32 Organische Chemie Teil II: Funktionelle Gruppen
Werbung

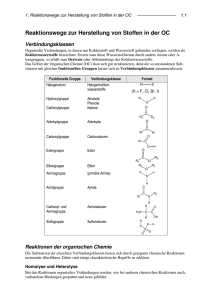
32 Organische Chemie Teil II: Funktionelle Gruppen 32 Organische Chemie Teil II: Funktionelle Gruppen 32.1 Halogenalkane. Nucleophile Substitution. Eliminierungsreaktionen · 513 32.2 Metallorganische Verbindungen · 516 32.3 Alkohole, Phenole und Thiole · 517 32.4 Ether · 520 32.5 Carbonyl-Verbindungen · 521 32.6 Carbonsäuren und ihre Derivate · 525 32.7 Amine und Carbonsäureamide · 533 32.8 Aminosäuren und Peptide · 535 32.9 Azo- und DiazoVerbindungen · 536 32.10 Heterocyclische Verbindungen · 537 Übungsaufgaben · 539 Schlüsselworte (s. Glossar) 2 Nucleophile Substitution Eliminierungsreaktion Grignard-Verbindung Heck-Reaktion Sonogashira-Kupplung Alkohole (primäre, sekundäre, tertiäre) β-Eliminierung Alkoholische Gärung Phenol Thiol 32 Ether Williamson-Synthese Halbacetal Acetal 512 Zusammenfassung / Funktionelle Gruppen sind Atomgruppierungen von Elementen außer C und H, die an ein Kohlenwasserstoff-Gerüst gebunden sind. In Halogenalkanen sind H-Atome eines Alkans durch Halogen-Atome substituiert. Die Halogen-Atome lassen sich durch nucleophile Substitutionsreaktionen gegen andere funktionelle Gruppen (z. B. OH-Gruppen) austauschen (SN1- und SN2-Reaktionen). Organolithium-, Organomagnesium- (Grignard-Verbindungen, R — MgX) und Organozink-Verbindungen entstehen aus Halogenalkanen und Lithium, Magnesium bzw. Zink. Sie reagieren leicht mit polaren Verbindungen und sind für Synthesereaktionen wertvoll. Organische Verbindungen von Übergangsmetallen treten als Zwischenstufen bei Reaktionen auf, bei denen Verbindungen dieser Metalle als Katalysatoren wirken. Ein Alkohol ist ein Derivat eines Alkans, in dem ein H-Atom durch eine OHGruppe substituiert ist. Ein Molekül eines mehrwertigen Alkohols enthält mehrere OH-Gruppen an verschiedenen C-Atomen. Alkohole können durch nucleophile Substitutionsreaktionen aus Halogenalkanen gewonnen werden. Meistens sind jedoch andere Synthesereaktionen wichtiger, zum Beispiel die alkoholische Gärung für Ethanol und die katalytische Hydrierung von Kohlenmonoxid für Methanol. Ein Aren, in dem eine OH-Gruppe an ein C-Atom des Benzol-Rings gebunden ist, nennt man Phenol. Thiole, R — SH, verhalten sich ähnlich wie Alkohole. Ein Ether hat die allgemeine Formel R1 — O — R2 (R1, R2: Alkyl- oder Aryl-Rest). Ether können aus Alkoholen durch Abspaltung von Wasser hergestellt werden. Cyclische Ether haben eine ringförmige Struktur. Dreigliedrige cyclische Ether sind — O, sind Carbonyl-Verbindungen. — O, und Ketone, R1R2C — Epoxide. Aldehyde, R — CH — Sie entstehen bei der Oxidation von primären bzw. sekundären Alkoholen. Die Doppelbindung der Carbonyl-Gruppe geht leicht Additionsreaktionen ein, zum Beispiel mit H2, HCN, Aminen oder Grignard-Verbindungen. Bei der Aldol-Addition geht eine Carbonyl-Verbindung eine Additionsreaktion mit einer anderen Carbonyl-Verbindung (oder mit sich selbst) ein. 1,3-Diketone zeigen die Erscheinung der Keto-Enol-Tautomerie. In einem Acetal sind zwei OR-Gruppen an dasselbe CAtom gebunden, in einem Halbacetal eine OH- und eine OR-Gruppe. Carbonsäuren (R — COOH) können durch Oxidation von Aldehyden, primären Alkoholen, Alkanen oder Alkenen hergestellt werden. Durch Wasserabspaltung erhält man daraus Carbonsäureanhydride (R — CO — O — CO — R). Durch Substitution der OH-Gruppe in der Carboxy-Gruppe durch ein Chlor-Atom entstehen Carbonsäurechloride (Acylchloride, R — CO — Cl). Sie eignen sich zur Einführung der AcylGruppe (RCO — ), z. B. bei der Friedel-Crafts-Reaktion. Carbonsäureester (R1 — CO — O — R2) entstehen aus Carbonsäuren oder Acylchloriden mit Alkoholen. Thiolester (R1 — CO — S — R2) übertragen Alkyl-Gruppen bei biochemischen Reaktionen. In Hydroxycarbonsäuren ist eine zusätzliche Hydroxy-Gruppe an die C-Atomkette der Carbonsäure gebunden; sie sind bei biochemischen Prozessen von Bedeutung. Acetessigester (H3C — CO — CH2 — COOC2H5), ein Oxocarbonsäureester, ist die Ausgangsverbindung für zahlreiche Synthesen. Amine sind Derivate des Ammoniaks, in denen ein, zwei oder drei H-Atome des NH3-Moleküls durch Alkyl- oder Aryl-Gruppen ersetzt sind. Sie sind schwache Basen. Sie können durch Reaktion von Ammoniak mit Halogenalkanen oder — durch Hydrierung von Nitrilen (R — C — — N) gewonnen werden. Anilin (C6H5 — NH2) ist ein aromatisches Amin, das durch Reduktion von Nitrobenzol (C6H5 — NO2) hergestellt wird. Carbonsäureamide (R — CO — NH2, Acylamine) werden aus Carbonsäurehalogeniden, -estern oder -anhydriden und Ammoniak hergestellt. Durch Wasserabspaltung gehen sie in Nitrile über. Ein Urethan (R1O — CO — NH — R2) ist Heruntergeladen von: Thieme E-Books & E-Journals. Urheberrechtlich geschützt. Übersicht 32.1 Halogenalkane. Nucleophile Substitution. Eliminierungsreaktionen Funktionelle Gruppen sind alle Atomgruppierungen aus Elementen außer C und H, die als Substituenten anstelle von Wasserstoff-Atomen an das Gerüst der Kohlenstoff-Atome eines Kohlenwasserstoffs gebunden sind. Außerdem werden Mehrfachbindungen zwischen C-Atomen als funktionelle Gruppen angesehen. Funktionelle Gruppen sind wesentlich für die Eigenschaften organischer Verbindungen verantwortlich. Auf ihrer Vielfalt beruht die Vielfalt in der organischen Chemie. 32.1 Halogenalkane. Nucleophile Substitution. Eliminierungsreaktionen Wie in Abschnitt 31.5 (S. 504) besprochen, können Halogenalkane aus Alkanen durch radikalische Substitutionsreaktionen oder aus ungesättigten Kohlenwasserstoffen durch Addition von Halogen oder Halogenwasserstoff hergestellt werden; vor allem Chlor- und Bromalkane werden so hergestellt. Eine weitere Synthesemöglichkeit, mit der sich insbesondere auch Iodalkane erhalten lassen, ist der Austausch der Hydroxy-Gruppe eines Alkohols durch das Halogen-Atom eines Halogenwasserstoffs. Diese Reaktion entspricht formal der Neutralisationsreaktion von Metallhydroxiden (Basen) mit Halogenwasserstoffsäuren, sie verläuft jedoch wesentlich langsamer, weil die OH-Gruppe eines Alkohols nicht als OH–-Ion vorliegt, sondern kovalent an ein Kohlenstoff-Atom gebunden ist. Der Zusatz einer Brønsted-Säure (z. B. Schwefelsäure) sorgt für eine Gleichgewichtseinstellung zugunsten des Halogenalkans. Für bestimmte Halogenkohlenwasserstoffe gibt es außerdem spezielle Syntheseverfahren. Zum Beispiel wird Tetrachlormethan aus Chlor und Kohlenstoffdisulfid erhalten. Letzteres wird wiederum aus Methan und Schwefeldampf gewonnen (vgl. Abschnitt 28.7, S. 437). Fluoralkane werden aus Chloralkanen durch Reaktion mit Fluorwasserstoff in Anwesenheit von Antimonpentachlorid erhalten. Halogenalkane, vor allem Chloralkane wie Dichlormethan (Methylenchlorid, CH2Cl2, Sdp. 40 °C), Trichlormethan (Chloroform, CHCl3, Sdp. 61 °C), Tetrachlormethan (Tetrachlorkohlenstoff, CCl4, Sdp. 77 °C) und 1,1,1-Trichlorethan (Methylchloroform, Cl3C — CH3, Sdp. 74 °C) sind gute Lösungsmittel und wurden als solche verwendet. Durch gesetzliche Verbote wird ihr Einsatz allerdings Carbonyl-Verbindung Aldehyd Keton Aldol-Addition Wittig-Reaktion Tautomerie Carbonsäure Acyl-Gruppe Carboxy-Gruppe Nitril Carbonsäureester Additions-Eliminierungs-Mechanismus Verseifung Thiolester Claisen-Kondensation Heruntergeladen von: Thieme E-Books & E-Journals. Urheberrechtlich geschützt. gleichzeitig Säureamid und Ester; es entsteht durch Reaktion eines Alkylisocya— O) mit einem Alkohol. Aminosäuren (R — CHNH — COOH) sind Carnats (R — N — —C — 2 bonsäuren mit einer Amino-Gruppe. Das Säureamid aus einer Aminosäure mit einer zweiten Aminosäure wird Peptid genannt. Polypeptide, in denen verschiedene Aminosäuren in genau definierter Abfolge aneinander gebunden sind, können unter Zuhilfenahme von Schutzgruppen aufgebaut werden. Aromatische Amine reagieren mit salpetriger Säure zu Diazonium-Salzen, z. B. – Benzoldiazoniumchlorid (C6 H5 —N+ 2 Cl ). Sie spalten leicht Stickstoff ab, wobei sehr reaktive Aryl-Kationen entstehen (z. B. C6 H+ 5 ), die mit jeder angebotenen Lewis-Base weiterreagieren. Man kann so zahlreiche substituierte aromatische Verbindungen synthetisieren. Diazonium-Salze können auch ohne Abspaltung von Stickstoff mit Phenolen oder aromatischen Aminen reagieren; dabei entste— N — R2; R1, R2 = Aryl-Reste), die gute Farbstoffe hen Azo-Verbindungen (R1 — N — sind. Heterocyclische Verbindungen bestehen aus ringförmigen Molekülen, die außer C-Atomen auch Atome anderer Elemente enthalten. Stickstoffhaltige Heterocyclen sind häufig vorkommende Bausteine in Substanzen biologischen Ursprungs oder mit besonderer biologischer Wirkung. Amin Carbonsäureamid Zwitter-Ion (Betain) Urethan Aminosäure Strecker-Synthese Peptid Schutzgruppe Azo-Verbindung Diazonium-Ion Heterocyclische Verbindung Heteroaren R — OH + HX → R — X + H2O Alkohol Halogenalkan 600 C CH4 + 4 S ! CS2 + 3 Cl2 → S— + 2 H2 S —C— —S Kohlenstoffdisulfid (Schwefelkohlenstoff) (Sdp: 46 C) CCl4 + S2 Cl2 Tetrachlormethan 32 HF + SbCl5 → SbCl4F + HCl 2× CCl4 + 2 SbCl4 F → CF2 Cl2 + 2 SbCl5 2 HF + CCl4 → CF2Cl2 + 2 HCl 513 Photochemische Spaltung eines Halogenalkans: hn CCl4 ! ∙CCl3 + ∙Cl Nucleophile Substitution: Nu– + R — X → Nu — R + X– Nu– = nucleophile Gruppe (das Nucleophil; Lewis-Base) X– = Abgangsgruppe (nucleofuge Gruppe) SN2-Mechanismus: SN1-Mechanismus: tert-Butylbromid 32 (tert-Butanol) 514 stark eingeschränkt, da einige von ihnen krebserregend sind und weil sie das Sauerstoff-Ozon-Gleichgewicht in der Stratosphäre stören (s. Abschnitt 27.9, S. 418). Wegen ihrer Flüchtigkeit geraten ihre Dämpfe in die Atmosphäre, und unter dem Einfluss der ultravioletten Strahlung der Sonne tritt Spaltung der C — Cl-Bindung zu sehr reaktionsfähigen Radikalen ein. Insbesondere können die sonst ungiftigen, gasförmigen Chlorfluorkohlenwasserstoffe (FCKW) umweltschädigend wirken. Vor allem das Dichlordifluormethan (CF2Cl2, Sdp. –30 °C) wurde als Treibmittel in Sprühdosen und als Kühlmittel in Kühlaggregaten verwendet. Als Ersatz in Kühlaggregaten wird jetzt 1,1,1,2-Tetrafluorethan — — CF ) bevorzugt; auch Propan (F3C — CH2F) und Tetrafluorpropen (H2C —CF 3 und Butan kommen zum Einsatz. Etliche Halogenalkane dürfen inzwischen nicht mehr oder nur mit Beschränkungen für spezielle Zwecke hergestellt werden, darunter Tetrachlormethan und Brommethan. Von Bedeutung sind die Halogenalkane (hauptsächlich Brom- und Chloralkane) vor allem für die synthetische Chemie. Neben der Friedel-Crafts-Reaktion zur Synthese von Alkylarenen (vgl. S. 508) spielen die nucleophilen Substitutionsreaktionen eine wichtige Rolle. Dabei wird die polare Bindung zwischen dem Kohlenstoff-Atom und dem elektronegativeren Substituenten X gelöst. Das Bindungselektronenpaar verbleibt bei der als Anion X– austretenden Gruppe X, während mit einem einsamen Elektronenpaar der eintretenden nucleophilen Gruppe Nu– eine neue Bindung geknüpft wird. Die oben genannte Synthese eines Halogenalkans aus einem Alkohol und einem Halogenwasserstoff ist ein Beispiel für eine nucleophile Substitution. In der Regel handelt es sich um reversible Gleichgewichtsreaktionen, die durch die Wahl der Konzentrationen der beteiligten Stoffe in die gewünschte Richtung gelenkt werden können (z. B. durch eine hohe Konzentration an nucleophilem Reagenz oder durch fortlaufende Entfernung des Reaktionsprodukts durch Destillation). Für den Mechanismus der nucleophilen Substitution gibt es zwei Grenzfälle: ● S 2-Reaktion (Synchron-Mechanismus). Als Beispiel nehmen wir die ReakN tion von Brommethan mit Hydroxid-Ionen. Das nucleophile Hydroxid-Ion, eine Lewis-Base, greift das Kohlenstoff-Atom an und verdrängt das BromidIon (ebenfalls eine Lewis-Base). Das Hydroxid-Ion greift auf der zum BromAtom rückwärtigen Seite an. In dem Maße, wie es sich dem Kohlenstoff-Atom nähert, beginnt sich eine kovalente Bindung zwischen dem O- und dem CAtom auszubilden. Gleichzeitig wird die C — Br-Bindung geschwächt bis das Bromid-Ion abgespalten ist. Im Übergangszustand sind beide nucleophilen Gruppen partiell an das Kohlenstoff-Atom gebunden. Die geometrische Anordnung der H-Atome wird bei der Reaktion invertiert indem die H-Atome so ähnlich umklappen wie ein Schirm in stürmischem Wind. Die Bezeichnung SN2 steht für nucleophile Substitution zweiter Ordnung, da die Reaktionsgeschwindigkeit sowohl von der Konzentration des Brommethans als auch von der der Hydroxid-Ionen abhängt. Am geschwindigkeitsbestimmenden Schritt, das ist die Bildung des Übergangszustands, ist das Zusammentreffen zweier Teilchen nötig. Die Reaktion ist bimolekular (vgl. Abschnitt 16.6, S. 259). ● S 1-Reaktion (Abspaltungs-Additions-Mechanismus). Im Falle von tertiären N Halogenalkanen wie tert-Butylbromid verhindern die drei Alkyl-Gruppen aufgrund ihrer räumlichen Ausdehnung den Angriff des Hydroxid-Ions von der dem Brom-Atom gegenüberliegenden Seite. Eine solche, durch den Platzbedarf der Substituenten bedingte Reaktionsbehinderung nennt man sterische Hinderung. In diesem Fall ist der erste Reaktionsschritt die Abspaltung eines Bromid-Ions unter Bildung eines Carbenium-Ions als Zwischenstufe. Dessen Bildung ist begünstigt, weil es sich um ein relativ stabiles tertiäres Carbenium-Ion handelt (vgl. S. 505). Darüber hinaus wird der SN1-Mechanismus durch polare Lösungsmittel begünstigt, da diese durch Solvatation das Carbenium-Ion stabilisieren können. Die Bildung des Carbenium-Ions ist der langsamste und damit geschwindigkeitsbestimmende Schritt. Die Gesamt- Heruntergeladen von: Thieme E-Books & E-Journals. Urheberrechtlich geschützt. 32 Organische Chemie Teil II: Funktionelle Gruppen 32.1 Halogenalkane. Nucleophile Substitution. Eliminierungsreaktionen reaktion verläuft nach einem Geschwindigkeitsgesetz erster Ordnung und ist nur von der Konzentration des tert-Butylbromids abhängig (vgl. Abschnitt 16.6, S. 260). Als starke Lewis-Säure lagert das Trimethylcarbenium-Ion schnell das nucleophile Hydroxid-Ion an. Da das Carbenium-Ion planar ist, kann die Anlagerung des Hydroxid-Ions von beiden Seiten erfolgen. Verglichen zur ursprünglichen Anordnung der Methyl-Gruppen in der tertiären Alkyl-Gruppe können die Methyl-Gruppen nach der Reaktion die gleiche Orientierung haben wie vorher, oder sie können umgeklappt sein. Dieser Aspekt ist bei chiralen Molekülen von Bedeutung (Abschnitt 33.1, S. 541). Weitere Beispiele für nucleophile Substitutionsreaktionen sind: Williamson-Synthese von Ethern (S. 520) Nitril-Synthese (S. 527) Amin-Synthese (S. 533) Heruntergeladen von: Thieme E-Books & E-Journals. Urheberrechtlich geschützt. Gute Nucleophile sind solche, die in einer SN2-Reaktion schnell reagieren. Die Nucleophilie ist ein qualitativer Begriff, der sich nach der Reaktionskinetik richtet und deshalb nicht mit der (thermodynamisch definierten) BrønstedBasizität gleichzusetzen ist. Außerdem wird die Reaktionsgeschwindigkeit stark vom Lösungsmittel beeinflusst. Ein Nucleophil, das über Wasserstoffbrücken solvatisiert ist, ist weniger reaktiv. Grobe Regeln sind: ● Weiche Lewis-Basen (Tab. 18.3, S. 287) sind stärker nucleophil als harte; ● Anionen sind stärker nucleophil als die entsprechenden ungeladenen Moleküle; ● mit zunehmender Elektronegativität nimmt die Nucleophilie eines Atoms ab; ● anders als in protischen Lösungsmitteln folgt die Nucleophilie in aprotischen Lösungsmitteln ungefähr der Basizität (z. B. F– > Cl– > Br– > I–); ● in aprotischen Lösungsmitteln verlaufen die Reaktionen schneller als in protischen; – ● die Abgangsgruppe X ist umso besser, je weniger basisch sie ist. Bei SN1-Reaktionen ist die Nucleophilie des eintretenden Nucleophils unerheblich, weil es am geschwindigkeitsbestimmenden Schritt nicht beteiligt ist. Nucleophilie in protischen Lösungsmitteln (H-Brücken bildend, z. B. H2O, ROH, CH3COOH; grobe Einteilung): RS– > CN– ≈ I– > HO– ≈ R3N > Br– ≈ NH3 > Cl– > RCOO– > H2O > F– > ROH E1-Mechanismus Eliminierungsreaktionen verlaufen ähnlich wie nucleophile Substitutionsreaktionen. Die nucleophile Gruppe wird jedoch nicht angelagert, sondern sie wirkt als Base und bewirkt die Ablösung eines Protons. Im Falle von tertiären Halogenalkanen ist bei der Eliminierung der erste Reaktionsschritt der gleiche wie bei der SN1-Reaktion, d. h. die monomolekulare Heterolyse zum Carbenium-Ion. Als Beispiel diene nochmals die Reaktion von tert-Butylbromid mit Hydroxid-Ionen. Der Reaktionsablauf in der Art, wie nebenstehend formuliert, wird E1-Mechanismus genannt. Er läuft bevorzugt dann ab, wenn das intermediär auftretende Carbenium-Ion relativ stabil ist (tertiäres Carbenium-Ion) und das Medium sauer oder neutral ist. In Anwesenheit von Basen (z. B. OH–) gewinnt dagegen der E2-Mechanismus, die bimolekulare β-Eliminierung, die Oberhand. Dabei verläuft die Bindungsbildung und -spaltung konzertiert (d. h. in einem Zug) wie beim SN2-Mechanismus. Im Unterschied zur SN2-Substitution reagiert die nucleophile Gruppe jedoch nicht mit dem C-Atom, das die Austrittsgruppe trägt, sondern mit einem Wasserstoff-Atom. Wie in den geschilderten Beispielen stammt das abgespaltene Proton meistens vom benachbarten, dem β-C-Atom. In diesem Fall spricht man von einer β-Eliminierung, und das Reaktionsprodukt ist ein Alken. Im Ergebnis ist die β-Eliminierung das Gegenstück zur Additionsreaktion. Nucleophile Substitutionsreaktionen und Eliminierungsreaktionen stehen oft in Konkurrenz zueinander. Bei der Substitutionsreaktion ist die Zahl der Teilchen vor und nach der Reaktion gleich groß. Bei der Eliminierungsreaktion ist die Zahl der Teilchen nach der Reaktion größer. Damit nimmt die Unordnung im System zu, die Reaktionsentropie ΔS ist positiv. Gemäß ΔG = ΔH – TΔS (Gleichung (21.9), Abschnitt 21.4, S. 328) begünstigt eine höhere Temperatur die Eliminierungsreaktion. E2-Mechanismus 32 515