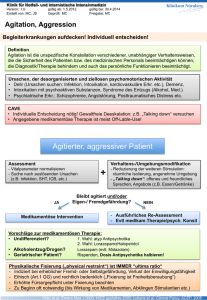
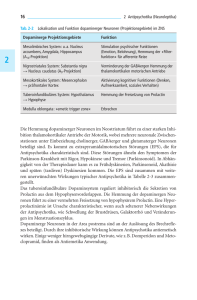
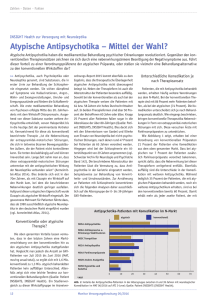
Arzneimittelinteraktionen in der Therapie mit Antipsychotika
Werbung

Diplomarbeit Arzneimittelinteraktionen in der Therapie mit Antipsychotika Unerwünschte Arzneimittelwirkungen in der Kombination verschiedener Antipsychotika untereinander und mit anderen Medikamentenklassen eingereicht von Ulrich Schneeberger Geb.-Datum.: 18.07.1984 zur Erlangung des akademischen Grades Doktor der gesamten Heilkunde (Dr. med. univ.) an der Medizinischen Universität Graz ausgeführt am Institut für Experimentelle und Klinische Pharmakologie unter der Anleitung von ao. Univ. Prof. Dr. Josef Donnerer Graz, am 22.01.2013 (Unterschrift) Eidesstattliche Erklärung Ich erkläre ehrenwörtlich, dass ich die vorliegende Arbeit selbstständig und ohne fremde Hilfe verfasst habe, andere als die angegebenen Quellen nicht verwendet habe und die den benutzten Quellen wörtlich oder inhaltlich entnommenen Stellen als solche kenntlich gemacht habe. Graz, am 22.01.2013 (Unterschrift) i Zusammenfassung Durch die quantitative Zunahme an zugelassenen Arzneimitteln und ihre teilweise notwendige Anwendung in Kombinationstherapien erlangt die Problematik der Arzneimittelinteraktionen im klinischen Alltag immer mehr Evidenz. In Österreich sind bis zu 7 % aller Krankenhauseinweisungen auf unerwünschte Arzneimittelnebenwirkungen zurückzuführen. Im geriatrischen Bereich liegt der Anteil mit 20 % wesentlich höher. Die Vielfalt an möglichen Interaktionen resultiert aus den pharmakodynamischen und pharmakokinetischen Eigenschaften der Arzneistoffe. Diese Diplomarbeit umfasst die aktuellen Ergebnisse einer Literaturrecherche (z. B. PUBMED, MEDLINE, OVID, Lehrbücher) zum Thema „Arzneimittelinteraktionen in der Therapie mit Antipsychotika“. Zur Veranschaulichung der Thematik und der klinischen Evidenz wurden als Beispiele für pharmakodynamische Interaktionen additive anticholinerge Nebenwirkungen, kardiovaskuläre Effekte (QT-Verlängerung), Exazerbation der Parkinson-Symptomatik (EPS) sowie die Gewichtszunahme und das metabolische Syndrom ausgewählt. Es werden die Arzneistoffe beschrieben, die in Kombination zu diesen klinischen Symptomen führen können. Weiters wird auf den induzierenden und inhibitorischen Effekt von Antipsychotika auf das Cytochrom P450 (CYP) eingegangen und auf die daraus resultierende Veränderung der Plasmaspiegel kombinativ eingenommener CYP-Substrate. Fazit: Wenn es der Therapiestandard erlaubt, sind Risikokombinationen von Arzneimitteln zu vermeiden. In Anbetracht der häufig vorkommenden Polypharmazie sowie der manchmal notwendigen klinischen Anwendung einer Kombination von Risikopräparaten sind die Veränderungen der Plasmaspiegel bzw. die Kontrolle dieser (Therapeutisches Drug Monitoring) der Schlüssel zur Vermeidung von möglicherweise schwerwiegenden Interaktionssymptomen. Diese Arbeit soll zur weiteren Sensibilisierung bezüglich der Problematik der Arzneimittelinteraktionen der Antipsychotika beitragen. ii Abstract Due to a quantitative increase in authorized medicinal drugs and their partially necessary application in combination therapy the problem with drug-drug interaction has become more and more evident in daily clinical practice. In Austria up to 7 % of all cases of hospitalization arise from undesirable side effects of medicinal drugs. In geriatrics the percentage is significantly higher (20 %) and the various possible interactions differ in terms of their pharmacodynamic and pharmacokinetic characteristics. This thesis covers current results of literature research (e.g. PUBMED, MEDLINE, OVID, medical textbooks) on the subject of drug-drug interaction in antipsychotics therapy. In order to demonstrate the issue and its clinical evidence additive anticholinergic side effects, cardiovascular effects (QT prolongation), exacerbation of Parkinson’s symptoms (EPS), weight gain and metabolic syndrome have been chosen as examples of pharmacodynamic interactions. Medicinal drugs which, when taken in combination, can cause these clinical symptoms will be described. Furthermore, the inductive and inhibitive effect of antipsychotic drugs on cytochromes P450 (CYP) as well as the resulting change in plasma levels of conjunctively used CYP substrates will be addressed. Conclusion: If standard treatment is available medicinal drug combinations which pose a risk should be avoided. Since polypharmacy is common clinical practice, high-risk drug combinations cannot always be avoided. The key to avoid possible serious symptoms of drug interaction is to detect the resulting changes in antipsychotic drug plasma levels or to perform an appropriate control of plasma concentrations ("therapeutic drug monitoring"). To summarize, this thesis should bring awareness to the issue of interaction of antipsychotic drugs. iii Inhaltsverzeichnis Eidesstattliche Erklärung Zusammenfassung Abstract Inhaltsverzeichnis Glossar und Abkürzungen Abbildungsverzeichnis Tabellenverzeichnis 1. Einleitung 1.1 Definition von Arzneimittelinteraktionen 1.2 Prinzipien der Pharmakokinetik 1.2.1 Resorption 1.2.2 Distribution 1.2.3 Metabolismus 1.2.4 Die Rolle der CYP-Enzyme 1.2.5 Elimination 1.2.6 Blutspiegel der Pharmaka 1.2.7 Therapeutisches Drug Monitoring 1.3 Prinzipien der Pharmakodynamik 1.3.1 Additive/synergistische/antagonistische Effekte an Rezeptoren 1.4 Vorstellung der Antipsychotikaklassen 1.4.1 Wirkungsmechanismen der Antipsychotika 1.4.2 Einteilung der Antipsychotika 1.4.2.1 Antipsychotische Wirkungsstärke („neuroleptische Potenz“) 1.4.2.2 Atypische Wirkkraft 1.4.2.3 Beispiele der Klassifizierung unter struktur-chemischen Gesichtspunkten 2. Material und Methoden 3. Ergebnisse – Resultate 3.1 Studien über pharmakodynamische Interaktionen 3.1.1 Additive anticholinerge Nebenwirkungen 3.1.2 Kardiovaskuläre Effekte 3.1.3 Exazerbation der Parkinson-Symptomatik sowie der extrapyramidalen Symptome (EPS) 3.1.4 Gewichtszunahme und das metabolische Syndrom 3.2. Studien über pharmakokinetische Interaktionen 4. Diskussion 4.1 Studien über pharmakodynamische Interaktionen 4.2 Studien über pharmakokinetische Interaktionen 5. Literaturverzeichnis i ii iii iv v vii viii 1 1 4 4 6 9 11 15 16 18 19 20 21 23 25 25 26 27 32 33 33 33 34 38 39 41 46 46 49 51 iv Glossar und Abkürzungen I II IKr % (A) ? + ++ +++ > 0 2 x tgl. 3 x tgl. 4 x tgl. 5-HT AAP Abb. ABC ABDA active moiety AGNP ATP BMC bzw. C Ca2+ cAMP case report CAVE chron. Cl KoMedikation Compliance Consensus CPZ CYP D d d. h. ED e.g. EKG EPS röm. für „Eins“ röm. für „Zwei“ Delayed Rectifier Prozent Konventionelle Antipsychotika mit ausgeprägten atypischen Eigenschaften keine Angaben geringgradig wirksam mittelgradig wirksam hochgradig wirksam mehr als keine Wirkung zweimal pro Woche dreimal pro Woche viermal pro Woche 5 Hydroxytryptamin (chemisches Kürzel: Serotonin) atypische Antipsychotika Abbildung ATP-binding cassette Bundesvereinigung Deutscher Apothekerverbände „aktiver Teil“ eines Arzneistoffes Consensus Guidelines for Therapeutic Drug Monitoring in Psychiatry Adenosintriphosphat BioMed Central beziehungsweise Kohlenstoff Kalzium (zweimal positiv geladen) zyklisches Adenosinmonophosphat Fallbericht lat. für „Warnung“ chronisch Chlor Zusatzmedikation Beschreibung der Einhaltung von Verhaltensmaßregeln, Gesetzen und Richtlinien durch PatientInnen lat. für „Übereinstimmung“ Chlorpromazin Cytochrom P (Pigment) Dopamin Tag das heißt Erhaltungsdosis zum Beispiel Elektrokardiogramm extrapyramidale Symptome et al. Evidenz F15063 F FDA FGA g ggf. H H hERG HP i.m. i.v. in vitro in vivo intrinsisch ion trapping KAP lat. L-Dopa LQTS M M. mACh MDR MEDLINE Meta-Analyse mg Minussymptomatik mL mol MP ms MTD N lat. für „und andere“ lat. für „das Herausscheinende“ Bezeichnung: AAP (in Entwicklung) Fluor Food and Drug Administration = Amerikanische Arzneimittelzulassungsbehörde first-generation antipsychotics Gramm gegebenenfalls Histamin Wasserstoff kardiale Kaliumkanäle hochpotent intramuskulär intravenös lat. für „im Glas“ bezeichnet man organische Vorgänge, die außerhalb eines lebenden Organismus stattfinden lat. für „im Lebendigen“ bezeichnet man in der Wissenschaft Prozesse, die im lebendigen Organismus ablaufen Aktivitätsbezeichnung: Intrinsische Aktivität: Maß für die Wirkstärke, die Zellfunktion zu ändern, die aus der Bindung eines Liganden an einen Rezeptor resultiert Bezeichnung der Anreicherung saurer Ionen in der Magenschleimhaut konventionelle Antipsychotika lateinisch Levodopa Long-QT-Syndrom Muskarin Morbus muskarinisches Acetylcholin Multi Drug Resistance Suchmaschine Zusammenfassung von Primäruntersuchungen zu Metadaten, die mit quantitativen, statistischen Mitteln arbeiten Milligramm Negativsymptomatik Milliliter Mol mittelpotent Millisekunden maximale Tagesdosis Stickstoff v ng NL NP O OH OVID p. o. PD P-gP pH PK PK Plussymptomatik Prodrug prospektiv PUBMED Nanogramm Neuroleptika niederpotent Sauerstoff Hydroxygruppe Suchmaschine per os pharmacodynamic P-Glykoprotein Pondus Hydrogenii = Maß für die Stärke der sauren bzw. basischen Wirkung einer wässrigen Lösung pharmacokinetic Negativer dekadischer Logarithmus der Dissoziationskonstante einer schwachen Säure; Säurekonstante Positivsymptomatik inaktiver oder wenig aktiver pharmakologischer Stoff, der erst durch Verstoffwechselung (Metabolisierung) im Organismus in einen aktiven Wirkstoff (Metaboliten) überführt wird Bezeichnung einer Studie, in der die Überprüfung der Hypothese der medizinischen oder psychologischen Wirksamkeit einer Behandlungsmethode geschieht, nachdem man zuvor die zu überprüfende Hypothese festgelegt hat. Dabei werden insbesondere die Daten gemäß der Hypothese erhoben Suchmaschine QT retrospektiv Review röm. S SGA SLV313 SPC SSR-181507 Steady-State t Tab. TD TdP tgl. Therapeutisches Drug Monitoring tmax U. S. UAW v2 x tgl. v x tgl. Wo x tgl. z. B. zN ZNS QT-Intervall (EKG-Parameter) Studienbezeichnung, in der man von der Gegenwart ausgehend die Vorgeschichte untersucht Bericht römisch Schwefel second-generation antipsychotics, novel antipsychotics Bezeichnung: AAP (in Entwicklung) summary of product characteristics Bezeichnung: AAP (in Entwicklung) Kumulationsgleichgewicht Zeit Tabelle Tagesdosis Torsade-de-pointes täglich Medikamentenüberwachung bzw. Medikamentenspiegelbestimmung Zeitmaximum United States unerwünschte Arzneimittelwirkungen verteilt auf zwei Einzeldosen verteilt auf mehrerer Einzeldosen Woche mehrmals täglich zum Beispiel zur Nacht (vor dem Schlafengehen) Zentralnervensystem vi Abbildungsverzeichnis Abb. 1: Formen der Interaktion 3 Abb. 2: Schematische Darstellung eines Plasmaspiegelverlaufes nach oraler Applikation 17 Abb. 3: Stoffbeschreibung Chlorpromazin 27 Abb. 4:Stoffbeschreibung Chlorprothixen 27 Abb. 5:Stoffbeschreibung Haloperidol 28 Abb. 6:Stoffbeschreibung Sulpirid 28 Abb. 7:Stoffbeschreibung Clozapin 29 Abb. 8:Stoffbeschreibung Olanzapin 29 Abb. 9:Stoffbescheibung Quetiapin 30 Abb. 10:Stoffbeschreibung Risperidon 30 Abb. 11: Stoffbeschreibung Aripiprazol 31 Abb. 12: Beispiele des erworbenen Long-QT-Syndroms (LQTS) 36 vii Tabellenverzeichnis Tab. 1: Neue Klassifikation der Interaktionen 2 Tab. 2: Einflussfaktoren der Resorption 5 Tab. 3: Einflussfaktoren der Distribution 6 Tab. 4: Induktoren und Inhibitoren von P-gP (ABCB1) 7 Tab. 5: Einteilung der enzymatischen Vorgänge in den Phasenreaktionen 9 Tab. 6: Die wichtigsten arzneistoffmetabolisierenden Cytochrom-P450-Enzyme (CYP) des Menschen und ihre relative Beteiligung bei der Verstoffwechselung der über CYP metabolisierten Arzneimittel in Prozent (%) 11 Tab. 7: Enzyme, die in den Abbau von Antipsychotika involviert sind 12 Tab. 8: Induktoren der CYP-Isoenzyme 13 Tab. 9: Inhibitoren der CYP-Isoenzyme, die pharmakokinetische Wechselwirkungen mit Antipsychotika hervorrufen können 14 Tab. 10: Empfohlene Plasmakonzentrationen von Psychopharmaka und literaturbasierte Empfehlungen zur Anwendung von therapeutischem Drug Monitoring 18 Tab. 11: Zielstrukturen, die durch Antipsychotika blockiert werden können und zu therapeutischer oder unerwünschter Wirkung führen 20 Tab. 12: Neuroleptika, Österreichische Arzneimittel im Taschenformat 22 Tab. 13: Rezeptorwirkungsprofile von Antipsychotika 23 Tab. 14: Dopaminerge Neuronensysteme im ZNS 24 Tab. 15: Unterteilung der Antipsychotika nach ihren „atypischen“ Eigenschaften 26 Tab. 16: Klinisch relevante Grenzwerte in Bezug auf das Auftreten von Arrhythmien 35 Tab. 17: Das MASTER-System nach Lemmer und Brune 50 viii 1. Einleitung 1.1 Definition von Arzneimittelinteraktionen In der pharmakologischen Therapie kommt es häufig zur gleichzeitigen Verordnung mehrerer Arzneimittel. Dadurch kann es zur gegenseitigen Beeinflussung der verabreichten Substanzen kommen. Das Erstpharmakon kann Einfluss auf das Zweitpharmakon nehmen oder umgekehrt, wodurch es zu einer Wirkungsverstärkung, Wirkungsabschwächung, Wirkungsverlängerung oder Wirkungsverkürzung kommen kann [1][2]. Die Ursachen der Wechselwirkungen (Interaktionen) liegen in den pharmakokinetischen und pharmakodynamischen Abläufen. Arzneimittelinteraktionen können in der pharmakologischen Therapie zu schwerwiegenden Zwischenfällen und unerwünschten Wirkungen bei PatientInnen führen. Durch erhobene Daten von Rivkin [3] konnte gezeigt werden, dass in Deutschland Interaktionen als unerwünschte Arzneimittelwirkungen (UAW) mitverantwortlich sind für ein Viertel der Hospitalisationen, die aufgrund von UAW notwendig werden, und sogar für die Hälfte der Intensivaufnahmen aufgrund von UAW. Umgekehrt konnte von Bertsche et al. [4] erhoben werden, dass die Vermeidung von Wechselwirkungen die UAW-Rate auf einer Intensivstation praktisch halbieren kann. Es wird angenommen, dass in Österreich bis zu 7 % aller Krankenhauseinweisungen aufgrund unerwünschter Arzneimittelnebenwirkungen erfolgen. In Bezug auf ältere PatientInnen ist anzumerken, dass im geriatrischen Bereich der Anteil mit 20 % wesentlich höher liegt. Die Wahrscheinlichkeit einer Entwicklung unerwünschter Arzneimittelinteraktionen und in der Folge einer Entwicklung von Arzneimittelnebenwirkungen steigt exponentiell mit der Anzahl der eingenommenen Wirkstoffe. Insbesondere bei geriatrischen PatientInnen besteht ein erhöhtes Risiko für Arzneimittelnebenwirkungen aufgrund altersassoziierter Veränderungen. Aus diesem Grund ist die Beachtung und Überprüfung der medikamentösen Therapie hinsichtlich möglicher pharmakologischer Interaktionen in der klinischen Praxis von großer Bedeutung. Im Gegensatz zu UAW sind erwünschte Interaktionen oft Voraussetzung für eine sinnvolle Therapie (z. B. Gabe von Antidoten bei Vergiftungen). Allein durch den Ausdruck 1 „Wechselwirkung“ können demnach keine Rückschlüsse auf die positive oder negative Wirkung von Interaktionen gezogen werden [5][1]. Angelehnt an Vorschläge, die von Hansten et al. [6] publiziert wurden, konnte eine Klassifikation der Interaktionen ins Leben gerufen werden, die sie nach den jeweils erforderlichen Maßnahmen einstuft. Die neue Klassifikation basiert auf dem jeweiligen Stand der Erkenntnis und bezieht Unsicherheiten bezüglich der tatsächlichen Bedeutung einer Wechselwirkung mit ein. Demnach werden folgende Einstufungen verwendet [7]: Tab. 1: Neue Klassifikation der Interaktionen Relevanz Neue Klassifikation 1. Schwerwiegende Folgen wahrscheinlich – kontraindiziert 2. Vorsichtshalber kontraindiziert 3. Überwachung bzw. Anpassung nötig 4. In bestimmten Fällen Überwachung bzw. Anpassung nötig Bei Risikofaktor: Überwachung/Anpassung 5. Vorsichtshalber überwachen 6. In der Regel keine Maßnahmen erforderlich. Erläuterungen Die beiden Arzneimittel dürfen nicht gleichzeitig angewandt werden, weil schwerwiegende Folgen dokumentiert sind. Die beiden Arzneimittel dürfen nicht gleichzeitig angewandt werden, weil schwerwiegende Folgen auf theoretischer Grundlage angenommen werden müssen. Hier sind in jedem Fall Maßnahmen erforderlich: Alternativarzneimittel, zeitliche Trennung der Einnahme, Dosisanpassung, Dosisbegrenzung, Überwachung auf unerwünschte Wirkungen Hier sind Maßnahmen erforderlich, wenn bestimmte Umstände vorliegen: z. B. Risikofaktoren, hohe Dosierung, bestimmte Reihenfolge der Anwendung, länger dauernde Therapie Die Interaktion ist theoretisch möglich, aber bislang nicht dokumentiert oder tritt nur in Einzelfällen auf, ohne dass Risikofaktoren bekannt sind, oder führt nur zu etwas verstärkten Nebenwirkungen. Adaptiert nach ABDATA Pharma-Daten-Service 2009 [8] Die Wahrscheinlichkeit einer Entwicklung unerwünschter Arzneimittelinteraktionen und in der Folge einer Entwicklung von Arzneimittelnebenwirkungen steigt exponentiell mit der Anzahl der eingenommenen Wirkstoffe. 2 In der psychopharmakologischen Therapie wird Leitlinien zufolge grundsätzlich eine medikamentöse Monotherapie zur effektiven Behandlung von psychiatrischen oder distinktiven Symptomen empfohlen. Jedoch wird dieser Empfehlung oftmals in der Praxis nicht entsprochen, wodurch es in der Regel zu einer Polypharmazie kommt. In einer vergleichenden Untersuchung von Daten unter anderem aus bayerischen Bezirkskrankenhäusern wurde von Katzendobler [9] ermittelt, dass psychiatrische PatientInnen, die eine stationäre Behandlung an Versorgungskrankenhäusern in Anspruch genommen hatten, in weniger als 30 % der Fälle eine psychopharmakologische Monotherapie erhielten. Zwei Drittel der PatientInnen wurden zwei bis vier Psychopharmaka verabreicht. In einer ähnlichen Analyse, die von Hausner et al. [10] durchgeführt wurde, erhob man, dass PatientInnen, die das Krankheitsbild der Schizophrenie aufwiesen, im Mittel mit drei und Depressionskranke mit vier Psychopharmaka behandelt wurden, wobei bei Männern die Anzahl der psychopharmakologischen Substanzen niedriger war als bei Frauen. Daraus kann man schließen, dass es bei den häufig vorkommenden Kombinationstherapien zur Beeinflussung der Wirkung der Medikamente untereinander kommt. Angesichts der großen Anzahl an Wirkstoffen sind deshalb Wechselwirkungen ein klinisch relevantes Problem [5][11]. Abb. 1: Formen der Interaktion Bei der Entscheidung, ob ein Arzneistoff zum Einsatz kommt, müssen die quantitativen Aspekte (pharmakokinetischen Eigenschaften) und die qualitativen Anforderungen (pharmakodynamischen Eigenschaften) einer Substanz bestimmt und berücksichtigt werden [11]. Ekins et al. 2007 [12] 3 1.2 Prinzipien der Pharmakokinetik Die Pharmakokinetik befasst sich mit der Frage, wie die Substanz in richtiger Konzentration und für ausreichend lange Zeit an den Wirkort (im Falle der Psychopharmaka in das zentrale Nervensystem – ZNS) gebracht werden kann, da zu hohe Konzentrationen zu einer unerwünschten Wirkungsentfaltung führen und zu niedrige Konzentrationen für eine therapeutische Wirkung nicht ausreichen können. Aufgrund des permanenten Stoff- und Energieaustausches des Organismus mit seiner Umwelt ist dieser als offenes oder Fließsystem anzusehen. Eine ausgeglichene Bilanz von Zu- und Ausfuhr wird als Fließgleichgewicht beschrieben [11][1]. Werden Pharmaka in dieses System eingebracht, folgen sie den anderen Biomolekülen in die verschiedenen Kompartimenten des Organismus. Dabei wird die Pharmakokinetik als die zeitliche Veränderung der Konzentration eines Arzneistoffes in verschiedenen Teilen des Organismus nach Applikation der jeweiligen Substanz verstanden. Zu den Teilprozessen der Pharmakokinetik gehören die Resorption (Aufnahme des Arzneistoffes), die Distribution oder Verteilung (Stofftransport vom Blut in die Gewebe/Kompartimente) und die Elimination der Arzneistoffe (Biotransformation, Metabolismus, Exkretion), die zu einer Konzentrationsabnahme im Organismus führt [11][1]. 1.2.1 Resorption Unter Resorption versteht man einen organischen Prozess, bei dem Substanzen bzw. Arzneistoffe über die Körperoberfläche oder von örtlich begrenzten Stellen im Körperinneren in die Blutbahn oder in das Lymphsystem aufgenommen werden, worauf die Verteilung (Distribution) im Gesamtorganismus einsetzt. Dies geschieht in Bezug auf Arzneistoffe hauptsächlich rein passiv durch Diffusion. Voraussetzung ist hierbei das Bestehen eines Konzentrationsgradienten [1]. Weitere Möglichkeiten des Transports umfassen die erleichterte Diffusion über nicht aktiven Transport (Carrier), aktiven Transport unter Verbrauch von Energie, Endozytose sowie die Diffusion oder Filtration über interzelluläre Spalten. Die Geschwindigkeit der Resorption und die Resorptionsquote (Verhältnis von resorbierter zu applizierter Menge) der Arzneistoffe hängen von mehreren Faktoren ab [11][1]: 4 Tab. 2: Einflussfaktoren der Resorption Einflussfaktoren Physikalische und chemische Eigenschaften (vor allem die Löslichkeit) Teilchengröße und spezifische Oberfläche Arzneiform und Hilfsstoffe Dosierung Applikationsart und Applikationsort Kontaktzeit mit Resorptionsfläche Größe der resorbierenden Fläche pH-Wert im Bereich der resorbierenden Fläche Integrität der Membranen Durchblutung des resorbierenden Organs Adaptiert nach Mutschler 2005 [1] Interaktionen der Resorption können durch eine Veränderung des Magen- oder Darm-pH entstehen. Dies kann beispielsweise durch die Gabe eines Erstpharmakons ausgelöst werden. Wird gleichzeitig mit einem Antazidum (Erstpharmakon) ein saurer oder basischer Arzneistoff verabreicht, kann es aufgrund der Erhöhung des pH-Werts im oberen Teil des Magen-Darm-Kanals zu einer Veränderung der Resorptionsquote des Wirkstoffs kommen [1]. Dieser Umstand ist für die Arzneimittelgruppe der Antipsychotika von Bedeutung, da diese meist eine basische funktionelle Gruppe, die geladen oder ungeladen sein kann, besitzen. Der saure pH-Wert im Magen verzögert bei den meisten basischen Psychopharmaka die Resorption. Der Ionisierungsgrad eines Arzneistoffes ist für die Durchwanderung der Lipidmembran von Bedeutung, da für den Übertritt der nicht ionisierte Zustand bevorzugt wird. Basische Arzneistoffe werden in ungeladener Form in die Zelle aufgenommen, jedoch bei intrazellulärem saurem pH-Wert protoniert. Durch die geladene Form des Medikamentes wird dessen Transport nach außen erschwert. Dieser Prozess wird als „ion trapping“ bezeichnet. Weitere Beeinflussung der Resorptionsquote eines Zweitpharmakons ist durch Verlängerung oder Verkürzung der Verweildauer im Magen-Darm-Kanal oder Komplexbildung möglich [11][1]. Antipsychotika werden nach oraler Applikation (parenterale Applikation bedingt in der Regel eine Erhöhung der Bioverfügbarkeit um das 4- bis 10-Fache) gut resorbiert. Ausnahme bilden Asenapin (Bioverfügbarkeit unter 2 %) und die substituierten Benzamide Sulpirid und Amisulprid, deren Bioverfügbarkeiten unter 50 % liegen. Weiters wird die Resorption von Ziprasidon durch gleichzeitige Nahrungsaufnahme nahezu verdoppelt und die Bioverfügbarkeit von Asenapin durch rasch folgende Flüssigkeitszufuhr deutlich reduziert [13]. 5 1.2.2 Distribution Unter Distribution versteht man den reversiblen Stofftransport von einem Teil des Organismus in einen anderen. Das Verteilungsgleichgewicht beschreibt dabei den Zustand konstanter Konzentrationsverhältnisse in den verschiedenen Teilen des Körpers. Aufgrund des Konzentrationsgefälles zwischen Blut und Gewebe kommt es zum Übertritt aus der Blutbahn ins Gewebe und zur Verteilung in verschiedene Kompartimente (Intrazellulärraum/Extrazellulärraum) des Gesamtorganismus. Die Distribution wird dabei durch die Eigenschaften des Organismus und des Arzneistoffes beeinflusst (Tab. 3) [1]. Tab. 3: Einflussfaktoren der Distribution Einflussfaktoren Eigenschaften des Organismus Durchblutung der Organe und Gewebe, Durchlässigkeit der Membranen, pH-Differenz von Plasma und Gewebe Molekülgröße, Bindung an Plasma-, Gewebe- und Eigenschaften des Arzneistoffes Erythrozytenproteine, Löslichkeit, chemische Eigenschaften Adaptiert nach Mutschler 2005; Riederer und Laux 2010 [1][11] In Abhängigkeit der Arzneistoffe von ihren physikochemischen Eigenschaften kann eine Gliederung hinsichtlich ihrer Verteilung in verschiedene Kompartimente vorgenommen werden. Pharmaka können demnach limitiert auf den Plasmaraum, Plasma und Extrazellulärraum oder Extra- und Intrazellulärraum verteilt werden. Makromolekulare (intravasal applizierte) Substanzen können den Plasmaraum nicht verlassen. Die Distribution der anderen Arzneistoffe ist abhängig vom Kapillaraufbau. Ein leichter Stoffaustausch erfolgt, wenn das Kapillarendothel des Gewebes größere und kleinere Poren (z. B. Leber, Pankreas) aufweist oder eine Basalmembran fehlt (z. B. Leber). Bei lückenlosen Endothelien und bei Vorhandensein einer Basalmembran sowie zusätzlichen Zellen auf den Kapillaren ist der Stoffaustausch erschwert [1]. Beispielsweise sind Hirnkapillaren dicht von Gliazellen überzogen und im Bereich der Plexus chorioidei (Bildung des Liquor cerebrospinalis) sind die zuführenden Kapillaren zum Liquorraum mit einem einschichtigen Epithel überzogen. Die Folge ist ein Permeationshindernis, welches auch als Blut-Hirn- bzw. Blut-Liquor-Schranke bezeichnet wird [1][11]. CAVE: Eine generelle Zunahme der Permeabilität ist bei entzündlichen Prozessen zu beobachten, wodurch auch Stoffe über die Blut-Hirn-Schranke diffundieren können, die unter physiologischen Bedingungen nicht in das ZNS eindringen könnten [1]. 6 Lipophile Substanzen können die Schranke gut überwinden, lipophobe Substanzen schlechter. Sie sind auf aktive Transportmechanismen (z. B. Aminosäuren) angewiesen. Andererseits können sogenannte ABC-Transporter (ABC = ATP-binding cassette) einen aktiven Transport von Pharmaka nach außen möglich machen und so zum Schutz von Organen bzw. des Organismus vor Fremdstoffen beitragen. Durch Hydrolyse des an die Transporter gebundenen ATP wird die nötige Energie für den aktiven Transport bereitgestellt. Ein wichtiger Vertreter dieser Membranproteine (ABC-Transporter) ist das P-Glykoprotein (P-gP), welches Substanzen aus der Zelle befördert und damit an Distribution und Elimination von Xenobiotika (Fremdstoffe) beteiligt ist. P-gP ist das Produkt des „Multi Drug Resistance (MDR1)“-Gens [1][11]. In Bezug auf Distribution und Elimination sind solche Arzneimitteltransporter neben dem Schutz des ZNS über die Blut-Hirn- bzw. Blut-Liquor-Schranke auch an der Schutzfunktion der Plazenta-Schranke beteiligt und können Interaktionen zwischen Pharmaka beeinflussen. Sie verfügen über spezifische Bindungsstellen, an denen es aufgrund von Affinität und Konzentrationsunterschieden von Arzneistoffen zu einer Verdrängungsreaktion unter den Substanzen kommen kann. Durch diese Konkurrenz wird bestimmt, welches Substrat transportiert und welches diesseits der Membran akkumuliert wird [2]. Hinsichtlich der Arzneimittelgruppe der Antipsychotika ist das P-gP von Bedeutung, da mehrere Antipsychotika Substrate von P-gP sind. Bei diesen Substraten handelt es sich beispielsweise um Risperidon und seinen Metaboliten 9-Hydroxy-Risperidon (identisch mit Paliperidon). Weitere Substrate von P-gP umfassen Amisulprid, Aripiprazol, Olanzapin und Perospiron. Clozapin und Quetiapin sind keine geeigneten Substrate von P-gP. In einer Untersuchung von Spina et al. [14] kam man zu dem Ergebnis, dass durch die Induktion von P-gP die Bioverfügbarkeit von ABCB1-Substraten herabgesetzt wird [11][15]. Tab. 4: Induktoren und Inhibitoren von P-gP (ABCB1) ABCB1-Induktoren ABCB1-Inhibitoren Carbamazepin Amiodaron Nelfinavir Johanniskraut Chinidin Ritonavir Rifampicin Ciclosporin Saquinavir Clarithromycin Spironolacton Erythromycin Tacrolimus Itraconazol Verapamil Ketoconazol Adaptiert nach Hafner et al. 2010 [2] 7 Im Gegensatz zur Induktion führt eine Inhibition von ABCB1 zu einer Erhöhung der Plasmakonzentration, einer Wirkungsverlängerung und Wirkungsverstärkung von ABCB1Substraten. Durch diesen Prozess können vor allem Pharmaka mit geringer therapeutischer Breite eine toxische Konzentration erreichen [2]. Neben den bereits erwähnten Beeinflussungsfaktoren ist die mögliche Eiweißbindung eines Stoffes relevant. Sie ist grundsätzlich reversibel und außer von den stofflichen Eigenschaften von pH-Wert des Plasmas und Lebensalter der PatientInnen abhängig. Beispielsweise nimmt bei einer Azidose der eiweißgebundene Anteil einiger Pharmaka ab und bei Neugeborenen ist die Proteinbindung geringer als bei Erwachsenen. Der Einfluss auf die Wirkstärke, -dauer und Elimination ergibt sich aus dem plasmaproteingebundenen Anteil eines Pharmakons, welcher nicht diffundieren kann und nicht der Biotransformation und Ausscheidung unterliegt. Dieser Teil stellt eine Speicherform dar, aus welcher bei einer Konzentrationserniedrigung der freien Form (keine Eiweißbindung) Pharmakonmoleküle zur Wiederherstellung des Gleichgewichts freigesetzt werden können [1]. Interaktionen der Verteilung bestehen, wenn mehrere Pharmaka im Blut um die Bindungsstellen der Plasmaeiweiße konkurrieren. Dieser Vorgang wird allerdings nur dann relevant, wenn Arzneistoffe mit hoher Proteinbindung, verhältnismäßig kleinem Verteilungsvolumen und geringer therapeutischer Breite betroffen sind. Diese Interaktion tritt auf, wenn einer der Arzneistoffe im oberen Milligramm- oder Grammbereich dosiert wird. Durch hochwirksame Arzneistoffe, von denen für einen therapeutischen Effekt nur geringe Dosierungen notwendig sind, wird dieser Typ der Interaktion vermieden, da ausreichend Bindungsstellen zur Verfügung stehen, auch wenn zwei gleichzeitig gegebene Wirkstoffe um identische Bindungsstellen konkurrieren [1]. Antipsychotika sind mit Ausnahme von substituierten Bezamiden äußerst lipophile Substanzen und daher im Plasma zu mehr als 90 % an Proteine gebunden. Die weniger lipophilen Benzamide weisen nur eine 20%ige Proteinbindung auf. Da nur der nicht an Plasmaprotein gebundene Anteil der Arzneistoffe für die Passage über die Blut-HirnSchranke zur Verfügung steht, können Antipsychotika mit Ausnahme der Benzamide die Blut-Hirn-Schranke gut überwinden und sich im ZNS anreichern [13]. 8 1.2.3 Metabolismus Für die renale Ausscheidung lipophiler Substanzen benötigt der Organismus Enzyme, die lipophile Fremdstoffe (Xenobiotika) in hydrophilere, leichter ausscheidbare Stoffe umwandeln. Je schneller dabei der Organismus fettlösliche Stoffe zu wasserlöslichen Verbindungen metabolisieren kann, desto höher ist die Eliminationsgeschwindigkeit lipophiler Substanzen [1]. Durch den Umbau zu hydrophileren Substanzen wird verhindert, dass Xenobiotika nach der glomerulären Filtration sogleich wieder aus den Nierentubuli rückresorbiert werden und sich im Fettgewebe anreichern. Dieser Vorgang nennt sich Biotransformation und findet vorwiegend in der Leber (hepatischer Metabolismus), aber auch in Darm, Niere, Lunge, Milz, Muskulatur, Haut und Blut statt. Die benötigten Enzyme der Biotransformation umfassen beispielsweise die Monooxygenase und Glukuronyltransferase, welche hauptsächlich in der Membran des endoplasmatischen Retikulums zu finden sind. Sie können Substrate unterschiedlicher chemischer Struktur umsetzen. Die Einteilung der enzymatischen Vorgänge erfolgt nach den durchgeführten Strukturveränderungen. Man spricht hierbei von Phase-Ι- und Phase-ΙΙ-Reaktion [1][11]. Tab. 5: Einteilung der enzymatischen Vorgänge in den Phasenreaktionen Reaktion Biomechanismen Phase-Ι-Reaktion Oxidative, reduktive und hydrolytische Veränderungen der Pharmakonmoleküle Phase-ΙΙ-Reaktion Konjugation eines Arzneistoffmoleküls bzw. eines in Phase Ι entstandenen Metaboliten an körpereigene Substanzen (z. B. aktivierte Glukuronsäure, Glyzin, SAdenosylmethionin) Adaptiert nach Riederer und Laux 2010 [11] Eine Konjugation in Phase ΙΙ kann von vorangehenden Phase-I-Reaktionen abhängig sein (Tab. 5). In der Phase-Ι-Reaktion ist neben Dioxygenasen, welche beide Atome des Sauerstoffs in das Xenobiotikum einführen, die mikrosomale Monooxygenase die bedeutendste Oxygenase für die oxidative Biotransformation von pharmakologischen Substanzen. Durch sie wird ein Sauerstoffatom von einem Sauerstoffmolekül getrennt und in das Xenobiotikum eingebaut, während das andere Sauerstoffmolekül zu Wasser reduziert wird. Reduktive Prozesse spielen während der Biotransformation eine untergeordnete Rolle [11]. 9 Das Einbauen bzw. Freilegen von funktionellen Gruppen ist Voraussetzung für die PhaseΙΙ-Reaktion und wird als Funktionalisierungsreaktion bezeichnet [16]. Die Phase-II-Reaktionen sind Konjugationsreaktionen, die durch Beteiligung spezifischer Transferasen ablaufen. Hierbei kommt es zu einer Koppelung von funktionellen Gruppen an z. B. sehr polare, negativ geladene endogene Moleküle. Die wichtigsten Phase-IIReaktionen umfassen die Glukuronidierung, Sulfatierung, Methylierung, Acetylierung und die Konjugation mit Aminosäuren und Glutathion. Die Einführung der funktionellen Gruppen unterliegt den Phase-I-Reaktionen und kann Voraussetzung sein für die Umwandlung von Arzneistoffen in Substrate für Phase-II-Reaktionen. Besitzt ein Arzneistoff bereits für eine Konjugation geeignete funktionelle Gruppen, kann eine direkte Konjugation ohne vorgeschaltete Phase-I-Reaktion stattfinden. Die Phase-II-Metaboliten sind meist unwirksam. Die entstandenen Konjugate sind sehr polar und somit auch gut wasserlöslich, wodurch eine schnelle renale und biliäre Ausscheidung möglich ist [16]. Diese Abfolge von Reaktionen ist bedeutend für die Elimination lipophiler Substanzen und stellt durch die Inaktivierung bzw. Wirkungsabschwächung von Metaboliten ein Entgiftungs- und Inaktivierungssystem dar. Phase-I-Metaboliten können selbst pharmakologisch wirksam sein oder der Metabolit stellt das eigentliche Wirkprinzip dar. Die Ausgangssubstanz wird hierbei als „Prodrug“ bezeichnet [16]. Grundsätzlich müssen alle durch Magen- oder Dünndarmschleimhaut resorbierte Pharmaka, bevor sie in den Herz-, Lungen- und Körperkreislauf gelangen können, die Leber passieren. Für die Wirksamkeit eines Pharmakons ist es von entscheidender Bedeutung, in welchem Ausmaß es zur Extraktion sowie biochemischen Umwandlung in der Leber und zur Metabolisierung im Magen-Darm-Kanal kommt. Dieser Vorgang wird als First-pass-Effekt bezeichnet [1]. Ein ausgeprägter First-pass-Metabolismus ist der wichtigste Grund für eine geringe orale Bioverfügbarkeit. Er erklärt, dass eine Substanz trotz 100%iger Resorption (Antipsychotika werden gut resorbiert [13]) nur eine orale Bioverfügbarkeit von wenigen Prozent aufweisen kann. Antipsychotika weisen einen besonders ausgeprägten First-passEffekt auf, wodurch eine geringe orale Bioverfügbarkeit bedingt sein kann. Zum Ausgleich kann hierbei die Dosis erhöht werden. Allerdings ist die Bioverfügbarkeit von interindividuellen oder auch alters- bzw. krankheitsbedingten Veränderungen der hepatischen Elimination beeinflusst, weshalb es zu einer starken Varianz der Plasmaspiegel bei Substanzen mit geringer Bioverfügbarkeit kommen kann [11]. 10 1.2.4 Die Rolle der CYP-Enzyme In Bezug auf Arzneimittelinteraktionen sind die von der metabolischen Interaktion betroffenen Enzyme zu besprechen. Die mikrosomale Monooxygenase, welche die Hämproteine Cytochrom P450 (CYP) enthält, zeichnet für die oxidativen Prozesse verantwortlich. Die Aufgabe dieser sogenannten CYP-Enzyme besteht darin, molekularen Sauerstoff in das Zielmolekül einzuführen. Aufgrund der unterschiedlichen Arzneimittelmetabolisierung ist hierbei die Einteilung der Isoenzyme in Klassen zu besprechen, basierend auf Homologien der Aminosäuresequenzen in Familien und Subfamilien. CYP-Enzyme der mikrosomalen Membranen sind verantwortlich für die Metabolisierung von Arzneimitteln, während die Klasse der CYP-Enzyme der inneren Mitochondrienmembran Einfluss auf die Steroidsynthese hat [11]. Tab. 6: Die wichtigsten arzneistoffmetabolisierenden Cytochrom-P450-Enzyme (CYP) des Menschen und ihre relative Beteiligung bei der Verstoffwechselung der über CYP metabolisierten Arzneimittel in Prozent (%) CYP-Enzyme CYP3A4 CYP2D6 CYP2C9/2C19 CYP1A2 Prozentanteil (%) 50 25 20 5 Adaptiert nach Hafner et al. 2010 [2] Der Hauptanteil der Expression von CYP-Enzymen findet zu 90–95 % in der Leber statt. Das am stärksten exprimierte Isoenzym ist das CYP3A4 (Tab. 6). Abgesehen von der Exprimiertheit des CYP3A4 ist auch seine Funktionalität bezüglich Interaktionen hervorzuheben, da durch diese CYP-Familie fast jedes 2. Arzneimittel abgebaut wird [11][2]. Die Expression von Isoenzymen kann stark variieren und ist von Genotyp, Alter, Lebensgewohnheiten (hohe CYP1A2-Aktivität bei Rauchern), Erkrankungen, Medikation und anderen Faktoren abhängig. Die breite und überlappende Substratspezifität der CYPEnzyme und die Konzentration des zu verstoffwechselnden Arzneistoffes haben Einfluss auf die Rolle der CYP-Enzyme. Aufgrund der Substratspezifität werden die meisten Psychopharmaka durch mehrere Isoenzyme abgebaut, allerdings gibt es auch Arzneimittel; die nur durch ein Isoenzym abgebaut werden [11]. 11 Tab. 7: Enzyme, die in den Abbau von Antipsychotika involviert sind Substanz (aktiver Enzyme Substanz (aktiver Metabolit) Metabolit) Amisulprid Mehr als 90 % werden nicht metabolisiert über die Niere ausgeschieden Olanzapin Aripiprazol (Dehydroaripiprazol) CYP2D6, CYP3A4 Paliperidon (= 9-HydroxyRisperidon) Asenapin N-Glucuronosyltransferase und CYP1A2 Perazin Benperidol Unklar Perphenazin Bromperidol Chlorpromazin Chlorprothixen Clozapin CYP3A4 CYP1A2, CYP2D6 CYP2D6 CYP1A2, CYP2C19, CYP3A4 CYP2D6 CYP2D6 Unklar Pimozid Pipamperon Prothipendyl Quetiapin Flupentixol Fluphenazin Fluspirilen Enzyme N-Glucuronosyltransferase, Flavinmonooxygenase, CYP1A2, CYP2D6 60 % werden nicht metabolisiert auf unterschiedliche Arten ausgeschieden CYP1A2, CYP2C19, CYP3A4, Flavinmonooxygenase, CYP1A2, CYP2C19, CYP2D6, CYP3A4 CYP1A2, CYP3A4 Unklar Unklar CYP3A4 Risperidon Sertindol Sulpirid CYP2D6, CYP3A4 CYP2D6, CYP3A4 Wird nicht metabolisiert über die Niere ausgeschieden Haloperidol CYP2D6, CYP3A4 Thioridazin CYP1A2, CYP2C19, CYP2D6, CYP3A4 Iloperidon CYP2D6, CYP3A4 Ziprasidon CYP3A4, Aldehydoxidase Levomepromazin CYP1A2, CYP2D6 Zotepin CYP1A2, CYP2D6, CYP3A4 Melperone Unklar Zuclopenthixol CYP2D6 Die wichtigsten Enzyme sind in Kursivschrift angegeben. Die Inhibition dieser Enzyme kann die Plasmakonzentration von Antipsychotika signifikant erhöhen; Induktion von CYP1A2 der CYP3A4 senkt die Plasmakonzentration. Adaptiert nach Hiemke et al. 2011 [17] Viele vor allem lipophile Xenobiotika mit langer Verweildauer in der Leber können eine vermehrte Enzymbildung in der Leber induzieren. Sie werden als Induktoren bezeichnet und unter anderen in einen Phenobarbital-, Methylcholanthren- und Rifampicin-Typ gruppiert. Die Enzyminduktion erfolgt durch Interaktion mit intrazellulären Rezeptoren [1]. 12 Tab. 8: Induktoren der CYP-Isoenzyme CYP Induzierende Substanz 1A2 Carbamazepin, Rifampicin Ernährung, pflanzliche Arzneistoffe und Lebensstil Zigarettenrauch, mit Holzkohle gegrilltes Essen, Brokkoli 2C19 Carbamazepin, Felbamat, Modafinil, Topiramat, Rifampicin, Johanniskraut, Phenytoin 2D6 Unbekannt Nicht ableitbar 3A4 Carbamazepin, Efavirenz, Dexamethason, Lovastatin, Oxybutynin, Rifabutin, Rifampicin, Johanniskraut (Hyperforin), Phenobarbital, Phenytoin, Primidon Für die in der Tab. aufgelisteten Inhibitoren bestehen Warnungen durch das SPC, in-vivo-Studien und Fallberichte, die zeigen, dass klinisch relevante Arzneimittelinteraktionen in Kombination mit Antipsychotika auftreten können. Adaptiert nach Hiemke et al. 2011; Spina et al. 2003; Pang et al. 2011; Sinz et al. 2008 [17][18][19][20] Durch die vermehrte Enzymbildung kommt es zu einem verstärkten Abbau und zu einer erhöhten Biotransformationsrate. Davon betroffen sind allerdings nicht nur die Enzyminduktoren, sondern auch alle anderen Arzneistoffe, die durch die entsprechenden Enzyme wie den Induktor abgebaut werden. Es kommt zu einer metabolischen Interaktion. Die Plasmahalbwertszeit der betroffenen Verbindungen wird verkürzt. Wird der Induktor abgesetzt, fällt die Abbaukapazität wieder. Dieser Effekt sowie die Stoffeigenschaften der Arzneistoffe sind für die medikamentöse Therapie wichtig. Sind die Abbauprodukte des Pharmakons aktiver als die Ausgangssubstanz, nimmt die Wirkung zu. Ist die Ausgangssubstanz aktiver, ist die Wirkung vermindert. Weiters klinisch relevant ist die Abnahme der Blutkonzentration eines Zweitpharmakons bei Gabe eines Enzyminduktors [1][11]. CAVE: Wird als Ausgleich die Dosis des Zweitpharmakons erhöht, kann bei Absetzung des Enzyminduktors ein eventuell gefährlicher Anstieg der Blutkonzentration des Zweitpharmakons die Folge sein – in dieser Situation darf man nicht vergessen, die Dosis wieder zu reduzieren [1]. Pharmaka können einerseits eine Enzyminduktion bewirken, andererseits können zahlreiche Arzneimittel die Biotransformationsprozesse hemmen und damit eine Wirkungsverlängerung und -steigerung anderer Substanzen hervorrufen. Die Enzyminhibition kann in der Weise erfolgen, dass ein Arzneimittel zu einer verminderten Synthese oder zu einem verstärkten Abbau von Enzymen des endoplasmatischen Retikulums führt oder dass es zwischen zwei oder mehreren Pharmaka zu einer 13 Konkurrenz um die Bindungsstelle der Enzyme und damit zu einer kompetitiven Hemmung kommt [1]. Tab. 9: Inhibitoren der CYP-Isoenzyme, die pharmakokinetische Wechselwirkungen mit Antipsychotika hervorrufen können CYP Inhibierende Substanz Ernährung, pflanzliche Arzneistoffe und Lebensstil 1A2 Cimetidin, Ciprofloxacin, Infektionen Enoxacin, Fluvoxamin, Norfloxacin, Perazin, Propafenon 2C19 Felbamat, Fluconazol, Fluvoxamin, Moclobemid, Omeprazol, Perazin 2D6 Bupropion, Cimetidin, Moclobemid, Norfluoxetin, Paroxetin, Pergolid, Amiodaron, Fluoxetin, Perphenazin, Propafenon, Propanolol, Quinidin, Levomepromazin, Melperon, Metoclopramid, Ritonavir, Ropinirol, Thioridazin 3A4 Bromocriptin, Cimetidin, Grapefruitsaft Cisaprid, Clarithromycin, Diltiazem, Erythromycin, Miconazol, Indinavir, Itraconazol, Ketoconazol, Nelfinavir, Ritonavir, Verapamil, Voriconazol, Mifepriston, Norfluoxetin, Saquinavir, Simvastatin, Troleandomycin Für die in der Tab. aufgelisteten Inhibitoren bestehen Warnungen durch das SPC, in-vivo-Studien und Fallberichte, die zeigen, dass klinisch relevante Arzneimittelinteraktionen in Kombination mit Antipsychotika auftreten können. Adaptiert nach Hiemke et al. 2011; Spina et al. 2003; Lynch und Price 2007 [17][18][21] Antipsychotika werden großteils extensiv hepatisch metabolisiert und durch das bereits erwähnte hepatische CYP-Enzym umgesetzt. Ausnahmen bilden nur die Benzamide Amisulprid und Sulpirid, welche zu 90 % unverändert renal ausgeschieden werden. Eine besondere Bedeutung bei der Metabolisierung antipsychotischer Substanzen wird den Isoenzymen CYP1A2, CYP2D6 und CYP3A4 zugesprochen. In Bezug auf die Arzneimittelgruppe der Antipsychotika sind Oxidationsprozesse wie die Hydroxilierung, die Demethylierung und andere von Bedeutung, wobei die Metaboliten weiter umgewandelt werden können oder teilweise auch bereits ausgeschieden werden. Größtenteils sind die Metaboliten pharmakologisch inaktiv. Die Ausnahme bilden einige pharmakologisch aktive Metaboliten, die zur biologischen und klinischen Wirkung beitragen. Zu diesen aktiven Metaboliten zählen Mesoridazin (Muttersubstanz Thioridazin), der Hydroxymetabolit von Haloperidol, 9-Hydroxy-Risperidon (Risperidon), 14 Dehydroaripiprazol (Aripiprazol) und mehrere N-demethylierte Metaboliten verschiedener Phenothiazine. Die Metabolisierung von trizyklischen Antipsychotika ist besonders umfangreich. Es kommt dabei zur Bildung von mehr als 100 verschiedenen Stoffwechselprodukten. Die Muttersubstanz von Haloperidol hat eine bedeutend höhere dopaminerge Aktivität als reduziertes Haloperidol, der einzige Metabolit des Haloperidols. Reduziertes Haloperidol wird möglicherweise zu Haloperidol zurückkonvertiert [13]. Eine hervorstechende Bedeutung haben die aktiven Hauptmetaboliten von Risperidon, 9Hydroxy-Risperidon (9-OH-Risperidon, Paliperidon). Während die Muttersubstanz und der Metabolit etwa äquipotent sind, unterscheiden sie sich in der Halbwertszeit stark voneinander. Während die Muttersubstanz mit einer Halbwertszeit von 3 Stunden eliminiert wird, kommt 9-OH-Risperidon auf eine Eliminationshalbwertszeit von ca. 20 Stunden, wodurch die klinische Wirkung hauptsächlich durch den Metaboliten (9-OHRisperidon) vermittelt wird. Aufschlüsse über den Metabolismus der Substanz und der Aktivität des Isoenzyms CYP2D6, wodurch Risperidon verstoffwechselt wird, gibt hierbei die Messung der Plasmakonzentration mit Einbezug der Summe von Muttersubstanz und Metabolit (active moiety) [13]. Ähnliche pharmakologische Charakteristik gilt auch für Dehydroaripiprazol, der Hauptmetabolit von Aripiprazol. Er weist eine Eliminationshalbwertszeit von über 90 Stunden auf. Diese ist damit noch etwas länger als die der Muttersubstanz [13]. 1.2.5 Elimination Die endgültige Ausscheidung (Elimination) eines Pharmakons kann überwiegend renal (mit dem Urin) oder biliär in das Intestinum (und in weiterer Folge über die Fäzes) erfolgen. Wie bei der Biotransformation kann es bei der Ausscheidung eines Pharmakons in Abhängigkeit von seinen physikalisch-chemischen Eigenschaften (Molekulargewicht, pKa-Wert, Löslichkeit) zur Abnahme der Wirkstoffkonzentration kommen. Die Geschwindigkeit und das Ausmaß der renalen Ausscheidung werden durch die glomeruläre Filtration, die tubuläre Rückresorption und die tubuläre Sekretion bestimmt. Im Gegensatz zur tubulären Rückresorption ist die glomeruläre Filtration von der Löslichkeit der Pharmaka unabhängig. Von einem eiweißgebundenen Wirkstoff wird nur der nichtgebundene Anteil filtriert. Die tubuläre Rückresorption ist für die meisten Arzneistoffe ein passiver Diffusionsprozess und neben den Löslichkeitseigenschaften der Pharmaka abhängig vom pKa-Wert des auszuscheidenden Pharmakons und pH-Wert des Urins. 15 Die tubuläre Sekretion ist im Gegensatz zur tubulären Rückresorption ein aktiver Prozess und von Transportsystemen abhängig. Organische Säuren, aber auch organische Basen können durch erleichterte Diffusion über ihre säure- bzw. basenspezifischen Carrier sezerniert bzw. in den Urin abgegeben werden. Es kann zur Interaktion zwischen einzelnen Substanzen kommen, wenn sie sich gegenseitig in ihrem Transport kompetitiv hemmen und eine Konkurrenz um die Bindung an Transporter entsteht. Die biliäre Ausscheidung ist abhängig vom Molekulargewicht der Substanz. Ein Molekulargewicht von mindestens 500 ist für die Ausscheidung einer Substanz über die Galle erforderlich, wobei der Transport aktiv oder passiv erfolgen kann. Die echte intestinale Ausscheidung von Pharmaka über eine Sekretion in das Darmlumen ist auch durch aktiven Transport mittels ABCTransportern möglich [1]. 1.2.6 Blutspiegel der Pharmaka Der Plasmaspiegel (oder auch Blutspiegel) beschreibt die Konzentration eines Stoffes im Blut. Er korreliert mit der pharmakologischen Wirkung und ist ein wichtiges Hilfsmittel, wenn die pharmakologische Wirkung nicht unmittelbar einsetzt oder nicht einfach bestimmt werden kann, wie es z. B. bei Antipsychotika zutreffen kann. Außerdem sind Blutspiegelwerte relativ einfach und exakt bestimmbar. In der Langzeittherapie dienen sie auch zur Überwachung der PatientInnen-Compliance. Aufgrund der geringen Größe des Plasma-Kompartimentes und der oftmals hohen Gewebeaffinität und Lipophilie von einigen Pharmaka ist oftmals trotz exakter Bestimmung des Plasmaspiegels nur der geringste Teil der verabreichten Dosis im Plasma als Plasmaspiegel nachzuweisen [1][11]. 16 Abb. 2: Schematische Darstellung eines Plasmaspiegelverlaufes nach oraler Applikation Riederer und Laux 2010 [11] In der Darstellung (Abb. 2) wird die Plasmakonzentration nach oral applizierter Dosis im zeitlichen Verlauf dargestellt. Nach Einnahme des Arzneimittels nimmt der Plasmaspiegel der Substanz langsam zu und erreicht bei ausreichender Dosierung den therapeutischen Bereich (Invasionsphase). In diesem Bereich kommt es zur therapeutischen Wirkung der applizierten Substanz. Die Evasionsphase beschreibt die Abnahme der Plasmakonzentration durch Eliminationsprozesse und ist für die Wirkdauer von essentieller Bedeutung [11]. Bei gleicher Dosierung von Antipsychotika können unterschiedliche Plasmakonzentrationen, entsprechend der individuellen Vielfalt der Menschen, gemessen werden. Die Bestimmung der Plasmaspiegel ist besonders bei PatientInnen wichtig, die bei mäßiger Dosierung unter ausgeprägten Nebenwirkungen leiden und bei denen es trotz sicherer Compliance zu keinem ausreichenden Ansprechen auf die antipsychotische Therapie kommt. Bei gleicher Dosierung von Antipsychotika werden extrem unterschiedliche Plasmakonzentrationen gemessen. Deswegen wurden als grobe Orientierungshilfe für die meisten Antipsychotika wirksame Plasmakonzentrationen definiert [13]. 17 Tab. 10: Empfohlene Plasmakonzentrationen von Psychopharmaka und literaturbasierte Empfehlungen zur Anwendung von therapeutischem Drug Monitoring Arzneimittel und aktiver Metabolit Empfohlene Plasmakonzentration in ng/mL (Consensus)1 Arzneimittel und aktiver Metabolit Empfohlene Plasmakonzentration in ng/mL (Consensus)1 Amisulprid 100–320 Paliperidon 20–60 Aripiprazol 150–250 Perazin 100–230 Benperidol 2–10 Perphenazin 0.6–2.4 Chlorpromazin 30–300 Pimozid 15–20 Chlorprothixen 20–200 Quetiapin 70–170 Clozapin 350–600 Risperidon 20–60 Fluphenazin 0.5–2 9-Hydroxy-Risperidon 20–60 Flupentixol >2 Sulpirid 200–1000 Haloperidol 5–17 Thioridazin 200–2000 Melperon > 50 Zotepin 12–120 Levomepromazin 15–60 Ziprasidon 50–120 Olanzapin 20–80 Zuclopentixol 4–50 1 Empfohlene Plasmakonzentrationen bezeichnen Arzneimittelkonzentrationen in Serum oder Plasma von PatientInnen im Steady State, bei denen nach derzeitigem Stand des Wissens mit höchster Wahrscheinlichkeit mit Therapieansprechen gerechnet werden kann. Adaptiert nach Baumann et al. 2004; Hiemke et al. 2005 [22][23] 1.2.7 Therapeutisches Drug Monitoring Hiemke et al. [17], Castberg et al. [24], Jerling et al. [25], Szegedi et al. [26], Silver [27] und Lu et al. [28] stimmen in ihren Publikationen überein, dass ein optimaler Blutspiegel durch Veränderung der Dosierung erreicht werden kann. Im Falle der Arzneimittelgruppe der Antipsychotika sollte die Adaptation der Dosierung durch Messung der Plasmakonzentration erfolgen [29]. 18 1.3 Prinzipien der Pharmakodynamik Die Pharmakodynamik befasst sich mit der Wirkung und den Wirkungsmechanismen von Pharmaka im menschlichen Organismus, wobei die entsprechenden Untersuchungen auf die Art (Wirkprofil, Wirkqualität) und den Ort der Wirkung, die Wirkstärke (Potenz) und die Wirksamkeit (Effektivität) fokussiert sind. Die betroffenen Wirkungsmechanismen umfassen meist die Bindung von Pharmaka an Proteine der Membranrezeptoren, Ionenkanäle, Transporter, Enzyme oder Transkriptionsfaktoren und Strukturproteine. Pharmakodynamische Wechselwirkungen sind die bei Weitem am häufigsten vorkommenden Interaktionen im Zuge einer Polypharmazie. Wirkt ein Arzneimittel am gleichen Rezeptor, Erfolgsorgan oder Regelkreis wie eine andere verabreichte Substanz, kann es zur Interaktion der Pharmaka kommen. Einerseits können sich daraus eine gleichsinnige Wirkung und eine Wirkungsverstärkung ergeben. Andererseits kann die Wirkung des einen Pharmakons agonistisch und die des anderen antagonistisch sein, wodurch der Effekt abgeschwächt wird. In der Behandlung von Intoxikationen ist dieser Effekt erwünscht [1][11]. 19 Tab. 11: Zielstrukturen, die durch Antipsychotika blockiert werden können und zu therapeutischer oder unerwünschter Wirkung führen Zielstruktur Therapeutischer Effekt Unerwünschte Wirkung Adrenozeptoren, α1 Unsicher bezüglich α1Adrenozeptoren im ZNS Unklar Orthostatische Hypotension, Reflextachykardien, Schwindel Begünstigt Tachykardien und den Antagonismus von antihypertensiven Effekten von Clonidin und Methyldopa EPS, Akathisie, Prolaktinfreisetzung, sexuelle Dysfunktion bei Männern, gestörte Thermoregulation, neuroleptisches Syndrom, durch Stimulation verursachte Übelkeit, Erbrechen, verstärkter Sexualtrieb Zu starke Sedierung, Delirium, Gewichtszunahme QT-Intervall-Prolongation, TdP, andere Arrhythmien Gestörte Akkommodation, trockener Mund, Sinustachykardien, Obstipation, Harnretention, Glaukom, kognitive Störungen, Delirium oder Krämpfe Erhöhtes Risiko für Diabetes mellitus Typ 2 Unklar Unklar Adrenozeptoren, α2 Dopamin-D2-Rezeptoren Antipsychotische Effekte, Besserung der Plussymptomatik Histamin-H1-Rezeptoren Sedierung und Schlaf Kardiale Kaliumkanäle (hERG) Keine Angaben Acetylcholin-M1-Rezeptoren Milderung von extrapyramidalen Nebenwirkungen Acetylcholin-M3-Rezeptoren Unklar Serotonin-5-HT2D-Rezeptoren Serotonin-5-HT2A-Rezeptoren Unklar Inhibition kann Angst, Minussymptomatik, ebenso Schlaf, Appetit beeinflussen Inhibition kann Angst, Minussymptomatik, ebenso Schlaf, Appetit beeinflussen Serotonin-5-HT2C-Rezeptoren Inhibition kann zu Gewichtszunahme führen und beeinflusst den Gastrointestinaltrakt sowie die Regelung der Körpertemperatur TdP: Torsade-de-pointes-Tachykardien (polymorphe ventrikuläre Tachyarrhythmien); EPS: extrapyramidale Symptome; unter Kombination von Antipsychotika mit anderen Substanzen können sich die Zielstrukturen überschneiden, wodurch pharmakodynamische Wechselwirkungen mit additivem oder antagonistischem pharmakologischen Effekt hervorgerufen werden können. Adaptiert nach Silvestre und Prous 2005; WenzelSeifert et al. 2011; Richelson und Souder 2000 [30][31][32] 1.3.1 Additive/synergistische/antagonistische Effekte an Rezeptoren Ein synergistischer Effekt kann vorliegen, wenn durch die Verabreichung von zwei oder mehreren Wirkstoffen der gemessene Effekt der Kombination größer ist als der Effekt der Einzelsubstanzen. Im Falle eines Antagonismus kommt es zur Abschwächung des pharmakologischen Effekts eines Pharmakons durch einen zweiten Wirkstoff [2]. 20 1.4 Vorstellung der Antipsychotikaklassen Antipsychotika stellen ein chemisch heterogenes Kollektiv an Arzneimitteln dar, welches einen antipsychotischen Wirksamkeitsfokus und verschiedene Nebenwirkungsspektren beinhaltet. Als Synonym wird historisch bedingt die Bezeichnung „Neuroleptikum“ verwendet. Die Disposition kann nach der chemischen Zusammensetzung, der antipsychotischen Effizienz („neuroleptische Potenz“) und den atypischen Eigenschaften erfolgen [33]. 21 Tab. 12: Neuroleptika (NL), Österreichische Arzneimittel im Taschenformat Neuroleptika (NL) Freiname Handelsname Dosierungen Typische NL Atypische NL Chlorprothixen Truxal Flupentixol Fluanxol Haloperidol Haldol Levomepromazin Nozinan Melperon Buronil 15–400 mg/d p. o.; 50–300 mg/d i.m. Individuell TD meist 5–15 mg p. o. in 2–3 ED 1–20 mg p. o. 2–3 x tgl.; 5–10 mg i.v./i.m., bis max. 60 mg/d Initial 25 mg 1–3 x tgl., steigern bis TD 150–300 mg, dann langsam reduzieren TD 125–150 mg p. o. 3 x tgl. Pimozid Orap 2–10 mg p. o. 1 x tgl. Prothipendyl Dominal 40–80 mg p. o. 2–4 x tgl. Sulpirid Dogmatil Tiaprid Delpral Zuclopenthixol Cisordinol 50–100 mg p. o. 3 x tgl.; Psychosen: initial 200–400 mg 3 x tgl., Erhaltungsdosis 200 mg 2–3 x tgl. Früdyskinesien: TD 150–400 mg; Spätdyskinesien: TD 300–800 mg; chron. Alkoholismus: TD 300–400 mg TD 6–40 mg p. o. Amisulprid Solian 50–800 mg p. o. v x tgl. Aripiprazol Abilify 15–30 mg p. o. 1 x tgl. Clozapin Leponex, Lanolept Olanzapin Aedon, Olanzapin Schizophrenie: initial 12,5 mg p. o. 1–2 x tgl., dann schrittweise (über 2–3Wo) auf 300 mg/d erhöhen, therapeutischer Dosisbereich 200– 450 mg/d, MTD 900 mg; Psychosen M. Parkinson: initial 12,5 mg zN, nach 2 Wo 50 mg zN, MTD 100 mg 5–20 mg p. o. 1 x tgl. Paliperidon Invega 3–12 mg p. o. 1 x tgl. 50 mg p. o. v 2 xtgl. d1, 100 mg p. o. v 2 x tgl. d2, 200 mg p. o. v 2 x tgl. d3, 300 mg p. o. v 2 tgl. d4; Erhaltung: 300–750 mg/d TD 2 mg 1d, 4 mg 2d, dann meist Risperidon Risperdal, 4–6 mg; Aggressivität: TD initial Risperdal Consta 0,25 mg 2 x tgl., dann 0,5–1 mg 2 x tgl.; Manie: 2 mg 1 x tgl., Dosisanpassung 2–6 mg tgl. 4 mg p. o. 1 x tgl., alle 4–5 Tage Sertindol Serdolect um 4 mg erhöhen, Erhaltungsdosis 12–20 mg 40–80 mg p. o. 2 x tgl. Ziprasidon Zeldox d: Tag; i.m.: intramuskulär; i.v.: intravenös; p. o.: per os; mg: Milligramm; TD: Tagesdosis; ED: Erhaltungsdosis; MTD: maximale Tagesdosis; Wo: Woche; zN: zur Nacht (vor dem Schlafengehen); tgl.: täglich; x tgl.: mehrmals täglich; v x tgl.: verteilt auf mehrere Einzeldosen; v 2 x tgl.: verteilt auf zwei Einzeldosen, 2 x tgl.: zweimal pro Tag; 3 x tgl.: dreimal pro Tag; 4 x tgl.: viermal pro Tag; Adaptiert nach Kähler et al. 2012 [34] Quetiapin Quetialan, Seroquel 22 1.4.1 Wirkungsmechanismen der Antipsychotika Tab. 13: Rezeptorwirkungsprofile von Antipsychotika AntiTriKlinische D1 psychotikum zyklisch Einteilung AAP AAP AAP KAP, HP KAP, HP KAP, NP 0 0 + 0 + ++ D3 5HT2 M1 α1 ++ +++ + +++ +++ + ++ +++ ++ ++ ++ + 0 ++ ++ ++ 0 ++ 0 0 0 0 0 + 0 + + + + + H1 0 + + 0 0 ++ + b + AAP ++ + ++ +++ +++ + ++ Clozapin + + KAP, HP ++ +++ +++ ++ 0 + + Flupentixol + KAP, HP ++ +++ ++ ++ 0 + + Fluphenazin – KAP, HP + +++ ++ + 0 0 0 Fluspirilen – KAP, HP + +++ + 0 0 ++ 0 Haloperidolb + KAP, NP 0 + + + ++ ++ ++ Levomepromazin – KAP, NP (A) 0 + + ++ 0 + + Melperonb + AAP ++ +++ + +++ +++ + ++ Olanzapinb + c – AAP 0 +++ + +++ 0 + + Paliperidon + KAP, MP 0 ++ ++ ++ + ++ ++ Perazin + + KAP, HP 0 +++ +++ ++ 0 ++ ++ Perphenazin b – KAP, HP 0 +++ +++ ++ 0 0 0 Pimozid – KAP, NP (A) 0 + + ++ 0 + 0 Pipamperon + KAP, NP ? + ? ? ? ? ? Prothipendyl + AAP + + + + 0 + ++ Quetiapin b – AAP 0 +++ + +++ 0 ++ + Risperidon – AAP ++ +++ + +++ 0 ++ 0 Sertindol – KAP, MP (A) 0 + +++ 0 0 0 0 Sulpirid + KAP, NP + ++ + ++ +++ +++ + Thioridazin – AAP + ++ ++ +++ 0 + ++ Ziprasidonb + KAP, MP/HP ++ +++ ++ 0 +++ +++ ++ Zuclopenthixol + Die Daten sind aus In-vitro-Rezeptoraffinitäten der Antipsychotika zusammengestellt und spiegeln daher nicht direkt die klinischen Wirkungen (in vivo) wider. Antipsychotika wirken primär als Antagonisten, d. h. blockierend an Neurotransmitterrezeptoren. Daneben werden durch höhere Konzentrationen Enzyme und Ionenkanäle gehemmt. a Partieller D2/D3-Agonist und 5-HT1A-Agonist; b D4-Antagonist; c 9-OH-Risperidon. KAP konventionelle Antipsychotika, AAP atypische Antipsychotika, HP hochpotent, MP mittelpotent, NP niederpotent, (A) KAP mit ausgeprägten atypischen Eigenschaften. Beschreibung: – nicht trizyklisch, + trizyklisch (betreffend die zweite Spalte); ? keine Angaben, 0 keine Wirkung, + geringgradig wirksam, ++ mittelgradig wirksam, +++ hochgradig wirksam; Adaptiert nach Benkert et al. 2011 [33] Amisulprid Aripiprazola, b Asenapin Benperidol Bromperidol Chlorprothixen – – – – – + D2 Die ursächliche Wirkungsweise und die Mechanismen der Wirkungsentfaltung sind nicht restlos geklärt. Eine Gemeinsamkeit der Antipsychotika besteht in der Dämpfung der 23 dopaminergen Überaktivität und der Blockade von D2-artiger Dopaminrezeptoren (Tab. 13) [33][35]. D1-artige (D1/5) Rezeptoren steigern die intrazelluläre Konzentration von cAMP. Hingegen kommt es durch D2-artige Rezeptoren zu einer Erniedrigung der intrazellulären cAMPKonzentration. Hierbei sind Affinitätsunterschiede der verschiedenen Antipsychotika zu den Dopaminrezeptorsubtypen (D1–5) zu bemerken (Tab. 13). Die Dopaminrezeptorsubtypen verteilen sich auf dopaminerge Neuronensysteme im ZNS. Tab. 14: Dopaminerge Neuronensysteme im ZNS Dem nigrostriatalen System unterliegt die Kontrolle der Motorik und es zeichnet dadurch für EPS verantwortlich. Das mesolimbische/mesokortikale System ist der vermutliche Hauptangriffsort und verantwortlich für die antipsychotische Wirkung. Das tuberoinfundibuläre System zeigt sich verantwortlich für neuroendokrinologische Nebenwirkungen, insbesondere den Prolaktinanstieg. Adaptiert nach Benkert et al. 2011 [33] Der typische Effekt von atypischen Antipsychotika beruht auf einer bevorzugten Beeinflussung der dopaminergen Neurotransmittersysteme in den mesolimbischen, mesokortikalen und nigrostrialen Bahnen. Ausschlaggebend sind hierbei die mesolimbische Bindungsselektivität, die 5-HT2-Rezeptorblockade und der partielle Agonismus an D2-artigen Dopaminrezeptoren. Ein zusätzlicher antagonisierender Effekt auf 5-HT2A-Rezeptoren und deren positiver Einfluss auf die Wirkung hinsichtlich der Minussymptomatik werden bei den meisten AAP diskutiert. Dies gilt auch für neuere Arzneistoffe wie Asenapin und Iloperidon, nicht aber für Amisulprid (Tab. 13) [35][33]. Durch manche Antipsychotika kommt es zusätzlich zu einer Blockade von 5-HT2(A, B, C)-, α1, α2, H1- und muskarinischen Acetylcholin(mACh)-Rezeptoren (M1–5). Weiters werden QT-Verlängerungen bei Iloperidon (5-HT2/D2-Rezeptorantagonist, ausgeprägter α2CRezeptorantagonismus) und Ziprasidon beobachtet. Einige Antipsychotika weisen Affinität zu 5-HT6- (Asenapin, Clozapin, Olanzapin, Sertindol, Ziprasidon, Zotepin, aber auch Chlorpromazin, Chlorprothixen, Fluphenazin) und 5-HT7-Rezeptoren (Clozapin, Pimozid, Quetiapin, Risperidon, Sertindol, Ziprasidon) auf. Die Wirkungsweise von Aripiprazol und anderen sich in der Entwicklung befindlichen AAP (SLV313, F15063, SSR-181507) besteht in einer partiellen dopaminagonistischen Wirkung an D2-artigen Rezeptoren („Dopamin-Stabilisierer“) und einem partiellen 24 agonistischen Effekt an serotonergen 5-HT1A-Rezeptoren bei antagonistischer Wirkungsweise an 5-HT2A- und 5-HT2C-Rezeptoren (Aripiprazol) [33][35]. „Der partielle Agonismus“ beruht auf der Toposelektivität des Aripiprazol. Bei zu niedriger dopaminerger Aktivität (Minussymptomatik) wirkt Aripiprazol als Agonist und bei hoher dopaminerger Aktivität (Plussymptomatik) als Antagonist. Es kommt durch Aripiprazol zu einer fast vollständigen D2-Rezeptorokkupation im nigrostrialen System, allerdings zu keiner kompletten Blockade der dopaminergen Neurotransmission aufgrund der etwa 20 bis 30%igen intrinsischen Aktivität an D2-artigen Dopaminrezeptoren. Als ursächlich für das Fehlen oder seltene Auftreten von EPS bei AAP verantwortlich diskutiert man eine Blockade der D4- und/oder 5-HT2A/C-Rezeptoren und eine Interaktion mit Subtypen von mACh-Rezeptoren. Für die überwiegend auf mesolimbische Neurone wirkenden Substanzen Clozapin, Olanzapin und Quetiapin wird regionalspezifisch ein Dopaminrezeptorantagonismus diskutiert [35][33]. 1.4.2 Einteilung der Antipsychotika 1.4.2.1 Antipsychotische Wirkungsstärke („neuroleptische Potenz“) Mit Unterstützung der „neuroleptischen Potenz“ werden Antipsychotika auf einer Größenordnung mit Chlorpromazin (CPZ) als Vergleichspunkt angeordnet. Einfließende präklinische und klinische Informationen bilden die Inhibition der D2-ähnlichen Dopaminrezeptoren und der dosisabhängige antipsychotische Einfluss. Betreffend die konventionellen Antipsychotika nimmt die neuroleptische Potenz Bezug auf den Grad der D2-Rezeptorhemmung [33]. Hochpotente Antipsychotika sind charakterisiert durch geringe Sedierung, starke antipsychotische Effektivität und signifikante extrapyramidale begleitende Wirkkraft bei geringer bis mittlerer Dosierung. Mittelpotente Antipsychotika weisen zum Unterscheid bei geringer bis mittelgradiger Dosierung eine mittelstarke antipsychotische Wirkungsweise sowie mäßig extrapyramidale Nebenwirkungen und mäßige Sedierung auf. Für niederpotente Antipsychotika sind eine ausgeprägte Sedierung, geringe extrapyramidale Nebenwirkungen und marginale antipsychotische Effizienz bei niedriger bis mittlerer Dosierung bezeichnend [35][33]. 25 Eine Korrelation zwischen neuroleptischer Potenz und EPS besteht nur bei niedrigen Dosen von KAP. Dieser Konnex ist für atypische Neuroleptika nicht zutreffend, da keine Verbindung zwischen antipsychotischer Wirkung der atypischen Neuroleptika und der Affinität zum D2-Rezeptor besteht [33]. CAVE: Es ist zu bemerken, dass es zu einer erheblichen Schwankungsbreite in der Dosierung von Antipsychotika bei der Akuttherapie sowie der Erhaltungstherapie schizophrener Psychosen kommt. Die Empfehlung für die Dosierung in der Akuttherapie mit 300–1000 CPZ-Einheiten und in der Erhaltungstherapie mit 300–600 CPZ differiert bei mehreren Autoren [33]. 1.4.2.2 Atypische Wirkkraft Tab. 15: Unterteilung der Antipsychotika nach ihren „atypischen“ Eigenschaften KAP, syn. typische/Typika?, herkömmliche Konventionelle Antipsychotika oder klassische Antipsychotika, firstgeneration antipsychotics, FGA AAP, syn. Atypika, neuere oder atypische Atypische Antipsychotika Antipsychotika der 2. Generation, novel antipsychotics, second-generation antipsychotics, SGA Adaptiert nach Benkert et al. 2011 [33] Die Unterscheidung zu den konventionellen Antipsychotika beruht auf einer relativ niedrigen Affinität zu D2-Rezeptoren und einer um den Faktor 20–100 höheren Affinität zu D4-, 5-HT2A-, α1-, Muskarin- und H1-Rezeptoren. Vergleichende Affinität zu 5-HT2ARezeptoren weisen Aripiprazol und Quetiapin auf. Gravierend höhere Affinität zu 5-HT2ARezeptoren zeigt sich bei atypischen Antipsychotika wie Risperidon, Ziprasidon und Olanzapin. Die Antagonisierung der 5-HT2A-Rezeptoren ist hierbei weit größer als die der D2-Rezeptoren. Ziprasidon und Aripiprazol haben außerdem die Funktion, als Agonisten am 5-HT2A-Rezeptor und Amisulprid als einzige Verbindung selektiv D2-, D3- und D4Rezeptoren zu antagonisieren und wie andere atypische Antipsychotika gegen Minussymptomatik wirksam zu sein [36]. Atypische Antipsychotika wurden auf der Basis entwickelt, möglichst viele Merkmale von Clozapin aufzuweisen. Die Gründe dafür liegen in der Effizienz gegen Plus- als auch gegen Minussymptomatik und den praktisch nicht vorhandenen extrapyramidalmotorischen Begleiterscheinungen [33][35][36]. 26 1.4.2.3 Beispiele der Klassifizierung unter struktur-chemischen Gesichtspunkten Konventionelle Antipsychotika Abb. 3: Stoffbeschreibung Chlorpromazin Chlorpromazin Substanzklasse: Phenothiazine Strukturformel: C17H19ClN2S Molekulargewicht: 318.86416 (g/mol) Auch bekannt unter: Largactil® National Center for Biotechnology Information, U.S. National Library of Medicine [37] Abb. 4: Stoffbeschreibung Chlorprothixen Chlorprothixen Substanzklasse: Thioxanthene Strukturformel: C18H18ClNS Molekulargewicht: 315.86022 (g/mol) Auch bekannt unter: Truxal® National Center for Biotechnology Information, U.S. National Library of Medicine [38] 27 Abb. 5: Stoffbeschreibung Haloperidol Haloperidol Substanzklasse: Butyrophenone Strukturformel: C21H23ClFNO2 Molekulargewicht: 375.864223 (g/mol) Auch bekannt unter: Haldol® National Center for Biotechnology Information, U.S. National Library of Medicine [39] Abb. 6: Stoffbeschreibung Sulpirid Sulpirid Substanzklasse: Benzamide Strukturformel: C15H23N3O4S Molekulargewicht: 341.42582 (g/mol) Auch bekannt unter: Dogmatil® National Center for Biotechnology Information, U.S. National Library of Medicine [40] 28 Atypische Antipsychotika Abb. 7: Stoffbeschreibung Clozapin Clozapin Strukturformel: C18H19ClN4 Molekulargewicht: 326.82326 (g/mol) Auch bekannt unter: Leponex® National Center for Biotechnology Information, U.S. National Library of Medicine [41] Abb. 8: Stoffbeschreibung Olanzapin Olanzapin Strukturformel: C17H20N4S Molekulargewicht: 312.4325 (g/mol) Auch bekannt unter: Zyprexa® National Center for Biotechnology Information, U.S. National Library of Medicine [42] 29 Abb. 9: Stoffbeschreibung Quetiapin Quetiapin Strukturformel: C21H25N3O2S Molekulargewicht: 383.5071 (g/mol) Auch bekannt unter: Seroquel® National Center for Biotechnology Information, U.S. National Library of Medicine [43] Abb. 10: Stoffbeschreibung Risperidon Risperidon Strukturformel: C23H29FN4O2 Molekulargewicht: 412.500363 (g/mol) Auch bekannt unter: Risperdal® National Center for Biotechnology Information, U.S. National Library of Medicine [44] 30 Abb. 11: Stoffbeschreibung Aripiprazol Aripiprazol Strukturformel: C23H27Cl2N3O2 Molekulargewicht: 448.38538 (g/mol) Auch bekannt unter: Abilify® National Center for Biotechnology Information, U.S. National Library of Medicine [45] In den Abb. 3 bis 11 sind Kohlenstoffverbindungen grau, Sauerstoffverbindungen rot, Stickstoffverbindungen blau, Chlorverbindungen grün, Fluorverbindungen hellgelb und Schwefelverbindungen gelb markiert. National Center for Biotechnology Information, U.S. National Library of Medicine [37][38][39][40]–[45] 31 2. Material und Methoden Diese Diplomarbeit wurde im Sinne einer Übersichtsarbeit auf Basis einer Recherche aktueller Literatur zum Thema „Arzneimittelinteraktion in der Therapie mit Antipsychotika“ aufgebaut. Es wurden dabei Lehrbücher der Pharmakologie, Pharmakologie und Toxikologie, Psychopharmakologie, Psychiatrie und ein Nachschlagewerk für Arzneimittel herangezogen. Weiters wurde über eine Datenbank (ABDA) versucht, Daten zu ermitteln. In Suchmaschinen wie beispielsweise PUBMED, MEDLINE oder OVID wurde unter anderem nach folgenden Begriffen gesucht: „antipsychotic drug interaction“ „antipsychotic drugs“ „drug-drug interaction“ „QT prolongation syndrom“ „CYP P450 induction/inhibition“ „pharmacodynamic/pharmacokinetic drug interaction“ „therapeutic drug monitoring“ Es wurden klinische Studien retrospektive Fall-Kontrollstudien, „case reports“ und Review-Artikel im Schwerpunktzeitraum von 2000 bis 2012 dafür ausgewählt, limitiert auf englischsprachige und deutsche Artikel. Diese Diplomarbeit besteht hauptsächlich aus einer persönlichen Auswahl an aktuellen Studien und soll dem Leser einen kurzen Überblick über die Thematik geben. 32 3. Ergebnisse – Resultate 3.1 Studien über pharmakodynamische Interaktionen Pharmakodynamische Interaktionen mit Antipsychotika können vergesellschaftet sein mit Symptomen oder Syndromen. Typische ungünstige Reaktionen auf diese Art der Wechselwirkung können EPS, metabolische Störungen, kognitive Beeinträchtigung, Delirium und QT-Prolongation umfassen [29]. 3.1.1 Additive anticholinerge Nebenwirkungen Nach Cancelli et al. [46] kann es durch eine synergistische Arzneimittelinteraktion von klassischen Antipsychotika (Chlorpromazin, Haloperidol, Promazin) in Kombination mit trizyklischen Antidepressiva und/oder Histamin-H1-Rezeptorantagonisten der 1. Generation durch einen additiven Effekt zu verstärkten peripheren und zentralen anticholinergen Nebenwirkungen wie Akkommodationsstörungen, Harnverhalten, Obstipation, Ileus, lebensbedrohlichen Arrhythmien oder auch zu Agitation, Reizbarkeit und Desorientierung kommen. Eine besondere Sensibilität zeigt sich hierbei bei älteren PatientInnen. In einer Untersuchung mit älteren PatientInnen zum Einfluss kumulativer arzneimittelinduzierter anticholinerger Effekte auf die Schwere eines Deliriums waren unter den 15 häufigsten anticholinerg wirksamen Arzneistoffen fünf Psychopharmaka (Haloperidol, Risperidon, Diazepam, Fluvoxamin, Thioridazin) und vier starke Opioide (Morphin, Codein, Fentanyl, Pethidin) [2][47]. Hervorzuheben sind additive Effekte bei Kombination von anticholinerg wirksamen Antipsychotika aus der Gruppe der Phenothiazine mit stark anticholinerg wirkenden trizyklischen Antidepressiva [47][48]. Neben der Funktion als Wiederaufnahmehemmer für Noradrenalin und/oder Serotonin aus dem synaptischen Spalt kommt es durch diese Antidepressiva auch zur Beeinflussung von zentralen postsynaptischen Rezeptoren wie muskarinergen Acetylcholinrezeptoren. Clozapin, Perphenazin, Chlorpromazin und Thioridazin sind in der Kombination mit Antidepressiva wie Amitriptylin und Trimipramin übermäßig anticholinerg wirksam. Alternativ weist das trizyklische Antidepressivum Desipramin geringe anticholinerge 33 Effekte auf. Entscheidend bei einer Wechselwirkung vermeidenden Therapie ist eine gezielte Substanzwahl [47][33]. Dem entspricht auch der Review-Artikel (Current Antipsychotics) im Handbook of Experimental Pharmacology von 2012. Eine gestörte Akkommodation, Sinustachykardie, Obstipation, Harnretention, Glaukom, beeinträchtigte Wahrnehmung, Delirium, Krampfanfälle oder ein trockener Mund sind typische klinische Auswirkungen bedingt durch eine Blockade von Acetylcholin(mACh)Rezeptoren (M1). Es gibt eine Reihe von Medikamenten unterschiedlicher Klassen, welche eine anticholinerge Wirkung aufweisen [29]. Weiters wurde kürzlich im Journal of the American Geriatrics Society eine Studie von Chew et al. [49] veröffentlicht, in der 107 Medikamente auf ihre anticholinerge Aktivität analysiert wurden. Es wurden Daten ermittelt, die belegen, dass unter den verschiedenen Antipsychotika Clozapin und Thioridazin eine ausgeprägte anticholinerge Wirkung zeigen. Weiters wurde in den Ergebnissen beschrieben, dass Chlorpromazin, Olanzapin und Quetiapin die zweitstärksten anticholinerg wirkenden Arzneimittel darstellen. Ferner besagen die Studienergebnisse, dass für in therapeutischen Dosen verabreichtes Aripiprazol, Haloperidol, Perphenazin, Risperidon und Ziprasidon keine anticholinerge Aktivität messbar war. In Bezug auf die anticholinerge Interaktion kam man zur Erkenntnis, dass die Kombination von einem Antipsychotikum und einem anderen Arzneimittel vermieden werden sollte, wenn beide Medikamente eine anticholinerge Wirkung aufweisen. Ein Beispiel dafür ist die kombinierte Gabe von Clozapin und Diphenhydramin [29]. 3.1.2 Kardiovaskuläre Effekte In Bezug auf kardiovaskuläre Effekte ist die Verlängerung des QT-Intervalls bzw. das angeborene LQTS zu behandeln. Die Kombination verschiedener Arzneimittel kann mit einer QT-Zeit verlängernden Wirkung einhergehen, wodurch das Risiko ventrikulärer Arrhythmien zunimmt [47]. QT-Verlängerung und Herzrhythmusstörungen Eine Vielzahl von Medikamenten, darunter auch die Arzneimittelgruppe der Antipsychotika, kann die myokardiale Erregungsrückbildung (Repolarisation) 34 beeinflussen, wodurch es zu einer pathologisch verlängerten frequenzkorrigierten QT-Zeit kommen kann. Durch die Hemmung repolarisierender Kaliumströme am Myokard kann eine QT-Verlängerung eintreten. Medikamente, die eine solche Nebenwirkung beinhalten und eine Hypokaliämie auslösen können, müssen als Risikofaktor in Bezug auf eine QTVerlängerung genannt werden [33]. Oftmals besteht auch eine hohe Konzentration des verabreichten Arzneimittels. Die verlängerte Repolarisation resultiert aus einem Kreislauf, in dem es zu einer Abnahme des Repolarisationsstroms durch Zunahme des Einwärtsstroms und Abnahme des Auswärtsstroms kommt. Dies geschieht durch den spezifischen Kaliumfluss. Die schnell wirksame Komponente des „Delayed Rectifier“ IKr spielt hierbei eine wichtige Rolle. Der Strom wird durch hERG fließendes Kalium verursacht, welche für eine Blockade durch verschiedene Substanzen wie psychotrope Arzneistoffe und verschiedene Antipsychotika empfindlich sind [29]. Als Folge des verlängerten QT-Intervalls und des damit verbundenen vermehrten Ca2+Einstroms und Instabilität der Repolarisation können proarrhythmische Effekte (TdP) auftreten, obwohl durch eine QT-Verlängerung tachykarde Rhythmusstörungen unterbrochen werden sollten [33]. Klinisch ist hierbei die Bestimmung von Grenzwerten bezüglich einer Risikoabschätzung für das Auftreten von Arrhythmien relevant. Tab. 16: Klinisch relevante Grenzwerte in Bezug auf das Auftreten von Arrhythmien Obere Grenzwerte QT-Zeit (ms) Männer 440 Frauen 450 Ein Wert > 500 ms und insbesondere > 600 ms bzw. eine Verlängerung um 60 ms im Vergleich zu einer Voruntersuchung bedeutet Gefahr von Arrhythmien! Adaptiert nach Benkert et al. 2011 [33] EKG- und Kaliumkontrollen unter allen Antipsychotika CAVE: Sorgfältige Beachtung der Ko-Medikation, regelmäßige EKG-Kontrollen (und Elektrolytkontrollen, v. a. Kalium) vor Beginn und während einer Behandlung mit Antipsychotika und bei PatientInnen, die ein erhöhtes Risiko für QT-Verlängerungen und TdP aufweisen, Bestimmung der Serumkaliumkonzentration, ggf. Korrektur einer Hypokaliämie, Anstreben der minimal therapeutisch wirksamen Dosis (QTVerlängerungen sind dosisabhängig) und eine medikamentöse Umstellung bei auftretenden 35 Pathologika (QT > 480 ms, medikamenteninduzierte Verlängerung > 60 ms) können zu einer Senkung der erhöhten kardiovaskulären Mortalität schizophrener PatientInnen beitragen. Weiters ist bei Vorliegen oder Auftreten kardialer Symptome immer eine kardiologische Abklärung notwendig [33]. Abb. 12: Beispiele des erworbenen Long-QTSyndroms (LQTS) Ein gemeinsames Merkmal ist eine Pause (grüner Stern), welche oftmals nach einer Extrasystole auftreten kann. Es entwickelt sich eine gestörte Repolarisation im folgenden Zyklus (roter Pfeil). A: Kontinuierliche Aufnahmen von einem 79jährigen Mann mit fortgeschrittener Herzerkrankung unter Dofetilid (Antiarrhythmikum)-Therapie. Nach dem abnormen QT-Intervall folgt 7 Schläge lang TdP. Bei diesem Patienten werden durch TdP nachhaltig monomorphe ventrikuläre Tachykardien aufgrund der zugrunde liegenden Herzerkrankung ausgelöst. B: TdP bei einem Patienten unter antipsychotischer Haloperidol-Therapie. C: TdP bei einem Patienten mit AV-Block. Die blauen Pfeile zeigen nonconducted Vorhofdepolarisationen. D: Deutlich abnormale postpausale Repolarisation bei einem Patienten mit fortgeschrittener Herzinsuffizienz. Solche ungeordneten Repolarisationen können ein erhöhtes Risiko für TdP darstellen. Roden et al. 2005 [50] Zu diesem Thema wurden kürzlich im British Journal of Clinical Pharmacology und im Deutschen Ärzteblatt zwei Arbeiten veröffentlicht. Nach van Noord et al. [51] und Wenzel-Seifert et al. [31] wurde bei einer großen Anzahl älterer und neuerer antipsychotisch wirkender Arzneistoffe über das Auftreten einer QTVerlängerung berichtet. Genauer beschreiben Wenzel-Seifert et al. nach einer Literatursuche Thoridazin und Ziprasidon als die Pharmaka mit dem höchsten Risiko für das Auftreten einer QT-Verlängerung und/oder das Auftreten von lebensbedrohlichen TdP. In Bezug auf Haloperidol besteht für sie eine signifikant erhöhte Gefahr bei intravenöser Verabreichung hoher Dosen. In einigen Fällen treten TdP in Verbindung mit neueren Antipsychotika wie Quetiapin und Amisulprid auf. Weiters kommen Wenzel-Seifert et al. zum Ergebnis, dass in der Regel nur die Anwesenheit von mehreren zusätzlichen Risikofaktoren (Alter > 65 Jahre, bestehende Herz-Kreislauf-Erkrankungen, Bradykardie, weibliches Geschlecht, Hypokaliämie, Hypomagnesiämie, eine supratherapeutische oder toxische Konzentration im Serum bzw. die gleichzeitige Gabe von anderen Arzneimitteln), 36 die zu einer verzögerten Repolarisation oder Störung des Arzneimittelstoffwechsels führen, das Auftreten einer QT-Verlängerung und/oder TdP ermöglicht. Als Fazit geben WenzelSeifert et al. an, dass ein sorgsamer Umgang in der Praxis mit psychotropen Substanzen unerlässlich ist und das EKG sowie der Elektrolythaushalt bei PatientInnen mit einer Psychopharmakatherapie regelmäßig überwacht werden müssen. Zu den weiteren Risikosubstanzen zählen beispielsweise Levomepromazin, Melperon, Pimozid und Sertindol. Diesbezüglich berichten Kannankeril und Roden [52] sowie Roden [53] im Current Opinion in Cardiology über den Zusammenhang zwischen einer QT-Verlängerung und einer Prolongation der ventrikulären Repolarisation, welche in schwerwiegenden Fällen zum plötzlichen Tod führen kann. In Verbindung mit einer QT-Verlängerung wurden in Veröffentlichungen aus dem Journal of Clinical Pharmacology mögliche kardiale Folgen beschrieben. Nach Tisdale et al. [54] können durch eine QT-Verlängerung TdP und Kammerflimmern hervorgerufen werden. Die Autoren kamen in ihrer multizentrischen, prospektiven Studie zu dem Ergebnis, dass die Empfindlichkeit bei PatientInnen mit einer bestehenden Herzinsuffizienz für das Auftreten einer medikamenteninduzierten QT-Verlängerung verstärkt wird, wodurch möglicherweise auch ein zusätzliches erhöhtes Risiko für substanzinduzierte TdP besteht. In Bezug auf die Genetik schreiben Roden und Viswanathan [50], Crumb et al. [55] und Unterecker et al. [56] unter anderem im Journal of Clinical Investigation, dass das Risiko einer QT-Verlängerung und möglicherweise schwerwiegender Komplikationen infolge einer Medikamentenverabreichung durch eine zugrunde liegende genetische Prädisposition beeinflusst sein kann, welche eine „reduzierte Repolarisationsreserve“ bedingt. Weiters bestehen zusätzliche Risikofaktoren. Zu diesen zählen Hypokalämie, Bradykardie, schwere Herzerkrankungen und auch das weibliche Geschlecht. Seit angenommen wird, dass substanzinduzierte QT-Verlängerungen konzentrationsabhängig sind, werden pharmkokinetische Interaktionen in Bezug auf die Pathogenese von LQTS immer relevanter. Bei Kombination von Antipsychotika, welche eine hERG-blockierende Wirkung haben und zu einer QT-Verlängerung führen, kann es bei gleichzeitiger Anwendung mit anderen QT-prolongierenden Medikamenten zu additiven oder sogar potenzierenden Effekten kommen. Aus diesem Grund hat die FDA dem „summary of product characteristics“ (SPC) eine Warnung beigefügt, um zu verhindern, dass atypische Antipsychotika wie Quetiapin in Verbindung mit anderen Substanzen wie Quinidin, 37 Procainamid, Amiodaron, Sotalol, Gatifloxacin oder Moxifloxacin gebracht werden. Auch für Lithium, welches gemeinsam mit Antipsychotika zur Anwendung kommt, muss die potenzielle Möglichkeit einer QT-Verlängerung berücksichtigt werden [29]. Im International Journal of Cardiology schreiben Letsas et al. [57], dass bei der Kombination von Arzneimitteln mit hERG-blockierenden Eigenschaften und pharmakokinetischen Wechselwirkungen eine besondere Gefahr hinsichtlich einer QTVerlängerung aufgrund der Kombination von pharmakodynamischen und pharmakokinetischen Interaktionen besteht. Solche Arzneimittelkombinationen und die Anwendung anderer Antipsychotika mit QT-Intervall prolongierenden Eigenschaften bedingen eine sorgfältige EKG-Überwachung [29]. 3.1.3 Exazerbation der Parkinson-Symptomatik sowie der extrapyramidalen Symptome (EPS) Das Ziel der Therapie des M. Parkinson ist es, die durch den Mangel an Dopamin hervorgerufene Symptomatik zu vermindern. Als Beispiel einer antagonistischen Arzneimittelinteraktion ist nach Jost et al. [58] bei der Therapie von Parkinson assoziierten psychiatrischen Symptomen (z. B. Psychosen) die gleichzeitige Gabe von Levodopa und klassischen Antipsychotika wie Haloperidol und Chlorpromazin zu vermeiden. Durch diese Substanzen kommt es zu einer eigenständigen Blockade von Dopamin-D2Rezeptoren und somit zur Antagonisierung des Effekts von Levodopa. Die Folge kann eine Exazerbation der Parkinson-Symptomatik bzw. EPS sein. Als Alternativen in der Anwendung bieten sich atypische Antipsychotika wie z. B. Clozapin an [2][48]. Zu diesem Zusammenhang wird auch im Review-Artikel (Current Antipsychotics) im Handbook of Experimental Pharmacology von 2012 berichtet. Hinsichtlich antagonistischer bzw. entgegengesetzter pharmakodynamischer Interaktionen sind Kombinationen von antipsychotischen Substanzen mit dopaminstimulierenden Arzneimitteln am relevantesten. Die Wirkung von Antipsychotika wird durch DopaminAgonisten antagonisiert und antipsychotische wirkende Substanzen schwächen den Effekt von dopaminstimulierenden Medikamenten. Wenn psychotische Symptome auftreten, können solche Kombinationstherapien bei an Parkinson erkrankten PatientInnen erforderlich sein. Dennoch sollte die Kombination mit Vorsicht angewandt werden. 38 Durch L-Dopa induzierte psychotische Symptome können durch Antipsychotika effektiv behandelt werden, zweckmäßiger bleibt aber die Behandlung durch eine Reduktion der LDopa-Dosis. [29]. Durch eine von Ghadirian et al. [59] durchgeführte Querschnittsstudie kam man zu dem Ergebnis, dass additive antidopaminerge Effekte meistens das Resultat einer Medikation mit mehr als einem Antipsychotikum sind. Symptome, die auf eine additive antidopaminerge Wirkung hinweisen, umfassen EPS, endokrine Auswirkungen, eine gestörte Thermoregulation und das neuroleptische Syndrom. Die Prävalenz des Parkinsonismus und der Spätdyskinesie ist unter der Kombinationstherapie mit verschiedenen Antipsychotika entsprechend erhöht. Das Risiko für die beschriebenen Symptome ist auch erhöht bei einer Ko-Medikation mit Lithium [29]. Nach Gross und Drescher [60] sowie Richelsen und Souder [32] ist es in Bezug auf die Therapieergebnisse plausibel, eine geringe Dosis einer anderen antidopaminergen Substanz, im Falle einer insuffizienten Unterdrückung der Plussymptomatik, durch das Steigern der antidopaminergen Wirkung hinzuzufügen. Dies kann unter Clozapin- oder Quetiapintherapie auftreten. Beide Substanzen weisen eine geringe Affinität zu Dopaminrezeptoren auf [29]. 3.1.4 Gewichtszunahme und das metabolische Syndrom Nach Heal et al. [61] besteht eine schwerwiegende Belastung der neuen Antipsychotika im Zusammenhang mit Gewichtszunahme und dem Auftreten eines metabolischen Syndroms. Nach Newcomer [62] und nach einer von Mitchell et al. [63] durchgeführten Meta-Analyse treten syndromspezifische Veränderungen am häufigsten unter Clozapin-, Olanzapin- und Thioridazineinnahme auf, sekundär bei der Einnahme von Paliperidon, Risperidon, Quetiapin und Sertindol. Ein geringes Risiko besteht bei der Verabreichung von Aripiprazol und Ziprasidon [29]. Baptista et al. [64] veröffentlichten in Current Drug Targets, dass das metabolische Syndrom eine Reihe von pathophysiologischen Veränderungen wie beispielsweise einen gestörten Glukosehaushalt und Lipidstatus umfasst. Die Gewichtszunahme kann in manchen Fällen zur Adipositas führen, welche vergesellschaftet ist mit einer erhöhten Mortalität. Bis jetzt gibt es nur eingeschränkte Belege dafür, ob sich eine Polypharmazie nachhaltig auf diese Nebenwirkungen auswirkt. 39 In einer im BMC Psychiatry Journal veröffentlichten Querschnittsstudie von Misawa et al. [65] wurde die Polypharmazie mit einer Monotherapie in Bezug auf das Auftreten solcher Nebenwirkungen [64] verglichen. Durch die neuen Daten kommt man zu dem Schluss, dass ein erhöhtes Risiko für die beschriebenen Nebenwirkungen bei Anwendung einer Polypharmazie besteht und ein geringeres bei der Anwendung einer Monotherapie [29]. 40 3.2. Studien über pharmakokinetische Interaktionen Von Cascorbi [66] wird in einem Review-Artikel (Current Antipsychotics) im Handbook of Experimental Pharmacology von 2012 beschrieben, dass pharmakokinetische Interaktionen Einfluss darauf nehmen, in welchem Ausmaß Substanzen ihren Wirkungsort erreichen können. Weiters können pharmakokinetische Wechselwirkungen während Absorption, Distribution, Metabolismus und Exkretion, also in jeder pharmakokinetischen Phase, auftreten. Interaktionen während der Absorption beeinflussen die Aufnahme von vor allem oral eingenommenen Antipsychotika aus dem Gastrointestinaltrakt in den Blutkreislauf. Cascorbi beschreibt in der Veröffentlichung einen Zusammenhang zwischen oral aufgenommenen antipsychotisch wirkenden Substanzen und Veränderung des pHWertes sowie der Motilität des Magen-Darm-Traktes. Weiters bezieht sich Cascorbi auf, für die Ausscheidung mitverantwortliche, spezifische Transporter und diesbezüglich auf den Einfluss auf die Wirkung von Antipsychotika [29]. Cascorbi kommt zu dem Ergebnis, dass Interaktionen auf Ebene der für die Ausscheidung zuständigen Transporter bzw. Transportsysteme möglich sind. Die Verfügbarkeit von Antipsychotika ist hierbei durch die Wirkung solcher Transporter in den endothelialen Zellen beschränkt [29]. Nach Moons et al. [15] werden über diese Transporter Substanzen nach erster Resorption in die Enterozyten wieder zurück in den Darm-Trakt transportiert (und eventuell sogar ausgeschieden). Der wichtigste Vertreter dieser Transporter ist das bereits gut charakterisierte P-gP. Risperidon, Paliperidon, Aripiprazol, Ziprasidon und mehrere typische Antipsychotika sind Substrate von P-gP. Für Moons ist bis jetzt jedoch unklar, ob P-gP-Inhibitoren wie Verapamil eine relevante klinische Wirkung auf die pharmakologische Aktivität von Antipsychotika haben [29]. Dürr et al. [67] weisen darauf hin, dass für P-gP-Induktoren wie Carbamazepin und Johanniskraut einige evidenzbasierte Daten vorhanden sind, die darauf hinweisen, dass die Verfügbarkeit von Risperidon und Palperidon durch diese Induktoren herabgesetzt werden könnte. Insgesamt sind sich Dürr et al. jedoch einig, dass nach dem aktuellen Wissensstand Wechselwirkungen während der Absorptionsphase nur geringe Relevanz bezüglich pharmakokinetischer Interaktionen mit Antipsychotika besitzen [29]. Die beschriebenen Zusammenhänge haben auch für die nach der Absorption folgende Distribution Gültigkeit. Im Organismus werden lipophile antipsychotische Substanzen über den Kreislauf verteilt und dabei an Plasmaproteine gebunden. Man war lange der Meinung, 41 dass die Verdrängung aus der Plasmaproteinbindung eine Zunahme der Substanzwirkung verursacht, wenn nur der ungebundene Teil pharmakologisch wirksam ist [29]. Benet und Hoener [68] wollen der Plasmaproteinbindung eine mindere Bedeutung beimessen. Sie sind der Ansicht, dass praxisbezogen die Plasmaproteinbindung nur eine untergeordnete Rolle spielt. Sie erklären in ihrer Veröffentlichung, dass die Extraktion von antipsychotischen Substanzen in das ZNS der effizienteste Prozess sei, da die Zielstrukturen hohe Affinität zu diesen Arzneistoffen aufweisen. Daher scheine es unwahrscheinlich, dass das Ausmaß der Plasmaproteinbindung die Wirksamkeit von Antipsychotika in vivo verändern kann [29]. CYP-Inhibitoren und Antipsychotika Nach Zhou [69] und Spina et al. [70] wird in Bezug auf pharmakokinetische Interaktionen mit Antipsychotika beschrieben, dass hepatische Cytochrom-P450-Enzyme im Phase-ΙMetabolismus sehr empfänglich seien. Unter mehreren Isoenzymen in der Leber sind CYP1A2, CYP2C19, CYP2D6 und CYP3A4 in der Praxis für den Abbau von antipsychotisch wirkenden Substanzen am relevantesten. Das CYP2D6 betreffend wurde nach Zhou in einer Reihe von klinischen Studien nachgewiesen, dass es signifikante Zusammenhänge zwischen dem CYP2D6-Genotyp und Steady-State-Konzentrationen von Perphenazin, Zuclopenthixol, Risperidon und Haloperidol gibt. Allerdings sind die Erkenntnisse über die Beziehung zwischen dem CYP2D6-Genotyp und Parkinsonismus bzw. Spätdyskinesien in der Behandlung mit herkömmlichen Antipsychotika widersprüchlich. Gründe dafür finden sich in einer geringen Stichprobenanzahl, der variablen CYP2D6-Metabolisierung und Ko-Medikation. Die Phänotypisierung und Genotypisierung scheinen bei der Vorhersage der Steady-StateKonzentrationen von einigen klassischen Antipsychotika nützlich zu sein, wobei die Nützlichkeit bezüglich einer Vorhersage eines klinischen Effektes noch erforscht werden muss. Therapeutic Drug Monitoring wird für viele Antipsychotika einschließlich Haloperidol, Chlorpromazin, Fluphenazin, Perphenazin, Risperidon und Thioridazin, welche durch CYP2D6 metabolisiert werden, empfohlen. Nach Zhou ist es möglich, eine therapeutische Überwachung und einen pharmakogenetischen Test für CYP2D6 in der klinischen Praxis zu verbinden [29]. Nach Hiemke et al. [17] unterscheiden sich die verschiedenen Antipsychotika hinsichtlich ihrer Substratspezifität (Tab. 7). 42 Die Induktion oder Inhibition der metabolischen Enzyme können die Elimination von antipsychotisch wirkenden Substanzen nachhaltig beeinflussen, wenn sie bevorzugt durch ein Enzym abgebaut werden. Dies trifft beispielsweise auf Clozapin zu. Dieser Arzneistoff wird primär durch CYP1A2 abgebaut. Aus diesem Grund können Veränderungen der CYP1A2-Aktivität den therapeutischen Plasmakonzentrationsbereich von Clozapin (350– 600 ng/mL) und somit die klinische Wirkung beeinflussen [22][23][29]. Die Faktoren, die zu einer Beeinflussung der Plasmakonzentrationen (Tab. 10) von Antipsychotika über eine Aktivitätsänderung der CYP-Enzyme (Tab. 7) führen, werden in Tab. 8 und 9 beschrieben. Zu den Inhibitoren, die CYP-Enzyme beeinflussen, welche in den Metabolismus von Antipsychotika eingreifen, gehören Perazin als Inhibitor des CYP1A2 und CYP2C19 sowie Levomepromazin, Melperon, Perphenazin und Thioridazin als Inhibitoren des CYP2D6. Ein Bespiel für eine pharmakokinetische Interaktion ist das Auftreten von EPS aufgrund einer erhöhten Plasmakonzentration unter der Behandlung mit Fluphenazin, welches ein bevorzugtes Substrat von CYP2D6 ist, wenn eine Ko-Medikation mit Paroxetin, einem CYP2D6-Inhibitor, erfolgt. Von Hiemke et al. [17] wurde in einem Update der „AGNP Consensus Guidelines for Therapeutic Drug Monitoring in Psychiatry“ der von Baumann et al. [22] und Hiemke et al. [23] beschriebene therapeutische Bereich der Plasmakonzentration von Fluphenazin von 0,5–2 ng/mL auf 1–10 ng/mL erweitert. Eine Konzentrationserhöhung über diesen Bereich kann zu vermehrt aufkommenden konzentrationsabhängigen Nebenwirkungen wie motorischer Symptomatik führen [29][31][49]. Wenn antipsychotisch wirkende Substanzen einen weiten sicheren Konzentrationsrahmen aufweisen (große therapeutische Breite), besteht für das Auftreten von ungünstigen Symptomen nach dem Hinzufügen eines Inhibitors ein geringeres Risiko als bei Antipsychotika mit einer geringen therapeutischen Breite. Eine Risikoreduktion kann durch die Minimierung solcher Kombinationen und durch die Adaptation der Dosierung unter Kontrolle der Plasmakonzentration erfolgen [29]. Bei Unterbrechung der Behandlung mittels des Inhibitors muss davon ausgegangen werden, dass der Entzug des Inhibitors zu einer Abnahme der Plasmakonzentration der Substanz führt, deren Elimination inhibiert wurde. Die Enthemmung birgt das Risiko einer Symptomverminderung oder Wirkungsverlust. Diesbezüglich ist es gleichsam hilfreich, eine Adaptation der Dosierung unter Kontrolle der Plasmakonzentration der antipsychotisch wirkenden Substanz durchzuführen [29]. 43 CYP-Induktoren und Antipsychotika Für Barbiturate wurde lange angenommen, dass bei chronischer Behandlung nur eine verstärkte Dosierung zum Erreichen der immer gleichen hypnotischen Wirkung erforderlich sei. Es wurde herausgefunden, dass dieser Effekt eine Folge von erhöhter Enzymaktivität in der Leber ist, welche das Ausmaß des Metabolismus und der Exkretion verstärkt [29]. Nach Ma et al. [71] und Zhu et al. [72] ist mittlerweile erwiesen, dass eine erhöhte Enzymaktivität das Resultat einer verstärkten Enzym-Protein-Synthese ist. CYP1A2 und CYP3A4 sind die wichtigsten Enzyme, die für eine Induktion empfänglich sind. Die wichtigsten Substrate von CYP1A2 sind Clozapin und Olanzapin. Für CYP3A4 ist Carbamazepin der wichtigste Induktor. Nach Castberg et al. [24] und Nickl-Jockschat et al. [73] ist Quetiapin ein empfängliches Substrat von CYP3A4. Durch Carbamazepin-Induktion kann die Plasmakonzentration von Quetiapin bis zu 90 % abnehmen, wodurch die Konzentration unter den therapeutischen Bereich fällt und in der Folge die Wirksamkeit abnimmt. Deshalb sollte diese Kombination vermieden werden. Von Hiemke et al. [17] wurde in einem Update der „AGNP Consensus Guidelines for Therapeutic Drug Monitoring in Psychiatry“ der von Baumann et al. [22] und Hiemke et al. [23] beschriebene therapeutische Bereich der Plasmakonzentration von Quetiapin von 70–170 ng/mL auf 100–500 ng/mL erweitert [29][31][49]. Nach Nakamura et al. [74] wäre es suffizienter, die Dosierung von Quetiapin zu adaptieren, um dadurch einen Wirkungsverlust zu vermeiden. Aripiprazol ist weniger empfindlich in Bezug auf enzyminduzierende Ko-Medikationen. Zigarettenrauch und seine Induktorqualitäten Polyzyklische aromatische Kohlenwasserstoffe sind Bestandteil von Zigarettenrauch und sind hierbei die stärksten Induktoren des CYP1A2 [29]. Bondolfi et al. [75], Cole et al. [76] und Lowe und Ackman [77] nehmen unter anderem in einer Veröffentlichung des Journal of Therapeutic Drug Monitoring stärker Bezug auf den Zusammenhang zwischen Induktion und Zigarettenkonsum. Bei unter Clozapin oder Olanzapin Behandlung stehenden PatientInnen kann es durch einen sofortigen Stopp des Zigarettenkonsums zu einer Intoxikation durch die Abnahme 44 der Elimination der Antipsychotika kommen. Hierbei steigen die Plasmakonzentrationen von Clozapin und Olanzapin weit über den therapeutischen Bereich von 350–600 ng/mL bzw. 20–80 ng/mL an [17][31][49]. Nach einer Dosisreduzierung kann die Symptomatik der Intoxikation wieder verschwinden. Dobrinas et al. [78] beschreiben bezüglich des Einflusses von Zigarettenrauch einen genetischen Zusammenhang [29]. Das Ausmaß der pharmakokinetischen Interaktion ist abhängig vom jeweiligen Genotyp. Die nach dem Rauchstopp beobachtete Abnahme der CYP1A2-Aktivität wird verursacht durch den CYP1A2*F-Genotyp. Wenn PatientInnen jedoch unter einer Ko-Medikation mit einem starken Inhibitor des CYP1A2 wie Ciprofloxacin stehen, wird der Rauchstopp, unabhängig vom Genotyp, die Pharmakokinetik nicht beeinflussen. Dies geschieht ab dem Zeitpunkt der Enzymblockierung durch das Antipsychotikum. CAVE: Für die Kombinationstherapie von Arzneimitteln mit Induktorqualitäten wird eine Überwachung der Plasmakonzentrationen und eine Adaptation der Dosierung nach einer Woche nach Beginn oder Beendigung der Einnahme des Induktors empfohlen [29]. Für Faber et al. [79] ist bei einem Abbruch einer Induktor-Behandlung mit Vorsicht vorzugehen. Wenn die Wirkung des Induktors ausfällt, kann die verstärkte Synthese des induzierten Enzyms auf ein Minimum der Expression fallen. Dadurch wird die Biotransformation eines Substrates wieder vermindert. Rosenzweig et al. [80] beziehen sich in einem Review auf die Ausscheidung. Bei Substanzen, die ausschließlich durch aktive Sekretion über die Nierentubuli ausgeschieden werden, könnten Interaktionen während der Exkretionsphase auftreten. Substanzen wie Amiodaron, Clarithromycin, Itraconazol, Propafenon oder Quinidin können diesen Prozess inhibieren. Bis jetzt sind allerdings nur mangelhafte Daten bezüglich Interaktionen mit Amisulprid und Sulpirid vorhanden. Die meisten antipsychotisch wirkenden Substanzen durchlaufen den Phase-Ι-Metabolismus und die nachfolgende Konjugation zu Glukuronsäure. Diese Arzneimittel sind nicht empfänglich für Interaktionen während des Exkretionsprozesses. 45 4. Diskussion 4.1 Studien über pharmakodynamische Interaktionen Veröffentlichungen zum Abschnitt Additive anticholinerge Nebenwirkungen Eine Therapie mittels kritischer Kombinationen von Medikamenten wäre durch die Verfügbarkeit von verifizierten Daten über die mögliche substanzspezifische Ausprägung anticholinerger Wirkung möglich. In diesem Zusammenhang kann die im Journal of the American Geriatrics Society kürzlich veröffentlichte Studie von Chew et al. [49] bei der klinischen Anwendung von Arzneimittelkombinationen und bei der Vermeidung ungünstiger Wechselwirkungen hilfreich sein, da in den Ergebnissen eine Abstufung bezüglich der anticholinergen Wirksamkeit verschiedener Antipsychotika vorgenommen wird. Die Evidenz solcher Daten spiegelt sich in einigen, sich mitunter entsprechenden, Veröffentlichungen wider. Cancelli et al. [46] sowie Strobach [47], Cascorbi [48] und Hafner et al. [2] stimmen in ihren Veröffentlichungen überein, dass die Kombination von einigen Antipsychotika vor allem mit trizyklischen Antidepressiva zu solchen kumulativen arzneimittelinduzierten anticholinergen Effekten führen kann. Cancelli et al. fügen außerdem an, dass Wechselwirkungen auch unter Kombination mit Histamin-H1-Rezeptorantagonisten der 1. Generation auftreten können. Wobei Cascorbi hinsichtlich möglicher Interaktionen die Gruppe der Phenothiazine in Kombination mit trizyklischen Antidepressiva hervorgehoben sehen will. In Bezug auf ein fortgeschritteneres Alter der PatientInnen besteht für Strobach nach einer Untersuchung ein Zusammenhang zwischen häufig verabreichten Antipsychotika in Verbindung mit verabreichten Analgetika und dem Schweregrad eines dadurch auftretenden Deliriums. Weiters stimmt der Review-Artikel (Current Antipsychotics) im Handbook of Experimental Pharmacology von 2012 [29] der Ansicht Strobachs hinsichtlich der übermäßig stark ausgeprägten anticholinergen Effekte bei einer Blockade von muskarinergen Acetylcholinrezeptoren zu. 46 Veröffentlichungen zum Abschnitt Kardiovaskuläre Effekte Betreffend eine Einschätzung zur Vermeidung des arzneimittelinduzierten Auftretens von QT-Prolongation und die richtige Anwendung bzw. die Vermeidung von ungünstigen Kombinationstherapien ist das im Review-Artikel (Current Antipsychotics) im Handbook of Experimental Pharmacology von 2012 [29] beschriebene „summary of product characteristics“ (SPC) der FDA zu nennen. Dieser Veröffentlichung wurde eine Warnung beigefügt, um zu verhindern, dass atypische Antipsychotika wie Quetiapin in Verbindung mit anderen Substanzen wie Quinidin, Procainamid, Amiodaron, Sotalol, Gatifloxacin oder Moxifloxacin gebracht werden. Dies gilt auch für Lithium. Ähnliche Warnungen wären auch für andere arzneimittelinduzierte Nebenwirkungen wünschenswert, da nach van Noord et al. [51] und Wenzel-Seifert et al. [31] mögliche QT-Verlängerungen nicht nur für atypische Antipsychotika wie Quetiapin in Kombination mit anderen Substanzen beschrieben werden. Dementsprechend wären auch für diese Kombinationen offizielle Warnungen von Vorteil. Die von Tisdale et al. [54] und van Noord et al. [51] beschriebenen Folgen (TdP, Kammerflimmern) könnten dadurch vermindert werden. Veröffentlichungen zum Abschnitt Exazerbation der Parkinson-Symptomatik sowie der extrapyramidalen Symptome (EPS) In den Veröffentlichungen von Jost et al. [58], Cascorbi [48] und Hafner et al. [2] bestehen Übereinstimmungen betreffend den Zusammenhang der Blockade von Dopamin-D2Rezeptoren und der Antagonisierung des Effekts von Levodopa. Ghadirian et al. [59] fügt Lithium als weiteres Risikomedikament für das Auftreten exrapyramidaler Symptomatik an. Weiters bestehen Unterschiede bezüglich der Behandlung. Dem Review-Artikel (Current Antipsychotics) im Handbook of Experimental Pharmacology von 2012 [29] zufolge können durch L-Dopa induzierte psychotische Symptome durch Antipsychotika effektiv behandelt werden, zweckmäßiger bleibt aber die Behandlung durch eine Reduktion der LDopa-Dosis. Gross und Drescher [60] sowie Richelsen und Souder [32] schlagen in ihren Veröffentlichungen vor, psychotische Symptome im Rahmen des M. Parkinson eher durch die Anwendung einer geringen Dosis einer anderen dopaminergen Substanz als L-Dopa zu behandeln. 47 Veröffentlichungen zum Abschnitt Gewichtszunahme und das metabolische Syndrom Eine Unterteilung hinsichtlich der Wahrscheinlichkeit des Auftretens von Gewichtszunahme und des Auftretens eines metabolischen Syndroms kann man aufgrund der erhobenen Daten von Newcomer [62] und Mitchell et al. [63] (Meta-Analyse) treffen. Dies ist, wie bereits im Zusammenhang mit der QT-Verlängerung und der additiven anticholinergen Wirkung von mir angesprochen, für die klinische Anwendung von Kombinationstherapien und für eine optimale Behandlung sinnvoll. Die Notwendigkeit einer solchen Unterteilung bzw. Zuordnung bezüglich des Potentials für ungünstige Wechselwirkungen (Baptista et al. [64]) während einer Kombinationstherapie mit mehreren Medikamenten wird durch die Ergebnisse einer veröffentlichten Querschnittsstudie von Misawa et al. [65] gestützt. Diese besagt, dass ein erhöhtes Risiko für die beschriebenen Nebenwirkungen bei Anwendung einer Polypharmazie im Gegensatz zu einer Monotherapie besteht. 48 4.2 Studien über pharmakokinetische Interaktionen Die Relevanz der für die pharmakokinetische Interaktionen verantwortlichen Prozesse ist für die praxisbezogene Anwendung von großer Bedeutung. In Bezug auf die Interpretation der Veröffentlichungen ergeben sich folgende Auffassungen zu den Themen Pg-P, Proteinbindung und CYP-Isoenzyme. Cascorbi [66] bezieht sich hinsichtlich möglicher Interaktionen unter anderem auf die für die Ausscheidung mitverantwortlichen spezifischen Transporter (P-gP). Auch Moons et al. [15] sehen eine Beteiligung des P-gP. Im Gegensatz zu Cascorbi lassen Moons et al. jedoch anklingen, sich über die klinische Relevanz von P-gP-Inhibitoren nicht im Klaren zu sein. Eine ähnliche Meinung lässt sich auch in der Veröffentlichung von Dürr et al. [67] ablesen, die eine Einflussnahme von P-gP-Induktoren auf die Verfügbarkeit von Antipsychotika beschreiben, aber auch klar die Relevanz dieser Einflussnahme während der Absorptionsphase anzweifeln. Ein ähnliches Relevanzproblem betrifft die Plasmaproteinbindung. Nach den Ergebnissen von Benet und Hoener [68] ist es unwahrscheinlich, dass das Ausmaß der Plasmaproteinbindung die Wirksamkeit von Antipsychotika in vivo verändern kann. Im Gegensatz zur Bedeutung des P-gP und der Proteinbindung besteht eine autorenübergreifende Übereinstimmung bezüglich der hohen Relevanz der CYP-Isoenzyme, deren substanzspezifischen Induktion und Inhibition sowie der Auswirkungen auf Plasmaspiegel und somit die Wirkung von Antipsychotika. Im Zusammenhang mit einer praxisbezogenen Anwendung der Plasmaspiegelkontrolle sollte diese als unterstützendes Werkzeug zur Detektion von kinetischen Interaktionen sowie zur Therapieüberwachung angesehen werden. Der praktische Nutzen ergibt sich aus Fällen, in denen entweder die PatientInnen nicht auf die standardisierte Dosis eines Medikamentes ansprechen bzw. es zu UAW kommt. Speziell bei Arzneistoffen mit einer geringen therapeutischen Breite kann es zu starken Schwankungen der Plasmaspiegel kommen. In Bezug auf die Medikamente mit einer geringen therapeutischen Breite kann die Kontrolle des Plasmaspiegels im Zuge einer notwendigen individuelleren Therapie für mehr Sicherheit und Benefit für die PatientInnen sorgen. Weiters können auch Begleiterkrankungen die Bestimmung der richtigen Dosis zum Erreichen eines therapeutischen Effektes eines Medikamentes erschweren. Auch bei indizierter Anwendung einer Polypharmazie, die das Risiko von Arzneimittelinteraktionen birgt, sind Kontrollen der Plasmakonzentrationen unabdingbar. Diese Faktoren und die exakte Bestimmbarkeit der Plasmakonzentration sprechen für die Plasmaspiegelkontrolle als 49 Mittel der Wahl zur pharmakologischen Therapieüberwachung und Steuerung. Generelle Leitlinien zur Vermeidung von Arzneimittelinteraktionen beschreiben Lemmer und Brune [81] in ihrem MASTER-System. Tab. 17: Das MASTER-System nach Lemmer und Brune M inimum an Arzneimitteln A lternative Medikamente einsetzen S tarte mit niedriger Dosis – langsame Dosissteigerung T itriere Dosierung nach Wirkung E rkläre den PatientInnen mögliche Probleme R egelmäßige Überwachung von PatientInnen und Arzneimittelwirkungen Adaptiert nach Lemmer und Brune 2007 [81] Diese Arbeit soll zur weiteren Sensibilisierung bezüglich der Problematik der Arzneimittelinteraktionen der Antipsychotika beitragen. 50 5. Literaturverzeichnis [1] Mutschler E, Geisslinger G, Kroemer HK, Ruth P, Schäfer-Korting M. Mutschler Arzneimittelwirkungen kompakt: Basiswissen Pharmakologie, Toxikologie. Stuttgart: Wiss. Verl.Ges.; 2005. [2] Hafner V, Grün B, Markert C, Czock D, Mikus G, Haefeli WE. [Drug interactions]. Internist (Berl). 2010 Mar;51(3):359-69; quiz 370. doi: 10.1007/s00108-009-2553-1. German. PubMed PMID: 20127303. [3] Rivkin A. Admissions to a medical intensive care unit related to adverse drug reactions. Am J Health Syst Pharm. 2007 Sep 1;64(17):1840-3. PubMed PMID: 17724366. [4] Bertsche T, Pfaff J, Schiller P, Kaltschmidt J, Pruszydlo MG, Stremmel W, Walter-Sack I, Haefeli WE, Encke J. Prevention of adverse drug reactions in intensive care patients by personal intervention based on an electronic clinical decision support system. Intensive Care Med. 2010 Apr;36(4):665-72. doi: 10.1007/s00134-010-1778-8. Epub 2010 Feb 9. PubMed PMID: 20143221. Lechleitner M. Educationen: Arzneimittelinteraktionen. Wiener Klinische Wochenschrift 2010; (1). [5] Lechleitner M. Educationen: Arzneimittelinteraktionen. Wiener Klinische Wochenschrift 2010; (1). [6] Hansten PD, Horn JR, Hazlet TK. ORCA: OpeRational ClassificAtion of drug interactions. J Am Pharm Assoc (Wash). 2001 Mar-Apr;41(2):161-5. PubMed PMID: 11297327. [7] Zagermann-Muncke P. Neue Interaktionsklassifikation: ABDA-Datenbank als Wegweiser im Wechselwirkungsdschungel. Pharmazeutische Zeitung (PZ) 2009; (2). Available from: URL:http://www.pharmazeutische-zeitung.de/index.php?id=18013. [8] ABDATA Pharma-Daten-Service eUdWVDAm. Einführung einer neuen Klassifikation für Interaktionen per 01.11.2009: Neue Klassifikation der Interaktionen; 2009. Available from: URL:http://www.pharmavista.net/downloads/company/download/D1_Neue_Klassifikation_IA.pdf. [9] Katzendobler NE. Verordnung von Antipsychotika und Antidepressiva an bayerischen Bezirkskrankenhäusern – eine vergleichende Untersuchung von Daten aus AMÜP-AGATE und dem Bezirkskrankenhaus Augsburg [Dissertation]. München: LMU; 2007. [10] Hausner H, Wittmann M, Hajak G, Haen E. Polypharmazie als geschlechtsspezifisches Phänomen in der Psychiatrie. Psychopharmakotherapie 2008; (15):21–3. [11] Riederer PF, Laux G. Grundlagen der Neuro-Psychopharmakologie: Ein Therapiehandbuch. Vienna: Springer-Verlag Vienna; 2010. [12] Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling. Br J Pharmacol. 2007 Sep;152(1):9-20. Epub 2007 Jun 4. Review. PubMed PMID: 17549047; PubMed Central PMCID: PMC1978274. [13] Gründer G, Benkert O. Handbuch der Psychopharmakotherapie. 2nd ed. Berlin: Springer; 2012. [14] Spina E, Avenoso A, Facciolà G, Salemi M, Scordo MG, Giacobello T, Madia AG, Perucca E. Plasma concentrations of risperidone and 9-hydroxyrisperidone: effect of comedication with carbamazepine or valproate. Ther Drug Monit. 2000 Aug;22(4):481-5. PubMed PMID: 10942191. 51 [15] Moons T, de Roo M, Claes S, Dom G. Relationship between P-glycoprotein and secondgeneration antipsychotics. Pharmacogenomics. 2011 Aug;12(8):1193-211. doi: 10.2217/pgs.11.55. Review. PubMed PMID: 21843066. [16] Aktories K, Förstermann U, Hofmann FB, Starke K, Forth W. Allgemeine und spezielle Pharmakologie und Toxikologie: Für Studenten der Medizin, Veterinärmedizin, Pharmazie, Chemie und Biologie sowie für Ärzte, Tierärzte und Apotheker. 9th ed. München: Elsevier, Urban & Fischer; 2006. [17] Hiemke C, Baumann P, Bergemann N, Conca A, Dietmaier O, Egberts K, Fric M, Gerlach M, Greiner C, Gründer G, Haen E, Havemann-Reinecke U, Jaquenoud Sirot E, Kirchherr H, Laux G, Lutz UC, Messer T, Müller MJ, Pfuhlmann B, Rambeck B, Riederer P, Schoppek B, Stingl J, Uhr M, Ulrich S, Waschgler R, Zernig G. AGNP consensus guidelines for therapeutic drug monitoring in psychiatry: update 2011. Pharmacopsychiatry. 2011 Sep;44(6):195-235. Review. PubMed PMID: 22053351. [18] Spina E, Scordo MG, D'Arrigo C. Metabolic drug interactions with new psychotropic agents. Fundam Clin Pharmacol. 2003 Oct;17(5):517-38. Review. PubMed PMID: 14703714. [19] Pang X, Cheng J, Krausz KW, Guo DA, Gonzalez FJ. Pregnane X receptor-mediated induction of Cyp3a by black cohosh. Xenobiotica. 2011 Feb;41(2):112-23. doi: 10.3109/00498254.2010.527021. Epub 2010 Oct 27. PubMed PMID: 20979450; PubMed Central PMCID: PMC3019262. [20] Sinz M, Wallace G, Sahi J. Current industrial practices in assessing CYP450 enzyme induction: preclinical and clinical. AAPS J. 2008 Jun;10(2):391-400. doi: 10.1208/s12248-0089037-4. Epub 2008 Aug 7. Review. PubMed PMID: 18686044; PubMed Central PMCID: PMC2751387. [21] Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007 Aug 1;76(3):391-6. Review. PubMed PMID: 17708140. [22] Baumann P, Hiemke C, Ulrich S, Eckermann G, Gaertner I, Gerlach M, Kuss HJ, Laux G, Müller-Oerlinghausen B, Rao ML, Riederer P, Zernig G; Arbeitsgemeinschaft für Neuropsychopharmakologie und Pharmakopsychiatrie. The AGNP-TDM expert group consensus guidelines: therapeutic drug monitoring in psychiatry. Pharmacopsychiatry. 2004 Nov;37(6):243-65. Review. PubMed PMID: 15551191. [23] Hiemke C, Baumann P, Laux G, Kuss HJ. Therapeutisches Drug Monitoring in der Psychiatrie. Psychopharmakotherapie 2005; (12):166–82. [24] Castberg I, Skogvoll E, Spigset O. Quetiapine and drug interactions: evidence from a routine therapeutic drug monitoring service. J Clin Psychiatry. 2007 Oct;68(10):1540-5. PubMed PMID: 17960969. [25] Jerling M, Lindström L, Bondesson U, Bertilsson L. Fluvoxamine inhibition and carbamazepine induction of the metabolism of clozapine: evidence from a therapeutic drug monitoring service. Ther Drug Monit. 1994 Aug;16(4):368-74. PubMed PMID: 7974626. [26] Szegedi A, Anghelescu I, Wiesner J, Schlegel S, Weigmann H, Härtter S, Hiemke C, Wetzel H. Addition of low-dose fluvoxamine to low-dose clozapine monotherapy in schizophrenia: drug 52 monitoring and tolerability data from a prospective clinical trial. Pharmacopsychiatry. 1999 Jul;32(4):148-53. PubMed PMID: 10505485. [27] Silver H. Fluvoxamine as an adjunctive agent in schizophrenia. CNS Drug Rev. 2001 Fall;7(3):283-304. Review. PubMed PMID: 11607044. [28] Lu ML, Lane HY, Chen KP, Jann MW, Su MH, Chang WH. Fluvoxamine reduces the clozapine dosage needed in refractory schizophrenic patients. J Clin Psychiatry. 2000 Aug;61(8):594-9. PubMed PMID: 10982203. [29] Hiemke C, Pfuhlmann B. Interactions and monitoring of antipsychotic drugs. Handb Exp Pharmacol. 2012;(212):241-65. doi: 10.1007/978-3-642-25761-2_10. PubMed PMID: 23129335. [30] Silvestre JS, Prous J. Research on adverse drug events. I. Muscarinic M3 receptor binding affinity could predict the risk of antipsychotics to induce type 2 diabetes. Methods Find Exp Clin Pharmacol. 2005 Jun;27(5):289-304. PubMed PMID: 16082416. [31] Wenzel-Seifert K, Wittmann M, Haen E. QTc prolongation by psychotropic drugs and the risk of Torsade de Pointes. Dtsch Arztebl Int. 2011 Oct;108(41):687-93. doi: 10.3238/arztebl.2011.0687. Epub 2011 Oct 14. Review. PubMed PMID: 22114630; PubMed Central PMCID: PMC3221427. [32] Richelson E, Souder T. Binding of antipsychotic drugs to human brain receptors focus on newer generation compounds. Life Sci. 2000 Nov 24;68(1):29-39. PubMed PMID: 11132243. [33] Benkert O, Hippius H, Fehr C, Gründer G, Hiemke C, Lange-Asschenfeldt C, Müller MJ, Regen F, Heiser P, Steiger A. Kompendium der psychiatrischen Pharmakotherapie. 8th ed. Berlin, Heidelberg, New York, NY: Springer Medizin; 2011. [34] Kähler CM, Kähler ST, Wiedermann CJ, Cima K, Feistritzer C, Pechlaner C, Kähler A. Das schlaue Büchlein: Österreichische Arzneimittel im Taschenformat. 7th ed. Kössen: pm-Verlag; 2012. [35] Rothenhäusler HB, Täschner KL. Kompendium Praktische Psychiatrie. Vienna: SpringerVerlag/Wien; 2007. [36] Graefe KH, Lutz W, Bönisch H, Hahn JM. Pharmakologie und Toxikologie. Stuttgart: Thieme; 2011. [37] National Center for Biotechnology Information. PubChem Compound: Chlorpromazine Compound Summary. Available from: URL:http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=2726&loc=ec_rcs. [38] National Center for Biotechnology Information. PubChem Compound: Chlorprothixene Compound Summary. Available from: URL:http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=667467&loc=ec_rcs. [39] National Center for Biotechnology Information. PubChem Compound: Haloperidol - Compound Summary. Available from: URL:http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=3559&loc=ec_rcs. [40] National Center for Biotechnology Information. PubChem Compound: Sulpiride - Compound Summary. Available from: URL:http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=5355. [41] National Center for Biotechnology Information. PubChem Compound: Clozapine - Compound Summary. Available from: URL:http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=2818. 53 [42] National Center for Biotechnology Information. PubChem Compound: Olanzapine - Compound Summary. Available from: URL:http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=4585&loc=ec_rcs. [43] National Center for Biotechnology Information. PubChem Compound: Quetiapine - Compound Summary. Available from: URL:http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=5002&loc=ec_rcs. [44] National Center for Biotechnology Information. PubChem Compound: Risperidon - Compound Summary. Available from: URL:http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=44402564. [45] National Center for Biotechnology Information. PubChem Compound: Aripiprazole Compound Summary. Available from: URL:http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=60795&loc=ec_rcs. [46] Cancelli I, Beltrame M, Gigli GL, Valente M. Drugs with anticholinergic properties: cognitive and neuropsychiatric side-effects in elderly patients. Neurol Sci. 2009 Apr;30(2):87-92. doi: 10.1007/s10072-009-0033-y. Epub 2009 Feb 20. Review. PubMed PMID: 19229475. [47] Strobach D. [Clinically significant drug-drug interactions between analgesics and psychotopics]. Med Monatsschr Pharm. 2012 Jul;35(7):245-54; quiz 255-6. Review. German. PubMed PMID: 22852275. [48] Cascorbi I. Drug interactions – principles, examples and clinical consequences. Dtsch Arztebl Int. 2012 Aug;109(33-34):546-55; quiz 556. doi: 10.3238/arztebl.2012.0546. Epub 2012 Aug 20. PubMed PMID: 23152742; PubMed Central PMCID: PMC3444856. [49] Chew ML, Mulsant BH, Pollock BG, Lehman ME, Greenspan A, Mahmoud RA, Kirshner MA, Sorisio DA, Bies RR, Gharabawi G. Anticholinergic activity of 107 medications commonly used by older adults. J Am Geriatr Soc. 2008 Jul;56(7):1333-41. doi: 10.1111/j.1532-5415.2008.01737.x. Epub 2008 May 26. PubMed PMID: 18510583. [50] Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest. 2005 Aug;115(8):2025-32. Review. PubMed PMID: 16075043; PubMed Central PMCID: PMC1180553. [51] van Noord C, Eijgelsheim M, Stricker BH. Drug- and non-drug-associated QT interval prolongation. Br J Clin Pharmacol. 2010 Jul;70(1):16-23. doi: 10.1111/j.1365-2125.2010.03660.x. Review. PubMed PMID: 20642543; PubMed Central PMCID: PMC2909803. [52] Kannankeril PJ, Roden DM. Drug-induced long QT and torsade de pointes: recent advances. Curr Opin Cardiol. 2007 Jan;22(1):39-43. Review. PubMed PMID: 17143043. [53] Roden DM. Drug-induced prolongation of the QT interval. N Engl J Med. 2004 Mar 4;350(10):1013-22. Review. PubMed PMID: 14999113. [54] Tisdale JE, Overholser BR, Wroblewski HA, Sowinski KM, Amankwa K, Borzak S, Kingery JR, Coram R, Zipes DP, Flockhart DA, Kovacs RJ. Enhanced sensitivity to drug-induced QT interval lengthening in patients with heart failure due to left ventricular systolic dysfunction. J Clin Pharmacol. 2012 Sep;52(9):1296-305. doi: 10.1177/0091270011416939. Epub 2011 Nov 1. PubMed PMID: 22045830. [55] Crumb WJ Jr, Ekins S, Sarazan RD, Wikel JH, Wrighton SA, Carlson C, Beasley CM Jr. Effects of antipsychotic drugs on I(to), I (Na), I (sus), I (K1), and hERG: QT prolongation, structure 54 activity relationship, and network analysis. Pharm Res. 2006 Jun;23(6):1133-43. Epub 2006 May 25. PubMed PMID: 16715368. [56] Unterecker S, Warrings B, Deckert J, Pfuhlmann B. Correlation of QTc interval prolongation and serum level of citalopram after intoxication – a case report. Pharmacopsychiatry. 2012 Jan;45(1):30-4. doi: 10.1055/s-0031-1286346. Epub 2011 Oct 12. PubMed PMID: 21993870. [57] Letsas KP, Sideris A, Kounas SP, Efremidis M, Korantzopoulos P, Kardaras F. Drug-induced QT interval prolongation after ciprofloxacin administration in a patient receiving olanzapine. Int J Cardiol. 2006 May 10;109(2):273-4. Epub 2005 Jun 1. PubMed PMID: 15935493. [58] Jost WH, Brück C. Drug interactions in the treatment of Parkinson's disease. J Neurol. 2002 Oct;249 Suppl 3:III/24-9. Review. PubMed PMID: 12522568. [59] Ghadirian AM, Annable L, Bélanger MC, Chouinard G. A cross-sectional study of parkinsonism and tardive dyskinesia in lithium-treated affective disordered patients. J Clin Psychiatry. 1996 Jan;57(1):22-8. PubMed PMID: 8543543. [60] Gross G, Drescher K. The role of dopamine D(3) receptors in antipsychotic activity and cognitive functions. Handb Exp Pharmacol. 2012;(213):167-210. doi: 10.1007/978-3-642-257582_7. PubMed PMID: 23027416. [61] Heal DJ, Gosden J, Jackson HC, Cheetham SC, Smith SL. Metabolic consequences of antipsychotic therapy: preclinical and clinical perspectives on diabetes, diabetic ketoacidosis, and obesity. Handb Exp Pharmacol. 2012;(212):135-64. doi: 10.1007/978-3-642-25761-2_6. PubMed PMID: 23129331. [62] Newcomer JW. Metabolic considerations in the use of antipsychotic medications: a review of recent evidence. J Clin Psychiatry. 2007;68 Suppl 1:20-7. Review. PubMed PMID: 17286524. [63] Mitchell AJ, Vancampfort D, Sweers K, van Winkel R, Yu W, De Hert M. Prevalence of Metabolic Syndrome and Metabolic Abnormalities in Schizophrenia and Related Disorders – A Systematic Review and Meta-Analysis. Schizophr Bull. 2011 Dec 29. [Epub ahead of print] PubMed PMID: 22207632. [64] Baptista T, Zárate J, Joober R, Colasante C, Beaulieu S, Páez X, Hernández L. Drug induced weight gain, an impediment to successful pharmacotherapy: focus on antipsychotics. Curr Drug Targets. 2004 Apr;5(3):279-99. Review. PubMed PMID: 15058313. [65] Misawa F, Shimizu K, Fujii Y, Miyata R, Koshiishi F, Kobayashi M, Shida H, Oguchi Y, Okumura Y, Ito H, Kayama M, Kashima H. Is antipsychotic polypharmacy associated with metabolic syndrome even after adjustment for lifestyle effects?: a cross-sectional study. BMC Psychiatry. 2011 Jul 26;11:118. doi: 10.1186/1471-244X-11-118. PubMed PMID: 21791046; PubMed Central PMCID: PMC3155482. [66] Cascorbi I. P-glycoprotein: tissue distribution, substrates, and functional consequences of genetic variations. Handb Exp Pharmacol. 2011;(201):261-83. doi: 10.1007/978-3-642-14541-4_6. Review. PubMed PMID: 21103972. [67] Dürr D, Stieger B, Kullak-Ublick GA, Rentsch KM, Steinert HC, Meier PJ, Fattinger K. St John's Wort induces intestinal P-glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin Pharmacol Ther. 2000 Dec;68(6):598-604. PubMed PMID: 11180019. 55 [68] Benet LZ, Hoener BA. Changes in plasma protein binding have little clinical relevance. Clin Pharmacol Ther. 2002 Mar;71(3):115-21. Review. PubMed PMID: 11907485. [69] Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part II. Clin Pharmacokinet. 2009;48(12):761-804. doi: 10.2165/11318070-000000000-00000. PubMed PMID: 19902987. [70] Spina E, Santoro V, D'Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008 Jul;30(7):1206-27. Review. PubMed PMID: 18691982. [71] Ma X, Idle JR, Gonzalez FJ. The pregnane X receptor: from bench to bedside. Expert Opin Drug Metab Toxicol. 2008 Jul;4(7):895-908. doi: 10.1517/17425255.4.7.895 . Review. PubMed PMID: 18624678; PubMed Central PMCID: PMC2535920. [72] Zhu BT. On the general mechanism of selective induction of cytochrome P450 enzymes by chemicals: some theoretical considerations. Expert Opin Drug Metab Toxicol. 2010 Apr;6(4):48394. doi: 10.1517/17425250903578642. Review. PubMed PMID: 20113197; PubMed Central PMCID: PMC2842473. [73] Nickl-Jockschat T, Paulzen M, Schneider F, Grözinger M. Drug interaction can lead to undetectable serum concentrations of quetiapine in the presence of carbamazepine. Clin Neuropharmacol. 2009 Jan-Feb;32(1):55. doi: 10.1097/WNF.0b013e31816a1cc6. PubMed PMID: 19471186. [74] Nakamura A, Mihara K, Nagai G, Suzuki T, Kondo T. Pharmacokinetic and pharmacodynamic interactions between carbamazepine and aripiprazole in patients with schizophrenia. Ther Drug Monit. 2009 Oct;31(5):575-8. doi: 10.1097/FTD.0b013e3181b6326a. PubMed PMID: 19701114. [75] Bondolfi G, Morel F, Crettol S, Rachid F, Baumann P, Eap CB. Increased clozapine plasma concentrations and side effects induced by smoking cessation in 2 CYP1A2 genotyped patients. Ther Drug Monit. 2005 Aug;27(4):539-43. PubMed PMID: 16044115. [76] Cole ML, Trigoboff E, Demler TL, Opler LA. Impact of smoking cessation on psychiatric inpatients treated with clozapine or olanzapine. J Psychiatr Pract. 2010 Mar;16(2):75-81. doi: 10.1097/01.pra.0000369968.80155.3f. PubMed PMID: 20511731. [77] Lowe EJ, Ackman ML. Impact of tobacco smoking cessation on stable clozapine or olanzapine treatment. Ann Pharmacother. 2010 Apr;44(4):727-32. doi: 10.1345/aph.1M398. Epub 2010 Mar 16. Review. PubMed PMID: 20233914. [78] Dobrinas M, Cornuz J, Oneda B, Kohler Serra M, Puhl M, Eap CB. Impact of smoking, smoking cessation, and genetic polymorphisms on CYP1A2 activity and inducibility. Clin Pharmacol Ther. 2011 Jul;90(1):117-25. doi: 10.1038/clpt.2011.70. Epub 2011 May 18. PubMed PMID: 21593735. [79] Faber MS, Jetter A, Fuhr U. Assessment of CYP1A2 activity in clinical practice: why, how, and when? Basic Clin Pharmacol Toxicol. 2005 Sep;97(3):125-34. Review. PubMed PMID: 16128905. [80] Rosenzweig P, Canal M, Patat A, Bergougnan L, Zieleniuk I, Bianchetti G. A review of the pharmacokinetics, tolerability and pharmacodynamics of amisulpride in healthy volunteers. Hum Psychopharmacol. 2002 Jan;17(1):1-13. Review. PubMed PMID: 12404702. 56 [81] Lemmer B, Brune K. Pharmakotherapie: Klinische Pharmakologie. 13th ed. Heidelberg: Springer; 2007. 57