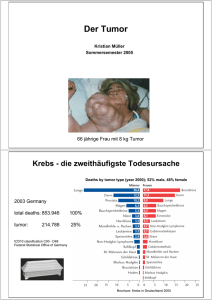
Endokrine PN gesamt.qxp_!GKM_04.qxd - cthomas
Werbung

www.cthomas-pathologie.de Endokrine und renale Paraneoplasien Literaturauswertung von C. Thomas E. Grom 2015 Herausgeber und Autor Prof. Dr. Carlos Thomas ehemaliger Direktor des Medizinischen Zentrums für Pathologie der Philipps-Universität Marburg. Institutsanschrift: Conradi Straße. 35043 Marburg Privatanschrift: Hopfengarten 16. 35043 Marburg-Bauerbach Tel: 06421-27647 Fax: 06421-27676 Email: [email protected] Homepage: www.cthomas-pathologie.de Wesentliche Teile dieses Beitrages stammen aus: Thomas, C., T. Windt und E. Grom: Hämatologische und endokrine Formen des paraneoplastischen Syndroms. Schattauer Verlag. Stuttgart – New York (1974). Thomas, C. (Hrgb.): Grundlagen der klinischen Medizin. Band 5. Gebert, G., C. Thomas: Endokrines System. Schattauer-Verlag. Stuttgart-New York (1992) Abbildungen auf dem Titelblatt: Links: knotige Hyperplasie der Nebennierenrinde beim ektopen Cushing Mitte: Fibrosarkom der Pleura bei paraneoplastischer Hypoglykämie Rechts: Fibrosarkom in der HE-Färbung Besonderer Hinweis: Die Medizin unterliegt einem fortwährenden Entwicklungsprozess, sodass alle Angaben, insbesondere zu diagnostischen und therapeutischen Verfahren, immer nur dem Wissensstand zum Zeitpunkt der Veröffentlichung des Beitrages entsprechen können. Hinsichtlich der an gegebenen Empfehlungen zur Therapie und der Auswahl sowie Dosierung von Medikamenten wurde die größtmögliche Sorgfalt beachtet. Gleich wohl werden die Benutzer aufgefordert, die Beipackzettel und Fachinformationen der Hersteller zur Kontrolle heranzuziehen und im Zweifelsfall einen Spezialisten zu konsultieren. Fragliche Unstimmigkeiten sollten bitte im allgemeinen Interesse dem Herausgeber mitgeteilt werden. Der Benutzer selbst bleibt verantwortlich für jede diagnostische oder therapeutische Applikation, Medikation und Dosierung. In diesem Beitrag sind eingetragene Wa© Prof. Dr. C. Thomas 2015 renzeichen (geschützte Warennamen) nicht besonders kenntlich gemacht. Es kann also aus dem Fehlen eines entsprechenden Hinweises nicht geschlossen werden, dass es sich um einen freien Waren Namen handelt. Das Werk mit allen seinen Teilen ist urheberrechtlich geschützt. Es wird kostenlos zur privaten Verwendung zur Verfügung gestellt. Jede Verwertung außerhalb dieser Bestimmung des Urheberrechtsgesetzes ist ohne schriftliche Zustimmung des Herausgebers unzulässig. Kein Teil des Werkes darf in irgendeiner Form ohne schriftliche Genehmigung des Herausgebers reproduziert werden. Das gilt insbesondere für Vervielfältigungen, Übersetzungen, Mikroverfilmungen und die Einspeicherung, Nutzung und Verwertung in elektronischen Systemen, dem Intranet und dem Internet. Einleitung ● Hoher Verbrauch durch den Tumor von physiologischen Stoffen. Für diese Annahme spricht die starke Verminderung von bestimmten Stoffen, die für Gerinnung (Verbrauchkoagulation, Thrombozytopenie und Fibrinogenmangel), Stoffwechsel (Tumorkachexie) oder Endokrinium (Insulinmangel bei Fibrosarkomen) wichtig sind. In einigen Fällen spricht man von einer Sequestration von Blutbestandteilen (Thrombozyten beim Kasabach-Merrit-Syndrom) bzw. eine endokrine Schwammwirkung bestimmter Neubildungen. I. Einleitung In den letzten Jahren sind zahlreiche funktionelle oder organische Störungen bei Tumorpatienten beobachtet worden, die man nicht auf die lokale Einwirkung der Geschwulst zurückführen konnte. Diese Krankheitsbilder traten meistens gleichzeitig mit dem Tumor auf, einige wurden aber schon vor bzw. erst längere Zeit nach der Geschwulst klinisch manifest. Statistische Untersuchungen sowie die Beobachtung, dass sich diese tumorbegleitenden Symptome nach der Behandlung des Neoplasmas zurückbildeten und bei einem Geschwulstrezidiv wieder auftraten, haben zu der Ansicht geführt, dass hier kein zufälliges Zusammentreffen von zwei primär unabhängigen Grundleiden vorliegen kann. Da die Pathogenese dieser Tumorsymptomatik nicht bekannt war, bezeichnete man sie als paraneoplastisches Syndrom. Dabei handelt es sich zwar um einen neuen Begriff (DennyBrown, 1948; Guichard, 1956; Uehlinger, 1957, 1966, Thomas, 1972, Thomas et al., 1974, 2002)), einschlägige Fälle sind aber schon im 19. Jahrhundert beschrieben worden (z. B. die atypischen Thrombosen bei okkulten Karzinomen: Trosseau, 1877). ● Bei einigen Paraneoplasien spielen immunologische Faktoren eine Rolle. Diese Möglichkeit wird besonders für die neurologischen und kutane Paraneoplasie diskutiert. ● Die Paraneoplasie ist Folge einer Tumorproduktion von Stoffen mit hormonähnlicher Wirkung. Dieser Mechanismus ist bei verschiedenen endokrinen Paraneoplasien nachgewiesen worden und gilt heute als gesicherter pathogenetischer Mechanismus. Zu diesem Nachweis haben moderne Untersuchungsmethoden von hoher Sensitivität und Sensibilität beigetragen. Eine besondere Aussagekraft gilt den Hormonmessungen vor (arteriell) und nach (venös) dem Tumor. Die Produktion dieser Stoffe wird auf eine Derepression bestimmter Gene im Rahmen der mitotischen Aktivität der Geschwulstzellen zurückgeführt. So können bestimmte Stoffe, die nur in der fetalen Periode vorkommen, auch beim Erwachsenen nachweisbar sein (α-Fetoprotein, CEA). Da die hormonähnlichen Verbindungen nicht vollständig mit den normalen Hormonen übereinstimmen, unterliegen sie nicht den üblichen regulatorischen Mechanismen (Feedback). So bleibst bei einer Überproduktion eine Suppression aus. Oft handelt es sich um Hormonvorstufen (IGF-2, PTHrP), die aber bereits hormonell aktiv sind. Das paraneoplastische Syndrom wurde von Boudin (1961, 1962) wie folgt definiert: »Pathologische Veränderungen, die an das Vorhandensein eines Karzinoms oder malignen Tumors gebunden sind, die aber bei einem Parallelverlauf nicht auf das Vorliegen von Metastasen zurückzuführen sind«. Heute kennen wir zahlreiche klinische Manifestationsformen eines paraneoplastischen Syndroms. Meist handelt es sich um Systemerkrankungen, manchmal bleiben sie auf ein Organ (Haut, Niere, Knochen) beschränkt. Es kommen neurologische, hämatologische und endokrine Störungen sowie Muskel-, Stoffwechsel-, Haut-, und Knochenerkrankungen vor. Mit der zunehmenden Zahl an Beobachtungen wird das paraneoplastische Syndrom zu einer »Modediagnose«; man sollte es aber von einfachen Tumorsymptomen (z. B. Fieber, Kachexie, Leukozytosen: Chisholm und Roy, 1971; Roth, 1973), Tumorsyndromen und Tumorsyntropien abgrenzen. Bei dem Tumorsyndrom wird die Symptomatik durch die Tumorzelle selbst hervorgerufen und stellt lediglich eine verstärkte Leistung der physiologischen Funktion des befallenen Organs dar (z. B. das Cushing-Syndrom beim Nebennierenrindenkarzinom). Bei den Tumorsyntropien kommen zwei eigenständige Krankheitsbilder synchron oder metachron vor und können durch Wechselbeziehungen den allgemeinen Krankheitsverlauf beeinflussen. Der Nachweis einer Paraneoplasie ist oft an anspruchsvolle diagnostische Untersuchungen gebunden. So sollten bei einer endokrinen Paraneoplasie die Konzentrationen an Hormonen bzw. hormonähnlichen Stoffen im Tumorgewebe sowie in dem zuführenden und abführenden Tumorblut bestimmt werden. Die biochemischen, histologischen, elektronenmikroskopischen und immunologischen Untersuchungsmethoden lassen sich aber in der Regel nur an frischem Material durchführen. Ähnliche Schwierigkeiten treten auch bei der Erfassung hämatologischer Störungen oder einer sensorischen Neuropathie auf (Thomas et al., 1972). Tumorkrankheit, Tumorsyndrom oder Paraneoplasie? Bei den ersten Publikationen über die Paraneoplasie wurde der nicht geklärte formalpathogenetische Zusammenhang – als wesentlicher Bestandteil der Begriffsbestimmung – in den Vordergrund gestellt. Dieser ist bei einigen Paraneoplasien auch heute noch weitgehend ungeklärt, bei anderen aber bereits gesichert. So ist z. B. bekannt, dass bestimmte Tumoren Substanzen mit hormonähnlicher Wirkung erzeugen können, die für die paraneoplastischen Befunde verantwortlich sind. So müssten letztere bereits als Bestandteil einer Tumorkrankheit bzw. eines Tumorsyndroms geführt werden. Trotzdem werden sie auch in den neuesten Publikationen immer noch als Paraneoplasie bezeichnet. Für die Eigenständigkeit der Diagnose »Paraneoplasie« spricht die Tatsache, dass diese oft vor dem Tumor klinisch manifest wird. Welche Erklärungen gibt es für einen pathogenetischen Zusammenhang zwischen Tumor und Praneoparaneoplasie? ● Das Zusammentreffen einer Neubildung und einer Paraneoplasie ist zufällig. Gegen diese Annahme sprechen heute mehrere Beobachtungen. So z. B. die Statistik. Auch das zeitliche Verhalten von Tumor und Neoplasie ist ein wichtiges Argument: Die Paraneoplasie wird mit dem Tumor klinisch manifest, bildet sich nach der Entfernung Geschwulst zurück und wird bei einem Rezidiv oder Metastasen wieder manifest. 1 Endokrine und renale Paraneoplasien Diese Arbeit stützt sich auf die Auswertung von 687 publizierten Fällen mit einer endokrinen Paraneoplasie. Sie wurden unter Berücksichtigung der allgemeinen Häufigkeit, der Alters- und Geschlechtsverteilung, der Lokalisation und Histologie des Primärtumors und der zeitlichen Wechselbeziehungen zwischen Tumor und Paraneoplasie ausgewerten. Dabei stellt sich besonders die Frage, ob die Erkennung eines paraneoplastischen Syndroms für die Diagnose (Frühdiagnose oder ein Hinweis auf die Lokalisation eines okkulten Karzinoms), Prognose und Therapie eines Geschwulstleidens von Bedeutung sein kann. ren mit endokriner Paraneoplasie geht – unter Berücksichtigung der Literaturangaben – aus folgender Zusammenstellung hervor: – 42% Lungenkarzinome (meist kleinzelliger Typ) – 11% Weichteiltumoren – 8% Leberkarzinome – 7% Pankreaskarzinome – je 4% Thymus- und Nierenkarzinome – 3% Nebennierenkarzinome – 21% andere Lokalisationen. Die zeitliche Korrelation zwischen Tumor und Paraneoplasie hängt an erster Stelle von der Neubildung (Art, Dignität und Lokalisation) ab. Bei einigen Tumoren (Pankreasund Nierenkarzinomen) wird die Diagnose meist spät gestellt. Nur in 15% der paraneopastischen Polygobulien wurde das Nierenkarzinom vor der Paraneopalsie nachgewiesen. In diesen Fällen kommt es zu einer frühen Manifestation der Paraneoplasie. In der Regel wurden Paraneoplasie und Tumor gleichzeitig bzw. die Neubildung früher festgestellt. II. Endokrine paraneoplastische Syndrome Die paraneoplastischen Endokrinopathien (ICD-C80.9) sind Überfunktionssyndrome auf dem Boden einer ektopen Hormonbildung, die weder örtliche, strukturelle noch ontogenetische Beziehungen zum physiologischen Bildungsort der Hormone aufweisen (Kracht, 1968). 1 Ektopes ACTH-Syndrom Heute kennen wir mehrere endokrine Störungen, die einen Tumor begleiten, gleichzeitig vor oder nach ihm klinisch manifest werden und dem Überfunktionssyndrom einer endokrinen Drüse entsprechen. Man unterscheiden folgende Formen eines endokrinen paraneoplastischen Syndroms: 1 ektopes ACTH-Syndrom, 2 extrapankreatische Hypoglykämie, 3 paraneoplastisches Hyperkalzämiesyndrom, 4 ektopes ADH-Syndrom, 5 paraneoplastisches Karzinoidsyndrom, 6 ektopes Gonadotropinsyndrom, 7 ektopes STH-Syndrom 8 multiple paraneoplastische Endokrinopathien, 9 ektopes TSH-Syndrom, 10 paraneoplastisches Zollinger-Ellison-Syndrom, 11 Erythropoetin-bildende Tumoren, 12 Renin-bildende Tumoren, 13 paraneoplastische Hormonbildung ohne klinische Manifestation. Synonyma: paraneoplastischer Hyperkortizismus, CushingSyndrom infolge eines nichtendokrinen Tumors, paraneoplastisches Cushing-Syndrom, sekundäres Cushing-Syndrom. Begriffsbestimmung Das ektope ACTH-Syndrom (ICD-E24.3) wurde erstmals von Brown (1928) beschrieben und ist das bekannteste endokrine Syndrom, das bei primär nichtendokrinen Tumoren vorkommt. Das klinische Bild des ektopen ACTH-Syndroms unterscheidet sich vom einfachen Cushing-Syndrom durch seine kurze Verlaufsdauer, seine abweichende Geschlechtsverteilung, die häufig auftretende hypokaliämische Alkalose sowie durch die Hyperpigmentierung. Eine Abgrenzung des ektopen ACTH-Syndroms gegenüber den klassischen endogenen Varianten erscheint daher gerechtfertigt (Bower., 1965; Jacobasch, 1967; Pfotenhauer und Kracht, 1967; Sachs, 1965, 1969; Bricaire and Leprat, 1969; Labhart, 1971; Albeaux Fernet, 1970; Flury et al., 1971; Knight et al., 1971; Mason, 1971; Saeger und Mitschke, 1973). Allgemeine Angaben über die Häufigkeit der endokrinen und renalen Paraneoplasien liegen nicht vor. Bei den ausgewerten Fällen handelte handelte es sich – in 34% der Fälle um ein ektopes ACTH-Syndrom, – in 26% der Fälle um eine extrapankreatische Hypoglykämie, – in 16% der Fälle um eine paraneoplastische Hyperkalzämie, – in 14% der Fälle um ein ektopes ADH-Syndrom und – in 10% um andere endokrine Paraneoplasien. Pathogenese Aufgrund der klinischen Befunde wurde eine Reihe von Hypothesen zur Erklärung der kausalen und formalen Pathogenese des ektopen ACTH-Syndroms aufgestellt: ● Zufälliges Zusammentreffen von Endokrinopathie und Tumor. Anfangs hielt man das gleichzeitige Auftreten der endokrinen Störung und eines neoplastischen Prozesses für zufällig (Brown, 1928). Weitere Untersuchungen und ein größeres Patientengut zeigten jedoch, dass bestimmte Tumorlokalisationen und -typen bevorzugt auftraten (Lunge, Mediastinum, Pankreas). Außerdem spracht eine deutliche Verschiebung der Relation zwischen Männern und Frauen gegen ein zufälliges Zusammentreffen. Das Pankreaskarzinom kommt nach Hegglin (1972) bei Männern 5-mal häu- Ein Tumor (meist eine maligne Neubildung) kann verschiedene Substanzen der Peptidkaskade produzieren: Prohormone, Hormone sowie Spaltprodukte. Die Produktion von Hormonvorstufen kann für eine abweichende Symptomatik verantwortlich sein. Die relative Häufigkeit der Primärtumo- 2 Ektopes ACTH-Syndrom figer vor als bei Frauen. Unter den zusammengestellten Fällen eines ektopen ACTH-Syndroms in Verbindung mit einem Pankreaskarzinom war das Geschlechtsverhältnis von Mann:Frau = 1:4. Pfotenhauer und Kracht (1967) gaben die Häufigkeit des ektopen ACTH-Syndroms im Sektions- und Operationsgut an Morbus Cushing bzw. Cushing-Syndrom (ohne Berücksichtigung kindlicher Cushing-Fälle) mit 5,7% an. So kann allein aus der Häufigkeit des Syndroms und der veränderten Geschlechtsspezifität ein rein zu fälliges Zusammentreffen der Endokrinopathie und des Tumors ausgeschlossen werden. Besondere Beachtung fanden Berichte, in denen eine bilaterale Nebennierenrindenhyperplasie, ein Hyperkortisolismus und ein Karzinom ohne klinische Symptome des ektopen ACTH-Syndroms nachgewiesen wurden. In den von Nichols und Gourley (1963) untersuchten Primärtumoren konnte eine Substanz festgestellt werden, die das Gewicht der Nebennieren von hypophysektomierten Ratten konstant hielt. Sie schlossen hieraus auf das Vorliegen von physiologisch unterschiedlich wirksamen ACTH-ähnlichen Substanzen: – eine Substanz, die der Hyperplasie der Nebennierenrinde verursacht (weight maintaining factor), – ein die Steroidsekretion auslösender Faktor, der die Cushing- Symptomatik induziert. – Bildung von Peptiden mit Corticotropin-ReleasingFactor ähnlicher Aktivität (CRF) durch den Tumor. Während in allen Fällen die ACTH-Sekretion durch Dexamethason nicht gehemmt ● Erhöhte Nebennierenrindenaktivität infolge einer Metastasierung des Tumors in die Nebenniere. Diese wurde bei 263 Fällen eines ektopen ACTH-Syndroms nur 50-mal beschrieben. Diese vergleichsweise geringe Zahl von Nebennierenrindenmetastasen und das Vorhandensein einer bilateralen Hyperplasie in allen Fällen, selbst in denen mit einseitiger Metastasierung, schließt einen pathogenetischen Zusammenhang aus. Es wurden mehrere Fälle beschrieben bei denen eine Reaktion auf Metopiron bestand (Liddle et al., 1962, 1963, 1964; Meador und Liddle, 1962; Miura et al., 1967; Strott et al, 1968; Jones et al., 1969). Metopiron ist ein 11-ß-Hydroxylasehemmer: Es senkt die Cortisolsyntheserate, steigert jedoch die Produktion von Desoxycorticosteron (Labhart, 197lc). Dieser für ein ektopes ACTH-Syndrom sehr ungewöhnliche Befund deutet darauf hin, dass das Hypothalamus-Hypophysen-Nebennierenrinden-System bei diesen Patienten intakt war. Es wurde nun die Möglichkeit diskutiert, dass gewisse Tumoren eine Substanz bilden, die zu einer gesteigerten ACTH-Sekretion durch die Hypophyse und dem klinischen Bild eines ektopen ACTH Syndroms führt. ● Die primäre Nebennierenrindenhyperplasie fördert die Karzinomentwicklung. In Übereinstimmung mit tierexperimentellen Untersuchungen beobachteten Scholz und Bahn (1959), dass exzessiv hohe Konzentrationen von adrenalen Steroiden sowohl die Entstehung als das Wachstum bestimmter epithelialer Zellen des primitiven Vorderdarmes fördern können. Brickner et al. (1961) wiesen jedoch darauf hin, dass in den Fällen eines Cushing-Syndroms mit langer Verlaufsdauer selten gleichzeitig extraadrenale und extrahypophysäre Karzinome beobachtet werden. ● Reaktiver Hyperkortizismus. De Gennes et al. (1962a) sahen in der gesteigerten Aktivität der Nebennierenrinde eine Reaktion des Organismus auf die Stresswirkung des Karzinoms. Ihre Ansicht wird durch die allgemein bekannte Beobachtung gestützt, dass bei Karzinomen aller Organe eine leichte Erhöhung der basalen Sekretionsrate der adrenalen Steroide bestehen kann (De Gennes et al., 1962a; Caranasos and Ruebner, 1963). Allein der sehr geringe Anteil des ektopen ACTH-Syndroms an der Gesamtzahl der Karzinomerkrankungen macht deutlich, dass eine unspezifische Stresswirkung des Karzinoms nicht als ursächlich für eine solch deutliche Nebennierenrindenüberfunktion angesehen werden kann. Upton und Amatruda (1971) gelang es, aus zwei Lungen- und zwei Pankreaskarzinomen und deren Metastasen eine Substanz zu isolieren, die gezielt die Freisetzung von ACTH aus der Hypophyse in vivo und in vitro steigerte. Aus dem Tumorgewebe wurde ein ACTH-ähnlicher Stoff isoliert. Keines der aus dem Tumor isolierten Peptide mit CRF-ähnlicher Aktivität zeigte einen direkten stimulierenden Effekt auf die Nebennierenrinde. Wird durch den Tumor eine Substanz mit CRF- und ACTHAktivität produziert, so kann die normalerweise durch den hohen ACTH-Gehalt im Plasma gebremste ACTH-Bildung in der Hypophyse erneut angekurbelt werden. Die hohen ACTH-Konzentrationen bedingen die gesteigerte Syntheseleistung der Nebennierenrinde, die sich morphologisch in einer bilateralen Hyperplasie manifestiert. ● Bildung und Freisetzung einer ACTH-ähnlichen Substanz durch den Tumor. Holub und Katz (1961) konnten eine ACTH-ähnliche Substanz in Tumormetastasen nachweisen. Meador und Liddle (1962) isolierten aus dem Primärtumor und den Metastasen von 5 Patienten ACTH-analoge Stoffe in einer Konzentration von 6 bis 125 mE/g Tumorgewebe. Biochemische Analysen der Hypophysen ergaben in 2 Fällen einen verminderten ACTH-Gehalt. Negative Dexamethason-Hemmungsteste bestätigten den autonomen Charakter der ACTH-Sekretion durch den Tumor. Die Steuerung der Hormonproduktion durch das Rückkoppelungssystem zwischen Hypothalamus, Hypophyse und der Nebennierenrinde war empfindlich gestört. Während Jarett und Lacy (1964) das Hypophysen- und TumorACTH als identisch ansahen, vertraten Felber et al. (1965) die Ansicht, dass dem Tumor-ACTH trotz seiner kortikotropen Wirkungen immunologisch andere Eigenschaften zukommen als dem hypophysären ACTH. Berücksichtigt man, dass bereits die Sequenz der ersten 18 Aminosäuren des aus 39 AS bestehenden Corticotropins eine kortikotrope Wirkung entfalten und die Krebszelle verschiedene ACTH-aktive Aminosäuresequenzen synthetisieren kann, so ist eine solche Abweichung sogar zu erwarten (Pfotenhauer und Kracht, 1967). Faktoren mit einer ACTH-Wirkung werden heute dem Vorläufermolekül Proopiomelanocortin (POMC) zugeordnet. Da diese Verbindung oft unvollständig synthetisiert wird, 3 Endokrine und renale Paraneoplasien ● Der häufigste Tumor ist das das kleinzellige Lungenkarzinom (55%), gefolgt von den Tumoren des Thymus und des Mediastinums (12%). Es folgen die Pankreastumoren (10,4%). Unter den Tumoren, die gelegentlich mit einer Nebennierenrindenüberfunktion einhergehen, sind das Schilddrüsenkarzinom (medullärer Typ), das Phäochromozytom, Karzinome der Leber, Gallenblase, Parotis, Ösophagus, Mamma, Cervix uteri, Ovar sowie die Karzinoide des Magen-Darm -Traktes zu nennen. kann die biologische Aktivität – und somit die klinische Manifestation – sehr unterschiedlich sein. Die formale Pathogenese der klinischen Symptome beim ektopen ACTH-Syndrom erklärt sich aus der Steroid-Überproduktion durch die Nebennierenrinde. Das Glucocorticoid Cortisol, dessen Konzentration unter allen Steroiden am deutlichsten erhöht ist, stimuliert die Gluconeogenese (Rapoport, 1969). Es führt zu einer negativen Stickstoffbilanz, einer verminderten Glucosetoleranz und zu eines erhöhtem Blutzucker. Die zuweilen sehr starke und an einen Morbus Addison erinnernde Pigmentierung der Haut kann sowohl durch die gleichzeitige Sekretion von MSH durch den Tumor (Ney et al., 1963; Steeno et al., 1965; O'Neal et al., 1968) als auch durch das ACTH, dessen melanozytenstimulierende Wirkung hinreichend bekannt ist, verursacht werden. Azzopardi und Whittaker (1968) stellten in den meisten der bisher publizierten Fällen Nachuntersuchungen an und kamen zu dem Ergebnis, dass allein die kleinzelligen Karzinome (kleinzellige Ca) sowie die bronchialen Karzinoide histologisch einwandfrei diagnostiziert wurden. So habe das von Rosenthal (1957) und Pfohl (1963) beschriebene Adenokarzinom weit größere Ähnlichkeit mit einem kleinzelligen Ca (strangförmig angeordnete Zellen) als mit einem Adenokarzinom. Die histologische Diagnose wurde häufig nur sehr vage gestellt, wie auch im Fall von Hills (1968), der ein »gut differenziertes, nicht muzinproduzierendes Adenokarzinom der Lunge« beschrieb, das zwar eine negative argentaffin Reaktion zeigte, aber dennoch im histologischen Bild eine gewisse Karzinoidähnlichkeit aufwies. Ebenso beschrieben Sachs et al. (1970) im elektronenmikroskopischen Bild eines lichtmikroskopisch eindeutigen Adenokarzinoms granuläre und vesikuläre Strukturen, die in ihrer Größe und Neigung sich traubenförmig anzuordnen, den von Karzinoiden und Inselzellen des Typs A her bekannten morphologischen Mustern ähnelten. Die sehr häufig beschriebenen Symptome einer extremen Muskelschwäche, passageren, mit Diuretika nicht zu beherrschenden Gesichts- und Knöchelödemen, Polyurie und Polydipsie sowie die sich schon sehr früh manifestierende hypokaliämische Alkalose lassen zunächst an das Vorliegen eines Conn-Syndroms denken. Die Hypokaliämie ist therapeutisch auch durch große, andauernde Kaliumgaben meist nicht zu beherrschen. Die Aldosteronsekretion wurde in den meisten daraufhin untersuchten Fällen als normal bzw. an der unteren Normgrenze gefunden (Balls et al., 1959; Allott und Skelton, 1960; Meador et al., 1962; Guinet et aJ., 1967; Biglieri et al., 1968). Strolt et al. (1968) wiesen auf die bronchialen Karzinoide und Adenome hin, die in 17 Fällen mit einem ektopen ACTH-Syndrom auftraten. In jüngster Zeit wird immer wieder aufgrund von elektronenmikroskopischen Untersuchungen die Ansicht vertreten, dass das kleinzellige Karzinom als eine hochmaligne Variante des bronchialen Karzinoids anzusehen sei (Siegenthaler et al., 1965; Hattor et al., 1968; Bensch et al., 1965, 1968). Die Tatsache, dass weder die Hypokaliämie noch der hohe Kaliumverlust im Urin durch den Aldosteronantagonisten Spironolacton beherrscht werden konnte, beweist, dass die Elektrolytstörung in diesen Fällen nicht durch das Aldosteron verursacht worden war. Cost (1963) machte vielmehr den erhöhten Corticosteronspiegel im Plasma für diese Elektrolytstörungen verantwortlich; Corticosteron nimmt eine Mittelstellung zwischen den Gluco- und Mineralocorticoiden ein. Diese Steroide wirken nun verstärkt auf die Membranen der distalen Tubulus- und Sammelrohrzellen und führen zu einer Intensivierung der dort ablaufenden Ionentransporte (Hökfelt, 1959; Hänze und Pierach, 1966). Mehrere Autoren wiesen auf die Parallelität zwischen Intensität der Steroidausscheidung und dem Grad der Hypokaliämie hin (Prunty et al., 1963; Bricaire et al., 1965). Eine Folge der zum Teil sehr ausgeprägten Hypokaliämie ist die Adynamie der Muskulatur, die als Frühsymptom in Verbindung mit einem Cushing-Syndrom zu einer intensiven Tumorsuche veranlassen sollte. ● Tumoren des Thymus (Karzinoide). Schon nach den ersten Berichten über Thymuskarzinome in Verbindung mit einem ektopen ACTH-Syndrom wurde insbesondere von Kracht und Hantschmann (1961) sowie Engel und Kahana (1963) darauf hingewiesen, dass in zahlreichen Fällen der Primärtumor zwar im Bronchialsystem lokalisiert, dort aber nicht mehr nachzuweisen war. Die Ähnlichkeit der histologischen Bilder von kleinzelligen Karzinomen und Thymomen wurde vermutet (Hills, 1968) und durch elektronenmikroskopische Untersuchungen von Kay und Willson (1970) bestätigt. Scholz und Bahn (1959) beschrieben ein rein epitheliales Karzinom mit gangähnlichen Strukturen, das eine auffällige Ähnlichkeit mit der Morphologie des bronchialen Karzinoids aufwies. ● Tumoren des Pankreas (Inselzelltumoren): Als weitere große Gruppe müssen die Tumoren des Pankreas genannt werden. Azzopardi (1968) betonte, dass auch bei diesen Tumoren häufig sehr unzureichende histologische Diagnosen gestellt wurden und mehr als 70% aller Tumoren des Pankreas in Verbindung mit einem ektopen ACTH-Syndrom histologisch Inselzellkarzinome waren. Es wurde auf die Schwierigkeit der histologischen Differenzierung und der strukturellen Ähnlichkeit mit dem kleinzelligen Karzinom der Lunge hingewiesen (Hallwright et al., 1964). Pathologische Anatomie Das pathologisch-anatomische Substrat beim ektopen ACTH-Syndrom besteht aus einer Nebennierenrindenhyperplasie, dem Vorkommen von Crooke-Zellen im Hypophysenvorderlappen und einem meist undifferenzierten Karzinom. 4 Ektopes ACTH-Syndrom Tumorlokalisation beim ektopen ACTH-Syndrom ● Tumoren der Schilddrüse: Hier wurden ebenfalls die anfänglich sehr abweichenden histologischen Diagnosen durch Nachuntersuchungen von Williams et al. (1968 b) korrigiert. Bei 12 der 13 Fälle lagen medulläre Schilddrüsenkarzinome mit typischem Amyloidstroma vor. Williams und Sandler wiesen schon 1965 darauf hin, dass diese Tumoren der Schilddrüse von den parafollikulären Zellen der Schilddrüse abstammen (sog. C-Zellen). Tierexperimentelle Untersuchungen erbrachten den Nachweis von 5-Hydroxytryptamin in parafollikulären Zellen bei Schafen (Falck et al., 1964). Moertel et al. berichteten über ein medulläres Schilddrüsenkarzinom, das mit einem Karzinoidsyndrom vergesellschaftet war. Weiterhin konnte bei den medullären Schilddrüsenkarzinomen häufig ein argyrophiles Verhalten nachgewiesen werden (Williams et al., 1968b). Aufgrund dieser Befunde scheint es sehr wahrscheinlich, dass auch die medullären Schilddrüsenkarzinome der Gruppe der Karzinoidtumoren zuzurechnen sind. Die erste Mitteilung über ein ektopes ACTH-Syndrom bei einem Blasentumor stammt von Roth und Stiller (1972). Histologisch entsprach der Tumor den von Evans (1966) angegebenen Kriterien eines entdifferenzierten Harnblasenkarzinoms mit erheblichem Pleomorphismus, pathologischen Mitosen und bizarren Riesezellen. Als weitere Komplikation bot der veröffentlichte Fall einen kardiovaskulären Symptomenkomplex, für den eine abakterielle Endokarditis polyposa fibrinosa und eine Thrombophlebitis caerulea sive migrans ohne entzündliche Reaktion typisch war. Die Unsicherheiten und Schwierigkeiten bei einer genauen histologischen Diagnose und Zuordnung zu einem definier- Eine abweichende Tumorlokalisation liegt bei den im Kindesalter auftretenden Fällen eines ektopen ACTH-Syn- ten Tumorbild fand sich auch bei anderen Geschwülsten. Viele Neubildungen (Parotis, Karzinoide und Karzinome des Magen-Darm-Traktes, Parathyroidea, Gallenblase) hatten eines gemeinsam: Alle diese epithelialen Tumoren stammen von Strukturen des primitiven Vorderdarmes ab, denen eine endokrine Potenz zugesprochen wird (Williams et al., 1968b; Azzopardi et al., 1968). 5 Endokrine und renale Paraneoplasien Thymustumoren beim Cushing-Syndrom Die pathologischen Veränderungen der Nebennierenrinde gehören als Kardinalbefund zur pathologischen Anatomie des ektopen ACTH-Syndroms. Das Nebennierengewicht (30-35 g) lag bei diesem Krankheitsbild im Durchschnitt über den Werten bei Morbus Cushing hypothalamisch-hypophysärer Genese mit bilateraler Hyperplasie. Histologisch bestand eine bilaterale, diffuse Rindenhyperplasie, wobei die normale Zonenaufteilung aufgehoben war. Die Rinde war vorwiegend faszikulär gestaltet. Daneben wurden auch knotige Rindenhypertrophien beobachtet (Huoson, B., 1962; Hänze und Pierach, 1966; Pfotenhauer und Kracht, 1967). droms vor. Während man bei Erwachsenen in mehr als der Hälfte aller Fälle Bronchialtumoren in Verbindung mit einem ektopen ACTH-Syndrom fand, fiel keiner der 14 Fälle bei Kindern und Jugendlichen in diese Gruppe. Dies ist jedoch nicht ungewöhnlich, da Tumoren des Bronchialsystems im frühen Lebensalter nur sehr selten vorkommen. Es finden sich dagegen Tumoren, die im weitesten Sinne von den Paraganglien ausgehen, die auch sonst bei Kindern und Jugendlichen gehäuft auftreten (Kracht, 1971; Omenn, 1971). Bei 78 nicht ausgewählten viszeralen Karzinomen wurden ACTH Bestimmungen im Tumorgewebe durchgeführt. 72 Fälle erbrachten negative, 6 positive Ergebnisse. Unter letzteren befand sich ein kleines Kind (Angaben über Alter und Geschlecht fehlen) mit einem Cushing-Syndrom und einem Nebennierenrindenkarzinom. Der ACTH-Nachweis war in einer Lungenmetastase geführt worden. In der makroskopisch unauffälligen Hypophyse fanden sich in mehr als 70% der untersuchten Fälle stark vermehrte basophile Zellen des ß-1-Typs (R-Zellen). Diese großen Zellen – hauptsächlich im zentralen Teil der Drüse lokalisiert – zeigten multilokuläre, zentrale zytoplasmatische Vakuolen, eine Hyalinisierung des peripheren Zytoplasmas und in einigen Bezirken gewisse zellspezifische Granulationen. Sie 6 Ektopes ACTH-Syndrom Pankreastumoren beim ektopen Cushing-Syndrom Ektopes ACTH-Syndrom. Klinische Symptome karzinom (Welbourn et al., 1971). Es fällt auf, dass es sich bei 57% der Fälle um Männer handelte, während sonst 80% der Cushing-Patienten weiblichen Geschlechts sind. werden nach dem Autor der Erstbeschreibung als CrookeZellen bezeichnet (Crooke, A. C., 1961; Kennedy, J. H., 1964; Morse et al., 1967, Sasano, 1969; Pfotenhauer und Kracht, 1967). Die Hyalinisierung dieser R-Zellen wird als eine Folge des exzessiv erhöhten, zirkulierenden Cortisols angesehen (Welbourn, 1969). Des weiteren konnte in vielen Fällen eine Übereinstimmung zwischen der Dicke der Nebennierenrinde und der Anzahl der Crooke-Zellen im HVL beobachtet werden (Sasano et al., 1969). Auf das Fehlen von chromophoben Zellen, denen die ACTH-Bildung im HVL zugeschrieben wird, wiesen Morse et al. (1967) hin. Das klinische Bild des ektopen ACTH-Syndroms ist durch das gleichzeitige Vorkommen von Tumorsymptomen und Zeichen des Hyperkortizismus gekennzeichnet. Gegenüber dem Cushing-Syndrom hypothalamisch-hypophysärer Genese weist es folgende Besonderheiten auf: – maligner Verlauf mit einer durchschnittlichen Lebenserwartung von 9 Monaten; – die klinische Cushing-Symptomatik kann fehlen; – schwere Elektrolytstörung in Form einer hypokaliämischen Alkalose; – häufig sehr ausgeprägte, an einen Morbus Addison erinnernde Pigmentierung. Klinik Bei der jüngsten Patientin handelte es sich um ein 9-jähriges Mädchen mit einem lnselzellkarzinom (Uei, 1968), die älteste Patientin war eine 84-jährige Frau mit einem Bronchial- 7 Endokrine und renale Paraneoplasien Tumorlokalisation beim ektopen ACTH-Syndrom bei Kindern ln rund 50% der Fälle fehlte die typische klinische CushingSymptomatik, obwohl die Cortisolsekretion eindeutig hoch und die Nebennierenrinden immer hyperplastisch waren. Als Erklärung für diese Befunde wird die kurze Verlaufsdauer angenommen, denn es war typisch, dass Patienten ohne das klinische Vollbild des Cushing-Syndroms in der Regel 3 Monate nach Auftreten der ersten Symptome der Krankheit verstarben (Pfotenhauer und Kracht, 1967; Labhart, 1971c). Cushing-Syndrom häufig ein langsam wachsender Tumor vorlag, der sehr wahrscheinlich auch wenig ACTH produzierte. In den meisten Fällen war ein derartiger Tumor im Mediastinum, Thymus, Pankreas oder Magen-Darm-Trakt (Karzinoid) lokalisiert. Hänze und Pierach (1966) sahen in der Kürze des klinischen Verlaufs keine Erklärung für das Ausbleiben der Cushing-Symptomatik, da ein iatrogen durch Steroidmedikation ausgelöstes Cushing-Syndrom bei hoher Dosierung in einigen Wochen bis wenigen Monaten entstehen könne. Beobachtungen von Camus et al. (1961) sowie Engel und Kahana (1963) ergaben, dass bei Patienten mit typischem 8 Ektopes ACTH-Syndrom Liddle et al. (1969) betonten, dass Patienten mit ektopem ACTH Syndrom immer an Gewicht verlieren, obgleich die Plasmaspiegel der stark anabolen Glucocorticoide teilweise exzessiv erhöht sind. Aufgrund dieses Gewichtsverlustes kommt es zu einer Umverteilung des Depotfettes, dem Abbau einer zentripetalen Adipositas; ebenso fehlen die für den hypophysären Cushing typischen Striae rubrae (Meador et al., 1962; Eastridge et al., 1965). war. In den von Lockwood (1958), Webster et al. (1959), Farant und Insley (1960) und Pfotenhauer und Kracht (1967) beschriebenen Fällen konnte die Hypersekretion von Aldosteron nicht die Ursache der Elektrolytstörung sein, da durch die Gabe von Spironolacton keine Wirkung erzielt wurde. Meador et al. (1962) fanden, dass die Ausscheidung von Kalium im Urin 50 mE/24 h trotz einer bestehenden schweren Hypokaliämie von weniger als 3 mEq/I überschritt. Die Hypokaliämie war therapeutisch auch durch größere, lang dauernde Verabreichung von Kalium meist nicht zu korrigieren (Pfotenhauer und Kracht, 1967; Liddle et al., 1969). Als ein weiteres Argument für die geringe klinische Manifestation wurde das bevorzugte Auftreten bei Männern angegeben: Virilisationserscheinungen bei einer Überproduktion von adrenalen Androgenen treten bei Frauen auffälliger hervor (Meador et al., 1962; Liddle et al., 1969). Eine Folge der Elektrolytstörungen waren die passageren Gesichts- und Knöchelödeme, die mit Diuretika nicht auszuschwemmen waren (Siegenthaler, 1967) und mit dem Ausbruch des akuten Krankheitsbildes sich zurückbildeten, ebenso die Polyurie, Polydipsie sowie die extreme muskuläre Adynamie. Diese Symptome traten in 30 bis 40% aller Fälle eines ektopen ACTH-Syndroms auf. Während man eine diabetische Stoffwechsellage nur in etwa 15% aller hypophysären Cushing-Fälle nachweisen kann (Labhart, 1971c), trat ein Diabetes beim ektopen ACTHSyndrom in ungefähr 90% aller Fälle auf (Eastridge et al., 1965; Liddle et al., 1969). Dabei handelte es sich um einen Steroiddiabetes, der nicht auf Insulinmangel, sondern auf die Überproduktion von Cortisol zurückging. Wie beim gewöhnlichen Diabetes wurde zu viel Glucose gebildet, aber im Gegensatz zu diesem war der periphere Verbrauch nicht gestört. Dieser Steroiddiabetes war zunächst gutartig, da er nicht zu hohe Blutzuckerwerte aufwies, stabil verlief und im allgemeinen nicht zu einer Ketose führte (Dfaot et al., 1968). Dieser Steroiddiabetes war durch Insulin kaum zu beeinflussen. Erst im fortgeschrittenen Stadium waren Blutzuckerwerte von 300 mg% und eine Glukosurie von 4 bis 9 g/24 h keine Seltenheit (Hänze und Pierach, 1966). Eine diabetische Ketose – sehr seltene Komplikation eines Steroiddiabetes bei einem ektopen ACTH-Syndrom – beschrieb Smith (1965). Auffallend war außerdem die starke Hyperpigmentierung, wie bei einem Addison-ähnlichen Bild, aber nur selten mit einer Schleihautbeteiligung auftrat (Steeno et al., 1965; Weizenecker und Burke, 1970). Untersuchungen von Abe et al. (1967) deuteten darauf hin, dass eine ektope ACTH-Bildung mit Freisetzung von MSH einherging. Neben der MSH-Aktivität des Tumor-ACTH wurden α-MSH und eine noch nicht identifizierte MSH-Komponente nachgewiesen, sodass mindestens 3 Tumormelanotropine vorkamen (Law et al., 1965; Steeno et al., 1965; Ney, 1965; O'Neal et al., 1968). Auf die melanozytenstimulierende Wirkung des ACTH wurde bereits im Abschnitt Pathogenese hingewiesen. Bei einem voll entwickelten paraneoplastischen CushingSyndrom wurden häufig psychische Veränderungen im Rahmen des endokrinen Psychosyndroms nachgewiesen. Apathische und erregte Stimmungslagen waren häufig, Übergänge in psychotische Episoden möglich. Es traten Verwirrungszustände, Halluzinationen sowie Wahnvorstellungen auf (Kaplan et al., 1949; Bleuler, 1954; Bricaire, 1965; Labhart, 1971c). Die Hypertonie war mit 65% (Eastridge et al., 1965: 50%) seltener als beim Cushing-Syndrom hypophysärer Genese (80%). Die Blutdruckwerte waren meistens nur wenig über dem Normwert erhöht. Die Pathogenese der Hypertonie ist noch ungeklärt: Sie muss auf die Cortisolüberproduktion zurückgeführt werden, da die Mineralocorticoidsekretion nicht erhöht waren. Cortisol hemmt die renale Natriumausscheidung und ist für die hypertone Wirkung des Noradrenalins an der Muskulatur der Arteriolen erforderlich (Eastridge et al., 1965; Labhart, 1971c). Eine besondere differenzialdiagnostische Bedeutung beim ektopen ACTH-Syndrom kommt den biochemischen Untersuchungen zu. In 70 der 263 Fälle wurden im Tumorgewebe und /oder Metastasen eine ACTH-ähnliche Substanz nachgewiesen. Lidd le et al. (1969) konnten bei 21 Fällen keinen Unterschied zwischen Hypophysen- und TumorACTH feststellen. Sie fanden bei ihren Patienten ACTHKonzentrationen von 0,27 bis 125 mE/g Tumorgewebe, die gemessen am ACTH-Gehalt der Hypophysen endokrin Gesunder wesentlich niedriger waren (Meador et al., 1962). Eine Osteoporose entwickelte sich nur selten und war wahrscheinlich auf die relativ geringe Überlebenszeit der Patienten zurückzuführen (Hubble, 1949; Siegenthaler, 1967; Uei et al., 1968). Das Vorkommen einer Amenorrhoe und der Striae rubrae wurde ebenfalls nur in vereinzelten Fällen beschrieben (McLetchie und Scott, 1942; Crooke, 1946; Scholz und Bahn, 1959). Die geringen Konzentrationen im Tumorgewebe erklären, warum mit relativ ungenauen Bestimmungsmethoden keine ACTH-Aktivität in vielen Tumoren festgestellt werden konnte. Der niedrigen ACTH-Konzentration im Tumor stand die große Tumormasse gegenüber, sodass man die absolute, in dem Malignom enthaltene Menge von ACTH-ähnlichen Verbindungen durchaus mit der Menge in der Hypophyse vergleichen kann (Hänze und Pierach, 1966). Die hypokaliämische Alkalose kann als Kardinalsymptom des ektopen ACTH-Syndroms angesehen und in ungefähr 75% aller Fälle bereits zu einem sehr frühen Zeitpunkt gefunden werden. Das Aldosteron spielte in diesem Zusammenhang keine Rolle, da die Sekretionsrate nicht erhöht 9 Endokrine und renale Paraneoplasien Die Plasmaelektrolyte veränderten sich im Sinne einer hypokaliämischen Alkalose, das Natrium war vermindert (125 mEq/l; O'Neal et al., 1968), normal (140 mEq/l ; Morse et al., 1967) oder auch erhöht (155 mEq/ l; Szijj et al., 1969). subtotale Adrenalektomie brachte meist keine befriedigende Ergebnisse; bei mehreren Fällen entwickelte sich erneut ein Cushing-Syndrom (McPhee, 1969; Camus et al., 1961; Engel und Kahana, 1963; Anderson und Glenn, 1966). Analysiert man die Ausscheidung der Steroidmetaboliten im Urin, so findet man deutlich erhöhte Cortisolwerte (bis zu 83 µg/100 ml, (normal:: 6 bis 25 µg/100 ml, Porter-Silber- Reaktion), die nach Brooks et al. (1963) noch höher liegen als beim hypothalamisch-hypophysären Cushing-Syndrom. Die 17-Hydroxycorticosteroide im Plasma waren stark erhöht, demzufolge auch die 17-Hydroxycorticoide und 17-Ketosteroide im Urin. Die Steigerung der Nebennierenrindenaktivität ging mit einem Verlust des normalen täglichen Ausscheidungsrhythmus einher. Die zeitliche Korrelation Tumor/Endokrinopathie ist in allen Varianten beschrieben worden: Das Bild der Nebennierenrindenüberfunktion konnte klinisch manifest werden, bevor der zugrunde liegende Tumor diagnostiziert wurde (Sobota und Reed, 1964; Reiche! und Moll, 1964; Steeno et al., 1965; Jensen et al., 1965). ln anderen Fällen wurde der Tumor vor Auftreten des ektopen ACTH-Syndroms nachgewiesen (Allott und Skelton, 1960; O'Riordan et al., 1966). Bei den meisten Patienten traten die Symptome der Endokrinopathie und des begleitenden Tumors gleichzeitig auf (Bagshawe, 1960). O'Riordan et al. (1966) beschrieben einen Fall, bei dem erst nach der Bestrahlung und operativen Entfernung des Primärtumors ACTH-sezernierende Metastasen zur Ausbildung eines Cushing-Syndroms führten. Die Ausscheidung von Testosteron und Androgenmetaboliten befanden sich im Normbereich, nur Holzmann et al. (1968) beschrieben bei einer 32-jährigen Frau mit einem ektopen ACTH-Syndrom, einem kleinzelligen Karzinom und starken Virilisierungserscheinungen eine um das 40- bis 100fach gesteigerte Testosteronausscheidung. Die Lebenserwartung beim ektopen ACTH-Syndrom reichte in der Regel von 30 Tage bis zu 2 Jahre (Labhart, 1971c), im Durchschnitt 9 Monate. Gelang es jedoch, den Tumor rechtzeitig zu diagnostizieren und radikal operativ zu entfernen, war die Lebenserwartung höher; in einigen Fällen wurde sogar eine Fünfjahresheilung erreicht (Cohen et al., 1960; Riggs und Spraguf, 1961; Anderson und Glenn, 1966; Steel et al., 1967 ; Morse et al., 1967; Welbourn et al., 1971; Vingerhoeds et al., 1971; Johnston und Waismann, 1971). Die Diagnose ektopes ACTH-Syndrom bietet keine größeren Schwierigkeiten, wenn sich bei einem Tumor Symptome eines Cushing-Syndroms entwickeln. Bei einem foudroyanten Verlauf eines ektopen ACTH-Syndroms ohne sicheren Tumornachweis kann man dagegen die Paraneoplasie nur vermuten: Eine rasch zunehmende Pigmentierung, eine hypokaliämische Alkalose und erhöhte Corticosteroidwerte im Plasma und Urin sind wichtige diagnostische Hinweise. Diese Befunde unterstreichen die Bedeutung einer frühen Diagnose des Tumors. Die Feststellung einer zunächst pathogenetisch unklaren hypokaliämischen Alkalose und muskulären Adynamie kann sich als wertvolles diagnostisches Leitsymptom für die Früherkennung eines ektop endokrin aktiven malignen Tumors erweisen. Differenzialdiagnostische Bedeutung kommt den Tests mit ACTH i.v., Dexamethason und Metopiron zu. Beim ACTHStimulations-Test wird die Ausscheidung der 17-Hydroxycorticosteroide und der 17-Hydroxycorticoidspiegel im Plasma während einer 8-stündigen ACTH-Dauerinfusion verfolgt (Martin und Hamman, 1966; Flury et al., 1971; Labhart, 1971C). Nur bei einem Teil der Patienten mit ektopem ACTH-Syndrom kam es im Verlauf dieser Tests zu einem Anstieg der Steroidproduktion sowie der Plasma und Urinkonzentration, bei den übrigen Fällen war die Nebennierenrinde schon maximal stimuliert (Hallwright et al., 1964; Jakobasch, 1967). 2 Extrapankreatische Hypoglykämie Aufgrund der unterschiedlichen pathologischen Anatomie und Lokalisation der Neubildungen unterscheidet man mehrere Syndrome, die nach den Autoren der Erstbeschreibung benannt werden: mesenchymale Tumoren beim Doege-Potter, Hepatome beim Nadler-Wolfer-Elliott-Syndrom, Tumoren der Nebennierenrinde beim Anderson-Syndrom und Pseudomyxome beim Rosenfeld-Syndrom. Karakteristisch für diese Paraneoplasie ist die Nüchtern-Hypoglykämie, die nicht auf einen Inselzelltumor zurückzuführen ist. Die Pathogenese wird durch eine Überproduktion von ICF-2 (Insulin like growth factor) erklärt. Nach radikaler chirurgischer Entfernung des Tumors bildeten sich häufiger die Zeichen des Hyperkortizismus – mit Ausnahme der Hyperpigmentierung – innerhalb kurzer Zeit zurück (Cohen et al., 1960; Riggs und Sprague, 1961; Kogut und Donell, 1961; Micic und Arsenijevm, 1963; Jarett et al., 1964; Livingston, 1965; Anderson und Glenn, 1966). Eine Strahlenbehandlung des Primärtumors war meist von geringer Bedeutung, da sie nicht auf die Metastasen einwirkte. Lediglich beim Fall von Anderson und Glenn (1966) war sie erfolgreich. 2.1 Doege-Potter-Syndrom Begriffsbestimmung Beim Doege-Potter-Syndrom handelt es sich um mesenchymale Tumoren, die durch hypoglykämische Anfälle klinisch charakterisiert sind. Eine bilaterale Adrenalektomie kam dann in Betracht, wenn die endokrinen Symptome bei noch geringer Ausdehnung des Primärtumors sehr ausgeprägt waren. Ob sie den Cortisolspiegel senkte, die Progression und Metastasierung des Tumors verzögerte, ist ungewiss (Wyss et al., 1971). Eine Pathogenese Die Pathogenese des Doege-Potter-Syndroms läßt sich nicht durch einen einzelnen pathogenetischen Mechanismus erklären. Die unterschiedlichen biochemischen Befunde verschie- 10 Extrapankreatische Hypoglykämie dener Autoren führten Laurent et al. (1971) auf die abweichenden Analysetechniken und Tests sowie auf die unterschiedlichen Bedingungen zurück, unter denen das zu untersuchende Material gewonnen wurde. auf das Fettgewebe von Ratten in vitro einen antilipolytischen Effekt zeigte. Sie folgerten hieraus, dass eine Lipolysehemmung die Glucoseoxidation begünstige und so zu einer Hypoglykämie führe. Allen Theorien zur Pathogenese gemeinsam ist, dass die mesenchymalen Tumoren als allein ursächlich für die Hypoglykämie angesehen werden (Meyer-Hofmann, G., 1960). Es werden folgende Mechanismen diskutiert: – Hypoglykämie bei einem metaplastischen Nesidioblastom, – Hypoglykämie infolge einer direkten mechanischen Wirkung des Tumors, – Hypoglykämie infolge einer primären Anomalie des Insulins sowie infolge der Produktion einer insulinähnlichen Substanz, – Hypoglykämie infolge einer primären Beeinflussung des Fettstoffwechsels, – Hypoglykämie aufgrund von Störungen im Glucosestoffwechsel. ● Primäre Störung des Glucosestoffwechsels. Exzessiver Glucoseverbrauch durch den Tumor. Diese Hypothese wurde von vielen Autoren mit folgenden Argumenten diskutiert: – hohes Gewicht der Tumoren, – Milchsäurekonzentration im Plasma deutlich erhöht (August und Hiatt, 1958; Barjon, 1961; Frerichs, 1970), – erhöhte Milchsäurekonzentration im Tumor (Barjon und Labauge, 1961; Laurent, 1969), – erhöhte Konzentration von hyperanabolen Substanzen (Glykogen, Aminosäuren u. a.) (Sellman et al., 1959; Laurent et al., 1971). Gegen diese Theorie wurde eingewandt: – Nicht alle Tumoren waren sehr groß (Scholz et al., 1957; Boshell et al., 1964), – die Tumoren entwickelten sich langsam (Lowbeer, 1961), – der Glucoseverbrauch durch die Tumoren war in vitro »mäßig« (Miller et al., 1959), – eine zu erwartende hyperplastische Reaktion des Pankreas lag nicht vor (Hayes et al., 1961). ● Metaplastisches Nesidioblastom. Skillern et al. stellten 1954 die Hypothese der Metaplasie von Tumoren, die von Inselzellen des Pankreas abstammen, auf. Es wurden in der Folgezeit verschiedene Fälle mitgeteilt, bei denen der immunchemisch bestimmte Plasmainsulinspiegel sehr hoch war (Oleesky et al., 1962; Samols, 1963). ● Die Fähigkeit eines Spindelzellsarkoms des Pankreas Insulin zu sezernieren, ist bekannt. Nach Skillern et al. (1954) kann eine fibrosarkomatöse Metastase eines derartigen Sarkoms die Störungen im Glucosestoffwechsel hervorrufen. Laurent et al. (1971) wiesen jedoch darauf hin, dass in einem Großteil der von diesen Autoren beschriebenen Fälle im Pankreas keine Hinweise für einen Primärtumor gefunden wurden. Die von Skillern aufgestellte Hypothese kann daher nur für extrem seltene Sonderfälle Gültigkeit haben. ● Sekretion einer insulinähnlichen Substanz durch den Tumor. Es wurde eine insulinähnlichen Substanz im Tumorgewebe angenommen, deren Aktivität jedoch durch Inhibitorsubstanzen unterdrückt wurde. Unter bestimmten Bedingungen könnte es zu einer Aktivierung dieser »lnsulinoide« kommen und somit zu Hypoglykämie (Roffo, 1927; August und Hiatt, 1958; Miller et al., 1959; Schonfeld et al., 1961; Steinke et al., 1962; Field et al., 1963; Floyd et al., 1963; Saeed et al., 1969). ● Direkte mechanische Wirkung des Tumors. Neben vielen anderen Autoren hatte insbesondere Layzer (1951) diese Hypothese nach Auswertung von 21 Fällen aufgestellt. – Einwirkung des Tumors auf den N. vagus, splanchnicus oder das Ganglion coeliacum; – Störung der autonomen Innervation der Leber, wodurch eine erhöhte Glykogenolyse bewirkt wird; – intrapankreatische venöse Stase, welche das Pankreas zu verstärkter Sekretion von Insulin stimuliert. – Primäre Anomalien des Insulins hinsichtlich Struktur und Stoffwechsel. Insulinähnliche Wachstumsfaktoren (TFG: insulin like growth factor) sind Polypeptide, die dem Insulin sehr ähnlich sind. Sie werden in Hepatozyten und anderen Gewebezellen produziert. Man unterscheidet zwei Varianten: – IGF-1 (Somatomedine C) wird in einer späten Phase der Zellentwicklung und -differenzierung – nach Stimulation durch das Wachstumshormon Somatotropin – produziert. Unter pathologischen Bedingungen wird es bei der Krebsentwicklung (verstärkte Zellproliferation und Unterdrückung der Apoptose) nachgewiesen. – IGF-2 (Somatomedine A) wirkt als Wachstumsfaktor bereits in der Fetalphase. Man hat es bei frühkindlichen Hirntumoren und Mammkarzinomen beobachtet. ● Primäre Störung im Fettstoffwechsel. Samols (1963) sah das Absinken der Konzentration von nicht veresterten Fettsäuren im Serum als Folge eines vom Tumor produzierten Insulins. Froesch et al. (1963) hingegen konnten experimentell nachweisen, dass die Konzentration der nicht veresterten Fettsäuren im Plasma durch eine künstlich hervorgerufene Hyperglykämie (Glucagon) nicht verändert wurde (Hayes et al., 1961; Laurent et al., 1971). Jakob et al. (1966) isolierten aus dem Serum eines Patienten eine Substanz, die Hohe, langandauernde Werte von IGF-1 und IGF-2 können für die Entstehung und Progression von Prostatakarzinomen von Bedeutung sein. 11 Endokrine und renale Paraneoplasien Bei 4 Patienten bestand zuvor ein Diabetes mellitus (Bousvaros, 1960; Nesbitt et al., 1958; Murphy, 1963; Vaissman, 1964), von denen 2 mit Insulin behandelt wurden. Mit der Entwicklung des Tumors »stabilisierte« sich der Diabetes bevor die Hypoglykämie klinisch manifest wurde. Als Begleitsymptome und Komplikationen wurden Gastro-DuodenalUlzera (Scholz et al., 1957; Nesbitt et al., 1958), eine paraneoplastische Acanthosis nigricans (Froesch, 1963) sowie eine hypertrophische pulmonale Osteoarthropatie Pierre-Marie beschrieben (Crawford, 1931; Barjon und Labauge, 1961; Fikenscher und Biom, 1963). Pathologische Anatomie Das pathologisch-anatomische Substrat des Doege-PotterSyndroms ist ein mesenchymaler Tumor. Tumorhistologie beim Doege-Potter-Syndrom Fibrosarkome 73 Leiomyosarkome 10 Lymphosarkome 4 Hämangioperiytome 3 immunoblastisches Lymphome 3 Rhabdomyosarkome 2 M. Hodgkin 1 keine Angabe 4 Insgesamt 100 Auffallend ist, dass bei etwa 10% der Fälle mit Doege-Potter Syndrom gleichzeitig auch eine Störung der Schilddrüsenfunktion vorlag. Bei 7 der 8 beschriebenen Fälle handelte es sich um Frauen. Barenghi (1965, 1966) berichtete über einen 37-jährigen Mann mit Gynäkomastie, Impotenz, hyperthyreoter Struma, Hypoglykämie und einem retroperitonealen Fibrosarkom. Dignität der Primärtumoren: Die Beurteilung der Malignität dieser Tumoren kann schwierig sein. Nach Auswertung der Literaturangaben kann man annehmen, dass in 75% der Fälle die Malignität sicher oder zumindest sehr wahrscheinlich war und nur in 13% ein gutartiger Tumor vorlag. Bei 12% der Neubildungen konnte über die Dignität keine sichere Aussage gemacht werden. Bei etwa 25% der Geschwülste lagen Lebermetastasen vor. Der Krankheitsverlauf hing von der Malignität des Tumors ab. Die Überlebenszeit wurde durch die Hypoglykämie nicht beeinflusst, wenn sie behandelt wurde (Laurent et al., 1971). Bei 45 Publikationen konnten folgende Angaben ermittelt werden: 19 Todesfälle, 4 Rezidive mit unbekanntem postoperativem Verlauf und 22 »geheilt«. Hier sei erwähnt, dass die Kontrollzeit dieser 22 geheilten Patienten weniger als 5 Jahre betrug. Das Volumen sowie das Gewicht der publizierten Tumoren zeigten große Abweichungen: Die schwerste Geschwulst wog 19,9 kg (Duncan und Schless, 1961), die leichteste 0,31kg (Crocker und Veith, 1965). Die Topografie dieser Neubildungen berücksichtigt folgende Lokalisationen: intrathorakal (24 Fälle), intraabdominal (39 Fälle), retroperitoneal (25 Fälle) und peripher (1 Fall). Bei den restlichen 11Fällen konnte die Tumorlokalisation anhand der Literaturangaben nicht bestimmt werden. 2.2 Nadler-Wolfer-Elliott-Syndrom Begriffsbestimmung Beim Nadler-Wolfer-Elliott-Syndrom handelt es sich vorwiegend um hepatozelluläre Karzinome, bei denen im Verlaufe der Erkrankung hypoglykämische Anfälle vorkommen. Nach Untersuchungen von McFadzean und Yeung (1969) kommt dieses Syndrom bei 27 bis 30% aller Hepatome vor. Klinik Im Vordergrund des klinischen Bildes des Doege-Potter-Syndroms kann sowohl die Tumorsymptomatik als auch die Hypoglykämie stehen. Der jüngste Patient war ein 14-jähriger Junge (Nissan et al., 1968), die älteste eine 83-jährige Frau (Rosselin et al., 1967). Geschlechtsverhältnis 1: 1. Bevorzugt befallen war die Altersgruppe zwischen dem 51. und dem 70. Lebensjahr (mehr als 50% der Fälle). Pathogenese Ein Tumor wird als Ursache der Hypoglykämie gedeutet. Die besondere topografische Lage dieser Neubildungen sollen – neben den bereits beim Doege-Potter-Syndrom diskutierten pathogenetischen Mechanismen – noch folgende berücksichtigt werden: – Störung der autonomen Leberinnervation, – stark erhöhter Glucoseverbrauch durch den Tumor, – Hyperinsulinismus (s. Doege-Potter-Syndrom), – Störung der Leberfunktion. Sehr oft standen die Tumorsymptome wie Gewichtsabnahme, Zunahme des Bauchumfangs bei intraabdominellen oder Atembeschwerden bei intrathorakalen Neubildungen im Vordergrund. Die Hypoglykämie trat häufig erst nach dem 2. oder 3. Tumorrezidiv auf (Skillern et al., 1954; Sellman et al., 1959; Leger et al., 1963; Subauste et al., 1965; Tugaye et al., 1965). Die von den Patienten beschriebenen Symptome (Schwächeanfall, Nachtschweiß, Ruhelosigkeit, Angstgefühle, Bewusstseinstrübung, Verschwinden dieser Symptome nach einer kalorienreichen Mahlzeit sowie die biochemisch nachweisbaren Blutzuckerwerte) erlaubten eine schnelle und sichere Diagnose einer Hypoglykämie. Dennoch wurden mehrere Patienten aufgrund der zentralnervösen Symptomatik zunächst in Neurologische Abteilungen eingewiesen (Silvis und Simon, 1956; Kuijer, 1961; Oleesky, 1962; Egeli et al., 1965). ● Klein und Klein (1959) sahen in der Störung der autonomen Leberinnervation die Ursache für eine gesteigerte Glykogenolyse, die infolge der ein geschränkten Leberfunktion zu einer Verarmung an Glucose führte. Mallet-Guy und Feroldi (1966) verneinten jedoch aufgrund experimenteller Untersuchungen die Bedeutung der autonomen Leberinnervation für die Mobilisierung von Leberglykogen. ● Stark erhöhter Glucoseverbrauch durch den Tumor. McFadzean und Yeung (1969) sahen im stark erhöhten Glucoseverbrauch durch den Tumor und in der verminderten 12 Extrapankreatische Hypoglykämie Nadler-Wolfer-Elliot-Syndrom. Unterschiedlicher Hypoglykämieverlauf Als unspezifische Nebenbefunde beschrieben Edmundson (1954) eine vorübergehende Monoplegie, Olmer et al. (1962) ein perforiertes Duodenalulkus. Die Prognose war infaust, da die Patienten innerhalb eines Jahres verstarben. Nachlieferung von Glucose (Zerstörung des restlichen Lebergewebes, Unterernährung) die Ursache der Hypoglykämie bei Tumoren des Typs A. Untersuchungen des Blutphosphorspiegels während einer experimentell erzeugten Hyperglykämie zeigten nur geringe Schwankungen. Dies weist auf einen nur geringen peripheren Verbrauch der Glucose hin. 2.3 Anderson-Syndrom Begriffsbestimmung Beim Anderson-Syndrom wird die paraneoplastische Hypoglykämie durch ein Nebennierenrindenkarzinom hervorgerufen. ● Hyperinsulinismus. Produktion einer insulinähnlichen Substanz durch den Tumor (vergl. Doege-Potter-Syndrom). ● Störung in der Leberfunktion. Nadler und Wolfer (1929) wiesen bereits auf den sehr geringen Glykogengehalt im Lebergewebe der Hepatompatienten hin. McFadzean und Yeung (1956) bestätigten diese »Dispersion des Leberglykogens« bei m Typ A. Bei den Tumoren des Typs B bestand eine zunehmende Reaktionsträgheit auf intravenös zugeführtes Glucagon. Bei der Obduktion konnten sie im geschwulstfreien Lebergewebe große Mengen von Glykogen (21,2 mg/g) nachweisen. McFadzean und Yeung (1969) sahen in dieser erworbenen Glykogenspeicherkrankheit einen zusätzlichen Faktor für die Entstehung der Hypoglykämie und einen wichtigen histochemischen Befund zur Differenzialdiagnose der beiden Tumortypen A und B. Von den 19 Fällen eines Tumors der Nebennierenrinde mit Hypoglykämie konnte bisher nur in 10 Fällen die Tumordiagnose eindeutig histologisch gesichert werden. Angaben über den biochemischen Nachweis stark erhöhter Ausscheidungen von 17-Ketosteroiden und 17-Hydroxysteroiden im Urin (mg/24 h) waren ein weiteres Indiz für das Vorliegen eines Tumors der Nebennierenrinde. Im Vordergrund des klinischen Bildes stand ein Cushing-Syndrom (Fettansatz am Stamm, Vollmondgesicht, Virilismus u. a.). Die Hypoglykämie trat in den meisten Fällen in den Hintergrund und wurde häufig erst nach komatösen Anfällen diagnostiziert. Pathologische Anatomie Das pathologisch-anatomische Substrat des Nadler-WolferElliott Syndroms war in den meisten Fällen ein hepatozelluläres Karzinom. Primäre mesenchymale Leberneubilungen oder Metastasen als Ursache eines Nadler-Wolfer-ElliottSyndroms wurden nur selten beobachtet. Die Leber war stark vergrößert; ihr Gewicht betrug bis zu 6,8 kg (lmperato und Lipton, 1965), bei keinem Fall lag es unter 2 kg. Pathogenese Als Ursache der Hypoglykämie beim Anderson-Syndrom wird der Nebennierenrindentumor angegeben. Neben den bereits beim Doege-Potter- sowie Nadler-Wolfer-Elliott Syndrom beschriebenen Hypothesen zur formalen Pathogenese der Hypoglykämie werden noch folgende Gesichtspunkte angeführt: Nach Eymontt et al. (1965) verbraucht nicht nur der Tumor in erhöhtem Maße Glucose, sondern der gesamte Organismus. Der Tumor soll in großen Mengen anabol wirkende Steroide produzieren. Der hohe Glucose-Verbrauch könnte bei sistierender oder zumindest weitgehend eingeschränkter Gluconeogenese nicht gedeckt werden. Kühnlein und Meythaler (1958) isolierten eine Substanz aus dem Tumor, die eine insulinähnliche Wirkung zeigte. Kreisberg et al. (1970) vertraten die Ansicht, dass diese Tumoren eine Substanz produzieren und freisetzen, die auf das periphere Gewebe und auf die Leber wie Insulin wirken, bisher aber nicht identifiziert werden konnte. Klinik Die Geschlechtsverteilung beim Nadler-Wolfer-Elliott Syndrom zeigt eine deutliche Geschlechtsprädisposition, denn in 80% der Fälle waren männliche Patienten betroffen. Zwischen dem 40. und 50. Lebensjahr lag der Häufigkeitsgipfel (30% aller berichteten Fälle). Die Zeitspanne zwischen dem Auftreten der HypoglykämieSymptome (s. Doege-Potter-Syndrom) und dem Tod des Patienten betrug nur in Ausnahmefällen mehr als ein Jahr (Imperato und Lipton, 1965). 13 Endokrine und renale Paraneoplasien Anderson-Syndrom Während in den bisher berichteten Fällen des Doege-Potterund des Nadler-Wolfer-Elliott-Syndroms die Hypoglykämiesymptome im Vordergrund standen, ist typisch für das Anderson-Syndroms, dass andere Begleitsymptome klinisch auffällig waren. Nur bei 2 Fällen beherrschte die Hypoglykämie die klinische Symptomatik (Schamaun, 1957; Kühnlein, Meythaler, 1958). In den anderen Fällen fielen neben den Tumorsymptomen und einer Hepatomegalie (nicht obligat) andere endokrine Symptome auf. Pathologische Anatomie Alle bisher bei diesem Syndrom gefundenen Tumoren der Nebennierenrinde erwiesen sich als maligne. Unsicherheit in der histologischen Diagnose der Nebennierenrindenkarzinome lag bereits in dem von Anderson (1930) beschriebenen Fall vor: »Karzinom der Nebennierenrinde, im histologischen Bild einem Hepatom ähnelnd«. Der Tumor zeigte ein infiltratives Wachstum (Kühnlein und Meythaler, 1958; Fonkalsrud et al., 1964) und setzte Metastasen in Lunge, Leber, regionäre Lymphknoten (Dohan et al., 1953). Bevorzugt war die linke Nebenniere befallen (8 von 10 Fällen). Das Tumorgewicht schwankte zwischen 330 g (Carpenter jr., 1953) und 3 kg (Williams, 1961), im Durchschnitt 2,2 kg. Die kontralaterale Nebenniere war in den meisten Fällen verkleinert (Fonkalsrud,1964). Die klinisch manifeste Hyperaktivität der Nebennierenrinde führte zur Ausbildung eines Cushing-Syndroms (Fettansatz am Stamm, Vollmondgesicht, Striae, Akne, Hirsutismus) und bei Frauen zu Virilisierungserscheinungen wie Hypertrophie der Klitoris (Broster und Patterson, 1948), Ausbleiben der Pubertät (Broster und Patterson, 1948) und Störungen der Menstruation (Eymontt et al., 1965). Klinik Es zeigte sich keine besondere Geschlechtsbevorzugung. In 4 Fällen waren Männer, in 6 Frauen betroffen. Auffallend ist, dass die Krankheit in den ersten Lebensjahrzehnten gehäuft auftrat (bis zum 30. Lebensjahr 60% aller Fälle). Der jüngste Patient war ein 14 Jahre altes Mädchen (Broster und Patterson, 1948), der älteste ein 48-jähriger Mann (Williams et al., 1961). Die ersten Zeichen einer sich entwickelnden Hypoglykämie traten erst im Endstadium der Tumorkrankheit auf. Die Gesamtdauer vom ersten Hypoglykämieanfall bis zur Operation, bzw. bei inoperablen Tumoren bis zum Tod, lag im allgemeinen unter 6 Monaten. Die Hypoglykämie trat nur im Nüchternzustand auf; sie war auch symptomlos (Dohan et 14 Extrapankreatische Hypoglykämie Hypoglykämie-induzierende epitheliale Tumoren – Rosenfeld-Syndrom her beschriebenen paraneoplastischen Hypoglykämie-Syndromen diskutierten Theorien und Hypothesen zur Pathogenese treffen auch für dieses Syndrom zu. al., 1953) oder führte zum Koma (Broster und Patterson, 1948). Die Prognose war meist infaust. In 6 Fällen (die nicht gesicherten Berichte mit eingeschlossen) erwies sich der Tumor als inoperabel. Keller et al. (1970) und Adamson et al. (1971) kamen aufgrund eigener Untersuchungen zu dem Ergebnis, dass bei den von ihnen beobachteten Fällen (Keller: Magenkarzinom; Adamson: malignes Karzinoid) ein hormonell aktiver Tumor vorlag, der durch Sekretion einer Insulin-ähnlichen Substanz zu den schweren Störungen des Glucosestoffwechsels geführt hatte. Bei den Pseudomyxomen (Rosenfeld-Syndrom) könnte nach Ansicht von Kreisberg et al. (1970) und Eymontt et al. (1965) ein exzessiver Glucoseverbrauch durch den Tumor bzw. den Gesamtorganismus die Entstehung der paraneoplastischen Hypoglykämie erklären. Im ersten von Rosenfeld (1949) berichteten Fall produzierte der Tumor etwa 9 kg Pseudomuzin. Für die Synthese einer solchen Menge von Pseudoschleim müssen große Energiemengen zur Verfügung gestellt werden. 2.4 Hypoglykämie-induzierende epitheliale Tumoren Begriffsbestimmung Neben den Nebennieren- und Lebergeschwülsten kommen gelegentlich auch andere epitheliale Neubildungen als Ursache einer paraneoplastischen Hypoglykämie infrage. Bemerkenswert ist diese endokrine Störung bei Pseudomyxomen, die zu einer syndromalen Koppelung (Rosenfeld-Syndrom) führt. Seit der ersten Beschreibung von Bielschowsky (1932) wurden weitere 16 Fälle bekannt, bei denen Tumoren von einer zwar erst spät auftretenden und kurz verlaufenden, aber sonst typischen Hypoglykämie begleitet wurden. Pathogenese Die Hypoglykämie wird auch bei diesem Fällen durch den begleitenden Tumor hervorgerufen. Die bereits bei den vor- 15 Endokrine und renale Paraneoplasien ● Produktion und Sekretion einer Vitamin-D-ähnlichen Substanz durch den Tumor. Bürki et al. (1963) untersuchten das Serum und Tumorgewebe eines Patienten mit Hypernephrom und paraneoplastischer Hyperkalzämie auf eine Vitamin-D-ähnliche Aktivität. Dabei fanden sie beim biologischen Test an der rachitischen Ratte eine Serumaktivität von 827 IE/100 ml, die um das Vierfache gegenüber der Norm gesteigert war. Auch das durch Sektion – nach vorausgegangener 6-tägiger Steroidmedikation – gewonnene Tumorgewebe zeigte im Rattenversuch eine zwar geringere, jedoch deutliche Vitamin-D-Aktivität. Unter kalziumarmer Kost und Prednisonbehandlung sank der Kalziumspiegel von 16,4 mg% auf l2,2 mg%. Die Glucocorticoide sind VitaminD-Antagonisten und hemmen in erster Linie die durch Vitamin D geförderte enterale Kalziumresorption. Pathologische Anatomie Die nachgewiesenen Geschwülste wiesen weder makroskopisch noch mikroskopisch Besonderheiten auf. Über Größe und Gewicht der Tumoren lagen nur vereinzelte Angaben vor. In einigen Fällen waren die Metastasen des Tumors größer als der Primärtumor (Bielschowsky, 1932; Derot et al., 1959). Am häufigsten wurden sie in den regionären Lymphknoten und der Leber beobachtet. Bei keinem Fall konnten Pankreasveränderungen festgestellt werden. Klinik Die Geschlechtsverteilung von 14 Fällen zeigt eine Bevorzugung des männlichen Geschlechts (2 : 1). Die Hypoglykämie zeigte keine Besonderheiten. Die ersten Zeichen der Störung traten meist erst nach einer längeren Nahrungskarenz auf. Bei einigen Fällen, in denen die Leber durch die Metastasen weitgehend zerstört war, trat die Hypoglykämie erst einige Tage vor dem Tode auf (Bielschowsky, 1932; Derot et al., 1959). Diese erst im Finalstadium der Erkrankung auftretende Hypoglykämie ist für das Rosenfeld Syndrom typisch. ● Die normale alkalische Serumphosphatase spricht für eine Vitamin-D-Aktivität und gegen eine parathormonähnliche Wirkung. ● Die manifeste Hypophosphatämie (1,6 mg%) ist durch eine gesteigerte Phosphaturie (erhöhte »tubular rejection fraction« für Phosphate) zu erklären. Sie kann wie bei der Vitamin-D Intoxikation durch die renale Wirkung VitaminD-ähnlicher Substanzen ·auftreten (Hennemann et al., 1956; Plimpton und Gellhorn, 1956; Bürki et al., 1963). 3 Das paraneoplastische Hyperkalzämie-Syndrom Von Interesse ist die von Erslev (1970) berichtete Beobachtung, dass gewisse Tumoren – insbesondere Mammakarzinome – 7-Dehydrocholesterol bilden können. Diese auch als Provitamin D (Ergosterin) bezeichnete Verbindung kann auch eine Hyperkalzämie hervorrufen. Begriffsbestimmung Mit der Bezeichnung »paraeoplastisches HyperkalzämieSyndrom« (ICD-E83.5) wird eine Reihe von Krankheitssymptomen bei Tumoren ohne osteolytische Skelettmetastasen zusammengefasst, die als direkte Folge einer Hyperkalzämie auftreten, keine wesentlichen morphologischen Veränderungen hervorrufen und nach einer Behandlung der Neubildung meistens wieder rasch verschwinden (Plimpton und Gellhorn, 1956; Watson, 1963; Omenn et al., 1969; Muggia, 1970; Labhart, 1971b; Neville, 1972; Ross, 1972). Die Tumorhyperkalzämie gehrt zu den häufigsten endokrinen Paraneopalsien. Man schätzt, dass sie bei 5% der Krebsleiden vorliegt. ● Produktion und Sekretion einer die Nebenschilddrüsen stimulierenden Substanz durch den Tumor. Klemperer (1923) versuchte die paraneoplastische Hyperkalzämie durch eine Hormonprodution im Tumor zu erklären. Er fand bei einer Patientin mit einem Mamma-Ca eine Hyperkalzämie, eine Nephrokalzinose sowie eine Hyperplasie der Nebenschilddrüsen und schloss daraus, dass der Tumor eine Substanz produziert, die die Hormonproduktion der Nebenschilddrüsen stimuliert. In den meisten Fällen waren die Nebenschilddrüsen normal oder atrophisch (Plimpton und Gellhorn, 1956; Azzopardi et al., 1968; u. a.). Eine subtotale (Stone et al., 1961; Lamberg, 1964; Dean et al., 1969; Menguy, 1969) oder totale Parathyroidektomie konnte die Hyperkalzämie nicht beseitigen. Es ist daher anzunehmen, dass die paraneoplastische Tumorwirkung nicht über eine Stimulierung der Nebenschilddrüsenfunktion zu erklären ist. Pathogenese Eine kausale Bedeutung wurde dem begleitenden Tumor beigemessen, da bei mehreren Fällen die klinischen und biochemischen Zeichen der paraneoplastischen Hyperkalzämie kurze Zeit nach der operativen Entfernung des Tumors verschwanden (Connor et al., 1956; Plimpton und Gellhorn, 1956; Turkington et al., 1966). Es wurden folgende Hypothesen diskutiert: ● Produktion und Sekretion von Parathormon oder einer PTH ähnlichen Substanz durch den Tumor. Nach Albright und Reifenstein (1948) kann eine paraneoplastische Hyperkalzämie durch die Bildung eines Stoffes mit Parathormonwirkung im Tumor entstehen. Plimpton und Gellhorn (1956) sowie Connor et al. (1956) beobachteten, dass sich die Hyperkalzämie bei einem Bronchialkarzinom ohne nachweisbare osteolytischen Skelettmetastasen nach der operativen Entfernung des Tumors zurückbildete. Bei einem Tumorrezidiv traten erneut die klinischen Symptome des paraneoplastischen Hyperkalzämie-Syndroms auf (Plimpton und ● Produktion und Sekretion einer vermehrt Kalziumbindenden Substanz durch den Tumor. Nach Plimpton und Gellhorn (1956) sowie Magnenat und Perret (1961) könnte der Tumor eine Substanz bilden, die Kalzium bindet. Dieser pathogenetische Mechanismus erscheint aber im Hinblick auf den normalen prozentualen Anteil des ionisierten Kalziums bei diesen Patienten unwahrscheinlich (Plimpton und Gellhorn, 1956; MGH, l957; Massaro und Owen, 1962). 16 Paraneoplastisches Hyperkalzämie-Syndrom – Gellhorn, 1956; Schatten et al., 1958; Smith et al., 1968; Menguy, 1969). Zunächst glaubte man, dass kleine Knochenmetastasen übersehen wurden. Häufiger lag eine verstärkte subperiostale Knochenresorption vor, obgleich die Nebenschilddrüse keinerlei Hinweise auf eine Überfunktion zeigte (Connor et al., 1956; MGH, 1963; Lamberg et al., 1964). Es ist daher anzunehmen, dass der Tumor eine oder mehrere Substanzen bildet, die direkt osteolytisch wirken (Smith et al., 1968). – – Buckle et al. (1970) untersuchten Tumorextrakte auf eine PTH-ähnliche Wirkung und kamen zu folgenden Ergebnissen: Das immunologische Verhalten der PTH-ähnlichen Substanz im Serum der V. renalis, die das Blut aus einem Nierenkarzinom drainierte, zeigte keine Unterschiede gegenüber dem Tumorextrakt. Immunologisch wiesen Tumorextrakt und menschliches PTH zwar keine völlige, aber doch weitgehende Übereinstimmung auf. Die PTH-Aktivität im Serum der A. und V. renalis ergab eine arteriovenöse Differenz von 1,93 ng/ml (1,93 ·10-1 g/m l). Nach Entfernung des Tumors fiel das PTH im Serum wieder auf Normalwerte ab (0,1ng/ml). Die nachgewiesenen PTH-Konzentrationen im Tumor lagen zwischen 2,2 µg/g Tumorgewebe (Buckle et al., 1970) und 13,3 µg/g (Tashjian et al., 1964). In einer Lungenmetastase bestimmten Goldberg et al. (1964) eine PTH-Aktivität von 39 µg/g-Metastasengewebe. Diese Konzentrationen waren sehr gering (Buckle et al., 1970). keine hyperplastischen Nebenschilddrüsen (objektivierbar durch Exploration der Halsorgane bzw. durch die Obduktion), keine Wirkung adrenaler Steroide auf die Höhe des Kalziumspiegels, Rückbildung der Hyperkalzämie nach einer Tumorresektion, erneutes Auftreten der Hyperkalzämie bei eeinem Tumorrezidiv. Die formale Pathogenese des Hyperkalzämie-Syndroms lässt sich durch die autonome Parathormonaktivität erklären. Beim paraneoplastischen Hyperkalzämie-Syndrom werden folgende Veränderungen beobachtet: – Erhöhung der Osteozyten- und Osteoblastenaktivität (Goldberg etal.,1964). Ein Zeichen hierfür war die wechselnd starke Erhöhung der alkalischen Phosphatase; eine Folge der Osteolyse war die vermehrte Ausschwemmung von Kalzium und Phosphor in den extrazellulären Raum und ins Serum. Am Skelett entstand ein erhöhter Umsatz des Kalziums und Phosphats (Pearson, 1968). – Der Phosphatspiegel im Serum war in den meisten Fällen niedrig (Tremblay und Ansell, 1964; Thomson, 1966). Die häufiger vorkommende Polyurie beruhte auf einer direkten Wirkung des Kalziums auf den distalen Tubulus, indem die Wasserreabsorption gehemmt und die »free-water-clearance« gefördert wurde. Die ADH-resistente Polyurie war stets mit einer im frühen Krankheitsverlauf nachweisbaren Hyposthenurie verbunden (MGH, 1963; Labhart, 1971). Die intestinale Kalziumresorption war erhöht. Die nur in vereinzelten Fällen beschriebenen Magenulzera bei einem paraneoplastischen Hyperkalzämie-Syndrom waren eine Sekundärerscheinung der Hyperkalzämie (Omenn et al., 1969; Moolten, 1970). Heute gilt als gesichert, dass es sich bei der parathormonähnlichen Substanz, um PTHrP (Parathyroid hormone related Protein) handelt. Dieser Stoff, der bei gesunden Erwachsenen nicht synthetisiert oder sezerniert wird, kann bei 80% der paraneoplastischen Hyperkalzämien nachgewiesen werden. Er zeigt in seiner Wirkung ähnliche Eigenschaften wie das Parathormon: Es stimuliert den Knochenabbau mit Freisetzung von Kalzium und verstärkt die renale Reabsorption im distalen Tubulus. Ferner spielt es eine Rolle in der Organentwicklung (Zahneruption, Mammaentwicklung). Ein echtes Parathormon wurde nur selten (Nieren-, Lunge-, Schilddrüsen- und Thymustumoren) nachgewiesen. Osteolysen (scharf gezeichnete Knochendefekte) und Osteoporosen (diffuser Kalziumabbau) spielen – im Rahmen des Knochenabbaus – eine wichtige Rolle bei der Hyperkalzämie. Typisch ist, dass einige maligne Tumoren (Nieren- und Lungenkarzinom) osteolytische Knochenmetastasen setzen. Dies trifft auch für das multiple Myelom zu. Da in diesen Fällen die Kalziumfreisetzung auf eine direkte Wirkung der Tumorzellen beruht, gehören diese Tumorosteolysen nicht zu einem paraneoplastischen Syndrom. Der Nachweis der Metastasen schließt aber nicht aus, dass die Tumorzellen eine hormonähnliche Aktivität zeigen können. Neuere Studien gehen auf die Wechselbeziehungen von Osteoblasen (Knochenabau) und Osteoklasten (Knochenabbau). Dabei konnte in den Tumoren zwei Faktoren nachgewiesen werden: Osteoprotergin (Osteoklasten-inhibierendes Protein) und Osteoprotergin-Ligand (Zytokin, das die Oseoklastenaktivität-stimuliert (Kasenwik und Schalboub, 2001). Pathologische Anatomie Das pathologisch-anatomische Substrat der paraneoplastischen Hyperkalzämie ist eine maligne, nicht von den Nebenschilddrüsen ausgehende Neubildung. Diese sind normal bzw. atrophisch (nur gelegentlich sekundär hyperplastisch). Ferner liegt ein eine subperiostale Knochenresorption unterschiedlicher Ausprägung vor. Bei Lymphompatienten wurde eine erhöhte Sekretion an 1,25-Dihydroxycholecalciferol beobachtet. Ferner fand man Stoffe mit knochenabbauender Wirkung (Interleukin 1, Interleukin 6, Tumornekrose-Faktor) Zusammenfassend stellte Sherwood et al. (1967) folgende Kriterien auf, die für die Bildung einer PTH-ähnlichen Substanz durch den Tumor sprechen: – Fehlen von osteolytischen Skeletmetastasen, – Hypophosphatämie, Tumorlokalisation: Die Lungen- (34,5%) und die Nierentumoren (22,7%) stellten mehr als die Hälfte aller Geschwül- 17 Endokrine und renale Paraneoplasien Tumorhistologie bei der paraneoplastischen Hyperkalzämie ste. Es folgen die Tumoren des Ovars und Pankreas mit je 8,1%. vor, die geringe tubuläre Phosphatreabsorption sprach jedoch für eine paraneoplastische Hyperkalzämie. Die histologische Auswertung ergab 21 Plattenepithelkarzinome unter den 38 Lungentumoren. Plattenepithelkarzinome fanden sich auch in Niere, Uterus, Vulva, Vagina, Penis, Harnblase, Ösophagus, Mundalveolarleiste und im Halsbereich. Somit wurden 31 der 110 Geschwülste als Plattenepithelkarzinome diagnostiziert. Eine paraneoplastische Hyperkalzämie kann auch bei tierexperimentellen Tumoren ohne osteolytische Knochenmetastasen beobachtet werden. Wilson et al. (1961) implantierten weißen Kaninchen das XV-2 Impfkarzinom; nach 3 Wochen entwickelte sich eine Hyperkalzämie und Hypophosphatämie ohne Knochenmetastasen. Zu de seltenen Beobachtungen zählen die Mitteilungen von Lytton et al. (1965) und Haauer et al. (1968). In dem von Lytton et al. (1965) beschriebenen Fall kamen im Verlauf von 15 Jahren 3 verschiedene primäre Neoplasien vor. Die Hy- Bei einem kleinzelligen Karzinom der Lunge (Snedecor und Baker, 1964) lagen zwar vereinzelte Wirbelkörpermetastasen 18 Paraneoplastisches Hyperkalzämie-Syndrom perkalzämie trat nach einem Nierenkarzinom auf. Hallauer et al. (1968) berichten über ein Lymphosarkom. das nach einem symptomfreien Intervall von 12 Jahren zu einer Hyperkalzämie führte. die Größe der Zellen und Zellkerne sowie der zytomorphologische Aufbau aus Hauptzellen, oxyphilen Zellen und anderen Zelltypen berücksichtigt werden (Seifert und Seemann, 1967). Galasko und Burn (1971) und Mavligit et al. (1971) beschrieben erstmalig Mammakarzinome mit einer paraneoplastischen Hyperkalzämie. Bei dem von Mavligit et al. (1971) mitgeteilten Fall lagen ausgedehnte Knochenmetastasen vor, doch konnte aus einer Lebermetastase des Tumors eine Substanz mit PTHAktivität isoliert werden. Es muss somit auch den Mammageschwülsten die Fähigkeit PTH-ähnliche Substanzen zu bilden zugesprochen werden. Als Veränderungen im Skelettsystem werden eine Osteoporose der Wirbelsäule (Plimpton und Gellhorn, 1956; Fry, 1962) sowie einzelne Solitärmetastasen des Tumors in Knochen und Wirbeln beschrieben. In keinem der Fälle konnte aufgrund einer Solitärmetastase die bestehende Hyperkalzämie erklärt werden. Die subperiostale Knochenresorption war im Röntgenbild als Kortikalisatrophie besonders an der Mittelphalange des Mittelfingers gut erkennen (Albrigh t et al., 1941; Connor et al., 1956; Lamberg et al., 1964). Diese Veränderung traten auch an der Kortikalis der Zahnalveolen auf und wurden als »Verlust der Lamina densa« bezeichnet (Labhart, 1971b). In einigen wenigen Fällen wurden Veränderungen an den Nieren im Sinne einer Nephrokalzinose beschrieben (Svane, 1964; Uehlinger, 1964; Keller et al., 1965). Die bisherigen Untersuchungen über den Funktionszustand der Epithelkörperchen bei paraneoplastischen Hyperkalzämien führten zu unterschiedlichen Ergebnissen. Bei den meisten Fällen wurden normale Nebenschilddrüsen beschrieben (Goldberg et al., 1964; O'Grady et al., 1965 u.a.), nur gelegentlich verkleinerte Drüsen (Samuellson und Werner, 1963) sowie vereinzelt Epithelkörperchenhyperplasien (MG H, 1957; 1964; Stone et al., 1961; Massaro und Owen, 1962). Die Frage, ob eine Erhöhung des Blutkalziumspiegels generell zu einer Inaktivierung der Nebenschilddrüsen führt, wird von den meisten Autoren verneint (Kracht, 1967a). Um eine histologische Aussage über den Aktivitätszustand der Epithelkörperchen zu machen, müssen das Gewicht der Nebenschilddrüsen, die Durchsetzung mit Fett- und Bindegewebe, Klinik Die Geschlechtsverteilung ergab eine leichte Verschiebung zugunsten des männlichen Geschlechts (3 : 2). Der Altersgipfel lag im 7. Dezennium. Nach Angaben von Carey (1966) findet man eine paraneoplastische Hyperkalzämie in 19 Endokrine und renale Paraneoplasien Paraneoplastische Hyperkalzämie. Klinik 6% aller Malignome ohne Skeletmetastasen, nach Locks (1962) in 7,5% der Fälle. Azzopardi (1969) beobachtete die Paraneoplasie in 16% der Lungenkarzinome ohne osteolytische Skelettmetastasen. Über die Häufigkeit bei anderen Organtumoren (z.B. Nieren, Ovar, Pankreas) liegen keine Angaben vor. Wahnerlebnissen. Vergleichende Untersuchungen ergaben, dass diese Reaktionsform erst bei einem Serumkalziumspiegel von mehr als 16 mg% auftrat (Kind, 1959). Diese psychische Veränderung bildeten sich – nach Beseitigung der Hyperkalzämie – rasch zurück (Plimpton und Gellhorn, 1956; Schatten et al., 1958; Breidahl und Ritschie, 1962; Cabau et al., 1968; Buchsbaum, 1969; Kissel et al., 1970). Die häufigsten und wichtigsten klinischen Symptome waren: Polydipsie – Nausea, Anorexie, Obstipation – Niere: Nykturie Polyurie – Zentralnervensystem: Lethargie Bewusstseinseintrübung Desorientierung Somnolenz – Koma In etwa 55% aller ausgewerteten Fälle lag der Serumkalziumspiegel über 14,5 mg% (normal: 9,0 bis 10,5 mg%). Der bisher höchste Kalziumwert wurde von Stone et al. (1961) mit 22,5% angegeben. Bei 56% der Patienten lag der Serumphosphorspiegel unter 3,0 mg% (normal: 3-3,5 mg%). Der niedrigste Wert lag bei 1,1mg% (Menguy, 1969). Als erstes renales Symptom fand sich meistens eine Nykturie, die in eine Polyurie überging. Eine häufige Folgeerscheinung war ein quälendes, durch keine Trinkmenge zu löschendes Durstgefühl. Neben der ausgeprägten Hyperkalzämie und Hypophosphatämie bestand eine deutliche Hyperkalziurie und Hyperphosphaturie. Die Phosphatclearance war erhöht (20,6 ml/min., Samuelsson und Werner, 1963), die tubuläre Phosphatreabsorption vermindert (Hallauer et al., 1968). Die alkalische Phosphatase war normal bis leicht erhöht, die BSG war in vielen Fällen stark beschleunigt (60 bis 100 mm/lh) Zu den gastrointestinalen Symptomen zählten Inappetenz und unmotivierte periodisch wiederkehrende Brechanfälle. Neurologischen Symptome waren die Muskelschwäche, die sich im Rahmen einer allgemeinen Schlaffheit, Asthenie und erhöhter Ermüdbarkeit bei körperlicher Anstrengung entwikkelten. Die Sehnenreflexe wurden als normal oder leicht abgeschwächt beschrieben (Kissei et al., 1970). In den meisten Fällen bestand auch eine Anämie mit Hämoglobinwerten von 9,1g% (Plimpton und Gellhorn, 1956; MG H 80, 1961; Stone et al., 1961; Goldberg et al., 1964; Buckle et al., 1970). Serumelektrophoretische Untersuchungen zeigten eine Verschiebung der Fraktionen zugunsten der α- und β-Globuline. Neben einer Hypoalbuminämie lag häufiger auch eine deutliche Hyperglobulinämie vor (Abouav, 1959; Snedecor und Baker, 1964; Turkington et al., 1966; Kofstad et al., 1967). Es wurden auch spontan auftretenden psychischen Störungen beobachtet (Kissel et al., 1970), die nach Untersuchungen von Bleuler (1954) bei fast jeder endokrinen Störung vorkommen können. Die chronische Form, das hirnfokale Psychosyndrom, wurde auf eine umschriebene Stoffwechselstörung des Gehirns zurückgeführt. Das klinische Bild war durch Veränderungen der Stimmung und der Antriebshaftigkeit geprägt. Bei der zweiten Form lag eine diffuse Hirnschädigung vor, die in den Rahmen des akuten exogenen Reaktionstyps gehörte. Klinisch äußerte sie sich in Form einer mehr oder weniger rasch einsetzenden Benommenheit oder Somnolenz mit Übergang in Präkoma und Koma oder einer Erregung bzw. Verwirrung mit Halluzinationen, Traum- und Der nicht-Protein-gebundene Stickstoff (NPN) lag im Normbereich und stieg erst terminal – als Ausdruck einer beginnenden Niereninsuffizienz – an (Loebel und Walkoff 1962; Goldberg et al., 1964). Zu den Nebenbefunden zählten die hypokaliämische Alkalose, die zwar nicht sehr ausgeprägt war, aber den Allge- 20 Ektopes ADH-Syndrom meinzustand des Patienten beeinträchtigte (MGH, 1963; Goldberg et al., 1964; Tashjian et al., 1964). Als formale Pathogenese nahm man eine »inadäquate Sekretion des antidiuretischen Hormons« an (Schwartz et al., 1957). Die Herkunft des ADH war aber unklar. Es wurden folgende Möglichkeiten diskutiert: Die zeitliche Korrelation zwischen dem ersten Auftreten der Symptome von Tumor und Hyperkalzämie war sehr unterschiedlich. Bei etwa einem Drittel aller Fälle wurde der Tumor vor der Hyperkalzämie röntgenologisch und durch Biopsie diagnostiziert (MGH, 1964; Keller et al., 1965). Bei 20% der Patienten wurden Tumor und Endokrinopathie gleichzeitig beobachtet. Bei ca. 50% aller Fälle konnte erst nach der Obduktion die Verdachtsdiagnose einer malignen Neubildung bestätigt werden. ● Mechanische Stimulierung hypothalamischer Kerne durch Gehirnmetastasen. In vielen Fällen wurden jedoch keine Gehirnmetastasen gefunden, sodass dieser Mechanismus sehr unwahrscheinlich war (Roberts, 1959; Amatruda et al., 1963; Williams, 1963; Delaere et al., 1965; de Sousa und Mach, 1965). ● Stimulation der Rezeptoren im Mediastinum über den N. vagus. Gegen diese Hypothese sprach die Tatsache, dass eine tumoröse Vagusinfiltration nicht obligat war. Bei der Differenzialdiagnostik der paraneoplastischen Hyperkalzämie sind ein Hyperparathyreoidismus, eine Vitamin-D-lntoxikation, ein Milch-Alkali-Syndrom (BurnettSyndrom), eine Sarkoidose, ein Plasmozytom, Hyperthyreoidismus, eine akute Osteoporose sowie eine metastatische Knochenschädigung auszuschließen (Anderson und Glenn, 1955; Ganzoni, 1964). Experimentell kann der Vagus durch lokale Instillation von Xylocain geblockt werden, ohne dass es zu einer signifikanten Änderung der Urinosmolarität kommt (Amalruda et al., 1963; Delaere et al., 1965). ● Produktion einer Substanz, die in der Lage ist, die Achse Hypothalamus-Hypophyse zu aktivieren. Diese Substanz konnte bisher noch nicht nachgewiesen werden. Die Prognose hing von dem Zeitpunkt ab, an dem die Tumordiagnose gestellt und eine Therapie eingeleitet wurde. Die durchschnittliche Überlebenszeit betrug bei den Lungentumoren 3,8 Monate. Turkington et al. (1966) berichteten über einen 3 Jahre, MGH (1957) über einen 4 Jahre dauernden Krankheitsverlauf. Bei den Nierentumoren lag die Überlebenszeit durchschnittlich bei 4,5 Monaten, ebenso bei den Tumoren der anderen Organe. ● »Reset« des Regelkreises. Eine Verminderung des Sollwertes im biologischen Regelkreis, ist sehr unwahrscheinlich, da der Harn auch bei einer geringen Plasmaosmolarität (220 mosm/kg) konzentriert sein kann (Schwartz et al., 1957; 1960; Delaere et al., 1965). ● Leberschädigung. ADH wird in der Leber abgebaut. Ist dieses Organ durch irgendeinen krankhaften Prozeß vorgeschädigt, so reichert sich das ADH – infolge des Gleichgewichtsverlustes zwischen Abbau und Produktion – im Plasma an. Diese Hypothese erklärt nicht die Fälle, bei denen eine voll funktionstüchtige Leber nachgewiesen wurde. 4 Das ektope ADH-Syndrom Synonyma: Schwartz-Bartter-Syndrom, Syndrom der inadäquaten Sekretion von ADH (Inappropriate secretion of antidiuretic hormone syndrome), paraneoplastische Hyponatriämie, SIADH. ● Sekretion und Freisetzung von ADH bzw. eines Polypeptides mit ADH-Wirkung. Die Produktion und Freisetzung von ADH durch den Tumor wurde erstmals von Schwartz und Bartter (1957) angenommen. Diese Hypothese wird durch folgende Beobachtungen unterstützt : – Die Bestrahlung des Tumors führte in einigen Fällen zu einem Abklingen der Symptome und zu einer Normalisierung des Serumnatriums (Bower et al., 1964). – Solitärmetastasen zerstörten den gesamten Hypophysenhinterlappen (HHL). Dies beweist, dass ein intakter HHL für die Entwicklung dieses Syndroms nicht notwendig ist. Obwohl dieser Befund nicht ausschließt, dass der HHL die Quelle des ADH war, blieben die Bemühungen, neurosekretorisches Material in der Regio supraoptica et paraventricularis des Hypothalamus mit einer Aldehyd-Fuchsin-Färbung nachzuweisen, ergebnislos (Bower et al., 1964). Begriffsbestimmung Die Bezeichnung ektopes ADH-Syndrom umfasst eine Reihe von klinischen Symptomen, die als direkte Folge der vom Tumor durch ektope ADH-Bildung verursachten Hyponatriämie anzusehen sind. Das Schwartz-Bartter Syndrom wurde bei zerebralen Erkrankungen (primären und metastatischen Hirntumoren, vaskulären Gehirnerkrankungen, Schädeltraumen und Mißbildungen), bei Stoffwechselstörungen wie Prophyrie mit Enzephalopathie und schließlich . auch idiopathisch – also ohne erkennbare Ursache – gefunden (Schwartz et al., 1957, 1960; Carter et al., 1961; Waldvogel et al. (1967). Pathogenese Bereits 1938 beobachteten Winkler und Crankshaw die Kombination von Hyponatriämie und Bronchialkarzinom. 20 Jahre später beschrieben Schwartz und Bartter ausführlich zwei Fälle. Es lag ein Zustand vor, wie nach Verabreichung hoher Dosen von Adiuretin (Leaf und Mamby, 1952). Amatruda et al. (1963) konnten erstmals aus dem Tumor eine Substanz mit einer antidiuretischen Wirkung von 70 bis 350 µU isolieren. Es gelang in der Folgezeit im Plasma und im Harn erhöhte ADH-Konzentrationen nachzuweisen (Lee et al., 1964; Barraclough, 1966; 1971; Faye et al., 1967; Mor- 21 Endokrine und renale Paraneoplasien Ektopes ADH-Syndrom. Gegenüberstellung von paraneoplastischem ADHSyndrom und Hypophysenhinterlappen-ADH-Wirkung (HHL-ADH) – nex et al., 1967; Edwards, 1971). Die pharmakologische Wirkung dieser antidiuretischen Substanz legte die Vermutung nahe, dass die vom Tumor sezernierte Substanz weitgehend mit dem Arginin-Vasopressin des HHL identisch war. In dem gleichzeitig vorliegenden kleinzelligen Ca konnte kein ADH nachgewiesen werden. Für die gesteigerte Natriurese werden folgende Faktoren verantwortlich gemacht: – vermehrte glomeruläre Filtration als Folge der primären Hypervolämie (de Sousa und Mach, 1965; Grantham et al., 1965; Bricaire et al., 1967). – Die Aldosteronsekretion ist durch die Hypervolämie vermindert. Mehrere Autoren verneinen jedoch eine Korrelation zwischen der Ausscheidung von Aldosteron und einer Natriurese (Cox et al., 1961; Jones et al., 1963; Clift et al., 1966). – Hemmung der tubulären Natrium-Reabsorption (Schwartz, 1957). Der Nachweis von ADH im Tumorgewebe beweist noch nicht, dass der Tumor das Hormon auch selbst gebildet hat. Unger et al. (1963) stellten die Hypothese auf, dass der Tumor selektiv das Hormon aus dem Plasma adsorbiert, konzentriert und später wieder abgibt. Diese Hypothese ist unwahrscheinlich, denn – die in diesem Tumor gefundene ADH-Konzentration war bis zu 10 000-mal größer als die des Plasmas (Marks, 1968). – Experimentelle Untersuchungen ergaben, dass die Rattenniere selbst bei extremer Stimulierung der Neurohypophyse nur 50% des zirkulierend en ADH aus dem Serum aufnehmen konnte (Ginsburg und Heller, 1953). Die formale Pathogenese des ektopen ADH-Syndroms ist demnach eine Folge einer erhöhten antidiuretischen Aktivität, die letztlich durch den Tumor hervorgerufen wird. 22 Ektopes ADH-Syndrom Ektopes ADH-Syndrom. Biochemische Befunde Ektopes ADH-Syndrom. Differenzialdiagnose zu Erkrankungen mit Hyponatriämie Ca als Ursache der Störung im Natrium- und Wasserhaushalt angesehen (MGH, Case 30, 1967). Pathologische Anatomie Bei 87 der 92 ausgewerteten Fälle eines Schwartz-BartterSyndroms lag ein Lungentumor vor. Ordnet man die Tumoren nach ihrem histologischen Bild, so erhält man folgende Verteilung: Bei 2 Patienten lagen gleichzeitig zwei Tumoren vor: ein kleinzelliges Ca und ein Plattenepithelkarzinom der Lunge (MGH Case 30, 1967) sowie ein kleinzelliges Ca und Adenokarzinom des Pankreas. Nur in den Extrakten des Pankreastumors ließ sich eine ADH-Aktivität nachweisen (Marks et al., 1968). Wenig differenzierte Adenokarzinome werden von Grantham et al. (1965), Tisher (1968) und Eastridge et al. (1968) beschrieben. Bei elektronenmikroskopischen Untersuchungen von zwei kleinzelligen Ca fanden sich neben Kernatypien reichlich Ribosomen und sekretorische Granula, deren Durchmesser zwischen 0,05 und 0,14µ schwankte (Faye et al., 1967). Whitelaw (1969) führte vergleichende Untersuchungen zwischen zwei kleinzelligen Ca durch von denen in einem Fall das Tumorgewebe eine ADH-Aktivität zeigte (40mU/mg). Er konnte keine Unterschiede im elektronenmikroskopischen Bild feststellen und kam zu dem Ergebnis, dass die Hormonproduktion eher als ein Ausdruck der Entdifferenzierung als einer Spezialisierung der Krebszelle anzusehen sei. Der häufigste Tumortyp ist das kleinzellige Ca. Oft wurde es als stark anaplastisches Karzinom beschrieben, sodass es gerechtfertigt erscheint, auch die anaplastischen Karzinome in diese Gruppe einzubeziehen und sie den höher differenzierten Tumoren der Lunge gegenüberzustellen. Beide Gruppen zusammen stellen etwa 83% der Gesamttumoren. Die Tumoren, insbesondere die Lungenkarzinome, metastasierten in die regionären Lymphknoten, Leber, Pankreas, Milz, Nebennieren und die Nn. vagi. Letztere waren in mehreren Fällen vom Tumor ummauert und zum Teil auch infil- Bei 4 Fällen wurde ein Plattenepithelkarzinom diagnostiziert (MGH 30, 1967; Mornex, 1969, 2; Giard, 1970). Bei einem Patienten wurde das gleichzeitig vorkommende kleinzellige 23 Endokrine und renale Paraneoplasien triert, sodass es lokal zu einer Entmyelinisierung kam. Diese Teilzerstörung des autonomen Nervensystems wurde als möglicher pathogenetischer Faktor von mehreren Autoren diskutiert (Schwartz, 1957, 1960; Roberts, 1959). 120 mEq/l (normal: 140 bis 142 mEq /l). Der niedrigste Wert betrug 100 mEq/l (Roberts, 1959). Neben der Hyponatriämie lag auch eine deutliche Hypochlorämie mit Durchschnittswerten von 85 mEq/l (normal: 103 mEq/l) vor. Die Kaliumund Bikarbonatkonzentration des Serums sowie der Hämatokrit waren normal. Die Plasmaosmolarität lag immer weit unter dem entsprechenden osmotischen Wert des Harns. Die Ausscheidung von Natrium im Urin war erhöht; Die Werte lagen im Durchschnitt zwischen 70 und 140 mEq/l, doch wurden auch Werte von 350 mEq/l beschrieben (Bower et al., 1964). Die glomuläre Filtrationsrate war normal oder leicht gesteigert (Schwartz et al., 1957; 1960). Die Clearancewerte lagen für Kreatinin, Inulin und PAH im Normbereich (Ivy, 1961; Williams, 1963). Auch die Aldosteronausscheidung im Urin war meist normal (Mornex et al., 1967; Roumagnoux et al., 1968), ebenso die Reninausscheidung (24 ng/ml, Mornex et al., 1967). Charakteristisch waren stark herabgesetzte Reststickstoffwerte, die in vielen Fällen unter 10 mg% lagen (Schwartz et al., 1960; Amatruda et al., 1963; Thorn, 1963). In den Nebennieren fanden sich meist nur kleine, versprengte Metastasen, die die Nebennierenfunktion nicht beeinträchtigten. Hirnmetastasen wurden bei mehreren Fällen beschrieben (siehe Schwartz et al., 1957). Eine Beteiligung der Hypophyse lag bei zwei Patienten vor, wobei in einem Fall eine Solitärmetastase den Hypophysenhinterlappen zerstört hatte (Bower et al., 1964). Klinik Da in mehr als 90% aller Fälle ein Lungentumor als Grundleiden vorlag, ergab die Alters- und Geschlechtsverteilung eine charakteristische Kurve. 62 der 70 Patienten waren Männer; der Häufigkeitsgipfel lag im 6. Dezennium. Der jüngste Patient war eine 28 Jahre alte Frau mit einem metastasierenden Melanom bei unbekanntem Primärtumor (Clinicopath. Conf., 1963b), der älteste war ein 75-jähriger Mann mit einem Adenokarzinom der Lunge (Grantham et al., 1965). Über die Häufigkeit des ektopen ADH-Syndroms bei Lungentumoren liegen folgende Berichte vor: Raymond et al. (1968) stellten bei 4 unter 155 Bronchialkarzinomen (2,5%) ein ektopes ADH Syndrom fest. Azzopardi et al. (1970) fanden unter 185 Karzinompatienten in 3 Fällen (1,6%) ein ektopes ADH-Syndrom. Die Diagnose des ektopen ADH-Syndroms wird in den meisten Fällen durch biochemische Untersuchungen gesichert. Wenn eine Exsikkose und Azotämie einerseits, Ödeme und Herzinsuffizienz andererseits ausgeschlossen werden können, die Hyponatriämie unter dieser Voraussetzung mit einem hypertonen oder ungenügend verdünnten Harn einhergeht und durch Einschränkung der Flüssigkeitszufuhr sich bessern lässt, dann liegt ein ektopes ADH-Syndrom vor (Eastridge et al., 1968; Labhart, 197la}. Die klinischen Symptome des ektopen ADH-Syndroms waren meist nicht sehr charakteristisch und traten häufiger sogar in den Hintergrund, sodass Schwartz et al. (1960) diese Fälle den asymptomatischen Hyponatriämieformen zuordneten. Die zeitliche Korrelation der Diagnosen Tumor bzw. ektopes ADH-Syndrom variierte stark. In den meisten Fällen wurden aber Tumor und das ektope ADH-Syndrom gleichzeitig nachgewiesen. Als typische Tumorzeichen wurden Husten, Hämoptoe, funktionelle Dyspnoe, Ermüdbarkeit, Schmerzen der oberen Extremität, des Schultergürtels und Rückens sowie ein deutlicher Gewichtsverlust beschrieben (Amatruda et al., 1963 ; Thorn und Transbol, 1963; Williams, 1963; Utiger, 1966; Eastridge et al., 1968). Nach Tumorbehandlung mit Röntgenstrahlen, Kobalt 60 (4500- 5000 R) und N-Lost-Präparaten wurde bei einigen Patienten eine Normalisierung der Natriumwerte beobachtet (Schwartz et al., 1957; 1960; Ivy, 1961; Amatruda et al., 1963; Thorn und Transbol, 1963 u.a.). Bei 8 Patienten wurde der Tumor operativ entfernt. Die Natriumwerte normalisierten sich vorübergehend, fielen aber nach dem Auftreten von Tumorrezidiven erneut ab. Die durch die Hyponatriämie verursachten Symptome traten erst bei Serumnatriumwerten unter 110 bis 115 mEq /I auf. Im Vordergrund standen häufig plötzliche Zeichen einer Wasserintoxikation: Anorexie, Nausea, Erbrechen, Ermüdung, mentale Konfusion, zeitliche und räumliche Desorientierung, Euphorie sowie Aggressivität oder Lethargie (Lindsey und Barnes, 1962; Amatruda et al., 1963; Thorn, 1963; Bricaire et al., 1967 ; Eastridge et al., 1968). Es fehlten dagegen die Zeichen einer Hypovolämie und Dehydratation sowie einer Hypotonie, ein verminderter Hautturgor, Oligurie und Azotämie. Die Achsel- und Schambehaarung waren normal, die Pigmentierung der Haut nur in wenigen Fällen verstärkt (Turner und Williams, 1962; Pfenninger et al., 1963). Als begleitende Komplikation wurden Duodenalulkus (Schwartz et al., 1957; Roberts, 1959), Fazialisparese (Schwartz, 1957 ), Dermatomyositis (Williams, 1963), Myasthenia (Hallpike et al., 1966), Hyperthyreoidismus (Nathan, 1970) und Porphyrie (Clinicopath. Conf., 1963) beschrieben. Der Verlauf wurde durch das Tumorgrundleiden bestimmt. Bei 43 Fällen konnten Angaben über Überlebenszeit und Beginn der Paraneoplasie ausgewertet werden. Bei den Lungentumoren betrug die Verlaufszeit durchschnittlich 6 Monate mit Schwankungen von einer Woche (Lebacq et al., 1964) bis zu l5 Monaten (Schwartz et al., 1957; Giard et al., 1970). Von entscheidender Bedeutung für die Diagnose und Differenzialdiagnose sind die biochemischen Befunde. Bei 57 von 70 Fällen lag der Serumnatriumspiegel bei bzw. unter 24 Paraneoplastisches Karzinoid-Syndrom Unmittelbare Todesursachen waren eine generalisierte Karzinomatose (Bower et al., 1964), respiratorische Insuffizienz, terminale Bronchopneumonie (Schwartz et al., 1957; Lindsey und Barnes, 1962; Williams, 1963). AA im Urin einhergingen (Warner et al., 1961; Parish et al., 1964 ). Während man früher das Karzinoidsyndrom als ein reines Überproduktionsyndrom des Serotonins ansah, wurden inzwischen Substanzen vom Typ der Kinine und neuerdings auch Prostaglandine gefunden, die die gleichen, aber wesentlich stärkeren Effekte als das Serotonin hervorrufen. Gemeinsam ist allen Substanzen ein synergistischer Effekt mit 5-HT. Es wird angenommen, dass das 5-HT neben seiner direkten Wirkung auf die Darmperistaltik hauptsächlich eine »Triggerfunktion« ausübt und einen Verstärkermechanismus für die Wirkung der anderen pharmakologisch aktiven Substanzen darstellt (Sjoerdsma et al., 1960; Sjoerdsma und Melmon, 1964; Oates et al., 1964 ; Wong und Melmon (1967 ). 5 Das paraneoplastische Karzinoidsyndrom Begriffsbestimmung Beim paraneoplastischen Karzinoidsyndrom wird die Endokrinopathie durch Tumoren hervorgerufen, die pathologischanatomisch keine Karzinoideigenschaften aufweisen. Klinisch treten die typischen Karzinoidsymptome auf, biochemisch liegt eine Überproduktion von 5-Hydroxytryptamin (5 HT, Serotonin) sowie von Precursorsubstanzen (5-Hydroxytryptophan (5 HTP) im Tumorgewebe vor. Die insbesondere bei Karzinoidtumoren auftretenden Herzveränderungen (Endokardfibrose und Pulmonalstenose) kamen beim paraneoplastischen Karzinoidsyndrom nicht vor. Häufiger wurden auch kombinierte Endokrinopathien beschrieben, bei denen das Karzinoidsyndrom zusammen mit einem CushingSyndrom oder einer Hypoglykämie auftrat (Arnett und Long, 1931; Harrison et al., 1957; Kähler und Heilmeyer, 1961; Moertel et al., 1965; Sayle et al., 1965). Pathologische Anatomie Pathologisch-anatomisches Substrat des paraneoplastischen Karzinoidsyndroms sind verschiedene Neoplasien unterschiedlicher Lokalisation, die nur eines gemeinsam haben: Es sind histologisch keine klassischen Karzinoide. Ordnet man die Tumoren nach Organen und histologischer Diagnose, so ergibt sich folgendes Bild: Bei den Lungentumoren handelte es sich in 6 Fällen um typische kleinzellige Ca mit spindelförmigen, kleinen hyperchromatischen Kernen und zahlreichen Mitosen. Metastasen fanden sich in den regionären Lymphknoten, Leber, Pankreas und vereinzelt im Gehirn. Als seltene Lokalisation der Metastasen wurden Pleura, Nebennierenrinde und Thyreoidea beschrieben (Williams und Azzopardi, 1960; Gowenlock et al., 1964; Azzopardi und Bellau, 1965; Kinloch et al., 1965; Majcher et al., 1966; Govindaraj, 1968). 2 Fälle waren undifferenzierte, anaplastische Karzinome mit pleomorphen Aneilen, die stellenweise plump gehäuft oder pseudoazinär angeordnet waren (Siegenthaler et al., 1965; Majcher et al., Fall 2, 1966). Pathogenese Beim atypischen paraneoplastischen Karzinoidsyndrom liegt eine Serotoninüberproduktion durch verschiedene Karzinome vor, die aber nicht zu den Karzinoiden gehören. Formalpathogenetisch wurde das Karzinoidsyndrom lange Zeit ausschließlich als ein Überfunktionssyndrom des Gewebshormons Serotonin angesehen. Es ist bekannt, dass Serotonin unter gewissen Bedingungen die Kapillaren erweitert (Erythem, Flush, Teleangiektasien), Hyper- und Tachypnoe, Bronchospasmen (Asthma) hervorruft und die Darmperistaltik verstärkt (Diarrhoe, Borborygmi). Es müssen aber auch noch andere Substanzen vasoaktiv wirken. Hierfür sprechen unter anderem folgende Befunde: – Eine intravenöse Injektion von 5 HT ruft keine typischen spontanen Flush-Attacken des Karzinoidsyndroms hervor (Page und Cubbin, 1956; Robertson et al., 1962; Levine und Sjoerdsma, 1963). – Es besteht eine geringe Korrelation zwischen freiem Plasmaserotoninspiegel und den Flushepisoden (Levine und Sjoerdsma, 1963). – Eine Infusion von Adrenalin bzw. Noradrenalin ruft bei Karzinoidpatienten einen typischen Flush-Anfall hervor, ohne dass hierbei eine Erhöhung des Plasmaserotoninspiegels weder in der V. hepatica noch in der A. brachialis nachgewiesen werden kann (Robertson et al., 1962; Peart et al., 1961). – Das Karzinoidsyndrom mit Flush-Episoden geht nicht unbedingt mit einer Erhöhung der 5-HIAA-Ausscheidung im Urin einher (Davis und Rosenberg, 1961; Davis, 1962). – Es wurde von metastasierenden Karzinoiden berichtet, die ohne das klinische Bild des Karzinoidsyndroms auszubilden, mit hoher Auscheidung von 5 HI- Bemerkenswert sind die als Nebenbefund beschriebenen hyperplastischen Nebennierenrinden (Azzopardi und Betlau, 1965; Kinloch et al., 1965). Bei zwei Fällen lag eine hypokaliämische Alkalose vor, die aber allein nicht zur Diagnose eines gleichzeitig vorliegenden Cushing-Syndroms berechtigte. Moertel et al. (1965) berichteten über ein primäres solides Karzinom der Schilddrüse mit Amyloidstroma und den klinischen Zeichen eines Karzinoidsyndroms. Anhand dieses Falles diskutierten sie die Hypothese einer Stammzelle, da sich die bekannten nichtkarzinoiden Primärtumoren mit Karzinoidsyndrom aus den entodermalen Aussackungen des primitiven Vorderdarmes entwickelten. Brown (1965) und Siegenthaler et al. (1965) wiesen auf die Schwierigkeiten bei der Differenzialdiagnose eines Karzinoms gegenüber dem Karzinoid hervor. Bei den ausgewerteten Fällen fiel die Versilberung nach Masson-Hamperl negativ aus, ebenso die Diazokupplungsreaktion, die SchmorlReaktion, die Versilberung nach Bodian und die Eigenfluoreszenz im Ultraviolettlicht. Allerdings ist zu berücksichtigen, dass zum Nachweis der argentaffinen Reaktion das Ge- 25 Endokrine und renale Paraneoplasien Typisches und atypisches Karzinoid-Syndrom Typisches und atypisches Karzinoid-Syndrom: Tumorhistologie webe vor 6 Stunden nach dem Tode in Formalin fixiert werden muss (Hedinger, 1962). Klinik Die Geschlechtsverteilung der ausgewerteten Fälle betrug nahezu 1:1. Ein deutlicher Häufigkeitsgipfel lag weder bei Männern noch bei Frauen vor. Das klinische Bild des paraneoplastischen Karzinoidsyndroms wurde im frühen und mittleren Krankheitsverlauf fast ausschließlich durch die Karzinoidsymptome beherrscht. Im Endstadium standen die meist unspezifischen Tumorsymptome im Vordergrund. Die Krankheitsdauer betrug im allgemeinen 2 bis 9 Monate, es sind jedoch Fälle beschrieben worden mit einem Verlauf von mehr als 13 Monaten. Die Häufigkeit, Intensität sowie Größe der befallenen Körperfläche variierten (Robertson, 1962). Es wurden eine zyanotische Komponente (Harrison et al., 1957; Gowenlock et al., 1964) sowie Flush-Attacken beschrieben, die sehr lange Zeit bestanden und sich gelegentlich zu einem Dauerflush entwickelten (Dengler, 1959; Kinloch et al., 1965). Die Flush-Episoden traten häufig postprandial oder nach Alkoholgenuss auf (Harrison et al., 1957; Mengel, 1963; Moertel, 1965). Sie begannen im Gesicht und griffen auf Hals, Nacken, Brust sowie die oberen Extremitäten über, seltener war die ganze Körperoberfläche betroffen. Bensch et al. (1965, 1968) und Hattori et al. (1968) fanden bei elektronenmikroskopischen Untersuchungen in Zellen eines kleinzelligen Ca Serotoningranula, wie sie auch in den argentaffinen Zellen und den bronchialen Schleimdrüsen gefunden werden. Sie beschrieben runde 800 bis 2000 Å im Durchmesser große serotoninhaltige Granula, die in den meisten Fällen von einer »unit-membrane« umgeben waren. Bei den anderen Lungenkarzinomtypen (Plattenepithelkarzinom, Adenokarzinom und großzelliges, undifferenziertes Karzinom) wurden trotz intensiver Suche keine Granula nachgewiesen. Obwohl die Serotoninkonzentration in den Krebszellen deutlich höher war als in den übrigen Zellen, fiel die Silberimprägnation in allen untersuchten Fällen negativ aus. Aufgrund dieser Ergebnisse wurde das kleinzellige Ca der Lunge als eine hochmaligne Variante des bronchialen Karzinoids gedeutet (Bensch et al., 1965, 1968; Siegenthaler et al., 1965; Hattori et al., 1968). 26 Ektopes Gonadotropin-Syndrom Wong und Melmon (1967) berichteten über die Augensymptome vor bzw. während dieser Flush-Attacken. Es handelte sich um eine initiale konjuktivale Injektion, Tränenfluss, Verengung der Netzhautarterien und nachfolgender Pigmentverschiebung. Gowenlock et al. (1964) sowie Peart et al. (1965) wiesen auf atypische Flush-Episoden mit leuchtend hellen Rötungen der Haut hin. Biochemische Untersuchungen des in diesem Falle vorliegenden kleinzelligen Ca ergaben, dass der Tumor nicht 5-Hydroxytryptamin (Serotonin), sondern die Vorstufe 5-Hydroxytryptophan sezerniert hatte. Nach Sandler und Snow (1961) können gewisse Tumoren (insbesondere das kleinzellige Ca ) kein 5-HTP speichern, besitzen andererseits eine nur sehr geringe oder fehlende Decarboxylaseaktivität. So wird das vom Tumor gebildete 5-HTP weder gespeichert noch durch Decarboxylierung in 5-HT abgebaut. und eine hypokaliämische Alkalose vor, sodass der Verdacht geäußert wurde, dass das vorliegende Bronchuskarzinom eine ACTH-ähnliche Substanz sezerniert hatte. Bei einem Patienten mit einem medullären Schilddrüsenkarzinom bestand außerdem ein Hyperparathyreoidismus aufgrund eines Epithelkörperchenadenoms (Moertel et al., 1965). Biochemisch wurden nur wenige Fälle gründlich untersucht. Zeitliche Korrelation: Bei 6 Patienten wurde zuerst der Tumor diagnostiziert, beim Fall von Brown (1965) wurde die Diagnose erst 4 Jahre später gestellt. Bei 3 dieser 6 Fälle bestanden weitere Endokrinopathien, die das klinische Bild beherrschten und eine frühere Tumordiagnose ermöglichten (Van der Sluys Veer et al., 1964; Brown, 1965; Moertel et al., 1965). Eine operative Entfernung des Tumors führte aber zu keiner Besserung des Krankheitsbildes, da bereits bestehende Lebermetastasen zur Entwicklung der klinischen Symptomatik eines Karzinoidsyndroms führten. Die Prognose war infolge der frühen Metastasierung in die Leber infaust. Bei den in der Literatur vereinzelt beschriebenen »brennenden Schmerzen der unteren Extremität« handelte es sich um Pellagra-ähnliche Symptome, die ebenfalls in die Gruppe der Hautsymptome des Karzinoidsyndroms gehören. Govindaraj (1968) sah die Ursache dieser Schmerzen bei Patienten mit einem Lungentumor in einer hypertrophischen pulmonalen Osteoarthropathie, die sich bereits vor der klinischen und radiologischen Manifestation entwickelt. 6 Das ektope Gonadotropin-Syndrom Als ein Frühsymptom des Karzinoidsyndroms ist die Diarrhoe anzusehen, die bei 17 der 20 Patienten beobachtet wurde. Sie trat im allgemeinen zwischen 5 und 10-mal täglich auf, gelegentlich sogar 30-mal (Azzopardi und Bellau, 1965; Dengler, 1959). Dabei handelte sich um wässrige, faulig riechende Stühle mit unverdauten Speiseresten, aber ohne Blutbeimengung. Zuweilen trat auch eine Steatorrhoe auf (Majcher et al., Fall 12, 1966). Laute Borborygmen waren die Manifestation für die auch röntgenologisch nachweisbare beschleunigte Darmmotorik charakteristisch. Zeitliche Korrelationen von Diarrhoen und Flush-Attacken wurden vereinzelt berichtet, scheinen aber nicht obligat zu sein. Bei dem von Driessens (1965) beschriebenen Fall fehlte die Diarrhoe, da eine vergrößerte Metastasenleber (9 kg) eine Teilokklusion des Darmes hervorgerufen hatte. Vorbemerkung Gonadotrope Hormone werden von der Hypophyse und der Plazenta sezerniert. Maligne Neubildungen dieser Organe können zu einer deutlichen Erhöhung des Gonadotropinspiegels im Plasma und Urin führen sowie zur Ausbildung – je nach Alter und Geschlecht – unterschiedlicher Krankheitsbilder. Es wurden Fälle beschrieben, bei denen Tumoren, die keine Ähnlichkeit zu den Hypophysentumoren oder Chorionkarzinomen zeigten, biochemisch nachweisbares Gonadotropin sezernierten und das klinische Bild einer durch Gonadotropinüberproduktion hervorgerufenen Endokrinopathie boten (Fusco und Rosen, 1966; Kosenow et al., 1967; Bernheim, 1968). Es lassen sich zwei Krankheitsbilder abgrenzen: 6.1 Gonadotropin-produzierende Lungentumoren Begriffsbestimmung Bei den Gonadotropin-produzierenden Lungentumoren handelte es sich meistens um anaplastische Karzinome, bei denen klinisch eine Gynäkomastie im Vordergrund stand. Biochemisch ließen sich hohe Gonadotropinwerte im Plasma und Harn quantitativ bestimmen. Das bei 7 Patienten beschriebene Asthma ist als Begleit- und Anfallssymptom anzusehen. Das typische Karzinoidsyndrom ist durch Herzveränderungen gekennzeichnet (insbesondere des rechten Herzens: Endokardfibrose, valvuläre Pulmonalstenosen, Trikuspidalinsuffizienz), die beim paraneoplastischen Syndrom bisher nur bei 2 Fällen beobachtet wurden. Arnett und Long (1931) beschrieben eine Pulmonalstenose, Peart et al. (1965) eine verdickte Trikuspidalis, in beiden Fällen betrug der klinische Verlauf 12 Monate. Die seltene Manifestation von Kardiopathien beim paraneoplastischen Syndrom wurde durch die Kürze der Krankheitsdauer erklärt. Bemerkenswert ist das gehäufte gleichzeitige Vorkommen von weiteren Endokrinopathien. Bei 3 Fällen lag synchron ein Cushing-Syndrom vor, bei 4 ein Diabetes mellitus mit Hyperglykämie, bei 2 ein Cushing-Syndrom, Diabetes mellitus und das paraneoplastisches Karzinoid-Syndrom gleichzeitig (Harrison et al., 1957; Sayle et al., 1965). In den von Azzopardi und Bellau (1965) und Kinloch et al. (1965) beschriebenen Fällen lagen hyperplastische Nebennierenrinden Pathogenese Bei dieser Paraneoplasie konnte durch Isolierung von Gonadotropin aus dem Tumorgewebe und durch den Nachweis von intrazellulär gespeichertem Gonadotropin in Tumorzellen der kausale Zusammenhang bestätigt werden (Fusco und Rosen, 1966; Becker et al., 1968; Swanson Beck et al., 1970). Weintraub und Rosen (1971) wiesen auf die Schwierigkeiten hin, die sich beim Nachweis eines kausalen Zusammenhangs zwischen Tumor und Endokrinopathie ergeben. Selbst nach Iso- 27 Ektopes Gonadotropin-Syndrom Gonadotropinproduzierende Lungentumoren. Biochemische Befunde Die histologischen Veränderungen der Hoden wurden anhand von Serienschnitten untersucht. Bei unterschiedlich langer Krankheitsdauer ergab sich ein abweichendes histologisches Bild. Die Tunica albuginea war stets verdickt. Bei nur kurzer Krankheitsdauer (wenige Monate) konnte eine geringe, bei einem längeren Verlauf (3 bis 3,5 Jahre) eine stärkere Hyperplasie der Leydig-Zwischenzellen und eine schwere Atrophie der Samenkanälchen nachgewiesen werden (Dailey und Marcuse, 1969). Der überwiegende Teil der Samenkanälchen war fibrosiert und hyalinisiert, einige wenige waren noch erhalten und von Sertoli-Zellen ausgekleidet; die Spermiogenese war stark eingeschränkt. Bei keinem Fall lag ein Hodentumor, ein infarziertes oder »ausgebranntes Chorionkarzinom« vor (Fusco und Rosen, 1966; Becker et al., 1968; Dailey und Marcuse, 1969). tersuchung von Probeexzisionen blieb zunächst ergebnislos. Im weiteren Verlauf vergrößerten sich die Mammae. Es waren leicht verschiebbare, im Durchschnitt 2,5 cm große feste Knötchen unterhalb der Mamille zu tasten. Die Hoden waren normal entwickelt und zeigten makroskopisch keine pathologischen Veränderungen. Bemerkenswert ist, dass sich die Gynäkomastie schon frühzeitig entwickelt hatte und somit als ein Frühsymptom anzusehen ist (Altschul, 1938; Castillo et al., 1941; Sirtori et al., 1957; Camiel et al., 1967). Biochemie: Die 17-Ketosteroidausscheidung war normal bis leicht erhöht. Bei kurzer Krankheitsdauer wurden stark erhöhte Östrogenwerte bis zu 15 µg/24 h gefunden (Fusco und Rosen, 1966; Paiman et al., 1967; Rosen et al., 1968), während sie bei längerem Krankheitsverlauf normal waren. Dieser Befund ist als Erschöpfung der endokrinen Zellen anzusehen, die trotz ununterbrochener Stimulierung durch das Gonadotropin nicht mehr in der Lage waren, Östrogen zu bilden. Die Gonadotropinausscheidung im Harn war in allen Fällen deutlich erhöht. Bei 5 von 9 Patienten konnte im Tumorgewebe Gonadotropin und bei 3 weiteren Patienten das »human chorionic somatomammotropin« nachgewiesen werden (Weintraub und Rosen, 1971). In 3 Fällen wurde der Serumgonadotropinspiegel bestimmt (Fusco und Rosen, 1966, Fall 4 und 4; Becker et al., 1968); in allen Fällen war er deutlich erhöht. Die histologische Untersuchung der Hypophyse, der Pinealis, Schilddrüse und Parathyreoidea zeigte keine pathologischen Befunde. Dailey und Marcuse (1969) beschrieben im Hypophysenvorderlappen viele große Zellen mit granulärem Zytoplasma sowie bläschenförmigen Kernen, die den »Schwangerschaftszellen« von Erdheim und Stumme (1909) entsprachen. Klinik Bei den ausgewerteten Fällen handelte es sich um 9 Männer und um eine 25-jährige Frau. Zu Beginn der Erkrankung wurden unspezifische Symptome wie Husten, Schwäche, Nachtschweiß und Schmerzen der oberen Extremität und im Brustbereich beobachtet. Im Röntgenbild waren im Anfangsstadium meist keine Veränderungen festzustellen, die Sputumdiagnostik war negativ. Auch die histologische Un- Tumordiagnose: Nur bei 3 Patienten konnte durch Thorakotomie die Tumordiagnose gestellt werden, bei den restlichen Fällen wurde sie erst durch die Obduktion gesichert. 28 Text bach et al., 1968) und in verschiedenen anderen Tumoren beobachtet (Weintraub und Rosen, 1971). lierung von Hormonen aus Tumorextrakten konnte nicht auf den Ursprung dieser Hormone geschlossen werden, da gelegentlich Tumoren als »Fallen« für Plasmahormone wirken (Unger et al., 1964 ; Weintraub und Rosen, 1971). Bei Patienten mit hoher Östrogenausscheidung kann ein Zusammenhang zwischen der Höhe des HCS-Spiegels im Serum und dem Grad der Gynäkomastie nachgewiesen werden. Untersuchungen des Tumorgewebes zeigten, dass das HCS intrazellulär lokalisiert war. Somit liegt die Annahme nahe, dass das HCS vom Tumor selbst synthetisiert wurde. Weitere Analysen ergaben, dass das Tumorgewebe Enzyme bildet, die als »Precursor-Substanzen« der Östrogensynthese wirkten. Zu den sichersten Beweisen einer ektopen Hormonbildung gehört der Nachweis einer arteriovenösen Differenz des Hormonspiegels nach Tumorpassage (Weintraub und Rosen, 1971). Faiman et al. (1967) konnten eine arteriovenöse Differenz eines FSH-ähnlichen Gonadotropins durch Anwendung einer immunchemischen Methode mit radioaktiv markierten Antikörpern während der Operation nachweisen. Becker et al. (1968) untersuchten bei ihrem Patienten den Einfluss hoher Dosen von Testosteron, Cortison und Stilböstrol auf die Höhe des Gonadotropinspiegels im Plasma und Urin. Sie fanden einen weitgehend konstant hohen Gonadotropinspiegel, der auch durch Bestrahlung des Tumors und Behandlung mit 2 antineoplastischen Substanzen kaum zu beeinflussen war. Pathologische Anatomie Es wurden 10 Gonadotropin-produzierende Lungentumoren ausgewertet. Ordnet man sie nach ihrem histologischen Typ, so handelte es sich um 6 großzellig-anaplastische Karzinome, 2 Plattenepithelkarzinome und um ein Adenokarzinom. Bei einem Fall lagen keine Angaben über den feingeweblichen Aufbau vor. Die unterschiedlich großen Tumoren waren auf einzelne Lungenlappen beschränkt (Fusco und Rosen, 1966) oder hatten die ganze Lunge befallen (Faiman et al., 1967; Dailey und Marcuse, 1969). Es fanden sich in den meisten Fällen kontralaterale Absiedelungen in der anderen Lunge (Hardy, 1960; Faiman et al., 1967; Dailey und Marcuse, 1969). Metastasen wurden außerdem in den regionären Lymphknoten, der Leber, im Gehirn, in den Nieren sowie in den Nebennieren gefunden. Bei den bisher beschriebenen Fällen wurden der gonadotropen Substanz luteotrope Eigenschaften zugeschrieben (Fusco und Rosen, 1966; Becker et al., 1968; Dailey und Marcuse, 1969): vergrößerte und zeitweilig sezernierende Mamma, Hypertrophie der Leydig-Zwischenzellen und Auftreten von Schwangerschaftszellen im Hypophysenvorderlappen (Dailey und Marcuse, 1969). Durch die langdauernde Stimulierung der Hoden und der Nebennierenrinde durch Gonadotropin wurde vermehrt Östrogen gebildet, das die morphologischen Mammaveränderungen hervorrief. Bei einem nur kurzen Krankheitsverlauf fand man eine hohe Östrogenausscheidung im Urin, eine nur geringe Vermehrung der Leydig-Zwischenzellen sowie das volle klinische und histologische Bild einer Gynäkomastie, das sich nach operativer Entfernung des Tumors wieder zurückbildete (Fusco und Rosen, 1966). Histologisch wurden beim großzelligen anaplastischen Karzinom pleomorphe oder polygonale Zellen beschrieben, die in synzytialen Strängen, Nestern oder Läppchen angeordnet waren und durch sinusoidale Räume, in denen Blut oder organisierte Thromben lagen, abgegrenzt wurden (Fusco und Rosen, 1966). Ein sehr ähnliches histologisches Bild wurde auch bei den Leberzellkarzinomen des hepatogenitalen Syndroms beschrieben. Bei 3 Geschwülsten wurden vielkernige Tumorriesenzellen beobachtet, die an ein Chorionkarzinom erinnerten. Dieser Tumortyp wurde jedoch nach einer späteren histologischer Nachuntersuchung durch mehrere Pathologen ausgeschlossen. Bei der Beobachtung von Becker et al. (1968), bei der eine Krankheitsdauer von 2 Jahren vorlag, waren die Östrogenausscheidung nicht erhöht, die Leydig-Zwischenzellen stark vermehrt. Alle diese Beobachtungen unterstreichen die Bedeutung der Östrogene für die Entstehung der Gynäkomastie. Die bereits erwähnten elektronenmikroskopischen Untersuchungen, denen zufolge kleinzellige Ca von den bronchialen Kulchitsky-Zellen abstammen sollen (Bensch et al., 1968) veranlassten Swanson Beck et al. (1970), auch die anaplastischen Karzinome zu untersuchen. Um eine eventuelle trophoblastenähnliche Zelldifferenzierung darstellen zu können, verwandter fluoreszenzoptisch markiertes »human chorionic somatomammotropin«. Bei keinem der Fälle wurde eine trophoblastenähnliche Differenzierung nachgewiesen, daher war eine echte ektope Hormonbildung durch den Tumor selbst anzunehmen. Ein weiteres Hormon, das »human chorionic somatomammotropin« (HCS, humanes Plazentalaktogen ) konnte inzwischen in einigen gonadotropinsezernierenden Lungenkarzinomen nachgewiesen werden. 1962 isolierten Josimovich und McLaren ein weiteres Trophoblastenhormon und nannten es zunächst »human placental lactogen«. Li et al. (1968) schlugen die heute allgemein übliche Bezeichnung »human chorionic somatomammotropin« (HCS) vor. HCS ist biochemisch ein Proteohormon, wird von der Plazenta gebildet und zeigt Eigenschaften, die dem Wachstumshormon der Hypophyse und dem Prolactin entsprechen (Josimovich und Atwood, 1964; Grumbach et al., 1968; Li et al., 1968; Samaan et al., 1968). Dieses Hormon wurde im Serum von Patienten mit einem Gonadotropin-sezernierenden, entdifferenzierten Lungenkarzinom (Weintraub und Rosen, 1969), einem Chorionkarzinom (Frantz et al., 1965; Grum- Die histologische Untersuchung des vergrößerten Brustgewebes ergab eine Hypertrophie und Ektasie der Drüsenschläuche sowie eine Hyperplasie des Gang-auskleidenden Epithels. Das interlobuläre Bindegewebe war stark vermehrt, das periduktale Gewebe fibrosiert und stellenweise hyalinisiert (Becker et al., 1968; Dailey und Marcuse, 1969). 29 Endokrine und renale Paraneoplasien Die Überlebenszeit nach den ersten klinischen Symptomen schwankte zwischen 2 Monaten und 3,4 Jahren. Die Prognose ist bis jetzt infaust. die durch Sinusoide mit sternzellähnlicher Endothelauskleidung voneinander abgegrenzt waren. Reeves et al. (1959) und Behrle et al. (1963) fanden Gallethromben zwischen den Tumorzellen. Metastasen wurden in den regionären Lymphknoten, Lungen, Nebennieren und beim Fall von Root et al. (1968) auch im linken Femur nachgewiesen. In allen Fällen stimmte das histologische Bild des Tumors mit den Metastasen überein. 1971 wurde von Grillo ein bisher einmaliger Fall beschrieben: Er berichtete über eine 25-jährige Nigerianerin mit 5 Monate anhaltenden Menometrorrhagien und ungewöhnlichen Absonderungen der Mammae. Ein hoher Gonadotropinspiegel im Harn deutete auf eine frühe Schwangerschaft. Eine Röntgenuntersuchung des Thorax ergab eine Verschattung im linken Untergeschoss der Lunge, die sich innerhalb von 5 Monaten in ihrer Größe verdoppelt hatte. Eine Thorakotomie ergab eine hämorrhagische Tumormasse im linken Unterlappen, die reseziert und histologisch als anaplastisches Karzinom diagnostiziert wurde. Postoperativ bildeten sich die klinischen Symptome innerhalb von 3 Wochen zurück. Zwei Monate nach der Lobektomie wurde die Patientin schwanger und nach einer komplikationslosen Schwangerschaft von einem gesunden Knaben entbunden. Dieser Fall ist bemerkenswert, da zum ersten Mal ein Gonadotropin-produzierender Tumor bei einer Frau diagnostiziert wurde. Die 2 Monate nach der Lobektomie eingetretene und voll ausgetragene Schwangerschaft beweist, dass keine Schäden im Hormonsystem geblieben waren. Klinik Klinisch standen beim hepatogenitalen Syndrom Lebervergrößerung und Pseudopubertas praecox im Vordergrund. Überdurchschnittliche Körpergröße, eine tiefe Stimme (oft bereits mit Stimmbruch), erste Anzeichen einer Schambehaarung, Vergrößerung des Penis bei relativ normaler Hodengröße sowie eine schon kräftig entwickelte Muskulatur wurden als häufigste Symptome beschrieben (Behrle et al., 1963; Hung et al., 1963; Kosenow et al., 1967; Root et al., 1968). Bei 3 von 9 Fällen wurde die Neubildung reseziert; bei 6 Patienten war der Tumor bereits inoperabel. Nach kombinierter Strahlen- und Zytostatikatherapie kam es zu einem leichten Rückgang der Schambehaarung, der 17-Ketosteroidausscheidung im Urin und des Lebertumors (Behrle et al., 1963; Hung et al., 1963; Thamdrup, 1965; Kosenow et al.,1967; Root et al.,1968). Bei keinem Patienten konnte eine Heilung erzielt werden. Die Überlebenszeit nach den ersten Symptomen schwankte zwischen 8 und 29 Monaten. 6.2 Das hepatogenitale Syndrom Begriffsbestimmung Bei diesem klinisch eigenständigen Krankheitsbild, bei dem ausschließlich männliche Patienten im Kindesalter betroffen waren, wurde eine Pseudopubertas praecox durch ein primäres Hepatoblastom hervorgerufen. Pathogenese Der Lebertumor wird als Ursache der gesteigerten Gonadotropinproduktion angesehen (Kosenow et al., 1967; Root et al., 1968). Der kausale Zusammenhang zwischen Tumor und Paraneoplasie wurde durch den Nachweis von Gonadotropin im Tumorgewebe bestätigt. Kosenow et al. (1967) und Root et al. (1968) identifizierten das Gonadotropin als LH -ähnliches Hormon. 7 Das ektope STH-Syndrom Begriffsbestimmung Beim ektopen STH-Syndrom handelt es sich um ein Krankheitsbild, das durch das gleichzeitige Vorliegen eines malignen Lungentumors und einer hypertrophen Osteoarthropathie (Pierre-Marie-Bamberger-Syndrom) gekennzeichnet ist. Hypertrophe Osteoarthropathien werden in ca. 4% (Yacoub, 1965), osteoartikuläre Symptome in bis zu 25% der Lungentumoren diagnostiziert (Bariety et al., 1964). Auch bei viszeralen Karzinomen sind diese Knochenveränderungen beschrieben worden (Hollis, 1967; Thibault, 1968). Steiner et al. (1968) bestimmten bei 8 Patienten mit Lungentumoren den Plasma-STH-Spiegel; nur in einem Fall war das STH im Plasma stark erhöht und mit dem klinischen Bild einer Osteoarthropathie vergesellschaftet. Sparagana et al. (1971) untersuchten das Tumorgewebe von 28 Lungenkarzinompatienten und stellten bei einem Adenokarzinom erhöhte STHWerte fest. Der Nachweis einer LH-ähnlichen Substanz stimmte auch mit der in fast allen Fällen beschriebenen Hyperplasie der Leydig-Zwischenzellen überein. Als Folge der Hyperplasie dieser Zellen kam es zu einer Überproduktion von Testosteron. Nach Ansicht von Behrle et al. (1963) sezernierte die Hypophyse das Gonadotropin und rief die Pubertas praecox hervor. Sie beobachteten, ebenso wie Reeves (1959) eine Vermehrung der basophilen Zellen in der Hypophyse, die als mögliche Gonadotropinbildner angesehen werden. Kosenow et al. (1967) gelang es erstmals aus dem Leberzelltumor eine Gonadotropin wirksame Substanz zu isolieren und eine luteinisierende Wirkung nachzuweisen. So kann der kausalpathogenetische Zusammenhang zwischen Hepatoblastom und Pseudopubertas praecox als gesichert gelten. Pathogenese Die Pathogenese der hypertrophen Osteoarthropathie ist noch weitgehend ungeklärt. Für einen humoralen Faktor sprechen die Befunde von Ginsburg und Brown (1961) – erhöhte Östrogenausscheidung bei 11 Fällen (darunter 10 Karzinomfälle) mit einer hypertrophen Osteoarthropathie – sowie Fusco und Rosen (1966), die unter Gynäkomastiefällen einen Patienten mit Gynäkomastie, hypertrophen Osteoarthropathie und Lungenkarzinom beschrieben. Pathologische Anatomie Pathologisch-anatomisch fand man Lebertumoren, die ein Gewicht von 2,6 kg erreichten (Kosenow et al., 1967). Das histologische Bild war in allen Fällen sehr einheitlich. In den unterschiedlich differenzierten, teils pseudoglomerulär und primitiv adenomatösen, malignen juvenilen Hepatoblastomen wurden mehrreihige Karzinomzellstränge beschrieben, 30 Ektopes STH-Syndrom Hepatogenitales Syndrom. Biochemische Befunde Die genaue Bestimmung einer venös-arteriallen Differenz einer Hormonkonzentration ist bei Lungentumoren äußerst schwierig, da man arterielles und venöses Blut sowohl aus den Vasa publica wie aus den Vasa privata der Lunge gleichzeitig gewinnen muss (Greenberg et al., 1972). Einen Hinweis auf einen möglichen kausalpathogenetischen Zusammenhang zwischen Tumor und einer hypertrophen Osteoarthropathie gaben die Befunde von Steiner et al. (1968), die bei 12 Patienten mit einem Lungenkarzinom und Trommelschlegelfingern den STH-Spiegel im Plasma bestimmten. Bei einem Patienten lag er mit 38 ng/ml weit über dem Normwert von 0 bis 9 ng/ml. Nach der operativen Entfernung des Tumors normalisierte sich der STH Spiegel im Plasma und die hypertrophe Osteoarthropathie bildete sich zurück. Ein weiterer Beweis für die Synthese einer STH-ähnlichen Substanz durch einen Tumor wurde von Greenberg et al. (1972) erbracht. Es gelang ihnen, Zellen aus einem anaplastischen Lungenkarzinom eines Patienten mit einer hypertrophen Osteoarthropathie 4 Monate in einer Kultur zu züchten. Das Kulturmedium enthielt radioaktiv markiertes 14C-Leucin, das von den Tumorzellen aufgenommen und in eine Substanz eingebaut wurde, die bei verschiedenen analytischen und immunchemischen Greenwood (1966) beobachtete bei Frauen mit Mammakarzinom vor und nach einer Mastektomie einen erhöhten Plasmaspiegel des somatotropen Hormons. Gleichzeitig war aber auch der Plasmahydrocortisonspiegel erhöht, sodass von einem »Stress-induzierten« STH-Anstieg gesprochen wurde. Bestimmungen ein übereinstimmendes Verhalten mit einer Standard-STH-Probe zeigte. Kreuzreaktionen mit HPL lagen nicht vor. Diese Befunde, die für einen kausalpathogenetischen Zusammenhang zwischen Tumor und hypertropher Osteoarthropathie sprechen, wurden durch die Beobachtung ergänzt, dass in praktisch allen Fällen die Knochenschmerzen sofort nach der operativen Entfernung des Tumors aufhörten und die gesamte Symptomatik der hypertrophen Osteoarthropathie innerhalb weniger Wochen vollständig zurückbildete. Cameron et al. (1969) wiesen mit immunchemischen Methoden im Tumorextrakt eine STH-ähnliche Substanz mit einer Aktivität von 169 ng/g-Tumorgewebe nach und konnten so experimentell die Ansicht von Stovin (1965), dass Tumorzellen auch langkettige Polypeptide wie ACTH und STH bilden können, bestätigen. Cameron et al. (1969) wiesen im nichttumorös infiltrierten Lungengewebe eine STH-Aktivität von 6,3 ng/g nach (Lungentumor: 169 ng/g). Durch immunhistochemische Untersuchungen mit FITC-markierten Antiseren (FITC = fluorensuinisothiocyanat) ließ sich fluoreszierendes Tumorgewebe darstellen. Bestimmte Zellgruppen zeigten auch eine Fluoreszenz mit FITC markierten Antiseren gegen menschliches plazentares Lactogen (HPL). Eine Bestimmung des STHSpiegels in der Pulmonalvene ergab 8,5 ng/ml, ein deutlich erhöhter Wert gegenüber 2,9 bis 5,9 ng/ml im übrigen Blut. Pathologische Anatomie Das pathologisch-anatomische Substrat des ektopen STH Syndroms sind der Tumor und die röntgenologisch nachweisbaren Knochenveränderungen. Bei den Tumoren handelte es sich bei den 8 ausgewerteten Fällen um Lungenkarzinome. Lymphknotenmetastasen wurden in 13 Fällen beschrieben. Die pathologischen Veränderungen der Knochen bestanden in einer schon röntgenologisch nachweisbaren pe- 31 Endokrine und renale Paraneoplasien Tumorhistologie beim ektopen STH-Syndrom riostalen Proliferation der langen Röhren-, Metakarpal- und Metatarsalknochen sowie der Phalangen von Händen und Füßen. Röntgenaufnahmen von Schädel und Sella turcica zeigten dagegen keine pathologischen Befunde. Die Knochenveränderungen erwiesen sich – nach operativer Entfernung des Tumors – als vollständig rückbildungsfähig. Pathogenese Auch bei diesen 21 ausgewerteten Fällen kann der kausale Zusammenhang zwischen Tumor und Endokrinopathien als gesichert angesehen werden. Pathologische Anatomie Ordnet man die Tumoren nach ihrer Lokalisation und Histologie, so ergibt sich die dargestellte Tabelle. Klinik Die Alters- und Geschlechtsverteilung entspricht einem Kollektiv mit Lungentumor. Das klinische Bild des ektopen STH-Syndroms wird durch die Symptomatik der hypertrophen Osteoarthropathie bestimmt. Kennzeichnend ist eine kolbige Auftreibung der Endphalangen mit uhrglasförmiger Wölbung der Nägel. In nahezu allen Fällen traten spontane Schmerzen in Hand- und Fußgelenken sowie an den benachbarten langen Röhrenknochen auf. Periphere therapieresistente Ödeme gehörten ebenfalls zum klinischen Bild der hypertrophen Osteoarthropathie (v. Wichert, 1971). Die Symptome der hypertrophen Osteoarthropathie traten vor der Tumormanifestation auf und wiesen auf einen möglichen malignen Lungenprozess hin. Klinik Die Klinik dieser multiplen Endokrinopathien unterscheidet sich nicht von der isolierten Endokrinopathie. In den meisten Fällen standen die Symptome einer bestimmten hormonellen Störung im Vordergrund, während die anderen klinischen Zeichen Nebenbefunde darstellten. Die Alters- und Geschlechtsverteilung zeigte keine wesentlichen Unterschiede. 9 Ektopes TSH-Syndrom Paraneoplastische Hyperthyreosen wurden erstmals 1956 von de Gennes und Bricaire publiziert. Als Primärtumoren wurden fast ausschließlich Chorionkarzinome und Tumoren aus versprengten Trophoblastkeimen beschrieben (de Gennes, 1956; 1962; Odell et al., 1963; Steigbigel, 1964; Hennen, 1966a; Winand et al., 1969). Um den strengen Kriterien für die Diagnose eines ektopen oder paraneoplastischen TSH-Syndroms genügen zu können, muss ein Tumor vorliegen, der keine histogenetischen Beziehungen zu trophoblastischem Gewebe besitzt. Außerdem muss aus dem Tumor TSH isoliert werden können, der Plasma-TSH-Spiegel bei der Tumorpassage eine arteriovenöse Differenz aufweisen und im gesamten deutlich erhöht sein. Biochemisch wurde die Diagnose eines ektopen STH-Syndroms durch die Bestimmung des STH-Spiegels vor und nach der Operation sowie durch den Nachweis der Synthese, Speicherung und Freisetzung von STH durch den Tumor bestätigt. Bei 6 Patienten führte die operative Entfernung des Tumors zu einer vollständigen Rückbildung der Symptome der hypertrophen Osteoarthropathie und bei einem zur völligen Genesung. Nur 1 Patient verstarb mehrere Wochen nach der Operation (Sparagana et al., 1971). So ist die Prognose im Vergleich zu den anderen endokrinen paraneoplastischen Syndromen relativ gut und wird im wesentlichen durch die frühe Diagnose des Grundleidens bestimmt. Hennen (1966 b/1967) beschrieb ein Bronchialkarzinom mit den Symptomen einer paraneoplastischen Hyperthyreose bei einem Mann. Im Tumor ließ sich TSH nachweisen und isolieren. Codaccioni und Jaquet (1971) berichteten über einen deutlich erhöhten TSH-Spiegel bei einer 45-jährigen Frau mit einem Mammakarzinom. 8 Multiple paraneoplastische Endokrinopathien Das ektope TSH-Syndrom unterscheidet sich von dem klassischen Bild einer Hyperthyreose durch eine nur gering ausgeprägte Symptomatik. Es fehlen Struma und Augensymptome. Auffallend sind eine Tachykardie und eine deutliche Begriffsbestimmung Gelegentlich werden maligne Tumoren von mehreren, gleichzeitig auftretenden Hormonstörungen begleitet. 32 Tumorkachexie Multiple paraneoplastische Endokrinopathien. Tumorhistologie Erhöhung des proteingebundenen Jods (PBJ) sowie der 24h-Jodaufnahme (Dowling et al., 1960). 13 Paraneoplastische Hormonbildung ohne klinische Wirkung 10 Paraneoplastisches ZollingerEllison-Syndrom Bei biochemischen und immunhistochemische Untersuchungen von bösartigen Neubildungen wurden verschiedene Hormone bzw. Stoffe mit hormonähnlicher Wirkung nachgewiesen, die aber – wegen der geringen Menge – nicht von klinischen Befunden begleitet wurden: – Prolaktin bei Lungen-, Nieren- und Pankreaskarzinomen. Nur bei einer besonders hohen Konzentration kam es einer Gynäkomastie oder Galaktorrhö. – Kalzitonin bei Lungen-, Mamma-, Kolon- und Pankreaskarzinomen. – Neurotensin bei Tumoren des endokrinen Pankreas – Bombesin (gastric releasing hormone) beim kleinzelligen Lungenkarzinom – Chromogranin bei neuroendokrinen Tumoren – HCG: Neben den klassischen HCG-produzierenden Neubildungen des Hodens (nichtseminomatöse Keimzelltumoren) und der Plazenta (Blasenmole) wurden vereinzelt hohe Werte bei einem malignen Melanom sowie bei Mamma-, Nieren-, und Lungenkarzinome festgestellt. Ein paraneoplastisches Zollinger-Ellison-Syndrom wurde bisher nur in Verbindung mit einem ektopen ACTH-MSHSyndrom als Mischendokrinopathie beobachtet (Law et al., 1965). Da die Gastrinbildung nicht nur auf den Magen beschränkt bleibt, kann sie bei den ulzerogenen Nicht-B-Zelladenomen des Pankreas nicht als ektop angesehen werden (Labhart, 1971b; Feurle et al., 1972). 11 Erythropoietin-bildende Tumoren Diese Tumorgruppe und das durch sie verursachte Krankheitsbild einer paraneoplastischen Polyglobulie wird unter den renalen Paraneoplasien beschrieben. 12 Renin-bildende Tumoren Hauger-Klevene veröffentlichte 1970 die Beobachtung über einen 38-jährigen Mann mit einem kleinzelligen Ca und einer hypokaliämischen Alkalose bei normalen Plasma-ACTH, Plasmacorticoid- und Natriumwerten. Die Alkalose wurde auf eine verstärkte Aldosteronsekretion zurückgeführt infolge einer Reninsekretion durch die Geschwulst. Die biochemische Untersuchung des Tumors zeigte eine Reninaktivität (28 ng/g-Tumorgewebe). Tumorkachexie Die Tumorkachexie – heute meist als Syndrom bezeichnet (CCS: cancer cachexia syndrome oder CACS: cancer anorexia–cachexia syndrome) – ist eine häufige Komplikation bei einem fortgeschrittenen Krebsleiden und ein wesentlicher Faktor als Todesursache. Diese Komplikation ist besonders häufig bei Kindern, älteren Menschen und bei Tumoren des 33 Endokrine und renale Paraneoplasien Klinik Wesentliche Zeichen einer Tumorkachexie sind Anorexie (nicht immer vorhanden), Entzündungen, Insulinresistenz und Atrophie der Muskulatur. Andere innere Organe sind meist vom Proteinabbau nicht betroffen. Der Gewichtsverlust kann im Rahmen einer Tumorkachexie – im Gegensatz zu einer gesunden Person – nicht durch verstärkte Nahrungsaufnahme kompensiert werden. Verdauungstrakts: Man rechnet das bis zu 50% der Krebspatienten betroffen sind. Beim Pankreaskarzinom sind es 80%. Eine Ausnahme macht das Mammakarzinom: Hier kommt es wesentlich seltener zu einer Tumorkachexie als letale Komplikation. Bis heute gibt es keine befriedigende Definition für die Begriffe Kachexie und Tumorkachexie. Häufig wird ein Verlust an Körpermaße als Kriterium angegeben. Dabei ist zu berücksichtigen, dass Kachexie durch Unterernährung und durch Tumor sich in einem wesentlichen Punkt unterscheiden: Bei der Tumorkachexie kommt es neben einem Verlust an Fettreserven auch zu einem Abbau der Muskulatur. III. Diskussion Allgemeine Häufigkeit endokriner paraneoplastischer Syndrome Aus der Literatur wurden klinische und laborchemische Befunde von 687 Patienten, die endokrine Störungen als Ausdruck eines paraneoplastischen Syndroms zeigten, ausgewertet. Es liegen noch keine Angaben über die allgemeine Häufigkeit des endokrinen paraneoplastischen Syndroms vor. Beschränkt man sich auf den häufigsten Tumor, das heißt auf das Bronchialkarzinom, so ergeben sich folgende Werte: 10 bis 15% aller Bronchialkarzinome gehen mit einem Hyperkalzämiesyndrom einher, 4 bis 5% mit einem ektopem ACTH-Syndrom und 2 bis 3% mit einem ektopem ADHSyndrom. Unter den Krebsleiden insgesamt sollen häufiger Zeichen einer paraneoplastischen Hyperkalzämie vorliegen. Die Tumorkachexie ist primär als eine Stoffwechselerkrankung einzuordnen. In fortgeschrittenen Stadien hat man aber häufiger Botenstoffe mit katabolischer Wirkung bevorzugt im Urin gefunden, die als Zytokine anderer Stoffwechselprodukte identifiziert werden konnten. Somit liegen Ähnlichkeiten gegenüber anderen endokrinen Paraneoplasien vor. Pathogenese Die Pathogenese einer Tumorkachexie ist sehr komplex. Als Folge des Krebsleidens werden Botenstoffe gebildet, die den katabolischen Stoffwechsel begünstigen, Fett aus dem Fettgewebe und Proteine in der Muskulatur abbauen. Ferner kommen noch andere Faktoren hinzu: eine herabgesetzte Nahrungsaufnahme sowie Maldigestion und Malabsorption. Auch Entzündungen sowie Blut- und Eiweißverlust aus ulzerierten Karzinomen spielen eine Rolle. Alters- und Geschlechtsverteilung beim endokrinen paraneoplastischen Syndrom Von 570 ausgewerteten Patienten waren 361 (63%) männlichen und 209 (37%) weiblichen Geschlechts. Die gesamte Altersverteilung läßt keine Unterschiede zwischen den Geschlechtern erkennen: Bei beiden lag der Häufigkeitsgipfel im 5. bis 7. Lebensjahrzehnt. 404 (71%) Patienten des gesamten Kollektivs waren 41 bis 70 Jahre alt. Männer und Frauen waren etwa im Verhältnis 2 : 1 betroffen. Wenn der Tumor eine bestimmte Größe erreicht und nicht mehr ausreichend mit Blut versorgt wird, kommt es zu Nekrosen. Dabei werden der Tumornekrosefaktor (TNF-α), weitere Zytokine (Interleukin-1, Interleukin-6) und andere Stoffwechselprodukte ausgeschüttet. Die Wirkung wurde experimentell an Mäusen bestätigt. Trotzdem wird bezweifelt, ob die genannten Stoffe die Hauptverantwortung für eine Tumorkachexie tragen. Häufigkeit der endokrinen Paraneoplasien Zu den häufigsten publizierten endokrinen Paraneoplasien gehören das ektope ACTH-Syndrom, die extrapankreatische Hypoglykämie und die paraneoplastische Hyperkalzämie. Zu selteneren Formen zählt das ektope STH-Syndrom, von dem nur 8 Fälle publiziert wurden. Das ektope ACTH-Syndrom (37,4%) und die extrapankreatischen Hypoglykämien (26,0%) machten zusammen 63,4% aller Paraneoplasien aus. Es folgten die paraneoplastische Hyperkalzämie (15,8% ) und das ektope ADH-Syndrom (13,7%). Tumoren sind in der Lage katabolische Botenstoffe zu produzieren. Insbesondere sind zwei Faktoren von Bedeutung: ● Der Lipid-mobilisierende Faktor ist ein Peptid, das die Lipolyse in Fettzellen stimuliert. Dieser Stoff kommt nur bei Krebspatienten mit Gewichtsverlust vor. ● Der Proteolyse-induzierende Faktor wurde aus dem Urin von Krebspatienten mit Tumorkachexie gewonnen. Er wurde nicht bei Krebsfällen ohne Gewichtsverlust bzw. bei Patienten ohne Krebs nachgewiesen. Im Tierversuch konnte der Abbau von Muskelmasse bestätigt werden. Kombinationen endokriner Erkrankungen beim paraneoplastischen endokrinen Syndrom Aus der Literatur wurden 14 Fälle ausgewertet, bei denen mehrere endokrine Störungen meta- oder synchron auftraten und nur durch einen Tumor hervorgerufen wurden. Am häufigsten wurde das ektope ACTH-Syndrom mit dem klinischen Bild eines Morbus Cushing – kombiniert mit anderen Endokrinopathien – beobachtet. In 8 Fällen war es mit einem Karzinoidsyndrom vergesellschaftet, in 4 Fällen mit einem ektopen ADH-Syndrom und in 2 Fällen mit einem paraneoplastischen Hyperkalzämiesyndrom. Bei 7 Patienten lag ein Als weitere Ursache einer Tumorkachexie wird der Krebsbedingte erhöhte Bedarf an Energie angeführt. Große Tumoren haben einen zusätzlichen Energiebedarf von 300 kcal/ Tag. Außerdem besteht ein erhöhter Bedarf an Glukose, die im Tumor zu Laktat abgebaut und in der Leber (über den Cori-Zyklus) wieder zu Glukose umgewandelt wird. Dieser Prozess beansprucht auch reichlich Energie. 34 Diskussion Häufigkeit der endokrinen Paraneoplasien unter den ausgewerteten Fällen Kombination verschiedener endokriner Paraneoplasien kleinzelliges Karzinom, bei 3 ein Inselzellkarzinom des Pankreas vor. 35 Endokrine und renale Paraneoplasien Endokrine Paraneoplasien unter Berücksichtigung der Tumorlokalisation Die paraneoplastische Polyglobulie wird unter den renalen Praneoplasien abgehandelt Tumorlokalisation beim paraneoplastischen endokrinen Syndrom Endokrine Paraneoplasien könnten als Frühsymptome ein Leitsymptom eines noch ungekannten Tumors sein. Es ist daher von diagnostischer Bedeutung, ob bei einzelnen Endokrinopathien bestimmte Tumorlokalisationen besonders häufig sind. Mit Ausnahme der extrapankreatischen Hypoglykämie überwogen bei allen anderen paraneoplastischen endokrinen Störungen die epithelialen Neubildungen. Die Lungenkarzinome stellten die häufigste Tumorgruppe dar. Sie wurden bei 287 Fällen beobachtet, das sind 42% aller Geschwülste und 50% aller Karzinome. Als zweithäufigstes Karzinom trat in 8% aller Fälle ein Leberkarzinom auf, ins- besondere beim Nadler-Wolfer-Elliott-Syndrom (87% aller Fälle) und bei dem durch ektope Gonadotropinbildung bei Knaben hervorgerufenen hepatogenitalen Syndrom. Pankreastumoren machten 7% aller Geschwülste aus, es folgten das Thymuskarzinom (4%), das nur in Verbindung mit einem ektopen ACTH-Syndrom gefunden wurde. Zu den häufigsten Sarkomen zählten die Fibrosarkome (11% aller Tumoren). Man fand sie in 72% der Doege Potter-Syndrome und in 4% der Nadler-Wolfer-Elliott- Syndrome. 36 Diskussion Relative Häufigkeitsangaben zur Tumorlokalisation bei endokrinen Paraneopasien Altersverteilung bei Tumoren mit und ohne endokrine Paraneoplasie die zeitliche Korrelation zwischen dem Auftreten bzw. der Diagnose einer endokrinen Störung und dem Tumor. Alters- und Geschlechtsverteilung der Tumoren mit und ohne paraneoplastische Endokrinopathie Die in der Literatur (Willis, 1967) angegebene Alters- und Geschlechtsverteilung der Neubildungen mit und ohne Paraneoplasie zeigten: Das Lungenkarzinom wies keine Abweichungen auf. Es trat in beiden Kollektiven vorwiegend im 6. Lebensjahrzehnt auf und war bei Männern 3,5- bis 5-mal häufiger. Die Altersverteilung der Lebertumoren zeigte keinen deutlichen Altersgipfel. Es kam im 1. Lebensjahrzehnt beim Gonadotropin-produzierenden hepatogenitalen Syndrom vor. Ein zweiter Gipfel fand sich im 5. Lebensjahrzehnt, bei dem auch die Lebertumoren ohne endokrine paraneoplastische Störungen vorkamen. Bei den Pankreas- und Nierentumoren mit Paraneoplasie zeigte die Altersverteilung eine leichte Vorverlegung der Kurven, das heißt, beide Neubildungen wurden wahrscheinlich früher diagnostiziert. Beim ektopen ACTH-Syndrom wurde bei 183 der 200 ausgewerteten Fälle die Endokrinopathie von dem Tumor diagnostiziert. Auch bei der extrapankreatischen Hypoglykämie wurde zunächst die Hypoglykämie (148 Fälle) und nur in 20 Fällen zuerst der Tumor nachgewiesen. Beim paraneoplastischen Hyperkalzämiesyndrom wurden etwa gleich häufig zuerst das endokrine Syndrom (48 Fälle) bzw. der Tumor (46 Fälle) erkannt. Dies gilt auch für das ektope ADH-Syndrom. In der Regel wurden zunächst die paraneoplastischen endokrinen Symptome und erst später die Neubildungen diagnostiziert. Somit ergibt sich die Frage, ob die endokrinen Symptome auf die Lokalisation des Tumors hinweisen können. Mit Ausnahme der extrapankreatischen Hypoglykämie muss bei allen anderen Paraneoplasien, insbesondere beim ektopen Gonadotropin und STH-Syndrom, zunächst an ein Bei der Auswertung der publizierten paraneoplastischen Endokrinopathien fanden sich bei 571 Fällen Angaben über 37 Endokrine und renale Paraneoplasien Zeitliche Korrelation zwischen Tumor- und Paraneoplasie-Manifestation Lungenkarzinom gedacht werden. Eine paraneoplastische Pubertas praecox bei Knaben bis zu 2 Jahren weist auf ein Leberkarzinom hin. ● Entwicklung aller peptidsezernierenden endokrinen Drüsen aus einer neuroektodermalen Stammzelle des primitiven Vorderdarmes. Prognose Die Prognose der paraneoplastischen Endokrinopathie hing vom Tumor und von dem Zeitpunkt der Tumordiagnose ab. In den meisten Fällen waren – mit Ausnahme der durch Fibrosarkom bedingten extrapankreatischen Hypoglykämie – die endokrinen Störungen präfinale Komplikationen des Geschwulstleidens, die innerhalb weniger Monate zum Tode führten. Während der Entwicklung eines Organismus unterziehen sich die aus der durch Teilung der omnipotenten Eizelle hervorgegangenen Zellen einer immer stärkeren Differenzierung. Diese Differenzierung resultiert aus einer Kombination von Induktion (Aktivierung von Strukturgenen) und Repression (Inaktivierung von Strukturgenen) durch basische Proteine, Histone und andere Substanzen und Stimuli. Am Ende dieser Entwicklung stehen hoch spezialisierte Zellen, die zwar ebenso wie die Eizelle sämtliche genetischen Informationen des Organismus besitzen, jedoch nur einen kleinen Teil hiervon verwirklichen (Oacob und Monod, 1961; Omenn et al., 1969). Diagnostische Kriterien einer endokrinen Paraneoplasie Bei den ersten Berichten über das Vorkommen von endokrinen Störungen und operativ bzw. autoptisch nachgewiesenen Tumoren, konnte nur aus der Rückbildung der endokrinen Symptomatik nach der Tumorentfernung auf einen kausalen Zusammenhang beider Krankheiten geschlossen werden. In der Folgezeit bestätigten die Bestimmungen der Hormonspiegel in Blut und Tumor diesen Zusammenhang. Heute stehen empfindliche Nachweismethoden auch für kleine Hormonkonzentrationen zur Verfügung. Ein interessantes Probleme der Tumorbiologie ist die Frage, wie eine entdifferenzierte Krebszelle mit ihren Stoffwechselanomalien und autonomem Wachstum die Synthesen von Proteinen mit einem Molekulargewicht von 30 000 (z. B. Gonadotropin) und mehr durchführen kann. Die früher geäußerte Ansicht, die Tumoren stellten nur eine Art »Hormonschwamm« dar, der die normal gebildeten Hormone speichert und vor dem Abbau schützt (Unger et al., 1963), kann bei der Dauer der Krankheitsbilder und den nachweisbaren hohen Syntheseraten bestimmter ektop gebildeter Hormone nicht mehr aufrechterhalten werden (Kracht, 1967b; Omen, 1970; Labhart, 1971b). Zwei Hypothesen stehen heute im Mittelpunkt der Diskussion: ● Aktivierung genetischer Informationen durch Derepression von Strukturgenen in der Krebszelle. Eine neoplastische Transformation kann als ein Verlust der Regulation des Zellwachstums angesehen werden. Die Tumorzelle dedifferenziert sich. Durch Derepressionen werden latent vorhandene Fähigkeiten freigelegt. Experimente von Gurdon (1968) bewiesen die Dedifferenzierung und Verwirklichung bisher unterdrückter Potenzen ursprünglich hochdifferenzierter Einzelzellen, induziert durch Wechselwirkungen mit dem Zytoplasma. Lange Zeit wurde die Frage diskutiert, ob eine einzige Zelle einen Dedifferenzierungsschritt vollzieht und eine Zelllinie (einen Klon von Krebszellen) erzeugt, der sich zu einem primären Tumor entwickelt und Tochtergeschwülste absiedelt, oder ob mehrere, verschiedene dedifferenzierte Zellen ein Karzinom erzeugen. Heute wird allgemein die erste Möglichkeit angenommen, und so erklärt es sich auch, dass eine einzige Krebszelle ein ganzes Spektrum von Hormonen ektop bilden kann (Waldenström, 1970). Mit der Annahme einer Derepression von Strukturgenen im Kern der Krebszelle ließen sich verschiedene Beobachtungen erklären: Pedersen beschrieb 1947 ein neues Serumprotein im Blut neugeborener Kälber, das er Fetuin nannte. 10 Jahre später gelang es Bergstrand und Czar (1957) dieses Fetuin im Serum menschlicher Föten nachzuweisen. Dieses Protein mit einem Molekulargewicht von 45 000 bis 75 000 wandert in der Elektrophorese als α-Globulin hinter dem Albumin und wird von der Leber gebildet. Dieses Eiweiß kommt normalerweise nur während des fötalen Lebens vor. Abelev et al. (1967) bewiesen, dass dieses Fetuin in Krebszellen der Leber von Mäusen gebildet wird, insbesondere in Karzinomen, die durch Dimethyl- 38 Diskussion Lokalisation des Primärtumors unter Berücksichtigung der Paraneoplasie aminoazobenzol hervorgerufen wurden. Tatarinow (1964) wies Fetuin auch in einer Reihe von menschlichen Hepatomen nach. Dieser Befund zeigt, dass ein fötales Protein, dessen Informationsmatrize während des späteren Lebens reprimiert ist, im Tumorgewebe dereprimiert werden kann und zur Synthese dieses Proteins führt. dall, 1969). Diese Vermutung wurde durch Untersuchungen von Hauger Klevene (1968) sowie Knight et al. (1971) gestützt, die nicht nur beim Vorliegen eines ektopen CushingSyndroms, sondern bei praktisch allen Lungentumoren ACTH im Tumor nachweisen konnten. Eine ähnliche Beobachtung betrifft ein embryonales Antigen, das im neoplastischen Gewebe und im Serum von Patienten mit Karzinomen des Gastrointestinaltraktes, der Leber und des Pankreas gefunden wurde (Gold, 1967). Dieses Antigen, das in erwachsenen Organen nur in diesen Karzinomen nachzuweisen war, findet sich aber als normaler Bestandteil von Darm, Leber und Pankreas des Föten (Studer et al., 1971). Ähnliche Befunde liegen von Benjamin et al. (1969) vor, der Patientinnen mit einem Endometriumkarzinom untersuchte. Diese Frauen waren meist hochgewachsen, adipös und litten an einem klinischen bzw. subklinischen Diabetes. Aus dieser Konstellation wurde die Verdacht abgeleitet, dass das Endometriumkarzinom mit einer abnormen, ektopen Sekretion des Wachstumshormons einhergehen könnte. Die STH-Konzentration im Blut erwies sich zwar als normal, doch wurde die STH-Sekretion durch eine intravenöse Glucoseinfusion nicht unterdrückt, sondern paradoxerweise gesteigert. Der Zusammenhang zwischen STH und Endometriumkarzinom ist noch ungeklärt, doch scheint auch hier ein Beweis vorzuliegen, dass bei gezielter Suche häufig bei einer malignen Neubildung Hinweise auf eine ektope Hormonbildung gefunden werden. Rucknagel und Chernoff (1955) wiesen das Vorkommen eines fötalen Hämoglobins im Blut Schwangerer nach. Zu nächst sah man dies als ein Zeichen des Austausches von fötalen und mütterlichen Erythrozyten in der Plazenta an. Spätere Untersuchungen von Bromberg et al. (1957) ergaben, dass auch bei Blasenmolen nach oder bei Schwangerschaften diese fötale Hämoglobin vorlag. Nach der Entfernung der Blasenmole konnte kein fötales Hämoglobin mehr nachgewiesen werden. Die Autoren sahen dies als Beweis dafür an, dass die Blasenmole eine Substanz produziert, die als Derepressor auf die für die Bildung von fötalen λ-Ketten des Hämoglobins notwendigen Matrizenbereiehe wirkte. Häufig wird die Frage gestellt, ob die ektope Hormonbildung eine Besonderheit des Tumorgewebes darstellt (Studer et al., 1971). Mindestens ein Beispiel ektoper Hormonsynthese in nicht neoplastischem Gewebe ist zweifelsfrei belegt: Es handelte sich um die Bildung des »Long acting thyroid stimulator« (LATS), eines Globulins mit Antikörpereigenschaften, in den Lymphozyten von Patienten mit einer Hyperthyreose. Dieses α-Globulin, das strukturell nicht mit dem normalen TSH der Hypophyse verwandt ist, hat die Eigenschaft, eine Überfunktion der Schilddrüse hervorzurufen. Diese Beobachtungen führen zu der Frage, ob nicht alle malignen Tumoren ektope Polypeptide bilden können, die jedoch aufgrund ihrer niedrigen Konzentration sich klinisch nicht manifestieren. Der Triggermechanismus dieses Phänomens müsste dann in seinem Wesen nach mit der neoplastischen Transformation und Progression verknüpft sein (Goo- 39 Endokrine und renale Paraneoplasien Unter den Paraneoplasien steht die ektope Hormonbildung im Vordergrund des Interesses, da die klinischen Bilder der Hormonüberproduktion, Zusammensetzung und Nachweismethoden vieler Hormone bereits bekannt sind. Es ist aber auch denkbar, dass andere klinische Manifestationen der Krebskrankheit auf – heute noch – unbekannte und vielleicht durch den Tumor selbst produzierte Stoffe zurückzuführen sind (Studer et al., 1971). 8 IV. Zusammenfassung 12 Nachdem in den ersten Berichten über endokrine Erkrankungen in Verbindung mit malignen Tumoren zunächst ein rein zufälliges Zusammentreffen angenommen wurde, konnte in späteren Berichten ein kausalpathogenetischer Zusammenhang zwischen der endokrinen Störung und einer Neoplasie gesichert werden. Diese klinischen Befunde wer den heute unter der Bezeichnung Paraneoplastisches Syndroms geführt. Die formale Pathogenese ist als Überfunktionssyndrom der betreffenden ektop sezernierten Hormone anzusehen. In dieser Literaturauswertung wurden 687 Fälle – nach Alter und Geschlecht, – nach pathologisch-anatomischen Befunden und – nach dem klinischen Erscheinungsbild ausgewertet. 13 9 10 11 Die multiplen paraneoplastischen Endokrinopathien: K und KT hängen von der dominanten paraneoplastischen Endokrinopathie ab Das ektope TSH-Syndrom: K: geringe Zeichen einer Hyperthyreose, hT: Lungenkarzinom Das paraneoplastische Zollinger-Ellison-Syndrom: K: Gastrinüberproduktion, Ulzera im Magendarmtrakt, hT: Gastrinome Erythropoetin-bildende Tumoren: K: Polyglobulie bei Erythropoetin-Überfunktion. hT : Nierenkarzinom Renin-bildende Tumoren: hypokaliämische Alkalose bei Renin-Überfunktion, hT: Lungenkarzinom Ektop gebildete Hormone bzw. Stoffe mit hormonähnlicher Wirkung ohne klinische Manifestation: K: Überproduktion von Prolaktin, Kalzitonin, Neurotensin, Bombesin, Chromogranin oder HCG, aber ohne oder mit nur geringer Symptomatik. hT: verschiedene Primärtumoren. ● Häufigkeit der Paraneoplasien und Tumoren. Das ektope ACTH-Syndrom wurde bisher als das häufigste paraneoplastische endokrine Syndrom beschrieben. Es folgen die extrapankreatische Hypoglykämie und die paraneoplastische Hyperkalzämie. Eine Kombination mehrerer endokriner Erkrankungen wurde nur vereinzelt beobachtet. Am häufigsten lag die Kombination eines ektopen ACTH-Syndroms und eines paraneoplastischen Karzinoidsyndroms vor. Der häufigste Tumor bei allen Endokrinopathien ist das Lungenkarzinom, besonders das kleinzellige Karzinom. Beim ektopen STH-Syndrom und einer Sonderform des ektopen Gonadotropinsyndroms lagen ausschließlich Lungentumoren vor. Beim hepatogenitalen Syndrom und beim Nadler-Wolfer Elliott-Syndrom kamen nur Lebertumoren vor. Fibrosarkome waren mit dem Doege-Potter-Syndrom vergesellschaftet, einer Sonderform der extrapankreatischen Hypoglykämie. Als paraneoplastisches endokrines Syndrom kamen vor: 1 Das ektope ACTH-Syndrom: K (klinische Hauptmanifestation): Cushing-Symptomatik. hT (häufigster Primärtumor): kleinzelliges Lungenkarzinom. 2 Die extrapankreatische Hypoglykämie mit den Sonderformen: 2.1 Doege-Potter-Syndrom: K: Hypoglykämie, hT: Fibrosarkome 2.2. Nadler-Wolfer-Elliott-Syndrom: K: Hypoglyämie, hT: hepatozelluläres Karzinom 2.3 Anderson-Syndrom: K: Hypoglykämie, hT: Nebennierenrindenkarzinom und 2.4 epitheliale Tumoren mit Hypoglykämie: verschiedene Primärtumoren. Beim Rosenfeld-Syndrom wurden Pseudomyxome des Ovars festgestellt. 3 Das paraneoplastische Hyperkalzämiesyndrom: K: Hyperkalzämie. hT: nicht-kleinzelligen Lungenkarzinome und hellzellige Nierenkarzinome. 4 Das ektope ADH-Syndrom: K: Hyponatriämie, hT : Lungenkarzinom 5 Das paraneoplastische Karzinoidsyndrom: K: Serotoninüberfunktion, Karzinoid-Symptome, hT: Kleinzellige Lungenkarzinome, Pankreaskarzinome. 6 Das ektope Gonadotropinsyndrom: K: gonadotrope Substanzen, human chorionic somatomamotropin. Gynäkomastie, hT: Lungenkarzinom, Hepatoblastom (hepatogenitales Syndrom mit Pseudopubertas praecox) 7 Das ektope STH-Syndrom: K: Osteoarthropathia Pierre-Marie-Bamberger-Syndrom, hT: nicht kleinzellige Lungenkarzinome ● Patienten mit einem endokrinen paraneoplastischen Syndrom ließen, abgesehen von den Pankreasneubildungen, keine signifikanten Unterschiede in der Geschlechtsverteilung erkennen. Die Kurve der Altersverteilung war etwas vorgezogen, sodass das Häufigkeitsmaximum meist ein Lebensjahrzehnt früher auftrat als bei den entsprechenden Tumoren ohne paraneoplastische Endokrinopathie. ● Zeitliche Beziehungen: Bei den meisten Fällen wurde zunächst – aufgrund der klinischen und biochemischen Befunde – die paraneoplastische Endokrinopathie diagnostiziert. So könnten die endokrinen Symptome Hinweise auf einen malignen Prozess sein und möglicherweise zu einer Frühdiagnose des Tumors führen. ● Die Prognose eines paraneoplastischen endokrinen Syndroms war bei den meisten ausgewerteten Fällen ungünstig. ● Zur kausalen Pathogenese der ektopen Hormonbildung werden vorwiegend zwei Hypothesen diskutiert: Nach der ersten Hypothese führt eine monoklonale Derepression von Genomabschnitten im Kern der Krebszelle zur Bildung von ektopen Polypeptiden. Die zweite Hypothese nimmt eine ge- 40 Renale Paraneoplasien einer Nephrektomie bildeten sich in 70% der Fälle die Symptome zurück und traten bei einem Tumorrezidiv wieder auf. Somit stellt das Stauffer-Syndrom einen Tumormarker dar. meinsame neuroektodermale Stammzelle aller endokrinen, peptidhormonsezierenden Drüsen an. Die Grundlagenforschung hat am Beispiel der ektopen Hormonbildung nicht nur weitere Kenntnisse über die Klinik der Tumorerkrankung erbracht, sondern auch zu einem tieferen Verständnis des Tumorstoffwechsels der entarteten Zelle geführt. Paraneoplastische Polyglobulie Begriffsbestimmung Die Polyglobulie ist durch eine Vermehrung der korpuskulären Blutelemente charakterisiert. Bei der primären oder essenziellen Form (Morbus Vaquez-Osler) liegt eine Vermehrung der Erythrozyten vor bei gleichzeitig gesteigertem Hämoglobingehalt mit Retikulo-, Leuko- und Thrombozytose. Von diesem Krankheitsbild »sui generis« lassen sich symptomatische Formen abgrenzen, das heißt, die Ursache ist bekannt. Sie sind außerdem durch die fehlende Leukound Thrombozytose gekennzeichnet. Diese symptomatischen Formen können als kompensatorische Polyglobulien (z. B. Sauerstoffmangel bei Herz- und Kreislaufinsuffizienz, Höhenglobulie) oder als Reizpolyglobulien (Stimulierung der Erythropoese durch verschiedene Noxen) vorkommen, sie können aber auch durch Tumoren – als paraneoplastische Polyglobulien – hervorgerufen werden. Diese Formen werden besonders bei Nieren- (Forsell-Syndrom), Gehirn- und Lebergeschwülsten nachgewiesen. V. Renale Paraneoplasien Die Niere kann die Ursache einer Paraneoplasie oder von einem extrarenalen Tumor ein paraneoplastisch betroffenes Organ sein. Nierentumoren kommen bei weniger als 4% der allgemeinen Paraneoplasien vor. 1 Nierentumoren als Ursache von Paraneoplasien Nierentumoren kommen bei verschiedenen Paraneoplasien vor. ● Endokrine Paraneoplasien: – Unter 565 ausgewerteten Fällen kam ein Nierentumor am häufigsten bei der paraneoplastischen Polyglobulie vor. Unter den 62 Fällen mit paraneoplastischer Polyglobulie waren 44 Nierenkarzinome. Meist handelte es sich um hochdifferenzierte hellzellige Karzinome (»Hypernephrome«). Nephroblastome wurde nur bei 2 Fällen beobachtet. Zweithäufigste Tumoren waren Leberzellkarzinome. – Unter weiteren endokrinen Paraneoplasien im Zusammenhang mit einem Tumor ist das paraneoplastische Hyperkalzämie-Syndrom zu nennen. In 25 Fällen war ein Nierenkarzinom (2 entdifferenzierte Karzinome und 2 Plattenepithelkarzinome im Nierenbecken) die Ursache der Paraneoplasie. Am häufigsten waren Lungenkarzinome mit 38 Fällen vertreten. Pathogenese Während die Ätiologie der Polycythaemia vera noch weitgehend unklar ist, kennen wir zahlreiche Krankheiten, die von einer symptomatischen Polyglobulie begleitet werden oder sie hervorrufen. In erster Linie kommt ein Sauerstoffmangel bei Herz- und Lungenerkrankungen infrage. Chemische Verbindungen wie Arsen, Phosphor oder Quecksilber, endokrine Störungen (z. B. Morbus Cushing), dienenzephale Prozesse (Enzephalitis) sowie Nephropathien (Hydronephrose und Nierenzysten (Rosse et al. 1963)) können die Erythropoese stimulieren und eine Polyglobulie induzieren. Zu den häufigsten Grundleiden einer paraneoplastischen Polyglobulie zählen Nieren-, Gehirn-, Lebertumoren sowie Myome des weiblichen Genitale. ● Andere Paraneoplasien. Ferner wurden nur Einzelbeobachtungen ermittelt: 1 Fall beim ektopen ACTH-Syndrom, 1 Fall bei einem Prolaktinom ohne nähere Angaben, 4 Fälle von leukämoider Reaktion (unter 565 hämatologischen Paraneoplasien), 4 Fälle von Dermatomyositis und 1 Fall von Dermatitis herpetiformis Duhring-Brocq (unter 704 kutanen Paraneoplasien) und 1 Fall mit limbischer Enzephalitis (unter 227 neuromuskulären Paraneoplasien) (Thomas et al., 1972 und 1974). Über formalpathogenetische Zusammenhänge zwischen Tumor und Polyglobulie liegen mehrere Hypothesen vor, bei denen ein erhöhter Erythropoetinspiegel, Störung zentralnervöser Regulationsmechanismen der roten Blutbildung, extramedulläre Blutbildung und Hypoxämie von Bedeutung sind. Weiter wurden diskutiert: eine tumorbedingte Stimulierung des Knochenmarks, Verlängerung der Erythrozytenlebensdauer(Babuna et al., 1959), Bildung abnormer renaler Proteine (Herbeuval et al., 1958) sowie kompensatorisch überschießende Hämatopoese bei chronischen Blutungen bzw. Knochenmarksmetastasen (Gross und Simon, 1963). ● Stauffer-Syndrom. Es handelt sich um eine Dysfunktion der Leber, die beim hellzelligen Nierenkarzinom vorkommt. Definitionsgemäß ist eine Metastasierung in die Leber auszuschliessen. Zu den biochemischen Befunden zählen eine Erhöhung der alkalischen Phosphatase und des indirekten Bilirubins sowie der γ-Globuline. Ferner kommt es zu einem Gewichtsverlust sowie zu Fieber. In der Leber finden sich die histopathologischen Zeichen einer unspezifischen Hepatitis. Diese Veränderungen treten fast ausschließlich beim Nierenkarzinom auf. Man nimmt an, dass Interleukine-6 und Prostaglandine eine Rolle spielen. Nach Über die Mechanismen, die zur Erhöhung des Erythropoetinspiegels im Blut führen, gehen die Ansichten der verschiedenen Autoren auseinander: Der Tumor soll selbst Erythropoetin oder eine Erythropoetin-ähnliche Substanz produzieren und durch fortdauernde Nekrobiose des Tumorgewebes freisetzen (Pietschmann u. Vormittag, 1970). Eine derartige Sekretion könnte von einem Nierenkarzinom 41 Endokrine und renale Paraneoplasien Paraneoplastische Polyglobulie: Klinik Paraneoplastische Polyglobulie: Labor (Bousser et al.,1959), von einem Leberkarzinom (Escobar u. Trobaugh, 1962), einem Leberhamartom (Josephs et al., 1962), einem zerebellaren Hämangiom (Waldmann et al., 1961), einem Nierenkarzinom mit einem zerebellaren Hämangiom64, einem zerebellaren Hämangioblastom (Ward et al., 1959), einem Lungen- und Nierenkarzinom (Donati et al., 1963) oder von einem Phäochromozytom stammen sowie sehr selten von einem Wilms-Tumor (Shalet et al, 1967). wurde von Fried et al. (Ösophagusfibrom), Giber et al. (Hypernephrom) sowie Escobar und Trobaugh (Leberkarzinom) diskutiert. Der Abbau der »erythropoetin producing substance (EPS)« soll durch bestimmte Stoffe, die vom Neoplasma abgegeben werden, verhindert werden: Jacobson (Leberkarzinom) sowie Brownstein und Schert (Hodgkin-Lymphom). Retroperitoneale Tumoren könnten durch Druck auf die Niere die Erythropoetinbildung anregen. Menzies nahm an, dass dies besonders bei Uterusmyomen vorkommt. Die Erythropoetin-Synthese könnte durch eine vom Tumor produzierte Substanz stimuliert werden. Diese Möglichkeit 42 Renale Paraneoplasien Mehrere Autoren sind der Annahme, dass ein im Zwischenhirn lokalisiertes Zentrum für die Regulation der Erythropoese verantwortlich sei. Seine Funktion könnte durch gewisse Tumoren des zentralen Nervensystems gestört werden: zerebellare Hämangiome (Haynal u. Graf, 1950, Schmid and French, 1965, Starr et al., 1958), Hypophysenadenome (Guilain et al., 1932), Glioblastoma multiforme (Meiner, 1932) sowie ein Stirnhirnsarkom (Salus, 1933). Zellveränderungen treten im allgemeinen nicht auf. Nur bei 7 der 41 ausgewerteten Fälle wurden histologisch Knochenmarksmetastasen gesichert. Bei weiteren 7 Patienten ergab die Knochenmarksbiopsie einen völlig normalen Befund. Klinik Die klinische und hämatologische Symptomatik der paraneoplastischen Polyglobulie ist zum größten Teil identisch mit der Polycythaemia vera. 80 Patienten waren männlichen, 27 weiblichen Geschlechts. Das Alter schwankte zwischen 10 und 77 Jahren, der Altersgipfel lag bei Männern zwischen dem 50. und 60., bei Frauen zwischen dem 40. bis 50. Lebensjahr. Es ist bemerkenswert, dass nephrogene Polyzythämien bei Nierenkarzinomen, Nierenzysten und Hydronephrosen vorkommen, beim Wilms Tumor aber nur selten diagnostiziert wurde. Kenny et al. beobachteten bei 11 von 12 Patienten mit einem Wilms-Tumor hohe Erythropoetinwerte vor der Nephrektomie, aber keine Zeichen einer Polyglobulie. Nach der operativen Entfernung der Geschwulst gingen die Erythropoetinwerte zurück und waren später bei 80% der Patienten mit Fernmetastasen wieder erhöht. Die Autoren konnten zwischen der Zytostatika- bzw. Strahlentherapie und den Erythropoetinwerten eine direkte Korrelation nachweisen. Diese Ergebnisse wurden auch von Murphy et al. Nilehn and Nilsson, 1966) bestätigt, die bei keinem Patienten mit einem Wilms-Tumor und hohen Erythropoetinwerten eine Polzythämie nachweisen konnten. Bei den klinisch geheilten Fällen normalisierte sich die Erythropoetinkonzentration im Serum kurz nach der Operation. Die klinische Symptomatik zeichnet sich durch eine Plethora der Haut und die tiefrote Verfärbung der Schleimhäute aus, die sowohl auf eine vermehrte Füllung der Blutgefäße, als auch auf eine periphere Zyanose zurückzuführen sind. Die Strömungsverlangsamung ist Folge der erhöhten Blutviskosität. Hepato- und Splenomegalie deuten auf eine Hyperämie dieser Organe hin, gelegentlich auch auf eine extramedulläre Blutbildung. Trotz hoher Thrombozytenzahl kann es zu einer Blutungsneigung kommen, die nach Marx Folge der übermäßigen Erythrozytenvermehrung ist. Erwähnenswert sind einige Einzelbeobachtungen wegen ihrer ungewöhnlichen klinischen Befunde. Damon et al. beschrieben zwei Fälle von Nierenkarzinom und Polyglobulie, die mit einem peptischen Magenulkus einhergingen (Ulzera werden bei Polycythaemia vera in ca. 10% der Fälle beobachtet). Jandl et al. berichteten über das gleichzeitige Vorkommen eines Leberzellkarzinoms, einer Polyglobulie und einer idiopathischen Hämochromatose. Becker et al. publizierten einen Fall, bei dem eine Blutdruckerhöhung über 140 mm Hg, Hämatemesis, Epistaxis, Melaena, Gingivorrhagie und Hautekchymosen vorlagen. Pochedly et al. beobachteten ein hellzelliges Nierenkarzinom bei einem 10 Monate alten männlichen Kind mit erhöhten Erythropoetinwerten, leukämischer Reaktion und Gerinnungsstörungen. Bei manchen Neoplasien entstanden extramedulläre Blutbildungsherde im Tumor selbst oder in seiner Umgebung. Dabei wurde die Möglichkeiten diskutiert: Der Nierentumor erhöht den Erythropoetinspiegel, er könnte die Niere und das perirenale Gewebe beeinflussen und dort zur Ausbildung von Blutbildungsherden führen (Roujeau and Abulker, 1933), oder der meist stark vaskularisierte Tumor war selbst der Sitz der Hämatopoese. Die Induktion dieser hämatopoetischen Aktivität ist noch weitgehend ungeklärt. Histologisch wurden Blutbildungsherde besonders bei Hämangioblastomen nachgewiesen (Umbach, 1954). Die hämatologischen Befunde der ausgewerteten Fälle stimmten – bis auf 2 Ausnahmen – mit denen der Polycythaemia vera überein. Es fehlten in der Regel lediglich die Leukozytose und die Thrombozytose, die bei ca. 75% der echten Polyzythämien zu beobachten sind. Bei der paraneoplastischen Polyglobulie wurde dagegen eine Thrombozytose nur in 12%, eine Leukozytose in 18% der Fälle nachgewiesen. Diese Befunde lassen auf eine weitgehend selektive Einwirkung des Tumors auf die Erythropoese schließen. Das klinische Bild wurde bei gutartigen, nicht rezidivierenden Grundleiden (Uterusmyom) in der Regel durch die Symptome der Polyglobulie bestimmt, die sich nach erfolgreicher Entfernung des Tumors zurückbildeten. In diesen Fällen war die Prognose günstig. Bei malignen Neubildungen (Hepatom) und rezidivierenden Hämangiomen vermischten sich die Polyglobuliesymptome mit denen des jeweils vorliegenden Neoplasmas. Der häufige letale Ausgang war fast immer unmittelbar auf die bösartige Geschwulst zurückzuführen. Bei kardiopulmonalen Erkrankungen regt eine Hypoxie die Erythropoese an. Auch eine tumorbedingte Hypoxie könnte Ursache einer paraneoplastischen Polyglobulie sein: Arteriovenöse Shunts in Tumoren rufen eine Hypoxie hervor (in Nierenkarzinomen und in Uterusmyomen (Josephs et al., 1962). Störungen der Atem- und Wasserregulationszentren im Rückenmark durch Tumordruck wurden von Starr et al. bei zerebellarem Hämangiom und von Holmes et al. bei Hirntumoren beschrieben. Letztlich könnten auch Störungen der Lungenfunktion durch große Myome bei kleinwüchsigen Frauen auftreten (Escobar and Trobaugh, 1962). Pathologische Anatomie Die pathologisch-anatomischen Veränderungen bei der Polyglobulie lassen sich vorwiegend im Knochenmark in Form einer erhöhten Proliferationsaktivität nachweisen. Das blutbildende Knochenmark ist zellreich, es überwiegen quantitativ die Erythrozytenvorstufen. Alle Reifungsstufen sind in gleichem Maße betroffen. In einem Drittel der untersuchten Fälle fand man gleichzeitig eine verstärkte Granulopoese oder eine Vermehrung der Megakaryozyten. Morphologische Tumoren, die von einer Polyglobulie begleitet werden Es fällt das gehäufte Auftreten von Nierengeschwülsten auf, insbesondere von hellzelligen Nierenkarzinomen, seltener von Leberzellkarzinomen, zerebellaren Gefäßtumoren und 43 Endokrine und renale Paraneoplasien Paraneoplastische Polyglobulie: Lokalisation der Primärtumoren (108 Fälle) 1.2 Nierenveränderungen bei extrarenalen Tumoren Myomen. Diese paraneoplastischen Polyzythämien kamen mit wenigen Ausnahmen nur bei Männern vor, während bei Frauen vorwiegend Myome auftraten. Andere Tumorlokalisationen stellen nur Einzelbeobachtungen dar: Thymom (Sundström, 1972), Phäochromozytome der Nebenniere (Waldman et al., 1961), maligner Knochentumor (Gross und Simon, 1963), Lungenkarzinom (Donati et al., 1963) und Plasmozytom (Rosenthal, 1955). Bei 59 paraneoplastischen Polyglobulien mit letalem Ausgang schwankte die manifeste Krankheitsdauer zwischen 5 Tagen und 14 Jahren (durchschnittlich 122 Monate). 1.2.1 Glomeruläre Veränderungen Die typische Manifestation ist eine schwere Proteinausscheidung von über 3,5 g/Tag. Es liegt das Bild eines nephrotischen Syndroms mit Ödemen, Hypoalbuminämie und Hyperlipidämie. Dieser Befund wurde bevorzugt bei über 40 Jahre alten Patienten beobachtet, bei denen das nephrotische Syndrom die Erstmanifestation darstellte. Histomorphologisch liegt meist eine membranöse Glomerulonephritis (seltener eine Minimal-change-Glomerulopathie) vor. Zu den häufigsten nachgewiesenen Geschwülsten zählen Magen-, Lungen- und Dickdarmkarzinome. Ferner treten diese Veränderungen gelegentlich auch bei Mammakarzinom, Hodgkin-Lymphom und beim malignem Melanom auf. Bei einer AL-Amyloidose manifestiert sich die Leichtkettennephropathie als noduläre Glomerulosklerose. Bei 107 Fällen konnte folgender zeitlicher Zusammenhang zwischen Tumor und Polyglobulie festgestellt werden: 37 Patienten zeigten zunächst klinische Zeichen einer Polyglobulie, 18 Tage bis 144 Monate später wurde die Geschwulst diagnostiziert (durchschnittlich nach 17,3 Monaten). Nur bei 15 Patienten wurde erst die Diagnose eines Tumors und später die der Polyglobulie gestellt (1 bis 238 Monate, durchschnittlich 51,9 Monate). Wechselbeziehungen zwischen Primärtumor und Paraneoplasie ließen sich auch bei operierten Patienten nachweisen: Nach der Nephrektomie bildete sich die paraneoplastische Polyglobulie zurück und bei Spätmetastasierung wurde sie wieder manifest (Wagner, 1962), 1.2.2 Tubulointerstitielle Nierenveränderungen Besonders beim Multiplen Myelom kommen diese Veränderungen vor und führen – unter dem Bild einer »Myelomniere« – letztlich zur Niereninsuffizienz. Große Mengen von Leichtketten werden glomerulär filtriert und schädigen die Tubulusepithelien. Gleichzeitig kommt es Präzipitaten in die Tubuluslichtung mit nachfolgender Obstruktion. Histomorphologisch lassen sich Eiweißzylinder in den Tubuluslich- 44 Renale Paraneoplasien phagozytose angenommen. Eine gesteigerte Blutstase bei Splenomegalie sowie toxische und hormonelle Beeinflussungen des Knochenmarks sollen ebenfalls eine Rolle bei der Induktion der Hämolyse spielen. tungen nachweisen. Bemerkenswert ist, dass es in der Umgebung dieser Eiweißzylinder zu einer Fremdkörperreaktion kommt. Risikofaktoren bei einer Myelomniere sind Dehydratation und Harnwegsinfekte. Analgetika und Röntgenkontrastmittel können auch eine Rolle spielen. Davidson diskutierten eine durch tumoröse Knochenmarksinfiltration ausgelöste Hämolyse: Durch den Fremdkörperreiz – als Folge der Tumorinfiltration – soll es zur Produktion qualitativ minderwertiger Erythrozyten kommen. 1.2.3 Abnorme Ablagerungen in den Nieren Beim Hyperkalzämie-Syndrom kommt es zu metastatischen Kalkablagerungen. Sie lassen sich makroskopische als weißliche Streifen im Nierenmark nachweisen. Histologisch sieht man hämatoxylinblaue Kalkablagerungen in den Glomeruli und im Zwischengewebe des Nierenmarks. Bei Neubildungen, die mit starkem Zellzerfall einhergehen (Leukämien, maligne Lymphome) treten Harnsäurepräzipitate auf, die mit einer Gichtniere vergleichbar sind. Sie lassen sich zunächst in den Tubuluslichtugen nachweisen. Nach einem Untergang von Tubulusepithelien kommen diese Harnsäurekristalle mit dem ortsständigem Zwischengewebe in Kontakt und rufen eine starke Fremdkörperreaktion hervor. Diese lässt sich histologisch eindrucksvoll im polarisierten Licht darstellen. Pathologische Anatomie Das blutbildende Knochenmark weist eine zelluläre Hyperplasie auf, insbesondere eine gesteigerte Erythropoese. Sie imponiert meistens als normoblastäre Hyperplasie und ist charakterisiert durch einen hohen Normoblastengehalt, seltener sind auch unreifere Vorstufen der roten Reihe vermehrt. Pathologisch veränderte Erythrozyten, wie z. B. Megaloblasten, Sphärozyten, werden nur vereinzelt beobachtet; Leuko- und Megakaryopoese entsprechen der Norm. Die histologische Untersuchung der Milz zeigt eine Hämosiderinspeicherung in Retikulumzellen und in Sinusendothelien der roten Pulpa, häufiger liegt auch eine retikuloendotheliale Hyperplasie vor. Die Sinus sind erweitert, die Endothelzellen vergrößert und abgerundet. Eine extramedulläre Blutbildung kommt nur bei schweren Hämolysen vor. 1.2.4 Mikrovaskuläre Nierenveränderungen Als hämatologische Paraneoplasie kann es bei verschiedenen malignen Primärtumoren (Lungen-, Pankreas-, Prostatakarzinom, malignen Lymphomen) zu einer thrombotischen Mikroangiopathie kommen. Dabei ist zu berücksichtigen, dass diese Veränderungen auch iatrogener Natur sein können (nach Mitomcyn- oder Cisplatin-Therapie). Zu den hämatologischen Paraneoplasien zählen auch Koagulopathien. Im Rahmen einer Verbrauchskoagulopathie kann es zu einer diffusen intrakapillären Thrombosierung aus Fibrin (DIC) kommen. Ähnliche feingewebliche Veränderungen zeigt die Leber. Einlagerungen von Hämosiderin finden sich in den KupfferSternzellen und in den Hepatozyten. In vereinzelten Fällen kam es auch hier zur extramedullären Erythropoese. Klinik Bei den ausgewerteten Fällen überwog leicht das männliche Geschlecht (65:54). Das Alter schwankte zwischen 2 Monaten und 84 Jahren, im Durchschnitt betrug es 49 Jahre. Der Altersgipfel lag bei den Männern im 6., bei Frauen im 4. bis 6. Dezennium. 1962 wurde der Begriff »mikroangiopathische hämolytische Anämie« von Brain et al. eingeführt. Sie beobachteten eine Mikrothrombosierung im Bereich kleinerer Arterien mit einem unvollständigen Verschluss der Gefäßlichtung. Bei dem Durchfluss durch diese eingeengte Lichtung sollen die Erythrozyten mechanisch geschädigt (Schistozytose) oder zerstört (Hämolyse) werden. Dieser Vorgang ist später von zahlreichen anderen Autoren bestätigt worden, und zwar sowohl beim Menschen (Zittoun et al., 1965) als auch im Experiment (Brain et al., 1962). Mikroangiopathische hämolytische Anämien wurden auch mit Thrombozytopenien beim Mammakarzinom oder bei einem verstärkten Fibrinogenkatabolismus nachgewiesen. Die Erythrozytopenie war zunächst Folge der vermehrten peripheren Destruktion und später Ausdruck der zunehmenden erythropoetischen Insuffizienz, wie sie auch bei langdauernden Hämolysen anderer Genese beobachtet wird. Der häufig positive Ausfall des Coombs Tests mit Nachweis von zirkulierenden (direkter Coombs-Test) bzw. zellständigen Antikörpern (indirekter Coombs-Test) lässt eine Zuordnung dieser Hämolyseformen in die Gruppe der durch autoaggressive Mechanismen verursachten Krankheitsbilder zu. Zu den klinischen Zeichen einer paraneoplastischen hämolytischen Anämie gehören folgende Symptome: – Ikterus ohne Pruritus, – Hepatosplenomegalie, – Anämie, – Retikulozytose, – Bilirubinämie, – positiver Coombs-Test, – Ikterus ohne Pruritus, – Sklerenikterus bis zum generalisierten Hautikterus, – Hepatomegalie Die mikroangiopathische hämolytische Anämie ist von dem hämolytisch urämischen Syndrom abzugrenzen, das vorwiegend bei Kindern vorkommt. Schulz und Dabels beschrieben ein hämolytisches urämisches Syndrom bei einem 72 Jahre alten Mann mit einem metastasierenden Prostatakarzinom, Hämolyse und Niereninsuffizienz. Im peripheren Blut wiesen sie Schistozyten nach. Eine weitere Ursache der paraneoplastischen Hämolyse könnte in einer tumorbedingten Stoffwechselstörung der Milz zu suchen sein. Es wird eine Hyperplasie der Milz und eine dadurch verstärkte Erythro- 45 Endokrine und renale Paraneoplasien Bei den mikroangiopathischen hämolytischen Anämien kommen Abweichungen der Erythrozytenform vor. Es werden Akanthozyten oder Kletterzellen («burr cells«), Schistozyten oder Fragmentozyten, Stahlhelmzellen («helmet cells«), Dreiecksformen (»triangular cells«) sowie Zahnraderythrozyten (»cog-wheel-cells«) beschrieben. Die Schistozyten sind als Formveränderungen der Erythrozyten infolge einer mechanischen Fragmentation anzusehen. Sie kommen nicht nur bei Tumoren vor, sondern auch nach Anwendung der Herz-Lungen-Maschine oder nach Implantation einer Herzklappenprothese. Zahnraderythrozyten sind bei Kindern mit einem Neuroblastom beschrieben worden. Nach Entfernung der Geschwülste bildeten sich diese Erythrozytenanomalien auch wieder zurück. durch das RHS entfernt. Ein verminderter Haptoglobinspiegel ist daher ein guter Indikator einer Hämolyse. Mit dem Stoffwechsel des Hämoglobin- und Nichthämoglobineisens bei Tumorpatienten beschäftigten sich die Arbeiten von Miller et al. (1956), Pribilla (1961) sowie von Heilmeyer und Keiderling (1959). Lockner (1960) stellte eine Verkürzung der Lebensdauer der Erythrozyten bei etwa 20% der Frauen mit einem Genitalkarzinom fest: 26,9 Tage bei unbehandelten Karzinomträgerinnen, 28,l Tage nach Behandlung und 34 Tage bei gesunden Kontrollfrauen. Über sideropenische Anämien mit retikuloendothelialer Siderose bei einem Nierenkarzinom berichteten Chatterjee und Maclellan (1968). Hypersideroblastosen, das heißt Siderozyten und Sideroblasten im Knochenmark und im peripheren Blut, sollen beim Di-Guglielmo Syndrom und bei Plasmozytomen vorkommen und zu dem Formenkreis der sideroachrestischen Anämien gehören. Bei einer intravasalen Hämolyse ist das Hämoglobin im Plasma vermehrt, wird an das Haptoglobin gebunden und 46 Literaturangaben Andreew, D., 1. Goranow, G. Krastinow: Hypoglykämie bei einem intra thorakal gelagerten Fibrom. Endokrinologie 38: 167-174 (1959). Ariel, 1. M.: Treatment of inoperable primary panceatic and liver cancer by intra-arterial administration of radioa ctive isotopes (Y 90 radiating microspheres). Ann. Surg. 162: 267-278 (1965). Arkless, H. A.: Coincidence of rhabdomyofibroma of the diaphragm idiopathic hypoglycemia, and retroperitoneal sarcoma. Med. Bull. Veterans' Adm. (Wash.) 19: 225-229 (1942). Arnett, J. H., Ch. F. Leng: A case of congenital stenosis of the pulmonary valve with late onset of cyanosis: death from carcinoma of the pancreas. Amer. J. med. Sei. 182: 212-217 (1931). Arnold, P., E. Merian, Ch. Maurer: Hypercorticisme paraneoplasique thymome malin. Strasbourg med. 20: 97-105 (1969). Aszkanazy, C. L., L. Jenkins, W. W. Simpson: Adrenal cortical carcinoma associated with hypoglycemia. Canad. med. Ass. J. 79: 482-484 (1958). Audet, B.: Hypoglycemie severe associee ä des tumeurs volumineuses d'origine non pancreatique. Lava! med. 35: 3349 (1964). August, J. Th., H. H. Hiatt: Severe hypoglycemia secondary to a non panceratic fibrosarcoma with insulin activity. New Engl. J. Med. 258: 17-20 (1958). Aurbach, G. D., J. T. Potts jr.: Parathyroid hormone. Amer. J. Med. 42: 1-8 (1967). Azzopardi, J. G., A. R. Beitau: Carcinoid syndrome, and oat cell carcinoma of the bronchus. Thorax 20: 393-397 (1965). Azzopardi, J. G., R. S. Whittaker: Bronchial carcinoma and hypercalcemia. J.clin. Path. 22: 718-724 (1969). Azzopardi, J. G., E. D. Williams: Pathology of nonendocrine tumors associated with Cushing's syndrom e. Cancer 22: 274-286 (1968). Azzopardi, J. G., E. Freeman, G. Poole: Endocrine and metabolic disorders in bronchial carcinoma. Brit. med.J. 4: 528529 (1970). Babuna, C., G. H. Gardner, R. R. Green: Erythrocytosis associated with a myomatous uterus. Amer. J. Obstet. Gynec. 77-2: 424-429 (1959). Bagshawe, K. D.: Hypokaliemia, carcinoma, and Cushing's syndrome. Lancet II: 284-287 (1960). Balls, K. F., J. T. L. Nicholson, H. L. Goodman, J. C. Touchstone:Functioning, islet-cell carcinoma of the pancreas with Cushing's syndrome. J. clin. Endocr. 19: 1134-1143 (1959). Barenghi,G., G. B. Cerutti: Sindrome ipoglicemica, ginecomastia e impotenza sessuale regredite dope aspotazione di sarcoma fusocellulare retro peritonea le. Rif.med. 79: 1382-1391 (1965). Barenghi, G.: Les hypoglycemies au cours des tumeurs extra-pancreatiques: diagnostic differentiel. Associatlon avec un e endocrinopathie. Problemes pathogeniques. J. ann. diabR Hötel Dieu 7: 183-196 (1966). Bariety, M., Ch. Coury, R. Rulliere: Les syndrom es paraneop lasiques dans le cancer bron chopulmon aire primitif. J. fran. Med. Chir. thor. 10: 19-68 (1964). VI. Literaturangaben Abe, K., D. P. Island, G. W. Liddle, N. Fleischer, W. E. Nicholson: Radio immunologic evidence for MSH (melanocyte stimulating hormone) in human pituitary, and tumor tissues. J. clin. Endocr. 27: 26-52 (1967). Abelev, G. J., J. V. Assecritova, N. A. Kraevsky, S. D. Perova, N. J. Percvodchikova: Embryonalserum globulin in cancer patients. Int. J. Cancer 2: 551-558 (1967). Abouav, j., St. B. Berkowitz, F. 0. Kolb: Reversible hypercalcemia in m asculinizing hypernephroid tumor of the ovary, report of a case. New Engl. J. Med. 260: 1057-1062 (1959). Adamson, A. R., D. G. Grahame-Smith, V. Bogomoletz, D. S. j. Maw, N. G. Rothnie: Malignant argentaffinoma with carcinoid syndrome and hypoglycemia. Brit. med. J. / : 9394 (1971). Akoun, G., Cl. Faye, Cl. Gharib, M.Thevenet, H. Brocard: Deux syndromes de Schwartz-Bartter (S. B.). Presse méd. 78: 2382 (1970). Alanis, B. F., J. F. Flanagan : Myopathy and hypercalc emia occuring with carcinoma of the kidney. J. A. M. A. 171: 2076-2080 (1959). Albeaux-Fernet, M., j.D. Romani, A. Kesseler, J. Barthelme: Tumeur maligne du thymus et syndrome de Cushing. Presse m d. 76:415-418 (1968). Albeaux-Fernet, M.: Le syndrome corticotrophique ectopique.Ann. Endocr. (Paris) 31: 12-24 (1970). Albright, F., Ch. H. Burnett, 0. Cope, W. Parson: Acute atrophy of hone (osteoporosis) simulating hyperparathyroidi sm.J.clin. Endocr.1; 711-716 (1941). Albright, F., E. C. Reifenstein jr.: Parathyroid glands, and metabolic hone disease. p. 93. Williams & Wilkins, Baltimore 1948. Allott, E. N., M. 0. Skelton: Increased adrenocortical activity associated with malignant disease. Lancet: 273-283 (1960). Altschul, A.: Gynecomastia in lung tumor associated with pulrnonary tuberculosis. N. Y. St. j. Med. 38: 637-640 (1938). Amatruda, Th. T. jr., P. J. Mulrow, J. C. Gallagher, W. H. Sawyer: Carcinoma of the lung with inappropriatc antid iurcsis. Demon,stration of an tidiuretic hormone like activity in tumor extract. New Engl. J. Med. 269: 544-549 (1963). Andersen, P.: Insulinom. Ugeskr. Laeg. 123: 727-728 (1961). Anderson, E. W. Haymaker : Cushing's syndrome. J. nerv. ment. Dis. 99: 511-520 (1944). Anderson, E. W., J. F. Glenn: Penile malignan cy, and hypercalcemi a. J. A. M. A. 192: 128-129 (1965). Anderson, E. E., J. F. Glenn: Cu shing's syndrome associated with anapla stic carcinoma of the thyroid gland. J. Urol. (Baltimore) 95: 1-4 (1966). Anderson, H. B.: A tumor of the adrenal gland with fatal hypoglycemia. Amer. J. med. Sei. N. S.180: 71-79 (1930). Anderson, 1. F., A. L. Webster, W. Wypkema: Primary malignant hepatoma associated with hypoglycaemi a. Case report with a review of the literature. S. Afr. med.J.41: 505-508 (1967). 47 Endokrine und renale Paraneoplasien Bernanke, D.: zit. nach Gelfman (1961). Bernheim, R.: Une nouvelle activit paran opla sique des cancers broncho pulmonaires: la decretion de gonadotrophines. Presse méd. 76: 2482 (1968). Berson, S. A., R. S. Yalow, G. D. Aurbach, J. T. Potts jr.: Immunoassay of bovine and human parathyroid hormone. Proc. nat. Acad. Sei. (Wash.) 49: 613-617 (1963). Berthelot, P., J. P. Benhamou, R. Fauvert: Hypercorticisme et cancer de l'uterus. Presse méd. 69: 1899-1902 (1961). Bielschowsky, F.: Zur Klinik und Pathologie der Spontanhypoglykämie. Klin. Wschr. 11: 1492-1494 (1932). Biglieri, E. G., P. E. Slaton, M. Schambelan, S. J..Kronfield: Hypermineralo corticoidism. Amer. Med. (Philad.) 45: 170-175 (1968). Billinghurst, J. R., A. K. Thould, 0. P. Galpin, J. M. Hinton: Carcinoma and Cushing's syndrome. Brit. med. J. II: 490493 (1961). Bleuler, E.: Endokrinologische Psychiatrie. Thieme, Stuttgart 1954. Bok, J.: Klinische-Pathologische Conferenties. Kwaadaarig gezwel met hoge uitscheiding van 17-Ketosteroiden in de urine. Ned. T. Geneesk. 99:3205-3213 (1955). Borm, D.: Hypernephroid es Carcinorn der linken N iere mit Zeichen eines primären Hyperparathyreoidismus. Endokrinologie 41: 291-297 (1961). Hornstein, P., J. P. Nolan, D. Bernanke: Adrenocortical hyperfunction in association with anaplastic carcinoma of the respiratory tract. New Engl. J. Med. 264: 363-371(1961). Boshell, B. R., J. J. Kirschenfeld, P. S. Soteres: Extrapancreatic insulin secreting tumor. New Engl.). Med. 270: 338-341 (1964). Boss, J. H.: Spontane Hypoglykämie als Folge eines nichtinsu linprodu zierenden Tumors der Nebennierenregion. Schweiz. Z. Path. 22: 232-242 (1959). Bour, H., Cl. Roy, M..Tutin, P. Pasquier, A. Fournier: A propos de l'association d'un cancer du rein d'une neuromyopathie et d'une hypercalcemie avec hypophosphoremie. Discussion d 'une hyperpara thyroidie paraneoplasique. Path. et Biol.14: 723-732 (1966). Bourgoigne, J., J. Cl. Dupont, R. Noiret: Association d'une pheo-chromocytome et d'un hypercorticisme du type Cushing avec hyperaldosteronurie. Ann. endocr. (Paris) 25: 269-284 (1964). Bourne, H. H., R. E. Tremblay, J. S. Ansell : Stupor, hypercalcemia, and carcinoma of the renal pelvis. New Engl. J. Med. 271: 1005-1006 (1964). Bousser, J., G. Mathe: Association du cancer epithelial et lymphomatose. Sem. Hôp. Paris 30: 821-837 (1954). Bousser, J., P. Tcherdakoff, J. C. Gabilan, D. Christo: A propos de 4 casde tumours renales ou cerebeleuses associees a une polyglobulie: Discussion des rapports possibles entreles deux affections. Sem. Hôp. Paris 35: 2563-2571 (1959). Bousvaros, G. A.: Hypoglycemia in metastatic fibrosarcoma of the liver. Brit. med. J. I: 836-838 (1960). Bower, B. F., D. M. Mason, P. H. Forsham: Bronchogenic carcinoma with inappropriate antidiuretic activity in plasma and tumor. New Engl. J. Med. 271: 934-938 (1964). Bower, B. F., G. S. Gordan: Hormonal effects of nonendocrine tumors. Ann. Rev. Med. 16: 83-ll8 (1965). Barjon, P., R. Labauge: Les tumeurs mesenchym ateu ses hypoglycemiantes. Presse méd. 69: 2635-2639 (1961). Barraclough, M. A.,J. J.Jon es, J.Lee: Production of vasopressin by anaplastic oat cell carcinoma of the bronch u s. Clin. Sei. 31: 135-144 (1966). Barraclough, M. A.: Inappropri ate secretion of antidiuretic hormone and potassium depletion. Proc. roy. Soc. Med. 64: 1069-1070 (1971). Barthelme, J., A. Kesseler, J. D. Romani, P. Renault, Phan Huu Trung, M. Albeaux-Fernet: Syndrome de Cushing cau se par une tumeur malign e du thymus secr etrice d'une corticotrophine. Ann. endocr. (Paris) 29: 263-268 (1968 a). Barthelme, J., J. L. Wiederkehr, A. Willie, W. Florange: Hypercorticismes paraneop lasiques et tumeurs bronchopulmonaires. Vie m ed. 49: 1569- 1578 (1968 b). Bauer, J.: zit. na ch Jores (1955). Becker, D.J., M. S.Sternberg, M. H. Kaiser, C. Gables: Hepatoma associated with hypoglycem ia, polycythemia, and hypercalcemia. J. A. M. A. 186: 1018-1019 (1963). Becker, K. L., J. Cottrell, Ch. F. Moore, J. L. Winnacker, M. J. Matth ews, S. Kat z: Endocrine studies in a pati ent wit h gonadotropin secreting bronchogeni c carcinoma. J. clin. Endocr. 28: 809-818 (1968). Beers, D.N., J.J.Morton: Primary carcinoma of the liver with hypoglycemia. Amer. J. Cancer 24: 51-55 (1935). Behrle, F. C., F. A. Mantz jr., R. L. Olson, J. C. Trombold: Virilization accompanying hepatoblastoma. Pediatrics 32: 265-271 (1963). Benichou, C., J. Hanoune : Un nouveau cas de syndrome de Schwartz Bartter. Le mode d'action de l'hormone ant id iuretique. Sem. Höp. (Paris) 43: 2506-2510 (1967). Benjamin, F., D. J. Casper, L. Sherman, H. D. Kolodny: Growth-hormone secretion in patients with endometrial carcinoma. New Engl. J. Med. 281: 1448-1452 (1969). Bensch, K. G., G. B. Gordon, L. R. Miller: Electron microscopic and bio chemical studieson the bronchialcarcinoid tumor.Cancer 18:592-602 (1965). Bensch, K. G., B. Corrin, R. Priente, H.Spen cer: Oat-cell carcinoma of the lung. Its origin and relationship to bronchial car cinoid. Cancer 22: 1163-1171 (1968). Bentley, P. J., F. R.Cobb: Neurohypophysea l hormon -like activity ofoat-cell carcinoma: actions on the toad bladder. J.clin. Endocr. 27: 1746-1748 (1967). Bercovitch, D. D., M. F. Levitt: Effects of saline loading on distal renal tubular sodium and water reabsorption. Proc. Soc. exp. Biol. N. Y. 125: 552-556 (1967). Bergenstal, D. M., R. Hertz, M. B. Lipsett, R. H. Moy: Chemotherapy of adrenocortical cancer with o, p DDD. Ann. intern. Med. 53: 672-682 (1960). Bergstrand, C. G., B. Czar: Paper electrophoretic study of human fetal serum proteins with demonstration of a new protein fraction. Scand. J. clin. Lab. Invest. 9: 277-286 (1957). Bergström, S., L. A. Carlson, L. Eklund, L. Oro: Cardiovascular and metabolic response to infusions of prostaglandin E 1 to simultaneous infusions of noradrenaline and prostaglandin E 1in man. Acta physiol. scand. 64: 332-339 (1965). 48 Literaturangaben Braband, H., T. Klemm: Nephrogene Polyzythämie (ForsellSyndrom ). Fortschr. Röntgenstr. 112: ll0-ll2 (1970). Bradley, J. E, J. D. Young, G. Lentz: Polycythemia secondary to pheochromocytoma. J. Urol. 86: 1-6 (1961). Bradley, J. E, J. D. Young, G. Lentz: Polycythemia secondary to pheochromocytoma. J. Urol. 86: 1-6 (1961). Brakier, T., H. Betz: Cancer primitif du foie et hypoglycemie. Acta clin.belg. 6: 140-152 (1951). Brain, M.C. J.V. Dacie, D.O.B Hourihane: Microangiopathic hemoyticm anemia: ithe possible rote of vascular lesions in pathogenesis. Brit. Haemat. 8, 358-374 (1962) Breidahl, H. D., B. C. Ritchie: Hypercalcaemia due to malignant ovarian tumor: a case report. Med. J. Aust. 49: 208210 (1962). Bricaire, H. D., R. Tourneur, J. Leprat, J. P. Luton: Syndrome de Cushing et cancer du pancreas. Aspect particulier des hypercorticismes paraneoplasiques. Sem. Hôp. (Paris) 41: 767-773 (1965). Bricaire, H. D., H. Saltiel, J. Hanoune: Le syndrome de Schwartz-Bartter: problemes pathophysiologiques. Presse méd. 75: ll-16 (1967). Bricaire, H. D., J. Leprat: Les syndromes de Cushing paraneoplasiques. Les cancer et les glandes endocrines. Problemes actuels d'endocrinologie et de nutrition. Expansion scientifique francraise. 179-187. Paris 1969. Bricker, N. S.: The control of sodium excretion with normal and reduced nephrom populations. The pre-eminence of third factor. Amer. J. Med. 43: 313-321 (1967). Brickner, Ph. W., M. Lyons, S. J. Landau: Cushing's syndrome associated with non-endocrine neoplasms. Amer. J. Med. 31: 632-639 (1961). Bromberg, Y. M., M. Salzberger, A. Abrahamov: Alkali resistant type of hemoglobin in women with molar pregnancy.Blood 12: ll22-1124 (1957). Brooks, R. V., J. Dupre, A. N. Gogate, 1. H.Mills, F. T. G. Prunty: Appraisal of adrenocortical hyperfunction: patients with Cushing's syndrome of "non-endocrine " tumors. J. clin. Endocr. 23: 725-736 (1963). Broster, L. R., J. Patterson: An unusual case of adrenal carcinoma with a note on the application of a new colour test. Brit. med. J. I: 781-782 (1948). Brown, J. D., D. B. Stone: Tobutamide-induced hypoglycemia. Amer. J. clin. Nutr. 15: 144-148 (1964). Brown, L.: Cushing's and malignant carcinoid syndromes from ovarian neoplasms. Arch. intern. Med. 115: 490-494 (1965). Brown, W. H.: A case of pluriglandular syndrome: diabetes of bearded women. Lancet /: 1022-1024 (1928). Browne, J. S. L., J.C. Beck, 1. Dyrenforth: Cushing's syndrome. Ciba Found. Colt. Endocr. 8: 505 (1955). Brownstein, M. H, H. S. Ballard: Hepatoma associated with erythrocytosis. Amer. J. Med. 40: 204-210 (1965). Bruggemeyer, C.D., G. Ramirez: Membranous nephropathy: a concern for malignancy. Amer. J. Kidney Dis. 9, 23-26 (1987) Buchsbaum, H. J.: Hypercalcemia associated with vulvar carcinoma. Gynaecologia 168: 203-208 (1969). Buckle, R., M:McMillan, Chr. Mallison: Ectopic secretion of parathyroid hormone by a renal adenocarcinoma in a patient with hypercalcemia. Brit. med. J. IV: 724-726 (1970). Bugaro, L., S. Malpiero: Su di un caso di ipoglicemia spontan.ea associata a carcinoma epatico primitivo. Friuli med. 17: 479-49 0 (1962). Burger, H. G., D. P. Cameron, K. J. Catt, A. Atk ins: Growth hormone secretion and osteoarthropathy in bronchogenic carcinoma. Aust. Ann. Med. 19: 73 (1970). Burgstedt, H. J.: Thymuskarzinom und Cushing-Syndrom. Mschr. Kinderheilk. 104: 395-398 (1955). Burkinshaw, J. H., D. O'Brien, J. D. H. Pendower: Cushing's syndrome associated with an islet-cell tumor of the pancreas in a boy aged 2 years. Arch. Dis. Childh. 42: 525531 (1967). Bürki, K., P. Cottier, W. Bandi, 1. Antener: Bestimmung einer Vitamin-D-Aktivität bei Hypernephrom mit Hypercalzämiesyndrom. Helv. med. Acta 30: 615-627 (1963). Burmeister, P., L. Bianchi, W. Klietmann, J. Torhorst: Paraneoplastisches Cushing-Syndrom bei primärem Leberkarzinom. Dtsch. med. Wschr. 93: 164-165 (1968). Cabau, A., C. Dubost, J. Leprat: Les hypercalcemies dites « paraneopla siques». A propos d'un cas simulant une hyperparathyroidie aigue. Rev. franc. Endocr. clin. 9: 213-228 (1968). Cameron, D. P., H. G. Burger, D. M. de Kretzer, K. J. Catt, J. B. Best: On the presence of immunoreactive growth hormone in a bronchogenic carcinoma. Austr. Ann. Med. 18: 143-146 (1969). Camiel, M. R., D. L. Benninghoff, L. L. Alexander: Gynecomastia associated with lung cancer. Dis. Chest. 52: 445451 (1967). Campbell, J. H., C. M. Pasquier, E. C. St. Martin, P. C. Worley: Hypernephroma associated with polycythemia and eczematoid dermatitis. J. Urol. 79: 12-15 (1958). Camus, J. L., A. Monsaigneon, G. Chomette: Syndrome de Cushing avec hyperplasie surrenalienne, surrenalectomie double,· develoment d'un thymome malin, hypophysectomie, regression du thymome. Presse méd. 69: 2266-2269 (1961). Cantarini, G., P. Dematte, G. B. Fontana: Sindrome di Cushing para neoplastica. Arch. ital. Pat. 11: 347-358 (1968). Caranasos, G., B. H. Ruebner: Adrenal weight and metastasis in bronchogenic carcinoma. Arch. Path. 76: 262-270 (1963). Carey, R. W., Th. G. Pretlow, E. Z. Ezdin li, J. H. Holland: Studies on the mechanism of hypoglycemia in a patient with massive intraperitoneal leiomyosarcoma. Amer. J. Med. 40: 458-469 (1966). Carey, V. C. 1.: Hypercalcemia and bronchial carcinoma. Amer. Rev. resp. Dis. 85: 258-260 (1962). Carey, V. C. 1.: The incidence of hypercalcemia in association with bronchogenic carcinoma. Amer. Rev. resp. Dis. 93: 584-586 (1966). Carpenter, L. C. jr.: zit. nach Laurent 1971. Carter, N. W., PI. C. Rector, D.W.Seldin :Hyponatr iemia in cerebral disease resulting from the inappropriate secretion of antidiuretic hormone. New Engl. J. Med. 264: 67-72 (1961). Castillo, E. B. del, F. A. de la Balze jr., J. Mambr ives: Ginecomastia y cancer del pum6n. Sem. med. 1: 1419-1432 (1941). 49 Endokrine und renale Paraneoplasien sis and management in 51 patients with a variety of disorders. Ann.Surg. 154: 491-508 (1961). Cost, W. S.: A. mineralocorticoid excess syndrome presumable due to excessive secretion of corticosterone. Lancet: 362-363 (1963). Costello, C.: Carcinoid syndrome control with a new antiserotonin agent. Case reports. Missouri Med. 63: 206-208 (1966). Coster, A. de, M. Payfa, R. Bellens, V. Conrad, P. A. Bastenie: Tumeur intrathoracique et hypoglycemie. J. franr;. Med. Chir. thor. 16: 191-199 (1962). Cottrell, J. C., K. L. Becker, Ch. F. Moore : Immunofluorescent studies in gonadotropin-secreting bronchogenic carcinoma. Amer. J. clin. Path.50: 422-427 (1968). Cox, J. R., P. J. Leonard, B. Singer:Effect of vasopressin on the volume of body fluid compartments and its relation to aldosterone excretion. Clin. Sci. 21: 205-219 (1961). Cox, M. L., R. D. Goruley, A. E.Kitabchi: Acinic cell adenocarcinoma of the parotid with ectopic production of adrenocorticotropic hormone.Amer. J. Med. 49: 529-533 (1970). Craven, J. L.: A pancreatic neoplasm presenting with hypercalcemia. Postgrad. med. J. 38: 583-586 (1962). Crawford, W. H.: Hypoglycemia with coma in a case of primary carcinoma of the liver.Amer. J. med.Sei. 181: 496502 (1931). Crocker, D. W., F. J. Veith : Mesodermal tumors associated with hypoglycemia. Review of literature and report of a case. Ann. Surg. 161: 418-427 (1965). Crooke, A. C.: Basophilism and carcinoma of the pancreas. J. Path.Bact. 58: 667-673 (1946). Crooke, A. C.: (1961) zit. nach J. H. Kennedy (1964). Dagnas, R : Le cancer primitif du foie ä forme hypoglycemia (thesis). Bordeaux 1959. Zit. nach Laurent 1971. Dailey, J. E., P. M. Marcuse: Gonadotropin secreting giant cell carcinoma of the lung. Cancer 24: 388-396 (1969). Damon, A., D. A. Holub, M. M. Melicow, A. C. Uson: Polycythemia and renal carcinoma: report of ten new cases, two with long hematologic remission following nephrectomy. Amer. J. Med. 25: 182-197 (1958). Dancot, H., P. Dustin : Syndrome de Cushing associe a un carcinome branchial (thymique?). Acta clin. belg. 3: 263275 (1948). David, N. J., J. V. Verner, P. L. Engel: The diagnostic spectrum of hypercalcemia. Amer. J. Med. 33: 88-110 (1962). Davis, R. B., J. C. Rosenberg : Carcinoid syndrome associated with hyper serotonnemia and normal 5-hydroxyindolacetic acid excretion. Amer. J. Med.30: 167-174 (1961). Davis, R. B., B. J. Kennedy: Carcinoid syndrome associated with adrenal hyperplasia. Arch. intern. Med.109: 192-200 (1962). Davidson, L.S.: Macrocytic haemolytic anemia. Quart. J. Med. 25, 543-578 (1932) Dean, A. C. B., A. T. Lambie, A. A. Shivas: Hypercalcaemia crisis and squamous carconoma of the renal pelvis. Brit. J. Surg. 56: 375-379 (1969). Delaere, J., R C. de Sousa, J. C. Rudler, R. S. Mach :A propos d'un cas de carcinome bronchique et syndrome de Schwartz-Bartter. Presse med. 73: 109-114 (1965). Castillo, E. B. del, E. Trucco, J. Manzuoli : Ma ladie de Cu shing et cancer du pancreas. Presse méd. 58: 783-785 (1950). Cheng Chao Ling, Liu Tze-Chün, Lin Tung-Ts'un, Li Ch'ouNung: Cushing's syndrome associated with bronchogenic carcinoma of the lungs. Report of a case with autopsy findings. Chin. med. J. 77: 260-261 (1958). Chochrane, W. A., W. W. Payne, M. J. Simpkiss, L. Woolf: Familial hypoglycemia precipitated by aminoacids. J. clin. Invest. 35: 4ll-422 (1956). Chowers, 1., Z. Eyal, N. J. Saltz: Cushing's syndrome associated with polypsof the colon. Digestion 3: 179-186 (1970). Christy, N. P.: Adrenocorticotrophic activity in the plasm a of patients with Cushing 's syndrome associated with pulmonary neoplasms. Lancet: 85-86 (1961). Christy, N. P., J. H. Laragh : Pathogenesis of hypokalemic alkalosis in Cushing's syndrome. New Engl. J. Med. 265: 1083-1088 (1961). Claxton, C. P. jr., H.T. McPherson, W.C. Sealy, W. G. Young: Hyponatriemia from inappropriate antidiuretic hormone elaboration in carcinoma of the lung. J. thorac. cardiovasc. Surg. 52: 331-341 (1966). Clift, G. W., F. E. Schletter, A. M. Moses, D. H. P. Streeten: Syndrom e of inappropriate vasopress in secretion. Arch. intern. Med. 118: 453-460 (1966). Clinicopathological Conference: Cushing's syndrome associated with a parotid gland tumor. Amer. J. Med. 34: 394406 (1963 a). Clinicopathological Conference: Malignant tumor, porphyria and inappropriate ADH secretion. Amer. J. Med. 35: 546559 (1963 b). Clinicopathological Conference: A case of carcinoma of the lung with endocrine changes. Brit. med. J.: 1492-1495 (1964). Clinicopathological Conference: A case of endocrine dysfunction with lung carcinoma. Brit. med. J.: 281-286 (1970). Codaccioni, J. C., Ph. Jaquet: Hyperhyroidie paraneoplasique avec T.S. H. plasmatique leve. Ann. endocr. 32: 553556 (1971). Cohen, R. B., G. D. Toll, B. Castleman: Bronchial adenoma in Cushing’s syndrome: their relation to thymomas and oat-cell carcinomas associated with hyperadrenocorticism.J. clin. Endocr.13: 812-817 (1960). Cohen, R. D., 1. P. Ross, A. D. Dayan: Metabolie studies in a case of a adrenocortical hyperfunction associated with carcinoma of the lung. J. clin. Endocr.24: 401-407 (1964). Cole, J. W., G. G. Bertino : Effect of chlorpromazine on excretion of 5-hydroxyindo leacetic acid in a patient with malignant carcinoid. Proc. Soc. exp. Biol. (N.Y.) 93: 100102 (1956). Coll, R., 1. Homer, Z. Kraiem, J. Gafni: Successful metyrapone therapy of the ectopic AC:TH-syndrome. Arch. intern. Med.121: 549-553 (1968). Connor, Th. D., W. C. Thomas jr., J. E. Howard: The etiology of hypercalcemia associated with lung carcinoma. J. clin. Invesl 35: 697 (1956). Cope, O., B.A.Barnes, B.Castleman, G.C.E. Mueller, S.J. Roth: Vicissitudes of parathyroid surgery: trials of diagno- 50 Literaturangaben Dengler, H.: Atypisches Karzinoidsyndrom mit vermehrter Ausscheidung von 5-Hydroxyindolessigsäure bei Pankreaskarzinom. Klin. Wschr. 37: 1245-1248 (1959). Drot, M., M. Goury-Lafont, M. Rathery, R. Roudier: L'hypoglycemie dans es tumeurs malignes du foie. Diabete 1: 25-31 (1959). Drot, M., P. Preychet, J. C. Chaput, D. Kleinknecht, P. Hirszel, G. Gschobroutsky: Etude clinique et biologique d'un cas d'hypercorticisme para neoplasique en rapport avec un nesidioblastome malin. Ann. endocr. 29: 765-780 (1968). Dexter, R. N., L. M. Fishman, R. L. Ney, G. W. Liddle: Inhibition of adrenal corticosteroid synthesis by aminotlutethimide :studies of the mechanism of action. J. clin. Endocr. 27: 473-490 (1967). Dobbins, W. T.: zit. nach Liddle (1965). Doege, K. W.: Pibrosarcoma of the mediastinum. Ann. Surg. 92: 955-960 (1930). Dohan, F. C., E. Rose, J. W. Eiman, E. M. Richardson, H. Zintel: Increased urinary estrogen excretion associated with adrenal tumors: report of four cases. J. clin. Endocr. 13: 415-428 (1953). Dolder, D. G. van : Spontane hypoglykemie bij een patient met een groot onggezwel. Ned. T. Geneesk. 105: 20302033 (1961). Dollinger, M. R., L. H. Ratner, Ch. A. Shamoian, B. D. Blakkbourne : Carcinoid syndrome associated with pancreatic tumour. Arch. intern. Med.120: 575-580 (1967). Donahower, G., 0. Schumacher, J. B. Hazard : two cases of medullary thyroid carcinom a causing Cushing's syndrome. J. clin. Endocr. 28: 1199-1204 (1968). Donati, R. M, M. McCarthy, R. D. Lange, N. 1. Gallagher: Erythrocythemia an neoplastic tumors. Ann. intern. Med. 58: 47-55 (1963). Dowling, J. Th., S. H. Ingbar, N. Freinkel: Iodine metabolism in hydatiform mole and choriocarcinoma. J. clin. Endocr. 20: 1-12 (1960). Dreyfus, G., G. Delzant, G. Chomette, E. Modigliani, R. Charles: Hypercorticisme paraneoplasique avec melanodermie et asthnie de type addisonien et revelateur d'un cancer thymique. Ann. m d. int. 121: 57-60 (1970). Driessens, J., A. Clay, L. Adenis, A. Demaille: Tumeur cervico-uterine et syndrome biologique de carcinoidose. Sem. Hop. Paris 12: 200-203 (1964). Duguid, J. B., A. M. Kennedy : Oat-cell-tumours of mediastinal gland. J. Path. Bact. 33: 93-99 (1930). Dun can, G. G., G. L. Schless: Massive retroperitoneal fibrosarcoma spontaneous hypoglycemia and generalized splanchnomegaly: report of a case. Metabolism 10: 200203 (1961). Dupont, B., J. H0yer, S. Borgeskor, j. Nerup: Plasma growth hormone and hypertrophic osteoarthropathy in carcinoma of the bronchus. Acta med. scand. 188: 25-30 (1970). Dyson, B.C.: Cushing's disease.Report of a case associated with carcinoma of the thyroid gland and cryptococosis. New Engl. J. Med. 261: 169-172 (1959). Eastridge, Ch. E., F. A. Hughes jr., J. L. Hamman: Cushing's syndrome in association with carcinoma of the respiratory tract. Ann. thorac. Surg. 1: 151-158 (1965). Eastridge, Ch. E., F. A. Hughes jr., M. L. Fields, J. R. Prather: Inappropriate antidiuretic hormone secretion in carci- noma of the lung. Dis. Chest. 54: 335-340 (1968). Edmondson, H. A., P. E. Steiner: Primary carcinoma of the liver. A study of 100 cases among 48 900 necropsies. Cancer 7: 462-503 (1954). Edmunds, A. W. B., K. C. McKeown, P. N. Coleman: Carcinoma of breast after adrenalectomy for Cushing's syndrome. Brit. med. J. / : 807-809 (1961). Edwards, C. R. W.: Measurement of plasma and urinary vasopressin by immunoassay. Proc. roy. Soc. Med. 64: 842844 (1971). Edwards, N. V.: Hyponatriemia and bronchial carcinoma. Brit. med. J. /:1547-1548 (1963). Egeli, E. S., S. Inceman, S. Devrim, E. Özdogan : Massif pleura tümörünebagli spontan hipoglisemi vak'sai. Türk. Tip. Cem. Mec. 31: 32-42 (1965). Elliott, Ch. A.: Hepatogenic hypoglycemia associated with primary liver-cell carcinoma. Trans. Ass. Amer. Phycns. 44: 121-124 (1929). Engel, P. L., L. Kahana: Cushing's Syndrome with malignant corticotropin producing tumor. Amer. J. Med. 34: 726-734 (1963). Engel, R.: Spontanhypoglykämie bei primärem Leberkrebs. Z. klin. Med. 141: 92-102 (1942). Erdheim, J., E. Strumme: Über die Schwangerschaftsveränderung der Hypophysis. Beitr. path. Anal. 46: 1-132 (1909). Erslev, A. J.: Anemia of chronic renal disease. Arch. intern. Med. 126: 774-780 (1970). Escobar, M. A., F. E. Trobaugh: Erythrocythemia in primary carcinoma of the liver. Arch. intern. Med. 110: 339-344 (1962). Escovitz, W. E., J. M. Reingold: Punctioning malignant bronchial carcinoid with Cushing's syndrome and recurrent sinus arrest. Ann. intern. Med. 54: 1248-1259 (1961). Euchenhofer, M., E. Streicher, H. Würz, K. A. Meurer, B. Steiner, P. Dürr, W. Kaufmann: Die hypokaliämische metabolische Alkalose. Dtsch. med.Wschr. 94: 1441-1447 (1969). Evans, R. W.: Histological appearances of tumors. 2nd Edition. Williams and Wilkins, Baltimore 1966. Eymontt, M. J., G. Gwinup, P. A. Kruger, D. E. Maynard, G. J. Hamwi: Cushing's syndrome wit h hypoglyc emia caused by adrenocortical carcinoma. J. clin. End ocr. 25: 4652 (1965). Paiman, Ch., J. A. Colwell, R. J. Ryan, J. M. Hershman, Th. W: Shelds: Gonadotropin secretion from a bronchogenic carcinoma. New Engl. J. Med. 277: 1395-1399 (1967). Falck, B., B. Larson, C. V. Mecklenburg, E. Rosengren, K. Svenaeus: On the presence of a second specific cell system in mammalian thyroid gland.Acta physiol. scand. 62: 491492 (1964). Parant, P. C., J. Insley: Increased adrenocortical activity and malignant disease. Lancet II : 434 (1960). Fast, B. B.: Hypoglycemia associated with massive intraabdominal mesenchymal tumor. Winnipeg clin. Quart. 15: 104-110 (1962). Faye, C., J. Chrétien, J. André Bo ugaran, R. Pariente,J.Chevalier, G. Brou et: Une nouvelle observation du syndrome de Sch.wartz-Bartter au cours d'un cancer anaplasique des 51 Endokrine und renale Paraneoplasien ne Regulationen des Calciumstoff wechsels -Spontan-Hypoglykämie, Glukagon.14.Symposium der deutschen Ges. f. Endokrinologie.132-145. Heidelberg, 7.-9.März 1968. Springer, 1969. Fry, L.: Pseudohyperparathyroidism with carcinoma of bronchus. Brit. med. J. 1: 301-302 (1962). Fusco, F. D., S. W. Rosen: Gonadotropin producing anaplastic large cell carcinomas of the lung. New Engl. J. Med. 275: 507-515 (1966). Gabcke, C.: Über einen Fall von primärem Spindelzellsarkom des Thymus. Inaugural Dissertation, Kiel 1896. Galasko, C. S. B., J. 1. Burn: Hypercalcaemia in patients with advanced mammary cancer. Brit. med.J./ :573-577 (1971). Ganzoni, A.: Hypercalcämiesyndrom. Schweiz. med. Wschr. 94: 855-861 (1964). Garfield, Cl. R., H. V. Belcher, J. S. Shuttleworth: Fibrous mesothelioma with hypoglycemia psychosis and coma. J. A. M. A. 181: 380-385 (1962). Gault, M. H., T. D. Kinsella: Carcinoma of Jung with adrenal hyperfunction and hypercalcemia treated by parathyroidectomy.Canad. med. Ass.J.92: 317-324 (1965). Gault, M. H., R. Bilefsky, T. D. Kinsella, A. Aronoff :Adrenocortical hyperfunction associated with bronchogenic carcinoma: report of five cases. Canad. med. Ass. J. 93: 1243-1249 (1965). Gelfman, N. A.: Bronchogenic carcinoma with Cushing's syndrome. A review with the report of an additional case. Amer. Rev. resp. Dis.83: 555-562 (1961). Gennes, L. de, H. Bricaire: Maladie de Basedow et affections malignes. Presse méd. 64: 2039 (1956). Gennes, L. de, H. Bricaire, J. Leprat: Les syndromes endocriniens paraneoplasiques et cancers viseraux. Presse méd. 70: 2035-2037 (1962 a). Gennes, L. de, H. Bricaire, J. Leprat: Les syndromes endocriniens para n oplasiques. II. Hyperthyroides et endocrinopathies diverses. Presse méd. 70: 2137-2139 (1962b). Geokas, M. C., J. Youl Chun, J. J. Dinan, 1. T. Beck: lsleteell carcinoma (Zollinger-Ellison-Syndrome) with fulminating adrenocortical hyperfunction and hypokaliemia. Canad. med. Ass. J. 93: 137-143 (1965). Giard, P., M.Paget, L. Croccel, J. Saout, Ph. Choteau, J. P. Leroy, A. Dutoit, M. Mahieu : Syndrome de SchwartzBartter et cancer pidermoide des bronches. J. Sei. méd. Lille 88: 517-519 (1970). Ginsberg, D. M.: Hypoglycemia associated with extrapancreatic neoplasms. Advanc. intern. Med. 12: 33-65 (1964). Ginsburg, J., J. B. Brown : lncreased oestrogen excretion in hypertrophic pulmonary osteoarthropathy.Lancet II: 12741276 (1961). Ginsburg, M., H. He!Jer: The clearance of injected vasopressin from the circulation and its fate in the body. J. Endocr. 9: 283-291 (1953). Gold, L., B. 1. Shnider: Some unusual syndromes associated with neoplastic disease. Ann. intern. Med.51: 890-896 (1959). Gold, Ph.: Circulating antibodies against carcinoembryonic antigens of the human digestive system. Cancer 20: 16631667 (1967). bronches. Probl mes physiopalho logiques concernant. L'hyponatr i mie, la natriur se et la r!\gulation de la s!\cretion d'aldosterone. Bull. Soc. méd. Hôp. Paris 118: 1191-1229 (1967). Felber, J. P., A.J. Moody, A. Villanueva, A. Vannotti: Détermination radioimmunologique de insuline et del ACTH en clinique Bull. Acad. Suisse méd. 21: 261-275 (1965). Ferenczy, A., T. Okagaki, ·R. M. Richart: Para-endocrine hypercalcemia in ovarian neoplasms. Cancer 27: 427-433 (1971). Feurle, G., H. Ketterer, W. Creutzfeldt: Zollinger-EllisonSyndrom. Med. Klin. 67: 37-46 (1972). Fichman, M. P., J. E. Bethune :The role of adrenocorticoids in the inappropriate anlidiuretic hormone syndrome. Ann. intern. Med. 68: 806-820 (1968). Field, J. B., H. Keen, Ph. Johnson, B. Herring: Insulin-like activity of nonpancreatic tumors associated with hypoglycemia. J. clin. Endocr. 23: 1229-1236 (1963). Fikenscher, D. Th., P. S. Biom: Hypoglykemie en fibrosarcoom. Med. T. Geneesk. 107: 1076-1083 (1963). Floyd, J. C. jr., L. Power, J. Rull, S. S. Fajans, J. W. Conn: Insulin and ILA in tissue extracts and plasma of patients with nonpancreatic tumors associated with hypoglycemia. Clin. Res. 11: 297 (1963). Flury, A., J. Müller, E. R. Froesch, A. Labhart: Das CushingSyndrom. Schweiz. med. Wschr. 101: 313-319 (1971). Fonkalsrud, E. W., R. B.Dilley, W.P. Longmire jr.: Insulin secreting tumors of the pancreas. Ann. Surg. 159: 730-741 (1964). Fontenaille, Ch., E. Mollet, J. Marchetti, M. Boulang. C. Huriet: Etude du systme renine-angiotensine-aldosterone dans 2 cas de syndrome de Schwartz-Bartter. Presse méd. 78: 965-968 (1970). Fälsch, E., P. Wahl, J.Drews, R. Rüfer: Untersuchungen zur tumorbedingten Hypoglykämie ohne Hyperinsulinismus. Helv. med. Acta 31: 545-552 (1964). Frank, E.: Thymuskarzinom mit Cushingschem Syndrom. Schweiz. med. Wschr. 75: 152-154 (1945). Frantz, A. G., M. T. Rabkin, H. Friesen: Human placental lactogen in choriocarcinoma of the male. Measurement by radioimmunoassay. J. clin. Endocr. 25: 1136-1139 (1965). Frerichs, H., B. Willms, W. Creuzfeldt: Further evidence for an inhibition of hepatic gluconeogenesis in hypoglycemia due to non-insulin-producing tumor. Horm. Metabol. Res.1: 89-90 (1969). Frerichs, H., B. Willms, H. Kasper, C. Creutzfeld, W. Creutzfeld: Con tribution to the pathogenesis of tumor hypoglycemia. Europ. J. clin. Invest. 1: 2-11 (1970). Friedman, M., P. Marshall-Jones, E. J. Ross: Cushing's Syndrome: adrenocortical hyperactivity secondary to neoplasms arising outside the pituitary-adrenal system. Quart. J. Med.35: 193-214 (1966). Friend, J.A. R., C. N. Haies: Spontaneous hypoglycemia and sarcoma. Acta endocr. (Kbh) 50: 233-238 (1965). Froesch, E. R., H. Bürgi, W. Ziegler, P. Bally, A. Labhart: Zur Pathogenese der tumorbedingten Hypoglykämie ohne Hyperinsulinismus. Schweiz. med. Wschr.93: 1250-1266 (1963). Froesch, E. R.,A. Jakob, A Labhart: Hypoglykämie bei extrapenlcreatischen Tu moren. Nebenschilddrüse und endokri- 52 Literaturangaben Goldberg, M. F., A. H. Tashjian, St. E. Order, G. ]. Dammin: Renal adenocarcinoma containing a parathyroid hormonelike substance and associated with marked hypercalcemia.Amer. J. Med. 36: 815-814 (1964). Goldberg, W. M., M. J. McNeil: Cushing's syndrome due to an ACTH producing carcinoma of the thyroid. Canad. med. Ass. J. 96: 1577-1579 (1967). Gonzales, F. M., G. L. Gold, B. J. Shnider:Gastrointestinal carcinoma and concomitant hypoglycemia.Ann. intern. Med. 58: 151-154 (1963). Goodall, C. M.: On para-endocrine cancer syndrome. Int. J. Cancer 4: 1-13 (1969). Gordon, Ph., Ch. E. Becker, G. S. Levey, J. Roth: Efficacy of aminoglutethimide in the ectopic ACTH-syndrome. J.clin. Endocr. 28: 921-923 (1968). Gordon, D.: The extrarenal manifestations of renal carcinoma. Canad. med. Ass. J. 88: 61-67 (1963). Govindaraj, M.: A case of oat-cell carcinoma of the lung with the carcinoid syndrome. Postgrad. med. J. 44: 815-816 (1968). Gowenlock, A. H., D. S. Platt, A. C. P. Cambell, K. G. Wormsley :Oat-cell carcinoma of the bronchus secreting 5-hydroxytryptophan. Lancet 1: 304-306 (1964). Göschke, H.: Pathophysiologie und medikamentöse Beeinflussung des Karzinoidsyndroms.Schweiz. med. Wschr. 97: 1548-1550 (1967). Grantham, J.J., R W. Brown, P. R. Schloerb: Asymptomatic hyponatriemia and bronchogenic carcinoma. The deletrious effects of diuretics. Amer. J. med. Sei. N. S.249: 273-279 (1965). Greenberg, P. B., C. Beck, T. J. Martin, H. G. Burger: Synthesis and release of human growth hormone from Jung carcinoma in cell culture. Lancet I: 350-352 (1972). Greenwood, F. C., V. H. T. James, B. F. Meggitt, J. D. Miller, P. H. Taylor: Paper read at the conference on factors influencing prognosis of breast cancer. Cardiff 1966, unpublished, zit. nach Steiner (1968). Grillo, I. A.: Endocrine manifestations of pulrnonary carcinoma in a nigerian. Brit. J. Cancer 25: 266-269 (1971). Gross, W., G. Simon: Polyzythämie bei maligner Schädeldachgeschwulst. Med. Welt 15: 1311-1314 (1963). Grumbach, M. M., L. S. Kaplan, J. J. Sciarra, 1. M. Burr: Chorionic growth hormone-prolactin (CGP): Secretion disposition, biologic activity in man, and postulated function as the "growth hormone" of the second half of pregnancy. Ann. N. Y. Acad.Sci.148: 501-531 (1968). Guillain, G., P. Lechelle, R. Garcin: La polyglobulie, avec ou sans erythrose, de certains syndromcs hypo-physotuberiens. Ann. Med. 91: 100-114 (1932). Guinet, P., R Mornex, A. Revol., J. Bertrand, A. Veyrat, G. Dorsit: Consid rations sur deux observations de maladie de Cushing avec syndrome d'alcalose hypokali mique. Rev. Lyon. M d. 16: 695-702 (1967). Gurdon, J. B.: Transplanted nuclei and cell differentiated. Sei. Amer.219: 24-35 (1968). Gülzow, M., A. Bienengräber: Spontane Hypoglykämie durch nicht vom Pankreas ausgehende Tumoren. Ärztl. Wschr.14: 513-519 (1959). Haggerty, J. F., M. X. Sullivan: Renine accumulation in tumor-bearing mice.Acta Un. int. Cancer 16: 1068-1073 (1960). Hall, Th., C.: Symptomatic hypokalemic alkalosis in hyperadreno-corticsm secondary to carcinoma of the prostate. Cancer 21: 190-192 (1968). Hallauer, W., J. Schirmeister, W. Oehlert: Hyperkalzämiesyndrom bei lymphoretikulärem Sarkom. Med. Klin. 63: 379-381 (1968). Hallpike, J. F., J. A. Morgan-Hughes: Dilutional hyponatriemia and myastnic syndrome in a patient with bronchial carcinoma. Brit. med. J. 11: 1573-1574 (1966). Hallwright, P. G., A. K. North, J. D. Reid: Pigmentation and Cushing's syndrome due to malignant tumor of the pancreas. J. clin. Endocr. 24: 496-500 (1964). Hardy, J. D.: Gynecomastia associated with lung cancer. J. A. M. A. 173: 1462-1465 (1960). Harrison, M. T., D. A. D. Montgomery, A. S. Ramsey, J. H. Robertson: Cushing's syndrome with carcinoma of the bronchus and with features suggesting carcinoid tumor. Lancet: 23-25 (1957). Hattori, Ch., M. Matsuda, R. Tatehshi, N. Tatsumi, T. Terazawa: Oat-Cell carcinoma of the lung containing serotonin granules. Gann 59: 123-129 (1968). Hauger-Klevene, J. H.: Asymptomatic production of ACTH. Radio immunoassay in squamous cell, oat cell,and adenocarcinoma of the lung. Cancer 22: 1262-1267 (1968). Hauger-Klevene, J. H.: High plasm a renin activity in an oatcell carcinoma: a renin-secreting carcinoma? Cancer 26: 1112-ll14 (1970). Hayes, D. M., Ch. L. Spurr, J. H. Felts, E. C. Miller: Von Recklinghausen's disease with massive intraabdominal tumor and spontaneous hypoglycemia: metabolic studies before and after perfusion of abdominal cavity with nitrogen mustard. Metabolism 10: 183-199 (1961). Haynal, F, F. Graf: The role of the hypophyseal-hypothalamine systcm in the pathogenesis of erythremia and symptomatic polycythemias. Acta med. scand. 139: 61-77 (1950). Hänze, S., C. A. Pierach: Paraneoplastischer Hypercorticismus. Klinische, biochemische und morphologische Befunde. Dtsch. med. Wschr. 91: 837-843 (1966). Hedinger, Chr.: Pathologische Anatomie des Carcinoidsyndroms. Verh. dtsch. Ges.inn. Med. 68: 182-194 (1962). Hegglin, R.: Differenzialdiagnose innerer Krankheiten. 12. Auflage. S. 617. Thieme, Stuttgart 1972. Heiskala, H., M. Gylling: Sarkoomaan liittyvä hypoglykemia. Duodecim. (Helsinki) 79: 543-546 (1963). Heiskala, H., M. Gylling: Hypoglycemia associated with sarcoma. Ann. Med. intern. Fenn. 52: 147-154 (1963). Heller, H., S. M. A. Zaidi: The neurohypophysis. Edited by H. Heller. P. 77. London 1957. Hemley, S. D., E. J. Arida, N. Finby: Cushing's syndrome associated with bronchogenic carcinoma. Radiology 80:.1116 (1963). Hendrix, T. R., M. Atkinson, J. A. Clifton, F. J. lngelfinger : The effect of 5-hydroxytryptamine on intestinal motor function in man.Amer. J. Med. 23: 886-893 (1957). Henneman, P. H.: zit. nach G. W. Liddle (1965). Henneman, P. H., E. F. Dempsey, E. F. Albright: Cause of hypercalciurie in sarcoid and its treatment with cortisone 53 Endokrine und renale Paraneoplasien Imperato, P. J., M. S. Lipton : Hypoglycemia in primary carcinoma of liver. N. Y. St. J. Med. 65: 2707-2710 (1965). Irmer, W., H. Daweke, J. Wedell, D. Grünklee, H. Schmitt, A. Jünemann: Zur Diagnostik und Therapie der Inselzelladenome. Dtsch. med. Wschr. 94: 1-9 (1969). lvy, H. K.: Renal sodium loss and bronchogenic carcinoma. Arch. intern. Med.108: 47-55 (1961). Jacob, F., J. Monod: On the regulation of gene activity. Cold Spring Harbor Symposium. Quart. Bio!. 26: 193-211 (1961). Jakob, A., N. Lauper, R. Flury, A. Labhart, E. R. Froesch: Pathognose de hypoglycemie d'origine tumorale. Aarhus 1966, 64 (1966), zit. nach Laurent (1971). Jacobasch, P.: Endokrine paraneoplastische Syndrome. Arch. Geschwulstforsch. 30: 245-264 (1967). Jarett, L., P. E. Lacy, D.M. Kipnis :Characterization by immuno-fluorescence of an ACTH-like substance in non-pituitary tumors from patients with hyperadrenocorticism. J. clin. Endocr. 24: 543-549 (1964). Jensen, M. K., L. Transbol, K. H. Olesen: Cancer og Cushing's syndrom. Nord. Med. 73: 197-224 (1965). Jepson, R. P., A. Jordan, M.J. Levell: Urinary steroid response to operation. Brit. J. Surg. 43: 390-395 (1955). Johnson, J. E.: zit. nach Liddle (1965) Johnston, W. H., J. Waisman: Carcinoid tumor of the vermiform appendix with Cushing's syndrome. Cancer 27: 681686 (1971). Jones, J.E., St. R. Shane, E.Gilbert, E.B. Flink: Cushing's syndrome induce by the ectopic production of ACTH by a bronchial carcinoid. J. clin. Endocr. 29: 1-5 (1969). Jones, N. F., M. A. Barraclough, 1. H. Mills: The mechanism of increased sodium excretion during water loading with 2,5% dextroso and vasopres sin. Clin. Sci. 25: 449-457 (1963). Josephs, B. N., G. Robbins, A. Levine: Polycythemia secondary to hamartoma of the liver. J. Amer. med. Ass. 179: 867-870 (1962). Josimovich, J. B., J. A. McLaren: Presence in the human placenta and term serum of a highly lactogenicsubstance immunologically related to pituitary growth hormone. Endocr. 71: 209-220 (1962). Josimovich, J. B., B. L. Atwood : Human placental lactogen (HPL), a trophoblastic hormone synergizing with chorionic gonadotropin and potentiating the anabolic effects of pituitary growth hormone. Am er. J. Obstet. Gynec. 88: 867-879 (1964). Kaplan, L. 1., L. Sokoloff, F. Murray, L. D.Stevenson: Sympathicoblastoma with metastases associated with the clinical picture of Cushing's syndrome. Report of a case. Arch. Neuro!. Psych iatr. (Chic.) 62: 696-698 (1949). Katsch, G., A. K. Focken: Insulinproduzierendes Malignom mit eigentümlichem Verlauf. Z. klin. Med. 153: 438-449 (1955). Kay, S., M. A. Willson: Ultrastructural studies of an ACTHsecreting tumor. Cancer 26: 445-452 (1970). Kaye, M.: An investigation into the cause of hyponatriemia in the syndrome of inappropriate secretion of antidiuretic hormone. Amer. J. Med. 41: 910-926 (1966). Kähler, H. J., L. Heilmeyer: Klinik und Pathophysiologie des Karzinoids und Karzinoidsyndroms unter besonderer Be- and sodium phytate. J. clin. lnvest 35: 1229 (1956). Hennen, G.: Detection of a thyroid stimulating factor in a chorio-cardnoma. occuring in a male. Arch. int. Physiol. Bioch. 74: 303-307 (1966 a). Hennen, G.: Thyrotropin-like factor in a non-endo crine cancer tissue. Arch. int. Physiol. Bioch. 74: 701-704 (1966b). Hennen, G.: Characterization of a thyroid-stimulating factor in a human cancer tissue. J. clin. Endocr. 27: 610-614 (1967). Herbeuval, R, G. Cuny, A. Larcan: Maladie de Vaquez et hypernephrome. Presse med. 65: 132-134 (1957). Hills, E. A.: Adenocarcinoma of the bronchus with Cushing's syndrome, carcinoid-syndrome, neuromyopathy, and urticaria. Brit. J.Dis.Chest 62: 88-92 (1968). Hines, R. E.: Hypoglycemia apparently due to retroperitoneal sarcoma. Med. Bull. Veteran's Adm. (Wash.) 20: 102-105 (1943). Holling, H. E.: Pu lmonary hypertrophic osteoarthropathy. Ann. intern. Med. 66: 232-234 (1967). Hollis, W. C.: Hypertrophie osteoarthropathy secondary to upper-gastrointestinal-tract neoplasm. Ann. intern. Med. 66: 125-130 (1967). Holten, C.: Hypoglycaemia-inducing tumour resembling spindle-cell sarcoma. Acta med. scand.157: 97-102 (1957). Holub, D. A., F. H. Katz: A possible etiologic link between Cushing's syndrom e and visceral malignancy. Clin. Res. 9: 194 (1961). Holtzmann, K., B. Runnebaum, F. Bahner, L. Zander: Ausscheidung von Testosteron und Epistosteron bei einem ektopischen ACTH-Syndrom. In: E. Klein (ed.): 13. Symposium der dtsch. Ges. für Endokrinologie. Würzburg, 2.-4. März 1967. Das Testosteron - Die Struma. S. 305-308. Springer, Berlin, Heidelberg, New York 1968. Horeau, J., J. Guenel. L. Bureau, M. Dubigeon: Le syndrome pseudo-hyperparathyroidien des tumeur sans mt!tastases osseuses. Presse méd. 73: 3057-3060 (1965). Horwitz, A., W. P. McKelway: Polycythemia associated with uterine myomas. J. Amer. med. Ass. 158: 1360-1361 (1955). Howard, J. E.: Differential diagnosis and therapy of spontaneou s hypoglycemia. Veterans Adm. techn. Bu ll.8: 119 (1955). Howard, E., P.C.Davis:Retroperitoneal hemangiopericytoma associated with hypoglycemia and masculinization. Delaware med. J. 31: 29-34 (1959). Hökfelt,B., B. Sjögren,Th. Falkheden: Steroid hormone production in a case of Cushing's syndrome with electrolyt changes simulating primary aldosteronism. Acta endocr. (Kbh) 31: 175-184 (1959). Hubble, D.: Cushing's syndrome and thymic carcinoma. Quart. J. Med. N.S. 18: 133-147 (1949). Hudson, B., J. Evans: Adrenocortical hyperplasia associated with bronchogenic carcinoma. J. clin. Endocr. 22: 494-500 (1962). Hymes, A. C., R, P. Doe: Adrenal function in cancer of the lung with and without Cushing's syndrome. Amer. J. Med.33: 398-407 (1962). 54 Literaturangaben rücksichtigung der Pharmakologie des 5-Hydroxytryp tamins. Ergebn. inn. Med. Kinderheilk N. F. 16: 292-550 (1961). Keating, F. R. jr., R. M. Wilder: zit. nach Laurent (1971). Keller, P., P. Hornung, D. Klemm: Paraneoplastische Spontanhypoglykämie. Therapiewoche 20: 3349-3351 (1970). Keller, R. T., 1. Goldschneider, P. W.Lafferty: Hypercalcemia secondary to a primary hepatoma. J. A. M. A.192: 112-114 (1965). Kennedy, J. H., M. J. Williams, Sh. C. Sommers: Cushing's syndrome and cancer of the lung. Pituitary Crooke cell hyperplasia in pulmonary oat cell carcinoma. Ann. Surg. 160: 90-94 (1964). Kenny, P. M., A. Stavrides, M. L. Voorhess, R. Klein : Cushing's syndrom associated with an adrenal neuroblastoma. A case in an infant with clinical, pathological, and adrenal cortical, and medullary hormone excretion studies. Amer. J. Dis. Child. 113: 611-615 (1967). Kepler, E. J.: Polyglandular dyscrasis involving abnormalities of sexual characteristics. Report of four cases. Proc. Mayo Clin.8: 102-110 (1933). Khaira, B. S.: Primary carcinoma of liver presenting as hypoglycemia. Brit. med. J. 1: 902-903 (1956). Kind, H.: Psychische Störungen bei Hyperparathyreodismus. Arch. Psychiat. Nervenkr.200: 1-11 (1959). Kinloch, J. D., J. N. Webb, D. Eccleston, J. Zeitlin: Carcinoid syndrome associated with oat cell carcinoma of bronchus. Brit. med.J. l : 1533-1535 (1965). Kipfer, K.: Spontane Hypoglykämie bei nicht-pankreatischen malignen Abdominaltumoren. Helv. med. Acta 24: 556-560 (1957). Kissel, P., M. Suc, J. M. André: Les hypercalcemies paraneoplasiques. Apropos de deux formes neuro-psychiques. Ann.méd. int 121: 51-55 (1970). Klein, H., S. P. Klein: Spontaneous hypoglycemia accociated with massive hepatoma. Arch. intern. Med.103: 273-278 (1959). Klemperer, P.:Parathyroid hyperplasia and bone destruction in generalized carcinomatosis. Surg.Gynec. Obstet. 36: 1115 (1923). Knight, R. A., J. G. Ratcliff e, G. M. Besser: Tumor ACTH concentrations in ectopic ACTH-syndrome and in control tissues. Proc. roy.Soc. Med. 64: 1266-1267 (1971). Kogut, M. D., G. N. Donell: Cushing's syndrome in association with renal ganglioneuroblastoma. Pediatrics 28: 566-577 (1961). Kofstad, J., 1. Prnyshov, E. Gjone, S. Blix: Pancreatic tumor with intractable watery diarrhoea, hypokaliemia, and hypercalcemia. Electrolyte balance studies. Scand. J. Gastroent. 2: 246-250 (1967). Kosenow, W., G. Feil, H. von Törne, J. R Bierich, M. Apostolakis: Sexuelle Frühreife durch primäres Lebercarcinom: .,Hepatogenitales Syndrom". Mschr. Kinderheilk. 115: 37-46 (1967). Kovach, R. D., L.H. Kyle: Cushing's syndrome and bronchogenic carcinoma. Amer. J. Med. 32: 981-988 (1958). Kracht, J., N. Hantschmann: Tumorsyntropien des CushingSyndroms. Acta endocr. (Kbh.) 38: 490-514 (1961). Kracht, J.: Ektopisch hormonbildende Tumoren. Dtsch. Ärz- tebl. 64: 1659-1660 (1967a). Kracht, J.: Pathologie der ektopisch hormonbildenden Tumoren. Verh. dtsch. Ges. inn. Med. 73: 488-495 (1967 b). Kracht, J.: Pathologie der ektopisch hormonbildenden Geschwülste. Med. Klin. 63: 41-46 (1968). Kracht, J.: Nebennierenpathologie des Cushing-Syndroms bei Kindern und Jugendlichen. Verh. dtsch. Ges. Path. S. 142-146. 55. Tagung. Fischer, 1971. Krapcho, J., C. F. Turk: Serotonin inhibitors. III. Compounds related to 2-(3-dimethylaminopropylthio)cinnamanilide. J. med. Chem. 9: 809-812 (1966). . Krecke, H., A. Linke, W. Müller: Spontane Hypoglykämie bei einem primären Leberkarzinom. Arch. klin. Med. 206: 102-108 (1959). Kreisberg, A., L. F. Pennington: Tumorhypoglycemia: a heterogenous disorder. Metabolism 19: 445-452 (1970). Kreisberg, R. A.,J. M. Hershman, J.G. Spenney, B. R. Boshell, L. P. Pennington: Biochemistry of extrapancreatic tumor hypoglycemia. Diabetes 19: 248-258 (1970). Krönke, E., G. W. Parade: Morbus Cushing bei Ovarialteratom. Z. klin. Med. 134: 698-718 (1938). Kuijjer, P. : A tumor of the lung with disturbances in the sugar metabolism. Arch.chir. Ned. 13: 81-96 (1961). Kühnlein, E., M. Meythaler: Extrapankreatische Tumoren mit schwerer Spontanhypoglykämie. Ärztl. Forsch. 12:189-205 (1958). Labhart, A., E. R. Proesch, W. Ziegler: Zur Diagnose und Therapie des Cushing-Syndroms. Schweiz. med. Wschr. 89: 44-53 (1959). Labhart, A.: Syndrom der Überproduktion von Vasopressin (Schwartz Bartter-Syndrom, inappropriate secretion of antidiuretic hormone). In : A. Labhart (ed.): Klinik der Inneren Sekretion. S. 58-66. Springer, Berlin, New York (1971a). Labhart, A.: Endokrine Überfunktionssyndrome bei ektopischer Hormonbildung (Paraneoplastische Syndrome). In: A. Labhart (ed.): Klinik der Inneren Sekretion. S. 10351038. Springer, Berlin, New York (1971b). Labhart, A.:Das Cushing-Syndrom. In:A. Labhart (ed.): Klinik der Inneren Sektion. 347-366.Springer, Berlin, New York (1971c). Lafferty, F. W.:Pseudohyperparathyroidism. Medicine 45: 247-260 (1966). Lamberg, B.A., R. Pelkonen, M. H. Frick:Hypercalcaemia in renal carcinoma. Report of a case. Acta med.scand. 176: 187-194 (1964). Lamotte, M., P. Haute.feuille, E. Martin, M. Bouteille, J.-M. Segresta, J.-L. Vilde: Hypoglycemie par tumeurs extrapancreatiques. Etude en microscopie optique et electronique. Surcharge glycogfoique du foie agressive. Presse méd. 74: 2321-2326 (1966). Landau, B. R., N. Wills, J. W. Craig, J. R. Leonards, T. Moriwaki: The mechani sm of hepatome - induced hypoglycemia. Cancer 15: 1188-1196 (1962). Landsberg, M.: Cushing's syndrome with carcinoma of the bronchus. Wis. med.J.59: 513-515 (1962). Laurent, J.: L'hypoglyc mie tumorale (Thesis). Nancy 1967. Laurent, J.: (1969) zit. nach Laurent (1971). Laurent, J., G. Debry, J. Floquet: Hypoglycemic tumors. 230 55 Endokrine und renale Paraneoplasien 25: 283-314 (1969). Lindsey, D. C., R. N. Barnes: Hyponatriemia probably due to inappropriate secretion of antidiuretic hormone associated with bronchogenic carcinoma. Sth. med. J. (Bgham, Ala.) 55: 337-340 (1962). Lipsett, M. B., W. D. OdelJ, L. E. Rosenberg, Th. A. Waldmann: Humoral syndromes associated with nonendocrine tumours. Ann. intern.Med. 61: 733-756 (1964). Livingston, W. C.: zit. nach Liddle (1965) Locks, M.: Incidence of hypercalcemia in patients with proved carcinoma of the lung. Lancet 82: 165 (1962). Lockwood, Ch. H.: Studies of adrenal cortical function in three cases of carcinoma. Canad. med. Ass. J. 79: 728-735 (1958). Loebel, A. S., Ch. S. Walkoff : Hypercalcemia in neoplastic disease without bone metastases. N. Y. Sl J. Med. 62: 101104 (1962). Lohrenz, F. N., G.St. Custer: ACTH-producing metastases from carcinoma of the esophagus.Ann. intern. Med. 62: 1917-1022 (1965). Loicq, A., J. Corvilan, P. Potvliege: Cancer bronchiqu e et hypercorticisme. Acta clin. belg. 17: 457-464 (1962). · Lowbeer, L.: Hypoglycemia-producing extrapancreatic neoplasms. Amer. J. clin. Path. 35: 233-243 (1961). Lucas, P. F.: Acute hypercalcemia from carcinomatosis without bone metastasis. Brit. med. J. I: 1330-1331 (1960). Luetscher, J. A.: Cause of Cushing's syndrome in patients with tumors arising from »non-endocrine« tissues. J. clin. Endocr. 22: 693-703 (1962). Luton, J. P., F. Girard, M. T. Pham-Huu, Trung, M. Binoux, J. Leprat, H. Bricaire: Thymome malin avec hypercorticisme. Etude clinique et biologiqe, pre- et postoperatoire. Ann. ME!d. int.121: 61-74 (1970). Luton,"j. P., Chaumerliac, C. Turpin, J. C. Valcke, Leprat, H. Bricaire: Les hypercorticismes paraneoplasiques. Sem. HOp. Paris 48: lll-120 (1972). Lytton, B., B. Rosof, J. S.Evans: Parathyroid hormon-like activity in a renal carcinoma producing hypercalcemia. J. Urol. (Baltimore) 93: 127-131 (1965). Mach, R. S., P. Rentchnick, A.-F. Müller, J. Lagier, H. C. Plattner: Syndrome humoral d'hypercorticisme avec alcalose et hypokaliémie par metastases surronaliennes de carcinomes bronchiques. Presse méd. 66: 437-438 (1958). MacNab, G. H., S. A. Moncrieff, M. Bodian: Primary malignant hepatic tumors in childhood. 168-174. Brit. Emp. Cancer Camp. 30th Ann. Rep. Eastbourn e, Sussex, Sumfield, and Day, LTD (1952). MacNaughton, M. C. .E., A. Priest: Hypoglycemia associated with retroperitoneal leiomyosarcoma. Lancet 1: 204205 (1960). Maesaka, J.K., S.K. Mittal, S. Fishbane: Paraneoplastic syndromes oft he kidney. Semin. Oncol. 24, 373-381 (1997) McPhee, L. W.: Cushing's syndrome associated with carcinoma of the bronchus. Brit. J. Surg. 46: 456-458 (1959). Magnenat, P., C. Perret: Pankreaskarzinom mit Hyperkalzämie. Gastroenterologia (Basel) 96: 197-202 (1961). Magnuss en, G.: (1941) zit. nach Laurent (1971). Meiner, E.: Hirngeschwulst und Polyglobulie. Schweiz. med. Wschr. 68: 338-339 (1936). p. Excerpta med. Amsterdam (1971). Law, D. H., G. W. Liddle, W. Scott, St. Tauber: Ectopic production of multiple hormones (ACTH, MSH, and Gastrin) by a single malignant tumor. New Engl. J. Med. 273: 292296 (1965). Lawrence, Ch.H.: Adrenal cortical tumor: a report of four cases. Ann. intern. Med. 11: 936-948 (1937). Layzer, H.: On the relation of functional hyperinsulinism to visceroptosis: report of twenty one (21) consecutive cases. Amer. J. Dig. 18: 300-303 (1951). Leaf, A., A. R. Mamby: An antidiuretic mechanism not regulated by extracellular fluid tonicity. J. clin. lnvest. 31: 60-71 (1952). Lebacq, E., R. Verberckmoes, P. Maldague: La secretion inappropie d'hormone antidiuretique dans des cas des tumeurs thoraciques anaplasiques de type oat-cell. Ann. endocr. 25: 361-373 (1964). Lebacq, E., E. Choche: Cancer bronchique »Oat-cell« et hypercorticisme sur nalien. Lille méd. 9: 151-157 (1964). Lee,J.,J.J.Jones, M.A.Barraclough: Inappropriate secretion of vasopressin. Lancet: 792-793 (1964). Leger, L., Ch. Sors, Cl. Dubost, M. Magdelaine, J. Lejeune, E. Rosenau, G. Lemaigre: Les hypoglycemie des tumeurs msenchymateuses extrapancreatiques. Presse méd. 71: 219-222 (1963). Lemnager, ]., M. Pays, M. Moulin, J. P. Bernard: Activit antidiuretiques au cours d'un cancer bronchique anaplasique. J. franc. méd. Chir. thor. 20: 775-788 (1966). Lem nager, J., M. Moulin: Le syndrome de Schwartz-Bartter et les troubles endocriniens au cours des cancers bronchopulmonaires. Cah. Coll. Méd. Hôp. Paris 10: 411-417 (1969). Levine, R. ]., A. Sjoerdsma: Pressor amines and carcinoid flush. Ann. intern. med. 58: 818-828 (1963). Lewis, J. M., N.B.Searle, G. L. Jordan jr.: Cushing's syndrome and bronchogenic carcinoma. Ann. intern. Med. 52: 1138-1146 (1960). Leyton, O H. M. Turnbull, A. B. Bratton: Primary cancer of the thymus with pluriglandular disturbance. J. Path. Bact 34: 635-660 (1931). Li, C. H., H. M. Grumbach, L. S. Kaplan, J. B. Josimovich, H. Friesen, K. J. Catt: Human chorionic somatomammotropin (HCS) proposed terminology for designation of placental hormone. Experientia (Basel) 24:1288 (1968). Liddle, G. W., D.Island, C.K. Meador: Regulation of corticotropin secretion in man. Recent Progr. Hormone Res. 18: 125-166 (1962). Liddle, G. W., D. P. Island, R. L. Ney, W. E. Nicholson, N. Shimizu: Nonpituitary neoplasmes and Cushing's syndrome. Arch. intern. Med. 111: 129-133 (1963). Liddle, G. W., J. R. Givens, W. E. Nicholson, D. P. Island: The ectopic ACTH-synd rome. Proc. Second Int. Congr. endocrin. Part II, 1064-1067, Excerpta med. Int. Congr.Series 83, Amsterdam 1964. Liddle, G. W., J. R. Givens, W. E. Nicholson, D. P. Island: The ectopic ACTH-syndrome. Cancer Res. 25: 1057-1061 (1965). Liddle,G. W. W. E.Nicholson, D. P. Island, D. N. Orth, K. Abe, St C. Lowder: Clinical and laboratory studies of ectopic humoral syndromes. Recent Progr. Hormone Res. 56 Literaturangaben Marks, V., W. H. R. Auld, J. B. Barr: Carcinoma of the stomach and other nonpancreatic lesions as causes of spontaneous hypoglycemia. Brit. J. Surg. 52: 925-928 (1965). Mars, H., O. P. Schumacher, L. J. McCormack: Intra-abdominal extrapancreatic neoplasm (leiomyosarcoma) associated with severe recurrent hypoglycemia. Report of a case. Cancer 20: ll55-ll60 (1967). Martin, M. M., B. L. Hamman: Patterns of urinary excretion of steroids in Cushing's syndrome. J. clin. Endocr. 26: 257267 (1966). Martin-Noel, P., B. Denis, J. Deschamps, D. Grunwald, M. Micoud, J. M. Mailion: Hypercorticisme et cancer du poumon. A propos d'une observation. J. méd. Lyon 49: 163176 (1968). Mason, A. St.: Reflections on Cushing's syndrome. Proc. roy.Soc. Med. 64: 749-752 (1971). Massaro, D. J., J. A. Owen jr.: Persistent hypercalcemia associated with bronchogenic carcinoma and primary chiefcell hyperplasia of the parathyroids.Amer. Rev. resp. Dis. 85: 727-734 (1962). Mavligit, G. M., J. L. Cohen, L. M. Sherwood : Ectopic production of parathyroid hormone by carcinoma of the breast. New. Engl. J. Med. 285: 154-156 (1971). McCuUagh, E. P., H. A. Tretbar: Treatment of Cushing's syndrome with Amphenome: report of two cases, one with probable thymoma. J. clin. Endocr. 18: 134-148 (1958). McDaniel, H. G., J. A. Pittman, S. R. Hili, W. R. Starnes: Adrenal cortical hyperfunction associated with bronchogenic carcinoma. Amer. J. Med. 35: 427-433 (1963). McDonald, R. A.: A study of 356 carcinoids of the gastrointestinal tract. Amer. J. Med. 21: 867-878 (1956). McDonald, R. A.: Primary carcinoma of the liver. Arch. intern. Med. 99: 266-279 (1957). McFadzean, A. J. S., T. T. Yeung: Hypoglycemia in primary carcinoma of the liver. Arch. intern. Med. 98: 720-731 (1956). McFadzean, A. J. S., R. T. T. Yeung: Further observations on hypoglycemia in hepatocellular carcinoma. Amer. J. Med. 47: 120-135 (1969). McLeod, C. E.: Report of an additional case of extrapancreatic tumor associated with hypoglycemia. Conn. med. 24: 458-460 (1960). McLetchie, N. G. B., L. D. W. Scott: Carcinoma of an adrenocortical rest associated with hypophyseal abnormality. J. Endocr. 3: 347-355 (1944). McMullen, F. F., H. H. Hanson: Excessive urinary 5-hydroxy-3-indole acetic acid in the absence of a metastatic carcinoid. Circulation 18: 883-886 (1958). McPeak, Ch. J.: Soft-part tumors producing hypoglycemia. Cancer management. ed. W. Cole Lippincott, Philadelphia 1968, 543-656 (1966). Meador, Cl. K., G. W. Liddle, D. P. Island, W. E. Nicholson, Ch. P. Lucas, J. G. Nuckton, J. A. Luetscher: Cause of Cushing's syndrome in patients with tumors arising from "nonendocrine" tissue. J. clin. Endocr. 22: 693-703 (1962). Mellgren, J.: Anterior pituitary in hyperfunction of adrenal cortex: an anatomical study with special reference to syndrome of Morgagni and notes on prostatic hypertrophy. Acta path. microbiol. scand. Suppl. 60: 96 (1945). Mellinkoff, S. M., P. A. Tumulty: Hepatic hypoglycemia. Its Mirsky, 1. A., G. Perisutti, D. Diengott: The hypoglycemic and insulinase inhibitory action of 1-tryptophan. Endocrinology 59: 369-379 (1956). Miura, K., Ch. Sasaki, J. Katsushima, T. Ohtomo, Sh. Sato, H. Demura, T. Torikai, N. Sasano: Pituitary-adrenocortical studies in a patient with Cushing's syndrome induced by thymoma. J. clin. Endocr. 27: 631-637 (1967). Miura, K., M. Lino, H. Demura: ACTH-secreting tumor. Jap. J. Cancer Clin.16: 463-468 (1970a). Miura, K., H. Demura, E. Sato, N. Sasano, N. Shimizu: A case of ACTH secreting cancer of the colon. J.clin. Endocr. 31: 591-595 (1970b). Miyabo, S., A. Fujimura, T. Matsuda, M. Murakami: Gastric cancer containing insulin and associated with hypoglycemia. Diabetes 17: 286-289 (1968). Moertel, Ch. G. O. H. Beahrs, L. B. Woolner, G. T. Tyce: »Malignant carcinoid syndrome« associated with noncarcinoid tumors. New Engl. J. Med. 273: 244-248 (1965). Moolten, L. S. E.: Hyperparathyroidism in carcinoma of the bronchus. J. med. Soc. N. J. 67: 474-476 (1970). Moretti, G., A. Broustet, J. Beylot, P. Bioulac, V. Veyret: Hypercorticisme surrfoalien et reticulosarcome. Sem. Hôp. Paris 48: 143-146 (1972). Mornex, R., Cl. Gharib, A. Veyrat, M. Dutruge, P. A. Byron, J. M. Magnin: Syndrome de Schwartz-Bartter par tumeur mlidiastinopulmonaire. Ann. Endocr. (Paris) 28: 391-399 (1967). Mornex, R., Cl. Gharib, J. Sassaro, F. Berthez ne: Les syndromes de Schwartz-Bartter d'origine paranlioplasiques. 189-216. H. P. Klotz (ed.): Les cancers et les glandes endocrines. Expansion scientifique 1969. Mahkota, St.: Spontane hipoglikemije pri obseznih tumorjih pljuc. Zdrav. Vestn.27: 326-330 (1958). Mahon, W. A., M. L. Mitchell, J. Steinke, M. S. Raben: Effect of human growth hormone on hypoglycemic states. New Engl. J. Med. 267: 1179 (1962). Maier, H. C., D. Barr: Intrathoracic tumors associated with hypoglycemia. J. thorac. Surg. 44: 321-329 (1962). Majcher, St. J., E. R. Lee, J. M. Reingold, J. Boyle, B. J. Haverback: Carcinoid syndrome in bronchogenic carcinoma. Arch. intern. Med. ll7: 57-63 (1966). Mallet-Guy, P., J. Feroldi: Physiologie des nerfs du foie. Rev. int. Hépat. 16: 9ll-925 (1966). Marks, L. J., A. E. Anderson, H. Liberman: Carcinoma of the lung associated with marked adrenocortical hyperplasia and adrenal hyper-responsivensess to ACTH in the absence of Cushing's syndrome. Ann. intern. Med. 54: 12431248 (1961). Marks, L. J., D. L. Rosenbaum, A. B. Russfield: Cushing's syndrome and corticotropin-secreting carcinoma of the lung. Ann. intern. Med. 58: 143-149 (1963). Marks, L. J., B. Berde, L. A. Klein, J. Roth, St. R. Goonan, D. Blumen, D. C. Nabsetz: Inappropriate vasopressin secretion and carcinoma of the pancreas. Amer. J. Med. 45: 967-974 (1968). Marks, V., E. Samols, R. Boltonin: Hyperinsulinism and Cushing's syndrome. Brit. med. J.I: 1419-1420 (1965). Marks, V. F., C. Rose: Hypoglycemia. Blackwell, Oxford 1965. 57 Endokrine und renale Paraneoplasien Morse, W. L, N. Kerenyi, D. H. Nelson: Prolonged hyperadrenocorticotrophism and pigmentation associated with bronchial carcinoid tumor. Canad.med. Ass. J. 96: 104-109 (1967). Muggia, F. M., H. O. Heinemann: Hypercalcemia associated with neoplastic disease. Ann. intern. Med. 73: 281-290 (1970). Murphy, R.: Hypoglycemia associated with an extrapancreatic neoplasm. Med. Clin. N. Amer. 47: 391-396 (1963). Mühe, E., K. T. Schriker, D. Raithel: Die Behandlung spontaner Hypoglykämien bei einem inoperablen Leberzellkarzinom durch Glucagon. Dtsch. med. Wschr.94: 1781-1785 (1969). Myers, W. P. L.: Clinical aspects and management of hypercalcemia. Med. Clin. N.Amer. 40: 871 (1956a). Myers, W. P. L.: Hypercalcemia in neoplastic disease. Cancer 9: 1135-1140 (1956b). Myers, W. P. L.: Cortisone in the treatment of hypercalcemia in neoplastic disease. Cancer 11: 83-88 (1958). Myers, W. P. L., Ch. K. Tashima, E. O. Rothschild: Endocrine syndrome associated with nonendocrine neoplasms. Med. Clin. N. Amer. 50: 763-778 (1966). Nabarro, J. D. N.: Case of bronchial carcinoma with Cushing's syndrome. In: A. S. Mason : Treatment of Cushing's syndrome by adrenalectomy. Proc. roy. Soc. Med.50: 766770 (1957). Nadler, W. H., J. A. Wolfer: Hepatogenic hypoglycemia associated with primary Iiver cell carcinoma. Arch.intern. Med.44: 700-710 (1929). Naess-Schmidt, T. E., K. Jorgensen: Hypoglykaemi forarsaget af ekstra pankreatiske tumorer. Nord.Med. 72: 14391442 (1964). Naess.-Schmidt, T. E., S. Jarnum, K. Jorgensen, F. Koch : Fatal hypoglycemia associated with retroperitoneal tumor. Acta med.scand.177:343-350 (1965). Nathan, E.: Hormonproducerende lungetumores. Ugeskr. Laeg. 132: 924- 927 (1970). Navarro, L. D.: Neoplasia hipoglucemiante extrahepática. Cirug. Ginec. Urol.19: 52-58 (1965). Neff, F. C., G. Tice, G. A. Walker, N. Ackerblad: Adrenal tumor in female infant with hypertrichosis, hypertension, overdevelopment of external genitalia, obesity, but absence of breast enlargement. Pseudo-sexual precocity. J. clin. Endocr. 2: 125-127 (1942). Nesbitt, K. W., J. T. Boswell, J. de Gonzales, S. Sarkisian: Malignant mesothelioma associated with hypoglycemia. J. clin. Path. 30: 148-157 (1958). Neville, A. M.: Ectopic production of hormones by tumours. Proc. roy. Soc. Med. 65: 55-59 (1972). Nevius, D. B., N. B. Friedman: Mesotheliomas and extraovarian thecomas with hypoglycemic and nephrotic syndromes. Cancer 12: 1263-1269 (1959). Ney, R. L., N. Shimizu, W. E. Nicholson, D. P. Island, G. W. Liddle: Correlation of plasma ACTH concentration with adrenocortical response in normal human subjects, surgical patients, and patients with Cushing's Syndrome.J. clin. Invest. 42: 1669-1667 (1963). Nichols, J., J. C. Warren, F. A. Mantz: ACTH-like excretion from carcinoma of the ovary. The clinical effect of DDD. J. A. M. A. 182: 713-718 (1962). occurence in congestive heart failure. New Engl. J. Med. 247: 745-750 (1952). Meloni, C. R., J. Tucci, J. J. Canary, L. H. Kyle : Cushing's syndrome due to bilateral adrenocortical hyperplasia caused by a benign adrenal medullary tumor. J. clin. Endocr. 26: 1192-1200 (1966). Melvin, K. E. W., A. H. Tashjian, C. E. Cassidy, J. R. Givens: Cushing's syndrome caused by ACTH and calcitonin secreting medullary carcinoma of the thyroid. Metabolism 19: 831-838 (1970). Menge, Ch. E.: Cutaneous manifestations of the malignant carcinoid syndrome. Ann. intern. Med. 58: 989-993 (1963). Menguy, R.: Pseudohyperparathyroidism due to malignant melanoma. Surg. Clin. N. Amer.49: 49-56 (1969). Metzler, C., E. B. Flink: Adrenal cortical hyperfunction associated with bronchogenic carcinoma. Lancet 76:242-244 (1956). Meyer-Hofmann, G., H. Schwarzkopf, H. Hartmann: Spontanhypoglykämien bei extrapankreatischen Tumoren. Dtsch. med. Wschr. 85: 2106- 2112 (1960). MGH-Case records: Case 39011. New Engl. J. Med. 248: 29-35 (1953). MGH-Case records: Case 39061. New Engl. J. Med. 248: 248-254 (1953). MGH-Case records: Case 41452. New Engl. J. Med. 253: 828-831 (1955). MGH-Ca se records: Case 43161. New Engl. J. Med. 256: 750-754 (1957). MGH-Case records: Case 44491. New Engl. J. Med. 259: 1128-1134 (1958). MGH -Case record s: Case 46451. New Engl. J. Med. 263: 965-973 (1960). MGH-Case records: Case 9-1961. New Engl. J. Med. 264: 242-248 (1961). MGH-Case records: Case 80-1961. New Engl. J. Med. 265: 953-957 (1961). MGH-Case records: Case 63-1963. New Engl.J. Med. 269: 801-808 (1963). MGH-Case records: Case 21-1964. New Engl. J. Med. 270: 898-906 (1964). MGH-Case records: Case 12-1967. New Engl. J.Med. 276: 629-634 (1967). MGH-Case records: Case 30-1967. New Engl. J. Med. 277: 147-154 (1967). Michael, C. A..: Pelvic fibroma causing recurrent attacks of hypoglycemia in a postmenopausal patient. Proc. roy. Soc. Med. 59: 835 (1966). Mii. R., M. Arsenijevm: Cushing's syndrome caused by mediastinaltumor? Lancet II: 436-438 (1963). Miller, D. R., R. E. Bolinger, D. Janigan, J. E. Crockett, St. Friesen: Hypoglycem ia due to nonpancreatic mesoderma l tumors: report of two cases. Arul. Surg. 150: 684-696 (1959). Miller, D. W. jr., Ch. F. Stewart, W. Rosner : Hypoglycemia caused by abdominal leiomyosarcoma. Amer. J. Surg.119: 754-757 (1970). Morrell, M. T.: Cushing's syndrome associated with bronchial carcinoma. Postgrad. med. J. 40: 614-615 (1964). 58 Literaturangaben Paraf, A., J. Rautureau, A. Benabadji: Hypoglycemie et cancer primitif du foie. Dosages hormonaux du diazoxyde. Rev. méd. chir. Mal. Foie 42:157-166 (1967). Parish, D. J., N. Crawford, A, T. Spencer: The secretion of 5-hydroxytryptamin by a poorly differentiated bronchial carcinoma. Thorax 19: 62-67 (1964). Parrish, J. A.: Hypokaliaemia, carcinoma and Cushing's syndrome. Lancet II: 603 (1960). Parsons, V., B. Rigby: Cushing's syndrome associated with adenocarcinoma of the ovary. Lancet II: 992-994 (1958). Paullada, J. J., A. Lisci-Garmilla, A. Gonzalés-Angulo, J. Jurado-Mendoza, M. Quijano-Narezo, L.. Gómez-Peralta, M. Doria-Medina: Hemangiopericytoma associated with hypoglycemia. Amer. J. Med. 44: 990-999 (1968). Paulson, G. S., J. J. Fechan, R. S. Westaby: Severe hypoglycemia due to mesothelioma arising in pleural cavity: case report. S. Dak. J. Med. Pharm. 14: 5-12 (1961). Pearson, Q, H.: Mineral metabolism abnormalities associated with cancer. Cancer management. Pp. 90-94. W.Cole Lippincott, Philadelphia 1968. Peart, W. S., T. M. Andrews, J.S. Robertson: Carcinoid syndrome: serotonin release induced with intravenous adrenaline or noradrenaline. Lancet 577-578 (1961). Peart, W. S., K. A. Porter, J. I.S. Robertson, M. Sandler, E. Baldock: Carcinoid syndrome due to pancreatic-duct neoplasm secreting 5-hydroxyptophan and 5-hydroxytryptamin. Lancet 239-242 (1965). Pedersen, J., F. Lund, J. Ringsted: Hypoglycaemia in the presence of massive fibrosarcoma (mesenchymoma). Acta endocr. 34: 148-156 (1960). Pedersen, K. W.: Ultracentrifugal and electrophoretic studies of fetuin. J. Phys. Colloid. Chem. 51: 164-171 (1947). Perkoff, G. T., E. L. Simons: Hypoglycemia in a patient with a fibromatous tumor. Arch. intern. Med. 112: 589-593 (1963). Pfeffer, K. H., W. Brandenburg, W. Frommhold: CushingSyndrom und Bronchialkarzinom mit funktionellen Störungen des Hypophysenvorderlappens. Dtsch. med. Wschr. 86: 1298-1302 (1961). Pfenninger, A., H. Micheli, A. F.Muller: Carcinome bronchique et secretion inappropriee d'hormone antidiuretique (Syndrome de Schwartz-Bartter). Helv. med. acta 30: 640644 (1963). Pfahl, R., R. P. Doe: Adrenal-pituitary studies in a patient with bronchogenic carcinoma and Cushing's syndrome. Ann. intern. Med. 58: 993-1002 (1963). Pfotenhauer, R., J. Kracht: Die extrahypophysären corticotropen Geschwülste. Endokrinologie 51: 23-53 (1967). Piante, M., Cl. Gharib, C. Barbe, A. Geiet, G. Aimard: A propos d'un nouveau cas de syndrome de Schwartz-Bartter avec manifestations neuropsychiques au cours d'un cancer broncho-pulmonaire anaplasique. J. Med. Lyon: 593-605 (1969). Pietschmann, H., W. Vormittag: Erythropoetingbestimmung bei Hyper nephrom mit Polyzythämie. Wien. inn. Med. 5: 217-226 (1970). Pinals, R. S., S. M. Krane: Medical aspects of renal carcinoma. Post-grad. med. J. 38: 507-519 (1962). Piper, W.: Cushing-Syndrom bei primärem ektopischem Chorionepitheliom der Leber. Internist 1: 420-424 (1960). Nichols, J., W. Gourley: Adrenal weight maintaining corticotropin in carcinoma of the lung. J. A. M.A. 185: 696698 (1963). Nilehn, J., E. M Nilsson: Coagulation studies in dffrent type of myeloma. Acta med. scand. suppl. 445, 194-199 (1966) Nissan, Sh., A. Bar-Maor, E. Shafrir: Hypoglycemia associated with extrapancreatic tumors. New Engl. J. Med. 278: 177-183 (1968). Noeinckx, F., R. Six, L. van Laethem: Tumeur maligne del'ovaire a effet hypercalcemiant et phosphaturique. Acta clin. belg. 17: 406-415 (1962). Norris, E. H.: Arrhenoblastoma: A malignant ovarian tumor associated with endocrinological effects. Amer. J. Cancer 32: 1-29 (1938). Oates, J. A., K. Melmon, A. Sjoerdsma, L. Gillespie, D. Mason: Release of a kinin peptide in the carcinoid syndrome. Lancet 1: 514-517 (1964). Odell, W. D., R. W. Bates, R. S. Rivlin, M. B. Lipsett, R. Hertz: lncreased thyroid function without clinical hyperthyroidism in patients with choriocarcinoma. J. clin. Endocr. 23: 658-664 (1963). O'Grady, A.S., L. J. Morse, J. B. Lee: Parathyroid hormonesecreting renal carcinoma associated with hypercalcemia and metabolic alkalosis. Ann. intern. Med. 63: 858-868 (1965). Oleesky, S., J. Bailey, E. Samols, D. Bilkus: A fibrosarcoma with hypoglycemia and high insulin-level. Lancet: 378380 (1962). Olmer, J., M. Mougin, R. Muratore, G. Calothy: La forme hypoglyclimique du cancer primitif du foie. Marseille méd.99: 108-112 (1962). Omenn, G. S., S. J. Roth, W. H. Baker: Hyperparathyroidism associated with malignant tumors of nonparathyroid origin. Cancer 24: 1004-1012 (1969). Omenn, G. S.: Ectopic polypeptide hormone production by tumors. Ann. intern. Med. 72: 136-138 (1970). Omenn, G. S.: Ectopic hormone syndromes associated with tumors in childhood. Pediatrics. 47: 613-622 (1971). O'Neal, L. W., D. M. Kipnis, S. A. Luse, P. E. Lacy, L. Jarett: Secretion of various endocrine substances by ACTH-secreting tumors. Gastrin, melanotropin, norepinephrine, serotonin, parathormone, vasopressin, glucagon. Cancer 21: 1219-1232 (1968). O'Neill, R. T., J. J. Mikuta: Reversible hypercalcemia in squamous cell carcinoma of the vagina. Report of a case. Obstet. and Gynec. 36: 458-461 (1970). O'Roirdan, J. L. H., G. P. Blanshard, A. Moxham, J. D. N. Nabarro: Corticotrophin-secreting carcinomas. Quart. J. Med. N. S. 35: 137-147 (1966). Ottenjann, R.: Akutes Hyperkalzämiesyndrom bei PankoastTumor. Med. Welt 14: 1342-1346 (1963). Page, J. H., W. McCubbin: Arterial pressure response to infused serotonin in normotensive dogs, cats, hypertensive dogs and man. Amer. J. Physiol. 184: 265-270 (1956). Pain, A.-K.: Hyponatriemia due to carcinoma of the bronchus. Brit. J. Pract. 25: 85-87 (1971). Papaioannou, A. N., Ch. J. McPeak: Rhabdomyosarcoma of the buttock producing hypoglycemia. Ann. Surg. 161: 553561 (1965). 59 Endokrine und renale Paraneoplasien with hepatoblastoma and sexual precocity. J. clin. Endocrin. 28: 1317-1322 (1969). Rosen,S. W., Ch. E. Becker. Sh.Schlaff, J.Easton, M. C. Gluck: Ectopic gonadotropin production before clinical recognition of bronchogenic carcinoma. New Engl. J.Med. 279: 640-641 (1968). Rosenberg, A. A.: Fulminating adrenocortica l-hyperfunction associated with islett-cell carcinoma of the pancreas: case report. J. clin. Endocr. 16: 1364-1373 (1956). Rosenberg, S. A.: (1961) zit. nach Laurent (1971). Rosenfeld, E. D.: Peritoneal pseudomyxoma. A report of four unusual cases. Arch. Path. 48: 255-272 (1949). Rosenthal, F. D.: Adrenocortical hyperactivity in a patient with bronchial carcinoma and diabetes mellitus. Brit. med. J. l: 139-141 (1957). Rosenthal R.L.: Multiple Myeloma associated with polycythemia, Report of four cases. Amer. J. med. sci. 218, 149154 (1955) Ross, E. J.: Hyponatremic syndrome associated with carcinoma of the bronchus. Quart. J. Med. 32: 297-320 (1963). Ross, E. J.: The cancer cell as an endocrine organ. Recent Advanc. Endocr. 293-327. 8th ed. Churchill, London 1968. Ross, E. J.: Some clinical aspects of the para-endocrine syndromes. Proc. roy. Soc. Med. 65: 59-60 (1972). Ross, L., E. Shelley: Mesonephric carcinoma of the ovary producing hypercalcemia. Amer. J. Obstet. Gynec. 100: 418-421 (1968). Rosse, W. P., T. A. Waldmann, P. Cohen: Renal cysts, erythropoietin, and polycythemia. Amer. J. Med. 34: 7681(1963). Rosselin, G., R. Assan, P. Reychet, J. Dolais, G. Tchobroutsky: Insuline, glucagon et hormone somatotrope plasmatiques dans cinq cas de tumeurs extra-pancreatiques hypoglycemiantes. Revue de la litterature. Presse méd. 75: 1045-1050 (1967). Rossman, E. M.: Mediastinal neurofibrosarcoma causing hypoglycemia. Arch. intern. Mcd. 104: 640-642 (1959). Roth, J., D. Stiller: Ektopisches ACTH-Syndrom bei einem Harnblasenkarzinom. Zbl. Chir. 97: 42-49 (1972). Rotolo, V.: Lipoglicemia nei tumori extrapancreatici. Minerva med. 62: 2723-2726 (1971). Roujeau, J., P. Aboulkcr: Cancer du rein, polycythemie et erythroblastose locale. Presse Med. 68: 156-158 (1960 (Donati et al., 1963)Haynal u. Grafd, 474 Roumagnoux, J., J. Durand, Cl. Gharib: Syndrome de Schwartz-Bartter avec manifestations neuropsychiques au cours de trois cancers ana plasiques du poumon. Lyon. méd. 35: 347-358 (1968). Ruch, W.: Estimation of antidiuretic hormmone in the urine of healthy subjects and patients with inappropriate secretion of vasopressin (Schwartz-Bartte r-Syndr om). Acta endocr. (Kbh.) 54: 113-121 (1967). Rucknagel, D. L., A. J. Chernoff: Immunologie studies of hemoglobin. III. Fetal hemoglobin changes in the circulation of pregnant women. Blood 10: 1092-1099 (1955). Sachs, B. A.: Endocrine disorders produced by non-endocrine malignant disorders. Bull. N. Y. Acad. Med.41: 10691086 (1965). Plimpton, C. H., A. Gellhorn: Hypercalcemia in malignant disease without evidence of hone destruction. Amer. J. Med. 21: 750-759 (1956). Portanova, R., H. Sachs: A specific adsorbent for vasopressin. The purification of labeled hormone. Endocrinology 80: 527-529 (1967). Porter, M. R.,V. K. Frantz: Tumors associated with hypoglycemia pancreatic and extrapancreatic. Amer. J. Med. 21: 944-961 (1956). Prunty, F. T. G., R. V. Brooks, J.Dupre, T. M. D. Gimilette, J. S. M. Hutchinson, R:R. McSwiney, 1. H. Mills: Adrenocortical hyperfunction and potassium metabolism in ptients with »non-endocrine« tumors and Cushing's syndrome. J. clin. Endocr. 23: 737-746 (1963). Rapoport,S. M.: Medizinische Biochemie. 5. Auflage. S. 760-761.VEB Verlag Volk und Gesundheit, Berlin 1969. Ratzenhofer, M.: Peyrters These von den peripheren endokrinen (parakrinen) Drüsen. Wien. klin. Wschr. 83: 222-231 (1971). Raymond, J. P., J. Chretien, J. Leprat, G. Brouet: Les manifestations endocriniennes paraneoplasiques des cancers bronchiques primitifs: la forme endocrinienne du cancer bronchique. J. franc. Med. Chir. thor. 22: 113-141 (1968). Reem, G. H., P. Vanamee: Electrolyte disturbances associated with cancer. J. chron. Dis.16: 737-755 (1963). Reeves, R. L., H. Tesluk, C. E. Harrison: Precocious puberty associated with hepatoma. J.clin.Endocrin.19: 1651-1660 (1959). Reichel,K., A. Moll: Bronchialkarzinom und Hyper'corticismus. Schweiz. med. Wschr. 94: 1068-1087 (1964). Renntchnik, P., A. F. Muller, J. Lagier, H. C. Plattner: Hypercorticisme provoqué par des métastases surrénaliennes d' un carcinoma bronchique. Helv. méd. Acta 24: 472-475 (1957). Revo A., J. Brun, P. Queneau: Les tumeurs malignes corticotropes a localisatio n pulmonaire et bronchique. Poumon Coeur 22: 121-146 (1966). Riggs, B. L. jr ., R. G. Sprague: Association of Cushing's syndrome and neoplastic disease.Arch. int. Med.108: 8593 (1961). Riordan, J. L. H., G. P. Blanshard, A. Moxham, J. D. N. Nabarro: Corticotrophin-secreting carcinomas. Quart. J. Med. N. S.35: 137-147 (1966). Roberts, H. J.: The syndrom e of hyponatriaemia and renal sodium loss probably resulting from inappropriate secretion of antidiuretic hormone. Ann. int. Med. 51: 1420-1426 (1959). Robertson, J. 1., W. S. Peart, T. M. Andrews: Mechanism of facial flushes in carcinoid-syndrome. Quart. J.Med. 31: 103-123 (1962). Robinson W.L., J.A. Mitas, R.W. Haerr. I.M. Cohen: Remission and exacerbation of tumors related nephrotic syndrom with treatment of the neoplasm. Cancer 15, 54-56 (1984) Roffo, A. H., L. M. Correa: On the presence of insulinoid in malignant tumors. J. Canc. Res.11: 126-129 (1927). Rogers, J. C. Th., J. H. Houseworth: Large fibrogenic tumors and hypoglycemia. J. A. M. A.178: 1132-1135 (1961). Root, A. W., A. M. Bongiovanni, W. R. Eberlein: A testicular-interstitial-cell stimulating gonadotroph in in a child 60 Literaturangaben Sachs, B. A.: Endocrine disorders associated with tumors of the lung. N. Y. St. J. Med. 69: 276-280 (1969). Sachs, B. A., N. Becker, A. E. Bloomberg, R. P. Grunwald: »Cure« of ectopic ACTH-syndrome secondary to adenocarcinoma of the lung. J. clin. Endocr. 30: 590-597 (1970). Sachsse, B., H. Blank: Funktionelle Hypoglykämie und organischer Hyperinsulinismus. Dtsch. med. Wschr. 84: 1679-1682 (1959). Sachsse, B.: Endogene Hypoglykämie infolge extrapankreatischer Tumoren. Med. Klin. 56: 714-715 (1961). Saeed, S. M., G. Fine, R. C. Horn jr.: Hypoglycemia associated with extra pancreatic tumor s. An immun ofluorescent study. Cancer 24: 158-166 (1969). Saegesser, F., J. Pettavel, P. Bougha, M. Muller: Tumeurs hypoglycemi antes extra-pancreatiques. Deux observations. Praxis 59: 876-880 (1970). Salus, F.: Zur zentral-nervösen Regulation des roten Blutbildes. Dtsch. Arch. klin. Med. 175: 214-220 (1933). Samaan, N., F. Lai, R. Fraser, R. B. Welbourn: Insulin assays in two cases of spontaneous hypoglycemia due to retroperitoneal mesothelioma. Brit. med. J. II: 195-198 (1965). Samaan, N., S. C. C. Yen, D. Gonzalez, 0. H. Pearson: Metabolic effects of placental lactogen (HPL) in man. J. clin. Endocr. 28: 485-491 (1968). Samols, E.: Hypoglycaemia in neoplasia. Postgrad. med. J. 31: 634-638 (1963). Samuelsson, S. M., L. Werner: Hepatic carcinoma simulating hyperparathyroidism. Acta med. scand.173: 539-547 (1963). Sandler, M., P. J. D. Snow: An atypical carcinoid tumour secreting 5-hydroxytryptophan. Lancet 1: 137-139 (1958). Sandler, M., P. J. Scheuer, P. J. Watt: 5-hydroxytryptophansecreting bronchial carcinoid-lumour. Lancet II: 19672069 (1961). Sasano, N., T. Fukuda, E. Satoh: Pathology of ectopic ACTH-syndrome with emphasis on pituitary Crooke cells and adrenocortical hyperplasia related to ACTH activities. in tumor tissues. Tohokou J. exp. Med. 99: 361-380 (1969). Sawyer, W. H.: Pharmacological characteristics of the antidiuretic principle in a bronchogenic carcinoma from a patient with hyponatremia. J. clin. Endocr. 27: 1497-1499 (1967). Sayle, B. A., P. A. Lang, W. O. Green jr., W. C. Bosworth, R. Gregory: Cushing's syndrome due to islet-cell carcinoma of the pancreas. Report of two cases: one with elevated 5-hydroxyindole acetic acid and complicated by aspergillosis. Ann. intern. Med. 63: 58-68 (1965). Schamaun,M., F. Deucher, S. Gablinger: Spontane Hypoglykämie bei großem Nebennierenrindentumor. Schweiz. med. Wschr. 87: 1348-1352 (1957). Schatten, W. E., A. G. Ship, W. J. Pieper, F. C. Bartter: Syndrome resembling hyperparathyroidism associated with squamous cell carcinoma. Ann. Surg. 148: 890-894 (1958). Schmid, J. R., L.A. French: Zerebellars Hämangioblastom mit Polyzythämie. Schweiz. med. Wschr. 85, 1274-1277 (1965) Schmidt, T.: Hepatoblastoma and hepatocarcinoma in infancy and childhood. Report of the pathology of eight cases from the university Hospital, Copenhagen. Acta path. microbiol. scand. (Suppl.) 212: 181-185 (1970). Schmidt, H. W., K. Schürholz: Hypoglykämische Anfälle durch intrathorakalen Tumor. Dtsch. med. Wschr. 86: 2231-2233 (1961). Sclfolz, D. A., L. B. Woolner, J. T. Priestley: Spontaneous hypoglycemia associated with fibrogenic tumor: report of two cases. Ann. intern. Med. 46: 796-807 (1957). Scholz, D. A., R. C. Bahn: Thymic tumors associated with Cushing's syndrome: review of three cases. Proc. Mayo Clin. 34: 433-441 (1959). Scholz, D. A., B. L. Riggs, R. C. Bahn, G. W. Liddle: Adrenocortical hyperfunction associated with a corticotrophinsecreting bronchogenic carcinoma: report of a case. Proc. Mayo Clin. 38: 45-51 (1963). Scholz, D. A., E. S. Horten, H. E. Lebovitz, D. 0. Ferris: Spontaneous hypoglycemia associated with an adrenocortical carcinoma. J. clin. Endocr. 27: 991-996 (1967). Schonfeld, A., D. Babott, K. Gundersen: Hypoglycemia and polycythemia associated with primary hepatoma. New Engl. J. Med. 265: 231-233 (1961). Schwartz, W. B., W. Sennet, S. Curelop, F. C. Barrter: A syndrome of renal sodium loss and hyponatriemia probably resulting from inappropriate secretion of antidiuretic hormone. Amer. J. Med. 23: 529-542 (1957). Schwartz, W. B., D. Tassel, Fr. C. Bartter: Further observation on hyponatriaemia and renal sodium loss probably resulting from inappropriate secretion of antidiuretic hormone. New Engl. J. Med. 262: 743-748 (1960). Seckel, H. P. G.: Postmortem hepatic glycogenolysis in hyperinsulinism and glycogen disease. J. clin. lnvest. 18: 723-731 (1939). Seftel, L. A., S. B. Gusberg: Hyperparathyroidism stated in gynecological malignancy. Review of the literature and report of a case. Obstet. and Gynec. 25: 693-697 (1965). Seifert, G., N. Seemann: Paraneoplastisches Hyperkalzämiesyndrom bei Ovarialkarzinom. Dtsch. med. Wschr. 92: 1104-1107 (1967). Seilmann, J. C., G. T. Perkoff, F. C. Null, J. R. Kimme!, F. H. Tyler: Hypoglycemia associated with massive intraabdominal mesothelial-cell sarcoma. New. Engl. J. Med. 260: 847-852 (1959). Shalet, M. F, R. M. Holder, T. R. Walters: Erythropoetin producing Wilm‘s tumor. J. Pediat. 70: 615-617 (1967). Shames, J. M., N. R. Dhurandhar, W. G. Blackard: Insulinsecreting bronchial carcinoid tumor with widespread metastases. Amer. J. Med. 44: 632-637 (1968). Sherwood, L. M., J. L. H. O'Riordan, G. D. Aurbach, J. T. Potts jr.: Production of parathyroid hormone by non-parathyroid tumors. J. clin. Endocr. 27: 140-146 (1967). Siegenthaler, D., W. Merki, H. U. Funk, Chr. Hedinger, A. Pletscher: Atypisches Karzinoidsyndrom bei kleinzelligem Bronchialkarzinom. Schweiz. med. Wschr. 95: 869-876 (1965). Siegenthaler, W.: Klinische Demonstrationen: 2. Paraneoplastisches Cushing-Syndrom bei Pflasterzellkarzinom der linken Lunge mit aus gedehnter Metastasierung. Schweiz. med. Wschr. 97: 230-233 (1967). Siegmund, H.: Cushing-Syndrom, Thymustumor und Landouzysche Tuberkulose. Dtsch. med. Wschr. 73: 33-36 61 Endokrine und renale Paraneoplasien Soutter, L., Sommers, A. S. Relman, Ch. P. Emerson: Problems in the surgical management of thymic tumors. Amer. Surg.146: 424-438 (1957). Sparagana, M., G. Phillips, C. Hoffman, L. Kucera: Ectopic growth hormone syndrome associated with lung cancer. Metabolism 20: 730-736 (1971). Spaulding, W. B., W. A. Oielle, A. G. Gorn all: Mineralocorticoid-like disturbance associated with adrenal metastases from a bronchogenic carcinoma. Ann. intern. Med. 42: 444-451 (1955). Sprague, R. G., A. B. Hayles, M. H. Power, H. L. Mason, W. A. Bennet: "Steroid Diabetes " and alkalosis associated with Cushing's syndrome. Report of a case, isolation of 17-hydroxycorticosterone (compound F) from urine and metabolic studies. J. clin. Endocr. 10: 289-306 (1950). Starr, G.F., F. Stroebel, T.P. Kearns: Polycythemia with papilledemaand infratentorial vascular tumors. Ann. intern. Med. 48, 978-986 (1958) Stauffer, J. M., G. E. Granville, S. W. Laws: Recurrent hypoglycemia and retroperitoneal fibrosarcoma. New Engl. J. Med. 265: 979-982 (1961). Steel, K., R. D. Baerg, D. O. Adams: Cushing's syndrome in association with a carcinoid tumor of the lung. J. clin. Endocr. 27: 1285-1289 (1967). Steeno, O., L. Lacquet, K. Delaere, J. Crabbe, P. de Moor: Striking hyperpigmentation in a case of Cushing's syndrome with a tumor of the lung. Acta clin. belg. 20: 128-135 (1965). Steigbigel, N. H., J. J. Oppenheim, L. M. Fishman, P. P. Carbone: Metastatic embryonal carcinoma of the testis associated with elevated plasma TSH like activity and hyperthyroidism. New. Engl. J. Med.271: 345-349 (1964). Stein, R. M., D. D. Bercovitch, M. F. Levitt: Dual effects of saline loading on renal tubul ar sodium reabsoption in the dog. Ame r. J. Physiol. 207: 826 (1964). Steiner, H., O. Dahlbäck, J. Waldenström: Ectopic growth hormone production and osteoarthropathy in carcinoma of the bronchus. Lancet 783-785 (1968). ' Steinke, J., J. S. Soeldner, A. E. Renold: lnsulin-like activity of extracts from large sarcomatous tumors associated with hypoglycemia. J. clin. Jnvest. 41: 1403 (1962). Stern, J. B.: Hypoglycemia associated with sarcoma. Lancet: 546-547 (1960). Stone, E., Ch. Waterhouse, R. Terry: Hypercalcemia of malignant disease: case report and a proposed mechanism of etiology. Ann. intern. Med. 54: 977-985 (1961). Stout, A. P.: Solitary fibrous mesothelioma of the peritoneum. Cancer 3: 820-825 (1950). Stovin, P. G. 1.: Syndromes of ectopic hormone production associated with pulmonary neoplasms. Amer. Rev. resp. Dis. 92: 484-488 (1965). Strott, Ch. A., Ch. A. Nugent, F. H. Tyler: Cushing's syndrome caused by bronchial adenomas.Amer. J. Med. 44: 97103 (1968). Stucki, R., P. de Werra: Les hyponatrimies. Praxis 58: 232-235 (1969). Studer, H., J. J. Staub, F. Wyss: Klinische und metabolische Fernwirkungen maligner Tumoren. Schweiz. med. Wschr. 101: 446-451 (1971). Subauste, C., R Calderon, L.A. Llerena, E. Carrion: Insulin (1948). Silverstein, M. N., K. G. Wakim, R. C. Bahn: Hypoglycemia associated with neoplasia. Amer. J. Med. 36: 415-423 (1964). Silverstein, M. N., G. Wakim, R. C. Bahn, R. H. Decker: Role of tryptophan metabolites in the hypoglycemia associated with neoplasia. Cancer 19:127-133 (1966). Silvis, R., D. S. Simon: Marked hypoglycemia associated with non-pancreatic tumors. New Engl. J. Med. 254: 1417 (1956). Sirtori, C., U. Veronesi: Gynecomastia: A review of 218 cases. Cancer 10: 645-654 (1957). Sjoerdsma, A., A. Oates, P. Saltzman, S. Udenfried: Serotonin synthesis in carcinoid patients. Its inhibition by α-methyl-DOPA with measurement of associated increases in urinary 5-hydroxytryptophan. New Engl. J. Med. 263: 585-588 (1960). Sjoerdsma, A., K. L. Melmon: The carcinoid spectrum. Gastroenterology 47: 10 -107 (1964). Skillern, P. G., L. }. McCormack, J. S. Hewlett, G. Crile: Hyperinsulinism due to islet-cell tumors simulating sarcoma. Diabetes 3: 133-140 (1954). Sluys Veer, J. van der, C. Choufoer, A. Querido: Metastasing islet-cell tumour of the pancreas associated with hypoglycaemia and carcinoid syndrome. Lancet: 1416-1418 (1964). Smilo, R. P., J. M. Earll, P. H. Forsham: Suppression of tumorous adrenal hyperfunction by aminoglutethim ide. Metabolism 16: 374-377 (1967). Smith, A. N., L. M. Nyhus, C. E. Dalgliesh, R. W. Dutton, B. Lennox, P. S.Mac Farlane: Further observations on endocrine aspects of argentaffinoma. Scot. med. II: 24-38 (1957). Smith, J. P., R. C. Boronow, L. More: Hypercalcemia accompanying ovarian mesonephroma without skeletal metastasis. Sth. med. J. (Bgham, Ala.) 61: 375-378 (1968). Smith, P. M.: Successful treatment of Cushing's syndrome secondary to an argentaffinoma.by bilateral adrenalectomy. Proc. roy. Soc. Med. 58: 573-576 (1965). Snapper, 1., W. C. Schraft jr ., D. M. Ginsberg: Severe hypoglycemia due to fibrosarcoma of the liver. Maandschr. Kindergeneesk. 32: 337-347 (1964). Snedecor, Ph. A., H. W. Baker: Pseudohyperparathyroidism due to malignant tumors. Cancer 17: 1492-1496 (1964). Sobota, J. T., R. J. Reed: Multiple bronchial adenoma, Cushing's syndrome and hypokalemic alkalosis. Dis. Chest. 46: 367-371 (1964). Sors, Ch., L. Uger, Cl. Dubost, G. Thomeret: Les mésotheliomes thoraciques hypoglycemiantes. Rev. Tuberc. (Paris) 26: 1268-1284 (1962). Sousa, R. C. de, J. Delaere, A. Thaon, J. C. Rudler, R. S. Mach: Le syndrome de Schwartz-Bartter: carcinoma du pumon avec sécretion inadequate d'hormone antidiuretique. Schweiz. med. Wschr. 94: 1805-1815 (1964). Sousa, R. C. de, M. Jenny: Hyponatremie par dilution dans un cas de carcinome pancréatique. Schweiz. med. Wschr. 94: 930-938 (1964). Sousa, R. C. de, R. S. Mach: Syndromes d'antidiurse inapproprie avec hyponatrémie par dilution. Actualités néphrol. Hôp. Necker. 283-309. Paris 1965. 62 Literaturangaben and insulin-like activity in tumor tissue and plasma of a patient with a fibrosarcoma associated with hypoglycemia. Metabolism 14: 881-884 (1965). Sundström, C.A.: A case of thymoma in association with erythrocytosis. Acta mirobiol. scand. Sec. A 80, 235-240 (1972) Svane, S.: Hypercalcemia in malignant disease without evidence of bone destruction. A case simulatig acute hyperparathyroidism. Acta med. scand.175: 353-357 (1964). Swanson Beck,, L. B. Porteous, J. L. Ullyot: Gonadotropinsecreting bronchial carcinoma: aberrant endocrine activity or trophoblastic differentation? Path. Bact. 101: 59-62 (1970). Szijj, L., Z. Csapó, F. A. Lázlo, K. Kovács: Medullary cancer of the thyroid gland associated with hypercorticism. Cancer 24: 167-173 (1969). Takaoka, Y.:(1965) zit. nach Laurent (1971). Tashjian, A. H., D. A. Ontjes, P. L. Munson: Iodacetate alkylation of bovine parathyroid hormone: effect on biological activity and structure. Fed. Proc. 22: 420 (1963). Tashjian, A. H. jr., L. Levine, P. L. Munson :lmmu nochemical identification of parathyroid hormone in non-parathyroid neoplasms associated with hypercalcemia. J. exp. Med. 119: 467-484 (1964). Tatarinov, J. S.: Presence of embryonal α-globulin in the serum of patient with primary hepatocellular carcinoma. Vop. med. Khim.10:90-91(1964). Taylor, D. M., A. W. Siemsen: Bronchogenic carcinoma simulating hyperparathyroidism. Arch. intern. Med.115: 6773 (1965). Tews, J. K., S. H. Carter, W. E. Stone: Chemical changes in the brain during insulin hypoglycemia and recovery. J. Neurochem. 12: 679-693 (1965). Thamdrup, E.: Precocious puberty in a boy with hepatoma. Acta endocr. (Kbh.) Suppl. 101: 23 (1965). Thannhauser, S.: Hypoglycemia in the early phase of adrenocortical carcinoma. J. clin. Endocr. 9: 791 (1949). Thibault, Ph.: Letaux d'hormone de coissance au cours du cancer bronchique avec dysacromélie. Presse méd. 76: 1505-1506 (1968). Thomas, C., W. Zengerling, H. Noetzel: Neurologische Formen des paraneoplastischen Syndroms. Schattauer-Verlag. Stuttgart – New York (1972). Thomas, C., R. Windt, E. Grom: Hämatologische und endokrine Foren des paraneoplastischen Syndroms. Schattauer-Verlag. Stuttgart – New York (1974). Thomas, C. (Hrsgb.). Bütter, R., C. Thomas. Allgemeine Pathologie. 3. Aufl. Schattauer-Verlag. Stuttgart – New York (2002). Thomas, C. (Hrsgb.): Grundlagen der klinischen Medizin. Band 6. Gröne, H.-J. T. Eisemhauer, B. Ulshöfer, G. Gebert, C. Thomas: Harnapparat & männliches Genitale. Schattauer-Verlag. Stuttgart – New York (1993). Thomas, C. (Hrsgb.): Grundlagen der klinischen Medizin. Band 5. G. Gebert, C. Thomas: Endokrines System. Schattauer-Verlag. Stuttgart – New York (1992). Thomas, C: Atlas der Infektionskrankheiten. Schattauer-Verlag. Stuttgart – New York (2010). Thomas, P., R Shields: Associated autonomic dysfunction and carcinoma of the pancreas. Brit. med. IV: 32 (1970). Thompson, Ch. M., D. J. Hilferty: Primary carcinoma of the liver (Cholangioma) with hypoglycemic convulsions. Gastroenterol. 20: 158-165 (1952). Thompson, G. S., L. Horwich, J. C. Davis: Carcinoma of bronchus and Cushing's syndrome. Lancet: 534-536 (1962). Thompson, K. W., L. Eisenhardt: Further considerations of the Cushing's syndrome. J. clin. Endocr. J: 445-452 (1943). Thomson, W. H. F., A. B. A. Karat: Hypercalcemia associated with adenocarcinoma of kidney without demonstrable bone lesions. Brit. med. J. II : 745-746 (1966). Thorn, G. W.: Case records of the Massachusetts General Hospital Case 34, New Engl. J. Med. 268: 1129-1139 (1963). Thorn, N. A., I. Transbol: Hyponatremia and bronchogenic carcinoma associated with renal excretion of large amounts of antidiuretic material. Amer. J. Med. 35: 257268 (1963). Thorne, M. G.: Cushing's syndrome associated with bronchial carcinoma. An enquiry into the relationship of this syndrome to neoplastic disease. Guy's hosp. Rep. 101: 251272 (1952). Thurman, W. G., H.Grabstald, P.H. Lieberman:Elevation of erythropoietin levels in association with Wilm‘s tumor. Arch. intern. Med. 117: 280-283 (1966). Tisher, C. C.: Correction of an inappropriate ADH-syndrome by tumor resection. Arch. intern. Med.121: 163-168 (1968). Tranquada, R. E., A. B. Sender, P. M. Beigelman: Hypoglycemia associated with carcinoma of the cecum and syndrome of testicular feminization. New Engl. J. Med. 266: 1302-1306 (1962). Tremblay, R. E., J. S. Ansell: Carcinoma of the kidney simulating hyperparathyroidism. J. Urol. (Baltimore) 91: 10-13 (1964). Tretbar, H. A., L. P. Cawley: Cushing's syndrome. J. Kans med. Soc. 64: 487-490 (1963). Tugaye, A., Ph. Daumet, Ch. Garnier, J. Mignot, R. Deuil: Hypoglycmie par tumeur mésenchymateuse thoracique. Diabete (Le Raincy) 13: 318-321 (1965). Turkington, R. W., J. K. Goldman, B. W. Ruffner, J. L. Dobson: Bronchogenic carcinoma simulating hyperparathyroidism. Cancer 19: 406-414 (1966). Turner, P., R. Williams: Unexplained steatorrhoea in the syndrome of hyponatriaemia and carcinoma of the bronchus. Brit. med. J. 1: 287-290 (1962). Uehlinger, E.: Hypercalcämie-Syndrome. Münch. med. Wschr. 106: 692-701 (1964). Uehlinger, E.: Paraneoplastische Syndrome. Almanach für ärztl. Fortbildung. S.17-44. Lehmann, München 1966. Uei, Y., U. Kirn, Y. Itatsu: An autopsy case of islet-cell carcinoma with Cushing's syndrome. Acta path. jap. 18: 333341 (1968). Ufer, J.: Zur Entstehung der Spontanhypoglykämie. Dtsch. med. Wschr. 69: 206-207 (1943). Umbach, W.: Vertebralisangiographie und Blutbild als diagnostisches Hilfsmittel zur Erkennung von Kleinhirnangioblastomen. Nervenarzt 25: 356-359 (1954). Unger, R. G., J. de V. Lochner, A. M. Eisentraut: Identifica- 63 Endokrine und renale Paraneoplasien Ward, A. A., E. L. Poltz, L. M. Knopp: Polycythemia associated with cerebellar hemangioblastoma. J. Neurosurg. 13: 248-257 (1956). Warner, R. R., P. A. Kirschner, G. M. Warner: Serotonin production by bronchial adenomas without carcinoid syndrome.J. A. M. A. 178: ll75- ll79 (1961). Warren, F. L.: Estimation of urinary 17-ketosteroids in the diagnosis of adrenal cortical tumors. Cancer Res. 5: 49-54 (1945). Watson, L.: Hypercalcemia and cancer. Postgrad. med. J. 39: 646-649 (1963). Webster, G. D.jr., J. C.Tou.chstone, M. Suzuki: Adrenocortical hyperplasia occuring with metastatic carcinoma of the prostate: report of a case exhibiting increased urinary aldosterone and glucocorticoid excretion. J. clin. Endocrin.19: 967-979 (1959). Weichert, R. F.: The neural ectodermal origin of the peptide secreting endocrine glands. A unifying concept for the etiology of multiple endocrine adenomatous and the inappropriate secretion of peptide hormones by nonendocrine tumors. Amer. J. Med. 49: 232-241 (1970). Weintraub, B. D., S. W. Rosen : Human placental lactogen production by undifferentiated carcinomas of the lung. Clin. Res. 17: 47 (1969). Weintraub, B. D., S. W. Rosen: Ectopic production of human chorionic somatomammotropin by nontrophoblastic cancers. J. clin. Endocr. 32: 94-101 (1971). Weizenecker, R., H. Burke:The ectopic ACTH syndrome. J. med. Ass. 57: 24-28 (1970). Welbourn, R. B.: (1965) zit. nach Laurent (1971) Welbourn, R. B.: Cushing's syndrome. A review of 50 patients in 15 years. Ann. roy. Coll. Surg. Engl. 44: 182-193 (1969). Welbourn, R. B., D. A. D. Montgomery, T. L. Kennedy: The natural history of treated Cushing•s syndrome. Brit. J. Surg. 58: 1-16 (1971). Werk, E. E. jr., L. J. Sholiton:Adrenocortical function in carcinoma of the lung. Cancer 13: 469-481 (1960). Werle, E., J. Kinine: J. mond. Pharm. (La Haye) 3: 291-312 (1968). Werte, E.: Chemie und Pharmakologie der Plasmakinine und ihre physiologische und pathologische Bedeutung. Med. Welt 21: 53-62 (1970). Whipple, A. O.: Pancreaticoduodenectomy for islet-cell carcinoma. A five year follow-up. Ann. Surg.121: 847-852 (1945). White,A. G.: Diabetes insipidus associated with edema. Report of a case with discussion of physiologic implication. New Engl.J.Med. 250: 633-636 (1954). Whitelaw, A. G.L.: Ultrastructure of an oat cell carcinoma of the bronchus producing an antidiuretic hormone.Brit. J. Cancer 23: 69-72 (1969). Whitney, J. E., C.G. Massey: Apparant insulin activity in a fibrosarcoma as sociated with spontaneous hypoglycemia. J.clin. Endocr.21: 541-547 (1961). Whitney, J. E., B. L. Heller : Increased insulin-like activity of the serum in a patient with spontaneous hypoglycemia associated with a retroperitoneal fibrosarcoma. Amer. J. Med. 30: 633-636 (1961). Wichert, P. v.: Paraneoplastische Syndrome. Problematik und tion of two hormones in a bronchogenic metastasis and the concept of tumor as a hormone spongue. J. Lab. clin. Med. 62: 1018 (1963). Unger, R. G.; J. de Lochner, A. M. Eisentraut: Identification of insulin and glucagon in a bronchogenic metastasis. J. clin. Endocr. 24: 823-831(1964). Upton, G. V., Th. T. Amatruda: Evidence for the presence of tumor peptides with corticotropin-releasing-factor activity in the ectopic ACTH syndrome. New Engl. J. Med. 285: 419-424 (1971). Urtega-Ballon, O.: (1949) zit. nach Laurent (1971) Utiger,R. D.: Inappropriate antidiuresis and carcinoma of the lung: detection of arginine vasopressin in the tumor extracts by immunoassay. J. clin. Endocr. 26: 970-974 (1966). Vague, Ph., Ch. Oliver, F. Laffargue, B. Argemi, P. Laval: Hypercortisolisme et hyperthyrocalcitonisme dans un cas de cancer «medullaire » hyroidien secretant ACTH et thyrocalcitonine. Ann. Endocr. (Paris) 32: 557-565 (1971). Vaissman, 1.: (1964) zit. nach Laurent (1971). Vingerhoeds, A. C. M., P. J. der Kinderen, J. H. H. Thijssen, F. Schwarz: Detection of an ACTH-secreting bronchial carcinoid tumour eighteen months after adrenalectomy for Cushing's syndrome. Acta endocr. 67:625-633 (1971). Vogel, M. D., F. R. Keating, R. C. Bahn: Acute Cushing's syndrome associated with bronchogenic carcinoma. Proc. Mayo Clin. 36: 387-393 (1961). Volk, B. W., M. G. Goldn er, B. Wainfeld: Spontaneous hypoglycemia with abdominal spindle-cell sarcoma. Geriatrics 15: 473-479 (1960). Volpe, R.,J. S.Evans, D. W. Clarke, N. Forbath, R. Ehrlich: Evidence favoring the sarcomatous origin of an insulin-like substance in a case of fibrosarcoma with hypoglycemia. Amer. J. Med. 38: 540-553 (1965). Vorherr, H., Sh. G. Massry, R. D. Utinger, Ch. R. Kleeman: Antidiuretic principle in malignant tumor extracts from patients with inappropriate ADH syndrome. J. clin. Endocrin. 28: 162-168 (1968). Wagner, K.: Sekundäre renale Polyzythämie bei hypernephroidem Nierenkarzinom. Krebsarzt 17: 210-214 (1962). Waldenström, J.:Maladies of derepression. Pathological, often monoclonal, derepression of protein forming templates. Schweiz. med. Wschr. 100: 2197-2206 (1970). Waldvogel, F., R. C. de Sousa. R. S. Mach: Intoxication a Leau due a une secretion inadequate d'hormone antidiuretique (syndrome de Schwartz Bartter) d'origine idiopathique.Schweiz. med. Wschr. 97: 929-938 (1967). Waldman, T. A., E. H. Levin, M.Baldwin: The association of polycythemia with a cerebellar hemangioblastoma. Amer. J. Med. 31: 318-324 (1961). Walser, M.: The separate effects of hyperparathyroidism, hypercalcemia of malignancy, renal failure, and acisosis on the state of calcium, phosphate, and other ions in plasma. J. clin. Invest. 41: 1454-1471 (1962). Walther, H.: Das Hyperkalzämiesyndrom beim Bronchialkarzinom ohne Skelettmetastasierung. Z. ges. inn.Med.25: 854-856 (1970). Wanebo, H. J., 1..Schlessinger, Ch. K. Tashima: Severe hypoglycemia associated with terminal lymphomas. Report of five cases. Cancer 19: 1451-1458 (1966). 64 Literaturangaben Williams, E. D., A. M. Morales, R. C. Horn: Thyroid carcinoma and Cushing's syndrome. J. clin. Path. 21: 129-135 (1968b). Williams, G. H., Ch. L. Crockett, W. S. Butler, K. R. Crispell:The coexistence of pheochromocytoma and adrenocortical hyperplasia. J. clin. Endocr. 20: 622-631 (1960). Zittoun, R., J. Bousse, F. Vachon, F. Berlin:, C. Sultan: A propos de deux cas d’anemie hemolytiquerapidement mortelle d’origme cancereuse.Nouv. Rev. franc. Hemat. 5: 857-862 (1965) . Bedeutung für Frühdiagnose und Therapie von Tumoren. Med. Klin. 66: 1461-1465 (1971). Wikman, J. H., J. D. McCracken : Hypoglycemia associated with adrenalcarcinoma. Amer. J. Surg. 110: 811-813 (1965). Williams, E. D., j.G. Azzopardi: Tumor of the lung and carcinoid syndrome. Thorax 15: 30-36 (1960). Williams, E. D., M. Sandler: The classification of carcinoid tumors.Lancet: 238-239 (1965). Williams, E. D., S. M. M. Karim, M. Sandler: Prostaglandin secretion by medullary carcinoma of the thyroid. Lancet I: 22-23 (1968a). Bemerkung: Autoren in diesen Literaturzitaten, die nicht im Text erwähnt werden, beziehen sich auf die publizierten und ausgewerteten Einzellfälle von Paraneoplasien in C. Thomas et al. (1974) 65