Nachrichten von der Suzuki-Reaktion
Werbung

Zuerst ersch. in: Nachrichten aus der Chemie 50 (2002), S. 1122-1127
1122
Valentin Wittmann
Neues von der Suzuki-Reaktion
Gut zwei Dekaden nach ihrer Entdeckung erfreut sich die Suzuki-Reaktion immer größerer
Beliebtheit. Neue Katalysatoren verkürzen den Zugang zu den Edukten und ermöglichen
die Reaktionsführung bei Raumtemperatur.
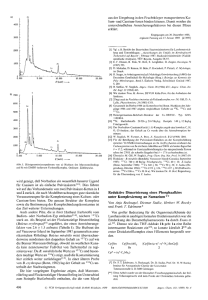
Abb. 1.
a) Die SuzukiMiyaura-Reaktion
und b) der zugrundeliegende Katalysezyklus. Die PdKomplexe sind
ohne Hilfsliganden
formuliert.
Die Knüpfung von C-C-Bindungen bildet das Fundament der organischen Synthese. Hierbei sind katalytische Varianten1) aus ökonomischer und ökologischer Sicht besonders bedeutend.
Die Suzuki-Miyaura- oder kurz
Suzuki-Reaktion,2) die zusammen
mit Heck- und Stille-Reaktion zu
den gebräuchlichsten Kreuzkupplungen zählt, wurde Ende der siebziger Jahre entwickelt und gestattet
die Kupplung von Aryl- und Alkenylhalogeniden, -triflaten und Enolphosphaten mit Aryl-, Alkenyl- und
Alkylboranen in Gegenwart einer
Base und eines Pd0-Katalysators
(Abbildung 1a). Im Gegensatz zu
Heck- und Stille-Reaktion sowie zu
anderen Kreuzkupplungen mit metallorganischen Verbindungen er-
laubt sie neben der Knüpfung von
(Csp2)-C(sp2)- auch die von
C(sp2)-C(sp3)-Bindungen. Diese Variante, bei der primäre Alkylborane
zum Einsatz kommen, nennt man
Eine
B-Alkyl-Suzuki-Reaktion.3)
weitere Stärke der Suzuki-Reaktion
liegt in den vielseitigen und zumeist
schonenden Verfahren zur Darstellung von Organoborverbindungen
sowie deren Eigenschaften. Insbesondere Boronsäureester sind unkompliziert zu handhaben und thermisch sehr stabil. Aufgrund ihrer
schwachen Nucleophilie sind sie mit
zahlreichen funktionellen Gruppen
kompatibel, was sie gerade zur Konstruktion komplexer Moleküle attraktiv macht. Einschränkend sind
in diesem Zusammenhang allerdings
die basischen Reaktionsbedingungen und die oft hohen Reaktionstemperaturen der Suzuki-Kupplung.
Schließlich schlägt die Toleranz der
Reaktion gegenüber Wasser sowie
die geringe Toxizität der Bor-Nebenprodukte positiv zubuche.
Jüngste Publikationen über eine
direkte Darstellung von Arylboronsäureestern
durch
katalytische
C-H-Aktivierung sowie neue Katalysatoren für die Suzuki-Reaktion waren der Anlass, diese Kreuzkupplung
zum Thema des „Blickpunkts“ zu
machen.
Der Katalysezyklus
Wie auch bei anderen Kreuzkupplungen besteht der Katalysezyklus2) (Abbildung 1b) aus der Abfolge von drei Prozessen: oxidative
Addition des Halogenids R1-X an
den Palladium(0)-Katalysator (1)
unter Bildung der Palladium(II)Spezies (2), Transmetallierung zur
Organopalladium(II)-Verbindung (3)
sowie reduktive Eliminierung unter
Bildung des Kupplungsprodukts
R1-R2 und Regeneration des Katalysators (1). Alkylhalogenide sind keine gebräuchlichen Substrate für die
Suzuki-Reaktion, da die oxidative
Addition mit ihnen langsam verläuft. Während die oxidative Addition und die reduktive Eliminierung
recht gut verstanden sind, ist über
den Mechanismus der Transmetallierung, die durch Zugabe einer Base
wie Natriumhydroxid oder -alkoxid
beschleunigt wird, weniger bekannt.
Die Base kann dabei an verschiedenen Stellen in die Reaktion eingreifen. Einerseits kann sie das Boran
2
1
R BY2, das selbst nicht mit R PdX
(2) reagiert, in den at-Komplex
[R2B(OR)Y2]– überführen. Hierdurch wird die Nucleophilie der organischen Gruppe R2 so weit erhöht,
dass eine Übertragung auf Palladium
unter Bildung von (3) erfolgt (Weg
A). Andererseits kann das Halogen X
in (2) durch Hydroxid oder Alkoxid
substituiert werden, wodurch der
hoch reaktive Palladium(II)-Komplex (4) gebildet wird. Dieser reagiert aufgrund seiner Basizität und
der hohen Oxophilie des Borans mit
letzterem ohne weitere Unterstützung durch Base zu (3) (Weg B).
Welcher Weg dominiert, ist abhängig von den Substituenten am Bor
und in vielen Fällen nicht geklärt.
Alkoxyreste am Bor beispielsweise
Konstanzer Online-Publikations-System (KOPS)
URL: http://www.ub.uni-konstanz.de/kops/volltexte/2008/5544/
URN: http://nbn-resolving.de/urn:nbn:de:bsz:352-opus-55448
1123
verringern dessen Lewis-Acidität
und damit die Tendenz zur Bildung
des at-Komplexes. In diesen Fällen
dominiert Weg B.
Abb. 2.
Anwendung der Suzuki-Reaktion zur
Totalsynthese von
a) Palytoxin4) und
b) CP-263,114.6)
Einsatz in der Naturstoffsynthese
Obwohl mechanistisch nicht in
allen Details verstanden, kam die
Suzuki-Reaktion in vielen Naturstoffsynthesen zum Einsatz. Ein
mittlerweile schon klassisches Beispiel ist die Verknüpfung der Fragmente (5) und (6) zu (7) im Verlauf
von Kishis Palytoxinsynthese (Abbildung 2a).4) In dieser Arbeit wurde
auch der positive Effekt der Base
Thalliumhydroxid auf die SuzukiReaktion beschrieben. Für die Herstellung von CP-263,114,5) einem
Pilzmetabolit, der sowohl die Squalensynthase als auch die Farnesyltransferase inhibiert, erarbeiteten
Danishefsky et al.6) eine Synthesesequenz unter Einsatz der B-AlkylSuzuki-Reaktion (Abbildung 2b).
Zunächst wurde das terminale Alken
(8) mit 9-BBN hydroboriert und anschließend an Vinyliodid (9) mit
[PdCl2(dppf)] als Katalysator gekuppelt. Katalysatoren mit derartigen Bisphosphin-Liganden haben
sich oft in Fällen bewährt, in denen
in der Spezies (3) Palladium an ein
sp3-C-Atom gebunden ist, das über
Wasserstoffatome in b-Stellung verfügt und in denen somit eine b-Hydrid-Eliminierung in Konkurrenz
zur reduktiven Eliminierung steht.
Der Bisphosphin-Ligand erzwingt
eine cis-Stellung der Reste R1 und R2
im quadratisch-planaren Komplex
(3). Hierdurch sowie aufgrund des
in derartigen Komplexen besonders
großen (P-Pd-P)-Winkels, der R1
und R2 einander näher bringt, wird die
reduktive Eliminierung begünstigt.
(9-BBN = 9-Borabicyclo[3.3.1]nonan,
dppf = 1,1'-Bis(diphenylphosphino)ferrocen, PMB =
p-Methoxybenzyl,
TBS = tert-Butyldimethylsilyl, Teoc =
2-(Trimethylsilyl)ethyloxycarbonyl)
Darstellung der Borane
Während sich Alkyl- und Alkenylborverbindungen leicht durch Hydroborierung von Alkenen und Alkinen erhalten lassen, ist die Darstellung von Arylborverbindungen aufArylboronsäureester
wendiger.7)
werden üblicherweise ausgehend
von Halogenaromaten über die entsprechenden Grignard-Verbindungen oder – besonders effizient8) –
Aryllithiumverbindungen und deren
Reaktion mit einem Trialkoxyboran
synthetisiert (Abbildung 3a). Die Arbeitsgruppen um Smith III9) sowie
Ishiyama, Miyaura und Hartwig,10)
die sich schon seit längerem mit der
Übergangsmetall-katalysierten Aktivierung von C-H-Bindungen beschäftigen, haben nun unabhängig
voneinander eine Abkürzung für
den aufgezeigten Weg gefunden. Mit
Iridium-Katalysatoren gelang es,
Benzolderivate in einem Schritt ohne Lösungsmittel in guten bis aus-
1124
Abb. 3.
a) Konventionelle
und b) Iridiumkatalysierte Borylierung von Aromaten nach Smith III.9)
c) Eintopfsequenz
aus Borylierung
und Suzuki-Reaktion.9) (COD =
1,5-Cyclooctadien,
dppe = 1,2-Bis(diphenylphosphino)ethan, Ind =
Indenyl, Pin =
-OC(Me)2C(Me)2O-)
Abb. 4.
Iridium-katalysierte
Borylierung von
Aromaten nach
Ishiyama et al.10)
(bpy = 2,2‘-Bipyridin, COE =
Cycloocten, dtbpy =
4,4'-Di-tert-butyl2,2'-bipyridin)
gezeichneten Ausbeuten in die entsprechenden Arylboronsäureester zu
überführen (Abbildungen 3 und 4).
Smith et al.9) führten die Reaktionen
mit Pinakolboran (12) in Gegenwart
von 2 Mol-% einer Mischung aus einem Iridium(I)-Vorläufer, meist
(4-1,5-Cyclooctadien)(5-indenyl)iridium, und einem Bisphosphin-Liganden, meist 1,2-Bis(diphenylphosphino)ethan (dppe) aus (Abbildung 3b). Für die Derivatisierung
von Benzol (11a) konnte die Dosierung des Katalysators bis auf 0,02
Mol-% reduziert werden. Nach einer
verlängerten Reaktionszeit von 61 h
betrug die Ausbeute hier immer
noch 90 %, woraus sich eine beachtliche Umsatzzahl (turnover number
= TON) von 4500 berechnet. Den
mechanistischen Vorstellungen der
Autoren zufolge ist der Iridium(III)Komplex (14) die für die C-H-Aktivierung verantwortliche Spezies.
Substituenten in (11) verhindern
aus sterischen Gründen die Zweitsubstitution in ortho-Stellung. Die
Iridium-katalysierte Borylierung ist
hoch chemoselektiv für ArylC-H-Bindungen. Viele andere funktionelle Gruppen, darunter benzylische C-H-Bindungen, C-HalogenBindungen, Arylether sowie Ester
sind stabil unter den Reaktionsbedingungen.
Iridium-Nebenprodukte
sollten also nicht bei Folgereaktionen
der Arylboronverbindungen stören,
was Smith et al. ermöglichte, die Borylierung im Eintopfverfahren mit einer Suzuki-Reaktion zu koppeln (Abbildung 3c). Zunächst wurde Dichlorbenzol (11b) zu (13b) boryliert
und dann mit 3-Bromtoluol in das Biphenyl (15) überführt, das in einer
Ausbeute von 80 % isoliert wurde.
Das Ishiyama-Miyaura-Hartwig10)
Team setzte Diboran (17) als Borylierungsreagenz in Verbindung mit
[{IrCl(COD)}2] und 2,2‘-Bipyridin
(bpy) ein, um Benzolderivate wie
(16a) – (16d) in die Arylboronsäureester (18a) – (18d) zu transformieren (Abbildung 4, Bedingung A). Die
Chemo- und Regioselektivitäten decken sich mit den Befunden von
Smith. Konkurrenzexperimente zeigten, dass elektronenarme Aromaten
reaktiver als elektronenreiche sind.
1125
Einer NMR- und GC/MS-Studie mit
deuteriertem Benzol zufolge ist der
Reaktion eine Induktionsphase vorgelagert, in der Cyclooctadien zu
Cycloocten reduziert wird. Daher ersetzte das Team Cyclooctadien im
Katalysatorvorläufer durch Cycloocten und verwendete 4,4‘-Di-tertbutyl-2,2‘-bipyridin (dtbpy) anstelle
von Bipyridin zur Steigerung der Löslichkeit. So wurde ein noch aktiverer
Katalysator erhalten, mit dem die
Borylierung zum ersten Mal bei
Raumtemperatur in weniger als fünf
Stunden gelang (Abbildung 4, Bedingung B). Bei 100 °C werden Umsatzzahlen von 8000 erreicht.
Schließlich glückte auch die Isolierung und Kristallisation der für die
C-H-Aktivierung vermutlich verantwortlichen
Iridium(III)-Spezies
(19), die stark dem von Smith postulierten Iridium(III)-Komplex (14)
ähnelt.
Neue Liganden für die SuzukiReaktion
Der vielfach bei Suzuki-Kreuzkupplungen eingesetzte Katalysator
[Pd(PPh3)4] ist mit einer Reihe von
Nachteilen behaftet. Der Komplex
ist luft- und lichtempfindlich, und
aufgrund seiner begrenzten Reaktivität sind oft hohe Katalysatormengen (1– 10 Mol-%) notwendig. Bei
Verfahren im industriellen Maßstab
bringt dies immense Kosten und
Probleme bei der Katalysatorrückgewinnung mit sich. Auch eignet
sich [Pd(PPh3)4] nur bedingt für
Abb. 5.
a) Einsatz von TrialkylphosphinLiganden zur
chemoselektiven
Suzuki-Reaktion
nach Fu et al.14)
b) Sterisch gehinderte Suzuki-Kupplung nach Buchwald et al.15) (Cy =
Cyclohexyl, dba =
trans,transDibenzylidenaceton)
Umsetzungen mit den ökonomisch
attraktiven, aber reaktionsträgen
Chloraromaten, z. B. zur Darstellung
von pharmazeutisch bedeutsamen
Biaryl-Derivaten. Die Suche nach
neuen elektronenreichen Liganden
für Palladium, die zu hoch reaktiven
Katalysatoren führen, 11) hat mittlerweile zu Kombinationen aus einer
PdII-Vorstufe und sterisch gehinderten basischen Liganden wie Tri-tertbutylphosphin, Dialkylarylphosphinen und N-heterocyclischen Carbe-
nen, aber auch zu Palladacyclen geführt. Man geht davon aus, dass die
PdII-Vorstufe unter Bildung von koordinativ ungesättigten Komplexen
mit 16 (PdL3), 14 (PdL2) und 12
Elektronen (PdL), die die eigentlichen Katalysatoren bilden, reduziert
wird.12) Im Folgenden werden einige
neuere Arbeiten dazu vorgestellt. 1126
Abb. 6.
Neue Pd-Katalysatoren für die Suzuki-Reaktion.16–18)
Einsatz des N-heterocyclischen Carbenkomplexes (30)
in einer Kreuzkupplung bei Raumtemperatur.19)
Auf der Suche nach einem allgemein anwendbaren Protokoll für
Suzuki-Reaktionen bei Raumtemperatur setzten Fu et al. die Trialkylphosphine PtBu3 und PCy3 ein.13)
Später fanden sie heraus, dass sich
unter bestimmten Bedingungen
Arylchloride neben Aryltriflaten
(und umgekehrt) umsetzen lassen,14) ein bemerkenswertes Ergebnis, da Aryltriflate normalerweise,
ähnlich Arylbromiden, wesentlich
reaktiver als die Chloride sind. So
ließ sich das Chlorphenyltriflat (20)
mit Boronsäure (21) in sehr guten
Ausbeuten wahlweise in das Triflat
(22) oder das Chlorbiphenyl (23)
überführen (Abbildung 5a). Buchwald et al. entwickelten eine Reihe
von Dialkylarylphosphin-Liganden,
die sich als sehr effizient bei Umsetzungen mit Arylchloriden bei Raum-
Gefährliche KaliumhydroxidMonohydrat-Kruste
Wir sind vor kurzem beauftragt
worden, ehemals im Handel erhältliches Kalium zu untersuchen,
dessen Manipulation und Handhabung vor einigen Jahren einen
tödlichen Unfall verursachte. Das
Kaliummetall wurde vakuumverpackt geliefert (eingeschweißt in
Polyethylen(PE), high-density,
0,2 mm Stärke) und nicht unter Öl,
wie sonst meist üblich. Der Unfall
(starke Explosion) ereignete sich,
als das Kalium in Petroleum-Benzin gegeben und bei ungefähr
90°C unter Argon geschmolzen
wurde. Das Metall war von einer
mehrere Millimeter starken gelblich-weißen Kruste bedeckt (10 –
70 Gew.-%!). Die Vermutung lag
nahe, dass die explosive Reaktion
des Kaliums mit dem organischen
Petroleum-Benzin auf Peroxid-Anteile in der Kruste – dafür gibt es
eine Reihe von Präzedenzfällen –
Kaliumbrocken (ca. 4 x 3 x 2 cm), überzogen mit einer gefährlichen Kruste aus Kaliumhydroxid-Monohydrat (KOH·H2O) und
Kaliumhyperoxid (KO2).
zurückzuführen sei. Jedoch zeigten
unsere vollständigen Untersuchungen durch chemische Analyse (Titration, ICP), IR- und
Raman-Spektroskopie, Massenspektrometrie, DTA-Analyse sowie
Röntgenpulver-Diffraktometrie,
dass die Kruste zu ca. 80 – 90 %
aus Kaliumhydroxid-Monohydrat
(KOH·H2O) und Kaliumhydroxid
(KOH) bestand und zu ca. 10 –
15 % aus Kaliumhyperoxid (KO2,
sowohl CaC2-Typ als auch NaCl-
temperatur erwiesen. Jüngst stellten
sie nun den Phenanthren-basierten
Liganden (26) vor,15) mit dem z. B.
die Kreuzkupplung von Mesitylbromid (24) und Boronsäure (25) zum
sterisch gehinderten tetra-ortho-substituierten Biphenyl (27) in einer
Ausbeute von 82 % gelingt (Abbildung 5b). Erwähnenswert im Zusammenhang mit den Buchwald-Liganden ist auch der von Beller et al.
beschriebene (1,6-Dien)palladium(0)monophosphin-Komplex (28) (Ab16)
bildung 6). Durch das Dien wird
ein molekular definierter Palladium(0)-Komplex mit genau einem
Phosphinliganden stabilisiert. Da
der mit Nebenreaktionen behaftete
Schritt zur Bildung koordinativ ungesättigter Pd0-Spezies entfällt, ist
dieser Katalysator effizienter als PdIIPräkatalysatoren.
Palladacyclen17) und N-heterocyclische Carbenkomplexe (NHCKomplexe)18) wie (29) bzw. (30)
sind relativ neue Klassen von Palladiumkatalysatoren, die sich nicht
Typ, Hochtemperaturphase). Die
Menge an Kaliumperoxid (K2O2)
lag unterhalb der Nachweisgrenze. Daraus folgern wir: Kalium
kann, wenn es in unzulänglicher
Weise gelagert wird, mit einer gefährlichen Peroxidkruste bedeckt
sein. Selbst wenn diese Kruste
fehlt, kann das Metall aber mit einer sehr gefährlichen Kruste aus
Kaliumhydroxid-Monohydrat
(KOH·H2O) überzogen sein, die
Wasser nach dem Schmelzen bei
ungefähr 110 – 140 °C freisetzt,
das mit dem flüssigen Kalium dann
explosiv reagieren kann. Fazit: Die
Vakuumverpackung von Kalium in
PE-Beuteln ist keine sichere und
empfehlenswerte Methode für die
Lagerung und den Transport dieses Materials.
Jürgen Evers, Thomas M. Klapötke,
Gilbert Oehlinger
Department Chemie,
Universität München (LMU)
[email protected]
1127
nur bei Suzuki-Reaktionen bewährt
haben. Beide zeichnen sich durch
hohe thermische Stabilität und große Umsatzzahlen aus. Die Palladacyclen durchlaufen vermutlich einen
PdII/PdIV-Katalysezyklus. Der NHCKomplex (30) ist das bisher aktivste
Kupplungsreagenz, um Chlorarene
in der Suzuki-Reaktion zu Biphenylen umzusetzen.19) So reagiert Chlortoluol (31) bei Raumtemperatur in
weniger als sechs Stunden mit Phenylboronsäure (32) in quantitativer
Ausbeute zu Biphenyl (33) (Abbildung 6).
Zusammenfassung und Ausblick
Wie die zahlreichen Anwendungen zeigen, ist die Suzuki-Reaktion
außerordentlich nützlich zur Konstruktion von Kohlenstoffgerüsten.
Gegenüber vergleichbaren Kreuzkupplungen zeichnet sie sich durch
eine einfache Zugänglichkeit der benötigten Borane, deren Kompatibilität mit zahlreichen funktionellen
Gruppen und die Toleranz gegenüber Feuchtigkeit aus. Die Iridiumkatalysierte Borylierung von Aromaten könnte für Arylboronsäureester
dieselbe Bedeutung erlangen wie die
Hydroborierung für Alkyl- und Alkenylborane. Nahe liegende Entwicklungen sind die weitere Verbesserung der Borylierungs- und Kreuzkupplungskatalysatoren, um zu
noch höheren Umsatzzahlen und
Umsatzfrequenzen, insbesondere bei
sterisch gehinderten Substraten, zu
gelangen sowie die Ausweitung der
B-Alkyl-Suzuki-Reaktion auf sekundäre oder sogar tertiäre Alkylborane.
1) F. Diederich, P. J. Stang, Metal-catalyzed
Cross-coupling Reactions, Wiley-VCH,
Weinheim, 1998.
2) Übersichten: N. Miyaura, Top. Curr.
Chem. 2002, 219, 11–59; W. A. Herrmann in Applied Homogeneous Catalysis with Organometallic Compounds,
Vol. 1, 2nd Ed. (Hrsg.: B. Cornils, W. A.
Herrmann), Wiley-VCH, Weinheim, 2002,
S. 591–598; J. Hassan, M. Sevignon, C.
Gozzi, E. Schulz, M. Lemaire, Chem. Rev.
2002, 102, 1359–1469; A. Suzuki, J. Organomet. Chem. 1999, 576, 147–168.
Zum Reaktionsmechanismus siehe K. Matos, J. A. Soderquist, J. Org. Chem. 1998,
63, 461 – 470.
3) S. R. Chemler, D. Trauner, S. J. Danishefsky,
Angew. Chem. 2001, 113, 4676–4701.
4) J. Uenishi, J.-M. Beau, R. W. Armstrong, Y.
Kishi, J. Am. Chem. Soc. 1987, 109, 4756
– 4758; R. W. Armstrong, J.-M. Beau, S. H.
Cheon, W. J. Christ, H. Fujioka, W.-H. Ham,
L. D. Hawkins, H. Jin, S. H. Kang, Y. Kishi,
M. J. Martinelli, W. W. McWhorter, Jr., M.
Mizuno, M. Nakata, A. E. Stutz, F. X. Talamas, M. Taniguchi, J. A. Tino, K. Ueda, J.-i.
Uenishi, J. B. White, M. Yonaga, J. Am.
Chem. Soc. 1989, 111, 7525 – 7530.
5) Ausführliche Übersicht zur Totalsynthese
der CP-Verbindungen: K. C. Nicolaou, P. S.
Baran, Angew. Chem. 2002, 114,
2800–2843.
6) D. Meng, S. J. Danishefsky, Angew. Chem.
1999, 111, 1582–1585.
7) A. Pelter, K. Smith, H. C. Brown, Borane
Reagents, Academic Press, New York,
1988; H. C. Brown, Organic Syntheses via
Boranes, Wiley, New York, 1975.
8) H. C. Brown, T. E. Cole, Organometallics
1983, 2, 1316.
9) J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka Jr., M. R. Smith III, Science 2002,
295, 305–308.
10) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi, J. F. Hartwig, J. Am.
Chem. Soc. 2002, 124, 390–391.
11) R. Stürmer, Angew. Chem. 1999, 111,
3509–3510.
12) C. Amatore, A. Jutand, Acc. Chem. Res.
2000, 33, 314–321.
13) A. F. Littke, G. C. Fu, Angew. Chem. 1998,
110, 3586–3587.
14) A. F. Littke, C. Dai, G. C. Fu, J. Am. Chem.
Soc. 2000, 122, 4020 – 4028.
15) J. Yin, M. P. Rainka, X.-X. Zhang, S. L. Buchwald, J. Am. Chem. Soc. 2002, 124,
1162–1163.
16) M. G. Andreu, A. Zapf, M. Beller, Chem.
Commun. 2000, 2475–2476.
17) Übersicht: W. A. Herrmann, V. P. W. Böhm,
C.-P. Reisinger, J. Organomet. Chem.
1999, 576, 23 – 41.
18) Übersicht: W. A. Herrmann, Angew.
Chem. 2002, 114, 1342–1363.
19) C. W. K. Gstöttmayr, V. P. W. Böhm, E.
Herdtweck, M. Grosche, W. A. Herrmann,
Angew. Chem. 2002, 114, 1421 – 1423.
Blickpunkt Synthese
will zur Beschäftigung mit neuen synthetischen Verfahren oder
eleganten und wichtigen Synthesen aus der Literatur anregen.
Die Rubrik erscheint von Februar
bis Dezember alle zwei Monate
und wird dieses Jahr von Dr. Valentin Wittmann, Institut für
Organische Chemie der Universität Frankfurt, betreut.