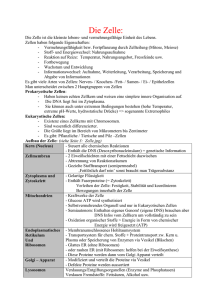
Immunologie und zelluläre Mikrobiologie B
Werbung

Immunologie und zelluläre Mikrobiologie B (LvNr. 300344) WS09 Skriptum GNU General Public License 1 – Allgemeine Prinzipien, Definitionen Mikroben sind infektiöse Einheiten, meist prokaryotische Organismen (Bacteria), aber auch eukaryotische Organismen und Viren. Im Vergleich zur Gesamtzahl der bekannten Spezies ist nur ein verschwindend geringer Anteil davon pathogen. Ein Pathogen ist eine Mikrobe, die fähig ist, dem Wirt Schaden zuzufügen. Die Definition eines Pathogens ist nutzlos, wenn darin nicht der Wirt vorkommt. Archaea sind apathogen. Neben Mikroben können auch Pilze, Flagellaten und Protozoen eine Pathogenese hervorrufen. Jeder Organismus (in diesem Zusammenhang sinnvollerweise der Mensch), der das Wachstum eines anderen Organismus durch Bereitstellung des Körpers als Habitat und Bereitstellung von Nahrstoffen unterstützt, kann Wirt für diesen Organismus sein. Bakterien können für immunkompromittierte und immunkompetente Wirte oder als Ergebnis einer überhöhten Immunantwort pathogen sein. Man unterscheidet Wechselbeziehungen zwischen Wirt und Mikrobe: Parasitismus, Commensialismus, Mutualismus. Die Interaktion mit Bakterien ist für multizelluläre Organismen meist neutral bis vorteilhaft. Parasitismus. Nachteile für eine Spezies überwiegen (Antibiose). Commensialismus. Eine Spezies hat Vorteile, die andere keine Nachteile (Probiose). Mutualismus. Beide Spezies profitieren, sie stehen in Abhängigkeit (Symbiose). Viele Körperregionen stehen mit der Außenwelt in Kontakt. Dort kolonisiert eine Normalflora die Oberfläche des Gewebes nach der Geburt. Nicht alle Orte sind gleich gut besiedelbar (Magen: ungünstiger pH-Wert). Körperregion Bakterienzahl Körperregion Bakterienzahl Auge/Nase 10<1-2 Dünndarm 105-7 Mund 109 Dickdarm 109-11 Haut 103 Urethra 106-7 Lunge 102 Magen 103-6 Die Normalflora stimuliert das Abwehrsystem zur Sekretion von IgA, verhindert die Kolonisierung durch Pathogene und ist Ort der Vitaminsynthese und Resorption von Steroiden. The gut epithelium is on the frontline of interactions with the resident microbiota and is a major effector of immune tolerance. It induces tolerance by sensing the number of bacteria and their distance from the epithelial surface using a combination of receptors such as Toll‐like receptors (TLRs) and NOD‐like receptors (NLRs) that sample pathogen‐associated molecular patterns (PAMPs). The gut epithelium then integrates this information and strategically responds to bacterial pathogens with inflammatory signals or to harmless commensals with immune tolerance signals (Cell, Volume 138, Issue 3, 7 August 2009, Pages 416‐420). Kommensale Bakterien haben eine verminderte Fähigkeit, dem Mucus von Schleimhäuten zu entkommen, sowie zur Adhäsion und Invasion der Epithelbarriere. Die geringe Endotoxizität ist das Ergebnis von penta-acyliertem Lipid A für gram-negative Bakterien. -1- Schleimhaut-Epithelien (mucosal epithelia) haben eine verminderte Fähigkeit zur Erkennung von pattern-associated molecular patterns (PAMPs), mit der Ausnahme von Krypten (crypt cells), da sie weniger Sensormoleküle exprimieren (TLR2, TLR4, TLR9, MD2, CD14) oder sequestrieren. Der Verteilung dieser Faktoren auf den Epithelzellen (basolaterale Expression von TLR5, erkennt Flagellin) wird auch eine immunregulierende Funktion zugesprochen. Schleimhäute enthalten Alarmsysteme zur Erkennung eindringender Pathogene. Unter dem Druck der Normalflora werden entzündungshemmende Pathways permanent induziert. Zum Beispiel führt die Aktivierung des nukleären Rezeptors PPAR-γ (peroxisome proliferator-activated receptor γ) zur Inhibierung des NF-κB-Pathways. Die Lamina propria enthält tolerogene dendritische Zellen, Macrophagen und regulatorische TZellen, die entzündungshemmende Cytokine wie Interleukin-10 und transforming growth factor-β als Antwort auf commensale Bakterien sezernieren. Im Intestinaltrakt finden sich in der Nähe epithelialer Stammzellen sogenannte Krypten, sackartige Vertiefungen, in denen sich Paneth-Zellen befinden, TLRs expriminert und normalerweise frei von Bakterien sind. Treten in eine Krypte Mikroben ein, kommt es zur NOD2-anhängigen Sekretion von Lysozymen, Peptidasen und Defensinen durch die Paneth-Zellen. Neutrophile Granulocyten sekretieren CXCL8. Am Eingang zu Krypten befinden sich Becherzellen, einzellige, becherförmige, Schleim (Mucin) produzierende Drüsen. Im gut associated lymphoid tissue (GALT) kommen Peyer-Plaques (peyer patches) vor, Aggregate aus 10-50 Lymphfollikeln. Sie sind mit einem speziellen Epithel bedeckt (M cells), welches Antigene sammelt und zu basolateralen antigen-präsentierenden Zellen transportiert. Bacteroides fragilis Polysaccharid A (PSA) schützt vor Colitis hervorgerufen durch das potentielle Pathogen Heliobacter hepaticus , welches ebenfalls in der Normalflora vorkommt (Nature 453, 2008, 602-604). PSA ist ein Cofaktor für dendritische Zellen. Inder Folge kommt es zu suboptimaler Effektor-T-Zell-Aktivierung und zur Aktivierung regulatorischer T-Zellen. Es existiert also ein fragiles Gleichgewicht, das von vielen Spezies getragen wird. -2- Positive und negative Funktionen der Normalflora: + - Stimulierung des Immunsystems Kreuzreaktionen Synthese von Vitaminen und Resorption von Steroiden Produktion cancerogener und/oder schädlicher Substanzen ( Heliobacter pylori ) Verhinderung der Kolonisierung durch Pathogene Infektionsquelle ( E.coli, C. difficile, C. albicans, S. epidermidis ) Die Normalflora ist eine Infektionsquelle, wenn die Epithelbarriere physisch verletzt wird (Wunde, Verbrennung) und so commensale Bakterien in Bereiche eindringen, die für sie nicht vorgesehen sind oder Teile der Normalflora durch Antibiotikaeinnahme vernichtet werden. Es werden mehr Krankheiten durch die Normalflora hervorgerufen als durch echte Pathogene. E. coli wird pathogen nach Schutzimpfung, C. difficile nach Antibiotikaeinnahme, C. albicans durch Immunsupression und S. epidermidis infiziert nach physischer Beschädigung des Epithels das Gewebe. Krankheiten verursacht durch die Normalflora sind Abszesse, Karies, Pharyngitis, Sepsis, Endocarditis, Lungenentzündung, Gastroenteritis, Peritonitis und urogenitale Entzündungen. KOCH's Postulate Wann kann ein Bakterium als Ursache für eine bestimmten Krankheit gehalten werden? Sind sie immer noch aktuell? 1. Das Bakterium soll in allen erkrankten Personen und Tieren, nicht aber in normalen, gesunden Individuen, gefunden werden. – Immunsuprimierte Patienten – niedrige Penetranz – asymptomatischer Zustand 2. Das Bakterium soll aus dem kranken Individuum isoliert und in Reinkultur gehalten werden können. – Nicht kultivierbare Mikroorganismen ( Treponema pallidum, Mycobacterium leprae ) – Veränderung der Virulenzfaktoren während der Kultur in vitro – Polymikrobische Infektionen 3. Die Reinokulierung dieser Reinkultur in gesunden Individuen soll die Symptome der ursprünglichen Krankheit hervorrufen. – Ethische Bedenken – Gleiche Symptome hervorgerufen von unterschiedlichen Bakterien 4. Dasselbe Bakterium sollte aus die nunmehr kranken Individuen isoliert werden und im Labor kultiviert werden. – Nicht kultivierbare Mikroorganismen ( Treponema pallidum, Mycobacterium leprae ) – Veränderung der Virulenzfaktoren während der Kultur in vitro – Polymikrobische Infektionen Die nächste GENeration - Wann können ein bakterielles Gen bzw. sein Produkt als notwendig für die Auslösung einer bestimmte Krankheit gehalten werden? 1. Gen bzw. Genprodukt sollen in den virulenten, nicht aber in den avirulenten Bakterienstämmen zu finden sein. 2. Die Zerstörung des Gens sollte die Virulenz des Bakteriums mildern - Die Expression des Gens bzw. des Genprodukts in avirulenten Stämmen sollte deren Virulenz steigern. -3- 3. Das Gen soll in vivo im Laufe der Krankheit exprimiert werden. 4. Die gegen das Genprodukt gerichtete Immunantwort sollte vor der Krankheit schützen. Virulenzfaktoren Die Virulenz von Bakterien wird in vivo und in vitro gemessen. Dabei wird bestimmt, wie viele Bakterien notwendig sind, um den Endpunkt (abhängig, welche Wirt-Parasit-Interaktion studiert werden soll, meistens Tod) zu errreichen. Je weniger Zellen ausreichen, desto virulenter ist ein Pathogen. Die in vivo Methode geht über Tierversuche, Tiere werden mit Pathogenen infiziert. Der Kolonisierung folgen Symptome bis zum Exitus. Es werden verschiedene Mengen verabreicht. Andere Methoden kommen ohne Tierversuche aus, in vitro Ansätze in Zellkulturen. Nachteilig wirkt sich aus, dass Zellen im Epithel immer polarisiert vorliegen und mit anderen Zellen wechelwirken, in der Kultur also nur isoliertes Verhalten sichtbar ist. Das versucht man in vitro Systemen zu umgehen. In Zellkulturen verändern sich Virulenzfaktoren auf zwei mögliche Arten. Diese Prinzipien werden zur Identifikation von Virulenzgenen herangezogen: gain of function – apathogener Organismus wird virulent. loss of function – Virulenzgen wird zerstört. In einem gain of funtion approach setzt man E. coli ein. Dazu wird ein apathogener Laborstamm mit DNA Fragmenten von beipielsweise Salmonella transformiert. Diejenigen E. coli Kolonien, die nun das Kulturgewebe, auf dem sie wachsen, invadieren, tragen die virulente Eigenschaft von Salmonella in sich. Einen loss of function approach führt man durch, wenn man mittels Transposons, die ein Resistenzgen (QR) enthalten, direkt in Salmonella einbringt. Jede Kolonie trägt nun ein Transposon, das jeweils an verschiedenen Stellen inseriert hat. Bei Inkubation auf einer Gewebekultur werden dann eine oder mehrere Kolonien ersichtlich, die nicht mehr virulent sind, in diesen hat das Transposon das Virulenzgen zerstört. Oft wird mehr als ein Gen zerstört, daher werden sie einzelen wieder eingeschleust, um das tatsächliche Virulenzgen zu bestimmen. Das neuerliche Einschleusen erfordert, dass man DNAFragmente kloniert hat. Zur Identifikation von sekretierten Virulenzfaktoren nutzt man PhoA-Fusionsproteine. PhoA, die alkalische Phosphatase, ist nur aktiv, wenn sie sich an der Zelloberfläche oder im Medium befindet. Virulenzgene werden vielfach nur in vivo exprimiert, in vivo Bedingungen sind sehr komplex, Konzentrationen von Spurenelementen (Fe), Wirtsfaktoren, pH-Wert, Temperatur, Osmolarität und O2-Gehalt aktivieren spezielle Promotoren. Darum wird in vivo expression technology (IVET), genutzt, um Gene zu finden, die nur im Wirt aktiv sind. Im Originalexperiment nach Mahan et al. (Mahan, Slauch und Mekalanos, 1993 Science 259:686) wurde der Vector piVET1 hergestellt. Er enthält eine Ampicillinresistenz, purA und lacZY mit einer BgIII-Schnittstelle upstream. PurA ist ein essentielles bakterielles Gen, in dessen Abwesenheit kein Adenin produziert werden kann und das hier als Selektionsmarker genutzt wird. Verdaute Salmonella typhimurium DNA wird in die Plasmide ligiert und in E. coli eingebracht. Mittels Konjugation erhalten ΔpurA Salmonellen die Vektoren, die Salmonellen werden auf Amp/Adenin-Medium inkubiert. Zellen, die Plasmid aufgenommen haben, injiziert man in Mäuse (Adenin arme Umgebung), der Rest geht am Ampicillin zu Grunde. Zellen mit intakten Promotoren upstream von purA überleben. Nach einigen Tagen entnimmt man die Milz der Mäuse und plattiert Salmonellen daraus auf X-Gal/Adenin/Amp. -4- Welche Kolonien zeigen nun wirtspezifische Promotoren an? Die weißen Kolonien. Alle haben intakte Promotoren, aber in den weißen Kolonien ist er am Medium nicht aktiv, sodass X-Gal nicht gespalten wird, im Wirt musste er allerdings aktiv sein, weil die Zelle sonst am Adenin-Mangel zu Grunde gegangen wäre. Eine weitere Methode zur Identifikation von Virulenzgenen ist signature-tagged mutagenesis. Dabei wird ein Pool an DNA signature tags in Transposons eingebracht, die zufällig im Genom integrieren und dabei die Funktion mancher Virulenzgene stören. Vorraussetzung ist, dass das Genom im Vorhinein sequenziert ist. Man weiß, welche signatures man einbringt, und vergleicht diese mit jenen, die man aus infiziertem Gewebe isoliert. Jene signatures, die dabei nicht mehr aufscheinen, haben mit Virulenzgenen interferiert (Henri L. Saenz and Christoph Dehio, Current Opinion in Microbiology 2005, 8:612 – 619). Bei der Begegnung eines Mikroorganismus mit dem Wirt kommt es zur Infektion, das ist aber nicht das gleiche wie eine Infektionskrankheit, also Symptome, die durch die Anwesenheit des Organismus entstehen. Ob eine Krankheit ausbricht hängt vom Zustand des Wirts ab. Folgende Ereignisse treten immer ein: • • • INFEKTION - Begegnung zwischen Mikroorganismus und Wirt KOLONISIERUNG ◦ Eintritt des Mikroorganismus in den Wirt ◦ Verbreitung des Mikroorganismus in dem Wirt ◦ Vermehrung des Mikroorganismus im Wirt Beschädigung des Wirtes durch den Mikroorganismus -5- • Endergebnis ◦ Wirt gewinnt ◦ Wirt und Mikroorganismus koexistieren ◦ Mikroorganismus gewinnt Man unterscheidet zwischen endogener und exogener Begegnung, endogene Keime stammen aus der Normalflora, exogene von außerhalb des Körpers. Das Wissen um die Art der Begegnung ist wichtig für die Ergreifung passender Präventionsmaßnahmen. Kontaktart Keim Quelle Prävention Inhalation Virus Aerosol Kontakt meiden Nahrung Bakterium Wasser, Speisen Hygiene, Minderung des Inokulums Sex Virus Partner Sozialverhalten, Kontakt meiden Insektenstich Protozoon Mücke Ausrottung des Vektors Wunde Normalflora Aseptische Technik Bakterium Weiters unterscheidet man fakultativ intrazelluläre (Salmonella, Shigella, Yersinia), obligat intrazelluläre (Rickettsia, Chlamydia, Coxiella) und extrazelluläre (Vibrio cholerae, Pseudomonas, enterotoxische E. coli (ETEC) ) Bakterien. Wachstum, Verbreitung im Wirt Bakterielle Keime verfolgen verschiedene Wachstums- und Verbreitungsstrategien. Bei Brucellus abortus folgt der Verbreitung des Keims im Wirt seine Vermehrung (in Placenta), während sich Staphylococcus zuerst vermehrt, um sich dann im Wirt auszubreiten. Clostridium tetani überspringt die Ausbreitungsphase, da es in der anaeroben Umgebung einer bedeckten Wunde bleiben muss, das Tetanospasmin wird sowieso über das Blut im Körper verbreitet. Im Zuge einer erfolgreichen Einnistung passen Bakterien ihre Wachstumsgeschwindigkeit an. Rasches Wachstum findet bei erstem Kontakt mit dem Epithel, um Rezeptoren zu überwinden, die einen Kontakt blockieren, bei primärer Einnistung im Gewebe, um Verluste auszugleichen, die durch heftige Immunabwehr entstehen, statt. Akute Symptome sind Folgen des raschen Wachstums. -6- Hat der Parasit das richtige Gewebe gefunden (tissue tropism), beginnt er mit dem Aufbau einer signifikanten Population, das Wachstum verlangsamt sich. Im chronischen Verlauf bleibt die Population annähernd konstant, die Stimulation des Immunsystems wird schwächer. Der carrier state ist für einen Parasit sehr vorteilhaft, dabei erreicht er eine resistente stationäre Phase, der Wirt weiß nicht, dass er infiziert ist. Am Ort der Infektion kommt es zum Kräftemessen zwischen Mikroorganismus und Immunsystem. Das Immunsystem versucht die Ausbreitung der Keime durch Blutgerinnung und Rekrutierung cytotoxischer Zellen zum Infektionsherd zu hemmen, Mikroorganismen setzen fibrinolytische Stoffe (Collagenasen, Hyaluronidasen), die Blutgerinnsel und extrazelluläre Matrix abbauen, und Toxine, die gegen Effektorzellen gerichtet sind, indem sie deren Plasmamembranen destabilisieren, ein (Leukocidine, Hämolysine, Phospholipasen). Ab jetzt beginnt die Schädigung des Wirts, die Ausschleusung toxischer Metabolite läuft an. Man unterscheidet bakterielle Toxine nach dem Wirkmechanismus: • • • • • Toxine, welche die Lyse von Wirtszellen verursachen ◦ Lipasen ( Clostridium perfringens α-Toxin ) ◦ Porenbildende Toxine ( Staphylococcus aureus α-Toxin, Streptolysin O ) Proteinsynthesehemmende Toxine ( Diphteria Toxin ) Pharmakologisch aktive Toxine ( Cholera Toxin, stürzt Metabolismus der Zelle ins Chaos ) Neurotoxine ( Botulinus-Toxin, Tetanospasmin ) Endotoxine ( Bestandteile der Keime selbst, werden bei Lyse wirksam ) Virulenz Virulenz kann man nur in Kombination mit dem Wirt beschreiben, ist multifaktoriell und situationsabhängig. Invasivität und Toxizität bestimmen die Virulenz. Invasivität ist die Fähigkeit, sich im Wirt zu vermehren, Toxizität gibt die Keimzahl an, ab der bereits ein Schaden für den Wirt entsteht. -7- Virulenzfaktoren von Salmonella: • Exo-(Enterotoxine) → sofortige Immunreaktion • Adhäsionsmoleküle → Bakterien bleiben lange genug im Wirt • Kapsel, LPS → verhindern Phagocytose und treiben Kolonisieung voran • Flagellum → Überwindung der Schleimschicht, Kontakt mit Zellen des Darmepithels • Proteine, die Fe binden ( in vivo limitierend ) Resistenz gegenüber Keimen hängt von vielen Faktoren ab: • Männer und Frauen haben mit unterschiedlichen Keimen Probleme • Gesundheitszustand: Leidet man an anderen chronischen oder akuten Krankheiten? • Immunstatus: Patienten in Chemotherapie, Immunsupression (HIV) • Extremer Stress • Genetische Faktoren: Prädisposition oder Resistenz gegenüber Infektionen ◦ Mycobacterium (Prädisposition) → IL12/23, IFN-γ Defizienz ◦ Human Papillomavirus (Prädisposition) → EVER1/EVER2 Defizienz ◦ HIV1 (Resistenz) → CCR5 Defizienz ◦ Norovirus (Resistenz) → FUT2 Defizienz Auch bei Bakterien bestimmen genetische Faktoren, ob der Wirt gegen sie immun ist. Ein Vorteil ist allerdings die hohe Plastizität des Genoms und die Geschwindigkeit der Replikation. Bakterien betreiben rasche Evolution über reduktive Deletionen (black holes), Akquisition neuer Funktionen mittels horizontalem Gentransfer (HGT), rearrangements oder Mutationen. Genetische Information für Virulenzfaktoren kann auf dem bakteriellen Chromosom liegen und über Plasmide, Bakteriophagen oder Transposons (liegen meist auf Plasmiden und inserieren ins Genom) in die Zelle gelangen. Mobile genetische Elemente führen beispielsweise zur Entstehung von E. coli Pathotypen. -8- Virulente Mikroorganismen verursachen die Zerstörung ihres Wirtes und verlieren somit ihre Nische. Daher sind erfolgreiche Parasiten solche, die ihren Wirt nicht oder nur gering beeinträchtigen (Normalflora), aber das Beste für eine Spezies ist nicht notwendiger Weise mit dem Besten für jedes einzelne Individuum gleichzusetzen (evolutionäre Theorie). Rapides Wachstum ermöglicht es, schneller als die Immunantwort zu sein, der Grenzwert der T-Zell Antwort wird schnell erreicht. Langsam wachsende Keime bleiben länger unbemerkt und sobald sie den Grenzwert erreichen, fällt auch die Immunantwort schwächer aus. Halten sich Immunantwort und Wachstum die Waage, liegt eine chronische Infektion vor. Die Transmission schnell wachsender Keime kann nur in einem kleinen Zeitfenster erfolgen, daher kann man gegen solche effizientere Vaccine herstellen als gegen solche, die langsam wachsen und den Wirt länger zu einer Infektionsquelle machen. Direkt übertragene Erreger sind meistens wenig virulent, sie können ohne den Wirt nicht überleben. Pathogene, die nicht permanent auf den Wirt angewiesen sind, haben höhere Virulenz. Dazu zählen: • • • • Vektor-übertragene (Yersinia pestis) Wasser-übertragene (Vibrio cholerae) Pathogene, die durch Krankenhaus-Personal übertragen werden (E. coli) Pathogene, die außerhalb eines Wirts überleben können (Mycobacterium tuberculosis) HIV ist ein Beispiel für einen wenig virulenten Erreger, der durch replicative fitness eine hohe Latenz erreicht. Das Virus befällt langlebige CD4+ Zellen, wodurch die Effizienz einer adaptiven Immunantwort sinkt, die hohe Mutationsrate torpediert zusätzlich die CTL-vermittelte Immunität. Infektionsroute ist abhängig vom Sozialverhalten, die notwendige direkte Übertragung (vertical transfer) zeichnet den Erreger als wenig virulent aus. Pathogenic fitness, viral load und replicative fitness hängen mit der HLA-Diversität zusammen, von der die Effizienz der CTL-Immunität abhängt. In einem Beispiel sei eine Person A mit HIV infiziert. Über Jahre sammeln sich Mutationen im HIV-Genom an, sodass die replicative fitness auf 4 steigt (dimensionsloser Index). Nun kommt es zu zwei Transmissionsereignissen. Im ersten wird eine Person X infiziert, dessen HLA-Allele denen von Person A stark ähneln. Daher ist der Virus resistenter gegen die CTL-vermittelte Immunität, die viral load steigt, der Erreger ist virulenter geworden (höhere pathogenic fitness). Im zweiten Ereignis wird eine Person B infiziert, dessen HLAAllele stark von denen von Person A abweichen. Die CTL-vermittelte Immunität ist effektiver, es -9- können Polymorphismen gegen die neuen Epitope entwickelt werden, der Erreger wird effektiver bekämpft. Im Vergleich zu Person A müsste die replicative fitness etwa 8 betragen, was aber nicht so schnell erreicht werden kann, die Virulenz des Erregers ist sehr gering, als Folge sinkt die pathogenic fitness und die viral load. Infektionskrankheiten führen auch zur Anpassung humaner Gene, beispielsweise von TRL4, einem Rezeptor des angeborenen Immunsystems. Die Mutation N299G führt zwar eine Prädisposition zu septischem Schock ein, vermindert aber das Risiko einer Malaria-Infektion (kommt nur in Afrika vor). T399I neutralisiert die negativen Auswirkungen von N299G (kommt hauptsächlich bei Europäern vor), steht aber im Verdacht, die Anfälligkeit für Herz- und Nierenerkrankungen zu steigern. Daraus schließt man, dass das menschliche Immunsystem in der Lage ist, sich langfristig auf bestimmte Lebensumstände einzustellen. 2 – Infektion, Adhesine als Virulenzfaktoren Das Substratum für die Adhäsion von Bakterien kann praktisch alles sein, eine Zelle, extrazelluläre Matrix, Plastik (Implantate!) oder inerte Oberflächen wie Zahnschmelz. Ein Biofilm beginnt zu entstehen, wenn einzelne Zellen reversibel mit einer Oberfläche assoziieren. Danach wirken Adhäsionsfaktoren, es kommt zur irreversiblen Adhäsion und Kolonisierung. Mikrokolonien produzieren hydratisierte Polymere, meist Saccharide, die die Kolonie umhüllen und Schutz vor Antikörpern und Antibiotika bieten. Im weiteren Reifungsprozess des Biofilms entstehen Makrokolonien, dreidimensionale Netzwerke, mit Kanälen, um Nährstoffe heran- und Metabolite wegzuschaffen sowie türmchenartige Strukturen zur Loslösung von Teilen der Kolonie. Meist coaggregieren mehrere Spezies, dabei kann es sogar zu organisierten Gemeinschaften mit funktioneller Heterogenität kommen. Bakterielle Biofilme ermöglichen Wachstum in einer feindlichen Umgebung und fördern die Propagation planktonischer Zellen. Einer großen Zahl chronischer bakterieller Infektionen liegen Biofilme zu Grunde, denen durch konventionelle Antibiotikatherapie nicht Herr zu werden ist. Meist bilden commensale Bakterien Biofilme. Biofilme setzen Antigene frei und stimulieren die Produktion von Antikörpern, diese können an die Verursacher aber nicht heran, vielmehr wird das umliegende gesunde Gewebe geschädigt (Phago- - 10 - cyten produzieren ROS wie Hypochlorit und Wasserstoffperoxid, wird in Außenschicht des Biofilms schneller neutralisiert als es diffundieren kann). Eine zweite Hypothese, die erklärt, warum Biofilme für antimikrobielle Substanzen wenig empfänglich sind, ist, dass die Zellen in einer stationären Phase überdauern und wenig Metabolismus haben, so kann zB Penicillin nicht wirken. Eine der am besten studierten Biofilm bildenden Spezies ist Pseudomonas aerunginosa. Sogenannte sad-Mutanten (surface attachment defective) zeigen, dass die Typ IV Pili Bedeutung bei der Bildung von Microkolonien haben (twitching). Bei Anhaftung werden die Gene algC, algD und algU exprimiert, was zur Bildung der Biofilmmatrix, in diesem Fall Alginat, führt. Danach vermitteln die Zellen ihre Anwesenheit im Quorum sensing mittels Acylhomoserinlacton (AHL) und man vermutet, dass die Differenzierung in reife Biofilme dadurch gestartet oder gesteuert wird. Es werden zwei Signalsysteme aktiviert: LasR-LasI (Expression extrazellulärer Virulenzfaktoren, Synthese von 3-Oxododecanoylhomoserinlacton) und RhlR-RhlI (Synthese von Butyrylhomoserinlacton). Die Dispersion von Einzelzellen oder Kolonieteilen wird vermutlich ebenfalls über ein Expressionsprogramm initiiert, wahrscheinlich wirken dabei Alginasen, ähnlich den cer-Genen von Rhodobacter sphaeroides. - 11 - Die Beschaffenheit und Spezifizität bakterieller Adhesine bestimmen: • • • den Wirt, der befallen wird (Speziestropismus) das Gewebe, in dem sich der Infektionsprozess abspielen wird (Gewebstropismus) die Zielzelle des Mikroorganismus (Cytotropismus) Adhäsion ermöglicht Kolonisierung, stimuliert aber auch die Infiltration und Aktivierung von und durch Immunzellen sowie Phagocytose. Deswegen laufen die ersten Interaktionen mit dem Wirt mit einem Sicherheitsabstand ab: Adhesine sind an polymeren Strukturen exprimiert, die aus einer Schutzschicht herausragen, die Erkennung und Phagocytose durch Immunzellen verhindert (zB M-Protein von Streptococcus). Adhesine in gram-negativen Bakterien sind unter anderem Typ1 Pili, P-Pili, TypIV pili und Curli, ähnliche Strukturen gibt es in gram-positiven. Adhesine vermitteln über Ligandeninteraktion verschiedene Signale, die die Invasion oder Entzündung des Gewebes beeinflussen. Interaktion mit Bestandteilen des Serums kann die Komplementanlagerung verhindern. Typ1 Pili und P-Pili werden in Chaperon/Usher Pathways synthetisiert. Die Translation der Monomere geschieht im Cytoplasma, es folgt Sekretion ins Periplasma über SecA/YEG und Bindung an Chaperon, welches die Wechselwirkung mit dem Usher vermittelt. Die tatsächlichen Adhesine sind Fim1 und PapG. TypIV Pili entstehen cotranslationell, das ungefaltete Polypeptid wird in statu nascendi ins Periplasma transloziert, Pre-Pilin-Peptidase spaltet das fertige Protein von einer N-terminalen Sequenz. Die weitere Assemblierung ist noch unklar, es sind einige Proteine und eine Sekretionspore involviert. Das Adhesin ist PilC. Curli sind Amyloid-Fibrillen, eine spezielle Quartärstruktur, in der viele β-sheets im rechten Winkel zur Achse des Fimbriums angeordnet sind. Curli sind Bestandteil der Matrix während dem sessilen Wachstum und haben keine Ligandenspezifität. Das Monomer CsgA wird von CsgG transloziert, zusammen mit CsgB bilden sich die Fibrillen. - 12 - Trimeric Autotransporter Adhesine (TAAs) bilden sehr stabile Translokatortrimere in der Außenmembran gram-negativer Bakterien. Sie haben eine head-stalk-anchor Architektur und vermitteln Kontakt zur ECM oder induzieren die Invasion von Zielzellen. Im Gegensatz zu Pili gram-negativer Bakterien werden die Untereinheiten von gram-positiven sortase assembled pili nach Sekretion kovalent durch Sortase (Transpeptidase) verbunden. Diese Abbildung zeigt die Interaktionen von Adhesinen mit ihren Liganden und Folgen davon. - 13 - P-Pilus Adhesine vermitteln die Anhaftung an Epithelzellen des Urogenitaltrakts (Pyelonephritis Adhesion Pilus, PAP, von E. coli). Der P-Pilus ist ein flexibler Stab mit helicaler Struktur bestehend aus verschiedenen Untereinheiten, welche in einem genetischen Cluster codiert sind. Die Untereinheiten werden Sec‐abhängig ins Periplasma sezerniert und in der korrekten Reihenfolge eingebaut dank der unterschiedlichen Affinität der Chaperon‐Untereinheit-Komplexe zum Usher. PapA PapH PapC PapD PapJ PapK PapE PapF PapG Stab, viele PapA bilden zusammen den Pilus-Stab Stab-Anker Usher Chaperon, bindet PapA und bringt es zu PapC ? Adaptor Spitze, daran binden PapF und PapG Adaptor Adhäsin, mehrere Klassen PapG erkennt Galabiose (D-Galactose-α(1-4)-D-Galactosid)-hältige Isorezeptoren von Erythrocyten, Nieren- und Blasenepithelien. Diese Isorezeptoren sind Glycosphingolipide, Globoside (Ceramid), die das Disaccharid Galabiose enthalten. Klasse I PapG-Adhesin Klasse II PapG-Adhesin Klasse III PapG-Adhesin → erkennt nur Galabiose auf Isorezeptoren der Gruppe 6 → erkennt Galabiose auf Isorezeptoren der Gruppe 5 und 6 (humane Pyelonephritis) → Galabiose auf Isorezeptoren der Gruppen 5 und 2 (humane Blasenentzündung) 95% der Menschen präsentieren eine Galabiose auf den Erythrozyten sowie den Epithelzellen der Blase, man spricht von der Blutgruppe P. Die restlichen 5% sind P-negativ und können nicht an Blasenentzündung erkranken. Streptococcus befällt unterschiedliche Schichten der Epidermis. Ist viel O2 vorhanden, exprimiert das Bakterium Adhesin F und bindet an Langerhans'sche Zellen. Findet es eine hohe CO2-Menge vor, wird Adhesin M exprimiert und Keratinocyten infiziert. Das Flagellum vereint mehrere Funktionen in sich, es erhöht die Beweglichkeit und das Vorankommen in mucösem Schleim (Vibrio cholerae), es wirkt als Adhesin (Pseudomonas aerunginosa) oder ermöglicht als Invasin das Eindringen in Wirtszellen (Yersinia enterocolitica). Kommt es zur Beschädigung oder Verlust des Flagellums entstehen Flagellin-Monomere, die von TRL5 erkannt werden. TLR5 befindet sich auf Monocyten, neutrophilen Granulocyten und auf Epithelzellen. Adhesine sind Angriffspunkte antimikrobieller Therapien, die die Anheftung der Keime zu blockieren suchen. Bei einer normalen Infektion binden bakterielle Lectine an Glykoproteine des Wirts. Verabreicht man nun ähnliche Substanzen (Glykoside), werden die Lectine damit blockiert, eine Anhaftung ist inhibiert. Alternativ können auch die Glykoproteine mit freien Lectinen kompetitiv inhibiert werden. - 14 - 3 – Invasion, Verbreitung im Wirt Ein Mikroorganismus kann seine Aufnahme in eine nicht-phagocytische Zelle erzwingen. Die Invasion ist ein aktiver Prozess, der vom Bakterium ausgeht, es stimuliert seine Aufnahme in nichtphagocytische Zellen. Das Ziel ist Eintritt in eine geschützte Nische oder das Überqueren von zellulären Barrieren. Das Bakterium beherrscht die biochemische Sprache der Wirtszelle. „Bakterien sind die besten Zellbiologen.“ - Stanley Falkow Dazu tricksen Bakterien die Signaltransduktion der Wirte aus. Signaltransduktion ist der biochemische Prozess, durch den Organismen (1) ihre Umgebung wahrnehmen können, (2) auf ihre Umgebung reagieren und sich adaptieren können (z.B. durch Veränderung des Genexpressionsmusters). Signaltransduktion wird implementiert durch (1) reversible Modifikation von Botenproteinen (Bindung eines Liganden oder kovalente Modifikation wie Phosphorylierung und Methylierung), (2) Bildung von Signaltransduktionskomplexen, (3) Lokalisierung von Signaltransduktionskomplexen. Zipper Mechanismus Yersinia pseudotuberculosis und Listeria monocytogenes besitzen Transmembranproteine, die Liganden der Zelladhäsion nachahmen. Die Interaktion zwischen bakteriellem Ligand und zellulärem Rezeptor täuschen eine Zell-Matrix-Verankerung vor. Es kommt zur Bildung einer Membraneinstülpung, die das Bakterium umschließt, was mit einem Rearrangement des Cytoskeletts einhergeht. Der Zipper-Mechanismus gliedert sich in drei Teilschritte: 1. Kontakt zwischen Rezeptor und bakteriellem Ligand. Rezeptor-clustering. 2. Bildung eines phagocytischen Bechers. Aktinpolymerisation und Membranerwerweiterung. 3. Abschnürung des cytosolischen Vesikels mit Bakterium darin. Aktindepolymerisation. Es sind drei Proteine bekannt, die jeweils eine andere Signalkaskade auslösen. In Yersinia interagiert Invasin mit Integrin, welches an der cytoplasmatischen β1-Kette FAK (focal adhesion kinase) rekrutiert. Zusammen mit Src, Rac1 und N-WASP sowie Phosphoinositid-3-Kinase (PI 3-K) und Arf6 kommt es zur Polymerisation von Actinfilamenten bzw. Änderung der Zusammensetzung der Phospholipide in der Zellmembran. Internalin, eine LRR-Protein ( lysine rich repeat ) von Listeria bindet an humanes E-Cadherin, welches normalerweise in homophilem Kontakt mit E-Cadherin einer weiteren Zelle adherens junctions etabliert. E-Cadherin bindet an seiner cytoplasmatischen Domäne Catenine (α, β, p120), die eine Rac-abhängige Actinpolymerisation einleiten. Myosin VIIa und sein Ligand Vezatin scheinen eine Rolle bei der Dynamik des phagocytischen Bechers zu spielen. InlB, ein LRR-Oberflächenprotein von Listeria, das auch in gelöster Form vorliegen und inlB Mutanten durch Bindung an die Zellwand das Eindringen in Säugerzellen ermöglichen kann, bindet an Met, eine Transmembrantyrosinkinase, dessen normaler Ligand HGF (hepatocyte growth factor) ist. Dadurch kommt es in einer Phosphorylierungskaskade durch PI 3-K zur Bildung von PIP3 aus PIP2 (Phosphoinositolbiphosphat) an der Kontaktstelle. Aktinpolymerisation wird von Rac-WAVEArp2/3 erledigt. Im Zuge der Phagocytose kommt es zur Akkumulation von Cofilin an die Filamente, welches die Depolymerisation katalysiert, sodass sich die Membran abschnürt. GTPasen der Rho-Familie (Rac1, Cdc42, RhoA) treiben Reorganisation des Cytoskeletts voran und haben im inaktiven Zustand GDP gebunden. Durch GEF (Guanidine Exchange Factor) wird GDP gegen GTP ausgetauscht, es folgt GTP-Hydrolyse mittels GAP (GTPase activating protein). - 15 - Cdc42 (Aktinpolymerisation) → Filopodien Rac1 (Aktinpolymerisation) → Lamellipodien RhoA (Filament-Bündelung) → Stressfasern Die Aktivierung von Integrin, normalerweise durch Interaktion mit extrazellulärer Matrix, führt zur Aktivierung von FAK und Src, welche GEFs aktivieren. Aktivierung von Rac1 / RhoA / Cdc42 löst dann die entsprechenden Änderungen des Cytoskeletts aus. Das Invasin-Gen von Yersinia wurde mit einer E. coli Genbank gefunden (Stanley Faltrow). Dabei wurde eine E. coli Kultur mit Fragmenten des Yersiniagenoms transformiert und auf Säugerzellen inkubiert. Säugerzellen wurden gründlich gewaschen, um außen anhaftende Bakterienzellen entfernen, es folgte eine sanfte Lyse der Säugerzellen. Inhalt wurde ausplattiert, anwachsende Kolonien stammten von intrazellulären E. coli, welche folglich das Yersinia-Invasin-Gen bekommen haben, um in die Zelle eindringen zu können. Trigger Mechanismus Salmonella und Shigella stellen den Wirt-Parasit-Kontakt über ein TypIII-Sekretionssystem her (TTSS). Dadurch injizieren die Bakterien Proteine, bakterielle Effektoren, in eukaryotische Zellen, wo mit Komponenten des Cytoskeletts der Wirtszelle interagieren können. Danach formieren sich makropinozytische Ausstülpungen (Filopodien, Lamellipodien) und überwachsen das Bakterium. - 16 - Aktindepolymerisation schließt die makropinozytischen Ausstülpungen über dem Bakterium. Der Trigger-Mechanismus gliedert sich in vier Phasen: • • • • pre-initiation stage ◦ Bakterielle Effektoren befinden sich im Cytoplasma des Pathogens, Chaperone verhindern vorzeitige Komplexbildung ◦ TTSS baut sich zusammen ◦ Warten auf Kontakt mit Wirtszelle interaction stage Salmonella haftet über verschiedene Adhesine an M-Zellen und CD18-Phagocyten. TTSS injiziert das SipB/C Translocon in die Membran der Wirtzelle. Shigella bindet über IpaB an CD44 (Hyaluronsäurerezeptor) des Wirts an diesen. Das Translocon heißt IpaB/C. actin rearrangement stage Das Pathogen erwirkt über lokales, aber massives Aktinrearrangement die Bildung einer macropinocytischen Tasche. Salmonella und Shigella sind da sehr ähnlich, beide bilden Filound Lamellipodien. In Shigella depolymerisiert VirA die Mikrotubuli, wodurch RhoA deaktiviert und Rac1 in Folge aktiviert wird. Actin wird abgebaut. SipC (Salmonella) und IpaC (Shigella) induzieren die Aktinpolymerisation, der Mechanismus dafür ist unbekannt. Die Bildung der Eintrittsfoci unterscheidet sich in den beiden Organismen. In Salmonella haben SopE1/SopE2 GEF-Aktivität auf Cdc42, Rac1 und RhoA. SopB/SopD, Phosphatidylinositol-Phosphatasen, bewirken die Loslösung der Aktinfilamente von der Membran. SipA stabilisiert Aktinfilamente. In Shigella aktiviert IpaC Cdc42 und Rac1, gefolgt von c-src, einer Tyrosinkinase, was zur Rekrutierung von Cortactin und massiver Aktinpolymerisation durch Arp2/3 führt. IpgD, ein SopB/SopD Homolog, verstärkt den Prozess. pocket closing In Salmonella hat SptP zwei Funktionen, (1) Tyrosinphosphatase-Aktivität auf MAPK, (2) GAP-Aktivität auf Cdc42 und Rac1 und wirkt somit antagonistisch zu SopE. In Shigella rekrutiert IpaA Vinculin ( fördert die Bildung von cell adhesion plaques ) und stimuliert die Actindepolymerisation. Anhaftung und Injektion finden immer an lipid rafts statt, da dort Rezeptoren (CD44, CD18) dichter beisammen stehen, die Lipidzusammensetzung stimmt und Signalmoleküle wie zum Beispiel die Tyrosinkinase Src in größerer Zahl vorhanden sind. Salmonella trägt zwei pathogenicity islands (PAIs), die Gencluster SPI-1 und SPI-2 (Salmonella pathogenicity island). PAIs sind durch einen 40%-Gehalt an G-C gekennzeichnet, sehr wenig für bakterielle DNA. SPI-1 codiert Gene für Invasion in Epithelzellen und Apoptose von Makrophagen: • TypIII-Sekretionssystem (TTSS) • SipA → stabilisiert Aktinfilamente • SopE → GEF-Aktivität für Cdc42 und Rac1 • SopB → Phosphatidylinositol-Polyphosphatase, stimuliert Aktin-Rearrangement • SptP → GAP-Aktivität auf Cdc42 und Rac1, wirkt SopE entgegen, bewirkt AktinDepolymerisation SPI-2 codiert Gene für das Überleben in Makrophagen, um in Leber und Milz verbreitet zu werden. - 17 - Während der Bildung der makropinocytischen Einbuchtung sind SopE und SopB aktiv. SopE(-) SopB(-) - Bakterien könnnen Epithelzellen nicht mehr befallen. SipA und SopE kooperieren in der Neugestaltung des Aktin-Cytoskeletts. SopE bindet T-Plastin und bringt es zur Invasionsstelle. SipA stabilisiert F‐Aktin und stimuliert die T‐Plastin‐vermittelte Bildung von Aktinfaser‐Komplexen. Beim Trigger-Mechanismus setzen Pathogene auch eigene GTPasen ein, die der WxxxE-Familie. Dazu gehören die Effektorproteine IpgB1 und IpgB2 von Shigella, SifA und SifB von Salmonella und Map, EspM sowie EspT von EPEC und EHEC. Diese imitieren GTPasen der Rho-Familie. Map imitiert Cdc42 (Filopodien), IpgB1 imitiert Rac1 (Lamellipodien) und IpgB2 imitert RhoA - 18 - (Stressfasern). Alle sind funktionelle, aber nicht strukturelle Imitate. Sie besitzen targeting Sequenzen, ähnlich den eukaryotischen PDZ-Domänen, die die Proteine an der Membran verankern (PDZ is an acronym combining the first letters of three proteins — post synaptic density protein (PSD95), Drosophila disc large tumor suppressor (DlgA), and zonula occludens-1 protein (zo-1) — which were first discovered to share the domain). Die Assemblierung des TypeIII-Sekretionssystems (T3SS, TTSS) gliedert sich in eine secabhängige und eine sec-unabhängige Phase. Die Lokalisierung der Plasmamembran- und Transperiplasmatischen Komponenten erfolgt sec-abhängig (PrgH, PrgK). An PrgH lagern sich cytoplasmatische Komponenten (InvA, SpaP, SpaQ, SpaR, SpaS) sowie ATPasen (InvC) an. Durch den PrgK-Kanal gelangen InvG und InvH zur Außenmembran, das Periplasma ist nun überbrückt, die sec-Abhängigkeit ist nicht mehr gegeben. Weitere Komponenten (IagB, PrgI, PrgJ, InvJ) vervollständigen die Struktur. Um vorzeitiges Durchschleusen von Effektoren zu verhindern, ist die Spitze und der cytoplasmatische Eingang des Kanals des TTSS in Yersinia mit YscP blockiert. Bei Kontakt wird zuerst YscP sezerniert, danach, das TTSS durchbohrt die Membran der Zielzelle, folgt die Sezernierung der Effektoren durch InvC. Das Prinzip findet sich auch beim Hook eines Flagellums. Dabei wird über FliK die Filamentassemblierung zeitlich reguliert. Regulation von Effektoren am Beispiel der Yop-Sezernierung über den Ysc/Lcr-Kanal in Yersinia. Im Standbymodus blockiert YopN(LcrE) zusammen mit LcrG den Ysc/Lcr-Kanal. Eine hohe cytoplasmatische Konzentration an LcrQ(YscM) und LcrH(SycD) reprimieren das Yop-Operon. Bei Kontakt mit der Wirtszelle, gibt YopN den Kanal frei, LcrQ diffundiert aus der Zelle, das YopOperon wird exprimiert und Yop sezerniert. Pathogene müssen sich in Geweben, in denen die Nährstoffkonzentration oft limitierend ist, vermehren können. Dazu synthetisieren sie spezifische Transportsysteme und/oder Rezeptoren. Beispiel Eisen: Essentiell für das Wachstum eukaryotischer Zellen und Bakterien. Siderophore werden sowohl vom Wirt als auch vom Bakterium produziert (Pyochelin, Yersiniabactin, Vibriobactin). Manche Bakterien können auch menschliche Siderophore (Hämoglobin, Transferrin, - 19 - Lactoferrin) oder Siderophore, welche von der Normalflora (Enterobactin, Anguibactin, Acinetobactin) produziert werden, binden und zum Eisentransport verwenden. 4 – Exo- und Endotoxine Toxine sind biologische Waffen, die dem Wirt Schaden zufügen. Man gliedert sie in: Exotoxine – Das sind extrazelluläre Proteine mit sehr hoher biologischer Aktivität, sie sind durch Hitze oder Säure denaturierbar, denaturierte Toxine werden als Impfstoff (Toxoide) eingesetz. Die Spezifität und Toxizität ist hoch. • • • Membranschädigende Toxine: Lipasen und porenbildende Toxine (α-Toxin, SLO) AB Toxine: bindungsspezifische und katalytisch aktive Domäne ◦ Proteinsynthese-hemmende Toxine (Diphteria Toxin) ◦ Pharmakologisch aktive Toxine (Cholera Toxin) ◦ Neurotoxine (Tetanospasmin, Botulinus Toxin) Superantigene Exotoxine sind auch ohne Bakterien schädlich: Wenn Bakterien, die Exotoxine produziert haben, in Nahrungsmitteln vorkommen, können die Exotoxine alleine dem Wirt schaden, selbst wenn die Bakterien den Verdauungstrakt ohne Ansiedelung verlassen. Exotoxine interferieren mit der Signaltransduktion, Verändern die Permeabilität der Membran und die Struktur des Cytoskeletts (Zonula adherens disassembly) sowie den intrazellulären Transport. Endotoxine – sind essentielle Bestandteile (LPS) der Bakterien selbst, diese erlangen bei Lyse toxische Wirkung. Sie sind nicht denaturierbar (Lipide), dafür haben sie geringe Spezifität und Toxizität. Membranschädigende Toxine – S. aureus α-Toxin Monomer (34 kDa) bindet an die Membran empfindlicher Zellen (Thrombocyten, Monocyten) durch einen spezifischen, aber noch nicht identifizierten, α-Toxin-Rezeptor. Die Untereinheiten bilden dann einen homoheptameren Ring mit einer zentralen Pore, durch die der Inhalt der Zelle nach außen gelangt. Dies verursacht wiederum eine entzündliche Reaktion im Gewebe. Membranschädigende Toxine – C. perfringens α-Toxin PDB-Code: 2AHL Spaltet als Phospholipase Phosphatidylcholin (Lecithin), welches sich in der Membran aller tierischer Zellen befindet. Die Ladung und Stabilität der Membran geht verloren. Bindet und zerstört in vivo Leukocyten. PDB-Code: 1CA1 - 20 - AB-Toxine haben mindestens zwei verschiedene Domänen, eine Untereinheit A hat katalytische Aktivität und ist Effektor des Toxins, eine Untereinheit B sorgt für spezifische Bindung an Zellen. Man unterscheidet zwischen einfachen AB-Toxinen, bestehend aus je einer A- und B-Untereinheit, und komplexen AB-Toxinen mit mehreren, möglicherweise unterschiedlichen B-Untereinheiten. So können mehrere Zelltypen affektiert werden! Aufnahme von AB-Toxinen wird über Rezeptor-vermittelte Endocytose bewerkstelligt. Die katalytische Untereinheit A ist in Endosom nicht wirksam – A wird ins Cytoplasma ausgeschleust und bindet dort an das Substrat. Selten wird A ohne Endocytose direkt in die Zelle eingeschleust und B gar nicht aufgenommen. B-Untereinheiten können als Impfstoff verwendet werden – Produktion von Anti-B Antikörpern. Neurotoxine sind Zn-Proteasen, ihre Substrate sind Sekretionsproteine in Synapsen. Neurotoxine werden als inaktives Zymogen (Proenzym, inaktive Enzymvorstufe) während der Sporen-Keimung synthetisiert, von bakteriellen oder Gewebsproteasen gespalten und danach endocytotisch aufgenommen. Neurotoxine sind aus einer schweren oder mehreren schweren Ketten B, zur Bindung an den neurospezifischen Rezeptor, und einer leichten Kette A, die katalytische Funktion hat, aufgebaut. Die Komponenten sind über Disulfid-Brücken zusammengehalten und werden durch Proteasen gespalten. Zwei schwere Ketten B binden an den neurospezifischen Rezeptor, es folgt Aufnahme in ein Endosom. Hier erfolgt Konformationsänderung durch sauren pH der schweren Kette (bildet Pore), die leichte Kette A wird aus dem Endosom in Cytosol translociert. Vertreter der Gattung Clostridium produzieren bei der Germination der Sporen starke Neurotoxine (LD50 0,1-1 ng/kg). Clostridien sind gram-positive, sporenbildende, obligat anaerobe Bakterien, zB. Clostridium tetani, Clostridium botulinum. Die Lokalisierung folgt dem vierstufigen Schema der Neurotoxine: 1. Neurospezifische Bindung an lipid rafts, reich an Cholesterol, Sphingolipid und Polysialogangliosid 2. Internalisierung und sorting (Bewegung der Vesikel über Mikrotubuli) 3. Translokation der leichten Kette mittels der schweren Kette(n) 4. Entfaltung der katalytischen Aktivität Clostridium-Toxine wirken proteolytisch auf bestimmte Membranproteine der sekretorischen Vesikel, welche zur Bindung an die synaptische Membran notwendig sind. Daher findet keine Ausschüttung von Neurotransmittern mehr statt. VAMP, Syntaxin und snap25 gehören zur Gruppe der SNARE-Proteine und bewerkstelligen die Fusion der Vesikel mit der Zellmembran. VAMP/Synaptobrevin Membranprotein acetylcholinhältiger Vesikel, stellt Kontakt zu Syntaxin und snap25 her Substrat für TeNT, BoNT serotyp B, D, F und G Syntaxin1 Partnermolekül in der Zellmembran der präsynaptischen Zelle BoNT serotyp C snap25 Adapterprotein Substrat für BoNT serotyp A und E - 21 - AB-Neurotoxine – Botulinus Toxin (BoNT) BoNT bleibt nach Internalisierung in der neuromuskulären Synapse hochaktiv, bereits ein Molekül kann alle Substrate in der Synapse spalten (reversibel)! Die leichte Kette inhibiert die Ausschüttung von Acetylcholin aus den synaptischen Vesikeln der Motorneuronen an der neuromuskulären Junction → Schlaffe Lähmung (Atemlähmung). PDB-Code 1F31 AB-Neurotoxine – Tetanus Toxin (TeNT, Tetanospasmin) TeNT wird nach der Internalisierung rückwärts im Axon des Motorneurons zu einem inhibitorischen Interneuron transportiert, wo es zur Transcytose kommt, also Freisetzung auf der einen und Aufnahme auf der anderen Seite des synaptischen Spalts. Die leichte Kette inhibiert die Ausschüttung von inhibitorischen Neurotransmittern aus den synaptischen Vesikeln der inhibierenden Interneurone → keine Inhibierung der Motorneurone → Verkrampfung. PDB-Code 1A8D Proteinbiosynthese-hemmende Toxine – Diphteria-Toxin Diphteriatoxin ist ein Vertreter von AB-Toxinen, das mit GProteinen wechselwirkt. Es entsteht in Corynebacterium diptheriae, welches das verantwortliche Gen, tox+, aber nicht von vornherein hat. Der Phage β bringt die genetische Information ein, das bakterielle dtxR reguliert tox+. Die Bindung erfolgt an einen HB-EGF (heparin-binding epidermal growth factor) ähnlichen Rezeptor. Der katalytische Teil ist ein Enzym, das ein ADP-Ribosyl auf Diphthamid, ein modifizierter Histidinrest des humanen Elongationsfaktors EF-2, von NAD überträgt, Nikotinamid bleibt über. Dadurch ist der Elongationsfaktor inhibiert, es kann keine RNA-Synthese mehr stattfinden und letztendlich stirbt die Zelle. Das Diphtherietoxin ist dabei so potent, dass PDB-Code 1DDT ein Molekül ausreicht, um eine Zelle zu töten. Es entstehen pro Stunde etwa 5000 Toxinmoleküle in einem einzigen Bakterium. EF2 (Elongationsfaktor 2), ein G-Protein, spaltet GTP in GDP, P i und Energie, die für Translokation der Peptidkette von der A-site zur P-site des Ribosoms genutzt wird. AB-Pharmakologisch aktive Toxine – Cholera-Toxin (CTX) Das Cholera-Toxin, produziert von Vibrio cholerae, ist ein 84 kDa Hexamer aus einer A- und 5 BUntereinheiten. Das Bakterium haftet an GM1, ein Gangliosid von Darmepithelzellen des Dünndarms, und konvertiert mit einer Neuraminidase weitere Ganglioside in GM1. - 22 - Die Untereinheit A wird in A1 und A2 gespalten. A1 wird von ARF zur basolateralen Membran gebracht, wo es ein stimulatorisches G-Protein ADP-ribosyliert, was zur Aktivierung der Adenylatcyclase führt. Damit geht die Erhöhung des cAMPSpiegels einher. PKA wird aktiviert, die Cl–-Sekretion hoch- und Na+-Resorption in Krypten herunterreguliert, die Na+/Cl– Resorption in Villuszellen herunterreguliert. Als osmotische Folge treten Unmengen an Wasser in den Dünndarm. Nur 15µg Choleratoxin reichen für 24 Liter Diarrhoe. Choleratoxin stört also den normalen Ionenhaushalt. PDB-Code 1XTC Die Produktion von Cholera‐Toxin hängt von der Infektion des Bakteriums mit zwei verschiedenen Phagen in einer bestimmten Reihenfolge ab. 1. 2. 3. 4. Infektion durch VPIΦ (vibrio pathenicity island phage), Akquirierung von tcpPH und toxT Ausbildung des toxin coregulated pilus (TCP), dem Rezeptor für den zweiten Phagen Infektion durch CTXΦ (cholera toxin phage) Insertion der CTX-Virulenzkassette ins bakterielle Genom und Toxinexpression CTXΦ bringt eine „Giftkassette“ mit, die neben dem eigentlichen Choleratoxin ctxA/B, noch andere Endotoxine (ace/zot) und Kolonisierungsfaktoren (cep – core encoding pilus) beinhaltet. Die ORFs von ctxA und ctxB überlappen, ctxB wird aber effizienter translatiert, so kommt es zum 5:1 Verhältnis im Holoenzym. AB-Pharmakologisch aktive Toxine – Pertussis-Toxin (PTX) Bordetella pertussis, ein gram-negativer Coccobacillus, verfolgt zwei andere Strategien, um den cAMP-Spiegel zu modulieren. Einerseits schwächt es die Wirkung inhibitorischer G-Proteine des Wirts (im Gegensatz zu Vibrio, welches die Wirkung von stimulierenden G-Proteinen fördert), andererseits bringt es eine eigene Adenylatcyclase (CyaA) ein, die kein AB-Toxin ist. PTX ist ein Hexamer aus 5 Untereinheiten, davon haben zwei Affinität zu Epithelzellen sowie Phagocyten, deren Wachstum durch Erhöhung des cAMP-Spiegels mittels Hemmung inhibitorischer G-Proteine gehemmt wird, die eukaryotische Adenylatcyclase bleibt als konstitutiv aktiv. Dann wird CyaA als inaktives Zymogen synthetisiert, welches durch Palmitoylierung aktiviert wird. CyaA besteht aus: 1. Katalytische Adenlylatcyclase-Domäne mit Calmodulin binding site 2. Hydrophobem Teil (Transmembran-Domänen) 3. Palmitoylierungsstelle zur Verankerung in die Membran 4. Ca2+ bindender Domäne 5 .Sekretorisches Signalpeptid - 23 - Der Rezeptor für CyaA ist β2-Integrin, ein Adhäsionsmolekül der Leukocyten, CyaA ist hauptsächlich gegen Makrophagen gerichtet. Ca2+-Bindung verursacht Strukturveränderungen, wodurch die AC-Domaine durch die Membran in die Zelle eingeschleust wird. Dort bindet es Calmodulin, welches die katalytische Aktivität aktiviert, sodass nur in der Zelle cAMP produziert wird. PDB-Code 1YRU Warum greifen Toxine in den cAMP-Haushalt ein? Ein erhöhter cAMP-Spiegel ist ein ApoptoseSignal, Zellen lysieren. Im Bakterium wird das Wachstum durch cAMP reguliert, sie gedeihen also in einer cAMP-reichen Umgebung besser. Superantigene Die bisherigen toxischen Wirkungen gehen direkt von Bakterien aus, Superantigene stammen zwar auch von Bakterien, hier bewirkt aber die Handhabung durch das Immunsystem den Schaden. Normalerweise nehmen APC Antigene auf, prozessieren sie und präsentieren sie an TH-Zellen. Diese tasten das in der MHCII-Furche liegende Proteinfragment mit α-und β-Einheit des TCR ab und entscheiden ob fremd oder nicht. Superantigene binden an der Außenseite des MHCII-Komplexes. TH-Zellen werden bei Interaktion zur IL-2 Ausschüttung angeregt, egal ob ein fremdes Antigen oder überhaupt ein Antigen gebunden war. Dadurch können im Prinzip all jene TH-Zellen aktiviert werden, die eine passende βvar-Region am TCR besitzen (etwa 20% aller T-Helfer-Zellen), und nicht nur die wenigen, die sonst für ein konventionelles Antigen spezifisch wären. Die Folgen ist ein Überschießen der Immunantwort in Form von: • • • Aktivierung zu vieler TH-Zellen Zu starke Proliferation Toxischer Schock: Fieber, Übelkeit, Erbrechen, Schock Superantigene können Autoimmunität und Immundepression hervorrufen. APC präsentieren im Normalfall auch manchmal Selbstantigene, TH-Zellen sprechen darauf dann aber nicht an. Ein Superantigen stellt jetzt aber eine stabile Verbindung zwischen MHC und TCR her, ob der TCR zum Antigen spezifisch ist, wird vernachlässigt. Die übermäßige Beanpruchung der T-Zellen führt nach einiger Zeit zu deren Tod, so stehen sie nicht mehr zur Bekämpfung späterer Infektionen zur Verfügung (die Immunkompetenz, Diversität an spezfischen TCRs sinkt), man spricht von Immundepression. Dieser Effekt kann aber Autoimmunität nach einiger Zeit ausgleichen. Man unterscheidet zwischen exogenen und endogenen Superantigenen. Die meisten Super-Antigene sind Exotoxine, manche werden aber von Retroviren codiert und werden von APC selbst exprimiert, man zählt diese dann zu den endogenen Superantigenen. Beispiele für Superantigene sind Enterotoxine von Staphylococcus (SEA, SEB, SEC1, SEC2, SEC3, SED, SEE), die im Darm wirksam sind und die Ursache von manchen Lebensmittelvergiftungen sind, toxic-shock-syndrome Toxin (TSS1), exfoliative-dermatitis Toxin (ExFT) oder pyrogene Exotoxine von Streptococcus (SPE-A, SPE-B, SPE-C, SPE-D → rheumatisches Fieber). - 24 - Endotoxine – LPS LPS befindet sich auf der Außenmembran von gram-negativen Bakterien, tragen zur strukturellen Integrität der Bakterien bei und schützen die Membran vor bestimmten Substanzen. LPS wird während des bakteriellen Wachstums und nach dem Tod des Bakteriums freigesetzt. Der toxische Teil des Moleküls wird durch das Lipid A dargestellt. Lipopolysacchariden sind komplexe, amphiphile Moleküle. Ihre chemische Komposition ist hochgradig variabel und kann in Lipid A, inner core, outer core und O-specific chain gegliedert werden (O-specific chain ist die Basis für die serologische Klassifizierung von Bakterienstämmen). Endotoxine – Lipoteichonsäure, Glykopeptide Die Zellwand von gram-positiven Bakterien enthält Glykopeptide sowie Teichonsäure. Diese Stoffe werden sowohl im Zuge des bakteriellen Wachstums als auch nach dem Tod der Bakteriums freigesetzt und wirken analog zum LPS. Die Eigenschaften von Exotoxinen und Endotoxinen im Vergleich: Eigenschaft Endotoxin Exotoxin Chemische Natur Lipopolysaccharid (10kDa) Protein (50-1000kDa) Lokalisierung Außenmembran Extrazellulär, diffundierend Denaturierung durch Kochen Nein Meistens Antigen Ja Ja Toxoid Nein Ja Effektivität Gering (>100µg) Extrem (1µg) Spezifität Gering Hoch Enzymatische Aktivität Nein Meistens Pyrogenität Ja Sporadisch Reaktion des Immunsystems auf Endotoxine Gram‐ und Gram+ Endotoxine binden an Rezeptoren auf der Oberfläche immunkompetenter Zellen. Dadurch können immunkompetente Zellen die Gefahr einer Infektion wahrnehmen. Toll‐likeRezeptoren (TLR) sind hochkonservierte Sensoren für die Bestandteile pathogener Mikroorganismen Die Bindung von Endotoxin an Monocyten und Makrophagen stimuliert die Produktion von Cytokinen wie TNF‐α, IL‐1, IL‐6 und IL‐8. Diese Stoffe regen wiederum Zielzellen an, deren normale Funktionen die Verteidigung des Wirts und die Zerstörung des Mikroorganismus sind (Entzündungsreaktion, Fieber, Immunantwort und Phagozytose). Die massive Freisetzung von Endotoxin kann wiederum ein Überschießen dieser Antwort verursachen, mit schweren Folgen für den Wirt (ARDS Acute respiratory disease syndrom, DIC disseminated intravascular clotting, MOSF multiple organ system failure). Die Bindung von Endotoxin an Rezeptoren und von IL‐8 an den IL‐8 Rezeptor auf der Oberfläche von Neutrophilen stimuliert die Freisetzung von Proteasen und toxischen Sauerstoff‐Radikalen. Diese Reaktion zerstört Bakterien, die sich in der Nähe befinden, sind aber auch für eukaryotische Zellen toxisch und können zu einer massiven Schädigung des umliegenden Gewebes führen. - 25 - TNF‐α und IL‐1 bewirken die Expression von Selektinen auf Endothelzellen und Spaltenbildung zwischen den Endothelzellen. Durch diese Spalten kann Diapedese stattfinden, dabei migrieren Immunzellen durch das Gewebe. Überdosierte Immunantworten führen zu septischem Schock (hohe Endotoxindosen führen zur Überproduktion von IL-1 und TNF- α durch Makrophagen) und toxischem Schock (geringe Dosen an Superantigen-Exotoxinen führen zur unspezifischen Aktivierung von 5 - 25% aller T H-Zellen, die resultierende Überproduktion von IL-2 führt zur Überstimulation von Makrophagen, die in Folge zuviel IL-1 und TNF-α produzieren). 5 – Immunantwort des Wirts Das Immunsystem hat drei Verteidigungslinien gegen Pathogene. • • • Epithelbarrieren: Haut und Schleimhäute, für rasche Entfernung von konterminiertem Mucus sorgen Tränenfluss, Urinfluss, Husten, Erbrechen, peristaltische Bewegung und Cilienschlag. Angeborenes Immunsystem: Immer in gleicher Intensität vorhanden, schnell, wenig spezifisch. Lysozyme, Magensäure, commensale Bakterien, Komplementprotein, Phagocytose. Adaptives Immunsystem: Langsamere Reaktion, aber spezifischer und zielgerichteter. Antikörper, Cytokine, Helfer-T-Zellen, cytotoxische T-Zellen, B-Zellen Komponenten und Zellen, die an einer inflammatorischen Reaktion beteiligt sind. • • • • • • • Makrophagen phagocytieren und töten Pathogene in peripheren Organen, produzieren inflammatorische Cytokine und sind APC, haben daher MHCII-Komplexe Neutrophile Granulocyten werden zu Entzündungsherden gelockt, phagocytieren und zerstören Mikroben Das Komplementsystem erhöht die inflammatorische Reaktion und bewirkt vermehrte Phagocytose und Lyse von Zellen Inflammatory Agents wie Bradykinin, Histamin und Prostaglandine wirken auf das Blutgefäßsystem und erhöhen Blutfluss und Durchlässigkeit Fibrin, aus seratösem Proenzym Fibrinogen hergestellt, sorgt für Gerinnung und lokale Begrenzung der Mikroorganismen Antikörper neutralisieren mikrobielle Toxine oder Viren, opsonisieren (bedecken) Pathogene und markieren sie somit zur Phagocytose Pyrogene, auch endogene Pyrogene wie IL-1, verursachen Fieber und wirken u.A. auf die Temperaturregulierung des Körpers (Hypothalamus) ein Der Effekt einer inflammatorischen Reaktion besteht darin, verschiedene Zellen und Komponenten zum Ort einer mikrobiellen Invasion zu bringen, welche direkte und indirekte Möglichkeiten bereitstellen, eine wirkungsvolle Verteidigung gegen eindringende Mikroorganismen zu etablieren. Neutrophile Granulocyten umfließen Fremdstoffe und zerstören sie mittels ROS (reaktive oxygen species) und anderen Radikalen im Phagolysosom. Makrophagen und Lymphocyten initiieren eine spezifische Immunantwort. Existierende Antikörper neutralisieren und opsonisieren mikrobielle Pathogene und/oder deren Toxine. Plasmakomponenten wie Lysozyme, Komplementproteine und Fibrin haben weitere antimikrobielle Eigenschaften. Das Komplementsystem macht 15% der Globinfraktion des Serums aus. Es besteht aus Proteinen - 26 - und Glykoproteinen, die als inaktive Zymogene von der Leber sezeniert werden und in inaktiver Form im Serum zirkulieren. Die Funktionen des Komplements sind: • • • • • • Opsonisierung (Markierung) von Antigenen (C3b) Anlocken von Phagocyten (C3a, C4a, C5a) Antigen-Antikörper-Komplexen (Immunkomplexe) entfernen (C3b, C4b) Aktivierung der inflammatorischen Reaktion: Vasodilatation, Extravasation von Lymphocyten (C3a, C4a, C5a) Aktivierung naiver B-Lymphocyten: Degranulation (C3a, C4a, C5a) Lyse von Zielzellen durch Porenbildung in der Membran von Bakterien oder Wirtszellen mit viralen Epitopen (MAC) Die Aktivierung des Komplements findet innerhalb einer regulierten enzymatischen Kaskade statt. Die Zymogene werden durch Spaltung einer inhibitorischen Domäne aktiviert. Das Zymogen eines Schritts wird zum Katalysator des nächsten. Die aktivierten Komponenten werden nach kurzer Zeit inaktiviert. Mindestens 3 Wege führen zur Komplementaktivierung, deren gemeinsames Ziel die Aktivierung der C5-Konvertase und des MAC (membrane attacking complex), der 70-100 Å große Poren bildet, sind. Serum C3 enthält eine labile Thioesterbindung, welche durch die Spaltung von C3a aktiviert wird. Dadurch kann C3b freie Hydroxyl‐ oder Aminogruppen in Zellmembranen binden. C3b wird von Sialinsäure, enthalten als Ligand in der Membran von Säugerzellen, inaktiviert. Die Zellwand von Bakterien und Hefe, sowie die Hülle mancher Viren, enthalten sehr wenig bis keine Sialinsäure. C3b und C4b werden als Opsonine, C3a, C4a und C5a als Anaphylatoxine bezeichnet. - 27 - Im klassischen Weg der Komplementaktivierung bindet C1 (Komplex aus C1r, C1q und C1s) an Antikörper-Antigen-Komplexe auf der Zelloberfläche eines Mikroorganismus. Daraufhin binden C4b und C2a und bilden zusammen die C3 Convertase. Die C3 Convertase spaltet C3 in C3a und C3b. C3b bindet nun an die antigenbedeckte Oberfläche des Mikroorganismus und opsonisiert sie. C3b bindet an die C3-Convertase und bildet so die C5-Convertase, die C5 zu C5a und C5b spaltet und aktiviert so die späten Schritte (C6-9, MAC). Komplementaktivierung nach dem klassischen Weg findet nur statt, wenn Antikörper an Antigen gebunden sind, da C1 sonst nicht an Fc binden kann. Frei gelöstes, ungebundenes IgM oder IgG haben kein für C1 zugängliches Fc. Vorraussetzung für den alternativen Weg der Komplementaktivierung ist die spontane Spaltung von C3 in C3a und C3b, was normalerweise im Plasma kontinuierlich mit niedriger Rate passiert. Wenn das Spaltprodukt C3b aber nicht unmittelbar an eine Mikrobe bindet, wird es inaktiviert. C3b bindet an die Mikroorganismus-Oberfläche, Faktor B bindet C3b, Faktor D spaltet B zu Bb. Bb und C3b bilden zusammen die C3-Convertase des alternativen Wegs, was zu verstärkter Spaltung von C3 zu C3a und C3b durch C3-Konvertase führt. C3b opsonisiert die Oberfläche des Mikroorganismus. C3 bindet an die C3-Convertase (C3bBb) und bildet so die C5-Convertase. Die Spaltung von C5 in C5a und C5b leitet weitere Schritte ein. Der alternative Weg ist phyolgenetisch älter, wurde aber später entdeckt. Der Lektinweg der Komplementaktivierung wird durch mikrobielle Polysaccharide ausgelöst, mannose-binding lectin (MBL) bindet Polysaccharide und aktiviert das Komplementsystem. MBL ist C1q strukturell ähnlich, aktiviert C1r/s oder MBP-associated serine esterase – beide spalten C4. Effektorfunktionen der Komplementaktivierung: • • • Entzündung wird eingeleitet durch Spaltprodukte C5a, C3a, C4a Aktivierung der späten Schritte des Komplementsystems durch C5 Convertase: Die C5Convertase spaltet C5 zu C5a und C5b. C5b bindet an C6-C7. C7 verankert den Komplex in der Membran. C8 kommt hinzu, lagert sich auch in die Membran ein. An diesen Komplex wird nun aus vielen C9 ein Kanal gebildet und eingelagert. Der gesamte Komplex bildet den membrane attacking complex (MAC). Durch den Kanal diffundieren Wasser und Ionen in die Zelle, die osmotische Anschwellung führt zum Platzen der Zelle. Zusätzlich treten Ca2+Ionen ein, eine hohe Ca2+-Konzentration führt zur Apoptose. Phagozytose der mit C3b opsonisierten Antigene (CR1- und Fc-Rezeptor vermittelt) - 28 - Komplementrezeptoren Immunstrategien bei verschiedenen Arten von Infektionen, Rolle des Komplementsystems Invasor Immunstrategie Mechanismus Beispiele VERMEHRUNG IN GEWEBSZELLEN Eintritt verhindern IgM, IgA, IgG Viren, Rickettsia Infizierte Zelle töten TC, NK, ADCC VERMEHRUNG IN PHAGOCYTEN Phagocyten aktivieren Phagocyten töten Lymphokine TC, NK, ADCC VERMEHRUNG EXTRAZELLULÄR Mikrobe töten C'-vermittelte Lyse Opsonisieren, IgG, IgM, C' Phagocytieren und Lyse Toxine neutralisieren IgG, IgM Viele Bakterien VERMEHRUNG EXTRAZELLULÄR, BINDUNG AN OBERFLÄCHEN ERFORDERLICH Anheftung verhindern Streptococci, E. coli, Neisseria IgA Viren, Mycobacterium tuberculosis Neutralisation von Fremdmaterial durch Neutrophile Granulocyten Neutrophile bilden chemotaktische Pseudopodien, mit denen Fremdantigene erstastet werden. Bei Kontakt wird der Fremdstoff (Immunkomplex, Mikroorganismus, Toxin) entweder in ein Phagosom internalisiert, welches mit einem Lysosom verschmilzt, um ein Phagolysosom zu bilden, oder es kommt zu extrazellulären Effektormechanismen (typischerweise bei viralen Epitopen auf Zellen oder bei Infektion von Zellen mit intrazellulären Mikroben). Ein Lysosom stellt eine sauerstoffabhängige Tötungsmaschinerie in Form von aggressiven Radikalen bereit: ROIs (reactive oxygen intermediates) wie O2– (Peroxidanion), HO• (Hydroxylradikal), H2O2 (Wasserstoffperoxid) und ClO– (Hypochloritanion), RNIs (reactive nitrogen intermediates) wie NO (Stickstoffmonoxid), NO2 (Stickstoffdioxid) und HNO2 (Nitriersäure) und andere wie NH2Cl - 29 - (Monochloramin). Degradiertes Material wird exocytotisch ausgeschieden sowie Teile davon auf MHCII präsentiert. Sezernierte Faktoren sind Defensine, TNF-α (nur bei Macrophagen) und hydrolytische Enzyme, die Fremdkörpern, aber auch dem Gewebe, stark zusetzen. 6 – Reaktion der Pathogene auf die Immunabwehr Mikrobielle Abwehr der Komplementeffektoren Viele Mikroorganismen besitzen Kapseln, bestehend aus Proteinen, Polysacchariden und/oder Glykoproteinen sowie Sialinsäure (Neisseria meningitidis) und Hyaluronsäure, die zur Inhibierung der C3-Konvertase beitragen. Dazu rekrutieren sie eukaryotische Komplementregulatoren: • • Faktor H inhibiert die Bindung von Faktor B an C3b Faktor I spaltet membrangebundenes C3b LPS kann mit Sialinsäure modifiziert (Bordetella, Haemophilus) oder sehr lang sein (E.coli, Salmonella), beides erschwert oder verhindert die Insertion des MAC in die Membran. Bakterielle Membranproteine können direkt mit dem MAC interagieren und dessen Membranintegration verhindern (Neisseria gonorrhoe, E.coli). Mikrobielle Elastase baut Bestandteile des Komplementsystems (C3a und C5a) ab (Pseudomonas aeruginosa). Viren, Pilze, Bakterien und Protozoen können Proteine sezernieren, die eukaryotische Komplementregulatoren nachahmen. Das M-Protein von Streptococcus hat eine konservierte Domäne, die Faktor H bindet, der Faktor B kompetitiv inhibiert, und als Folge Faktor I, der C3b in inaktives iC3b spaltet, rekrutieren kann. Die Komplementaktivierung bleibt aus, der räumliche Einfluss des Faktors H an der konservierten Domäne verhindert die Bindung von vorhandenen Antikörpern gegen die variable Domäne. Daneben ist der N-Terminus negativ geladen, was eine elektrische Abstoßung der Phagocyten bewirkt. Bakterielle Interaktion mit Phagocyten Kapseln verhindern Phagocytose, da membrangebundenes C3b verdeckt wird und der CR1-Rezeptor dieses nicht erkennen kann (Streptococcus pneumoniae). Streptococcus pyogenes hat außerdem das M-Protein und Hyaluronsäure im Peptidoglykan, wodurch eine chemotaktische Repulsion für Phagocyten etabliert wird. Streptolysin (Streptococcus pyogenes) führt zur Entladung der Lysomen ins Cytoplasma – Phagocyten gehen zu Grunde. - 30 - Leukocidin (Staphylococcus aureus) hat den gleichen Effekt wie Streptolysin. Protein A , ein mikrobieller Fc-Rezeptor, bindet in Staphylococcus aureus Immunglobuline an deren Fc-Domäne, und zwar so, dass spezifische sowie unspezifische Antikörper nutzlos werden. Dadurch wird außerdem der Zugang von weiteren Antikörpern, die möglicherweise Epitope erkennen, sterisch versperrt. Mehrere Bakterien haben Immunglobin-bindende Proteine und umgehen somit die Phagocytose. Immunglobulin Spezies Protein IgG1, 2, 4 (IgM, A, E) Staphylococcus aureus Protein A IgG1, 2, 3, 4 Gruppe C und G Streptococcen Protein G IgG1, 2, 3, 4 Gruppe A Streptococcen Verschiedene, FcRA76, Protein H IgA1, 2 Gruppe A Streptococcen Protein Arp IgA1, 2 Gruppe B Streptococcen (β Antigen) Protein Bac IgD Haemophilus influenzae Protein D κ Peptococcus magnus Protein L Yersinia klinkt sich in die Signaltransduktion des Wirts ein und kommt durch den Zipper-Mechanismus in M-Zellen (microfold cells) des Dünndarms. M-Zellen fangen Antigene aus dem Lumen des Dünndarms, um sie via Transcytose an Antigen-präsentierende Zellen auf der anderen Seite weiterzuleiten. Yersinia nützt ebenfalls den Vorgang der Transcytose, bewirkt via Zipper-Mechanismus Aufnahme in M-Zellen und das Durchschleusen durch die Epithelbarriere. Auf der anderen Seite trifft das Bakterium auf Makrophagen, in die Yersinia Faktoren zur AktinDepolymerisation injiziert. Der Makrophage hat keine Kontrolle mehr über sein Cytoskelett und kann das Bakterium nicht umfließen. Die Infektion der Makrophagen führt letzten Endes zur Apoptose. • YopE → GAP für Rac1, Cdc42 und RhoA. Unwirksammachung der GTPasen. • YopH → Phosphatase, die Cas-pY inhibiert. GEFs für Rac1, Cdc42 und RhoA bleiben inaktiv. • YopT → Wirkt direkt auf RhoA, keine Stressfasern. Salmonella typhimurium, Shigella flexneri und Legionella pneumophila entkommen der Phagocytose indirekt, da sie sich aus Phagocyten wieder befreien, indem sie den Phagocyten töten. Diese Pathogene induzieren Caspase-1 Aktivierung, IL1β und IL-18 Ausschüttung sowie raschen Zelltod. Verschiedene bakterielle Faktoren wie monomeres Flagellin führen zur Oligomerisierung von inaktivem IPAF zum IPAF-Inflammosom, welches Caspase-1 aktiviert. - 31 - Listeria monocytogenes entkommt aus dem Phagosom bevor dieses mit Lysosom fusioniert. Listeria lysiert die Phagosom-Membran mithilfe von Phospholipase C, bevor das Phagosom mit dem Lysosom fusioniert und gelangt mit Hilfe von actA in die nächste Zelle. In aktivierten Makrophagen fusioniert das Lysosom mit dem Phagosom so schnell, dass das Bakterium schon getötet wird, bevor es ausbrechen kann. Die Migration von einer Zelle zur nächsten geschieht durch Einwachsen. Listeria gelangt via Zipper und Internalin zuerst in die erste Zelle und wandert dann mit Hilfe eines Aktin-Kometen über eine lange Membranausbuchtung, die in die nächste Zelle hineinragt, in diese und wächst so in die nächste Zelle hinein. In der neuen Zelle angekommen hat das Bakterium nun eine doppelte Membran um sich herum, diese Doppelmembran-Vakuole wird durch Lecithinase lysiert. So gelangt Listeria in andere Zellen, ohne den Interzellulärraum zu passieren. Die Aktin-Kometen bestehen aus vielen, zu einem langen Strang quervernetzten kurzen Aktinfilamenten. Das polymerisierende Ende ist zum Bakterium gerichtet (positives Ende). Die Polymerisation der Filamente findet an einer bestimmten Stelle auf der Oberfläche des Bakteriums statt. Das Bakterium wird von den wachsenden Filamenten, die selbst stationär im Cytosol bleiben, weggeschoben und somit fortbewegt. Das multifunktionelle Protein ActA initiiert die Aktin-Polymerisation. Es hat Sequenzhomologien zu Vinculin und Zyxin. - 32 - Profilin rekrutiert Actinmonomere (Profilaktin), das ActA-Dimer verarbeitet die Monomere in das wachsende Filament und schiebt somit die Zelle vorwärts. Die Quervernetzung wird mit α-Actinin und Fimbrin erreicht, die Stabilisierung der Struktur über Cofilin. Mycobacterium tuberculosis ist ein krummes Stäbchen, das resistent gegen Säure, Lauge und Austrocknung ist. Die Zellwand enthält komplexe Glycolipide (Lipidoarabinomannan). Es vermehrt sich in vitro extrem langsam (18-24 Stunden) und kann in Phagocyten überleben, indem es die Fusion von Phagosom und Lysosom verhindert. - 33 - Mycobacterium stoppt die phagosomale Reifung auf einer frühen Stufe. Phagosomen enthalten in der Membran die GTPase Rab5. Mycobacterium hält die Ca2+-Konzentration gering, sodass CaMKII und PI3K (durch Man-LAM inhibiert) nicht aquiriert werden können, die Vorraussetzung für EEA1 (early endosomal antigen 1) ist. EEA1, ein Rab5-Effektor, ermöglicht die Rekrutierung von Rab7, eine andere GTPase, aus Fusion mit einem weiteren Endosom, die schlussendlich die Fusion mit dem Lysosom vermittelt. (Nature Reviews Microbiology 2/2004, 189-202, doi:10.1038/nrmicro840) Zusammenfassung der Interaktionen mit Phagocyten Bakterium Art der Interaktion Mechanismus Streptococcus pyogenes Entdeckung durch Phagocyten verhindern, Dem Umfließen widerstehen, Phagocyten töten, Chemotaxis der Neutrophilen verhindern Hyaluronsäurekapsel, M Protein an Fimbrien, Streptolysin entleert Lysosomen, Streptolysin chemotakt. Schreckstoff Staphylococcus aureus Opsonisierte Phagocytose verhindern, Phagocyten töten, Tötung durch Radikale widerstehen Protein A blockiert Fc, Polysaccharidkapsel (manche), Leokocidin entleer Lysosomen, Carotinoide, Catalase und SuperoxidDismutase detoxifizieren Radikale Yersinia spp. Phagocytose verhindern Virulenzfaktoren sezerniert durch TTSS Salmonella typhimurium Phagocyten töten, Überleben innerhalb von Phagocyten Virulenzfaktoren (SPI-1) induzieren Apoptose, Resistenz gegen pH, ROS, Defensin (SPI-2) Listeria monocytogenes Dem Phagosom entkommen Listeriolysin, Phospholipase-C lysieren die Phagosomenmembran Mycobacteria spp. Tötung und Verdau widerstehen Zellwandkomponenten detoxifizieren aggressive Agentien, Acidifizierung des Phagolysosoms verhindern Wozu töten Invasoren Wirtszellen? Parasit-Wirt-Interaktionen in Bezug auf Zelltod sind sehr komplex. Erfolgreiche Infektionen erfordern ein sensibles Gleichgewicht an pro- und antiapoptotischen Strategien sowohl von Seiten des Parasits als auch des Wirts. Pathogene induzieren den Tod von Immunzellen, um die Abwehrmechanismen des Wirts zu schwächen, das Töten von Phagocyten verhindert das Entfernen von Mikroben aus dem Gewebe. Der Tod von epithelialen oder endothelialen Zellen ermöglicht das Eindringen in tiefere Gewebsschichten eines Organs und in den Blutkreislauf. Systemische Infektionen, Sepsis und septischer Schock sind die Folge. Andererseits inhibieren manche Mikroben den Zelltod für eine erfolgreiche Infektion. Für obligat intrazelluläre Parasiten ist ein lebensfähiger Wirt überlebensnotwendig. Rickettsia rickettsii verhindert Apoptose durch Stimulation des NF-κB-Pathways. Apoptose – Selbstmordprogramm eukaryotischer Zellen Apoptose ist ein morphologisch genau definierbarer Prozess, der essentiell für den korrekten Ablauf der Organbildung sowie immunologischer Reaktionen ist. Es ist eine von vier Varianten des programmierten Zelltods: Pyroptose, Apoptose, Oncose und Autophagie. Davon muss Necrose klar abgegrenzt werden, da Necrose nicht vorprogrammiert ist. - 34 - Apoptose geht von Zelle selbst aus, benötigt Energie und folgt einem genau definierten Ablauf: Chromatin-Kompaktierung und Segregation, Konzentrierung des Cytoplasmas, Schrumpfen der Zelle, Aufteilung (blobbing) in viele kleine Vesikel (apoptotic bodies), welche von Phagocyten phagocytiert werden. Während der Necrose kommt es zu Chromatin-Verklumpung, Anschwellen der Organellen, Disintegration, Platzen der Zelle sowie Auflösung der Zellmembran und Freiwerden des intrazellulären Inhalts, was eine inflammatorische Reaktion auslöst. Cystein‐Aspartat Proteasen (CASPasen), eine Protease-Familie, sind die Effektormoleküle der Apoptose. Cystein katalysiert im aktiven Zentrum die Proteolyse zwischen Aspartet und einer Aminosäure X im Substrat. Caspasen liegen im Cytosol als Proenzym vor, man unterscheidet Initiator- und Effektorcaspasen. Initiatorcaspasen spalten die Precursor der Effektorcaspasen und aktivieren diese somit nach Kontakt mit zB mikrobiellen Produkten. Caspasen wirken auch in der Entzündungsregulation: Caspase-1 spaltet pro-Interleukin-1 zu aktivem Interleukin-1. Caspase-1 ist Effektormolekül der Pathogen-vermittelten Pyroptose und wird beispielsweise von SipB und IpaB stimuliert. Caspase-1 schädigt die DNA, öffnet Poren in der Membran, wodurch die Osmoregulation fällt, die Zelle schwillt an, platzt und gibt cytosolische Komponenten frei. Mikrobielle Abwehr von IgA Sekretorische IgAs verhindern die Kolonisierung der Schleimhäute, sie prädominieren in den Körperflüssigkeiten, welche die Schleimhäute benetzen. Sie werden von Plasmazellen im subepithelialen Gewebe in großen Mengen (5‐15g/Tag) produziert. Sekretorische IgAs enthalten eine sekretorische Komponente (J Kette), mit der sie während des Transports durch die Epithelzellen der Schleimhäute als Dimer verbunden bleiben. IgA‐Dimere binden an einen Poly-Ig‐Rezeptor auf der basolateralen Membran der Epithelzelle und werden durch Rezeptor‐vermittelte Endocytose internalisiert. Der Rezeptor wird nach dem Transport zur apikalen Membran enzymatisch gespalten, wodurch die sekretorische IgAs ins Lumen freigesetzt werden können. Neisseria gonorrhoe, ein gram-negativer Coccobacillus, besiedelt den Genitaltrakt. Es entgeht der Erkennung durch IgA durch Änderung der Sequenz ihres Pilins mittels Genkonversion (unidirektionale Rekombination) und Synthese einer Protease, die die hinge-Domäne von IgA spaltet. Bei der Genkonversion rekombiniert das aktuelle Pilin-Gen mit einer promotorlosen silent-Variante. Streptococcus, gram-positiv, ändert die Sequenz des exponierten N-Terminus seines M-Proteins durch Deletion, der C-Terminus zeigt wenig Variation. Trypanosoma brucei, ein Protozoon und Ursache der Schlafkrankheit, stellt periodisch neue Varianten seines umhüllenden Glykoproteins VSG (variable surface glycoprotein) mittels Genduplikation und Translokation her. Es existieren viele VSG-Varianten im Genom und rund jede Woche wird eine neue Variante in die expression site kopiert. Die Antikörperproduktion muss von neuem anlaufen. Der Influenza-Virus setzt auf sein hypervariables Haemagglutinin, bedingt durch Punktmutationen induziert durch seine fehleranfällige RNA-abhängige Polymerase. So kommt es zum antigenic drift. Wird eine Zelle von zwei Influenza-Stämmen gleichzeitig infiziert, kann zum reassortment, also einer Neuverteilung der 8 ssRNA-Strängen, also der Genome, kommen. So variieren Antigene noch stärker. - 35 - Mikrobielle Interaktion mit dem Adaptiven Immunsystem Mikroben haben sogar Wege gefunden, die T-Zell-vemittelte Immunantwort zu intervenieren. CTLs (cytotoxic T cells) erkennen MHCI-Komplexe, die fast jede Körperzelle mit Zellkern auf ihrer Oberfläche trägt. Wird eine somatische Zelle von einem Virus befallen, so exprimiert diese die viralen Antigene auf dem MHCI-Komplex als Signal, getötet werden zu müssen, um der Vermehrung des Virus vorzubeugen. EBV, hCMV und HSV verhindern den Abbau endogener Antigene sowie die Assemblierung mit dem MHCI. Adenoviren und hCMV verhindern die MHCI-Synthese und seine Lokalisierung an der Zellmembran, indem sie die Reifung im ER, Golgi-Apparat oder sekretorischem Vesikel aufhalten. Daneben exprimiert mCMV ein MHCI-ähnliches Molekül, das kein Antigen bindet, um in einem Decoy-Mechanismus die Anwesenheit viraler Antigene zu verschleiern. Adenoviren und EBV exprimieren Apoptose-Inhibitoren, um im Wirt lange unerkannt zu bleiben. Mikroben sind in der Lage, sich der Kommunikation mittels Cytokinen, lösliche Signale immunkompetenter Zellen, zu bedienen, um die Macrophagen-Aktivierung zu unterbinden Die relative Konzentration von IL‐12 und IL‐4 in der Anfangsphase einer Infektion bestimmt die Reaktion des Wirtes. Inhibition der Makrophagen‐Aktivierung wird durch Eingriff in die Signaltransduktion erreicht, das Ziel ist, die DTH (delayed type hypersensitivity) zu unterbinden: • • • EBV produziert ein Homolog des Cytokins IL‐10 Poxviren kodieren für Cytokinrezeptor‐Homologe (IFNγ, TNF-α, und IL‐1) Bestandteile der Zellwand von Mycobakterien interferieren mit IFNγ - 36 - 7 – Friendly Fire Pathogenese muss nicht immer von Invasoren ausgehen, eine unpassende Immunantwort kann ebenfalls beträchtlichen Schaden anrichten: Lang anhaltende Entzündungsreaktion führt zu somatischem Zellschaden. Die von Makrophagen produzierten Cytokine (TNF‐α, IL‐1, IL‐6, IL‐8 und PAF) dienen zur Verteidigung des Wirts und zur Zerstörung des Mikroorganismus, (Entzündungsreaktion, Fieber, Immunantwort und Phagocytose). Die massive Freisetzung von LPS kann wiederum ein Überschiessen dieser Antwort verursachen, mit schweren Folgen für den Wirt (ARDS Acute respiratory disease syndrom; DIC disseminated intravascular clotting; MOSF multiple organ system failure). Eine ernste Gefahr geht vom molecular mimicry seitens der Mikroben aus. Diese Taktik vermindert die Immunantwort vielleicht nicht, macht aber auch wirtspezifische Strukturen zum Ziel. Ein Beispiel sind rheumatische Herzerkrankungen in Folge einer Streptokokken-Infektion, verursacht durch Antikörper gegen Meromyosin, die eine Kreuzreaktion auslösen. Die Immunantwort auf Superantigene schadet dem Körper mehr als es für ihn Vorteile bringt. Eine DTH-Reaktion baut sich in zwei Phasen auf, der sensitization phase und der effector phase. In der ersten Phase, werden TDTH-Zellen, meistens TH1-Zellen oder manchmal CD8+-Zellen, durch APC wie Macrophagen und Langerhans-Zellen stimuliert. Aktivierte TDTH-Zellen schütten IFNγ und TNF‐α aus und mobilisieren ruhende Macrophagen, die daraufhin MHCII- und TNF-Rezeptoren exprimieren sowie Sauerstoffradikale und Stickoxide produzieren. Zwei Pathogene, die eine DTH-Reaktion hervorrufen, sind Mycobacterium tuberculosis und Mycobacterium leprae. Bei einer Tuberkulose siedeln sich Bakterien in der Lunge an und können auch in Phagosomen von Macrophagen überleben und sich dort ungestört vermehren. Schließlich stirbt der Macrophagen, freigesetzte Bakterien werden durch andere Macrophagen aufgenommen. Infizierte Macrophagen produzieren Cytokine, die andere Phagocyten wie Monocyten, Macrophagen und Neutrophile anlocken. Es kommt zur Bildung eines Granuloms (klassische Tuberkuloseläsion). In Granulomen werden Mycobakterien von Macrophagen und TDTH-Zellen umstellt, um einer weiteren Ausbreitung vorzubeugen. In der Mitte befindet sich nekrotisierendes Gewebe, rundherum aktivierte Macrophagen sowie TDTH-Zellen. In 90% der Fälle heilen die Granulomen aus und verkalken, in 10% der Fälle wird die Infektion chronisch oder die Granulomen reißen auf, die Bakterien verbreiten sich im Wirt und es kommt zur extrapulmonaren Tuberkulose. Lepra-Patienten zeigen zwei unterschiedliche Krankheitsbilder. Bei Lepra tuberculoides verursacht eine starke zelluläre Antwort gegen infizierte Macrophagen die Bildung von Granulomen bei niedriger Anzahl von Mycobakterien. Gute Überlebenschance, beträchtliche Haut- und Nervenschäden. Lepra lepromatosa wird mit einer starken humoralen Immunantwort bekämpft, die zelluläre Immunantwort ist eher schwach, die Folge ist Hypergammaglobulinämie und geringe Granuloma-Bildung. Horrende Populationen von Bakterien entgehen den humoralen Antikörpern, weil sie sich im Inneren der Macrophagen aufhalten. Schlechte Überlebensprognose, Veränderung von Knochen und Knorpeln, extensive Nervenschäden. - 37 - 8 – Übungsfragen Was sind Pathogene? Wie entwickeln sich Biofilme am Beispiel von Pseudomonas? Durch welche Interaktion kann E.coli Pyelonephritis hervorrufen? Wie gelangen invasive Bakterien in Wirtszellen am Beispiel Salmonella? Was sind Neurotoxine und wie wirken Neurotoxine aus Clostridium? In welchen Kreislauf sind GEFs und GAPs involviert, wie funktioniert er, was bewirkt er? Auf welche Weise wirken BoNT und TeNT? Welchen Vorteil ziehen Pathogene aus der Regulierung des cAMP-Spiegels, wie machen sie das? Vergleiche die Eigenschaften von Endo- und Exotoxinen! Wie bringen Superantigene das Immunsystem aus dem Gleichgewicht? Wie reagiert das Immunsystem auf Endotoxine? Wie und wodurch wird das Komplementsystem aktiviert? Welche bakteriellen Spezies setzen welche Strategien ein, um der Phagocytose zu entgehen? Wie interagieren Mikroben mit der adaptiven Immunantwort? - 38 - 9 – Quellen 1→ War and peace at mucosal surfaces doi:10.1038/nri1499 http://www.nature.com/nri/journal/v4/n12/full/nri1499.html Genomic fluidity and pathogenic bacteria: applications in diagnostics, epidemiology and intervention (2008) doi:10.1038/nrmicro1889 http://www.nature.com/nrmicro/journal/v6/n5/full/nrmicro1889.html The race between infection and immunity: how do pathogens set the pace? (2009) doi:10.1016/j.it.2008.11.001 http://www.cell.com/trends/immunology/fulltext/S1471-4906(08)00273-1 Is HIV-1 evolving to a less virulent form in humans? (2007) doi:10.1038/nrmicro1594 http://www.nature.com/nrmicro/journal/v5/n2/full/nrmicro1594.html 2→ Bacterial Biofilms: A Common Cause of Persistent Infections (1999) doi:10.1126/science.284.5418.1318 http://www.sciencemag.org/cgi/reprint/284/5418/1318.pdf Bacterial Adhesins in Host‐Microbe Interactions (2009) doi:10.1016/j.chom.2009.05.011 http://linkinghub.elsevier.com/retrieve/pii/S1931312809001784 The developmental model of microbial biofilms: ten years of a paradigm up for review doi:10.1016/j.tim.2008.11.001 http://www.cell.com/trends/microbiology/fulltext/S0966-842X(09)00005-5 3→ Bacterial Invasion: The Paradigms of Enteroinvasive Pathogens doi:10.1126/science.1090124 http://www.sciencemag.org/cgi/content/full/304/5668/242 Salmonellae interplay with host cells doi:10.1038/nrmicro1788 http://www.nature.com/nrmicro/journal/v6/n1/full/nrmicro1788.html Regulation of actin cytoskeleton dynamics by Arf-family GTPases doi:10.1016/j.tcb.2008.02.002 http://www.cell.com/trends/cell-biology/fulltext/S0962-8924(08)00069-X Bacterial Guanine Nucleotide Exchange Factors SopE-Like and WxxxE Effectors doi:10.1128/IAI.01250-09 http://iai.asm.org/cgi/content/full/78/4/1417 Structural insights into host GTPase isoform selection by a family of bacterial GEF mimics doi:10.1038/nsmb.1647 http://www.nature.com/nsmb/journal/v16/n8/full/nsmb.1647.html - 39 - Genetics and Assembly Line Enzymology of Siderophore Biosynthesis in Bacteria doi: 10.1128/MMBR.66.2.223-249.2002 http://www.ncbi.nlm.nih.gov/pmc/articles/PMC120789/ 4→ The journey of tetanus and botulinum neurotoxins in neurons (2003) doi:10.1016/S0966-842X(03)00210-5 http://linkinghub.elsevier.com/retrieve/pii/S0966842X03002105 Botulinum neurotoxin B recognizes its protein receptor with high affinity and specificity (2006) doi:10.1038/nature05387 http://www.nature.com/nature/journal/v444/n7122/full/nature05387.html 5→ The Complement System doi: http://www.profelis.org/amc/vorlesungen/immunologie/komplementsystem.html Human complement component 5 (C5) http://www.bio.davidson.edu/COURSES/Immunology/Students/spring2006/Fiser/C5.html 6→ Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation doi:10.1038/nri1997 http://www.nature.com/nri/journal/v7/n1/full/nri1997.html Interplay between mycobacteria and host signalling pathways doi:10.1038/nrmicro840 http://www.nature.com/nrmicro/journal/v2/n3/full/nrmicro840.html 7→ Delayed Type Hypersensitivity: Current Theories with an Historic Perspective doi: http://dermatology.cdlib.org/DOJvol5num1/reviews/black.html - 40 -