Wiederholt generalisierte Lymphadenopathie, akutes
Werbung

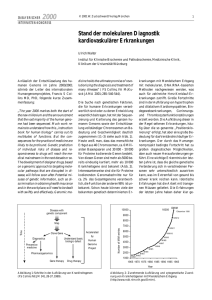
IgG4-Syndrom Generalisierte Lymphadenopathie, akutes Nierenversagen und Anämie bei einer 65-jährigen Frau Jodocy D, Grießhammer M, Kellner U, Reinbold WD, Radermacher J, Lakomek HJ. Einführung ● Immunoglobin-G4 assoziierte Erkrankungen werden zunehmend als eine Einheit mit analogen Eigenschaften unklarer Ätiologie(1,2) verstanden: – Anschwellen der betroffenen Gewebe – Lymphoplasmozytäre Gewebsinfiltration mit v.a. IgG4-positiven Plasmazellen und T-Lymphozyten, welche meist mit Fibrose und obliterativer Phlebitis sowie milder Gewebseosinophilie einhergehen – ● Diagnostik ● ● Erhöhtes Serum-IgG4 (>135mg/dl) Diskussion Ig-G4 (Serum): 339mg/dl [Normbereich 30 – 210] ● Bildgebung A Fälle von IgG4-Erkrankung mit dialysepflichtigem akutem Nierenversagen und hämolytischer Anämie sind bislang in der Literatur nicht beschrieben. C ● ● ● Eine 65-jährige Patientin wird mit Übelkeit, Erbrechen, Durchfall und Exsikkose ins auswärtige Krankenhaus eingewiesen. A .Axillär vermehrte Lymphknoten bis 10mm Größe. B. Paraaortal im Bereich des Truncus coeliacus und A. Mesenterica superior multiple teils bis 2,3cm große Lymphknoten. C. Die vergrößerten Nieren zeigen beidseitig eine ödematöse Umgebungsreaktion auf. ● Übernahme mit dialysepflichtigem akutem Nierenversagen (Kreatinin 10,1 mg/dl; Harnstoff 190 mg/dl; Kalium 5,6 mmol/l) Zudem fallen eine IgG-übermittelte Coombs-positive autoimmun hämolytische Anämie (Hb 88 g/l) und Thrombopenie (76 g/l) auf. ● – Autoimmun Pankreatitis Typ I – IgG4 abhängige sklerosierende Cholangitis – Mikulicz Erkrankung – Dacryoadenitis – Küttner Tumor – Sialadenitis – Inflammatorisch orbitaler Pseudotumor – Ormond Erkrankung – retroperitoneale Fibrose – Aortitis und Periaortitis – chronisch sklerosierend – Riedel Thyreoiditis – Tubulointerstitielle Nephritis und Glomerulonephritis – Hypophysitis – Pachymeningitis B Historie und Anamnese ● IgG4-assoziierte Erkrankungen: (bei 6090% aller Patienten tritt >1 davon auf)(1,3,4) ● Nierenbiopsie: schwergradige floride plasmazellreiche nichteitrige interstitielle Nephritis. Immunhistochemisch 11 IgG4 positive Plasmazellen pro HPF. Knochenmark-Ausstrich ● Zwei Jahre zuvor erfolgte die Abklärung einer generalisierten Lymphadenopathie (LK maximal 2,1 cm) und Splenomegalie (14 cm). Mittels immunhistopathologischer Aufarbeitung von zervikalem, präkavalem, inguinalem und peritonealem Lymphknotengewebe konnte ein Ausschluß von Lymphomerkrankungen oder Morbus Castleman erfolgen. IgG4-assoziierte Lymphadenopathie(5) – Meist begleitend zu anderen Manifestationen von IgG4-assoziierten Erkrankungen – LK meist nicht größer als 2cm – Betreffen mehrere LK-Regionen IgG4-assoziierte tubulointerstitielle Nephritis(6) – Begleitend zu anderen Manifestationen von IgG4-assoziierten Erkrankungen – Meist keine klinische Symptomatik – Lymphoplasmazytäre Infiltration von IgG4-positiven Plasmazellen und begleitende Fibrose-Entstehung Therapie und Verlauf ● Hyperzelluläres Knochenmark, Lymphozytenvermehrung, angeregte Erythropoese sowie Vermehrung (bis 20%) von Plasmazellen des kleinen Zelltyps. → reaktive KMVeränderungen mit Lympho- und Plasmozytose. ● ● LK-IgG4-Immunhistochemie ● Lymphknoten-Histologie mit Nachweis einer gut erhaltenen Grundstruktur – Sinus, Lymphfollikel und Mantelzone, regelrechte Schichtung der Keimzentren. Zusammen mit der Immunhistochemie beschrieben als reaktive Veränderungen bei chronisch unspezifischer Lymphadenitis. ● Die Patientin klagt über Mundtrockenheit, Taubheitsgefühle beider Unterarme und Hände sowie allgemeines Krankheitsgefühl mit Schwäche. Kein Gewichtsverlust, kein Nachtschweiß, keine Übelkeit. Optimale Therapie nicht etabliert(7) 1. Wahl: Steroide – 40mg/Tag oral bis zur klinischen Besserung, danach Ausschleichen über 2 Monate bis zum Absetzen Bei Therapieversagen oder Rezidiv – Azathioprine bis zu 2 mg/kg KG/Tag – Mycophenolatmofetil 750mg 2xtgl. – Rituximab 1000mg (an Tagen 1+15) ➔Die 65-jährige Patientin zeigte unter Steroiden eine rasche Regredienz der Nierenfunktion und Hämolyse; Besserung des subjektiven Krankheitsgefühls. Vermehrte Lymphozyten mit Expression des Immunglobulin-G4. ➔Komplette Remission der generalisierten 1. Stone JH, Zen Y, Deshpande V. IgG4-realted disease. N Engl J Med 2012; 366:539 2. Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010; 17:303. 3. Khosroshahi A, Stone JH. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol 2011; 23-57. 4. Stone JH, Khosroshahi A, Deshpande V, et al. IgG4-related disease: recommendations for the nomenclature of this condition and its individual organ system manifestations. Arthritis Rheum 2012 5. Cheuk W, Yuen HK, Chu SY, et al. Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol 2008; 32:671. 6. Saeki T, Nishi S, Imai N, et al. Clinicopathological characteristics of patients with IgG4-related tubulointerstitial nephritis. Kidney Int 2010; 78:1016. 7. Khosroshahi A, Stone JH. Treatment approaches to IgG4-related systemic disease. Curr Opin Rheumatol 2011; 23:67-71. Lymphadenopathie im CT nach 9 Monaten und Absetzen der Steroidtherapie.