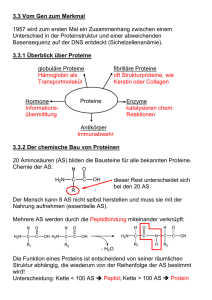
Unterschiedliche Funktionen von SRP und SecA beim Transport
Werbung

Aus dem Institut für Biochemie und Molekularbiologie der Albert-Ludwigs-Universität Freiburg i. Br. Unterschiedliche Funktionen von SRP und SecA beim Transport eines bakteriellen Proteins INAUGURAL-DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg i. Br. Vorgelegt 2001 von Christoph Neumann-Haefelin geboren in Freiburg i. Br. Dekan: Prof. Dr. rer. nat. M. Schumacher Erster Gutachter: Prof. Dr. med. Matthias Müller Zweiter Gutachter: PD Dr. med. Darius Moradpour Jahr der Promotion: 2003 Für meine Eltern Die Ergebnisse dieser Arbeit wurden veröffentlicht in: Christoph Neumann-Haefelin, Ute Schäfer, Matthias Müller and Hans-Georg Koch (2000) SRP-dependent co-translational targeting and SecA-dependent translocation analyzed as individual steps in the export of a bacterial protein. The EMBO Journal 19, pp. 6419-6426 Außerdem wurde im Rahmen dieser Arbeit veröffentlicht: Hans-Georg Koch, Thomas Hengelage, Christoph Neumann-Haefelin, Juan MacFarlane, Hedda K. Hoffschulte, Karl-Ludwig Schimz, Bernd Mechler and Matthias Müller (1999) In vitro studies with purified components reveal signal recognition particle (SRP) and SecA/SecB as constituents of two independent protein-targeting pathways of Escherichia coli. Molecular Biology of the Cell 10, pp 2163-2173 Inhaltsverzeichnis Inhaltsverzeichnis 1 ZUSAMMENFASSUNG 1 2 EINLEITUNG 2 2.1 2.2 2.2.1 2.2.2 2.3 2.3.1 2.3.2 2.4 2.4.1 2.4.2 2 4 4 5 6 6 7 7 7 2.5 2.5.1 2.5.2 2.6 Überblick: Proteintransport an der Innenmembran von Escherichia coli Transportsignale Signalsequenz sekretorischer Proteine Signal-Anker- und Stopp-Transfer-Sequenzen polytoper Innenmembranproteine Erkennung der naszierenden Ketten am Ribosom Sekretorische Proteine werden von Trigger factor erkannt Polytope Innenmembranproteine werden von SRP erkannt Targeting an das SecYEG-Translokon SecB und SecA sind am Targeting sekretorischer Proteine beteiligt SRP und sein Rezeptor FtsY sind am Targeting polytoper Innenmembranproteine beteiligt Translokation und Integration am SecYEG-Translokon Translokasefunktion des SecYEG-Translokons Integrasefunktion des SecYEG-Translokons Kooperieren SecA/SecB und SRP beim Transport bestimmter Substrate? 10 12 13 13 14 3 ZIELSETZUNG 16 4 MATERIAL UND METHODEN 17 4.1 4.1.1 4.1.2 4.1.3 4.1.4 4.1.5 Material Chemikalien Geräte und Materialien Antibiotika Escherichia coli - Stämme Plasmide Klonierung von pMomp2 Molekularbiologische Methoden DNA-Isolierung DNA-Restriktionsenzymverdau und Agarosegelelektrophorese SDS-Polyacrylamid-Gelelektrophorese Western Transfer Herstellung von Bestandteilen des zellfreien Systems Bestandteile des zellfreien Transkriptions-Translations-Systems Zytosolische Translationsfaktoren (CTF) Ribosomen Initiationsfaktor 2 17 17 17 18 19 19 19 22 22 22 23 25 26 26 26 28 28 4.2 4.2.1 4.2.2 4.2.3 4.2.4 4.3 4.3.1 Inhaltsverzeichnis 4.4 4.4.1 4.4.2 4.4.3 4.4.4 4.4.5 4.4.6 4.4.7 4.4.8 4.4.9 4.4.10 4.4.11 T7-RNA-Polymerase Membranvesikel Inside-Out Innenmembranvesikel (INV) Harnstoffextrahierte Vesikel (HINV) Translokations- und Insertionsfaktoren Ffh-Aufreinigung Versuche im zellfreien System zum Proteintransport in Escherichia coli Gekoppelte Transkription-Translation im zellfreien System Synthese ribosomenassoziierter naszierender Ketten (RANCs) Puromycin-Behandlung Protease-Resistenz-Test Differentialzentrifugation über zweistufigen Saccharosegradienten Quervernetzung mit DSS Isolierung ribosomenassoziierter naszierender Ketten (RANCs) Isolierung von Innenmembranvesikeln TCA-Fällung Immunpräzipitation SDS-PAGE und Autoradiographie 28 28 28 30 30 30 32 32 34 34 35 35 36 36 36 36 37 38 5 ERGEBNISSE 39 5.1 Momp2: Ein Hybridprotein aus Mannitpermease (MtlA) und Outer membrane protein A (OmpA) Momp2 wird effizient in „Inside-out“-Innenmembranvesikel (INV) transloziert Momp2 wird am Ribosom sowohl von Ffh als auch von Trigger factor erkannt Die Translokation von Momp2 in HINV benötigt nur SecA und SecB, jedoch nicht SRP Eine gestörte Interaktion zwischen SecA und SecY beeinträchtigt die Translokation, nicht jedoch die Membranassoziation von Momp2 Quervernetzung von Momp2-RANCs mit Membrankomponenten: Kotranslationale Interaktion zwischen Momp2 und SecY Das kotranslationale Targeting von Momp2 erfolgt SRP-abhängig Nach dem SRP-abhängigen Targeting werden SecA und SecB zur Translokation von Momp2 benötigt 4.3.2 4.3.3 5.2 5.3 5.4 5.5 5.6 5.7 5.8 39 40 42 46 48 51 53 54 6. DISKUSSION 56 6.1 Momp2: Ein Hybridprotein aus der Signal-Anker-Sequenz von MtlA und dem signalsequenzlosen OmpA als Modellprotein zum Proteintransport in Escherichia coli 56 Inhaltsverzeichnis 6.2 6.3 6.4 6.5 6.10 Die Signal-Anker-Sequenz reicht für die Erkennung durch SRP aus Trigger factor erkennt den reifen Anteil sekretorischer Proteine SRP und Trigger factor konkurrieren um naszierende Ketten Die Signal-Anker-Sequenz ermöglicht ein kotranslationales, SRP-abhängiges Targeting von Momp2 Die Translokation der periplasmatischen Domäne von Momp2 erfordert SecA Der Membrantransport von Momp2 erfolgt in drei Schritten, bei denen SRP und SecA unterschiedliche Rollen übernehmen Der Membrantransport von Innenmembranproteinen mit großen periplasmatischen Domänen erfordert eine Kooperation zwischen SRP und SecA Unterschiedliche Mechanismen bei der Integration von Innenmembranproteinen Ausblick 7. LITERATUR 69 8. ANHANG 78 8.1 8.2 Abkürzungen Plasmidkarten von pDMB, pOmpA-SpeI, p717MtlA-B, pMtlA-SpeI-2 und Momp2 Sequenzen von ompA, mtlA und momp2 78 6.6 6.7 6.8 6.9 8.3 DANKSAGUNG LEBENSLAUF 56 57 58 59 61 63 64 67 68 80 82 1 1 1 Zusammenfassung Zusammenfassung Escherichia coli besitzt als gramnegatives Bakterium eine Zellhülle aus Innenmembran (Zytoplasmamembran), Periplasma und Außenmembran. Proteine dieser Kompartimente müssen nach ihrer Synthese im Zytosol an ihre Bestimmungsorte transportiert werden. Für das Targeting (Zielsteuerung) ungefalteter Proteine an die Innenmembran und ihren anschließenden Transport sind im wesentlichen zwei unterschiedliche Transportwege beschrieben. Entscheidend für das duale Targeting ist dabei die spezifische Erkennung der jeweiligen Substrate während ihrer Synthese am Ribosom: Polytope Innenmembranproteine werden anhand ihrer Signal-Anker-Sequenzen vom Signal recognition particle (SRP) erkannt; ihr Membrantargeting und ihre Integration in die Innenmembran erfolgen kotranslational und werden von SRP und seinem Rezeptor FtsY vermittelt. Sekretorische Proteine (lösliche periplasmatische Proteine und Außenmembranproteine) werden am Ribosom hingegen von Trigger factor gebunden; ihr Membrantargeting und ihre Translokation durch die Innenmembran werden erst posttranslational von SecA und SecB vermittelt. Bisher war jedoch umstritten, ob SecA auch an der Integration bestimmter Innenmembranproteine beteiligt ist. Daher wurde in dieser Arbeit der Membrantransport eines Hybridproteins aus dem polytopen Innenmembranprotein Mannitpermease (MtlA) und dem sekretorischen Outer membrane protein A (OmpA) in einem zellfreien Transkriptions-Translations-System aus Escherichia coli untersucht. Das Hybridprotein wurde durch die Fusion der ersten SignalAnker-Sequenz von MtlA mit dem reifen Proteinteil von OmpA, also dem signalsequenzlosen OmpA, hergestellt. Diese Konstruktionsweise legt folgende Membrantopologie nahe, die experimentell bestätigt wurde: Während die von MtlA stammende Signal-Anker-Sequenz das Protein in der Innenmembran verankert, wird der von OmpA stammende Proteinanteil als periplasmatische Domäne durch die Innenmembran transloziert. Die von MtlA stammende Signal-Anker-Sequenz ermöglichte die Erkennung des Hybridproteins durch SRP sowie das durch SRP und seinen Rezeptor FtsY vermittelte kotranslationale Targeting: Im Gegensatz zu ribosomenassoziierten naszierenden Ketten von OmpA wurden ribosomenassoziierte naszierende Ketten des Hybridproteins nicht nur mit Trigger factor, sondern auch mit SRP quervernetzt. Weiterhin wurden nur in Anwesenheit von SRP und FtsY ribosomenassoziierte naszierende Ketten des Hybridproteins als Ausdruck eines kotranslationalen Targetings mit SecY quervernetzt, das die zentrale Komponente der am Proteintransport beteiligten Membranpore ist. Im Anschluß an dieses SRP-abhängige Targeting erforderte die Translokation der von OmpA stammenden periplasmatischen Domäne jedoch SecA: Die Translokation in Inside-out-Innenmembranvesikel (INV) wurde durch SRP und FtsY nicht erreicht, sondern erforderte SecA. SRP und SecA haben beim Transport dieses Hybridproteins also unterschiedliche Funktionen: SRP und FtsY vermitteln das kotranslationale Targeting und die Integration der Signal-Anker-Sequenz, während SecA die Translokation der periplasmatischen Domäne vermittelt. Diese Kooperation zwischen SRP und SecA ist wahrscheinlich auch beim Membrantransport bestimmter authentischer Innenmembranproteine in Escherichia coli nötig, die wie das hier untersuchte Hybridprotein ausgedehnte periplasmatische Domänen enthalten. 2 Einleitung 2 2 Einleitung 2.1 Überblick: Proteintransport an der Innenmembran von Escherichia coli Escherichia coli besitzt als gramnegatives Bakterium eine Zellhülle aus Innenmembran (Zytoplasmamembran), Periplasma und Außenmembran. Proteine dieser Kompartimente müssen nach ihrer Synthese im Zytosol an ihre Bestimmungsorte transportiert werden. Im folgenden werden die Vorgänge näher beschrieben, die im Zytosol und an der Innenmembran stattfinden. Hier lassen sich drei Schritte des Proteintransports voneinander abgrenzen: Zunächst werden die zu transportierenden Proteine im Zytosol spezifisch erkannt. Im zweiten Schritt werden sie an die Innenmembran herangeführt. Dieser Vorgang wird als Targeting (Zielsteuerung) bezeichnet. Schließlich werden Innenmembranproteine in die Lipidschicht integriert; lösliche periplasmatische Proteine sowie Außenmembranproteine müssen hingegen durch die Innenmembran transloziert werden. Sie werden daher entsprechend der Nomenklatur bei Eukaryonten als sekretorische Proteine bezeichnet. Diese beiden unterschiedlichen Vorgänge, nämlich Integration der Innenmembranproteine sowie Translokation sekretorischer Proteine, erfordern unterschiedliche Erkennungs-, Targeting- und Transportmaschinerien (Abb. 2.1). Die Selektion für den jeweiligen Transportweg erfolgt dabei bereits während einer frühen Phase der Proteintranslation am Ribosom: Sekretorische Proteine werden – ebenso wie zytosolische – von dem ribosomenassoziierten Chaperon Trigger factor gebunden; erst nach beendeter Translation und Loslösung vom Ribosom erfolgt das SecA- und SecB-abhängige Targeting. Sekretorische Proteine benutzen für ihre Translokation durch die Innenmembran das SecYEG-Translokon als Membranpore; dieser Vorgang erfolgt posttranslational und erfordert die ATPase-Aktivität von SecA sowie den Protonengradienten der Innenmembran. Polytope Innenmembranproteine werden hingegen von dem Signal recognition particle (SRP) gebunden, das aus dem Protein Ffh (Fifty-four homologue) und einer 4,5S RNA besteht. SRP erkennt die Innenmembranproteine anhand ihrer langen hydrophoben Signal-Anker-Sequenzen. Anschließend erfolgt durch die Interaktion zwischen SRP und seinem Rezeptor FtsY das Targeting an das SecYEG-Translokon; dieses vermittelt die kotranslationale Integration in die Innenmembran. Die Integrasefunktion des SecYEG-Translokons erfordert im Gegensatz zu seiner Translokasefunktion nur die SecY- und SecE-Untereinheiten, nicht aber SecG, und zusätzlich YidC. Drei weitere Transportwege stehen offenbar nur wenigen Proteinen offen: Das PhagenMantel-Protein M13 procoat protein benötigt offenbar nur YidC für seine ansonsten von Proteinen unabhängige Membraninsertion (Samuelson et al., 2000). Die erst kürzlich identifizierte und noch wenig charakterisierte TAT-Maschinerie (TAT für Twin arginin translocation) vermittelt den Membrantransport einiger Redoxfaktor-enthaltender Proteine in vollständig gefaltetem Zustand (Berks et al., 2000). Toxine wie z.B. Hämolysin werden mit 2 Einleitung 3 Hilfe von ABC-Transportern (ABC für ATP binding cassette) über die Innen- und Außenmembran ins Zelläußere transportiert (Binet et al., 1997). In den folgenden Kapiteln werden die einzelnen Schritte im SecA/SecB-abhängigen Transport sekretorischer Proteine und im SRP-abhängigen Transport polytoper Innenmembranproteine einander gegenübergestellt. Signalpeptidase N G G SecA G E E N C N Sec Y E Yid C E N FtsY SRP SRP ADP + Pi C SecA FtsY µH + C ATP SecB E SecA SecA Yid C Sec Y Sec Y Sec Y Sec Y N N SecB C N SRP TF SRP A TF B Abb. 2.1: Membrantransport von sekretorischen Proteinen (A) und Innenmembranproteinen (B) in Escherichia coli A: Sekretorische Proteine werden am Ribosom von Trigger factor (TF) erkannt; ihr Targeting an das SecYEG-Translokon findet posttranslational statt und wird von SecA und SecB vermittelt. SecA transloziert sekretorische Proteine in ATP-abhängigen Zyklen durch die Innenmembran; bei diesen Zyklen dreht sich die Membrantopologie von SecG um. Der Protonengradient Innenmembran + (∆µH ) unterstützt die Translokation. Die Signalpeptidase spaltet der die Signalsequenz nach ihrer Translokation ab. B: Polytope Innenmembranproteine werden kotranslational von SRP erkannt; dieses vermittelt zusammen mit seinem Rezeptor FtsY das kotranslationale Targeting an das SecYEG-Translokon. Bei der Integration in die Innenmembran sind nur SecY und SecE, nicht jedoch SecG, sowie YidC beteiligt. (Abb. modifiziert nach Müller et al., 2000) 2 Einleitung 4 2.2 Transportsignale Bereits 1975 schlugen Blobel und Dobberstein in ihrer ”Signalhypothese” vor, daß in allen Zellen sekretorische Proteine anhand N-terminaler Signale erkannt werden. In der Tat konnten sowohl für sekretorische Proteine als auch für polytope Innenmembranproteine Transportsignale identifiziert werden. 2.2.1 Signalsequenz sekretorischer Proteine Bakterielle sekretorische Proteine werden in der Regel als Vorläuferproteine (Präkursorproteine) synthetisiert, die am N-Terminus eine abspaltbare Signalsequenz tragen. Erst im Laufe ihrer Translokation wird diese Signalsequenz durch die Signalpeptidase, ein integrales Membranprotein der Innenmembran, entfernt. Die Signalsequenz ist 18 bis 26 Aminosäuren lang und setzt sich aus drei Regionen zusammen: Der positiv geladenen, Nterminalen N-Region, der hydrophoben H-Region sowie der eher polaren, C-terminalen CRegion, welche die Schnittstelle für die Signalpeptidase enthält (von Heijne, 1990). In Abb. 2.2 ist exemplarisch die Signalsequenz des Outer membrane protein A (OmpA) dargestellt. OmpA MKK TAIAIAVALAGFATV AQA N C H MtlA MSSDIKIKVQSFGRFLSNMVMPNI GAFIAWGIITALFIPTGWLP Signal-Anker-Sequenz Amphiphile Helix Abb. 2.2: Signalsequenz von OmpA (oben) und Signal-Anker-Sequenz mit vorangehender amphiphiler Helix von MtlA (unten) Oben: Die N-Region (3 AS) der OmpA-Signalsequenz enthält zwei positiv geladene Lysine (K), die H-Region (15 As, gelb hinterlegt) besteht aus hydrophoben Aminosäuren und die C-Region (3 As) enthält an den Positionen –1 und –3 vor dem reifen OmpA Alanine (A), eine Aminosäure mit kleinen, neutralen Seitenketten. Dieses Motiv wird von der Signalpeptidase erkannt, sie spaltet die Signalsequenz ab (blauer Pfeil). Unten: Die N-terminale Signal-Anker-Sequenz (20 As, gelb hinterlegt) besteht aus hydrophoben Aminosäuren; ihr geht eine amphiphile Helix voraus, die ebenfalls eine positive Nettoladung trägt: Sie enthält zwei positiv geladene Lysine (K), ein positiv geladenes Arginin (R), aber nur ein negativ geladenes Aspartat (D). Die N-Region ist 1-5 Aminosäuren lang und enthält die basischen Aminosäuren Lysin oder Arginin. Diese positive Ladung ist für die korrekte Orientierung der Signalsequenz in der Innenmembran verantwortlich: Die negativ geladenen Kopfgruppen der Membranphospholipide halten die N-Region auf der zytosolischen Seite fest (de Vrije et al., 1990); dies wird vermutlich durch das elektrochemische Membranpotential (auf der 5 2 Einleitung zytosolischen Seite negativ) unterstützt (von Heijne, 1986a). Die hydrophobe H-Region ist 7 bis 15 Aminosäuren lang und bildet eine α-Helix aus, mit der die Signalsequenz in der Innenmembran verankert wird. Die Translokationseffizienz ist von der Hydrophobizität und Länge der H-Region abhängig (Chou und Kendall, 1990; Doud et al., 1993). Die C-Region ist 3-7 Aminosäuren lang und enthält die Schnittstelle für die Signalpeptidase: Die Aminosäuren an den Positionen -1 und -3 vor der Schnittstelle tragen kleine, neutrale Seitenketten. Die Prozessierung des Vorläuferproteins ist für seine Translokation jedoch nicht essentiell: Nach einer Mutation in der Schnittstelle bleibt das Vorläuferprotein über seine Signalsequenz in der Membran verankert. Die C-Region der Signalsequenz sowie der NTerminus des reifen Proteins sind neutral oder negativ geladen: Basische Aminosäuren in dieser Region stören die korrekte Orientierung der Signalsequenz in der Innenmembran und behindern dadurch die Translokation (Li et al., 1988). Obwohl alle sekretorischen Proteine Signalsequenzen enthalten, wurde auch eine Translokation signalsequenzloser Proteine unter bestimmten Voraussetzungen beobachtet (Derman et al., 1993; Flower et al., 1994; Prinz et al., 1996). Umgekehrt führt die artifizielle Ausstattung von Proteinen mit einer Signalsequenz nicht immer zu deren Translokation (Lee et al., 1989). Dies läßt darauf schließen, daß auch der reife Proteinanteil, also das Protein ohne seine Signalsequenz, eine wesentliche Rolle beim Transport dieser Proteine spielt. Hiermit stimmt überein, daß sowohl Trigger factor als auch SecB auch an nicht-sekretorische Proteine binden. Durch genetische Analysen wurden secA-, secY-, secE- und secG-Mutanten gefunden, die einen durch eine mutierte Signalsequenz bedingten Translokationsdefekt aufheben (Bieker et al., 1990; Schatz und Beckwith, 1990; Bost und Belin, 1997). Daraus wurde zunächst geschlossen, daß diese vier Sec-Proteine direkt mit der Signalsequenz interagieren. Biochemisch konnte solch eine Interaktion jedoch nur mit SecA gezeigt werden (Miller et al., 1998). 2.2.2 Signal-Anker- und Stopp-Transfer-Sequenzen polytoper Innenmembranproteine Polytope Innenmembranproteine tragen im Gegensatz zu sekretorischen Proteinen keine abspaltbare Signalsequenz. Statt dessen sind sie mit einer sich abwechselnden Folge von Signal-Anker- und Stopp-Transfer-Sequenzen ausgestattet. Diese sind hydrophober und mit ca. 20 Aminosäuren länger als die H-Region der Signalsequenz sekretorischer Proteine. In Abb. 2.2 wird die erste Signal-Anker-Sequenz der Mannitpermease (MtlA) mit der Signalsequenz von OmpA verglichen. Die Signal-Anker- und Stopp-Transfer-Sequenzen bilden jeweils α-Helices aus, die die Innenmembran als transmembrane Domänen durchspannen. Die korrekte Membranorientierung ergibt sich aus der “positiv-inside rule”: Signal-Anker-Sequenzen sind N-terminal, Stopp-Transfer-Sequenzen jedoch C-terminal von basischen Aminosäuren flankiert (von Hejine, 1986b). Experimentell läßt sich die Membranorientierung durch einen Tausch der Ladungsverteilung umdrehen (von Heijne, 1989). 2 Einleitung 6 2.3 Erkennung der naszierenden Ketten am Ribosom Sekretorische Proteine werden posttranslational durch die Innenmembran transloziert, während polytope Innenmembranproteine kotranslational in diese integriert werden. Dies macht eine spezifische Erkennung der naszierenden Ketten schon während einer frühen Phase ihrer Synthese am Ribosom notwendig: Naszierende Ketten sekretorischer Proteine müssen von Chaperonen gebunden und an ihrer Faltung gehindert werden, da die Proteine für ihre Translokation zumindest teilweise entfaltet vorliegen müssen (Randall und Hardy, 1986). Polytope Innenmembranproteine sind sehr hydrophob und daher im wässrigen Milieu des Zytosols besonders aggregationsgefährdet (Ahrem et al., 1989). Ihre kotranslationale Integration erfordert daher einen raschen Transport der naszierenden Ketten zum SecYEGTranslokon, bevor die Translation weit fortgeschritten ist. 2.3.1 Sekretorische Proteine werden von Trigger factor erkannt Sekretorische Proteine werden während ihrer Synthese am Ribosom von Trigger factor gebunden. Trigger factor ist ein 48 kD großes, nicht essentielles Protein, das in allen bisher untersuchten Bakterien vorkommt (Bukau et al., 2000). Seinen Namen erhielt Trigger factor aufgrund der Beobachtung, daß er den Membrantransport von OmpA ”triggert”, indem er das Vorläuferprotein pOmpA in einer translokationskompetenten, lose gefalteten Konformation hält (Crooke und Wickner, 1987). Trigger factor bindet vermutlich nahe dem Ausgang des Peptidkanals an die große Ribosomenuntereinheit und ist sowohl ein Chaperon als auch eine Peptidyl-Prolyl-cis/trans-Isomerase (PPIase). Trigger factor besteht aus drei Domänen: Die N-terminale Domäne vermittelt die Bindung ans Ribosom; die zentrale Domäne besitzt PPIase-Aktivität und vermittelt die Substratbindung; die Funktion der C-terminalen Domäne ist bisher nicht bekannt (Stoller et al., 1996; Hesterkamp et al., 1997; Scholz et al., 1998). Trigger factor interagiert in vitro mit naszierenden Ketten aller bisher getesteten sekretorischen und zytosolischen Proteine (Hesterkamp et al., 1996; Valent et al., 1995,1997; Beck et al., 2000). Dabei dient die Signalsequenz sekretorischer Proteine offenbar nicht als primäre Bindestelle für Trigger factor: Trigger factor bindet auch an naszierende Ketten mit stark modifizierten Signalsequenzen (Valent et al., 1997); seine Bindestelle(n) ist (sind) also im reifen Proteinanteil enthalten. Die Interaktion von Trigger factor mit naszierenden Ketten sekretorischer Proteine verhindert das Binden von SRP: Nur in Abwesenheit von Trigger factor kann SRP mit geringer Affinität an naszierende Ketten sekretorischer Proteine binden (Beck et al., 2000). Außerdem verhindert Trigger factor das kotranslationale Binden von SecB und SecA: Nach der Beendigung der Translation und Freisetzung der Polypeptidkette vom Ribosom dissoziiert Trigger factor von der Polypeptidkette und SecB und SecA können binden (Beck et al., 2000). Trigger factor könnte also zwei unterschiedliche Rollen bei der Translokation sekretorischer Proteine erfüllen: Einerseits dient Trigger factor als Chaperon und hält sein Substrat in teilweise ungefalteter Konformation, bevor SecB diese Rolle übernehmen kann. Andererseits 7 2 Einleitung könnte Trigger factor naszierende Ketten sekretorischer Proteine vor der Bindung durch SRP schützen; die Bindung naszierender Ketten durch Trigger factor führt jedoch per se nicht zur Translokation: Auch zytosolische Proteine werden von Trigger factor erkannt und gebunden (Hesterkamp et al., 1996; Valent et al., 1995 und 1997). Außerdem führt die Depletion von Trigger factor nicht zu einem Defekt in der Proteintranslokation (Guthrie und Wickner, 1990). 2.3.2 Polytope Innenmembranproteine werden von SRP erkannt Im Gegensatz zu sekretorischen Proteinen werden naszierende Ketten polytoper Innenmembranproteine nicht von Trigger factor gebunden (Beck et al., 2000). Statt dessen werden sie vom Signal recognition particle (SRP) erkannt und gebunden (Einzelheiten zu SRP siehe 2.4.2). Dabei erkennt SRP seine Substrate anhand ihrer N-terminalen SignalAnker-Sequenzen (Valent et al., 1997; de Gier et al., 1998; Beck et al., 2000). Diese sind im Vergleich zu den Signalsequenzen sekretorischer Proteine deutlich länger und hydrophober; dementsprechend können auch sekretorische Proteine von SRP erkannt werden, wenn die Hydrophobizität ihrer Signalsequenz artifiziell erhöht wird (Valent et al., 1997; Lee und Bernstein, 2001). Ob SRP neben der N-terminalen auch die weiter C-terminal folgenden Signal-Anker-Sequenzen erkennt, ist bisher nicht geklärt. Anschließend erfolgt durch die Interaktion zwischen SRP und seinem Rezeptor FtsY das Targeting an das SecYEGTranslokon. 2.4 Targeting an das SecYEG-Translokon 2.4.1 SecB und SecA sind am Targeting sekretorischer Proteine beteiligt Nach beendeter Translation werden die sekretorischen Vorläuferproteine vom Ribosom freigesetzt. Dabei verlieren sie auch ihren Kontakt mit dem ribosomenassoziierten Chaperon Trigger factor (Beck et al., 2000). Die Vorläuferproteine müssen jetzt zum Translokon in der Innenmembran transportiert werden (Targeting); dabei müssen sie weiterhin an ihrer Faltung gehindert werden. Bei bestimmten Vorläuferproteinen übernimmt SecB die Aufgabe als Chaperon (Collier et al., 1988; Lecker et al., 1990) und kooperiert mit SecA beim Targeting dieser Vorläuferproteine (Hartl et al., 1990; Swidersky et al., 1990). SecB ist ein nicht-essentielles Protein, das nur in gramnegativen Bakterien vorkommt. Die 17 kD großen Untereinheiten bilden Homotetramere (Driessen, 2001). Vor kurzem konnte die Kristallstruktur von SecB aus Haemophilus influenzae bestimmt werden (Abb. 2.3; Xu et al., 2000). Das Tetramer ist als ein Dimer aus Dimeren organisiert und hat eine rechteckige Form. Auf beiden Seiten befindet sich ein langgestreckter Kanal, der zwei unterschiedliche Peptidbindestellen enthält. Die eine Bindestelle ist eine tiefe Spalte, die vermutlich hydrophobe und aromatische Peptidregionen erkennt; die zweite Bindestelle ist eine flache Rinne, die vermutlich ausgedehnte Peptidregionen mit hohem Gehalt an basischen Aminosäuren bindet. Mithilfe einer Peptidbibliothek wurde ein entsprechendes SecB- 2 Einleitung 8 Bindemotiv auf sekretorischen Proteinen ermittelt, das neun Aminosäuren lang und reich an aromatischen und basischen Aminosäuren ist (Knoblauch et al., 1999). Um die Peptidbindestellen auf beiden Seiten des SecB-Tetramers zu besetzen, wickeln sich die sekretorischen Proteine vermutlich als langgestreckte Polypeptidkette um das Chaperon herum. Tatsächlich wurden auf sekretorischen Proteinen bis zu 460 Aminosäuren lange Bereiche identifiziert, die von SecB gebunden werden (Smith et al., 1997). Da SecB in vitro auch an zytosolische Proteine bindet, ergibt sich die Frage, wie seine Spezifität für sekretorische Proteine in vivo entsteht. SecB erkennt Peptidsequenzen, die sich bei gefalteten Proteinen typischerweise im Inneren befinden, und bindet daher bevorzugt an ungefaltete Polypeptide (Knoblauch et al., 1999). Die Signalsequenz sekretorischer Proteine verlangsamt deren Faltung (Park et al., 1988); nach dem “Kinetic partitioning model” wird dadurch die Bindung von SecB möglich; nicht-sekretorische Proteine hingegen falten schneller und verbergen potentielle SecB-Bindemotive in ihrem Inneren (Hardy und Randall, 1991). Die Signalsequenz selbst ist jedoch keine primäre SecB-Bindestelle (Randall et al., 1990). Auch die SecA-Bindestelle auf SecB konnte mit Hilfe der Kristallstruktur näher charakterisiert werden (Abb. 2.3C): Die konservierten Aminosäuren Asp20, Glu24, Leu75 und Glu77, die schon in früheren Arbeiten als SecA-Bindestelle identifiziert wurden (Kimsey et al., 1995; Fekkes et al., 1998), liegen auf flachen Ebenen auf beiden Seiten des SecB-Tetramers. Diese Ebenen sind negativ geladen. Auch die 13 C-terminalen Aminosäuren von SecB sind an der SecA-Bindung beteiligt (Volkert et al., 1999). Sie bilden in der Kristallstruktur α-Helices in der Form von sehr beweglichen Armen. A B C Abb. 2.3: Struktur von SecB A SecB ist ein Homotetramer (Untereinheiten A-D); es ist als Dimer aus Dimeren organisiert. Die 13 C-terminalen Aminosäuren bilden jeweils α-Helices in der Form sehr beweglicher Arme (Pfeile). Der Verlauf des Peptidbindekanals ist schwarz schraffiert. B Der Peptidbindekanal (Ein- bzw. Ausgang mit Pfeilen markiert) enthält zwei unterschiedliche Bindestellen: Bindestelle 1 ist eine tiefe Spalte; Bindestelle 2 ist eine flache Rinne. C Die schon früher als SecA-Bindestelle identifizierten Aminosäuren von SecB (Pfeile) liegen auf einer polaren Ebene. Die rote Kolorierung spiegelt das Maß an Polarität wieder. Das SecBTetramer ist in (C) im Vergleich zu (A) und (B) um 90° rotiert. (Abb. aus Xu et al., 2000) 9 2 Einleitung Der zweite Targetingfaktor für sekretorische Proteine ist SecA. Er ist für das Membrantargeting sekretorischer Proteine unabdingbar (Hartl et al., 1990; Swidersky et al., 1990). SecA ist ein essentielles Protein, das seine Biosynthese auf der Stufe der Translation autoreguliert; die 102kD-Untereinheiten bilden Homodimere (Economou, 1998; Manting und Driessen, 2000). SecA-Homologe wurden in allen Bakterien sowie in Chloroplasten beschrieben, nicht jedoch in Archäen und Eukaryonten außerhalb der Chloroplasten. SecA besteht aus zwei Domänen. Die 70 kD große N-Domäne hat ATPase-Aktivität; sie enthält eine hochaffine sowie eine niedrigaffine Nukleotidbindestelle (Mitchell und Oliver, 1993). Zwischen den beiden Nukleotidbindestellen liegt die Bindestelle für das Vorläuferprotein (Kimura et al., 1991). Die 30 kD große C-Domäne ist für die Dimerisierung von SecA und für die Regulation seiner ATPase-Aktivität verantwortlich (Hirano et al., 1996) und vermittelt die Interaktion mit SecY (Snyders et al., 1997) und mit Membranphospholipiden. Die 22 Cterminalen Aminosäuren bilden die Bindestelle für SecB. Die Bindestelle ist hoch konserviert: Sie enthält die basischen Aminosäuren Arginin und Lysin sowie ein CysteinHistidin-Motiv, das ein zweiwertiges Zinkion koordiniert (Fekkes et al., 1999); dadurch wird eine optimale Bindung an die negativ geladene Bindeebene von SecB möglich. SecA liegt zur Hälfte löslich im Zytosol vor, die andere Hälfte ist mit der Innenmembran assoziiert (Müller et al., 2000). Diese Membranassoziation wird durch SecY und Phospholipide vermittelt. Mögliche Bindestellen für SecA auf Vorläuferproteinen sind bisher noch wenig charakterisiert. Sowohl die Signalsequenz als auch Regionen im reifen Proteinteil scheinen hieran beteiligt zu sein (Miller et al., 1998). Es konnte jedoch gezeigt werden, daß SecA unabhängig von SecB an Vorläuferproteine binden kann (Beck et al., 2000). Daher müssen für SecB und SecA individuelle Bindestellen existieren. In mehreren Arbeiten wurde die Bindung von SecA (Chun und Randall, 1994) und SecB (Kumamoto und Francetic, 1993; Randall et al., 1997) an ribosomenassoziierte naszierende Ketten gezeigt. Dem widersprechen jedoch neuere Daten, nach denen die Polypetidketten erst nach ihrer Ablösung vom Ribosom mit SecA und SecB interagieren. Solange die naszierende Kette am Ribosom gebunden ist, werden ihre Bindestellen für SecA und SecB offenbar durch das ribosomenassoziierte Chaperon Trigger factor blockiert (Beck et al., 2000). Dementsprechend können SecA und SecB das Membrantargeting sekretorischer Proteine erst nach deren Loslösung vom Ribosom übernehemen (Behrmann et al., 1998). Es bleibt umstritten, ob SecA in seiner löslichen Form oder in seiner membrangebundenen Form den Komplex aus Vorläuferprotein und SecB bindet: Im ersten Modell bindet lösliches SecA zunächst den Komplex aus Vorläuferprotein und SecB und bindet erst dann an das SecYEG-Translokon (Hoffschulte et al., 1994). Im zweiten Modell hat SecA bereits an das SecYEG-Translokon gebunden und dient als Rezeptor für den Komplex aus Vorläuferprotein und SecB (Fekkes et al., 1997). Jedenfalls spielt die Signalsequenz bei der Bildung des großen Komplexes aus Vorläuferprotein, SecB, SecA und SecYEG eine wesentliche Rolle (Fekkes et al., 1998). Sobald SecA ATP bindet, dissoziiert SecB von diesem großen Komplex 2 Einleitung 10 (Fekkes et al., 1997). SecA bildet hingegen mit seiner ATPase-Aktivität den Motor des Translokationsvorgangs. 2.4.2 SRP und sein Rezeptor FtsY sind am Targeting polytoper Innenmembranproteine beteiligt Das Signal recognition particle (SRP) und sein Rezeptor (SR für SRP-Rezeptor) existieren in allen drei Domänen des Lebens, also in Bakterien, Archäen und Eukaryonten. Sie wurden erstmals in Säugerzellen entdeckt und vermitteln hier das kotranslationale Targeting sowohl von sekretorischen Proteinen als auch von Membranproteinen an das Endoplasmatische Retikulum (Keenan et al., 2001). Das Säuger-SRP besteht aus einer 7SL RNA sowie sechs Proteinuntereinheiten, die nach ihrem Molekulargewicht benannt sind. Sein Rezeptor SR besteht aus einer α-Untereinheit, die die Bindestelle für SRP enthält, sowie einer β-Untereinheit, die als Membrananker dient. Sobald bei der Synthese von sekretorischen Proteinen oder Membranproteinen die Signalsequenz auf der Oberfläche des Ribosomen erscheint, wird diese von SRP erkannt: SRP bindet über seine Komponenten SRP54 und 7SL RNA sowohl an die Signalsequenz als auch an das Ribosom. Im Anschluß an die Erkennung der naszierenden Kette bewirken SRP9 und SRP14 eine Translationsarretierung; diese beiden Komponenten sind über eine spezielle Domäne der 7SL RNA, der Alu-Domäne, mit den anderen SRP-Komponenten verknüpft. Anschließend erfolgt das Targeting der arretierten naszierenden Kette durch die Bindung von SRP54 an den SRP-Rezeptor SR. Sowohl SRP54 als auch SRα haben stark homologe GTPase-Domänen; diese durchlaufen einen typischen Zyklus: Die Bindung von SRP54 an SRα erfolgt im nukleotidfreien Zustand; anschließend binden beide GTP. Hierauf wird die Signalsequenz auf den Sec61-Komplex, das Homolog zum SecYEG-Komplex, übertragen. Die anschließende GTP-Hydrolyse führt zur Dissoziation der Bindungspartner, die schnell in den nukleotidfreien Zustand zurückkehren (Rapiejko und Gilmore, 1997). Auch SRβ trägt eine GTPase-Domäne, deren Funktion allerdings noch unklar ist. In Escherichia coli existieren Homologe zu SRP54, der 7SL RNA und SRα, nämlich das Fifty-four homologue (Ffh), die 4,5S RNA sowie FtsY (Keenan et al., 2001). Alle drei SRP/SR-Komponenten sind essentielle Zellbestandteile. Ffh wird nach seinem eigenen Molekulargewicht auch P48 genannt. Es besteht wie das eukaryote homologe Protein aus drei Domänen: Die Funktion der N-Domäne ist bisher nicht bekannt. Die G-Domäne ist eine GTPase vom Ras-Typ und für die Bindung an FtsY verantwortlich (Freymann et al., 1997). Die M-Domäne bindet die Domäne IV der 4,5S RNA. Zusammen bilden sie den hoch konservierten Kern von SRP, dessen Kristallstruktur vor kurzem bestimmt wurde (Keenan et al., 1998; Batey et al., 2000): Eine tiefe Grube dient durch hydrophobe und elektrostatische Wechselwirkungen als Erkennungsstelle für Signal-Anker-Sequenzen (Abb. 2.4). Die 4,5S RNA ist mit 114 Nukleotiden wesentlich kleiner als ihr Säugerhomolog (über 300 Nukleotide). FtsY enthält wie das eukaryote homologe Protein eine N- sowie eine G-Domäne, 2 Einleitung 11 welche ebenfalls eine GTPase vom Ras-Typ ist und die SRP-Bindung vermittelt (Montoya et al., 1997). Zusätzlich verfügt FtsY über eine negativ geladene Region am N-Terminus, die ADomäne. In Escherichia coli wurde bisher kein homologes Protein zu SRβ identifiziert, das in Säugerzellen als Membrananker dient. Wahrscheinlich bindet FtsY sowohl über Phospholipide, als auch über ein bisher nicht identifiziertes Membranprotein, eventuell das Translokon, an die Membran. Dabei spielt die A-Domäne eine wichtige Rolle (Millman et al., 2001). Signalerkennungsgrube A B Abb. 2.4: Die M-Domäne von Ffh und die Domäne IV der 4,5S RNA bilden zusammen den Kern von SRP A Oberflächendarstellung des Komplexes aus der M-Domäne von Ffh (rot) und der Domäne IV der 4,5S RNA (blau). Hydrophobe Aminosäuren in der Signalerkennungsgrube sind gelb dargestellt, die angrenzenden Nukleotide grün. B In diesem Modell ist eine Signal-Anker-Sequenz (grüner und blauer Zylinder) in der Signalerkennungsgrube gebunden. Ffh ist als rotes Band dargestellt, die 4,5S RNA als schwarze Kette. Der positiv geladene N-Terminus der Signal-Anker-Sequenz zeigt zum negativ geladenen Rückrat der 4,5S RNA (in diesem Bereich ist die 4,5S RNA rot eingezeichnet). (Abb. aus Batey et al., 2000 und Walter et al., 2000) Als mögliche Substrate für das bakterielle SRP wurden polytope Innenmembranproteine identifiziert. Diese sind besonders hydrophob und aggregieren daher in vitro in der Abwesenheit von Membranlipiden schnell (Ahrem et al., 1989). Ein kotranslationales Targeting durch SRP könnte daher ihre Aggregation im wässrigen Milieu des Zytosols verhindern. In der Tat konnte gezeigt werden, daß die Integration mehrerer polytoper Innenmembranproteine von SRP abhängt (MacFarlane und Müller, 1995; Seluanov und Bibi, 1997; Tian et al., 2000). Um Proteine auf ihre Abhängigkeit von unterschiedlichen Targetingfaktoren zu untersuchen, wurde ein zellfreies System aus Escherichia coli-Extrakten entwickelt, das sowohl frei von SecA und SecB als auch von SRP und FtsY ist. Hierin konnte gezeigt werden, daß ein polytopes Membranprotein für seine Integration in Innenmembranvesikel alle drei SRP/SR-Komponenten benötigt, nicht jedoch SecA oder SecB. Für den Transport eines sekretorischen Proteins hingegen werden SecA und SecB, nicht jedoch die SRP/SR-Komponenten benötigt (Koch et al., 1999). 2 Einleitung 12 Das SRP-abhängige Targeting in Bakterien scheint dem in Säugerzellen in vielen Punkten zu entsprechen: Auch das Targeting in Bakterien erfolgt kotranslational (Beck et al., 2000) und erfordert eine Interaktion zwischen Ffh, FtsY und Membranlipiden; die Bindung von Ffh an FtsY erfolgt dabei in Abwesenheit von Nukleotiden, während die Dissoziation von Ffh und FtsY die Bindung von GTP (Valent et al., 1998; Scotti et al., 1999) sowie dessen Hydrolyse (Miller et al., 1994) erfordert. Bisher ist in Escherichia coli jedoch im Gegensatz zu Säugerzellen keine Translationsarretierung beobachtet worden. Für die SRP-Komponenten SRP9, SRP14 und die Alu-Domäne der 7SL RNA, die in Säugerzellen die Translationsarretierung bewirken, finden sich in Escherichia coli keine Homologe. Ein weiterer Unterschied zum eukaryotischen System besteht darin, daß der bakterielle SRPRezeptor FtsY sowohl löslich als auch membranassoziiert vorliegt. Ob eine funktionelle Bindung von Ffh auch an lösliches FtsY erfolgen kann, ist noch umstritten (Zelazny et al., 1997; Valent et al., 1998). 2.5 Translokation und Integration am SecYEG-Translokon Obwohl die posttranslationale Translokation sekretorischer Proteine und die kotranslationale Integration polytoper Innenmembranproteine zwei grundlegend unterschiedliche Vorgänge sind, benutzen beide die gleiche Membranpore, das SecYEG-Translokon (Koch et al., 1999). Wesentliche Unterschiede bestehen jedoch in der Energetisierung der Transportvorgänge sowie in den beteiligten Domänen und Kofaktoren des Translokons. In vivo liegen die integralen Membranproteine SecY, SecE und SecG als Komplex vor und bilden eine Membranpore, das SecYEG-Translokon (Manting und Driessen, 2000). SecY und SecE sind essentielle Proteine mit zehn bzw. drei transmembranen Domänen. SecG ist ein nicht-essentielles Protein mit zwei transmembranen Domänen. In Abwesenheit von SecE wird SecY von der Protease FtsH abgebaut (Akiyama et al., 1996). SecY und SecE bilden auch ohne SecG einen funktionellen Komplex (Akimaru et al., 1991); dabei sind multiple Kontaktstellen zwischen SecY und SecE nachgewiesen worden (Manting und Driessen, 2000). Durch die Interaktion von SecE-Molekülen unterschiedlicher SecYEG-Komplexe kommt es zu deren Oligomerisierung (Kaufmann et al., 1999). Wahrscheinlich liegt SecYEG in seiner inaktiven Form als Dimer vor und bildet in seiner aktiven Form als Translokon ein Tetramer (Manting et al., 2000). Neben SecYEG gibt es noch einen weiteren Komplex, der von den kotranskribierten Proteinen SecD, SecF und YajC gebildet wird. SecD- und secF-Deletionsmutanten sind kaum lebensfähig; YajC ist hingegen nicht essentiell. Der SecDFYajC-Komplex liegt in nur sehr wenigen Exemplaren pro Bakterienzellen vor; es ist bisher unklar, ob dieser Komplex mit SecYEG nur transient assoziiert oder einen stabilen Holokomplex bildet. 13 2 Einleitung 2.5.1 Translokasefunktion des SecYEG-Translokons Für die Translokation sekretorischer Vorläuferproteine in Proteoliposomen sind in vitro SecA, ATP und isolierte SecY-SecE-Komplexe ausreichend (Akimaru et al., 1991). In vivo wird zusätzlich SecB für das Targeting zahlreicher Vorläuferproteine benötigt. Die Rollen von SecB und SecA beim Targeting sekretorischer Vorläuferproteine an das SecYEG-Translokon wurden bereits oben beschrieben. Dieses Targeting führt zur Bindung eines Komplexes aus Vorläuferprotein, SecA und SecB an das SecYEG-Translokon. Daneben stellt SecA jedoch auch den Motor der Translokation dar (Economou und Wickner, 1994; Economou 1998): Nach der Bindung von ATP an SecA dissoziiert SecB von dem Komplex und SecA inseriert tief in die Membran. Bei dieser Membraninsertion nimmt SecA einen N-terminalen Abschnitt des Vorläuferproteins inklusive der Signalsequenz mit; diese kann hierauf von der Signalpeptidase abgespalten werden. Die Hydrolyse des gebundenen ATPs führt zur Deinsertion von SecA; durch wiederholte ATP-abhängige Insertions-Deinsertions-Zyklen kann SecA so das gesamte Protein in Abschnitten von 20 bis 40 Aminosäuren durch die Membran “hindurchfädeln”. Die Länge des je Zyklus translozierten Abschnittes wird durch hydrophobe Abschnitte und Sekundärstrukturen wie zum Beispiel Disulfidbrücken vermindert (Uchida et al., 1995; Sato et al., 1997; van der Wolk et al., 1997). Neben der ATP-Hydrolyse dient auch der Protonengradient der Innenmembran der Energetisierung der Translokation (Swidersky et al., 1990). Die Rolle der einzelnen Translokonkomponenten bei der Translokation sind erst teilweise bekannt. SecY interagiert während der Translokation direkt mit SecA und seinem Substrat. Der Austausch einer einzelnen Aminosäure in der C-terminalen zytosolischen Domäne in der secY205-Mutante verhindert die Insertion von SecA und führt somit zu einer starken Beeinträchtigung der Translokation (Matsumoto et al., 1997). Auch SecE ist für die Translokation essentiell; hierfür sind jedoch die dritte transmembrane Domäne und die angrenzende zytosolische Schleife ausreichend (Schatz et al., 1991). SecG wird für die Translokation in Proteoliposomen nicht benötigt; bei niedrigen Temperaturen oder in Abwesenheit des Protonengradienten ist es jedoch für Zellwachstum und Proteintranslokation essentiell (Hanada et al., 1996). Während der Membraninsertion von SecA dreht sich die Membrantopologie von SecG um (Nishiyama et al., 1996); möglicherweise ist diese Inversion der SecG-Topologie mit dem Insertions-Deinsertions-Zyklus von SecA gekoppelt und unterstützt diesen. Über die Rollen von SecD, SecF und YajC bei der Translokation gibt es erst vage Vorstellungen; secD- und secF-Mutanten zeigen Translokationsdefekte, yajCMutanten jedoch nicht. 2.5.2 Integrasefunktion des SecYEG-Translokons Auch naszierende Ketten verschiedener Innenmembranproteine können mit SecY quervernetzt werden (Valent et al., 1998; Scotti et al., 1999; Beck et al., 2000). Nach der Blockierung des SecYEG-Translokons ist nicht nur die Translokation sekretorischer Proteine, 2 Einleitung 14 sondern auch die Integration polytoper Innenmembranproteine beeinträchtigt (Koch et al., 1999). Dies zeigt, daß das SecYEG-Translokon auch als Integrationspore für polytope Innenmembranproteine dient. Bei der Translokation bzw. Integration sind jedoch unterschiedliche Domänen von SecY beteiligt: Die kältesensitive Mutante secY40 reagiert stark sensitiv auf reduzierte SRP-Spiegel und zeigt reduzierte Integrationsraten, jedoch keinen Translokationsdefekt (Newitt und Bernstein, 1998). Eine Mutation in der C-terminalen zytosolischen Domäne von SecY (secY205) stört die Interaktion mit SecA (Matsumoto et al., 1997); hierdurch wird die Translokation stark eingeschränkt, während die Integration nicht betroffen ist (Koch und Müller, 2000). Neben SecY erfordert die Integration polytoper Innenmembranproteine auch SecE: Konditionelle secE-Mutanten sowie secY24-Mutanten, deren Interaktion mit SecE gestört ist, zeigen sowohl einen Translokations- als auch einen Integrationsdefekt (Koch und Müller, 2000). Wie oben beschrieben, spielt SecG wahrscheinlich bei den InsertionsDeinsertions-Zyklen von SecA eine Rolle. Da SecA an der kotranslationalen Integration polytoper Innenmembranproteine nicht beteiligt ist, sollte dies folglich auch für SecG zutreffen. In der Tat wurde in einer secG-Mutante ein Translokationsdefekt, jedoch keine Beeinträchtigung der Integration polytoper Innenmembranproteine beobachtet (Koch und Müller, 2000). Somit ist auch verständlich, daß Eukaryonten SecY- und SecE-, nicht jedoch SecG-homologe Proteine haben: In Eukaryonten gibt es auch kein SecA. Vor kurzem wurde beobachtet, daß auch YidC an der Integration SRP-abhängiger Innenmembranproteine beteiligt ist (Scotti et al., 2000; Houben et al., 2000). YidC ist ein essentielles integrales Membranprotein, daß zu Oxa1p in Mitochondrien und Albino3 in Chloroplasten homolog ist. YidC scheint auf die Integration von Membranproteinen spezialisiert zu sein: Auch die Insertion des M13 procoat protein, die nicht auf die Hilfe anderer Proteine angewiesen ist, benötigt YidC (Samuelson et al., 2000). Die Integration von Innenmembranproteinen findet im Gegensatz zur Translokation sekretorischer Proteine kotranslational statt (Beck et al., 2000). Dabei geht - wie schon früher in Säugerzellen beobachtet - das Ribosom auch in Bakterien einen engen Kontakt mit dem Translokon ein und dichtet es dadurch vermutlich ab (Prinz et al., 2000). 2.6 Kooperieren SecA/SecB und SRP beim Transport bestimmter Substrate? In den vorangehenden Kapiteln wurden die SecA/SecB-abhängige Translokation sekretorischer Proteine und die SRP-abhängige Integration polytoper Innenmembranproteine einander gegenübergestellt. In mehreren Arbeiten wurde jedoch gezeigt, daß neben SRP auch SecA am Membrantransport bestimmter Innenmembranproteine beteiligt ist. Die dabei untersuchten Proteine besitzen neben transmembranen Domänen zusätzlich ausgedehnte periplasmatische Domänen, die durch die Innenmembran transloziert werden müssen (Details siehe 6.8). Bisher war die Rolle von SecA beim Transport dieser Proteine umstritten: 15 2 Einleitung Einerseits wurde die Interaktion von SecA mit Innenmembranproteinen als unspezifische Interaktion gedeutet (Manting und Driessen, 2000), andererseits wurde vorgeschlagen, daß SecA die Translokation der großen periplasmatischen Domänen katalysiert (Scotti et al., 1999; Dalbey et al., 2000); ein weiteres Modell ging davon aus, daß SecA ein obligater Bestandteil des Translokons ist und auch bei der Integration transmembraner Domänen eine essentielle Funktion hat (Qi und Bernstein, 1999). 3 Zielsetzung 3 16 Zielsetzung Für das Targeting ungefaltener Proteine an die Innenmembran und ihren anschließenden Transport sind in Escherichia coli im wesentlichen zwei unterschiedliche Wege beschrieben (Abb. 2.1): Das Membrantargeting und die Translokation sekretorischer Proteine werden durch Trigger factor, SecA und SecB vermittelt, während das Membrantargeting und die Integration polytoper Innenmembranproteine durch das Signal recognition particle (SRP) sowie seinen Rezeptor FtsY vermittelt werden. Entscheidend für dieses duale Targeting ist dabei die spezifische Erkennung der jeweiligen Substrate, die während ihrer Synthese am Ribosom stattfindet: Sekretorische Proteine werden anhand ihrer reifen Proteinanteile durch Trigger factor erkannt; ihre Signalsequenz scheint dabei eine untergeordnete Rolle zu spielen. Polytope Innenmembranproteine werden hingegen anhand ihrer Signal-Anker-Sequenz(en) durch SRP erkannt. Dieses Konzept zweier individueller Transportmechanismen beruht im wesentlichen auf Arbeiten mit dem sekretorischen SecA/SecB-abhängigen Protein Outer membrane protein A (OmpA) sowie dem SRP-abhängigen polytopen Innenmembranprotein Mannitpermease (MtlA) (Koch et al., 1999; Beck et al., 2000). Es wird jedoch durch Arbeiten in Frage gestellt, denen zufolge sowohl SRP als auch SecA an der Integration bestimmter Innenmembranproteine beteiligt sind (Traxler und Murphy, 1996; Valent et al., 1998; Qi und Bernstein, 1999; Tian et al., 2000). Ziel dieser Arbeit war es daher, in dem für OmpA und MtlA etablierten zellfreien System zu klären, ob für die Integration bestimmter Innenmembranproteine sowohl SRP als auch SecA benötigt werden. Gegebenenfalls sollten die unterschiedlichen Funktionen von SRP und SecA bei der Integration dieser Innenmembranproteine untersucht werden. Als Modellprotein wurde ein Hybridprotein aus der ersten Signal-Anker-Sequenz von MtlA sowie dem signalsequenzlosen OmpA gewählt. Da die Signal-Anker-Sequenz von MtlA die Erkennungssequenz für SRP darstellt, war auch eine Erkennung des Hybridproteins Momp2 durch SRP zu erwarten. So konnte geprüft werden, ob SRP alleine zur Integration von Momp2 führt oder ob hierzu zusätzlich SecA benötigt wird. Dazu sollten das Hybridprotein sowie OmpA und MtlA als ausschließlich SecA/SecB- bzw. SRP-abhängige Kontrollproteine in einem zellfreien Transkriptions-Translations-System aus E. coli synthetisiert werden; anschließend sollte ihre Interaktion mit unterschiedlichen Targeting- und Transportfaktoren in Quervernetzungsversuchen untersucht und die Bedingungen für ihren Transport in Innenmembranvesikel durch unterschiedliche Methoden (Protease-Resistenz-Test und Differentialzentrifugation) charakterisiert werden. 17 4 4 Material und Methoden Material und Methoden 4.1 Material 4.1.1 Chemikalien S-Methionin / 35S-Cystein Markierungsmischung; „Enhanced Chemiluminescence ECL Westernblotting System“ Baker: Polyethylenglykol 6000 Boehringer Mannheim: Alkalische Phosphatase; Ampicillin; Aprotinin; ATP; Chloramphenicol; CTP; E64 Proteaseinhibitor; GTP; Kanamycinsulfat; Kreatinphosphat; Kreatinphoshatkinase; Leupeptin; PMSF; Proteinase K; RNaseH (Ribonuklease H); Tetracylin; UTP Caltag Laboratories: Ziege-anti-Kaninchen-Antikörper, Meerrettich-Peroxidase-gekoppelt DIFCO: Bacto-Agar für Agarplatten BRL Life Technologies: 1-kb-DNA-Standard; gefärbte Molekulargewichtsmarker, niedrigbzw. hochmolekular ICN: Saccharose (ultra rein) Kleeblatt: Milchpulver New England BioLabs: Restriktionsenzyme; T4-DNA-Polymerase; T4-DNA-Ligase Merck: Ammoniumperoxodisulfat; Bromphenolblau Pharmacia: Protein-A-Sepharose Promega: RNasin RNase-Inhibitor Pierce: DSS Roth: 30 % Acrylamid / 0.8 % Bisacrylamid; BSA; DTT; EDTA; Essigsäure; Ethanol; Ethidiumbromid; Glukose; Glycerin; Glycin; Harnstoff; HCl; Hefeextrakt; Imidazol; IPTG dioxanfrei; Isobuthanol; Isopropanol; Kaliumacetat; KH2PO4; K2HPO4; KOH; Magnesiumacetat; MgCl2; NaCl; NaOH; Pepton aus Casein; Ponceau S; SDS; TCA; Tris Base ultra rein; Triton X100 Serva: Agarose für DNA-Elektrophorese; Brilliant Blau G 250 Sigma: 18 L-Aminosäuren (ohne Methionin und Cystein); DMSO rein; Fruktose; Phosphoenolpyruvat; Puromycin; Putrescin; Spermidin; TEMED; Triethanolamin; Tween 20; Xylencyanol DNA-Oligonukleotide wurden von Dr. G. Igloi am Institut für Biologie III der Universität Freiburg synthetisiert. Amersham: 35 4.1.2 Geräte und Materialien Abimed Gilson: Agfa: Bachhofer: Peristaltikpumpe Minipuls 2 Röntgenfilmentwickler Curix 60 UV Transilluminator 312 nm 4 Material und Methoden 18 Beckman: Airfuge-Ultrazentrifuge mit Rotor A-100/30; Ultrazentrifuge TL100; Rotor TLA 100.2; Ausschwingrotor SW28; Rotor 50.2 Ti Biorad: Geltrockner 583; Western-Transfer-Apparatur Trans-BlotTM Cell Eppendorf: Thermomixer 5436 Fuji: Röntgenfilme (Medical X-Ray) Heraeus: Tischzentifuge Biofuge pico Herolab: Geldokumentation Easy Lab Hettich: Kühlzentrifuge Universal 16R Hoefer: Gradientenmischer Kontes Glass: Dounce Homogenisator Kontron: Ultrazentrifuge TGA-65; Ausschwingrotor TST 41.14 Kontron / Hermle: Kühlzentrifuge Centrikon H-401; Rotoren A6.9 und A8.24 BRL Life Technologies: Agarosegelelektrophorese-Apparatur Horizon 11-14 Molecular Dynamics: Phosphoimager mit Expositionskassetten und Software Image Quant Pharmacia: FPLC-Anlage (Pumpe LKB Pump P-500, Kontrollgerät LKB Controller LCC-500 Plus, Ventil LKB Motor Valve MV-7, UVMonitor Single Path Monitor UV-1, Schreibgerät LKB REC-2, Fraktionssammler LKB RediFrac); Photometer Ultraspec 3000; pHMeter 766 Calimatic; Elektrophorese-Netzgerät EPS 500 / 400; 5 ml HiTrap Chelating Säule; 5 ml HiTrap Desalting Säule; Qiagen: Plasmid Maxi Kit Roth: Spritzenfilter 0.2 µm Schleicher und Schüll: Nitrozellulosemembran; Whatman 3MM Filterpapier SCM Aminco: French Pressure Zelle Institutswerkstatt: SDS-PAGE-Apparatur 4.1.3 Antibiotika Antibiotikum Konzentration der Stammlösung gelöst in Verdünnungsfaktor Endkonzentration Ampicillin (Amp) 100 mg / ml H2O 1 : 1000 100 µg / ml Chloramphenicol (Cm) 35 mg / ml Ethanol 1 : 1000 35 µg / ml Kanamycin (Kan) 50 mg / ml H2O 1 : 1000 50 µg / ml Tetracyclin (Tet) 5 mg / ml 50 % Ethanol 1 : 250 20 µg / ml Die Antibiotika-Stammlösungen wurden über Filter (0.2 µm) steril filtriert. 4 Material und Methoden 19 4.1.4 Escherichia coli - Stämme Stamm Beschreibung Resistenz Referenz MRE600 RNase-negativ Cammack und Wade, 1965 CTF MC4100 araD139 ∆(argF-lac) U169 rpsL150 relA1 flbB3501 deoC1 ptsF25 rbsR Silhavy et al., 1984 CTF DNA-Isolierung (pDMB; pMomp2) XL1-Blue supE44 hsdR17 recA1 endA1 gyrA46 thi relA1 lacF‘ [proAB+ lacIq lacZ∆M15 Tn10(tetr)] Tet Bullock et al., 1987 DNA-Isolierung (p717MtlA-B) N4156 pAra14FtsY’ polA end thy gyrA pAra14-FtsY‘ (ftsY unter Kontrolle des araB Promotors) Amp Luirink et al., 1994 FtsY--CTF AD179 MC4100∆ompT Akiyama und Ito, 1990 Wildtyp-INV TY1 ompT::kan, secY205 Kan, Tet Matsumoto et al., 1997 secY205-INV BL21(DE3) pLysS F ompT hsdSB(rBmB) gal dcm (DE3) pLyS Cm Ffh-Aufreinigung [Novagen] Verwendung 4.1.5 Plasmide • pET-19b-P48 (G. Eisner, unveröffentlicht): ffh-10His unter T7-Promotor-Kontrolle in pET-19b [Novagen] • pDMB (Behrmann et al., 1998): ompA unter T7-Promotor-Kontrolle in pGEM-3Z [Promega] • p717MtlA-B (Beck et al., 2000): mtlA unter T7-Promotor-Kontrolle in pKSM717 (Maneewannakul et al., 1994) Klonierung von pMomp2 Die Klonierung des Plasmids pMomp2 wurde von Dr. U. Schäfer durchgeführt (Plasmidkarten und DNA-Sequenzen siehe 8.2 bzw. 8.3). Zunächst wurden im Plasmid pDMB hinter der Signalsequenz von OmpA sowie im Plasmid p717MtlA-B hinter der ersten Signal-Anker-Sequenz von MtlA SpeI-Schnittstellen eingefügt. Hierzu wurde eine ortsspezifische Mutagenese mittels PCR in zwei Schritten in modifizierter Form (Beck, 2000) nach Dilsiz und Crabbe (1994) durchgeführt (Abb. 4.1). Im Fall von pDMB wurden in der ersten PCR der mutagene Primer OmpA66 mit der SpeISchnittstelle sowie OmpA168 als Primer verwendet; in der zweiten PCR kamen ein T7Promotor-Primer sowie das Produkt aus der ersten PCR zum Einsatz (Sequenzen der Primer siehe Tabelle). Das hierbei entstandene Produkt wurde mit EcoRI und BstXI geschnitten und in das mit alkalischer Phosphatase dephosphorylierte, ebenfalls mit EcoRI und BstXI geschnittene Plasmid pDMB zurückkloniert. Das resultierende Plasmid wurde pOmpA-SpeI genannt. 4 Material und Methoden -167 -145 20 1 76 ompA PT7 -146 EcoRI 67 Matrize: pDMB 117 BstXI 92 OmpA66-SpeI 150 169 Primer 1. PCR OmpA168 -167 -148 -344 Primer 2. PCR Produkt der 1. PCR T7-PromotorPrimer 1 -302 139 Matrize: p717MtlA-B mtlA PT7 -31 NcoI 130 154 254 NcoI MtlA129-SpeI 262 283 Primer 1. PCR MtlA282 -344 -325 T7-PromotorPrimer Produkt der 1. PCR Primer 2. PCR Abb. 4.1: Einführung der SpeI-Schnittstellen in pDMB und p717MtlA-B durch ortspezifische Mutagenese. Gezeigt sind die Lokalisation der verwendeten Primer und die Schnittstellen der Restriktionsenzyme im Bezug auf das ompA- bzw. mtlA-Gen. Abb. 4.2: Klonierung von pMomp2 aus pOmpA-SpeI und p717MtlA-SpeI-2 pOmpA-SpeI wurde mit PstI geschnitten, die überstehenden 3‘-Enden mit T4-DNA-Polymerase entfernt und das linearisierte Plasmid darauf mit SpeI geschnitten; p717MtlA-SpeI-2 wurde mit KpnI geschnitten, die überstehenden 3‘-Enden mit T4-DNA-Polymerase entfernt und das linearisierte Plasmid darauf mit SpeI geschnitten. Anschließend wurde das 3707 Bp Fragment von p717MtlASpeI-2 mit dem 1059 Bb Fragment von pOmpA-SpeI mit Hilfe der T4-DNA-Ligase ligiert und das resultierende Plasmid pMomp2 genannt. 4 Material und Methoden 21 Im Fall von p717MtlA-B wurden in der ersten PCR der mutagene Primer MtlA129 mit der SpeI-Schnittstelle sowie MtlA282 als Primer verwendet; in der zweiten PCR kamen wiederum ein T7-Promotor-Primer sowie das Produkt aus der ersten PCR als Primer zum Einsatz. Das hierbei entstandene Produkt wurde mit NcoI geschnitten und in das dephosphorylierte, ebenfalls mit NcoI geschnittene Plasmid p717MtlA-B zurückkloniert. Das resultierende Plasmid wurde p717MtlA-SpeI-2 genannt. Anschließend wurden die Plasmide pOmpA-SpeI sowie p717MtlA-SpeI-2 mit SpeI sowie einem zweiten Restriktionsenzym stromabwärts (3‘) von ompA bzw mtlA geschnitten, und die gewünschten Fragmente zu pMomp2 ligiert (Abb. 4.2). Hierzu wurde pOmpA-SpeI mit PstI geschnitten, die überstehenden 3‘-Enden mit T4-DNA-Polymerase entfernt, und das linearisierte Plasmid darauf mit SpeI geschnitten. p717MtlA-SpeI-2 wurde mit KpnI geschnitten, die überstehenden 3‘-Enden mit T4-DNA-Polymerase entfernt, und das linearisierte Plasmid darauf ebenfalls mit SpeI geschnitten. Schließlich wurde das 3,7 kb Fragment von p717MtlA-SpeI-2 mit dem 1 kb Fragment von pOmpA-SpeI mit Hilfe der T4DNA-Ligase ligiert. Das resultierende Plasmid wurde pMomp2 genannt. Es enthält momp2 unter T7-Promotor-Kontrolle. momp2 wurde sequenziert und die erwartete Basenfolge bestätigt. DNA-Oligonukleotide für die Klonierung von pOmpA-SpeI sowie p717MtlA-SpeI-2: Oligonukleotid Sequenz 5’ 3’ Bemerkung OmpA66 CCGAAAGATACTAGTTGGTACA CTGG hybridisiert an Bp 67-92 von ompA Einführung einer SpeI-Schnittstelle nach der Signalsequenz in ompA (N26 zu T26; T27 zu S27) OmpA168 CCAGTTGGTTTTCATGGGTC komplementär zu Bp 169 bis 150 von ompA MtlA129 CCGAACGAGACTAGTGCGAAGC hybridisiert an Bp 130-154 von mtlA TGG Einführung einer SpeI-Schnittstelle nach der ersten transmembranen Helix in mtlA (T47 unverändert; L48 zu S48) MtlA282 GCATGTCTGCGCCGACGATAAC komplementär zu Bp 283 bis 262 von mtlA T7-PromotorPrimer (Promega) TAATACGACTCACTATAGGG hybridisiert an Bp 1 bis 20 des T7Promotors 4 Material und Methoden 22 4.2 Molekularbiologische Methoden 4.2.1 DNA-Isolierung Die Zellen wurden in 250 ml LB-Medium (10 g Pepton, 10 g NaCl und 5 g Hefeextrakt auf 1 l Wasser) übernacht bei 37°C im Schüttler angezogen, wobei die den jeweiligen Resistenzen entsprechenden Antibiotika zugegeben wurden: • pDMB im Stamm XL1-Blue: Tetracyclin (Resistenz auf F1-Plasmid kodiert), Ampicillin (Resistenz auf pDMB kodiert) • p717MtlA-B im Stamm MC4100: Ampicillin (Resistenz auf p717MtlA-B kodiert) • pMomp2 im Stamm XL1-Blue: Tetracyclin (Resistenz auf F1-Plasmid kodiert), Ampicillin (Resistenz auf pMomp2 kodiert) Die Isolierung der Plasmid-DNA erfolgte mit Hilfe des Qiagen Plasmid Maxi Kit nach dem mitgelieferten Protokoll. Das luftgetrocknete DNA-Pellet wurde in 500 µl TE-Puffer resuspendiert und die DNA durch Zugabe von 50 µl 3 M Kaliumacetat, pH 5.5 und 1 ml Ethanol 30 min bei -70°C gefällt. Anschließend wurde die DNA durch 15minütige Zentrifugation bei 14 000 Rpm und 4°C in der Kühlzentrifuge Universal 16R sedimentiert und nach einmaligem Waschen mit 70 % Ethanol in 100 µl TE-Puffer resuspendiert. TE-Puffer: 10 mM 1 mM Tris / HCl pH 8.0 EDTA pH 8.0 Zur Kontrolle wurde die isolierte Plasmid-DNA mit zwei plasmidspezifischen Restriktionsenzymen verdaut und die erwartete Größe der Fragmente in der Agarosegelelektrophorese verifiziert. Die DNA-Konzentration wurde über die Messung der Absorption bei 260 nm bestimmt (zur Messung wurde die DNA 1 : 500 mit H2O verdünnt; eine A260 von 1 entspricht 50 µg/ml). 4.2.2 DNA-Restriktionsenzymverdau und Agarosegelelektrophorese Der Verdau der Plasmid-DNA fand nach folgendem Schema mit einem oder mehreren geeigneten Restriktionsenzymen bei 30minütiger Inkubation bei 37°C statt: DNA (1-2 µg/µl) 1 µl Restriktionsenzyme (10 u/µl) je 1 µl mitgelieferter10x Puffer 2 µl H2O ad 20 µl Anschließend wurden 2 µl 10x DNA-Ladelösung zugegeben und die Proben über ein 0.8%iges Agarosegel bei 100 mA aufgetrennt. Als Laufpuffer wurde 1x TAE-Puffer verwendet. Die DNA-Banden konnten im UV-Licht sichtbar gemacht und über ein EDVgestütztes Photosystem dokumentiert werden. 23 10x DNA-Ladelösung: 50 % 0.05 % 0.05 % Glycerin Bromphenolblau Xylencyanol 50x TAE-Puffer: 242 g 57 ml 100 ml ad 1 l Tris Base Essigsäure 0.5 M EDTA pH 8.0 H2O 4 Material und Methoden Herstellung des 0.8%igen Agarosegels: Zu 80 ml 1x TAE-Lösung wurden 0.64 g Agarose hinzugefügt, das Gemisch in der Mikrowelle erhitzt und dann mit 10 µl Ethidiumbromidlösung (10 mg/ml in H2O) versetzt. Die heiße Lösung wurde in die Elektrophoreseapparatur eingefüllt. 4.2.3 SDS-Polyacrylamid-Gelelektrophorese Die Auftrennung denaturierter Proteine erfolgte durch SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) nach Laemmli (1970). Dabei kamen Gele mit einer PolyacrylamidKonzentration von 13 bzw. 15 % zur Anwendung. 13%iges Trenngel: 30 ml 30 % Acrylamid, 0.8 % Bisacrylamid 14 ml 2 M Tris / HCl pH 8.8 280 µl 25 % SDS ad 70 ml H2O Direkt vor dem Gießen wurden zugegeben: 28 µl TEMED 400 µl 10 % Ammoniumperoxodisulfat 15%iges Trenngel: 35 ml 30 % Acrylamid, 0.8 % Bisacrylamid weiter wie bei 13%igem Trenngel. Zur optimalen Auftrennung von naszierenden Ketten und ihren Quervernetzungsprodukten wurden 7 - 17%ige Gradientengele gegossen. Dabei wurden 30 ml einer 7%igen Acrylamidlösung und 30 ml einer 17%igen Acrylamidlösung mittels Gradientenmischer gemischt und durch Überschichtung in die Gelapparatur gegossen. 7 %ige Lösung: 7 ml 6 ml 120 µl ad 30 ml 17%ige Lösung: 17 ml 6 ml 120 µl 7 ml - 30 % Acrylamid, 0.8 % Bisacrylamid 2 M Tris / HCl pH 8.8 25 % SDS 2.5 M Saccharose H2O Direkt vor dem Gießen wurden zu beiden Lösungen jeweils 10 µl TEMED und 100 µl 10% Ammoniumperoxodisulfat zugegeben. 4 Material und Methoden Sammelgel: 24 3.35 ml 30 % Acrylamid, 0.8 % Bisacrylamid 2.4 ml 0.5 M Tris / HCl pH 6.8 3.75 ml 2.5 M Saccharose 80 µl 25 % SDS ad 20 ml H2O Direkt vor dem Gießen wurden zugegeben: 8 µl TEMED 150 µl 10 % Ammoniumperoxodisulfat Neben den in SDS-PAGE-Ladepuffer denaturierten Proben wurden als Längenstandards vorgefärbte Molekulargewichtsmarker in einer Probentasche des Polyacrylamidgels aufgetragen. SDS-PAGE-Ladepuffer: Lösung 1: 0.2 M Tris Base 0.02 M EDTA, pH 7.5 Lösung 2: 8.3 % SDS 83.3 mM Tris Base 29.2 % Glycerin 0.03 % Bromphenolblau jeweils frisch bzw. in Aliquots bei -20°C gelagert: 0.5 ml Lösung 1 0.4 ml Lösung 2 0.1 ml 1 M DTT Die Gele liefen übernacht bei einer Stromstärke von 15 mA, wobei mit SDS-PAGELaufpuffer gepuffert wurde. SDS-PAGE-Laufpuffer: 5x Tris-Glycin-Puffer: 400 ml 5x Tris-Glycin-Puffer 8 ml 25 % SDS ad 2 l H2O 150 g Tris 720 g Glycin ad 5 l H2O Nach beendetem Lauf konnten die Gele zur Sichtbarmachung der Proteinbanden in Coomassie-Blau-Lösung gefärbt und anschließend mit Fixierlösung entfärbt werden. Coomassie-Blau-Lösung: 3.125 g 1250 ml 250 ml 1000 ml Fixierlösung: Brilliant Blau G 250 technischer Ethanol Essigsäure H2O 10 % Essigsäure 35 % Ethanol 25 4 Material und Methoden Zur Autoradiographie wurden die Gele meist ohne Färbung 30 min in Fixierlösung inkubiert, 15 min gewässert und anschließend bei 55°C (Gradientengele) bzw. 60°C (13 und 15 %ige Gele) 2 h unter Vakuum getrocknet. 4.2.4 Western Transfer Western Transfer diente vor allem dazu, den Gehalt an Translokations- bzw. Insertionsfaktoren (SecA, SecB, Ffh, FtsY, SecY) in den verschiedenen Komponenten des zellfreien Systems zu bestimmen. Die Proben wurden zunächst durch SDS-PAGE aufgetrennt und anschließend auf eine Nitrozellulosemembran transferiert. Dazu wurden folgende Materialien 5 min in Transferpuffer äquilibriert und dann in folgender Reihenfolge ohne Luftblasen in die Blottaparatur eingesetzt: Minuspol Pluspol Saugschwamm Whatman-Papier SDS-PAGE-Gel Nitrozellulosemembran Whatman-Papier Saugschwamm Transferpuffer: 600 ml 600 ml 2 ml ad 3 l 5x Tris-Glycin-Puffer (siehe 4.2.3.) technischer Ethanol 25 % SDS H2O Der Transfer erfolgte 2 h bei 80 V und 400 mA. Zur Kontrolle wurden die auf die Nitrozellulosemembran transferierten Proteinbanden kurz mit Ponceau-S-Lösung (0.2 % Ponceau S, 3 % TCA) angefärbt und mit H2O wieder entfärbt. Alle anschließenden Schritte fanden unter leichtem Rütteln statt. Zunächst wurde die Membran 2 h mit Milchpulver (5 % Milchpulver in TTBS) blockiert und dann mit TTBS gewaschen (nach dem Abspülen der Milchpulverlösung 3 x 10 min auf dem Rüttler). 10x TBS: 1M 9% Tris / HCl pH 7.5 NaCl TTBS: 0.1 % Tween-20 in 1x TBS Daraufhin wurde mit dem gewünschte Antiserum in folgender Verdünnung in 1 % BSA / TTBS für 1 h inkubiert: 4 Material und Methoden Antiserum (vom Kaninchen) αFfh αFtsY αSecA αSecB αTrigger factor αSecY 26 Verdünnung 1 : 2500 1 : 2500 1 : 5000 1 : 5000 1 : 10000 1 : 2500 Herkunft K.-L. Schimz K.-L. Schimz K.-L. Schimz K.-L. Schimz E. Deuerling M. Müller Nach nochmaligem Waschen (wie oben beschrieben) wurde der 2. Antikörper (antiKaninchen Antikörper von der Ziege, Meerrettich-Peroxidase-gekoppelt) in der Verdünnung 1 : 5000 in 1 % BSA, 20 mM MgCl2 in TTBS zugegeben und nochmals für 1 h inkubiert. Nach weiteren Waschschritten (wie oben beschrieben) wurde zur Detektion der Antikörpermarkierten Banden das „Enhanced Chemiluminescence ECL Westernblotting System“ verwendet. Dazu wurde ein 1:1 Gemisch der beiden ECL-Lösungen auf die durch vorsichtiges Tupfen mit Whatman-Filterpapier abgetrocknete Membran gleichmäßig verteilt und nach einminütigem Einwirken wieder mit Whatmann-Filterpapier entfernt. Durch Auflegen eines Röntgenfilmes für ca. 10 - 20 s und anschließende Filmentwicklung konnten die lumineszierenden Banden sichtbar gemacht werden. 4.3 Herstellung von Bestandteilen des zellfreien Systems 4.3.1 Bestandteile des zellfreien Transkriptions-Translations-Systems Zytosolische Translationsfaktoren (CTF) Die Zytosolischen Translationsfaktoren (CTF) wurden nach dem Protokoll von Müller und Blobel (1984b) entweder aus dem RNAse-freien E. coli-Stamm MRE600 oder aus dem E. coli-Stamm MC4100 gewonnen. Vier mal je 1 Liter S-150-Medium wurde mit je 10 ml einer Übernachtkultur angeimpft und die Zellen bei 37°C bis zu einer OD580 von 1.6 angezogen. S-150-Medium: 753 ml H2O mit 10 g Hefeextrakt (MRE600 / MC4100) 41 ml 1 M KH2PO4 166 ml 1 M K2HPO4 41 ml 25 % D-Glukose Die Lösungen wurden jeweils einzeln autoklaviert. Alternativ konnten FtsY-depletierte CTF aus dem E. coli-Stamm N4156 pAra14-FtsY’ gewonnen werden, in dem das ftsY-Gen unter der Kontrolle eines Arabinosepromotors steht. Dabei kam ein abgeändertes S-150-Medium zur Anwendung, dem außerdem Ampicillin zugegeben wurde. Da FtsY in E. coli ein essentielles Protein darstellt, wurde die ÜbernachtVorkultur in Anwesenheit von 0.2 % Arabinose zur Induktion des ftsY-Gens angezogen. 27 S-150-Medium: (für FtsY─-CTF) 4 Material und Methoden 984 ml 8 g Pepton, 5 g Hefeextrakt und 5 g NaCl in Wasser gelöst +100 µl 10 N NaOH 16 ml 25 % D-Fruktose Diese beiden Lösungen wurden einzeln autoklaviert. Alle folgenden Schritte erfolgten auf Eis bzw. bei 4°C. Nach einem Waschschritt wurden die Zellen durch 10minütige Zentrifugation bei 7 000 Rpm im Rotor A6.9 geerntet. Anschließend wurden die Zellen in S30-Puffer im Verhältnis 1 ml Puffer je 1 g Feuchtgewicht resuspendiert und bei drei Durchläufen durch die French Press bei 8 000 Psi aufgeschlossen. S30-Puffer: 50 mM 50 mM 15 mM 1 mM 0.5 mM Triethanolaminacetat pH 7.5 Kaliumacetat pH 7.5 Magnesiumacetat DTT PMSF (in Ethanol frisch gelöst) Durch 30minütige Zentifugation bei 15 000 Rpm im Rotor A8.24 (30 000 x g) konnten Zelltrümmer und ganze Zellen entfernt werden; der Überstand wurde nochmals 2.5 h bei 45 000 Rpm im Rotor 50.2Ti (150 000 x g) zentrifugiert. Der hierbei entstandene Überstand war frei von Membranen und Ribosomen und wurde S-150 genannt. Aus dem Pellet konnten die Ribosomen weiter aufgereinigt werden (siehe unten). Jeweils 6 ml einer 10 bzw. 30%igen (w/w) Saccharoselösung (10.38 g bzw. 33.81 g Saccharose /100 ml in CTF-Puffer) wurden im Gradientenmischer gemischt und mithilfe einer langen Kanüle in Zentrifugenröhrchen unterschichtet. CTF-Puffer: 50 mM 50 mM 5 mM 1 mM Triethanolaminacetat pH 7.5 Kaliumacetat pH 7.5 Magnesiumacetat DTT Der so entstandene Saccharosegradient wurde 1 h bei 4°C äquilibriert; anschließend wurde je Zentrifugenröhrchen 1 ml des S-150 auf den Gradienten aufgetragen. Diese wurden 20-24 h bei 39 000 Rpm im Ausschwingrotor TST 41.14 mit freiem Auslaufen zentrifugiert. Die Zentrifugationsröhrchen wurden von unten mit einer Nadel punktiert und die Gradienten dann langsam mit Hilfe der Pumpe Minipuls 2 (Geschwindigkeit 130 Einheiten) abgezogen; unter Aufzeichnung der Absorption bei 280 nm wurden ca. 20 Fraktionen à 600 µl (23 Tropfen im Pharmacia Fraktionssammler LKB RediFrac) gesammelt. Sofern die Absorptionsmuster unterschiedlicher Gradienten sich entsprachen, wurden die jeweils gleichen Fraktionen gepoolt und auf Aktivität im zellfreien Transkriptions-Translations-System getestet und die aktiven Fraktionen (meist Fraktionen 12-14) wiederum gepoolt. 4 Material und Methoden 28 Ribosomen Die Ribosomen wurden nach dem Protokoll von Müller und Blobel (1984b) aus dem Pellet gewonnen, das bei der Herstellung des S-150 anfiel (siehe oben). Um ribosomenassoziierte Proteine zu entfernen, wurden die Ribosomen mit Hochsalz gewaschen. Alle Schritte wurden wiederum auf Eis bzw. bei 4°C durchgeführt. Die Ribosomen wurden in Hochsalzpuffer resuspendiert und dann durch einstündige Zentrifugation bei 90 000 Rpm im Rotor TLA 100.2 erneut pelletiert. Hochsalzpuffer: 50 mM 1M 5 mM 1 mM Triethanolaminacetat pH 7.5 Kaliumacetat pH 7.5 Magnesiumacetat DTT Das durchsichtige Ribosomenpellet war von einer gelblichen Membranschicht überlagert, die manuell entfernt werden konnte. Das Ribosomenpellet wurde in 1 ml Hochsalzpuffer je Liter Ausgangskultur resuspendiert. Je 200 µl wurden auf einen 10-40%igen (w/w) Saccharosegradienten aufgetragen (10.38 g bzw. 47.06 g Saccharose je 100 ml Lösung in CTF-Puffer (siehe oben)) und 17 h bei 29 000 Rpm im Rotor TST 41.14 zentrifugiert. Die Gradienten wurden in Fraktionen à 500 µl gesammelt, wobei die Absorption bei 254 nm aufgezeichnet wurde (Absorption durch die rRNA). 25 µl jeder Fraktion wurden über SDS-Polyacrylamid-Gelelektrophorese aufgetrennt und mit Coomassie angefärbt. Die ribosomenhaltigen Fraktionen konnten anhand ihres charakteristischen Bandenmusters identifiziert werden (meist Fraktionen 1-9). Die ribosomenhaltigen Fraktionen wurden gepoolt, die Ribosomen nochmals pelletiert (1 h bei 90 000 Rpm im Rotor TLA 100.2) und in CTF-Puffer resuspendiert. Initiationsfaktor 2 Der Initiationsfaktor 2 wurde von H. Hoffschulte nach dem Protokoll von Müller und Blobel (1984a) aufgereinigt. T7-RNA-Polymerase Die T7-RNA-Polymerase wurde von R. Helde aufgereinigt (Beck, 2000). 4.3.2 Membranvesikel Inside-Out Innenmembranvesikel (INV) Die Herstellung der INV folgte weitgehend dem Protokoll von Müller und Blobel (1984a). Zur Präparation von Wildtyp-INV wurde der E. coli-Stamm AD179 (∆ompT) verwendet. Vier mal je 1 Liter INV-Medium wurde mit je 20 ml einer Übernacht-Vorkultur angeimpft und die Zellen bei 37°C im Schüttler bis zu einer OD580 von 1.6 angezogen. 29 INV-Medium: 4 Material und Methoden 753 ml H2O mit jeweils 10 g Hefeextrakt und Pepton 41 ml 1 M KH2PO4 166 ml 1 M K2HPO4 41 ml 25 % D-Glukose Die Lösungen wurden jeweils einzeln autoklaviert. SecY205-INV wurden aus dem Stamm TY1 gewonnen; dabei wurde dem INV-Medium Arabinose statt Glukose zugesetzt. Alle folgenden Schritte wurden auf Eis bzw. bei 4°C durchgeführt. Die Zellen wurden zunächst durch 10minütige Zentrifugation bei 7 000 Rpm im Rotor A6.9 geerntet und dann nach einem Waschschritt in Puffer A im Verhältnis 1 ml Puffer je 1 g Feuchtgewicht resuspendiert. Puffer A: 50 mM 250 mM 1 mM 1 mM 0.5 mM Triethanolamin pH 7.5 Saccharose EDTA DTT PMSF (in Ethanol frisch gelöst) Bei drei Durchläufen durch die French Press (8 000 Psi) wurden die Zellen aufgeschlossen, Zelltrümmer und ganze Zellen wurden durch 5minütige Zentrifugation bei 5 000 Rpm im Rotor A8.24 entfernt. Der Überstand wurde für 2 h bei 40 000 Rpm (150 000 x g) im Rotor 50.2Ti zentrifugiert. Das hierbei entstehende Rohmembranpellet (bestehend aus Innenmembranen, Außenmembranen sowie Ribosomen) wurde mit Hilfe eines Dounce Homogenisators in Puffer A resuspendiert; anschließend wurden die Innenmembranen durch einen Saccharosestufengradienten von den anderen Bestandteilen abgetrennt. Hierzu wurden 12 ml 0.77 M mit 12 ml 1.44 M und schließlich mit 10 ml 2.02 M Saccharoselösung (jeweils in Puffer A) in einem Zentrifugenröhrchen untergeschichtet und der so entstandene Stufengradient für 1 h bei 4°C äquilibriert. Nach Überschichtung von ca. 2 ml Rohmembranen je Gradient wurde für 16 h bei 25 000 Rpm im Ausschwingrotor SW28 zentrifugiert. Die Innenmembranen befanden sich hiernach als gelbe Bande zwischen der 0.77 M und 1.44 M Saccharoselösung; die Zentrifugenröhrchen wurden seitlich mit einer Kanüle punktiert und die gelbliche Lösung abgezogen. Diese wurde etwa vierfach mit 50 mM Triethanolamin pH 7.5 verdünnt, anschließend wurden die INV durch zweistündige Zentrifugation bei 40 000 Rpm im Rotor 50.2Ti pelletiert und in INV-Puffer resuspendiert. INV-Puffer: 50 mM 1 mM 250 mM Triethanolamin pH 7.5 DTT Saccharose Das INV-Sediment, das aus 4 l Kultur gewonnen wurde, wurde in ca. 1 ml Puffer resuspendiert, um eine ausreichend hohe INV-Konzentration zu erhalten (Die OD bei 280 nm 4 Material und Methoden 30 sollte über 30 liegen; die Absorptionsmessung erfolgte in einer 100fach mit 2% SDS verdünnten Suspension). Harnstoffextrahierte Vesikel (HINV) Um die INV mit einer Endkonzentration von 6 M Harnstoff zu extrahieren, wurden 100 µl INV mit 400 µl 7.5 M Harnstoff-Puffer vermischt und für 1 h auf Eis inkubiert (Helde et al., 1997). Harnstoff-Puffer: 7.5 M 50 mM 250 mM Harnstoff Triethanolamin pH 7.5 Saccharose Anschließend wurden die 500 µl Vesikelsuspension durch 200 µl 750 mM Saccharosekissen (750 mM Saccharose, 50 mM Triethanolamin pH 7.5) 15 min bei 70 000 Rpm und 4°C im Rotor TLA 100.2 sedimentiert. Zur Entfernung eventueller Harnstoffreste wurde das Pellet in 100 µl INV-Puffer (siehe oben) resuspendiert und nochmals über ein 750 mM Saccharosekissen pelletiert. Schließlich wurde das Pellet in 50 µl INV-Puffer resuspendiert. 4.3.3 Translokations- und Insertionsfaktoren aufgereinigt von Protokoll Konzentration SecA K. Beck und D. Trescher Helde et al.,1997 1 mg / ml SecB H. Hoffschulte Hoffschulte et al., 1994 2 mg / ml F1-ATPase H. Hoffschulte Müller et al., 1987 1 mg / ml FtsY H. Hoffschulte Koch et al., 1999 (modifiziert nach Luirink et al., 1994) 1 mg / ml Die F1-ATPase wurde in 50 mM Tris pH 8.0, 2 mM EDTA pH 8.0 aufgenommen. Alle anderen Proteine wurden in CTF-Puffer (siehe 4.3.1) aufgenommen. Ffh-Aufreinigung Zur Aufreinigung von Ffh wurde der Vektor pET-19b-P48 im E.coli-Stamm BL21(DE3) pLysS verwendet (G. Eisner, unveröffentlicht). Auf diesem Vektor wurde das ffh-Gen hinter eine Sequenz aus 10 Histidin kloniert. Es steht unter der Kontrolle der T7-RNA-Polymerase, die durch IPTG induziert bzw. durch Glucose reprimiert wird. Die ersten Reinigungsschritte bis zum S-30 wurden ebenfalls von G. Eisner durchgeführt. Eine Vorkultur wurde in LBMedium mit Ampicillin, Chloramphenicol und 0.4 % Glucose übernacht angezogen. Mit je 10 ml dieser Übernachtkultur wurden zwei mal je 1 l LB-Medium mit Ampicillin und Chloramphenicol angeimpft. Die Zellen wurden bei 30°C bis zu einer OD580 von 0.45 angezogen. Nach der Zugabe von je 0.5 ml 1 M IPTG zur Induktion des ffh-Gens wurden die Zellen bis zu einer OD580 von 1.35 weiter angezogen. 31 LB-Medium: 10 g 10 g 5g 100 µl ad 1 l 4 Material und Methoden Pepton NaCl Hefeextrakt 10 N NaOH H2O Alle folgenden Schritte erfolgten auf Eis bzw. bei 4°C. Die Zellen wurden durch 10minütige Zentrifugation bei 7 000 Rpm im Rotor A6.9 geerntet und in 25 ml Startpuffer resuspendiert. Startpuffer: 50 mM 300 mM Kaliumphoshat-Puffer pH 7.2 NaCl Anschließend wurden die Zellen mit Proteaseinhibitoren versetzt (siehe Tabelle) und bei drei Durchläufen durch die French Press bei 8 000 Psi aufgeschlossen. Durch eine 30minütige Zentrifugation bei 15 000 Rpm im Rotor A8.24 (30 000 x g) entstand der S-30 als Überstand. Eine 5 ml Pharmacia HiTrap Chelating Säule wurde mit Hilfe der FPLC-Anlage zunächst mit 10 ml H2O (0.5 ml / min bei allen folgenden Schritten) gespült und dann mit 20 ml Startpuffer äquilibriert. Der S-30 wurde nochmals mit Proteaseinhibitoren versetzt, dann wurden 2 x 3 ml S-30 nacheinander auf die Säule aufgetragen. Nach dem Auftragen der ersten 3 ml wurde die Säule mit Startpuffer gespült. Proteaseinhibitor Stammlösung Verdünnung Endkonzentration Aprotinin 2 mg / ml in H2O 1 : 1000 2 µg / ml Leupeptin 0.5 mg / ml in H2O 1 : 1000 0.5 µg / ml E64 10 mg / ml in 50 % Ethanol 1 : 1000 10 µg / ml PMSF 100 mM in Ethanol 1 : 100 1 mM Anschließend wurde erst mit Startpuffer und dann mit 150 mM Imidazol-Puffer (150 mM Imidazol in Startpuffer) jeweils solange gespült, bis kein bei 280 nm absorbierendes Material mehr eluiert wurde. Danach wurde mit 300 mM Imidazol-Puffer (300 mM Imidazol in Startpuffer) das Ffh-10His eluiert. Dabei wurde möglichst eng fraktioniert: Nur die 3 ml Eluat mit der höchsten Absorption bei 280 nm wurden sofort weiterbehandelt. Andernfalls wurde ein Ausfallen des Ffh-10His in der wässrigen Lösung beobachtet. Nachdem eine 5 ml Pharmacia HiTrap Desalting Säule mit CTF-Puffer (siehe 4.3.1) äquilibriert worden war, wurden 1.5 ml Ffh-10His-Eluat mit Hilfe einer sterilen Spritze aufgetragen; durch das anschließende Auftragen von 2 ml CTF-Puffer wurde das Ffh-10His in CTF-Puffer eluiert. Durch sofortige Zugabe von 1 Volumen Glycerin und Lagerung bei -20°C wurde das Ausfallen des Ffh-10His verhindert. Die Ffh-Konzentration lag bei 0.1 mg/ml. 4 Material und Methoden 32 4.4 Versuche im zellfreien System zum Proteintransport in Escherichia coli 4.4.1 Gekoppelte Transkription-Translation im zellfreien System Die Proteinsynthese erfolgte durch gekoppelte Transkription-Translation im zellfreien System (Koch et al., 1999). Das zellfreie System enthielt an biologischen Komponenten Zytosolische Translationsfaktoren (CTF), Ribosomen, Initiationsfaktor 2 sowie T7-RNA-Polymerase. Zusätzlich wurden Nukleotide, Aminosäuren und ein ATP-regenerierendes System (Kreatinphosphat, Kreatinphosphatkinase und Phosphoenolpyruvat) benötigt. Durch Zugabe von Spermidin, Putrescin, DTT (zum Schutz vor Oxidation), dem RNase-Inhibitor RNasin (zum Schutz der RNA vor unspezifischem Verdau) sowie Phosphoenolpyruvat wurde die Synthese optimiert. Als Puffersystem diente 40 mM Triethanolaminacetat pH 7.5, bei DSSQuervernetzungsexperimenten hingegen 40 mM HEPES pH 7.5. Die optimalen Salzbedingungen waren von dem jeweils sythetisierten Protein abhängig; in der Regel war eine effiziente Synthese bei 70 mM K+ und 8 mM Mg++ möglich. Durch Zugabe des Gens des zu synthetisierenden Proteins in Form von Plasmid-DNA unter T7-Promotor-Kontrolle sowie radioaktiv markierter Aminosäuren (35S-Met und 35S-Cys) wurde während 30minütiger Inkubation bei 37°C das gewünschte radioaktiv markierte Protein synthetisiert. Ein 25-µl-Ansatz des Transkriptions-Translations-Systems setzte sich im einzelnen folgendermaßen zusammen: Stammlösung Endkonzentration µl / Ansatz Komponente 10fach Kompensationspuffer (siehe unten) 2.5 25 µl Endvolumen H2O Polyethylenglycol 40 % w/v 3.2 % w/v 2 18 Aminosäuren (ohne Met, Cys) je 1 mM je 0.04 mM 1 Putrescin 200 mM 8 mM 1 DTT 200 mM 2 mM 0.25 50 mM je 10 mM 200 mN 2.5 mM je 0.5 mM 1.25 Phosphoenolpyruvat 200 mM 5 mM 0.625 Kreatinphoshat 500 mM 8 mM 0.4 Kreatinphosphatkinase 10 mg / ml 40 µg / ml 0.1 NTP-Mix: ATP GTP, UTP, CTP KOH (zur Neutralisation) Zytosolische Tranlstionsfaktoren (CTF) Ribosomen 3* 7.5 µM * 90 nM * Initiationsfaktor 2 0,3 * 0,5 * Plasmid-DNA 1 mg / ml * 40 µg / ml * 1* RNasin RNase-Inhibitor 40 U / µl 0.32 U / µl 0.2 T7-RNA-Polymerase 20 U / µl 0.24 U / µl 0.3 weitere Komponenten je nach Fragestellung 35 S-Met, 35S-Cys 10 µCi / µl 0.28 µCi / µl 0.7 * variierte je nach Präparation und mußte daher für jede Präparation neu titriert werden. 4 Material und Methoden 33 Dabei wurden für n Syntheseansätze jeweils (n+1) Ansätze zusammen pipettiert und diese erst nach Zugabe der radioaktiv markierten Aminosäuren in Aliquots aufgeteilt. Je nach Fragestellung wurden dem Transkriptions-Translations-Systems weitere Komponenten zugesetzt (siehe 4.4.2, 4.4.4 und 4.4.5). Diese konnten entweder vor dem Aliquotieren allen Ansätzen oder erst nach dem Aliquotieren einzelnen Ansätzen zugesetzt werden. In jedem Fall mußten jedoch auch diese Komponenten bei der Berechnung des Endvolumens berücksichtigt werden. Um die gewünschten Puffer- und Salzbedingungen zu erreichen, mußten die Ionenkonzentrationen der Puffer berücksichtigt werden, in denen die biologischen Komponenten aufgenommen waren. Daher wurde zunächst ein 10fach „Kompensationspuffer“ berechnet und pipettiert. Das folgende Rechenbeispiel geht von einer Synthese mit 3 µl CTF, 0.3 µl Ribosomen, 0.5 µl Initiationsfaktor 2, 0.5 µl INV und 2 µl SecA aus: T K Mg Sper gewünschte Endkonzentration: mM 40 70 8 0.8 entspricht pro 25-µl-Ansatz: (1) nmol 1000 1750 200 20 in biologischen Komponenten sind bereits enthalten: 3 µl CTF (T50K50Mg5) 0.3 µl Ribosomen (T50K50Mg5) 0.5 µl Initiationsfaktor 2 (K350) 0.5 µl INV (T50) 2.0 µl SecA / SecB / FtsY (T50K50Mg5) µl Ffh (T25K25Mg2.5) Σ (2) nmol nmol nmol nmol nmol nmol nmol 150 15 150 15 175 15 1.5 25 100 ____ 290 100 ____ 440 10 ____ 26.5 je 25 µl Ansatz müssen noch hinzugefügt werden: (3) = ∆ (1) – (2) nmol 710 1310 173.5 20 jeder µl von 2.5 µl 10fach Kompensationspuffer muß enthalten: (4) = (3) / 2.5 nmol 284 524 69.4 8 Stammlösung (5) M 1M 4M 1M 0.1 M für 0.25 ml 10fach Kompensationspuffer muß an Stammlösung zugefügt werden: (6) = (4) x 0.25 / (5) µl 71 32.8 17,4 20 T Triethanolaminacetat pH 7.5 Mg Magnesiumacetat K Kaliumacetat pH 7.5 Sper Spermidin H2 O 108.8 ( 250) 4 Material und Methoden 34 4.4.2 Synthese ribosomenassoziierter naszierender Ketten (RANCs) Die Synthese von ribosomenassoziierten naszierenden Ketten (RANCs) wurde mit einer nach Haeuptle et al. (1986) modifizierten Methode durchgeführt (Beck et al., 2000). Vor Start der Synthese im zellfreien System wurden DNA-Oligonukleotide zugegeben, deren Sequenz zur mRNA des zu synthetisierten Proteins komplementär waren. In der TranskriptionsTranslations-Reaktion hybridisieren diese DNA-Oligonukleotide mit der mRNA; die Endonuclease RNaseH erkennt diese Hybride und verdaut den hybridisierten mRNA-Teil. Die so verkürzte mRNA trägt an ihrem 3‘-Ende kein Stoppkodon, so daß das Ribosom am Ende der Translation die Polypeptidkette nicht freisetzen kann und ribosomenassoziierte naszierende Ketten (RANCs) entstehen. Für die Synthese der RANCs wurden folgende DNA-Oligonukleotide in den genannten Konzentrationen verwendet: Oligonukleotid Sequenz 5’ 3’ Synthetisiertes Polypeptid MtlA 189 ACCGTGGTTAATGGC naszierende Ketten von MtlA bis GTTGTT L189 (MtlA-189) 3 µg / 25 µlAnsatz OmpA β5 TAAACGTTGGATTTA naszierende Ketten von pOmpA bis GTGTC A125 (pOmpA-125) naszierende Ketten von Momp2 bis A146 (Momp2-146) 3 µg / 25µlAnsatz 4 µg / 25µlAnsatz OmpA β8 GGAGCTGCCTCGCCC naszierende Ketten von pOmpA bis TGACC F191 (pOmpA-191) naszierende Ketten von Momp2 bis F212 (Momp2-212) 3 µg / 25µlAnsatz 4 µg / 25µlAnsatz OmpA 280 GGAGATCAGGTAAT CAACAAC 3 µg / 25µlAnsatz 4 µg / 25µlAnsatz anti-10SaRNA TTAAGCTGCTAAAGC 10Sa-RNA-antisenseGTAGTTTTCGTCGTT Oligonukleotid TGCGACTA naszierende Ketten von pOmpA bis S280 (pOmpA-280) naszierende Ketten von Momp2 bis S301 (Momp2-301) Konzentration 0,25 µg / 25µlAnsatz RNaseH war bereits in den CTF in ausreichendem Maße vorhanden, nur bei Verwendung von CTF aus dem Stamm MC4100 wurden 0.6 U RNaseH zugesetzt. Außerdem wurden 0.25 µg eines 10Sa-RNA-antisense-Oligonukleotids dem Reaktionsansatz zugegeben; dieses antisense-Oligonukleotid hybridisiert mit der 10Sa RNA und unterdrückt so ihre Funktion. Die 10Sa RNA löst synthetisierte Polypeptide ohne Stoppkodon vom Ribosom los, so daß diese verdaut werden können (Hanes und Plückthun, 1997; Keiler et al., 1996). 4.4.3 Puromycin-Behandlung Zur Loslösung der naszierenden Ketten vom Ribosom konnten diese mit Puromycin behandelt weren. Dazu wurden nach der Synthese 2 µl 11mM Puromycin, mit KOH auf pH 6.5 titriert, je 25 µl Syntheseansatz zugegeben und für weitere 15 min bei 37°C inkubiert. 35 4 Material und Methoden 4.4.4 Protease-Resistenz-Test Um die Translokation bzw. Insertion der im zellfreien System synthetisierten Proteine in Innenmembranvesikel zu untersuchen, wurden dem jeweiligen Syntheseansatz nach 5 min entweder INV oder HINV zugegeben und die Synthese dann 30 min bei 37°C fortgesetzt. Die optimale Menge an Vesikeln mußte für jede Vesikelpräparation durch Titration im zellfreien System neu ermittelt werden. Bei Verwendung von HINV mußten zur Wiederherstellung der Translokationsbzw. Integrationskompetenz gereinigte Translokationsbzw. Integrationsfaktoren zugegeben werden. Die dabei verwendeten Konzentrationen betrugen: SecA SecB F1-ATPase Ffh FtsY 80 ng / µl 40 ng / µl 40 ng / µl 2 ng / µl 20 ng / µl Da die Wildtyp-CTF inaktives FtsY enthielt, das bei der „Reaktivierung“ von HINV durch zugegebenes FtsY störte, wurden bei der Verwendung von HINV FtsY─-CTF verwendet. Im Anschluß an die 30minütige Synthese wurde die Hälfte eines Doppelansatzes (50 µl) direkt mit TCA gefällt, die andere Hälfte wurde zunächst mit 25 µl Proteinase K (1 mg / ml) für 20 min bei 25°C inkubiert, anschließend wurde auch diese Probe mit TCA gefällt. Um eine vollständige Inaktivierung der Proteinase K zu erreichen, wurden mit Proteinase K behandelte Proben nach Zugabe der TCA zunächst 10 min auf 56°C erhitzt und erst dann auf 4°C abgekühlt. 4.4.5 Differentialzentrifugation über zweistufigen Saccharosegradienten Die Assoziation von im zellfreien System synthetisierten Proteinen mit Innenmembranvesikeln wurde durch die Auftrennung des Versuchsansatzes in unterschiedliche Fraktionen mittels eines zweistufigen Saccharosegradienten untersucht. Im Anschluß an die Synthese wurden jeweils Doppelansätze (50 µl) auf einen SaccharoseStufen-Gradienten aufgetragen, der aus je 50 µl einer 0.77 M und einer 1.44 M Saccharoselösung (jeweils in 50 mM Triethanolamin pH 7.5; 50 mM Kaliumacetat pH 7.5; 8 mM Magnesiumacetat) durch Unterschichtung hergestellt worden war. Nach 30minütiger Zentrifugation bei 30 Psi in der Beckman Airfuge wurde der Gradient in drei Fraktionen aufgeteilt: Die oberen 75 µl enthielten als Überstand die zytosolischen Komponenten des zellfreien Systems; die unteren 75 µl waren die Membranfraktion: Sie enthielten an der Interphase zwischen den beiden Saccharoselösungen die Membranvesikel. Diese Fraktionen wurden jeweils mit TCA gefällt. Aggregierte, während der Zentrifugation pelletierte Proteine wurden in 25 µl SDS-PAGE-Ladepuffer (siehe 4.2.3) durch 5minütiges Rütteln resuspendiert und durch 5minütige Inkubation bei 95°C denaturiert. 4 Material und Methoden 36 4.4.6 Quervernetzung mit DSS Zur Quervernetzung von im zellfreien System synthetisierten naszierenden Ketten mit ihren Bindungspartnern wurde das homobifunktionelle, membrangängige Quervernetzungsreagenz DSS verwendet. Nach der 30minütigen Synthese (dabei diente HEPES statt Triethanolaminacetat zur Pufferung, siehe oben) wurden die Reaktionsansätze mit 1/10 Volumen 25 mM DSS in DMSO gemischt und weitere 20 min bei 25°C inkubiert. Anschließend wurde die Quervernetzungsreaktion durch Zugabe von Tris / HCl pH 7,5 mit einer Endkonzentration von 50 mM und 15minütiger Inkubation bei 25°C abgestoppt und die Probe entweder TCA-gefällt oder zur Identifizierung der Quervernetzungsprodukte mit spezifischen Antiseren immunpräzipitiert. 4.4.7 Isolierung ribosomenassoziierter naszierender Ketten (RANCs) Zur Isolierung der ribosomenassoziierten naszierenden Ketten wurden diese durch ein Saccharosekissen pelletiert. Hierzu wurden die Proben auf 200 µl Saccharose-Kissen aufgetragen und 60 min bei 70 000 Rpm im Rotor TLA 100.2 bei 4°C zentrifugiert. Anschließend wurden die RANCs in 10 µl INV-Puffer (siehe 4.3.2) je Syntheseansatz resuspendiert. Saccharose-Kissen: 580 mM 40 mM 70 mM 6 mM 1 mM 0.1 mM 8 mM 5 mM Saccharose HEPES pH 7.5 Kaliumacetat pH 7.5 Magnesiumacetat DTT Spermidin Putrescin Phosphoenolpyruvat 4.4.8 Isolierung von Innenmembranvesikeln Im Anschluß an die Quervernetzung von ribosomenassoziierten naszierenden Ketten (RANCs) mit SecY von Innenmembranvesikeln wurden die Vesikel über ein Saccharosekissen isoliert, um die Spezifität und Sensitivität der folgenden Immunpräzipitation zu erhöhen. Hierzu wurden die Proben auf 200 µl Saccharosekissen (750 mM Saccharose, 50 mM Triethanolaminacetat pH 7.5) aufgetragen und durch 15minütige Zentrifugation bei 70 000 Rpm im Rotor TLA 100.2 bei 4°C pelletiert. Anschließend wurden die Vesikel in 100 µl INV-Puffer (siehe 4.3.2) ohne DTT resuspendiert. 4.4.9 TCA-Fällung Zur TCA-Fällung wurden die Proben mit jeweils 1 Volumen 10 % TCA versetzt und mindestens 30 min bei 4°C inkubiert. Anschließend wurde das gefällte Material durch 5 min Zentrifugation bei 13000 Rpm in der Tischzentrifuge „Biofuge pico“ pelletiert; nach Absaugen des Überstandes wurde das Pellet durch 5minütiges Rütteln in 25 µl SDS-PAGELadepuffer (siehe 4.2.3) gelöst und 5 min bei 95°C denaturiert. 37 4 Material und Methoden 4.4.10 Immunpräzipitation Zunächst wurden pro Immunpräzipitation 5 mg Protein-A-Sepharose in 1 ml Tritonpuffer aufgenommen und zum Quellen 5 min auf Eis inkubiert. Tritonpuffer: 1% 0.9 % 25 mM 5 mM Triton X-100 NaCl Tris / HCl pH 6.8 EDTA pH 8 Durch einminütige Zentrifugation mit 13000 Rpm in der Tischzentrifuge „Biofuge pico“ wurde die Protein-A-Sepharose pelletiert; nach dem Absaugen des Überstandes und einem Waschschritt mit 1 ml Tritonpuffer wurde die Protein-A-Sepharose schließlich in 1 ml Tritonpuffer aufgenommen und das spezifische Antiserum (vom Kaninchen) hinzugefügt: Antiserum αTF αFfh αSecY µl pro Immunpräzipitation 10 µl 15 µl 20 µl Die folgende einstündige Inkubation bei 4°C erfolgte auf einem Überkopfschüttler, so daß das Sedimentieren der Protein-A-Sepharose verhindert wurde. Nach zwei weiteren Waschschritten mit jeweils 1 ml Tritonpuffer wurden die Antikörper-ProteinA-SepharoseKomplexe wiederum in 1 ml Tritonpuffer aufgenommen. Die Proteine von vierfachen Syntheseansätzen (100 µl) wurden durch 5minütige Inkubation bei 95°C in 1 % SDS denaturiert, mit den in Tritonpuffer gelösten Antikörper-ProteinASepharose-Komplexen gemischt und anschließend für 1 h bei 4°C auf dem Überkopfschüttler gedreht. Für die Immunpräzipitation von SecY fand die Denaturierung mit 1 % SDS 10 min bei 37°C statt und die anschließende Inkubation mit Protein-A-Sepharose wurde übernacht durchgeführt. Schließlich wurden die Proben zweimal mit 1 ml Tritonpuffer gewaschen (jeweils zweiminütige Zentrifugation bei 13 000 Rpm in der Biofuge), durch einen Waschschritt mit 1 ml Äquilibrierungspuffer (25 mM Tris / HCl pH 6.8) an die Puffer- bzw. Salzbedingungen des SDS-PAGE-Ladepuffers angepaßt, in 30 µl SDS-PAGE-Ladepuffer (siehe 4.2.3) aufgenommen, 5 min gerüttelt, 7 min bei 95°C inkubiert und nach einminütiger Zentrifugation (zur Sedimentierung der nicht auf das SDS-PAGE-Gel aufzutragenden Protein-A-Sepharose) auf ein SDS-PAGE-Gel aufgetragen. Die Denaturierung der SecYImmunpräzipitate erfolgte in SDS-PAGE-Probenpuffer ohne DTT, um die Reduktion der Immunglobuline gering zu halten. Dies war notwendig, weil die schweren Ketten der Immunglobuline im SDS-PAGE-Gel auf etwa gleicher Höhe wie Quervernetzungsprodukte zwischen SecY und naszierenden Ketten von MtlA bzw. Momp2 laufen und dies zu unerwünschten Interferenzerscheinungen führt. 4 Material und Methoden 38 4.4.11 SDS-PAGE und Autoradiographie Im Anschluß an die TCA-Fällung bzw. Immunpräzipitation wurden die in SDS-PAGELadepuffer denaturierten Proteine über SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) aufgetrennt (siehe 4.2.3). Die Sichtbarmachung der radioaktiven Proteinbanden erfolgte mittels Phosphoimager / Image Quant (Molecular Dynamics), wobei die getrockneten Gele in den Filmkasetten übernacht, bei Quervernetzungsexperimenten 1-3 Wochen exponiert wurden. Die Quantifizierung der Banden wurde ebenfalls mit dem Programm Image Quant durchgeführt. Abb 4.3 faßt die verschiedenen Versuche im zellfreien System zusammen. Transkription-Translation im zellfreien System • Plasmid-DNA •Nukleosidtriphosphate (NTP) •ATP-regenerierendes System (PEP, CP, CPK) •T7-Polymerase • Ribosomen • Initiationsfaktor 2 • Zytosolische Translationsfaktoren (CTF) • Aminosäuren inclusive 35S-Methionin und 35S-Cystein Je nach Fragestellung: •Inside-out-Innenmembranvesikel (INV) oder harnstoffgewaschene Vesikel (HINV) • aufgereinigte Translokationsfaktoren (SecA, SecB, Ffh, FtsY) • DNA-Oligomere zur Synthese naszierender Ketten Inkubation bei 37°C Evtl. Puromycin-Behandlung zur Loslösung der naszierenden Ketten vom Ribosom Quervernetzung mit DSS kovalente Kopplung miteinander interagierender Proteine TCA-Fällung Immunpräzipitation Protease -Resistenz-Test Differentialzentrifugation über zweistufigen Saccharosegradienten Auftrennung in Nicht transportierte Proteine werden von Proteinase K verdaut TCA-Fällung Überstand (lösliche Proteine) Membranfraktion Pellet (aggregierte Proteine ) TCA-Fällung Auftrennung der Proteine über SDS-Gelelektrophorese, Nachweis der synthetisierten, radioisotopmarkierten Proteine durch Szintillations-Abbildungsverfahren (Phosphor-Imager) =>Nachweis von Protein-ProteinInteraktionen =>Nachweis erfolgreicher Translokation bzw. Integration => Nachweis von MembranTargeting der Proteine Abb. 4.3: Versuche im zellfreien System zum Membrantransport von Escherichia coli 5 Ergebnisse 39 5. Ergebnisse 5.1 Momp2: Ein Hybridprotein aus Mannitpermease (MtlA) und Outer membrane protein A (OmpA) Das Hybridprotein Momp2 wurde durch die Fusion von amphiphiler Helix und erster SignalAnker-Sequenz von MtlA mit dem signalsequenzlosen OmpA hergestellt: Das Outer membrane protein A (OmpA) wird als Vorläuferprotein (pOmpA) synthetisiert; nach seiner Translokation durch die Innenmembran wird die Signalsequenz durch die Signalpeptidase abgespalten, so daß das reife OmpA ins Periplasma freigesetzt wird (Abb. 5.1A). Mannitpermease (MtlA) ist ein polytopes Innenmembranprotein mit N-terminaler amphiphiler Helix, sechs transmembranen α-Helices sowie einer großen C-terminalen zytoplasmatischen Domäne (Abb. 5.1B; Sugiyama et al., 1991). Die erste transmembrane α-Helix von MtlA dient als Signal-Anker-Sequenz und wird von SRP erkannt (Beck et al., 2000). Bei der Konstruktion von Momp2 wurde die 21 Aminosäuren lange Signalsequenz sowie 5 weitere Aminosäuren von pOmpA durch die N-terminalen 47 Aminosäuren von MtlA ersetzt. Dieser Bereich von MtlA besteht aus der amphiphilen Helix (24 Aminosäuren), der ersten SignalAnker-Sequenz (20 Aminosäuren) sowie 3 zusätzlichen Aminosäuren (Abb. 5.1C). pOmpA 346 As 27 26 21 47 48 Periplasma Momp2 367 As MtlA 637 As Signalpeptidase 27 47 44 Innenmembran 25 24 334 Zytosol A B C Abb. 5.1: Membrantopologie von pOmpA, MtlA und Momp2 A. OmpA wird als Vorläuferprotein (pOmpA) synthetisiert; nach seiner Translokation durch die Innenmembran wird die Signalsequenz (grün, 21 As) durch die Signalpeptidase abgespalten, so daß das reife OmpA ins Periplasma freigesetzt wird. B. Das Innenmembranprotein MtlA besteht aus einer N-terminalen amphiphilen Helix (blau, 24 As), sechs transmembranen α-Helices (die erste transmembrane α-Helix ist rot eingezeichnet und besteht aus den As 25-44) sowie einer großen zytoplasmatischen Domäne am C-Terminus (As 334-637). C. Momp2 ist ein Hybridprotein aus amphiphiler Helix (blau) und erster transmembraner α-Helix (rot) von MtlA sowie einem signalsequenzlosen OmpA. Bei seiner Konstruktion wurden die ersten 26 As von pOmpA durch die ersten 47 As von MtlA ersetzt. As = Aminosäuren 5 Ergebnisse 40 Die Konstruktionsweise von Momp2 legt die im Diagramm gezeigte Membrantopologie von Momp2 nahe: Die amphiphile Helix bleibt auf der zytosolischen Seite, die Signal-AnkerSequenz vermittelt eine Membranverankerung, und der von OmpA stammende Proteinanteil ragt in das Periplasma hinein. Diese Topoplogie wurde, wie weiter unten ausgeführt, experimentell bestätigt. Somit entspricht Momp2 einer Reihe von authentischen Innenmembranproteinen in Escherichia coli, die aus einer großen periplasmatischen Domäne bestehen, welche durch eine α-Helix in der Membran verankert ist. 5.2 Momp2 wird effizient in „Inside-out“-Innenmembranvesikel (INV) transloziert Zunächst sollte geprüft werden, ob das Hybridprotein Momp2 in Innenmembranvesikel transportiert werden kann: Die Translokationseffizienz* von Momp2 sollte mit der Translokationseffizienz von pOmpA bzw. der Integrationseffizienz von MtlA verglichen werden. Hierzu wurde die Synthese der drei Proteine in einem zellfreien System mit einem Protease-Resistenz-Test gekoppelt: Die Proteine wurden in einem zellfreien TranskriptionsTranslations-System aus E. coli synthetisiert und dabei durch die Zugabe von 35S-Methionin und 35S-Cystein radioaktiv markiert. Dem Syntheseansatz wurden „Inside-out“Innenmembranvesikel (INV) zugegeben. Diese sind im Vergleich zu den Verhältnissen in der E. coli-Zelle invertiert: Die zytosolische Seite der Innenmembran zeigt nach außen, die periplasmatische Seite hingegen nach innen. Außerdem enthalten die INV alle Targetingfaktoren (SecA, SecB, SRP und den SRP-Rezeptor FtsY) in ausreichenden Mengen. Sekretorische Proteine können daher durch die Innenmembran in die INV transloziert werden, während Innenmembranproteine in die Lipidschicht der INV integriert werden können. Im Anschluß an die Synthesephase wurden die Versuchsansätze mit Proteinase K inkubiert. Diese konnte alle Proteine außerhalb der INV verdauen. Sekretorische Proteine, die in die INV transloziert worden waren, waren hingegen vor Proteinase K geschützt. Auch die transmembranen und periplasmatischen Anteile integraler Innenmembranproteine wurden durch die Lipidschicht der INV vor einem Proteaseverdau geschützt; große zytosolische Anteile wurden jedoch verdaut, so daß kleinere Proteinfragmente (membrangeschützte Fragmente) entstanden. Schließlich wurden die Proteine mit TCA gefällt und mittels SDSGelelektrophorese aufgetrennt. Die synthetisierten, radioaktiv markierten Proteine wurden durch ein autoradiographisches Verfahren (Phoshoimaging) sichtbar gemacht. Abb. 5.2A zeigt diesen Protease-Resistenz-Test für pOmpA. In Abwesenheit von INV entstand nur das Vorläuferprotein pOmpA, das durch Proteinase K so gut wie vollständig verdaut wurde (Spuren 1 und 2). Nach Zugabe von INV wurde ein großer Teil des * Beim Membrantransport von Momp2 wird die Signal-Anker-Sequenz in die Innenmembran integriert, die große periplasmatische Domäne jedoch durch die Innenmembran transloziert. Vereinfachend wird der Membrantransport von Momp2 hier als Translokation bezeichnet. 5 Ergebnisse 41 Vorläuferproteins durch die Abspaltung seiner Signalsequenz durch die Signalpeptidase zum reifen OmpA „prozessiert“ (Spur 3); das reife OmpA war proteaseresistent (Spur 4). Daß auch das Vorläuferprotein pOmpA teilweise proteaseresistent war, war nicht zu erwarten; dieses Phänomen wird jedoch in vitro regelmäßig beobachtet und ist möglicherweise auf eine im Vergleich zu den Verhältnissen in der E. coli-Zelle verminderte Signalpeptidaseaktivität in den INV zurückzuführen. Auch MtlA wurde in Abwesenheit von INV nahezu vollständig von Proteinase K verdaut (Abb. 5.2B, Spuren 1 und 2); in Anwesenheit von INV entstand beim Proteinase-K-Verdau ein membrangeschütztes Fragment (Spuren 3 und 4): Die amphiphile Helix sowie die zytosolische C-terminale Domäne von MtlA liegen auf der Außenseite der INV und konnten daher von Proteinase K verdaut werden (vergleiche Abb. 5.1B). Abb. 5.2C zeigt den Protease-Resistenz-Test für das Hybridprotein Momp2: In der Negativkontrolle ohne INV wurde Momp2 durch Proteinase K vollständig verdaut (Spuren 1 und 2). In Anwesenheit von INV entstand ein nur etwas kleineres membrangeschütztes Fragment (Spuren 3 und 4). Die vergleichsweise geringe Größenabnahme von Momp2 nach Proteinase-K-Behandlung läßt darauf schließen, daß nur die amphiphile Helix für einen Proteaseverdau zugänglich ist. Die Signal-Anker-Sequenz ist hingegen in die schützenden Membranlipide integriert und der reife OmpA-Anteil von Momp2 ragt in die INV hinein (vergleiche Abb. 5.1C). Die Translokationseffizienz von Momp2 entsprach in etwa der Translokationseffizienz von pOmpA bzw. der Integrationseffizienz von MtlA. + INV + PK + + + INV + PK + + MtlA INV 3 4 MtlAMGF 1 A 3 4 + PK Momp2MGF OmpA 2 + + Momp2 pOmpA 1 + 2 3 1 2 4 B C Abb. 5.2: Membrantransport von pOmpA, MtlA und Momp2 in einem zellfreien System pOmpA, MtlA und Momp2 wurden in einem zellfreien Transkriptions-Translations-System aus E. 35 35 coli synthetisiert und dabei durch die Verwendung von S-Methionin und S-Cystein radioisotopmarkiert. Die Synthese fand jeweils in Abwesenheit (Spuren 1 und 2) bzw. in Anwesenheit (Spuren 3 und 4) von INV statt. Anschließend wurde je ein Aliquot direkt mit TCA gefällt (Spuren 1 und 3), ein weiteres Aliquot wurde zuvor mit Proteinase K (PK) behandelt (Spuren 2 und 4). Die Proteine wurden über SDS-Gelelektrophorese aufgetrennt; die radioisotopmarkierten Proteine wurden (Phosphoimaging) sichtbar gemacht. MGF = membrangeschütztes Fragment durch ein autoradiographisches Verfahren 5 Ergebnisse 42 5.3 Momp2 wird am Ribosom sowohl von Ffh als auch von Trigger factor erkannt Aus früheren Berichten ist bekannt, daß ribosomenassoziierte naszierende Ketten (RANCs) des sekretorischen Proteins pOmpA von Trigger factor, nicht jedoch von Ffh gebunden werden (Beck et al., 2000). Dabei bevorzugt Trigger factor offenbar das reife OmpA als Bindestelle, während die Signalsequenz eher eine untergeordnete Rolle zu spielen scheint (Valent et al., 1997). RANCs des polytopen Innenmembranproteins MtlA werden hingegen von SRP, nicht jedoch von Trigger factor gebunden. SRP bindet dabei mindestens an die erste Signal-Anker-Sequenz von MtlA (Beck et al., 2000). Das Hybridprotein Momp2 enthält also einerseits mit der Signal-Anker-Sequenz von MtlA eine Bindestelle für SRP, andererseits aber mit dem reifen OmpA auch die vermutliche(n) Bindestelle(n) für Trigger factor. Dies ließ erwarten, daß sowohl SRP als auch Trigger factor an RANCs von Momp2 binden würden. Dies sollte durch Quervernetzungsexperimente geprüft werden. Hierzu wurden im zellfreien Transkriptions-Translations-System ribosomenassoziierte, naszierende Ketten von pOmpA, MtlA und Momp2 nach einer Methode von Haeuptle et al. (1986) synthetisiert (Abb. 5.3): Dem Syntheseansatz wurden DNA-Oligonukleotide zugegeben, deren Sequenz zu einem Abschnitt der mRNA des zu synthetisierten Proteins komplementär war. In der Transkriptions-Translations-Reaktion hybridisieren diese DNAOligonukleotide mit der mRNA; die Endonuklease RNAseH erkennt diese Hybride und verdaut den hybridisierten mRNA-Teil. Die so verkürzte mRNA trägt an ihrem 3‘-Ende kein Stoppkodon, so daß das Ribosom am Ende der Translation die Polypeptidkette nicht freisetzen kann und ribosomenassoziierte naszierende Ketten (RANCs) entstehen. Diese imitieren eine Momentaufnahmen einer Polypeptidkette in statu nascendi. T7 Promotor Plasmid Transkription 5' antisense DNAOligonukleotid 5' 3' mRNA endogene 3' RNaseH 5' 3' Translation verkürzte mRNA 5' ribosomenassoziierte naszierende Kette Abb. 5.3: Synthese ribosomenassoziierter naszierender Ketten (RANCs) Dem Transkriptions-Translations-Ansatz werden „antisense“ DNA-Oligonukleotide zugefügt, die mit der mRNA hybridisieren. RNaseH erkennt diese Hybride und verdaut den hybridisierten mRNATeil. Die so verkürzte mRNA trägt an ihrem 3‘-Ende kein Stoppkodon, so daß das Ribosom am Ende der Translation die Polypeptidkette nicht freisetzen kann. (Abb. modifiziert nach Beck, 2000) Im Anschluß an die Synthese der RANCs wurden deren Bindungspartner durch Quervernetzung mit dem homobifunktionellen, membranpermeablen Quervernetzungsreagenz Disuccinimidyl-Suberat (DSS) identifiziert. DSS besteht aus Suberinsäure (COOH-(CH2)6COOH), die an beiden Enden mit N-Hydroxysuccinimid (NHS) verestert ist (Abb. 5.4A). 43 5 Ergebnisse A B pH 7 - 9 Abb. 5.4: Strukturformel (A) und Reaktionsschema (B) von DSS DSS reagiert im neutralen Bereich mit primären Aminen unter Abspaltung von NHS und Ausbildung einer Amidbindung (Abb. 5.4B). Somit ermöglicht es die kovalente Koppelung eng benachbarter Polypeptide, wobei v.a. die ε-Amine der Lysin-Seitenketten als Reaktionspartner dienen. Das Suberat-Molekül kann einen Abstand von bis zu 11.4 Å zwischen zwei miteinander quervernetzten Polypeptiden überbrücken. Im Anschluß an die Quervernetzung konnten die Versuchsansätze mit spezifischen Antisera immunpräzipitiert werden, um die einzelnen Quervernetzungspartner zu identifizieren. In Abb. 5.5 wird zunächst das Verhalten von 125 Aminosäuren langen RANCs von pOmpA (pOmpA-125) sowie 189 Aminosäuren langen RANCs von MtlA (MtlA-189) demonstriert. Für pOmpA-125 entstand als DSS-Quervernetzungsprodukt eine prominente Doppelbande, welche mit einem spezifischen Antiserum gegen Trigger factor immunpräzipitiert werden konnte (Spuren 2 und 3). Trigger factor war also im zellfreien System bereits enthalten. Das Auftreten einer Doppelbande als Quervernetzungsprodukt zwischen pOmpA und Trigger factor ist bereits früher beobachtet worden (Beck et al., 2000); die untere Bande entspricht mit ihrem Molekulargewicht von etwas mehr als 70 kD der Summe von pOmpA-125 (14 kD) und Trigger factor (das 48 kD große Protein zeigt im SDS-Polyacrylamidgel ein aberrantes Laufverhalten und läuft bei 58 kD; Hesterkamp et al., 1996). Das Entstehen der oberen Bande ist nicht endgültig geklärt; mögliche Ursachen für das veränderte Laufverhalten sind: (1) Die RANCs sind in ihrer Länge heterogen, (2) unterschiedliche Lysine von Trigger factor oder den RANCs reagieren mit DSS, so daß ein unterschiedliches Laufverhalten resultiert, oder (3) Trigger factor wird an mehreren Bindestellen mit den RANCs quervernetzt, so daß ein „Knäuel“ mit schlechteren Laufeigenschaften entsteht. Nach Zugabe des SRP-Proteins Ffh konnte kein weiteres Quervernetzungsprodukt nachgewiesen werden (Spuren 6-8). Für MtlA189 konnte in Abwesenheit von Ffh kein prominentes Quervernetzungsprodukt beobachtet werden (Spuren 10 – 12). Erst nach Zugabe von Ffh wurde eine deutliche Quervernetzungsbande sichtbar, die in ihrem Molekulargewicht von etwas weniger als 70 kD der Summe von MtlA-189 (19 kD) und Ffh (48 kD) entsprach und auch mit spezifischem anti-Ffh-Serum immunpräzipitiert werden konnte (Spuren 14 und 16). Beim Quervernetzen von Momp2-146 entstand ebenfalls eine Doppelbande, die aufgrund ihrer Größe und durch 5 Ergebnisse 44 Immunpräzipitation mit anti-Trigger-factor-Serum als Quervernetzungsprodukt von Momp2146 mit Trigger factor identifiziert werden konnte (Spuren 18 und 19). Nach Zugabe von Ffh entstand ein weiteres Quervernetzungsprodukt, welches mit anti-Ffh-Serum immunpräzipitiert werden konnte (Spuren 22 und 24). Die Quervernetzungsintensität zwischen Momp2-146 und Trigger factor nahm durch die Zugabe von Ffh deutlich ab (vergleiche Spuren 19 und 23). pOmpA-125 Ffh DSS IP α * MtlA-189 + + + + + + + TF Ffh TF Ffh Momp2-146 + + + + + + + TF Ffh + + + + + + TF Ffh TF Ffh + TF Ffh 221 __ __97 __70 x TF x x Ffh * * MtlA * * x x * * * *x x Momp2 pOmpA MtlA-189 pOmpA-125 Momp2 -146 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 __46 __29 __19 __14 21 22 23 24 Abb. 5.5: Momp2-RANCs können sowohl mit Ffh als auch mit Trigger factor quervernetzt werden pOmpA-125, MtlA-189 und Momp2-146 wurden in Ab- bzw. Anwesenheit von Ffh im zellfreien System synthetisiert; jeweils ein Reaktionsansatz (25 µl) wurde direkt mit TCA gefällt (Spuren 1,5,9,13,17 und 21), der Rest wurde mit DSS quervernetzt und die RANCs über ein Saccharosekissen isoliert; hiervon wurden wiederum ein Reaktionsansatz mit TCA gefällt (Spuren 2,6,10,14,18,22) und jeweils vierfache Reaktionsansätze mit anti-Trigger-factor- (Spuren 3,7,11,15,19,23) bzw. anti-Ffh-Antikörpern (Spuren 4,8,12,16,20,24) immunpräzipitiert. Dieses Experiment zeigt, daß sowohl SRP als auch Trigger factor an naszierende Ketten von Momp2 binden. Eine funktionelle Interaktion zwischen RANCs und Ffh kann durch die Zugabe des SRP-Rezeptors FtsY zusammen mit Innenmembranen aufgelöst werden (Valent et al., 1998; Scotti et al., 1999). Daher sollte im folgenden überprüft werden, ob auch im Fall von Momp2 die Zugabe von FtsY und Innenmembranen die Dissoziation von Ffh von den naszierenden Ketten bewirkt. Zunächst wurden die Versuchsbedingungen mit naszierenden Ketten von MtlA etabliert (Abb. 5.6 oben): MtlA-189 wurde in Abwesenheit (Spuren 1 und 2) bzw. in Anwesenheit (Spuren 3 - 12) von Ffh synthetisiert und anschließend mit DSS quervernetzt. Nur in Anwesenheit von Ffh entstand ein Quervernetzungsprodukt, das mit seinem Molekulargewicht von etwas weniger als 70 kD dem oben beschriebenen Quervernetzungsprodukt von MtlA-189 und Ffh entsprach (x in Spur 4). Waren bei der 5 Ergebnisse 45 Synthese INV mit ihrem endogenen FtsY-Gehalt anwesend, verschwand das Quervernetzungsprodukt zwischen der naszierenden Kette und Ffh (Spur 6); durch Zugabe der FtsY-freien HINV oder von FtsY alleine konnte dieser Effekt nicht erzielt werden (Spuren 8 und 10). Erst durch die gleichzeitige Zugabe von aufgereinigtem FtsY und HINV wurde die Fähigkeit, Ffh von MtlA-189 abzulösen, weitgehend wieder hergestellt (Spur 12). Dies zeigt, daß FtsY und Innenmembranen erforderlich sind, um die Interaktion zwischen Ffh und naszierenden Ketten von MtlA zu beenden. INV Vesikel Ffh HINV HINV + FtsY DSS + + + + + + + + + __97 __70 X =MtlA-189 x Ffh ? x MtlA x x __46 __29 __ 19 __14 MtlA-189 __97 __70 X =Momp2-146 x Ffh ? x x x __46 Momp2 __29 __ 19 __14 Momp2146 1 2 3 4 5 6 7 8 9 10 11 12 Abb. 5.6: Die Dissoziation von Ffh von MtlA- bzw. Momp2-RANCs erfordert FtsY und Innenmembranen MtlA-189 und Momp2-146 wurden ohne Zugabe (Spur 1 und 2) bzw. nach Zugabe von Ffh (Spur 312) im zellfreien System synthetisiert. Zusätzlich waren in einzelnen Ansätzen INV (Spuren 5 und 6), HINV (Spuren 7 und 8 sowie 11 und 12) und FtsY (Spuren 9-12) bei der Synthese anwesend. Im Anschluß an die Synthese wurde jeweils ein Reaktionsansatz (25 µl) direkt mit TCA gefällt (ungeradzahlige Spuren), ein weiterer Reaktionsansatz wurde zuvor mit DSS quervernetzt (geradzahlige Spuren). Die in Spur 2 mit „?“ markierte Bande bei ca. 70 kD entspricht einem unspezifischen Syntheseprodukt, das durch DSS-Quervernetzung verstärkt wurde; es trat zwar in allen Versuchen auf, wird bei den in diesem Versuch geringen Syntheseraten jedoch besonders deutlich. 5 Ergebnisse 46 Derselbe Effekt wurde auch für Momp2-146 beobachtet (Abb. 5.6 unten): Die Ablösung von Ffh von der naszierenden Momp2-Kette erforderte ebenfalls sowohl FtsY als auch HINV. Aufgrund geringer Syntheseraten von Momp2-146 in diesem Versuch und des hohen Hintergrundes sind die Ergebnisse in diesem Versuch jedoch nur bedingt aussagekräftig. Die beiden beschriebenen Quervernetzungsexperimente zeigen einerseits, daß sowohl SRP als auch Trigger factor kotranslational an Momp2 binden. Andererseits schien die Dissoziation von Ffh von den Momp2-RANCs nach Zugabe von FtsY und Innenmembranen einen ersten Hinweis dafür zu liefern, daß die Interaktion von SRP mit naszierenden Ketten von Momp2 von funktionellem Charakter ist. Bei dem sekretorischen Protein pOmpA führt die Bindung von Trigger factor an die naszierenden Ketten zu einer späteren Interaktion mit SecA und SecB. Bedeutet demnach die Bindung von Trigger factor an naszierende Ketten von Momp2, daß auch Momp2 zu einem späteren Zeitpunkt mit SecA und SecB interagiert? Wenn ja, welches sind dann die spezifischen Aufgaben von SRP bzw. Trigger factor, SecA und SecB beim Membrantransport von Momp2? Diese beiden Fragen sollten in den nächsten Versuchen geklärt werden. 5.4 Die Translokation von Momp2 in HINV benötigt nur SecA und SecB, jedoch nicht SRP In Abb. 5.2 wurde bereits gezeigt, daß Momp2 in „Inside-out“-Innenmembranvesikel (INV) transloziert werden kann. Da die INV alle Targetingfaktoren (SecA, SecB, Ffh und FtsY) in ausreichenden Mengen enthalten, geht aus diesem Versuch jedoch nicht hervor, welche Targetingfaktoren hierfür tatsächlich nötig sind. Dies sollte im folgenden Versuch durch die Verwendung von harnstoffgewaschenen Vesikeln (HINV) getestet werden. Durch die Behandlung mit 6 M Harnstoff werden die Targetingfaktoren SecA, SecB, Ffh und FtsY entfernt. Auch das verwendete zellfreie Transkriptions-Translations-System ist frei von diesen Targetingfaktoren (Koch et al., 1999). Im Rahmen eines Protease-Resistenz-Tests wurden zunächst pOmpA, MtlA und Momp2 in der Abwesenheit von Vesikeln synthetisiert. Bei dem anschließenden Verdau mit Proteinase K wurden alle drei Substrate wieder beihnahe vollständig verdaut (Abb. 5.7, Spuren 1 und 2). Nach der Zugabe von INV hingegen wurden die bereits in Abb. 5.2 gezeigten proteaseresistenten Spezies beobachtet, die für alle drei Substrate eine erfolgreiche Translokation bzw. Integration anzeigen (Spuren 3 und 4). Durch die Verwendung der harnstoffgewaschenen Vesikel (HINV) wurde der Transport aller drei Substrate nahezu vollständig unterbunden (Spuren 5 und 6). Entsprechend früheren Ergebnissen (Koch et al., 1999), konnte die Translokation von pOmpA durch SecA, SecB und die F1-Untereinheit der F1F0-ATPase wiederhergestellt werden (Spuren 7 und 8), während die Integration von MtlA von der Zugabe der SRP-Komponente Ffh und des SRP-Rezeptors FtsY abhängig war (Spuren 9 und 10); die Abhängigkeit der Integration von 4,5S RNA kann in dem verwendeten zellfreien System nicht demonstriert werden, da die 4,5S RNA in dem verwendeten 5 Ergebnisse 47 Zellextrakt in ausreichendem Maße enthalten war (Koch et al., 1999). Trotz der weiter oben demonstrierten Bindung von SRP an naszierende Ketten von Momp2, konnte die Translokation von Momp2 in HINV durch die Zugabe von Ffh und FtsY nicht wiederhergestellt werden (Spuren 9 und 10); vielmehr war die Zugabe von SecA, SecB und der F1-Untereinheit der F0F1-ATPase notwendig und ausreichend (Spuren 7 und 8). INV HINV + + SecA, SecB, F1 + + + + + + + + + 44 1,1 40 2 22 24 + Ffh, FtsY PK + + pOmpA OmpA % Translokation 1,3 45 1,5 MtlA MtlA-MPF 1 % Integration 24 2 Momp2 Momp2-MPF 1 % Integration 1 Abb. 5.7: Momp2 2 34 3 benötigt 4 für 0,6 5 seine 6 32 7 0,9 8 Translokation 9 in 10 28 11 12 harnstoffgewaschene Innenmembranvesikel (HINV) nur SecA, SecB und die F1-ATPase pOmpA, MtlA und Momp2 wurden in Abwesenheit von Innenmembranvesikeln (Spuren 1 und 2), in Anwesenheit von INV (Spuren 3 und 4) oder in Anwesenheit von HINV (Spuren 5 - 12) im zellfreien System synthetisiert. Außerdem wurden entsprechend der Beschriftung SecA, SecB, F1-ATPase, Ffh und FtsY bei der Synthese zugegeben. Anschließend wurde jeweils ein Reaktionsansatz (25 µl) direkt mit TCA gefällt (ungeradzahlige Spuren), ein weiterer Reaktionsansatz wurde zuvor mit Proteinase K (PK) behandelt (geradzahlige Spuren). Zur Bestimmung der Translokations- bzw. Integrationseffizienz wurde die Radioaktivität in den einzelnen Proteinbanden mit Hilfe des Programmes Image Quant ermittelt. Die Translokationseffizienz von OmpA entspricht dem Verhältnis der OmpA- und pOmpA-Banden nach Protease-K-Verdau zu den OmpA- und pOmpABanden vor Protease-K-Verdau. Die Integrationseffizienz von MtlA entspricht dem Verhältnis von MtlA-MGF zu MtlA. Dieser Wert wurde dann mit 1.5 multipliziert, da MtlA-MGF nur noch 16 der ursprünglich 24 Methionine enthält. Die Translokationseffizienz von Momp2 entspricht dem Verhältnis von Momp2-MGF zu Momp2. MGF = membrangeschütztes Fragment. 5 Ergebnisse 48 Die Translokation von Momp2 ist in vitro also nicht von SRP abhängig und kann auch in Abwesenheit von SRP stattfinden. Da in vivo die Synthese von Momp2 jedoch immer in Anwesenheit von SRP stattfindet, ist zu erwarten, daß dann auch eine Bindung von SRP an naszierende Ketten von Momp2 erfolgt, wie dies weiter oben durch DSS-Quervernetzung von Ffh an RANCs von Momp2 gezeigt wurde. Diese Bindung scheint über die Bindung von Trigger factor zu dominieren, da bei Zugabe von Ffh die Quervernetzungsbande mit Trigger factor deutlich schwächer wurde (Abb. 5.5). Außerdem bewirkten FtsY und Innenmembranen eine Dissoziation von SRP vom SRP-RANC-Komplex (Abb. 5.6). Diese Ergebnisse sprechen für eine funktionelle Rolle von SRP bei dem Membrantargeting von Momp2. Die Rolle von SRP beim Export von Momp2 zu analysieren, machte daher einen anderen experimentellen Ansatz erforderlich. 5.5 Eine gestörte Interaktion zwischen SecA und SecY beeinträchtigt die Translokation, nicht jedoch die Membranassoziation von Momp2 Hierzu kamen in den folgenden Versuchen INV eines SecY-Mutanten-Stammes zum Einsatz. Die secY205-Mutante unterscheidet sich vom Wildtyp durch den Austausch einer einzigen Aminosäure (Tyrosin zu Aspartat) in der C-terminalen, zytoplasmatischen Domäne von SecY. Hierdurch ist die Interaktion zwischen SecA und SecY gestört, wodurch die Translokation sekretorischer Proteine stark beeinträchtigt wird (Matsumoto et al., 1997). Im Gegensatz dazu ist die Integration polytoper Innenmembranproteine nicht betroffen, da diese auf eine funktionelle SecA-SecY-Interaktion nicht angewiesen sind (Koch und Müller, 2000). Somit stellten die secY205-INV ein ideales Werkzeug dar, um die Beteiligung von SecA und SecB bzw. SRP an Targeting und Transport von Momp2 zu untersuchen. Durch Verwendung der secY205-INV im Protease-Resistenz-Test sollte zunächst geprüft werden, ob eine gestörte SecA-SecY-Interaktion den Membrantransport von Momp2 beeinträchtigt; zusätzlich sollte durch Differentialzentrifugation in Anwesenheit der secY205-INV untersucht werden, ob trotz gestörter SecA-SecY-Interaktion ein Membrantargeting von Momp2 durch SRP zustande kommen kann. Die secY205-INV wurden zunächst auf die Translokationseffizienz von pOmpA bzw. die Integrationseffizienz von MtlA hin untersucht. Dabei wurden frühere Ergebnisse bestätigt (Abb. 5.8): Die Translokationseffizienz von pOmpA verringerte sich im Fall der secY205INV auf ein Fünftel des Wildtypniveaus (Spuren 5 und 6 im Vergleich zu Spuren 3 und 4); die Integrationseffizienz von MtlA hingegen zeigte bei Wildtyp-INV und secY205-INV keinen Unterschied. Momp2 verhielt sich auch in diesem Versuchsansatz wie pOmpA: Die Translokationseffizienz sank bei Verwendung der secY-205-INV auf etwa ein Viertel des Wildtypniveaus. 5 Ergebnisse 49 Wildtyp INV + PK secY205 + + pOmpA OmpA % Translokation 4 50 10 1 28 27 1 46 12 MtlA MtlA-MGF % Integration Momp2 Momp2-MGF % Translokation 1 2 3 4 5 6 Abb. 5.8: SecY205-INV erlauben die Integration von MtlA, nicht jedoch die Translokation von pOmpA und Momp2 pOmpA, MtlA und Momp2 wurden in Abwesenheit von Innenmembranvesikeln (Spuren 1 und 2), in Anwesenheit von Wildtyp-INV (Spuren 3 und 4) oder in Anwesenheit von secY205-Mutanten-INV (Spuren 5 und 6) im zellfreien System synthetisiert. Anschließend wurde jeweils ein Reaktionsansatz (25 µl) direkt mit TCA gefällt (ungeradzahlige Spuren), ein weiterer Reaktionsansatz wurde zuvor mit Proteinase K (PK) behandelt (geradzahlige Spuren). MGF = membrangeschütztes Fragment Damit zeigt der Protease-Resistenz-Test in zwei unterschiedlichen Versuchen, nämlich zum einen bei der Wiederherstellung der Translokationskompetenz von HINV und zum anderen beim Vergleich zwischen Wildtyp-INV und secY205-INV, daß eine vollständige Translokation von Momp2 SecA-abhängig ist. Um den Targetingmechanismus von Momp2 näher zu untersuchen, wurde die Differentialzentrifugation der Translationsprodukte über einen zweistufigen Saccharosegradienten eingesetzt (Abb. 5.9). Hierbei wurden wiederum pOmpA, MtlA und Momp2 ohne Vesikel (Spuren 1-3), in der Anwesenheit von Wildtyp-INV (Spuren 4 – 6) sowie in der Anwesenheit von secY205-INV (Spuren 7 – 9) synthetisiert. Anschließend erfolgte die Auftrennung der Syntheseansätze in einen lösliche Anteile enthaltenden Überstand, eine Membranfraktion sowie ein Pellet aus aggregiertem Material. Für alle drei Substrate war eine deutliche Verschiebung vom Überstand zur Membranfraktion durch die Zugabe von Wildtyp-INV zu beobachten (vergleiche Spuren 1/2 und 4/5), was ihre Membranassoziation ausdrückt. Beim Einsatz der secY205-INV wurde die 5 Ergebnisse 50 Membranassoziation von pOmpA gegenüber den Wildtyp-INV deutlich reduziert, während die Membranassoziation von MtlA durch die Verwendung der secY205-INV nicht betroffen war (vergleiche Spuren 4/5 und 7/8). In diesem Versuchsansatz verhielt sich Momp2 nun in gleicher Weise wie MtlA: Die Membranassoziation von Momp2 wurde durch die secY205Mutation ebenfalls nicht beeinträchtigt. - INV Fraktion Üst Wildtyp Mem Pel Üst secY205 Mem Pel Üst Mem Pel pOmpA OmpA % der Probe 69 12 20 30 46 24 50 19 31 42 8 50 13 47 40 8 49 43 48 18 33 8 87 5 12 74 13 1 2 3 4 5 6 7 8 9 MtlA % der Probe Momp2 % der Probe Abb. 5.9: Im Gegensatz zu pOmpA assoziieren MtlA und Momp2 mit secY205-INV pOmpA, MtlA und Momp2 wurden in Abwesenheit von Innenmembranvesikeln (Spuren 1 - 3), in Anwesenheit von Wildtyp-INV (Spuren 4 - 6) oder in Anwesenheit von secY205-Mutanten-INV (Spuren 7 - 9) im zellfreien System synthetisiert. Anschließend wurde je ein Doppelansatz (50 µl) über einen zweistufigen Saccharosegradienten in Überstand (Spuren 1,4 und 7), Membranfraktion (Spuren 2,5 und 8) sowie Pellet (Spuren 3, 6 und 9) aufgetrennt. Üst = Überstand; Mem = Membranfraktion; Pel = Pellet Somit decken sich für pOmpA und MtlA die Ergebnisse von Protease-Resistenz-Test und Differentialzentrifugation mit secY205-INV: Sowohl Translokation als auch Targeting von pOmpA werden durch die gestörte SecA-SecY-Interaktion behindert; Integration bzw. Targeting der MtlA werden hierdurch jedoch nicht beeinflußt. Anders verhält es sich für Momp2: Die geringe Translokationseffizienz bei Verwendung von secY205-INV, nachgewiesen durch Proteaseresistenz, spricht dafür, daß eine vollständige Translokation von Momp2 ohne eine produktive SecA-SecY-Interaktion nicht stattfinden kann; die Membranassoziation von Momp2 ist von dieser SecA-SecY-Interaktion jedoch unabhängig. Dies läßt auf ein SecA-unabhängiges Targeting von Momp2 schließen. 51 5 Ergebnisse 5.6 Quervernetzung von Momp2-RANCs mit Membrankomponenten: Kotranslationale Interaktion zwischen Momp2 und SecY Wie aus früheren Arbeiten bekannt ist, interagieren integrale Innenmembranproteine wie MtlA kotranslational mit Bestandteilen des Translokons (Valent et al., 1998; Beck et al., 2000). Im Gegensatz dazu binden sekretorische Proteine wie pOmpA erst posttranslational an die Membran; das Membrantargeting von pOmpA setzt die Loslösung des Proteins vom Ribosom voraus (Behrmann et al., 1998). Dementsprechend können ribosomenassoziierte naszierende Ketten (RANCs) von pOmpA auch nicht mit SecY, der Hauptkomponente des Translokons, quervernetzt werden; hierfür ist vielmehr die Loslösung vom Ribosom sowie die artifizielle Schaffung eines Translokationsintermediats durch das Anhängen eines größeren Moleküls an den C-Terminus von pOmpA erforderlich, um das pOmpA-Intermediat im Translokon zu arretieren (Beck et al., 2000). Im folgenden sollte untersucht werden, ob Momp2 kotranslational mit dem Translokon interagiert und Momp2-RANCs daher mit SecY von INV quervernetzt werden können. In dem in Abb. 5.10 dargestellten Versuch wurde zunächst die kotranslationale Interaktion zwischen MtlA und SecY bestätigt: Nach der Synthese von MtlA-189 in Anwesenheit von INV trat nach anschließender Inkubation mit DSS eine zusätzliche prominente Bande auf, die in ihrer Größe von ca. 50 kD einem Quervernetzungsprodukt zwischen MtlA-189 (19 kD) und SecY (das 49 kD große Protein zeigt im SDS-Polyacrylamidgel ein aberrantes Laufverhalten und läuft bei 35 kD, Ito 1984) entsprach; dieses Quervernetzungsprodukt konnte auch mit einem spezifischen SecY-Antiserum immunpräzipitiert werden (Spur 4). Auch RANCs von Momp2 in drei verschiedenen Längen (Momp2-146, Momp2-212 und Momp2-301) konnten mit SecY von INV quervernetzt werden (Spuren 8,12 und 16); die Interaktion von Momp2212 mit SecY war aufgrund geringerer Syntheseraten nur schwach zu erkennen. 5 Ergebnisse 52 MtlA-189 + + INV DSS Momp2-146 + + + Momp2-212 Momp2-301 + + + + + + + + + + + 221 97 > < x SecY 72 > < MtlA > Momp2 Momp2-301 < > < 46 < > 29 Momp2-212 20 MtlA-189 Momp2-146 14 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 97 72 > IP α SecY > < > > < < 46 < Abb. 5.10: Momp2-RANCs unterschiedlicher Länge können mit SecY von INV quervernetzt werden. MtlA-189, Momp2-146, -212 und -301 wurden jeweils in Ab- oder Anwesenheit von INV im zellfreien System synthetisiert. Anschließend wurden jeweils ein Reaktionsansatz (25 µl) direkt mit TCA gefällt (ungeradzahlige Spuren), der Rest mit DSS quervernetzt. Hiervon wurde ein einfacher Reaktionsansatz wiederum mit TCA gefällt (geradzahlige Spuren, obere Bildhälfte); aus den restlichen Ansätzen wurden die Vesikel über ein Saccharosekissen isoliert (zu Ansätzen, die bisher keine Vesikel enthielten, wurden vor der Zentrifugation INV zur Gleichbehandlung zugegeben) und dann 15fache Ansätze mit anti-SecY-Antikörpern immunpräzipitiert (Spuren im unteren Bildteil). Um den kotranslationalen Charakter des Membrantargetings von Momp2 zu unterstreichen, wurde Momp2-146 in Anwesenheit von INV synthetisiert; anschließend wurde ein Aliquot direkt mit DSS quervernetzt, das andere zuvor mit Puromycin behandelt (Abb. 5.11). Das Antibiotikum Puromycin löst Polypeptidketten vom Ribosom ab; nach der Loslösung von Momp2-146 vom Ribosom war die Quervernetzung mit SecY deutlich schwächer (siehe auch Abb. 5.12, Spur 7). 5 Ergebnisse 53 INV Puromycin DSS + + + + + + IP α SecY Momp2-146 x SecY 46 1 2 3 Abb. 5.11: Die Quervernetzung von Momp2-146 mit SecY von INV verschwindet weitgehend nach Ablösung der Ribosomen durch Puromycin Momp2-146 wurde in Abwesenheit (Spur 1) bzw. in Anwesenheit von INV (Spuren 2 und 3) im zellfreien System synthetisiert. Ansatz Nr. 3 wurde im Anschluß an die Synthese mit Puromycin behandelt, um die Ribosomen von den naszierenden Ketten abzulösen. Die Proben wurden mit DSS quervernetzt; anschließend wurden die Vesikel über ein Saccharosekissen isoliert (zu Ansatz Nr.1, der bisher keine Vesikel enthielt, wurden vor der Zentrifugation INV zur Gleichbehandlung zugegeben) und dann 15fache Ansätze mit anti-SecY-Antikörpern immunpräzipitiert. Somit sprechen zwei Beobachtungen gegen ein SecA-abhängiges Targeting von Momp2: Zum einen kann das Targeting von Momp2 unabhängig von einer funktionellen SecA-SecYInteraktion stattfinden, was mit Hilfe der Assoziation an secY205-INV gezeigt wurde; zum anderen erfolgte das Targeting von Momp2 kotranslational; ein kotranslationales, durch SecA vermitteltes Targeting kann jedoch ausgeschlossen werden (Behrmann et al., 1998). Die Ähnlichkeit zwischen dem Targeting von MtlA und Momp2 lassen demnach eher ein SRPabhängiges Targeting auch von Momp2 vermuten. 5.7 Das kotranslationale Targeting von Momp2 erfolgt SRP-abhängig Um zu prüfen, ob das kotranslationale Targeting von Momp2 tatsächlich von SRP abhängig ist, wurden Momp2-RANCs in Anwesenheit unterschiedlicher Translokationsfaktoren synthetisiert und mit SecY von harnstoffgewaschenen Vesikeln (HINV) quervernetzt (Abb. 5.12). Wie oben beschrieben, waren die Targetingfaktoren SecA, SecB, Ffh und FtsY durch die Harnstoffbehandlung entfernt worden. Hierdurch wurde die kotranslationale Interaktion zwischen Momp2-146 und SecY deutlich reduziert, die Quervernetzungseffizienz sank auf ein Drittel des Ausgangswertes (vergleiche Spuren 2 und 3). Die Interaktion zwischen Momp2-146 und SecY konnte ausschließlich durch Ffh und FtsY (Spur 4) wiederhergestellt werden, die Quervernetzungseffizienz übertraf sogar den Wert für INV. Die Zugabe von SecA, SecB und F1-ATPase zeigte hingegen keinen wesentlichen Effekt (Spur 5). Bei der gleichzeitigen Zugabe aller Targetingfaktoren wurden Ffh und FtsY offenbar in ihrer Funktion gehemmt (Spur 6). Dieser Effekt war in dem zellfreien System schon früher beobachtet worden; eine Interpretation ist auf der Basis der bisherigen Daten jedoch schwierig. Die Behandlung eines Aliquots mit Puromycin vor der Quervernetzung diente wiederum dem Nachweis, daß das Targeting kotranslational erfolgt: Nach der Loslösung der 5 Ergebnisse 54 naszierenden Kette vom Ribosom war eine Quervernetzung auch hier nicht mehr möglich (Spur 7). Vesikel - INV HINV Ffh, FtsY HINV HINV + + + SecA, SecB, F1 HINV + + Puromycin DSS HINV + + + + + + + + IP α SecY Momp2-146 x SecY 46 Quervernetzungs<1 effizienz in % 100 35 135 41 89 2 2 3 4 5 6 7 1 Abb. 5.12: Nur in Anwesenheit von Ffh und FtsY können Momp2-RANCs effizient mit SecY von Innenmembranvesikeln quervernetzt werden. Momp2-146 wurde in Anwesenheit von Vesikeln (Spur 1), in Anwesenheit von INV (Spur 2) oder in Anwesenheit von HINV (Spuren 3-7) im zellfreien System synthetisiert. Außerdem wurden SecA und SecB (Spuren 5 und 6) oder Ffh und FtsY (Spuren 4, 6 und 7) bei der Synthese zugegeben. Die Proben wurden mit DSS quervernetzt; anschließend wurden die Vesikel über ein Saccharosekissen isoliert (zu Ansatz Nr. 1, der bisher keine Vesikel enthielt, wurden vor der Zentrifugation INV zur Gleichbehandlung zugegeben) und dann 15fache Ansätze mit anti-SecYAntikörpern immunpräzipitiert. Ansatz Nr. 7 wurde vor der DSS-Quervernetzung mit Puromycin behandelt, um die Ribosomen von den naszierenden Ketten abzutrennen. Die Quervernetzungseffizienz für INV wurde gleich 100 % gesetzt. Durch den geschilderten Versuch wird klar gezeigt, daß das kotranslationale Targeting von Momp2 durch SRP vermittelt wird. Die Tatsache, daß die Interaktion zwischen Momp2-146 und SecY durch die Verwendung von HINV nicht vollständig unterbunden wird, legt nahe, daß unter den gewählten In-vitro-Bedingungen ein gewisses Maß an Membrantargeting auch ohne Targeting- und Transportfaktoren möglich ist. Ein entsprechendes Phänomen war zuvor schon bei Arbeiten zum Proteintransport in Säugerzellen beobachtet worden; es wurde auf die unphysiologisch hohen Konzentrationen an RANCs und Translokons im In-vitro-System zurückgeführt (Neuhof et al., 1998; Raden und Gilmore, 1998). 5.8 Nach dem SRP-abhängigen Targeting werden SecA und SecB zur Translokation von Momp2 benötigt Durch die Membranassoziaton von Momp2 an secY205-INV (Abb. 5.9) sowie die Quervernetzung von Momp2-RANCs mit SecY von HINV (Abb. 5.12) konnte gezeigt 55 5 Ergebnisse werden, daß das kotranslationale Targeting von Momp2 nicht von SecA/SecB abhängig ist, sondern durch SRP vermittelt wird. Für die vollständige Translokation von Momp2 waren jedoch SecA/SecB notwendig; dies wurde durch die Reaktivierung von HINV (Abb. 5.7) sowie den Effekt der secY205-Mutation auf die Translokation in secY205-INV im ProteaseResistenz-Test (Abb. 5.8) gezeigt. Im Anschluß an diese Doktorarbeit wurde von H.-G. Koch eine Methode etabliert, mit der das Membrantargeting von Momp2 und seine anschließende Translokation als einzelne Schritte im Transport von Momp2 untersucht werden konnten. Zunächst wurde das Targeting von Momp2 untersucht: RANCs von Momp2 wurden in der Anwesenheit von HINV und unterschiedlichen Targetingfaktoren synthetisiert; anschließend wurden die HINV durch ein Flotationsverfahren als Membranfraktion isoliert. Hierbei bestätigten sich die weiter oben beschriebenen Ergebnisse: Nur in Anwesenheit von Ffh und FtsY assoziierten die Momp2RANCs mit HINV und wurden mit diesen in der Membranfraktion isoliert. Dann wurde die Translokation von Momp2 untersucht: Die Membranfraktion wurde mit Puromycin inkubiert, um die naszierenden Ketten von den Ribosomen loszulösen. Gleichzeitig wurde einem Aliquot SecA, SecB und die F1-ATPase zugesetzt. Durch die folgende Inkubation mit Proteinase K wurde die Translokation in die HINV geprüft. Auch hierbei wurden die weiter oben beschriebenen Ergebnisse bestätigt: Das Targeting naszierender Ketten von Momp2 führte per se noch nicht zur Proteinase-K-Resistenz. Für die Translokation naszierender Ketten von Momp2 in die HINV und die daraus resultierende Proteinase-K-Resistenz waren SecA/SecB nötig. Die im Rahmen dieser Arbeit gewonnenen Ergebnisse zeigen, daß der Transport von Momp2 in zwei Schritten erfolgt: Das Membrantargeting von Momp2 erfolgt kotranslational und SRP-abhängig. Die anschließende Translokation von Momp2 hingegen erfordert SecA und SecB. 6 Diskussion 6. 56 Diskussion 6.1 Momp2: Ein Hybridprotein aus der Signal-Anker-Sequenz von MtlA und dem signalsequenzlosen OmpA als Modellprotein zum Proteintransport in Escherichia coli In vorangehenden Arbeiten wurden individuelle Transportmechanismen für das sekretorische Protein Outer membrane protein A (OmpA) sowie das polytope Innenmembranprotein Mannitpermease (MtlA) identifiziert (Koch et al., 1999; Beck et al., 2000): Das Membrantargeting und die Translokation von OmpA werden durch Trigger factor, SecB sowie SecA vermittelt, während das Membrantargeting und die Integration von MtlA durch das Signal recognition particle (SRP) sowie seinen Rezeptor FtsY vermittelt werden (Abb. 2.1). Das auf diesen Arbeiten beruhende Konzept zweier individueller Transportmechanismen für sekretorische Proteine bzw. Innenmembranproteine wird jedoch durch Arbeiten in Frage gestellt, denen zufolge sowohl SRP als auch SecA an der Integration bestimmter Innenmembranproteine beteiligt sind (Einzelheiten siehe 6.8). Daher sollte in dieser Arbeit geklärt werden, ob für die Integration bestimmter Innenmembranproteine sowohl SRP als auch SecA benötigt werden und welche unterschiedlichen Funktionen SRP und SecA dabei übernehmen. Als Modellprotein wurde ein Hybridprotein aus der ersten Signal-Anker-Sequenz von MtlA sowie dem signalsequenzlosen OmpA gewählt. Da die Signal-Anker-Sequenz von MtlA die Erkennungssequenz für SRP darstellt, war auch eine Erkennung des Hybridproteins Momp2 duch SRP zu erwarten. So konnte geprüft werden, ob SRP alleine zur Integration von Momp2 führt oder ob hierzu zusätzlich SecA benötigt wird. Der OmpA-Anteil von Momp2 bildet eine periplasmatische Domäne, die über die von MtlA stammende Signal-Anker-Sequenz in der Membran verankert ist (Abb. 5.1C). Damit gleicht Momp2 in seiner Membrantopologie einer Reihe authentischer Innenmembranproteine, wie zum Beispiel dem FtsQ, einem an der Zellteilung beteiligten Protein (Abb. 6.2A). Naszierende Ketten von FtsQ konnten sowohl mit SRP als auch mit SecA quervernetzt werden, woraus geschlossen wurde, daß beide Faktoren am Targeting und der Integration von FtsQ beteiligt sind (Valent et al., 1998). Momp2 ist im Vergleich zu FtsQ als Modellprotein besonders geeignet: Sein Targeting und Transport kann direkt mit den ausschließlich SecA/SecB- bzw. SRP-abhängigen Kontrollproteinen OmpA und MtlA verglichen werden. 6.2 Die Signal-Anker-Sequenz reicht für die Erkennung durch SRP aus In früheren Arbeiten wurden die Signal-Anker-Sequenzen verschiedener Innenmembranproteine als Erkennungssequenz für SRP identifiziert: (1) Die Quervernetzung von SRP an naszierende Ketten von FtsQ mit dem Quervernetzer DSS konnte durch das Einfügen zusätzlicher Lysine in die Signal-Anker-Sequenz von FtsQ deutlich gesteigert werden (Valent et al., 1997). Die Aminogruppe von Lysin ist der bevorzugte Reaktionspartner für DSS. (2) Ffh konnte „ortsspezifisch“ mit der Signal-Anker-Sequenz in naszierenden 57 6 Diskussion Ketten von MtlA quervernetzt werden (Beck et al., 2000). Hierfür wurde Tmd-Phe in die Signal-Anker-Sequenz von MtlA eingebaut; Tmd-Phe ist ein Aminosäureanalogon, das durch UV-Bestrahlung als Quervernetzungsreagenz aktiviert werden kann. (3) Das SRPunabhängige Phagen-Mantel-Protein M13 procoat protein wurde SRP-abhängig, wenn die Hydrophobizität seiner ersten transmembranen Domäne artifiziell erhöht wurde (de Gier et al., 1998). Im Einklang mit der Beobachtung, daß SRP seine Substrate anhand ihrer Signal-AnkerSequenzen erkennt, konnten naszierende Ketten von MtlA, nicht jedoch naszierende Ketten des sekretorischen Proteins pOmpA mit Ffh quervernetzt werden (Beck et al., 2000; Abb. 5.5, Spuren 6/8 und 14/16). Bisher war jedoch nicht bekannt, ob neben der hydrophoben SignalAnker-Sequenz noch weitere Eigenschaften der Innenmembranproteine für die Erkennung durch SRP nötig sind. In dieser Arbeit wurde gezeigt, daß naszierende Ketten von Momp2 im Gegensatz zu denen von pOmpA mit Ffh quervernetzt werden können (Abb. 5.5, Spuren 22 und 24): Erst die Fusion von OmpA mit der Signal-Anker-Sequenz von MtlA ermöglicht also die Erkennung durch SRP. Damit konnte erstmals gezeigt werden, daß eine lange und hydrophobe SignalAnker-Sequenz für die Erkennung eines Innenmembranproteins durch SRP ausreicht; andere für Innenmembranproteine spezifische Eigenschaften sind hierfür offenbar nicht nötig. Inzwischen wurde dieses Ergebnis durch die Arbeit einer anderen Arbeitsgruppe bestätigt: Neben OmpA wurde auch das sekretorische Protein Maltose binding protein (MBP) von SRP erkannt, nachdem seine Signalsequenz durch die erste Signal-Anker-Sequenz des polytopen Innenmembranproteins Acriflavine resistance protein B (AcrB) ersetzt wurde (Lee und Bernstein, 2001). 6.3 Trigger factor erkennt den reifen Anteil sekretorischer Proteine Naszierende Ketten von Momp2 konnten nicht nur mit Ffh, sondern auch mit Trigger factor quervernetzt werden (Abb. 5.5, Spuren 18 und 19). Da Trigger factor an pOmpA, nicht aber an MtlA-Sequenzen bindet (Beck et al., 2000; Abb. 5.5, Spuren 10 und 11), erkennt Trigger factor Momp2 offenbar am reifen Proteinanteil von OmpA. Daß die Signalsequenz für die Erkennung sekretorischer Proteine durch Trigger factor keine Rolle spielt, steht mit früheren Beobachtungen in Einklang: (1) Trigger factor besitzt keine Substratspezifität für sekretorische Proteine, sondern bindet auch an naszierende Ketten zytosolischer Proteine (Valent et al., 1995 und 1997; Hesterkamp et al., 1996). (2) Trigger factor bindet auch dann an sekretorische Proteine, wenn deren Signalsequenz stark modifiziert wurde. So konnten in der Signalsequenz der Alkalischen Phosphatase (PhoA) 10 Aminosäuren durch Leucin (stark hydrophob) oder Alanin (wenig hydrophob) ersetzt werden, ohne daß die Quervernetzungseffizienz mit Trigger factor beeinflußt wurde (Valent et al., 1997). (3) Trigger factor bindet auch dann an naszierende Ketten des Außenmembranporins PhoE, wenn diese ohne Signalsequenz synthetisiert wurden (Valent et al., 1997). (4) Vor kurzem wurden 6 Diskussion 58 mit Hilfe einer Peptidbibliothek Bereiche im reifen OmpA, nicht jedoch in seiner Signalsequenz, als Erkennungssequenzen für Trigger factor identifiziert (Schaffitzel, 2001). 6.4 SRP und Trigger factor konkurrieren um naszierende Ketten Sowohl SRP als auch Trigger factor binden an ribosomenassoziierte naszierende Ketten. Sie unterscheiden sich jedoch hinsichtlich ihrer Substratspezifität: So wird das sekretorische Protein pOmpA nur von Trigger factor, nicht jedoch von SRP gebunden (Beck et al., 2000; Abb. 5.5, Spuren 6-8). Das Innenmembranprotein MtlA wird hingegen von SRP, nicht jedoch von Trigger factor gebunden (Beck et al., 2000; Abb 5.5, Spuren 14-16). Auch eine andere Arbeitsgruppe untersuchte die Substratspezifität von SRP und Trigger factor (Valent et al., 1995, 1997): In einer ersten Arbeit (Valent et al., 1995) wurden ribosomenassoziierte naszierende Ketten in Weizenkeimlysat synthetisiert, aufgereinigt und dann in E.coli-Lysat mit interagierenden Proteinen quervernetzt. Dabei wurden in Einklang mit den oben beschriebenen Ergebnissen verschiedene sekretorische Proteine, nicht jedoch die Innenmembranproteinen FtsQ und Signalpeptidase (Lep) mit Trigger factor quervernetzt. Im Widerspruch zu oben beschriebenen Ergebnissen wurden jedoch nicht nur Innenmembranproteine, sondern auch sekretorische Proteine mit SRP quervernetzt. Dies könnte auf die Synthese im heterologen Weizenkeimlysat zurückgeführt werden. In einer späteren Arbeit (Valent et al., 1997) wurde ein homologes E. coli-System verwendet. Erstaunlicherweise wurden in diesem System nicht nur sekretorische Proteine, sondern auch FtsQ und Lep mit Trigger factor quervernetzt. Hingegen konnten unter den normalen Konzentrationen an SRP und Trigger factor nur Innenmembranproteine, nicht jedoch sekretorische Proteine mit SRP quervernetzt werden. Dies unterstützt die Hypothese, daß die Quervernetzung zwischen SRP und sekretorischen Proteinen in der früheren Arbeit auf die Synthese im Weizenkeimlysat zurückgeführt werden kann. Offenbar konkurrieren SRP und Trigger factor um naszierende Ketten sekretorischer Proteine: In einem Trigger factor-depletierten System konnte SRP auch an pOmpA binden (Beck et al., 2000). Unter physiologischen Verhältnissen könnte diese Bindung von SRP an sekretorische Proteine durch Trigger factor verhindert werden. Außerdem verhindert Trigger factor, daß SecB und SecA an die naszierende Kette binden, bevor diese vom Ribosom freigesetzt wurde (Beck et al., 2000). Wahrscheinlich können sekretorische Proteine nur wesentlich langsamer durch das Translokon transloziert werden, als die Proteintranslation am Ribosom fortschreitet. Ein kotranslationales Targeting sekretorischer Proteine würde daher SecYEG-Translokons blockieren. Diese liegen in der Zelle jedoch in relativ geringer Anzahl vor und könnten somit den limitierenden Faktor beim Proteintransport darstellen. Die Rolle von Trigger factor könnte daher darin liegen, ein kotranslationales Targeting sekretorischer Proteine, sei es durch SRP, sei es durch SecB und SecA, zu verhindern. Das Membrantargeting und die Integration von polytopen Innenmembranproteinen müssen hingegen kotranslational erfolgen, da diese besonders hydrophob sind und nach ihrer 59 6 Diskussion vollständigen Synthese im Zytosol schnell aggregieren würden (Ahrem et al., 1989). Auch von Valent et al. (1995) konnte eine Konkurrenz zwischen SRP und Trigger factor um die naszierenden Ketten sekretorischer Proteine demonstriert werden; nur nach SRP-Depletion konnte Trigger factor auch an FtsQ und Lep quervernetzt werden. In der späteren Arbeit (Valent et al., 1997) wurde dieses Modell allerdings verändert: Zwar konnte in hohen Konzentrationen zugefügtes SRP auch mit sekretorischen Proteinen quervernetzt werden; die Quervernetzungseffizienz von Trigger factor wurde dadurch jedoch nicht beeinflußt, was eher gegen eine Konkurrenz zwischen SRP und Trigger factor spricht. In dieser Arbeit wurde das Hybridprotein Momp2 auf seine Interaktionen mit SRP und Trigger factor hin untersucht. Momp2 enthält einerseits mit der Signal-Anker-Sequenz von MtlA eine Bindestelle für SRP, andererseits mit dem reifen Proteinanteil von OmpA auch Bindestellen für Trigger factor. Waren in einem Syntheseansatz Ffh und Trigger factor anwesend, dann konnten beide Proteine mit naszierenden Ketten von Momp2 quervernetzt werden (Abb. 5.5, Spur 22-24). Dieses Ergebnis kann jedoch auf zwei unterschiedliche Weisen interpretiert werden: (1) Ffh und Trigger factor können gleichzeitig an dieselbe naszierende Kette binden, ohne sich dabei gegenseitig zu behindern. (2) Ffh und Trigger factor konkurrieren beim Binden an die naszierende Kette; dies könnte sogar so weit führen, daß entweder nur Ffh oder nur Trigger factor an eine einzelne Momp2-Kette binden kann. Durch die Zugabe von SRP nahm die Quervernetzungseffizienz mit Trigger factor deutlich ab (Abb. 5.5, vergleiche Spuren 19 und 23). Dies scheint auf eine Konkurrenz der beiden Proteine hinzuweisen. 6.5 Die Signal-Anker-Sequenz ermöglicht ein kotranslationales, SRPabhängiges Targeting von Momp2 Das Targeting von naszierenden Ketten an das Endoplasmatische Retikulum von Säugerzellen erfolgt kotranslational. Dabei sorgen die SRP-Untereinheiten SRP9 und SRP14 für eine Arretierung der Translation, bis der nötige Kontakt zwischen Ribosom und Translokon hergestellt ist. In Bakterien wurde bisher weder eine Arretierung der Translation selbst nachgewiesen, noch konnte eine SRP-Untereinheit identifiziert werden, die hierfür verantwortlich sein könnte. Daher erscheint es fraglich, ob ein kotranslationales Targeting in Bakterein überhaupt möglich ist. Eine Lösung dieses Problems könnte darin liegen, daß die Synthese von Innenmembranproteinen in Bakterienzellen in unmittelbarer Nachbarschaft der Innenmembran stattfindet (Lynch und Wang, 1993; Ma et al., 1994) und ein kotranslationales Targting daher auch ohne Translationsarretierung möglich ist. Es konnte gezeigt werden, daß naszierende Ketten von MtlA nur solange mit dem SecYEG-Translokon interagieren, wie sie mit dem Ribosom assoziiert sind (Beck et al., 2000), was für ein kotranslationales Targeting spricht. Bisher wurde jedoch nicht gezeigt, daß dieses kotranslationale Targeting von SRP vermittelt wird. In dieser Arbeit wurde gezeigt, daß das Targeting von Momp2 an SecY kotranslational 6 Diskussion 60 erfolgen kann. So konnten ribosomenassoziierte naszierende Momp2-Ketten mit SecY quervernetzt werden (Abb. 5.10, Spuren 8, 12 und 16). Nach der Loslösung der naszierenden Ketten vom Ribosom durch das Antibiotikum Puromycin konnten diese hingegen nicht mehr mit SecY quervernetzt werden (Abb. 5.11, Spur 3). Ribosomenassoziierte naszierende pOmpA-Ketten binden hingegen nicht an Membranvesikel; sie müssen zuvor von den Ribosomen abgelöst werden (Behrmann et al., 1998). Es ist also die von MtlA stammende Signal-Anker-Sequenz, die das kotranslationale Targeting von Momp2 an SecY möglich macht. Im Anschluß wurde gezeigt, daß dieses kotranslationale Targeting von Momp2 an SecY SRPabhängig ist: Die Quervernetzung der ribosomenassoziierten Momp2-Ketten an SecY von harnstoffgewaschenen Membranvesikeln konnte nur durch SRP, nicht aber durch SecA/SecB wiederhergestellt werden (Abb. 5.12). Analog dazu war die Bindung von Momp2 an secY205Membranvesikel durch die gestörte SecA-SecY-Interaktion in diesen Vesikeln nicht betroffen (Abb. 5.9, vergleiche Spuren 4/5 und 7/8), so daß eine SecA-abhängige Membranassoziation ausscheidet. Somit konnte erstmals gezeigt werden, daß das SRP-abhängige Targeting von Innenmembranproteinen kotranslational erfolgt. Erstaunlicherweise konnten ribosomenassoziierte Momp2-Ketten auch in Abwesenheit von Targeting- und Transportfaktoren mit geringer Effizienz an SecY quervernetzt werden (Abb. 5.12, Spur 3). Ein entsprechendes Phänomen war schon zuvor beobachtet worden, nämlich ein SRP-unabhängiges Targeting an das Endoplasmatische Retikulum von Säugerzellen. Dieses Targeting war jedoch nicht funktionell (Jungnickel und Rapoport, 1995) und wurde auf die hohen Konzentrationen an naszierenden Ketten und Translokons im Invitro-System zurückgeführt (Neuhof et al., 1998; Raden und Gilmore, 1998). Wahrscheinlich ermöglichen auch hier im zellfreien System aus E. coli die hohen Konzentrationen an ribosomenassoziierten naszierenden Momp2-Ketten und SecYEG-Komplexen deren unspezifische Interaktion. Außerdem binden Ribosomen nicht nur über naszierende Polypeptidketten an Translokons: Auch nicht translatierende Ribosomen binden über ihre ribosomale RNA (rRNA) direkt an Translokons (Prinz et al., 2000). Wie oben dargelegt, ist das kotranslationale Targeting von Momp2 SRP-abhängig. Trotzdem ergab ein Protease-Resistenz-Test, daß Momp2 in vitro auch in Abwesenheit von SRP in Innenmembranvesikel transloziert werden kann (Abb. 5.7, Spuren 7/8). Dies entspricht der Beobachtung, daß bestimmte Proteine, die unter physiologischen Verhältnissen Substrate von SRP sind, in Abwesenheit von SRP auch alternative Targetingmechanismen, vermutlich SecB und SecA, nutzen können: Fusionsproteinen aus der ersten transmembranen Domäne verschiedener Innenmembranproteine und dem sekretorischen Protein Alkalische Phosphatase (PhoA) wurden auf ihre Integration hin untersucht. Die Funktion von SRP wurde dabei durch die Expression eines dominanten, lethalen ftsY-Allels blockiert. Nur ein Teil der Fusionsproteine wurde dadurch betroffen (Newitt et al., 1999). Möglicherweise ist dabei die Hydrophobizität der transmembranen Domänen und somit die Anfälligkeit der Proteine zur 61 6 Diskussion Aggregation im Zytosol ausschlaggebend. Von Lee und Bernstein (2001) wurden Fusionsproteine zwischen der ersten Signal-Anker-Sequnez des Innenmembranproteins AcrB und den sekretorischen Proteinen OmpA bzw. Maltose binding protein (MBP) untersucht. Die Integration dieser beiden Fusionsproteine wurde durch die Depletion von Ffh verhindert. Diese unterschiedlichen Ergebnisse zeigen, daß bestimmte Proteine in Abwesenheit von SRP vermutlich SecB und SecA als alternative Targetingmechanismen nutzen können, andere hingegen auf SRP angewiesen sind. Momp2 scheint zu der ersten Gruppe zu gehören: Wahrscheinlich wird Momp2 in Abwesenheit von SRP am Ribosom nur von Trigger factor gebunden; das Targeting erfolgt dann anschließend durch SecA und SecB. Möglicherweise wird die von MtlA stammende Signal-Anker-Sequenz dabei als alternative Signalsequenz erkannt. Dieses Targeting erfolgt jedoch auf alle Fälle posttranslational, da das kotranslationale Targeting an SecY nur duch SRP vermittelt werden kann (Abb. 5.12). SRP ist ein essentieller Zellbestandteil. Daher erfolgt in vivo die Proteinsynthese immer in Anwesenheit von SRP; die starke Bindung von SRP an ein naszierendes Innenmembranprotein, wie hier für Momp2 beobachtet (Abb. 5.5, Spuren 22 und 24), sollte dann zum kotranslationalen Targeting führen. 6.6 Die Translokation der periplasmatischen Domäne von Momp2 erfordert SecA Obwohl die Bindung von SRP an Momp2 zum Membrantargeting führt, kann die vollständige Translokation von Momp2 nicht durch SRP vermittelt werden; für diesen Schritt wird SecA benötigt. Dies wurde in dieser Arbeit durch zwei Experimente gezeigt: Die Translokation von Momp2 in harnstoffgewaschene Innenmembranvesikel konnte durch SRP nicht vermittelt werden; hierfür mußte SecA/SecB zugegeben werden (Abb. 5.7). Zusätzlich war die Translokation von Momp2 in secY205-Mutantenvesikel, bei denen die Interaktion von SecA mit SecY gestört ist, stark beeinträchtigt (Abb. 5.8). SecA ist vermutlich für die Translokation von Momp2 essentiell, da es durch seine ATPabhängigen Insertions-Deinsertions-Zyklen den „Motor“ für die Translokation des von OmpA stammenden Proteinanteils, also der periplasmatischen Domäne, bildet. In mehreren Arbeiten wurde gezeigt, daß SecA an die Signalsequenz von sekretorischen Proteinen bindet (Miller et al., 1998). Hieraus wurde gefolgert, daß SecA seine Substrate an der Signalsequenz erkennt. Für Momp2 kann dies jedoch nicht der Fall sein, da Momp2 statt der Signalsequenz eines sekretorischen Proteins die Signal-Anker-Sequenz eines Innenmembranproteins enthält. Daß SecA dennoch mit Momp2 interagieren kann, läßt sich nur damit erklären, daß SecA auch den reifen Proteinanteil von OmpA erkennt. Dies steht mit dem von Economou und Wickner (1994) entwickelten Modell in Einklang, nach dem SecA sekretorische Proteine durch repetitive Insertions-Deinsertions-Zyklen durch das Translokon fädelt. Beim ersten Zyklus könnte SecA dabei an die Signalsequenz binden; diese wird jedoch schon früh während der Translokation durch die Signalpeptidase abgespalten (van der Wolk et al., 1997) und steht bei 6 Diskussion 62 den folgenden Insertions-Deinsertions-Zyklen zur Substratbindung durch SecA nicht mehr zur Verfügung. Folglich müßte SecA nicht nur an die Signalsequenz sekretorischer Proteine, sondern auch an diverse Regionen innerhalb des reifen Proteins binden. In den hier vorgestellten Experimenten wurde zusammen mit SecA auch immer SecB zugegeben. SecB hat beim Targeting sekretorischer Proteine zwei unterschiedliche Aufgaben: Zum einen verhindert es als Chaperon die Faltung sekretorischer Polypeptidketten. Zum anderen ist SecB zusammen mit SecA am Targeting sekretorischer Proteine beteiligt. An der Translokation selbst ist SecB jedoch nicht beteiligt: SecB dissoziiert vor dem ersten Insertions-Deinsertions-Zyklus von dem Komplex aus Vorläuferprotein und SecA (Fekkes et al., 1997). Das Targeting von Momp2 erfolgt kotranslational und wird von SRP vermittelt. Daher ist es gut vorstellbar, daß SecB für den Transport von Momp2 entbehrlich ist: Die Targetingfunktion von SecB wird von SRP übernommen und seine Chaperonfunktion wird beim kotranslationalen Targeting wahrscheinlich nicht benötigt. Ein entsprechendes Ergebnis wurde für Fusionsproteine der sekretorischen Proteine OmpA bzw. Maltose binding protein (MBP) erzielt: Die Translokation der beiden Proteine wurde durch die Fusion mit der ersten Signal-Anker-Sequenz des Innenmembranproteins AcrB SRP-abhängig (siehe oben); sie war aber nicht mehr SecB-abhängig. Die Abhängigkeit dieser Fusionsproteine von SecA wurde dabei nicht untersucht (Lee und Bernstein, 2001). Aus den in dieser Arbeit beschriebenen Experimenten kann nicht geschlossen werden, ob die Translokation der periplasmatischen Domäne von Momp2 ko- oder posttranslational erfolgt. Die Bindung von Trigger factor an naszierende Ketten von pOmpA verhindert das kotranslationale Binden von SecA; erst nach der Freisetzung der Polypeptidkette vom Ribosom und damit auch von Trigger factor kann SecA binden (Beck et al., 2000). Im Fall von Momp2 wäre jedoch auch ein kotranslationales Binden von SecA denkbar: Trigger factor kann vermutlich nicht an eine naszierende Kette binden, an die zuvor schon SRP gebunden hat (siehe 6.4). Bei der kotranslationalen Translokation ins Endoplasmatische Retikulum von Säugerzellen bindet das translatierende Ribosom an das Translokon und dichtet dadurch die Membranpore ab; dieser enge Kontakt bleibt vermutlich während der gesamten Translokation bestimmter Proteine bestehen (Crowley et al., 1993 und 1994). Auch in Escherichia coli wurde eine Bindung des Ribosoms an das Translokon nachgewiesen (Prinz et al., 2000). Es ist vorstellbar, daß das Ribosom bei der kotranslationalen Integration ausschließlich SRPabhängiger Innenmembranproteine auch in Escherichia coli das Translokon abdichtet. Die Translokation der periplasmatischen Momp2-Domäne erfordert jedoch die Insertion und Deinsertion von SecA in das Translokon (vergleiche Economou und Wickner, 1994). Der Ribosom-Translokon-Kontakt müßte also so dynamisch sein, daß er diese großen Konformationsänderungen von SecA zuläßt. Außerdem konnte Momp2 auch dann transloziert werden, wenn das kotranslationale, SRP-abhängige Targeting in der Abwesenheit von SecA stattfand und SecA erst anschließend zugegeben wurde (siehe 5.8). Der Ribosom-TranslokonKontakt müßte also auch so flexibel sein, daß er den Zugang von zwei SecA-Untereinheiten 63 6 Diskussion (je 102 kD) zum Translokon ermöglicht. In der Tat konnte bei Säugerzellen beobachtet werden, daß bei der Translokation bestimmter Proteine ein regulierter Zugang vom Zytosol zum Translokon besteht (Hedge und Lingappa, 1996). Die andere Möglichkeit ist, daß SecA erst posttranslational an Momp2 bindet und dann die Translokation der periplasmatischen Domäne katalysiert. Welche zusätzlichen Komponenten des Translokons neben SecY und SecE für den Membrantransport von Momp2 notwendig sind, wurde in dieser Arbeit nicht untersucht. Zwei mögliche Kandidaten sind YidC und SecG: YidC spielt bei der Topogenese von transmembranen Domänen eine Rolle (Scotti et al., 2000; Houben et al., 2000; Beck et al., 2001) und könnte an der Integration der Signal-Anker-Sequenz von Momp2 in die Lipiddoppelschicht beteiligt sein, während SecG bei den Insertions-Deinsertions-Zyklen von SecA mitwirkt (Nishiyama et al., 1996) und die Translokation der periplasmatischen Domäne von Momp2 unterstützen könnte. 6.7 Der Membrantransport von Momp2 erfolgt in drei Schritten, bei denen SRP und SecA unterschiedliche Rollen übernehmen Die oben beschriebenen Beobachtungen lassen darauf schließen, daß der Membrantransport von Momp2 in drei Schritten abläuft, bei denen SRP und SecA unterschiedliche Rollen übernehmen (Abb. 6.1): Sobald die N-terminale Signal-Anker-Sequenz von Momp2 aus dem Peptidtunnel auf der Oberfläche des Ribosoms auftaucht, wird sie von SRP erkannt und gebunden (Abb. 6.1A). Anschließend führt die Interaktion zwischen SRP und seinem Rezeptor FtsY zum kotranslationalen Targeting an das Translokon (Abb. 6.1B). Schließlich erfolgt die Translokation der periplasmatischen Domäne; nur dieser Vorgang erfordert SecA (Abb. 6.1C). Es ist bisher nicht geklärt, ob die Translokation der periplasmtischen Domäne ko- oder posttranslational erfolgt. Mindestens SecY und SecE werden als Komponenten des Translokons benötigt; wahrscheinlich unterstützt zusätzlich SecG die Insertions-DeinsertionsZyklen von SecA. YidC könnte an der Integration der Signal-Anker-Sequenz in die Lipidphase beteiligt sein. 6 Diskussion 64 Translokon Translokon FtsY SecA SRP N ? SRP A B C Abb. 6.1: Der Membrantransport von Momp2 erfolgt in drei Schritten, bei denen SRP und SecA unterschiedliche Rollen übernehmen A Sobald die N-terminale Signal-Anker-Sequenz von Momp2 auf der Oberfläche des Ribosoms auftaucht, wird sie von SRP erkannt und gebunden. B Die Interaktion zwischen SRP und seinem Rezeptor FtsY führt zum kotranslationalen Targeting an das Translokon. C Nur die Translokation der periplasmatischen Domäne von Momp2 erfordert SecA. Ob dies kooder posttranslational erfolgt, ist bisher nicht geklärt (durch das ? am Ribosom symbolisiert). Wahrscheinlich wirken neben SecY und SecE als Komponenten des Translokons auch SecG bei der Translokation der periplasmatischen Domäne und YidC bei der Integration der Signal-AnkerSequenz in die Lipidphase mit. 6.8 Der Membrantransport von Innenmembranproteinen mit großen periplasmatischen Domänen erfordert eine Kooperation zwischen SRP und SecA In mehreren Arbeiten wurde gezeigt, daß sowohl SRP als auch SecA am Membrantransport bestimmter Proteine in Escherichia coli beteiligt sind. Dabei handelte es sich um Innenmembranproteine, die wie Momp2 über ausgedehnte periplasmatische Domänen verfügen: FtsQ, ein an der Zellteilung beteiligtes Protein, hat N-terminal einen 25 Aminosäuren langen zytosolischen Abschnitt, eine 25 Aminosäuren lange transmembrane Domäne sowie eine 225 Aminosäuren große periplasmatische Domäne (Abb. 6.2A). Damit ist FtsQ strukturell Momp2 sehr ähnlich. Die Integration von FtsQ ist SRP-abhängig: Sie wird durch Mutationen im Gen ffs, welches die 4,5S RNA von SRP kodiert, gestört (Tian et al., 2000). Außerdem können naszierende Ketten von FtsQ mit Ffh und SecA quervernetzt werden (Valent et al., 1998). Die Signalpeptidase I (Lep) ist ein Innenmembranprotein mit zwei transmembranen Domänen. Am C-Terminus folgt eine 247 Aminosäuren lange periplasmatische Domäne (Abb. 6.2B). Die Integration von Lep ist SecA- (Wolfe et al., 1985) und SRP-abhängig (de Gier et al., 1996), jedoch nicht SecB-abhängig (de Gier et al., 1998). Dementsprechend konnten naszierende Ketten von Lep mit SecA und Ffh quervernetzt 6 Diskussion 65 werden (Valent et al., 1998). Maltosepermease (MalF) durchspannt die Innenmembran mit acht transmembranen Domänen; zwischen der dritten und vierten transmembranen Domäne liegt eine 183 Aminosäuren lange periplasmatische Domäne (Abb. 6.2C). Die Integration von MalF konnte durch Natriumazid verhindert werden, einem Inhibitor von SecA (Traxler und Murphy, 1996). In einem genetischen Ansatz konnte gezeigt werden, daß sowohl Mutationen in secA als auch in ffs, welches die 4,5S RNA von SRP kodiert, die Integration von MalF stören (Tian et al., 2000). Das Acriflavine resistance protein B (AcrB) hat zwölf transmembrane Domänen; auf die erste sowie die siebte transmembrane Domäne folgen periplasmatische Domänen von 311 bzw. 315 Aminosäuren Länge (Abb. 6.2D). Die Integration von AcrB wurde sowohl durch eine SecA-Depletion (Qui und Bernstein, 1999) als auch durch eine Mutation in ffs behindert (Tian et al., 2000). 311 247 315 183 225 Periplasma 3 33 6 31 13 6 13 Innenmembran Zytosol FtsQ 275 As Lep 514 As MalF 514 As AcrB 1049 As A B C D Abb. 6.2: FtsQ, Lep, MalF und AcrB sind Innenmembranproteine mit großen periplasmatischen Domänen. Dargestellt ist die Membrantopologie nach Swissprot-Analyse. Die Zahlen bezeichnen die Länge der periplasmatischen Domänen in Aminosäuren. Große periplasmatische Domänen sind rot gezeichnet. As = Aminosäuren Bisher war umstritten, welche Rolle SecA beim Transport dieser Proteine spielt. So wurde vorgeschlagen, daß die Quervernetzung von SecA mit naszierenden Ketten von FtsQ und Lep nicht auf einer spezifischen Interaktion, sondern auf der Nähe am gemeinsamen Bindepartner SecY beruhen könnte (Manting und Driessen, 2000). Andere Arbeiten schienen eher dafür zu sprechen, daß SecA als obligater Bestandteil des Translokons sowohl für die Integration transmembraner Domänen als auch die Translokation periplasmatischer Domänen essentiell ist (Qi und Bernstein, 1999). Ein drittes Modell ging davon aus, daß SecA die Translokation der großen periplasmatischen Domänen katalysiert (Scotti et al., 1999; Dalbey et al., 2000, Müller et al., 2000). Dieses Modell basierte auf der Beobachtung, daß das Targeting und die Integration kurzer naszierender Ketten von FtsQ kotranslational stattfindet und nicht von SecA, sondern nur von SRP abhängig ist (Scotti et al., 1999). Mit diesem Modell stimmen 6 Diskussion 66 auch Ergebnisse von Untersuchungen zu Lep überein: Die Membrantopologie von Lep wurde durch eine Verschiebung der Ladungsverteilung invertiert; dadurch wurde die große Cterminale periplasmatische Domäne ins Zytosol verlegt. Dieses Konstrukt, Lep-inv, war zwar noch SRP-abhängig (de Gier et al., 1996), jedoch nicht mehr SecA-abhängig (von Heijne, 1989). In dieser Arbeit konnten anhand des Modellproteins Momp2 die unterschiedlichen Funktionen von SRP und SecA beim Transport eines Proteins mit einer transmembranen Domäne und einer großen periplasmatischen Domäne eindeutig geklärt werden. Dabei übernimmt SRP das Targeting an das SecYEG-Translokon, während SecA in einem späteren Stadium für die Translokation der großen periplasmatischen Domäne benötigt wird. Die oben zusammengestellten Ergebnisse zu Innenmembranproteinen mit großen periplasmatischen Domänen lassen darauf schließen, daß das hier für Momp2 entwickelte Modell auf diese Gruppe von Innenmembranproteinen mit großen periplasmatischen Domänen übertragen werden kann. Da Innenmembranproteine mit kleinen periplasmatischen Abschnitten SecAunabhängig, solche mit großen periplasmatischen Domänen jedoch SecA-abhängig in die Membran integrieren, ist es vorstellbar, daß ab einer bestimmten kritischen Größe dieser periplasmatischen Bereiche SecA für die Translokation benötigt wird. So wurde schon früher vermutet, daß für die Translokation von periplasmatische Domänen bis zu einer Länge von 60-70 Aminosäuren SecA entbehrlich ist (von Heijne, 1989). Während bei einfach strukturierten Proteinen wie Momp2, FtsQ und Lep sowohl eine ko- als auch eine posttranslationale Translokation der periplasmatischen Domänen denkbar ist, sind die Verhältnisse bei komplexer strukturierten Proteinen wie MalF und AcrB komplizierter: Bei diesen folgen auf die periplasmatischen Domänen weitere transmembrane Domänen. Es erscheint plausibel, daß auch die Integration dieser C-terminalen transmembranen Domänen kotranslational erfolgen muß, um ihre Aggregation zu verhindern. Auch hier sind zwei unterschiedliche Mechanismen denkbar: (1) Sowohl die Integration der transmembranen Domänen als auch die Translokation der periplasmatischen Domänen erfolgt kotranslational. Integration und Translokation könnten dann in der Reihenfolge der Synthese aufeinander folgen (Integration–Translokation–Integration). (2) Die periplasmatischen Domänen verbleiben so lange im Zytosol, bis alle transmembranen Domänen kotranslational integriert sind und die Polypeptidkette vom Ribosom freigesetzt ist; erst dann transloziert SecA die periplasmatischen Domänen. In beiden Fällen müßte der Ribosomen-Translokon-Kontakt ein hohes Maß an Flexibilität besitzen. 67 6 Diskussion 6.9 Unterschiedliche Mechanismen bei der Integration von Innenmembranproteinen Aus den Ergebnissen dieser Arbeit sowie den oben dargestellten Beobachtungen zu anderen Innenmembranproteinen ergibt sich ein Modell, demzufolge die Integration von Innenmembranproteinen in Escherichia coli durch drei unterschiedliche Mechanismen erfolgen kann (Tabelle 6.1): (1) Die Integration des M13 procoat protein benötigt neben YidC offenbar keine weiteren Proteine. (2) Die Integration von Innenmembranproteinen, die nur über kurze periplasmatische Domänen verfügen, erfordern die löslichen Komponenten SRP und FtsY sowie ein Translokon aus SecY und SecE. Wahrscheinlich ist YidC an der Integration der transmembranen Domänen in die Lipidschicht beteiligt. Solche Proteine sind z.B. MtlA, Lactosepermease (LacY) und SecY. Die größten periplasmatischen Domänen dieser Proteine sind 22, 18 bzw. 34 Aminosäuren lang. Diese Arbeit bekräftigt frühere Ergebnisse, denen zufolge SecA für die Integration dieser Innenmembranproteine nicht benötigt wird (MacFarlane und Müller, 1995; Koch et al., 1999; Beck et al., 2000). SecA wird nur für den Membrantransport einer dritten Gruppe von Innenmembranproteinen benötigt: (3) Innenmembranproteine mit großen periplasmatischen Domänen (siehe Abb. 6.2) benötigen zusätzlich SecA für die Translokation der großen periplasmatischen Domänen. Hieran könnte auch SecG beteiligt sein. Integration abhängig von: Innenmembranprotein: 1 • YidC • M13 procoat protein 2 • SRP/FtsY • Mannitpermease (MtlA) • SecYE, evtl. YidC • Lactosepermease (LacY) • SecY 3 • SRP/FtsY • Maltosepermease (MalF) • SecA • Acriflavine resistance protein B (AcrB) • SecYE, evtl. SecG, YidC • Signalpeptidase I (Lep) • FtsQ Tabelle 6.1: Die Integration von Innenmembranproteinen in Escherichia coli kann durch drei unterschiedliche Mechanismen erfolgen 6 Diskussion 68 6.10 Ausblick In dieser Arbeit konnte am Beispiel des Hybridproteins Momp2 der grundsätzliche Integrationsmechanismus für Innenmembranproteine mit langen periplasmatischen Domänen aufgezeigt werden. Mit Momp2 und den hier vorgestellten Methoden können in Zukunft weitere Detailfragen zum Proteintransport in Escherichia coli geklärt werden: In dieser Arbeit wurde gezeigt, daß die Translokation der langen periplasmatischen Domäne von Momp2 SecA/SecB benötigt. Es stellt sich jedoch die Frage, ob SecA für die Translokation ausreicht, oder ob SecB immer anwesend sein muß. Da nur solche Innenmembranproteine SecA für ihre Integration benötigen, die über große periplasmatische Domänen verfügen, könnte mit unterschiedlich langen naszierenden Ketten von Momp2 getestet werden, bis zu welcher Länge der periplasmtischen Domänen die Proteine auch ohne SecA integriert werden können. Außerdem bleibt zu überprüfen, ob SecA die Translokation der periplasmatischen Domänen bereits kotranslational oder aber erst posttranslational vermittelt. 69 7 7 Literatur Literatur Ahrem B, Hoffschulte HK, Müller M (1989) In vitro membrane assembly of a polytopic, transmembrane protein results in an enzymatically active conformation. J Cell Biol 108: 1637-1646 Akimaru J, Matsuyama S, Tokuda H, Mizushima S (1991) Reconstitution of a protein translocation system containing purified SecY, SecE, and SecA from Escherichia coli. Proc Natl Acad Sci USA 88: 6545-6549 Akiyama Y, Ito K (1990) SecY protein, a membrane-embedded secretion factor of E. coli, is cleaved by the ompT protease in vitro. Biochem Biophys Res Commun 167: 711-715 Akiyama Y, Kihara A, Tokuda H, Ito K (1996) FtsH (HflB) is an ATP-dependent protease selectively acting on SecY and some other membrane proteins. J Biol Chem 271: 3119631201 Batey RT, Rambo RP, Lucast L, Rha B, Doudna JA (2000) Crystal structure of the ribonucleoprotein core of the signal recognition particle. Science 287: 1232-1239 Beck K (2000) Quervernetzungsstudien früher Translokationsintermediate in Escherichia coli. Dissertationsschrift an der Fakultät für Biologie, Albert-Ludwigs-Universität Freiburg Beck K, Wu LF, Brunner J, Müller M (2000) Discrimination between SRP- and SecA/SecBdependent substrates involves selective recognition of nascent chains by SRP and trigger factor. EMBO J 19: 134-143 Beck K, Eisner G, Trescher D, Dalbey RE, Brunner J, Müller M (2001) YidC, an assembly site for polytopic Escherichia coli membrane proteins located in immediate proximity to the SecYE translocon and lipids. EMBO rep 2: 709-714 Behrmann M, Koch HG, Hengelage T, Wieseler B, Hoffschulte HK, Müller M (1998) Requirements for the translocation of elongation-arrested, ribosome-associated OmpA across the plasma membrane of Escherichia coli. J Biol Chem 273: 13898-13904 Berks BC, Sargent F, Palmer T (2000) The Tat protein export pathway. Mol Microbiol 35: 260-274 Bieker KL, Phillips GJ, Silhavy TJ (1990) The sec and prl genes of Escherichia coli. J Bioenerg Biomembr 22: 291-310 Binet R, Létoffé S, Ghigo JM, Delepelaire P, Wandersman C (1997) Protein secretion by gram-negative bacterial ABC-transporters - a review. Gene 192: 7-11 Blobel G, Dobberstein B (1975) Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. II. Reconstitution of functional rough microsomes from heterologous components. J Cell Biol 67: 835-862 Bost S, Belin D (1997) prl mutations in the Escherichia coli secG gene. J Biol Chem 272: 4087-4093 Bukau B, Deuerling E, Pfund C, Craig EA (2000) Getting newly synthesized proteins into shape. Cell 101: 119-122 7 Literatur 70 Bullock WO, Fernandez JM, Short JM (1987) BioTechniques 5: 376 Cammak KA, Wade HE (1965) The sedimentation behaviour of ribonuclease-active and inactive ribosomes from bacteria. J Biochem 96: 671-680 Chou MM, Kendall DA (1990) Polymeric sequences reveal a functional interrelationship between hydrophobicity and length of signal peptides. J Biol Chem 265: 2873-2880 Chun SY, Randall LL (1994) In vivo studies of the role of SecA during protein export in Escherichia coli. J Bacteriol 176: 4197-4203 Collier DN, Bankaitis VA, Weiss JB, Bassford PJ Jr. (1988) The antifolding activity of SecB promotes the export of the E. coli maltose-binding protein. Cell 53: 273-283 Crooke E, Wickner W (1987) Trigger factor: a soluble protein that folds pro-OmpA into a membrane-assembly-competent form. Proc Natl Acad Sci USA 84: 5216-5220 Crowley KS, Reinhart GD, Johnson AE (1993) The signal sequence moves through a ribosomal tunnel into a noncytoplasmic aqueous environment at the ER membrane early in translocation. Cell 73: 1101-1115 Crowley KS, Liao S, Worrell VE, Reinhart GD, Johnson AE (1994) Secretory proteins move through the endoplasmic reticulum membrane via an aqueous, gated pore. Cell 78: 461-4 471 Dalbey RE, Chen M, Jiang F, Samuelson JC (2000) Understanding the insertion of transporters and other membrane proteins. Curr Opin Cell Biol 12: 435-442 de Gier JW, Mansournia P, Valent QA, Phillips GJ, Luirink J, von Heijne G (1996) Assembly of a cytoplasmic membrane protein in Escherichia coli is dependent on the signal recognition particle. FEBS Lett 399: 307-309 de Gier JW, Scotti PA, Saaf A, Valent QA, Kuhn A, Luirink J, von Heijne G (1998) Differential use of the signal recognition particle translocase targeting pathway for inner membrane protein assembly in Escherichia coli. Proc Natl Acad Sci USA 95: 14646-14651 de Vrije GJ, Batenburg AM, Killian JA, de Kruijff B (1990) Lipid involvement in protein translocation in Escherichia coli. Mol Microbiol 4: 143-150 Derman AI, Puziss JW, Bassford PJ Jr., Beckwith J (1993) A signal sequence is not required for protein export in prlA mutants of Escherichia coli. EMBO J 12: 879-888 Dilsiz N, Crabbe MJC (1994) A high-yield modification of mutation by overlap extension using three primers. Anal Biochem 222: 510-511 Doud SK, Chou MM, Kendall DA (1993) Titration of protein transport activity by incremental changes in signal peptide hydrophobicity. Biochemistry 32: 1251-1256 Driessen AJ (2001) SecB, a molecular chaperone with two faces. Trends Microbiol 9: 193196 Economou A, Wickner W (1994) SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78: 835-843 Economou A (1998) Bacterial preprotein translocase: mechanism and conformational dynamics of a processive enzyme. Mol Microbiol 27: 511-518 71 7 Literatur Fekkes P, van der Does C, Driessen AJ (1997) The molecular chaperone SecB is released from the carboxy-terminus of SecA during initiation of precursor protein translocation. EMBO J 16: 6105-6113 Fekkes P, de Wit JG, van der Wolk JP, Kimsey HH, Kumamoto CA, Driessen AJ (1998) Preprotein transfer to the Escherichia coli translocase requires the co-operative binding of SecB and the signal sequence to SecA. Mol Microbiol 29: 1179-1190 Fekkes P, de Wit JG, Boorsma A, Friesen RH, Driessen AJ (1999) Zinc stabilizes the SecB binding site on SecA. Biochemistry 38: 5111-5116 Flower AM, Doebele RC, Silhavy TJ (1994) PrlA and prlG suppressors reduce the requirement for signal sequence recognition. J Bacteriol 176: 5607-5614 Freymann DM, Keenan RJ, Stroud RM, Walter P (1997) Structure of the conserved GTPase domain of the signal recognition particle. Nature 385: 361-364 Guthrie B, Wickner W (1990) Trigger factor depletion or overproduction causes defective cell division but does not block protein export. J Bacteriol 172: 5555-5562 Haeuptle M-T, Frank R, Dobberstein B (1986) Translation arrest by oligonucleotides complementary to mRNA coding sequences yields polypeptides of predetermined length. Nucleic Acids Res 14: 1427-1448 Hanada M, Nishiyama K, Tokuda H (1996) SecG plays a critical role in protein translocation in the absence of the proton motive force as well as at low temperature. FEBS Lett 381: 25-28 Hanes J, Plückthun A (1997) In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 94: 4937-4942 Hardy SJ, Randall LL (1991) A kinetic partitioning model of selective binding of nonnative proteins by the bacterial chaperone SecB. Science 251: 439-443 Hartl FU, Lecker S, Schiebel E, Hendrick JP, Wickner W (1990) The binding cascade of SecB to SecA to SecY/E mediates preprotein targeting to the E. coli plasma membrane. Cell 63: 269-279 Hegde RS, Lingappa VR (1996) Sequence-specific alteration of the ribosome-membrane junction exposes nascent secretory proteins to the cytosol. Cell 85: 217-228 Helde R, Wiesler B, Wachter E, Neubuser A, Hoffschulte HK, Hengelage T, Schimz KL, Stuart RA, Müller M (1997) Comparative characterization of SecA from the alpha-subclass purple bacterium Rhodobacter capsulatus and Escherichia coli reveals differences in membrane and precursor specificity. J Bacteriol 179: 4003-4012 Hesterkamp T, Hauser S, Lütcke H, Bukau B (1996) Escherichia coli trigger factor is a prolyl isomerase that associates with nascent polypeptide chains. Proc Natl Acad Sci USA 93: 44374441 Hesterkamp T, Deuerling E, Bukau B (1997) The amino-terminal 118 amino acids of Escherichia coli trigger factor constitute a domain that is necessary and sufficient for binding to ribosomes. J Biol Chem 272: 21865-21871 Hirano M, Matsuyama S, Tokuda H (1996) The carboxyl-terminal region is essential for SecA dimerization. Biochem Biophys Res Commun 229: 90-95 7 Literatur 72 Hoffschulte HK, Drees B, Müller M (1994) Identification of a soluble SecA/SecB complex by means of a subfractionated cell-free export system. J Biol Chem 269: 12833-12839 Houben EN, Scotti PA, Valent QA, Brunner J, de Gier JL, Oudega B, Luirink J (2000) Nascent Lep inserts into the Escherichia coli inner membrane in the vicinity of YidC, SecY and SecA. FEBS Lett 476: 229-233 Ito K (1984) Identification of the secY (prlA) gene product involved in protein export in Escherichia coli. Mol Gen Genet 197: 204-208 Jungnickel B, Rapoport TA (1995) A posttargeting signal sequence recognition event in the endoplasmic reticulum membrane. Cell 82: 261-270 Kaufmann A, Manting EH, Veenendaal AK, Driessen AJ, van der Does C (1999) Cysteinedirected cross-linking demonstrates that helix 3 of SecE is close to helix 2 of SecY and helix 3 of a neighboring SecE. Biochemistry 38: 9115-9125 Keenan RJ, Freymann DM, Walter P, Stroud RM (1998) Crystal structure of the signal sequence binding subunit of the signal recognition particle. Cell 94: 181-191 Keenan RJ, Freymann DM, Stroud RM, Walter P (2001) The signal recognition particle. Annu Rev Biochem 70: 755-775 Keiler KC, Waller PRH, Sauer RT (1996) Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271: 990-993 Kimsey HH, Dagarag MD, Kumamoto CA (1995) Diverse effects of mutation on the activity of the Escherichia coli export chaperone SecB. J Biol Chem 270: 22831-22835 Kimura E, Akita M, Matsuyama S, Mizushima S (1991) Determination of a region in SecA that interacts with presecretory proteins in Escherichia coli. J Biol Chem 266: 6600-6606 Knoblauch NT, Rüdiger S, Schönfeld HJ, Driessen AJ, Schneider-Mergener J, Bukau B (1999) Substrate specificity of the SecB chaperone. J Biol Chem 274: 34219-34225 Koch HG, Hengelage T, Neumann-Haefelin C, MacFarlane J, Hoffschulte HK, Schimz KL, Mechler B, Müller M (1999) In vitro studies with purified components reveal signal recognition particle (SRP) and SecA/SecB as constituents of two independent proteintargeting pathways of Escherichia coli. Mol Biol Cell 10: 2163-2173 Koch HG, Müller M (2000) Dissecting the translocase and integrase functions of the Escherichia coli SecYEG translocon. J Cell Biol 150: 689-694. Kumamoto CA, Francetic O (1993) Highly selective binding of nascent polypeptides by an Escherichia coli chaperone protein in vivo. J Bacteriol 175: 2184-2188 Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685 Lecker SH, Driessen AJ, Wickner W (1990) ProOmpA contains secondary and tertiary structure prior to translocation and is shielded from aggregation by association with SecB protein. EMBO J 9: 2309-2314 73 7 Literatur Lee C, Li P, Inouye H, Brickman ER, Beckwith J (1989) Genetic studies on the inability of beta-galactosidase to be translocated across the Escherichia coli cytoplasmic membrane. J Bacteriol 171: 4609-4616 Lee HC, Bernstein HD (2001) The targeting pathway of Escherichia coli presecretory and integral membrane proteins is specified by the hydrophobicity of the targeting signal. Proc Natl Acad Sci USA 98: 3471-3476 Li P, Beckwith J, Inouye, H (1988) Alteration of the amino terminus of the mature sequence of a periplasmic protein can severely affect protein export in Escherichia coli. Proc Natl Acad Sci USA 85: 7685-7689 Luirink J, ten Hagen-Jongman CM, van der Weijden CC, Oudega B, High S, Dobberstein B, Kusters R (1994) An alternative protein targeting pathway in Escherichia coli: studies on the role of FtsY. EMBO J 13: 2289-2296 Lynch AS, Wang JC (1993) Anchoring of DNA to the bacterial cytoplasmic membrane through cotranscriptional synthesis of polypeptides encoding membrane proteins or proteins for export: a mechanism of plasmid hypernegative supercoiling in mutants deficient in DNA topoisomerase I. J Bacteriol 175: 1645-1655 Ma D, Cook DN, Pon NG, Hearst JE (1994) Efficient anchoring of RNA polymerase in Escherichia coli during coupled transcription-translation of genes encoding integral inner membrane polypeptides. J Biol Chem 269: 15362-15370 MacFarlane J, Müller M (1995) The functional integration of a polytopic membrane protein of Escherichia coli is dependent on the bacterial signal-recognition particle. Eur J Biochem 233: 766-771 Maneewannakul S, Maneewannakul K, Ippen-Ihler K (1994) The pKSM710 vector casette provides tightly regulated lac and T7lac promotors and stratagies for manipulating N-terminal protein sequences. Plasmid 31: 300-307 Manting EH, van der Does C, Remigy H, Engel A, Driessen A (2000) SecYEG assembles into a tetramer to form the active protein translocation channel. EMBO J 19: 852-861 Manting EH, Driessen AJM (2000) Escherichia coli translocase: the unravelling of a molecular machine. Mol Microbiol 37: 226-238 Matsumoto G, Yoshihisa T, Ito K (1997) SecY and SecA interact to allow SecA insertion and protein translocation across the Escherichia coli plasma membrane. EMBO J 16: 6384-6393 Miller A, Wang L, Kendall DA (1998) Synthetic signal peptides specifically recognize SecA and stimulate ATPase activity in the absence of preprotein. J Biol Chem 273: 11409-11412 Miller JD, Bernstein HD, Walter P (1994) Interaction of E. coli Ffh/4.5S ribonucleoprotein and FtsY mimics that of mammalian signal recognition particle and its receptor. Nature 367: 657-659 Millman JS, Qi HY, Vulcu F, Bernstein HD, Andrews DW (2001) FtsY binds to Escherichia coli inner membranes via interactions with phosphatidylethanolamine and membrane proteins. J Biol Chem 276: 25982-25989 7 Literatur 74 Mitchell C, Oliver D (1993) Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol Microbiol 10: 483-497 Montoya G, Svensson C, Luirink J, Sinning I. Crystal structure of the NG domain from the signal-recognition particle receptor FtsY. Nature. 1997 Jan 23;385(6614):365-8 Müller M, Blobel G (1984a) In vitro translocation of bacterial proteins across the plasma membrane of Escherichia coli. Proc Natl Acad Sci USA 81: 7421-7425 Müller M, Blobel G (1984b) Protein export in Escherichia coli requires a soluble activity. Proc Natl Acad Sci USA 81: 7737-7741 Müller M, Fisher RP, Rienhofer-Schweer A, Hoffschulte HK (1987) DCCD inhibits protein translocation into plasma membrane vesicles from Escherichia coli at two different steps. EMBO J 6: 3855-3861 Müller M, Koch HG, Beck K, Schäfer U (2000) Protein traffic in bacteria: multiple routes from the ribosome to and across the membrane. Prog Nucleic Acid Res Mol Biol 66: 107-157 Neuhof A, Rolls MM, Jungnickel B, Kalies KU, Rapoport TA (1998) Binding of signal recognition particle gives ribosome/nascent chain complexes a competitive advantage in endoplasmic reticulum membrane interaction. Mol Biol Cell 9: 103-115 Newitt JA, Bernstein HD (1998) A mutation in the Escherichia coli secY gene that produces distinct effects on inner membrane protein insertion and protein export. J Biol Chem 273: 12451-12456 Newitt JA, Ulbrandt ND, Bernstein HD (1999) The structure of multiple polypeptide domains determines the signal recognition particle targeting requirement of Escherichia coli inner membrane proteins. J Bacteriol 181: 4561-4567 Nishiyama K, Suzuki T, Tokuda H (1996) Inversion of the membrane topology of SecG coupled with SecA-dependent preprotein translocation. Cell 85: 71-81 Park S, Liu G, Topping TB, Cover WH, Randall LL (1988) Modulation of folding pathways of exported proteins by the leader sequence. Science 239: 1033-1035 Prinz A, Behrens C, Rapoport TA, Hartmann E, Kalies KU (2000) Evolutionarily conserved binding of ribosomes to the translocation channel via the large ribosomal RNA. EMBO J 19:1900-1906 Prinz WA, Spiess C, Ehrmann M, Schierle C, Beckwith J (1996) Targeting of signal sequenceless proteins for export in Escherichia coli with altered protein translocase. EMBO J 15: 5209-5217 Qi HY, Bernstein HD (1999) SecA is required for the insertion of inner membrane proteins targeted by the Escherichia coli signal recognition particle. J Biol Chem 274: 8993-8997 Raden D, Gilmore R (1998) Signal recognition particle-dependent targeting of ribosomes to the rough endoplasmic reticulum in the absence and presence of the nascent polypeptideassociated complex. Mol Biol Cell 9: 117-130 75 7 Literatur Randall LL, Hardy SJ (1986) Correlation of competence for export with lack of tertiary structure of the mature species: a study in vivo of maltose-binding protein in E. coli. Cell 46: 921-928 Randall LL, Topping TB, Hardy SJ (1990) No specific recognition of leader peptide by SecB, a chaperone involved in protein export. Science 248: 860-863 Randall LL, Topping TB, Hardy SJ, Pavlov MY, Freistroffer DV, Ehrenberg M (1997) Binding of SecB to ribosome-bound polypeptides has the same characteristics as binding to full-length, denatured proteins. Proc Natl Acad Sci USA 94: 802-807 Rapiejko PJ, Gilmore R (1997) Empty site forms of the SRP54 and SRα GTPases mediate targeting of ribosome-nascent chain complexes to the endoplasmic reticulum. Cell 89: 703713 Samuelson JC, Chen M, Jiang F, Moller I, Wiedmann M, Kuhn A, Phillips GJ, Dalbey RE (2000) YidC mediates membrane protein insertion in bacteria. Nature 406: 637-641 Sato K, Mori H, Yoshida M, Tagaya M, Mizushima S (1997) Short hydrophobic segments in the mature domain of ProOmpA determine its stepwise movement during translocation across the cytoplasmic membrane of Escherichia coli. J Biol Chem 272: 5880-5886 Schaffitzel E (2001) In vitro Analysen der Interaktion von Chaperonen mit naszierenden Polypeptidketten. Dissertationsschrift an der Medizinischen Fakultät der Albert-LudwigsUniversität Freiburg Schatz PJ, Beckwith J (1990) Genetic analysis of protein export in Escherichia coli. Annu Rev Genet 24: 215-248 Schatz PJ, Bieker KL, Ottemann KM, Silhavy TJ, Beckwith J (1991) One of three transmembrane stretches is sufficient for the functioning of the SecE protein, a membrane component of the E. coli secretion machinery. EMBO J 10: 1749-1757 Scholz C, Mucke M, Rape M, Pecht A, Pahl A, Bang H, Schmid FX (1998) Recognition of protein substrates by the prolyl isomerase trigger factor is independent of proline residues. J Mol Biol 277: 723-732 Scotti PA, Valent QA, Manting EH, Urbanus ML, Driessen AJ, Oudega B, Luirink J (1999) SecA is not required for signal recognition particle-mediated targeting and initial membrane insertion of a nascent inner membrane protein. J Biol Chem 274: 29883-29888 Scotti PA, Urbanus ML, Brunner J, de Gier JW, von Heijne G, van der Does C, Driessen AJ, Oudega B, Luirink J (2000) YidC, the Escherichia coli homologue of mitochondrial Oxa1p, is a component of the Sec translocase. EMBO J 19: 542-549 Seluanov A, Bibi E (1997) FtsY, the prokaryotic signal recognition particle receptor homologue, is essential for biogenesis of membrane proteins. J Biol Chem 272: 2053-2055 Silhavy TJ, Berman ML, Enquist LW (1984) Experiments with gene fusion, Cold Spring Harbor Laboratory Press, New York, pp XI-XII Smith VF, Hardy SJ, Randall LL (1997) Determination of the binding frame of the chaperone SecB within the physiological ligand oligopeptide-binding protein. Protein Sci 6: 1746-1755 7 Literatur 76 Snyders S, Ramamurthy V, Oliver D (1997) Identification of a region of interaction between Escherichia coli SecA and SecY proteins. J Biol Chem 272: 11302-11306 Stoller G, Tradler T, Rucknagel KP, Rahfeld J-U, Fischer G (1996) An 11.8 kDa proteolytic fragment of the E. coli trigger factor represents the domain carrying the peptidyl-prolyl cis/trans isomerase activity. FEBS Lett 384: 117-122 Sugiyama JE, Mahmoodian S, Jacobson GR (1991) Membrane topology analysis of Escherichia coli mannitol permease by using a nested-deletion method to create mtlA-phoA fusions. Proc Natl Acad Sci USA 88: 9603-9607 Swidersky UE, Hoffschulte HK, Müller M (1990) Determinants of membrane-targeting and transmembrane translocation during bacterial protein export. EMBO J 9: 1777-1785 Tian H, Boyd D, Beckwith J (2000) A mutant hunt for defects in membrane protein assembly yields mutations affecting the bacterial signal recognition particle and Sec machinery. Proc Natl Acad Sci USA 97: 4730-4735 Traxler B, Murphy C (1996) Insertion of the polytopic membrane protein MalF is dependent on the bacterial secretion machinery. J Biol Chem 271: 12394-12400 Uchida K, Mori H, Mizushima S (1995) Stepwise movement of preproteins in the process of translocation across the cytoplasmic membrane of Escherichia coli. J Biol Chem 270: 3086230868 Valent QA, Kendall DA, High S, Kusters R, Oudega B, Luirink J (1995) Early events in preprotein recognition in E. coli: interaction of SRP and trigger factor with nascent polypeptides. EMBO J 14: 5494-5505 Valent QA, Degier JWL, von Heijne G, Kendall DA, Tenhagenjongman CM, Oudega B, Luirink J (1997) Nascent membrane and presecretory proteins synthesized in Escherichia coli associate with signal recognition particle and trigger factor. Mol Microbiol 25: 53-64 Valent QA, Scotti PA, High S, de Gier JW, von Heijne G, Lentzen G, Wintermeyer W, Oudega B, Luirink J (1998) The Escherichia coli SRP and SecB targeting pathways converge at the translocon. EMBO J 17: 2504-2512 van der Wolk JP, de Wit JG, Driessen AJ (1997) The catalytic cycle of the Escherichia coli SecA ATPase comprises two distinct preprotein translocation events. EMBO J 16: 7297-7304 Volkert TL, Baleja JD, Kumamoto CA (1999) A highly mobile C-terminal tail of Escherichia coli protein export chaperone SecB. Biochem Biophys Res Commun 264: 949-954 von Heijne G (1986a) Net N-C charge imbalance may be important for signal sequence function in bacteria. J Mol Biol 192: 287-290 von Heijne G (1986b) The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans-membrane topology. EMBO J 5: 3021-3027 von Heijne G (1989) Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature 341: 456-458 von Heijne G (1990) The signal peptide. J Membr Biol 115: 195-201 77 7 Literatur Walter P, Keenan R, Schmitz U (2000) SRP – where the RNA and membrane worlds meet. Science 287: 1212-1213 Wolfe PB, Rice M, Wickner W (1985) Effects of two sec genes on protein assembly into the plasma membrane of Escherichia coli. J Biol Chem 260: 1836-1841 Xu Z, Knafels JD, Yoshine K (2000) Crystal structure of the bacterial protein export chaperone SecB. Nat Struct Biol 7: 1172-1177 Zelazny A, Seluanov A, Cooper A, Bibi E (1997) The NG domain of the prokaryotic signal recognition particle receptor, FtsY, is fully functional when fused to an unrelated integral membrane polypeptide. Proc Natl Acad Sci USA 94: 6025-6029 8 Anhang 8 78 Anhang 8.1 Abkürzungen A Amp Abb. As ATP Bp BSA Cm CP CPK CTF CTP DMSO DNA DSS DTT E. coli EDTA Ffh GTP HEPES HINV INV IP (α) IPTG Kan kD MGF mRNA Momp2 MtlA NTP OD OmpA PAGE PCR PEP PK PMSF pOmpA psi RANC RNA Rpm S SDS SR SRP Absorption Ampicillin Abbildung Aminosäure(n) Adenosintriphosphat Basenpaar(e) Rinderserumalbumin Chloramphenicol Kreatinphosphat Kreatinphosphatkinase Zytosolische Translationsfaktoren Cytidintriphosphat Dimethylsulfoxid Desoxyribonukleinsäure Dissuccinimidylsuberat Dithiotreitol Escherichia coli Ethylendiamintetraacetat Fifty-four homolgue Guanosintriphosphat N-(2-Hydoxyethyl)piperazin-N‘-(2-Ethan-Sulfonsäure) harnstoffgewaschene INV inside-out Innenmembranvesikel Immunpräzipitation (gegen) Isopropyl-β-D-thiogalactosid Kanamycin Kilodalton membrangeschütztes Fragment messenger RNA Hybridprotein aus der ersten Signal-Anker-Sequenz von MtlA und dem signalsequenzlosen OmpA Mannitpermease Nukleosidtriphosphat Optische Dichte Outer membrane protein A Polyacrylamid-Gelelektrophorese Polymerasekettenreaktion Phosphoenolpyruvat Proteinase K Phenylmethylsulfonylfluorid Vorläuferprotein des Outer membrane protein A pounds per square inch ribosomenassoziierete naszierende Kette Ribonukleinsäure Rotationen pro Minute Svedberg-Koeffizient Natriumdodecylsulfat SRP-Rezeptor Signal recognition particle 79 TCA TEMED Tet Tmd-Phe Tris U UTP UV w/w Trichloressigsäure N, N, N‘, N‘-Tetramethylethylendiamin Tetracylin L-4‘-(3-[triflouromethyl]-3H-diazirin-3yl)phenylalanin Trihydroxymethylaminomethan Unit(s) Uridin-5‘-Triphosphat ultraviolett weight per weight (Gewicht pro Gewicht) 8 Anhang 8 Anhang 80 8.2 Plasmidkarten von pDMB, pOmpA-SpeI, p717MtlA-B, pMtlA-SpeI-2 und Momp2 Abb. 8.1: Plasmidkarte von pDMB bzw. pOmpA-SpeI Angegeben ist jeweils das erste Nukleotid der Restriktionsenzymschnittstelle. Die in pOmpA-SpeI eingefügte SpeI-Schnittstelle ist rot eingezeichnet. Abb. 8.2: Plasmidkarte von p717MtlA-B bzw. pMtlA-SpeI-2 Angegeben ist jeweils das erste Nukleotid der Restriktionsenzymschnittstelle. Die in pMtlA-SpeI-2 eingefügte SpeI-Schnittstelle ist rot eingezeichnet. 81 Abb. 8.3: Plasmidkarte von pMomp2 Angegeben ist jeweils das erste Nukleotid der Restriktionsenzymschnittstelle. 8 Anhang 8 Anhang 82 8.3 Sequenzen von ompA, mtlA und momp2 ompA in pDMB EcoRI TAATACGACTCACTATAGGGCGAATTCTTACATCGCCAAGGGTGCTCGGCATAAGC T7-Promotor T7-PromotorCGAAGATATCGGTAGAGTTAATATTGAGCAGATCCCCCGGTGAAGGATTTAACCGT Primer GTTATCTCGTTGGAGATATTCATGGCGTATTTTGGATGATAACGAGGCGCAAAAA Signalsequenz Signalsequenz OmpA66 mit SpeISchnittstelle BstXI M K K T A I A I A V A L A G F A ACTAG T A T S OmpA168 OmpAβ5 OmpAβ8 OmpA280 Abb. 8.4: Sequenz von ompA in pDMB Die in pOmpA-SpeI geänderten Nukleotide und Aminosäuren sind rot angegeben. T V A Q 8 Anhang 83 mtlA in p717MtlA-B T7-Promotor TAATACGACTCACTATAGGGGAATTGTGAGCGGATAACAATTCCCCTCTAGCGCGTATTCAGG T7-PromotorCTGACCCTGCGCGCTGCGCAGGGCTTTATTGATTCCATTTTTACACTGATGAATGTTCCGTTG Primer CGCTGCCCGGATTACAGGGGAATTAATTCTCACTCATTAGGCACCCCAGGCTTTACACTTTAT GCTTCCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGAAT TAATTCTTACACTTAGTTAAATTGCTAACTTTATAGATTACAAAACTTAGGAGGGTTTTTACC NcoI ATGGGGGATCCATAAGAAGGGGTGTTTTT Membrandomänen MtlA129 mit SpeISchnittstelle G A F I A W G I I T A L F I P T L V G P M I T Y L L A G I I G M P L A S I F L G G M T G N I A G V C AC TAGT G W L P P L L I G Y T S T G G NcoI MtlA282 I L A I L A F L G I G P I N F M V V H D M L MtlA189 V V E F P T A L T I L G G G L A A A M A V S F V V S A I L 8 Anhang 84 Abb. 8.5: Sequenz von mtlA in pDMB Die in p717MtlA-SpeI-2 geänderten Nukleotide und Aminosäuren sind rot angegeben. 8 Anhang 85 momp2 in pMomp2 T7-Promotor TAATACGACTCACTATAGGGGAATTGTGAGCGGATAACAATTCCCCTCTAGCGCGTATTCAGG T7-PromotorCTGACCCTGCGCGCTGCGCAGGGCTTTATTGATTCCATTTTTACACTGATGAATGTTCCGTTG Primer CGCTGCCCGGATTACAGGGGAATTAATTCTCACTCATTAGGCACCCCAGGCTTTACACTTTAT GCTTCCGGCTCGTATAATGTGTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGAAT TAATTCTTACACTTAGTTAAATTGCTAACTTTATAGATTACAAAACTTAGGAGGGTTTTTACC NcoI ATGGGGGATCCATAAGAAGGGGTGTTTTT 1. Membrandomäne SpeI G G W L A F I P OmpAβ5 OmpAβ8 OmpA280 Abb. 8.6: Sequenz von momp2 in pMomp2 A W G I I T A L F I P T Danke ! Herrn Prof. Matthias Müller danke ich herzlich für die Aufnahme in seine Arbeitsgruppe, vor allem für die freundliche Atmosphäre und die ständige Hilfsbereitschaft bei der experimentellen Arbeit und der Niederschrift dieser Arbeit. Bei Herrn Privatdozent Dr. Darius Moradpour bedanke ich mich für die freundliche Übernahme des Zweitgutachtens. Ganz besonders danke ich Dr. Hans-Georg Koch für die hervorragende Betreuung im Labor. Sein Einsatz und seine Hilfe bei der Konzeption und Durchführung der Versuche hat ganz besonders zum Gelingen dieser Arbeit beigetragen. Vielen Dank für die gute Atmosphäre bei unserer Zusammenarbeit! Für die herzliche Aufnahme, die ständige Hilfsbereitschaft und zahlreiche gemeinsame Unternehmungen danke ich der ganzen Arbeitsgruppe Müller. Mein besonderer Dank gilt Dr. Ute Schäfer für die Klonierung des Hybridproteins Momp2. Der F.-F.-Nord-Stiftung danke ich für die Unterstützung meiner Arbeit durch ein Forschungsstipendium. Ganz besonders herzlich möchte ich mich bei meinen Eltern bedanken, die mir eine sorgenfreie Ausbildung ermöglichen und mich auch sonst in allem unterstützen. Dir, liebe Elke, gilt mein herzlichstes Dankeschön. Durch Dein Verständnis und Deine Hilfe hast Du mir auch für diese Arbeit die nötige Kraft gegeben! Lebenslauf Name: Christoph Neumann-Haefelin Geburtsdatum: 21.10.1977, Freiburg im Breisgau Eltern: Dr. Christiane Neumann-Haefelin, Ärztin, z.Z. nicht berufstätig Dr. Dieter Neumann-Haefelin, Universitätsprofessor für Virologie Geschwister: Tobias, 34 Jahre, Assistenzarzt Daniel, 27 Jahre, Wirtschaftsingenieur Schulbildung: 1983-1987 1987-1996 1996 Weiherhof-Grundschule, Freiburg Friedrich-Gymnasium, Freiburg Abitur mit Mathematik, Latein, Physik und Geschichte als Prüfungsfächern Militär-/Zivildienst entfällt, da meine beiden Brüder bereits entsprechende Dienste geleistet haben. Studium: Seit 10/1996 9/1998 3/2000 8/2000 9/2000 2/2001 3/2001 Medizinstudium, Albert-Ludwigs-Universität Freiburg Physikum 1. Staatsexamen Famulatur in Chirurgie, Missionsärztliche Klinik Würzburg Famulatur in Innerer Medizin, Katharinen-Krankenhaus Frankfurt Famulatur in Radiologie, Institut für Radiologische Diagnostik Freiburg Famulatur in Augenheilkunde, Universitäts-Augenklinik Freiburg Dissertation: Seit 10/1998 Experimentelle Doktorarbeit bei Prof. Dr. med. M. Müller, Institut für Biochemie und Molekularbiologie, Universität Freiburg Stipendien: Seit 12/1996 Stipendiat der Studienstiftung des deutschen Volkes 4-12/1999 Forschungsstipendium der F.-F.-Nord-Stiftung Freiburg, den 20. August 2001