Artikel Dr. Stehr pdf - NCL
Werbung

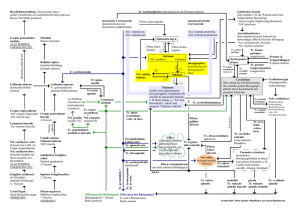
orthoptik - pleoptik 32/2009 Aus der NCL-Stiftung Leitung Forschung: Dr. F. Stehr Haben Sie auch schon mal eine Neuronale Ceroid-Lipofuszinose „übersehen“?* Frank Stehr Zusammenfassung Die Neuronalen Ceroid-Lipofuszinosen (NCL) stellen die häufigsten neurodegenerativen Erkrankungen des Kindesalters dar. Dennoch kommt es immer noch zu Fehldiagnosen, wie z. B. Retinitis pigmentosa. Für den Augenarzt, der zugleich der Erstdiagnostizierende ist, ist vor allem die juvenile NCL von Interesse. Wichtig ist dabei, im Fall einer rasch zunehmenden Sehminderung bei einem Kind im Einschulalter und elektrophysiologisch gesicherter tapetoretinaler Degeneration, an NCL zu denken und eine Kontrolluntersuchung nach spätestens einem Jahr oder sogar eher zu vereinbaren (RÜTHER 2006). Zu Beginn muss die Erkrankung nicht zwangsläufig mit einem erkennbaren geistigen Abbau verbunden sein. Schlüsselwörter: Neuronale Ceroid-Lipofuszinose, lysosomale Speicherkrankheit, BattenSpielmeyer-Vogt, Schießscheibenmakulopathie, Retinitis pigmentosa, Autofluoreszenz, Neurodegeneration Have you ever overlooked a neuronal ceroid lipofuscinosis? Summary The neuronal ceroid lipofuscinoses (NCLs or Batten disease) are the collectively most common neurodegenerative disorders of childhood. Each form presents with visual failure, but the juvenile form is the most important for ophthalmologists as they are usually the first to see these children. However, misdiagnoses such as Retinitis pigmentosa are frequent and it is important to have a second visual examination within a year, especially in case of sudden visual decline in children who are starting school. Tapetoretinal degeneration can be confirmed by electrophysiological means and when coupled with mental decline, Batten disease may often be the underlying reason (RÜTHER 2006). Keywords: Batten-Spielmeyer-Vogt disease, Neuronal ceroid lipofuscinosis, retinitis pigmentosa, bull’s eye maculopathy, autofluorescence, lysosomal storage disease, neurodegeneration * In Auszügen als Vortrag gehalten anlässlich der Regionaltagung Hamburg/Schleswig-Holstein des Berufsverbands der Orthoptistinnen Deutschlands e.V. in Hamburg am 27.09.2008 1 Einleitung Die Neuronalen Ceroid-Lipofuszinosen (NCLs) stellen die häufigsten neurodegenerativen Erkrankungen des Kindes- und Jugendalters dar. 1826 wurden die NCLs das erste Mal von dem norwegischen Arzt Dr. Otto Christian Stengel beschrieben. Weiter führende Beschreibungen erfolgten durch den britischen Neurologen BATTEN, der 1903 von zwei Geschwistern berichtete, die eine progressive Makuladystrophie in Kombination mit einer cerebralen Degeneration aufwiesen. In den folgenden Jahren beschäftigten sich mehrere Forscher mit diesen Stoffwechselkrankheiten, wie zum Beispiel Walter Spielmeyer und Heinrich Vogt. Diese weltweit vorkommenden genetischen Erkrankungen des Gehirns und der Augen weisen eine Inzidenz von 1 : 25.000 Lebendgeborenen auf (ZEMAN 1976). In Deutschland kommen jedes Jahr ca. 10 - 15 neue NCL-Fälle hinzu. Ihnen liegt, bis auf eine seltene adulte Form, eine autosomal-rezessive Vererbung zugrunde. Der Gendefekt verursacht auffällige Ablagerungen in den Lysosomen, den Recyclinghöfen der Zellen, daher werden diese Krankheitsbilder auch zu der Gruppe der lysosomalen Speicherkrankheiten gezählt. Diese Ablagerungen, die v. a. in Neuronen vorkommen, haben unter dem Elektronenmikroskop eine charakteristische Erscheinung (Abb. 1) und können daher zur Diagnostik herangezogen werden. Da jedoch bereits eine Vielzahl der betroffenen Gene identifiziert worden ist, wird die Diagnostik heutzutage in der Regel durch molekulargenetische Untersuchungen vervollständigt. Mindestens zehn verschiedene genetische NCL-Formen sind bekannt, die in unterschiedlichem Alter auftreten und mit CLN1 – CLN10 bezeichnet werden (Abb. 2). Somit kommt der molekulargenetischen Untersuchung eine große Bedeutung bei. Neben der intrazellulären Schnittmenge, dem wachsartigen Speichermaterial Ceroid-Lipofuszin, tritt auch bei allen NCL-Formen eine Kombination von Krankheitssymptomen auf: Visusverlust – Demenz – Epilepsie. Diese Symptome können in Abhängigkeit von der NCL-Form in unterschiedlicher Reihenfolge auftreten. Die einzelnen Formen unterscheiden sich auch im Alter der Patienten bei Beginn der Krankheit. Fast allen NCL-Formen ist gemeinsam, dass die Patienten zunächst vollkommen gesund und normal entwickelt sind. Für die Orthoptik und die Augenärzte ist vor allem die juvenile Form (Spielmeyer-Vogt, Batten disease, CLN3) von Bedeutung. Abb. 1 Elektronenmikroskopische Aufnahme einer JNCL-Patienten-Zelle. Es sind charakteristische Fingerabdruck-ähnliche Ablagerungen in den Lysosomen zu sehen. Die Hauptproteinkomponente dieser Ablagerungen ist die Untereinheit c der mitochondrialen ATP-Synthase (Quelle: Hans-Hilmar Goebel). 2 Abb. 2 NCL-Genfamilie: Bisher sind zehn NCL-Gene postuliert bzw. zum großen Teil bereits charakterisiert worden. Die CLN-Gene kodieren für lysosomale Enzyme sowie für putative Membranproteine, deren Funktion noch nicht eindeutig geklärt ist. CLN1, CLN2 und CLN10 kodieren die Palmitoylprotein-Thioesterase (PPT1), Tripeptidyl-Peptidase (TPP1) bzw. Cathepsin D. Die meisten Mutationen in diesen Genen führen zu der infantilen, klassischen spät-infantilen und der kongenitalen NCL-Form. Gendefekte in den CLN3 und CLN9 verursachen einen juvenilen Verlauf. CLN5-CLN8 sind variante Formen der spät-infantilen NCL. CLN4 ist für eine seltene adulte Form verantwortlich. Krankheitsverlauf und betroffenes Gen der juvenilen NCL (JNCL): Die juvenile NCL ist gekennzeichnet durch den nach einem charakteristischen Zeitplan (Abb. 3) ablaufenden Verlust zunächst der Sehkraft, dann der geistigen Fähigkeiten, der Beweglichkeit und eine Neigung zu epileptischen Anfällen (KOHLSCHÜTTER 1988). Im Einschulalter zeigen die juvenilen Patienten ihr erstes Symptom: eine Sehverschlechterung. Bis zu diesem Zeitpunkt waren es vollkommen gesunde Kinder. Die Visusminderung und die bald folgende Einengung des Gesichtsfelds führen rasch – innerhalb von ein bis drei Jahren – zur vollständigen Erblindung. NCL ist nicht nur eine Erkrankung des Auges, denn schleichend findet parallel ein geistiger Abbau statt. Das gesamte Zentrale Nervensystem ist betroffen (Abb. 4). Die schulischen Leistungen sinken, beginnend mit Problemen im Bereich des abstrakten Denkens. Besonders das Rechnen fällt den Schülern schwerer. Das Kurzzeitgedächtnis ist zunehmend eingeschränkt. Das Langzeitgedächtnis bleibt im Verhältnis dazu recht lange erhalten. Sehbehindertenpädagogen sind häufig die ersten, die auf die Zeichen der Krankheit aufmerksam werden. Nach zwei bis vier Jahren, nachdem die Sehverschlechterung startete, treten häufig die ersten epileptischen Anfälle auf, meist vom großen generalisierten (Grand-mal-)Typ (KOHLSCHÜTTER 2005). Im Verlauf der fortschreitenden Hirnatrophie gehen bereits erworbene kognitive und motorische Fähigkeiten verloren. Das Gangbild der Kinder wird immer auffälliger. Eine anfängliche Ungeschicklichkeit weitet sich zu einer gestörten Koordination der Bewegungsabläufe aus. Der Gang wird immer kleinschrittiger und es fällt den Patienten immer schwerer, gerade zu stehen. Wie Parkinson-Patienten stehen die Jugendlichen leicht vornüber gebeugt und ihre Knie sind eingeknickt. Die hinzu 3 kommenden spastischen Lähmungen und Myoklonien führen zum Einsatz eines Rollstuhls, später zur vollständigen Bettlägerigkeit. Aufgrund von Schluckbeschwerden werden die Patienten mittels PEG-Sonde enteral ernährt. Die Pubertät zeichnet sich durch ein psycho-organisches Syndrom aus. Aufgrund des Sprachverlustes und schwerer Angst- und Panikzustände, die durch Halluzinationen hervorgerufen werden können, kommt es zu schweren psychischen Problemen wie Aggressionen und Depressionen. Das unaufhaltsame Fortschreiten dieser Krankheit führt dazu, dass die jungen Erwachsenen nicht älter als 20 bis maximal 30 Jahre alt werden (WARBURG 1982). Abb. 3 Der durchschnittliche Krankheitsverlauf eines juvenilen NCL-Patienten. Abb. 4 Aufgrund des krankhaften Absterbens der Nervenzellen ist das Gehirn eines juvenilen NCL-Patienten deutlich reduzierter im Volumen als das eines gesunden Individuums (Quelle: Taina Autti). Das juvenile NCL-Gen (CLN3) wurde 1995 auf dem Chromosom 16 (p12.1-11.2) lokalisiert (THE INTERNATIONAL BATTEN DISEASE CONSORTIUM 1995) und somit der Grundstein für eine molekulargenetische Diagnostik gelegt. Der Gendefekt führt zum Verlust eines lysosomalen Membranproteins mit noch unbekannter Funktion. Antikörper gegen das entsprechende Prote4 in haben in den retinalen Neuronen der Maus eine gegenüber den corticalen Neuronen sehr frühe Expression nachgewiesen (MUNROE 1997). Dieses könnte auf eine wichtige Funktion von CLN3 in der Retina hinweisen und der Grund dafür sein, warum die Kinder als erstes erblinden. Augenärztliche Diagnostik: Sehverlust bei Schulanfängern Fehlinterpretationen der Symptome sowie die ärztliche Unkenntnis der Krankheit führen zu Fehldiagnosen, so dass häufig erst Jahre später die korrekte Diagnose erfolgt. Dem Augenarzt kommt eine bedeutende Rolle für die frühe Erkennung der JNCL zu, da die Verschlechterung der Sehkraft als Initialsymptom gilt. Die häufigsten Fehldiagnosen sind Retinitis pigmentosa und der Morbus Stargardt. Auffällig ist bei der JNCL ein schnell progressiver Verlust des Visus infolge einer Makulopathie im Alter von fünf bis acht Jahren. Es folgt eine rasche Degeneration von Netzhaut und Pigmentepithel. Zuerst geht das zentrale Sehen verloren (Zentralskotom), gefolgt von Farbsehstörungen und anschließender Nachtblindheit. In nur ein bis drei Jahren sind die Kinder bereits vollständig erblindet (SEELIGER 1997). Die Untersuchung von SPALTON 1980 zeigte bei 7 von 26 JNCL-Kindern ein sog. „Vorbeischauen“ („Overlooking“). Die Kinder wurden gebeten, den Untersucher direkt anzuschauen. Daraufhin blickten sie ihm nicht ins Gesicht, sondern häufig aufgrund der exzentrischen Fixation oberhalb der Makula scheinbar über ihn hinweg, einige jedoch auch an ihm vorbei (Abb. 5) (KOHLSCHÜTTER 2001). Ein möglicher Grund für die meist oberhalb der Makula anzutreffende Fixation könnte eine erhöhte Phototoxizität der JNCL-Netzhaut sein, da die inferiore Retina am intensivsten dem Licht ausgesetzt ist. Die superiore Retina ist daher möglicherweise später betroffen. Abb. 5 Ein auffälliges „Vorbeischauen“ (Overlooking) von Tim, dem Sohn des Stiftungsgründers. Die Funduskopie zeigt eine Schießscheibenmakulopathie (Bull’s eye) (Abb. 6) sowie stark verdünnte retinale Gefäße. Auch Pigmentverschiebungen und ein sog. „Pfeffer-und-Salz-Fundus“ treten häufig auf. Außerdem kommt es zu einer sich schnell entwickelnden konzentrischen Gesichtsfeldeinschränkung (RÜTHER 2006). Dieses sind die ersten Hinweise auf eine hereditäre Netzhautdegeneration. Auf jeden Fall sollte ein Ganzfeld-Elektroretinogramm durchgeführt 5 werden. Dieses ist bei JNCL-Kindern meist vermindert und im Verlauf rasch nicht mehr nachweisbar (COLLINS 2006). Abb. 6 Augenhintergrund eines juvenilen NCL-Patienten mit deutlicher schießscheibenartiger Strukturierung der Makula (Quelle: RÜTHER). Bei plötzlichem Visusabfall im Einschulalter und elektrophysiologisch gesicherter tapetoretinaler Degeneration in Verbindung mit einem geistigen Abbau sollte an NCL gedacht und eine Kontrolluntersuchung nach spätestens einem Jahr vereinbart werden (RÜTHER 2006). Weiter führende Untersuchungen wie z. B. ein EEG können Hinweise auf eine cerebrale Veränderung geben (PAMPIGLIONE 1977). Ein Blutausstrich sollte unter dem Lichtmikroskop von einer erfahrenen Person auf vakuolisierte Lymphozyten hin untersucht werden (Abb. 7), die einen wichtigen Hinweis auf eine zugrunde liegende JNCL geben. Abb. 7 Ein Blutausstrich weist unter dem Lichtmikroskop einen vakuolisierten Lymphozyten auf. Diese Untersuchungsmethode sollte zur Diagnosestellung einer JNCL hinzugezogen werden (Quelle: KOHLSCHÜTTER). 6 Eine frühe und korrekte Diagnose ist sehr wichtig, obwohl noch keine Therapie vorhanden ist, da die Familien von einer genetischen Prädisposition wissen sollten (Familienberatung). Prämorbide Diagnostik bei jüngeren Geschwistern und pränatale Diagnostik sind möglich. Auch werden Fehlbehandlungen vermieden und die Ausnutzung palliativer Behandlungsmöglichkeiten wird gewährleistet. Sportliche Betätigungen helfen, vor allem das Tandem Radfahren und auch Schwimmen. Ausblick: Der Krankheitsverlauf ist weder zu verzögern noch zu stoppen. Um diese Situation zu ändern, gründete 2002 Dr. Frank Husemann, dessen Sohn Tim an der juvenilen NCL leidet, die Stiftung National Contest for Life (NCL-Stiftung). Ziel der Stiftung ist es, durch Forschungsförderung und -vernetzung sowie gezielte Aufklärungsarbeit bei relevanten Ärztegruppen NCL zu heilen. Augen- und Kinderärzte werden durch Fortbildungsmaßnahmen und Übersichtsartikel über NCL informiert, um die Rate an Fehldiagnosen zu reduzieren. Die Stiftung fördert die wissenschaftliche Therapieentwicklung. Hierzu werden Forscher unterschiedlicher Disziplinen miteinander vernetzt. Viel versprechende Forschungsansätze werden mit Doktorandenstipendien gefördert. Aktuell fördert die NCL-Stiftung die ersten Schritte der Entwicklung einer Gentherapie, um die genetische Krankheitsursache direkt zu bekämpfen. Zuerst sollen geeignete Genfähren kloniert und an Zell- und Tiermodellen auf ihre Effektivität hin getestet werden. Die frühzeitige Diagnose ist der Grundstein für eine erfolgreiche Therapie. In Regensburg befindet sich die Entwicklung eines Gen-Chips für die Diagnose von erblichen Netzhauterkrankungen im experimentellen Stadium. Falls der Chip routinemäßig zum Einsatz kommen sollte, ist es das Ziel, dass bei einer RP-Verdachtsdiagnose vor der Beprobung des Chips eine einfache Polymerase-Kettenreaktion auf die CLN3-Hauptdeletion durchgeführt wird. 96% der Patienten weisen diese charakteristische Deletion auf, 74% auf beiden Chromosomen und 22% auf einem Chromosom (MUNROE 1997). Dieser konkrete Ansatz soll helfen, die Fehldiagnose RP zu vermeiden. Literatur BATTEN FE Cerebral degeneration with symmetrical changes in the maculae in two members of a family. Trans Ophthalmol Soc UK, 23: 386-390 (1903) COLLINS J, HOLDER GE, HERBERT H, ADAMS GG Batten disease: features to facilitate early diagnosis. Br J Ophthalmol, 90: 1119-1124 (2006) KOHLSCHÜTTER A Band 3 der Schriftenreihe Theorie und Praxis der Blinden- und Sehbehindertenpädagogik, Buchtitel NCL. Zur Lebenssituation von blinden Kindern und Heranwachsenden mit einer unheilbaren Abbauerkrankung. Beiträge aus Pädagogik, Therapie und Medizin. Herausgeber Hartmut Schlegel (2001) KOHLSCHÜTTER A, LAABS R, ALBANI M Juvenile neuronal ceroid lipofuscinosis (JNCL): quantitative description of its clinical variability. Acta Paediatr Scand, 77: 867-872 (1988) KOHLSCHÜTTER A, GOEBEL H-H, SCHULZ A, LUKACS Z Die neuronalen Ceroid-Lipofuszinosen. Dtsch Ärztebl 102: 284-288 (2005) MUNROE PB, MITCHISON HM, O’RAWE AM, ANDERSON JW, BOUSTANY R-M, LERNER TJ, TASCHNER EM, DE VOS N, BREUNING MH, GARDINER RM, MOLE SE Spectrum of mutations in the Batten disease gene, CLN3. Am J Hum Genet, 61: 310-316 (1997) 7 PAMPIGLIONE G, HARDEN A So-called neuronal ceroid lipofuscinosis. Neurophysiological studies in 60 children. J Neurol Neurosurg Psychiatry, 40: 232-330 (1977) RÜTHER K, GAL A, KOHLSCHÜTTER A Relevanz ophthalmologischer Diagnostik für die Betreuung von Patienten mit juveniler neuronaler Ceroid-Lipofuszinose. Klin Monatsbl Augenheilkd, 223: 542-544 (2006) SEELIGER M, RÜTHER K, APFELSEDT-SYLLA E, SCHLOTE W, WOHLRAB M, ZRENNER E Juvenile neuronal ceroid lipofuscinosis (Batten-Mayou) disease. Ophthalmologic diagnosis and findings. Ophthalmologe 94: 557-562 (1997) SPALTON DJ, TAYLOR DSI, SANDERS MD Juvenile Batten’s disease: an ophthalmological assessment of 26 patients. Brit J of Ophthalm 64: 726-732 (1980) THE INTERNATIONAL BATTEN DISEASE CONSORTIUM Isolation of a novel gene underlying Batten disease, CLN3. Cell 82: 949-957 (1995) WARBURG M The natural history of Jansky-Bielschowsky’s and Batten’s disease. In: Armstrong D, Koppang N, Rider JA, editors, Ceroid-Lipofuscinosis (Batten’s disease), Amsterdam: Elsevier Biomedical Press: 35-44 (1982) ZEMAN W The neuronal ceroid-lipofuscinoses. Prog Neuropathol 3: 203-223 (1976) Anschrift des Autors: Dr. Frank Stehr NCL-Stiftung Holstenwall 10 20355 Hamburg E-Mail: [email protected] Homepage: www.ncl-stiftung.de 8