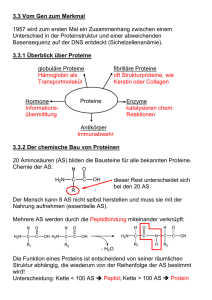
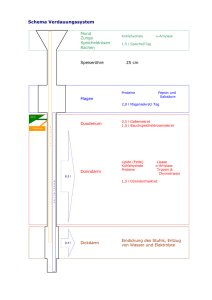
ZIP - FreiDok plus
Werbung

Aus dem Department of Molecular and Cell Biology University of California, Berkeley und dem Institut für Pathologie Albert-Ludwigs-Universität Freiburg i. Br. Charakterisierung des deubiquitinierenden Enzyms USP4 Inaugural-Dissertation zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg i. Br. Vorgelegt 2011 von Jakob Neubauer geboren in Berlin 1 Dekan: Prof. Dr. med. Dr. h.c. mult. Hubert Blum Erstgutachter: Prof. Dr. med. Martin Werner Zweitgutachter: Prof. Dr. med. Ralph Wäsch Jahr der Promotion: 2011 2 Inhaltsverzeichnis: 1. Einleitung 8 1.1. Ubiquitin 8 1.2. Ubiquitinierung 8 1.3. Deubiquitinierung 11 1.4. Spliceosom 15 1.5. Zellzyklus 16 1.6. Zielsetzung der Arbeit 20 2. Materialien 22 2.1. Geräte 22 2.2. Software 23 2.3. Chemikalien 23 2.4. Proteine 24 2.4.1. Proteine 24 2.4.2. Primärantikörper 25 2.4.3. Sekundärantikörper 25 2.5. DNA 25 2.6. Zellen 25 2.6.1. Bakterienstämme 25 2.6.2. Humane Zelllinien 25 2.6.3. Insektenzelllinien 25 2.7. Kits 25 2.8. Puffer 26 2.9. Verbrauchsmaterialien 27 3. Methoden 28 3.1. Molekularbiologische Methoden 28 3.1.1. Herstellung chemisch kompetenter E. coli 28 3.1.2. Transformation chemisch kompetenter E. coli 28 3.1.3. Plasmidpräparation 29 3.1.4. DNA Konzentrationsmessung 29 3 3.1.5. Polymerase Kettenreaktion 30 3.1.6. Mutations-PCR 31 3.1.7. Agarosegelelektrophorese 31 3.1.8. Sequenzierung 32 3.1.9. Klonieren 32 3.2. Proteinbiochemische Methoden 33 3.2.1. Natriumdodecylsulfat-PolyacrylamidGelelektrophorese (SDS-PAGE) 33 3.2.2. Coomassie-Färbung 34 3.2.3. Silberfärbung 35 3.2.4. Western Blot 35 3.2.5. Autoradiographie 36 3.2.6. Aufreinigung von MBP-markierten Proteinen aus Bakterien 36 3.2.7. Aufreinigung von His6-markierten Proteinen aus Bakterien 37 3.2.8. Expression und Aufreinigung von Proteinen aus Insektenzellen 38 3.2.9. In vitro Transkription und Translation 39 3.2.10. Immunpräzipitation 39 3.2.11. Pulldown 39 3.2.12. Herstellung von Zelllysaten 40 3.2.13. Deubiquitinierungsassay 40 3.2.14. Ubiquitinierungsassay 41 3.2.15. Degradationsassay 41 3.2.16. Massenspektrometrie 41 3.3. Zellbiologische Methoden 42 3.3.1. Immunfluoreszenz 42 3.3.2. Proteinextraktion 42 3.3.3. Zellkultur 43 4. Ergebnisse 44 4.1. Bindungspartner von USP4 44 4.2. Katalytische Aktivität von USP4 52 4.3. Lokalisation von USP4 58 4 4.4. Degradation von USP4 62 5. Diskussion 64 5.1. Bindungspartner von USP4 64 5.2. Katalytische Aktivität von USP4 66 5.3. Funktionen von USP4 im Zellzyklus 67 5.4. Lokalisation von USP4 70 6. Zusammenfassung 72 7. Literaturverzeichnis 73 8. Danksagung 78 5 Abkürzungen APC/C anaphase promoting complex/cyclosome APS Ammoniumpersulfat ATM Ataxia teleangiectasia mutated protein ATR ATM und Rad3 related BubR1 Budding uninhibited by benzimidazoles 1 Cdh Cadherin Cdc20 Cell-division cycle protein Cdk Cyclin dependent kinase Chk Check protein CYLD Cylindromatosis protein ddH2O Doppelt destilliertes H20 DMEM Dulbecco's Modified Eagle Medium DMSO Dimethylsulfoxid DPBS Dulbecco's phosphate buffered saline DUB Deubiquitinierendes Enzym EDTA Ethylenglykol-bis(aminoethylether)-N,N,N’N’-Tetraessigsäure Emi1 Early mitotic inhibitor 1 FANCD2 Fanconi anemia, complementation group D2 FBS fetales Rinderserum HA Hämagglutinin Anker HeLa Zervixcarzinomzelllinie der Patientin Henrietta Lacks HEPES N-(2-Hydroxyethyl)piperazin- N'-(2-ethansulfonsäure) His6 Histidin (6 aufeinanderfolgende Histidine) IP Immunpräzipitation IPTG Isopropyl-ß-D-thiogalactopyranosid JAMM Jab1/MPN Domäne des Metalloenzymmotifs kbp Kilo Basen Paare Mad2 Mitotic arrest deficient 2 MBP Maltose bindendes Protein MJD Machado-Joseph Disease mM milli Mol mRNA Messenger RNA 6 NEM N-Ethylmaleinimid NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells OTU Ovarian tumor Domäne PMSF Phenylmethylsulfonylfluorid PRP3 Pre mRNA processing factor 3 PRP4 Pre mRNA processing factor 4 PRP4B Pre mRNA processing factor 4 homolog B/ Serin-Threonin Kinase Rb Retinoblastom-Protein RT Raumtemperatur SAP Shrimp alkaline Phosphatase SART3 Squamous cell carcinoma antigen recognized by T-cells 3 SDS Natriumdodecylsulfat siRNA small interfering Ribonukleinsäure snRNA small nuclear Ribonukleinsäure snRNP small nuclear Ribonukleoprotein TEMED N,N,N',N'-Tetramethylethylenediamine TRIM21 Tripartite motiv containing 21 TRIS 2-Amino-2-(hydroxymethyl)-propan-1,3-diol UAF1 Usp1-associated factor 1 UBA Ubiquitin associated UbcH10 E2 Enzym UCH Ubiquitin carboxyl-terminal hydrolase UIM Ubiqutin interacting motif USP4 Ubiquitin specific protease 4 WB Western Blot Wnt Wingless integration site 1 7 1. Einleitung Die Erforschung molekularbiologischer Mechanismen wie der Mitose oder der Proteinbiosynthese hat einen entscheidenden Beitrag zum Verständnis der Pathogenese vieler Erkrankungen geleistet. Dadurch konnte unter anderem in der Onkologie die Therapie von Erkrankungen weiterentwickelt werden. Auch in Bezug auf das Protein Ubiquitin wurden pathogenetisch relevante Daten gesammelt (Hershko & Ciechanover, 1998; Nakayama & Nakayama, 2006). So ist Ubiquitin zum Beispiel ein wichtiger Regulator im Bereich der DNA-Reparatur, die eine pathogenetische Bedeutung für die Fanconi Anämie und familliären Brustkrebs hat (Kennedy & D'Andrea, 2005). 1.1. Ubiquitin Ubiquitin ist ein Protein mit 76 Aminosäuren, das ubiquitär in allen eukaryoten Zellen vorkommt. Nach seiner Entdeckung 1975 erkannte man in den frühen 1980er Jahren, dass die kovalente Verbindung von Ubiquitin mit anderen Proteinen ein Proteolysesignal darstellt. Seither konnte in vielen Gebieten der Molekularbiologie eine Bedeutung von Ubiquitin als Regulationssignal nachgewiesen werden. 1.2. Ubiquitinierung Die kovalente Konjugation von Ubiquitin an Zielproteine wird als Ubiquitinierung bezeichnet. Diese posttranslationelle Modifikation stellt für die Zelle ein Signal dar, das bei Proteindegradation, Zellzyklusregulation, DNA-Reparatur, Endozytose oder Signalkaskaden eine wichtige Rolle spielt. Ubiquitinierung kann die Stabilität, Funktion und Lokalisation eines anderen Proteins verändern. Der Vorgang der Ubiquitinierung beinhaltet mehrere nacheinander geschaltete enzymatische Reaktionen (Abb 1-1 A). Mindestens drei Enzymklassen sind an der Katalyse beteiligt: Ein Ubiquitin aktivierendes Enzym E1, ein Ubiquitin konjugierendes Enzym E2 und eine Ubiquitinligase E3 (Hershko & Ciechanover, 1998; 8 Hicke, 2001). Fakultativ können auch Kettenelongationsfaktoren (E4) an der Synthese beteiligt sein. Das humane Genom kodiert für zwei Ubiquitin-spezifische E1-, 38 E2- (Ye & Rape, 2009) und etwa 600 bis1000 E3-Enzyme (Jin et al, 2008). Zuerst kommt es zu einer Aktivierung des freien Ubiquitins durch das E1-Enzym. In einer ATP-abhängigen Reaktion wird ein Thioester zwischen dem Cterminalen Glycin des Ubiquitins und dem aktiven Cystein des E1 gebildet. Dadurch wird die Aminosäure für einen nukleophilen Angriff präpariert. Im zweiten Schritt wird das derart aktivierte Ubiquitin auf das Cystein des aktiven Zentrums eines E2-Enzyms übertragen. E1 ist in den Zellen geringer konzentriert als alle E2-Enzyme zusammen. Da die Aktivierungsreaktion im Vergleich zur Ubiquitinierung der Substrate schneller abläuft, ist die Verfügbarkeit von aktiviertem Ubiquitin trotzdem gesichert. Das für beladenes E1 hochaffine E2Enzym kann nach Aufnahme von Ubiquitin mit mehreren unterschiedlichen E3Ligasen in einer Kaskade reagieren. Mit Hilfe der E3-Ligase kommt es dann zur kovalenten Übertragung des Ubiquitins auf das Substratprotein (Pickart, 2001). Der Mechanismus der Übertragung von Ubiquitin unterscheidet sich bei den einzelnen E3-Klassen (Hochstrasser, 2006). Die E3-Ligasen mit einer RingFinger-Domäne stellen lediglich den Kontakt zwischen E2 und dem Substrat her (Vander Kooi et al, 2006), was den Wechsel des Ubiquitins vom E2 direkt auf das Zielprotein zur Folge hat. Bei Ligasen mit einer HECT-Domäne hingegen wird das Ubiquitin auf ein in der Sequenz konserviertes Cystein übertragen und von dort aus weitergegeben. In den meisten Fällen wird das Ubiquitin mit seinem Cterminalen Glycin auf die ε-Aminogruppe eines Lysins im Substrat übertragen (Kerscher et al, 2006). 9 Abbildung 1-1: Schematische Darstellung der Ubiquitinierung: A. Ubiquitin (Ubi) wird unter Verbrauch von ATP durch das E1-Enzym aktiviert und auf ein E2-Enzym übertragen. Mit Hilfe von Ring-E3-Ligasen erfolgt dann die Übertragung des Ubiqutins auf das Substrat (S). E2Enzyme können das Ubi auch auf HECT-E3-Ligasen übertragen, die dann die Übertragung auf das Substrat vornehmen. E3-Ligasen katalysieren die Bindung des Substrates über bestimmte Erkennungssignale, sogenannte Degrons, und vermitteln so die Substratspezifität. Degrons stellen Aminosäureabfolgen in den Polypeptidsequenzen der Substrate dar. Beispiele dafür sind D-Box, KEN-Box und TEK-Box, die ein Degron für die E3-Ligase APC/C darstellen (Jin et al, 2008; Pfleger & Kirschner, 2000). Degrons können durch Phosphorylierung entstehen oder maskiert werden (Hershko & Ciechanover, 1998). Bei der Ubiquitinierung wird das Substrat entweder mit einem oder mehreren Ubiquitinmoleküle verbunden. Werden mehrere übertragen, so kann dies durch Monoubiquitinierung verschiedener Orte des Substrats oder durch Konjugation von Ubiquitinketten erfolgen (Hochstrasser, 2006). Die Monoubiquitinierung spielt eine Rolle bei der Ubiquitinierung von Histonen und damit auch bei der Regulation der Transkription (Hicke, 2001). Monoubiquitinierung ist für viele membranständige Proteine ein Signal zur Endozytose (Mosesson et al, 2008) und reguliert die DNA-Reparatur, was der entscheidende pathogenetische Faktor bei der Fanconi Anämie und dem familiären Brustkrebs ist (Kennedy & D'Andrea, 2005). 10 Das Ubiquitinmolekül besitzt in der eigenen Polypeptidsequenz sieben Lysine (K), die potentiell zur Bildung von Ketten genutzt werden können. Dadurch entstehen Ubiquitinketten unterschiedlicher Konformation. In Hefe konnten alle Verbindungen in vivo nachgewiesen werden (Peng et al, 2003). In humanen Zellen wurde diese Vielfalt bis jetzt nicht beobachtet. Hier vermitteln über K48 verbundene Ketten mit einer Länge von mindestens vier Ubiquitinen den Abbau des jeweiligen Proteins durch das Proteasom (Thrower et al, 2000). Auch K11Ketten, übertragen durch die E3-Ligase APC/C, führen zur Degradation der Substrate (Jin et al, 2008). K63 verbundene Ketten hingegen haben eine Bedeutung in der Vermittlung von Bindungspartnern (Adhikary et al, 2005). Ubiquitinketten können nach der Anheftung an das Substrat durch weitere Proteine verändert und so verlängert werden. In der Zelle existieren Kettenelongationsfaktoren, die auch als E4 bezeichnet werden. Diese Enzyme besitzen oft eine U-Box-Domäne, die eine strukturelle Ähnlichkeit zur RingFinger-Domäne aufweist. Es ist also vorstellbar, dass E4-Kettenelongationsfaktoren mit E3-Enzymen wie E3-Enzyme mit E2-Enzymen funktionieren und so lange Ubiquitinketten katalysieren können (Hochstrasser, 2006). Ubiquitinierung bedeutet die kovalente Bindung von Ubiquitin an andere Proteine und wird durch ein komplexes System von Enzymen katalysiert. Das übertragene Ubiquitin stellt ein Signal dar, das Einfluss auf den Abbau oder auch auf Bindungspartner des ubiquitinierten Proteins hat. Ubiquitinierung ist damit ein Werkzeug der Zelle, um wichtige zelluläre Prozesse zu regulieren. 1.3. Deubiquinierung Neben der Verknüpfung von Ubiquitin und Proteinen über die oben beschriebene Proteinkaskade gibt es auch Enzyme, die an Proteine gebundenes Ubiquitin abspalten können. Diese deubiquitinierenden Enzyme (DUBs) sind Proteasen und katalysieren die Spaltung der Isopeptidbindung zwischen Ubiquitin und dessen Substrat (Abb. 1-2 A.). Die Reaktion wird als Deubiquitinierung bezeichnet. 11 Ubiquitin wird in den Zellen als lineares Polyubiquitin oder in Verbindung mit einem ribosomalen Protein (Proubiquitine) translatiert. DUBs spalten das Polymer und das Konstrukt und generieren auf diese Weise überhaupt erst das frei verfügbare Ubiquitinmonomer. Sie regulieren den zellulären Pool an freiem Ubiquitin ebenfalls durch Spaltung von freien Ubiquitinketten und von unspezifischen Ubiquitinverbindung mit kleinen zellulären Nukleophilen (Reyes-Turcu et al, 2009). Eine weitere Funktion von DUBs ist es, durch katalytische Spaltung konjugierter Ketten oder Monomere eine Ubiquitinierung von Zielproteinen rückgängig zu machen. Auf diese Weise können sie z.B. die Degradation von Proteinen durch das Proteasom verhindern. Im menschlichen Genom finden sich Gene für ungefähr 100 DUBs (Nijman et al, 2005). Die unterschiedlichen Enzyme lassen sich nach der Ähnlichkeit ihrer katalytischen Domänen in fünf Gruppen einteilen, wobei vier Gruppen Papain-ähnliche Cystein-Proteasen sind: Es gibt vier Ubiqitin C-terminale Hydrolasen (UCH), 58 Ubiquitin spezifische Proteasen (USP), 14 ovarian tumor domain (OTU) und fünf Josephin domain (MJD) DUBs. Die fünfte Gruppe beinhaltet 14 Metalloproteasen mit einer Zink komplexierenden Domäne, die auch als JAMM-Domäne bezeichnet wird (Nijman et al, 2005). Die meisten deubiquitinierenden Enzyme besitzen einen negativ geladenen Bereich in ihrer katalytischen Domäne, um den C-terminalen Teil des Ubiquitin zu binden. Auch weisen viele DUBs weitere Ubiquitin bindende Bereiche auf (UBA; UIM), die eine Erklärung für die unterschiedliche Spezifität der Enzyme auf verschiedene Ubiquitinketten bieten können. Die Bindung von Ubiquitin an das DUB hat in jedem Fall eine Ummodellierung der Proteinstruktur zur Folge, wodurch das DUB erst seine katalytische Aktivität erhält (Hu et al, 2002). 12 Abbildung 1-2: Schematische Darstellung der Deubiquitinierung: A. Das ubiquitinierte Substrat (S) wird an das DUB oft über ein Adaptorprotein (z.B. UAF1) gebunden. Das DUB spaltet Ubiquitin (Ubi) von dem Substrat ab und entlässt die beiden Bindungspartner. Die große Anzahl der DUBs macht eine hohe Substratspezifität der Enzyme wahrscheinlich. Diese wird zum einen durch die Ubiquitinbindung (s.o.) aber auch durch die Erkennung von Teilen des ubiquitinbeladenen Substrats sichergestellt. Darüber hinaus sind DUBs oft erst im Komplex mit anderen Proteinen aktiv, die auch zur Substratbindung beitragen (Ventii & Wilkinson, 2008). Dies trifft z.B. auf USP1 zu, das erst UAF1 (USP1 associated factor 1) binden muss, um FANCD2 zu deubiquitinieren und so in den DNA Reparatur Signalweg der Fanconi Anämie eingreifen zu können (Cohn et al, 2007). Posttranslationelle Modifikationen wie die Phosphorylierung, Sumoylierung und Ubiquitinierung können die Aktivität der DUBs regulieren. Dies ist z.B. der Fall bei USP34, das nach ionisierender Bestrahlung von Zellen durch ATM (Ataxia teleangiectatica mutated) phosphoryliert wird und so an der Regulation des G1Arrests beteiligt ist (Mu et al, 2007). Nicht selten binden DUBs auch E3-Ligasen und regulieren sich gegenseitig (Song & Rape, 2008). Auch USP4 bindet das E3Enzym TRIM21, das auch als Sjörgren Syndrom Antigen A/ SSA/ Ro52 bekannt ist, und deubiquitiniert es. Daneben wird USP4 durch TRIM21 ubiquitiniert. Die beiden Enzyme regulieren so gegenseitig ihren Ubiquitinierungsstatus (Wada & Kamitani, 2006; Wada et al, 2006). 13 Die Deubiquitinierung stellt also wie auch die Ubiquitinierung eine komplex gesteuerte Reaktion dar. Diese Reaktion greift in viele zelluläre Prozesse ein. Das beinhaltet unter anderem Proteintransport, Kinaseaktivierung, Proteindegradation, DNA Reparatur und Regulation des Zellzyklus (Song & Rape, 2008). Interessant ist auch, dass Mikroorganismen Gene für DUBs besitzen. Da Prokaryoten über das Ubiquitin-System aber nicht verfügen, könnten diese DUBs folglich eine Bedeutung als Pathogenitätsfaktoren haben (Chen et al, 2009; Le Negrate et al, 2008). Mutationen und Veränderungen im Expressionsmuster von DUBs konnten im Rahmen von mehreren Krankheiten beobachtet werden. So wurden in dem DUB UCHL1 Mutationen nachgewiesen, die entweder für Morbus Parkinson prädestinieren oder gegen diesen protektiv wirken. Eine Überexpression von USP6 durch Translokationen mit Vorschaltung fremder Promotoren konnte in aneurysmatischen Knochenzysten und selten auch in Sarkomen (Ewing Sarkom, Osteoblastom, Myofibrom) nachgewiesen werden (Singhal et al, 2008). Mutationen des Tumorsuppressors CYLD, der als DUB den NF-κB Signalweg inhibiert, führen zum Krankheitsbild der familiäre Zylindromatose. Die Patienten entwickeln multiple Hauttumoren an Kopf und Hals (Stegmeier et al, 2007). Auch USP4 besitzt onkogene Eigenschaften. Überexpression von USP4 bewirkte eine tumorigene Transformation von NIH3T3-Zellen (Gupta et al, 1994). Sein Expressionsmuster in Tumoren weist auf eine Rolle von USP4 bei der Tumorentstehung hin. Daten zu drei Tumorentitäten liegen vor: In Adenokarzinomen der Lunge und Nebenniere konnte eine erhöhte Expression von USP4 nachgewiesen werden (Gray et al, 1995; Velazquez-Fernandez et al, 2005). Für kleinzellige Bronchialkarzinome ist die Publikationslage widersprüchlich. Eine Arbeitsgruppe berichtet über erhöhte mRNA Expression von USP4 in Gewebeproben aus kleinzelligen Bronchialkarzinomen (Gray et al, 1995). Dagegen wurde von anderer Seite eine erniedrigte Konzentration von USP4-mRNA und Protein in Zelllinien des kleinzelligen Bronchialkarzinoms festgestellt (Frederick et al, 1998). 14 Es gibt Daten, dass USP4 an Proteine aus der Retinoblastom-Proteinfamilie bindet (Blanchette et al, 2001). Bis jetzt konnte jedoch keine funktionelle Relevanz dieser Bindung nachgewiesen werden. In einer weiteren Publikation wurde USP4 als Spieler im Wnt Signalweg identifiziert (Zhao et al, 2009). Auf der Suche nach relevanten Bindungspartnern konnten in USP4-PulldownVersuchen mit HeLa Micha Zellextrakten die Proteine SART3 und PRP3 detektiert werden. Beide Proteine binden an das Spliceosom und sind wichtig für korrektes Splicing (Trede et al, 2007; Wang et al, 1997). SART3 hat darüber hinaus eine Funktion als Recyclingfaktor der snRNPs (small nuclear ribonucleoprotein) U4/U6 (Damianov et al, 2004; Trede et al, 2007). Die Bindung des deubiquitinierenden Enzyms USP4 an Komponenten des Spliceosoms ist sehr interessant, da es Daten gibt, nach denen Ubiquitin für den Aufbau des Spliceosoms wichtig ist (Bellare et al, 2008). 1.4. Spliceosom Die meisten Vorläufer-Transkripte in eukaryoten Zellen enthalten Introns (intervening sequences) und Exons (expressed sequences), wobei nur Exons die Grundlage für die Translation durch das Ribosom darstellen. Die VorläuferTranskripte werden im Zellkern modifiziert, was unter anderem das Anhängen eines 5´-Cap, eines poly-A-Schwanzes und das Entfernen der Introns durch Splicing beinhaltet (Epstein, 2003). Splicing wird im Zellkern durch einen großen Komplex aus snRNAs (small nuclear RNAs) und deren assoziierte Proteine, small nuclear Ribonucleoprotein particles (snRNPs), durchgeführt. Dieser Komplex aus fünf snRNPs wird als Spliceosom bezeichnet. Die Interaktionen zwischen snRNPs untereinander und zwischen snRNPs und Introns sind komplex und verändern sich im Verlauf des Splicingprozesses. Diese dynamischen Veränderungen, die vor allem redundante und multiple Intron-Erkennungsereignisse beinhalten, werden durch RNA-abhängige ATPasen ermöglicht (Brow, 2002). 15 Splicing wird eingeleitet durch die Bindung des U1-snRNP an das konservierte 5´-Ende des Introns. U2-snRNP bindet im Intron an die weiter 3´-liegende Verzweigungsstelle, welche ein Adenosinnukleotid beinhaltet (branch point recognition). Erst danach werden auch U4/U6-U5 snRNPs hinzugefügt, wobei U4 und U6 über Basenpaarung fest aneinander gebunden sind. Nun folgt die Umformung des inaktiven Komplexes in den aktiven Komplex, welcher den ersten Phosphoryl-Transfer auf das Adenosinnukleotid in der Verzweigungsstelle katalysiert. Danach kommt es zur Trennung von U4 und U6 snRNPs, wobei U6snRNP das U1-snRNP vom 5´-Ende verdrängt und den zweiten PhosphorylTransfer vermittelt. Danach fällt das Spliceosom auseinander, und die so entstandene mRNA wird entlassen. Das Intron bleibt in Lassogestalt zurück (Epstein, 2003; Wahl et al, 2009). Die geordnete Umformung der einzelnen Komponenten des Spliceosomes während des Splicings ist komplex und wird hauptsächlich von den Interaktionen zwischen den snRNA assoziierten Proteinen angetrieben. Die Steuerung dieser Interaktionen erfolgt auch durch Phosphorylierung, Azetylierung und Ubiquitinierung (Bellare et al, 2008; Wahl et al, 2009). Die Bedeutung des Splicings für die Herstellung von translationsfähigen Transkripten macht auch seine Rolle bei der Zellzykluskontrolle deutlich. Wie das Ubiquitin-System z.B. für den Abbau von Cyclinen von Bedeutung ist, so ist Splicing für die Produktion von Cyclinen essentiell. Damit ist es ein Mechanismus, der den reibungslosen Ablauf des Zellzyklus gewährleistet. 1.5. Zellzyklus Zellen entstehen durch Zellteilung. Für diesen Vorgang vollzieht die Zelle eine streng geordnete Reihe von Prozessen, welche die Verdopplung ihrer Bestandteile und die anschließende Teilung in zwei Tochterzellen zur Folge hat. Die wichtigste Aufgabe des Zellzyklus ist die Weitergabe der genetischen Information an die nächste Zellgeneration. Um die genetische Identität bei der mitotischen 16 Teilung zu gewährleisten, muss die DNA der Zellen möglichst fehlerlos dupliziert und verteilt werden. Der Zellzyklus wird in vier Phasen eingeteilt (Abb. 1-3 A). Die Zelle befindet sich nach der Zellteilung in der G1-Phase (Gap1). Von dort aus kann sie in einen Ruhezustand übergehen, der als G0-Phase bezeichnet wird. Alternativ tritt sie in die S-Phase (Synthese) ein, in welcher die Verdopplung der Chromosomen erfolgt. Zwölf Stunden benötigt die Säugerzelle für die S-Phase. Dann folgt die G2-Phase, nach der letztendlich die M-Phase (Mitose) eintritt. Hier vollzieht sich die Verteilung der Chromosomen auf die Tochterzellen und die Teilung des Zytoplasmas, die auch als Zytokinese bezeichnet wird. Für die Mitose benötigt die Zelle ungefähr eine Stunde. Abbildung 1-3: Schematische Darstellung des Zellzyklus: A. Eine Zelle durchläuft den Zyklus von der G1-Phase (Gap) über die S-Phase (Synthese) und die G2-Phase (Gap) bis zur M-Phase (Mitose). Dabei passiert sie mehrere Check- und Restriktionspunkte, die einen geordneten Ablauf des Zellzyklus gewährleisten. Die M-Phase lässt sich in mehrere Abschnitte einteilen: Während der Prophase kommt es zur Kondensation der Chromosomen, die mit ihren SchwesterChromatiden über Cohesin-Komplexe verbunden sind. Die Kernmembran löst sich in der Prometaphase auf, und die Chromatiden werden an ihren Kinetochoren mit dem Spindelapparat verbunden. In der Metaphase werden die Schwester-Chromatiden jeweils von Mikrotubuli gegenüberliegender Zentromere 17 des bipolaren Spindelapparates gebunden und in der Äquatorialebene angeordnet. Nach der Spaltung einer Cohesin-Untereinheit in der Anaphase kommt es zur Trennung der Schwesterchromatiden, die zu entgegengesetzten Spindelpolen gezogen werden. In der Telophase vollzieht sich dann die Zytokinese und die Bildung zweier neuer Zellkerne. Für die Produktion ihrer Proteine und Organellen benötigen die Zellen weitere Zeit. Dafür existieren im Zellzyklus die G1-Phase zwischen M- und S-Phase und die G2-Phase zwischen S- und M-Phase. Die G1-, S- und G2-Phasen werden zusammen auch als Interphase bezeichnet und der Mitose als Phase der Zellteilung gegenübergestellt. Der komplexe Ablauf des Zellzyklus wird streng reguliert. Dies geschieht hauptsächlich durch Kontroll-Hürden, sogenannte Check- und Restriktionspunkte. Der erste Restriktionspunkt befindet sich am Ende der G1-Phase und ist für die Initiierung der Chromosomenverdopplung verantwortlich. Nur wenn die intra- und extrazellulären Bedingungen es zulassen, wird der Start für die Zellteilung freigegeben. Dafür wird das Rb-Protein hyperphosphoryliert und gibt dadurch E2F aus seiner Bindung frei. E2F wirkt als Transkriptionsfaktor und führt zur Expression von Proteinen wie cyclinE und cyclinA, die für den Eintritt in die SPhase nötig sind. Vom Beginn der S-Phase bis zum G2/M-Checkpunkt erfolgt die Regulation des Zellzyklus auch durch Cyclin-abhängige Kinasen (Cyclin dependent kinases [Cdks]), die während des Zellzyklus konstant exprimiert werden. Ihre Aktivität oszilliert während des Zellzyklus jedoch stark, da Cdks nur aktiv sind, wenn sie an Cycline gebunden vorliegen. Cycline sind Proteine, die während jedes Zellzyklus zu bestimmten Zeitpunkten synthetisiert und proteasomal degradiert werden. So kommt es zur zyklischen Änderung der Phosphorylierung von Proteinen, die wichtige Ereignisse des Zellzyklus regulieren (Alberts B., 2008; Malumbres & Barbacid, 2009). Zusätzlich gibt es im Zellzyklus Checkpunkte, die sensitiv auf DNA-Schäden reagieren. Diese befinden sich am Ende der G1-Phase (G1/S-Checkpunkt), in 18 der S-Phase (Intra-S-Checkpunkt) und am Übergang von G2- zu M-Phase (G2/M-Checkpunkt). DNA-Schäden verursachen die Bindung und Phosphorylierung von ATM und ATR, deren nachgeschaltete Signalkaskaden unter anderem über Chk1 einen Zellzyklusarrest verursachen (Seiler et al, 2007). Ein weiterer Checkpunkt befindet sich in der Mitose zwischen Meta- und Anaphase. Hier wird die Trennung der Chromatiden durch den Spindelcheckpunkt inhibiert, solange nicht alle Chromosomen über ihre Kinetochore mit Mikrotubuli verbunden sind. Die Regulation des Checkpunktes ist komplex: Die Proteine BubR1, Mad2 und Bub3 werden ab der Prometaphase an den Kinetochoren aktiviert und halten die Ring-Finger E3-Ligase Cdc20-APC/C (anaphase promoting complex or cyclosome) in einer inaktiven Konfiguration (Herzog et al, 2009). Erst wenn auch das letzte Kinetochore durch Mikrotubuli gebunden ist und eine Spannung zwischen den Spindelpolen aufgebaut werden kann, wird der Checkpunkt abgeschaltet und Cdc20-APC/C im Gegenzug aktiviert (Logarinho & Bousbaa, 2008). Cdc20-APC/C katalysiert die Ubiquitinierung und damit den proteasomalen Abbau von Securin, einem Inhibitor der Separase. Die Separase spaltet daraufhin Cohesin, das die Schwester-Chromatiden verbindet, und ermöglicht so die Trennung der Chromatiden und deren Verteilung auf zwei Tochterzellen. APC/C führt auch zum Abbau mitotischer Cycline (cyclinB), wodurch der Zellzyklus mit Aufbau der Kernmembran, Dekondensation der Chromosomen, Zytokinese und Zerfall der Spindel fortgeführt und die M-Phase abgeschlossen wird (Cleveland et al, 2003). Dysregulation des Spindelcheckpunktes hat die genomische Instabilität der Zellen zur Folge (Engelbert et al, 2008; Wasch & Engelbert, 2005). Um katalytisch aktiv zu sein und seine Substrate zu erkennen, benötigt APC/C die Cofaktoren Cdc20 oder Cdh1. APC/C kann immer nur einen der beiden Cofaktoren binden, was im Zellzyklus streng reguliert wird. Eine Phosphorylierung von APC/C durch Cdk1 führt zur Bindung von Cdc20. Andererseits wird die Bindung von Cdh1 an APC/C durch Phosphorylierung mit Cdk2-cyclinA verhindert. Durch Dephosphorylierung zerfällt Cdc20-APC/C in der späten M19 Phase, und Cdc20 wird durch Cdh1-APC/C abgebaut. So ist Cdc20-APC/C in der frühen M-Phase und Cdh1-APC/C in der späten M-Phase und der G1-Phase aktiv (Meyer, 2008). Eine Hemmung von Cdh1-APC/C am Ende der G1-Phase erfolgt dann wieder durch Phosphorylierung von Cdh1. Dies wird über cyclinA vermittelt, das als Substrat des Cdh1-APC/C durch Anwesenheit des APC/C Inhibitors Emi1 und der Degradation eines mit APC/C interagierenden E2 UbcH10 akkumulieren kann (Reimann et al, 2001). 1.6. Zielsetzung der Arbeit Ausgehend von der Idee, dass es DUBs geben könnte, welche den Spindelcheckpunkt regulieren, führte Michael Rape ein siRNA-screen gegen alle humanen DUBs durch. Der Screen wurde in HeLa-Zellen (Zervixkarzinom) ausgeführt, die mit dem Inhibitor des Mikrotubiliabbaus Paclitaxel, dem Inbibitor der Mibrotubulipolymerisation Nocodazol oder dem Kinesininhibitor Monastrol behandelt wurden. Auf diese Weise wurde der Aufbau des Spindelapparates gestört und der Spindelcheckpunkt aktiviert. Man erwartet einen Arrest der Zellen in der Metaphase. Fällt nun durch siRNA ein wichtiger Regulator des Spindelcheckpunktes weg, so ist davon auszugehen, dass die Zellen trotz Inhibition der Mikrotubuli in die Interphase übertreten, was als Spindelcheckpunkt-Bypass bezeichnet wird. Für die siRNA gegen USP4 konnte ein solcher Bypass beobachtet werden. Es war also wahrscheinlich, dass USP4 in die Regulation des Spindelcheckpunktes eingreift. Wir wollten herausfinden, auf welche Art und Weise USP4 auf den Zellzyklus Einfluss nimmt. Dafür interessierten uns die Bindungspartner von USP4. Mit SART3 und PRP3 hatten wir zwei interessante Kandidaten gefunden. Wir wollten die genauen Bindungsverhältnisse untersuchen und nach weiteren Partnern suchen. Der nächste wichtige Punkt war die deubiquitinierende Aktivität von USP4. Unser Ziel war es, die Aktivität von USP4 in vitro nachzuweisen und zu analysieren. Uns interessierte, ob USP4 seine Aktivität autark oder nur unter Bindung von 20 weiteren Partnern erlangt. Wir wollten auch wissen, ob USP4 eine Präferenz für Ubiquitinketten einer bestimmten Lysinverbindung hat. Als entscheidend für das Verständnis der Regulation des Spindelcheckpunktes durch USP4 schien es uns, Substrate von USP4 zu finden. Die im Pulldown entdeckten Bindungspartner SART3 und PRP3 waren hierfür erste Anhaltspunkte. Weiter interessierte uns die Lokalisation von USP4 in der Zelle. In der Literatur gab es kontroverse Daten. Wir wollten verstehen, welche Faktoren Einfluss auf die Lokalisation von USP4 nehmen. Von der Untersuchung dieser Fragen erhofften wir uns Aufschlüsse über die regulatorische Rolle von USP4 im Zellzyklus und über seine mögliche Bedeutung für die Tumorentstehung. 21 2. Materialien 2.1. Geräte Autoklav Blotkammer Eismaschine Filmentwickler Consolidate Ind. MK2 Mini Trans-Blot cell Stills and Sterilizers Biorad Hoshizaki Konica SRX-101 Diagnostic Imaging Inc. Fluoreszensmikroskop MZ FL 2 Leica Geltrockner Model 583 Biorad Heizblock Thermomixer Eppendorf Inkubator NAPCO 8000 DH Thermo Electron Corp. Inverses Mikroskop Invertoskop 40 C Zeiss Kondensator Model 494 Isco TM Power Pac HC Biorad Magnetrührer Fisher Stirrer Fisher Scientific PCR- Maschienen PTC-200 MJ-Research TM MyCycler Biorad pH-Meter SevenEasy Mettler Toledo Rotator Mini Rotator Glas-Col Scanner Epson Perfection V700 Epson Schwenker Orbital Shaker Bellco Biotech. Rocker BioModell 110A Denville Scientific Inc. SDS-Gelelektrophoresekammer Vertical unit R. Shadel Inc. Mini Protean 2 Biorad Spektrometer Untraspec 3 Pharmacia Nanodrop Thermo Scientific Sterile Zellkulturbank Logic Labconco Storage-Phosphor-Scanner Typhoon 9400 Molecular Dynamics Storage-Phosphor-Screen PhosphorImager Biorad Ultraschall Modell W185D Ultrasonic TM Vakuummaschine HydroTech Biorad Waage Mettler P1200 VWR AB104 Mettler Toledo Wasseraufbereiter Milli Q Synthesis A10 Millipore Wasserbad Isotemp 220/202 Fisher Scientific Zentrifuge 5415C/ 5417C/ 5415R Eppendorf Sorvall Legend Micro Thermo Scientific 21R RC-3B Sorvall Instruments 22 2.2. Software: DNA Absorptionsmessung Datenbanken Sequenzvergleich Bildbearbeitung Textverarbeitung Nanodrop Software Pubmed (www.pubmed.com) BLAST (www.ncbi.nlm.nih.gov/BLAST) ClustalW2 (www.ebi.ac.uk/clustalw) Adobe Photoshop 7.0.1 Adobe Illustrator 10.0.3 ImageJ Microsoft Word X for Mac 2.3. Chemikalien Acrylamide (Ultra Pure Proto Gel) National Diagnostics Adenosintriphosphat Agar Agarose (Low-EEO/MultiPurpose/Molecular Biology Grade) Ampicillin Amylose Resin APS Borsäure Calciumchlorid Cellfectin Chemilumineszens Luminol Chloramphenicol Coomassie Brilliant Blue R-250 DMEM DMSO DTT dNTPs DPBS Essigsäure Ethanol Ethidiumbromid EDTA FBS FBS Tetrazyklin depletiert Glycerol Glycin Hefeextrakt HEPES Imidazol IPTG Isopropanol Kaliumacetat Kaliumchlorid Kaliumhydrogenphosphat Sigma Fisher Scientific Fisher Scientific Fisher Scientific NEB EMD Chemicals EMD Chemicals Fisher Scientific Invitrogen Immobilon Fisher Scientific Pierce Biotechnology Gibco Sigma Invitrogen NEB Gibco Fisher Scientific Gold Shield Chemical Corp. Fisher Scientific Fisher Scientific HyClone HyClone Fisher Scientific MP Biomedicals EMD Chemicals Fisher Scientific Fisher Scientific Apex Fisher Scientific Fisher Scientific Fisher Scientific Fisher Scientific 23 Kanamycin Magnesiumchlorid Maganchlorid Methanol Milchpulver Natriumchlorid Natriumhydrogenphosphat Natriumhydroxid Nickel-Nitrilotriacetat-Agarose Nocodazol Ponceau S Precision Plus Protein Standard Protease Inhibitor (Pallets) Rubidiumchlorid S35-Methionin SDS Thymidin Triton X-100 Trypsin/EDTA 0,5% Trypton Shrimp alkalische Phospatase TEMED TRIS Tween 20 Fisher Scientific EMD Chemicals Sigma Ficher Scientific Nestle Fisher Scientific Fisher Scientific Fisher Scientific Qiagen Sigma Sigma Biorad Roche Alfa Aesar MP Biomedicals Fisher Scientific Sigma Fisher Scientific Gibco BD USB Corp. Biorad Fisher Scientific Fisher Scientific 2.4. Proteine 2.4.1. Proteine allgemein His APC10 AscI DpnI His E1 (aus Insektenzellen) FseI His Importin alpha C-terminal Lysozym NotI PfuUltra™ High Fidelity DNA Polymerase His p31 SalI SAP Taq-Polymerase T4-Ligase His UbcH5c Ubiquitin XbaI XhoI 24 Michael Rape, UC Berkeley NEB NEB Adam Williamson, UC Berkeley NEB Ling Song, UC Berkeley Sigma NEB Stratagene/ Agilent Ling Song, UC Berkeley NEB VWR Scientific Fisher Scientific NEB Hermann-Josef Meyer, UC Berkeley Boston Biochem Roche NEB 2.4.2. Primärantikörper Kaninchen polyklonal Anti-myc (NEQKLISEEDL-C) Maus monoklonal Anti-HA Maus monoklonal Anti-Ubiquitin FL Santa Cruz Roche Santa Cruz 2.4.3. Sekundärantikörper Ziege anti Maus mit Peroxidase Ziege anti Kaninchen mit Peroxidase Ziege anti Maus mit Alexa488 Ziege anti Kaninchen mit Cy3 Sigma Amersham Molecular Probes Molecular Probes 2.5. DNA 1kb-DNA-ladder Oligonucleotide Invitrogen Operon 2.6. Zellen 2.6.1. Bakterienstämme DH5alpha XL1-blue BL21 Rosetta 2.6.2. Humane Zelllinien HeLa Micha HeLa S3 U2OS host cell line 2.6.3. Insektenzelllinien SF9 2.7. Kits TNT SP6 Quick Coupled Transcription/ Translation System Promega QIAGEN Miniprep Kit Qiagen QIAGEN Midiprep Kit Qiagen QIAquick Gel Extraction Kit Qiagen Flp-InTM T-RExTM Core Kit Invitrogen 25 2.8. Puffer LB-Medium 10g Typton 5g yeast extract 10 g NaCl in 1 l ddH2O TBF1 70 mM KAc 100 mM RbCl2 10 mM CaCl2 50 mM MnCl2 15% Glycerol pH 6,5 TBF2 10 mM MOPS 75 mM CaCl2 10 mM RbCl2 15% Glycerol pH 6,5 Isolationslösung 1 15 mM Tris 10 mM EDTA 100 ng/ml RNase A pH 8,0 Isolationslösung 2 0,2 N NaOH 1% SDS 10x TBE 108 g Tris 55 g Borsäure 40 ml 500 mM EDTA 1l ddH2O pH 8,0 5x Ladepuffer TBE Puffer 20% Glycerol 0,01% Bromphenolblau 2x SDS-Ladepuffer 62,5 mM Tris pH 6,8 2% SDS 25% Glycerol 0,01% Bromphenolblau 5% beta-Mercaptoethanol SDS-Laufpuffer 25 mM Tris 193 mM Glycin 0,1% SDS Transferpuffer 25 mM Tris 193 mM Glycin 20% Methanol 26 2.9. Verbrauchsmaterialien Blotpapier Dialysekammern Film Kulturschalen Nitrocellulosemembran Proteinaufreinigungssäulen Schreibwaren Biorad Thermo Scientific Kodak Greiner GE Biorad Barker Hall Storeroom 27 3. Methoden 3.1. Molekularbiologische Methoden 3.1.1. Herstellung chemisch kompetenter E. coli Die Aufnahme von freier DNA durch E. coli wird als Transformation bezeichnet. Für diese Transformation werden Bakterien benötigt, die in der Lage sind, DNA in größerem Ausmaß aus ihrer Umgebung aufzunehmen. Solche Bakterien werden als kompetent bezeichnet. Die Herstellung kompetenter Bakterien erfolgte mit einer Übernachtkultur des jeweiligen Stammes. Danach wurden 1,5 l LB-Medium (1% Trypton, 0,5% yeast extract, 1% NaCl) angeimpft und bis zu einer OD600 von 0,4 bis 0,6 bei 37ºC mit 180 U/min geschwenkt. Die Suspension wurde 5 min auf Eis gelagert und 10 min bei 4000 U/min und 4ºC zentrifugiert. Das Pellet wurde in TBF1-Puffer (70 mM KAc, 100 mM RbCl2, 10 mM CaCl2, 50 mM MnCl2, 15% Glycerol, pH 6,5) resuspendiert und 90 min auf Eis inkubiert. Danach wurde 5 min bei 4000 U/min und 4ºC zentrifugiert, und die Bakterien wurden in TBF2-Puffer (10 mM MOPS, 75 mM CaCl2, 10 mM RbCl2, 15% Glycerol, pH 6,5) aufgenommen. Aliquots wurden mit flüssigem Stickstoff schockgefroren und bei –80ºC gelagert. 3.1.2. Transformation chemisch kompetenter E. coli Die Übertragung freier DNA auf kompetente Bakterien erfolgte mit Hilfe eines Hitzeschocks (Hanahan, 1983). 25 µl Bakteriensuspension und 1 µl PlasmidDNA wurden zuerst 30 min bei 4ºC, anschließend 1 min bei 42ºC und dann 5 min auf Eis inkubiert. Danach wurden 250 µl LB-Medium dazugegeben und die Probe 2 Stunden in dem 37ºC Wasserbad gelagert. Die Suspension konnte mit autoklavierten Glaskugeln auf selektivem Nährboden (1% Typton, 0,5% yeast extract, 1% NaCl, 1,5% Agar mit unterschiedlichen Antibiotika in folgenden Konzentrationen: 100 µg/ml Ampicillin, 25 µl/mg Kanamycin, 34 µg/ml 28 Chloramphenicol) ausplattiert oder für das Animpfen von Flüssigkulturen genutzt werden. 3.1.3. Plasmidpräparation Mit dem QIAGEN Miniprep Kit und dem QIAGEN Midiprep Kit wurden Plasmide <10 kbp aus Übernachtkulturen transformierter Bakterien aufgereinigt. Dabei wurden zelluläre Bestandteile und bakterielle DNA ausgefällt und die PlasmidDNA auf Anionenaustauschersäulen gegeben. Wegen der Phosphatgruppen bindet hier die DNA, kann gereinigt und eluiert werden. Die Aufreinigung fand gemäß Herstellervorgaben statt. Für die Aufreinigung größerer Plasmide >10 kbp wurde das Zellpellett von 5 ml Übernachtkultur transformierter Bakterien in 1 ml Isolationslösung 1 (15 mM Tris, 10 mM EDTA, 100 ng/ml RNase A, pH 8,0) aufgenommen und gevortext. Danach wurde 1 ml Isolationslösung 2 (0,2 N NaOH, 1% SDS) dazu gegeben, 5 min bei RT inkubiert und schließlich 250 µl 3 M KAc (pH 5,5) ergänzt. Die Probe wurde 10 min auf Eis gelagert, 10 min bei 14000 U/min zentrifugiert, und der Überstand mit 750 µl Isopropanol gemischt. Eine erneute Zentrifugation (10 min bei 14000 U/min) wurde durchgeführt, der Überstand durch 500 µl Ethanol ausgetauscht und ein letztes Mal zentrifugiert (10 min bei 14000 U/min). Der Alkohol wurde abgenommen, und das Pellet getrocknet. Die Auflösung erfolgte in EB-Buffer der QIAGEN Kits. 3.1.4. DNA Konzentrationsmessung Die Konzentration von DNA in wässriger Lösung wurde durch Lichtabsorption bei einer Wellelänge von 260 nm an dem Gerät Nanodrop durchgeführt. Nach dem Lambert Beer Gesetz lässt sich mit dem Absorptionskoeffizienten e so die Konzentration von DNA in der jeweiligen Flüssigkeit errechnen. Die Errechnung erfolgte durch die zugehörige Software. 29 3.1.5. Polymerase Kettenreaktion Die Polymerase Kettenreaktion (polymerase chain reaction = PCR) ist eine Methode zur exponentiellen Vervielfältigung von DNA-Fragmenten durch eine Polymerase. Für die Reaktion benötigt das Enzym Oligonukleotide, welche spezifisch an die Anfangs- und komplementäre Endsequenz des Fragments binden. Die Oligonukleotide werden als Primer bezeichnet und bilden den jeweiligen Anfang der zu synthetisierenden Sequenz. Die Amplifikation erfordert drei verschiedene Schritte. Bei der 1) Denaturierung wird die doppelsträngige DNA durch Erhitzen in Einzelstränge getrennt, so dass die Primer beim 2) Annealing das DNA-template binden können und so die 3) Elongation der Sequenz katalysiert werden kann. Dieser Zyklus wird wiederholt und hat die exponentielle Amplifikation der von den Primern umschlossenen Sequenz zu Folge. Bei dem Design der Primer wurden folgende Kriterien beachtet: - mehr als 18 bp lang - T Annealing [=2x(A/T)+4x(G/C)-4] zwischen 57ºC und 60ºC - GC-Anteil der Sequenz über 40% Danach wurde den Primern am 5´-Ende die Erkennungssequenz des jeweils gewünschten Restriktionsenzyms angehängt. Die Reaktionen wurden in 50 µl Ansätzen mit dem Pfu-Puffer (Stratagene) durchgeführt. Wir nahmen 100 ng Ausgangsplasmid und ergänzten dieses mit Desoxynukleotidmix (0,5 mM) und Primern (0,5 µM). Der Reaktion wurden zeitweise auch DMSO (10%) und BSA (0,1 mg/ml Endkonzentration im Mix) zugegeben. Nach Hinzufügen von 5 U Pfu-Ultra-Polymerase wurde die Reaktion in einer PCR-Maschine gestartet. In bestimmten Fällen wurde Pfu-UltraPolymerase durch die Taq-Polymerase ergänzt. Die Denaturierung wurde initial 1 min und bei jedem folgenden Zyklus 30 s bei 94ºC durchgeführt. Dann folgte das Annealing 30 s bei 48-58ºC abhängig von dem jeweiligen Primerpaar. Die Elongation wurde 1 min pro 1 kbp bei 68ºC 30 durchgeführt. Nach 25 Zyklen wurde auf 4ºC abgekühlt und das Amplifikationsprodukt mit Gelelektrophorese analysiert. 3.1.6. Mutations-PCR Das Einbringen von spezifischen Mutationen in ein kloniertes Gen wurde mit Primern durchgeführt, welche diese Mutation enthielten und bei der PCR als Ausgangspunkt für die Amplifikation des gesamten Plasmides genutzt wurden. Für das Primerdesign wurde auf folgende Punkte geachtet: - Anzahl (n) der bp 25-45 - GC-Gehalt von über 40% - 3´-Ende auf C oder G - Tm [=81,5+0,41x(%G/C)–(675/n)-%mismatch] über 78ºC Der 20 µl PCR-Mix in Pfu-Puffer setzte sich aus 50 ng Ausgangsplasmid, 0,5 mM Desoxynukleotidmix, 0,25 µM Primer und 5 U Pfu-Ultra-Polymerase zusammen. Nach 2 min Denaturierung bei 94ºC wurden 20 PCR-Zyklen durchgeführt von jeweils 30 s bei 94ºC, 30 s bei 50-55ºC (abhängig von den jeweiligen Primern) und 1 min pro 1 kbp bei 68ºC. Anschließend wurden die PCR-Produkte 90 min bei 37ºC mit 20 U Dpn1 inkubiert, um die Ausgangsplasmide zu verdauen. 25 µl kompetente XL1-blue E. coli wurden mit 5 µl PCR-Produkt transformiert und auf selektivem Nährboden ausplattiert. Aus hier gewachsenen Kolonien wurden die Plasmide extrahiert und mittels Sequenzierung auf die gewünschte Mutation hin überprüft. 3.1.7. Agarosegelelektrophorese Zur Trennung von in der Größe differierenden DNA-Fragmenten wurde die Elektrophorese in dem Galactosepolymer Agarose durchgeführt. Durch die negative Ladung der Phosphordiester-Zucker-Kette wandert die DNA in einem angelegten elektrischen Feld durch die Trennmatrix. Dabei wandern kleinere 31 Fragmente schneller als größere, wodurch eine Auftrennung der Größe nach erfolgt. Agarose wurde in 0,5 bis 1,5 Gewichtprozent in 1x TBE-Puffer (10x TBE-Puffer: 108 g Tris, 55 g Borsäure, 40 ml 500 mM EDTA, 1 l ddH2O, pH 8,0) aufgelöst und in einer Mikrowelle aufgekocht. Danach wurde 0,5 µg/ml Ethidiumbromid hinzugegeben und abgekühlt. Die DNA wurde 5:1 mit dem 5x Ladepuffer (1x TBE-Puffer, 20% Glycerol, 0,01% Bromphenolblau) gemischt und in die Geltaschen pipettiert. Für die Elektrophorese wurde Gleichstrom mit 100-150 V (8 V / cm Elektrodenabstand) angelegt. Die Analyse erfolgte unter Ultraviolettlicht der Wellenlänge 366 nm, welches das an die DNA bindende Ethidiumbromid sichtbar macht. Die Extraktion von DNA aus dem Gel erfolgte mit dem QIAquick Gel Extraction Kit nach den Angaben des Herstellers. 3.1.8. Sequenzierung Plasmid-DNA wurde durch die University of California Berkeley DNA Sequencing Facility 303 Barker Hall (http://mcb.berkeley.edu/barker/dnaseq/index.html) sequenziert. Die Proben wurden mit 0,5 µg DNA und 3,2 pmol Primer in 13 µl Gesamtvolumen angesetzt. Als Primer wurden unter anderem für den SP6 Promoter: 5'-GATTTAGGTGACACTATAG-3' und für den T7 Promoter: 5'TAATACGACTCACTATAGGG-3' verwendet. 3.1.9. Klonieren Um ein DNA-Fragment in einen Vektor einzusetzen, wurde mit Restriktionsverdau und Ligation gearbeitet. Die Technik wird als Klonieren bezeichnet. Restriktionsendonukleasen schneiden DNA spezifisch an den für das jeweilige Enzym charakteristischen, meist palindromischen Sequenzen. Plasmid-DNA und PCR-Produkte wurden im Rahmen der für die jeweiligen Enzyme vom Hersteller empfohlenen Bedingungen verdaut. Danach erfolgte entweder eine Gelelektro32 phorese plus Gelextraktion oder einfach nur eine PCR-Aufreinigung. Für die PCR-Aufreinigung wurde das QIAquick Gel Extraction Kit verwendet. Die Religation linearisierter Vektor-DNA wurde durch Dephosphorylierung bei 90 min und 37ºC mit SAP verhindert. Die Ligation oder Verbindung von Insert und Vektor wurde durch Inkubation mit der Ligase des Bakteriophagen T4 durchgeführt. Unter ATP-Verbrauch katalysiert das Enzym die Phosphordiesterverbindung zwischen der 3´OH-Gruppe des Zuckers und der 5´OH-Gruppe des Phosphatendes. Dabei wurden Vektor und Insert in einem stöchiometrischen Verhältnis von 1:1 bis 1:5 eingesetzt und zwischen 3 Stunden und bis über Nacht bei RT mit T4-Ligase inkubiert. 3 µl des Ansatzes wurden zur Transformation von 25 µl DH5alpha-Bakterien verwendet. Der Selektion auf Agar folgte die Kultivierung in LB-Medium und die Extraktion der Plasmid-DNA. Die Plasmide wurden verdaut und bei erwartetem Bandenmuster in der Gelelektrophorese sequenziert. 3.2. Proteinbiochemische Methoden 3.2.1. Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-PAGE) PAGE trennt Moleküle abhängig von deren Ladung und molarer Masse im Trennmedium Polyacrylamid auf. SDS-PAGE wird verwendet, um Proteine nur ihrer molaren Masse nach aufzutrennen. Dabei werden Sekundär- und Tertiärstruktur der Proteine durch Anlagerung von SDS und Spaltung der Thioesterund Disulfidgruppen mit beta-Mercaptoethanol zerstört. Durch die stark negative Ladung der im Überschuss zugegebenen SDS-Moleküle wird die Eigenladung der Proteine irrelevant in Bezug auf Wandereigenschaften in einem elektrischen Feld. So erfolgt die Auftrennung allein aufgrund der unterschiedlichen molaren Masse der jeweiligen Proteine. Diese kann mit Hilfe von Markern abgeschätzt werden. Die Polyacrylamidgele wurden in eindimensionalen Minigel- oder Maxigelformaten der Firma Biorad hergestellt. Die Polymerisation erfolgte zwischen Acrylamid und N,N´-Methylenbisacrylamid unter Zugabe des Radikalstarters 33 Ammoniumpersulfat und des Elektronenüberträgers TEMED. Mit einer Acrylamidkonzentration von 8-15% wurde zuerst das Trenngel gegossen, dessen glatte Überfläche durch Überspritzen mit Ethanol sichergestellt wurde. Darüber wurde das Sammelgel infundiert und mit einem Gelkamm modelliert. Beispiele für die Zusammensetzung von Trenngel und Sammelgel sind: Trenngel 8% Sammelgel ddH2O 6,9 ml 3,4 ml 30% Acrylamidmix 4,0 ml 0,83 ml 1,5 M Tris 3,8 ml (pH 8,8) 0,63 ml (pH 6,8) 10% SDS 0,15 ml 0,05ml 10% APS 0,15 ml 0,05 ml TEMED 0,009 0,05 ml Die Proteinproben wurden 1:1 mit 2x SDS-Ladepuffer (62,5 mM Tris pH 6,8, 2% SDS, 25% Glycerol, 0,01% Bromphenolblau, 5% beta-Mercaptoethanol) vermischt, 5 min bei 95ºC inkubiert und in die Geltaschen pipettiert. Die Elektrophorese wurde bei 100 V (Mini) beziehungsweise 300 V (Maxi) mit SDSLaufpuffer (25 mM Tris, 193 mM Glycin, 0,1% SDS) durchgeführt. 3.2.2. Coomassie-Färbung Zum Nachweis von Proteinen wurde das SDS-Gel 2 Stunden in 0,05% Coomassie Blue Stain (0,5 g Coomassie Blue R250, 250 ml Isopropanol, 100 ml Essigsäure, mit ddH2O auf 1 l) geschwenkt und anschließend in 10% Essigsäure bis zum gewünschten Grad entfärbt. Coomassie Blue detektiert unspezifisch alle Proteine. 34 3.2.3. Silberfärbung Zuerst wurde das Gel 10 min in SF-Lösung 1 (40 ml Methanol, 13,5 ml Formaldehyd, 46,5 ml ddH2O) fixiert. Danach wurde es zweimal 5 min in ddH2O gewaschen. Anschließend wurde es 1 min in SF-Lösung 2 (0,02 g Na2S2O3, 100 ml ddH2O) inkubiert, um danach 20 s mit ddH2O abgespült zu werden. Die Färbung erfolgte 10 min in der SF-Lösung 3 (0,1 g Silbernitrat, 100 ml ddH2O), die Entwicklung bis zum gewünschten Färbegrad in der SF-Lösung 4 (3 g Na2CO3, 50 µl Formaldehyd, 2 ml SF Lösung 2, 98 ml ddH2. Die Reaktion wurde mit kristalliner Zitronensäure gestoppt. 3.2.4. Western Blot Die in dem Gel durch SDS-PAGE aufgetrennten Proteine wurden mit einem elektrischen Feld auf Nitrocellulose- oder PVDF-Membran transferiert. Der Nachweis erfolgte über selektive Primärantikörper und Peroxidase gekoppelte Sekundärantikörper. Der Transfer wurde im Nasstankverfahren bei Kühlung auf 4ºC im Eisbad mit Transferpuffer (25 mM Tris, 193 mM Glycin, 20% Methanol) durchgeführt. PVDFMembranen wurden 1 min in reinem Methanol präinkubiert. Der Aufbau von der Anode zur Kathode erfolgte auf diese Art: Schwamm – 2 Whatman Papiere – Membran – Gel – 2 Whatman Papiere – Schwamm. Über 2 Stunden wurde eine Spannung von 70V angelegt. Zur Kontrolle der erfolgreichen Übertragung der Proteine auf die Membran wurde eine Färbung mit Ponceau S über wenige Minuten durchgeführt. Anschließend wurde mit ddH2O entfärbt. Für die Detektion mittels Immunoblot wurden die Membranen zuerst mit 5% Milchpulver TBS/T (50 mM Tris, pH 7,4, 150 mM NaCl, 2 mM KCL, 0,05% TritonX100) für eine Stunde geblockt und dann bei 4ºC über Nacht mit dem in 5% Milchpulver TBS/T verdünntem Primärantikörper inkubiert. Nach Waschen mit TBS/T wurde der Sekundärantikörper bei RT für 90 min hinzugegeben. Die Membran wurde noch einmal gewaschen und die 35 Chemilumineszenz-Reaktion wurde mit Luminol durchgeführt. Luminol wird an der Peroxidase des Sekundärantikörpers zum Luminolperoxid umgewandelt, das beim Zerfall durch Film detektierbares Licht absondert. Für die Detektion von Ubiquitin wurden die Membranen nach dem Transfer zwischen Whatman Papier in ddH2O gelagert und autoklaviert. Dies erfolgte 30 min im Flüssig-Modus und danach 15 min im Trocken-Modus. Die Blockierung der Membran und die Verdünnung der Antikörper erfolgten in 20% FBS TBS/T. 3.2.5. Autoradiographie Gele mit 35S-markierten Proteinen wurden über Nacht in Fixierlösung (20% Ethanol, 6% Essigsäure) geschwenkt. Nach dem Trocknen der Gele 2 Stunden bei 76ºC unter Vakuum wurden diese einem Storage-Phosphor-Image-Screen exponiert. Die radioaktive Strahlung regte lokal die Metallsalze des Screens an. Eine Stimulation mit niederenergetischem Licht der Wellenlänge 600 nm führte zur Emission der Energie in Form von Licht, welches mit einem Typhoon-Gerät detektiert werden konnte. 3.2.6. Aufreinigung von MBP-markierten Proteinen aus Bakterien Zuerst klonierten wir Gene in pMAL, was eine n-terminale Markierung der resultierenden Proteine mit dem 42 kDa großen Maltose bindenden Protein zur Folge hatte. Wir transformierten E. coli der Stämme Rosetta (DE3) oder BL-21 mit den klonierten Konstrukten für eine selektive Übernachtkultur. 1,5-6 l LB-Medium wurden mit dieser Kultur angeimpft und bis zum Erreichen einer OD600 von 0,4-0,6 bei RT oder 37ºC geschüttelt. 0,5 mM IPTG wurde dazugegeben und die Kultur über Nacht bei RT inkubiert. Die Zellen wurden 20 min bei 4000 U/min und 4ºC zentrifugiert und mit PBS gewaschen. Nach erneutem zehnminütigen Zentrifugieren wurde 12,5 bis 50 ml 36 Lyse-Puffer (20 mM Tris pH 7,5, 500 mM NaCl, 1 mM DTT, 0,2 mg/ml Lysozym) hinzugegeben und 30 min auf Eis inkubiert. Mit Ultraschall wurden die Bakterien lysiert und anschließend 30 min bei 10000 U/min und 4ºC zentrifugiert. Der Überstand wurde auf mit 0,5-2 ml MBP-Puffer (20 mM Tris pH 7,5, 500 mM NaCl) gewaschenen Amylose-Resin gegeben und zusammen 3 Stunden bei 4ºC rotiert. Anschließend wurde 1 min bei 1000 U/min und 4ºC zentrifugiert, zweimal mit Waschpuffer 1 (20 mM Tris pH 7,5, 500 mM NaCl, 1 mM DTT, 0,01% TritonX 100) gewaschen, die Suspension in eine Säule übertragen und mit Waschpuffer 2 (20 mM Tris pH 7,5, 500 mM NaCl, 1 mM DTT) übergossen. Die Elution der an Agarose gebundenen Proteine wurde in 0,5 ml Schritten mit dem Elutionspuffer (20 mM Tris pH 7,5, 500 mM NaCl, 1 mM DTT, 10 mM Maltose) durchgeführt. Die Proteinkonzentration der einzelnen Proben wurde mittels Bradford-Reagenz und SDS-PAGE mit nachfolgender Coomassie-Färbung abgeschätzt. Gepoolte Dialyse wurde über Nacht bei 4ºC gegen 2 mM DTT PBS (137 mM NaCl, 2,7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 pH7,4) durchgeführt. 3.2.7. Aufreinigung von His6-markierten Proteinen aus Bakterien Zur Aufreinigung wurden die Gene der Proteine in pET28 und pETduet kloniert, was eine n-terminale Markierung mit sechs Histidin Aminosäuren zur Folge hatte. Wir transformierten E.coli der Stämme Rosetta (DE3) oder BL-21 mit den klonierten Konstrukten für eine selektive Übernachtkultur. 1,5-6 l LB-Medium wurde mit dieser Kultur angeimpft und bis zum Erreichen einer OD600 von 0,4-0,6 bei RT oder 37ºC geschüttelt. 0,5mM IPTG wurde dazugegeben und die Kultur über Nacht bei RT inkubiert. Die Zellen wurden 20 min bei 4000 U/min und 4ºC zentrifugiert und mit PBS gewaschen. Nach erneutem zehnminütigen Zentrifugieren wurde 12,5 bis 50 ml Lyse-Puffer (50 mM NaH2PO4, 500 mM NaCl, 10 mM Imidazol, 0,2 mg/ml Lysozym, pH 8,0) hinzugegeben und 30 min auf Eis inkubiert. Nach der Lyse der Zellen mit Ultraschall wurde der Suspension TritonX 100 auf 0,01% (v/v) hinzugefügt. Anschließend wurde 30 min bei 10000 U/min und 4ºC zentrifugiert. 37 Der Überstand wurde auf 1-4 ml Ni-NTA Agarose pipettiert und zusammen 3 Stunden bei 4ºC rotiert. Anschließend wurde 2 min bei 2000 U/min und 4ºC zentrifugiert, zweimal mit Lysepuffer mit 0,01% TritonX 100 gewaschen, die Suspension in eine Säule gegossen und erneut mit Lysepuffer ohne Triton übergossen. Die Elution erfolgte in 0,5 ml Schritten in 1,5 ml Tubes die jeweils mit 1 µl 1M DTT versehen worden waren (Elutionspuffer: 50 mM NaH2PO4, 500 mM NaCl, 200 mM Imidazol). Die Proteinkonzentration der einzelnen Proben wurde mittels Bradford-Reagenz und SDS-PAGE mit nachfolgender Coomassie-Färbung abgeschätzt. Gepoolte Dialyse wurde über Nacht bei 4ºC gegen 2mM DTT PBS (137 mM NaCl, 2,7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 pH 7,4) durchgeführt. 3.2.8. Expression und Aufreinigung von Proteinen aus Insektenzellen Um Proteine in eukaryoten Zellen zu exprimieren und danach aufzureinigen, wurden Insektenzellen mit Baculoviren infiziert, wobei die Viren die genetische Information für das jeweils entsprechende Gen trugen. Zuerst wurde die cDNA des Proteins in den Vektor pFastBac kloniert, was eine Kombination mit His6- und Flag-Tags zur Folge hatte. Mit dem Konstrukt wurden E. coli vom Stamm DH10Bac transformiert. In den Zellen fand die Einfügung des Vektors in eine 135 kbp Bacmid DNA statt, die wiederum für die Transfektion von SF9 Insektenzellen mit Cellfectin verwendet wurde. Die Bacmid DNA enthielt die Information für ein Baculovirus, das nach Transfektion in den Insektenzellen hergestellt wurde und mit der darauf folgenden Lyse der jeweiligen Zelle in das Nährmedium abgegeben wurde. So wurden alle weiteren Insektenzellen mit dem infektiösen Überstand der transfizierten Kultur 4-6 Tage inkubiert. Nach 5 min Zentrifugation bei 1500 U/min wurden die Zellen mit PBS (137 mM NaCl, 2,7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7,4) gewaschen und mit dem doppelten Eigenvolumen Lysepuffer (20 mM Tris, pH 7,5, 150 mM NaCl, 1% NP40) übergossen. Anschließend wurden sie 20 Mal in einem Glaszylinder gestampft und 15 min bei 10000 U/min und 4ºC zentrifugiert. Der Überstand wurde auf mit PBS gewaschene Ni-NTA-Agarose gegeben und nach Zugabe der gleichen Menge Waschpuffer (50 mM NaPO4, 500 mM NaCl, 20 mM Imidazol) 3 38 Stunden bei 4ºC inkubiert. Danach wurde die Agarose dreimal mit Waschpuffer gewaschen und auf eine Säule gegeben. Die Elution erfolgte mit Elutionspuffer (50 mM NaPO4, 500 mM NaCl, 250 mM Imidazol) in 500 µl Schritten. Gepoolte Dialyse erfolgte über Nacht bei 4ºC gegen 2 mM DTT PBS. 3.2.9. In vitro Transkription und Translation Das TNT SP6 Quick Coupled Transcription/ Translation System enthält neben Retikulozytenlysat auch noch eine RNA-Polymerase, einen Ribonukleaseinhibitor und alle Aminosäuren bis auf Methionin. Die Zugabe von 35 S-Methionin zu dem Reaktionsmix macht die Produktion von radioaktiv markierten Proteinen möglich. Dazu wurden 4 µl 35S-Methionin (10 mCi/ml) und 21 µl TNT Quick Master Mix mit 2,5 µg DNA in Form von pCS2 Vektoren mit SP6 Promotoren zusammen pipettiert und 90 min bei 30ºC inkubiert. 3.2.10. Immunpräzipitation Um bestimmte Proteine aus Proteingemischen oder Zellextrakten aufzureinigen, wurden auch Antikörper benutzt. Die Zellen wurden gesammelt und in IP-Puffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 5 mM KCl, 0.1% Tween 20, 2 mM DTT, Protease Inhibitor Cocktail von Roche) lysiert. Die Lysate wurden mit myc-Agarose (SantaCruz), Flag-Agarose (Sigma) oder HA-Matrix (Roche) 4 Stunden bei 4ºC inkubiert. Danach wurden die Körnchen gewaschen, eluiert oder in weiteren Assays verwendet. 3.2.11. Pulldown Die Bindung von Proteinen in vitro wurde mit Pulldown-Versuchen untersucht. MBP- oder His6-markierte Proteine wurden mit 20 µl Körner (Amylose Resin bzw. Ni-NTA-Agarose) und 300 µl MBP Bindungspuffer (20 mM HEPES pH 7,5, 5 mM KCl, 1,5 mM MgCl2, 0,1% Tween20, 1x BSA, 45 mM NaCl) bzw. His39 Bindungspuffer (20 mM HEPES pH7,5, 5 mM KCl, 1,5 mM MgCl2, 0,1% Tween20, 1x BSA, 45 mM NaCl, 5 mM Imidazol) 2 Stunden bei 4ºC rotiert. Danach wurde der Überstand abgesaugt und mit SB (20 mM HEPES pH 7,5, 5 mM KCl, 1,5 mM MgCl2) gewaschen. Nach erneuter zweistündiger Inkubation der beladenen Körner auf dem Rotator in MBP Bindungspuffer mit einem weiteren Protein wurde erneut mit SB gewaschen und mit 2xSDS-Ladepuffer eluiert. Als Negativkontrolle diente für MBP-Versuche solitäres MBP und für die HisVersuche His6-markiertes APC10. Die Analyse erfolgte über SDS-PAGE und weitere Nachweisverfahren für Proteine (s.o.). 3.2.12. Herstellung von Zelllysaten HeLa S3 Zellen wurden in Drehinkubatoren kultiviert und unter Einfluss von Thymidin (24 Stunden) und Nocodazol (12 Stunden) in der Prometaphase synchronisiert. Für G1-Extrakte wurden die Zellen ohne Nocodazol vier weitere Stunden kultiviert. Die Zellen wurden in Lysepuffer (20 mM HEPES pH 7,5, 5 mM KCl, 1,5 mM MgCl2, 1 mM DTT, 1,3 mM ATP, 15 mM Kreatinphosphat, 1 Tablette Protease Inhibitor auf 10 ml) zweimal schockgefroren, wieder aufgetaut und anschließend zehnmal durch eine Nadel der Stärke 25 G 5/8 gepresst. Nach Zentrifugation wurde der Überstand aliquotiert und bei –80ºC eingefroren (Williamson et al, 2009). 3.2.13. Deubiquitinierungsassay Für die Untersuchung der deubiquitinierenden Eigenschaften von USP4 verwendeten wir aus Insektenzellen aufgereinigtes His6-USP4 und aus E. coli (Rosetta) gewonnenes His6-USP4. Als Substrate standen verschiedene Ubiquitinketten und zuvor ubiquitinierte Proteine zur Verfügung. Die Reaktionen erfolgten unter Zugabe von 1,3 mM ATP, 10 mM DTT in UBABPuffer (25 mM Tris, 50 mM NaCl, 10 mM MgCl2). Für die Negativkontrolle wurde NEM (N-Ethylmaleimid) verwendet, welches die Cysteinprotease USP4 inhibiert. 40 Die Auswertung wurde durch anti-Ubiquitin Western Blot oder Autoradiographie vorgenommen. 3.2.14. Ubiquitinierungsassay In 1,3 mM ATP, 1 mg/ml Ubiquitin, 10 mM DTT UBAB-Puffer (25 mM Tris, 50 mM NaCl, 10 mM MgCl2) wurde unter Zugabe von His E1 (aus Insektenzellen), unterschiedlichen E2s (aus E. coli) und E3s die Ubiquitinierung von in vitro hergestellten Substraten bei 30ºC untersucht. 3.2.15. Degradationsassay Mitotische Zellextrakte wurden mit UbcH10 und p31 versetzt und mit radioaktiv gelabelten Proteinen inkubiert. Die Reaktion wurde für jedes Protein bei 0 min, nach 120 min und nach 120 min unter Hinzufügung von Emi1 und Securin gestoppt. Die Analyse erfolgte über SDS-Gelelektrophorese und Autoradiographie. 3.2.16. Massenspektrometrie Proteingemische wurden von unserem Labor auf Eis gelagert an das University of California Berkeley Proteomics and Mass Spectrometry Laboratory (http://qb3.berkeley.edu/pmsl/home.htm) ausgehändigt. Dort wurden die Proben verdaut und massenspektrometrisch analysiert. 3.3. Zellbiologische Methoden 3.3.1. Immunfluoreszenz Die auf Glasträgern (zuvor 30 min im Trockenmodus autoklaviert) bis zu 80% Konfluenz kultivierten HeLa Micha Zellen wurden mit TBS (50 mM Tris, pH 7,4, 41 150 mM NaCl, 2 mM KCl) gewaschen. Nach der 20 minütigen Behandlung mit dem Fixierpuffer (890 µl TBS, 10 µl 10% TritonX 100, 100 µl Fomaldehyd [37%]) wurden die Zellen erneut mit TBS gewaschen und 10 min mit dem Permeabilitätspuffer (960 µl Abdil, 40 µl 10% TritonX 100) inkubiert. Danach wurden sie 30 min mit Abdil (TBS, 2% BSA) blockiert und 2 Stunden mit 1:100 Primärantikörper (anti-myc oder anti-HA) in Abdil benetzt. Es folgte fünfmaliges Waschen mit Abdil und erneute Inkubation mit 1:150 Sekundärantikörper (Ziegeanti-Maus oder Ziege-anti-Kaninchen) in Abdil. Schließlich wurde dreimal in Abdil gewaschen, 10 min mit Dapi (5 µl DAPI in 1 ml Abdil) inkubiert, zweimal mit TBS gewaschen und der Objekträger fixiert. Die Objektträger wurden auf dem Mikroskop Olympus IX71 mit einem 63-fach vergrößernden Öl-Objektiv mikroskopiert. Es wurden auf jedem Objektträger mindestens 3 unabhängige areale analysiert. Repräsentative Bereiche wurden abgelichtet. 3.3.2. Proteinextraktion Die Zellen eines Wells mit 3 cm Durchmesser wurden mit 0,5 ml 1xTrypsin von der Kulturschale abgelöst und mit 0,5 ml Medium in 1,5 ml Behältern 5 min bei 5000 U/min zentrifugiert. Es wurde mit PBS gewaschen und danach 50 µl 2xSDS-Ladepuffer (62,5 mM Tris pH 6,8, 2% SDS, 25% Glycerol, 0,01% Bromphenolblau, 5% beta-Mercaptoethanol) hinzugegeben. Nach 10 min Inkubation bei 95ºC wurde 1 min bei 13000 U/min zentrifugiert und der Überstand abpipettiert. 3.3.3. Zellkultur HeLa Micha Zellen wurden in DMEM mit 10% FBS ohne Antibiotoka kultiviert. Transfektionen wurden mit TransIT-LT1 nach Herstellerangaben durchgeführt. Mit dem Flp-InTM T-RExTM Core Kit der Firma Invitrogen wurden mit vorhandenen U2OS host cells stabil transfizierte und durch Tetracyclin induzierbar exprimierende U2OS stable cell lines hergestellt. U2OS host cells wurden in DMEM mit 10% Tetracyclin depletiertem FBS und den Antibiotika Zeocin und 42 Blasticidin kultiviert. U2OS stable cell lines wurden in DMEM mit 10% Tetracyclin depletiertem FBS und den Antibiotika Hygromycin B und Blasticidin kultiviert. Die Expression wurde mit 0,2 ng/µl Doxycyclin induziert. 43 4. Ergebnisse 4.1. Bindungspartner von USP4 Um mehr über die Funktion von USP4 in der Zellzyklusregulation zu erfahren, wollten wir Proteine finden, die mit USP4 interagieren. Dafür hatte Eun Joo Song ein N-terminal mit MBP markiertes ∆297-963 USP4 aus E. coli Kulturen aufgereinigt und mit Zellextrakten aus HeLa-Zellen inkubiert. ∆297-963 USP4 ist der Teil des Proteins ohne die katalytische UCH-Domäne (Ubiquitin carboxyl-terminal hydrolase). Die an ∆297-963 USP4 gebundenen Proteine wurden gelelektrophoretisch getrennt. Es konnten zwei spezifische Banden identifiziert werden, die sich in der Massenspektroskopie als SART3 und PRP3 erwiesen. Es war aber unklar, ob USP4 diese Kandidaten direkt bindet oder die Bindung über andere Partner vermittelt wird. Um zu erfahren, wie SART3 und PRP3 mit USP4 interagieren, wurde die Bindung dieser Proteine in vitro in Pulldown-Versuchen nachgestellt. Dafür wurden die cDNA von USP4, SART3 und PRP3 in pMAL- und pET28-Vektoren kloniert und die jeweiligen Proteine aus transformierten E. coli Kulturen aufgereinigt. Die so entstandenen Proteine weisen am N-Terminus entweder ein Maltose bindendes Protein (MBP des pMAL-Vektors) oder eine Kette von sechs Histidinen (His6 des pET28-Vektors) auf. Das MBP bindet an Amylosekörnchen die aus einer Suspension durch Zentrifugation abgetrennt werden können. Für die Versuche inkubiert man das MBP-gekoppelte Protein „A“ mit den Amylosekörnchen. Danach wird der potentielle Bindungspartner „B“ dazugegeben. Man wäscht die Körnchen und eluiert die MBP-Proteine von den Körnchen. Anschließend trennt man das Eluat durch SDS-PAGE und analysiert es durch Coomassie-Färbung. Hat der Bindungspartner „B“ gebunden, so sieht man eine Bande in der Positivprobe. Um unspezifische Bindungen der anderen Proteine an MBP auszuschließen, wird für jeden Ansatz auch eine Negativkontrolle mit MBP allein pipettiert. 44 Wir konnten zeigen, dass MBP-USP4 direkt an SART3 bindet (Abb. 4-1 A. lanes 2+6/ B.). Jedoch war keine Bindung zwischen MBP-USP4 und PRP3 detektierbar (Abb. 4-1 B. lane 6). MBP-SART3 hingegen bindet sowohl His6-USP4 als auch His6-PRP3 (Abb. 4-1 C. lanes 2+4). MBP-PRP3 wiederum bindet nur an His6SART3, nicht aber an His6-USP4 (Abb. 4-1 D). Aufgrund dieser Ergebnisse gehen wir davon aus, dass USP4 direkt mit SART3 interagiert. PRP3 bindet nicht an USP4. PRP3 bindet jedoch an SART3 und wird so in den Komplex integriert. Abb. 1 E stellt diese Bindungsverhältnisse schematisch dar. Unsere Experimente geben keine Hinweise dafür, dass USP4 und PRP3 um die gleichen Bindungsareale auf SART3 konkurrieren. Die Bindung beider Partner an SART3 ist sowohl getrennt als auch in Kombination etwa gleich stark (Abb. 4-1 C. lanes 2+4 verglichen mit lane 6). Gegen eine konkurrierende Bindung spricht auch, dass His6-USP4 in Anwesenheit von His6-SART3 an MBP-PRP3 gebunden wird (Abb. 4-1 D. lane 6). Wenn man von diesem Modell ausgeht, stellt sich als nächste Frage, welche Abschnitte der Proteine miteinander interagieren. Dafür wurden mittels PCR Deletionsmutanten der beteiligten Proteine hergestellt und in Vektoren kloniert. Beim Design der Mutanten haben wir uns an der Proteindomänenstruktur der Bindungspartner orientiert. USP4 besitzt drei konservierte Domänen. Die DUSP(domain in ubiquitin specific proteases), die DUF- (domain of unknown function) und die UCH- (ubiquitin carboxyl-terminal hydrolase) Domäne (Abb. 4-2 B). Wir erstellten Deletionsmutanten, denen jeweils eine der oben genannten Domänen fehlte und untersuchten das Bindeverhalten dieser Mutanten in PulldownVersuchen. 45 Abbildung 4-1: USP4 bindet PRP3 über SART3 in vitro (MBP-Pulldown). A. USP4 bindet SART3: MBP/ MBP-USP4 wurde mit Histidin-markierten SART3 und PRP3 inkubiert. Proteine wurden mit Coomassie-Färbung sichtbar gemacht. SART3 aber nicht PRP3 bindet direkt an USP4. Bei Inkubation mit SART3 und PRP3 zusammen kann PRP3 (auf gleicher Höhe wie das Degradationsprodukt von SART3) nachgewiesen werden (lane 6). Die Bindung von PRP3 an USP4 wird also über SART3 vermittelt. B. USP4 bindet PRP3 über SART3: MBP/ MBP-USP4 wurden abwechselnd mit Histidin- und radioaktiv markierten SART3 und PRP3 inkubiert. Proteine wurden mit Autoradiographie und Coomassie-Färbung nachgewiesen. Die Bindung von PRP3 an USP4 wird über SART3 vermittelt (lane 8). C. SART3 bindet USP4 und PRP3: MBP/ MBPSART3 wurden mit Histidin-markierten USP4 und PRP3 inkubiert. Proteine wurden mittels Coomassie-Färbung dargestellt. SART3 kann USP4 und PRP3 gleichzeitig binden. D. PRP3 bindet SART3: MBP/ MBP-PRP3 wurde mit Histidin-markierten SART3 und USP4 inkubiert. Proteine wurden mit Coomassie-Färbung detektiert. Da MBP-PRP3 und His6-SART3 die gleiche Größe haben, erkennt man die Bindung nur durch die Verstärkung der Bande in Lane 2 und 6. USP4 wird nur in Anwesenheit von SART3 gebunden. E. Schematische Darstellung: Bindungsverhältnisse zwischen USP4, SART3 und PRP3. (Teile der Grafik von smart.emblheidelberg.de) 46 Mit den Vektoren stellten wir durch in vitro Transkription und Translation radioaktiv markierte USP4-Mutanten her. Diese wurden in Pulldown-Experimenten mit MBP-SART3 inkubiert. Das Eluat der Amylasekörnchen wurde mittels SDS-PAGE und Autoradiographie ausgewertet. Wir konnten zeigen, dass die UCH-Domäne keinen Einfluss auf das Bindeverhalten von USP4 zu SART3 nimmt. Verglichen mit dem Wildtyp USP4 sahen wir bei ∆287-963 USP4 in vitro keine Einschränkung der Interaktion (Daten nicht gezeigt). Anders jedoch führt die Entfernung der DUSP- oder DUF-Domäne zu einer starken Abschwächung der Interaktion zwischen USP4 und SART3. USP4-Deletionsmutanten ∆216-310 USP4 wiederum zeigt ein ungestörtes Bindeverhalten (Abb. 4-2 A.). Wir gehen also davon aus, dass Abschnitte innerhalb der sich überschneidenden DUSPund DUF-Domänen von USP4 die Bindung zu SART3 herstellen. Abbildung 4-2: USP4 bindet SART3 über DUF- und DUSP-Domänen in vitro. A. M B P Pulldown: MBP/ MBP-SART3 wurden mit mehreren Mutanten von radioaktiv markiertem USP4 inkubiert. Starke Bindung konnte nur für Mutanten (∆287-963) USP4 und (∆216-310) USP4 gezeigt werden. Die Bindung der USP4-Mutanten mit Deletion der DUF- oder DUSP-Domänen war deutlich abgeschwächt. B. Schematische Darstellung von USP4 und seine Bindung zu SART3: Neben der DUF- (domain of unknown function) und DUSP- (domain in ubiquitin specific proteases) Domäne besitzt USP4 auch die katalytische UCH- (ubiquitin carboxyl-terminal hydrolase) Domäne. USP4 bindet SART3 im Bereich der DUF- und DUSP-Domäne. (Teile der Grafik von smart.embl-heidelberg.de) 47 Den gleichen experimentellen Ansatz wie bei USP4 verwendeten wir auch bei SART3. SART3 hat eine Vielzahl von Proteindomänen (Abb. 4-3 B). Die RRM(RNA recognition motiv) als auch die HAT- (half a tetratricopeptide repeat) Domäne kommen in vielen RNA-bindenden Proteinen vor. Wir generierten Deletionsmutanten von SART3 für bestimmte Domänengruppen. Die Mutanten wurden radioaktiv in vitro hergestellt und ihr Bindeverhalten an MBP-USP4 und MBP-PRP3 in Pulldown-Experimenten untersucht. Die Analyse erfolgte über SDS-PAGE und Autoradiographie. Eine Deletion innerhalb des Aminosäurenabschnittes 287-704 (HAT 4-6-, HAT 7und coiled coil-Domäne) verhindert die Interaktion der SART3-Mutanten mit USP4 (Abb. 4-3 A.). Nur die SART3-Mutanten, welche den Abschnitt 287-704 erhalten haben, können USP4 binden. Das lässt darauf schließen, dass dieser Bereich für die Bindung von SART3 an USP4 benötigt wird. Die Entfernung der Domänen HAT 1-3 oder HAT 4-6 führt zur Unterbrechung der Bindung von SART3-Mutanten an PRP3 (Abb. 4-3 A). Für die Bindung von SART3 an PRP3 sind also die Aminosäuren 121-520 wichtig. Obwohl unsere Experimente (Abb. 4-1 C + D und 4-4 A) keinen Hinweis auf eine konkurrierende Bindung von USP4 und PRP3 an SART3 liefern konnten, sind die Bindungsbereiche der beiden Proteine auf SART3 offensichtlich überlappend. Wahrscheinlich erfolgt die tatsächliche Bindung über kleinere Bereiche, die für eine korrekte Faltung jedoch eine größere Proteinstruktur benötigen. Es wäre auch vorstellbar, dass die Bindungsstellen von SART3 von auf Ebene der Primärstruktur auseinanderliegenden Abschnitten geformt werden 48 Abbildung 4-3: Bindung von deletiertem SART3 an PRP3 und USP4 in vitro. A. MBPPulldown, daneben schematische Darstellung der SART3-Deletionsmutanten: Die Bindung von rekombinantem MBP/ MBP-PRP3/ MBP-USP4 an verschiedenen Mutanten von radioaktiv markiertem SART3 wurde in vitro untersucht und mit Autoradiographie detektiert. Für die Bindung von PRP3 an SART3 ist ∆1-120; ∆521-963 SART3 suffizient, für die Bindung an USP4 ist es ∆1286; ∆705-963 SART3. B. Schematische Darstellung von SART3 und seiner Bindung zu USP4 und PRP3: SART3 besitzt sieben HAT- (half a tetratricopeptide repeat) Domänen und zwei RRM(RNA recognition motiv) Domänen, die an RNA binden können. SART3 bindet mit den ersten sechs HAT-Domänen an PRP3. Über den Bereich der Aminosäuren 287-704 wird die Bindung zu USP4 vermittelt. (Teile der Grafik von smart.embl-heidelberg.de) 49 PRP3 verfügt über eine PWI- (Domäne in Splicing Faktoren) und eine PRP3Domäne (Abb. 4-4 C). Wir stellten Deletionsmutanten von PRP3 her, die jeweils nur eine dieser Domäne beinhalten. Die entsprechenden Proteine wurden MBPmarkiert aus transformierten Bakterienkulturen aufgereinigt. In MBP-PulldownVersuchen inkubierten wir diese PRP3-Mutanten mit in vitro translatierten und radioaktiv markierten SART3 und PRP4B. Das Eluat wurde mit SDS-PAGE und Autoradiographie analysiert. Nur die PRP3-Mutante mit PRP3-Domäne bindet an SART3 (Abb. 4-4 B. lane 10). Auch die Bindung an den Splicing Faktor PRP4B wird über diesen Teil des Proteins hergestellt (Abb. 4-4 B. lane 11). Darüber hinaus bindet PRP4B auch an SART3 (Abb. 4-4 A). PRP4B ist eine Kinase. Die Bindungen von PRP4B an SART3 und PRP3 sind nicht abhängig von der Kinaseaktivität des Proteins, da eine katalytisch inaktive Variante des Proteins (K893M)PRP4B in vitro das gleiche Bindeverhalten zeigt (Daten nicht gezeigt). Sowohl für SART3 als auch für PRP3 sind Mutationen beschrieben, die mit bestimmten Erkrankungen assoziiert sind. Für die Mutation V519M in SART3 wurde ein Zusammenhang mit Aktinischer Keratose aufgezeigt (Zhang et al, 2005). In PRP3 scheinen zwei Mutationen in Verbindung mit der Retinitis pigmentosa aufzutreten (Gonzalez-Santos et al, 2008). Es interessierte uns, ob diese Mutationen veränderte Bindungseigenschaften der Proteine zur Folge haben. Mittels Mutagenese-PCR etablierten wir die Mutationen in Vektoren, welche die Gene trugen. Wir stellten die Proteine radioaktiv markiert in vitro her und inkubierten sie mit MBP-USP4 und MBP-PRP3 bzw. MBP-SART3. Das Eluat der Pulldown-Versuche wurde mit SDS-PAGE und Autoradiographie analysiert. Die Mutanten unterschieden sich in ihrem Bindeverhalten grundsätzlich nicht von den Wildtypen (Abb. 4-5 A + B). Lediglich die Bindung von (V519M)SART3 an USP4 scheint in diesem Versuch latent abgeschwächt (Abb. 4-5 B. lane 5). 50 Abbildung 4-4: PRP3 bindet SART3 und PRP4B über PRP3-Domäne in vitro. A. MBPPulldown: MBP/ MBP-USP4 inkubiert mit radioaktiv markiertem PRP3 und 4B alternierend mit und ohne Zugabe von His6-SART3. Detektion der radioaktiven Proteine erfolgte mit Autoradiographie. PRP3/4B binden über SART3 an USP4. B. MBP-Pulldown: MBP/ MBP-PRP3 (N-90)/ MBP-PRP3 (91-300)/ MBP-PRP3 (301-C) wurden mit radioaktiv markiertem SART3 und PRP4B inkubiert. Detektion der radioaktiven Proteine erfolgte mit Autoradiographie. Bindung konnte nur für MBP-PRP3 (301-C) gezeigt werden. C. Schematische Darstellung von PRP3 und seiner Bindung zu SART mit Domänenlegende: PRP3 bindet SART über seine PRP3-Domäne. (Teile der Grafik von smart.embl-heidelberg.de) D. Schematische Darstellung der Bindungsverhältnisse zwischen SART3, PRP3, PRP4B und USP4. 51 Abbildung 4-5: Bindung von PRP3 und SART3-Mutanten in vitro. A. MBP-Pulldown: MBP und MBP-SART3 wurden mit radioaktiv markiertem PRP3-Wildtyp und PRP3-Mutanten inkubiert. Gebundene Proteine wurden mit Autoradiographie detektiert. Es wurde kein Unterschied im Bindeverhalten der PRP3-Mutanten zu SART3 zu detektiert. B. MBP-Pulldown: MBP, MBP-USP4 und MBP-PRP3 wurden mit radioaktiv markiertem SART3 und (V591M)SART3 inkubiert. Radioaktive Proteine wurden mit Autoradiographie dargestellt. Es liegt kein relevanter Unterschied im Bindeverhalten von (V591M)SART3 im Vergleich zum Wildtyp vor. 4.2. Katalytische Aktivität von USP4 Um die katalytische Aktivität von USP4 in vitro analysieren zu können, benutzten wir unterschiedliche kommerziell erworbene Ubiquitinketten und ubiquitinierte Proteine. Eine Kette von mit sechs Histidinen markiertem USP4 wurde aus Baculovirus-infizierten Insektenzellen aufgereinigt. Die Ubiquitinketten wurden mit USP4 in unterschiedlichen Konzentrationen in UBAB-Puffer inkubiert. Um einen unspezifischen Abbau der Ubiquitinketten auszuschließen, wurde eine Negativkontrolle mit NEM (N-Ethylmaleinimid) durchgeführt. NEM inhibiert spezifisch Cysteinproteasen wie USP4. Die Reaktion wurde mit SDS gestoppt und die Probe mit SDS-PAGE aufgetrennt. Die weitere Analyse erfolgte mit Western Blot, wobei die Nitrozellulosemembranen nach dem Transfer wegen Antikörperspezifität autoklaviert werden mussten. 52 Abbildung 4-6: USP4 schneidet K48 und K63 verknüpfte Ubiquitinketten. A.-F. Unterschiedliche Ubiquitinketten wurden 30 Minuten bei Raumtemperatur ohne und mit ansteigenden Mengen von aus Insektenzellen aufgereinigtem His6USP4 und der höchsten Konzentration von His6USP4 mit NEM inkubiert. Die Ubiquitinketten wurden mit Western Blot analysiert. A. Ubiquitinketten mit 3-7 Monomeren über Lysin 48 verbunden (K48-Ubi(3-7)) verdaut durch USP4. B . Ubiquitinketten mit 3-7 Monomeren über Lysin 63 verknüpft (K63-Ubi(3-7)) wurden von USP4 verdaut. C . Ubiquitinketten mit 5 Monomeren über Lysin 48 verknüpft (K48-Ubi(5)) wurden mit USP4 verdaut. D . Ubiquitinketten mit 5 Monomeren über Lysin 63 verknüpft (K63-Ubi(5)) durch USP4 verdaut. E. + F. Alternierend über Lysin 48 und 63 verknüpfte Ubiquitinketten mit 4 Monomeren (K63-K48-K63-Ubi(4) und K48-K63-K48-Ubi(4)) wurden durch USP4 verdaut. G . Input von USP4 entsprechend der Versuche A.-F. Proteine wurden mit Coomassie-Färbung analysiert. 53 In der linken Spalte der Western Blots erkennt man den Input an Ubiquitinketten. Der Nachweis von Fragmenten zeigt sich korrespondierend mit einem Verlust des Inputs in den rechts folgenden Spalten. Das bedeutet, dass die Ketten hier verdaut werden. Wir konnten so nachweisen, dass USP4 in vitro sowohl K48 als auch K63 verknüpfte Ubiquitinketten spaltet (Abb. 4-6 A + B). Dabei fällt auf, dass die K63 verknüpften Ketten von USP4 insgesamt besser abgebaut werden können als K48 verknüpfte Ketten. Eine Ausnahme stellen K48-Ubi (4 + 2) dar, die von USP4 offensichtlich sehr gut verdaut werden können. Während man in Abb. 4-6 B bei K63 Ketten einen ungefähr vergleichbaren Verdau aller Kettengrößen sieht, so kann man in Abb. 4-6 A praktisch keinen Abbau der Ketten K48Ubi(5 + 6) erkennen. Die Präferenz für K63 Ketten wird auch in den Versuchen mit Ubiquitinpentameren sichtbar (Abb. 4-6 C + D). USP4 katalysiert bereits bei einer Konzentration von ca. 2 mg/ml den kompletten Verdau der K63 Ketten (Abb. 4-6 D. lane 4), wohingegen die K48 Ketten nur teilweise verdaut werden (Abb. 4-6 C. lane 4). Bei den Experimenten mit Ubiquitinketten, die abwechselnd über K48 und K68 verknüpft sind, spaltet USP4 sowohl K48- als auch K63-Verknüpfungen (Abb. 4-6 E + F). Aus diesen in vitro Versuchen lässt sich ableiten, dass USP4 in der Lage ist, sowohl K48 als auch K63 Ubiquitinketten zu verdauen, wobei es eine Präferenz für K63 verknüpfte Ketten besitzt. Wir wussten, dass USP4 an SART3 und TRIM21 bindet. Uns interessierte, ob die Bindung dieser Proteine an USP4 einen Einfluss auf seine Aktivität hat. Wir inkubierten Ubiquitinketten mit USP4 in unterschiedlichen Konzentrationen. Dabei wurde jedem zweiten Ansatz TRIM21, SART3 oder SART3 und PRP3 hinzugefügt. Die Proben wurden mit SDS-PAGE aufgetrennt und mit Western Blots analysiert. In jeder Konzentration war der Verdau der Ubiquitinketten mit und ohne Bindungspartner etwa gleich stark. Auf die katalytische Aktivität von USP4 gegen freie Ubiquitinketten hat die Bindung von TRIM21, SART3 oder SART3 und PRP3 also keinen Einfluss (für K63-Ubi(3-7): Abb. 4-7 A; für K48Ubi(3-7): nicht gezeigt). 54 Abbildung 4-7: Bindungspartner von USP4 haben keinen Einfluss auf katalytische Aktivität. A. Ubiquitinketten mit 3-7 Monomeren über Lysin 63 verknüpft wurden mit aus Insektenzellen gewonnenem His6-USP4 in unterschiedlichen Konzentrationen verdaut. Jeder zweiten Probe wurden Bindungspartner von USP4 zugegeben (TRIM21, SART3 oder SART3+PRP3). Negativkontrolle wurde mit NEM durchgeführt. Die Analyse erfolgte mit Western Blot gegen Ubiquitin. Mit und ohne Bindungspartner zeigt USP4 die gleiche katalytische Aktivität. B. Ubiquitinketten mit 3-7 Monomeren über Lysin 48 und 63 verknüpft wurden mit aus transfizierten Bakterien aufgereinigten His6-USP4 Mutanten verdaut. Ubiquitin wurde mit Western Blot detektiert. His6-USP4 und Mutanten wurden mit Coomassie-Färbung sichtbar gemacht. Während die Mutanten ∆DUSP(∆27-125) und ∆DUF(∆82-216) ähnliche Aktivität wie der Wildtyp aufweisen, zeigt die ∆ND(∆216-310) Mutante keine katalytische Aktivität. C. An SART3 gebundene deubiquitinierende Aktivität schneidet präferentiell K63 verknüpfte Ubiquitinketten. Stabil Flag-SART3 exprimierende Zelllinie wurde zu einem Teil mit- und zum anderen Teil ohne Nocodazol inkubiert. In den Zelllysaten beider Ansätze wurde eine IP gegen Flag durchgeführt. Die gewaschenen Körnchen wurden bei Raumtemperatur 15 Minuten mit Ubiquitinketten und wahlweise NEM inkubiert. Reaktion wurde mit SDS Puffer gestoppt. Ubiquitin wurde im Western Blot analysiert. Die an SART3 gebundene deubiquitinierende Aktivität spaltet eher K63 verknüpfte Ketten. 55 Uns interessierte nun, ob die deubiquitinierende Aktivität von USP4 zellzyklusabhängig reguliert wird. Dafür benutzten wir eine Zelllinie, die stabil Flag-SART3 exprimiert. Einen Teil der Zellen arretierten wir mit Nocodazol in der Mitose. In Extrakten der nicht-arretierten und arretierten Zelllinie führten wir eine Immunpräzipitation gegen Flag durch, um die an Flag-SART3 gebundene deubiquitinierende Aktivität von USP4 zu untersuchen. Wir inkubierten das Eluat der IP mit K48- und K63-verknüpften Ubiquitinketten. Es ließ sich eine spezifische deubiquitinierende Aktivität nachweisen, die präferentiell K63-verknüpfte Ubiquitinketten schneidet (Abb. 4-7 C. lanes 5 + 7 verglichen mit lanes 1 + 3). Die Aktivität unterscheidet sich nicht wesentlich zwischen den mit Nocodazol arretierten und den nicht-arretierten Zellen (Abb. 47 C. lane 5 verglichen mit lane 7). In diesem Versuch lässt sich also kein Hinweis auf eine zellzyklusabhängige Regulation der deubiquinierenden Aktivität von USP4 finden. Um zu erfahren, welche USP4-Domänen für die Katalyse notwendig sind, reinigten wir auch Deletionsmutanten von USP4 aus transformierten E. coli auf, und führten Deubiquitinierungsassays durch. Wir inkubierten K48- und K63verknüpfte Ubiquitinketten mit den Deletionsmutanten. Wir detektierten mittels SDS-PAGE und Western Blot. Sowohl die ∆DUSP (∆27-125 USP4) als auch die ∆DUF (∆82-216 USP4) Mutanten waren katalytisch vergleichbar aktiv mit dem Wildtyp USP4. Allein die ∆ND (∆216-310 USP4/ ∆no domain) Form zeigte keine enzymatische Aktivität (Abb. 4-7 B). Es könnte also sein, dass der Abschnitt ND für die Aktivität von USP4 von Bedeutung ist. Die Domänen DUF und DUSP sind für diese Aktivität jedenfalls nicht essentiell. Wir waren auch sehr interessiert daran, mögliche Substrate von USP4 zu finden. Um zu sehen, ob USP4 PRP3 oder an PRP3 gebundene Proteine deubiquitiniert, überexprimierten wir mit Hämagglutinin (HA)-Epitop verbundenes PRP3 in HeLaZellen und führten eine IP gegen HA durch. Auf den IP-Beads führten wir dann ein Deubiquitinierungsexperiment durch. Wir eluierten mit SDS, trennten das Proteingemisch mit SDS-PAGE auf und analysierten mit Western Blot gegen Ubiquitin und das HA-Antigen (Abb. 4-8 A.). 56 In den Spalten, denen aktives USP4 zugefügt wurde, sehen wir eine Abschwächung im Ubiquitinsignal der oberen Produkte und dafür monomeres Ubiquitin (Abb. 4-8 A. lanes 2+4). Dies lässt darauf schließen, dass USP4 Ubiquitin von den in der IP detektierten Proteinen abschneidet. USP4 deubiquiniert also Proteine, die in der IP an HA-PRP3 gebunden haben und wahrscheinlich auch HA-PRP3 selber (Abb. 4-8 A). Die Deubiquitinierung erfolgte auch in Abwesenheit von rekombinantem SART3 (Abb. 4-8 A. lane 2). Dies lässt sich vielleicht damit erklären, dass HA-PRP3 in der IP bereits mit SART3 beladen war. In parallelen Experimenten konnte in HeLa-Zellen nachgewiesen werden, dass USP4 PRP3 in vivo deubiquitiniert. Dahingegen wurde SART3 in den Versuchen nicht durch USP4 deubiquitiniert (Daten von Eun Joo Song, Postdoc in der Arbeitsgruppe von Michael Rape, UC Berkeley). Abbildung 4-8: USP4 deubiquitiniert in vivo und in vitro ubiquitinierte Proteine: A. Mit einem HA-Anker verbundenes PRP3 wurde in HELA Zellen überexprimiert. 48 Stunden nach Transfektion wurden die Zellen geerntet. In dem Zelllysat wurde eine IP gegen HA-Anker durchgeführt. Die IP-Körnchen wurden gewaschen und in Puffer mit aus Insektenzellen aufgereinigtem His6-USP4, wahlweise NEM und His6-SART3 inkubiert. Im Western Blot wurden Ubiquitin und HA detektiert. USP4 spaltet (mit und ohne SART3) an PRP3 direkt oder indirekt gebundenes Ubiquitin ab. B. Radioaktives TRIM 21 und Mutanten wurden im Ubi-Mix autoubiquitiniert und dann wahlweise mit USP4 und USP4+NEM inkubiert. Ein Nachweis erfolgte mit Autoradiographie. USP4 deubiquitiniert autoubiquitiniertes Wildtyp TRIM21. TRIM21 ∆bbox wird von USP4 nicht deubiquitiniert. TRIM21 ∆pry und ∆spry werden von USP4 nur schwach deubiquitiniert. 57 Der USP4-Bindungspartner TRIM21 ist eine E3-Ligase und ubiquitiniert sich selbst. USP4 deubiquitiniert Wildtyp TRIM21. Uns interessierte, ob bestimmte Abschnitte in TRIM21 für eine Deubiquitinierung durch USP4 wichtig sind. Wir stellten TRIM21 und Deletionsmutanten radioaktiv her, induzierten Autoubiquitinierung und inkubierten mit USP4. In der ersten Spalte erkennt man den Input des radioaktiv markierten TRIM21. In der zweiten Spalte dann das autoubiquitinierte TRIM21, das wesentlich größer ist. Die dritte Spalte zeigt das deubiquitinierte TRIM21 und die vierte ist eine Negativkontrolle mit NEM. Dieses Schema wurde für den Wildtyp und alle Mutanten durchgeführt (Abb. 4-8 B). Es ist gut zu erkennen, dass eigentlich nur der Wildtyp stark deubiquitiniert wird. Die Deletion der pry- und spry-Domänen führen zu einer Abschwächung der deubiquitinierenden Wirkung von USP4. Die Deletion der bbox-Domäne unterbindet die Deubiquitinierung durch USP4 (Abb. 4-8 B. lane 7). Die Bindungen von TRIM21 und ∆bboxTRIM21 an USP4 sind in vitro ähnlich stark (Daten nicht gezeigt). Es könnte sein, dass die bbox für eine korrekte Interaktion zwischen USP4 und TRIM21 wichtig ist. Sie könnte auch eine Funktion für die korrekte Faltung des Proteins besitzen. 4.3. Lokalisation von USP4 Um weitere Bindungspartner von SART3 und damit eventuell weitere Substrate von USP4 oder andere für die Funktion von USP4 wichtige Proteine finden zu können, stellten wir eine induzierbar stabil Flag-SART3 exprimierende Zelllinie in U2OS Zellen (Osteosarkom) her. Wir züchteten mehr als 120 große Petrischalen der Zelllinie und führten in dem Lysat der Zellen eine IP gegen Flag durch (Abb. 4-9 A.). Nach detergenzfreiem Waschen wurden die Bindepartner durch Massenspektrometrie bestimmt (Abb. 4-9 B.). Das Ergebnis zeigte viele unpezifische Bindungspartner des Zellgerüsts. Interessant fanden wir in dieser Liste aber das Importin alpha, da es uns Aufschlüsse über die Zelllokalisation von USP4 geben konnte. 58 Abbildung 4-9: SART3 bindet an Importin alpha: A. anti-Flag IP im Zelllysat von U2OS Zellen und U2OS Zellen, die Flag-SART3 stabil exprimieren. Stabil Flag-SART3 exprimierende Zelllinie und Kontrollzelllinie U2OS host cells wurden in jeweils 120 großen Petrischalen herangezüchtet und lysiert. In dem Lysat wurde die IP gegen Flag durchgeführt. 10% des Eluats wurden auf dieses 10%-SDS-Gel geladen und mit Silberfärbung sichtbar gemacht. B. Mögliche Bindungspartner von SART3: 90% des Eluats der IP wurde zur massenspektrometrischen Analyse verwendet. Proteine, die mit mehr als 2 Fragmenten nachgewiesen werden konnten, wurden an uns übermittelt. Diese sind hier aufgelistet absteigend nach dem Grad der Sequenzerkennung. C. SART3 und PRP3 binden an Importin alpha C-terminal in vitro: MBP Pulldown: MBP/ MBP-PRP3/ MBP-USP4/ MBP-SART3 wurden mit Importin alpha CT inkubiert und gewaschen. Proteine wurden mit Coomassie gefärbt. Bindung an Importin alpha CT konnte für PRP3 und SART3 nachgewiesen werden. D. Bindung von Importin alpha an SART3 inhibiert die Bindung von USP4 an SART3 nicht: MBP Pulldown: MBP/ MBP-SART3 wurden mit und ohne Importin alpha CT und danach mit radioaktivem USP4 inkubiert. Proteine wurden mit CoomassieFärbung und radioaktives USP4 mit Autoradiographie sichtbar gemacht. Bindung von USP4 an Importin alpha CT SART3 ist vergleichbar mit der Bindung von USP4 an SART3 . 59 Für die in vitro Versuche benutzten wir den C-terminalen Teil von Importin alpha (Importin alpha CT), da der N-terminale Abschnitt die Bindung von Importin alpha an andere Proteine in Abwesenheit von Importin beta inhibiert (Lange et al, 2007). Wir untersuchten die Bindung von Importin alpha CT an die MBPmarkierten SART3, PRP3 und USP4 in MBP-Pulldown-Versuchen. Das Eluat wurde mit SDS-PAGE und Coomassie-Färbung analysiert. Eine relevante Bindung von Importin alpha CT konnten wir für MBP-PRP3 und MBP-SART3 an Importin alpha CT in vitro zeigen (Abb. 4-9 C. lanes 2 + 4). Für MBP-USP4 war keine Bindung detektierbar (Abb. 4-9 C. lane 3). Als Nächstes interessierte uns, ob USP4 an SART3 binden kann, auch wenn SART3 an Importin alpha gebunden ist. Dazu inkubierten wir radioaktiv markiertes USP4 mit MBP-SART3 und mit MBP-SART3, das zuvor mit Importin alpha CT beladen worden war. Die Bindung von USP4 an MBP-SART3 und MBP-SART3Importin alpha CT ist in vitro äquivalent (Abb. 4-9 D. lane 3+4). Die nächste Frage war, ob USP4 über SART3 an Importin alpha gebunden werden kann. Dazu nahmen wir MBP-Importin alpha CT und inkubierten es mit radioaktivem USP4 und wahlweise His6-SART3. Wir konnten zeigen, dass USP4 über SART3 an Importin alpha CT in vitro gebunden werden kann (Abb. 4-10 A). Aus dem Versuch geht auch hervor, dass His6-USP4 die Bindung von SART3 an Importin alpha nicht beeinflusst. SART3 bindet in und ohne Anwesenheit von His6-USP4 gleich stark an Importin alpha (Abb. 4-10 A. lanes 7+8). Die in vitro Versuche legen also nahe, dass USP4 über SART3 an Importin alpha gebunden werden könnte und so in den Nukleus gelangt. Uns interessierte nun, ob diese Bindung auch in vivo eine Relevanz hat. Dazu exprimierten wir USP4 in HeLa-Zellen in verschiedenen Kombinationen und analysierten die Lokalisation des Proteins mit Immunfluoreszenz. Wir exprimierten in einem weiteren Ansatz HA-NLS-USP4, das N-terminal die Nukleuslokalisations-Sequenz des Semian-Virus 40 trägt, in HeLa-Zellen. Darüber hinaus coexprimierten wir HA-USP4 und myc-SART3 in einem dritten Ansatz. Die Zellen wurden 48 Stunden nach der Transfektion fixiert. Primärantikörper gegen HA und myc, Alexa-488- und Cy3- gekoppelte Sekundärantikörper sowie DAPI für die 60 Kerngegenfärbung wurden für die Immunfluoreszenz verwendet. Die Abbildung 4-10.B zeigt Färbungen von repräsentativen Zellen. USP4 ist in Abwesenheit von SART3 in der gesamten Zelle und vorwiegend im Zytoplasma lokalisiert. Dagegen sehen wir für das mit einer artifiziellen NLS ausgestattete USP4 eine klare nukleäre Lokalisation des Proteins. Das legt den Schluss nahe, dass der Transport von USP4 nicht über eigene NLS vermittelt wird, da sonst auch der Wildtyp im Zellkern lokalisiert sein müsste. Bei der Coexpression von HA-USP4 und myc-SART3 kommt es zur Colokalisation der beiden Proteine im Zellkern mit Betonung der Kernmembran (Abb. 4-10 B). Abbildung 4-10: USP4 wird durch Bindung an SART3 in den Zellkern transportiert: A. USP4 bindet Importin alpha CT über SART3: MBP-Pulldown: MBP und MBP-Importin alpha CT wurden mit His6-SART3 und radioaktivem USP4 inkubiert. Proteine wurden mit CommassieFärbung und Autoradiographie detektiert. Bindung von USP4 konnte nur in Anwesenheit von SART3 aufgezeigt werden. Ebenso wurden MBP und MBP-Importin alpha CT mit His6-USP4 und radioaktivem SART3 inkubiert. Proteine wurden mit Commassie-Färbung und Autoradiographie detektiert. Die Bindung von SART3 an Importin alpha scheint durch die Anwesenheit von USP4 sogar noch verstärkt zu werden. B. Lokalisation von USP4 in vivo abhängig von SART3. Immunfluoreszenz: obere Reihe: Überexpression von HA-USP4 in HeLa-Zellen: Zytoplasmatische Lokalisation von USP4. mittlere Reihe: Überexpression von HA-NLS-USP4 (mit der Nukleus-lokalisations-Sequenz von SV40 Virus) in HeLa Zellen: klare nukleäre Lokalisation von HA-NLS-USP4. Untere Reihen: Co-Überexpression von HA-USP4 und myc-SART3 in HeLaZellen: deutliche nukleäre Lokalisation von SART3 und auch USP4. 61 Wir vermuten also, dass USP4 gekoppelt an SART3 in den Zellkern transferiert wird. Wildtyp USP4 allein wird nicht in den Zellkern gebracht, da die endogene SART3 Konzentration wahrscheinlich nicht dafür ausreicht. Die in vitro Ergebnisse werden also durch die Daten in vivo unterstützt, dass USP4 über die Bindung an SART3 in den Zellkern transportiert werden kann. 4.4. Degradation von USP4 Um zu erfahren, ob USP4 oder mit USP4 assoziierte Proteine Substrate von APC/C sind, führten wir in vitro Degradationsversuche durch. Dafür wurden die radioaktiven Proteine für zwei Stunden in mitotischen Extrakten unter Zugabe von UbcH10 und p31 bei 30ºC inkubiert. Wir führten jeweils drei Reaktionen durch, wobei die erste Reaktion sofort gestoppt wurde. Die Zweite wurde nach zwei Stunden gestoppt. Der dritten Reaktion wurden zu Beginn Emi1 und Securin zugegeben, um spezifisch die Aktivität von APC/C gegen das potentielle Substrat zu vermindern. Diese Reaktion wurde ebenfalls nach zwei Stunden gestoppt (Williamson et al, 2009). Wie bereits erwähnt, hatten wir eine Assoziation von PRP3 und PRP4B an USP4 über SART3 zeigen können. Wir vermuteten eine Assoziation von USP4 mit Rae1, Lsm2 und Nup98. Für Rae1 und Nup98 konnte gezeigt werden, dass sie für den Aufbau des Spindelapparates wichtig sind (Blower et al, 2005). Wir testeten alle diese Proteine auf Degradation in mitotischen Extrakten. Keiner der Kandidaten wurde in den mitotischen Extrakten (Abb. 4-11 A.) und G1-Extrakten degradiert. Die veränderte Bandenhöhe von Nup98 lässt sich am ehesten durch eine Phosphorylierung erklären, wobei die verantwortliche Kinase wahrscheinlich Substrat des APC/C ist (Abb. 4-11 A. right blot lane 7-9). Weder USP4 noch eines der assoziierten Proteine wurde in vitro durch APC/C reguliert. TRIM21 ist die einzige bekannte E3-Ligase, die USP4 ubiquitiniert und damit seine Stabilität beeinflusst (Wada & Kamitani, 2006). 62 Abbildung 4-11: USP4 wird nicht degradiert in mitotischen Zellextrakten: A. Degradationsversuch: Radioaktiv markiert wurden Proteine 0h, 2h und 2h unter Zugabe von Emi1 und Securin bei 30ºC in mitotischen Extrakten inkubiert. Die Reaktion wurde unter Zugabe von SDS gestoppt, und die Proteine wurden mit Autoradiographie analysiert. APC/C spezifische Degradation kann nur für die Positivkontrolle Cyclin A detektiert werden. 63 5. Diskussion Michael Rape und Mitarbeiter konnten in einem siRNA Screen gegen deubiquitinierende Enzyme zeigen, dass der Wegfall von USP4 eine Dysregulation des Zellzyklus zur Folge hat. Die Zellen, denen durch Inhibition des Spindelapparates eine korrekte Mitose nicht möglich war, konnten bei siRNA-Depletion von USP4 den physiologischen Spindelcheckpunkt passieren (Song et al, 2010). Sie zeigten den dafür typischen Phänotyp von multinukleären Zellen und lobulierten Zellkernen. USP4 wird also für die Regulation des Spindelcheckpunktes benötigt. Bei Transfektion in Zelllinien zeigte USP4 onkogene Eigenschaften, was eine Rolle von USP4 bei der Tumorigenese wahrscheinlich macht (Gray et al, 1995). Es stellte sich also nun die Frage, wie USP4 in die Regulation des Zellzyklus eingreift. 5.1. Bindungspartner von USP4 Deubiquitinierende Enzyme benötigen oft andere Proteine als Bindungspartner, um katalytisch aktiv zu sein, Substrate zu binden und auch die zelluläre Lokalisation zu verändern (Reyes-Turcu et al, 2009). Deshalb haben wir zuerst nach neuen Bindungspartnern von USP4 gesucht. Dabei stießen wir auf SART3, das in Zellen mit dem Spliceosom assoziiert ist und für das Spliceosom die Funktion eines Recyclingfaktors hat (Medenbach et al, 2004). Wir konnten zeigen, dass USP4 in vitro eine starke Bindung zu SART3 eingeht (Abb. 4-1). Eun Joo Song, Postdoc in der Arbeitsgruppe von Michael Rape an der UC Berkeley, konnte in IP-Versuchen mit Zelllysaten von HeLa-Zellen eine Bindung von USP4 an SART3 nachweisen (Song et al, 2010). USP4 und SART3 weisen in der Immunfluoreszenz eine Colokalisation im Kern der Interphasezellen auf (Abb. 4-10 B). Die Redundanz des Ergebnisses in den unterschiedlichen Systemen lässt auf eine hohe Signifikanz der Daten schließen. Zusammenfassend scheint SART3 ein fester Bindungspartner von USP4 zu sein. In vitro konnten wir mit Mutanten von USP4 und SART die Bindungsabschnitte der beiden Proteine füreinander ermitteln. USP4 bindet SART3 in seinem N64 terminalen Bereich im Abschnitt der DUSP- und DUB-Domänen (Abb. 4-3 A). Diese Teile des Proteins unterscheiden USP4 grundlegend von anderen deubiquitinierenden Enzymen. Wir können also annehmen, dass SART3 nicht unspezifisch an viele unterschiedliche DUBs bindet. Das Protein SART3 besteht wie USP4 aus 963 Aminosäuren. Es benötigt für die Bindung an USP4 die Aminosäuren 287 bis 704. Dieser mittlere Teil des Proteins bindet an USP4 (Abb. 4-2 A). In einer chinesischen Familie wurde ein Zusammenhang zwischen der Aktinischen Keratose und der Mutation V519M in SART3 festgestellt (Zhang et al, 2005). Diese Mutation liegt in dem Bereich von SART3, der an USP4 bindet. Wir konnten in vitro zeigen, dass diese Mutation die Bindung von SART zu USP4 nicht wesentlich abschwächt und also kein molekularbiologisches Korrelat der Erkrankung darstellt (Abb 4-5 A). Auf der Suche nach Bindungspartnern von USP4 war initial auch PRP3 als Kandidat gefunden worden. PRP3 bindet USP4 aber nicht direkt, sondern nur über SART3 (Abb. 4-1). SART3 bindet mit einem weiter N-terminal liegenden Abschnitt (Aminosäuren 121-486) an PRP3 (Abb. 4-2 A). Die Bindungsareale von SART3 für USP4 und PRP3 überschneiden sich. Trotzdem zeigt sich in den Experimenten keine konkurrierende Bindung der beiden Proteine an SART3 (Abb. 4-1 C). Es ist also wahrscheinlich, dass die tatsächlichen Bindungsareale wesentlich kleiner sind. Eine Erklärungsmöglichkeit wäre, dass die primärstrukturelle Integrität der größeren Proteinabschnitte für die korrekte Faltung und damit Funktion von SART3 wichtig ist. Möglich wäre aber auch, dass die Bindungsflächen von mehreren Abschnitten des Proteins gebildet werden, die in der Aminosäurensequenz nicht nebeneinander liegen. PRP3 ist wie SART3 auch ein Protein, das mit dem Spliceosom assoziiert ist. PRP3 bindet an SART3 über seine C-terminale PRP3-Domäne. Auch diese Domäne ist im Menschen nur in PRP3 vertreten, was die Spezifität der Bindung zwischen SART3 und PRP3 wahrscheinlich macht. Abbildung 5-1 A veranschaulicht schematisch die infolge der in vitro Versuche angenommenen Bindungsverhältnisse zwischen den Proteinen USP4, SART3 und PRP3 (Abb. 5-1 A). Eun Joo Song konnte diese Bindungsverhältnisse auch mit IP-Experimenten in HeLaZellextrakten nachweisen. 65 Abbildung 5-1: Schematische Darstellung der Bindungsverhältnisse zwischen USP4, SART3 und PRP3: A. Markiert sind die Bereiche der Proteine, die in in vitro Versuchen essentiell für die Bindung zwischen den Partnern sind. (Teile der Grafik von smart.embl-heidelberg.de) 5.2. Katalytische Aktivität von USP4 Neben den Bindungsverhältnissen von USP4 interessierte uns auch seine katalytische Aktivität, um mehr über die Funktionen von USP4 zu erfahren. Wir exprimierten humanes USP4 in eukaryoten SF9 Insektenzellen und reinigten dieses Protein für unsere in vitro Experimente auf. Wir konnten zeigen, dass USP4 effektiv K48 und K63 verknüpfte Ubiquitinketten spaltet (Abb. 4-6 A + B). Dabei ließ sich eine Präferenz für K63 verknüpfte Ketten feststellen (Abb. 4-7 C). K48 verknüpfte Ubiquitinketten mit fünf oder sechs Monomeren wurden von USP4 kaum verdaut (Abb. 4-6 A + C). Die Bindungspartner SART3 und TRIM21 haben in vivo keinen Einfluss auf die katalytischen Eigenschaften von USP4 (Abb. 4-7 A). Allerdings scheint der Proteinabschnitt mit den Aminosäuren 216-310 (ND) für die katalytische Aktivität von USP4 von Bedeutung zu sein. ND ist jedoch kein Teil der konservierten UCH-Domäne, die in anderen Proteinen für die deubiquitinierende Aktivität verantwortlich ist. Dafür enthält sie Bereiche mit low complexity und intrinsic disorder, welche die Bindung an andere Faktoren vermitteln können. Wir konnten allerdings keinen Bindungsfaktor finden, der die Aktivität von USP4 beeinflusst. Es wäre möglich, dass dieser Bereich für die korrekte Faltung der UCH-Domäne wichtig ist. Der Wegfall dieser Sequenz 66 könnte so eine Fehlfaltung der UCH-Domäne zur Folge haben, die ihre Funktion dann nicht mehr korrekt wahrnehmen könnte. Über K48 verknüpfte Ubiquitinketten haben in der Zelle vor allem die Aufgabe, Proteine für die Degradation durch das Proteasom zu markieren. K63-Ketten wiederum haben unterschiedliche regulatorische Funktionen vor allem in der Signaltransduktion (vgl. Einleitung). K48-Ketten weisen eine eher geschlossene Konformation auf, wohingegen K63-Ketten offen konformiert sind und so z.B. andere Proteine an das markierte Protein koppeln können (Datta et al, 2009). Die in vivo Ergebnisse machen aber klar, dass USP4 die Regulation im Zellzyklus nicht als Gegenspieler einer Degradationsubiquitinierung durchführen muss, sondern auch in regulatorische Systeme eingebunden sein kann. 5.3. Funktionen von USP4 im Zellzyklus In siRNA Experimenten zeigte sich, dass USP4 an der Regulation des Spindelcheckpunktes beteiligt ist. Der Spindelcheckpunkt wird durch die Aktivität der E3Ligase APC/C vermittelt. Wir wollten also herausfinden, ob USP4 ein Substrat von APC/C ist und auf diese Weise an der Regulation des Checkpunktes beteiligt ist. Wir untersuchten die Ubiquitinierung von USP4 als auch anderer mit USP4 assoziierter Proteine in vitro. USP4 und seine Partner sind keine Substrate von APC/C (Abb. 4-11 A). Wir mussten also weitere Möglichkeiten erwägen, welche Funktion USP4 in der Zelle hat. Spannend war nun, dass die von uns gefundenen Bindungspartner von USP4 Teile des Spliceosoms darstellen. Für das katalytisch inaktive DUB USP39 gibt es bereits Daten, dass es mit dem Spliceosom interagiert und den Zellzyklus über korrekt gesplicete mRNA von Aurora B reguliert (van Leuken et al, 2008). Darüber hinaus gibt es auch Hinweise dafür, dass Ubiquitin eine Rolle in der Regulation des Spicings spielt. Ubiquitin wurde zusammen mit Komponenten des Spliceosoms aufgereinigt. Splicingfaktoren wurden ubiquitiniert vorgefunden (Bellare et al, 2008). Außerdem besitzt der Splicingfaktor PRP19 eine UboxDomäne, was ihn als potentielles E3-Enzym denkbar macht. 67 Um herauszufinden, ob USP4 eine Funktion auf das Spliceosom ausübt interessierte uns, ob USP4 Komponenten des Spliceosoms deubiquitiniert. Wir überexprimierten HA-PRP3 in HeLa-Zellen und führten eine IP gegen HA durch. Auf den Beads führten wir in vitro Deubiquitinierungsversuche durch. Wir konnten zeigen, dass USP4 Proteine deubiquitiniert, die in dieser IP aufgereinigt wurden (Abb. 4-8 A). Dabei handelt es sich sicher um PRP3 und wahrscheinlich auch um weitere Proteine des Spliceosoms. Die Deubiquitinierung fand auch ohne Zugabe von SART3 statt, das den Kontakt zwischen USP4 und PRP3 herstellt. Auch dies lässt vermuten, dass abgesehen von PRP3 in der IP auch andere Proteine (in jedem Fall SART3) aufgereinigt wurden. Die Arbeitsgruppe von Eric Sontheimer konnte zeigen, dass auch PRPF8, ein weiterer Splicingfaktor (U5 snRNP), ubiquitiniert wird (Bellare et al, 2008). Es könnte also sein, dass auch PRPF8 durch USP4 deubiquitiniert wird. Eun Joo Song führte in vivo den Beweis der Deubiquitinierung von PRP3 durch USP4 durch. USP4, eine katalytisch inaktive Variante von USP4 und HA-PRP3 wurden in HeLa-Zellen alternierend überexprimiert, und die Zellextrakte wurden mit Western Blot gegen HA analysiert. Hier zeigte sich, dass PRP3 in HeLaZellen ubiquitiniert wird und dass USP4 PRP3 spezifisch deubiquitiniert (Song et al, 2010). Während meiner Zeit im Labor von Michael Rape an der UC Berkeley hatte ich versucht, PRP3 auf die unterschiedlichsten Weisen in vitro zu ubiquitinieren, ohne dass sich das erwartete Ergebnis zeigte. Dies gelang Eun Joo Song schließlich mit PRPF19, das mittels IP aus HeLa-Zellextrakten gewonnen wurde. Sie konnte in vitro zeigen, dass PRP3 von PRPF19 ubiquitiniert und von USP4 deubiquitiniert wird. Dabei katalysiert PRPF19 K63 verknüpfte Ubiquitinketten (Song et al, 2010). Dieses stimmt mit der katalytische Präferenz von USP4 für diese Kettenform überein (Diskussion s.o.). In Hefeextrakten konnte gezeigt werden, dass Ubiquitin für die Funktion des Spliceosoms notwendig ist. Bei Hemmung der Ubiquitinierung durch die Ubiquitinmutante I44A erlag das Splicing in den Extrakten, und die U4/U6.U5 tri snRNP wurden destabilisiert (Bellare et al, 2008). Es ist durchaus denkbar, dass die Ubiquitinierung von PRP3 für die Stabilität der U4/U6.U5 tri snRNP wichtig ist. USP4 wird durch SART3 in den Komplex eingebracht (Abb. 4-1). SART3 hat 68 die Funktion eines Recyclingfaktors für das Spliceosom (Medenbach et al, 2004). Die nahe liegende Schlussfolgerung wäre, dass die von PRPF19 und USP4 regulierte Ubiquitinierung von PRP3 den Aufbau des Spliceosoms beeinflusst und zum regelhaften Ablauf der Konformationsänderungen während des Splicings beiträgt. PRPF8 ist Teil der U5 snRNP und hat eine JAMM-Domäne, die bekanntlicherweise Ubiquitin bindet (Bellare et al, 2008). PRP3 ist Teil der U4 snRNP im Spliceosom. Während des Splicings liegen U4 und U5 snRNPs teilweise aneinander gebunden vor. Es wäre nun denkbar, dass durch Ubiquitinierung von PRP3 die Affinität von PRPF8 zu PRP3 verstärkt wird. Eun Joo Song konnte Daten generieren, die dieses Konzept nahe legen (Song et al, 2010). Mit Proteinen, die von mir aus SF9 Insektenzellen und E. coli aufgereinigt wurden, konnte Julie Aspen, Postdoc in dem Labor von Donald Rio an der UC Berkeley, in vitro Splicingexperimente durchführen. Sie gab USP4 zusammen mit SART3 zu nukleären Zellextrakten und untersuchte die Aktivität des Splicings. Dabei konnte sie zeigen, dass die Aktivität des Spliceosoms durch einen Überschuss an USP4 und SART3 inhibiert wird (Song et al, 2010). Auch diese Daten legen eine entscheidende Rolle von USP4 in der Regulation des Splicings nahe. Es ist also wahrscheinlich, dass USP4 an der Regulation des Spindelcheckpunktes über das Splicing beteiligt ist. USP4 könnte für das korrekte Splicen von mRNAs, deren Genprodukte für den Spindelcheckpunkt wichtig sind, benötigt werden. Shannon Werner, Postdoc in der Arbeitsgruppe von Michael Rape an der UC Berkeley, konnte in Experimenten zeigen, dass die mRNAs von alphaTubulin und Bub1 bei Hemmung von USP4 in viel geringerer Zahl gesplicet werden. Wenn aber das Monomer der Spindel alpha-Tubulin und das für die Regulation des Spindelcheckpunktes wichtige Bub1 nicht adäquat hergestellt werden kann, dann wird klar, warum das Ausschalten von USP4 zum Spindelcheckpunkt-Bypass führen kann. 69 5.4. Lokalisation von USP4 Da USP4 mit dem Spliceosom interagiert würde man eine nukleäre Lokalisation von USP4 in den Zellen vermuten. Tatsächlich ist das Protein in verschiedenen Zelllinien aber unterschiedlich verteilt. Während HeLa-Zellen (Zervixkarzinom) eine rein nukleäre Lokalisation von USP4 zeigen, weisen HepG2 Zellen (Hepatozelluläres Karzinom) eine zytoplasmatische Lokalisation auf. Saos-2 (Osteosarkom) und MDA-MB-231 (Mammakarzinom) zeigen sowohl nukleäre als auch zytoplasmatische Verteilung für USP4 (Soboleva et al, 2005). USP4, das in HeLa-Zellen überexprimiert wurde, lokalisierte sich zytoplasmatisch (Abb. 4-10 B). Erst die Anheftung eines artifiziellen nukleären Importsignales (SV40) an USP4 führte zum Transport von USP4 in den Nukleus. Denselben Effekt hatte auch die Co-Überexpression von SART3 (Abb. 4-10 B.). Es wurde berichtet, dass USP4 ein nukleäres Import- (NLS) und ein nukleäres Exportsignal (NES) besitzt. Der nukleäre Import von USP4 sei über die Bindung von Importin alpha/beta vermittelt (Soboleva et al, 2005). Die Bindung von Importin alpha an die NLS sei stark, die Bindung an das gesamte Protein jedoch ausgesprochen schwach (Soboleva et al, 2005). Die Bindung von USP4 an Importin beta oder alpha konnten wir in vitro nicht nachweisen. Dagegen bindet SART3 gut an Importin alpha (Abb. 4-9 C). Eine Rekrutierung von USP4 an Importin alpha über SART3 ist möglich (Abb. 4-10 A). Auf der anderen Seite liegt das mögliche NES (Aminosäuren 133-141) von USP4 im Bereich der Bindungsstelle zu SART3. Es wäre also denkbar, dass SART3 für den nukleären Transport von USP4 verantwortlich ist (Abb. 5-2 A). Eventuell kommt es durch die Bindung von SART3 an USP4 zur Maskierung der NES von USP4, was die Akkumulation von USP4 im Zellkern ermöglichen würde. Die Unterschiede in der zellulären Verteilung von USP4 bei den einzelnen Zelllinien könnten sich eventuell aus der unterschiedlichen Expressionsrate von SART3 in diesen Zellen erklären. Auch die Aktivität der Nemo like Kinase soll zur nukleären Lokalisation von USP4 beitragen (Zhao et al, 2009). Es gibt bis jetzt keine Daten, die dieses Phänomen erklären könnten. 70 Abbildung 5-2: Modellzeichnung des angenommenen nukleären Transportes von USP4: A. USP4 bindet im Zytosol an SART3, das über seine Bindung an Importin alpha in den Zellkern transportiert wird. Hier kann USP4 an SART3 gebunden seine Funktion im Splicing wahrnehmen. Es wurde gezeigt, dass USP4 in unterschiedlichen Tumorentitäten über- und unterexprimiert vorliegt. Bei einigen dieser Erkrankungen werden Spindelgifte wie Taxane therapeutisch eingesetzt. Es wäre sehr interessant zu erfahren, ob das Ansprechen auf die Therapie mit der Expression von USP4 korreliert. Nach unserem Konzept müssten Patienten mit deletiertem USP4 schlecht auf Chemotherapie mit Taxanen ansprechen, da sie den Spindelcheckpunkt umgehen können. Diesen Patienten könnte man eine nebenwirkungsreiche Therapie ersparen und anderen Patienten eine molekularbiologisch begründete, zielgerichtete Therapie anbieten. Darüber hinaus sollten die genauen Auswirkungen der Ubiquitinierung und Deubiquitinierung von PRP3 und deren Bedeutung für den Ablauf des Splicings untersucht werden. Die Suche nach neuen Substraten von USP4 muss auch weiterhin fortgesetzt werden. 71 6. Zusammenfassung In einem siRNA-Screen gegen deubiquitinierende Enzyme konnten Michael Rape und Mitarbeiter nachweisen, dass USP4 an der Regulation des Zellzyklus beteiligt ist. Zellen, die mit Spindelgiften behandelt worden waren, verloren bei Hemmung von USP4 die Fähigkeit, den Spindelcheckpunkt zu aktivieren. Wir konnten nachweisen, dass USP4 allein und ohne Bindungspartner eine deubiquitinierende Aktivität aufweist. USP4 verdaut als Protease K48- und K63Ubiquitinketten, wobei eine Präferenz für K63-Ubiquitinverbindungen besteht. Die USP4-Bindungspartner TRIM21 und SART3 haben keinen Einfluss auf diese katalytische Aktivität. USP4 bindet an SART3, das ein Protein des Spliceosoms ist. SART3 bindet an Importin alpha und vermittelt so den nukleären Transport von USP4. Über SART3 stellt USP4 auch die Verbindung zu PRP3 her. PRP3 ist ein Substrat von USP4. Es ist sehr wahrscheinlich, dass die Deubiquitinierung von PRP3 und anderen Proteinen des Spliceosoms die komplexen Proteininteraktionen während des Splicings beeinflusst. Unser Erklärungsmodell sagt aus, dass eine siRNA Hemmung von USP4 zu einer Dysregulation des Splicings führt. Die daraus folgenden geringeren Konzentrationen korrekt gespliceter mRNAs und entsprechender Genprodukte könnten im Verlust des Spindelcheckpunktes resultieren. Experimente von Shannon Werner unterstützen diese These (Song et al, 2010). Es ist also wahrscheinlich, dass USP4 das Ansprechen von Tumoren auf eine Therapie mit Spindelgifte verändert. Die klinische Relevanz dieses Effektes sollte erforscht werden. Die Ergebnisse dieser Dissertation sind teilweise in die Publikation „The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome“ in der Zeitschrift Genes and Development eingeflossen. 72 7. Literaturverzeichnis Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, Bernard S, Quarto M, Capra M, Goettig S, Kogel U, Scheffner M, Helin K, Eilers M (2005) The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 123(3): 409-421 Alberts B. JA, Lewis J., Raff M., Roberts K., Walter P. (2008) The Cell Cycle In Molecular biology of the cell., Alberts B. JA, Lewis J., Raff M., Roberts K., Walter P. (ed), Vol. 1, 5. edn, 17, pp 1053-1115. New York, NY: Garland Science Bellare P, Small EC, Huang X, Wohlschlegel JA, Staley JP, Sontheimer EJ (2008) A role for ubiquitin in the spliceosome assembly pathway. Nat Struct Mol Biol 15(5): 444-451 Blanchette P, Gilchrist CA, Baker RT, Gray DA (2001) Association of UNP, a ubiquitin-specific protease, with the pocket proteins pRb, p107 and p130. Oncogene 20(39): 5533-5537 Blower MD, Nachury M, Heald R, Weis K (2005) A Rae1-containing ribonucleoprotein complex is required for mitotic spindle assembly. Cell 121(2): 223-234 Brow DA (2002) Allosteric cascade of spliceosome activation. Annu Rev Genet 36: 333-360 Chen X, Chou CY, Chang GG (2009) Thiopurine analogue inhibitors of severe acute respiratory syndrome-coronavirus papain-like protease, a deubiquitinating and deISGylating enzyme. Antivir Chem Chemother 19(4): 151-156 Cleveland DW, Mao Y, Sullivan KF (2003) Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112(4): 407-421 Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D'Andrea AD (2007) A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell 28(5): 786-797 Damianov A, Schreiner S, Bindereif A (2004) Recycling of the U12-type spliceosome requires p110, a component of the U6atac snRNP. Mol Cell Biol 24(4): 1700-1708 Datta AB, Hura GL, Wolberger C (2009) The structure and conformation of Lys63-linked tetraubiquitin. J Mol Biol 392(5): 1117-1124 Engelbert D, Schnerch D, Baumgarten A, Wasch R (2008) The ubiquitin ligase APC(Cdh1) is required to maintain genome integrity in primary human cells. Oncogene 27(7): 907-917 73 Epstein J (2003) RNA processing and translation. In Human Molecular Biology, Epstein J (ed), Vol. 1, 1 edn, 4, pp 96-114. Cambridge: Cambridge University Press Frederick A, Rolfe M, Chiu MI (1998) The human UNP locus at 3p21.31 encodes two tissue-selective, cytoplasmic isoforms with deubiquitinating activity that have reduced expression in small cell lung carcinoma cell lines. Oncogene 16(2): 153165 Gonzalez-Santos JM, Cao H, Duan RC, Hu J (2008) Mutation in the splicing factor Hprp3p linked to retinitis pigmentosa impairs interactions within the U4/U6 snRNP complex. Hum Mol Genet 17(2): 225-239 Gray DA, Inazawa J, Gupta K, Wong A, Ueda R, Takahashi T (1995) Elevated expression of Unph, a proto-oncogene at 3p21.3, in human lung tumors. Oncogene 10(11): 2179-2183 Gupta K, Chevrette M, Gray DA (1994) The Unp proto-oncogene encodes a nuclear protein. Oncogene 9(6): 1729-1731 Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166(4): 557-580 Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67: 425-479 Herzog F, Primorac I, Dube P, Lenart P, Sander B, Mechtler K, Stark H, Peters JM (2009) Structure of the anaphase-promoting complex/cyclosome interacting with a mitotic checkpoint complex. Science 323(5920): 1477-1481 Hicke L (2001) Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2(3): 195-201 Hochstrasser M (2006) Lingering mysteries of ubiquitin-chain assembly. Cell 124(1): 27-34 Hu M, Li P, Li M, Li W, Yao T, Wu JW, Gu W, Cohen RE, Shi Y (2002) Crystal structure of a UBP-family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell 111(7): 1041-1054 Jin L, Williamson A, Banerjee S, Philipp I, Rape M (2008) Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell 133(4): 653-665 Kennedy RD, D'Andrea AD (2005) The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev 19(24): 2925-2940 Kerscher O, Felberbaum R, Hochstrasser M (2006) Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol 22: 159-180 74 Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, Corbett AH (2007) Classical nuclear localization signals: definition, function, and interaction with importin alpha. J Biol Chem 282(8): 5101-5105 Le Negrate G, Faustin B, Welsh K, Loeffler M, Krajewska M, Hasegawa P, Mukherjee S, Orth K, Krajewski S, Godzik A, Guiney DG, Reed JC (2008) Salmonella secreted factor L deubiquitinase of Salmonella typhimurium inhibits NF-kappaB, suppresses IkappaBalpha ubiquitination and modulates innate immune responses. J Immunol 180(7): 5045-5056 Logarinho E, Bousbaa H (2008) Kinetochore-microtubule interactions "in check" by Bub1, Bub3 and BubR1: The dual task of attaching and signalling. Cell Cycle 7(12): 1763-1768 Malumbres M, Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9(3): 153-166 Medenbach J, Schreiner S, Liu S, Luhrmann R, Bindereif A (2004) Human U4/U6 snRNP recycling factor p110: mutational analysis reveals the function of the tetratricopeptide repeat domain in recycling. Mol Cell Biol 24(17): 7392-7401 Meyer H (2008) Katalysemechanism im Ubiquitin/ Proteasom-System. Medizinische Hochschule Hannover(Diplomarbeit) Mosesson Y, Mills GB, Yarden Y (2008) Derailed endocytosis: an emerging feature of cancer. Nat Rev Cancer 8(11): 835-850 Mu JJ, Wang Y, Luo H, Leng M, Zhang J, Yang T, Besusso D, Jung SY, Qin J (2007) A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. J Biol Chem 282(24): 17330-17334 Nakayama KI, Nakayama K (2006) Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 6(5): 369-381 Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123(5): 773-786 Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, Gygi SP (2003) A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21(8): 921-926 Pfleger CM, Kirschner MW (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev 14(6): 655-665 Pickart CM (2001) Mechanisms underlying ubiquitination. Annu Rev Biochem 70: 503-533 75 Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, Jackson PK (2001) Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 105(5): 645-655 Reyes-Turcu FE, Ventii KH, Wilkinson KD (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 78: 363-397 Seiler JA, Conti C, Syed A, Aladjem MI, Pommier Y (2007) The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and DNA fiber analyses. Mol Cell Biol 27(16): 5806-5818 Singhal S, Taylor MC, Baker RT (2008) Deubiquitylating enzymes and disease. BMC Biochem 9 Suppl 1: S3 Soboleva TA, Jans DA, Johnson-Saliba M, Baker RT (2005) Nuclear-cytoplasmic shuttling of the oncogenic mouse UNP/USP4 deubiquitylating enzyme. J Biol Chem 280(1): 745-752 Song EJ, Werner SL, Neubauer J, Stegmeier F, Aspden J, Rio D, Harper JW, Elledge SJ, Kirschner MW, Rape M (2010) The Prp19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev 24(13): 1434-1447 Song L, Rape M (2008) Reverse the curse--the role of deubiquitination in cell cycle control. Curr Opin Cell Biol 20(2): 156-163 Stegmeier F, Sowa ME, Nalepa G, Gygi SP, Harper JW, Elledge SJ (2007) The tumor suppressor CYLD regulates entry into mitosis. Proc Natl Acad Sci U S A 104(21): 8869-8874 Thrower JS, Hoffman L, Rechsteiner M, Pickart CM (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J 19(1): 94-102 Trede NS, Medenbach J, Damianov A, Hung LH, Weber GJ, Paw BH, Zhou Y, Hersey C, Zapata A, Keefe M, Barut BA, Stuart AB, Katz T, Amemiya CT, Zon LI, Bindereif A (2007) Network of coregulated spliceosome components revealed by zebrafish mutant in recycling factor p110. Proc Natl Acad Sci U S A 104(16): 6608-6613 van Leuken RJ, Luna-Vargas MP, Sixma TK, Wolthuis RM, Medema RH (2008) Usp39 is essential for mitotic spindle checkpoint integrity and controls mRNAlevels of aurora B. Cell Cycle 7(17): 2710-2719 Vander Kooi CW, Ohi MD, Rosenberg JA, Oldham ML, Newcomer ME, Gould KL, Chazin WJ (2006) The Prp19 U-box crystal structure suggests a common dimeric architecture for a class of oligomeric E3 ubiquitin ligases. Biochemistry 45(1): 121-130 Velazquez-Fernandez D, Laurell C, Geli J, Hoog A, Odeberg J, Kjellman M, Lundeberg J, Hamberger B, Nilsson P, Backdahl M (2005) Expression profiling of 76 adrenocortical neoplasms suggests a molecular signature of malignancy. Surgery 138(6): 1087-1094 Ventii KH, Wilkinson KD (2008) Protein partners of deubiquitinating enzymes. Biochem J 414(2): 161-175 Wada K, Kamitani T (2006) UnpEL/Usp4 is ubiquitinated by Ro52 and deubiquitinated by itself. Biochem Biophys Res Commun 342(1): 253-258 Wada K, Tanji K, Kamitani T (2006) Oncogenic protein UnpEL/Usp4 deubiquitinates Ro52 by its isopeptidase activity. Biochem Biophys Res Commun 339(3): 731-736 Wahl MC, Will CL, Luhrmann R (2009) The spliceosome: design principles of a dynamic RNP machine. Cell 136(4): 701-718 Wang A, Forman-Kay J, Luo Y, Luo M, Chow YH, Plumb J, Friesen JD, Tsui LC, Heng HH, Woolford JL, Jr., Hu J (1997) Identification and characterization of human genes encoding Hprp3p and Hprp4p, interacting components of the spliceosome. Hum Mol Genet 6(12): 2117-2126 Wasch R, Engelbert D (2005) Anaphase-promoting complex-dependent proteolysis of cell cycle regulators and genomic instability of cancer cells. Oncogene 24(1): 1-10 Williamson A, Jin L, Rape M (2009) Preparation of synchronized human cell extracts to study ubiquitination and degradation. Methods Mol Biol 545: 301-312 Ye Y, Rape M (2009) Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol 10(11): 755-764 Zhang ZH, Niu ZM, Yuan WT, Zhao JJ, Jiang FX, Zhang J, Chai B, Cui F, Chen W, Lian CH, Xiang LH, Xu SJ, Liu WD, Zheng ZZ, Huang W (2005) A mutation in SART3 gene in a Chinese pedigree with disseminated superficial actinic porokeratosis. Br J Dermatol 152(4): 658-663 Zhao B, Schlesiger C, Masucci MG, Lindsten K (2009) The Ubiquitin Specific Protease 4 (Usp4) Is a New Player in the Wnt Signalling Pathway. J Cell Mol Med 77 8. Danksagung Mein herzlicher Dank richtet sich an Herrn Professor Dr. Martin Werner für die Betreuung der Doktorarbeit an der Universität Freiburg. Bei Herrn Professor Dr. Ralph Wäsch bedanke ich mich herzlich für die Übernahme des Zweitgutachtens dieser Doktorarbeit. Besonders möchte ich mich bei Herrn Professor Dr. Michael Rape bedanken, dass er mir die Möglichkeit gegeben hat, in seiner Arbeitsgruppe an der University of California Berkeley so viel zu lernen. Seine Ratschläge, Ideen und sein Verständnis haben mir bei der Arbeit sehr geholfen. Den Mitarbeitern in dem Labor möchte ich für Ihre unschätzbare Unterstützung danken. Adam Williamson, Herrmann-Josef Meyer, Kate Wickliffe, Ling Song, Lingyan Jin, Sudeep Banerjee, Xining Zhu und vor allem Eun Joo Song haben die Forschung überhaupt erst möglich gemacht. Sehr herzlich möchte ich mich auch bei Frau PD Dr. Silke Lassmann für Ihre Geduld und große Hilfe in vielen Schritten der Promotion bedanken. Den Mitarbeitern im Labor der Pathologie Freiburg, Anja Schöpflin und Corinna Herz, bin ich ebenfalls zu Dank verpflichtet. Des Weiteren möchte ich mich noch bei der Studienstiftung des deutschen Volkes bedanken, ohne deren großzügige finanzielle und ideelle Unterstützung der Aufenthalt in Berkeley, CA, nicht möglich gewesen wäre. Meiner Familie danke ich für die erfahrene Unterstützung und Motivationsspende. 78