Ferienkurs Experimentalphysik 4
Werbung

Ferienkurs Experimentalphysik 4
Vorlesung 3
Atome im Magnetfeld, Mehrelektronensysteme
Florian Lippert & Andreas Trautner
29.08.2012
Inhaltsverzeichnis
1 Atome im externen Magnetfeld
1.1 Elektronenspin-Resonanz . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.2 Zeeman-Effekt . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1.3 Paschen-Back-Effekt . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
1
1
2
4
2 Das
2.1
2.2
2.3
2.4
2.5
.
.
.
.
.
5
5
6
7
7
9
3 Mehrelektronensysteme
3.1 Hundsche Regeln . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
9
9
Heliumatom
Grundlagen und Ortswellenfunktion . . . . . .
Spinwellenfunktion . . . . . . . . . . . . . . .
Gesamtwellenfunktion . . . . . . . . . . . . .
Termschema und Spektoskopische Notation . .
Auswahlregeln für elektrische Dipolübergänge
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
.
Abbildungsverzeichnis
1
2
3
4
5
Spin Energieniveaus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Anomaler (links) und normaler Zeeman-Effekt (rechts). . . . . . . . . . .
Paschen-Back-Effekt der Feinstruktur am Beispiel von Natrium. . . . . .
Qualitatives Helium Termschema. Eingezeichnet sind einige erlaubte spektrale Übergänge. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Orbitalbesetzung des Grundzustandes von Phosphor und Schwefel. . . . .
2
4
5
8
10
1 Atome im externen Magnetfeld
Bisher haben wir die Struktur eines Atoms in Abwesenheit externer Felder untersucht.
Nun wollen wir das Verhalten von Atomen in einem externen Magnetfeld genauer betrachten. Wie immer koppelt das Magnetfeld an magnetische Momente, in diesem Fall
die magnetischen Momente der Konstituenten des Atoms. Hierbei spielen je nach größe
des externen Magnetfeldes im Vergleich zu internen magnetischen Wechselwirkungen im
Atom verschiedene einzelne und zusammengesetzte magnetische Momente eine Rolle.
Genannt seien hier die magnetischen Momente des Kerns und des Elektrons selbst, sowie die magnetischen Momente von Bahn- bzw. Gesamtdrehimpuls der Elektronenhülle.
1.1 Elektronenspin-Resonanz
In der vorangegangenen Vorlesung haben wir gesehen, dass jedem Drehimpuls in der
klassischen Vorstellung eines Kreisstromes auch ein magnetisches Moment zugeordnet
werden kann. Für einen Bahndrehimpuls ergab sich
µ
~l =
q ~
l,
2m
(1)
Wobei q die eletrische Ladung und m die Masse des betrachteten Teilchens ist. Dieser Zusammenhang gilt etwas allgemeiner auch für den Spin als Eigendrehimpuls von
Elementarteilchen. Hierbei tritt jedoch ein zusätzlicher Faktor, der sogennante LandéFaktor gl auf, der in der hier verwendeten, semi-klassichen Behandlung des Spins weder
hergeleitet noch verstanden werden kann. In der Quantenelektrodynamik kann dieser
Faktor aus elementaren Überlegungen mit beeindruckender Genauigkeit hergeleitet werden. Für das magnetische Moment des Elekronenspins gilt:
µ
~ s = gs
µB
q
~s = −gs ~s.
2m
~
(2)
Hier haben wir im letzten Schritt die Ladung des Elektrons −e sowie das Bohrsche
Magneton
µB =
~e
eV
≈ 5, 8 · 10−5 ,
2me
T
(3)
eingesetzt. Für Elektronen gilt in guter Näherung gs ≈ 2. Wie immer ist der Erwartungswert des magnetischen Momentes durch die Eigenwerte des zugehörigen Operators gegeben. Aus vorangegangen Betrachtungen wissen wir bereits, dass für einen Drehimpulsoperator das Betragsquadrat des Drehimpulses (im Gegensatz zu den einzelnen
Richtungskomponenten) immer eine gute Quantenzahl ist. Für das magentische Moment
ergibt sich daher:
µ 2
B
2
ˆ
hµ
~ i = gs
h~sˆ2 i = gs2 µ2B s(s + 1).
(4)
~
1
Und für die Projektion des magnetischen Momentes auf die z-Achse
hµ̂z i = gs
µB
hŝz i = gs µB ms ,
~
(5)
wobei ms = ± 21 für s = 12 gilt.
Die potentielle Energie eines Dipols (magnetisches Moment) in einem externen Magnet~ ist im Allgemeinen gegeben durch
feld B
~ = −µz |B|
~
E = −~µ · B
(6)
wobei wir für das letzte Gleichheitszeichen unser Koordinatensystem mit der z-Achse entlang des Mangetfeldes gewählt haben. Für ein Elektron in einem
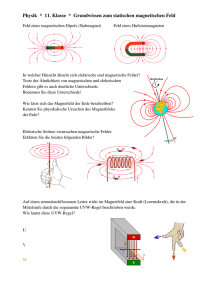
externen Magnetfeld ergeben sich also je nach Ausrichtung des Spins zwei Energieniveaus
1
~
E = ± gs µB |B|,
2
(7)
~ linear im
mit der Energiedifferenz ∆E = gs µB |B|
Betrag des Magnetfeldes. Hierdurch lässt sich mit
Abb. 1: Spin Energieniveaus
∆E = ~ωL die Lamorfrequenz ωL definieren, das
ist die Frequenz, deren Photonen mit dem Zweizustandssystem resonant wechselwirken (ESR), d.h. den Spin umklappen würden.
ωL := gs
µB ~
|B|.
~
(8)
1.2 Zeeman-Effekt
Wie bereits behandelt setzt sich der Gesamtdrehimpuls und somit das gesamte magnetische Moment der Elektronen in einem Atom aus Bahn- sowie Eigendrehimpuls
(Spin) zusammen. Ohne externes Magnetfeld wechselwirken also das magnetische Moment des Spins und das magnetische Moment des Bahndrehimpulses miteinander und
erzeugen ein gesamt-magnetisches Moment (L-S-Kopplung). Als Zeeman-Effekt bezeichnet man im allgemeinen das Aufspalten der Energieniveaus in der Elektronenhülle eines Atoms in einem externen Magnetfeld Bext Bint , d.h. das externe Magnetfeld ist
nicht stark genug die Wechselwirkung des Spin-magnetischen Momentes mit dem magnetischen Moment des Bahndrehimpulses aufzubrechen und wechselwirkt stattdessen
mit dem magnetischen Moment des Gesamtdrehimpulses. Das heißt die Energieniveaus
werden im wesentlichen durch die Spin-Bahn-Kopplung dominiert, die Aufspaltung aufgrund des externen Magnetfeldes erzeugt hierzu lediglich eine kleine Störung die wir
hier behandeln wollen. Wir haben in der vorangegangenen Vorlesung bereits die Hy-
2
perfeinstrukturaufspaltung (HFS) diskutiert, der Effekt hier ist völlig analog dazu bis
auf das dass magnetfeld mit dem der Gesamtdrehimpuls J wechselwirkt nicht das vom
Kern erzeugte Magnetfeld, sondern ein externes ist! Für das magnetische Moment des
Gesamtdrehimpulses gilt
µB ~
~ ≈ − µB L
~ + 2S
~ = − µB J~ + S
~ .
gL L + gs S
µ
~J = µ
~L + µ
~S = −
(9)
~
~
~
Insbesondere ist das zum Gesamtdrehimpuls gehörige magnetische Moment nicht parallel
zu demselbigen,
~
µ
~ J ∦ J.
(10)
Eine natürliche Wahl der Quantisierungsachse (insbesondere auch ohne externes Magnetfeld) für µ
~ J ist deshalb J~ selbst. Wie immer interessiert uns zur Berechnung der
zusätzlichen Energie der Erwartungswert des magnetischen Momentes projeziert auf die
Quantisierungsachse, in diesem Fall
!
~ · J~ + 2S
~ · J~
µJ · J~
(µL + µS ) · J~
µB L
.
(11)
µJ J =
=
=−
~
~
~
~
|J|
|J|
|J|
~ · J~ benutzen wir den üblichen Trick
Für die Berechnung der Skalarprodukte vom typ L
~ = J~ − L,
~ da wir so die Erwartungswerte aus den quadrierten Drehimund quadrieren S
pulsoperatoren berechnen können. Es ergibt sich
~2
1 h ~2 ~ 2 ~ 2 i
J +L −S i=
[J(J + 1) + L(L + 1) − S(S + 1)]
2
2
h
i
2
~ = h 1 J~2 − L
~2 + S
~ 2 i = ~ [J(J + 1) − L(L + 1) + S(S + 1)] ,
hJ~ · Si
2
2
~ =
hJ~ · Li
h
(12)
und damit
hµJ iJ = −µB
3J(J + 1) − L(L + 1) + S(S + 1)
p
.
J(J + 1)
(13)
Für den Fall des Zeeman-Effektes Bext Bint ist auch trotz des externen Feldes, J~ die
dominierende Richtung des Magnetfeldes. Somit präzediert µ
~ J wesentlich schneller um
~
~
J als um B (die Lamor-Frequenz der Präzession steigt linear mit der Magnetfeldstärke).
Das heißt für den Zeeman-Effekt zählt lediglich das zeitliche Mittel von µ
~ J , also dessen
~
Projektion auf J. Das effektive (zeitl. gemittelte) magnetische Moment ist demnach
~J =
µ̄
µB ~
µ
~ J · J~ J~
= −gJ
J,
~ |J|
~
~
|J|
3
(14)
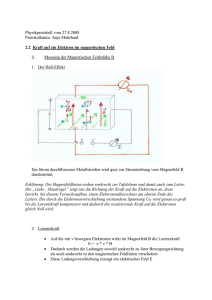
Abb. 2: Anomaler (links) und normaler Zeeman-Effekt (rechts).
wobei wir den Landé-Faktor des Gesamtdrehimpulses
gJ := 1 +
J(J + 1) + S(S + 1) − L(L + 1)
,
2J(J + 1)
(15)
definiert haben. Damit ergibt sich in einem externen Magnetfeld, welches wir wie gewohnt
entlang der z-Achse wählen, eine Energieaufspaltung
~ = − hµ̄J i |B|
~ = mJ gJ µB |B|,
~
~Ji · B
E = − hµ̄
z
(16)
mit −J ≤ mJ ≤ J. Beachte, dass die Energieniveaus in diesem Fall nicht äquidistant
~ von gJ und somit indirekt von L und S abhängt.
aufspalten da ∆E = gJ µB |B|
Diesen Allgemeinfall bezeichnet man irreführenderweise als anormalen Zeeman-Effekt
obwohl es der Regelfall ist.
Für den Spezialfall abgeschlossener Elektronenschalen addiert sich S wie wir sehen werden immer zu 0 und somit gilt J = L damit gJ = 1 und mJ = mL und folglich
~ und die Energieniveaus spalten unabhängig von L äquidistant auf. Das
∆E = mL µB |B|
ist der sogenannte normale Zeeman-Effekt.
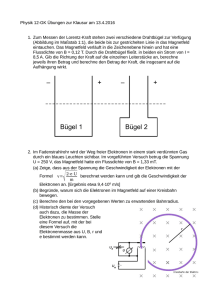
1.3 Paschen-Back-Effekt
Im genau anderen Grenzfall Bext Bint spielt die interne Wechselwirkung der magnetischen Momente von Bahn- und Eigendrehimpuls aufgrund der geringeren Stärke keine
Rolle mehr, d.h. es kommt nicht zur Bildung eines Gesamtdrehimpulses (man sagt, der
Gesamtdrehimpuls ist keine gute Quantenzahl mehr). In diesem Fall wechselwiren die
4
Abb. 3: Paschen-Back-Effekt der Feinstruktur am Beispiel von Natrium.
magnetischen Momente von Bahndrehimpuls und der Spin jeweils unabhängig voneinander mit dem externen Magnetfeld. Das ist der sogenannte Paschen-Back-Effekt. Die
Wechselwirkungsenergie ergibt sich dann einfach aus der Summer der beiden Energien
~ −µ
~ = (mL + 2mS )µB |B|.
~
E = −~µL · B
~S · B
(17)
Die Größenordnung für den Übergang zwischen Zeeman- und Paschen-Back-Effekt ist
natürlich im Bereich der internen Magnetfeldstärke und ungefähr von der Größenordnung
Bint ∼ 1T. Deise Größe hängt aber, wie bei bereits bei der Spin-Bahn-Kopplung gesehen
stark von der Kernladung ab (geht ∝ Z 4 ). Die Beschreibung des Übergangsbereiches
Bext ≈ Bint ist theoretisch wesentlich komplizierter aber prinzipell möglich.
2 Das Heliumatom
2.1 Grundlagen und Ortswellenfunktion
Im Folgenden soll nun das Heliumatom als einfachstes Beispiel für Mehrelektronensysteme diskutiert werden. Der Hamiltonoperator für zwei Elektronen im Zentralpotential
des Kerns ist gegeben als
Z e2
~2 2
Z e2
e2
1
~2 2
∇r1 −
+ −
∇r2 −
+
.
Ĥ = Ĥef f + ĤW W = −
2me
4π0 r1
2me
4π0 r2
4π0 |~r1 − ~r2 |
(18)
Für Helium gilt hier natürlich Z = 2. Die potentielle Energie der zwei Elektronen Wechselwirkung ĤW W verletzt die Radialsymmetrie des Potentials. Hierdurch wird eine exakte
analytische Rechnung unmöglich. In erster Näherung wollen wir daher die Wechselwirkung der Elektronen untereinander vernachlässigen. In diesem fall Separiert die Wel-
5
lenfunktion für das Zweielektronensystem zu einem Produkt von zwei EinzelktronenWellenfunktionen
Ψ(r1 , r2 ) = Ψa1 (r1 )Ψa2 (r2 ).
(19)
Die Einelektronen Wellenfunktionen sind dann einfache Wasserstoff Wellenfunktionen
(mit Z = 2). Gleichung (19) ist allerdings noch nicht ganz komplett. Wenn wir uns an
die 1. Vorlesung erinnern wird klar, dass wir, da die beiden Elektronen identische Teilchen
sind, das Verhalten unter Austausch der beiden Elektronen berücksichtigen müssen. Es
gibt wie damals gesehen, dann zwei Möglichkeiten für die korrekte Ortswellenfunktion
1
Ψ± (r1 , r2 ) = √ (Ψa1 (r1 )Ψa2 (r2 ) ± Ψa1 (r2 )Ψa2 (r1 )) .
2
(20)
Hierbei bezeichnen a1 und a2 die Quantenzahlen n, l, m den Spin betrachten wir später.
Dieses sogenannte Zentralfeldmodell liefert die Bindungsenergien mit Abweichungen zum
Experiment von etwa 30%. Korreturen z.B. mittels Störungstheorie und Einführung
einer effektiven Kernladung sind einfach möglich, sollen hier aber nicht weiter behandelt
werden.
2.2 Spinwellenfunktion
Wichtiger für das qualtitative Verständnis des Helium Termschemas ist die Berücksichtigung
des Elektronenspins respektive der fermionischen Natur der Elektronen und deshalb des
Pauli-Prinzips. Die Spins beiden Hüllenelektronen s1,2 = 21 , mS1 ,S2 = ± 12 Koppeln und
der Gesamtspin der Elektronenhülle ergibt sich per Drehimpulsaddition der Einzelelektronenspins
S = |s1 − s2 |, ..., s1 + s2 = 0, 1.
(21)
Die Spinprojektion ergibt sich wie üblich als einfache Addition der Einzelprojektionen
M = m1 + m2 . Für den Zustand des Gesamtspins |S, M i ergeben sich die Möglichkeiten
1
Singlet |0, 0i = √ (|↑↓i − |↓↑i)
2
|1, 1i = |↑↑i
1
|1, 0i = √ (|↑↓i + |↓↑i) .
Triplet
2
|1, −1i = |↓↓i
(22)
Hier wurde auf der rechten Seite der Gleichungen die Zerlegung in die Spinzustände
up ↑ und down ↓ der einzelnen Elektronen gezeigt. Insbesondere sieht man, das bei einem Vertauschen der beiden Elektronen in der Spinwellenfunktion der Triplet Zustand
symmetrisch (positives Vorzeichen) und der Singlet Zustand anti-symmetrisch transformieren.
6
2.3 Gesamtwellenfunktion
Die Gesamtwellenfunktion setzt sich nun zusammen als Produkt von Orts- und Spinanteil
Ψges (r1 , r2 ) = Ψ± (r1 , r2 ) |S, M i .
(23)
Da die Elektronen identische Fermionen sind, muss die Gesamtwellenfunktion antisymmetrisch unter dem Austausch der Elektronen sein (Pauli-Prinzip) daraus ergibt sich
(
Ortswellenfunktion
)
)
(
anti-symmetrisch
symmetrisch
.
⇔ Spinwellenfunktion
symmetrisch
anti-symmetrisch
(24)
Es ist beispielsweise damit sofort klar, dass für den Heliumgrundzustand (1s2 )“ mit
”
beiden Elektronen in einem 1s Orbital (n1 = n2 = 1, l1 = l2 = 0, ml1 = ml2 = 0)
die Ortswellenfunktion symmetrisch ist und somit als Spinwellenfuntkion nur das antisymmetrische Singlet |0, 0i in Frage kommt.
2.4 Termschema und Spektoskopische Notation
Um Mehrelektronensysteme diskutieren zu können führen wir die sogenannte Spektroskopische notation ein um den Zustand einer Elektronenhülle zu beschreiben. Prinzipell
ist jeder quantenmechnaische Zustand durch die Angabe aller Quantenzahlen (in unserem Beispiel {n1 , l1 , ml1 , s1 , ms1 } und {n2 , l2 , ml2 , s2 , ms2 } ) vollständig charakterisiert.
Wesentlich relevanter ist jedoch die Angabe des Gesamtbahndrehimpulses L, Gesamtspins S und des Gesamtdrehimpulses J der Elektronenhülle, welche wir in der spektroskopischen Notation schreiben als
n2S+1LJ .
(25)
Die Gesamtdrehimpulse(bzw. Spin) ergeben sich wie gewohnt aus der Drehimpulsaddition der einzelnen Drehimpulse(Spins)
L = |l1 − l2 |, ..., l1 + l2
S = |s1 − s2 |, ..., s1 + s2
J = |L − S|, ..., L + S,
(26)
und n gibt die Hauptquantenzahl des höchstangeregten Elektrons an. Für L schreibt
man außerdem im Regelfall nicht die Zahl sondern das zugehörige Schalensymbol (L =
0, 1, 2, 3, 4, 5, .. ↔ L = S, P, D, F, G, H, ..). Die sogenannte Multiplizität 2S + 1 ist die
Anzahl an Feinstrukturniveaus für den jeweiligen Zustand. Für den (1s2 ) Zustand gilt
wie oben bereits festgestellt l1 = l2 = 0, ml1 = ml2 = 0 und damit L = 0, S = 0 und
J = 0 (der S = 1 Zustand ist wie oben diskutiert durch das Pauli-Prinzip verboten) und
damit als spektroskopisches Symbol 1 S0 . Liegt hingegen Helium im angeregten Zustand
7
(1s1p) vor, so ist l1 = 0, l2 = 1, ml1 = 0, ml2 = 0, ±1, s1 = s2 =
Möglichkeiten
(
0, J = 1 ⇒ 21P1
L = 1, S =
1, J = 0, 1, 2 ⇒ 23P0 , 23P1 , 23P2 .
1
2
und es gibt die
(27)
Wir sehen, dass im Gegensatz zum Grundzustand in angeregten Zuständen auch Triplet
Spinkonfigurationen erlaubt sind. Ein wesentlicher Punkt fehlt noch zum Verständnis
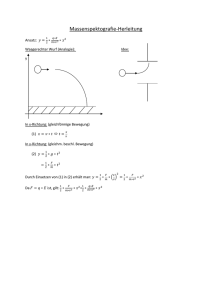
Abb. 4: Qualitatives Helium Termschema. Eingezeichnet sind einige erlaubte spektrale
Übergänge.
des Helium Termschemas: Da Übergänge zwischen zwei Niveaus in der Atomhülle dominant durch elektrische Dipolwechselwirkung stattfinden sind nicht beliebige Übergänge
möglich (siehe nächster Abschnitt). Bei Dipolübergängen gilt insbesondere ∆S = 0,
d.h. der Gesamtdrehimpuls kann sich nicht ändern und Übergänge zwischen Zuständen
mit unterschiedlichem S sind verboten ( Interkombinationsverbot“). Das führt zu einer
”
Aufspaltung des Helium Termschemas in zwei Teile, solche mit S = 1 ( Ortho-“) und
”
solche mit S = 0 ( Parahelium“). Aufgrund der Symmetrie-Eigenschaft der Spinwel”
lenfunktion ergibt sich direkt die Symmetrie der Ortswellenfunktion, diese ist demnach
für Para(Ortho)-Helium immer (anti-)symmetrisch. Da eine antisymmetrische Ortswellenfunktion automatisch größeren mittleren Abstand zwischen den beiden Elektronen
bedeutet, liegen Zustände im Orthohelium immer energetisch niedriger als vergleichbare
Zustände des Paraheliums.
8
2.5 Auswahlregeln für elektrische Dipolübergänge
Grundsätzlich gelten für elektrische Dipolübergänge in der Atomhülle folgende Auswahlregeln (die Regeln ergeben sich aus dem Übergangsmatrixelement des Dipoloperators
und werden hier ohne Herleitung angegeben)
∆l = ±1
∆m = 0, ±1 und damit
∆s = 0
∆L = ±1
∆J = 0, ±1 (J = 0 6→ J = 0)
∆S = 0.
(28)
3 Mehrelektronensysteme
Was am Beispiel des Heliumatoms eingeführt wurde gilt Grundsätzlich auch für größere
Atome. Die Gesamtzahl an Elektronen in einer Hauptschale (n = const.) beträgt 2n2 .
Um den Grundzustand komplexer Atome zu bestimmen gibt es die sog. Hund’schen
Regeln die auf der Minimierung der Gesamtenergie basieren sowie dem Pauli-Prinzip
basieren.
3.1 Hundsche Regeln
1. Volle Schalen und Unterschalen tragen nicht zum Gesamtdrehimpuls bei und müssen
deshalb bei der Bestimmung des Grundzustandes nicht berücksichtigt werden.
2. Die Orbitale der Unterschalen werden möglichst mit parallelen Spins besetzt (parallele Spins sind energetisch günstiger wegen der daraus resultierenden
asymmeP
trischen Ortswellenfunktion). S ist das sich hieraus ergebende
ms .
3. Die Orbitale der Unterschalen werden so besetzt,
P dass große ml -Werte zuerst besetzt werden. L ist das sich hieraus ergebende | ml |.
4. J ist |L − S|, wenn die Unterschalde halb oder weniger als halb besetzt ist und
L + S sonst.
Zur Verdeutlichung zwei Beispiele:
Phosphor (Z = 15) hat die Form (Ne)(3s)2 (3p)3 . Die 3s Unterschale ist also abgeschlossen. Die Spins in der 3p schale werden gemäß Regel 2 parallel ausgerichtet zu S = 23 . Da
die Schale genau halb gefüllt ist ergibt sich L = 0. Für den Gesamtdrehimpuls bleibt
deshalb keine andere Möglichkeit als J = 23 . Somit folgt das Spektroskopische Symbol
des Phosphor Grundzustandes als 34S3/2 .
Schwefel (Z = 16) hat die Form (Ne)(3s)2 (3p)4 . Als Gesamtspin ergibt sich S = 1, da
sich die Spins in dem vollständig gefüllten Orbital gemäß des Pauli-Prinzips anti-parallel
ausrichten müssen. Der Bahndrehimpuls ergibt sich aus Regel 3 zu L = 1. Gemäß Regel
9
Abb. 5: Orbitalbesetzung des Grundzustandes von Phosphor und Schwefel.
4 ergibt sich dann aufgrund der mehr als halb gefüllten Unterschale der Gesamtdrehimpuls als maximale Kombination von S + L zu J = 2. Das Spektroskopische Symbol des
Schwefelgrundzustandes ist also 33P2 .
10