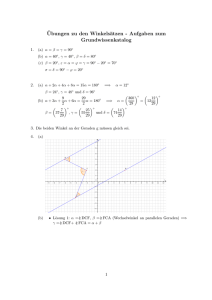
Skript zum Chem. Praktikum - Philipps
Werbung

Skript zum Chemischen Praktikum für Studierende der Biologie und Humanbiologie Fachbereich Chemie Philipps-Universität Marburg 4. Februar 2016 Zusammenfassung Dies ist die zweite revidierte Fassung des Skripts zum organischen Teil des Chemischen Praktikum für Studierende der Biologie und Humanbiologie. Es entstand im Rahmen einer völligen Neubearbeitung vorhandener Materialien und unter Einarbeitung neuer Gesichtspunkte (Sicherheit, Umweltverträglichkeit der Versuche etc.) und wurde von Dr. Jürgen Müller in Zusammenarbeit mit Dr. Ulrich Wolf verfasst. Sollten Sie in dieser Ausgabe Fehler finden (was zu vermuten ist) oder sollten Sie Anregungen zu Versuchen oder zum Praktikum haben (was zu hoffen ist), geben Sie diese bitte bei Dr. Philipp Reiß ab <[email protected]>. Zur Beachtung: Dieses Skript ersetzt nicht das zum Praktikum angebotene Seminar! Der Inhalt der theoretischen Kapitel kann von dem Stoff des Seminars abweichen. Es wird vorausgesetzt, dass sich der Praktikant auch in den gängigen Lehrbüchern über die theoretischen Hintergründe informiert. Inhaltsverzeichnis 1 Informationen zum Praktikum 2 Das Vorpraktikum 2.1 Wichtige Handgriffe im Labor (1. Woche) . . . 2.1.1 Entsorgung . . . . . . . . . . . . . . . 2.1.2 Umgang mit dem Bunsenbrenner . . . 2.1.3 Umgang mit Reagenzgläsern . . . . . . 2.1.4 Herstellen und titrieren von Lösungen . 2.1.5 Filtrieren und zentrifugieren . . . . . . 2.1.6 Umgang mit dem Scheidetrichter . . . 5 . . . . . . . . . . . . . . . . . . . . . 3 Allgemeine und anorganische Chemie 3.1 Themen des 1. Kolloquiums . . . . . . . . . . . . . 3.2 Lösungen und Mischungen (2. Woche) . . . . . . . 3.2.1 Versuch: Lösen von Kupfersulfat . . . . . . . 3.2.2 Versuch: Lösen von Kaliumchlorid . . . . . . 3.2.3 Versuch: Lösungsenthalpie . . . . . . . . . . 3.2.4 Versuch: Biomoleküle in Lösung . . . . . . . 3.3 Verteilungsgleichgewichte . . . . . . . . . . . . . . . 3.3.1 Versuch: Extraktion . . . . . . . . . . . . . 3.3.2 Versuch: Diffusion . . . . . . . . . . . . . . . 3.3.3 Versuch: Dialyse von Riboflavin/Hämoglobin 3.4 Chem. Gleichgewichte, Massenwirkungsgesetz . . . 3.4.1 Versuch: Das Kalkgleichgewicht . . . . . . . 3.4.2 Versuch: Iodstärke-Reaktion . . . . . . . . . 3.5 Säure/Base-Reaktionen und Puffer (3. Woche) . . . 3.5.1 Versuch: Saure und basische Salze . . . . . . 3.5.2 Versuch: Titration . . . . . . . . . . . . . . 3.5.3 Versuch: Titrationskurve . . . . . . . . . . . 3.5.4 Analyse 1: Bestimmung von HCl . . . . . . 3.5.5 Versuch: Puffer . . . . . . . . . . . . . . . . 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9 9 9 10 12 13 15 17 . . . . . . . . . . . . . . . . . . . 19 19 21 27 28 29 30 32 33 35 36 37 41 44 45 50 51 54 56 58 INHALTSVERZEICHNIS 3.6 3.7 3.8 3.9 Themen des 2. Kolloquiums . . . . . . . . . . . . . . . . . . . Redoxreaktionen (4.–5. Woche) . . . . . . . . . . . . . . . . . 3.7.1 Versuch: Metallionen, Spannungsreihe . . . . . . . . . . 3.7.2 Versuch: Lokalelement . . . . . . . . . . . . . . . . . . 3.7.3 Versuch: pH-Abhängigkeit des Redoxpotentials . . . . 3.7.4 Versuch: Wasserstoffperoxid als redoxamphoteres System Komplexe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.8.1 Versuch: Cu-Komplexe . . . . . . . . . . . . . . . . . . 3.8.2 Versuch: Fe-Komplexe . . . . . . . . . . . . . . . . . . 3.8.3 Versuch: Gleichgewicht von Kupferkomplexen . . . . . 3.8.4 Versuch: Magnesium im Spinat . . . . . . . . . . . . . Analytische Bestimmungen . . . . . . . . . . . . . . . . . . . . 3.9.1 Analyse 2: Bestimmung von NaCl durch Ionenaustauscher . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.9.2 Analyse 3: Photometrische Bestimmung von Phosphat 3 62 64 70 71 72 74 75 78 80 81 83 85 85 89 4 Organische Chemie 93 4.1 Themen des 3. Kolloquiums: . . . . . . . . . . . . . . . . . . . 93 4.2 Einführung . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95 4.2.1 Versuch: Betrachtung an Molekülmodellen . . . . . . . 105 4.3 Substitution, Eliminierung, Addition (6.–7. Woche) . . . . . . 109 4.3.1 Versuch: SN1 - und SN2 -Substitution: t-Butylchlorid . . . 111 4.3.2 Versuch: Eliminierung . . . . . . . . . . . . . . . . . . 115 4.3.3 Versuch: Addition an Doppelbindungen . . . . . . . . . 117 4.4 Carbonylverbindungen, Acidität (8.–9. Woche) . . . . . . . . . 120 4.4.1 Versuch: Säurestärke organischer Verbindungen . . . . 125 4.4.2 Versuch: Aldolkondensation . . . . . . . . . . . . . . . 126 4.4.3 Versuch: Präparative Darstellung eines Esters . . . . . 129 4.4.4 Versuch: Fettverseifung . . . . . . . . . . . . . . . . . . 133 4.4.5 Versuch: Decarboxylierung . . . . . . . . . . . . . . . . 134 4.5 Identifizierung organischer Stoffe (10. Woche) . . . . . . . . . 138 4.5.1 Versuch: Nachweis von Alkoholen . . . . . . . . . . . . 139 4.5.2 Versuch: Nachweis von Aminen . . . . . . . . . . . . . 141 4.5.3 Versuch: Nachweis von Aldehyden und Ketonen . . . . 142 4.5.4 Analyse 4: Organische Substanzen . . . . . . . . . . . . 143 4.6 Themen des 4. Kolloquiums . . . . . . . . . . . . . . . . . . . 146 4.7 Zucker/Kohlenhydrate (11.–12. Woche) . . . . . . . . . . . . . 147 4.7.1 Versuch: Aufbau von Molekülmodellen . . . . . . . . . 154 4.7.2 Wahlpflichtversuch: Fehlingsche Probe (1/6) . . . . . . 156 4.7.3 Wahlpflichtversuch: Probe nach Tollens (2/6) . . . . . 157 4.7.4 Wahlpflichtversuch: Reduz. Wirkung von D-Glucose (3/6)159 4 INHALTSVERZEICHNIS 4.7.5 4.8 Wahlpflichtversuch: Reakt. v. Glucose mit Methylenblau (4/6) . . . . . . . . . . . . . . . . . . . . . . . . . 160 4.7.6 Wahlpflichtversuch: Red. v. Kaliumpermanganat durch Glucose (5/6) . . . . . . . . . . . . . . . . . . . . . . . 162 4.7.7 Wahlpflichtversuch: Holzverzuckerung (6/6) . . . . . . 163 4.7.8 Versuch: Prüfung von Saccharose mit Fehlingscher Lösung . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164 4.7.9 Versuch: Inversion von Saccharose . . . . . . . . . . . . 165 4.7.10 Versuch: Nachweis von Vitamin C . . . . . . . . . . . . 166 4.7.11 Versuch: Pentaacetylderivat eines Monosaccharids . . . 169 4.7.12 Wahlpflichtversuch: Herstellung von Stärkekleister (1/4) 171 4.7.13 Wahlpflichtversuch: Säurehydrolyse der Stärke (2/4) . . 172 4.7.14 Wahlpflichtversuch: Enzymatische Hydrolyse von Stärke (3/4) . . . . . . . . . . . . . . . . . . . . . . . . . . 173 4.7.15 Versuch: Untersuchung von Nahrungsmitteln auf Stärke (4/4) . . . . . . . . . . . . . . . . . . . . . . . . . . 174 Aminosäuren und Proteine (13.–14. Woche) . . . . . . . . . . 176 4.8.1 Versuch: Titration von Glycin . . . . . . . . . . . . . . 178 4.8.2 Versuch: Isoelektrischer Punkt und Löslichkeit von Casein . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182 4.8.3 Versuch: Gewinnung von Casein . . . . . . . . . . . . . 184 4.8.4 Versuch: Aussalzen von Casein . . . . . . . . . . . . . . 185 4.8.5 Versuch: Hippursäure . . . . . . . . . . . . . . . . . . . 186 4.8.6 Versuch: Einwirkung von Natriumhydroxidlösung auf Proteine . . . . . . . . . . . . . . . . . . . . . . . . . . 188 4.8.7 Versuch: Proteinbestimmung durch Biuretreaktion . . . 189 4.8.8 Analyse 5: Trennung und Identifizierung eines Aminosäuregemisches . . . . . . . . . . . . . . . . . . . . . . 190 Kapitel 1 Informationen zum Praktikum Aufbau des Skriptes Dieses Skript beinhaltet alle Versuchsvorschriften der im Biologen-Praktikum zu absolvierenden Versuche. Die Versuche sind jeweils für eine Praktikumswoche zusammengestellt und können nur in der dafür vorgesehenen Woche durchgeführt werden. Die benötigten Chemikalien werden nur für die jeweilige Praktikumswoche bereitgestellt. Zu Beginn eines jeden Themenblocks finden Sie eine kurze Zusammenfassung der wichtigsten theoretischen Fakten. Diese Zusammenfassung kann und soll ein Lehrbuch nicht ersetzen. Es ist unbedingt notwendig, dass Sie sich in den Lehrbüchern ihrer Wahl über die jeweiligen Themen informieren. Dies kann anhand der angegebenen Stichwörter geschehen. Jede Versuchsvorschrift besteht aus sechs Teilen: Stichworte: Hier stehen alle Stichworte, die dem Praktikanten helfen sollen, in den einschlägigen Lehrbüchern die theoretischen Aspekte des Versuchs nachschlagen zu können. Allgemeines: Hier werden die für den Versuch wichtigen theoretischen Hintergründe in kurzen Worten beschrieben. Diese Ausführungen können nicht das Lehrbuch ersetzen, sondern sind nur als eine mehr oder weniger vollständige Zusammenfassung zu verstehen. Chemikalien: Für den Versuch benötigte Chemikalien. Durchführung: Eine möglichst klare Beschreibung der auszuführenden Tätigkeiten. 5 6 KAPITEL 1. INFORMATIONEN ZUM PRAKTIKUM Aufgaben: An dieser Stelle sind alle Aufgaben zusammengefasst, welche der Praktikant zu bearbeiten und in seinem Laborjournal zu protokollieren hat. Entsorgung: Vorgehensweise bei der Entsorgung und Name des Sammelgefäßes. Vorgehensweise im Praktikum Zu Anfang eines jeden Praktikumstages bespricht der Assistent kurz die nötigen theoretischen Grundlagen der Versuche und behandelt die bei der Vorbereitung aufgekommenen Fragen der Praktikanten. Der Assistent ist gehalten diese Fragen zu beantworten, aber auch den Grad der Vorbereitung der Praktikanten zu beurteilen. Nur wer gut vorbereitet ist, kann qualifizierte Fragen stellen. Kolloquien In diesem Skript werden am Anfang des entsprechenden Themenblocks die Stichworte des dazugehörenden Kolloquiums aufgelistet. Der Praktikant hat damit einen Anhaltspunkt für die Vorbereitung auf das Kolloquium. Das Kolloquium wird immer vor dem jeweiligen Themenblock durchgeführt. Die Versuche des Themenblocks können nur nach erfolgreicher Teilnahme an dem Kolloquium durchgeführt werden. Während der Kolloquien werden auch Fragen zur Sicherheit im Labor gestellt. Laborjournal und Versuchsprotokolle Das Laborjournal besteht aus einem gebundenen Schreibbuch der Größe A4 oder A5. Jedes Protokoll ist dem Assistenten spätestens am nachfolgenden Praktikumstag zur Begutachtung vorzulegen. Genügt das Protokoll den Anforderungen, unterschreibt der Assistent das Versuchsprotokoll. Ein Protokoll sollte folgendermaßen aufgebaut sein: • Überschrift: Nummer und Name des Versuchs. • Kurze Beschreibung der Tätigkeit mit Reaktionsgleichungen (falls möglich). • Lösungen der verschiedenen Aufgaben. 7 Das Protokoll sollte handschriftlich abgefasst sein. Anfang und Ende eines Versuchsprotokolls sollten klar erkennbar sein. Am Ende des Praktikums wird der Assistent die Laborjournale einsammeln und abschließend auf Vollständigkeit prüfen und bewerten. Hinweise auf Gefahren Bei Handelsprodukten sind Hinweise auf die Eigenschaften des Gefahrstoffes den aufgedruckten Gefahrensymbolen und Gefahrenbezeichnungen zu entnehmen. Die dazugehörigen, ebenfalls auf der Verpackung befindlichen, Gefahrenhinweise (R-Sätze) und Sicherheitsratschläge (S-Sätze) sind zu beachten. E Explosiv O F Brandfördernd Leichtentzündlich T+ Sehr giftig Xn Gesundheitsschädlich F+ Hochentzündlich Xi Reizend C Ätzend T Giftig N Umweltgefährlich Alle chemischen und biologischen Stoffe, deren Ungefährlichkeit nicht zweifelsfrei feststeht, sind als Gefahrstoffe anzusehen und entsprechend zu behandeln. Arbeiten mit Gefahrstoffen, bei denen diese in die Atemluft eintreten können, sind möglichst im geschlossenen Abzug durchzuführen. Beim Umgang mit Gefahrstoffen muss ein geeigneter Augenschutz (Schutzbrille) getragen werden. In Bereichen, in denen mit chemischen Stoffen gearbeitet wird, ist ständig eine Schutzbrille zu tragen. Der Kontakt von Gefahrstoffen mit der Haut oder der Kleidung ist grundsätzlich zu vermeiden. Bei Arbeiten mit Gefahrstoffen ist geeignete Arbeitsschutzkleidung (Laborkittel) zu tragen. Gegebenenfalls sind geeignete Schutzhandschuhe aus beständigem Gummi oder Kunststoff zu benutzen. Essen, Trinken, Rauchen oder Schnupfen ist in Bereichen, in denen mit Gefahrstoffen gearbeitet wird, verboten. 8 KAPITEL 1. INFORMATIONEN ZUM PRAKTIKUM Wird Kleidung durch unvorhergesehene Zwischenfälle mit Gefahrstoffen stark verschmutzt oder durchtränkt, ist sie sofort auszuziehen. Vor der Aufnahme von Speisen sind die Hände gründlich mit geeigneten Reinigungsmitteln zu waschen. Lesen Sie gründlich die in ihrem Arbeitsplatz befindlichen Broschüren zur Arbeitssicherheit durch. Informieren Sie sich über die Sicherheitseinrichtungen im Labor (Notduschen, Augenduschen, Feuerlöscher, Verbandkästen). Kapitel 2 Das Vorpraktikum 2.1 Wichtige Handgriffe im Labor (1. Woche) Dieses Vorpraktikum soll ihnen die während des Praktikums benötigten Handgriffe und Techniken im Labor näherbringen. Der Assistent wird ihnen Aufbau und Umgang mit den Geräten vor jeder Aufgabe zeigen und auf die sicherheitsrelevanten Aspekte eingehen. Danach führen Sie die Aufgaben selbständig unter Aufsicht des Assistenten aus. 2.1.1 Entsorgung Der Assistent zeigt Ihnen die sich im Labor befindlichen verschiedenen Abfallgefäße. Blaue oder schwarze Tonne: Für trockene Feststoffe, beschmutzte Glasabfälle, trockene Filterpapiere, u. ä.; bitte nur trockene (feste) Chemikalien oder Gegenstände einwerfen. Brauner oder blauer Metallbehälter: Organische Lösungsmittel wie Chloroform, Petrolether u. ä. Die Lösungen bitte vorher mit dem pHPapier auf pH-Neutralität überprüfen. In diesen Kanister dürfen nur neutrale Lösungen entsorgt werden. Kunststoffkanister: Wässrige Schwermetallabfälle. Die Lösungen bitte vorher mit dem pH-Papier auf pH-Neutralität überprüfen. In diesen Kanister dürfen nur neutrale Lösungen entsorgt werden. 9 10 2.1.2 KAPITEL 2. DAS VORPRAKTIKUM Umgang mit dem Bunsenbrenner Bunsenbrenner und Teclubrenner benutzt man im Labor zum kurzzeitigen Erhitzen kleinerer Gefäße wie Reagenzgläser, Bechergläser oder Porzellanschalen. Auch einfache Glasbearbeitung wird mit diesen Brennern durchgeführt. Vor Inbetriebnahme des Brenners wird seine Luftzufuhr geschlossen. Danach wird er an den gelben Hahn am Labortisch angeschlossen (auf festen Sitz des Schlauches achten). Die Gaszufuhr am Brenner und der Gashahn werden geöffnet und die Flamme gezündet. Der Brenner brennt mit einer leuchtenden, gelben Flamme. Mit Öffnen der Luftzufuhr wird die Flamme blau. Bei voller Luft- und Gaszufuhr besteht die Brennerflamme aus mehreren Zonen. Die Spitze des inneren Kegels ist der heißeste Punkt der Flamme. Dort können Temperaturen bis 1500˚C auftreten. Das Innere dieses Kegels ist demgegenüber eine relativ kühle Zone. Dort befindet sich noch nicht verbranntes Gas-Luft-Gemisch. Aufgabe 2.1 Die Zonen der Brennerflamme Lassen Sie sich die korrekte Bedienung des Brenners vorher vom Assistenten zeigen. Die unterschiedlichen Temperaturen der Flammenzonen lassen sich mit Hilfe eines Magnesiastäbchens zeigen. Halten Sie ein Magnesiastäbchen in die verschiedenen Zonen der Brennerflamme. Es wird nur über der Spitze des inneren Kegels hell aufleuchten. Versuchen Sie dasselbe auch mit einem Stück Draht (Vorsicht Holzklammer verwenden!!). Entsorgung: Benutzte Magnesiastäbchen oder Draht können nach dem Abkühlen in den Hausmüll unter dem Waschbecken gegeben werden. 2.1. WICHTIGE HANDGRIFFE IM LABOR (1. WOCHE) 11 Aufgabe 2.2 Glasbearbeitung Glasrohr zerschneiden Lassen Sie sich ein Glasrohr vom Assistenten aushändigen und schneiden Sie ein etwa 20–30 cm langes Stück ab. Dazu ritzen Sie es an der gewünschten Stelle mit einer Ampullensäge auf einer Seite ein, befeuchten die Stelle mit etwas Wasser, legen beide Daumen, geschützt durch ein Tuch oder den Laborkittel, gegenüber der Kerbstelle an und brechen das Glas unter etwas Druck ziehend“ auseinander. Die scharfkantigen Bruchenden werden unter ständi” gem Drehen in der heißen Brennerflamme rundgeschmolzen. Das noch heiße Glas unter Fächeln abkühlen lassen, nicht sofort auf die kalte Tischplatte legen oder in kaltes Wasser tauchen. Glasrohr zu einem rechten Winkel biegen Das Glasrohr wird in der heißen Flamme unter ständigem Drehen auf einer längeren Strecke (3–5 cm) erweicht und dann außerhalb der Flamme gleichmäßig zu einem rechten Winkel gekrümmt; dabei dürfen keine Verwindungen oder scharfen Knicke entstehen. Glasrohr zuschmelzen Ein anderes Stück Glasrohr wird am Ende erhitzt und mit einer Pinzette eine Spitze ausgezogen, die man abschneidet und abgerundet zuschmilzt. An dem zugeschmolzenen Stück lässt sich durch Schmelzen und Blasen eine Glaskugel formen. Pipetten und Kapillaren ziehen Ein etwa 20 cm langes Glasrohr, das an beiden Enden rundgeschmolzen ist, wird in der Mitte unter Drehen gleichmäßig bis zum Erweichen erhitzt. Außerhalb der Flamme zieht man das Rohr gleichmäßig und kräftig, aber nicht zu schnell auseinander, bis die gewünschte Form und Länge erreicht ist. Zieht man dagegen das erhitzte Glasrohr außerhalb der Flamme rasch und kräftig auseinander, so entsteht ein langes Kapillarrohr, das man in etwa 7 cm lange Stücke brechen und zum Applizieren kleinster Tröpfchen auf Chromatogramme, Objektträger oder dergleichen benutzen kann. Rührstab herstellen Schneiden Sie ein ca. 20–30 cm langes Stück Glasstab wie oben beschrieben zu und runden Sie die Enden in der Brennerflamme ab. Formen Sie aus einem Ende des Stabes eine kleine Platte. Sie erhalten damit einen Rührund Mischstab (siehe Aufgabe 4). Entsorgung: Saubere Glasabfälle werden in den im Labor bereitstehenden Glasabfallbehälter entsorgt. 12 2.1.3 KAPITEL 2. DAS VORPRAKTIKUM Umgang mit Reagenzgläsern Das Reagenzglas ist ein Gefäß, in dem kleine Mengen Flüssigkeit erhitzt werden können. Beim Erhitzen passiert es leicht, dass durch einen Siedeverzug die heiße Flüssigkeit herausspritzt und in ungünstigen Fällen Haut und Gesicht verbrüht oder gar verätzt. Darum darf beim Erhitzen das Reagenzglas mit der Öffnung nie auf einen Menschen zeigen. Lösungen sollten generell im Abzug erhitzt werden. Aufgabe 2.3 Erhitzen im Reagenzglas Chemikalien: Nicht mehr als ca. 2 g Natriumchlorid (NaCl) pro Praktikant. Füllen Sie in ein Reagenzglas etwa drei Finger hoch entionisiertes Wasser. Versuchen Sie durch schrittweises Einfüllen und Schütteln zuerst bei Raumtemperatur das Salz zu lösen. Prüfen Sie dabei mit der Hand die Temperatur des Reagenzglases. Welche Veränderungen bemerken Sie? Lässt sich das Salz nicht mehr weiter lösen (nach ca. 2–3 min. schütteln), erwärmen Sie das Reagenzglas über dem Brenner. Dabei halten Sie das Reagenzglas mit der Öffnung immer von sich und anderen weg und schwenken es mit Hilfe der Reagenzglasklammer über dem Brenner bis sich das Salz löst. Danach geben Sie unter Erwärmen weiter Natriumchlorid in kleinen Portionen zu. Die Lösung darf beim Erwärmen nie zur Ruhe kommen, da sonst ein Siedeverzug (plötzliches Aufkochen und Herausspritzen) erfolgen kann. Das Erhitzen von Reagenzgläsern sollte daher generell nur im Abzug erfolgen. Lässt sich das Salz auch in der Hitze nicht mehr lösen, stellen Sie das Reagenzglas in den Ständer und lassen es 15 min. abkühlen. Was ist zu beobachten? Entsorgung: Die Lösung kann im Ausguß entsorgt werden. 2.1. WICHTIGE HANDGRIFFE IM LABOR (1. WOCHE) 13 Aufgabe 2.4 Mischen Chemikalien: NaHCO3 , Methylrot, 1 M Essigsäure Im Reagenzglas versetzt man nur 2–3 ml der gesättigten Natriumhydrogencarbonat-(NaHCO3 )-Lösung mit 2 Tropfen Indikator und fügt wiederum 1 M Essigsäure in kleinen Anteilen bis zum Umschlag zu. Durch schütteln des Reagenzglases kann man leicht und sauber völlige Durchmischung erzielen. Die Mengen wurden dem Reagenzglas angepasst. Achten Sie in den folgenden Reagenzglasversuchen immer auf die vernünftige Menge, da nur so sauberes Arbeiten möglich ist. Entsorgung: Die Lösung kann im Ausguss entsorgt werden. 2.1.4 Herstellen und titrieren von Lösungen Bei dieser Aufgabe geht es darum, den richtigen Umgang mit skalierten Glasgeräten wie Mess- und Vollpipetten, Büretten und Messzylindern zu üben. Um typische Fehler zu vermeiden, lesen Sie sich die folgenden Ausführungen gut durch. Pipetten In ihrem Arbeitsplatz finden Sie zwei Arten von Pipetten. Die Messpipette ist durchgehend skaliert. Mit ihr lassen sich verschiedene Volumina pipettieren. Die Vollpipette hat ein definiertes Volumen. Mit ihr kann man nur das auf der Pipette angegebene Volumen pipettieren. Sie ist dafür aber auch die genauere Pipette. Sollen also genau 20 ml pipettiert werden, wie es bei einer Titration oft der Fall ist, so ist hier die 20 ml-Vollpipette das Gerät der Wahl. Beim Entleeren der Pipette legt man die Auslaufspitze an die Wand des Gefäßes, lässt die Pipette ruhig ausfließen und streift sie zuletzt an der Gefäßwand ab. Die kleine Flüssigkeitsmenge, die in der Pipette zurückbleibt, ist bei der Eichung berücksichtigt. Die Pipette darf daher nicht ausgeblasen werden. Der Peleusball Die Pipetten werden mit einem Peleusball bedient. Nie mit dem Mund pipettieren!! Lassen Sie sich die korrekte Bedienung des Peleusballs vom Assistenten vorführen. Der Peleusball wird vorsichtig oben auf die Pipette gesteckt. Dabei ist unbedingt darauf zu achten, dass der Pipettenschaft dabei das Ventil (2) nicht beschädigt. Der Peleusball wird durch drücken des oberen Ventils (1) und gleichzeitiges zusammendrücken des Ballonteils entlüftet. Durch drücken des 14 KAPITEL 2. DAS VORPRAKTIKUM unteren Ventils (2) wird die Pipette mit dem Ballonteil des Peleusballs in Verbindung gebracht. Durch den Unterdruck im Ballonteil wird die Flüssigkeit in die Pipette gesaugt. Es ist in jedem Fall zu vermeiden, dass Flüssigkeit in den Peleusball kommt. Dies kann den Peleusball unbrauchbar machen. Das seitliche Ventil (3) dient zum Belüften der Pipette. Wird dieses Ventil gedrückt, so läuft die Pipette aus. Die Bürette Büretten sind graduierte Rohre, die gewöhnlich 25–50 ml fassen. Zur Ablesung bestimmt man den Teilstrich der Bürette, der den unteren Meniskus der Flüssigkeit tangiert. Ist die Bürette mit einem blauen Schellbachstreifen ausgestattet, so zeigt das an der Meniskusunterseite entstehende Dreieck den Ablesewert an der Graduierung an. Bauen Sie die Bürette nach den Anweisungen des Assistenten auf. Achten Sie darauf, dass Sie nur die dafür vorgesehenen Bürettenklammern verwenden. Die Bürette sollte gerade hängen und das Küken leicht drehbar sein. Glasschliffküken sollten mit etwas Schlifffett geschmiert werden. Nach dem Gebrauch der Bürette müssen die Küken vom Schlifffett gereinigt werden. Die weißen Teflon-Küken dürfen dagegen nicht mit Schlifffett behandelt werden. Die verwendeten, skalierten Glasgeräte dürfen nach dem Säubern nicht im Trockenschrank getrocknet werden, da sie sonst durch das Erhitzen ihre Genauigkeit einbüßen. Sie müssen luftgetrocknet, oder mit der zu messenden Lösung vorher mehrfach gut ausgespült werden. 2.1. WICHTIGE HANDGRIFFE IM LABOR (1. WOCHE) 15 Aufgabe 2.5 Herstellen und titrieren von Lösungen Chemikalien: NaOH-Plätzchen, 1 M HCl, Phenolphtalein Lösen Sie in einem 100 ml Becherglas 4 g Natriumhydroxid in ca. 50 ml entionisiertem Wasser und geben Sie danach diese Lösung in einen 100 ml Messkolben. Spülen Sie mit entionisiertem Wasser nach und füllen Sie den Messkolben auf 100 ml auf. Die Bürette wird mit Hilfe eines kleinen Trichters bis etwas über die Nullmarke mit 1 M Salzsäure (HCl) aufgefüllt. Nach dem Herausnehmen des Trichters wird der Meniskus durch Ablassen genau auf die Nullmarke eingestellt. Pipettieren Sie mit der genaueren Vollpipette jeweils 20 ml der selbst hergestellten Natriumhydroxidlösung in drei 100 ml Bechergläser. Geben Sie ca. 3 Tropfen Phenolphtaleinlösung in jedes Becherglas. Titrieren Sie nun unter Anweisung des Assistenten den Inhalt der drei Bechergläser bis zum Farbumschlag. Versuchen Sie auf den Tropfen genau den Verbrauch an Salzsäure zu messen. Liegt der Verbrauch bei 20 ml, so haben Sie beim Herstellen der Natriumhydroxidlösung gut gearbeitet. Erkennen Sie Nachteile dieser Titrationsmethode? Entsorgung: Sammeln Sie die übriggebliebenen Natriumhydroxid(NaOH)-Lösungen und Salzsäure-Lösungen und geben Sie diese sehr vorsichtig (!) in ein großes Becherglas zusammen. Rühren Sie die Lösung mit einem Glasstab (Abzug, Schutzbrille) und prüfen Sie den pH-Wert (Definition s. S. 47) mit dem Indikatorpapier, indem Sie mit dem Glasstab einen Tropfen auf das pH-Papier geben. Wird das Indikatorpapier rot, so ist die Lösung sauer. Wird es blau, so ist sie alkalisch. Zeigt es eine grüne Farbe oder bleibt es gelblich, so ist der pH-Wert der Lösung im neutralen Bereich (pH 6–7). In diesem Fall kann die Lösung in dem Abguss entsorgt werden. Hat die Lösung einen alkalischen oder sauren pH-Wert, lassen Sie sich bitte vom Assistenten zeigen, wie Sie die Lösung neutralisieren. 2.1.5 Filtrieren und zentrifugieren Der Büchnertrichter Neben dem normalen Filtrieren mit einem Glastrichter und einem gefalteten Filterpapier gibt es im Labor noch die Möglichkeit, mit dem Büchnertrichter (Nutsche) zu arbeiten. Der Büchnertrichter wird, mit einer Dichtung versehen, auf eine spezielle dickwandige Saugflasche gesteckt und diese an eine Vakuumpumpe (Membranpumpe, Wasserstrahlpumpe) angeschlossen. Hierfür finden Sie an ihrem Arbeitsplatz extra dicke Vakuumschläuche. Der Büchnertrichter besitzt einen durchlöcherten Porzellanboden. Auf diesen Boden wird ein passendes, rundes Filterpapier gelegt und mit etwas Flüssigkeit 16 KAPITEL 2. DAS VORPRAKTIKUM angefeuchtet. Wird die Vakuumpumpe betätigt, so bewirkt der Unterdruck in der Saugflasche einen Saugeffekt durch das Filterpapier. Die Zentrifuge Eine weitere Möglichkeit, feste von flüssigen Phasen zu trennen, ist das Zentrifugieren. Die Zentrifuge unterstützt mit Hilfe der Zentripetalkraft das Absetzen der festen Körper in einer Lösung. Gerade bei sehr feinen Teilchen hat die Zentrifuge gegenüber dem Filterpapier Vorteile, da ein Filter bei kleinen Teilchen leicht verstopfen kann. Zum Zentrifugieren befinden sich in ihrem Arbeitsplatz spezielle Zentrifugengläser aus dickwandigem Glas oder PVC. Sie laufen unten spitz zu. Benutzen Sie bitte nur diese Zentrifugengläser, wenn Sie etwas zentrifugieren wollen. Da sich eine Zentrifuge mit hoher Geschwindigkeit dreht, ist es, um eine Unwucht zu vermeiden, wichtig, dass sich immer zwei Zentrifugengläser mit möglichst identischem Gewicht (bzw. gleich hohem Flüssigkeitsstand) gegenüberliegend in der Halterung der Zentrifuge befinden. Beim starten der Zentrifuge sollte man zuerst mit einer kleinen Drehzahl beginnen. Zeigt die Zentrifuge keine Unwucht (ruckartiges Bewegen der Zentrifuge), so kann man die Drehzahl langsam erhöhen. 2.1. WICHTIGE HANDGRIFFE IM LABOR (1. WOCHE) 17 Aufgabe 2.6 Fällen,filtrieren und zentrifugieren eines Niederschlags Zweiergruppe Chemikalien: BaCl2 -Lösung, 1 M H2 SO4 Füllen Sie ein 100 ml Becherglas mit etwa 50 ml entionisiertem Wasser. Lösen Sie darin 2 g Bariumchlorid (BaCl2 ). Tropfen Sie mit der Pasteurpipette solange 1 M Schwefelsäure hinzu, bis sich kein weißer Niederschlag mehr bildet. Filtrieren Sie danach mit dem Büchnertrichter die Lösung und zentrifugieren Sie das Filtrat anschließend. Hat der Filter wirklich alle Teilchen des entstandenen Bariumsulfat-(BaSO4 )-Niederschlages zurückgehalten? Tropfen Sie in die zentrifugierte Lösung weitere zwei Tropfen 1 M Schwefelsäure (H2 SO4 ). Bildet sich wieder ein Niederschlag, wiederholen Sie das Zentrifugieren und Zutropfen so lange, bis sich kein Niederschlag mehr bildet. Die obige Methode eignet sich sehr gut zum quantitativen Ausfällen von Substanzen. Entsorgung: Die Lösung wird in den Behälter für wässrige Schwermetalle gegeben. Der Niederschlag wird auf dem Filterpapier getrocknet und in die Feststoffabfälle gegegeben. 2.1.6 Umgang mit dem Scheidetrichter Der Scheidetrichter wird im Labor zum Trennen von zwei flüssigen, nicht miteinander mischbaren Phasen benutzt. Er wird nach dem Befüllen mit einem PVC-Stopfen (nach Möglichkeit kein Glasstopfen) geschlossen. Der Inhalt wird durch kräftiges schütteln durchmischt. Dabei wird der Scheidetrichter mit einer Hand an dem Hahnstück gehalten, während die andere Hand den Stopfen gut festhält. Beim Durchmischen von Lösungen kann aufgrund des Dampfdruckes des Lösungsmittels oder durch Gasbildung infolge einer Reaktion der Innendruck beim Schütteln steigen. Um ein plötzliches Herausspringen des Stopfens zu vermeiden, sollte man zwischendurch den Scheidetrichter mit dem Stopfen nach unten halten und über den Hahn entlüften. Danach kann man den Scheidetrichter wieder in seinen Haltering zurückhängen. Arbeiten Sie mit dem Scheidetrichter bitte immer im Abzug! 18 KAPITEL 2. DAS VORPRAKTIKUM Aufgabe 2.7 Ausschütteln mit dem Scheidetrichter Chemikalien: NaHCO3 , I2 /KI-Lösung, Petrolether Der Versuch wird im Abzug durchgeführt. Es darf bei diesem Versuch kein Gasbrenner oder elektrisches Gerät im Abzug oder in der Nähe in Betrieb sein, da Sie mit einem brennbaren Stoff arbeiten. Lösen Sie in einem Becherglas 9 g Natriumhydrogencarbonat (NaHCO3 ) in etwa 100 ml Wasser und geben Sie etwa 3–5 ml Iod-Kaliumiodid-Lösung hinzu. Bringen Sie nach Anweisung des Assistenten den Haltering für den Scheidetrichter im Abzug an. Füllen Sie die Lösung in einen kleinen Scheidetrichter und geben Sie 100 ml Petrolether (Vorsicht brennbar!!) und 20 ml 0.1 M Essigsäure hinzu. Verschließen Sie den Scheidetrichter mit einem PVC-Stopfen und schütteln Sie kräftig. Vorsicht Gasbildung! Belüften Sie mehrmals. Nachdem Sie ca. 2 min. die Lösung gut durchgeschüttelt haben, hängen Sie den Scheidetrichter in den Haltering. Nachdem sich die Phasen getrennt haben, werden Sie bemerken, dass sich auch der Petrolether (obere Phase) violett gefärbt hat. Diese Farbe rührt von den Iod-Molekülen (I2 ) her, die Sie vorher mit der Iod-Kaliumiodid-Lösung in die wässrige Phase gegeben haben. Da die Iodmoleküle sich viel besser in Petrolether lösen als in Wasser, haben sie die Phasengrenze überschritten und sich in der Petrolether-(organischen)Phase angereichert. Das Herauslösen einer Substanz aus einer Phase in die andere nennt man auch Extraktion. In diesem Fall ist es eine Flüssig/FlüssigExtraktion. Entsorgung: Beide Phasen werden in den Behälter für organische Lösungsmittelabfälle gegeben. Kapitel 3 Allgemeine und anorganische Chemie 3.1 Themen des 1. Kolloquiums Es folgt eine Zusammenstellung der wichtigsten Themen in Stichworten. Es wird empfohlen, die Stichworte in den einschlägigen Lehrbüchern nachzuschlagen. Periodensystem der Elemente (PSE) Aufbau, Hauptgruppenelemente, Alkali-, Erdalkalimetalle, Halogene, Nebengruppenelemente, Elektronegativitätsverteilung im PSE) Stoffe, Lösungen und Mischungen Definition der Stoffmenge und Konzentration: NA , n, M , Molarität, c, Umrechnungen, Einheiten Ursache chemischer Reaktionen und Zustandsänderungen: ∆G, ∆H, T , ∆S, Formeln, endotherm, exotherm, endergonisch, exergonisch, Wann verlaufen Reaktionen freiwillig ab?, usw. Intermolekulare Kräfte: Ionenbindung, Dipolkräfte, Ionengitter, kovalente Bindung, Induktionskräfte, Dispersionskräfte/van-der-Waals Kräfte, induzierte Dipole, Formeln, usw. 19 20 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Lösungen und Mischungen: wässrige/nicht wässrige Lösungen, Lösungsenthalpie, ähnliches löst sich in ähnlichem“, polare und unpolare Stoffe, usw. ” Löslichkeitsprodukt: Formelherleitung, Lösungsgleichgewicht, Gleichgewichtskonstante, gesättigte Lösung, Löslichkeit, gleichioniger Zusatz, Kalkgleichgewicht, usw. Verteilungsgleichgewichte: Diffusion, Dialyse, von welchen Parametern abhängig?, Brownsche Molekularbewegung“, Extraktion, Nernstscher Ver” teilungssatz usw. Säuren, Basen und Puffer Das Massenwirkungsgesetz (MWG): Reaktionsgeschwindigkeiten, Geschwindigkeitskonstanten, K, usw. Protonenübertragungen: Säure-Base-Begriff nach Brönsted und nach Lewis, Donator, Akzeptor, Proton, Hydrid-Ion, molekularer Wasserstoff, Wasserstoff-Atom, Säurerest, Protolyse, amphoter, Ampholyt, konjugierte Säure/Base, Definition des pH-Wertes, usw. Starke und schwache Säuren und Basen: Dissoziationsgleichgewicht, pK-Wert, Dissoziationsgrad, Definition starke/schwache Säure, usw. Pufferlösungen: Puffergleichung, Herstellung, Puffer in der Natur, usw. Säure-Base-Titrationen, Titrationskurven: Erklärung der Titrationskurven, Äquivalenzpunkt, Neutralpunkt, Pufferbereich, Indikatoren, Titer, usw. Außerdem: Stichworte, Themen und Ausführung der Versuche 3.2.1 bis 3.5.5. 3.2. LÖSUNGEN UND MISCHUNGEN (2. WOCHE) 3.2 21 Lösungen und Mischungen (2. Woche) Stichworte bitte in den Lehrbüchern nachschlagen: Periodensystem der Elemente, relative Atommasse, Avogadro-Loschmidt-Konstante, Molarität, Stöchiometrie, Chemische Bindung, Atombindung, Ionenbindung, Elektronegativität, Kristallgitter, Hydratation, Wasserstruktur, zwischenmolekulare Kräfte, van-der-Waals Kräfte, Wasserstoffbrücken, endotherm, exotherm, Wärmetönung, Gibbs-Helmholtz-Gleichung, Enthalpie, Entropie, Freie Enthalpie ∆H, ∆S, ∆G Stöchiometrie Reaktionen zwischen Stoffen gehorchen den Gesetzen der Stöchiometrie: Mit einem Molekül Salzsäure (HCl) reagiert 1 Molekül Natriumhydroxid (NaOH), mit einem Molekül Schwefelsäure (H2 SO4 ) aber reagieren 2 Moleküle Natriumhydroxid (NaOH) unter Neutralisation zu Wasser. HCl + NaOH → H2 O + NaCl H2 SO4 + 2NaOH → 2H2 O + Na2 SO4 Definitionen der Stoffmenge und Konzentrationen Entscheidend für das Ausmaß und die Geschwindigkeit chemischer Prozesse ist die Zahl der (pro Volumeneinheit) vorhandenen Moleküle, ihre Stoffmenge bzw. Konzentration. Alle quantitativen Angaben in der Chemie werden daher primär auf die Anzahl der Moleküle bezogen und nicht auf die Masse in Gramm. Die Stoffmenge n Die Einheit der Stoffmenge n ist das Mol (Einheitssymbol mol). 1 mol ist die Stoffmenge, die ebenso viele Teilchen enthält wie in 12 g des reinen Kohlenstoffisotops 12 C Atome enthalten sind. Diese Zahl ist bekannt und wird Avogadrosche (NA ) oder Loschmidtsche Konstante genannt. NA = 6.022 × 1023 mol−1 In einem mol sind immer NA Teilchen des Stoffes vorhanden. Da sich die Einheit mol nur auf die Zahl, nicht aber auf die Art der Teilchen bezieht, muss die Art der Teilchen immer angegeben werden. 22 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Die molare Masse M Die molare Masse M (Molmasse) ist die Masse der Stoffmenge 1 mol. Ihre Einheit ist g/mol. Für die Masse eines Teilchens kann die Einheit Dalton (Da) verwendet werden. 1 Da ist 1/12 der Masse des reinen Kohlenstoffisotops 12 C (Standardmasse, 1.6602 × 10−24 g). Diese Art der Bezeichnung ist bei höhermolekularen biochemischen Substanzen gebräuchlich, beispielsweise für Hämoglobin m = 64.500 Da (64, 5 kDa). Es gilt M = mNA . Relative molare Masse Mr Im Periodensystem der Elemente ist die relative molare Masse Mr als Massenzahl an jedem Element angegeben. Sie ist dimensionslos. Relative Atomoder Molekülmassen (früher Molekular- bzw. Atomgewicht, Formelgewicht) werden häufig angegeben (z. B. in Tabellen, Katalogen, auf Chemikalienflaschen u. dergl.). Die molare Masse eines Moleküls wird ermittelt durch die Addition der relativen Atommassen seiner Komponenten. Es gilt dann Molare Masse M = Mr · g/mol. Beziehung zwischen Stoffmenge n, Masse m und molare Masse M Die Stoffmenge n steht mit der Masse m und der molaren Masse M in folgender Beziehung: g m mol = n= M g · mol−1 Die Stoffmengenkonzentration c Die chemisch eindeutige und vorgeschriebene Angabe einer Stoffmengenkonzentration c(X) eines gelösten Stoffes X bezieht sich auf seine Stoffmenge n(X) in mol, die in 1 Liter Lösung enthalten sind: c(X) = n(X) , VLösung wobei VLösung das Volumen der Lösung ist. Die Konzentration eines Stoffes X wird schriftlich in der Form c(X) ausgedrückt. Das selbe bedeutet die ebenfalls noch übliche Schreibweise in eckigen Klammern [X]. Die vollständige, DIN-gerechte Angabe einer Stoffmengenkonzentration, z. B. für Salzsäure mit dem Gehalt 1 mol/l, ist c(HCl) = 1 mol · L−1 . 3.2. LÖSUNGEN UND MISCHUNGEN (2. WOCHE) 23 In der Praxis wird auch die Ausdrucksweise Molarität benutzt: 1 mol gelöster Stoff pro Liter Lösungsmittel heißt 1 molar, abgekürzt 1 M. Diese Bezeichnung findet sich häufig auf Lösungsmittelflaschen, z. B. 1 M HClbedeutet eine 1 molare Lösung von HCl-Gas in Wasser. Verwechseln Sie aber niemals die Molarität mit dem Einheitenzeichen M mit der Stoffmenge mol oder der molaren Masse mit dem Formelsymbol M! Es sind noch viele weitere Definitionen der Konzentration üblich: Molarität, c Löslichkeit Prozentgehalt mol/l Lösung g Substanz/100 g Lösungsmittel g Substanz in 100 g Lösung oder in 100 ml Lösung (%Vol ) ppm (parts per million) 1/106 (=1 ˆ µg/g =1 ˆ µg/ml =1 ˆ mg/l =1 ˆ g/m3 ppb (parts per billion) 1/109 (=1 ˆ µg/kg =1 ˆ µg/l =1 ˆ mg/m3 ) Stoffmengenkonzentration (Symbol ceq ) von Äquivalenten; Normalität (Einheit N) Diese Konzentrationsangabe wird heute nicht mehr verwendet. Da sie aber noch in einigen älteren Büchern und auf Chemikalienflaschen verwendet wird, wird sie an dieser Stelle angesprochen. Beispiel: 1 mol H2 SO4 dissoziert in wässriger Lösung in 2 mol Protonen (H+ ) und 1 mol Sulfat-Ionen (SO4 2− ): H2 SO4 → 2H+ + SO2− 4 Das Äquivalent der entsprechenden Menge an Schwefelsäure, die in einem Liter Lösung ein Mol Protonen abgibt, wird als Äquivalentkonzentration bezeichnet. ceq (H2 SO4 ) = 2c(H2 SO4 ) = 1mol · l−1 Das bedeutet, dass eine 0.5 molare Lösung von H2 SO4 eine Äquivalentkonzentration von 1 mol·l−1 hat. Eine solche Lösung bezeichnete man früher als 1 normale Lösung. Beachten Sie bitte, dass die Normalität einer Lösung für verschiedene Stoffe eine vollkommen unterschiedliche Aussage über ihre Molarität macht. So ist im Gegensatz zur obigen Rechnung z. B. eine 1 molare Lösung von HCl ebenfalls 1 normal! In beiden Fällen findet man auf Chemikalienflaschen oft die Bezeichnung 1 N Lösung. 24 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Die Dichte D Die Dichte D eines Stoffes ist die Masse des Stoffes pro Volumeneinheit, ausgedrückt in g/cm3 (1 cm3 = 1 ml) bzw. kg/l; der Zahlenwert wird häufig dimensionslos geschrieben. Dichte von Wasser bei 4˚C = 1.0 g/cm3 , bei 20˚C = 0.998 g/cm3 . Massenanteil (= Massengehalt) In Mischungen wird der Massenanteil w(X) der Komponente X definiert als: w(X) = m(X) mges mges = Masse der gesamten Mischung. Beispiel: Man mischt 90 g Wasser mit 10 g Natriumchlorid (NaCl) (= 100 g Mischung). Dann ist w(NaCl) = 10 g(NaCl) = 0.1 100 g w ist dimensionslos. Die Angabe des Gehaltes in Prozent, ergibt sich daraus mit 100 % · w(X). Im obigen Beispiel ist 100 % · w(NaCl) = 10 % mit w(NaCl) = 0.1. Wird der Gehalt einer Lösung oder Mischung in Prozent angegeben, so bezieht sich das immer auf die Massenprozent. Beziehen sich die Angaben auf die Volumenprozent, so wird das immer angegeben (z. B. eine Flasche Wein enthält 11 %Vol. Alkohol). Der Lösungsvorgang Salze (Ionenverbindungen) lösen sich in polaren Medien unter Ausbildung von Ion-Dipol-Beziehungen. Nicht-ionische, aber polare Verbindungen dagegen gehen in polaren Medien Dipol-Dipol-Beziehungen mit Lösungsmittelmolekülen ein, die ihre Mischung oder Lösung begünstigen. In Substanzen die eine OH-Gruppe besitzen (Wasser, Alkohole, Zucker) spielt besonders die gegenseitige Bildung von Wasserstoffbrücken zwischen den Molekülen des Lösungsmittels und denen des gelösten Stoffes eine Rolle. Unpolare Stoffe dagegen lösen sich nicht, oder nur zu einem geringen Teil in polaren Lösungsmitteln. Die intermolekularen Kräfte zwischen unpolaren Molekülen beschränken sich auf van-der-Waals Kräfte. Merke: Ähnliches löst sich in Ähnlichem: Polare Stoffe in polaren Lösungsmitteln, unpolare Substanzen in unpolaren Lösungsmitteln. 3.2. LÖSUNGEN UND MISCHUNGEN (2. WOCHE) 25 Enthalpie Chemische Vorgänge, z. B. das lösen eines Stoffes in einem Lösungsmittel oder eine chemische Reaktion können entweder freiwillig ablaufen (spontan) oder indem man dem System von außen Energie zuführt. In diesem Kapitel werden Sie Prozesse kennenlernen, die unter Energieabgabe in die eine und unter Energieaufnahme in eine andere Richtung ablaufen können. Es gibt aber auch Prozesse, die nur in eine bestimmte Richtung ablaufen können: Lässt man z. B. einen Ball aus einer bestimmten Höhe auf eine Fläche fallen, wird er mehrmals von dieser Fläche abprallen, dabei nach und nach Energie in Form von Wärme an die eigenen Moleküle und die der Fläche abgeben und schließlich liegenbleiben. Führt man jedoch umgekehrt den Molekülen der Fläche und des Balls Wärmeenergie zu, wird keinesfalls der Umkehrprozess stattfinden und der Ball plötzlich zu springen anfangen. Die Thermodynamik untersucht die Energieänderungen in Systemen und die sich daraus ergebenden Konsequenzen für Vorgänge in diesen Systemen. Sie beschäftigt sich dazu detailliert mit den Energiezuständen der betrachteten Systeme. Der erste Hauptsatz der Thermodynamik besagt, dass Energie weder erzeugt, noch vernichtet werden kann. Wenn bei einer chemischen Reaktion Energie frei wird, bedeutet dies, dass diese Energie vorher schon irgendwie in dem System enthalten gewesen sein muss. Man spricht deshalb von der sogenannten Inneren Energie U . Wenn an einem System Arbeit geleistet wird oder Wärme zugeführt wird, muss sich diese Innere Energie ändern. Führen wir dem System z.B. eine unendlich kleine Wärmemenge dq zu, ohne dass wir eine Volumenänderung oder eine andere Form von Arbeit an dem System erlauben, so ändert sich die Innere Energie um den Betrag dU = dq (konstantes Volumen!). Allerdings erfolgen die meisten Vorgänge im Labor nicht unter der Voraussetzung konstanten Volumens, sondern konstanten Drucks. Das hat zur Folge, dass bei der Wärmezufuhr nicht die gesamte Wärmeenergie in Innere Energie umgewandelt wird, sondern ein Teil für Volumenarbeit pdV aufgewendet werden muss: dU + pdV = dq (konstanter Druck!). Dies kann man zusammenfassen zu dH = dqp , wobei der Index p in dqp anzeigen soll, dass es sich um die bei konstantem Druck geflossene Wärmemenge handelt. Diese Wärmemenge hat man 26 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE dadurch mit der Änderung einer anderen Systemgröße, die Enthalpie H des Systems, gleichgesetzt. Ihre Änderung bei einem bestimmten Vorgang entspricht nach der obigen Formel also der bei diesem Vorgang aufgenommenen oder abgegebenen Wärmeenergie bei konstantem Druck. Sie hängt mit der Inneren Energie zusammen nach H = U + pV und ist für die Chemie von außerordentlicher Bedeutung. Alle denkbare Vorgänge sind gekennzeichnet durch die Enthalpie des betrachten Systems vor (H1 ) und nach (H2 ) dem Vorgang. Die Änderung der Enthalpie bei einem beliebigen Vorgang ist also ∆H = H2 − H1 . Wenn z. B. bei einer chemischen Reaktion Energie (z. B. in Form von Wärme) frei wird ist die sogenannte Reaktionsenthalpie ∆Hr < 0 und man spricht von einer exothermen Reaktion. Wird Energie verbraucht, ist ∆Hr > 0 und man spricht von einer endothermen Reaktion. Bei Lösungsvorgängen kann man dementsprechend anhand von Lösungsenthalpien exotherme und endotherme Lösungsvorgänge unterscheiden. Anhand dieser Betrachtung kann man beurteilen, ob ein bestimmter gedachter Vorgang überhaupt stattfinden kann: die Gesamtenergie des Sytems muss erhalten bleiben. Andererseits kann man erklären, warum bei manchen Vorgängen Energie aufgewendet werden muss, bei anderen aber Energie frei wird. Nimmt z. B. die Enthalpie der gelösten Moleküle eines Salzes gegenüber der kristallinen Form ab, wird sich die Lösung bei Auflösen des Salzes erwärmen, da die freigewordene Energie im System erhalten bleiben muss. Ist die Enthalpie der gelösten Moleküle jedoch höher als im Kristall, muss Energie in Form von Wärme von den Molekülen des Lösungsmittels abgegeben werden und die Lösung kühlt sich ab. Es kann jedoch noch nicht erklärt werden, warum andere Vorgänge, wie in dem oben erwähnten Beispiel das spontane Springen eines Balls auf einer heißen Fläche, trotz Energiezufuhr nicht stattfinden werden, obwohl sie aufgrund des 1. Hauptsatzes der Thermodynamik durchaus erlaubt wären. Diese Frage wird in einem späteren Kapitel mit Hilfe des 2. Hauptsatzes der Thermodynamik geklärt werden. 3.2. LÖSUNGEN UND MISCHUNGEN (2. WOCHE) 3.2.1 27 Versuch: Lösen von Kupfersulfat Stichworte: Kristallgitter, Kristallwasser, Hydration, Wärmetönung, Enthalpie (∆H), Entropie (∆S), Freie Enthalpie (∆G), endotherm, exotherm Allgemeines: Das blaue Kupfersulfat enthält eine gewisse Menge sogenanntes Kristallwasser. Das darin enthaltene Cu2+ -aqua-Ion ist für die blaue Farbe verantwortlich. Entzieht man ihm dieses Kristallwasser, so wird es farblos (bzw. weiß). Man spricht dann auch von wasserfreiem Kupfersulfat. Enthält ein Salz Kristallwasser, so gibt man dieses in folgender Weise in dessen Summenformel an: BaCl2 · 2H2 O. Das Salz Bariumchlorid enthält also im Kristall zwei Moleküle Wasser auf ein Molekül Bariumchlorid, d. h. das Molverhältnis in diesem Kristall zwischen BaCl2 und H2 Obeträgt 1:2. Die Wassermoleküle sind in dem Kristallgitter eingebaut. Chemikalien: CuSO4 · xH2 O Durchführung: Es wird auf einer Waage (Genauigkeit mind. ±0.1 g; Assistent befragen!) 1 g CuSO4 · xH2 Oin einer kleinen Porzellanschale abgewogen. Dabei sollte auch das Gesamtgewicht der Porzellanschale notiert werden. Die Porzellanschale wird mittels Dreifuß und Drahtnetz über dem Bunsenbrenner erhitzt. Dabei sollte man mit dem Bunsenbrenner unter der Porzellanschale solange fächeln, bis sich das Kupfersulfat von blau bis schmutzig“ ” weiß verfärbt hat. Nach dem Abkühlen wird nun zügig die Gewichtsdifferenz bestimmt. Sie gibt das Gewicht des Kristallwassers an. Jetzt lässt sich mit Hilfe der Atommassentabelle im Anhang oder eines Periodensystems errechnen, wieviel mol Kristallwasser pro mol CuSO4 abgegeben wurden. Das Molverhältnis und X können jetzt berechnet und protokolliert werden. Das wasserfreie CuSO4 wird nun in ein Reagenzglas gefüllt und in 3 ml Wasser gelöst. Mit einem Thermometer bestimmt man die Temperaturveränderung. Aufgaben: a.) Finden Sie heraus, wieviel Kristallwasser blaues Kupfersulfat enthält. Vervollständigen Sie die Formel: CuSO4 · xH2 O. (Wie groß ist x?) 28 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE b.) Protokollieren und erklären Sie eine evtl. auftretende Temperaturdifferenz beim Lösen von entwässertem Kupfersulfat. Entsorgung: Die Kupfersulfatlösung wird in den Behälter für wässrige Schwermetalle gegeben. Ende Versuch 3.2.1 3.2.2 Versuch: Lösen von Kaliumchlorid Stichworte: Siehe vorheriger Versuch. Allgemeines: Löst man verschiedene Salze in Wasser, so stellt man fest, dass sich je nach gelöstem Salz die Temperatur der Lösung ändert. Bei Salzen die sich endotherm lösen, kühlt sich die Lösung ab. Der Lösungsprozess dieser Salze hat eine positive Lösungsenthalpie ∆H. Salze die sich exotherm lösen, haben eine negative Lösungsenthalpie ∆H: Die Lösung erwärmt sich. Diese Eigenschaft der Salze macht man sich bei Kältemischungen zunutze. Beispielsweise kann man durch Mischen von wasserhaltigem Kalziumchlorid (CaCl2 · 6H2 O) und Eis eine Temperatur von -55˚C erreichen. Chemikalien: KCl Durchführung: 25 ml Wasser werden im Erlenmeyerkolben auf ihre Temperatur geprüft, dann 10 g Kaliumchlorid (KCl) zugefügt und rasch unter Rühren mit dem Glasstab gelöst. Die Temperatur wird mit dem Thermometer verfolgt. Aufgaben: Protokollieren Sie die Temperaturänderung. Welches Vorzeichen hat ∆H bei diesem Prozess? Ist er endotherm oder exotherm? Entsorgung: Ausguss. Ende Versuch 3.2.2 3.2. LÖSUNGEN UND MISCHUNGEN (2. WOCHE) 29 3.2.3 Versuch: Lösungsenthalpie Boxweise Stichworte: Enthalpie, Lösungsenthalpie, exotherm, endotherm Allgemeines: Die Lösungsenthalpie ist diejenige Energiemenge, welche beim lösen eines Stoffes in einem Lösungsmittel (z. B. eines Salzes in Wasser) bei konstantem Druck abgegeben oder aufgenommen wird. Erwärmt sich die Lösung beim Lösungsvorgang, so wird Energie in Form von Wärme frei (∆H < 0). Man spricht dann von einem exothermen Lösungsvorgang. Kühlt sich die Lösung ab, wird Wärmeenergie für den Lösungsvorgang verbraucht (∆H > 0). Man spricht dann von einem endothermen Lösungsvorgang. In diesem Versuch soll untersucht werden, ob die Lösungsenthalpie eines Salzes von dessen schon gelöster Konzentration abhängt. Chemikalien: NH4 NO3 Durchführung: Die im Folgenden beschriebenen Schritte müssen zügig hintereinander durchgeführt werden, um die Messfehler durch Temperaturausgleich mit der Umgebung möglichst klein zu halten. Es ist ratsam, den Versuch dementsprechend vorzubereiten. Die Portionen sollten schon vorher abgewogen werden und die unten beschriebene Tabelle im Laborjournal vorbereitet sein. a.) In ein 250 ml Becherglas werden 150 ml entionisiertes Wasser gefüllt. Danach ist die Anfangstemperatur zu ermitteln. Jetzt werden 15 g Ammoniumnitrat hinzugegeben und durch Rühren mit dem Glasstab komplett aufgelöst. Direkt nach dem Auflösen wird die Temperatur gemessen. Insgesamt werden auf diese Weise 10 Portionen Ammoniumnitrat (15 g) nacheinander aufgelöst und die Temperatur jeweils kurz nach dem Auflösen gemessen. b.) 80 g Ammoniumnitrat werden in das 250 ml Becherglas gefüllt und aufgelöst. Nach dem Auflösen wird auch jetzt wieder die Temperatur gemessen. Danach werden wie oben 5 Portionen (10 g) Ammoniumnitrat nacheinander aufgelöst und die Temperaturen kurz nach dem Auflösungsvorgang ermittelt und protokolliert. 30 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Aufgaben: Die jeweils gemessenen Temperaturen werden in die folgende Tabelle eingetragen und die Temperaturunterschiede ∆T errechnet. Verändern sich diese, so ist die Lösungsenthalpie konzentrationsabhängig. Beispiel: NH4 NO3 [g] Temperatur ˚ [C] Temperaturdifferenz ∆T ˚ [C] 0 +22.3 15 +13.9 30 +8.7 - 8.4 usw. 45 +5.7 60 +2.9 75 +0.6 90 -1.2 105 -2.2 120 -2.8 135 -2.9 150 -3.1 Entsorgung: Behälter für wässrige Schwermetallabfälle. Ende Versuch 3.2.3 3.2.4 Versuch: Biomoleküle in Lösung In diesem Versuch soll das Lösungsverhalten eines Proteins bei verschiedenen Bedingung beobachtet und erklärt werden. Stichworte: Reversible und irreversible Prozesse, Primär- und Sekundärstruktur bei Proteinen, Denaturierung, saure, basische und neutrale Salze Allgemeines: Ob Proteine in Wasser löslich sind, hängt nicht zuletzt von ihrer Sekundärstruktur ab. Wird diese verändert, so kann sich das Lösungsverhalten drastisch ändern. Vergleichen Sie das rohe Eiweis mit dem in der Pfanne gebratenen. Das eine löst sich recht gut in Wasser, das andere ist nicht mehr in Lösung zu bringen. Der Prozess ist nicht mehr umkehrbar ohne die Moleküle zu zerstören, also irreversibel. Chemikalien: Globulin oder Albumin, 0.9 % NaCl, NH4 SO4 , Ethanol Durchführung: Eine Spatelspitze (ca. 20–30 mg) des Proteins Globulin oder Albumin wird in einem Reagenzglas in 3 ml physiologischer Kochsalzlösung (0.9% NaCl-Lsg.) gelöst und danach zu etwa gleichen Teilen auf drei Reagenzgläser verteilt. Reagenzglas 1: Zugabe von 1 g Ammoniumsulfat (NH4 SO4 ) 3.2. LÖSUNGEN UND MISCHUNGEN (2. WOCHE) 31 Reagenzglas 2: Zugabe von 3 ml Ethanol (CH3 CH2 OH) Reagenzglas 3: Kurzzeitiges Erhitzen über dem Bunsenbrenner auf ca. 80– 90˚C. Aufgaben: Protokollieren Sie Ihre Beobachtung und erklären Sie diese. Falls sich Niederschläge gebildet haben, so prüfen Sie, ob sich diese wieder auflösen. Ist der Vorgang beim erhitzen (Reagenzglas 3) reversibel? Entsorgung: Ausguss. Ende Versuch 3.2.4 32 3.3 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Verteilungsgleichgewichte Stichworte (nachschlagen): Extraktion, Nernstscher Verteilungssatz, Verteilungskoeffizient, Diffusion, osmotischer Druck, thermische Bewegung von Molekülen (Brownsche Molekularbewegung), Geschwindigkeitsverteilung, Aggregatzustände Entropie In Lösungen herrschen ebenso wie im Gaszustand keine statischen Verhältnisse, sondern die Moleküle besitzen kinetische Energie und befinden sich in ständiger, regelloser Bewegung (Brownsche Molekularbewegung). Sie ist abhängig von der Masse der Teilchen und der Temperatur. In solchen ungeordneten Systemen ist die sogenannte Entropie S anschaulich leicht verständlich: Sie ist ein Maß für die Unordnung in einem System. Ohne weitere Grundannahmen lässt sich ihr Betrag nicht absolut festlegen. Leichter ist es aber, ihre Änderung quantitativ zu erfassen. Wird einem bestimmten System (in dem also die Masse der Teilchen festgelegt ist) eine Energiemenge q in Form von Wärme zugeführt, so wird sich die Entropie erhöhen, weil auch die ungeordnete Bewegung der Teilchen zunehmen wird. Diese Änderung ist aber bei niedriger Temperatur größer als bei hohen Temperaturen: ∆S = q/T In Mischungen, in denen zunächst räumlich getrennt unterschiedlich hohe Stoffkonzentrationen existieren, oder an der Grenze zweier Phasen (z. B. Flüssigkeit/Gas oder zwei miteinander nicht mischbare Flüssigkeiten) führt dies freiwillig und ohne äußere Energiezufuhr zur Einstellung dynamischer Gleichgewichte. Solche reversiblen Prozesse ohne chemische Stoffänderung sind in der Natur und in lebenden Zellen ebenso häufig und wichtig wie in der Technik (Verdunstung und Kondensation; Diffusion, Osmose, Dialyse; Extraktion, Destillation). Bei den folgenden Versuchen überlege man sich jeweils, wie die beobachteten Effekte zustande kommen und wie die Energieund Entropieänderungen sein müssen. Die Extraktion Die Extraktion ist eine auf einem selektiven Lösevorgang beruhende Trennmethode, die als Fest-Flüssig-Extraktion oder als Flüssig-Flüssig-Extraktion durchgeführt werden kann. Bei der Fest-Flüssig-Extraktion werden Komponenten durch bestimmte Extraktionsmittel aus einem Feststoffgemisch gezielt 3.3. VERTEILUNGSGLEICHGEWICHTE 33 aufgenommen. Diese Extraktion kann z. B. zur Gewinnung von Naturstoffen aus Biomaterial verwendet werden. Bei der Flüssig-Flüssig-Extraktion wird eine Flüssigkeit aus einem Flüssigkeitsgemisch oder ein gelöster Stoff aus einer Lösung abgetrennt. Wird eine Substanz zwischen zwei wenig oder nicht mischbaren flüssigen Phasen A und B verteilt, so stellt sich ein Lösungsgleichgewicht ein, d. h. das Konzentrationsverhältnis der Substanz in beiden Phasen stellt sich entsprechend der Löslichkeit in dem jeweiligen Lösungsmittel ein. Der Nernstschen Verteilungssatz beschreibt dieses Verhältnis anhand der Konzentrationen c(Phase 1) und c(Phase 2) des Stoffes in den beiden Phasen mit dem Verteilungskoeffizienten α: c(Phase 1) =α c(Phase 2) 3.3.1 Versuch: Verteilung von Iod zwischen zwei Phasen, Extraktion Stichworte: Nernstscher Verteilungssatz, Phasen, Extraktion Allgemeines: Wasser und Chloroform sind praktisch nicht miteinander mischbar. Werden beide in ein Gefäß gegeben, bilden sich schnell zwei Schichten: eine wässrige und eine organische Phase. Da die Dichte von Chloroform höher ist als die von Wasser, schwimmt in diesem Falle die wässrige auf der organischen Phase. Mischt man andererseits z. B. Benzin mit Wasser, so schwimmt die organische auf der wässrigen Phase, da die Dichte des Benzins geringer ist als die von Wasser. Iod löst sich in Chloroform wesentlich besser als in Wasser. Damit es sich überhaupt nennenswert in Wasser löst, wird zu dem Iod das Salz Kaliumiodid (KI) hinzugegeben (Iod/KI-Lösung). In beiden Phasen ist Iod mit unterschiedlicher Konzentration c löslich. Quantitativ lässt sich das mit dem Nernstschen Verteilungskoeffizienten α ausdrücken. Nernstscher Verteilungssatz: c(Phase 1) =α c(Phase 2) Chemikalien: Chloroform, Iod/KI-Lösung, KI 34 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Durchführung: Dieser Versuch darf nur unter dem Abzug durchgeführt werden!! a.) Reversibler Phasenübertritt: 1 ml der ausgegebenen Iod/KI-Lösung wird mit Wasser auf das 20-fache verdünnt. In ein Reagenzglas pipettiert man nun 5 ml dieser Lösung und 5 ml Chloroform (CHCl3 ). Das Reagenzglas wird kräftig geschüttelt. Was beobachten Sie? Vorsicht !!! Das Reagenzglas nicht mit dem Daumen verschließen! Chloroform ist gesundheitsschädlich und darf nicht mit der Haut in Berührung kommen! Die organische (untere) Phase wird nun mit einer Pasteurpipette abgesaugt und in ein anderes Reagenzglas überführt. Nach Zugabe von 5 ml Wasser und einiger Kristalle KI, wird wieder kräftig geschüttelt. Protokollieren Sie auch hier ihre Beobachtungen. b.) Wirksamkeit vielfacher bzw. einmaliger Extraktion: In zwei Reagenzgläser werden je 5 ml der Iod/KI-Lösung pipettiert. Reagenzglas 1: Es wird dreimal mit je 5 ml Chloroform extrahiert. Die letzte Fraktion (Chloroformphase) wird in einem Reagenzglas zum Vergleich aufbewahrt. Reagenzglas 2: Es wird solange mit jeweils 1 ml Chloroform extrahiert, bis die Chloroformphase in etwa die gleiche Färbung aufweist wie die Vergleichsprobe. Aufgaben: a.) Protokollieren Sie ihre Beobachtung und erklären Sie diese. Beschreiben Sie in eigenen, kurzen Worten was passiert ist. b.) Protokollieren Sie ihre Beobachtungen und vergleichen Sie den Chloroformverbrauch der beiden Extraktionen bis zum Erreichen desselben Extraktionsgrades. Welche Extraktion war effektiver? Entsorgung: Organische Lösungsmittelabfälle Ende Versuch 3.3.1 3.3. VERTEILUNGSGLEICHGEWICHTE 3.3.2 35 Versuch: Diffusion Stichworte: Diffusion, Brownsche Molekularbewegung Allgemeines: In zwei ursprünglich getrennten, aber mischbaren Phasen verschwindet im Laufe der Zeit durch Diffusion die Phasengrenze und das Konzentrationsgefälle, auch ohne mechanisches Vermischen der Phasen z. B. durch Umrühren. Dies geschieht aufgrund der temperaturabhängigen Braunschen Molekularbewegung. Chemikalien: Riboflavin (Vitamin B2 ), Glycerin/Wasser(1:1)-Lösung Durchführung: Ein Reagenzglas wird ca. 3 cm hoch mit der ausgegebenen Riboflavin-Lösung gefüllt. Danach unterschichtet man vorsichtig mit Hilfe einer Pasteurpipette die Riboflavin-Lösung mit der ausgegebenen Glycerin/Wasser-(1:1)-Lösung, indem man die Pasteurpipette bis auf den Grund des Reagenzglases bringt und vorsichtig auslaufen lässt. Dabei sollten sich die beiden Phasen möglichst nicht miteinander mischen. Stellen Sie das Reagenzglas an einen ruhigen Ort und bewegen Sie es nach Möglichkeit nicht mehr. Aufgaben: Beobachten Sie die Phasengrenze und die Färbung über einen längeren Zeitraum. Protokollieren und erklären Sie ihre Beobachtungen. Nimmt die Entropie bei diesem Prozess zu oder ab? Entsorgung: Wässrige Schwermetallabfälle Ende Versuch 3.3.2 36 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE 3.3.3 Versuch: Dialyse von Riboflavin/Hämoglobin Boxweise Stichworte: Dialyse, semipermeable Membran Allgemeines: Sind zwei mischbare Lösungen durch eine semipermeable Membran getrennt, so kann ein Ausgleich von Konzentrationsunterschieden durch Diffusion nur für solche Teilchen erfolgen, die die Membran wegen ihrer passenden Größe oder Struktur passieren können. Die Dialyse ist eine biochemische Methode zum Entfernen kleiner Moleküle aus Lösungen von Makromolekülen. Auch in den Körperzellen finden ähnliche Prozesse aufgrund der semipermeablen Zellmembranen statt. Chemikalien: Riboflavin (0.1 mg/ml), Hämoglobin (10 mg/ml), 0.9 % NaCl-Lösung, Dialyseschlauch Durchführung: Lassen Sie sich vom Assistenten ein ca. 10 cm langes Stück Dialyseschlauch geben. Dieser wird dann an einem Ende verknotet. Der Dialyseschlauch muss vor Gebrauch erst ca. 5 min. in einem Becherglas mit Wasser vorquellen. In diesen vorgequollenen Dialyseschlauch wird nun die ausgegebene Lösung aus Riboflavin (gelb und niedermolekular) und Hämoglobin (braunrotes Protein) in 0.9 %iger NaCl-Lsg. gegeben und das offene Ende mit einem Draht oder Bindfaden knapp über dem Flüssigkeitsstand abgebunden. Der gefüllte Dialyseschlauch wird jetzt in ein mit Wasser gefülltes Becherglas gegeben und über einige Stunden beobachtet. Aufgaben: Protokollieren Sie ihre Beobachtungen und erklären Sie diese. Entsorgung: Die Lösungen können in den Ausguss entsorgt werden. Der Dialyseschlauch kann in den normalen Hausmüll gegeben werden. Ende Versuch 3.3.3 3.4. CHEM. GLEICHGEWICHTE, MASSENWIRKUNGSGESETZ 3.4 37 Chemische Gleichgewichte, Massenwirkungsgesetz Stichworte: Massenwirkungsgesetz, Löslichkeitsprodukt, Gleichgewichtskonstante und Freie Energie ∆G, Prinzip von Le Chatelier Freie Energie Im letzten Kapitel haben Sie die Entropie als ein Maß für die Unordnung eines Systems kennengelernt. Sie ist, genauso wie die Innere Energie oder die Enthalpie eine Zustandsgröße und beschreibt genau wie diese einen Zustand des betrachteten Systems. Während die Innere Energie und die Enthalpie bestimmte Energiezustände beschreiben, kann man die Entropie als ein Maß für die Qualität der Energie des Systems ansehen. Mit detaillierten Betrachtungen lässt sich zeigen, dass die Entropie des Weltalls immer nur zunehmen kann. Die gleiche Aussage kann man auf abgeschlossene Systeme beziehen, die keinerlei thermischen Kontakt zu ihrer Umgebung haben. In einem offenen System kann die Entropie durchaus auch abnehmen, wie etwa im menschlichen Körper. Gleichzeitig muss aber die Entropie in der Umgebung des Systems, wie z. B. unserer Umwelt, zunehmen. Neben der Energieerhaltung ist diese Zunahme der Unordnung oder Abnahme der Energiequalität ein weiteres Kriterium für die Möglichkeit, ob ein bestimmter Vorgang stattfinden kann. Für Systeme mit konstantem Volumen bzw. konstantem Druck kann man die Änderungen der Inneren Energie und der Enthalpie deshalb sinnvoll mit der Änderung der Entropie zusammenfassen: dA = dU − T dS für Systeme bei konstantem Volumen und konstanter Temperatur und dG = dH − T dS für Systeme bei konstantem Druck und konstanter Temperatur. Durch diese Änderungen sind dann auch wieder zwei neue Zustandsgrößen definiert, die sogenannte Freie Energie und die Freie Enthalpie: A = U − T S Freie Energie (Helmholtz-Funktion) G = H − T S Freie Enthalpie (Gibbs-Funktion) Für die Chemie wichtiger ist die Freie Enthalpie, da sie sich auf Reaktionen und Vorgänge bezieht, die bei konstantem Druck ablaufen. Solche Vorg¨ange laufen genau dann spontan ab, wenn ∆ G < 0. Man spricht dann von einem exergonischen Vorgang. Vorg¨ange mit ∆ G > 0 laufen nicht spontan ab und werden als endergonisch bezeichnet. 38 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Das Massenwirkungsgesetz (MWG): In einer Reaktion, in der Edukte und Produkte miteinander im Gleichgewicht stehen, d. h. sowohl Edukte wie auch Produkte nach Beendigung der Reaktion in merklicher Konzentration nachweisbar sind, gilt das Massenwirkungsgesetz. Hat sich das Gleichgewicht eingestellt, erfolgt also keine Konzentrationsänderung der beteiligten Stoffe mehr, so lässt sich das Gleichgewicht durch die Gleichgewichtskonstante K ausdrücken. Reaktionsgleichung: A+B* )C+D Massenwirkungsgesetz für obige Reaktion im Gleichgewicht: K= c(C)c(D) Produkte = Edukte c(A)c(B) In Worten ausgedrückt: Das Produkt der Konzentrationen der Produkte dividiert durch das Produkt der Konzentrationen der Edukte ist im Gleichgewicht konstant. Für Systeme im Gleichgewicht, also auch für diese Gleichgewichtsreaktion, gilt das Prinzip von Le Chatelier: Übt man auf ein im Gleichgewicht befindliches System durch Änderung der Bedingungen (z. B. Temperatur, Druck, Konzentration) einen Zwang aus, so verschiebt sich das Gleichgewicht derart, dass es dem Zwang ausweicht. Wird nun beispielsweise die Konzentration von D erhöht, indem wir etwas D von außen zugeben (Temperatur bleibt konstant), so weicht das System gemäß diesem Prinzip aus: Auch die Konzentration der Edukte muss sich erhöhen, damit K konstant bleibt. Das kann nur dadurch geschehen, dass C und D solange miteinander reagieren, bis das Verhältnis der Konzentrationen von Produkten und Edukten wieder den konstanten Wert erreicht hat. K ist im obigen Fall dimensionslos. Bei einer Reaktion mit anderer Stöchiometrie, wie z. B. A+B* )C hat K die Einheit mol−1 ·l. Um dimensionslose Gleichgewichtskonstanten zu bekommen, formuliert man das Massenwirkungsgesetz besser unter Verwendung der Standardkonzentration. Sie ist definiert als cst = 1 mol · l−1 . Geht man allgemein von einer Reaktion mit beliebigen stöchiometrischen Faktoren aus, so kann die Reaktionsgleichung für eine allgemeine Gleichgewichtsreaktion als aA + bB * ) cC + dD 3.4. CHEM. GLEICHGEWICHTE, MASSENWIRKUNGSGESETZ 39 formuliert werden. Die stöchiometrischen Faktoren a, b, c und d sind in der Regel kleine Zahlen. Man definiert: c∗ (X) = c(X)/cst Diese Quotienten c∗ (X) sind dimensionslos. Damit wird K unter Einbeziehung der Standardkonzentration immer dimensionslos: K= c∗ (C)c · c∗ (D)d c∗ (A)a · c∗ (B)b K und ∆G: Auch für Gleichgewichtsreaktionen gelten die thermodynamischen Kriterien für den Ablauf von Vorgängen. Wenn im Laufe einer spontan ablaufenden Gleichgewichtsreaktion immer mehr Edukte abreagieren, wird sich dies auch auf die Änderung der Freien Enthalpie auswirken: Sie wird für die Hinreaktion immer kleiner werden. Für die Rückreaktion wird sie in der gleichen Zeit immer größer werden. Im Gleichgewichtszustand sind dann beide gleich groß, nur mit unterschiedlichem Vorzeichen. Insgeamt ändert sich dann die Freie Enthalpie des Systems nicht mehr, die Reaktion kommt scheinbar zum Stillstand (tatsächlich laufen aber Hin- und Rückreaktion mit der gleichen Reaktionsgeschwindigkeit ab!). Bis dahin muss bei der Reaktion ein bestimmter Betrag an Freier Enthalpie freigeworden sein. Bezieht man diese Freie Enthalpie auf einen definierten Standardzustand, steht sie mit der dimensionslosen Gleichgewichtskonstanten K im Zusammenhang: ∆G = −RT ln K Dabei ist ∆G die Freie Standard Enthalpie, R = 8.134×10−3 kJ·mol−1 ·K−1 die Gaskonstante und T die absolute Temperatur in Kelvin (z. B. 0˚C = 273.15 K). Das Löslichkeitsprodukt: Gibt man zu einer Substanz nur soviel Lösungsmittel, dass sich nicht alles auflöst, so bleibt das Ungelöste als Bodenkörper zurück, über dem sich, wenn das System im Gleichgewicht ist, die gesättigte Lösung der Substanz befindet. Man kann auch hier das MWG formulieren. Aa Bb sei ein Salz, das in seine Ionen Ai+ und Bj− dissoziiert. Reaktionsgleichung der Dissoziation: Aa Bb → aAi+ + bBj− 40 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE MWG: c(Ai+ )a c(Bj− )b K= c(Aa Bb ) Bei vorhandenem Bodenkörper kann man c(Aa Bb ) als konstant ansehen. Man kann deshalb das Produkt (Aa Bb ) · K zur Konstanten Lp zusammenfassen. Lp wird als Löslichkeitsprodukt bezeichnet. Es gilt dann: c(Ai+ )a c(Bj− )b = Lp Dimension: mola+b · l−(a+b) Die Dimension von Lp ist durch die Zahlen a und b festgelegt. Unter Verwendung von c∗ kann man auch ein dimensionsloses Lp definieren. Beispiele zur Anwendung des Löslichkeitsproduktes: 1. Silberchlorid Silberchlorid (Ag Cl) ist ein schwerlösliches Salz: Lp(Ag Cl) = c(Ag+ ) · c(Cl− ) = 1.7 × 10−10 mol2 · l−2 . In der gesättigten Lösung ist also die Konzentration an Silber-Ionen: p c(Ag+ ) = c(Cl− ) = 1.7 × 10−10 mol2 · l−2 = 1.3 × 10−5 mol · l−1 . Stellt man eine gesättigte Lösung von Ag Cl in einer NaCl-Lösung mit c(NaCl) = 0.1 mol/l her, so ist c(Cl− ) fast gleich 0.1 mol/l. Die Silberionen-Konzentration berechnet sich nach dem Ansatz: c(Ag+ ) · 0.1 mol · l−1 = 1.7 × 10−10 mol2 · l−2 zu c(Ag+ ) = 1.7 × 10−9 mol · l−1 , d. h. in dieser Lösung sinkt die Silberionen-Konzentration um vier Zehnerpotenzen. Das System hat gemäß dem Prinzip von Le Chatelier reagiert. 2. Bleichlorid Bleichlorid (PbCl2 ) ist nicht ganz so schwerlöslich wie Silberchlorid (Ag Cl). Lp(Pb Cl2 ) = c(Pb2+ ) · c(Cl− )2 = 2.0 × 10−5 mol3 · l−3 Da aufgrund der Summenformel PbCl2 nach der Dissoziation für jedes BleiIon (Pb2+ ) zwei Chlorid-Ionen (Cl− ) existieren, gilt: c(Cl− ) = 2 · c(Pb2+ ) bzw. 0.5 · c(Cl− ) = c(Pb2+ ) 3.4. CHEM. GLEICHGEWICHTE, MASSENWIRKUNGSGESETZ 41 Die Konzentration an Blei-Ionen berechnet sich aus dem Ansatz: c(Pb2+ ) · (2 · c(Pb2+ ))2 = 2.0 × 10−5 mol3 · l−3 p 3 2+ c(Pb ) = 0.5 × 10−5 mol3 · l−3 = 1.7 × 10−2 mol · l−1 . Die Konzentration an Chlorid-Ionen ist doppelt so hoch und berechnet sich zu: c(Cl− ) = 3.4 × 10−2 mol · l−1 . Löst man PbCl2 in einer Natriumchlorid-(NaCl)-Lösung mit c(NaCl) = 0.1 mol/l, sinkt die Konzentration an Blei-Ionen. Zur Vereinfachung nimmt man an, dass c(Cl− ) = 0.1 mol/l. Dann ist wegen c(Pb2+ ) · (0.1)2 mol2 · l−2 = 2.0 × 10−5 mol3 · l−3 die Konzentration an Blei-Ionen c(Pb2+ ) = 2 × 10−3 mol/l, d. h. ca. eine Zehnerpotenz geringer als ohne zusätzliche Chlorid-Ionen. Exakt kann man diese Konzentration mit folgendem Ansatz berechnen: c(Pb2+ ) · (2 · c(Pb2+ ) + 0.1 mol · l−1 )2 = 2.0 × 10−5 mol3 · l−3 . Die Lösung dieser Gleichung 3. Grades ergibt: 1.85×10−3 mol/l. Dieses Ergebnis zeigt, dass die oben durchgeführte Vereinfachung gerechtfertigt ist. Erst wenn der Zahlenwert Lp ≥ 10−3 wird, führt die Vereinfachung zu merklichen Abweichungen. Die Löslichkeit Die Löslichkeit ist definiert als Gramm (g) gelöste Substanz in 100 Gramm (g) Lösungsmittel. Beispiel: eine gesättigte Lösung von KClO4 in Wasser (bei 20˚C) ist 1.7 %ig. 100 g dieser Lösung enthalten also 98.3 g Wasser und 1.7 g KClO4 . Die Löslichkeit beträgt also 1.75 g/100 g Wasser. Will man c(KClO4 ) dieser Lösung berechnen, so benötigt man die Dichte der Lösung; sie ist D = 1.0105 g/ml. Mit M (KClO4 ) = 138.55 g/mol erhält man c(KClO4 ) = 0.124 mol/l. Das Löslichkeitsprodukt berechnet sich daraus zu: Lp = 1.54 × 10−2 mol2 · l−2 . 3.4.1 Versuch: Das Kalkgleichgewicht 42 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Stichworte: Löslichkeitsprodukt, Kohlensäure, Prinzip von Le Chatelier Allgemeines: Carbonat (CO3 2− ) lässt sich aus Kohlendioxid und HydroxidIonen herstellen. CO2 + 2OH− → CO2− 3 + H2 O Kalk bzw. Calciumcarbonat (CaCO3 ) ist das Calciumsalz der Kohlensäure (H2 CO3 ). In wässriger Lösung besteht folgendes Gleichgewicht: * Ca2+ + CO2− 3 ) CaCO3 Das Anion CO3 2− steht wiederum in einem Gleichgewicht mit physikalisch gelöstem Kohlendioxid (CO2 ) und Hydrogencarbonat (HCO3 − ) (Kalkgleichgewicht): CO32− + CO2 + H2 O * ) 2HCO− 3 Bringt man also CO2 in eine Kalklösung ein, so verringert sich aufgrund der Bildung von Hydrogencarbonat die Carbonat-(CO3 2− )-Konzentration. Das Löslichkeitsprodukt Lp von CaCO3 wird unterschritten. Nach dem Prinzip des kleinsten Zwanges von Le Chatelier geht aus dem Bodenkörper gerade soviel CaCO3 in Lösung, bis das Produkt aus Carbonat und Calziumionen wieder den Wert des Löslichkeitsproduktes Lp von Calciumcarbonat besitzt. In der unbelebten Natur tritt diese Reaktion z. B. in Kalkgebirgen (Tropfsteinbildung) auf. Aber auch lebende Organismen wie Muscheln oder Schnecken bedienen sich unter anderem dieser Reaktion um Kalkgehäuse zu bilden. Chemikalien: Ca(OH)2 , Trockeneis Durchführung: a.) Eine Spatelspitze Calciumhydroxid (Ca(OH)2 ) wird in einem 100 ml Becherglas in ca. 50 ml Wasser unter starkem Schütteln gelöst. Die Lösung wird vom unlöslichen Rest in einen 250 ml Erlenmeyerkolben abfiltriert oder mit dem Büchnertrichter abgesaugt. In einen weiteren Erlenmeyerkolben geben Sie etwas Trockeneis und verschließen diesen mit einem durchbohrten Stopfen, in dem ein gebogenes, an der Spitze etwas ausgezogenes Glasrohr steckt. Dieses Glasrohr wird nun in die Lösung getaucht und der Erlenmeyerkolben mit dem Trockeneis mit Hilfe eines 30–40˚C warmen Wasserbades erwärmt. 3.4. CHEM. GLEICHGEWICHTE, MASSENWIRKUNGSGESETZ 43 Der einsetzende CO2 -Strom wird nun so lange in die klare Lösung eingeleitet, bis eine starke Trübung einsetzt. b.) Ein Teil der erhaltenen trüben Lösung wird etwa ein Finger hoch in ein Reagenzglas gefüllt und auch hier CO2 eingeleitet, bis die Trübung sich wieder aufgelöst hat. Aufgaben: a.) Was verursachte die Trübung durch Einleiten von CO2 ? Formulieren Sie die Reaktionsgleichung. b.) Welche Reaktion führte zur Aufklarung der Lösung? Formulieren Sie die Reaktionsgleichung. Entsorgung: Ausguss. Ende Versuch 3.4.1 44 3.4.2 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Versuch: Temperaturabhängigkeit der Gleichgewichtsreaktion von Iod mit Stärke in wässriger Lösung Stichworte: exotherme u. endotherme Reaktionen, Prinzip von Le Chatelier, Iodstärke-Reaktion Allgemeines: Iod reagiert mit Stärkelösung unter intensiver Blaufärbung, die durch die Ausbildung einer Einschlussverbindung (Iodstärke) verursacht wird. Es bildet sich ein Gleichgewicht aus, welches bei Raumtemperatur auf der Seite des Produktes liegt. Iod + Stärke * ) Iodstärke Wird nun die Temperatur geändert, verschiebt sich das temperaturabhängige Gleichgewicht gemäß dem Prinzip von Le Chatelier in Richtung der Edukte. Anhand der Blaufärbung lässt sich gut erkennen, in welche Richtung sich das Gleichgewicht verschiebt. Chemikalien: Verdünnte Stärkelösung (0.5 %), verd. I2 /KI-Lsg. (ca. 0.3 %) Durchführung: In zwei Reagenzgläser werden je 10 ml verdünnte Stärkelösung und einige Tropfen verdünnte Iod-Kaliumiodidlösung gegeben. Eines der beiden Reagenzgläser wird nun in einem Wasserbad, bestehend aus einem mit ca. 180 ml Wasser gefüllten 250 ml Becherglas, mit Hilfe einer Heizplatte langsam auf ca. 60˚C erwärmt (Farbeffekt ?). Beim Erreichen dieser Temperatur wird das Reagenzglas aus dem Wasserbad genommen und neben das als Vergleich dienende zweite Reagenzglas gestellt. Tritt mit fortschreitender Abkühlung ein Farbeffekt ein? Aufgaben: Beobachten und protokollieren Sie eventuell eintretende Farbeffekte und versuchen Sie diese zu erklären. Formulieren Sie eine Hin- und Rückreaktion und geben Sie an, welche exotherm und welche endotherm ist. Entsorgung: Organische Lösungsmittelabfälle. Ende Versuch 3.4.2 3.5. SÄURE/BASE-REAKTIONEN UND PUFFER (3. WOCHE) 3.5 45 Säure/Base-Reaktionen und Puffer (3. Woche) Stichworte: Säure-Base-Begriff nach Brönstedt, Säure-Base-Begriff nach Lewis, Ionenprodukt Kw des Wassers, pH-Wert, Säurestärke, pK-Wert, Dissoziation, Dissoziationsgrad, Pufferlösungen, Salzhydrolyse Säurestärke, Ks -Wert und pH-Wert Die Übertragung eines Protons H+ von einem Molekül auf ein anderes ist eine der einfachsten, schnellsten und häufigsten chemischen Reaktionen. Protonenübertragungen treten ein, wenn ein Molekül aus einer bestimmten Bindung A—H den Wasserstoff ohne Bindungselektronen (eben als H+ ) freisetzen kann und ein anderer Stoff das H+ -Ion an einer bestimmten Struktur B wieder bindet: Der erste Stoff ist eine Säure, der zweite eine Base: AH + B * ) A− + BH+ Säure/Base-Definition nach Brönstedt: Säure = Protonendonator (gibt Protonen ab) Base = Protonenakzeptor (nimmt Protonen auf) Die Tendenz einer Säure ihren Wasserstoff als H+ -Ion (Proton) abzugeben, ist von Säure zu Säure unterschiedlich. Man unterscheidet starke und schwache Säuren. Ein Zahlenwert, der die Stärke einer Säure beschreibt, ist der Ks -Wert (bzw. pKa - oder pKs -Wert). Der Ks -Wert ist die Gleichgewichtskonstante der Dissoziation der Säure in Wasser: AH + H2 O * ) A− + H3 O+ Ks = c∗ (A− ) · c∗ (H3 O+ ) c∗ (AH) · c∗ (H2 O) Beachten Sie die Verwendung standardisierter Konzentrationen (s. S. 39). Hat die Dissoziationskonstante Ks der Säure einen großen Wert, so liegt das Gleichgewicht der Dissoziation auf der rechten Seite, d. h. es handelt sich um eine starke Säure. Kleine Dissoziationskonstanten dagegen weisen auf eine schwache Säure hin; das Gleichgewicht liegt links, der größte Teil der Säure liegt undissoziiert vor. Statt des Ks -Wertes wird oft der pKs -Wert pKs = − log Ks 46 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE als Maß für die Stärke von Säuren verwendet. Kleine oder negative pKs -Werte zeigen an, dass die Säure stark ist, große Werte, dass sie schwach ist. Da es auch verschieden starke Basen gibt, lässt sich auf analoge Weise auch ein Kb bzw. pKb -Wert definieren (siehe Lehrbücher). Das aus einer Säure HA durch Deprotonierung entstehende Anion A− kann H+ wieder aufnehmen und ist daher definitionsgemäß eine Base, nämlich die konjugierte Base“ von HA; ebenso entsteht aus der Base B durch ” Protonierung die konjugierte Säure“ BH+ . An Säure-Base-Reaktionen sind ” daher stets zwei Säure-Base-Paare beteiligt. Der pKs -Wert einer Säure und der pKb -Wert ihrer konjugierten Base hängen in wässriger Lösung wie folgt zusammen: pKs + pKb = 14 Ks · Kb = 10−14 Ostwaldsches Verdünnungsgesetz Eine für schwache Säuren oder Basen ebenfalls interessante Größe ist der Dissoziationsgrad oder Protolysegrad α: α= protolysierte Teilchen gelöste Teilchen vor Protolyse Ist C die Gesamtkonzentration vor der Protolyse, so kann man im Falle einwertiger Säuren das MWG schreiben: 2 α αC · αC =C Ka = C − αC 1−α q Ks 2 Ist α klein, so kann vereinfacht werden zu Ks = Cα bzw. α = . C Dieses sog. Ostwaldsche Verdünnungsgesetz sagt aus, dass der Dissoziationsgrad mit zunehmender Verdünnung der Lösung (abnehmender Konzentration) größer wird. Ionenprodukt des Wassers In wässrigen Systemen muss in die Beschreibung von Säure-Base-Reaktionen die Eigenschaft des Wassers einbezogen werden, sowohl als Säure wie als Base fungieren zu können ( amphotere Natur“, Ampholyt“). In einer Gleichge” ” wichtsreaktion protoniert ein Molekül Wasser ein zweites zum HydroxoniumIon H3 O+ und es entsteht zugleich das Hydroxid-Ion OH− . H2 O + H 2 O * ) H3 O+ + OH− (Autoprotolyse) 3.5. SÄURE/BASE-REAKTIONEN UND PUFFER (3. WOCHE) 47 übliche Kurzform: H2 O * ) H+ + OH− Diese Gleichgewichtsreaktion lässt sich wieder durch eine Gleichgewichtskonstante K beschreiben. K= c(H+ ) · c(OH− ) c(H2 O) Diese hat jedoch bei Normaltemperatur einen sehr kleinen Wert. Es sind nur sehr wenige der neutralen Wassermoleküle protoniert bzw. deprotoniert. Daher bezieht man die praktisch konstante Konzentration des Wassers (55 mol/l) in die Konstante K mit ein und formuliert das sog. Ionenprodukt des Wassers Kw . Kw = c(H+ ) · c(OH− ) = 10−14 mol2 · l−2 (bei 25˚C und Normaldruck; bei 50˚C ist Kw = 5.5 × 10−14 mol2 · l−2 ). Demnach sind die Konzentrationen von H3 O+ und OH− in reinem, entionisiertem Wasser je 10−7 mol · l−1 . Diese Konzentrationen sind gering aber durchaus messbar. Gibt man nun eine Säure (z. B. HCl) in reines Wasser, so werden viel mehr Wassermoleküle protoniert, c(H3 O+ ) wird größer als 10−7 mol · l−1 . Eine Base dagegen (z. B. NH3 ) deprotoniert weitere amphotere Wassermoleküle, c(OH− ) nimmt zu und die H3 O+ -Ionen-Konzentration sinkt wegen der Konstanz von Kw unter 10−7 mol · l−1 . pH-Wert Auf diesem Zusammenhang beruht die Definition des pH-Wertes als Maß der Säure- (genauer: Protonen (H+ )- oder Hydroxonium-Ionen (H3 O+ )-) Konzentration einer Lösung. Um den Umgang mit negativen Exponenten zu vermeiden, wird definiert: pH = − log(c∗ (H+ )) Merke: Der pH-Wert ist der negative dekadische Logarithmus der (Standard)-Protonenkonzentration. Analog kann man auch für Basen definieren: pOH = − log(c∗ (OH− )) Mit dem Ionenprodukt des Wassers gilt: pH + pOH = 14 48 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Puffer Liegt neben einer schwachen Säure auch ihr Salz in derselben Lösung vor, z. B. wenn die Säure partiell mit Base neutralisiert wurde, so muss in der Ableitung des pH-Wertes auch die Konzentration von A− berücksichtigt werden: c∗ (HA) c∗ (HA) c∗ (H+ ) = Ks · ∗ − bzw. pH = pKs − log ∗ − c (A ) c (A ) Gleichung nach Henderson-Hasselbalch: pH = pKs + log c∗ (A− ) c∗ (HA) Ist das Verhältnis zwischen A− und HA 1:1 gilt: pH = pKs . In Pufferlösungen derartiger Zusammensetzung ändert sich der pH-Wert bei begrenztem Zusatz weiterer Säure oder Base kaum, weil entweder die Salzionen A− zusätzliche Protonen zur undissoziierten Säure HA abfangen, oder HA-Moleküle zusätzliche Base unter Bildung von A− neutralisieren. Pufferlösungen dienen daher im Labor zur Aufrechterhaltung eines definierten pH-Wertes, und sie kommen auch in der Natur in großem Maße vor. Enthalten Lösungen Salze, deren Ionen konjugierte Base oder Säure einer schwachen Säure bzw. Base sind (zum Beispiel Natriumacetat oder Natriumcarbonat als Salz der schwachen Essigsäure bzw. Kohlensäure; Ammoniumsulfat als Salz der schwachen Base Ammoniak), so ist deren pH-Wert nicht 7, weil die Ionen selbst Protolysereaktionen eingehen ( Salzhydrolyse“): Acetat” oder Carbonatlösungen enthalten die Anionen der entsprechenden schwachen Säuren als Base und reagieren schwach alkalisch, Ammoniumsalz-Lösungen enthalten das Ammonium-Ion NH4 + als Säure und sind schwach sauer. Der pH-Wert einer solchen Lösung ist: 14 + pKs + log(c∗ (A− )) Salze schwacher Säuren: pH = 2 pKs − log(c∗ (A− )) Salze schwacher Basen: pH = 2 Beispiele: 1 mM (millimolare) Soda-Lösung (Na2 CO3 ), pKs = 10.2; pH = 10.6. 0.5 M Ammoniumsulfat-((NH4 )2 SO4 )-Lösung, pKs = 9.2; pH = 4.6. Säure-Base-Titration, Titrationskurve Werden Säuren und Basen zusammengebracht, so reagieren sie äußerst rasch miteinander unter Neutralisation: Protonen und OH− -Ionen treten unter Abgabe von Neutralisationswärme zu Wasser zusammen, während die entsprechenden Gegenionen nebeneinander als Salz“ in der Lösung verbleiben. ” 3.5. SÄURE/BASE-REAKTIONEN UND PUFFER (3. WOCHE) 49 Säure + Base → Wasser + Salz Erfolgt die Neutralisation schrittweise durch Zutropfen einer Maßlösung“ ” bekannter Konzentration der einen Komponente zu einer vorgelegten Probe der anderen Komponente, so spricht man von einer Titration. Die graphische Darstellung des pH-Wertes während der Titration ist die Titrationskurve. In der Mischung ändert sich der pH-Wert je nach erreichtem Mengenverhältnis der Komponenten zwischen dem Titrationsgrad 0 und 1 (= stöchiometrische Äquivalenz) in einer charakteristischen Weise. Bei Titration mit einer starken Base unterscheidet sich der Titrationsverlauf und die Titrationskurve einer starken und einer schwachen Säure sehr markant. Im zweiten Fall entsteht bei partieller Neutralisation zuerst ein Puffergemisch und im Bereich pH = pKs ist die pH-Änderung nur gering. Beachten Sie den pH-Wert-Unterschied der Lösungen am Äquivalenzpunkt! pH pH Umschlagbereich Phenolphtalein Umschlagbereich Phenolphtalein Äquivalenzpunkt 7 7 Neutralpunkt Umschlagbereich Methylrot Pufferbereich (pH=pKS ) 0.5 1 Titrationsgrad 0.5 1 Titrationsgrad Säure-Base-Titrationen werden häufig analytisch angewandt. Man kann titrimetrisch eine unbekannte Säuremenge mit Hilfe einer bekannten Basenmenge (= Volumen Maßlösung) bestimmen oder umgekehrt ( Alkalimetrie“, ” Acidimetrie“), oder man kann durch Aufnahme einer Titrationskurve die ” charakteristischen pKs -Werte einer Substanz ermitteln. Indikatoren Titrationen erfordern neben der genauen Volumenmessung in Büretten eine verlässliche Anzeige des Endpunktes (Äquivalenzpunktes), an dem bereits bei kleiner Zugabe von Titrationslösung der größte pH-Sprung auftritt. In 50 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE diesem Praktikum benutzen wir pH-Indikatoren zur Endpunktbestimmung, die jeweils bei einem bestimmten pH-Wert (der ihrem eigenen pKs -Wert entspricht) ihre Farbe ändern ( umschlagen“). Dabei muss allerdings je nach Art ” der zu titrierenden Probe ein Indikatorfarbstoff passenden Umschlagbereichs gewählt werden. Häufig gebrauchte Farbindikatoren für Säure-Base-Titrationen sind: • Methylrot (Azofarbstoff): Umschlag zwischen pH 4.4 und 6.2 (rot → gelb). • Phenolphtalein (Triphenylmethanfarbstoff): bei pH 8 bis 10 (farblos → rot). 3.5.1 Versuch: Saure und basische Salze Stichworte: Dissoziation, Salzhydrolyse, Säure-Base-Begriff nach Brönstedt Allgemeines: Salze sind immer aus einem Kation M+ (zumeist ein Metallion oder z. B. NH4 + ) und einem Anion X− aufgebaut. Man kann sie als Reaktionsprodukte einer Reaktion (Neutralisation) zwischen einer Säure HX und einer Base MOH auffassen. HX + MOH → MX + H2 O Lösungen von Salzen können je nach gelöstem Salz verschiedene pH-Werte annehmen. Dies wird deutlich, wenn man die Dissoziation eines Salzes in Wasser in einer Reaktionsgleichung formuliert: MX + H2 O → M+ + OH− + H+ + X− Formal haben sich die Säure und die Base wieder gebildet. Der pH-Wert der Lösung hängt nun von der jeweiligen Stärke der Säure bzw. Base ab. Sind also die pKs - bzw. pKb -Werte der entsprechenden Säure bzw. Base bekannt, so lässt sich der pH-Wert der Salzlösung vorhersagen. Chemikalien: NaCl, Natriumacetat, NH4 Cl 3.5. SÄURE/BASE-REAKTIONEN UND PUFFER (3. WOCHE) 51 Durchführung: In je einem Reagenzglas werden eine Spatelspitze Natriumchlorid (NaCl), Natriumacetat (NaAc oder CH3 COONa) und Ammoniumchlorid (NH4 Cl) in etwa drei Finger breit entionisiertem Wasser gelöst. Prüfen Sie mit dem pH-Papier die pH-Werte der verschiedenen Lösungen. Aufgaben: Geben Sie an, welche Lösung basisch, neutral und sauer reagiert. Interpretieren Sie die von Ihnen gemessenen pH-Werte, indem Sie für jedes Salz die formale Reaktionsgleichung für das Auflösen unter Einbeziehung von Wasser (H2 O) formulieren. Entsorgung: Ausguss Ende Versuch 3.5.1 3.5.2 Versuch: Herstellung und Titration von 1 M NaOH und Essigsäure Boxweise Stichworte: Säure-Base-Reaktionen, Titer, Äquivalenzpunkt, starke und schwache Säuren und Basen Allgemein: Die hier herzustellenden Lösungen werden noch in späteren Versuchen benötigt. Jede Box stellt nur jeweils eine Lösung Natronlauge und Essigsäure her und beschriftet diese mit der Boxnummer. Natriumhydroxid (NaOH) ist eine feste Substanz und wird oft in Form kleiner Plätzchen oder Schuppen geliefert. In Wasser gelöstes Natriumhydroxid (NaOH) bezeichnet man auch als Natronlauge. Der stark basische Charakter rührt von den Hydroxid-Ionen (OH− ) her, die bei der Dissoziation von Natriumhydroxid in Wasser entstehen. Natriumhydroxid dissoziert vollständig. Das Dissoziationsgleichgewicht liegt nahezu völlig auf der Seite der Dissoziationsprodukte. Daher ist Natriumhydroxid eine starke Base. Festes Natriumhydroxid ist sehr hygroskopisch (wasseranziehend). Lässt man es auch nur kurze Zeit an der Luft stehen, wird es schmierig. Es wird daher auch zum Trocknen von Gasen benutzt. 52 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE NaOHreagiert mit dem Kohlendioxid (CO2 ) der Umgebungsluft. Kohlendioxid löst sich in Wasser und reagiert mit diesem in einer Gleichgewichtsreaktion zu Kohlensäure (H2 CO3 ). CO2 + H2 O * ) H2 CO3 Die Kohlensäure (H2 CO3 ) reagiert nun in einer Säure-Base-Reaktion mit Natriumhydroxid (NaOH). H2 CO3 + 2NaOH → Na2 CO3 + 2H2 O Da Kohlendioxid in der Laborluft und im zur Natronlaugeherstellung verwendeten Wasser vorhanden ist, verringert sich die Konzentration von festem und gelöstem Natriumhydroxid aufgrund der Bildung von Na2 CO3 mit der Zeit. Daher sollte man beim Abwiegen der NaOH-Plätzchen etwa 1 % mehr abwiegen, um den späteren Verlust durch die Reaktion mit CO2 abzufangen. Da der genaue Verlust nicht bekannt ist, muss man nach der Herstellung der Natronlauge den Titer (oder auch Faktor genannt) ermitteln. Hierzu titriert man die etwa 1 molare Natronlauge mit einer genau eingestellten 1 molaren Salzsäurelösung (HCl). Neutralisation von Natriumhydroxid mit Salzsäure: NaOH + HCl → NaCl + H2 O Ist die Natronlauge genau 1 molar, so wird der Verbrauch von Salzsäure genau so hoch ausfallen wie die vorgelegte Menge an Natronlauge. Ist sie etwas schwächer konzentriert, so wird der Verbrauch an Salzsäure geringer sein. Der Verbrauch der Salzsäure gibt also die tatsächliche Konzentration der Natronlauge an. Titriert man z. B. 10 ml einer etwa 1 molaren Natronlauge mit einer genau 1 molaren Salzsäure, so ist der theoretische Verbrauch 10 ml 1 M HCl. Der tatsächliche Verbrauch sei aber z. B. 9.2 ml. Für den Titer (Faktor) gilt dann: t= 9.2 ml VP = = 0.92 VT 10 ml VP = praktischer Verbrauch VT = theoretischer Verbrauch Um die wahre Konzentration der Natronlauge anzugeben, muss man nun die ungefähre Konzentration 1 mol/l mit dem Titer 0.92 multiplizieren. Die Konzentration der Natronlauge ist also nur 0.92 mol/l. Da die Konzentration der Natronlauge aber nach einer gewissen Zeitspanne wieder abgenommen haben kann, sollte man möglichst kurz vor Benutzung 3.5. SÄURE/BASE-REAKTIONEN UND PUFFER (3. WOCHE) 53 der Natronlauge den Titer ermitteln und ihn auf das Vorratsgefäß schreiben. Die für die Analyse ausgegebene Natronlauge sollte stets mit einem aktuellen Titer versehen sein. Chemikalien/Geräte: NaOH-Plätzchen, 1 M HCl (Titriplex o. ä.), Eisessig, 1 l Messkolben mit PVC-Stopfen (evtl. in der Geräteausgabe ausleihen) Durchführung/Aufgaben: Herstellen einer etwa 1 molaren NaOH: Es soll 1 mol NaOHin einem Liter Wasser gelöst werden. Wieviel g Natriumhydroxid (+ ca. 1 %) müssen eingewogen werden? Nach dem Abwiegen werden die Natriumhydroxid-(NaOH)-Plätzchen zügig in einen 1 l Messkolben gegeben und zunächst nur mit ca. 50 ml Wasser gelöst. Nach dem Auflösen wird der Messkolben bis zu Eichmarke mit Wasser gefüllt. Hierbei ist zu beachten, dass der untere Meniskus der Wasseroberfläche genau auf dem Eichstrich liegt. Nach dem Auffüllen wird der Kolben mit einem PVC-Stopfen (ein Glasstopfen würde mit der Zeit festbacken) verschlossen und die Lösung durch mehrfaches Umkippen des Messkolbens (der Assistent zeigt Ihnen, wie es geht) gut durchmischt. Vor Gebrauch einer solchen Lösung sollte diese immer gut durchmischt werden, da sich nach einer gewissen Zeit das schwerere Natriumhydroxid im unteren Bereich absetzt und somit ein Konzentrationsgradient entsteht. Der Titer dieser nur ungefähr 1 molaren Natronlauge wird nun durch Titration mit einer genau 1 molaren Salzsäure (HCl) ermittelt. Titrationsvorgang: Die Bürette wird gemäß der Anleitung des Assistenten aufgebaut (siehe Vorpraktikum). Sie darf nicht unter Spannung stehen und das Hahnkücken sollte leichtgängig sein. Glaskücken müssen ausreichend mit Schlifffett gefettet sein. Teflonkücken (weiß) dürfen dagegen nicht gefettet werden. Sollte die Bürette nicht sauber und trocken, sondern z. B. noch feucht sein, wird sie mit ca. 10 ml der 1 molaren Salzsäure zweimal gespült. Die saubere Bürette wird nun mittels eines kleinen Trichters bis über die Nullmarke mit der Salzsäure gefüllt. Nach dem Befüllen sollte keine Luftblase in der Bürette vorhanden sein und der Trichter entfernt werden. Die überschüssige Salzsäure wird bis zur Nullmarke abgelassen. 20 ml der zu titrierenden Natronlauge werden mit einer 20 ml Vollpipette in einen 250 ml Erlenmeyerkolben (oder Becherglas) pipettiert. Dabei hält 54 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE man die Spitze gegen die Gefäßwand und lässt die Pipette auslaufen (nicht ausblasen). Ein kleiner Rest wird in der Pipettenspitze bleiben. Zur Erhöhung des Titrationsvolumens und um kleine Spritzer von der Glaswand zu spühlen, werden mit der Spritzflasche etwa 20 ml entionisiertes Wasser hinzugegeben. Warum hat das keinen Einfluss auf das Ergebnis? 2–3 Tropfen Phenolphtaleinlösung werden hinzugegeben. Die Lösung färbt sich daraufhin rot. Die Titration wird nun begonnen und so lange fortgesetzt, bis sich die Lösung entfärbt. Während des Titrierens sollte die Säure nie als zusammenhängender Strahl aus dem Hahnkücken treten, sondern nur als Tropfenfolge. Der Erlenmeyerkolben sollte kontinuierlich leicht geschwenkt werden. Die Titration wird zur Sicherheit vier mal mit je 20 ml wiederholt. Der Mittelwert aller gelungenen Titrationen wird nun berechnet und damit der Titer der Natronlauge bestimmt. Herstellen einer 1 molaren Essigsäure: Konzentrierte Essigsäure (Eisessig) besteht nicht zu 100 % aus Essigssäure. Bei der Herstellung einer genau 1 molaren Essigsäure wird daher genau wie bei der Natronlauge vorgegangen. Etwa 60 ml konzentrierte Essigsäure entsprechen in guter Näherung etwa 1 mol Essigsäure. Diese werden in einem 1 l Messkolben mit Wasser verdünnt und dieser genau auf 1 l aufgefüllt (siehe oben). In die saubere Bürette wird nun die vorher genau eingestellte Natronlauge eingefüllt (siehe oben) und vier Portionen á 20 ml der Essigsäure in einem 250 ml Erlenmeyerkolben vorgelegt. Als Indikator wird wieder Phenolphtalein benutzt. Es wird jeweils bis zur schwachen Rosafärbung titriert. Sie darf durch Schütteln nicht mehr verschwinden. Aus dem Verbrauch lässt sich der Titer der Essigsäure bestimmen. Entsorgung: Neutralisieren und danach Ausguss Ende Versuch 3.5.2 3.5.3 Versuch: Titrationskurve Zweiergruppe 3.5. SÄURE/BASE-REAKTIONEN UND PUFFER (3. WOCHE) 55 Stichworte: Starke und schwache Säuren, Pufferbereich, Äquivalenzpunkt, Neutralpunkt, Umschlagsbereich eines Indikators Allgemeines: Bei der Titration einer schwachen Säure mit einer starken Base oder umgekehrt, zeigt der Kurvenverlauf des pH-Wertes, aufgetragen über der Menge des zugetropften Volumens, einen typischen Verlauf. An dieser Kurve lassen sich verschiedene Punkte und Bereiche erkennen. Der Neutralpunkt liegt grundsätzlich immer bei pH = 7. Am Äquivalenzpunkt ist das Molverhältnis zwischen Säure und Base 1:1. Der Umschlagbereich ist vom verwendeten Indikator abhängig. Universalindikatoren (wie z. B. pH-Papier) sind eine Mischung aus mehreren Indikatoren. Dadurch erstreckt sich deren Umschlagsbereich über den gesamten pH-Bereich wässriger Lösungen. Der Umschlagbereich von Phenolphtalein liegt zwischen pH 8 und pH 10. Chemikalien: 1 M NaOH (selbst hergestellt), 1 M Essigsäure (selbst hergestellt), Spezialindikatorpapier Durchführung: 20 ml der selbst hergestellten 1 molaren (Titer?) Essigsäure werden mit insgesamt 40 ml der selbst hergestellten 1 molaren Natronlauge (Titer?) titriert bis der pH-Wert weit im alkalischen Bereich liegt. Dabei geben Sie schrittweise 0.5 ml der Natronlauge hinzu und messen dann den pH-Wert. Achten Sie darauf, dass das verwendete Indikatorpapier auch für den jeweiligen pH-Bereich geeignet ist. Aufgaben: Die erhaltenen pH-Werte tragen Sie in einem Diagramm gegen das Volumen der zugegebenen Maßlösung auf. Nach Zugabe von 40 ml sollten Sie in etwa eine Kurve erstellt haben, die einer Titrationskurve einer schwachen Säure und starken Base gleicht (siehe Lehrbücher). Interpretieren Sie die Kurve und zeichnen Sie den Neutralpunkt, Äquivalenzpunkt, Umschlagbereich von Phenolphtalein und den Pufferbereich ein. Was gilt im Pufferbereich? Warum wäre Phenolphtalein für diese Titration auch ein geeigneter Indikator, obwohl er erst im alkalischen Bereich umschlägt? Entsorgung: Neutralisieren, danach Ausguss Ende Versuch 3.5.3 56 3.5.4 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Analyse 1: Bestimmung von HCl Stichworte: Titration, Indikatoren Allgemeines: Eine Salzsäurelösung mit einen Ihnen unbekannten Konzentration soll mit der ausstehenden Natronlauge titriert und die Konzentration von ihnen berechnet werden. Stellen Sie bitte einen 100 ml Messkolben mit PVC-Stopfen auf den von dem Assistenten vorgesehenen Platz. Der Messkolben sollte mit folgenden Daten beschriftet sein: Nachname, Vorname: Analyse: 1 Versuch: 1 (Bei einer Falschansage des Ergebnisses ist ein zweiter Versuch mit einer anderern Analysenlösung möglich) Box Nr.: Saal: Am nächsten Kurstag können Sie sich den Messkolben mit etwas Salzsäure befüllt abholen. Chemikalien: Voreingestellte, ausgegebene NaOH Durchführung: Der mit etwas Salzsäure befüllte Messkolben wird mit entionisiertem Wasser vorsichtig exakt bis zur Eichmarke gefüllt. Die Lösung wird durch Schütteln gut durchmischt und in 4 Portionen von 20 ml mit der ausstehenden Natronlauge (Titer beachten!!!) titriert. Dabei wird jede Portion im Erlenmeyerkolben auf ca. 50–100 ml verdünnt und mit Methylrot als Indikator (3–5 Tropfen der ausstehenden Lösung) versetzt. Methylrot zeigt im sauren Bereich eine rote und im alkalischen Bereich eine gelbe Farbe. Aufgaben: Der mittlere Verbrauchswert der Titrationen wird nun zur Berechnung der Menge an eingewogener Salzsäure in mg herangezogen. Die zulässige Fehlergrenze liegt bei ±1 %. Folgende Überlegungen müssen angestellt werden, um die Berechnung durchführen zu können: Wieviel mmol NaOH wurden verbraucht? 3.5. SÄURE/BASE-REAKTIONEN UND PUFFER (3. WOCHE) 57 Die Konzentration der verwendeten Natronlauge ist z. B. cNa OH = t · 0.1 mol · l−1 t = Titer Für die weitere Berechnung ist es vorteilhaft, die Konzentration in mmol/ml anzugeben. Mit dem verbrauchten Volumen an Natronlauge (VNa OH in ml) ergibt sich für die verbrauchte Stoffmenge nNa OH : nNa OH [mmol] = cNa OH [mmol · ml−1 ] · VNa OH [ml] In den vorgelegten 20 ml Salzsäurelösung befand sich die gleiche Stoffmenge HCl. Es gilt daher: nNa OH [mmol] = nH Cl [mmol] Für die Konzentration der Salzsäure in mmol/ml gilt: cH Cl [mmol · ml−1 ] = nH Cl [mmol] 20 ml Die Gesamtstoffmenge in den 100 ml beträgt demnach: nH Cl [mmol] = nH Cl [mmol] · 100 ml 20 ml Da die eingewogene Menge an HCl in mg gesucht ist, muss die Stoffmenge nH Cl /100 ml mit Hilfe der molaren Masse MH Cl in die Masse mH Cl /100 ml (in mg) umgerechnet werden. mH Cl = nH Cl · MH Cl [mg · mmol−1 ] Zusammengefasst in eine Formel ergibt sich für mH Cl in mg: mH Cl [mg] = MH Cl [mg · mmol−1 ] · VNa OH [ml] · cNa OH [mmol · ml−1 ] · t · 100 ml 20 ml Entsorgung: Neutralisieren, Ausguss Ende Versuch 3.5.4 58 3.5.5 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Versuch: Herstellung von Puffern (pH-Wert vom Assistenten vorgegeben) Stichworte: Puffer, pKs -Wert, Henderson-Hasselbalch-Gleichung Allgemeines: Pufferlösungen sind in der Lage, ihren pH-Wert bei begrenztem Zusatz von Säure oder Base konstant zu halten. Um die Protonen, die bei Zugabe einer Säure frei werden, abfangen zu können, muss die Pufferlösung eine Base enthalten. Damit der pH-Wert auch gegenüber einer basischen Verunreinigung unverändert bleibt, muss auch eine Säure in der Lösung vorhanden sein. Meist stellt man Pufferlösungen aus einer schwachen Säure und ihrem Alkalisalz, also aus einer Säure und ihrer konjugierten Base, her. Dissoziation einer Säure (HA) in Wasser zu ihrer konjugierten Base (A− ): HA + H2 O * ) H3 O+ + A− Bei Zusatz von A− wird das Gleichgewicht nach links verschoben. Es entsteht kaum noch A− aus der Säure. Die Säure liegt jetzt hauptsächlich undissoziert vor. Die auf diese Weise gebundenen Protonen bilden jetzt ein Reservoir zum Abfangen von basischen Teilchen wie z. B. OH− . Je nach der gewählten Säure bzw. Base vermag die Lösung in einem ganz bestimmten pH-Bereich zu puffern. Für die obige Reaktion lässt sich die Gleichgewichtskonstante nach dem Massenwirkungsgesetz formulieren: HA * ) H+ + A− c∗ (H+ ) · c∗ (A− ) Ks = c∗ (HA) c∗ (HA) c∗ (H+ ) = Ks · ∗ − c (A ) Der pH-Wert einer solchen Lösung kann mit der Henderson-HasselbalchGleichung berechnet werden: c∗ (A− ) c∗ (HA) pH = pKs + log ∗ oder auch pH = pKs − log ∗ − c (HA) c (A ) Liegen A− und HA im Molverhältnis 1:1 vor, gilt: pH = pKs 3.5. SÄURE/BASE-REAKTIONEN UND PUFFER (3. WOCHE) 59 Aufgaben: Sie haben die Aufgabe, einen basischen Ammoniakpuffer (pHBereich = 8.3–10.1) und einen sauren Acetatpuffer (pH-Bereich = 3.8–5.6) herzustellen. Der geforderte pH-Wert wird ihnen vom Assistenten vorgegeben. Protokollieren Sie ihre Berechnungen. Chemikalien: Natriumacetat, 1 molare Essigsäure, NH4 Cl, 10 % AmmoniakLösung Durchführung: Man berechnet mit der Henderson-Hasselbalch-Gleichung und dem bekanntem pKs -Wert das für den gewünschten pH-Wert erforderliche Verhältnis von Base und Säure (bzw. Salz und Säure). Unter Berücksichtigung der gewünschten Molarität und Gesamtmenge der Pufferlösung errechnet man die Einwaage der festen Substanzen (zumeist das Salz) und das Volumen der flüssigen Komponenten (zumeist die Säure). Acetatpuffer: Es soll 100 ml eines 0.5 molaren Acetatpuffers aus eingewogenem Na-Acetat und 1 molarer Essigsäure mit dem geforderten pHWert hergestellt werden. Das abgewogene Salz und die Säure werden zuerst gemischt und dann auf das Endvolumen aufgefüllt (100 ml Messkolben). Der fertige Puffer wird in ein 100 ml Becherglas gefüllt. Der Assistenten misst den pH-Wert mit einem pH-Meter. (pKs -Wert Essigsäure: 4.8) Ammoniakpuffer: Es sollen 50 ml eines 2 molaren Ammoniakpuffers mit dem geforderten pH-Wert hergestellt werden. Abgewogenes Ammoniumchlorid (NH4 Cl) wird in der berechneten Menge einer 10 %igen Ammoniaklösung gelöst und auf das geforderte Volumen (Messzylinder) aufgefüllt. (pKs -Wert Ammoniak: 9.3) Entsorgung: Neutralisieren, Ausguss Beispiel-Rechenweg: 100 ml 0.5 M Acetatpuffer pH = 4.2 Acetat-Ionen = Ac− ; CH3 COOH= HAc; pKs = 4.8 Für die Berechnung der Konzentrationen der Pufferkomponenten kann man zwei Ansätze wählen. Für den ersten Ansatz kann man die Henderson-Hasselbalch-Gleichung für einstufige Puffer auch ohne standardisierte Konzentrationen schreiben: pH = pKs + log c(Ac− ) c(HAc) 60 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Einsetzen der Werte für pH und pKs : c(Ac− ) c(HAc) − c(Ac ) ⇒ − 0.6 = log c(HAc) c(HAc) ⇒ 100.6 = c(Ac− ) 4.2 = 4.8 + log (3.1) Für den zweiten Ansatz schreibt man das Massenwirkungsgesetz ebenfalls mit nicht standardisierten Konzentrationen: ⇒ c(H+ ) · c(Ac− ) Ks = c(HAc) c(HAc) c(H+ ) = Ks · c(Ac− ) Einsetzen der Werte für Ks und die H+ -Konzentration: c(HAc) c(Ac− ) c(HAc) = c(Ac− ) 10−4.2 = 10−4.8 · ⇒ 100.6 (3.2) Beide Wege liefern für das Verhältnis der Essigsäure- und Acetat-IonenKonzentration das gleiche Ergebnis: Jede Mischung aus Essigsäure und AcetatIonen im Verhältnis 100.6 : 1 ist ein Puffer mit der geforderten H+ -Konzentration! Als weitere Bedingung soll die Konzentration des Puffers c(Ac− ) + c(HAc) = 0.5 mol · l−1 = 0.05 mol · (100 ml)−1 (3.3) betragen. Durch einsetzen von (3.2) für die Essigsäurekonzentration in (3.3) ergibt sich: c(Ac− ) + 100.6 c(Ac− ) = 0.05 mol · (100 ml)−1 0.05 ⇒ c(Ac− ) = mol · (100 ml)−1 1 + 100.6 0.05 = mol · (100 ml)−1 4.98 = 0.01 mol · (100 ml)−1 3.5. SÄURE/BASE-REAKTIONEN UND PUFFER (3. WOCHE) 61 Die molekulare Masse von Natriumacetat ist MNa Ac = 82.034 g · mol−1 . Um 0.01 mol zu erhalten, müssen also 0.01 · 82.034 g = 0.82 g abgewogen werden. Mit Hilfe von (3.3) kann man auch die benötigte Menge an Essigsäure berechnen: c(HAc) = 0.05 − 0.01 mol · (100 ml)−1 = 0.04 mol · (100 ml)−1 Die verwendete Essigsäure hat eine Konzentration von c(HAc) = 1 mol · l−1 = 0.1 mol · (100 ml)−1 = 0.04 mol · (40 ml)−1 d. h. es werden 40 ml benötigt. Ende Versuch 3.5.5 , 62 3.6 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Themen des 2. Kolloquiums Es folgt eine Zusammenstellung der wichtigsten Themen in Stichworten. Es wird empfohlen, die Stichworte in den einschlägigen Lehrbüchern nachzuschlagen. Redoxreaktionen Definitionen: Oxidation, Reduktion, Elektronenakzeptor/Donator, Oxidations/Reduktionsmittel usw. Oxidationszahlen: Definition von Oxidationszahlen, pH-Abhängigkeit, Regeln zur Berechnung Oxidationszahlen usw. Aufstellen von Redoxgleichungen: Regeln, Redox-Äquivalenz, Stöchiometrie usw. Galvanische Elemente und Elektrolyse: Vorgänge an den Elektroden, Anode, Kathode, Daniell-Element, Potentialdifferenz, Vergleich: Galvanisches Element und Elektrolyse, ∆G, Überspannung, Definition Anion/Kation usw. Spannungsreihe, Standard-Reduktionspotentiale: Redoxkette, elektrochemische Zelle, Eigenspannung ∆E, Normal-/Standardwasserstoffelektrode (Aufbau?), Welche Redoxreaktion läuft an welcher Elektrode ab?, Wie kommt die Spannungsreihe zustande und wozu ist sie gut?, Welche Voraussagen sind mit ihr zu machen?, Elektronegativitäten usw. Nernstche Gleichung: Konzentrations- und pH-Abhängigkeit des Redoxpotentials, Zusammenhang ∆G und ∆E, Funktion und Bedeutung der einzelnen Teile der Nernstschen Gleichung usw. Übergangsmetalle und Komplexverbindungen Elektronenkonfiguration, Atomorbitale, d-Orbitale, Besetzungsregeln, essentielle Funktionen der Übergangsmetalle in der Natur (Fe- und Cu-Komplexe) usw. Komplex- od. Koordinationsverbindungen: Definition Komplex-/Koordinationsverbindungen, Liganden, Zentralatom, mehrzähnige Liganden, Nomenklaturregeln usw. 3.6. THEMEN DES 2. KOLLOQUIUMS 63 Geometrie und Isomerie von Komplexen: Definition Isomerie, geometrische Grundkörper, Koordinationszahl, Spiegelebene, cis (Z)/trans(E) usw. Stabilität von Komplexen: Komplexbildungskonstante, ionische-/kovalente Natur der Bindungen, Definition Lewis-Säure/Base, harte/weiche LewisSäuren/Basen usw. Chelatkomplexe: Definition Chelatkomplexe, Glycin, EDTA, Struktur usw. Quantitative Analyse: Bestimmung von NaCl durch Ionenaustausch: Funktionsweise des Ionenaustauschers sauer/basisch usw. Phosphatbestimmung: Funktionsweise eines Photometers, Kolorimetrie, Photometrie, Lambert-Beersches Gesetz, Kalibrierkurve usw. Außerdem: Stichworte, Themen und Ausführung der Versuche 3.7.1 bis 3.9.2. 64 3.7 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Redoxreaktionen (4.–5. Woche) Stichworte: Reduktion und Oxidation, Oxidationszahl, Spannungsreihe, Definition edler und unedler Metalle, Redoxpotential, Normalpotential, Nernstsche Gleichung, Galvanische Elemente, Lokalelement, Daniell-Element, Elektrolyse Neben den Säure-Base-Reaktionen gibt es in der Chemie eine zweite häufige und wichtige Klasse von Austauschprozessen, die Redoxreaktionen. Bei ihnen werden Elektronen übertragen und nicht Protonen wie zwischen Säuren und Basen. Die Abgabe von Elektronen aus Atomen, Molekülen oder Ionen wird als Oxidation bezeichnet. Die Aufnahme von Elektronen durch Atome, Moleküle oder Ionen heißt Reduktion. Ein Elektronenakzeptor wirkt als Oxidationsmittel, ein Elektronendonator ist ein Reduktionsmittel. Beispiele: Na → Na+ + e− Oxidation Cl2 + 2e− → 2Cl− Reduktion Ein Natriumatom geht durch Entfernen eines Elektrons in ein Natriumkation über und ist daher ein Reduktionsmittel. Ein Chlormolekül geht durch Aufnahme von zwei Elektronen in zwei Chloridionen über und ist daher ein Oxidationsmittel. Die Triebkraft für diese Elektronenverschiebung liegt in der Tendenz der Elemente durch Abgabe (bei Natrium) bzw. Aufnahme von Elektronen (im Falle von Chlor) energetisch günstige Teilchen mit abgeschlossener Elektronenkonfiguration zu bilden. Durch die Coulomb-Wechselwirkungen der geladenen Teilchen im Festkörper (Kristall) wird ein zum Teil erheblicher Energiegewinn gegenüber den ungeladenen Teilchen erzielt. Zwei Spezies wie Na und Na+ , die sich nur durch die Anzahl ihrer Elektronen unterscheiden, nennt man ein korrespondierendes Redoxpaar, in Kurzschreibweise: Na/Na+ . Wie Protonen sind auch Elektronen hochreaktive Teilchen und unter üblichen chemischen Bedingungen in freier Form nicht existenzfähig. Daher treten Oxidationen und Reduktionen nur gemeinsam auf, an einer ReduktionsOxidations-Reaktion (Redoxreaktion) sind zwei Redoxpaare beteiligt: Es gibt bei einer chemischen Reaktion keine Oxidation ohne Reduktion und umgekehrt. Oxidationszahlen Einfache Elektronenübergänge zwischen Elementen und ihren Ionen sind an deren unterschiedlichen Eigenschaften i. a. leicht zu erkennen. Allerdings gibt 3.7. REDOXREAKTIONEN (4.–5. WOCHE) 65 es auch viele Fälle, in denen Elemente mehrere Redoxpaare bilden (z. B. Mangan (Mn) oder Eisen (Fe)), die man differenzieren muss. Werden schließlich bei einer Reaktion kovalente Bindungen gebrochen und neu geknüpft, ist oft schwierig festzustellen, ob eine Redoxreaktion vorliegt oder nicht. Bei der Reaktion zwischen molekularem Wasserstoff (H2 ) und molekularem Sauerstoff (O2 ) zu Wasser (H2 O) werden beispielsweise symmetrische, homöopolare Bindungen gelöst und stark polare O—H-Bindungen gebildet. Dies ist als Elektronentransfer, also Redoxreaktion, zu betrachten, weil in H2 O die Bindungselektronen stärker von O als von H angezogen werden. Um Redoxreaktionen leichter zu erkennen und Redoxgleichungen aufzustellen, ist der Begriff der Oxidationszahl oder Oxidationsstufe von Nutzen. Die Oxidationszahl in einem Ion oder Molekül entspricht der Ladung, die das betrachtete Atom besäße, wenn die Elektronen der von ihm ausgehenden Bindung völlig dem jeweils elektronegativeren Bindungspartner zugeordnet werden. Man denkt sich fiktiv ein Teilchen aus völlig ionisierten Atomen zusammengesetzt. Regeln und Beispiele (siehe auch Lehrbücher): a.) Die Atome in Elementen (Metalle, Nichtmetalle wie Wasserstoff (H2 ), Stickstoff (N2 ), Sauerstoff (O2 ), Chlor (Cl2 )) haben die Oxidationszahl Null. b.) Bei einfachen Atom-Ionen in salzartigen Verbindungen ist die Oxidationszahl gleich der Ionenladung. NaCl: Na+ (Oxidationszahl +I) Cl− (Oxidationszahl -I) CaCl2 : Ca2+ (Oxidationszahl +II) Cl− (Oxidationszahl -I) FeS: Fe2+ (Oxidationszahl +II) S2− (Oxidationszahl -II) c.) In kovalenten Verbindungen werden die Bindungselektronen gedanklich dem elektronegativeren Atom zugeteilt, die entstandenen Ladungen entsprechen den Oxidationszahlen. d.) Die höchste positive Oxidationszahl kann nicht höher sein als die Gruppennummer (im Periodensystem der Elemente, PSE) des Elementes. Die niedrigste negative Oxidationszahl kann nicht kleiner sein als die Gruppennummer minus 8. Aufstellen von Redoxgleichungen (siehe auch Lehrbücher) a.) Reaktanden und Produkte, die an der Reduktion und Oxidation beteiligt sind, sind als erstes alle anzugeben; für sie werden die betreffenden Oxidationszahlen ermittelt. 66 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE b.) Das Zahlenverhältnis, in dem Reduktionsmittel und Oxidationsmittel miteinander reagieren, wird bestimmt, indem die OxidationszahlZunahme und die Oxidationszahl-Abnahme balanciert werden: die Zahl der abgegebenen und aufgenommenen Elektronen muss gleich sein. c.) Die Summe der Ionenladungen und die Anzahl anderer Atome auf beiden Seiten der Gleichung wird ausgeglichen. Für den Ausgleich von Ionenladungen dienen in wässriger Lösung H+ - und OH− -Ionen. Beispiel: Das starke Oxidationsmittel Kaliumpermanganat (KMnO4 ) reagiert in sauerer Lösung mit Iodid (I− ). Es oxidiert Iodid zu Iod (I2 ) und wird dabei zu Mangan(II)-salzen reduziert. Man formuliert zuerst die Reaktionsgleichung ohne stöchiometrische Koeffizienten: 2+ − MnO− + I2 4 + I → Mn Um die stöchiometrischen Koeffizienten der Gleichung zu ermitteln, formulieren wir die jeweiligen Teilgleichungen, ermitteln die Anzahl der jeweils wechselnden Elektronen und bilden das kleinste gemeinsame Vielfache. 2+ − Reduktion MnO− 4 + 5e → Mn Oxidation 2I− → I2 + 5e− 2+ − Gesamtreaktion 2MnO− 4 + 10I → 5I2 + 2Mn (mal 2) (mal 5) Da die Reaktion in saurer Lösung stattfindet, wird der Ladungsausgleich mit Protonen (H+ ) durchgeführt. 2+ − + 2MnO− + 8H2 O 4 + 10I + 16H → 5I2 + 2Mn Galvanische Elemente und Elektrolyse (siehe auch Lehrbücher) Taucht man einen Zinkstab in eine Kupfersulfatlösung, so scheidet sich auf ihm metallisches Kupfer ab. Es findet eine Redoxreaktion statt: Zn + Cu2+ → Zn2+ + Cu Zink reduziert die Kupferionen zu metallischem Kupfer und wird dabei selbst zu Zinkionen oxidiert. Die zwischen dem Reduktionsmittel Zink und dem Oxidationsmittel Cu(II)-Ionen übertragenen Elektronen stellen einen elektrischen Strom dar, der von der Potentialdifferenz (Spannung) zwischen den Redoxpaaren Zn/Zn2+ und Cu/Cu2+ herrührt. 3.7. REDOXREAKTIONEN (4.–5. WOCHE) 67 Die Potentialdifferenz ∆E kann allerdings in dieser einfachen Versuchsanordnung nicht bestimmt und die Elektronenübertragung selbst nicht beobachtet werden. Da Elektronen jedoch mit Hilfe elektrischer Leiter über weite Entfernungen transportiert werden können, ist es möglich, den Oxidations- und Reduktionsvorgang einer Redoxreaktion räumlich zu trennen. Eine solche Anordnung ist ein galvanisches Element, in dem die zwischen zwei Redoxpaaren herrschende Potentialdifferenz gemessen werden kann. Im sog. Daniell-Element taucht im linken Reaktionsraum (Halbelement, Halbzelle) ein Zinkstab in eine Lösung, die Zn2+ und SO4 2− -Ionen enthält. Im rechten Reaktionsraum befindet sich ein Kupferstab, der in eine CuSO4 Lösung eintaucht. Wenn die beiden Metallstäbe (Elektroden) durch einen äußeren Leiter verbunden werden, gehen aus der Zinkelektrode Zn2+ -Ionen in die ZnSO4 -Lösung über. Die überschüssigen Elektronen fließen über den Leiter zur Kupferelektrode und reagieren dort mit den in der CuSO4 -Lösung befindlichen Cu2+ -Ionen, die sich als metallisches Kupfer am Cu-Stab abscheiden. Durch diese Vorgänge entsteht in der Lösung der linken Zelle ein Überschuss, in der rechten Zelle ein Mangel an positiven Ladungen. Durch Wanderung von Sulfationen aus der rechten in die linke Zelle durch eine für sie durchlässige Zwischenwand (Diaphragma) erfolgt Ladungsausgleich: Es liegt ein geschlossener Stromkreis vor. Zinkelektrode (Oxidation) Zn → Zn2+ + 2e− Kupferelektrode (Reduktion) Cu2+ + 2e− → Cu Gesamtreaktion (Redoxreaktion) Zn + Cu2+ → Zn2+ + Cu Weitere Ausführungen über die Elektromotorische Kraft und Akkumulatoren entnehmen Sie bitte den Lehrbüchern. 68 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Elektroden Kathode (negativ): Elektronen abgebende Elektrode. Bei Stromdurchgang wandern Kationen an die Kathode und können hier elementar abgeschieden werden (kathodische Reduktion). Anode (positiv): Elektronen aufnehmende Elektrode. Bei Stromdurchgang wandern Anionen zur Anode. An der Anode können Stoffe — das Anodenmaterial, Anionen oder Wasser — oxidiert werden (anodische Oxidation). Spannungsreihe, Standard-Reduktionspotentiale Ein System wie das galvanische Element, in dem zwei räumlich getrennte Redoxpaare über einen metallischen Leiter verbunden sind, wird auch als Redoxkette oder elektrochemische Zelle bezeichnet. Die zwischen den Elektroden einer Redoxkette im stromlosen Zustand messbare Eigenspannung ∆E ist die Differenz zwischen den Potentialen der beteiligten Redoxpaare. Die Absolutwerte der Potentiale können nicht gemessen werden, sondern nur die Potentialdifferenzen zwischen zwei Elektroden. Um trotzdem die Potentiale verschiedener Redoxpaare miteinander vergleichen zu können, wird das Potential eines Bezugsredoxsystems als Null definiert. Misst man die Potentiale aller anderen Redoxpaare gegen diese Vergleichselektrode unter vergleichbaren Bedingungen, so ergibt sich eine Skala relativer Potentialwerte, die sog. Spannungsreihe (s. unten). Als Vergleichselektrode dient die Normal- oder Standardwasserstoffelektrode mit dem Redoxpaar 2H+ /H2 . Sie besteht aus einer Platinelektrode, die unter Normal- oder Standardbedingungen (Temperatur 298 K = 25˚C; Konzentration 1 mol/l) in eine H+ -Ionen-haltige Lösung taucht (1 N HCl) und von Wasserstoffgas unter einem Druck von 1013 mbar (1 atm) umspült wird. Der am Platin adsorbierte molekulare Wasserstoff bildet mit Protonen ein Redoxpaar (2H+ /H2 ). An der Normalwasserstoffelektrode läuft die folgende Redoxreaktion ab: 2H+ + 2e− → H2 Das Potential eines Redoxpaares Ox + ne− → Red (bei 25˚C, alle Konzentrationen 1 mol/l) in Kombination mit der Wasserstoffelektrode ist sein Normal- oder Standard-Reduktionspotential E . Die Potentiale von Redoxpaaren, aus denen freiwilig Elektronen frei werden, haben ein negatives Vorzeichen. Beispielsweise gehen aus dem Redoxpaar Zn/Zn2+ 3.7. REDOXREAKTIONEN (4.–5. WOCHE) 69 Zinkionen in Lösung und die Elektronen reduzieren an der Wasserstoffelektrode Protonen zu Wasserstoff; das Standardpotential von Zn2+ + 2e− → Zn ist E = −0.76 V. Umgekehrt besitzen Redoxpaare, zu deren oxidierter Form Elektronen aus Wasserstoff hinfließen, ein positives Standardpotential, wie beispielsweise in Cu2+ + 2e− → Cu mit E = +0.34 V. Charakteristische Standard-Reduktionspotentiale E (weitere finden Sie in Lehrbüchern und Tabellenwerken) sind: Natrium Na+ /Na Aluminium Al3+ /Al Zink Zn2+ /Zn Eisen(II) Fe2+ /Fe Blei Pb2+ /Pb Wasserstoff 2H+ /H2 Kupfer Cu2+ /Cu Eisen(III) Fe3+ /Fe2+ Silber Ag+ /Ag Sauerstoff O2 /2O2− Wasserstoffperoxid H2 O2 /H2 O -2.71 -1.69 -0.76 -0.44 -0.13 0.00 +0.34 +0.77 +0.80 +1.23 +1.78 (Von oben nach unten nimmt die Oxidationskraft zu) Die Spannung eines Galvanischen Elementes bei Standardbedingungen ist gleich der Differenz der Standardpotentiale der beiden Halbelemente mit höherem und tieferem Potential, also ∆E = E1 − E2 . Wichtig: Aus der Kenntnis von Standard-Reduktionspotentialen kann man vielfach vorhersagen, welche Stoffe von welchen anderen (zumindest prinzipiell) oxidiert bzw. reduziert werden können. 70 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE 3.7.1 Versuch: Metallionen, Spannungsreihe Zweiergruppe Stichworte: Spannungsreihe, edle und unedle Metalle Allgemeines: Wird ein Metall in eine Metallsalzlösung eines edleren Metalls eingetaucht, so scheidet sich das edlere Metall aufgrund seines edleren Charakters an dem unedlen Metall ab. Im umgekehrten Fall passiert dies nicht. Auf diese Weise lässt sich herausfinden, ob ein Metall edler ist als das andere. Löst sich ein Metall in Säure unter Gasbildung (Wasserstoff H2 ) auf, so ist das ein Hinweis darauf, dass es ein negatives Redoxpotential besitzt. Chemikalien: 1 molare HCl, Ag NO3 , CuSO4 , FeSO4 , Mg Cl2 , ZnSO4 , Mg -Band, Mg -Späne, Cu-Band, Cu-Späne, Zink-Granalien, Eisenpulver, Eisennagel, Stangenzink, Zinnfolie Durchführung: a.) In fünf 100 ml Bechergläsern (hohe Form) bereiten Sie jeweils durch Einrühren einer Spatelspitze Salz in ca. 5 ml Wasser eine Lösung der folgenden Salze: Ag NO3 , CuSO4 , FeSO4 , Mg Cl2 , ZnSO4 . Tauchen Sie nacheinander in jede Lösung: Magnesiumband, Eisennagel, Kupferband und Stangenzink. Becherglas: Metallsalz: Cu: Fe: Mg : Zn: 1 Ag NO3 2 CuSO4 3 FeSO4 4 Mg Cl2 5 ZnSO4 b.) In fünf Reagenzgläser werden fünf verschiedene Metalle gegeben und ca. zwei Finger hoch mit einer 1 molaren Salzsäure (HCl) aufgefüllt. Reagenzglas: Metall: 1 2 3 eine Zinkgranalie Eisenpulver Magnesium- Kupferspäspäne ne 4 5 Zinnfolie 3.7. REDOXREAKTIONEN (4.–5. WOCHE) 71 Aufgaben: a.) Protokollieren und interpretieren Sie ihre Beobachtungen, indem Sie in einer Tabelle eine relative Spannungsreihe von den verwendeten Metallen anfertigen. b.) Beobacheten Sie das jeweilige Metall und ziehen Sie Schlüsse aus ihren Beobachtungen. Entsorgung: Anorganische Schwermetallabfälle Ende Versuch 3.7.1 3.7.2 Versuch: Lokalelement Boxweise Stichworte: Agar, Phenolphtalein, Berliner Blau, Spannungsreihe Allgemeines: Unedle Metalle werden in einer Elektrolytlösung oxidiert und lösen sich auf. Ihre positiven Ionen gehen dabei in Lösung und lassen die Elektronen im Metall zurück. Wenn sie dabei von einem edleren Metall berührt werden, wird dieser Prozess beschleunigt, da das edlere Metall die zurückbleibenden Elektronen aufnimmt. Dabei lädt sich das edlere Metall negativ auf. Agar ist ein Gemisch aus Agarose und Agaropectin. Es besteht aus verschiedenen Polysacchariden. In der Mikrobiologie wird es häufig als Träger für Bakterien- und Pilzkulturen benutzt, da es nur von einigen marinen Bakterien zersetzt wird. Agar wird als Pulver geliefert und bei 100˚C in Wasser gelöst. Danach bleibt er bis etwa 45˚C flüssig. Kühlt er weiter ab, erstarrt er zu einer zähen, dann festen, geleeartigen Masse. Chemikalien: 0.01 M NaCl, Agar, K3 Fe(CN)6 -Lösung (1 %), alkoholische Phenolphtaleinlösung (1 %), Eisenagel, Kupferdraht Durchführung: 0.01 mol NaCl wird mit 1 g Agar, 3 ml 1 %iger K3 Fe(CN)6 Lsg., 0.5 ml 1 %iger alkoholischer Phenolphthaleinlösung und mit Siedesteinchen in einem 250 ml Becherglas mit 100 ml Wasser erhitzt, bis sich der Agar 72 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE gelöst hat. Die heiße Lösung wird in eine Petrischale gegossen und etwas abgekühlt. In diese Lösung werden vorsichtig zwei saubere Eisennägel gelegt, von denen einer mit einem Kupferdraht umwickelt wurde. Die Petrischale sollte jetzt nicht mehr bewegt werden. Decken Sie die Petrischale ab, da sonst die Agar-Lösung vorzeitig austrocknen kann. Anmerkung: K3 Fe(CN)6 ergibt mit Fe2+ -Ionen eine tiefblaue Färbung. Dieses Berliner Blau“ ist ein Nachweis ” für gelöste Fe2+ -Ionen. Aufgaben: Interpretieren Sie die auftretende Färbung mit ihren eigenen Worten. Was zeigt die rote Farbe an? Woher kommen die angezeigten Teilchen? Entsorgung: Agar eintrocknen lassen. Danach Feststoffabfälle. Eisennagel säubern und zurückgeben. Ende Versuch 3.7.2 3.7.3 Versuch: pH-Abhängigkeit des Redoxpotentials Stichworte: Oxidationszahlen, Redoxreaktion, Oxidationsmittel, Reduktionsmittel, Nernstsche Gleichung, Redoxpotential, Standardbedingungen Allgemeines: Für die Abhängigkeit zwischen dem Potential E eines Redoxpaares und den Konzentrationen der beteiligten Stoffe hat W. Nernst aus thermodynamischen Überlegungen die Gleichung E = E − 2.3 RT [Red] log nF [Ox] abgeleitet, in der R = 8.314 J·K−1 mol−1 die Gaskonstante, T die Temperatur und F = 96484.56 C · mol−1 die Faradaykonstante ist. Bei 25˚C lassen sich die Konstanten zusammenfassen: E = E − 0.059 [Red] log n [Ox] 3.7. REDOXREAKTIONEN (4.–5. WOCHE) 73 E gibt das Redoxpotential bei Standardbedingungen an. Der negative zweite Term bezieht sich auf die Konzentrationsabhängigkeit. n ist die Anzahl der bei dem Prozess bewegten Elektronen. An vielen Redoxprozessen sind auch Protonen beteiligt: Red * ) Ox + ne− + mH+ Ihre Konzentration ist ebenfalls für die Berechnung von E wichtig. Die pHabhängige Nernstsche Gleichung lautet: E = E − [Red] m 0.059 log − 0.059 · · pH n [Ox] n Unter Standardbedingungen (pH = 0) verschwindet der dritte Term. Unter physiologischen Bedingungen (pH = 7) hat der dritte Term mitunter 0 einen großen Einfluss auf E. Normalpotentiale bei pH 7 werden E genannt und oft in der Biochemie verwendet. Sie dürfen nicht mit E verwechselt werden. Chemikalien: KMnO4 (Kristalle), konz. H2 SO4 , NaOH-Plätzchen, H2 O2 Lösung (3 %) Durchführung: Zwei Reagenzgläser werden mit jeweils 5 ml Wasser gefüllt und darin ein paar kleine Kristalle Kaliumpermanganat (KMnO4 ) aufgelöst. Reagenzglas 1: Drei Tropfen konz. Schwefelsäure werden hinzugegeben. Reagenzglas 2: Zwei Plätzchen Natriumhydroxid (NaOH) (Vorsicht! Nicht mit bloßen Händen anfassen!) werden hinzugegeben und gelöst. In beide Reagenzgläser wird eine 3 %ige Wasserstoffperoxid-Lösung bis zum Ende der Gasentwicklung zugetropft. In Reagenzglas 1 entsteht Mangansulfat (MnSO4 ) und in Reagenzglas 2 entsteht Braunstein (MnO2 ). Aufgaben: Stellen Sie für beide Prozesse exakte Reaktionsgleichungen, unter Einschluss von H+ - und OH− - Ionen, auf und geben Sie dabei die Oxidationszahlen und die Anzahl der übertragenen Elektronen an. Unter welchen Bedingungen ist Kaliumpermanganat (KMnO4 ) das schwächere Oxidationsmittel? Interpretieren Sie den Einfluss des pH-Wertes anhand der Nernstschen Gleichung. Entsorgung: Neutralisieren, wässrige Schwermetallabfälle Ende Versuch 3.7.3 74 3.7.4 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Versuch: Wasserstoffperoxid als redoxamphoteres System Stichworte: Nernstsche Gleichung, Amphoterie, Disproportionierung Allgemeines: Wasserstoffperoxid (H2 O2 ) kann sowohl als Oxidationsmittel oxidierend wirken als auch als Reduktionsmittel reduzierend wirken. H2 O2 + 2e− + 2H+ → 2H2 O H2 O2 → O2 + 2H+ + 2e− (oxidierend) (reduzierend) Es ist in wässriger Lösung bei Raumtemperatur metastabil. Erst bei Energiezufuhr beginnt es sich in Sauerstoff und Wasser zu zersetzen. Chemikalien: H2 O2 -Lösung (3 %), saure KI-Lösung (einige Kristalle KIin wenig Wasser + 2 ml 1 M H2 SO4 ), CHCl3 , saure KMnO4 -Lösung (einige Kristalle in 5 ml Wasser + 2 ml 1 M H2 SO4 ), MnO2 Durchführung: Drei Bechergläser (50 oder 100 ml) werden mit jeweils ca. 5 ml 3 %iger H2 O2 -Lösung gefüllt. Becherglas 1: Zugabe von ca. 5 ml saurer Kaliumpermanganat-Lösung. Becherglas 2: Zugabe von saurer Kaliumiodid-Lösung. Mit 2 ml Chloroform (CHCl3 ) unterschichten. Becherglas 3: Zugabe von einer kleinen Spatelspitze Braunstein (MnO2 ). Aufgaben: Protokollieren und interpretieren Sie ihre Beobachtungen und stellen Sie für jede der drei Reaktionen vollständige Reaktionsgleichungen auf. Welche Oxidationszahl hat Sauerstoff in Wasserstoffperoxid (H2 O2 )? Welche der drei Redoxreaktionen ist eine Disproportionierung? Als was dient die Spatelspitze Braunstein (MnO2 ) in Becherglas 3? Entsorgung: Neutralisieren, wässrige Schwermetallabfälle. Ende Versuch 3.7.4 3.8. KOMPLEXE 3.8 75 Komplexe Stichworte: Übergangsmetalle, Besetzung von d- und f-Orbitalen, LewisSäure, Lewis-Base, Farbe von Komplexverbindungen, Koordinationszahl, Geometrie und Stereochemie von Komplexen, Chelatkomplexe, Struktur von Hämin, Chlorophyll und Vitamin B12 Weiterführend: Ligandenfeldtheorie, VB-Theorie, Diamagnetismus und Paramagnetismus In Atomen der schwereren Elemente ab Ordnungszahl 20 können nach Besetzung der kernnahen s- und p-Orbitale insgesamt fünf d-Orbitale mit maximal 10 Elektronen gefüllt werden, ehe ab Element 31 (Gallium) wieder p-Zustände zur Besetzung kommen. Zwischen der 2. und der 3. Hauptgruppe existieren daher 10 Nebengruppen“ mit sog. Übergangsmetallen. ” Die Zahl von d-Elektronen in den energetisch höherliegenden d-Orbitalen bestimmt die chemischen und physikalischen Eigenschaften dieser Übergangsmetalle. Gemeinsame Merkmale und charakteristische Unterschiede zu den Hauptgruppenelementen sind ihr Auftreten in verschiedenen Oxidationsstufen, die starke Tendenz zur Bildung von Komplexen, in denen Liganden zusätzliche Elektronenpaare für unbesetzte d-Orbitale beisteuern, und das häufigste Vorkommen farbiger sowie paramagnetischer Ionen und Verbindungen. Angesichts ihrer chemischen Vielfalt ist es nicht erstaunlich, dass eine Reihe von Übergangsmetallen essentielle biologische ( bioanorganische“) Funk” tionen übernehmen. Einige wichtige Beispiele sind Mangan: Eisen: Cobalt: Nickel: Kupfer: Wasserabspaltung im Photosystem II der grünen Pflanzen Sauerstofftransport, Redoxsystem in Cytochromen Reaktives Zentrum im Vitamin B12 Mikrobielle Bildung von Methangas und Wasserstoff Redoxsystem in der Atmungskette (Cytochromoxidase) Hämocyanin (sauerstoffträger bei Wirbellosen, z. B. Mollusken) Zink: Bestandteil einer Reihe von Enzymen Molybdän: Katalyse der Stickstofffixierung durch Nitrogenase Neben den bekannten Metallen Eisen, Kupfer und Zink sind viele weitere Übergangsmetalle technisch wichtig, wie Titan, Vanadium, Chrom, Mangan und Nickel in Legierungen, Nickel und die Platinmetalle (Rhodium, Palladium, Platin) als Katalysatoren, Silber und Gold als Edelmetalle hoher Leitfähigkeit. Das flüssige Quecksilber mit seinen meist toxischen Verbindungen 76 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE nimmt eine Sonderstellung in der Chemie der Übergangsmetalle ein und wird hier nicht behandelt. Komplex- oder Koordinationsverbindungen Ein Komplex entsteht, wenn sich verschiedene, an sich auch allein stabile Atome, Ionen oder Moleküle miteinander koordinieren“ und stöchiometrisch ” sowie geometrisch wohldefinierte neue Verbindungen höherer Ordnung bilden. Komplexbildung erfolgt zwischen Teilchen mit Elektronenlücken“ (d. h. mit ” einer kleineren Zahl von Elektronen als zum Erreichen der nächsten stabilen abgeschlossenen Elektronenkonfiguration erforderlich) und anderen Teilchen (Liganden), die über freie Elektronenpaare verfügen: Bei Kombination beider erreicht das System einen energetisch günstigeren Zustand. Komplexbildung ist nicht auf Übergangsmetalle beschränkt, aber wegen der im Allgemeinen nicht voll besetzten d-Orbitale dort besonders ausgeprägt. Ein Komplex besteht aus einem Zentral-Atom oder Zentral-Ion als Koordinationszentrum und der Ligandenhülle. Das Koordinationszentrum ist typischerweise ein Metallion, als Liganden dienen zumeist Anionen oder Neutralmoleküle mit freien Elektronenpaaren. Die Anzahl der Bindungen zwischen dem Zentralteilchen und seinen Liganden bezeichnet man als Koordinationszahl (KZ) des Zentralteilchens. Liganden mit mehr als einer Koordinationsstelle (d. h. freie Elektronenpaare oder negative Ladungen) heißen mehrzähnige- oder Chelatliganden. Einige der wichtigsten Liganden und Chelatliganden sind das sechszähnige EDTA, Oxalat (zweizähnig; zwei Carboxylat-Gruppen), Ethylendiamin (zweizähnig, zwei Amino-Gruppen mit einem freien Elektronenpaar), Acetylacetonat (zweizähnig; zwei CarboxylatGruppen), Wasser und Ammoniak. Zeichnen Sie zur Übung bei den folgenden Liganden die zur Komplexbildung benötigten freien Elektronenpaare ein. O O O NH3 H Ammoniak H Wasser NH2 -O Ethylendiamin Oxalat O CH3 CH3 N O O O O Acetylacetonat NH2 O- O N O O O-O Ethylendiamintetraacetat = EDTA Wird ein Metall komplexiert, so unterscheiden sich die Eigenschaften des Komplexes oft drastisch von denen des freien Metall-Ions. Dasselbe gilt für die Liganden. 3.8. KOMPLEXE 77 Häufig sind Farbänderungen zu beobachten: Eine Lösung von Kupfersulfat in Wasser ist schwach hellblau, mit Ammoniak wird die Lösung tiefblau, bei Zusatz von Chloridionen ändert sich dagegen die Farbe der Lösung über Grün nach Gelb. In den Lösungen sind unterschiedliche Komplexionen enthalten: ([Cu(H2O)6]2+ (hellblau), [Cu(NH3)4]2+ (tiefblau), [CuCl2(H2O)4] (gr¨un), [CuCl4]2− (gelb) Ionen können durch Komplexbildung bzgl. ihres Löslichkeitsverhaltens und ihrer Reaktivität maskiert“ werden. Während bekanntlich Ag+ -Ionen ” durch Chlorid als Ag Cl ausgefällt werden, reagieren die Komplexionen [Ag(NH3 )2 ]+ nicht mit Chlorid unter Fällung. Ag+ -Ionen werden als Oxidationsmittel (E = +0.8 V) leicht zu elementarem Silber reduziert, nicht aber in Gegenwart von Cyanid als Cyanokomplex [Ag(CN)2 ]2− , der ein negatives Redoxpotential aufweist (E = −0.31V). Kationsäuren Lösungen von Metall-aquakomplexen wie [Fe(H2O)6]3+ reagieren stark sauer (sie sind sog. Kationsäuren“), weil Wassermoleküle in der Ligandensphäre ” polarisiert werden und Protonen abspalten. Informieren Sie sich in den Lehrbüchern über die Geometrie und Isomerie von Komplexen. Nomenklaturregeln zur Benennung von Komplexen • Ist die Komplexverbindung ein Salz, so wird zuerst das Kation ge-nannt. Im [Ag(NH3)2]Cl wird daher zuerst der kationische Komplex [Ag(NH3)2] + und dann das Gegenion Cl− geschrieben. • In den Namen der Komplex-Ionen oder -Moleküle werden die Liganden vor dem zentralen Matallion aufgeführt, verschiedene Ionen oder neutrale Moleku¨le als Liganden in alphabetischer Reihenfolge. So heißt das komplexe Kation [Cr(H2O)4Cl2]+ Tetraquadichloridochrom(III). • Anionische Liganden enden auf -o, während neutrale Liganden in der Regel die Moleku¨lnamen tragen. Fu¨r einige wichtige Liganden lauten die Bezeichnungen: N3− azido, Br− bromido, CN− cyanido, F− fluorido, HO− hydroxido, CO32− carbonato, C2O42− (Oxalat) oxalato, S2− thio, NH3 ammin, CO carbonyl, H2O aqua (häufig wird aber auch aquo benutzt). • Die Zahl der Liganden einer Sorte wird durch griechische Zahlworte (di-, tri-, tetra-, penta-, hexa-) angegeben. Enthalten die Namen der Liganden schon solche Präfixe, dann werden die Vorsilben bis-, tris-, tetrakis 78 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE usw. verwendet. So lautet der Name für den Komplex [Co(en)3 ]Cl3 Tris(ethylendiamin)-cobalt(III)chlorid. • Ist der Komplex ein Anion, so wird die Endung -at benutzt. So heißt z. B. die Verbindung K4 [Fe(CN)6 ] Kalium-hexacyanoferrat(II), Na[Al(OH)4 ] heißt Natriumtetrahydroxoaluminat(III). 3.8.1 Versuch: Cu-Komplexe Stichworte: Komplexbildungskonstante, Liganden, Stabilität von Komplexen Allgemeines: Kupfer bildet, wie viele andere Übergangsmetalle, mit negativ geladenen Ionen oder einigen Lewis-Basen mit einem freien Elektronenpaar farbige Komplexe. So entsteht mit Wasser der bläuliche TetraquaKomplex [Cu(H2 O)4 ]2+ , mit Clorid (Cl− ) der grüne Tetrachloro-Komplex [CuCl4 ]2− und mit Ammoniak (NH3 ) der blaue Tetramin-Komplex [Cu(NH3 )4 ]2+ . Die Bildung eines Komplexes ist zumeist eine Gleichgewichtsreaktion. Für eine Komplexbildungsreaktion wie: M + 6L * ) ML6 (M = Metallion, Zentralatom; L = Ligand) lässt sich auch anhand des Massenwirkungsgesetzes (MWG) eine Komplexbildungskonstante formulieren: K= [ML6 ] [M] · [L]6 Die Bildung eines Komplexes hängt demnach auch stark von der Konzentration der Liganden ab. Die Aminosäure Glycin bildet mit Kupfer einen stabilen Komplex. Glycin ist ein zweizähniger“ Chelatligand. Er kann hier als Modell für viele im ” Körper wichtige Komplexe dienen. Die beiden Zähne bestehen zum einen aus dem negativen Sauerstoff und dem freien Elektronenpaar des Stickstoffs. O _ [Cu(H2O)]2+ + 2 NH2 O O O- - NH2 + Cu2+ H2N -O O 2 H2O 3.8. KOMPLEXE 79 Chemikalien: CuCl2 · H2 O, NaCl, verd. NH3 , CuSO4 -Lösung (1 g CuSO4 + 10 ml H2 O) Durchführung: a.) Ligandenaustausch: In einem Reagenzglas wird 1 g Kupferchlorid (CuCl2 · 2H2 O) in ca. 6 ml Wasser gelöst. Die leicht blaue Farbe der Lösung lässt sich durch Zugabe von einigen Spatelspitzen Natriumchlorid (NaCl) in Grün überführen. Durch Verdünnen tritt die leicht blaue Farbe wieder auf. Zu 5 ml dieser Lösung wird verd. Ammoniak (NH3 ) zugetropft. Nach jedem Tropfen wird das Reagenzglas geschüttelt. Das sich anfänglich bildende, feste Kupferhydroxid (Cu(OH)2 ) löst sich nach weiterer Zugabe wieder auf, und es entsteht bei Ammoniak-Überschuss ein löslicher Komplex. b.) Diglycinokupfer(II)-Komplex: 0.5 g Glycin werden in 10 ml Wasser unter vorsichtigem Erhitzen über dem Bunsenbrenner gelöst. Die pH-Werte dieser Lösung und der Kupfersulfatlösung (CuSO4 ) werden mit Indikatorpapier gemessen. In einem Reagenzglas werden jeweils 3 ml der Kupfersulfat-Lösung und der Glycin-Lösung vereinigt und der pH-Wert erneut gemessen. Um die Stabilität des entstandenen Komplexes zu demonstrieren, wird durch Zutropfen von Natriumhydroxid (NaOH) versucht, den Komplex zu zerstören. Das Ausfallen“ des fe” sten Cu(OH)2 würde den Zerfall des Komplexes anzeigen. Man tropft Natriumhydroxid (NaOH) zu, bis die Lösung schwach alkalisch ist (Indikatorpapier). Aufgaben: a.) Protokollieren Sie ihre Beobachtungen und interpretieren Sie diese, indem Sie zu jedem Prozess eine Reaktionsgleichung formulieren. Welcher grüne Komplex bildet sich nach Zugabe von Natriumchlorid (NaCl)? Warum löst er sich nach Verdünnen mit Wasser wieder auf? Welcher lösliche Kupferkomplex bildet sich nach Zugabe von Ammoniak (NH3 )? b.) Interpretieren sie den gemessenen pH-Wert nach der Vereinigung von Kupfersulfat-Lösung (CuSO4 ) und Glycin-Lösung. Konnte der Komplex durch Zugabe von Natriumhydroxid (NaOH) zerstört werden? Entsorgung: Wässrige Schwermetallabfälle Ende Versuch 3.8.1 80 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE 3.8.2 Versuch: Fe-Komplexe Zweiergruppe Stichworte: Kationsäure, Autooxidation, Chelatliganden Allgemeines: Bioanorganische Komplexe des Eisens (Hämin, Ferredoxin, Ferritin u. a.) spielen in der Biochemie eines lebenden Organismus eine tragende Rolle. In wässriger Lösung werden Fe3+ -Ionen von sechs Wassermolekülen zum Hexaqua-Komplex [Fe(H2 O)6 ]3+ komplexiert. Durch die hohe Ladung des Zentralatoms werden die Liganden so polarisiert, dass es recht leicht ist, eines der Wasserstoffatome als H+ an die Umgebung abzugeben. Der Komplex reagiert sauer. Man spricht hier von einer Kationsäure. [Fe(H2 O)6 ]3+ → [Fe(H2 O)5 (OH)− ]2+ + H+ Fe2+ ist zwar in festen Salzen stabil, wird aber durch Autooxidation in wässriger Lösung nach gewisser Zeit zu Fe3+ . In Lösung liegen daher zumeist Fe3+ -Ionen vor. Eisenionen bilden mit Thiocyanat (SCN− ) einen sehr farbstarken, roten Komplex. Selbst kleine Mengen Eisen können damit sichtbar gemacht werden. Diese Reaktion eignet sich daher als Nachweisreaktion für Eisen. Mit o-Phenantrolin, einem zweizähnigen Chelatliganden, ergibt sich ebenfalls ein tiefrot gefärbter 3:1 Komplex. Auch diese Reaktion eignet sich als Nachweisreaktion. Allerdings lassen sich durch diese Nachweise nur freie Eisenionen in Lösung nachweisen. Chemikalien: FeCl3 , NH4 SCN, FeSO4 , K3 [Fe(CN)6 ], K4 [Fe(CN)6 ], o-Phenantrolin-Lösung (2 % in Ethanol gelöst), 2 M H2 SO4 Durchführung: a.) Kationsäure: Messen Sie den pH-Wert einer FeCl3 -Lösung (0.5 g FeCl3 in 10 ml Wasser). Stellen Sie eine frische Eisensulfat-Lösung (FeSO4 ) aus ca. 0.5 g Eisensulfat und 10 ml Wasser her. Messen Sie den pH-Wert sofort 3.8. KOMPLEXE 81 nach der Herstellung und dann einige Zeit später. b.) Eisennachweis: Reagenzglas 1: zwei Finger breit frische FeSO4 -Lösung + 1 Spatelspitze Ammoniumthiocyanat (NH4 SCN) Reagenzglas 2: zwei Finger breit frische FeCl3 -Lösung + 1 Spatelspitze Ammoniumthiocyanat (NH4 SCN) Reagenzglas 3: zwei Finger breit Kaliumhexacyanoferrat-(III)-(K3 [Fe(CN)6 ])Lösung (rotes Blutlaugensalz) + 1 Spatelspitze Ammoniumthiocyanat (NH4 SCN) Reagenzglas 4: zwei Finger breit Kaliumhexacyanoferrat-(II)-(K4 [Fe(CN)6 ])Lösung (gelbes Blutlaugensalz) + 1 Spatelspitze Ammoniumthiocyanat (NH4 SCN) Die gleiche Nachweisreihe wird mit o-Phenantrolin-Lösung anstelle von Ammoniumthiocyanat (NH4 SCN) wiederholt. Aufgaben: a.) Protokollieren Sie die gemessenen pH-Werte, und erklären Sie eine eventuelle Änderung dieser Werte. b.) Protokollieren Sie die gemachten Beobachtungen, und vergleichen Sie die beiden Nachweisreaktionen. Gibt es Unterschiede? Erklären Sie, warum der Nachweis bei manchen eisenhaltigen Substanzen nicht erfolgreich war. Entsorgung: Neutralisieren, wässrige Schwermetallabfälle Ende Versuch 3.8.1 3.8.3 Versuch: Gleichgewicht von Kupferkomplexen Zweiergruppe Stichworte: Hydration, Aqua-Ionen, Komplexbildungsgleichgewicht, Chloridnachweis mit Silbernitrat 82 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Allgemeines: Komplexgleichgewichtsreaktionen eignen sich wegen der oft auffälligen Farbreaktionen gut für eine Veranschaulichung der Temperaturund Konzentrationsabhängingkeit von Gleichgewichtsreaktionen. Kupferchlorid zeigt in wässriger Lösung folgendes Gleichgewicht: [Cu(H2 O)4 ]2+ +4Cl− * ) [CuCl4 ]2− + H2 O (blau) (gelb) Das blaue Kupfertetraqua-Ion [Cu(H2 O)4 ]2+ steht im Gleichgewicht mit dem gelben, negativ geladenen Kupfertetrachlorokomplex [CuCl4 ]2− . Die Farbe der Lösung gibt also die Lage des obigen Gleichgewichtes an. Dieses Gleichgewicht lässt sich gemäß dem Massenwirkungsgesetz für diese Reaktion durch Zugabe oder Entzug von Chloridionen (Cl− ) beinflussen: [CuCl4 ]2− · [H2 O] K= 4 [Cu(H2 O)4 ]2+ · Cl− Durch Zugabe von Silbernitrat (Ag NO3 ) bildet sich mit den Chloridionen (Cl− ) das schwerlösliche, weiße Silberchlorid (Ag Cl). Diese Reaktion ist zum Nachweis und zur Verringerung der Konzentration von Chloridionen (Cl− ) geeignet. Chemikalien: 1 M CuCl2 -Lösung (17 g CuCl2 · 2H2 O in 100 ml Wasser), konz. HCl, konz. NH3 , 1 M Ag NO3 -Lösung Durchführung: 50 ml einer 1 M Kupferchlorid-(CuCl2 )-Lösung werden gleichmäßig auf 4 Reagenzgläser verteilt. Die Lösung in Glas 1 ist blaugrün und nähert sich beim Verdünnen mit Wasser dem himmelblauen Farbton einer Kupfersulfatlösung. Zugabe von konzentrierter Salzsäure in Glas 2 führt zur Farbänderung nach Hellgrün, welche beim Erwärmen in einen Braunton übergeht. In Glas 3 setzt man konzentrierte Ammoniaklösung bis zur tiefen Blaufärbung zu, die bei Verdünnung einen helleren Ton annimmt. Glas 4 wird zunächst wie Glas 2 mit konzentrierter Salzsäure behandelt, fügt man 1 M Silbernitrat (Ag NO3 ) Lösung im Überschuss hinzu, erhält man einen weißen Niederschlag, und die Farbe wechselt nach Blaugrün. Aufgaben: Protokollieren Sie Ihre Beobachtungen, und formulieren Sie für jeden der 4 Prozesse eine Reaktionsgleichung. Erklären Sie die Prozesse in eigenen, kurzen Worten anhand des Massenwirkungsgesetzes. Warum nimmt die Lösung in Reagenzglas 1 beim Verdünnen die Farbe einer Kupfersulfatlösung an? Entsorgung: Neutralisieren, wässrige Schwermetalle Ende Versuch 3.8.3 3.8. KOMPLEXE 83 3.8.4 Versuch: Magnesium im Spinat Boxweise Stichworte: Chelatligand, Zentralatom, Chlorophyll, Titangelb, organische Farbstoffe Allgemeines: Ein Chelatligand ist ein Molekül, welches gleich mehrere Zentren besitzt, die entweder durch Ladung oder durch ein freies Elektronenpaar eine Komplexbildung zu einem Zentralatom aufbauen können. Schlagen Sie dazu die entsprechenden Kapitel in den einschlägigen Lehrbüchern nach. Der Pflanzenfarbstoff Chlorophyll ist ein Chelatkomplex mit Magnesium (Mg ) als Zentralatom. In höheren Pflanzen liegt ein Gemisch aus blaugrünem Chlorophyll a (ca. 75 %) und gelbgrünem Chlorophyll b vor. Diese unterscheiden sich nur an dem Rest R (siehe Abbildung). R N _ Mg \ N _ N O O O OC20H39 C2H5 \N OMe Chlorophyll a: R = CH3 Chlorophyll b: R = CHO Das Zentralatom Magnesium ist hier von vier Stickstoffatomen mit freien Elektronenpaaren umgeben. Diese bilden die vier Zähne des Chelatliganden. Setzt man das Magnesium frei, so lässt es sich mit einem typischen Magnesiumtest mit Titangelb nachweisen. SO3Na S N _ SO3Na _ _ N N _ N S H N _ Als einen Vertreter chlorophyllhaltiger Pflanzen benutzen wir im diesem Versuch Spinat. Er enthält Magnesium, welches unter anderem auch im Chlorophyll gespeichert ist. 84 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Der grüne Chlorophyll-Komplex ist hier im Gegensatz zu den vorrangegangenen farbigen Komplexen nicht aufgrund der Wechselwirkung der dZustände farbig. Vielmehr liegt die grüne Farbe an dem konjugierten System der Doppelbindungen. Auch die Farbigkeit des Titangelb-Moleküls beruht auf diesem Effekt. Aufgrund des Umfanges dieses Themas kann Ihnen an dieser Stelle nur empfohlen werden, sich dieses Wissen mit Hilfe eines Lehrbuches anzueignen (siehe auch organischer Teil). Es ist biologisch durchaus von Interesse. Chemikalien: Spinat, 4 M HCl, 2 M NaOH, pH-Papier, Titangelb-Lösung (0.05 %) Durchführung: 10 g kleingeschnittener Spinat (tiefgekühlt und aufgetaut) werden mit 10 ml 4 M Salzsäure (HCl) in einem 250 ml Erlenmeyer-Weithalskolben über dem Bunsenbrenner (Dreifuß, Drahtnetz) im Abzug einmal kurz aufgekocht. Zur Vermeidung von Siedeverzügen werden einige Siedesteine in die wässrige Aufschlämmung der Spinatblätter gegeben (Vorsicht vor Spritzern!). Danach wird die Suspension durch ein Faltenfilter gegeben und das klare Filtrat durch tropfenweise Zugabe von 2 M Natronlauge unter Prüfung mit pH-Papier neutralisiert. Dabei kann sich die Lösung leicht trüben. Die neutralisierte Lösung wird auf etwa 100 ml verdünnt und ungefähr 2 ml der verdünnten Lösung in ein Reagenzglas gegeben. In das Reagenzglas wird zusätzlich die gleiche Menge 2 M NaOH sowie 2–3 Tropfen der 0.05 %igen Titangelblösung gegeben und mit einem Glasstab umgerührt. Rotfärbung oder ein roter Niederschlag zeigen die Anwesenheit von Mg2+ im Hydrolysat des Spinats an. Aufgaben: Protokollieren Sie die einzelnen Verfahrensschritte in kurzen Worten und stellen Sie Vermutungen darüber an, warum diese nötig waren, um das Magnesium zu isolieren. Entsorgung: Reste neutralisieren und in den Ausguss entsorgen Ende Versuch 3.8.4 3.9. ANALYTISCHE BESTIMMUNGEN 3.9 85 Analytische Bestimmungen 3.9.1 Analyse 2: Bestimmung von NaCl durch Ionenaustauscher Zweiergruppe Stichworte: Massenwirkungsgesetz, Säure/Base-Titration, Sulfonsäuregruppe Allgemeines: Die hier verwendeten Ionenaustauscher bestehen aus einem Glasrohr mit einem Auslauf an einem Ende. Das Glasrohr ist mit dem Ionenaustauscherharz befüllt. Wird nun eine Natriumchloridlösung (NaCl) eingefüllt, so kommt nach kurzer Zeit am Auslauf nur noch Salzsäure (HCl) heraus. Die am Austauscherharz vorhanden H+ -Ionen wurden mit den Na+ -Ionen der Lösung ausgetauscht und aus der Säule ausgespült“. ” Kationenaustauscher tauschen spezifisch alle positiven Metallionen (z. B. Na+ ) gegen Protonen (H+ ) aus. Sie bestehen aus einem Harz welches die feste, immobile Phase darstellt. An diesem Harz sind saure funktionelle Gruppen chemisch gebunden, wie z. B. Sulfonsäure-Gruppen. Diese tragen acide(saure) Wasserstoff-Atome. Lässt man nun eine wässrige Salzlösung (Wasser ist die mobile Phase) durch den Kationenaustauscher fließen, so geben die Sulfonsäuregruppen die Protonen ab und binden die Natriumionen an sich. Harz Harz O S O H O + Na+ - H+ O S O Na+ O Anionenaustauscher tauschen spezifisch alle negativ geladenen Anionen aus. Sie haben funktionelle Gruppen mit Zentren, welche in der Lage sind, die Anionen an sich zu binden und dafür Hydroxidionen (OH− ) abzugeben. Auf diese Weise lässt sich eine Natriumsulfat-(Na2 SO4 )-Lösung in eine Natriumhydroxid-(NaOH)-Lösung überführen. 86 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Neben Reinigungsfunktionen (Herstellung von entionisiertem Wasser) haben Ionenaustauscher auch eine analytische Bedeutung. Die Konzentration einer Natriumchlorid-(NaCl)-Lösung lässt sich durch Umwandeln in eine Salzsäure-(HCl)-Lösung und anschließende Titration ermitteln. Die durch die Titration ermittelte Konzentration der Salzsäure-(HCl)-Lösung, muss dieselbe sein, wie die Konzentration der Natriumchlorid-(NaCl)-Lösung. Chemikalien: Ionenaustauscher, 4 M HCl Durchführung: Diese Analyse wird in Zweiergruppen durchgeführt. Probenabgabe: Beschriften Sie einen 100 ml Messkolben mit folgenden Daten (siehe auch Analyse 1): Praktikant 1: Nachname, Vorname: Analyse: 2 Box Nr.: Saal: Praktikant 2: Nachname, Vorname: Analyse: 2 Box Nr.: Saal: Im Gegensatz zur Analyse 1 wird hier, bei einer Falschansage, nur in Ausnahmefällen eine weitere Probe für den Praktikanten ausgegeben. Am darauffolgenden Tag sind die gefüllten Proben abzuholen. Vorbereiten der Säule: Lassen Sie sich den Aufbau und Befüllung sicherheitshalber vom Assistenten genau beschreiben. In eine extra ausgegebene Glassäule wird unten ein Pfropf aus Glaswolle gestopft. Benutzen Sie dabei Haushaltshandschuhe und stellen Sie sicher, dass Sie möglichst wenig von der Glaswolle einatmen. Der Pfropf kann mit einem langen Glasstab vorsichtig unten in der Säule festgedrückt werden. Er dient dem Zurückhalten des Ionenaustauscherharzes. Spannen Sie die Säule senkrecht in die dafür vorgesehene Halterung. Lassen Sie den korrekten Sitz vom Assistenten überprüfen. Bringen Sie ein Stück Gummischlauch an dem Auslauf der Säule an und verschließen Sie diesen mit einem Quetschhahn. Der Schlauch sollte so bemessen sein, dass er bis etwa zu einem Drittel in einen 250 ml Erlenmeyerkolben hineinragen kann (Aufbau siehe Bild). 3.9. ANALYTISCHE BESTIMMUNGEN 87 Bei geschlossenem Quetschhahn wird zunächst etwas Wasser hineingegeben, so dass die Glaswolle davon bedeckt ist. Mit dem Glasstab wird die Glaswolle solange gedrückt, bis sich keine Luftblasen in ihr befinden. Bei geöffnetem Quetschhahn wird das Ionenaustauscherharz eingeschlämmt (Vorsicht, enthält noch einen Rest Salzsäure). Dabei ist darauf zu achten, dass sich keine Luftblasen bilden. Diese lassen sich durch vorsichtiges Umrühren mit einem Glasstab beseitigen. Nachdem der Ionenaustauscher bis etwa 2 cm unter der Auslaufhöhe des Rohres gleichmäßig eingeschlämmt wurde, füllen Sie ihn mit entionisiertem Wasser (kein Leitungswasser!!) und spülen Sie ihn damit so lange, bis die am Schlauch austretenden Tropfen neutrale Reaktion zeigen (pH-Papier). Die Säule darf ab jetzt nicht mehr trockenlaufen“. Der ” Flüssigkeitsstand darf also nicht mehr unter den Stand der Harzbefüllung fallen. Benutzen Sie beim Durchspülen ein großes Becherglas und neutralisieren Sie die Lösung anschließend. Diese kann danach im Ausguss entsorgt werden. Der Ionenaustauscher ist jetzt gebrauchsfertig. Lassen Sie die Flüssigkeit bis kurz über die Befüllung auslaufen und schließen Sie den Quetschhahn. Spülen Sie den Inhalt ihres Analysekolbens quantitativ in die Ionenaustauschersäule. Benutzen Sie zum Ausspülen des Analysekolbens eine PVCSpritzflasche mit entionisiertem Wasser. Achten Sie aber darauf, dass Sie nur so viel Wasser wie unbedingt nötig benutzen. Das Aufbringen der Analysenlösung sollte möglichst in einem Schritt und konzentriert erfolgen. Der 88 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Quetschhahn wird jetzt gerade so weit geöffnet, das höchstens ca. ein Tropfen in der Sekunde austreten kann. Lassen Sie etwa 50 ml auf diese Weise in ein Auffanggefäß laufen, bevor Sie einen sauberen 250 ml Erlenmeyerkolben unter den Auslaufschlauch stellen. Prüfen Sie dabei immer wieder den pHWert der Lösung (pH-Papier). Sollte dieser sich zum sauren hin verändern, stellen Sie sofort einen sauberen 250 ml Erlenmeyerkolben als Auffanggefäß auf. Füllen Sie die Säule immer wieder mit entionisiertem Wasser auf, damit sie nicht trockenläuft. Lassen Sie so lange die Lösung durchlaufen, bis die Tropfen wieder neutrale Reaktion zeigen (pH-Papier). Danach wird die so aufgefangene Salzsäurelösung mit einer 1 M Natronlauge (NaOH) (wird ausgegeben) mit bekanntem Titer titriert und der Gehalt an Salzsäure in mol bestimmt (siehe Titrationen). Hierbei wird der gesamte Inhalt des Erlenmeyerkolbens bei nur einer Titration titriert. Als Indikator wird Phenolphthalein verwendet. Regenerieren des Austauscherharzes: Zum Entfernen der Natriumionen aus dem Autauscherharz wird durch einen Überschuss an Säure das Gleichgewicht verschoben. Es bildet sich wieder Natriumchlorid, welches ausgeschwemmt wird. Dazu lässt man ca. 100 ml 4 M Salzsäure (HCl) langsam durch den Austauscher laufen und spült solange mit entionisiertem Wasser nach, bis die austretenden Tropfen eine neutrale Reaktion mit pH-Papier zeigen. Der Austauscher kann jetzt wieder in das Vorratsgefäß gegeben werden. Soll der Autauscher von einer anderen Gruppe benutzt werden, so ist er folgendermaßen zu beschriften: Regeneriert am:. . . von:. . . Aufgaben: Berechnen Sie anhand des Verbrauches bei der Titration den Gehalt an NaCl in ihrem Analysekolben in mg. Protokollieren Sie ihre Vorgehensweise, Berechnungen und das Ergebnis. Machen Sie bei Ihrem Assistenten eine Ergebnisansage. Es sind zwei Ansagen möglich. Entsorgung: Lösungen neutralisieren, Ausguss. Der regenerierte Ionenaustauscher wird in das Vorratsgefäß zurückgegeben. Ende Versuch 3.9.1 3.9. ANALYTISCHE BESTIMMUNGEN 89 3.9.2 Analyse 3: Photometrische Bestimmung von Phosphat Boxweise Stichworte: Photometrie, Lambert-Beersches-Gesetz, Extinktion, Extinktionskoeffizient, Photometer, Kalibrierkurve Allgemeines: Die Photometrie wird sehr häufig zur Bestimmung der Konzentration einer Lösung herangezogen. Hierbei wird der Zusammenhang zwischen Lichtabsorption und Konzentration ausgenutzt. Dieser Zusammenhang lässt sich durch das Lambert-Beersche-Gesetz beschreiben. Eine farbige Lösung absorbiert in einem bestimmten Wellenlängenbereich Licht. Die nicht absorbierten Wellenlängen (Farben) des aus der Lösung austretenden oder reflektierten Lichtes erzeugen im Auge des Betrachters einen Farbeindruck. Auch vermeintlich farblose Lösungen absorbieren Licht bestimmter Wellenlängen. Da diese Wellenlängen des Lichts aber für den Menschen nicht sichtbar sind, entsteht für uns kein Farbeindruck. Ist bekannt, welche Wellenlänge am besten von einem Stoff absorbiert wird, so lässt sich diese Wellenlänge für eine photometrische Konzentrationsmessung nutzen. Ist eine Substanz in einem Lösungsmittel gelöst und wird ein Lichtstrahl geeigneter Wellenlänge hindurchgesandt, so wird um so mehr Licht von der Lösung absorbiert, je konzentrierter diese ist. Der austretende Strahl hat eine geringere Intensität I als der eintretende Strahl I0 . I0 Lösung I I ist um so kleiner, je größer die Konzentration der Lösung und die Schichtdicke d (Weg des Lichtes durch die Lösung) ist. Zur Durchlässigkeit D stehen diese Größen in folgender Beziehung: D= I = 10−εcd I0 Der molare, dekadische Extinktionskoeffizient ε ist wellenlängenabhängig und stoffspezifisch. Für völlige Lichtabsorption ist D = 0, während bei völliger Durchlässigkeit D = 1 ist. Aus praktischen Gründen wird diese Beziehung zum Lambert-Beerschen Gesetz umgeformt. Es gibt die Beziehung der Extinktion E zu den Größen d (Schichtdicke in cm), ε (Extinktionskoeffizient in cm2 /mmol) und c (Konzentration in mmol/ml) an: E = log I0 = εcd I 90 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Die Extinkion E ist dimensionslos und geht von Null (keine Lichtabsorption) bis unendlich (völlige Lichtabsorption). Messbar ist i. a. der Bereich 0 bis 2. Lambert-Beersches Gesetz: Bei konstanter Schichtdicke d sind die gemessene Extinktion E und die Konzentration c einer Lösung einander direktproportional. Für c = 1 mol/l und d = 1 cm wird E gleich ε. Ist ε bekannt, so genügt eine Extinkionsmessung zur Berechnung von c. Oft ist aber für komplizierte oder empirische Farbreaktionen (Phosphat-, Zucker-, Proteinbestimmung) ε nichtbekannt oder E ist stark von den Messbedingungen abhängig. In diesen Fällen erstellt man mit Lösungen bekannter Konzentration eine Kalibrierkurve. Die Konzentrationen der Kalibirierlösungen sollten in dem vermuteten Konzentrationsbereich der zu messenden Probe liegen, um die Genauigkeit der Interpolation zu erhöhen. Wird die Extinktion E bei einer konstanten Wellenlänge mit verschiedenen bekannten Konzentrationen gemessen, so resultiert eine Kalibrierkurve mit einem linearen Bereich. Innerhalb von diesem Bereich lässt sich durch Interpolation die Konzentration einer Probe ermitteln. !"#%$'&(#*)+ Aufbau eines Photometers: Ein Photometer besteht aus einer Lichtquelle, einem Monochromator (Farbfilter, Prisma oder Gitter) zur Erzeu- 3.9. ANALYTISCHE BESTIMMUNGEN 91 gung einfarbigen Lichts einer bestimmten Wellenlänge, einem Küvettenraum für die Probe, einer Photozelle zum Messen der Lichtintensität und einem Anzeigegerät (Galvanometer). Lichtquelle Anzeige Photozelle Monochromator Küvette Filter, Prisma oder Gitter Chemikalien: Ammoniummolybdat, 2 M H2 SO4 , FeSO4 , P-Standardlösung (1.0 µmol P/ml) Durchführung: Probenabgabe: Ein 100 ml Kolben wird wie folgt beschriftet: Namen, Vorname: Analyse: 3 Box Nr.: Saal: Die Beschriftung sollte auf einem geügend großen Stück Papier mit einem Loch erfolgen und am Meßkolben angebracht werden. Reagenzlösung: 1.7 g Ammoniummolybdat in 50 ml 2 M Schwefelsäure (H2 SO4 ) in einem 100 ml Messkolben lösen, kurz vor Gebrauch mit 1 g Eisen(II)sulfat (FeSO4 ) versetzen und auf 100 ml auffüllen. Probe: Sie erhalten die Probe in einem 100 ml Messkolben. Diesen füllen Sie auf 100 ml mit entionisiertem Wasser auf und mischen die Lösung. Kalibrierkurve und Messung: Jede Gruppe erstellt eine eigene Kalibrierkurve, um systematische und Gerätefehler möglichst auszuschließen und um gleiche Messbedingungen zu garantieren. Das genaue Arbeiten bei der Vorbereitung der Proben für die Kalibrierkurve wirkt sich stark auf das Ergebnis aus. Vier saubere Reagenzgläser bereithalten und P-Standardlösung (1.0 µmol P/ml) hineinpipettieren. 92 KAPITEL 3. ALLGEMEINE UND ANORGANISCHE CHEMIE Reagenzglas 1: 1.0 ml P-Standardlösung + 2.5 ml Reagenzlösung + 1.5 ml dest. Wasser Reagenzglas 2: 1.5 ml P-Standardlösung + 2.5 ml Reagenzlösung + 1 ml dest. Wasser Reagenzglas 3: 2.0 ml P-Standardlösung + 2.5 ml Reagenzlösung + 0.5 ml dest. Wasser Reagenzglas 4: 2.5 ml P-Standardlösung + 2.5 ml Reagenzlösung Reagenzglas 5: 1.0 ml der Probenlösung + 2.5 ml Reagenzlösung + 1.5 ml dest. Wasser Alle Reagenzgläser sollten jetzt mit 5 ml Lösung gleich hoch befüllt sein. Nach 5 min. sollten die Lösungen zusammen mit der Probe zügig am Photometer gemessen werden. Dazu werden die Lösungen am Photometer in die Messküvette gefüllt und die Extinktion gegen eine Wasserprobe gemessen, um die Absorption des Lösungsmittels (Wasser) von der Messung auszuschließen. Die Quarzküvetten sind Präzisionsgeräte und müssen sorgfältig behandelt werden. Sie dürfen nur an den milchigen bzw. rauhen Seiten angefasst werden! (Ein Küvettenpaar kostet etwa 1000,- DM). Außen an der Küvette dürfen keine Tropfen hängen. Anweisungen des Assistenten beachten. Aufgabe: Die gemessene Extinktion der Lösungen 1–4 wird über µmol Phosphor aufgetragen. Die Konzentration der Probe wird mit Hilfe der Kalibrierkurve ermittelt. Entsorgung: Wässrige Schwermetallabfälle. Ende Versuch 3.9.2 Kapitel 4 Organische Chemie 4.1 Themen des 3. Kolloquiums: Es folgt eine Zusammenstellung der wichtigsten Themen in Stichworten. Es wird empfohlen, die Stichworte in den einschlägigen Lehrbüchern nachzuschlagen. Organische Chemie Funktionelle Gruppen, Namensgebung organischer Verbindungen (Nomenklatur), räumliche Struktur, Stereoisomerie in organischen Molekülen (Chiralität, Konfiguration, Konformation, Konstitution, R/S-Nomenklatur, E/ZIsomerie). Reaktionsmechanismen Elektrophile, nucleophile Substitutionsreaktionen (SN1 - und SN2 -Reaktionen, Übergangszustände, Inversion bzw. Walden Umkehr, Carbeniumionen), Eliminierung (isolierte und konjugierte Doppelbindungen), Additionsreaktionen (π-Komplex, Bromoniumion usw.) Aromaten Definition von Aromaten, konjugierte Doppelbindungen, mesomere Grenzformeln (Resonanzformen), Hückel Regel usw. Alkohole primäre, sekundäre, tertiäre Alkohole usw. 93 94 KAPITEL 4. ORGANISCHE CHEMIE Amine primäre, sekundäre, tertiäre Amine, usw. Carbonylverbindungen Ketone und Aldehyde: Carbonylgruppe, Polarität der Carbonylgruppe, Carbeniumion, Carbanion, Aldolkondensation, usw. Carboxylgruppe Carbonsäuren und Carbonsäurederivate (Anhydrid, Säurechlorid, Säureamid), Acidität und Mesomerie der Carboxylgruppe, Mechanismus der Esterbildung mit Carbonsäuren und Säurechloriden, Verseifung, Fette und Fettsäuren, Decarboxylierung, Keto-Enol-Tautomerie (Definition Tautomerie, Mesomerie, Enolform) usw. Redoxreaktionen Definition Oxidation/Reduktion bei organischen Molekülen, Hydrition, ( Oxi” dationsreihe“: Alkohol → Aldehyd/Keton → Carbonsäure), Dehydrierung und Hydrierung usw. Acidität Mesomeriestabilisierung, induktiver Effekt, Struktureinflüsse auf die C-H Acidität, Alkoholat, Thiolat usw. 4.2. EINFÜHRUNG 4.2 95 Einführung Stichworte bitte in Lehrbüchern nachschlagen: Atomorbitale, Hybridisierung, Einfachbindung, Nomenklatur, Isomere, Funktionelle Gruppen, Cyclohexanring (axial, equatorial), Konstitution, Doppelbindungen, konjugierte Doppelbindungen, Dreifachbindung, Stereochemie, optische eigenschaften von chiralen Molekülen, CIP-Regeln, R/S-Nomenklatur, D/L-Nomenklatur, Enantiomere, Diastereomere, Newman-Projektion, Fischer-Projektion, Heteroatome. Die organische Chemie ist die Chemie des Kohlenstoffs in seinen zahlreichen Verbindungen. Kohlenstoff nimmt eine Sonderstellung unter den Elementen ein. Kein anderes Element kann so viele verschiedene Verbindungen mit sich selbst eingehen. Entsprechend seiner Stellung im Periodensystem (Ordnungszahl 6, Elektronenkonfiguration: [He] s2 p2 ) besitzt er eine nichtabgeschlossene Elektronenkonfiguration mit vier Aussen- oder Valenzelektronen.Diese Valenzelektronen befinden sich in s- und p-Orbitalen. Durch Hybridisierung ( Vermischung“) dieser s- und p-Orbitale zu den tetraedrischen ” sp3 -, trigonal-planaren sp2 - und linearen sp-Hybridorbitalen resultieren für die Chemie des Kohlenstoffs typische Eigenschaften: • Kohlenstoffatome gehen miteinander, mit Wasserstoff und mit anderen Atomen ( Heteroatome“ wie N, O, P, S) stabile kovalente Einfach” und/oder Mehrfachbindungen ein. • Kohlenstoffatome können sich unter Bildung von Ketten und Ringen (auch mit Heteroatomen) aneinanderreihen. Kein anderes Element ist dazu so befähigt wie Kohlenstoff. • Für einen bestimmten Satz von Atomen (z. B. C3 H8 O) bestehen mehrere Anordnungsmöglichkeiten, die verschiedenen Molekülen mit unterschiedlichen physikalischen und chemischen Eigenschaften entsprechen (Isomere). 96 KAPITEL 4. ORGANISCHE CHEMIE Kohlenstoffgerüste und funktionelle Gruppen Organische Moleküle lassen sich unter zwei Aspekten betrachten: 1.) Das Kohlenstoffgerüst (kurze oder lange Kette, Ringe, usw.) bestimmt maßgeblich die Größe und räumliche Struktur. 2.) Die an das Kohlenstoffskelett gebundenen funktionellen Gruppen sind überwiegend für die Reaktivität und chemischen Eigenschaften verantwortlich. Funktionelle Gruppen sind unterscheidbare Molekülteile mit Heteroatomen oder Mehrfachbindungen zwischen Kohlenstoffatomen, deren chemische Natur weitgehend unabhängig vom Gesamtmolekül eine bestimmte Reaktionsweise vorgibt. Allerdings beeinflussen sich Kohlenstoffgerüst und Stellung einer funktionellen Gruppe im Molekül gegenseitig. Wenn ein Molekül mehrere verschiedene funktionelle Gruppen trägt, hat es auch mehrere, chemisch unterschiedliche Reaktionsmöglichkeiten, deren Eintreten und relative Bedeutung von den jeweiligen Reakionsbedingungen und -partnern abhängen. Der unpolare Charakter der Alkylreste R (Kohlenwasserstoffketten oder Ringe) spiegelt sich im Verhalten der Kohlenwasserstoffe wider. Dagegen bedingt die unterschiedliche Elektronegativität des CAtoms und der mit ihm verknüpften Heteroatome einer funktionellen Gruppe mehr oder weniger polare Eigenschaften eines Moleküls. Sowohl die Kohlenstoffgerüste als auch die funktionellen Gruppen dienen zur Klassifizierung und Benennung (Nomenklatur) organischer Verbindungen. Grundlage für die Benennung organischer Moleküle ist das NomenklaturRegelwerk der IUPAC (International Union of Pure and Applied Chemistry). Für altbekannte und gängige Substanzen werden aber daneben weiterhin Trivialnamen verwendet. Informieren Sie sich in den Lehrbüchern über die Benennung organischer Moleküle und Molekülteile. 4.2. EINFÜHRUNG 97 Eine Auswahl an funktionellen Gruppen: R NH2 Amin R NO2 Nitroverbindung R CN Cyanoverbindung O R S O H O Sulfonsäure Halogenverbindung Alkohol R X X = F, Cl, Br, I R O H Thiol R S H Ether R O R O R H Aldehyd O Keton R R O R O H Carbonsäure Carbonsäurederivate: O O Carbonsäureanhydrid R R O O R Cl Carbonsäurehalogenid O R Carbonsäureamid R=Alkylrest NH2 Wasserstoffbrückenbindung Die Hydroxyl-Gruppe -OH nimmt wegen ihrer Fähigkeit zur Ausbildung starker Wasserstoffbrücken eine Sonderstellung ein. Aufgrund der besonders 98 KAPITEL 4. ORGANISCHE CHEMIE starken Polarisierung der OH-Bindung fungiert sie sowohl als H-Brücken” Donator“ (starke positive Teilladung δ+ am H-Atom) als auch als H-Brücken” Acceptor“ (starke negative Teilladung δ− am O-Atom). In vereinfachter Form wird die Ausbildung von H-Brücken wie folgt dargestellt: O H O H O H R R R Auf starke H-Brücken ist auch der hohe Siedepunkt des Wassers zurückzuführen. Eine eminent wichtige Rolle spielt auch die Ausbildung von HBrücken mit NH− und OH-Gruppierungen als Donator“ und N- oder O” Atomen als Acceptor“ in den Desoxyribonucleinsäuren und den Ribonucle” insäuren so wie zwischen -NH und Carbonyl-O-Atomen in den Proteinen. CH-Gruppierungen können dagegen nur in Ausnahmefällen als vergleichsweise sehr schwache H-Brücken-Donatoren wirken. Hydrophobe und hydrophile Molekülteile In Analogie zu den unpolaren Kohlenwasserstoffen, speziell den Alkanen, bezeichnet man auch unpolare Alkylreste als hydrophob oder lipohil, während polare Gruppen—insbesondere solche, die starke H-Brücken bilden können— als hydrophil bezeichnet werden. So lässt sich das unterschiedliche Verhalten der einzelnen Verbindungen beim Versuch des Lösens in Wasser bzw. Cyclohexan aufgrund der entweder hydrophilen oder hydrophoben Eigenschaften der Moleküle erklären. Im Ethanolmolekül überwiegt z. B. der hydrophile Charakter der OH-Gruppe. Deshalb ist der Lösungsvorgang in Wasser stark exergonisch, und Wasser und Ethanol bilden in jedem Verhältnis homogene Gemische. Dagegen dominiert im Pentanolmolekül mit seinem größeren Alkylrest der hydrophobe Charakter, und dadurch wird seine Löslichkeit in Wasser stark herabgesetzt, so dass bei den hier benutzten Mengenverhältnissen zwei Phasen auftreten. Isomerie Isomere Die Summenformel einer Verbindung zeigt nur die Anzahl der vorhandenen Atome an. Die Konstitution oder Konstitutionsformel beschreibt zusätzlich die Art und Reihenfolge, in der die Atome miteinander verknüpft sind. Es ist manchmal möglich, Atome in mehr als einer Konstitution zusammenzufügen. Man nennt Verbindungen, die zwar dieselbe Summenformel, jedoch 4.2. EINFÜHRUNG 99 verschiedene Konstitutionen haben, Konstitutionsisomere. Die Bezeichnung Isomer leitet sich vom griechischen isos meros, d. i. gleiche Teile, ab. Ein Beispiel für Konstitutionsisomere sind die verschiedenen Isomere des Pentans. Stereoisomere besitzen dieselbe Konstitution, jedoch verschiedenen räumlichen Bau. Die Konfiguration, bei gegebener Konstitution, beschreibt die räumliche Anordnung der Atome um ein starres (Doppelbindung oder Ringsysteme, cis-trans-Isomerie) oder chirales Strukturelement. Stereoisomere werden eingeteilt in Enantiomere und Diastereomere und, unabhängig davon, in Konformations- und Konfigurations-Isomere. Enantiomere verhalten sich wie Bild und Spiegelbild, sie sind chiral, d. h. von ihrem Spiegelbild verschieden. Typische chirale Objekte sind Hände, Füße, Handschuhe, Schuhe, Schrauben, Wendeltreppen etc. und natürlich auch die DNA-Doppelhelix. Einen chiralen Gegenstand erkennt man immer an der Inkongruenz (Deckungsungleichheit) mit seinem Spiegelbild. Das Spiegelbild der linken Hand ist eine rechte Hand. Man kann diese beiden Hände jedoch auf keinerlei Weise zu einer räumlichen Deckung bringen. Deswegen spricht man oft auch von der Händigkeit, wenn man die Chiralität meint. Ein Molekül, in dem ein C-Atom von vier verschiedenen Substituenten umgeben ist, ist chiral. Diese C-Atome nennt man auch Chiralitätszentren oder asymmetrisch substituierte C-Atome. Achirale Objekte oder Moleküle besitzen keine Händigkeit. Sie sind deckungsgleich mit ihrem Spiegelbild. COOH COOH H3C * H H C2H5 * CH C2H5 3 S R Spiegel Enantiomerenpaare haben unter achiralen Bedingungen (z. B. achirales Lösungsmittel/Reaktionspartner) die gleichen physikalischen Eigenschaften, 100 KAPITEL 4. ORGANISCHE CHEMIE unter chiralen Bedingungen verschiedene, weil dabei Diastereomere entstehen. Enantiomere können unter achiralen Bedingungen physikalisch oder chemisch nicht getrennt werden. Diastereomere verhalten sich nicht wie Bild und Spiegelbild. Hat eine Verbindung mehrere Chiralitätszentren, so können sich Enantiomeren- und Diastereomerenpaare bilden. Ein Enantiomer dieser Verbindung erhält man durch Umkehr der absoluten Konfiguration aller Chiralitätszentren. Kehrt man nicht alle, sondern nur einige um, so erhält man Diastereomere. Diastereomere haben unter allen Bedingungen verschiedene physikalische Eigenschaften. Sie können physikalisch oder chemisch voneinander getrennt werden. Konfigurationsisomere, genauso wie Konstitutions-Isomere, können nur durch die Aufhebung und Neuknüpfung von kovalenten Bindungen ineinander umgewandelt werden. H O H OH O H HO H H Achiraler Aldehyd H O * OH HO * H OH H O H H H OH H OH H OH H OH H O H D-Glycerinaldehyd H O OH H H D-Glycerinaldehyd L-Glycerinaldehyd Enantiomere O H D-Glycerinaldehyd H H H O H H OH HO H OH H OH H H H H OH OH H D-Erythrose D-Threose Diastereomere R- und S-Nomenklatur nach Cahn-Ingold-Prelog-Regeln (CIP) Die absolute Konfiguration eines Enantiomeren wird durch die Bezeichnung R bzw. S angegeben. Zur Bestimmung der absoluten Konfiguration eines asymmetrischen Atoms werden seine Substituenten entsprechend den CIPRegeln nach ihren Prioritäten geordnet. Das Molekül wird so gedreht, dass der Substituent niedrigster Priorität nach hinten zeigt. Wenn dann die übrigen Substituenten in aufsteigende Priorität im Uhrzeigersinn zueinander stehen, liegt ein R-konfiguriertes Atom vor. Im entgegengesetzten Fall handelt es sich um ein S-konfiguriertes Atom. CIP-Regeln: Die Regeln werden in ihrer Reihenfolge nacheinander angewandt, bis eine 4.2. EINFÜHRUNG 101 Unterscheidung der Substituenten möglich ist. 1.) Ordnungszahl: Ein Substituent, der mit einem Atom höherer Ordnungszahl an das asymmetrische Atom gebunden ist als ein anderer, hat höhere Priorität. 2.) Atommasse: Ein Substituent, der mit einem Atom höherer Atommasse an das asymmetrische Atom gebunden ist als ein anderer, hat höhere Priorität. 3.) Substitutionsgrad: Erlauben die ersten beiden Regeln keine Differenzierung durch die direkt an das asymmetrische Atom gebundenen Atome, so werden die jeweils nächsten Substituentenatome herangezogen, bis eine Differenzierung möglich wird. 4.) Mehrfachbindungen: Doppel- und Dreifachbindungen werden in der Weise behandelt, dass man von einer Verdoppelung bzw. Verdreifachung der Atome der Mehrfachbindung ausgeht. HS HO R H CH3 Cl R CH3 R S Br S Cl S D R H COOH O OH OH R OH E/Z- und cis-, trans-Isomere Während Einfach-Bindungen acyclischer Moleküle frei drehbar sind, ist eine Rotation in cyclischen Molekülen und um Mehrfachbindungen eingeschränkt. Daher muss bei substituierten Alkenen und cyclischen Verbindungen die relative Konfiguration berücksichtigt werden, durch die die Lage der Substituenten relativ zueinander angegeben wird. Die relative Konfiguration wird in eindeutigen Fällen durch die Bezeichnungen cis und trans beschrieben. 102 KAPITEL 4. ORGANISCHE CHEMIE cis-2-Buten trans-2-Buten trans-1,3-Dimethylcyclopentan cis-1,3-Dimethylcyclopentan Bei höhersubstituierten Alkenen sind die Bezeichnungen cis und trans nicht mehr eindeutig. Man benutzt dann die Bezeichnungen E (entgegen) und Z (zusammen). Dazu wird an jedem Ende der Doppelbindung der Substituent der höchsten Priorität bestimmt. Liegen beide Substituenten höchster Priorität auf der gleichen Seite der Doppelbindung, wird die relative Konfiguration als Z bezeichnet, liegen sie auf entgegengesetzten Seiten der Doppelbindung, ist die relative Konfiguration E. Die Prioritäten der Substituenten werden nach den Cahn-Ingold-Prelog-Regeln (CIP) bestimmt, die auch zur Bestimmung der absoluten Konfiguration benutzt werden und sich primär an den Ordnungszahlen, am Atomgewicht und am Substitutionsgrad der Substituenten orientieren. H Cl H3C Br F H5C2 E Li CH3 H3CS CH3 NH2 OMe Z E D H Cl2B CH3 F SO3H H3C C6H5 E Z Konformations-Isomere lassen sich durch Rotation um Einfachbindungen ineinander umwandeln (isomerisieren). Konformationen sind alle räumlichen Anordnungen eines Moleküls, die durch Rotationen um Einfachbindungen entstehen. Dreht man z. B. die Methylgruppen im Ethanmolekül um die C–CEinfachbindung, so erhält man unendlich viele Konformationen dieses Moleküls. 4.2. EINFÜHRUNG 103 Bei Cyclohexan-Ringen treten durch Drehung der C–C-Einfachbindungen auch Konformationsisomere auf. Die Substituenten lassen sich hierbei in axiale (ax) und equatoriale (eq) Substituenten einteilen. Durch Drehung der C–CEinfachbindungen lassen sie sich ineinander überführen. ax eq eq ax X X Y Y X Y X Y Y X Y Y X X Y Y X X X X Y Y Y X Informieren Sie sich in Lehrbüchern über: Fischer-Projektion, NewmanProjektion und D-, L-Nomenklatur. Optische Aktivität Ein Enantiomer ist in der Lage, die Schwingungsebene von linear polarisiertem Licht zu drehen; die Substanz ist optisch aktiv. Der Drehwinkel kann mit Hilfe eines Polarimeters gemessen werden. Der Betrag der Drehung wird durch die äusseren Faktoren Wellenlänge des verwendeten Lichts, Schichtdicke und Konzentration der Probe, Temperatur und Lösungsmittel beeinflusst. Bei vorgegebener Temperatur und Wellenlänge (zumeist die Natrium D-Linie) kann aus der gemessenen Drehung, der Konzentration und der Schichtdicke die spezifische Drehung als charakteristische Eigenschaft einer Verbindung ermittelt werden. Spiegelbildisomere drehen die Schwingungsebene jeweils um den gleichen Betrag, jedoch in entgegengesetzte Richtungen. Wird die Ebene im Uhrzeigersinn gedreht, ist die Substanz rechtsdrehend“. ” Das linksdrehende“ Enantiomer dreht die Ebene um den gleichen Betrag ” nach links. Die Drehrichtung wird mit einem +“ für rechtsdrehend und ei” nem −“ für linksdrehend vor dem Substanznamen angegeben. Die Konfigu” ration eines Enantiomers lässt keine Aussage darüber zu, in welche Richtung polarisiertes Licht gedreht wird, d. h. man kann der absoluten Konfigurationsangabe R oder S keine Information bezüglich der Drehrichtung entnehmen. Liegt ein 1:1-Gemisch vom Enantionerenpaar vor (Racemat), ist die Probe optisch inaktiv, da sich die Drehrichtungen gegenseitig aufheben. 104 KAPITEL 4. ORGANISCHE CHEMIE Aromaten Wenn mehrere C=C-Doppelbindungen in einer Kette mit Einfachbindungen alternieren ( konjugierte Doppelbindungen“), so ergibt sich ein gegen” über isolierten Doppelbindungen verändertes π-Elektronensystem. Die πElektronenwolken erstrecken sich teilweise auch über die Einfachbindungen und führen zu einem merklichen (aber nicht völligen!) Bindungsausgleich. Liegt aber eine bestimmte Zahl solcher konjugierter Doppelbindungen in einem ebenen Ring, so erreicht das π-Elektronensystem eine völlig gleichmäßige Elektronendichte (Delokalisierung) und gleichmäßige Bindungslängen aller Bindungen. Der klassische aromatische Kohlenwasserstoff Benzol mit 6 π-Elektronen ist daher nicht als Cyclohexatrien mit 3 Einfach- und 3 Doppelbindungen aufzufassen, sondern besitzt vollständig delokalisierte Bindungen: Aromatische Verbindungen sind formal durch Mesomerie energetisch stabilisiert. Die Strichformeln geben nur fiktive Grenzstrukturen wieder, der wahre Zustand liegt in der Mitte. Er wird durch einen Kreis im Sechsring angedeutet. Die Strichformeln werden dennoch oftmals aus zeichnerischen und didaktischen Gründen verwendet. Beachte: Der Mesomeriepfeil ←→, der zwischen zwei fiktiven Gebilden steht, hat nichts mit einem Gleichgewichtspfeil * ) zu tun! Aromatischer Charakter und Mesomerie sind nicht auf Benzol beschränkt. Nach einer Regel von Hückel gilt er für alle cyclischen, planaren Systeme mit n = 0, 1, 2, . . . π-Elektronen. Auch freie Elektronenpaare von Heteroatomen können Teil des π-Systems sein, wie z. B. in Pyrol: _ N H Neben aromatischen Kohlenwasserstoffen sind substituierte Aromaten wie Phenol oder Anilin wichtige und häufige Verbindungen. In ihnen verhalten 4.2. EINFÜHRUNG 105 sich die funktionellen Gruppen anders als in aliphatischen Verbindungen, weil die freien Elektronenpaare von O- oder N-Atomen in Wechselwirkung mit dem aromatische System treten. Dies kann durch zusätzliche Grenzstrukturen beschrieben werden: O OH - O O - -H+ +H+ - Phenol, ein aromatischer Alkohol, hat andere Eigenschaften und Reaktivität als ein aliphatischer Alkohol, Anilin, ein aromatisches Amin andere Eigenschaften als ein aliphatisches Amin (siehe Azofarbstoffe). Aber auch die Reaktionsweise des aromatischen Ringes wird von Substituenten durch Mesomerie- und induktive Effekte wesentlich mitbestimmt. Informieren Sie sich in Lehrbüchern über Erstsubstituenteneinflüsse und dirigierende Wirkungen dieser Erstsubstituenten auf Angriffe von Zweitsubstituenten. 4.2.1 Versuch: Betrachtung an Molekülmodellen Boxweise Anhand von Molekülmodellen kann man die Konstitutionen und Konformationen von Molekülen nachahmen. Anhand der Modelle lassen sich die stereochemischen Problemstellungen dreidimensional darstellen. Geräte: MINIT Molekülbaukasten. Durchführung: Da dem Praktikum nur ein Molekülbaukasten zur Verfügung steht, wird in den ersten zwei Wochen des Organikteils immer jeweils eine Gruppe (Box) von Praktikanten für diesen Versuch eingeteilt. Diese Gruppe löst die Aufgaben und stellt die Molekülmodelle dem Assistenten vor. Der Versuch dauert ca. 15–30 min. Aufgaben: Konstitutions-Isomere: Bauen Sie mit Hilfe des Molekülbaukastens die möglichen Konstitutionsisomere von C5 H12 . Protokollieren Sie die Konstitutionsformeln und benennen sie die Konstitutionsisomere nach IUPAC. Wieviele Konstitutionsisomere sind möglich? 106 KAPITEL 4. ORGANISCHE CHEMIE Chiralität, Enantiomere: Das Modell eines sp3 -hybridisierten Kohlenstoffatoms (C-Tetraeder) wird mit vier verschiedenen Substituenten (vier verschiedenfarbigen Kugeln) versehen. An einem zweiten C-Tetraeder werden die gleichen vier Kugeln so angebracht, dass sich die Modelle wie Bild und Spiegelbild verhalten. Versuchen Sie, durch Drehen die beiden Modelle zur Deckung zu bringen, also herauszufinden, ob sie identisch sind. Was muss geschehen damit sich die beiden Moleküle wieder zur Deckung bringen lassen? Bauen Sie Modelle für zwei enantiomere Aminosäuren, z. B. Alanin, auf und überzeugen Sie sich davon, dass sie sich wie Bild und Spiegelbild verhalten aber nicht deckungsgleich sind. O H2N OH OH O H H NH2 CH3 CH3 L-Alanin HO H3C D-Alanin O O OH H NH2 H H2N CH3 S-Alanin R-Alanin Konformation: Bauen Sie Sechsringe aus: • sp3 -hybridisierten Kohlenstoffatomen (C-Tetraeder) • sp2 -hybridisierten Kohlenstoffatomen (trigonal-planar) Kennzeichnen Sie die Substituenten des Sechsringes aus sp3 -hybridisierten Kohlenstoffatomen in der Sesselform als axial und äquatorial, indem Sie für jede Sorte eine andere Farbe benutzen. Wie lassen sich diese ineinander überführen? Warum ist die Wannenform ungünstiger als die Sesselform? Bauen Sie ein Molekül-Modell von n-Butan und drehen Sie dieses an der zentralen Bindung so, dass gestaffelte Konformationen entstehen, welche Energieminima darstellen. Zeichnen Sie diese Zustände in der sog. Newman-Projektion. 4.2. EINFÜHRUNG 107 Cis- und trans-Isomerie: 1.) Bauen Sie Modelle für Malein- und Fumarsäure. Welche hat cis-, und welche trans-Konfiguration (vgl. Citronensäure-Cyclus des Stoffwechsels)? Veranschaulichen Sie sich an den Modellen, welche der beiden Säuren unter Wasseraustritt ein inneres Anhydrid (Fünfring) bilden kann. Wie kann eine Umwandlung des einen Isomeren in das andere erreicht werden? 2.) Cis- und trans-disubstituierte Cycloalkane: Bauen Sie ein monosubstituiertes Cyclopentan oder Cyclohexan auf und daraus diastereomere, disubstituierte Cycloalkane. E/Z-Isomerie: Der Sehvorgang beginnt mit einer cis-trans-Isomerisierung. Wie sind E- u. Z-Konfiguration definiert? Bauen sie das Molekül 2-Buten einmal als E-Isomer und einmal als Z-Isomer. Komplexe Moleküle (Steroide): Zur Stoffgruppe der Steroide gehören zahlreiche biologisch wichtige Verbindungen, die in der Natur weit verbreiteten Verbindungen wegen ihrer Wirkungsvielfalt für die Biologie von hohem Interesse. Bauen sie mit Hilfe des Molekülbaukastens die hier gezeigten Moleküle dreidimensional auf. Achten Sie dabei auf die Stereozentren. Wieviele Chiralitätszentren besitzen die Moleküle? Obwohl Estradiol und Cortison das gleiche Grundgerüst besitzen, erfüllen sie im Körper ganz unterschiedliche Aufgaben. In der zweidimensionalen Darstellung sehen sich beide Moleküle sehr ähnlich. Die dreidimensionale Darstellung unterstreicht die Unterschiede besser. Estradiol: Ein weibliches Sexualhormon. CH3 OH H H H H HO Estradiol / Östradiol 108 KAPITEL 4. ORGANISCHE CHEMIE Cortison: Ein Stereoidhormon der Nebennierenrinde. Es wird vielseitig therapeutisch eingesetzt. HO CH3 O H H3C H O Cortison Ende Versuch 4.2.1 H O OH 4.3. SUBSTITUTION, ELIMINIERUNG, ADDITION (6.–7. WOCHE) 109 4.3 Substitution, Eliminierung, Addition (6.– 7. Woche) Stichworte bitte nachschlagen: Nucleophil, Elektrophil, Substitution, SN1 und SN2 , Walden-Umkehr, Addition, Bromoniumion, cis-/trans-Isomerie, Eliminierung, Regel nach Markownikow In diesem Kapitel geht es um die drei wichtigsten Reaktionstypen in der organischen Chemie: Substitution, Eliminierung und Addition. Fast jede Reaktion in der organischen Chemie lässt sich, mehr oder weniger, einer der drei Reaktionsarten zuordnen. Um die Mechanismen dieser Reaktionstypen verstehen zu können, müssen zuerst ein paar Grundbegriffe erklärt werden. Nucleophile (nucleophil = kern“liebend) ” Wegen der unterschiedlichen Elektronegativität von C und Br ist die C-BrBindung polarisiert. Man deutet dies dadurch an, dass auf dem entsprechenden Kohlenstoffatom eine positive Partialladung eingezeichnet wird. Man kann erwarten, dass ein derartiges elektronenarmes Kohlenstoffatom bevorzugt von Reagentien angegriffen wird, die über ein nicht bindendes Elektronenpaar verfügen. Dieses kann genutzt werden, um eine Bindung mit dem Kohlenstoffatom auszubilden, wodurch dessen Elektronenmangel ausgeglichen wird. Diese Reagentien mit einem nichtbindenden Elektronenpaar heißen Nucleophile: - OH OEt H2O NH3 Elektrophile (elektrophil = elektronenliebend) Elektronenarme Reagentien wie die hier gezeigten nennt man Elektrophile. + H + NO2 δ+ " Br δ− Br " Alkohole und Amine Kohlenstoffatome werden nach der Anzahl der direkt gebundenen C-Atome unterschieden: An primären Kohlenstoffatomen ist ein weiteres Kohlenstof- 110 KAPITEL 4. ORGANISCHE CHEMIE fatom gebunden, an sekundäre sind zwei, an tertiäre drei und an quartäre vier weitere Kohlenstoffatome gebunden. Entsprechend lassen sich drei Arten von Alkoholen unterscheiden. Je nach dem, ob die Alkoholgruppe (-OH) an einem primären (endständigen), einem sekundären oder einem tertiären C-Atom gebunden ist spricht man von primären, sekundären oder tertiären Alkoholen. Auch bei Aminen ist der Substitutionsgrad (Anzahl der direkt an das N-Atom gebundenen C-Atome) ein wichtiges Unterscheidungsmerkmal. Hier bestimmt allerdings die Anzahl der am Stickstoff-Atom gebundenen C-Atome die Bezeichnung. R + R N R R R N| R _ R NH2 R NH _ primär sekundär R R tertiär quartiär Carbeniumionen Im Verlauf vieler organischer Reaktionen bilden sich intermediär Carbeniumionen. Das sind Moleküle mit positiv geladenen C-Atomen, die im Laufe der Reaktion ein Elektronenpaar an ein anderes Teilchen abgegeben haben. Die Stabilität eines Carbeniumions hängt von dem Substitutionsgrad des jeweiligen C-Atoms ab. Je mehr Alkylgruppen an dem positiv geladenen CAtom sitzen, um so stabiler ist das Carbeniumion. Die Alkylgruppen haben einen leichten elektronenliefernden (donierenden) Effekt (Hyperkonjugation und induktiver Effekt: siehe Lehrbücher). Damit wird die positive Ladung zum Teil auf Nachbaratome verlagert. H R C H Primär R R C H sekundär R R C R tertiär Auch die Art des Lösungsmittels hat einen großen Einfluss auf die Stabilität eines Carbeniumions. Da ein Carbeniumion ein geladenes Teilchen ist, wird es durch ein polares Lösungsmittel besser solvatisiert. 4.3. SUBSTITUTION, ELIMINIERUNG, ADDITION (6.–7. WOCHE) 111 Zusammengefasst: Carbeniumionen sind um so stabiler, je höher ihr Substitutionsgrad und je polarer das Lösungsmittel ist. 4.3.1 Versuch: SN1 - und SN2 -Substitution: t-Butylchlorid Stichworte: monomolekular, bimolekular, Racemat, Inversion Allgemeines: Eine Kohlenstoff-Halogenverbindung ist polarisiert. Die positive Partialladung am C-Atom ermöglicht einen Angriff durch Nucleophile. Eine typische Reaktion der Halogenalkane RX ist deshalb die nucleophile Substitution. RX + Y− → RY + X− Die austretende Gruppe X, häufig auch als Abgangsgruppe bezeichnet, wird durch ein Nucleophil Y− mit freiem Elektronenpaar (eine Lewis-Base) ersetzt (substituiert). Dabei nimmt die austretende Gruppe das bindende Elektronenpaar mit und die neu eintretende Gruppe stellt beide Elektronen der neuen Bindung. Mechanismus: Nucleophilen Substitutionen (SN ) können nach zwei unterschieldichen Reaktionsmechanismen ablaufen: die monomolekulare Substitution (SN1 ) und die bimolekulare Substitution (SN2 ). SN1 : Die Reaktionsgeschwindigkeit der idealen SN1 -Reaktion ist nur von der Konzentration des Eduktes abhängig. Sie verläuft über ein CarbeniumIon und wird gefördert a.) durch polare Lösungsmittel mit guten Solvatationseigenschaften und b.) durch alle Strukturparameter, die ein Carbenium-Ion stabilisieren können, also z. B. hohe Alkylsubstitution des Reaktionszentrums. Tertiäre Kohlenstoffatome sind typische Zentren, an denen eine Substitution nach SN1 -Mechanismus stattfindet. Die SN1 -Reaktion an einem chiralen Zentrum bewirkt eine, zumindest teilweise, Racemisierung (Bildung eines racemischen Gemisches beider 112 KAPITEL 4. ORGANISCHE CHEMIE Enantiomere), weil sich das Nucleophil von zwei Seiten an die Carbeniumzwischenstufe annähern kann. Im Idealfall ist keine der beiden Seiten bevorzugt und man erhält ein Racemat (Gemisch beider Enantiomere, so dass sich die optischen Eigenschaften gegenseitig aufheben). R' A Y R' R R'' -x- Y - X R' A B + C R'' R Y R'' R - R' B R R'' Y SN2 : Die Reaktionsgeschwindigkeit der SN2 -Reaktion ist sowohl von der Konzentration des Eduktes als auch von der des Nukleophils abhängig. Der Eintritt des Nucleophils und die Abspaltung der Abgangsgruppe erfolgen im Idealfall gleichzeitig. Die SN2 -Reaktion an einem asymmetrischen Kohlenstoffatom führt zu einer Konfigurationsinversion, die als Walden-Umkehr bekannt ist. R H R' +Y X R Y R' -X H X - R Y R' H Übergangszustand Die SN2 -Reaktion ist damit im Idealfall ein Beispiel für eine stereospezifische Reaktion (siehe Lehrbücher). Sie wird durch niedersubstituierte Reaktionszentren (sek. C-Atome) und unpolare Lösungsmittel gefördert. Beispiel: Substituiert man jeweils an einem prim., sek., und tert. Alkohol die OH-Gruppe durch ein Chlorid (Cl− ), so lässt sich im Hinblick auf den Reaktionsmechanismus folgendes Schema formulieren. 4.3. SUBSTITUTION, ELIMINIERUNG, ADDITION (6.–7. WOCHE) 113 primärer Alkohol: H R OH H SN2 1. + HCl 2. - H2O H Cl R H sekundärer Alkohol: R SN1 1. + HCl 2. - H2O R R R polares LM Cl OH H R SN2 R unpolares LM Cl tertiärer Alkohol: R R OH R R SN1 R 1. + HCl 2. - H2O Cl R Chemikalien: tert. Butanol (falls in festem Zustand, durch Erwärmen des Gefäßes im Wasserbad (30-40˚C) in flüssigen Zustand bringen), konz. HCl, ges. NaCl-Lösung, ges. NaHCO3 -Lsg., CaCl2 . Durchführung: 0.1 mol tert. Butanol werden mit 0.5 mol konzentrierter Salzsäure (HCl) (achten Sie auf die Molarität der ausstehenden konzentrierten Salzsäure) in einem 100 ml Scheidetrichter vereinigt und durch sanftes Schwenken (ohne Aufsetzen des Stopfens) gründlich gemischt. Nach einer Minute wird der Scheidetrichter mit einem PVC-Stopfen verschlossen, und einige Minuten unter häufigem Belüften durch den Hahn geschüttelt. Vorsicht HCl-Gas entweicht!! (Abzug!!). Nach Beendigung der Reaktion (10 min.) lässt man die Phasen sich entmischen und trennt die organische Phase (oben) von der wässrigen. Man wäscht die organische Phase zunächst mit 25 ml gesättigter Natriumchlorid-(NaCl)-Lösung ( Aussalzen“), dann mit 25 ml gesättig” ter Natriumhydrogencarbonat-(NaHCO3 )-Lösung. Dazu wird die organische 114 KAPITEL 4. ORGANISCHE CHEMIE Phase in dem Scheidetrichter mit der Waschlösung durchmischt und wieder getrennt. Das Waschen mit der Natriumhydrogencarbonat-(NaHCO3 )Lösung verursacht heftige Kohlendioxid-(CO2 )-Entwicklung und Schaumbildung, darum wird auch hier zunächst der Scheidetrichter offen geschwenkt und dann unter häufigem Belüften geschüttelt. Zum Schluss wird die organische Phase mit 20 ml Wasser gewaschen. Die etwas trübe Flüssigkeit wird in einem Rundkolben über wasserfreiem Kalziumchlorid (CaCl2 ) getrocknet, indem man eine Spatelspitze des Salzes in den Kolben gibt und nach fünfminütigem Schwenken die organische Phase wieder abdekandiert oder filtriert. Beilsteinprobe: Der Assistent führt mit der organischen Phase die sog. Beilsteinprobe durch. Dazu entfacht er in einem Abzug (frei von brennbaren Chemikalien!) einen Brenner und erhitzt einen Kupferdraht. Diesen Kupferdraht taucht er in die Lösung und hält danach den mit der organischen Phase benetzten Draht in die Brennerflamme. Ist eine organische Halogenverbindung anwesend, so leuchtet die Flamme grün auf, weil aus dem Kupferdraht und dem aus der organischen Halogenverbindung freigesetzten Halogenwasserstoff flüchtige Kupferhalogenide entstehen, die sich wiederum in der heißen Flamme teilweise in ihre Atome zersetzen und somit das in der Hitze angeregte Kupferatom seine grüne Spektralfarbe aussendet. Ein Gegentest mit dem Edukt tert. Butanol sollte keine grüne Flammenfärbung zeigen. Aufgaben: Protokollieren Sie in kurzen Worten ihre Vorgehensweise und formulieren Sie die Reaktionsgleichung mit Übergangsprodukt. Nach welchem Mechanismus verläuft die Reaktion und warum? Entsorgung: Organische Lösungsmittelabfälle Ende Versuch 4.3.1 4.3. SUBSTITUTION, ELIMINIERUNG, ADDITION (6.–7. WOCHE) 115 4.3.2 Versuch: Eliminierung Stichworte: Eliminierung, Doppelbindung, Aromaten Allgemeines: X H R Y R H H - -x - R H H R Eliminierungsreaktionen treten—oft in Konkurrenz zur Substitution— ein, wenn eine Abgangsgruppe X ein Molekül verlässt, wodurch ein Carbeniumion entsteht, und anschließend ein Nucleophil als Protonenakzeptor (Brönsted-Base) wirkt und vom β-C-Atom (das dem betrachteten positiv geladenen C-Atom benachbarte C-Atom) ein Proton übernimmt. Da sich Nucleophilie und Basizität verschiedener Teilchen Y im allgemeinen nicht gleichsinnig ändern und von den Reaktionsbedingungen (Lösungsmittel, pHWert) abhängen, ist man in der Lage, SN - oder Eliminierungsreaktionen selektiv zu begünstigen. Wichtige Methoden zur Einführung von Doppelbindungen in Moleküle durch Eliminierung sind z. B. die Dehydratisierung von Alkoholen durch Säure oder die Halogenwasserstoffabspaltung aus Alkylhalogeniden durch Basen, die geringe Nucleophilie zeigen. Die Einführung von Doppelbindungen in organische Moleküle durch Abspaltung von Wasser (Dehydratisierung) wird durch Säuren erleichtert, die die nicht sehr reaktive OH-Funktion protonieren und so ihren Austritt als H2 O einleiten. Jedoch muss auch eine Base zur Übernahme des H vom benachbarten C-Atom vorhanden sein. OH -H2O Cyclohexanol Sdp. 160-161°C Cyclohexen Sdp. 83°C Chemikalien: Cyclohexanol, KHSO4 , Kaliumpermanganat-Lösung. 116 KAPITEL 4. ORGANISCHE CHEMIE Durchführung: Man mischt in einem 100 ml-Rundkolben 15 g Cyclohexanol mit 15 g fein gemörsertem Kaliumhydrogensulfat (KHSO4 ) und setzt einen kleinen Destillationsaufsatz (wird vom Assistenten ausgegeben) auf. Die Apparatur muss vor der Inbetriebnahme vom Assistenten begutachtet werden. Der Vorlagekolben wird mit Eis gekühlt, indem man ihn in ein Becherglas mit Eiswasser taucht. Der Reaktionskolben wird mit einem passenden Heizpilz bis zum Sieden erhitzt (Siedesteine zugeben) bis das gebildete Cyclohexen und Wasser (aber nicht Cyclohexanol) überdestillieren. Cyclohexanol hat einen höheren Siedepunkt als Cyclohexen. Steigt die Temperatur am Destillationsvorstoß an, so ist dies ein Zeichen, dass jetzt nicht umgesetztes Cyclohexanol überdestilliert. Brechen Sie an dieser Stelle die Destillation ab. Bei Beendigung der Destillation sollte noch etwas Flüssigkeit im Kolben vorhanden sein. Trennen Sie mit einer Pasteurpipette die beiden Phasen des Destillats und führen Sie mit der organischen Phase die Baeyerprobe durch. Die wässrige Phase wird verworfen (organische Lösungsmittelabfälle). Baeyer-Probe auf Alkene: Zum Nachweis von Doppelbindungen eignet sich diese Reaktion mit KMnO4 . Auf diese Weise lässt sich feststellen, ob sich auch wirklich eine Doppelbindung gebildet hat. Doppelbindungen (ausser die formalen Doppelbindungen in Aromaten) entfärben Permanganat-Lösung, wobei über cyclische Mangansäureester 1,2Dialkohole (Glykole) und niedere Oxidationsstufen von Mangan entstehen. Bei dieser Addition werden beide C-Atome der Doppelbindung von der gleichen Seite angegriffen (cis-Addition). Aus Cycloalkenen entstehen cis-1,2Diole. 4.3. SUBSTITUTION, ELIMINIERUNG, ADDITION (6.–7. WOCHE) 117 +MnO4- +H2O + O O Mn O O OH OH niedere Ox. Stufen von Mangan z.B. MnO2 Glycol (1,2-Diol) Durchführung der Baeyer-Probe: Ein Mikroreagenzglas wird ca. ein Finger hoch mit dem Alken gefüllt und die ausstehende Permanganat-Lösung mit einer Pasteurpipette zugetropft. Entfärbt sich die rote PermanganatLösung, so liegt eine Doppelbindung vor. Toluol besitzt drei Doppelbindungen. Machen sie die gleiche Probe mit Toluol. Aufgaben: Protokollieren Sie kurz Ihre Vorgehensweise und Beobachtungen. Formulieren Sie die Reaktionsgleichung mit allen Einzelschritten und, nach Möglichkeit, Oxidationszahlen. Welche Rolle spielt KHSO4 ? Welche Reaktion zeigt Toluol bei der Baeyer-Probe und warum? Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.3.2 4.3.3 Versuch: Addition an Doppelbindungen Stichworte: π-Komplex, Bromoniumion Allgemeines: Doppel- und Dreifachbindungen sind elektronenreiche Stellen eines Moleküls, da neben der σ-Einfach-Bindung die π-Bindung vorhanden ist. Daher werden sie bevorzugt von Elektrophilen (Lewis-Säuren) angegriffen. Die Auflösung einer π-Bindung unter Anlagerung zweier neuer Substituenten heißt elektrophile Addition AE . Biologisch wichtige Reaktionen von Olefinen (Alkenen) sind z. B. die Hydrierung (H2 -Addition zu gesättigten Verbindungen), Hydratisierung zu Alkoholen, HCl-Addition zu Alkylhalogeniden, Säureanlagerung zu Estern, Bromierung zu Dibromiden. Der Mechanismus ist zweistufig: 118 KAPITEL 4. ORGANISCHE CHEMIE _ | Br _ _ Br| _ R H H R' Alken Br δR H Br δ+ H R + Br H R' H R' π-Komplex + _ | Br _ | Bromoniumion Br H R R' H Br 1,2-Dibromalkan Das unpolare, aber polarisierbare Brommolekül Br2 richtet sich in der Nähe einer Doppelbindung so aus, dass das positive Ende des induzierten Dipols sich zur elektronenreichen Doppelbindung orientiert (π-Komplex). In dieser Situation kann das Brom heterolytisch ionisieren unter Bildung des Bromid-Ions Br− . Das formal zurückbleibende, aber nicht frei auftretende Kation Br+ lagert sich an die Doppelbindung zu einem Bromonium-Ion“ ” an. Aus dem Alken (elektronenreich, Nucleophil) ist ein Kation mit Elektronenmangel (Elektrophil) geworden, das von dem in Lösung befindlichen Bromid-Ion von der Seite angegriffen wird, die nicht vom Brom abgeschirmt wird (trans-Addition). Chemikalien: Cyclohexen, Brom/Eisessig (1:1) (10%), 2 M NaOH Durchf¨uhrung: (Vorsicht beim Umgang mit Brom!! Einatmen und Haut-kontakt vermeiden! Thiosulfat-Lösung bereithalten) 1 ml des ausstehenden oder selbst hergestellten Cyclohexens wird in einem Reagenzglas unter Umschütteln in kleinen Portionen mit der ausstehenden Brom/Eisessig-Lösung versetzt (zum Schluss tropfenweise), bis die Bromlösung nicht mehr entfärbt wird. Dann versetzt man die Mischung mit 2 M NaOH, bis sich das ölige, terpentinähnlich riechende Dibromid abzuscheiden beginnt. Es bilden sich zwei Phasen. Stellen Sie fest, welche Phase wässrig ist und entsorgen sie diese (organische Lösungsmittelabfälle). Das Produkt wird mit NaOH neutralisiert. Zum Bromnachweis kann der Assistent die Beilsteinprobe ausführen. Aufgaben: Protokollieren Sie kurz Ihre Vorgehensweise und Beobachtungen. Formulieren Sie die Reaktionsgleichung mit allen Einzelschritten. 4.3. SUBSTITUTION, ELIMINIERUNG, ADDITION (6.–7. WOCHE) 119 Entsorgung: Organische Lösungsmittelabfälle. Ende Versuch 4.3.3 120 4.4 KAPITEL 4. ORGANISCHE CHEMIE Carbonylverbindungen, Acidität (8.–9. Woche) Stichworte: Carbonyl-Gruppe, α-H-Atome, C-H-Acidität, Mesomerie, Resonanzstabilisierung, Keto-Enol-Tautomerie, Tautomere, Elektronegativität, I-Effekte Aldehyde und Ketone Carbonylverbindungen enthalten eine Carbonylgruppe C=O. Ist mindestens ein Wasserstoffatom direkt an das Carbonylkohlenstoffatom gebunden, handelt es sich um Aldehyde. In Ketonen sind zwei Kohlenstoffatome an die Carbonylgruppe gebunden. Die IUPAC-Nomenklatur kennzeichnet aliphatische Aldehyde mit dem Suffix (Endung) -al, das an den jeweiligen Kohlenwasserstoffnamen angehängt wird. Für aliphatische und cycloaliphatische Ketone wird die Endung -on verwendet. Ketone werden häufig auch durch die Namen beider organischer Reste und die Endung -keton benannt (z. B. Ethylmethylketon). Aus historischen Gründen tragen zahlreiche Aldehyde und Ketone Trivialnamen, die sehr gebräuchlich sind (z. B. Formaldehyd, Aceton). Ist die Aldehyd- oder Ketogruppe nicht die Hauptfunktionalität der Verbindung, so wird das Präfix Oxo- zur Namensgebung verwendet. Ist die Aldehydgruppe der Substituent eines Ringgerüstes, so wird dieser mit dem Suffix -carbaldehyd bezeichnet. Die Reste -CHO und -COCH3 heißen Formyl- bzw. Acetylgruppe. Die Carbonylgruppe besteht aus einer kurzen und stabilen C=O-Doppelbindung. Das Kohlenstoffatom ist sp2 -hybridisiert, so dass die Bindungswinkel am Carbonylkohlenstoffatom ungefähr 120˚ betragen und die Carbonylgruppe, sowie die daran direkt gebundenen Atome eine Ebene bilden. Die unterschiedliche Elektronegativität von Sauerstoff und Kohlenstoff führt zu einer starken Polarisierung der Carbonylgruppe, die durch die mesomere Grenzformeln oder durch Partialladungen beschrieben werden kann. O + _ O _| od. δ+ δO Die Polarität der Carbonylgruppe prägt die physikalischen und chemischen Eigenschaften der Carbonylverbindungen. Die Siedepunkte der Aldehyde und Ketone liegen höher als die vergleichbarer Kohlenwasserstoffe aber tiefer als die vergleichbarer Alkohole. Die H-Atome am benachbarten (α)-CAtome sind leicht acid (sauer). Starke, nichtnucleophile Basen können daher 4.4. CARBONYLVERBINDUNGEN, ACIDITÄT (8.–9. WOCHE) 121 ein α-Proton abstrahieren. Dabei entstehen Enolat-Anionen oder Enolate, die über die Carbonylgruppe resonanzstabilisiert sind. O HC 3 H + Base H - H-Base H O O 3HC H - - 3HC H H H Enolat Die Reprotonierung kann nun an zwei Positionen erfolgen: am Kohlenstoffatom, unter Rückbildung der Carbonylverbindung, oder am Sauerstoffatom, unter Bildung eines Enols. Die Keto- und Enolform sind Tautomere, die in saurer oder basischer Lösung miteinander im Gleichgewicht (KetoEnol-Tautomerie) stehen. Das Gleichgewicht liegt meist weit auf der Seite der Carbonylverbindung, kann aber zur Enol-Form verschoben werden, wenn diese durch andere Faktoren begünstigt ist. So wird z. B. im 2,4-Pentandion (Acetylaceton) die Enolform durch Wasserstoffbrückenbindungen in einem sechsgliedrigen Ring stabilisiert. Darüberhinaus entstehen bei der Verschiebung zur Enolform konjugierte Doppelbindungen die eine Mesomeriestabilisierung (Delokalisierung von π-Elektronen) ermöglichen. H-Brücke O H3C H O H Keton O CH3 H H3C O CH3 H Enol Acidität Alkohole und Phenole Wie Wasser sind auch Alkohole und Phenole schwache Säuren. Die ihnen gemeinsame Hydroxygruppe ist ein Protonendonor und dissoziiert in gleicher Weise wie Wasser: 122 KAPITEL 4. ORGANISCHE CHEMIE * HO− + H-Base Wasser: HO–H + Base ) Alkohol: RO–H + Base * ) RO− + H-Base Die konjugierte Base eines Alkohols ist ein Alkoxid-Ion bzw. Alkoholat. Methanol und Ethanol haben fast dieselbe Säurestärke wie Wasser. tButanol z. B. ist jedoch eine erheblich schwächere Säure, während Phenol um den Farktor 106 stärker sauer ist als Ethanol! Wie können diese großen Unterschiede erklärt werden, wo doch jedesmal eine Hydroxygruppe der Protonendonor ist? Der Hauptgrund für die höhere Acidität von Phenolen gegenüber Alkoholen ist die Möglichkeit der Resonanzstabilisierung (Mesomerie) im Phenoxid- bzw. Phenolat-Anion, die im entsprechenden Alkoxid-Anion nicht gegeben ist. O - O O - - Die Ladung kann hier delokalisiert werden und somit ein Energiegewinn für das System erziehlt werden. Beim Alkoxid- bzw. Alkoholat-Anion bleibt sie am Sauerstoff lokalisiert. Carbonsäuren Carbonsäuren sind wesentlich acider (sauer) als Alkohole. Im CarboxylatAnion (COO− ) ist die negative Ladung mesomeriestabilisiert. _ O O _| _ O _ | O Beide Sauerstoffatome sind gleichmäßig an der Ladungsverteilung beteiligt. Die Ladungsverteilung durch Mesomeriestabilisierung bedeutet, dass das Carboxylat-Anion gegenüber dem Alkoholat-Anion weit stabiler ist. Dementsprechend ist seine Tendenz, sich aus der korrespondierenden Säure zu bilden, viel größer. Bei Alkoholen wie auch bei Carbonsäuren erhöht sich die Acidität durch induktive Effekte von elektronenziehenden Substituenten wie z. B. Halogenen. So reagieren Trifluoressigsäure und α-halogenierte Alkohole wesentlich stärkere Säuren als ihre nichthalogenierten Formen. 4.4. CARBONYLVERBINDUNGEN, ACIDITÄT (8.–9. WOCHE) 123 Ergänzung zur Acidität von organischen Verbindungen: Acidität und Elektronegativität (E.N.) Säure pKs H3C-H ca. 50 H2N-H 33 - HO-H 16 - F-H 3 - - korresp. Base H3C| H2N| HO| F| E.N. N:3,0 O:3,5 F:4,0 C:2,5 Acidität und Delokalisierung (Mesomerie) H H H H H Säure H H H H H H H _- H H H H H 16 H H H Korresp. Base H 43 ca. 50 pKs H H H H _- H H H H H -_ H Durch Abgabe eines Protons entsteht ein Aromat. - - _- H H H Aromat Allyl-Anion - - Cyclopentadienid-Anion Acidität und Delokalisierung + Elektronegativität H Säure H H H H ca.20 43 pKs O O O O O H H H ca.5 ca.11 H H O O O O _O O _O -_ O O H H korresp. Base _- _- _- H H -_ H H -_ O H Allyl-Anion EnolatAnion EnolatAnion des Stoffwechsels Vergleichen Sie den pKs-Wert der Essigsäure (ca. 5) mit dem von Ethanol (ca. 17). Worauf beruht der Unterschied? H 124 KAPITEL 4. ORGANISCHE CHEMIE Oxidation und Reduktion Alkohole, die mindestens ein Wasserstoff am sauerstoffgebundenen C-Atom tragen, können durch Oxidation in eine Carbonylverbindung umgewandelt werden. Primäre Alkohole liefern zunächst Aldehyde, dann Carbonsäuren. Aus sekundären Alkoholen entstehen Ketone. H R OH H prim. Alkohol Ox. H R O Aldehyd Ox. OH R O Säure Die Oxidation von Alkoholen in biologischen Systemen nennt man Dehydrierung. Das dabei beteiligte Enzym ist die Alkoholdehydrogenase (ADH). Desweiteren wird ein Coenzym benötigt, welches den formal entstehenden Wasserstoff (H2 ) bindet. In der Regel ist dies Nicotinamid-adenindinucleotid (NAD+ /NADH + H+ ) (siehe Lehrbücher der Physiologie und Biochemie). Die Oxidation eines primären Alkohols zu einem Aldehyd und unter Umständen weiter zu einer Carbonsäure ist verbunden mit einer Änderung der Oxidationszahl (Oxidationsstufe) des Kohlenstoffatoms, an dem die Reaktion stattfindet. Oxidation und Reduktion sind oft Begleiterscheinungen von Substitutionen, Additionen oder Eliminierungen. Allgemein wird die Oxidation eines Atoms definiert als Verlust von Elektronen, die Reduktion als Gewinn von Elektronen. In der organischen Chemie bezieht sich die Oxidation, beziehungsweise Reduktion, auf Kohlenstoffatome mit kovalenten Bindungen. Der Überschuss oder Unterschuss an Elektronen eines Kohlenstoffatoms gegenüber seinem elementaren Zustand mit vier Valenzelektronen (Oxidationszahl = 0) wird nach Art der gebundenen Atome bestimmt. Ein elektronegatives Atom erhöht die Oxidationszahl des Kohlenstoffs, denn es zieht die Bindungselektronen zu sich. Ein elektropositiveres Atom, auch Wasserstoff, erniedrigt die Oxidationszahl, denn die Bindungselektronen werden dem Kohlenstoff zugeschlagen. Bei einer Kohlenstoff-Kohlenstoff-Bindung werden die Bindungselektronen geteilt. An dieser Stelle sei nochmals darauf hingewiesen, dass die Oxidationszahlen nur formale Grössen sind. 4.4. CARBONYLVERBINDUNGEN, ACIDITÄT (8.–9. WOCHE) 4.4.1 125 Versuch: Säurestärke organischer Verbindungen Stichworte: Mesomerie, Stabilisierung von Ladungen Allgemeines: Organische Verbindungen können Wasserstoff als Proton abgeben (auf Basen übertragen), wenn folgende Effekte wirksam werden: a.) Mesomeriestabilisierung: Mesomerie, die eine Verteilung der Ladung über mehrere Atome erlaubt. Essigsäure z. B. ist acider als Methanol. Zwar wird bei beiden eine O–H-Bindung heterolytisch gespalten, für das entstehende Acetat-Ion gibt es jedoch, im Gegensatz zum MethanolatIon (Methoxid-Ion), zwei mesomere Grenzformeln. Methanol: H3CO-H + H2O H3CO - + + H3O Essigsäure: O _ O _ H + H2O _ O _| O _ O _| + + H3O O b.) Elektronegativität: Je elektronegativer das Atom ist, das das entsprechende Proton trägt, desto bereitwilliger gibt es das Proton ab. Chemikalien: Ethanol, Acetylaceton, Essigsäure, Trichloressigsäure, Phenolphtalein-Lösung, 1 M NaOH Durchführung: Prüfen Sie folgende Verbindungen auf ihre Säurenatur (pH-Papier): Ethanol, Acetylaceton, Essigsäure, Trichloressigsäure. O Et O Ethanol O O Cl3C OH Acetylaceton OH Essigsäure O Trichloressigsäure 126 KAPITEL 4. ORGANISCHE CHEMIE Versuchen Sie, den pH-Wert der Substanz direkt zu ermitteln (Indikatorpapier); dann machen Sie Wasser + 1 Tropfen Phenolphtalein-Lösung mit der kleinsten Menge 1 M Natriumhydroxid-(NaOH)-Lösung alkalisch, geben die Substanz zu und prüfen auf Entfärbung. Aufgaben: Protokollieren Sie die gemessenen pH-Werte und interpretieren Sie diese anhand von Reaktionsgleichungen unter Berücksichtigung evtl. entstehender mesomerer Grenzformeln. Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.4.1 4.4.2 Versuch: Aldolkondensation Stichworte: C-H-Acidität, Ladungsstabilisierung durch Mesomerie, Carbanionen, Enolat-Ion, C–C-Verknüpfungsreaktionen Allgemeines: Ketone wie z. B. Aceton (= Propanon) oder Aldehyde sind in der Lage, in Gegenwart von starken Basen (OH− -Ionen) zu einem geringen Teil in Enolat-Ionen überzugehen. Ein Enolat-Ion ist ein starkes Nucleophil, das über zwei nucleophile Zentren, das α-C-Atom und das O-Atom verfügt. Dieses Enolat-Ion greift mit seinem freien Elektronenpaar am C-Atom das Carbonyl-C-Atom eines Neutralmoleküls an. Das so gebildete Anion nimmt vom Wasser ein Proton auf, wobei ein Aldol oder Ketol entsteht. Da die Enolat-Ionen durch diese Reaktion ständig verbraucht werden, bilden sich im Rahmen des Säure-Basen-Gleichgewichts laufend Enolat-Ionen nach, bis die Reaktion abgeschlossen ist. In Abhängigkeit von den Bedingungen können entweder Aldole oder Ketole isoliert werden (Verknüpfung zweier Moleküle unter Bildung einer C–C-Einfachbindung), oder die Reaktion kann in einem Zug unter Wasserabspaltung bis zu α, β-ungesättigten (Bildung einer C=CDoppelbindung durch Kondensation) Carbonyl-Verbindungen führen. In der Reaktion eines Aldehyds mit einem Keton addiert im allgemeinen das Enolat des Ketons an den Aldehyd. Wegen der Aldol-Bildung trägt die Reaktion den Namen Aldol-Reaktion. 4.4. CARBONYLVERBINDUNGEN, ACIDITÄT (8.–9. WOCHE) H O H2C -_ H2C | Base H H2C H H Enolat-Ion δO δ+ H _ O _| O Aldoladdition: H3C 127 + _H2C H +H2O - OH O - _ H |O _ H3C O _ HO _ H H H3C H O 3-Hydroxybutanal (Aldol) Aldolkondensation: ( OH-katalysiert) H HO H3C _ |O H H H + _ | O| H - _ | OH _ -H2O HO H3C H H H -OH H3C O H Grundsätzlich könnte natürlich das Enolat-Ion die Carbonyl-Komponente auch über das O-Atom angreifen. Das dabei entstehende Produkt wäre jedoch thermodynamisch ungünstiger. Chemikalien: Benzaldehyd, Aceton, Ethanol, ges. Natriumhydroxid-Lösung Durchführung: In einem Reagenzglas gibt man zu etwa 2 ml Benzaldehyd (C6 H5 –CHO) 1 ml Aceton und als Lösungsmittel ca. 4 ml Ethanol. Durch Zugabe von 1 ml gesättigter Natriumhydroxid-Lösung wird die Reaktion gestartet. Nach einigen Minuten reibt man die Reagenzglaswand (innen) vorsichtig mit einem Glasstab und lässt das Reagenzglas ca. 10 min. im Ständer stehen. Das kristallin ausgefallene Reaktionsprodukt wird auf einem Hirschtrichter abgesaugt, mit einigen ml Ethanol nachgewaschen und durch Umkristallisation aus Ethanol gereinigt. Bestimmen Sie den Schmelzpunkt des Produkts (er sollte bei 112˚C liegen). Umkristallisieren: Die Substanz wird zunächst mit einer zur vollständigen Auflösung nicht ausreichenden Menge des Lösungsmittels erhitzt. Da normalerweise die Löslichkeitskurve in der Nähe des Lösungsmittelsiedepunkts steil ansteigt, sollt man beim Umkristallisieren immer bis zum Sieden erhitzen. Man gibt dann vorsichtig so lange Lösungsmittel nach, bis sich in 128 KAPITEL 4. ORGANISCHE CHEMIE der Siedehitze alles aufgelöst hat. Danach lässt man die Lösung langsam auf Raumtemperatur abkühlen und filtriert die entstandenen Kristalle ab. Bestimmung der Schmelztemperatur in der Kapillare Die fein pulverisierte, gut getrocknete Substanz wird in einer 2–4 mm hohen Schicht in ein etwa 1 mm weites, einseitig zugeschmolzenes Kapillarröhrchen gebracht. Dazu taucht man die Kapillare in die Substanzprobe und klopft das Pulver vorsichtig auf den Kapillarboden bzw. lässt das Röhrchen mehrfach durch ein langes, senkrecht auf einer harten Unterlage stehendes Glasrohr fallen. Das sogenannte Schmelzpunktröhrchen wird im einfachsten Fall mit einem Gummiring oder durch Ankleben des oberen Endes mit einem Klebeband an einem Thermometer befestigt. Die Substanzprobe muss sich dabei in Höhe der Quecksilberkugel des Thermometers befinden. Das Thermometer wird nun in ein mit Silikonöl gefülltes 100 ml Becherglas getaucht. Das Becherglas wird mit Hilfe einer Heizplatte langsam erhitzt (4–6˚C pro min.) bis das Pulver in der Kapillare zu schmelzen beginnt. Beim Erhitzen ist es notwendig, mit einem speziellen Rührer (wird mit dem Ölbad ausgegeben) die gleichmäßige Durchmischung des Öls, und damit der Temperatur, sicherzustellen. Aufgaben: Stellen Sie zunächst die Bruttoreaktionsgleichung für die Kondensation (Wasserabspaltung) zwischen zwei Mol Benzaldehyd und einem Mol Aceton auf. Welche der beiden Verbindungen wirkt als CH-acide Kom- 4.4. CARBONYLVERBINDUNGEN, ACIDITÄT (8.–9. WOCHE) 129 ponente? Versuchen Sie dann, die einzelnen Schritte der Reaktion zu formulieren. Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.4.2 4.4.3 Versuch: Präparative Darstellung eines Esters Boxweise Stichworte: Azeotrope, Ester & Ether, Säureanhydride Allgemeines: Ester sind in ihren Eigenschaften sehr typisch für die organische Chemie. Sie sind z. B. wasserunlöslich, flüchtig und meist stark riechend; sie kommen als Fette und Wachse in der Natur vor haben aber auch technische Bedeutung als Lösungs- und Extraktionsmittel. Ester entstehen aus zwei verschiedenen Substanzklassen, einer Carbonsäure und einem Alkohol mit den funktionellen Gruppen -COOH bzw. -OH durch Kondensation unter Abspaltung von Wasser. O R OH + HO R' O Kat. R Kat. + H2O O R' Die Esterbildung ist durch eine einfache Reaktion (Hydrolyse, Versei” fung“) wieder rückgängig zu machen. Hin- und Rückreaktionsschritte sind normalerweise langsam und müssen durch Katalysatoren beschleunigt werden. 130 KAPITEL 4. ORGANISCHE CHEMIE O R +H+ R OH OH + OH _ HO _ R' R' _ H +O _ R OH _ | OH _ _ | OR' _ H R O+ H | OH _ -H2O + OH R O OH R R OR' -H+ OH + OR' Ester Um präparativ eine hohe Ausbeute an Ester zu erreichen, muss man das Gleichgewicht nach rechts verschieben, indem man z. B. den Alkohol im Überschuss einsetzt und/oder das entstehende Wasser durch wasserentziehende Mittel oder apparative Maßnahmen (Abdestillieren, Wasserabscheider) aus dem Reaktionsgemisch entfernt. Günstiger ist die Umsetzung eines Säureanhydrids mit Alkohol, weil dann kein Wasser entsteht und die Rückreaktion nicht eintritt. Auch hier wirkt Säurekatalyse beschleunigend. Wasserabscheider: Informieren Sie sich in Lehrbüchern über Azeotrope. Die Azeotropbildung kann man ausnutzen, um einen Stoff—hier Wasser— aus einem Gemisch herauszuschleppen“. Man setzt der zu trocknenden Sub” stanz einen Stoff zu, der mit Wasser ein Azeotrop bildet und mit Wasser in der Kälte nicht mischbar ist, z. B. Toluol und erhitzt in der Apparatur (s. Abb.) die Mischung zum Sieden. Das Wasser geht mit dem Toluol azeotrop über und scheidet sich beim Abkühlen in Tropfen aus, die im graduierten Rohr des Wasserabscheiders nach unten sinken. Auf diese Weise ist das Ende der Wasserabscheidung leicht zu erkennen sowie die Wassermenge messbar. Bei chemischen Umsetzungen, bei denen Wasser entsteht, kann man daher den Fortgang der Reaktion gut beobachten. Durch dauernde Entfernung des Reaktionswassers wird darüber hinaus das Gleichgewicht im gewünschten Sinne verschoben. 4.4. CARBONYLVERBINDUNGEN, ACIDITÄT (8.–9. WOCHE) 131 Der Wasserabscheider sollte einen Tag vor dem geplanten Versuch, von einem Praktikanten aus der jeweiligen Laborbox, bei der Glasausgabe des Fachbereiches ausgeliehen werden. Durchführung: Jede Box stellt beide Ester her. 1.) Essigsäureisopentylester aus Essigsäure und Isopentanol Chemikalien: Eisessig, 3-Methyl-1-butanol (Isopentanol), p-Toluolsulfonsäure-monohydrat, Toluol, ges. NaHSO4 -Lösung, MgSO4 O H3C OH Essigsäure + HO CH3 CH3 3-Methyl-1-butanol -H2O CH3 O + H H3C O CH3 Essigsäureisopentylester 132 KAPITEL 4. ORGANISCHE CHEMIE Eine Lösung von 10 ml Eisessig (konz. Essigsäure), 5 ml Isopentanol (Isoamylalkohol, 3-Methyl-1-butanol) und 0.3 g p-Toluolsulfonsäuremonohydrat in 30 ml Toluol wird in einem 100–250 ml Rundkolben mit Wasserabscheider 1 h unter Rückfluss gekocht (Siedesteine und Heizpilz verwenden). Die Apparatur wird gemäß den Anweisungen des Assistenten aufgebaut und muss vor Inbetriebnahme von dem Assistenten begutachtet werden. Nach dem Abkühlen wird die organische Phase im Scheidetrichter mit 20 ml Wasser, 2 × 20 ml gesättigter Natriumhydrogencarbonatlösung (Vorsicht: CO2 -Entwicklung) und erneut 2 × 20 ml Wasser gewaschen. Nach dem Trocknen mit wasserfreiem Magnesiumsulfat (MgSO4 ) und Abdestillieren des Lösungsmittels am Rotationsverdampfer (Assistent) erhält man den Ester als farbloses Öl. 2.) Acetylsalicylsäure (ASS, Aspirin“) aus Salicylsäure und Essigsäurean” hydrid Chemikalien: Essigsäureanhydrid, wasserfreie Salicylsäure, konz. H2 SO4 , 50%ige Essigsäure O COOH + OH Salicylsäure O O H3C O OH O H+ CH3 -CH3COOH O Me Essigsäureanhydrid Acetylsalicylsäure In einem 100–250 ml Rundkolben gibt man 5 ml Essigsäureanhydrid 3.5 g wasserfreie Salicylsäure und anschließend 5 Tropfen konzentrierte Schwefelsäure. Man mischt den Inhalt des Kolbens und erwärmt mit dem Wasserbad 30 min. auf 50–60˚C (Rückflusskühler verwenden). Die auf Raumtemperatur abgekühlte Reaktionsmischung gießt man auf ca. 75 ml Wasser, schüttelt zur Hydrolyse des überschüssigen Säureanhydrids, kühlt dann in Eis und isoliert das ausgefallene Produkt. Es kann aus 50%iger Essigsäure umkristallisiert werden. Aufgaben: Protokollieren Sie ihre Vorgehensweise in kurzen Worten, formulieren Sie die Reaktionsgleichung möglichst in allen Teilschritten (Mechanismen) und berechnen Sie die Ausbeute an Produkt. Bestimmen Sie den Schmelzpunkt der selbst hergestellten Acetylsalicylsäure. 4.4. CARBONYLVERBINDUNGEN, ACIDITÄT (8.–9. WOCHE) 133 Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.4.3 4.4.4 Versuch: Fettverseifung Stichworte: Säure und Basenkatalyse der Verseifung, Aufbau natürlicher Fette, gesättigte & ungesättigte Fettsäuren, Weiterführend: Phospholipide, Aufbau von Zellmembranen. Allgemeines: Katalysiert man, vom Ester ausgehend, die Gleichgewichtseinstellung (Rückreaktion) nicht durch Säure, sondern hydrolysiert mit Alkalihydroxiden, so entstehen neben Alkohol die Alkalisalze der Fettsäuren und das Gleichgewicht wird völlig zu den Endprodukten verschoben. Fette sind Ester von langkettigen Carbonsäuren und dem dreiwertigen“ Alkohol ” Glycerin. O Na + O -O R O R O H2C HC O R' H2C O R'' OH 3 NaOH H2C HC OH H2C OH O Triglycerid O + Na + R' -O O Na + R'' -O Glycerin Fettsäure-Na-Salz R, R’ und R” können, aber müssen nicht identisch sein. Fette: Ester des dreiwertigen“ Alkohols Glycerin CH2 OH–CHOH–CH2 OH ” mit 3 Molekülen langkettiger Fettsäuren ( Triglycerid“). ” Seifen: Alkalisalze der langkettigen Fettsäuren. 134 KAPITEL 4. ORGANISCHE CHEMIE Wachse: Ester langkettiger Fettsäuren mit langkettigen Fettalkoholen“, z. B. ” Bienenwachs = Palmitinsäure-myricylester C15 H31 CO–O–C30 H61 . Chemikalien: Speiseöl, NaOH, EtOH, CaCl2 Durchführung: Man mischt in einem 250 ml Erlenmeyerkolben 5 g Speiseöl mit einer Lösung von 5 g NaOH in 20 ml Wasser/Ethanol 1:1 und erwärmt vorsichtig 15 min. zu schwachem Sieden. Verdampfender Alkohol wird ggf. ersetzt. Nach dem Erkalten bleibt als Produkt eine halbfeste Masse aus Natriumsalzen der Fettsäuren (Kernseifengeruch) und Glycerin zurück. Lösen Sie eine kleine Probe der Seife und messen sie den pH-Wert mit Indikatorpapier. Durch Zugabe von CaCl2 -Lösung fällt flockige Kalkseife aus und die Schaumbildung geht zurück. Der Hauptteil der Seife wird mit Hilfe des Büchnertrichters vorsichtig mit kleinen Wasserportionen alkalifrei gewaschen und getrocknet. Aufgaben: Protokollieren Sie Ihr Vorgehen und stellen Sie die Reaktionsgleichung auf. Welche Beobachtung machen Sie während des Auflösens einer kleinen Probe der Seife? Welchen pH-Wert hat die Seifenlösung und warum? Warum bildet sich bei Zugabe von CaCl2 -Lösung ein weißer Niederschlag? Welche Formel hat Kalkseife? Entsorgung: organische Lösungsmittelabfälle. Ende Versuch 4.4.4 4.4.5 Versuch: Decarboxylierung und oxidative Decarboxylierung Stichworte: Keto-/Enoltautomerie, Hydroxysäuren, β-Ketocarbonsäuren, Weiterführend: Bedeutung von Brenztraubensäure (Pyrovarsäure) und Decarboxylierung in biologischen Systemen. Allgemeines: Decarboxylierung (Abspaltung von CO2 ) normaler Carbonsäuren erfordert hohe Temperaturen. Unter milden Bedingungen decarboxylieren β-Ketocarbonsäuren wie Acetessigsäure (die daher in reiner Form nicht 4.4. CARBONYLVERBINDUNGEN, ACIDITÄT (8.–9. WOCHE) 135 isolierbar ist), da ein cyclischer Übergangszustand über ein Enol die Reaktion erleichtert: H O H3C H O O O H3C + CO2 CH2 Enol Acetessigsäure (eine ß-Ketocarbonsäure) O CH3 H3C Aceton Oxidative Decarboxylierung kommt bei Hydroxycarbonsäuren vor. Diese lassen sich leicht zu Ketosäuren dehydrieren, die dann wie oben spontan decarboxylieren. Die folgende Reaktion gibt es (etwas komplizierter) auch im Stoffwechsel: Ox. HO H3C HO OH Äpfelsäure OH OH OH O O O O O O Oxalsäure + CO2 O Brenztraubensäure Nachweis von CO2 im Gärröhrchen: Durch Auflösen von Bariumhydroxid Ba(OH)2 wird eine gesättigte Ba(OH)2 -Lösung hergestellt. Nachdem sich nicht gelöstes Ba(OH)2 am Reagenzglasboden abgesetzt hat, wird dekantiert (obere klare Lösung abschütten) und damit eine klare, gesättigte Ba(OH)2 -Lösung gewonnen. Diese wird wie gezeigt in ein Gärröhrchen gefüllt. Wird nun CO2 -haltiges Gas von unten durch das Gärröhrchen geblasen, so trübt sich die klare Lösung infolge von Bariumcarbonat-(BaCO3 )-Bildung. BaCO3 ist schwerlöslicher als Ba(OH)2 und fällt daher als weißer, feiner Niederschlag aus. 136 KAPITEL 4. ORGANISCHE CHEMIE Durchführung: a.) Decarboxylierung von Acetessigsäure: Chemikalien: 1 M NaOH, Acetessigester, Ba(OH)2 -Lösung, 2 M H2 SO4 Mischen Sie in einem kleinen Erlenmeyerkolben 1 ml Acetessigsäureethylester mit 3 ml 1 M Natriumhydroxid-(NaOH)-Lösung und erwärmen den Erlenmeyerkolben auf einer Heizplatte bis der typische Estergeruch verschwunden ist (ca. 5–10 min.). Danach geben Sie 5 ml 2 M Schwefelsäure (H2 SO4 ) hinzu und verschließen das Reagenzglas mit einem durchbohrten Gummistopfen. Durch den Gummistopfen wird ein Gärröhrchen gesteckt, welches mit einer wässrigen, klaren Ba(OH)2 Lösung gefüllt ist. Nach anfänglichem Aufkochen entwickelt sich CO2 , das mit Ba(OH)2 im Gärröhrchen nachgewiesen wird. b.) Oxidative Decarboxylierung von Äpfelsäure: Chemikalien: Äpfelsäure, FeSO4 , H2 O2 -Lösung (30%), Ba(OH)2 -Lösung In einem Reagenzglas werden 0.2 g Äpfelsäure und 0.1 g Eisensulfat FeSO4 in 5 ml Wasser gelöst. 1 ml dieser Lösung wird als Vergleich (Blindprobe) in ein zweites Reagenzglas gefüllt. Zu der restlichen Lösung gibt man mit einer Pasteurpipette 2 Tropfen H2 O2 -Lösung (30%) (Vorsicht! Bei Berührung mit der Haut sehr ätzend und schmerzhaft) und verschließt das Reagenzglas unverzüglich mit einem Gärröhrchen (s. o.). Falls nach 5 Minuten noch kein erkennbarer Gasstrom durch 4.4. CARBONYLVERBINDUNGEN, ACIDITÄT (8.–9. WOCHE) 137 das Gärröhrchen blubbert, fügt man weitere 2 Tropfen H2 O2 -Lösung zu. Sobald die CO2 -Entwicklung beendet ist, erhitzt man 5 min. auf 60˚C im Wasserbad (mit Wasser gefülltes 250 ml Becherglas auf einer Heizplatte) zur Verkochung (d. h. Zerstörung) des überschüssigen H2 O2 . Anschließend versetzt man Probe und Blindprobe mit je 1 ml Dinitrophenylhydrazin-Lösung (Nachweis auf Ketone und Aldehyde). Aufgaben: Protokollieren Sie Ihr Vorgehen und formulieren Sie die Reaktionsgleichungen mit allen Teilschritten. Was beobachten Sie bei der Zugabe von Dinitrophenylhydrazin-Lösung? Begründen Sie die Beobachtung. Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.4.5 138 4.5 KAPITEL 4. ORGANISCHE CHEMIE Identifizierung organischer Stoffe (10. Woche) Stichworte: Struktur und Derivate der Alkohole, Amine, Aldehyde und Ketone, Carbonsäureamide Stoffwechselprodukte, unbekannte Naturstoffe oder Produkte organischer Reaktionen, müssen identifiziert werden. Diese Aufgabe ist wegen der Vielfalt organischer Substanzen und Häufigkeit von Isomeren viel komplizierter als in der anorganischen Analyse. Wichtige Möglichkeiten sind: 1.) Chemische Derivatisierung zur Feststellung funktioneller Gruppen Da die Unterschiede in Eigenschaften und Reaktivität organischer Verbindungen besonders stark von ihren funktionellen Gruppen abhängen, ist man bestrebt, zunächst die Zugehörigkeit zu einer Substanzklasse (Alkohole, Amine, Aldehyde, Ketone, Carbonsäuren usw.) festzustellen. Dies ist möglich durch chemische Umsetzungen, die spezifisch und vollständig alle Vertreter einer Klasse in gut definierte, möglichst kristalline Derivate überführen (z. B. Alkohole → Ester). Gelingt eine solche Derivatisierung“, so beweist sie die Zugehörigkeit zur Substanzklasse, ” und die physikalischen Daten des Derivates (Schmelzpunkt) erlauben eine endgültige Identifizierung mit Hilfe von Tabellenwerten. Typische Beispiele sind die Analyse von Alkoholen, Aminen und Carbonylverbindungen. 2.) Vergleich mit authentischen Substanzen bekannter Struktur Wenn für eine unbekannte Substanz nur eine begrenzte Zahl von Möglichkeiten in Frage kommt, so genügt–unter Verzicht auf chemische Umwandlung—manchmal der Vergleich einer leicht zu messenden Eigenschaft (Spektrum, optische Aktivität, chromatographisches Verhalten) mit denen von mehreren Vergleichssubstanzen. Natürliche Fettsäuren, Zucker oder Aminosäuren werden so identifiziert. Die Methode stellt keinen strengen Strukturbeweis dar und muss durch Nachweise nach der 1. oder 3. Art ergänzt werden, wenn Verdacht auf andersartige, von den Vergleichssubstanzen nicht erfasste Strukturen besteht. 3.) Spektroskopische Instrumentalanalyse In organischen Molekülen mit ihren zahlreichen Bindungen ist es möglich, durch Bestrahlung in langwelligen (niederenergetischen) Spektralbereichen bestimmte Schwingungen, magnetische Änderungen u. dergl. 4.5. IDENTIFIZIERUNG ORGANISCHER STOFFE (10. WOCHE) 139 nur an einzelnen Bindungen oder Atomen anzuregen, so dass die beobachteten Absorptionssignale Strukturdetails der Moleküle direkt anzeigen. Hierzu zählen vor allem die Methode der kernmagnetischen Resonanz (NMR, siehe auch Kernspin-Tomographie), die wir wegen des apparativen Aufwands nicht ausführen, und die Infrarotspektroskopie (IR), die funktionelle Gruppen und ggf. Molekülgerüste an kleinen Proben und ohne Vorarbeiten zu analysieren gestatten. Meist wird man in komplizierten Fällen zwei oder mehr Wege bis zur endgültigen Strukturaufklärung beschreiten müssen. Die Tendenz geht, u. a. wegen der im Naturstoffbereich oft sehr kleinen vorhandenen Substanzproben, zur Instrumentalanalyse, doch behalten auch einfache chemische und chromatographische Nachweise ihren Wert. Auftrennung oder Derivatisierung der Aminosäuren in einem Proteinhydrolysat im Mikromaßstab üben wir später. Vorversuche Im Folgenden werden Ihnen verschiedene Nachweisreaktionen für Alkohole, Amine und Ketone/Aldehyde vorgestellt. Testen Sie die Nachweisreaktionen jeweils mit den ausstehenden Substanzen und machen Sie damit Blindversuche, um die Nachweisreaktionen kennenzulernen. Danach lassen Sie sich eine Analysensubstanz geben und analysieren die Inhaltsstoffe mit Hilfe der folgenden Nachweisreaktionen. 4.5.1 Versuch: Nachweis von Alkoholen Stichworte: Struktur und Eigenschaften von Alkoholen (primär, sekundär, tertiär), Veresterung mit Säurechloriden Allgemeines: Rekapitulieren Sie die Eigenschaften von Alkoholen. Warum sind die niederen Vertreter wasserlöslich, die höheren nur begrenzt mit Wasser mischbar? Chemikalien: Ammoniumcer(IV)nitrat, 2 M HNO3 , Pyridin, 3,5-Dinitrobenzoylchlorid, 6 M HCl, NaHCO3 -Lösung, Ethanol 140 KAPITEL 4. ORGANISCHE CHEMIE Durchführung: Identifizierung der Stoffgruppe: Alle Alkohole sowie Hydroxysäuren und Hydroxyaldehyde geben unter Ligandenaustausch einen roten Komplex mit der gelborangefarbenen Lösung von Ammoniumcer(IV)nitrat. [Ce(NO3 )6 ]2− + R–OH → [Ce(OR)(NO3 )5 ]2− + H+ + NO− 3 Vom Reagenz (10 g (NH4 )Ce(NO3 )6 in 25 ml 2 M Salpetersäure) werden 0.5 ml mit 3 ml Wasser in einem Reagenzglas verdünnt, 4–5 Tropfen der zu prüfenden Substanz zugegeben und geschüttelt. Bis auf Phenole (intensive Dunkelfärbung, evtl. Niederschlag) und Amine (evtl. Ausfällung) wird der Alkoholnachweis durch andere organische Substanzen nicht gestört. Individuelle Identifizierung: Alkohole identifiziert man als schwerlösliche Ester der 3,5-Dinitrobenzoesäure. Zur quantitativen Umsetzung verestert man nicht mit der Säure, sondern dem reaktionsfähigeren Säurechlorid. O2N O2N O Cl + + H-Base + Cl O R O2N O2N 3,5-Dinitrobenzoesäure O |Base HO R Alkohol 0.5 ml Alkohol werden in einem 50 ml-Rundkolben unter Eiskühlung mit 3 ml Pyridin und 2 g 3,5-Dinitrobenzoylchlorid vermischt und unter Verwendung eines Rückflusskühlers 10 Minuten auf dem Wasserbad auf 80˚C erhitzt. Die Mischung wird (zur Hydrolyse des überschüssigen Säurechlorids) auf 50 ml Eiswasser gegossen und tropfenweise mit 6 M Salzsäure HCl auf pH = 5 gebracht. Den Niederschlag filtriert man mit dem Büchnertrichter ab, wäscht mit Natriumhydrogencarbonat-(NaHCO3 )-Lösung und trocknet auf Filterpapier (nicht im Trockenschrank!). Das Produkt ist aus Ethanol umzukristallisieren und der Schmelzpunkt des Derivates zu bestimmen. Anhand des Schmelzpunktes kann auf den Alkohol geschlossen werden. Aufgaben: Führen Sie die Nachweisreaktionen mit verschiedenen Alkoholen aus. Protokollieren Sie Ihr Vorgehen und formulieren Sie die Reaktionsgleichungen. Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.5.1 4.5. IDENTIFIZIERUNG ORGANISCHER STOFFE (10. WOCHE) 4.5.2 141 Versuch: Nachweis von Aminen als Benzamid Stichworte: Struktur und Eigenschaften von Aminen, Basizität von Aminen Allgemeines: Amine sind basische Verbindungen, die sich durch Substitution der Wasserstoffatome durch organischen Resten von NH3 abgeleitete Verbindungen. Man unterscheidet hier: _ R N R H R N| R H H N\ + R N R R tertiäres Amin sekundäres primäres R R R quartäres Ammoniumsalz Beispiele: C2H5 _ NH2 | NH Ethylamin Et Et N\ Et HO Cholin Triethylamin Piperidin CH3 CH3 N+ OH CH3 Amine reagieren ähnlich wie Alkohole als Nucleophil mit elektrophilen Reaktionszentren, wie z. B. X–C=O von Carbonsäurederivaten. Die primären und sekundären Amine identifiziert man durch Umsatz mit Benzoylchlorid zu substituierten Benzoesäureamiden (kurz Benzamide). O R + Cl Base O + HN HCl N R R R Chemikalien: 2 M KOH, 0.5 ml Benzoylchlorid (frisch destilliert) Durchführung: 0.5 g Amin werden in 10–15 ml 2 M Kaliumhydroxid(KOH)-Lösung gelöst, mit 0.5 ml Benzoylchlorid versetzt und kräftig geschüttelt. Der Benzoylzusatz wird 1–2 mal wiederholt, bis der Amingeruch verschwunden ist. Das Produkt wird mit dem Büchnertrichter abgesaugt, mit 142 KAPITEL 4. ORGANISCHE CHEMIE Wasser gewaschen und aus Ethanol/Wasser (1:1) umkristallisiert. Nach dem Trocknen der Substanz wird der Schmelzpunkt bestimmt. Anmerkung: Nicht in Gegenwart von Alkoholen ausführen: Esterbildung als Konkurrenzreaktion! Aminosäuren reagieren in gleicher Weise wie Amine unter Bildung von N-Benzoylaminosäuren. Aufgaben: Führen Sie die Nachweisreaktionen mit verschiedenen Aminen aus. Protokollieren Sie Ihr Vorgehen und formulieren Sie die Reaktionsgleichungen. Sind auch tertiäre Amine mit dieser Methode nachzuweisen? Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.5.2 4.5.3 Versuch: Nachweis von Aldehyden und Ketonen als Dinitrophenylhydrazone Stichworte: Struktur und Eigenschaften von Carbonylverbingungen, Elektrophilie des Carbonyl-C-Atoms Allgemeines: Zum Nachweis von Aldehyden und Ketonen eignen sich besonders schwerlösliche C=N-Derivate der C=O-Gruppe. Sie entstehen durch primäre Addition von nucleophilen NH2 -Gruppen geeigneter Reagenzien und Wasserabspaltung. O + _ _ N OH _ _ _ H2N OH _ -H2O Oxime Hydroxylamin O + _ NH2 _ H2N _ _ N N H O H O Semicarbazid O + H _ H2N N _ _ NH2 Semicarbazone NO2 O2N 2,4-Dinitrophenylhydrazin _ H N N _ NO2 O2N 2,4- Dinitrophenylhydrazon 4.5. IDENTIFIZIERUNG ORGANISCHER STOFFE (10. WOCHE) 143 Chemikalien: Dinitrophenylhydrazin, Phosphorsäure (85%), Ethanol Durchführung: Reagenz: (Steht normalerweise im Labor aus) Als Reagenz wird 1 g Dinitrophenylhydrazin in 10 ml konz. Phosphorsäure (85%) unter Erwärmen gelöst (Wasserbad) und mit 10 ml Ethanol verdünnt, ggf. durch Glasfritte filtrieren. Hautkontakt vermeiden! Man löst je 5 Tropfen Substanz in 1 ml Ethanol, tropft Reagenz zu und beobachtet die ausfallenden Niederschläge (ggf. Eiskühlung). Zur Identifizierung einer unbekannten Substanz geht man von ca. 0.5 g Substanz aus, wäscht den Niederschlag mit Wasser und kristallisiert aus Ethanol oder Essigester um. Nicht im Trockenschrank trocknen! Bestimmen Sie den Schmelzpunkt. Aufgaben: Führen Sie die Nachweisreaktionen mit verschiedenen Aldehyden und Ketonen aus. Protokollieren Sie Ihr Vorgehen und formulieren Sie die Reaktionsgleichungen. Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.5.3 4.5.4 Analyse 4: Organische Sustanzen (Alkohole, Aldehyde, Ketone, Amine) Allgemeines: Versehen Sie ein Blatt Papier mit einem Loch, so dass ein Reagenzglas hindurchpasst und beschriften Sie diese wie folgt: Kurs Nr.: Analyse Nr.: 4 Namen, Vorname: Box Nr.: Saal: Stecken Sie ein mit einem Stopfen verschlossenes Reagenzglas durch das Loch und stellen Sie das Reagenzglas in den vom Assistenten aufgestellten 144 KAPITEL 4. ORGANISCHE CHEMIE Halter für die Analysen. Am nächsten Kurstag können Sie ihr Reagenzglas gefüllt wieder entgegenehmen. Sie erhalten jeweils eine organische Substanz, die Sie zunächst nach ihrer Substanzklasse (funktionelle Gruppe) zuordnen und ihrem Assistenten ansagen“ sollen. Bei richtiger Ansage“ der funktionellen Gruppe identifi” ” zieren Sie dann die individuelle Verbindung mit Hilfe der vorangegangenen Nachweisverfahren. Durchführung: Erste Orientierung: Aussehen, Geruch, pH-Wert. Auf keinen Fall aber Geschmacksprobe!! Gruppenzugehörigkeit: Mit 1/3 der Analysensubstanz nach Löslichkeitsverhalten und qualitativen Tests entscheiden. Identifizierung: Mit restlicher Analysensubstanz das entsprechende Derivat herstellen. Siehe vorangegangene Nachweisverfahren. Entsorgung: organische Lösungsmittelabfälle. 4.5. IDENTIFIZIERUNG ORGANISCHER STOFFE (10. WOCHE) Schmelzpunktliste: Amine Propylamin Isobutylamin Benzylamin Diethylamin Morpholin Glycin Alanin Alkohole Methanol Ethanol 1-Propanol 2-Propanol 1-Butanol 1-Pentanol Cyclohexanol Benzylalkohol Aldehyde und Ketone Formaldehyd Chloral (Hydrat) n-Butyraldehyd Benzaldehyd Pinakolon Cyclohexanon Aceton Ende Versuch 4.5.4 145 Fp. = Schmelzpunkt (Schmp.) CH3 CH2 CH2 NH2 CH3 CH2 CH(CH3 )NH2 C6 H5 CH2 NH2 (C2 H5 )2 NH C4 H9 NO NH2 CH2 COOH NH2 CH(CH3 )COOH CH3 OH CH3 CH2 OH CH3 CH2 CH2 OH CH3 CH(OH)CH3 CH3 (CH2 )3 OH CH3 (CH2 )4 OH C6 H11 OH C6 H5 CH2 OH HCHO CCl3 -CHO CH3 CH2 CH2 CHO C6 H5 -CHO CH3 -CO-C(CH3 )3 C6 H10 O CH3 -CO-CH3 Fp. Benzamid 84˚C 57˚C 105˚C 42˚C 75˚C 187˚C 166˚C Fp. Dinitrobenzoat 109˚C 93˚C 74˚C 122˚C 64˚C 46˚C 112˚C 112˚C Fp. Dinitrophenylhydrazon 166˚C 131˚C 122˚C 237˚C 125˚C 162˚C 128˚C 146 4.6 KAPITEL 4. ORGANISCHE CHEMIE Themen des 4. Kolloquiums Es folgt eine Zusammenstellung der wichtigsten Themen in Stichworten. Es wird empfohlen, die Stichworte in den einschlägigen Lehrbüchern nachzuschlagen. Kohlenhydrate Monosaccharide, Disaccharide, Polysaccharide, Oligosaccharide, glycosidische Bindung, räumlicher Bau und Stereochemie der Kohlenhydrate, Fischer-Projektion, Haworth-Projektion, Pyranose, Furanose, Glucose, Halbacetale, Acetale, Enantiomere, Epimere, Mutarotation, Oxidation und Reduktion von Monosacchariden, reduzierende Zucker. Aminosäuren und Proteine Aminosäuren: Definition und Struktur von Aminosäuren, Chiralität, Zwitterionen, isoelektrischer Punkt IP, Titrationskurve bei Aminosäuren, pKs Werte von Aminosäuren, D/L- und R/S-Nomenklatur, Dünnschichtchromatografie, chromatographische Trennung (Rf -Werte) usw. Peptide und Proteine: Bildung, Struktur und chemisches Verhalten der Peptidbindung, Di- und Tripeptide usw. 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 4.7 147 Zucker/Kohlenhydrate (11.–12. Woche) Stichworte: Chiralität in Sacchariden, Fischer-Projektion, D/L-Nomenklatur, cyclische Strukturen (Halbacetale) der Monosaccharide und deren Konformations-Isomere, Epimere, Anomere, Haworth-Projektion, glycosidische Bindungen, Polysaccharide, Struktur von Cellulose und Amylose (Stärke) Kohlenhydrate haben in allen Organismen lebenswichtige Aufgaben zu erfüllen. Sie entstehen aus Kohlendioxid und Wasser durch Photosynthese in grünen Pflanzen. Die bekanntesten Kohlenhydrate sind Stärke und Cellulose sowie die unter dem Sammelbegriff Zucker zusammenfassbaren Monosaccharide Glucose, Galaktose und Fructose sowie die Disaccharide Saccharose, Maltose und Lactose. Cellulose ist das wichtigste Baumaterial der Pflanzen. Sie wird zur Verseifung der Zellwände, zur Bildung von Fasern und zur Verfestigung des Holzgewebes benötigt. Stärke ist ein Reservekohlenhydrat. Die Pflanze benutzt Stärke als Energie- und Rohstoffspeicher. Viele Pflanzen, zum Beispiel die Zuckerrübe und das Zuckerrohr, produzieren große Mengen an Saccharose, dem handelsüblichen Zucker, heute noch der Hauptsüßstoff unserer Speisen und ein wichtiges Nahrungsmittel. Das Monosaccharid Glucose ist als Blutzucker die unmittelbare Energiequelle des Säugerorganismus. In Kombination mit Phosphorsäure und mit heterocyclischen Basen finden sich die Monosaccharide Ribose und 2-Desoxyribose im genetischen Material, den Nucleinsäuren. Viele andere Zucker sind Bestandteile von Antibiotika und Coenzymen, von Knorpelgewebe, Haut und Knochen, vom Aussenskelett der Crustaceen und den Zellwänden der Bakterien. Die chemischen Reaktionen der Kohlenhydrate umfassen die zweier funktioneller Gruppen, der Carbonylgruppe und der Hydroxygruppe. Die grobe Einteilung von Kohlenhydraten erfolgt im allgemeinen nach Molekülgröße in Monosaccharide (aus einem Zuckerbaustein), Oligosaccharide (bis zu zehn Zuckereinheiten) und Polysaccharide (bis zu mehreren tausend Zucckereinheiten). Der Ausdruck Saccharid kommt vom lateinischen saccharum (Zucker) und deutet darauf hin, dass die meisten niedermolekularen Kohlenhydrate süß schmecken. Monosaccharide Man klassifiziert die Monosaccharide nach der Anzahl ihrer Kohlenstoffatome (Triose, Tetrose, Pentose, Hexose, Heptose, usw.) und nach der Art der Carbonylgruppe. Wenn es sich um einen Aldehyd handelt, nennt man die Verbindung allgemein Aldose, ist es ein Keton, wird sie Ketose genannt. Bei- 148 KAPITEL 4. ORGANISCHE CHEMIE de Konstitutionsmerkmale kann man zusammenfassen und etwa von einer Aldopentose oder einer Ketohexose sprechen. Glycerinaldehyd steht in wässriger, alkalischer Lösung mit 1,3-Dihydroxyaceton im Gleichgewicht. Es handelt sich hierbei um eine Keto-Enol-Tautomerie mit einem Endiol als tautomeres Zwischenprodukt. H O H CH2OH OH H O * OH OH CH2OH CH2OH CH2OH Endiol 1,3-Dihydroxyaceton eine Keto-triose Glycerinaldehyd eine Aldo-triose Fischer Projektion und D/L-Nomenklatur Zur strukturellen Darstellung der Monosaccharide wird häufig die FischerProjektion verwendet. Dabei wird die längste Kohlenstoffkette senkrecht angeordnet, das am höchsten oxidierte Kohlenstoffatom steht oben. Die waagerechten Linien stellen Bindungen dar, die auf den Betrachter hin gerichtet sind, senkrechte Linien weisen von ihm weg. Die Stereozentren der Zucker können durch die R,S-Nomenklatur eindeutig beschrieben werden. Dennoch wird häufig ein älteres System zur Klassifizierung verwendet: Die Zuordnung zur D- oder L-Reihe. Für die Zuordnung ist das unten stehende asymmetrische (chirale) C-Atom der Kohlenstoffkette verantwortlich, d. h. das asymmetrische Kohlenstoffatom, das am weitesten von der Aldehyd- bzw. Ketogruppe entfernt ist. Zeigt in der Fischer-Projektion die OH-Gruppe dieses C-Atoms nach links, gehört der Zucker zur L-Reihe, weist sie nach rechts, gehört er zur D-Reihe. Die D- und L-Form eines Zuckers, z. B. D-(-)-Erythrose und L-(+)-Erythrose, verhalten sich an allen asymmetrischen Zentren wie Bild und Spiegelbild, sie sind Enantiomere. 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) CHO CHO H * OH HO * H H * OH HO * H CH2OH CH2OH 1 CHO 1 CHO H 2 HO 3 * H H 4 * OH H 5 * OH * OH HO 2 HO 3 * H H 4 * OH H 5 * OH L-(+)-Erythrose * H 6 CH OH 2 6 CH OH 2 Spiegel D-(-)-Erythrose 149 D-Glucose D-Mannose Enantiomere Epimere Stereoisomere, die sich nur durch die Konfiguration an C-2 unterscheiden, wie z. B. D-Glucose und D-Mannose, werden als Epimere bezeichnet. Die Drehrichtung einer Verbindung für linear polarisiertes Licht wird durch (+) bzw. (-) angegeben und steht mit der Zugehörigkeit zur D- oder L-Reihe in keinem Zusammenhang. Halbacetal Die Fischer-Projektion ist natürlich nur eine anschauliche und eindeutige Schreibweise für Moleküle. Die wirklich vorliegende dreidimensionale Struktur kann man aus ihr nicht ohne weiteres erkennen. Schreibt man die Formeln mit den echten Bindungswinkeln, so erkennt man folgendes: CHO H H OH HO HO H H OH H OH CH2OH OH H C C OH H CHO C OH C HOC2H H Diese Schreibweise lässt deutlich werden, dass sich das C-1-Atom und die OH-Gruppe von C-5 (bzw. C-4) sehr nahe beieinander befinden. Analog dem Mechanismus der Aldolkondensation (s. o.) bildet sich ein Sechserring (bzw. Fünferring), und zwar ein sogenanntes Halbacetal (der genaue Mechanismus dieser Reaktion s. u.). 150 KAPITEL 4. ORGANISCHE CHEMIE CH2OH H O OH H H HO HO H H OH Schreibt man dieses Halbacetal ohne Keilbindungen, das C-1-Atom rechts, den Halbacetal-Sauerstoff rechts oben und zeichnet unter Vernachlässigung der echten Bindunswinkel die OH-Gruppen (und H-Atome) nur nach oben oder unten, so kommt man zur Haworth-Formel. Die Fischerprojektion wird dabei nach rechts umgelegt. Rechts stehende OH-Gruppen werden nach unten zeigend gezeichnet und links stehende OH-Gruppen nach oben. Die Formelzeichnung cyclischer Zucker kann in der Fischer-, Haworth- oder Sesselprojektion erfolgen. CHO H 1 H 2 HO 3 H 4 H 5 wird immer nach oben zeigend gezeichnet 6 CH OH 2 5 OH H OH O OH O 4 OH HO OH 3 H 4 1 6 2 1 3 OH O 5 HO HO 2 OH OH 6 CH OH 2 Haworth-Projektion Fischer-Projektion Sesselkonformation Bei Monosacchariden führt die intramolekulare Addition der Hydroxygruppe von C-4 oder C-5 der Aldosen an das Carbonyl-C-Atom also zur Bildung von cyclischen Halbacetalen. Die fünfgliedrigen Halbacetale werden als Furanosen und die sechsgliedrigen als Pyranosen bezeichnet. (Diese Bezeichnungen leiten sich vom Pyran bzw. Furan ab) O O Tetrahydropyran Tetrahydrofuran 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 151 Mechanismus der Reaktion: Bei dem nucleophilen Angriff des HydroxylSauerstoffes an das Carbonyl-C-Atom entsteht ein neues chirales Zentrum. Der nucleophile Angriff des Hydroxyl-O-Atoms auf das Carbonyl-C-Atom der Aldehyd-Gruppe kann von zwei Seiten erfolgen. Die erhaltenen Diastereomere unterscheiden sich nur durch die Konfiguration am Halbacetal-C-Atom und werden Anomere genannt. Das neue Chiralitätszentrum ist das anomere Zentrum. Hat das Halbacetalkohlenstoffatom die S-Konfiguration, bezeichnet man das Diastereomere als α-Anomer, bei R-Konfiguration als β-Anomer. OH OH O β OH OH HO HO 5 HO HO O O HO H β-D-Glucose O + R Aldehyd Alkohol O OH α OH OH R O H HO HO α-D-Glucose D-Glucose al-Form _ HO _ R' OH H R' Halbacetal Die OH-Gruppe des Halbacetals (α oder β) bezeichnet man als glycosidische OH-Gruppe. Bindungen, an denen sie beteiligt ist, nennt man glycosidische Bindungen. Mutarotation In wässriger Lösung besteht ein Gleichgewicht zwischen den Anomeren und der nichtcyclischen Form. Durch Einstellung geeigneter Kristallisationsbedingungen ist es möglich, die reine α- oder β-Pyranoseform der Glucose zu erhalten. Bringt man die reinen Anomere wieder in Lösung, so ergibt sich nach kurzer Zeit wieder ein Gleichgewicht. Die Umwandlung wird durch die Veränderung der spezifischen Drehung der Lösung angezeigt. Das Phänomen wird als Mutarotation bezeichnet. 152 KAPITEL 4. ORGANISCHE CHEMIE HO HO HO O HO HO OH O HO HO OH OH α-D-Glucopyranose OH O HO HO OH OH H β-D-Glucopyranose D-Glucose Oligosaccharide Oligosaccharide sind Kohlenhydrate, die glycosidisch aus zwei bis zehn Monosacchariden zusammengesetzt sind. Bei der Verknüpfung der Saccharidbausteine gibt es grundsätzlich zwei Möglichkeiten: Zum einen können die Hydroxygruppen der beiden anomeren C-Atome unter Wasserabspaltung (Kondensation) zwei Vollacetale mit einem gemeinsamen Brückensauerstoff ausbilden (1,1-glycosidische Bindung). Zum anderen kann eines der beiden halbacetalischen C-Atome mit einer der alkoholischen Hydroxygruppe des anderen Saccharids reagieren (1,2 oder 1,3 oder 1,4 usw. glycosidische Bindung). Dies hat zur Folge, dass eine reaktive Halbacetalgruppierung erhalten bleibt. Zucker ohne halbacetalische Hydroxylgruppe zeigen keine Mutarotation und wirken auf Fehlingsche Lösung nicht reduzierend. Man unterscheidet deshalb auch zwischen reduzierenden und nichtreduzierenden Zuckern. OH OH OH O HO HO α OH OH + HO β R O R' Halbacetal OH O -H2O + + H O R'' R O R' -H2O Acetal Alkohol OH OH OH HO HO OH OH O OH OH HO Saccharose β-D-Fructofuranosyl-α-D-glucopyranosid β-D-Fructose _ HO _ R'' O O HO α-D-Glucose _ OH _ HO HO OH O 1 HO O OH 4 O OH 1,4-glycosidische Bindung in α-Lactose OH 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 153 Polysaccharide Polysaccharide sind aus wesentlich mehr Bausteinen aufgebaut, prinzipiell sind alle Kombinationen möglich und viele kommen auch in der Natur vor (z. B. Glycokalyx). Die beiden prominentesten Vertreter wurden oben schon erwähnt, Cellulose und Amylose (Stärke). Beide Polysaccharide sind ausschließlich aus D-Glucose-Einheiten aufgebaut und unterschieden sich im Grunde nur durch einen wesentlichen Punkt: CH2OH O H O H CH2OH O H OH H H OH O H OH H O H H OH n Verknüpfung β-glycosidisch ⇒ Faltblatt ⇒ Cellulose CH2OH CH2OH O H O H H H OH H H H O OH H O O H OH H OH n Oxidation Einfache Zucker können je nach Oxidationsmittel und Bedingungen zu unterschiedlichen Produkten reagieren, wie z. B. zu Aldonsäuren. Ein wichtiges Beispiel für eine Aldonsäure ist Ascorbinsäure (Vitamin C). 154 KAPITEL 4. ORGANISCHE CHEMIE HO OH H O O H HO OH L-Ascorbinsäure (Vitamin C) Reduktion von Monosacchariden Aldosen und Ketosen lassen sich zu Zuckeralkoholen (Alditole) reduzieren. Die Alditole aus Hexosen, die Hexite, sind gut kristallisierende, süß schmeckende Verbindungen, die beispielsweise Verwendung als Zuckeraustauschstoffe (Diabetikerzucker) finden. Die wichtigsten Hexite sind Dulcit, D-Mannit (Bestandteile des Mannas“) und D-Sorbit. ” CH2OH CH2OH CH2OH OH HO H H HO H HO H HO HO H H H H OH CH2OH Dulcit H OH H OH H OH OH H OH CH2OH D-Mannit CH2OH D-Sorbit 4.7.1 Versuch: Aufbau von Molekülmodellen (Stereochemische Aspekte der Zuckerchemie) Boxweise Stichworte: Allgemeines: Die Vorgehensweise ist die gleiche wie bei Versuch 4.2.1. Mit Hilfe des MINIT-Molekülbaukasten-Systems bauen Sie sich Molekülmodelle verschiedener Verbindungen und diskutieren die entsprechenden stereochemischen Aspekte. 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 155 C-Atome = schwarze, O-Atome = rote, H-Atome = weiße Bausteine, CH- und OH-Bindungen sowie C=O-Doppelbindungen sollten durch kleinere, C-C- und C-O-Einfachbindungen durch längere Verbindungsstücke markiert werden. Für die Carbonyl-C-Atome sind die dreibindigen und für die Carbonyl-O-Atome die einbindigen Bausteine zu verwenden. Durchführung und Aufgaben: a.) Bauen Sie Modelle für D- und L-Glycerinaldehyd auf und vergewissern Sie sich, dass die beiden Formen (Bild und Spiegelbild) nicht miteinander zur Deckung zu bringen sind! Wie müssen die beiden Moleküle im Raum orientiert werden, damit man ihre Zuordnung (D-Form oder L-Form) treffen kann? Schreiben Sie die Formeln beider Verbindungen in der Fischer-Projektion auf! Welche der beiden möglichen Formen nach der R,S-Nomenklatur (Roder S-) entspricht der D-Form? b.) Man fügt jeweils drei weitere H–C–OH Einheiten hinzu, so dass zwei D-Glucose-Moleküle in ihrer offenen Form entstehen. (Dabei muss die Stellung der OH-Gruppe an C-2 des ursprünglichen L-Glycerinaldehyds korrigiert werden!). Man beachte dabei, dass eine räumliche Orientierung des Moleküls, wie sie die Fischer-Projektion zur Festlegung der Konfiguration an den einzelnen C-Atomen vorschreibt, zu einer quasiringförmigen Anordnung des Moleküls führt. Aus den beiden identischen Molekülen bildet man einerseits α-D-Glucose, andererseits β-DGlucose. Dabei muss das dreibindige sp2 -C-Atom durch ein vierbindiges sp3 -C-Atom ersetzt werden, und anstelle kürzerer Verbindungsstücke müssen längere eingefügt werden. Zweckmäßigerweise zeichne man sich die Formeln der ringförmigen Formen vorher auf. Die beiden Isomeren liegen genau wie Cyclohexan in der Sesselform vor. Um welche Art von Stereoisomerie handelt es sich bei diesem Isomerenpaar? Sind die beiden Formen energetisch gleichwertig oder nicht? Wenn nicht, welche ist die energieärmere (stabilere) Form und warum? Man vergleiche die Formeln von D-Glucose und D-Galactose (offene Form). An welchem C-Atom der Modelle müssten H und OH vertauscht werden, um α-D-Glucose und β-D-Glucose in α-D- bzw. β-D-Galactose zu überführen? c.) Man forme das Modell der β-D-Glucose ebenfalls in α-D-Glucose um. Dann verbinde man die beiden α-D-Glucose-Moleküle zum Disaccharid 156 KAPITEL 4. ORGANISCHE CHEMIE Maltose! (Vorher Formel aufschreiben!) An welchen Positionen müssen die beiden Moleküle miteinander verbunden werden? Welche OHGruppe wird bei der unter Wasserabspaltung ablaufenden Bildung der Maltose tatsächlich abgespalten und warum? d.) Nachdem das Modell der Maltose wieder in zwei α-D-Glucosemoleküle zerlegt worden ist, fügt man diese zum Disaccharid Trehalose zusammen! An welchen Stellen müssen die beiden Moleküle jetzt verbunden werden (vorher Formel aufschreiben!)? Maltose und Trehalose verhalten sich bei der Reduktionsprobe völlig unterschiedlich, obwohl sie aus den gleichen Grundbausteinen bestehen. Erklären Sie das! Ende Versuch 4.7.1 Wahlpflichtblock 1 Von jedem Praktikanten sind vier der folgenden sechs Versuche durchzuführen. Innerhalb einer Laborbox-Gruppe sind insgesamt alle Versuche des Wahlpflichtblocks durchzuführen. 4.7.2 Wahlpflichversuch: Reduktion von Fehlingscher Lösung durch Glucose (Fehlingsche Probe) (1/6) Stichworte: Reduzierende Zucker Allgemeines: Die Fehlingprobe ist aufgrund der leicht zu beobachtenden Farbänderungen ein wichtiger Nachweis auf reduzierende Zucker. In der folgenden chemischen Gleichung sind an Stelle der Kupfertartrat-KomplexIonen der Fehlingschen Lösung nur die Kupfer(II)-Ionen angegeben. Die Glucose wird dabei zu Gluconsäure beziehungsweise zu Gluconat-Ionen oxidiert. O 2 Cu2+ + CH2OH-(CHOH)4 blau + rot + CH2OH-(CHOH)4 - H O Cu2O 5 OH O - + 3H2O 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 157 Chemikalien: 0.5 M Glucose-Lösung, Fehlingsche Lösung I und II, mitgebrachte Früchte- und Gemüseproben Durchführung: In einem Reagenzglas werden 6 ml eines Gemisches von Fehlingscher Lösung I und II mit 3 ml Glucoselösung im Wasserbad unter dauerndem Schütteln zum Sieden erhitzt (Vorsicht! Neigung zum Siedeverzug!). Untersuchung von Früchten und Gemüse auf Zucker: Untersuchen Sie die selbst mitgebrachten Früchte- und Gemüseproben auf reduzierende Zucker. Aus einem Apfel, einem Stück Karotte oder einigen Beeren ist ein Brei herzustellen. Zwei Spatelspitzen Brei werden nach Zugabe von etwa 5 ml Wasser in einem Reagenzglas zum Sieden erhitzt. Anschließend ist ein Gemisch aus 2 ml Fehlingscher Lösung I und 2 ml Fehlingscher Lösung II mit 3 ml des wässrigen Obstauszuges zum Sieden zu erhitzen. Aufgaben: Protokollieren Sie ihre Vorgehensweise und stellen Sie die Reaktionsgleichung mit Oxidationszahlen auf. Beschreiben Sie ihre Beobachtungen. Ergänzen Sie in der obigen Reaktionsgleichung die freien Elektronenpaare und geben Sie die Oxidationszahlen an. Warum kann Glucose reduzierend wirken? Entsorgung: anorganische Schwermetallabfälle Ende Versuch 4.7.2 4.7.3 Wahlpflichtversuch: Silberspiegel (Probe nach Tollens) (2/6) Stichworte: reduzierende Zucker Allgemeines: Eine weitere Nachweisreaktion ist die Probe nach Tollens. Hierbei werden Silberionen zu elementarem Silber reduziert, welches sich mit etwas Glück und Geschick als Silberspiegel“ an der Glaswand des Reagenz” glases niederschlägt. 158 KAPITEL 4. ORGANISCHE CHEMIE Im Fall der Glucose lautet die Reaktionsgleichung: O 2 [Ag(NH3)2]NO3 + CH2OH-(CHOH)4 H + H2O Glucose 2 Ag + O CH2OH-(CHOH)4 O - + NH4+ + 2 NH4NO3 + NH3 Ammoniumgluconat Chemikalien: 0.5 M Glucoselösung, ammoniakalische Silbernitratlösung Durchführung: Verwenden Sie für diesen Versuch ein neues Reagenzglas. Es darf keine Spur von Verunreinigung oder Fett an der Glaswand haften. Füllen Sie ca. 5 ml der ausstehenden Silbernitat-(AgNO3 )-Lösung in ein neues Reagenzglas. Geben Sie ca. 5 ml der 0.5 M Glucoselösung hinzu und erhitzen sie das Reagenzglas vorsichtig unter Drehen im ca. 90˚C heißen (kurz vor dem Sieden) Wasserbad, bis sich an der Wandung ein Silberspiegel abscheidet. Aufgaben: Protokollieren Sie ihre Vorgehensweise und stellen Sie die Reaktionsgleichung mit Oxidationszahlen und freien Elektronenpaaren auf. Beschreiben Sie ihre Beobachtungen. Warum kann Glucose reduzierend wirken? Warum muss die Silbernitratlösung ammoniakalisch sein? Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.7.3 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 4.7.4 159 Wahlpflichtversuch: Reduzierende Wirkung von D-Glucose gegenüber Triphenyltetrazoliumchlorid (3/6) Stichworte: organische Farbstoffe, konjugierte Systeme und Farbigkeit von Molekülen Allgemeines: Zum Nachweis von reduzierenden Zuckern lässt sich auch die Oxidation mit Triphenyltetrazoliumchlorid verwenden. N N N N + Cl - + ? e- N N H + ? H+ farblos + HCl N N rot Chemikalien: Triphenyltetrazoliumchlorid, Glucose Durchführung: 1 ml Triphenyltetrazoliumchlorid-Lösung wird im Reagenzglas zum Sieden erhitzt (Wasserbad) und dann mit etwa dem gleichen Volumen Glucoselösung versetzt. Aufgaben: Protokollieren Sie ihre Vorgehensweise und stellen Sie die Reaktionsgleichung auf. Beschreiben Sie ihre Beobachtungen. Warum kann Glucose reduzierend wirken? Wieviel Elektronen werden ausgetauscht? Entsorgung: organische Lösungsmittelabfälle. Ende Versuch 4.7.4 160 4.7.5 KAPITEL 4. ORGANISCHE CHEMIE Wahlpflichtversuch: Reaktion von Glucose mit Methylenblaulösung (4/6) Stichworte: organische Farbstoffe, konjugierte Systeme und Farbigkeit von Molekülen Allgemeines: Methylenblau wird durch basische Glucoselösung zu einer farblosen Leukoverbindung reduziert, wobei Glucose zur Gluconsäure oxidiert wird. Die farblose Leukoverbindung des Methylenblaus lässt sich durch Luft wieder oxidieren. H N Cl (H3C)2N S+ Methylenblau N(CH3)2 - + 2H (H3C)2N N + S HCl N(CH3)2 Leukomethylenblau Wegen der leichten Reduzierbarkeit dient Methylenblau als Wasserstoffakzeptor für biochemische Redoxprozesse. Methylenblau wird nicht nur in der Textilfärberei verwendet, sondern besitzt außerdem die Fähigkeit, die graue Substanz im peripheren Nervensystem selektiv anzufärben (Ehrlich, 1885). Man bezeichnet dieses Verfahren als Vitalfärbung, da sie am lebenden Organismus durchführbar ist. Methylenblau zählt somit zu den Vitalfarbstoffen“. ” Chemikalien: Glucose, Methylenblaulösung, 3 M NaOH Durchführung: In einem Erlenmeyerkolben werden etwa 20 g Glucose in 100 ml Wasser gelöst und mit so viel Methylenblaulösung versetzt, bis die Lösung kräftig blau gefärbt ist. Dann sind 15 ml Natriumhydroxid-(NaOH)Lösung hinzuzufügen. Nun wird diese Lösung auf einer Heizplatte auf 30–40˚C erwärmt. Wenn die Farbe verschwunden ist, schütteln Sie den Erlenmeyerkolben kräftig. Wiederholen Sie diese Prozedur mehrmals. Aufgaben: Protokollieren Sie ihre Vorgehensweise und stellen Sie die Reaktionsgleichung auf. Beschreiben Sie ihre Beobachtungen. Warum kann man das Farbphänomen auch nach mehrmaligem Erhitzen beobachten? 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.7.5 161 162 4.7.6 KAPITEL 4. ORGANISCHE CHEMIE Wahlpflichtversuch: Reduktion von Kaliumpermanganat durch Glucose (5/6) Stichworte: Glucose, reduzierende Zucker Allgemeines: Das Oxidationsmittel Kaliumpermanganat KMnO4 wird durch Glucose zu braunem Mangan(IV)-oxidhydrat MnO(OH)2 reduziert. O ? CH2OH-(CHOH)4 + ? MnO4 - H + H2O Glucose O ? CH2OH-(CHOH)4 O - + ? MnO(OH)2 + H+ Gluconat-Ionen Chemikalien: 0.5 M Glucose-Lösung, Kaliumpermanganat-Lösung (6%) Durchführung: Etwa 5 ml Glucoselösung werden in einem Reagenzglas mit 3 Tropfen Kaliumpermanganat-(KMnO4 )-Lösung versetzt und geschüttelt. Aufgaben: Protokollieren Sie ihre Vorgehensweise und stellen Sie die Reaktionsgleichung mit Oxidationszahlen auf. Beschreiben Sie Ihre Beobachtungen. Warum kann Glucose reduzierend wirken? Ergänzen Sie in der obigen Reakionsgleichung die freien Elektronenpaare. Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.7.6 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 4.7.7 163 Wahlpflichtversuch: Holzverzuckerung (6/6) Stichworte: Glycosidische Bindungen Allgemeines: Die Cellulose des Holzes wird durch starke Säuren bis zu niedermolekularen Kohlenhydraten hydrolytisch abgebaut. Diese lassen sich dann anhand der Fehlingschen Probe nachweisen. CH2OH CH2OH O O OH O OH + H+ + H2O O OH ?? O OH n Beispiel für glycosidische Bindung in Polysacchariden wie Cellulose Chemikalien: Feines Holzmehl, konz. HCl, Haines Reagenz (2 g CuSO4 , 15 g Glycerin, 150 g 5%ige KOH-Lösung) Durchführung: Etwa 1 g feines Holzmehl wird mit 8 ml konzentrierter Salzsäure gut verrieben und etwa 15 min. im Abzug stehengelassen. Dann ist das Stoffgemisch in einem Becher mit 50 ml Wasser zu versetzen und zum Sieden zu erhitzen. Nach 15 min. werden 3–4 ml Lösung in ein Reagenzglas filtriert. Das Filtrat ist mit Natriumhydroxidlösung bis zur basischen Reaktion zu versetzen und mit 4 ml Haines Reagenz zum Sieden zu erhitzen. Aufgaben: Protokollieren Sie ihre Vorgehensweise und beschreiben Sie ihre Beobachtungen. Versuchen Sie anhand einer Reaktionsgleichung die Hydrolyse von Cellulose zu niedermolekularen Kohlenhydraten in saurem Medium zu erklären. Entsorgung: Neutralisieren und organische Lösungsmittelabfälle Ende Versuch 4.7.7 164 4.7.8 KAPITEL 4. ORGANISCHE CHEMIE Versuch: Prüfung von Saccharose mit Fehlingscher Lösung Stichworte: nichtreduzierende Zucker, Saccharose Allgemeines: Saccharose ist der Zucker schlechthin. Sie kommt in der Zuckerrübe und im Zuckerrohr in Anteilen bis 25% vor. Ihre technische Gewinnung erfolgt hieraus in großem Maßstab. Saccharose ist ein β-D-Fructofuranosylα-D-glucopyranosid. Die anomeren Kohlenstoffatome der α-D-Glucopyranose (C-1) und der β-D-Fructofuranose (C-2) sind glycosidisch verbunden. HOH2C HO HO O 1 H H OH OH HO O CH2OH O 2 CH2OH Saccharose Chemikalien: Saccharose (Rohrzucker), Fehlingsche Lösung I und II Durchführung: In einem Reagenzglas werden 2 ml Fehlingsche Lösung I mit 2 ml Fehlingscher Lösung II gemischt. Dann sind in einem zweiten Reagenzglas etwa 0.5 g Saccharose in etwa 2 ml Wasser zu lösen, anschließend mit Fehlingscher Lösung zu versetzen und unter dauerndem Schütteln im siedenden Wasserbad zu erhitzen. Machen Sie dieselbe Probe mit Haines Reagenz. Aufgaben: Protokollieren Sie ihre Vorgehensweise und beschreiben Sie ihre Beobachtungen. Ist Saccharose ein reduzierender Zucker? Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.7.8 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 4.7.9 165 Versuch: Inversion von Saccharose Stichworte: Invertzucker, optische Drehung Allgemeines: Bei Einwirkung verdünnter Säuren wird Saccharose durch Hydrolyse in Glucose und Fructose gespalten: H+ C12H22O11 + H2O C6H12O6 Saccharose HOH2C HO HO + D(+)-Glucose C6H12O6 D(-)-Fructose O H H OH OH HO O CH2OH O + H+ H2O CH2OH Saccharose HOH2C HO HO O HO H OH α-Glucopyranose + HO HO O OH OH CH2OH β-Fructopyranose Bemerkung: Die Saccharoselösung ist rechtsdrehend (optisch). Das entstehende Stoffgemisch von D(+)-Glucose und D(-)-Fructose ist jedoch linksdrehend, da die D(-)-Fructose stärker nach links als D(+)-Glucose nach rechts dreht. Der Vorgang der Hydrolyse wird wegen der Umkehrung der Drehung als Inversion und das entstehende Gemisch als Invertzucker bezeichnet. Chemikalien: Saccharose (Rohrzucker), 1 M H2 SO4 , 3 M NaOH-Lösung, Haines Reagens, Unitestpapier Durchführung: In einem 250 ml Becherglas werden 2 g Saccharose in 20 ml Wasser gelöst, mit 2 ml 1 M Schwefelsäure (H2 SO4 ) versetzt und im Wasserbad zum Sieden erhitzt. Nach 5 min. ist zum Stoffgemisch bis zur basischen Reaktion Natriumhydroxid-(NaOH)-Lösung zuzufügen. Dann werden 10 ml Haines Reagenz zugegeben. 166 KAPITEL 4. ORGANISCHE CHEMIE Aufgaben: Protokollieren Sie ihre Vorgehensweise, beschreiben Sie ihre Beobachtungen und formulieren Sie die Reaktionsgleichung. Warum gelingt hier die Reduktion von Haines Reagenz? Erklären sie mit Formeln die Umwandlung des Fünfringes aus der Saccharose in den Sechsring der β-Fructopyranose. Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.7.9 4.7.10 Versuch: Nachweis von Vitamin C mit Tillmans Reagenz Stichworte: Wasserstoffbrücken, Enole, Vitamine. Weiterführend: Aufbau wichtiger Vitamine (A, B, C, E) und deren Funktion als Coenzyme. Allgemeines: Mensch sowie Affe, Meerschweinchen und einige andere Tierarten benötigen die L(+)-Ascorbinsäure (Vitamin C) als Vitamin, weil ihnen verschiedene Enzyme zur körpereigenen Synthese fehlen. Die Endiolgruppierung der Ascorbinsäure besitzt ein großes Redoxpotential, das für den Ablauf physiologischer Reduktions- und Oxidationsprozesse benötigt wird. HO OH H O O _ H H O \O H H-Brücken L-Ascorbinsäure (Vitamin C) Die biochemischen Wirkungen der Ascorbinsäure beruhen auf einer Beteiligung am mikrosomalen Elektronentransport sowie an zahlreichen Hydroxylierungsreaktionen. 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 167 Bei Vitamin C-Avitaminose (Skorbut) werden Symptome beobachtet, die das mesemchymale Gewebe betreffen, z.Ḃ. Blutungen in der Muskulatur und Schmerzen in den Extremitäten. Die Resistenz gegen Infektionen lässt stark nach. Der tägliche Vitamin C-Bedarf von ca. 50–100 mg kann durch den Verzehr pflanzlicher Lebensmittel, die zum Teil reich an Vitamin C sind, wie z. B. Hagebutten, schwarze Johannisbeeren, Citrusfrüchte, Paprika gedeckt werden. Auch Kartoffeln sind mit einem Gehalt von 3–30 mg/100 g eine wichtige Vitamin C-Quelle. Ascorbinsäure ist im neutralen oder alkalischen Milieu, bei erhöhter Temperatur und in Gegenwart von Schwermetallionen sehr oxidationsempfindlich dies kann zu Vitamin C-Verlusten beim Zubereiten der Speisen führen. In der Lebensmitteltechnologie wird Ascorbinsäure in immer größerem Umfang als Antioxidationsmittel zur Stabilisierung von Lebensmitteln eingesetzt. Die Oxidation von Fetten und Ölen kann z. B. durch Zusatz von fettlöslichem Ascorbylpalmitat verhindert werden. In diesem Versuch wird L-Ascorbinsäure (AS) aus dem entsprechend vorbereiteten Untersuchungsmaterial mit Oxalsäure-Lösung extrahiert und anschließend mit blauem 2,6-Dichlorphenolindophenol (DI) zur Dehydroascorbinsäure (DAS) umgesetzt. HO Cl OH H O O H HO + O OH N O Cl DI (blau) AS HO Cl HO N OH H H O - + O O Cl DI-Leukoform farblos Beispiele für Untersuchungsmaterialien: • frisches Obst und Gemüse • Frucht- und Gemüsesäfte • Konservenprodukte O H O DAS - 168 KAPITEL 4. ORGANISCHE CHEMIE • Multivitaminpräparate, Bonbons • Wurstwaren wie z. B. BiFi (hier wird Vit. C als Konservierungsmittel eingesetzt) Chemikalien: Tillmanns-Reagenz bzw. DI-Lösung (2,6-Dichlorphenolindophenol als Natriumsalz-Dihydrat C12 H6 Cl2 NNaO2 × 2H2 O, L(+)-Ascorbinsäure C6 H8 O6 , Oxalsäure C2 H2 O4 × 2H2 O, Oxalsäure-Lösung (2%) Durchführung: Probenaufarbeitung: Flüssigkeiten wie Frucht- und Gemüsesäfte werden mit Oxalsäure-Lösung verdünnt und, wenn nötig, filtriert. Feste Lebensmittelproben wie Früchte und Gemüse werden grob zerkleinert, unter Zusatz von Oxalsäure-Lösung homogenisiert und filtriert. Sonstige Vitamin-C-haltige, feste Lebensmittel werden in Oxalsäure-Lösung gelöst, zur Ausfällung von Proteinen mit etwas Trichloressigsäure versetzt und filtriert. Nachweis: Die möglichst klare und nicht zu farbige Probenlösung wird nun ca. zwei Finger hoch in ein Reagenzglas gefüllt und einige Tropfen Tillmans-Reagenz hinzugetropft. Die Entfärbung von Tillmanns-Reagenz zeigt die Anwesenheit von Vitamin-C an. Tillmans-Reagenz ist bei Raumtemperatur und bei Sonnenlicht nicht sehr lange haltbar! Aufgaben: Protokollieren Sie Ihr Vorgehen. Warum liegt Vitamin-C in der Enolform vor? Warum entfärbt sich das Tillmanns-Reagenz? (Antwort freigestellt). Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.7.10 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 4.7.11 169 Versuch: Darstellung des Pentaacetylderivats eines Monosaccharids Stichworte: Acetylierung, Anhydride, Katalysatoren Allgemeines: Monosaccharide lassen sich durch Derivatisierung identifizieren. Bei der Acetylierung der Zucker mit Acetanhydrid setzt man saure oder basische Katalysatoren, z. B. Zinkchlorid, konz. Schwefelsäure, wasserfreies Natriumacetat oder Pyridin, zu. Die Hexosen gehen dabei in Pentaacetate und die Pentosen in Tetraacetate über, z. B. entsteht aus Glucose die Pentaacetylglucose. Diese Acetate reduzieren Fehlingsche Lösung nicht, d. h., sie enthalten keine freie Aldehydgruppe, treten aber in α- und β-Form auf. Welches Anomer vorwiegend auftritt, hängt entscheidend vom Katalysator ab, wie folgendes Beispiel zeigt: HOH2C HO HO Ac = CH3CO- (Ac)2O, NaOAc AcOH2C AcO AcO OH O O HO H,OH OH α,β-Form D-Glucose (Ac) O, ZnCl O β OAc OAc β-D-Glucopyranose-pentaacetat OH 2 AcOH2C AcO AcO H,OH OH α,β-Form D-Galactose 2 O α Ac O OAc α-D-Glucopyranose-pentaacetat Chemikalien: Glucose, Galactose, Natriumacetat (wasserfrei), Essigsäureanhydrid Durchführung: Lassen Sie sich vom Assistenten eine Zuckerprobe geben. Ihre Aufgabe ist es nun, herauszufinden, ob es sich um Glucose oder Galaktose handelt. Erhitzen Sie 4 g dieser Probe mit 2 g wasserfreiem Natriumacetat und 20 g Essigsäureanhydrid in einem 100 ml Rundkolben mit aufgesetztem Rückflusskühler (die Apparatur muss vor dem Gebrauch vom Assistenten begutachtet werden) bis zur Lösung und halten dann noch 10 Minuten bei leichtem 170 KAPITEL 4. ORGANISCHE CHEMIE Sieden. Danach gießt man die Reaktionsmischung in 250 ml Eiswasser und rührt das sich abscheidende Öl bis zur Verfestigung. Man lässt noch 10 Minuten unter gelegentlichem Umrühren stehen, saugt die feste Masse mit dem Büchnertrichter ab, wäscht mit ca. 100 ml Wasser nach und kristallisiert aus Ethanol um, indem man die Masse in einem 100 ml Rundkolben, der mit einem Rückflusskühler zu versehen ist, in möglichst wenig Ethanol heiß löst (Heizpilz) und danach die Lösung in einem Eisbad abkühlt. Der dabei entstehende Niederschlag wird mit dem Hirschtrichter abgesaugt und auf gleiche Weise wie zuvor nochmals kristallisiert. Eine kleine Probe der abgesaugten und mit etwas Ethanol nachgewaschenen Substanz wird zwischen Filterpapier trockengepresst, dann wird eine Schmelzpunktbestimmung durchgeführt. Je nach dem welcher Zucker ausgegeben wurde, ist entweder Pentaacetyl-β-DGlucose (Schmp. 131–134˚C) oder Pentaacetyl-β-D-Galactose (Schmp. 142˚C) durch Veresterung der OH-Gruppen von Glucose oder Galactose entstanden. Aufgaben: Protokollieren Sie ihr Vorgehen bei der Reaktion und formulieren Sie die Reaktionsgleichung. Dem Assistenten ist anzugeben, welcher Zucker vorgelegen hat. Um welche Art von Isomeren handelt es sich bei den beiden möglichen Produkten? Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.7.11 Wahlpflichtblock 2 Von jedem Praktikanten sind zwei der folgenden vier Versuche durchzuführen. Innerhalb einer Laborbox-Gruppe sind insgesamt alle Versuche des Wahlpflichtblocks durchzuführen. 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 171 4.7.12 Wahlpflichtversuch: Herstellung von Stärkekleister (1/4) Boxweise; wird für die nachfolgenden Versuche gebraucht! Stichworte: viskose, kolloide Lösungen Allgemeines: Die Stärke ist das Assimilationsprodukt der grünen Pflanzenzellen. Unter Assimilation versteht man die Überführung des Kohlendioxids der Luft in Stärke unter katalytischer Mithilfe des grünen Pflanzenfarbstoffs Chlorophyll und gleichzeitiger Einwirkung des Sonnenlichts (Photosynthese). Bei diesem z. T. noch ungeklärten Prozess gibt die Pflanze molekularen Sauerstoff ab. Hierdurch entstehen auf der Erde jährlich etwa 100 Milliarden Tonnen Stärke. Die biochemisch erzeugte Stärke lagert sich in Form von kleinen, weißen Stärkekörnern in den Chloroplasten“ ab. Ein direkter Transport der Stär” ke innerhalb der Pflanze ist nicht möglich, da die Pflanzenmembranen für Kolloide undurchlässig sind. Die Pflanze hat aber die Fähigkeit, die durch Assimilation entstandene Stärke enzymatisch über Maltose zu Glucose abzubauen. Die Glucose kann in der Pflanze wandern und in bestimmten Depots, vor allem in den Wurzelknollen (Kartoffeln) und Samen (Getreidekörnern), wieder zur Reservestärke“ aufgebaut werden. ” Die Stärkekörner bestehen aus Amylopektin (etwa 80%) und Amylose (etwa 20%), die sich in ihren chemischen und physikalischen Eigenschaften unterscheiden. Das Verhältnis Amylopektin:Amylose ist abhängig von der Pflanzenart; z. B. gibt es Maissorten, die über 98% Amylopektin enthalten. Amylose besteht sterisch aus einer spiralförmigen Helix mit 6 D-Glucoseeinheiten pro Windung. Ihre relative Molekülmasse schwankt zwischen 17.000–225.000, das entspricht einer Sequenz von etwa 100–1400 Glucoseeinheiten innerhalb der Kette. OH HO HO O α OH OH O O α HO OH OH O O Amylose HO n OH α H OH 172 KAPITEL 4. ORGANISCHE CHEMIE In kaltem Wasser ist Stärke nicht löslich. Beim Erwärmen im Wasser setzt bei etwa 50˚C eine starke Quellung ein. Schließlich bilden die Stärkekörner einen durchscheinenden Stärkekleister. Die Verkleisterungstemperatur ist für die einzelnen Stärkearten verschieden, sie liegt etwa zwischen 60˚C und 80˚C. Der Stärkekleister ist eine kolloide Lösung der Stärke. Chemikalien: Stärke Durchführung: Etwa 5 g Stärke (evt. etwas mehr) werden mit etwa 20 ml kaltem Wasser zu einem dünnen Brei angerührt, der anschließend unter ständigem Umrühren in 100 ml siedendes Wasser zu gießen ist. Es entsteht eine viskose Lösung, der Stärkekleister. Aufgaben: Protokollieren Sie Ihr Vorgehen. Entsorgung: Ausguss Ende Versuch 4.7.12 4.7.13 Wahlpflichtversuch: Säurehydrolyse der Stärke (2/4) Stichworte: Haines Reagenz, Hydrolyse von Polysacchariden Allgemeines: Stärkekleister reagiert nicht mit Haines Reagenz, während nach der Säureeinwirkung ein ziegelroter Niederschlag ausfällt. Beim Sieden des Stärkekleisters mit einer verdünnten starken anorganischen Säure wird die Stärke hydrolytisch zu Glucose abgebaut. Die entstandene Glucose reduziert in basischer Lösung Haines Reagenz zu Kupfer(I)-oxid. Chemikalien: Stärkekleister, 3 M HCl, 3 M NaOH, Haines Reagens 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 173 Durchführung: In einem Becherglas wird ein Gemisch von etwa 10 ml Stärkekleister, 10 ml Wasser und 2 ml 3 M Salzsäure (HCl) etwa 10 min. zum Sieden erhitzt. Nach dem Abkühlen ist Natriumhydroxid-(NaOH)-Lösung bis zur basischen Reaktion (pH-Papier) zuzusetzen und 5 ml Haines Reagenz zuzufügen. Dann werden 5 ml nicht behandelter Stärkekleister mit 5 ml Haines Reagenz zum Sieden erhitzt. Aufgaben: Protokollieren Sie Ihr Vorgehen und stellen Sie die Reaktionsgleichungen auf. Entsorgung: Nach dem Neutralisieren Abguss Ende Versuch 4.7.13 4.7.14 Wahlpflichtversuch: Enzymatische Hydrolyse von Stärke (3/4) Stichworte: Iod-Stärke-Komplex, Iod-Kaliumiodid-Lösung, Enzyme, Abbau glycosidischer Bindungen Allgemeines: Der Mundspeichel enthält das Enzym Ptyalin, das Stärke über Dextrin zu Maltose hydrolytisch abbaut. Die Abnahme der Viskosität des Kleisters beruht auf der fortschreitenden hydrolytischen Spaltung der Stärke. HOH2C HO HO O HOH2C OH O HO O H OH OH Maltose (α-Form) Die in der Stärke enthaltene Amylose ist aus α-1,4-glycosidisch verknüpften Glucosepyranosemolekülen aufgebaut. Amylosemoleküle sind zu spiralförmigen Ketten angeordnet. Der Stärkenachweis mit Iod/Iodid beruht darauf, 174 KAPITEL 4. ORGANISCHE CHEMIE dass I5 − -Ionen von der Helix der Amylosemoleküle umhüllt werden und so eine blaue Einschlussverbindung bilden. Chemikalien: Stärkekleister, I2 /KI-Lösung, Haines Reagens Durchführung: In 8 kleinen Reagenzgläsern werden je 10 ml Wasser mit je 1 Tropfen Iod-Kaliumiodidlösung geschüttelt. Die Iodlösungen dürfen nur schwach gelbbraun gefärbt sein. Anschließend werden in einem großen Reagenzglas etwa 10 ml Stärkekleister mit reichlich Speichel vermischt und in einen Becher gestellt, der 40˚C warmes Wasser enthält. Die Temperatur soll keinesfalls höher sein. Sofort zu Beginn und dann jeweils nach 30 Sekunden werden etwa 0.5 ml des stärkehaltigen Gemisches in je ein Reagenzglas mit Iodlösung getropft. Nachdem keine Farbreaktion mit der Iodlösung mehr zu beobachten ist, wird das restliche Stoffgemisch mit 4 ml Haines Reagenz zum Sieden erhitzt. Aufgaben: Protokollieren Sie Ihr Vorgehen und Ihre Beobachtung. Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.7.14 4.7.15 Versuch: Untersuchung von Nahrungsmitteln auf Stärke (4/4) Stichworte: Stärkegehalt von Nahrungsmitteln, Iod-Stärke-Reaktion Allgemeines: Stärke kann man aus vielen Lebensmitteln durch Auskochen gewinnen. Durch die Behandlung mit siedendem Wasser bildet sich aus der Stärke Stärkekleister, der nach Abkühlung auf Zimmertemperatur mit Iodlösung die charakteristische blaue Färbung ergibt. Chemikalien: Iod-Kaliumiodid-Lösung, verschiedene Nahrungsmittel wie: Kartoffel, weiße Bohnen, Getreidekörner, Brot, Mehl, Haferflocken und Nudeln 4.7. ZUCKER/KOHLENHYDRATE (11.–12. WOCHE) 175 Durchführung: Die Nahrungsmittel sind zunächst zu zerkleinern. Von einer Kartoffel wird etwas abgeschabt, Getreidekörner sind in der Reibschale mit dem Pistill zu zerdrücken. Kleine Stoffmengen dieser Nahrungsmittel werden in Reagenzgläser gegeben und mit je etwa 5 ml Wasser zum Sieden erhitzt und dann auf Zimmertemperatur abgekühlt. Dann ist je ein Tropfen Iod-Kaliumiodid-Lösung zuzusetzen. Färbt sich die Lösung blau, so ist Stärke nachgewiesen. Aufgaben: Protokollieren Sie Ihr Vorgehen und Ihre Beobachtungen. Entsorgung: Ausguss Ende Versuch 4.7.15 176 4.8 KAPITEL 4. ORGANISCHE CHEMIE Aminosäuren und Proteine (13.–14. Woche) Stichworte: Zwitterionen, isoelektrischer Punkt Aminosäuren sind Carbonsäuren, die eine Aminogruppe im Molekül enthalten. In der Natur sind α-Aminosäuren am häufigsten, deren Aminogruppe sich am Kohlenstoffatom C-2 α-ständig zur Carboxylgruppe befindet. _ NH2 R β α O OH α-Aminosäure R β α O NH _ 2 OH β-Aminosäure Aminosäuren sind die Bausteine hochmolekularer Naturstoffe, der Proteine (Molekulargewicht >10.000 g/mol). Obwohl es mehr als fünfhundert unterschiedliche, natürliche Aminosäuren gibt, bestehen die Proteine aller Organismen zum größten Teil aus nur zwanzig verschiedenen Aminosäuren. Diese werden auch als proteinogene Aminosäuren bezeichnet. Acht dieser Aminosäuren, die essentiellen Aminosäuren, muss der Mensch mit seiner Nahrung zu sich nehmen, da sie nicht von seinem Stoffwechsel synthetisiert werden können. In allen häufig vorkommenden Aminosäuren besitzt das Stereozentrum an C-2 die S-Konfiguration. Die einfachste α-Aminosäure, Glycin, ist achiral. Durch ihre Carboxylund Aminogruppe sind Aminosäuren zugleich sauer und basisch. Sie liegen als zwitterionische Ammoniumcarboxylate (Zwitter-Ion) vor. In wässriger Lösung bilden sich je nach pH-Wert unterschiedliche Säure-Base-Gleichgewichte unter Beteiligung der funktionellen Gruppen aus. Der Ladungszustand von Aminosäuren wird durch den pH-Wert der Lösung beeinflusst. Je nach pHWert liegen Aminosäuren als Kation, Zwitterion oder Anion vor. Am Isoelektrischen Punkt (IP) liegt die Aminosäure ganz überwiegend als Zwitterion vor und ist nach aussen hin neutral. 4.8. AMINOSÄUREN UND PROTEINE (13.–14. WOCHE) _ NH2 R + +H+ O + NH3 R O -H+ |O _| |O _| pKs2 - +H+ R NH3 O -H+ - OH Kation In saurem Milieu pKs1 IP Zwitter-Ion Anion In basichem Milieu 177 Der IP jeder Aminosäure liegt bei einem charakteristischen Wert (bei neutralen Aminosäuren in der Nähe des Neutralpunktes). Für saure Aminosäuren liegt der IP bei niedrigerem, für basische Aminosäuren bei höherem pH-Wert. Aminosäuren können entsprechend ihrer funktionellen Gruppen wie Amine oder wie Carbonsäuren reagieren. Die Carboxylgruppe von α-Aminosäuren lässt sich auf dem üblichen Weg mit Alkoholen verestern. Über die Ester sind auch Amide zugänglich. Eine wichtige Farbnachweisreaktion für Aminosäuren ist die mit Ninhydrin. Mit Ausnahme von Prolin und Hydroxyprolin reagieren alle natürlichen α-Aminosäuren mit Ninhydrin unter Bildung eines intensiv violett gefärbten Anions. Nur das Stickstoffatom der Aminosäure findet sich im Reaktionsprodukt, das nach einem komplizierten Mechanismus entsteht. _ NH2 O OH + OH O _ | O| O R |O _| - O _ N O -CO2 -3H2O + R H O O Aminosäuren können sich durch Bildung von Amidbindungen zwischen einer α-Aminogruppe und einer Carboxylgruppe unter Wasseraustritt verbinden. Auf diese Weise können sehr viele Aminosäuren zu Proteinen polymerisieren. Eine zwischen zwei Aminosäuren geknüpfte Amidbindung nennt man auch Peptidbindung (Lehrbücher: Mesomerie der Peptidbindung, teilweiser Doppelbindungscharakter der C–N-Bindung, keine freie Drehbarkeit). Eine Verbindung aus zwei amidartig verknüpften Aminosäuren nennt man Dipeptid. 178 KAPITEL 4. ORGANISCHE CHEMIE R' O _ N R N-Terminus + O |O _| - NH3 H C-Terminus Peptidbindung Man schreibt eine Peptidkette im allgemeinen so, dass die freie Ammoniumgruppe links, die freie Carboxylatgruppe rechts steht. Die entsprechenden Aminosäuren nennt man je nachdem N-terminale beziehungsweise Cterminale Aminosäure. Cystein Thiole und Disulfide können durch Oxidation bzw. Reduktion ineinander überführt werden. Auf diese Weise wird auch die Aminosäure Cystein in Cystin umgewandelt. Die reversible Bildung der Disulfidbrücke, die auch unter physiologischen Bedingungen erfolgt, ist wichtig für die Raumstrukturbildung von Proteinen und findet unter anderem im Friseurhandwerk Anwendung (Dauerwellen). O O - _ SH _ O Ox. Red. - O + NH3 Cystein 4.8.1 + _ _ S _ S _ NH2 O - O + NH3 Cystin Versuch: Titration von Glycin Stichworte: amphoteres Verhalten, Pufferbereiche, pKs -Werte von Aminosäuren, IP 4.8. AMINOSÄUREN UND PROTEINE (13.–14. WOCHE) 179 Allgemeines: Da Aminosäuren formal eine saure Carboxyl-(-COOH)-Gruppe und eine basische Amin-(-NH2 )-Gruppe besitzen, existieren für jede Aminosäure auch mehrere pK-Werte. O H2N OH _ H2N O + H3N O |O _| - OH Anion Kation O + H3N |O _| - Glycin (liegt so nie vor!) Zwitterion Bemerkung: In der Realität liegt eine Aminosäure nie in der oben gezeigten Weise vor! Aminosäuren liegen entweder als Kation (Ammoniumform), Anion (Carboxylatform) oder als Zwitterion vor! Titriert man Aminosäuren (z. B. Glycin) mit starken Säuren bzw. Basen, so zeigt sich das amphotere Verhalten der Aminosäuren gegenüber Säuren und Basen. Im Sauren—Titration mit HCl—(hier fungiert die Aminosäure in ihrer Ammoniumform als Base) zeigt sich das gleiche Bild, wie bei einer Titration einer schwachen Base mit einer starken Säure. Man erkennt einen Pufferbereich beim ersten pKs -Wert der Aminosäure. Am isoelektrischen Punkt (IP) liegt die Aminosäure als Zwitterion vor. Im Basischen—Titration mit NaOH—(hier fungiert die Aminosäure als Carboxylat-Anion als Säure) bietet sich das gleiche Bild wie bei einer Titration einer schwachen Säure mit einer starken Base. Man erkennt auch hier beim zweiten pKs -Wert einen Pufferbereich. Der pH-Sprung (Wendepunkt) am jeweiligen Äquivalenzpunkt der Kurven ist sehr schwach ausgeprägt (Pfeile). 180 KAPITEL 4. ORGANISCHE CHEMIE In der Praxis kann man daher Aminosäuren mit Säure oder Base und den üblichen Indikatoren nicht titrieren. Ein Ausweg ist die Titration in Gegenwart von Formaldehyd (wässrige Lösung: Formalin), mit dem die Aminogruppe zur Methylol- oder Methylenverbindung kondensiert: R O H O + H3N R H Aminosäure O O H H - + N OH Methylol-Verbindung O H R H2C Formaldehyd - H2O O N OH Methylen-Verbindung 4.8. AMINOSÄUREN UND PROTEINE (13.–14. WOCHE) 181 Bei dieser Aminosäuretitration nach Sörensen“ ist die Basizität des Stick” stoffs durch die Stubstitution stark herabgesetzt und man titriert die Verbindung wie eine normale, undissoziierte Carbonsäure mit gut erkennbarem Äquivalenzpunkt. Chemikalien: kristallines Glycin, Phenolpthalein (1% in Ethanol), 0.5 M NaOH, Formalin (Formaldehyd in Wasser), Na2 CO3 Durchführung: Man löst im Erlenmeyerkolben 0.02 mol kristallines Glycin in 100 ml Wasser, versetzt mit 1 ml Phenolpthalein und beginnt, mit 0.5 M Natriumhydroxid-(NaOH)-Lösung zu titrieren. Vergleichen Sie das verbrauchte Volumen NaOH-Lösung bei Indikatorumschlag mit der theoretisch benötigten Menge. Nun gibt man 10 ml Formalin zu (das zur Neutralisation von Ameisensäure als Verunreinigung zuvor mit festem Na2 CO3 neutralisiert wurde), beobachtet den Indikator und titriert erneut bis zum Umschlag. Aufgaben: Protokollieren Sie Ihr Vorgehen und Ergebnisse. Stellen Sie die Reaktionsgleichungen auf. Ist der letzte Umschlag der richtige Äquivalenzpunkt? Welchen Effekt beobachten Sie bei Zugabe des Formaldehyds und welche Ursache hat er? Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.8.1 182 4.8.2 KAPITEL 4. ORGANISCHE CHEMIE Versuch: Isoelektrischer Punkt und Löslichkeit von Casein Stichworte: Globuline, Strukturproteine Allgemeines: Eine der—auch biochemisch—wichtigen Eigenschaften von Proteinen ist ihre Löslichkeit in Wasser. Bestimmend dafür sind Art und räumliche Verteilung der Aminosäure-Seitenreste R, deren hydrophober oder hydrophiler, saurer oder basischer Charakter. Aus Gründen der Hydrationsenthalpie und der Gesamtentropie (Molekül plus Lösungsmittel) haben globuläre Proteinmoleküle normalerweise die hydrophoben Seitenreste im Inneren und die polaren nach aussen hin angeordnet: O + HO NH3 - O O + NH3 OH CH3 + H3N OH CH3 + H3N CH3 HO CH3 HO - HO O O O Isoelektrischer Punkt Nettoladung = 0 niedriger pH, positiv geladen - O O NH2 OH CH3 H2N CH3 HO - O O hoher pH, negativ geladen Der Ionisierungszustand der sauren und basischen Reste wird vom pHWert der Lösung bestimmt. Am Isoelektrischen Punkt (IP) des Proteins be- 4.8. AMINOSÄUREN UND PROTEINE (13.–14. WOCHE) 183 sitzt das Molekül die Nettoladung Null. Hier kann neben Solvation der Reste ein anderer Effekt dominieren: Da die Moleküle sich nicht mehr—wie bei vom IP abweichenden pHWerten, bei denen sie alle gleichsinnig geladen sind—voneinander abstoßen, neigen sie zur Aggregation unter intermolekularer Wechselwirkung zwischen positiver und negativer Ladung. Am IP besitzen daher die meisten Proteine ein Löslichkeitsminimum, sie fallen aus oder flocken aus“ (in der Regel in ” amorpher, nicht kristalliner Form). Da in den IP eines Proteins die Ladungsbeiträge aller im Molekül vorhandenen ionisierten Gruppen (mit unterschiedlichen pK-Werten) eingehen, ist er für ein Protein im Gegensatz zu Aminosäuren nicht zu berechnen, sondern muss experimentell bestimmt werden. Kennt man ihn, so kann er zur Isolierung von Proteinen durch Ausfällen benutzt werden. Chemikalien: Casein, 1 M NaAc Durchführung: Bereiten der Casein-Lösung: 0.4 g Casein werden in 10 ml 1 M Natriumacetatlösung unter schwachem Erwärmen gelöst und mit Wasser auf 100 ml im Messkolben aufgefüllt. Zur Herstellung von Essigsäure-Acetatpuffer werden folgende Essigsäurelösungen in Reagenzgläsern angesetzt: Reagenzglas 0.1 M Essigsäure (ml) Wasser (ml) Casein-Lösung (ml) pH-Wert Trübung 1 0.1 8.9 1 2 0.3 8.7 1 3 0.6 8.4 1 4 1 8 1 5 2 7 1 6 4 5 1 7 6 3 1 8 10 1 9 15 1 Trübung: 1=keine, 2=erkennbare, 3= starke, 4=sehr starke. Die abweichenden Volumina in Reagenzglas 8 und 9 seien vernachlässigt. Aufgaben: Protokollieren Sie Ihr Vorgehen und Ergebnisse. Berechnen Sie den pH-Wert der Pufferlösungen und bestimmen Sie den isoelektrischen Punkt durch Vergleich der Trübung in den einzelnen Puffern. Casein ist in reinem Wasser zunächst unlöslich, bei schwach alkalischem pH löslich. Welche Art von Seitenresten R erwarten Sie daher in größerer Zahl im Molekül? Entsorgung: Abfälle neutralisieren, danach organische Lösungsmittelabfälle Ende Versuch 4.8.2 184 4.8.3 KAPITEL 4. ORGANISCHE CHEMIE Versuch: Gewinnung von Casein aus Milch (Eiweißgerinnung durch Säuren) Boxweise Stichworte: Denaturierung von Proteinen (reversibel/irreversibel), Koagulation Allgemeines: Casein ist zu etwa 3% in der Kuhmilch als Calciumsalz enthalten und fällt am isoelektrischen Punkt (etwa pH = 4.7) durch Koagulation aus. Chemikalien: Magermilch, konz. Essigsäure (Eisessig) Durchführung: Etwa 500 ml Magermilch sind mit 3–5 ml konz. Essigsäure anzusäuern und mit dem Glasstab gut umzurühren. Der entstehende Niederschlag wird mit dem Büchnertrichter abfiltriert, gründlich mit Wasser gewaschen. Zum Trocknen können die Produkte des ganzen Saales vom Assistenten gesammelt und in einem sog. Exikkator unter Vakuum getrocknet werden. Das gewonnene Casein kann in einem der folgenden Versuche verwendet werden. Aufgaben: Protokollieren Sie Ihr Vorgehen und stellen Sie die Ausbeute an Casein aus 500 ml Milch durch Auswiegen der Trockenmasse fest. Wieviel Casein hätten Sie erhalten müssen, wenn die Milch 3% Casein enthält? Entsorgung: Ausguss Ende Versuch 4.8.3 4.8. AMINOSÄUREN UND PROTEINE (13.–14. WOCHE) 4.8.4 185 Versuch: Aussalzen von Casein Stichworte: Denaturierung von Proteinen (reversibel/irreversibel), Koagulation Allgemeines: Die isoelektrische Ausfällung gelingt nicht überall, oder sie kann bei pH-Werten beiderseits pH 6–8 empfindliche Proteine denaturieren. Dagegen ist es fast immer möglich, Proteine durch Ammoniumsulfat auszu” salzen“. In sehr hohen Konzentrationen (bei Sättigung 760 g (NH4 )2 SO4 pro Liter Wasser; die Lösung ist ca. 4 M) entziehen die Salzionen dem Protein die Hydrathülle völlig, das Protein fällt aus. Chemikalien: Casein, 0.1 M Na-Acetat, (NH4 )2 SO4 Durchführung: 0.4 g Casein werden in 10 ml 0.1 M Na-Acetat unter schwachem Erwärmen gelöst. Anschließend wird die Lösung zentrifugiert. 3 ml der überstehenden, klaren Caseinlösung werden mit 7 ml einer gesättigten (NH4 )2 SO4 -Lösung versetzt. Die Lösung trübt sich durch ausgefallenes Casein. Das gefällte Casein wird abzentrifugiert. Prüfen Sie, ob sich der Niederschlag wieder in Wasser auflöst. Aufgaben: Protokollieren Sie Ihr Vorgehen und Ihre Beobachtungen. Ist der Aussalzungsprozess reversibel? Warum ist er oft dem Ausfällen durch Ansäuern vorzuziehen? Entsorgung: Ausguss Ende Versuch 4.8.4 186 4.8.5 KAPITEL 4. ORGANISCHE CHEMIE Versuch: Hippursäure Stichworte: Peptidbindung, primär-, sekundär-, teriär-Struktur von Proteinen Allgemeines: Soll in der Aminogruppe einer Aminosäure ein H-Atom durch eine Acylgruppe ersetzt werden, wie das z. B. zur Knüpfung einer Peptidbindung nötig ist, so tritt eine Reaktion nur ein, wenn: • die Aminogruppe nicht protoniert ist (-NH2 , nicht -NH3 + ) und • ein reaktionsfähiges Säurederivat (z. B. Chlorid) verwendet wird. O + H3N O Benzoesäure OH Zwitter-Ion O- (Kein N-Nucleophil) (Würde Aminogruppen protonieren) O O H2N Benzoylchlorid Cl Glycin-Anion O- -HCl O HO O N H Hippursäure Hier wird Hippursäure (Benzoylglycin) als Modell für die Knüpfung einer Peptidbindung hergestellt. Bemerkung: Im tierischen Organismus können nicht-physiologische aromatische Verbindungen wie Benzoesäure mit Aminosäuren konjugiert“ d. h. ver” bunden und dann ausgeschieden werden, so z. B. die erstmals beim Pferd beobachtete Hippursäure. 4.8. AMINOSÄUREN UND PROTEINE (13.–14. WOCHE) 187 Chemikalien: Glycin, 2 M NaOH, Benzoylchlorid, Diethylether, 0.5 M NaOH, Phenolphthalein Durchführung: a.) Herstellung von Hippursäure: Man löst in einem 100 ml Erlenmeyerkolben 2 g Glycin in 10 ml Wasser, setzt einige Tropfen 2 M NaOH zu und tropft unter Rühren nach und nach 8 g Benzoylchlorid zu. Die Lösung muss durch zutropfen von NaOH dauernd alkalisch gehalten werden (insgesamt etwa 30 ml 2 M NaOH) und wird am Schluss solange geschüttelt, bis der Geruch nach Benzoylchlorid verschwunden ist. Nach Ansäurern mit konzentrierter Salzsäure fällt ein Gemisch aus Benzoesäure und Hippursäure aus. Das Produkt lässt man 15 Minuten stehen und saugt es mit einer Nutsche ab. Es wird zwischen Filterpapier getrocknet und in 10 ml Ether (Scheidetrichter) zur Entfernung der Benzoesäure geschüttelt. Danach wird wieder abgesaugt. Die rote Hippursäure kristallisiert man aus heißem Wasser um. Überprüfen Sie die Reinheit ihres Produktes anhand des Schmelzpunktes (Schmp. 187˚C). b.) Titration von Hippursäure: Man löst 0.01 mol Hippursäure (C6 H5 – CONHCH2 COOH) unter Erwärmen in Wasser und titriert mit 0.5 M NaOH gegen Phenolphthalein bis zum Umschlagspunkt. Aufgaben: Protokollieren Sie Ihr Vorgehen und Ihre Beobachtungen. Stellen Sie die Reaktionsgleichung auf. a.) Warum führt die Kombination R–COOH + + NH3 –CH2 –COOH nicht zur Reaktion? b.) Vergleichen Sie den NaOH-Verbrauch mit der dem theoretischen Verbrauch und der Titration des Glycins. Was können Sie aus diesem Ergebnis über die Basizität des Stickstoffs in der N-Acylverbindung aussagen? Ist es für die Eigenschaften von Peptiden nötig, die -CO– NH-Peptidbindungen als basische Gruppe zu berücksichtigen? Entsorgung: Restliches Benzoylchlorid vorsichtig mit Eiswasser hydrolysieren. Organische Lösungsmittelabfälle. Ende Versuch 4.8.5 188 4.8.6 KAPITEL 4. ORGANISCHE CHEMIE Versuch: Einwirkung von Natriumhydroxidlösung auf Proteine Stichworte: Spaltung der Peptidbindung Allgemeines: Proteine (Eiweiße) sind gegenüber Basen sehr empfindlich. Sie werden in basischem, wie auch in stark saurem Milieu hydrolytisch gespalten. Dabei wird für jede aufgelöste Peptidbindung ein H2 O-Molekül verbraucht. Die saure Hydrolyse wird oft zu analytischen Zwecken angewandt. Die alkalische Hydrolyse eignet sich dafür nicht, da viele Aminosäuren sich dabei verändern. Die menschliche und tierische Haut und einzelne Textilien sind vor Einwirkung von Lösungen mit basischer Reaktion zu schützen. Da Schafwolle aus Eiweiß (Keratinen) besteht, darf sie nicht mit basisch reagierenden Waschmitteln gereinigt werden. Chemikalien: 3 M NaOH-Lösung, Schafwolle, Menschenhaar, Casein Durchführung: In Reagenzgläsern werden kleine Proben z. B. Schafwolle, Menschenhaar und Casein mit je 5–7 ml 3 M Natriumhydroxid-(NaOH)Lösung unter dauerndem Schütteln im Wasserbad zum Sieden erhitzt. Aufgaben: Protokollieren Sie Ihr Vorgehen und Ihre Beobachtungen. Entsorgung: Mit Eisessig neutralisierte Lösung kann in den Ausguss gegeben werden. Ende Versuch 4.8.6 4.8. AMINOSÄUREN UND PROTEINE (13.–14. WOCHE) 4.8.7 189 Versuch: Proteinbestimmung durch Biuretreaktion Stichworte: Chelatkomplexe, Komplexbindungen Allgemeines: Kupfer-Ionen geben in alkalischer Lösung mit allen Substanzen, die mindestens zwei Peptidbindungen enthalten, violett gefärbte Komplexe. R O O N N/ K+,Na+ Cu2+ N O /N O R Die Reaktion ist nach der analogen Komplexbildung mit dem dimeren Harnstoff Biuret“ NH2 –CO–NH–CO–NH2 benannt. ” Chemikalien: Proteinlösung (auch Milch), 3 M NaOH-Lösung, 0.5 M Kupfer(II)sulfatlösung Durchführung: Etwa 3 ml Proteinlösung werden, in einem Reagenzglas, mit 3 ml Natriumhydroxid-(NaOH)-Lösung und 3–4 Tropfen Kupfer(II)Sulfatlösung kräftig geschüttelt. Aufgaben: Protokollieren Sie Ihr Vorgehen und Ihre Beobachtungen. Entsorgung: organische Lösungsmittelabfälle Ende Versuch 4.8.7 190 KAPITEL 4. ORGANISCHE CHEMIE 4.8.8 Analyse 5: Trennung und Identifizierung eines Aminosäuregemisches Stichworte: Dünnschichtchromatographie (DC), Chromatographie, Kapillareffekt, stationäre und mobile Phase Allgemeines: Grundlage: Nernstscher Verteilungskoeffizient. Chromatographische Verfahren beruhen alle auf dem gleichen Prinzip, nämlich der unterschiedlichen Verteilung der zu trennenden Stoffe zwischen einer stationären und einer beweglichen (mobilen) Phase. Ein chromatographisches System besteht daher aus zwei nicht miteinander mischbaren Phasen, von denen sich die eine an der anderen vorbeibewegt. Das Trennprinzip besteht darin, dass unterschiedliche Stoffe mit der stationären und der mobilen Phase unterschiedlich starke Wechselwirkungen eingehen. Bei der Dünnschichtchromatographie (DC) wird die stationäre Phase als dünne Schicht auf einen geeigneten Träger, z. B. eine Glasplatte, Polyesteroder Aluminiumfolie, aufgebracht. Auf dieser Schicht wird das Substanzgemisch durch Elution mit einem Lauf- oder Fließmittel (in der DC häufig die Bezeichnung für die mobile Phase) getrennt. Das Laufmittel transportiert die einzelnen Komponenten des Substanzgemisches je nach Löslichkeit und/oder Adsorptionsverhalten unterschiedlich weit. Die Laufstrecke der Substanz, angegeben als Rf -Wert (Retentions-Faktor), wird dann zu ihrer Identifizierung benutzt. Der Rf -Wert ist das Verhältnis des zurückgelegten Weges der Substanz und des Laufmittels (Lösemittel). Rf = Weg der Substanz Weg des Laufmittels Der Rf -Wert ist immer kleiner als 1 und ist für jede Substanz unter gleichen Bedingungen eine reproduzierbare, konstante Kenngröße. 4.8. AMINOSÄUREN UND PROTEINE (13.–14. WOCHE) 191 Laufmittelfront Weg des Laufmittels Weg der Substanz + Substanz + VergleichsSubstanz Chemikalien: Glycin, Isoleucin, Lysin, Fließmittel (n-Butanol/Eisessig/Wasser 4:1:1) Durchführung: Befüllen sie eine DC-Kammer (oder ein 250 ml Becherglas, das mit einer Petrischale oder Aluminiumfolie abgedeckt werden kann) etwa 0.5–1 cm hoch mit Fließmittel, stellen Sie ein Filterpapier hinein und verschließen sie die Kammer. Das Filterpapier unterstützt die schnelle Einstellung des Verdampfungsgleichgewichts in der DC-Kammer. Auf einer DC-Karte wird 1.5–2 cm vom unteren Rand entfernt ganz vorsichtig mit Bleistift eine Startlinie gezogen, auf der im gleichen Abstand 4 Startpunkte markiert werden. Mit Hilfe der Kapillaren werden nun die Lösungen der drei bekannten, ausstehenden Aminosäuren und die unbekannte Analysenlösung auf die markierten Punkte aufgetragen. Dazu muss die Kapillare mit der Lösung ruhig und senkrecht auf der DC-Karte aufgesetzt werden, die Lösung darf nicht auf die DC-Karte aufgetropft werden. Die aufgetragenen Flecke sollten einen Durchmesser von nicht mehr als 2–3 mm haben. Danach wartet man, bis die Lösung auf der Karte getrocknet ist. Man stellt die DC-Karte in die Kammer und verschließt diese sofort wieder. Dabei muss die DC-Karte etwa 0.5 cm in das Fließmittel eintauchen, auf keinen Fall aber dürfen die aufgetragenen Flecken in das Fließmittel eintauchen. Durch die Wirkung der Kapillarkräfte steigt das Fließmittel auf der DC-Karte nach oben. Kurz bevor die Fließmittelfront das obere Ende der DC-Karte erreicht (ca. 1.5 Stunden), nimmt man die DC-Karte aus der 192 KAPITEL 4. ORGANISCHE CHEMIE Kammer, markiert die Fließmittelfront mit einem Bleistift und wartet, bis das Fließmittel verdampft ist. Danach wird die DC-Karte mit Ninhydrinlösung besprüht und kurz (!) mit der Metallseite auf eine erhitzte Heizplatte gelegt. An den Stellen, bis zu denen die Aminosäuren gewandert sind, bilden sich violette Flecken. Aufgaben: Protokollieren Sie Ihr Vorgehen und Ihre Beobachtungen. Bestimmen Sie die Rf -Werte der 3 Aminosäuren. Welche Aminosäuren haben in der Analysenlösung vorgelegen? Entsorgung: Feststoffabfälle, organische Lösungsmittel Ende Versuch 4.8.8