Molekulare und zellbiologische Mechanismen der
Werbung

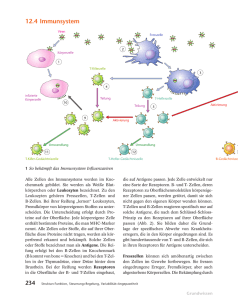
589_641_BIOsp_0609.qxd 606 17.09.2009 10:19 Uhr Seite 606 W I S S E N SCH AFT Antigenpräsentation Molekulare und zellbiologische Mechanismen der Kreuzpräsentation ACHMET IMAM CHASAN, CHRISTIAN KURTS, SVEN BURGDORF INSTITUTE FÜR MOLEKULARE MEDIZIN UND EXPERIMENTELLE IMMUNOLOGIE (IMMEI), UNIVERSITÄTSKLINIKUM BONN Kreuzpräsentation ist erforderlich für eine Immunität gegen Viren und Tumoren und für Impfungen. Wir konnten erstmals die molekularen und zellbiologischen Mechanismen der Kreuzpräsentation eines Modellantigens aufklären und damit unser Verständnis des Immunsystems fundamental erweitern. Cross-presentation is required for immunity against viruses and tumors and for vaccinations. We have clarified the molecular and cell-biological mechanisms of cross-presentation of a model antigen, which fundamentally extends our understanding of adaptive immunity. ˚ Abb. 1: Molekulare Mechanismen der Antigenpräsentation. Endogene Antigene werden vom Proteasom abgebaut (1), durch TAP ins ER transportiert und auf MHC I beladen (2). Pinozytierte oder SR-aufgenommene Antigene gelangen in Lysosomen (3) zur ausschließlichen Beladung auf MHC II (4). MR-aufgenommene Antigene gelangen in stabile frühe Endosomen (5), von wo sie zum proteasomalen Abbau ins Zytoplasma gebracht werden (6). Die so entstandenen Peptide werden durch endosomales TAP wieder in die Endosomen gebracht (7), um dort auf MHC I beladen zu werden (8). Entstehung einer adaptiven Immunantwort ó Zur Bekämpfung von Krankheitserregern und Tumoren bedient sich das Immunsystem unterschiedlicher Mechanismen. T-Lymphozyten (T-Zellen) spielen bei der Regulation und Ausführung dieser Prozesse eine wichtige Rolle. Während CD4+-T-Helfer-Zellen die Funktionen von anderen Immunzellen, wie z. B. Makrophagen und B-Zellen, regulieren, können zytotoxische CD8+-T-Zellen infizierte Zellen oder Tumorzellen spezifisch abtöten. Um ihre Aktivität ausüben zu können, müssen T-Zellen zunächst aktiviert werden. Diese Aufgabe übernehmen die dendritischen Zellen (DC) [1], die Komponenten von Erregern (Antigene) im Gewebe aufnehmen können. Die Erkennung dieser Erreger löst in den dendritischen Zellen einen Reifungsprozess aus, der ihre Migration in den Lymphknoten initiiert. Dort können die DC die verarbeiteten Antigene den T-Zellen präsentieren und somit Antigen-spezifische T-Zellen aktivieren. Da dies die Hauptfunktion dendritischer Zellen ist, werden sie auch professionelle Antigenpräsentierende Zellen (APZ) genannt. Zur Aktivierung von T-Zellen werden die von den DC aufgenommenen Antigene abgebaut. Die so entstandenen Peptidfragmente werden anschließend an MHC(major histocompatibility)-Moleküle gebunden. Diese Peptid-MHC-Komplexe werden an die Zelloberfläche befördert, wo sie von T-Zellen erkannt werden können. Zytotoxische CD8+-T-Zellen erkennen antigene Peptide, die an MHC-IMoleküle gebunden sind; CD4+-T-Helfer-Zellen erkennen Peptide auf MHC-II-Molekülen. Neben Antigenen, die extrazellulär aufgenommen werden, können auch endogene Antigene, das heißt Proteine, die von der präsentierenden Zelle selbst synthetisiert wurden, präsentiert werden. Dies geschieht in fast jeder Körperzelle und führt dazu, dass ein Bild des gesamten Zellinhalts auf den Zelloberflächen vorliegt. Sobald eine Veränderung, z. B. aufgrund einer Infektion oder malignen Entartung, auftritt, kann diese BIOspektrum | 06.09 | 15. Jahrgang 589_641_BIOsp_0609.qxd 17.09.2009 10:19 Uhr Seite 607 607 durch (APZ-aktivierte) spezifische CD8+-zytotoxische T-Zellen erkannt werden. Diese zytotoxischen T-Zellen können dann in den infizierten oder entarteten Zellen den programmierten Zelltod (Apoptose) auslösen. Unser Forschungsschwerpunkt ist die Präsentation extrazellulärer Antigene auf MHCI-Molekülen an CD8+-zytotoxische T-Zellen, auch Kreuzpräsentation genannt. Die Kreuzpräsentation spielt eine zentrale Rolle bei der Aktivierung von zytotoxischen T-Zellen gegen Virus-infizierte Zellen oder Tumoren. Zudem hat sie eine wichtige Bedeutung bei der Aufrechterhaltung der Immuntoleranz gegen körpereigene Proteine [2, 3]. Die Aufklärung der molekularen und zellbiologischen Mechanismen der Kreuzpräsentation ist von zentralem Interesse. Die Art der Antigenaufnahme entscheidet über die Art der Antigenpräsentation Der erste Schritt der Antigenpräsentation ist die Antigenaufnahme. Die Aufnahme von lös- BIOspektrum | 06.09 | 15. Jahrgang lichen Antigenen durch APZ erfolgt über unterschiedliche Mechanismen. Unspezifische Aufnahme geschieht durch Pinozytose, wobei kleine Mengen an extrazellulärer Flüssigkeit mitsamt den darin gelösten Stoffen nach Ausstülpung der Zellmembran aufgenommen werden können. Zudem können über Rezeptor-vermittelte Endozytose bestimmte Antigene, die von einem bestimmten Rezeptor erkannt werden, von der APZ internalisiert werden. In früheren Studien konnten wir zeigen, dass der Weg, über den das Antigen von der DC aufgenommen wird, die Art der Präsentation bestimmt [4, 5]. So wurde das Modellantigen Ovalbumin (OVA), wenn es über den Mannose-Rezeptor (MR) aufgenommen wurde, ausschließlich auf MHC I an CD8+-T-Zellen präsentiert, wohingegen OVA, welches von Scavenger-Rezeptoren (SR) bzw. über Pinozytose aufgenommen wurde, auf MHC II an CD4+-T-Zellen präsentiert wurde. Zudem konnten wir zeigen, dass beide Wege der Antigenpräsentation räumlich von- einander abgegrenzt sind und in unterschiedlichen Zellorganellen stattfinden. Während ein Antigen, das für die Präsentation an CD4+-T-Zellen bestimmt ist, rasch in Lysosomen vorliegt, gelangt ein Antigen, das an CD8+-T-Zellen präsentiert und vom MR zur Kreuzpräsentation aufgenommen wird, in eine separate Population von Endosomen, den sogenannten stabilen frühen Endosomen (stable early endosomes, SEE). Diese verharren im Stadium eines frühen Endosoms und reifen nicht zum Lysosom aus. Molekulare Mechanismen der Antigenpräsentation im stabilen frühen Endosom In einer aktuellen Studie haben wir die Mechanismen der Antigenverarbeitung zur Kreuzpräsentation im stabilen frühen Endosom untersucht [6]. Mehrere Arbeitsgruppen konnten bereits zeigen, dass im Rahmen der Kreuzpräsentation die Antigene nach deren Internalisierung aus den Endosomen in das Zytoplasma transportiert werden müssen [5, 7, 8]. 589_641_BIOsp_0609.qxd 608 17.09.2009 10:19 Uhr Seite 608 W I S S E N SCH AFT Dort werden diese Antigene anschließend vom zytoplasmatischen Proteasom abgebaut. Lange wurde angenommen, dass die so entstandenen Peptidfragmente in dem MHC-IPräsentationsweg für endogene Antigene weiterprozessiert werden. Endogene Antigene werden nach proteasomalem Abbau von einem Transporter namens TAP in das endoplasmatische Retikulum (ER) gebracht, wo die Peptide auf MHC-I-Moleküle beladen werden [9]. Anschließend gelangen die MHC-IPeptid-Komplexe über den sekretorischen Weg zur Zelloberfläche. Wir konnten erstmals für ein Modellantigen zeigen, dass aus Antigenen entstandene Peptide für die Kreuzpräsentation nicht in das ER zur Beladung auf MHC I transportiert werden, sondern dass die Beladung in den Endosomen selbst stattfindet [6]. Bei der Untersuchung der oben erwähnten stabilen frühen Endosomen konnten wir den TAPTransporter in der endosomalen Membran detektieren, was eine Unabhängigkeit von der Antigenbeladung im ER vermuten ließ. Um zwischen Peptidbeladung im ER und in den Endosomen zu unterscheiden, haben wir in enger Zusammenarbeit mit der Gruppe von Prof. Tampé der Universität Frankfurt versucht, den TAP-Transporter Endosomenspezifisch zu inhibieren. Dies gelang uns durch die Kopplung von dem spezifischen TAP-Inhibitor US6 an Transferrin. Die Kopplung an Transferrin bewirkte, dass US6 spezifisch in die frühen Endosomen gebracht wurde und somit TAP ausschließlich in den Endosomen, nicht aber im ER inhibierte. Der Einsatz dieser US6-Transferrin-Komplexe in Kreuzpräsentationsversuchen zeigte, dass endosomales TAP für die Kreuzpräsentation essenziell ist und die Peptide zur Beladung in die Endosomen gebracht werden [6]. Außerdem konnten wir erstmals Peptidbeladene MHC-I-Moleküle mittels Immunfluoreszenz in diesen Endosomen nachweisen. Hierzu verwendeten wir den Antikörper 25-D1.16 [10], welcher das kreuzpräsentierte OVA-Peptid (SIINFEKL) gebunden an MHC I erkennt. Anhand von Untersuchungen mit dem Arzneimittel Primaquin konnten wir weiterhin zeigen, dass ohne Zugabe von Primaquin die Peptid-MHC-I-Komplexe nach der Beladung in den Endosomen auf direktem Wege zur Zelloberfläche transportiert wurden, wo sie von CD8+-T-Zellen erkannt werden konnten. Primaquin inhibiert den Transport von Endosomen zur Zelloberfläche und verhinderte demzufolge bei dessen Zugabe die Kreuzprä- sentation [6]. Die Präsentation von endogenen Antigenen, deren Beladung im ER stattfindet, blieb dagegen unverändert durch Zugabe von Primaquin. Auch dies schließt eine Beladung im ER für die Kreuzpräsentation aus. Zusammenfassend konnten wir zeigen, dass die MHC-I-vermittelte Präsentation von endogenen Antigenen, die Kreuzpräsentation und die MHC-II-vermittelte Präsentation in unterschiedlichen Zellkompartimenten stattfindet (Abb. 1). Die Beobachtung, dass die Mechanismen der Antigenaufnahme entscheiden, welche Art der Immunantwort induziert wird, könnte wichtige Implikationen für die Entwicklung von Impfstoffen gegen Viren oder Tumoren haben, die ja darauf abzielen, zytotoxische T-Zellen zu aktivieren. So könnte man, wenn man einen Impfstoff derart modifiziert, dass er über den MR in die APZ gelangt, eine erhöhte Kreuzpräsentation bewirken, die eine verbesserte Aktivierung von zytotoxischen T-Zellen ermöglicht, welche Tumor- oder Viren-infizierte Zellen abtöten. ó Literatur [1] Mellman I, Steinman RM (2001) Dendritic cells: specialized and regulated antigen processing machines. Cell 106:255–258 [2] Kurts C, Heath WR, Carbone FR et al. (1996) Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med 184:923–930 [3] den Haan JM, Lehar SM, Bevan MJ (2000) CD8(+) but not CD8(–) dendritic cells cross-prime cytotoxic T cells in vivo. J Exp Med 192:1685–1696 [4] Burgdorf S, Lukacs-Kornek V, Kurts C (2006) The mannose receptor mediates uptake of soluble but not of cell-associated antigen for cross-presentation. J Immunol 176:6770–6776 [5] Burgdorf S, Kautz A, Bohnert V, Knolle PA et al. (2007) Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 316:612–616 [6] Burgdorf S, Scholz C, Kautz A et al. (2008) Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol 9:558–566 [7] Norbury CC, Chambers BJ, Prescott AR et al. (1997) Constitutive macropinocytosis allows TAP-dependent major histocompatibility complex class I presentation of exogenous soluble antigen by bone marrow-derived dendritic cells. Eur J Immunol 27:280–288 [8] Ackerman AL, Giodini A, Cresswell P (2006) A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 25:607–617 [9] Abele R, Tampe R (2004) The ABCs of immunology: structure and function of TAP, the transporter associated with antigen processing. Physiology 19:216–224. [10] Song R, Porgador A, Harding CV (1999) Peptide-receptive class I major histocompatibility complex molecules on TAPdeficient and wild-type cells and their roles in the processing of exogenous antigens. Immunology 97:316–324 Korrespondenzadresse: Dr. Sven Burgdorf Prof. Dr. Christian Kurts Institute für Molekulare Medizin und Experimentelle Immunologie (IMMEI) Sigmund-Freud-Straße 25 D-53105 Bonn Tel.: 0228-28711038 Fax: 0228-28711052 [email protected] [email protected] www.ukb.uni-bonn.de/IMMEI AUTOREN Sven Burgdorf Achmet Imam Chasan Jahrgang 1976. 1993–1998 Bio-Ingenieur-Studium in Leuven, Belgien. 1998–2000 Wissenschaftlicher Mitarbeiter im Max-Planck-Institut für Züchtungsforschung, Köln und MPB Cologne GmbH. 2000–2003 Doktorarbeit im Institut für Genetik, Bonn bei Prof. Dr. Scheidtmann. 2004–2008 Postdoc im IMMEI, Bonn bei Prof. Dr. Kurts. Seit 2008 Arbeitsgruppenleiter im IMMEI, Bonn. Jahrgang 1980. 2001– 2007 Biologiestudium an der Universität Bonn. Seit 2007 Promotion am IMMEI, Bonn. Christian Kurts Jahrgang 1964. 1985–1991 Medizin- und Physikstudium in Göttingen, Promotion in Immunologie. 1991–1995 und 1998–2000 Assistenzarzt und wissenschaftlicher Mitarbeiter, medizinische Hochschule Hannover. 1995–1998 Wissenschaftlicher Mitarbeiter am WEHI, Melbourne, Australien. 2000–2003 AG-Leiter am Uniklinikum der RWTH Aachen. 2003–2009 Professur für Molekulare Immunologie, Uniklinikum Bonn. 2009 Direktor des dortigen Instituts für experimentelle Immunologie. BIOspektrum | 06.09 | 15. Jahrgang