Genetik - NCL
Werbung

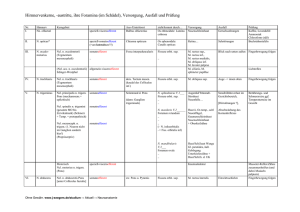
Leitthema Ophthalmologe 2010 · 107:612–615 DOI 10.1007/s00347-009-2107-x Online publiziert: 9. Juni 2010 © Springer-Verlag 2010 M.N. Preising · B. Lorenz Klinik und Poliklinik für Augenheilkunde, Universitätsklinikum Gießen und Marburg GmbH, Standort Gießen Genetik der neuronalen Zeroidlipofuszinosen Aspekte der humangenetischen Beratung Lysosomale Speicherkrankheiten stellen eine breit gefächerte Gruppe von Stoffwechselerkrankungen dar. Ihnen gemein ist die Ansammlung von Stoffwechselendprodukten in den lysosomalen Kompartimenten. Diese Ablagerungen belasten den zellulären Stoffwechsel und führen über kurz oder lang zur Degeneration der Zellen. Neuronale Zellen sind dabei oft empfindlicher als andere Körperzellen, wodurch vermehrt Beeinträchtigungen im Bereich der Hirnfunktionen, der Wahrnehmung und der motorischen Funktionen auftreten. Eine Untergruppe der lysosomalen Speicherkrankheiten sind die neuronalen Zeroidlipofuszinosen. Die Vererbung und die humangenetische Beratung der Betroffenen dieser – mit wenigen Ausnahmen – sich sehr früh manifestierenden Erkrankungen ist Thema der folgenden Übersichtsarbeit. Die neuronalen Zeroidlipofuszinosen (NCL) sind eine Gruppe von seltenen progredienten neurodegenerativen Erkrankungen. Die weltweite Prävalenz liegt bei 1:100.000. In den USA oder Skandinavien erreicht die Prävalenz bis zu 1:12.500 [6]. Die NCL sind eine Gruppe von lysosomalen Speicherkrankheiten, die sowohl Zerebrum, Zerebellum und Hirnstamm als auch wichtige afferente und efferente Nervenbahnen betreffen [1]. Dies führt in erster Linie zu Epilepsien, Krampfanfällen, motorischen und kognitiven Störungen sowie Persönlichkeitsstörungen, betrifft aber auch, bis auf wenige Aus- 612 | Der Ophthalmologe 7 · 2010 nahmen (CLN4 und CLN8 [1, 4]), den N. opticus und die Netzhaut. Die Erkrankung ist klinisch heterogen und wird anhand des zeitlichen Beginns der Symptomatik eingeteilt (. Tab. 1, s. Krohne et al. in dieser Ausgabe). Der Visusverlust aufgrund einer Netzhautdegeneration ist in den meisten Fällen eines der ersten Symptome und bei der juvenilen NCL (JNCL) sogar die Einstiegsdiagnose. Damit kommt dem Augenarzt eine wichtige Funktion bei der Früherkennung dieser Erkrankungen zu. NCL zeichnen sich durch Ablagerungen von Protein-Lipid-Komplexen in den lysosomalen Kompartimenten aller Zellen aus [4, 10]. Diese Ablagerungen bestehen aus einer Proteinkomponente und autofluoreszentem lipophilem Material. In den meisten Fällen stellt die Untereinheit c der mitochondrialen ATP-Synthase die Proteinkomponente [1, 7]. In diesen Fällen stellen sich die Ablagerungen in den lysosomalen Kompartimenten als lineare Profile im elektronenmikroskopischen Bild dar. Die Linien dieser Profile können dabei geradlinig (RFP), kurvenförmig (CLP) oder ähnlich Fingerabdrücken (FPP) sein [1, 7]. Bei anderen NCLFormen, v. a. bei jenen mit sehr frühem Beginn, finden sich Saposine (sphingolipidaktivierende Proteine: SAP-A, SAPD) als Hauptanteil der Proteinkomponente. In diesen Fällen zeigt das elektronmikroskopische Bild einen granulären osmiophilen Inhalt für die lysosomalen Ablagerungen ([1, 7], . Tab. 1). Mit Ausnahme des CLN6- und des CLN8-Genprodukts sind alle bislang bekannten NCL-Gen- produkte im Lysosom selbst aktiv. Die genaue Funktion ist allerdings nicht bekannt [1]. Es werden Beteiligungen am Einwärts­ transport von z. B. Arginin in die lysosomalen Kompartimente und bei der pHRegulation in den Lysosomen diskutiert. Auch eine Beteiligung am Abbau von Proteinen innerhalb der Lysosomen wird immer wieder angeführt [2]. Wenn die Diagnose einer NCL aufgrund der klinischen Symptome durch den Augenarzt oder Neuropädiater gestellt wurde (s. Krohne et al. in dieser Ausgabe), folgt eine elektronenmikroskopische Untersuchung an Blutzellen oder anderem Biopsiematerial zur Beschreibung der lysosomalen Ablagerungen. Die­ se Ergebnisse dienen dem Humangenetiker als Entscheidungsgrundlage für die weiterführende molekulargenetische Diagnostik. Derzeit sind 10 Genorte beschrieben, denen 8 Gene zugeordnet wurden (. Tab. 1). Dabei ist die JNCL mit CLN3 als mutationstragendem Gen eine der häufigsten Formen und die kongenitale NCL (KNCL) mit Cathepsin D (CTSD) als mutationstragendem Gen die seltenste Form. CLN5 bis CLN8 wurden initial als lokale Formen in definierten Populationen angesehen. Zwischenzeitlich konnten aber Mutationen in den ursächlichen Genen bei Patienten anderer Populationen nachgewiesen werden, sodass auch hier von einer weiteren Verbreitung auszugehen ist. CLN1, CLN2 und CLN3 sind die am häufigsten betroffenen Gene [1]. Dabei ist zu berücksichtigen, dass bei CLN3 bei 85% der Patienten dieselbe 1-kb-Deletion zwischen Exon 6 und Exon 9 vorliegt und zu- Zusammenfassung · Abstract sätzlich noch weitere 40 Mutationen bekannt sind [1, 5]. CLN1 und CLN2 zeigen etwa gleich viele Mutationen. Dabei zeigt sich, dass für bestimmte Populationen lokal gehäuft bestimmte Mutationen vorkommen. Dies korreliert mit der erhöhten Prävalenz der NCL in verschiedenen Populationen und der lokalen Häufung für CLN5–8-Mutationen und liegt an der Struktur verschiedener ethnischer Gruppen in diesen Populationen. Für die genetische Beratung in Deutschland sind diese lokalen Mutationen von nachgeordneter Bedeutung und kommen erst zum Tragen, wenn ein entsprechender Patient eine Beratung wünscht. Viel wichtiger für die genetische Beratung in Deutschland ist die 1-kb-Deletion im CLN3-Gen, da sie etwa 85% der JNCL-Patienten in Deutschland betrifft. Eine derartige Häufung von Fällen mit einer einzigen Mutation erlaubt einen vereinfachten Zugang und eine beschleunigte Testung. Heterozygote Fälle müssen zwar in einem zweiten Schritt auf das vollständige Gen untersucht werden, aber der Vor­ ausschluss über die 1-kb-Deletion hat eine hohe Trefferquote. >Die NCL wird meistens autosomal-rezessiv vererbt Abgesehen von einzelnen Patienten mit CLN4 wird die NCL autosomal-rezessiv vererbt. Daher besteht formal für die Geschwister eines Patienten ein 25%iges Risiko, ebenfalls betroffen zu sein. Zu spontanen Mutationsereignissen oder uniparentalen Disomien (UPD), die eine Vererbung außerhalb der Mendel-Formalgenetik bedingen, liegen in der Literatur keine Angaben vor. Eltern betroffener Kinder steht nach Identifikation der ursächlichen Mutation die pränatale Diagnostik offen. Dabei sind die klinische Diagnose und die mikroskopische Diagnostik Vorraussetzung für eine zielgerichtete molekulargenetische Diagnostik. Als rezessive Erkrankung ist die NCL ein gutes Ziel für gentherapeutische Behandlungen. Die verschiedenen anatomischen Veränderungen sind allerdings schwierig aus einer einzelnen Anwendung heraus zu behandeln. Der Zugang zu ihnen ist erschwert, da sie einerseits im gesamten Körper und über al- Ophthalmologe 2010 · 107:612–615 DOI 10.1007/s00347-009-2107-x © Springer-Verlag 2010 M.N. Preising · B. Lorenz Genetik der neuronalen Zeroidlipofuszinosen. Aspekte der humangenetischen Beratung Zusammenfassung Die neuronalen Zeroidlipofuszinosen sind autosomal-rezessiv vererbte Erkrankungen der Nervenzellen. Sie werden zu den lysosomalen Speicherkrankheiten gerechnet und zeichnen sich durch Akkumulation von Protein-Lipid-Komplexen in den lysosomalen Kompartimenten aller Körperzellen aus. Diese Ablagerungen führen zu degenerativen Vorgängen im Nervensystem, speziell im Groß- und Kleinhirn und den zu- und abführenden Hirnnerven. Die Erkrankung führt, mit einer spät manifestierenden Ausnahme, zu einem Verlust rezeptiver, kognitiver und motorischer Funktionen in der 1. Lebensdekade und somit derzeit unweigerlich zu einem frühen Tod in der 3. bis 4. Dekade. Derzeit wurden 10 Gen­orte identifiziert, denen 8 bekannte Gene zugeordnet sind. Der spezifische Phänotyp und die Prognose sind abhängig vom ursächlichen Gen, ohne dass eine gezielte GenotypPhänotyp-Korrelation möglich ist. Letzteres liegt an dem fehlenden Wissen um die Funktion der einzelnen betroffenen Genprodukte. Wir fassen in dieser Übersicht die bekannten genetischen Informationen zu den neuronalen Zeroidlipofuszinosen zusammen und kommentieren die Möglichkeiten der therapeutischen Intervention. Schlüsselwörter Juvenile Zeroidlipofuszinose · Adulte Zeroid­ lipofuszinose · Lysosomale Speicherkrankheiten · Autosomal-rezessiv · Morbus Batten Genetics of neuronal ceroidlipofuscinoses. Aspects of genetic counseling Abstract Neuronal ceroid lipofuscinoses are autosomal recessive inherited disorders of neuronal cells. Neuronal ceroid lipofuscinoses belong to the lysosomal storage disorders and are characterized by accumulation of proteinlipid complexes in the lysosomal compartments of all somatic cells. This debris causes degenerative activities in the nervous system, especially in the cerebrum, the cerebellum and the afferent and efferent cranial nerves. With one exception of adult onset the disorder causes the loss of receptive, cognitive and control function in the first decade of life and an early death by the age of 20. Currently 10 loci are known which correlate to 8 genes. The genotype related phenotype and the correlated prognosis depend on the underlying gene and type of mutation. The genotype phenotype correlation is hampered by a lack of knowledge on the function of the mutant gene products. In this review we summarize the known genetic data on neuronal ceroid lipofuscinoses and comment on therapeutic approaches. Keywords Juvenile ceroidlipofuscinoses · Adult ceroidlipofuscinoses · Lysosomal storage disorders · Autosomal recessive · Batten disease Der Ophthalmologe 7 · 2010 | 613 Leitthema Tab. 1 Neuronale Zeroidlipofuszinosen Lokalisation Gen/OMIM des Genprodukts Entreza HUGOb LSDBc Kongenital bis frühe 11p15.5 Schulzeit/nach Stunden bis zu wenigen Wochen oder bis zur 2. Dekade Lysosom CTSD/116840 1509 2529 SAP-A, SAP-D/GROD 0,5.–1,5. LJ/6.–15. LJ 1p32 Lysosom (keine Vakuolen [3]) PPT1/600722 5538 9325 cln1 ATP-Synthase Untereinheit c/CLP ATP-Synthase Untereinheit c/RLP, CLP, FPP ATP-Synthase Untereinheit c/RLP, CLP, FPP ATP-Synthase Untereinheit c/RLP, CLP, FPP ATP-Synthase Untereinheit c/CLP 2.–4. LJ/? 11p15.5 Lysosom TPP1/607998 1200 2073 cln2 4.–7. LJ/14.–36. LJ 13q21.1–q32 Lysosom CLN5/608102 1203 2076 cln5 1,5.–8. LJ/5.–12. LJ 15q21–23 ER CLN6/606725 54982 2077 cln6 2.–7. LJ/? 4q28.1–q28.2 Lysosom MFSD8/611124 256471 28486 5.–10. LJ/13.–? LJ 8p23 ER CLN8/607837 2055 2079 cln8 ATP-Synthase Unterein- 5.–10. LJ/2.–3. LD heit c/FPP, granulär ATP-Synthase Unterein- heit c/CLP (FPP, GROD) 16p12.1 Lysosom CLN3/607042 1201 2074 cln3 ? ATP-Synthase Unterein- heit c ? 1202 2075 ? Erkrankung/Trivialname/OMIM Speicherprotein/Struk- Beginn/Lebenserwartur der Ablagerungen tung [1, 7] Kongenital (KNCL) CLN10/610127 SAP-A, SAP-D/GROD Infantil (INCL) CLN1/Santavuori-Haltia/256735 Spät infantil (vLINCL) CLN2/Jansky-Bielschowsky/204500 CLN5 / Finnische Variante/256731 CLN6/Costa Rica Variante/601780 CLN7/Türkische Variante/610951 CLN8/Northwestern Epilepsievariante/610003 Juvenil (JNCL) CLN3/Batten-SpielmeyerSjögren-Vogt/204200 CLN9/609059 Adult (ANCL) CLN4a (rezessiv)/Morbus Kuf/162350 CLN4b (dominant)/Parry/204300 Chromosomale Lokalisation cln7 ER endoplasmatisches Retikulum, GROD granuläre osmiophile Ablagerungen, CLP kurvenförmige Profile, FPP Fingerabdruckprofile, RLP geradlinige Profile, LJ Lebensjahr, LD Lebensdekade, SAP sphingolipidaktivierende Proteine. ahttp://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene. bhttp://www.genenames.org/data/hgnc_data.php?hgnc_id=[geneID]. chttp://www.ucl.ac.uk/ncl/[page link].shtml. le Zellarten verteilt sind und andererseits besonders massiv im Gehirn auftreten, das gegen­über dem restlichen Körper abgeschottet ist. Außerdem ist die Tatsache, dass bei Auftreten der Symptome bereits große Schäden an irreparablen Geweben vorhanden sind und dass die Erkrankung sehr früh in der Kindheit einsetzt, kontraproduktiv. Eine heilende medikamentöse Therapie gibt es derzeit nicht (s. auch Rüther et al. und Steinfeld et al. in dieser Ausgabe). Die Behandlung der Patienten dient lediglich der Linderung der Beschwerden (Krämpfe, epileptische Anfälle, Persönlichkeitsveränderungen oder Depressionen), sodass der fatale Ausgang der Erkrankung derzeit nicht abwendbar ist. 614 | Der Ophthalmologe 7 · 2010 Aspekte der genetischen Beratung Die Diagnose einer NCL stellt den Anfang eines Betreuungsnetzwerks dar. In der Regel besteht dieses Netzwerk aus 3 Säulen: 1.dem Ophthalmologen als initialem Diagnostiker der frühen Symptome, 2.dem Humangenetiker, der die klinische Diagnose molekulargenetisch sichert bzw. dem klinischen Chemiker, der die klinische Diagnose biochemisch sichert, 3.dem Neuropädiater, der die Betreuung der Patienten und eine mögliche Therapien übernimmt. Die genetische Beratung umfasst dabei die Besprechung des Erbgangs, der als autoso- mal-rezessiver Erbgang auf dem Funktionsverlust beider Genkopien beruht. Charakteristisch ist, dass beide Geschlechter gleich häufig betroffen sind und die Erkrankung durch beide Geschlechter weitergegeben wird. Die rezessive Wirkung der defekten Genkopien führt zu symptomfreien heterozygoten Mutationsträgern, wie an den in der Regel nicht betroffenen Eltern der NCL-Patienten zu sehen ist. Damit liegt das Risiko für ein Elternpaar, die beide heterozygote Mutationsträger sind, ein betroffenes Kind zu zeugen, jeweils bei 25%. Dieses Risiko ist für jedes weitere Kind identisch. 50% der Nachkommen sind statistisch gesehen symptomlose, gesunde heterozygote Mutationsträger und weitere 25% der Nachkommen tragen keine veränderte Genko- Fachnachrichten pie und sind somit gesund. Oft ist die Familienplanung zum Auftreten der ersten Symptome bei den betroffenen Nachkommen bereits abgeschlossen, sodass den Eltern der Betroffenen keine Gelegenheit bleibt, die Erkenntnisse aus der molekulargenetischen Diagnostik für ihre eigene Familienplanung zu nutzen. Dem Humangenetiker bleibt nach der Diagnosesicherung und der Aufklärung über den Erbmodus nur die Überweisung an den Neuropädiater zur Behandlung der Symptomatik, da er selbst therapeutisch nicht tätig ist. An wenigen Standorten in Deutschland besteht die Möglichkeit, die genetische Beratung durch den Diagnostiker zu leisten, da die beteiligten Ophthalmologen oder Neuropädiater selbst über eine entsprechende Zusatzausbildung verfügen. >Eine Bündelung von Diagnostiker, Berater und Therapeut ist erstrebenswert Da ohnehin nur wenige hoch spezialisierte Labore die molekulare Diagnostik anbieten, ist eine Bündelung von Diagnostiker, Berater und Therapeut in einer Person unter Nutzung eines der vorhandenen diagnostischen Speziallabors erstrebenswert, auch wenn die Anfangssymptomatik häufig primär nicht zu einer Konsultation des behandelnden Facharztes führt. Letztlich müssen der Patient und seine Familie die schwerwiegende Diagnose ohne aktuelle kausale Therapiemöglichkeiten bewältigen. Neben den spezia­ lisierten Ärzten können dabei v. a. auch Patientenorganisationen helfen. Korrespondenzadresse M.N. Preising Klinik und Poliklinik für Augenheilkunde, Universitätsklinikum Gießen und Marburg GmbH, Standort Gießen Friedrichstraße 18, 35392 Gießen [email protected] Interessenkonflikt. Der korrespondierende Autor gibt an, dass kein Interessenkonflikt besteht. Literatur 1. Jalanko A, Braulke T (2009) Neuronal ceroid lipofuscinoses. Biochim Biophys Acta 1793(4):697–709 2. Kyttala A, Lahtinen U, Braulke T et al (2006) Functional biology of the neuronal ceroid lipofuscinoses (NCL) proteins. Biochim Biophys Acta 1762(10):920–933 3. Mitchison HM, Hofmann SL, Becerra CHR et al (1998) Mutations In The Palmitoyl-Protein Thioesterase gene (PPT CLN1) causing juvenile neuronal ceroid lipofuscinosis with granular osmiophilic deposits. Hum Mol Genet 7(2):291–297 4. Mole SE, Williams RE, Goebel HH (2005) Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics 6(3):107–126 5. Rothberg PG, Ramirez-Montealegre D, Frazier SD et al (2004) Homogeneous polymerase chain reaction nucleobase quenching assay to detect the 1kbp deletion in CLN3 that causes Batten disease. J Mol Diagn 6(3):260–263 6. Santavuori P (1988) Neuronal ceroid-lipofuscinoses in childhood. Brain Dev 10(2):80–83 7. Siintola E, Lehesjoki AE, Mole SE (2006) Molecular genetics of the NCLs – status and perspectives. Biochim Biophys Acta 1762(10):857–864 8. Siintola E, Partanen S, Stromme P et al (2006) Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 129(Pt 6):1438–1445 9. Steinfeld R, Reinhardt K, Schreiber K et al (2006) Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet 78(6):988–998 10. Williams RE, Aberg L, Autti T et al (2006) Diagnosis of the neuronal ceroid lipofuscinoses: an update. Biochim Biophys Acta 1762(10):865–872 Sehen Depressive alles grau? Depression und Melancholie werden in Kunst und Literatur schon immer mit visuellen Begriffen umschrieben: Grau und Schwarz sind die Farben, die für Melancholie oder Depressivität stehen. Dass sich hinter diesen Sprachbildern auch eine empirische Wirklichkeit versteckt, hat nun eine Arbeitsgruppe am Universitätsklinikum Freiburg in Zusammenarbeit von Psychiatrie und Psychotherapie und Augenheilkunde gefunden. Schon bei früheren Untersuchungen fanden sie heraus, dass depressive Menschen Schwarz-Weiß-Kontraste schlechter wahrnehmen als Gesunde. In ihrer aktuellen Studie untersuchten die Freiburger Wissenschaftler mittels einer objektiven elektrophysiologischen Methode (quasi ein EKG der Netzhaut) die Antwort der Netzhaut auf alternierende Schachbrettmuster mit unterschiedlichen Kontrasten bei Depressiven und Gesunden. Es zeigten sich hoch signifikante Unterschiede: Depressive Menschen haben dramatisch kleinere Antwortamplituden auf der Netzhaut. Sollten sich diese Untersuchungsbefunde in weiteren Studien bestätigen, stünde mit dieser Methode ein Verfahren zu Verfügung, mit dem auf objektive Art und Weise der eigentlich subjektive Zustand der Depression gemessen werden könnte. Dies könnte weit reichende Auswirkungen nicht nur auf die Depressionsforschung, sondern auch auf die Diagnose und Therapie von depressiven Zuständen haben. Literatur: Bubl E, Kern E, Ebert D et al (2010) Seeing Gray When Feeling Blue? Depression Can Be Measured in the Eye of the Diseased. Biological Psychiatry Epub ahead of print Fazit für die Praxis Nicht selten ist es der Ophthalmologe, der die Zeichen der neuronalen Lipofuszinose als erster diagnostiziert. Daher ist es notwendig die Aufmerksamkeit für die Symptomatik dieser seltenen, aber fatalen Erkrankungen bereits bei den niedergelassenen Augenärzten zu schärfen, um die wenigen therapeutischen Optionen frühzeitig nutzen und die beteiligten Personen frühzeitig beraten zu können. Quelle: Universitätsklinikum Freiburg, www.uniklinik-freiburg.de Der Ophthalmologe 7 · 2010 | 615