T - Chemie Unibas
Werbung

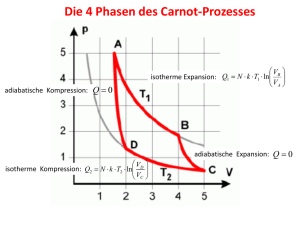
Der Zweite Hauptsatz der TD- Lernziele o Einleitung o Entropie (Definition, Entropie Clausius‘sche Ungleichung) als Zustandsfunktion, die o Der Zweite Hauptzatz der TD o Die Entropieänderungen bei speziellen Prozessen o Der Dritte Hauptsatz der TD o Die Freie Energie und die Freie Enthalpie o Freie Standardreaktionsenthalpien o Die Verbindung von Erstem und Zweitem Hauptsatz (Die Fundamentalgleichung) o Die Eigenschaften der Freien Energie, und der Freien Enthalpie P. Atkins, J. de Paula, “Physikalische Chemie”, Wiley-VCH Verlag GmbH& Co, 2013, 3.1-3.3. G. Wedler, H-J. Freund, „Lehrbuch der Physikalischen Chemie“, Wiley-VCH Verlag GmbH& Co, 2013, 1.1.18-1.1.22. Einleitung Unser Ziel: wann und warum Umwandlungen freiwillig ablaufen? physikalische und chemische Vorgänge: - die freiwillig – von selbst- ablaufen - die nicht freiwillig ablaufen. z. B. Tw Tk Tw > Tk Tg Freiwillige Prozesse Freiwillige Prozesse = Prozesse die mit einer naturgegebenen Neigung, von selbst abzulaufen. Wichtig: in der Praxis können diese Prozesse stattfinden oder auch nicht. ? Der Zweite Hauptsatz der TD (Kelvin Formulierung) : Ein Prozess, bei dem lediglich Wärme aus einem Reservoir entnommen und vollständig in Arbeit umgewandelt wird, ist unmöglich. Die Richtung freiwilliger Prozesse Freiwillige Prozesse sind immer mit einer Disipation von Energie verbunden. z. B. Die Richtung freiwilliger Zustandsänderung für einen Ball der den Boden entlang springt. - bei jedem Aufprall Ekin Ethermisch Ungeordnete Bewegung von Molekülen Die Richtung freiwilliger Prozesse Die ungeordnete Energieverteilung ist nicht von selbst in eine ordnente Bewegung verwandelt. z. B. - Ein Gas zieht sich nicht freiwillig auf ein kleineres Volumen zusammen. - Ein kalter Gegenstand wird nicht spontan wärmer als die Ungebung. - Ein Ball liegt auf einem wärmeren Untergrund (a). ? Das Streben zur grosserer Unordnung ist die treibende Kraft jeglicher Veränderung (zur Verteilung von Energie und Materie). Der Carnot’sche Kreisprozess Wir benötigen ein ideales Gas. 1. Reversible, isotherme Expansion V1, T1 V2, T1 V2 > V1 dU = 0 dT = 0 QT1 = Wärmemenge wird aus dem Wärmebad entnommen. V2 3. Reversible, isotherme Kompression V3, T2 V4, T2 V3 > V4 dU = 0 dT = 0 QT1 = Wärmemenge wird an die kalte Umgebung abgegeben. V3 V4 V1 2. Reversible, adiabatische Expansion V2, T1 V3, T2 V3 > V2 und T2 < T1 dQ = 0 γ −1 γ −1 T 1V 2 = T 2V 3 4. Reversible, adiabatische Kompression V4, T2 V1, T1 V4 > V1 und T2 < T1 dQ = 0 γ −1 γ −1 T 1V 1 V4 V3 V2 = T 2V 4 V1 Der Carnot’sche Kreisprozess Geleisteten Volumenarbeiten ergeben sich als Fläche zwischen den Isothermen und Adiabaten: V2 W 1 = − nRT 1 ln V1 W 2 = nC V , m (T 2 − T1 ) V3 V4 W 4 = nC V , m (T1 − T 2 ) W 3 = nRT 2 ln Die gesamte Arbeit : W = nR ln V2 (T 2 − T1 ) = nR ln V 2 ∆ T V1 V1 W <0 V1 < V2 und T2 < T1 Wärmekraftmaschine Der Carnot’sche Kreisprozess Die Aenderung der Inneren Energie bei dem gesamten Kreisprozess: ∆ U = nc V , m (T 2 − T1 ) + nc V , m (T1 − T 2 ) = 0 Bei den Isothermen ∆U = 0 Bei den adiabatischen Schritten eintretenden Aenderungen kompensieren. Q = nRT 1 ln V2 V3 = V1 V4 V V2 − nRT 2 ln 3 V1 V4 Q = nR ln V2 (T1 − T 2 ) > 0 V1 Das Arbeitgas entnimmt einem Thermostaten hoher Temperatur eine Wärmemenge und leisted eine Arbeit (gibt auch eine restliche Wärmemenge an einen Thermostaten tieferer Temperatur). Wärmekraftmaschine / Wärmepumpe Kann man einen höheren Wirkungsgrad mit einer anderen Maschine erreichen? Maschine dann als Wärmepumpe benützen, um einen CarnotProzess anzutreiben! T1 Annahme: Der Wirkungsgrad von A ist höher als der, der Maschine B ! W QT1 QT1 w‘‘ Q T Q QT2 2 QT2 T 2 T2 Da der Wirkungsgrad der Maschine A höher ist, wird von A mehr Arbeit produziert als von der Maschine B aufgenommen wird, um die Wärmemenge QT1 wieder dem warmen Reservoir zuzuführen!‘ X Wärmekraftmaschine Die Wärmekraftmaschine : - das Arbeitsgas entnimmt dem wärmeren Reservoir die Wärmemenge QT1, - leistet die Arbeit W - führt dem kleineren T Reservoir die Wärmemenge T1 QT2 = QT1 – W zu QT1 Das Carnotische Kreisprozess: - Aufnahme einer QT1 von einem Thermostaten (T1) - Abgabe einer QT2 an einem Thermostaten (T2) Arbeit ist gewonnen ! QT2 T2 Die Wärmekraftmaschine Wärmepumpe Der Prozess läuft in entgegengesetzter Richtung: Die Wärmepumpe: - das Arbeitsgas entnimmt dem kälteren Reservoir die Wärme QT2 - pumpt sie mit kleinerem Arbeitsaufwand w‘ (als gesamte Wärmemenge QT1) in das wärmere Reservoir Eine gegenüber der ursprünglichen Wärmemenge QT1 - um den Arbeitsbetrag w‘ verminderte Wärmemenge - fliesst zurück ins wärmere Reservoir! Wirkungsgrad der Wärmekraftmaschine Wirkungsgrad η der Wärmekraftmaschine = geleistete Arbeit geteilt durch die aufgenommene Wärme η= w QT1 V2 −nR(T1 − T2 ) ln w V1 T1 − T2 T η= = = = 1− 2 V T1 T1 QT1 nRT1 ln 2 V1 Da 0 < T2 < T1 0<η<1 Carnot’scher Kreisprozess Bei diesem Kreisprozess würde Wärme in Arbeit umgewandelt, ohne dass irgendwelche Veränderungen eintreten Widerspruch zur Erfahrung. 2. Hauptsatz der TD: Es gibt keine Wärmekraftmaschine mit höherem Wirkungsgrad, als den Carnot-Prozess! An dem reversibel durchgeführten Carnot‘schen Kreisprozess lässt sich eine interessante Funktion ableiten! Reduzierte Wärme Betrachte: Mit der Umgebung ausgetauschte Wärmemenge beim Carnot‘schen Prozess (Verhältnis, der mit der Umgebung ausgetauschten Wärmemenge) V nRT1 ln 2 Q V1 T1 − T1 = = QT2 T2 V nRT2 ln 3 V4 d.h. V2 = nRln T1 V1 QT1 V4 V2 und = nRln = −nRln T2 V3 V1 QT2 Q QT1 QT2 = + =0 ∑ T1 T2 rev T Die Summe der reduzierten Wärmen Q/T ist null beim Carnot‘schen-Prozess! Ist die reduzierte Wärme eine Zustandsfunktion? Wenn Q/T eine Zustandsfunktion ist, muss ihre Aenderung unabhängig vom Weg sein! z. B. Carnot’scher Kreissprozess 1+2 A Zustand p1, V1, T1 C Zustand p2, V3, T2 p2, V2, T2 3+4 Q V2 = nRln ∑T V1 1+2 Q V V ∑ T = −nRln V3 = −nRln V2 4 1 3+4 Die reduzierte Wärme ist eine Zustandsfunktion Beliebige Kreissprozesse Man kann jeden beliebigen reversiblen Kreisprozess näherungsweise in eine Anzahl von Carnot-Prozessen zerlegen. Zweckmässiger Weisse führt man statt summieren eine Integration durch ∫ dQrev =0 T oder ∫ dQirr >0 T Q/T ist ein Mass für die Irreversibilität eines Vorgangs! Die Entropie Die Entropie (S) ist ein Mass für die Disipation der Energie bei einem bestimmten Prozess. Entropie ist eine Grösse, die die Richtung freiwillig (spontan) ablaufender Vorgänge vorhersagt! Der Begriff Entropie = tropé (Wendung) und entrépein (umwenden, eine Richtung geben) Die Entropieformulierung beschreibt welche der erlaubten Zustandsänderung freiwillig ablaufen Bei einer freiwilligen Zustandsänderung eines abgeschlossenen Systems: ∆Sgesamt > 0 TD Definition der Entropie Die Uebertragung von Wärme: thermische Bewegung der Teilchen der Umgebung. Die Uebertragung von Energie in Form von Arbeit: geordnete Bewegung der Teilchen der Umgebung. dQrev = dS T Thermodynamische Definition der Entropie: Für endliche Zustandsänderung: E dQrev T A ∆S = ∫ TD Definition der Entropie z. B.: freie Expansion eines Gases Gesamtenergie (Innere Energie) bleibt konstant, aber die Verteilung der Gasmoleküle ändert sich! ‚Qualität‘ der Energie nimmt ab! ‚Spontane Vorgänge sind eine Folge des Strebens der Welt nach grösserer Unordnung‘ Einheit: [S ] = JK-1 S ist eine extensive Eigenschaft. Entropie z. B. Beim schmelzen von Eis: die geordnete Eiskristallstruktur wird in eine regellose Bewegung einzelner Wassermoleküle überführt. Energie und Entropie vom Getränk werden auf die Wassermoleküle des Eiswürfels übertragen. Eigenschaften der S: Die Entropie bleibt nur dann unverändert, wenn die Prozesse reversibel verlaufen (für ein Kreisprozess). Reale Zustandsänderungen sind immer mit Energieverlusten (z. B. durch Reibung) verbunden, wodurch sich die Entropie erhöht. Eine Verringerung der Gesamtentropie in einem geschlossenen System ist nicht möglich. Mikroskopische Interpretation der Entropie Die Energie eines Atoms oder Moleküls kann nur bestimmte Werte annehmen die Energienivaus T > 0 → die Molekule einer Probe sind auf alle verfügbaren Energienivaus verteilt Es ist nicht möglich, den Energiezustand eines einzelnen Moleküls zu verfolgen Aussage über die mittlere Anzahl der Moleküle in jedem Zustand Mikroskopische Interpretation der Entropie Boltzman Verteilung die mittlere Anzahl der Molekule in jedem Zustand Population der Moleküle – für jeden Zustand T=0 Nur der Zustand niedrigster Energie ist besetzt T ↗ Mehr Zustande sind für die Moleküle erreichbar Ni = N exp (-Ei /kBT) / ∑ exp (-Ei /kBT) Anzahl der Moleküle einer Probe (N Moleküle) → die Energiezustand Ei besetzen (bei T – System in thermischem Gleichgewicht) kB = 1,3807·10-23 J/K (die Boltzmann Konstante) Mikroskopische Interpretation der Entropie Boltzmann Formel Die Verbindung zwischen der Verteilung der Moleküle auf die Energienivaus und die Entropie S = k ln (W) W – die Anzahl der Mikrozustände (die unterschiedlichen Möglichkeiten, die Moleküle bei gegebener Gesamtenergie eines Systems auf die zur Verfügung stehenden Energienivaus sich verteilen). Mikroskopische Interpretation der Entropie Die Entropie eines Makrozustands → statistisches Gewicht W (die Anzahl seiner Mikrozustände). n Molekule auf zwei Raumhälften: n1 – einer Hälfte, n2 – andere Hälfte. n ist ein ganzes Mol (6,0220·1023 Mol-1 ) und Bei gleichmässiger Aufteilung: Stirling Formel: Damit wird die Entropie nach der Expansion Weitere PCi Vorlesung ? Aktuelle Endekungen Der TD Zeitpfeil beruht auf dem Zweiten Hauptsatz der TD: Die Zukunft ist die Zeitrichtung, in der die Entropie zunimmt. Ein interessanter Punkt ist, dass dieser Zeitpfeil im thermodynamischen Gleichgewicht nicht existiert: Für einen Gleichgewichtszustand gibt es keine thermodynamisch definierte Vergangenheit und Zukunft; der Gleichgewichtszustand ist sozusagen zeitlos. Die Zukunft ist die Richtung des grösseren Universums (die Entropie nimmt zu!). 17.03.2014: Astrophysiker haben in der Kosmischen Hintergrundstrahlung erstmals Hinweise auf Gravitationswellen endeckt (Kosmische Inflation) Entropie: Zustandsfunktion Entropie: Zustandsfunktion, die anzeigt, ob ein Zustand von einem anderen aus, spontan erreichbar ist. Entropie ist ein Bindeglied zwischen makroskopischer und mikroskopischer Welt! 2. Hauptsatz: Ein Prozess, bei dem lediglich Wärme aus einem Reservoir entnommen und vollständig in Arbeit umgewandelt wird, ist unmöglich! 2. Hauptsatz: Bei einer freiwilligen Zustandsänderung nimmt die Entropie eines abgeschlossenen Systems zu! Die Clausius’sche Ungleichung Reversible Prozessbedingungen → mehr Energie (in Form von Arbeit) als bei irreversiblen Prozessen - dwrev ≥ - dw → dw – dwrev ≥ 0 Die Aenderung der U zwischen A und E: d U = dQ + dw = dQrev + dwrev dQrev – dQ = dw – dwrev ≥ 0 dQrev/T ≥ dQ/T d S = dQrev/T dQ = 0 (abgeschlossenes System) ← dS ≥ dQ/T dS ≥ 0 Die S eines abgeschlossenen Systems freiwilligen Zustandsänderung kann nicht abnehmen (der zweite Hauptsatz der TD). Entropieänderung bei speziellen Prozessen Freiwillige Abkühlung 1. Einem warmen Reservoir wird eine Wärmemenge entzogen → sinkt seine Entropie. (Führt man dieselbe Wärmemenge zu) 2. Einem kalten Reservoir → steigt dessen Entropie um einen grösseren Wert. Daher nimmt die Entropie während des Prozesses insgesamt zu, und dieser läuft freiwillig ab. dS = dQ/Tw dQ dS = dQ/Tk Entropieänderung bei speziellen Prozessen Freiwillige Abkühlung dS ≥ dQw/Tw + dQk/Tk dS = dQ/Tw dQw = - dQk dS ≥ -dQk/Tw + dQk/Tk = dQk (1/Tk – 1/Tw) dQ dQk > 0 dS ≥ 0 → die Abkühlung ist immer freiwillig dS = dQ/Tk Nimmt S während des Prozesses insgesamt zu Der Prozess läuft freiwillig ab. Entropieänderung bei speziellen Prozessen Isotherme Expansion Die logarithmische Zunahme der Entropie eines idealen Gases bei der isothermen Expansion. ∆U = Qrev + wrev = 0 ⇒ Qrev = - wrev dS = dQrev /T E 1 Q ∆S = ∫ dQrev = rev T A T Isotherme Expansion → wrev = - nRT ln(VE/VA) ∆S = nR ln VE VA S ist eine Zustandsfunktion → die Gleichung kann für reversible/irreversible Zustandsänderung angewendet werden. Entropieänderung bei speziellen Prozessen Phasenübergänge Sieden / Erstarren: Ttrans: Eine T wo sich zwei Phasen (bei 1 bar) miteinander im Gleichgewicht befinden Wärmeaustausch zwischen beiden Phasen → reversibel p = konstant Q = ∆trans H ∆ S = ∆transH / Ttrans Exotherme Phasenübergänge: ∆trans H < 0 ∆S<0 ( bei diesen Phasenübergängen können Energie und Materie lokalisiert werden) Endotherme Phasenübergänge: ∆trans H > 0 ∆S>0 (Energie und Materie werden verteilt) Entropieänderung bei speziellen Prozessen Temperaturänderungen E dQrev T A ∆S = ∫ S kann berechnet werden (bei einer beliebigen T) E S(TE) = S(TA) + z.B. p konstant dQrev ∫A T dQrev = cpdT E S(TE) = S(TA) + ∫ c p dT A T (keine Expansionarbeit) + cp konstant S(TE) = S(TA) + cp ln(TE/TA) Entropieänderung bei speziellen Prozessen Temperaturänderungen Analog (Austauch von cp gegen cv) gilt die Gleichung für konstantes Volumen Die Entropie eines Systems der Temperatur T → kann auf seine Entropie bei T = 0 zurückgeführt werden cp bei verschiedenen T gemessen und das Integral auswertet Man muss die jeweilige Phasenübergängsentropie ∆trans H/T addieren. Entropieänderung bei speziellen Prozessen Temperaturänderungen Beispiel: Eine Substanz schmilzt bei TSm und siedet bei Ts Die Entropie entspricht der Summe aus der Fläche unter der oberen Kurve (bei cp/T = f(T)) und den Entropien aller durchlaufenen Phasenumwandlungen S (T ) = S (0 ) + TSm ∫ 0 TS c p (l ) c p (g ) c p (s ) ∆ Sm H ∆ H dT + + ∫ dT + V + ∫ dT T TSm T T T S TSm TS T Phasenübergänge müssen berücksichtigt werden.