dampfdruck einer Flüssigkeit gleich
dem auf der Flüssigkeit wirkenden Druck
eines anderen Gases (z. B. Luft auf Wasser) ,
so bilden sich auch im lnnem der Flüssigkeit
Dampfblasen ; die Flüssigkeit siedet. Wird
der auf der Flüssigkeitsoberfläche liegende
Druck erhöht, dann steigt der Siedepunkt.
Dieser Effekt wird bei einem Dampfkochtopf
ausgenützt. Wird der Druck erniedrigt, so fällt
der Siedepunkt, so daß beispielsweise Wasser
in großen Höhen deutlich
unterhalb
8 = 100 oc kocht. Die Temperaturabhängigkeit des Siedepunkts wird aus der Dampfdruckkurve (Bild 3-45) erkennbar.
198
3. Thermodynamik
Eine Verdampfung in offener Umgebung ist
eine Verdunstung. Da der Dampf ständig wegtransportiert wird, kann sich kein Phasengleichgewicht bilden, so daß große Mengen
Flüssigkeit verdunsten können. Die aufzuwendende Verdampfungswärme wird zum Teil
der Flüssigkeit entzogen, die sich deshalb abkühlt ( Verdunstungskälte).
sprechend niedrigen Drücken und Temperaturen statt. Diesen Vorgang kann man bei Normaldruck bei Kohlensäureschnee (Trockeneis)
beobachten.
3.4.3.4. Koexistenz dreier Phasen
Der Verlauf der Phasengrenzen zwischen den
drei Aggregatzuständen fest, flüssig und gasförmig in Abhängigkeit von Druck, Temperatur
und Volumen wird durch ein Zustands3.4.3.3. Gleichgewicht zwischen fester
diagramm
beschrieben. Bild 3-47 a zeigt dieund flüssiger Phase
ses dreidimensionale "Gebirge': Bild 3-47 b das
Auch zwischen flüssiger und fester Phase be- p, T-Zustandsdiagramm und Bild 3-47 c das
steht ein Gleichgewicht. Die Schmelztempe- p, T-Zustandsdiagramm für Kohlendioxid. Beratur ist wie bei der Phasenumwandlung flüssonders wichtig sind die Gleichgewichtsgesig- gasförmig nach der Clausius-Clapeyronbiete (Koexistenzgebiete). Die grauen Fläschen Gleichung vom Druck abhängig. Diese
chen in Bild 3-47 a zeigen die GleichgewichtsSchmelzdruckkurve beschreibt die für das
gebiete zwischen Festkörper und Flüssigkeit
Gleichgewicht zwischen fester und flüssiger
(I) , Flüssigkeit und Gas (2) sowie Festkörper
Phase maßgebenden Wertepaare von Schmelzund Gas (3). Außerdem ist der kritische
druckpr und Temperatur T:
Punkt K ersichtlich. Das Flüssigkeitsgebiet
wird oberhalb des kritischen Drucks Pk durch
dpr
die kritische Isotherme Tk gegen das Gasgebiet
(3- 124)
abgegrenzt (gestrichelte rote Linie in Bild
dT
3-47). Die Begrenzungshyperbel am rechten
Hierbei ist D.H ms die molare Schmelzenthal- Bildrand gibt den Übergang zum idealen Gas
an. Am kritischen Punkt K für Kohlendioxid
pie, V~ 1 bzw. v~est das Molvolumen der flüssigen bzw. festen Substanz und T die Schmelz- betragen die Werte für die Zustandsgrößen
temper::;ttur. Die Volumenänderung V~ 1 - v~est Pk = 75 bar und Tk = 304,2 K. An der Sublimationsdruckkurve
von
Kohlendioxid
beim Ubergang vom festen in den flüssigen
läßt
sich
der
Vorgang
der
Subiimation
bei
Zustand ist wesentlich geringer als vom gasförmigen in den flüssigen Zustand. Deshalb Normaldruck zeigen. für den Normdruck
zeigen die Schmelzdruckkurven einen steile- Pn = 1,0 13 bar ergibt sich im Gleichgewicht
aus der Sublimationsdruckkurve die Temperen Anstieg als die Dampfdruckkurven (Bild
3-47). In den meisten Fäll en ist das Volumen ratur T = l95K (.9=- 78°C). Bei dieser
.des festen Körpers v~est kleiner als das Flüs- Temperatur findet ein direkter Übergang vom
festen in den gasförmigen Zustand statt (Subsigkeitsvolumen V~ 1 , so daß die Schmelzlimation).
Im p, T-Zustandsdiagramm gibt es
druckkurve mit zunehmender Temperatur
einen
einzigen
Punkt Tr, in dem die feste, flüssteigt. Bei Wasser dagegen ist das Eisvolumen
Phase im Gleichgewicht
sige
und
gasförmige
größer als das Flüssigkeitsvolumen (Anomalie
stehen.
Er
wird
Tripelpunkt
genannt. Die Kodes Wassers). Dann wird nach GI. (3-124) die
existenz von drei Phasen tritt nur bei einer
Steigung der Schmelzdruckkurve dprld T newohldefinierten Temperatur auf, weshalb der
gativ. Dies hat zur Folge, daß die Schmelztemperatur mit zunehmendem Druck sinkt, so' Tripelpunkt zur Temperaturdefinition geeignet
daß Eis bei gleichbleibender Temperatur ist. Der Tripelpunkt des Wassers ist der Fundurch Druckerhöhung schmilzt. Dieser Effekt damentalpunkt für die Temperaturskala nach
Kelvin. Er liegt bei der Temperatur TT, =
macht Eissportarten, z. B. Schlittschuhlaufen,
273,16 K, der Druck beträgt PTr = 612 Pa. Für
möglich: Infolge des Drucks schmilzt das Eis;
Kohlendioxid betragen die Werte TTr = 216,6 K
wird der Druck weggenommen, dann geund
PTr = 0,52 MPa (Bild 3-47 c).
friert der Wasserfilm wieder.
Der Übergang vom festen in den gasförmigen
Aggregatzustand (Sublimieren) . findet bei ent-
Befinden sich in einem Gefaß mehrere Phasen, dann sind die Zustandsvariablen Druck
3.4. Zustandsänderungen realer Gase
199
p
Bild 3-47. Zustandsdiagramm. a) Dreidimensionales p, V,T-Diagramm (schematisch), b) zweidimensionales
p,T-Diagramm (schematisch).
p Druck, V Volumen, T absolute Temperatur, Tr Tripelpunkt, K kritischer Punkt, 1, 2, 3 Gleichgewichtsgebiete
und Temperatur nicht voneinander unabhängig.
Die Anzahl der Freiheitsgrade f, d. h. die Anzahl der physikalischen Zustandsgrößen, die
frei variiert werden können, sind durch die
Gibbssche Phasenregel gegeben:
f=k+2-P.
(3-125)
1
Es bedeuten hierbei k die Anzahl der unabhängigen chemischen Komponenten und P die
Anzahl der Phasen. Für reines Wasser ist
k = I. Liegt nur eine Phase vor (z. B. die
Gasphase), dann ist P = 1, und es gibt!= 2
Freiheitsgrade. Dies bedeutet, daß die Temperatur und der Druck unabhängig vonein..:
ander variieren können. Liegen aber zwei
Phasen gleichzeitig vor (z. B. entlang der
Dampfdruckkurve), so gibt es nur noch einen
Freiheitsgrad (f = I) ; beispielsweise ist dann
nur die Temperatur unabhängig variierbar.
Im Tripelpunkt liegen alle drei Phasen nebeneinander vor (P = 3). In diesem Fall gibt
es keinen Freiheitsgrad mehr (f = 0), d. h., ·
die physikalischen Zustandsgrößen Druck p
und Temperatur T sind festgelegt.
3.4.4. Dämpfe und Luftfeuchtigkeit
Die Berechnung und Auslegung von Luftzuständen (Konditionierung) ist ein wichtiges
Arbeitsfeld der Klimatechnik und Luft das
technisch wichtigste Dampf-Gas-Gemisch.
Wenn in der Luft Wasserdampf enthalten ist,
200
3. Thermodynamik
MPa
10 2
.§'
~
~
(j
Pk
2
l:j
flüssig
~
-l
10
l?k
Erstarren
&
fest
Tk
= 7,38 MPa
= 304,2 K
= 466,1 k~
m
K
Cl.
~
(..)
:J
0
Kondensieren
PT,= 0,52 MPa
TT,= 216,6 K
..);0
..:§
---#'
s-"
10-1
gasförmig
>:-<:;
~0
'1>
---0.;;if~
170
Bild 3-47c.
Desublimieren
200
230
260
absolute Temperatur T
290
K
320
p, T-Diagrammfor Kohlendioxid.
liegt feuchte Luft vor. Die Aufgabe der Klimatechnik besteht darin, Luftmassen zu befeuchten oder zu trocknen. Nach Bild 3-48
gibt es hierfür drei Möglichkeiten:
- Mischung von Lufimassen,
- Wärmezu- bzw. -abfuhr und
- Wasserzu- bzw. -abfuhr.
Diese Konditionierungskonzepte für Luft werden beispielsweise zur Lösung folgender Aufgaben eingesetzt:
- Auslegung von stationären Klimaanlagen,
- Auslegung der Klimatisierung von Verkehrsmitteln (air condition in Bussen und
Flugzeugen) sowie
- Auslegung von Produktionshallen zur Kunststoffverarbeitung. (Einige Kunststoffe geben
nach zu feuchter Verarbeitung Wasser ab.
Dann schrumpft das Kunststoffteil, es ist
nicht mehr maßhaltig.)
Die zahlenmäßigen Angaben in den folgenden Gleichungen sind auf den Normdruck
(Pn = l ,013 · l 0 5 Pa) bezogen und für den in
der Klimatechnik üblichen Temperaturbereich
zwischen 9 = - 10 °C und 9 = + 40 oc näherungsweise gültig.
Druck der feuchten Luft
Der Druck PFL der feuchten Luft wird unmittelbar an einem Barometer abgelesen (Abschn.
2.11.2.1) und setzt sich nach dem Daltonschen
Gesetz aus der Summe der Partialdrücke
(Druck der trockenen Luft PTL und Druck des
Wasserdampfes p 0 ) zusammen: PFL = PTL +PD·
3.4. Zustandsänderungen realer Gase
~
Befeuchten
Trocknen
Mischung
von
Luftmassen
Zufuhr
feuchter
Luft
Zufuhr
trockener
Luft
Wärme zubzw. -abfuhr
Temperaturabsenkung
Temperaturerhöhung
Wasserzubzw. -abfuhr
Wasserzufuhr
(durch Einsprühen)
Wasserentzug
(durch Abkühlen
unter Taupunkt)
201
g
Bild 3-48.
Aufgaben der Klimatechnik und ihre technische Realisierung.
Absolute Luftfeuchtigkeit
Die absolute Luftfeuchtigkeit rp. ist der Quotient aus der Masse des in der Luft enthaltenen Wasserdampfes m 0 und dem Volumen
der feuchten Luft VFL:
mo
'Pa=--.
(3-126)
VFL
das Beschlagen abgekühlter Spiegel (Taupunktsspiegel) zur Feuchtemessung.
Die fortlaufende Messung der Temperatur
und der relativen Luftfeuchtigkeit ist für die
Überwachung von technischen und baulichen
Anlagen von Bedeutung (z. B. Telefonzentralen oder Kunstausstellungen). Sie kann mit
Thermo-Hygrographen gemäß Bild 3-49 erfolgen.
Relative Luftfeuchtigkeit
Die relative Luftfeuchtigkeit rp ist der Quotient
aus dem Partialdruck des Wasserdampfes Po
und dem Sättigungsdampfdruck des Wasserdampfes Ps (bei der jeweiligen Temperatur):
Po
rp=-.
Ps
(3-127)
1
(Der Wert wird manchmal noch mit 100 multipliziert, und die relative Luftfeuchtigkeit rp
in Prozent angegeben.) Je nachdem, ob die
relative Luftfeuchtigkeit rp < I, rp = I oder
rp > I ist, ist die Luft ungesättigt, gesättigt
oder übersättigt.
Physikalische Effekte, die stark abhängig von
der Feuchtigkeit sind, dienen zur Messung
und Regelung der relativen Luftfeuchtigkeit.
Früher wurde vorwiegend die Längenänderung hygroskopischer Stoffe zur Messung herangezogen. In Feuchtesensoren modernerer
Art nutzt man die Änderung von elektrischen
Eigenschaften (z. B. Widerstands- oder Kapazitätshygrometer), die vom Sättigungsgrad
der Luft abhängige Abkühlung befeuchteter
Thermometer (Aspirationspsychrometer) oder
Bild 3-49. Thermo-Hydrograph.
Werkphoto: Lufft
Feuchtegrad
Unter dem Feuchtegrad x versteht man den
Quotienten aus der Masse des Wasserdampfes
m 0 und der Masse der trockenen Luft mTL:
mo
x=--.
mTL
(3-128)
202
3. Thermodynamik
Der Feuchtegrad kann mit der all gemeinen
Gasgleichung p V= m Ri T in Druckverhältnisse umgerechnet werden; dabei ist für
trockene Luft RiTL = 287 J/kg Kund für Wasserdampf Rio= 462 J/kg K zu setzen.
Dichte der feuchten Luft
Die Dichte der feuchten Luft f>FL setzt sich
aus der Dichte der trockenen Luft f>TL und des
Dampfes Qo 1usammen: f>FL = f>TL + Qo. Wird
das allgemc1ne Gasgesetz verwendet, so ist
f>TL = PTLIRiTL T und Qo = PDIRm T. Nach
dem Da/tonschen Gesetz (GI. (3-119)) ist
PTL = PFL- Po, so daß sich für die Dichte der
feuchten Luft ergibt
- I (PFL -PD
f>FL-T
RiTL
+ -PD-)
Rm
(3-129)
·
Da R i o größer als R iTL ist, ergibt sich nach
GI. (3-129) , daß feuchte Luft leichter ist als
trockene.
Spezifische Enthalpie feuchter Luft
Die spezifische Enthalpie (h = H Im [kJ/kg])
der feuchten Luft hFL ist die Summe aus der
spezifischen Enthalpie der trockenen Luft hTL
und der mit dem Feuchtegrad x multiplizierter spezifischen Enthalpie des Wasserdampfes
ho, also
(3-130)
Setzt man für T0 = 273, 15 K die Enthalpie
willkürlich gleich null, dann gilt nach GI.
(3-52) für die spezifische Enthalpie der trockenen Luft hTL = CpTL (T- T 0 ) und für die des
Wasserdampfes unter Berücksichtigung der
spezifischen Verdampfungsenthalpie 11hv des
Wassers h 0 = Cpo (T- T0 ) + 11hv.
Für klimatechnische Berechnungen geeigneter
ist das Mo//ier-Diagramm (R. MoL LIER, 1863
bis 1935), eine graphische Darstellung der Zusammenhänge von GI. (3~Ü8) bis (3-1 30) zwischen der Temperatur 9 der spezifischen En-
Feuchtegrad x
2
4
6
8
10
12
14
16
18
oc
35
30
25
<!::>
.....,:; 15
Q)
0.
E
Q)
1-
5
0
- 5
-10
-15
1
Bild 3-50. h,x-Diagramm nach M ollier fo r feuchte
Luft beim Druckp = 1013 hPa (VDJ~R ich tlinie 2067,
Blatt 3) .
q> relative Feuchte
Die roten Linien beziehen sich auf Beispiel 3.4-2.
3.5. Wärmeübertragung
thalpie h der fe uchten Luft, der relativen
Luftfeuchtigkeit rp und dem Feuchtegrad x.
Üblicherweise erstellt man das Mollier-D iagram m für Normald ruck gemäß Bild 3-50.
Beispiel
3.4-2: Gegeben sind m = 50 kg feuc hte Luft vom
Umgebungsdruck p = 1,013 · 10 5 Pa mit einer Temperatur 9 = 35 °C und einer relativen Luftfeuchtigkeit cp 1 = 0,5 (50%). Berechnet werden soll die
Wärmemenge, die dieser Luftmasse zu entziehen
ist, um als neuen Luftzustand eine Temperatur
9 2 = 20 °C bei einer relativen Luftfeuchtigkeit von
cp = I (100%) zu erzielen. Ferner soll bestimmt werden, welche Kondenswassermenge hierbei anfällt.
Lösung:
In Bild 3-50 ist dieser Vorgang rot eingezeichnet.
Der Luftzustand I hat einen Feuchtegrad von
x 1 = 17,5 g/kg und eine spezifische Enthalpie von
h 1 = 80 kJ/kg. Da der Feuchtegrad sich bis zur rela~
tiven Luftfeuchtigkeit von cp = 100% nicht ändert,
wird im h, x-Diagramm eine senkrechte Wegstrecke
zurückgelegt. Entlang der Sättigungslinie verläuft der
Prozeß weiter bis zum Zustand 2. Dieser hat einen
Feuchtegrad x 2 = 14,6 g/kg und eine spezifische
Enthalpie h2 =57 kJ/kg. Daraus läßt sich die Kondenswassermenge ~mH,O = ~x m berechnen, wobei
~x = x 1 - x 2 = 2,9 g/kg ist. Somit errechnet sich
~mH ,o = 2,9 ·50 g = 145 g Kondenswasser. Für die
abgeführte Wärmem enge gilt
~H
= (h 2 - h 1) m =
-
kJ
23 - · 50 kg
kg
= -
!550 kJ.
3.5. Wärmeübertragung
Durch die Trennwand zwischen thermodynamischen Systemen mit unterschiedlichen Temperaturen und damit unters'chiedlichen ki netischen Energien wird vom System höherer
Temperatur Wärme an das System mit niedri gerer Temperatur abgegeben. Der Wärmedurchgang läßt sich gemäß Bild 3-51 in die
drei Übertragungsmechanismen Wärmeleitung,
Konvektion und Wärmestrahlung einteilen. In
F estkö:pern tritt nur Wärmeleitung in Form
einer Ubertragung der Schwingungsenergien
benachbarter Moleküle und der kinetischen
Energien der Leitungselektronen in Stoßprozessen auf (Abschn. 9.3. 1). In Flüssigkeiten
kommt es auch ohne von außen aufgeprägter
Zwangsströmung zu Strömungen erwärmter
Teilmengen, zur freien Konvektion. Wird die
Flüssigkeit durch äußere Druckkräfte in Bewegung versetzt, so wird dieser Wärmetransportmechanismus als er::wungene Konvektion
bezeichnet. In stehenden Flüssigkeiten bestimmt die Wärmeleitung den Wärmetransport. Mit Ausnahme dünner ruhender Gasschichten, in denen die Wärmeleitung nicht
vernachlässigbar ist, dominieren in Gasen die
Konvektion und die Wärmestrahlung zwischen
den Wänden des Gasvolumens. Im Vakuum
ist der Wärmetransport durch Wärmestrahlung der einzige Wärmeübertragungsmechanismus.
Konvektion
Wärmeleitung
203
Wärmestrahlung
m
V
e-
ß1
62
61
e-
Energieübertragung gekoppelter
Gitterschwingungen
(Phononentransport) und durch
bewegliche Ladungsträger
(freie Elektronenbewegung)
Bild 3-51.
c('·,
m
W ärm eübertragung durch die
f reie oder erzw ungene Strömung
von Materi e (Massent ransport)
Wärmeü bertragungsmechanismen
··H·,
W ärmeübertragung durch
elektromagnetische Strahlung
(Photonentransport)