Genetische und Physiologische Grundlagen der Achromatopsie
Werbung

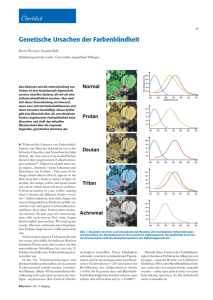
Genetische und Physiologische Grundlagen der Achromatopsie Susanne Kohl Molekulargenetisches Labor Universitäts-Augenklinik Tübingen 1. Achromatopsie-Symposium Maritim-Hotel Gelsenkirchen 21. April 2007 Überblick Grundlagen der Genetik Physiologie des Sehens Achromatopsie Welche Ursachen kennen wir ? Molekulargenetisches Labor der Universitäts-Augenklinik Mindestens 170 erbliche Erkrankungen mit Auswirkungen auf das Sehsystem Bis heute allein für die erblichen NetzhautErkrankungen 132 Gene identifiziert Mit was arbeiten wir? DNA = Träger der Erbsubstanz Genom: Gesamtheit der vererbbaren Information einer Zelle (ca. 25.000 Gene) Chromosom: kurzer p-Arm, langer q-Arm Begriffe in der Genetik Gen: DNA-Abschnitt, der die Grundinformationen für die biologisch aktive Botenmolekül enthält, die wiederum für ein funktionelles Produkt (Protein) kodiert Gen-Locus: physikalische Position eines Gens im Genom bzw. auf dem Chromosom Allel: Merkmalsausfertigung / Ausprägungsform eines Gens (z.B. blonde oder braune Haare, Gen-Defekt oder Wildtyp = gesund) Gene – Allele 46 Chromosomen: 2 x 23 Paare = 2 x 22 = 44 Autosomen, +2 = Geschlechts-Chromosomen Frau 46, XX / Mann 46, XY Daher hat jeder gesunde Mensch von allen autosomalen Genen zwei Kopien = zwei Allele, eine vom Vater, und eine von der Mutter Ausnahme: X- und Y-Chromosom X-chromosomale (geschlechtsgebundene) Vererbung Nur männliche Betroffene haben keine betroffenen Kinder, aber geben die Anlage an alle ihre Töchter weiter Männliche Nachkommen haben kein Risiko die Erkrankung zu ererben Weibliche (gesunde) Überträgerinnen haben ein 50% Risiko, die Anlage an ihre Töchter zu vererben 50% Risiko, dass ihre Söhne Anlage ererben und erkranken werden Beispiel für X-chromosomale (geschlechtsgebundene) Vererbung Autosomal dominante Vererbung Geschlechtsunabhängig Betroffene in jeder Generation Jeder Anlageträger erkrankt 50% Risiko, betroffene Kinder zu bekommen für jeden Anlageträger Beispiel für autosomal dominante Vererbung Autosomal rezessive Vererbung Beide Eltern sind gesund, aber Anlageträger Meist keine Betroffenen in früheren Generationen Betroffene müssen je ein defektes Allel von Vater und Mutter ererben, d.h. beide Kopien sind defekt 25% Risiko für weitere betroffene Kinder 50% der Kinder sind wieder Anlageträger, aber gesund 25% der Kinder sind keine Anlageträger Autosomal rezessive Vererbung (2) Gesunde Anlageträger haben nur minimal erhöhtes Risiko für betroffene Kinder Betroffener hat ebenfalls nur minimal erhöhtes Risiko, kranke Kinder zu bekommen Ausnahme: Konsanguinität = Blutsverwandtschaft bei Partnerwahl Beispiele für autosomal rezessive Vererbung Vom Gen zum Protein DNA GAG TAT CAT CAA CCT ATG GAG TAT CAT CAA CCT ATG Besteht aus den vier Basen A, G, C und T Je 3 Basen kodieren für eine Aminosäure Protein Besteht aus einzelnen Bausteinen, den 20 verschiedene Aminosäuren Vom Gen zum Protein: Mutation – Austausch einer Aminosäure DNA GAG TAT CAT CAA CCT ATG Aminosäuren Ein Basenaustausch = Mutation in der DNA führt zu einer veränderten Aminosäure und damit zu einem veränderten Proteins GAG TAT CAT CAA CCT AAG Vom Gen zum Protein: Mutation – verkürztes Protein DNA GAG TAT CAT CAA CCT ATG Aminosäuren Mutation in der DNA führt zum Abbruch der Proteinsynthese und zu einem verkürzten Protein GAG TAA T CAT CAA CCTATG STOP Beispiel zwei verschiedener Mutationen bei Patienten mit Achromatopsie Wildtyp Sequenz homozygot c.887C>T Arginin-283-Tryptophan Wildtyp Sequenz heterozygot c.888G>A Arginin-283-Glutamin Auge – Netzhaut – Photorezeptoren Das Photorezeptor-System Stäbchen Sehen in der Dämmerung / Dunkelheit Stäbchen Zapfen Sehen bei hellen Leuchtintensitäten Farbensehen Zapfen Das Stäbchen-System Dämmerungssehen und Sehen in der Dunkelheit Maximale LichtSensitivität Dichte an Stäbchen in der mittleren Peripherie am Höchsten Das Zapfen-System Tagessehen Farbensehen, Sehschärfe und höchste Auflösung Höchste Dichte im Zentrum der Netzhaut > Fovea = gelber Fleck: Stelle des schärfsten Sehens Dichte der Zapfen nimmt zur Peripherie hin ab Das Farbensehen Drei verschiedene Typen von ZapfenPhotorezeptoren: S (shortwave = kurzwelligsensitiver)-Typ: „blau“ M (middlewave = mittelwelligsensitiver)-Typ: „grün“ L (longwave = langwelligsensitiver)-Typ: „rot“ Achromatopsie Synonyme: Stäbchen-Monochromasie Hemeralopie = Tagblindheit Komplette Farbenblindheit Häufigkeit: 1 : 30.000 Erbgang: autosomal rezessiv Achromatopsie - Klinik Photophobie (Blendempfindlichkeit) Nystagmus (Augenpendeln /Augenzittern) Fehlendes Farbensehen Stark reduzierte Sehschärfe 0.1 - 0.2 keine Zapfen-Antwort im ERG (=Elektroretinogramm) Kongenital (angeboren bzw. frühkindlich) Stationär Differential-Diagnose: Blauzapfen-Monochromasie Stark reduzierte Sehschärfe (0.2-0.3) Photophobie Nystagmus (nicht immer) Stark eingeschränkte Farbwahrnehmung X-chromosomaler Erbgang = nur männliche Betroffene Mutationen und Rearrangements der Rot-/Grün-Opsin-Gene auf dem X-Chromosom Die Physiologischen Grundlagen der Achromatopsie Phototransduktion = Sehkaskade Phototransduktion - Sehkaskade Licht Photopigment Transducin Phosphodiesterase [cGMP] sinkt CNG-Kanal schließt Hyperpolarisation Genetische Ursache der Blauzapfen-Monochromasie Photopigment für Rot- bzw. GrünZapfen Genetische Ursachen der Achromatopsie Transducin – GNAT2-Gen GNAT2-Gen Transducin Mutationen im GNAT2-Gen (Transducin) auf Chromosom 1 Fast alle Mutationen erzeugen stark verkürzte Transducin-Proteine Wahrscheinlich gar kein Transducin vorhanden ! Komplette Achromatopsie (1 Ausnahme) Tiermodell in der Maus: Zapfen sind vorhanden, aber leere Hüllen Keine Degeneration Genetische Ursachen der Achromatopsie Phosphodiesterase – PDE6C-Gen GNAT2 PDE6C Phosphodiesterase Mutationen im PDE6C-Gen (Phosphodiesterase) auf Chromosom 10 Alle bekannten Mutationen erzeugen entweder Stark verkürzte Phosphodiesterase-Proteine oder Treffen Aminosäuren, von denen man weiß, dass sie essentiell wichtig für die Funktion sind Immer komplette Achromatopsie Tiermodell in der Maus: Zapfen sind am Anfang vorhanden, aber nicht funktionell Und degenerieren später schnell Genetische Ursachen der Achromatopsie CNG-Kanal – CNGA3-Gen und CNGB3-Gen CNGA3 CNGB3 CNG-Kanal GNAT2 PDE6C Der Zapfen-Photorezeptor CNG-Kanal Tetramer aus CNGA3 (kanalbildende Untereinheit) und CNGB3 (modulierende Untereinheit) des CNG-Kanals hochkonservierte Kanal (6 TransmembranDomänen, Pore und cGMPBindestelle) Mutationsspektrum in CNGA3 z Missense z Nonsense z Deletion z Insertion Mutationen im CNGA3-Gen auf Chromosom 2 Die meisten Mutationen AminosäureAustausche und treffen Aminosäuren, von denen man weiß, dass sie essentiell wichtig für die Funktion sind Sehr konserviertes Protein (~90% identisch in allen Tierarten) Meistens komplette Achromatopsie Manchmal inkomplette Achromatopsie Tiermodell in der Maus: Ebenfalls: Zapfen sind anfangs vorhanden, sind aber funktionslos Und degenerieren dann schnell Mutationsspektrum in CNGB3 Mutationen im CNGB3-Gen auf Chromosom 8 Die meisten Mutationen erzeugen Stark verkürzte Kanal-Protein oder Nur wenige Aminosäure-Austausch-Mutationen Meistens komplette Achromatopsie Besonderheit: Eine sehr häufige Mutation, welche bei ca. 40% aller AchromatopsiePatienten für die Erkrankung verantwortlich ist (c.1148delC) Natürliches Tiermodell in Hunden: Vergleichbar mit PDE6C und CNGA3 Pingelapese blindness Entdeckung des ACHM3-Locus anhand von Familien mit Achromatopsie von der Südsee-Insel Pingelap (Pingelapese Blindness) Hohe Inzidenz (1:10) durch Gendrift und Inzest nach Typhoon im 17. Jahrhundert „Die Insel der Farbenblinden“ von Oliver Sachs Zusammenfassung CNGB3 - Gen auf Chromosom 8q21 = ACHM3-Locus ca. 50% aller Achromatopsie-Patienten 75% dieser Patienten tragen c.1148delC-Mutation CNGA3 - Gen auf Chromosom 2q11 = ACHM2-Locus ca. 25% aller Achromaten GNAT2 - Gen auf Chromosom 1p13 = ACHM4-Locus Nur in ~ 2 % aller Patienten mit Achromatopsie PDE6C - Gen auf Chromosom 10q24 ~ 2 % aller Achromatopsie-Patienten Weiterführende Untersuchungen Funktionelle Untersuchungen der gefundenen Mutationen in den einzelnen Proteinen, um das Verständnis über die Funktionsweise dieser Proteine zu verbessern Studieren der Tiermodelle, um die Prozesse der Erkrankung zu verstehen (Gen-)Therapeutische Ansätze Molekulargenetische Untersuchung EDTA-Blut vom Patienten (10ml), und wenn möglich auch von den Eltern Zeitlich aufwendige Untersuchung: ~ 8 - 10 Wochen bis Befundbericht Abrechnung über GKV möglich, nicht budget-belastend (Überweisungsschein Muster 6) Abrechnung über PKV zunehmend schwierig! Das Molekulargenetische Labor der Universitäts-Augenklinik Tübingen Forschungslabor: Optikus-Atrophie Glaukom Zapfen- und ZapfenStäbchen-Dystrophien Achromatopsie Neue Krankheits-Gene Funktion der Proteine Krankheitsmechanismen Das Molekulargenetische Labor der Universitäts-Augenklinik Tübingen Service-Einheit: RetDis-Datenbank Archivierung von DNA, RNA und Zelllinien von Patienten ~ 16.000 Proben verknüpft mit Patienten- bzw. Familienakten für erbliche Augenerkrankungen (Stammbäume, klinische Daten) Das Molekulargenetische Labor der Universitäts-Augenklinik Tübingen Service-Einheit: DNA-Diagnostik Autosomal dominante Retinitis pigmentosa Autosomal dominante Optikus-Atrophie Lebers hereditäre Optikus-Neuropathie Kongenitales Glaukom X-chromosomale Blauzapfen-Monochromasie Autosomal rezessive Achromatopsie Das Molekulargenetische Labor der Universitäts-Augenklinik Tübingen Vielen Dank für Ihre Aufmerksamkeit !