Die Fontan – Operation
Werbung

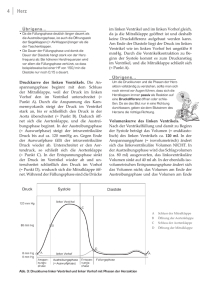
HERZKIND e.V. Die Fontan -Operation Die Fontan – Operation von Dr. Kozlik-Feldmann, Prof. Netz, Klinikum Großhadern Kinderherzzentrum Die Fontan Operation (Fontan und Baudet 1971), welche eine Verbindung zwischen rechter Vorkammer und Lungenschlagader unter Umgehung einer Herzkammer schafft, wurde ursprünglich zur Kreislauftrennung bei der Trikuspidalklappenatresie durchgeführt. Mittlerweile wird diese Operationstechnik bzw. Idee bei verschiedenen Herzfehlern angewendet bei denen keine 2 funktionstüchtigen Herzkammern zur Verfügung stehen • Trikuspidalatresie • Mitralatresie (inkl. Hypoplastisches Linksherzsyndrom) • Double-inlet ventricle • Herzen mit Isomerie der Vorkammern • Pulmonalatresie mit intaktem Ventrikelseptum • Ebstein Malformation • Andere Trikuspidalatresie Bei der Trikuspidalatresie fehlt die rechtsseitige atrioventrikuläre Verbindung, also die AV-Klappe zwischen dem rechten Vorhof und dem darunterliegenden Ventrikel. Die zum rechten Vorhof gehörende AV-Klappe ist verschlossen. Während bei der weitaus häufigeren, klassischen Trikuspidalatresie die Anlage der rechtsseitigen AV-Klappe völlig fehlt, ist bei der als Trikuspidalatresie mit imperforierter Klappe bezeichneten Form eine Klappenanlage zwar vorhanden, diese hat jedoch eine atretische („imperforierte“) Membran. Zumeist findet sich nur ein rudimentärer rechter Ventrikel. Diese verkümmerte rechte Kammer zeigt keinerlei Verbindung zu einem der beiden Vorhöfe und kommuniziert über ein Foramen interventriculare ausschließlich mit dem linken Ventrikel. Dieses Verbindungsloch kann manchmal zu klein sein oder im Laufe der Zeit zu klein werden, so dass ein reduzierter Blutstrom zur Lungenschlagader mit vermehrter Zyanose (Blausucht) des Kindes resultiert. Die Stellung der großen Arterien kann normal sein, so dass die Aorta hinten und rechts, die Pulmonalarterie vorn und links liegt oder aber es besteht eine Malposition der großen Arterien. Sowohl eine d-Malposition als auch eine l-Malposition kommen vor. Entsprechend der Stellung der großen Arterien werden folgende Typen unterschieden: Typ I: normale Stellung der großen Arterien Typ II: d-Malposition der großen Arterien Typ III: l-Malposition der großen Arterien Oft besteht eine Pulmonalstenose bzw. sogar eine Pulmonalatresie, die bei normaler Stellung der großen Arterien durch eine restriktive Kommunikation zwischen dominantem und rudimentären Ventrikel verstärkt werden kann. Die Beurteilung der Pulmonalarterie ist für das weitere therapeutische Vorgehen (z.B. Fontan-Operation) entscheidend, da eine Fontan-Operation nur bei einem niedrigen Druck in der Pulmonalarterie vorgenommen werden kann. Wegen der Bedeutung des pulmonalarteriellen Druckes für das weitere therapeutische Vorgehen werden Trikuspidalatresien deshalb nach dem Ausmaß der Pulmonalstenose eingeteilt: Typ A: Pulmonalatresie Typ B: Pulmonalstenose Typ C: keine Stenose zur Lungenstrombahn Beim Typ A muß die Lungendurchblutung zunächst über einen Ductus Botalli erfolgen (Die Kinder bekommen nach erkennen des Herzfehlers eine Prostaglandin E1 Dauerinfusion, damit der Ductus aufbleibt) und erhalten dann operativ einen Shunt (künstl. Kurzschlussverbindung) zur Sicherung der Lungendurchblutung. Beim Typ C besteht eine gesteigerte Lungendurchblutung, so dass eine Bändelung der Pulmonalarterie indiziert ist, um den hohen Blutdruck in der Lunge zu senken (Normalerweise beträgt dieser 1/6 des Körperblutdrucks, ohne Stenose entspricht er in dieser Situation dem Körperblutdruck). Schließlich ist die Verbindung zwischen beiden Vorhöfen (Vorhofseptumdefekt) deshalb von hämodynamischer BeSeite 1 von 6 HERZKIND e.V. Die Fontan -Operation deutung, weil sie den einzigen Auslass aus dem rechten Vorhof darstellt. Heute wird ein zu kleiner Vorhofseptumdefekt als ungünstig angesehen, weil die rechtsatriale Überdehnung vermutlich zu späteren Rhythmusstörungen disponiert. Deshalb sollte der Gradient am Vorhofseptum nicht höher als 8 mmHg sein. Schließlich muß eine Insuffizienz (Undichtigkeit) der (einzigen) AV-Klappe vor einer etwaigen Fontan-Operation ausgeschlossen werden, da diese zu einer Stauung in die Lunge führt. Mitralatresie Bei der Mitralatresie fehlt eine Verbindung zwischen dem linken Vorhof und dem linken Ventrikel. Zwei grundlegend unterschiedliche Formen der Mitralatresie müssen unterschieden werden: • • die Mitralatresie mit Ventrikelseptumdefekt, das hypoplastische Linksherzsyndrom Beim hypoplastischen Linksherzsyndrom ist das Ventrikelseptum intakt, so dass eine Mitralatresie dazu führt, dass sich der linke Ventrikel nicht entwickelt. Fast immer besteht hier gleichzeitig eine Aortenatresie. Dagegen kann sich der linke Ventrikel bei der Mitralatresie mit Ventrikelseptumdefekt entwickeln, weil er sich über diesen Ventrikelseptumdefekt sowohl füllen als auch entleeren kann. Morphologisch ähnelt die Mitralatresie mit Ventrikelseptumdefekt der Trikuspidalatresie. Nach einer Fontan-Operation bildet die rechte Herzkammer zusammen mit der linken (die zu klein oder normal groß sein kann) die Hauptkammer. Ist hier die Verbindung zwischen den beiden Kammern restriktiv (zu eng), ist in diesem Fall allerdings der Blutstrom zur Körperschlagader behindert mit der Gefahr, daß es nach der Fontan-Operation zu einem gravierenden Blutstau zur Lunge kommt. Der Ursprung der großen Arterien ist variabel, wie bei der Trikuspidalatresie besteht oft eine Pulmonalstenose oder Pulmonalatresie. Ein Vorhofseptumdefekt ist auch hier essentiell. Hypoplastisches Linksherzsyndrom Das hypoplastische Linksherzsyndrom bezeichnet ein Spektrum an Fehlbildungen der Aorten- und Mitralklappe. Beide Klappen können stenotisch bis atretisch sein, eine Klappenanlage ist jedoch in jedem Fall vorhanden. Im Extremfall besteht eine Atresie der Aorten- und Mitralklappe, so dass sich der linke Ventrikel weder füllen noch entleeren kann. Häufiger ist die Aortenklappe atretisch und die Mitralklappe extrem stenosiert, verdickt und dysplastisch. Unabhängig von diesen anatomischen Variationen ist der linke Ventrikel beim hypoplastischen Linksherzsyndrom hypoplastisch und zeigt meist eine Endokardfibroelastose. Die Perfusion des Systemkreislaufes erfolgt nicht vom linken Ventrikel aus, sondern durch den rechten Ventrikel über einen offenen Ductus Botalli. Die Aorta ascendens ist hypoplastisch, sie wird rückwärts vom Ductus aus perfundiert und versorgt auch die Koronargefäße. Schließlich findet sich immer ein Defekt im Vorhofseptum, über den das pulmonalvenöse Blut abfließen kann. Therapeutisch können Neugeborenen mit hypoplastischem Linksherzsyndrom nur 2 Wege angeboten werden, neben der Herztransplantation bleibt als Palliation die sogenannte Norwood-Operation die letztendlich eine Variation der Fontan-Operation ist. Neben der nötigen Rekonstruktion einer ausreichend großen Körperschlagader ist zu beachten, dass ausschließlich eine funktionsfähige rechte Herzkammer zur Verfügung steht. Pulmonalatresie mit intaktem Ventrikelseptum Bei der Pulmonalatresie mit intaktem Ventrikelseptum ist die Pulmonalarterie nicht durchgängig, so daß der rechte Ventrikel kein Blut auswerfen kann. Die Atresie ist meist membranös, d.h. eine Klappenanlage ist vorhanden. Seltener besteht eine muskuläre Atresie, bei der die Entfernung von rechter Herzkammer zur Lungenschlagader langstreckig ist. Der einzige Auslass aus dem rechten Ventrikel bei der Pulmonalatresie mit intaktem Ventrikelseptum ist eine immer bestehende Trikuspidalinsuffizienz. Oft besteht eine Dysplasie der Trikuspidalklappe im Sinne einer Ebstein-Malformation: die Trikuspidalklappe ist dann in den rechten Ventrikel vorverlagert. Diese Fehlbildung gibt es auch ohne Pulmonalatresie, sie kann eine ähnliche Hämodynamik wie bei Pulmonalstenose oder -atresie bedingen. Da die Trikuspidalinsuffizienz die einzige Möglichkeit für den rechten Ventrikel darstellt, Blut auszuwerfen, entwickelt sich der rechte Ventrikel nicht normal und bleibt hypoplastisch. Als Faustregel gilt, je ausgeprägter die Trikuspidalinsuffizienz, desto größer ist der rechte Ventrikel. Das Spektrum der Größe des rechten Ventrikels reicht von einem kaum auffindbaren Restlumen (ähnlich dem hypoplastischen Linksherzsyndrom) bis zu einer fast normalen Ventrikelgröße (wie beim normalen Herzen). Das venöse Blut erreicht über einen Defekt im Vorhofseptum den linken Vorhof, die Durchblutung der Lungenstrombahn ist ductusabhängig. Oft finden sich Fehlbildungen der Koronargefäße, insbesondere kommen Myokardsinusoide vor. Myokardsinusoide sind embryonale Kommunikationen zwischen dem rechten Ventrikel und dem Koronargefäßsystem. Da der Druck im rechten Ventrikel regelmäßig höher als im Körperkreislauf ist, wird venöses Blut aus dem rechten Ventrikel in das KoSeite 2 von 6 HERZKIND e.V. Die Fontan -Operation ronargefäßsystem ausgeworfen. Zusätzlich zur Koronarperfusion mit untersättigtem Blut finden sich oft an den Verbindungen zwischen Myokardsinusoiden und Koronargefäßen Stenosen, so dass eine Ischämie des Myokards (Sauerstoffmangel des Herzmuskels) auftritt. Bei einer hypoplastischen rechten Kammer bleibt zur Kreislauftrennung nur eine Fontan-Operation. Double inlet ventricle Double inlet ventricle sind Herzfehler mit praktisch fehlender Kammerscheidewand (2 AV-Klappen füllen eine Herzkammer aus der beide Schlagadern entspringen). Da eine künstliche Septierung hämodynamisch sehr unbefriedigend ist (fehlende Muskelmasse im Septum zum Gegendruckaufbau zur Hinterwand der linken Kammer, bleibt zumeist nur der Weg einer Fontan-Operation zur Kreislauftrennung). Bei Fehlen einer Pulmonalstenose muss das Lungengefäßsystem zunächst durch ein Lungenarterien-Bändchen vor hohem Druck geschützt werden. Die Fontan-Operation Die Grundidee der Fontan-Operation ist die Schaffung eines Blutstromes zum Lungenarteriensystem ohne eine dazwischenliegenden Pumpkammer. Dazu wurde die Lungenschlagader (Pulmonalis) von der Herzkammer abgetrennt, der Stumpf zur Herzkammer vernäht, das „Ohr“ der rechten Vorkammer abgeschnitten und die Lungenschlagader an dieser Stelle mit dem Vorhof verbunden. Ursprünglich wurde an die Verbindungsstelle zwischen Vorhof und aufgenähter Pulmonalarterie eine prosthetische Herzklappe angebracht. Dies ist heute völlig verlassen worden, es gibt aber eine ganze Reihe von Modifikationen der Fontan-Operation: "Evolution" der Fontan-Operation Operation/Modifikation Autoren Fontan (mit prothetischer Klappe) Fontan und Baudet 1971 Kreutzer (Verbindung VorhofPulmonalisbifurkation) Kreutzer et al. 1973 Bjork (Anastomose Vorhof- rechter Ventrikel) Bjork et al. 1979 Aortopulmonale Konnektion bei Subaortenstenose Waldmann et al. 1988 Totaler cavopulmonaler Bypass de Leval et al. 1990 Bidirektionale Cavopulmonale Anastomose (Zweizeitige Fontan-Operation) Bridges et al. 1990 "Fenestrierter Fontan" (ein kleiner Vorhofdefekt wird belassen) Bridges et al. 1990 Extrakardialer Fontan (Tunnel außerhalb des Vorhofs) Marcelletti et al. 1990 Damit eine Fontan-Operation mit hoher Wahrscheinlichkeit funktioniert, wurden bereits zu Beginn der Fontan-Ära Kriterien aufgestellt, die möglichst von den Patienten erfüllt werden sollen (siehe Tabelle). Kritrien für eine Fontan-Operation (n. R. Freedom) • • • • • Normaler Sinusrhythmus Normale, nicht behinderte Zuflüße aus Körper- und Lungenvenen Normaler Lungengefäßwiderstand (Mittlerer Lungendruck max. bei 15 - 20 mmHg) Keine höhergradigen Pulmonalarterienstenosen Relation Pulmonalarterie zur Aorta > 0,75 Seite 3 von 6 HERZKIND e.V. • • • • Die Fontan -Operation „Normale“ Ventrikelfunktion (Bei rechten Ventrikeln kaum objektiv zu beurteilen) Keine signifikante Undichtigkeit der systemischen atrio-ventrikulären Herzklappe Normale diastolische Funktion des Ventrikels („Steifheit“ bei der Füllung) Optimales Alter: unsicher, neuerdings wird bei günstiger Anatomie und Hämodynamik ein Alter zwischen 2- 4 Jahren bevorzugt* * In unserem Hause wird ein sog. zweizeitiges Vorgehen bevorzugt. Hierbei erfolgt als erster Schritt im Alter zwischen 1-2 Jahren ein Hemi-Fontan (Bild A/B: zur besseren Übersicht wurde ein Zwischenstück der Körperschlagader weggelassen). Hierbei wird zunächst die Pulmonalarterie von der Herzkammer abgesetzt und auf das „Dach“ des rechten Vorhofs aufgenäht. Durch einen entsprechend in die Vorkammer eingenähten Kunststoff-Patch wird erreicht, dass nur das Blut der oberen Körperhälfte zur Pulmonalarterie gelangt und das Blut der unteren Körperhälfte, gemischt mit dem sauerstoffreichen Blut der Lungenvenen, weiterhin zur Herzkammer gelangt und wieder in den Körper ausgeworfen wird. Dies hat den Vorteil, dass zunächst nur etwas mehr als 50% des Blutes „passiv“ ohne Herzkammer durch die Lunge muss und die Herzkammer (die um diese Blutmenge beim aktiven Auswurf entlastet wird) sich bis zum zweiten Operationsschritt erholen kann. Die Intensivliegezeit ist nach diesem Schritt kurz. Die Kinder bleiben nach diesem ersten Operationsschritt weiterhin zyanotisch. Beim zweiten Operationsschritt (nach ca. 1 Jahr) wird dann der Patch in der Vorkammer durch einen prothetischen Tunnel ersetzt, so dass auch das Blut der unteren Körperhälfe zur Lunge gelangt (Bild C/D). Die Operation ist relativ kurz und da die Kreislaufsituation nicht so dramatisch geändert wird wie bei Total-Fontan ist die postoperative Adaptation nicht so langwierig. Mit anderen Worten: Die Gesamtliegezeit auf der Intensivstation wird durch das zweizeitige Vorgehen verkürzt! Bei Kindern über 12-15 kg Körpergewicht wird heute häufig der extrakardiale Tunnel bevorzugt, weil dieser ohne HerzLungenmaschine zu etablieren ist! Das Gesamtrisiko der Fontanoperation hängt davon ab, wie gut die Kriterien von den Patienten erfüllt werden. Nach den Beobachtungen von Mair et al. gilt zum Beispiel folgende Beziehung zwischen Überlebenswahrscheinlichkeit und Lungengefäßwiderstand: Seite 4 von 6 HERZKIND e.V. PVR (U/m*2) Die Fontan -Operation Frühsterblichkeit (% ) Gesamtsterblichkeit (% ) <2 3 15 2,1 - 3 11 44 3,1 - 4 50 50 Beziehung zwischen präoperativem Lungenwiderstand (PVR) und der Sterblichkeit nach Fontan-Operation bei Patienten mit Double-inlet left ventricle Ähnliche Zahlen gibt es von derselben Arbeitsgruppe für Patienten mit einer Trikuspidalatresie. Hier wurde ein Index der Mayo-Klinik berechnet, der den möglichen Lungenfluss vorausberechnet: PVR (U/m*2) + [LVEDP/Qpl+Qsl]** Gesamtsterblichkeit (%) 1,1 - 2,0 5 2,1 - 3,0 6 3,1 - 4,0 14 4,1 - 5,0 27 >5,0 57 Sterblichkeit nach Fontan-Operation bei Trikuspidalatresie verglichen mit einem präoperativen Katheter-Index (**LVEDP = Druck in der Kammer nach deren Füllung, QpI = Flussvolumen pro Minute durch die Lunge, QsI = Flussvolumen pro Minute zum Körper) Zum Vergleich zu diesen Daten hier die natürliche Sterblichkeit bei einer Trikuspidalatresie (ohne Therapie): · · 1. Jahr: 10 - 20 % bis 15 J.: 50% In den Zentren mit Kinderkardiochirurgie liegen die Frühsterblichkeiten zwischen 2 und 6%. Beim zweizeitigen Vorgehen (1. Hemifontan, 2. Fontan) liegen die Zahlen mehr im unteren Bereich dieser Spanne. Das sind Zahlen, die auch von Deutschen Zentren erreicht werden. Von den vor 15 Jahren operierten Patienten leben heute noch 55 bis 80% (je nach Serie) mit eigenem Herzen (nach Freedom). Als Spätoption für Patienten mit Fontan-Zirkulation, bei denen der Systemventrikel versagt oder aufgrund einer chronischen Rückstauung des Blutes zum Körper ein starker Eiweißverlust über den Darm auftritt, bleibt die Herztransplantation. Dass aber ein zu langes Zögern mit operativen Schritten kein Gewinn für die Patienten bringt, zeigen folgende Zahlen von Franklin et al. (Zahlen aus Toronto und London): Von den 40% der Patienten, die die Fontan-Kriterien aus irgendeinem Grund nicht erfüllten, starben 48% oder verschlechterten sich derart, dass an eine Fontan-Operation überhaupt nicht mehr zu denken war. In dieser Patientenserie war die Überlebensrate nach dem 1. Jahr 72%, nach 5 Jahren 53% und nach 10 Jahren 46%. Diese Zahlen zeigen, dass es junge Patienten gibt, die die Kriterien knapp nicht erfüllen, sich durch zuwarten aber desolat verschlechtern. Bei diesen Patienten ist eine frühzeitige Operation zu erwägen! Im 3. Jahrzehnt nach Einführung der Fontan-Operation muss also überdacht werden, in welcher Art Eingangskriterien für diese Prozedur aufrecht erhalten werden und wo die Grenzwerte sind. Allein schon durch die verfeinerten Operationstechniken haben aber heute die Patienten wesentlich bessere Chancen als in den Pionierzeiten. Die Lebensqualität nach einer Fontanoperation ist im allgemeinen gut, wenn auch vom Herzen nicht das gleiche Herzzeitvolumen wie beim Gesunden (Auswurf in l/min) erbracht werden kann. Die meisten Patienten müssen zumindest durch Digitalis medikamentös unterstützt werden. Es gibt aber Berichte ehrgeiziger Patienten wie zum Beispiel den Seite 5 von 6 HERZKIND e.V. Die Fontan -Operation einer Schwimmerin, die mit bestehender Fontan-Zirkulation an den Para-Olympics teilnahm. Ansonsten sind die Patienten den normalen Belastungen des Alltags gewachsen. Ein bedeutsames Risiko, zumindest in den ersten Monaten nach der Fontan-Operation, besteht durch eine möglich Thrombenbildung an dem intraatrialen Tunnel. Dies kann auf der systemischen Seite zu Hirnembolien führen. Das Risiko hierfür wird mit etwa 2,6% angegeben. Wir führen daher eine Blutgerinnungshemmung mit Cumarin für mindestens ein halbes Jahr durch. Bei Patienten mit echokardiographisch erfassbaren Risikostellen in den Vorkammern wird dann eine Dauerprophylaxe mit Acetylsalicylsäure (z.B. Aspirin ®), so z.B. bei extrakardialem Tunnel, oder bei besonderen Risikofaktoren mit Marcumar durchgeführt. Bezüglich dieser Problematik verfährt aber praktisch jedes Zentrum anders. Die Richtlinien zur Endokarditisprophylaxe sind lebenslang zu beachten! Dr. Kozlik-Feldmann, Prof. Netz Klinikum Großhadern Kinderherzzentrum Wir danken für die freundliche Genehmigung zum Nachdruck. Weitere interessante Informationen finden Sie unter www.kinderkardiologie-muenchen.de Seite 6 von 6