Zellen und Zellenorganelle
Werbung

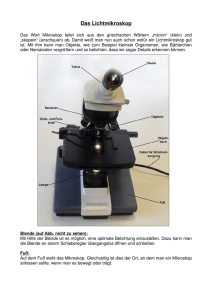
Das Lichtmikroskop Griechisch: mikrôs – klein skopein – betrachten Geschichte des Lichtmikroskops „Erfinder“: Hans & Zacharias Janssen um 1595 © http://www.amuseum.de Galileo Galilei © http://www.amuseum.de Robert Hooke © http://www.amuseum.de Antoni van Leeuwenhoek © http://www.amuseum.de Mikroskope von Leeuwenhoek - eigentlich ein Lupes - Kristallinse im Metalrahmen - Vergrösserung von 50X bis 300X Auflösungsvermögen: 1/1000 mm Die wichtigsten Entdeckungen von Leeuwenhoek Mikroorganismen Chloroplasten rote Blutkörperchen Spermien Das Licht Licht: im engeren Sinne die für das menschliche Auge sichtbare elektromagnetische Strahlung mit Wellenlängen zwischen 380 und 780 nm (sichtbares Licht), im weiteren Sinne der Wellenlängenbereich zwischen etwa 100 nm und 1 mm (optische Strahlung), der auch die Ultraviolett- und Infrarotstrahlung umfasst. Licht hat Eigenschaften von Teilchen (besteht sich aus Photonen) wie auch von Wellen Monochromatisches Licht: Licht mit Strahlen die eine einzige, bestimmte Wellenlänge haben Polychromatisches Licth: besteht sich aus einer Mischung unterschiedlicher Farben, ein Gemisch aus vielen Wellenlängen. Sichtbares Licht: 380-780 nm Das Licht Merkmale des Lichtes Wellenlänge (λ): der kleinste Abstand zweier Punkte gleicher Phase einer Welle bezeichnet (in nm). Frequenz: die Anzahl von Schwingungen pro Sekunde. Die Einheit der Frequenz ist das Hertz (1/s), kurz Hz Amplitude (bzw. Schwingungsweite): die maximale Auslenkung (Elongation) der Schwingungswellen Komponenten des sichtbaren Lichtes Warum benutzt man Mikroskop bei der Medizin? Die häufigsten Untersuchungsobjekte am Mikroskop: - Zellen - Geweben Proben können nativ oder gefärbt untersucht werden. Größenverhältnisse Zellen Organellen Moleküle mm – cm makroskopisch sichtbar 0,2 mm Auflösungsgrenze des bloßen Auges 20 µm 2 µm 0,2 µm 20 nm 2 nm 0,2 nm Atome Auflösungsgrenze des Lichtmikroskops Auflösungsgrenze des Elektronenmikroskops Durchschnittsgrösse der Zellen Prokaryotische Zelle: 2-4 µm Eukaryotische Zelle: 10- 30 µm Die funktionellen Einheiten des Lichtmikroskops Weg des Lichtes im Mikroskop Okular Zwischenbild Objektiv Objekt Kondensor Schärferegulierung Lichtquelle Die Vergrösserung des Mikroskops Die Vergrösserung des Mikroskops (Vm): Vm = Vob X Vok Vob: Vergrösserung des Objektivs Vok: Vergrösserung des Okulars Okular: 10-20 x Vergrösserung Marke der Farbekorrektion Marke der Immersion Objektiv: 4-100 x Vergrösserung Vergrösserung des Objektivs mechanische Tubuslänge (mm) Wert der numerischen Apertur Empfohlene Deckglasdichte Marke der speziellen Bestimmung Immersionsmarke Frontlinse des Objektivs Abbildungsfehler von optischen Linsen Sphärische Aberration Die sphärische Aberration ist ein sogenannter Zonenfehler, der bei sphärischen Linsen mit größerem Durchmesser und stärkerer Krümmung auftritt. Randstrahlen werden stärker als achsennahe Strahlen gebrochen und bilden einen Brennpunkt näher an der Linse. So entsteht eine Abweichungskreis. Chromatische Aberration Entsteht wegen der Dispersion. Blaues Licht wird stärker gebrochen als rotes so daß sich die Brennweiten unterscheiden. Mit weißem Licht erzeugte Bilder bekommen dann Farbsäume. Den Abstand zwischen dem roten und dem blauen Brennpunkt bezeichnet man als axiale chromatische Aberration. Brechzahl An der Grenzfläche zweier Medien mit unterschiedlichem Brechungsindex wird Licht gebrochen und reflektiert. Dabei nennt man das Medium mit dem höheren Brechungsindex das optisch dichtere. Reflexion Vakuum Medium c cM Brechung Lichtgeschwindigkeit bei 20º C und 584 nm Material Vakuum Luft (1 atm) Wasser Augenlinse Ethylalkohol Quarzglas Flintglas Zederöl n 1 1,00027 1,333 1,34 1,361 1,459 1,613 1,510 c absolute Brechzahl: n cM 1 Trockenobjektiv Immersionsobjektiv Frontlinse Immersionsflüssigkeit Deckglas Objektglas Wir lassen eine Flüssigkeit zwischen dem Deckglas und der Frontlinse tropfen, welches eine Brechzahl höher als die Luft hat, und wir lassen die Frontlinse ins Flüssigkeitropfen eintauchen. Solche Linsen (meisstens 100X Vergrösserung) nennen wir Immersionslinsen. Immersion=Eintauchen Immersionsflüssigkeit: Am häufigsten benutzen wir Zederöl. Die Ölobjektive nennen wir dementsprechend Ölimmersions-, oder homogene Immersionsobjektive (HI). Wenn auf dem Objektiv WI steht, dass heisst das wir destilliertes Wasser zwischen dem Deckglas und der Frontlinse tropfen müssen. Trockenobjektiv Ölimmersionsobjektiv Lichtstrahl 3 wird nach dem Austritt aus dem Deckglas so stark gebrochen, dass er nicht mehr in die Frontlinse des Objektivs gelangt. Die Brechzahl von Deckglas und Immersionsöl sind fast identisch. Lichtstrahl 3 wird nach dem Austritt aus dem Deckglas nicht merklich gebrochen und gelangt in die Frontlinse des Objektivs. Auflösungsvermögen des Mikroskops Das Auflösungsvermögen eines Objektivs ist davon abhängig, wie viel Licht von einer Struktur des Präparates in das Objektiv gelangt. Diese Lichtmenge ist nun wiederum abhängig vom sogenannten Öffnungswinkel des entsprechenden Objektivs. Je größer der Öffnungswinkel ist, desto besser löst ein Objektiv die Details eines Präparates auf. Längenmessung am Mikroskop Objektmikrometer: Objektträger mit Mikrometerteilung Okularmikrometer: Strichplatte ins Okular eingelegt Längenmessung am Mikroskop Haare Skala des Objektmikrometers Skala des Okularmikrometers Skala des Okularmikrometers Mikroskop in Forschung video Fortgeschrittene Mikroskopie Fluoreszenz-Mikroskopie Human Zellen DNA microtubules F-actin Foto eines Fluoreszenzmikroskops Hier ist ein handelsübliches Fluoreszenzmikroskop abgebildet. Zu erkennen sind die zwei Lichtquellen, eine für Durchlicht- eine für Fluoreszenzmikroskopie. Die Objektive von Fluoreszenzmikroskopen müssen UV-gängig sein, wenn Fluorochrome mit UVAnregung verwendet werden sollen. Solche Objektive tragen die Bezeichnung FLUO. Die orangene Platte unterhalb der Okulare schützt die Augen vor UV-Licht. Der Kasten rechts neben dem Mikroskop ist das Netzgerät für die Epifluoreszenzbeleuchtung. Grundlagen der Fluoreszenzentstehung Bei der Absorption von Licht einer bestimmten Wellenlänge (=Anregungslicht) ist bei verschiedenen Molekülen eine gleichzeitige Emission von Licht mit größerer Wellenlänge beobachtbar. Dieses Verhalten (Absorption von kurzwelligem Licht, Emission von längerwelligem Licht) wird als Fluoreszenz bezeichnet. Fluoreszenz ist die spontane Emission von Licht beim Übergang eines elektronisch angeregten Systems in einen Zustand niedrigerer Energie. Grundlagen der Fluoreszenzentstehung Fluorochromen Viele Fluorochrome haben aromatische Ringsysteme, deren delokalisierte Elektronen in bindenden p-Orbitalen für die Entstehung von Fluoreszenz wichtig sind. Chinin (quinine) war eine der ersten Substanzen, an denen Fluoreszenz untersucht worden ist (Herschel, 1845). Chinin wird durch UVLicht angeregt und erzeugt eine schwache, bläuliche Fluoreszenz in Tonic Water, das mit Chinin versetzt ist. Fluorescein und Rhodamin spielen eine große Rolle bei der Fluoreszenz-Immunhistochemie. POPOP wird für Szintillationszähler verwendet und Acridin-Orange eignet sich als DNAFarbstoff. Coumarin-Derivate werden in vielen Bereichen verwendet, zB für ELISA-Tests. Fluorochromen Anregungslicht und Fluoreszenz-Filtrierung Im herkömmlichen Mikroskop werden Fluoreszenz-Erscheinungen überstrahlt und sind deshalb nicht erkennbar. Deshalb muss man durch eine geschickte Auswahl von Filtern dafür sorgen, dass nur das Fluoreszenz-Licht an der Bildentstehung teilnimmt. Anregungsfilter haben eine möglichst hohe Transmission für die Wellenlängen, welche die Fluoreszenz anregen - insbesondere längerwellige Strahlung wird von diesen Filtern zurückgehalten und gelangt deshalb nicht zum Präparat. Das Anregungsfilter befindet sich im beleuchtenden Strahlengang vor dem Präparat. Sperrfilter haben eine hohe Transmission für die energieärmeren (längeren) Wellenlängen des Fluoreszenzlichts - das energiereiche Anregungslicht wird möglichst vollständig eliminiert. Das Sperrfilter befindet sich im abbildenden Strahlengang - also optisch nach dem Präparat. Aufbau des Fluoreszenzmikroskops Fluorochromen Immunfluoreszenz Mikroskopie Immunfluoreszenz Mikroskopie Prinzip der konfokalen Mikroskopie Beim konventionellen Lichtmikroskop wird das Lampenlicht durch die Kondensorlinse auf das Objekt fokussiert, und das vom Objekt ausgehende Licht wird durch die Objektivlinse in die Zwischenbildebene fokussiert. Das so entstehende Bild wird durch die Okularlinse betrachtet. Nicht nur Licht aus der Brennebene des Objektivs (hier rot dargestellt) sondern auch unfokussiertes Licht aus Bereichen außerhalb der Brennebene (hier blau und grün dargestellt) erreicht bei diesem Mikroskop das Auge. Durch die Überlagerung von fokussiertem und unfokussiertem Licht ist die räumliche Auflösung des konventionellen Mikroskops eingeschränkt. Prinzip der konfokalen Mikroskopie Beim konfokalen Mikroskop wird Licht, das nicht aus der Brennebene des Objektivs kommt, ausgeblendet. Die einfachste Konstruktion ist hier gezeigt: Die Kondensorlinse wird durch eine Linse ersetzt, die der Objektivlinse identisch ist. Die Ausleuchtung des Objekts wird durch eine Lochblende (A) beschränkt, die auf dem Objekt scharf abgebildet wird. Eine zweite Lochblende (C) beschränkt das Sichtfeld auf einen Punkt. Durch den symmetrischen Aufbau dieses Systems sind beide Blenden und ein Punkt des Objekts in der Brennebene der Linsen konfokal. Der Durchmesser der Blenden wird so klein gewählt, daß Licht aus Bereichen des Objekts, die nicht in der Brennebene liegen, nicht in die Apertur der Blende C fallen und damit ausgeblendet werden (hier: grüne und blaue Strahlen). In den Photomultiplier (PMT) gelangt deshalb nur Licht aus der Brennebene des Objekts. Prinzip der konfokalen Mikroskopie Im Unterschied zum konventionellen Mikroskop erzeugt das konfokale Mikroskop also zunächst nur einen Bildpunkt, der allerdings genau einen Punkt aus der Brennebene des Objektivs darstellt. Um eine vollständiges Bild des Objekts zu erhalten, muß das Objekt Punkt für Punkt gerastert (gescannt) werden. Bei der hier gezeigten Anordnung geschieht das dadurch, daß das Objekt jeweils eine kleine Strecke verschoben wird, bevor der nächste Punkt vom Photomultiplier registriert wird ("stage scanning"). Die dabei gesammelten Bildpunkte werden dann von einem Rechner zu einem vollständigen Bild zusammengesetzt. Bei den meisten modernen Konfokalmikroskopen wird nicht das Objekt bewegt, sondern ein Laserstrahl wird Punkt für Punkt über das Objekt geführt, und das Bild entsteht durch digitale Verarbeitung im Rechner: Laserscanning Konfokalmikroskop. Am Beispiel einer Fluoreszenzdoppelfärbung ist hier der Vorteil der konfokalen Mikroskopie gezeigt. Bei dieser Zelle, die sich in der Meta-/Anaphase der Zellteilung befindet, ist die Plasmamembran mit einem rotfluoreszierenden Antikörper markiert, der Spindelapparat mit einem grünfluoreszierenden. Links das Bild mit dem konventionellen Mikroskop: Am Rand der Zelle sieht man Membranfärbung, die wie ein Schleier das ganze Bild überlagert. Die Darstellung der Spindelfasern ist unscharf, aber besonder intensiv in der Kinetochor-Region. Rechts: Die selbe Zelle in konfokaler Optik. Die seitliche Plasmamembran ist scharf dargestellt ("optischer Schnitt" durch Ausblenden unfokussierten Lichtes) und die Fasern des Spindelapparates sind zu erkennen. Mikroskopie in Forschung Mikroskopie in Forschung video Mikroskopie in Forschung video Fluoreszenz- und konfokale Mikroskopie Vergleich des Konfokalen- und Fluoreszenzmikroskop Pflanzenzellen: Zwiebelepidermis Präparation der Zwiebelschuppenepidermis von Allium cepa Legen Sie die Schuppenepidermis auf einen Objektträger, versehen Sie es mit Wasser und Deckglas. Beobachten Sie die Probe. Lassen Sie 10%-ige Essigsäure in die Probe diffundieren und beobachten Sie die erscheinenden, fixierten Zellkerne. Färbung der Probe: Fixierung: 60% (V/V) Ethanol, 30% Chloroform, 10% Eisessigsäure, 20 Min., Waschen mit Hahnwasser. Färbung: 0.5% Methylgrün gelöst in 0.1 M Acetat-Puffer, pH4, 10 Min., Waschen mit 50% Ethanol, 5 Min. Legen Sie die gefärbte Zwiebelschuppenepidermis auf einen Objektträger und beobachten sie es im Mikroskop.