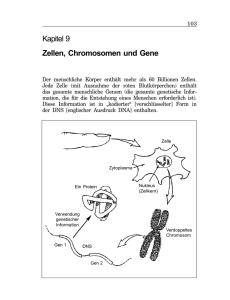
Mehrdimensionale in-cell & SAR by NMR
Werbung

Mehrdimensionale in-cell & SAR by NMR-Studien zur funktionellen Charakterisierung der kleinen GTPase Rheb Dissertation zur Erlangung des Grades Doktor der Naturwissenschaften (Dr. rer. nat.) der Fakultät für Chemie und Biochemie an der Ruhr-Universität Bochum vorgelegt von Katharina F. G. Jockers, M. Sc. Biochem. Bochum 2012 Die vorliegende Arbeit wurde im Zeitraum von Oktober 2008 bis September 2012 am Lehrstuhl für Biochemie II in der Arbeitsgruppe Biomolekulare Spektroskopie von Herrn Prof. Dr. Raphael Stoll unter dessen wissenschaftlicher Aufsicht an der Fakultät für Chemie und Biochemie der RuhrUniversität Bochum angefertigt. Tag der Disputation: 14.12.2012 Vorsitz: Prof. Dr. Martin Muhler Referent: Prof. Dr. Raphael Stoll Korreferent: Prof. Dr. Peter Bayer Danke.... Ich danke Herrn Prof. Dr. Raphael Stoll für die Vergabe dieses Themas, die freundliche Aufnahme in seine Arbeitsgruppe, die hervorragende Betreuung sowie die ständige Bereitschaft zur Diskussion. Ebenso danke ich ihm für die vielen Freiheiten, die er mir bei der Erstellung dieser Arbeit eingeräumt hat und das mir entgegengebrachte Vertrauen. Herrn Prof. Dr. Peter Bayer danke ich für die unkomplizierte, kompetente und freundliche Übernahme des Zweitgutachtens sowie für die Nutzung der NMR-Spektrometer an der Universität Duisburg-Essen. Herrn Prof. Dr. Jürgen Scherkenbeck danke ich für die sehr gute Kooperation und die ständige Bereitschaft zur Diskussion. Peter Düppe danke ich für die Bereitstellung der Liganden sowie Diskussion der Ergebnisse. Der Arbeitsgruppe Proteininteraktionen von Herrn Prof. Dr. Christian Herrmann danke ich für die Möglichkeit, Ressourcen der Arbeitsgruppe zu nutzen und für viele Diskussionen (auch über das Thema dieser Arbeit hinaus). Herrn Prof. Dr. Guiscard Seebohm danke ich für die Bereitstellung von Frosch-Oozyten sowie die kompetente Diskussionen rund um das in-cell NMR-Thema. Weiter danke ich Herrn Prof. Dr. Rolf Heumann, dass ich die Ressourcen des Lehrstuhls Biochemie II nutzen durfte und für die ständige Diskussionsbereitschaft. Veena Nambiar Potheraveedu danke ich für die Durchführung erster Zelltests. Dr. Gerrit Paefcke danke ich für die Bereitstellung des Farnesyltransferase-Klons. Der Arbeitsgruppe von Dr. Dirk Wolters danke ich für die durchgeführten ESI-Messungen. Danken möchte ich dem SFB642, der Deutschen Krebsforschung sowie der Research School der RuhrUniversität Bochum, die dieses Promotionsvorhaben finanziell unterstützt haben. Besonderer Dank geht auch an Hans-Jochen Hauswald, Martin Gartmann und Gregor Barchan, die mir immer mit Rat und Tat zur Seite standen, wenn das NMR-Spektrometer nicht so wollte wie ich. Danken möchte ich auch Stefanie Pütz, Dr. Florian Brüsselbach sowie Katja Gonschorek für die freundliche Aufnahme in ihr Büro im Zuge der Umzugs- und Umbaumaßnahmen. Den Doktoranden der Graduiertenschule des SFB642 danke ich für die vielen Diskussionen und die tolle Atmosphäre während den Summer Schools und sonstigen Veranstaltungen. Ein besonderer Dank geht auch an meine Masterstudenten Philipp Kotthoff, Katrin Koclega und Miriam Schöpel für ihre kompetente und engagierte Hilfe bei vielen Experimenten. Danken möchte ich den ehemaligen und aktuellen Mitarbeitern der Arbeitsgruppe Biomolekulare Spektroskopie. Meinen Eltern, meiner Oma, Dr. Gerd Kock und meinen Freunden danke ich für ihre stetige Unterstützung während der Promotionsphase und die Entfernung der vielen „so“ aus dieser Arbeit. So! Einszweidrei, im Sauseschritt läuft die Zeit, wir laufen mit (Julchen- Wilhelm Busch) Für die weltbeste und verrückteste Familie INHALTSVERZEICHNIS INHALTSVERZEICHNIS 1 EINLEITUNG 1 1.1 1 GTP-BINDENDE PROTEINE 1.1.1 RAS-SUPERFAMILIE 1 1.1.2 G-PROTEINE: STRUKTURELLE MERKMALE 2 1.1.3 BIOCHEMIE UND REGULATION DER GTPASEN DER RAS-SUPERFAMILIEN 4 1.1.4 REGULATION VON MTOR DURCH KLEINE GTPASEN 10 1.1.5 RHEB- EINE BESONDERE KLEINE GTPASE 13 1.2 18 IN-CELL NMR-SPEKTROSKOPIE 1.2.1 PROKARYOTISCHE ZELLEN 19 1.2.2 EUKARYOTISCHE ZELLEN 22 1.3 25 VOM SCREENING ZUM VALIDIERTEN HIT 1.3.1 FBS ZUR IDENTIFIZIERUNG VON LIGANDEN VON K-RAS 28 1.4 30 NMR-SPEKTROSKOPIE 2 ZIELSETZUNG UND GLIEDERUNG 32 3 MATERIAL UND METHODEN 33 3.1 MATERIAL 33 3.1.1 CHEMIKALIEN 33 3.1.2 VERWENDETE GERÄTE 38 3.1.3 VERWENDETE PLASMIDE 39 3.1.4 PROTEINMARKER 39 3.1.5 MEDIEN UND PUFFER 40 3.1.6 PUFFER UND LÖSUNGEN FÜR SDS-PAGE 43 3.2 44 METHODEN 3.2.1 MOLEKULARBIOLOGISCHE METHODEN 44 3.2.2 PROTEINBIOCHEMISCHE METHODEN 45 3.2.3 BESTIMMUNG DER PROTEINKONZENTRATION NACH BRADFORD 48 3.2.4 NUKLEOTIDAUSTAUSCH 48 I INHALTSVERZEICHNIS 3.2.5 PROTEINPRÄPARATION FÜR DIE NMR-SPEKTROSKOPIE 48 3.2.6 HERSTELLUNG VON E. COLI-LYSAT 48 3.2.7 HERSTELLUNG VON OOZYTEN-EXTRAKT 49 3.2.8 INJEKTION VON PROTEIN IN XENOPUS-OOZYTEN 49 3.2.9 HERSTELLUNG VON LIPID-VESIKELN 49 3.2.10 LIPID-BINDUNGSTEST 50 3.2.11 ESI-MS 50 3.3 SCREENING DER SUBSTANZBIBLIOTHEKEN 50 3.3.1 BESTIMMUNG DER DISSOZIATIONSKONSTANTEN 51 3.4 COMPUTERGESTÜTZTE ARBEITEN 52 3.4.1 VERWENDETE COMPUTERPROGRAMME 52 3.5 NMR-EXPERIMENTE 54 3.5.1 DAS 1D 1H-EXPERIMENT 54 3.5.2 ZWEIDIMENSIONALE EXPERIMENTE 55 3.6 PROZESSIERUNG VON NMR-DATEN 57 3.7 PULSPROGRAMME 58 4 ERGEBNISSE-TEIL I 59 4.1 PROTEINPRÄPARATION VON RHEB 59 4.2 AUFNAHME VON EIN- UND MEHRDIMENSIONALEN NMR-SPEKTREN 60 4.2.1 DAS 1D 1H-NMR-EXPERIMENT 60 4.2.2 DAS 2D 1H-15N-HSQC-EXPERIMENT 62 4.3 64 IN-CELL NMR-SPEKTROSKOPIE 4.3.1 E. COLI-ZELLEN 65 4.3.2 XENOPUS-OOZYTEN 68 4.4 FARNESYLIERUNG VON RHEB 70 4.4.1 IN VITRO FARNESYLIERUNG 71 4.4.2 IN VIVO FARNESYLIERUNG 71 4.4.3 ESI-MS 73 4.4.4 KOSEDIMENTATIONSANALYSE 74 4.4.5 NMR-SPEKTROSKOPISCHE CHARAKTERISIERUNG VON FARNESYLIERTEM RHEB 75 5 DISKUSSION - TEIL I 77 II INHALTSVERZEICHNIS 5.1 IN-CELL NMR-SPEKTROSKOPIE 77 5.1.1 E. COLI-ZELLEN 77 5.1.2 XENOPUS-OOZYTEN 81 5.2 FARNESYLIERUNG VON RHEB 6 ERGEBNISSE - TEIL II 82 85 6.1 1 H-15N-SOFAST-HMQC-SPEKTRUM 85 6.2 SCREENING DER SUBSTANZBIBLIOTHEK 86 6.2.1 AUFBAU EINER SUBSTANZBIBLIOTHEK 87 6.3 88 VERWENDETE SUBSTANZGEMISCHE 6.3.1 GEMISCH 1 89 6.3.2 GEMISCH 2 94 6.3.3 GEMISCH 3 97 6.3.4 GEMISCH 4 100 6.3.5 GEMISCH 5 103 6.3.6 GEMISCH 6 105 6.3.7 GEMISCH 7 108 6.4 BIPHENYL- UND PHENOLDERIVATE 111 6.5 BISPHENOL A 115 6.6 ANALYSE DER BESTEN HITS 116 6.6.1 MODELLIERUNG MIT HADDOCK 116 6.6.2 4,4´-BIPHENOL 117 6.6.3 1-(3,4-DICHLOROPHENYL)PIPERAZIN 122 6.6.4 4,4'-DIHYDROXYBENZOPHENON 124 6.6.5 BISPHENOL A 125 6.7 127 BESTIMMUNG DER DISSOZIATIONSKONSTANTEN 7 DISKUSSION - TEIL II 129 7.1 AUFBAU UND SCREENING DER SUBSTANZBIBLIOTHEK 129 7.2 MODELLIERUNG MIT HADDOCK 130 7.2.1 VERGLEICH DER UNTERSCHIEDLICHEN KOMPLEXE 132 7.3 133 VERGLEICH MIT RAS III INHALTSVERZEICHNIS 8 ZUSAMMENFASSUNG 137 9 CONCLUSION 139 10 AUSBLICK 141 11 ABKÜRZUNGSVERZEICHNIS 142 12 ABBILDUNGSVERZEICHNIS 148 13 TABELLENVERZEICHNIS 151 14 LITERATURVERZEICHNIS 152 15 ANHANG 168 15.1 AIR-TABELLE FÜR DIE HADDOCK-MODELLIERUNG 168 15.2 ÄNDERUNGEN HADDOCK-RUN 168 IV 1 Einleitung 1 EINLEITUNG 1.1 GTP-BINDENDE PROTEINE Die Hydrolyse von Guanosintriphosphat (GTP) spielt eine zentrale Rolle in einer Vielzahl von biochemischen Prozessen. So werden die Proteinbiosynthese, das Wachstum, die Zelldifferenzierung und die Sinneswahrnehmung über einen solchen Prozess reguliert. Darüber hinaus werden die Zytoskelettorganisation sowie eine Vielzahl von Transportprozessen kontrolliert1. Zur katalysierten Umsetzung von Guanosintriphosphat zu Guanosinmonophosphat (GDP) und anorganischem Phosphat (Pi) bedarf es in der Zelle Guaninnukleotid-bindender Proteine (GNBPs). Anhand von Strukturmerkmalen und der Sequenz lassen sich GNBPs in zwei Klassen einteilen2: die translation factors (TRAFAC) und die signal recognition particle, MInD, BIoD (SIMIBI). Zur Gruppe der TRAFACs gehören Translationsfaktoren der Proteinbiosynthese, heterotrimere G-Proteine, Proteine der Ras-Superfamilie sowie die ATPasen Myosin und Kinesin. Alle Mitglieder dieser Klasse weisen ein mindestens sechssträngiges ɴ-Faltblatt auf, dessen zweiter Strang antiparallel orientiert ist. Weiter zeichnen sich alle TRAFACs durch einen hochkonservierten Serin- oder Threoninrest im Loop zwischen Strang 2 und 3 aus. Strukturell lassen sich SIMIBI-Proteine von TRAFAC-Proteinen sehr leicht abgrenzen, da sie nur ein paralleles, siebensträngiges ɴ-Faltblatt aufweisen. Zu dieser Klasse gehören signal recognition particle (SRP) GTPasen, MinD-like ATPasen, welche besonders für Proteinlokalisiation, Chromosomenteilung und Membrantransport zuständig sind sowie eine Gruppe von Kinasen oder Phosphattransferasen. Bevor durch Röntgenstrukturanalyse und NMR-Spektroskopie ein Zugang zu der Struktur der GNBPs ermöglicht wurde, erfolgte eine Einteilung anhand ihrer Funktion. 1.1.1 RAS-SUPERFAMILIE In Eukaryoten wurden bisher über 150 verschiedene Mitglieder aus der Ras-Superfamilie der kleinen GNBPs identifiziert3. Die Ras-Superfamilie wird traditionell in fünf Untergruppen4 gegliedert: ඵ Ras-Familie (u. a. H-, K-, N-Ras, Ral, Rheb, Rap1B) ඵ Rho-Familie (u. a. RhoA, RhoB, Rac1, Rho, CDC42) ඵ Rab-Familie (u. a. Rab1A, Rab1B) ඵ Ran-Familie (u. a. Ran, Gsp1 in Hefe) ඵ Sar1/Arf-Familie (u. a. Arf1, Sar1a, Arl1) 1 1 Einleitung Durch seine zentrale Rolle bei der Entstehung vieler humaner Tumore steht besonders Rat sarcoma (Ras) im Fokus der aktuellen Forschung. Hierbei sind Mutationen in den drei humanen Ras-Genen (H-, K- und N-Ras) für etwa 30 % der menschlichen Tumore verantwortlich. Zum Beispiel finden sich bei etwa 75 % der Pankreaskarzinome Mutationen im Kodon 12 von K-Ras5. Ras-Proteine regulieren eine Vielzahl von zellulären Signalwegen, nämlich das Zellwachstum, die Differenzierung sowie die Apoptose. Die Aktinorganisation, der Zellzyklus und die Genexpression werden von Mitgliedern der Ras homology (Rho) Familie kontrolliert6. Die größte Familie stellen die Ras-like proteins in brain (Rab)-Proteine mit 61 Mitgliedern dar7. Diese GTPasen regulieren den intrazellulären Vesikeltransport und leiten das Trafficking der Proteine zwischen verschiedenen Organellen des endozytotischen und sekretorischen Signalwegs8. Die Klasse der Ras-like nuclear (Ran) Proteine ist die am häufigsten in der Zelle vorkommende kleine GTPase und ist für den Zellkerntransport von RNA und Proteinen zuständig9. Die Mitglieder der ADPribosyltion factor (ARF) Familie sind, wie auch die Rab-Proteine, an der Regulation des vesikulären Transports beteiligt10. 1.1.2 G-PROTEINE: STRUKTURELLE MERKMALE Alle G-Proteine sind sich, trotz vielfältiger Funktionen, strukturell sehr ähnlich. Gemeinsam ist allen Proteinen die sogenannte G-Domäne11. Diese Domäne weist eine universelle Struktur sowie einen universellen Mechanismus zur Nukleotidbindung und -hydrolyse auf. Durch das Vorhandensein eines gemischten sechssträngigen ɴ-Faltblatts, welches von beiden Seiten von fünf ɲ-Helices umgeben wird, lassen sich G-Proteine als ɲ,ɴ-Proteine beschreiben. Die Mitglieder der Rho-Familie enthalten zusätzlich eine ɲ-Helix von 13 Aminosäuren. Die Proteine Arf, Arl und Sar1 weisen eine zusätzliche aminoterminale Verlängerung auf, welche für die Membrananbindung und -interaktion notwendig ist. Allen GNBPs sind einige konservierte Sequenzmotive gemeinsam, welche die Nukleotidbindetasche umgeben und teilweise auch bei ATPasen zu finden sind. Das G1-Motiv, welches auch P-Loop oder Walker-A-Motiv genannt wird, ist zur Bindung des Phosphats wichtig. Dabei wird die Bindung zƵŵɲƵŶĚɴ-WŚŽƐƉŚĂƚŚĂƵƉƚƐćĐŚůŝĐŚĚƵƌĐŚ,ĂƵƉƚŬĞƚƚĞŶŬŽŶƚĂŬƚĞƐƚĂďŝůŝƐŝĞƌƚ͕ ǁŽďĞŝĚĂƐɶ-Phosphat durch ein konserviertes >LJƐŝŶ ŬŽŽƌĚŝŶŝĞƌƚ ǁŝƌĚ͘ Ğƌ >ŽŽƉ ǀĞƌďŝŶĚĞƚ ĚĂƐ ŚLJĚƌŽƉŚŽďĞ ɴ-&ĂůƚďůĂƚƚ ;ɴϭͿ ŵŝƚ einer amphiphatisĐŚĞŶɲ-Helix. Das folgende, auch als switch I bezeichnete G2-Motiv beinhaltet den N-dĞƌŵŝŶƵƐ ĚĞƐ njǁĞŝƚĞŶ ɴ-Faltblatts und den vorhergehenden Loop. In diesem Loop gibt es bei Bindung von GTP eine große Konformationsänderung, was maßgeblich durch die Orientierungsänderung des konservierten Threonins hervorgerufen wird. Zusammen mit dem Serin 2 1 Einleitung aus G1 koordinieren die Seitenketten dieses Threonins das für die GTP-Hydrolyse wichtige Magnesium-Ion, welches seinerseits ĚĂƐ ɴ- ƵŶĚ ɶ-Phosphat des GTPs koordiniert. Mitglieder aus einer GTPase-Familie weisen im G2-Motiv eine sehr hohe Sequenzähnlichkeit auf. Das in allen GTPasen konservierte DXXG-Motiv wird auch als G3-Motiv oder switch II bezeichnet. Das invariable Aspartat bindet indirekt über ein verbrückendes Wassermolekül an das Magnesium-Ion. ĂƐɶ-Phosphat des GTPs wird zusätzlich von einem Glycin über eine Wasserstoffbrücke stabilisiert. Bei den verschiedenen Nukleotidzuständen ändert sich auch in dieser Region sehr stark die Konformation des Proteins. Vor der Konsensussequenz (N/T)(K/Q)xD des G4-Motivs liegen vier hydrophobe oder apolare Aminosäuren. Durch das Aspartat in dieser Region wird die Guaninbase über zwei Wasserstoffbrücken stabilisiert. Durch Ausbildung von Wasserstoffbrücken zwischen Asparagin und Lysin aus der G4-Region zu Aminosäuren aus dem P-Loop erfolgt eine weitere Stabilisierung des Nukleotids. Eine eher indirekte Stabilisation erfährt das Nukleotid durch Interaktion des ɴϲ-Faltbaltts ƵŶĚĚĞƌɲϱ-Helix, welche im G5-Motiv lokalisiert sind, mit Resten aus G4. Die Strukturmotive G4 und G5 sind aufgrund ihrer Bindung zum Nukleotid verantwortlich für die Spezifität der Nukleotidbindung. Um verschiedene heterotrimere G-Proteine miteinander zu vergleichen, wird die G-Domäne von Ras als minimale Untereinheit und alle anderen als darauf aufbauende Variationen angesehen11. B Abbildung 1.1: Struktur von Guaninnukleotid-bindenden Proteinen und Wechselwirkungen zum Nukleotid. A In der Cartoon-Darstellung der minimalen G-Proteine (Ras-Protein) sind die konservierten Sequenzelemente sowie die switch-Regionen, das Nukleotid und das Magnesium-Ion in verschiedenen Farben abgebildet. Die Termini sind verkürzt und 11 durch N und C kenntlich gemacht . B Die Wechselwirkungen der konservierten Aminosäuren zum Nukleotid (hier GTPAnalogon GppNHp) sind anhand des Ras-Proteins dargestellt (mc = Peptidhauptkette)12. 'ɲ-Proteine, die ein Molekulargewicht von etwa 40-45 kDa aufweisen, besitzen neben diesem zentralen Motiv der G-Domäne einige weitere Domänen, ǁŝĞ ĞŝŶĞ ɲ-Helix sowie einige LoopRegionen. Das humane Guanylat-bindende Protein 1 (hGBP1), welches zur Dynaminfamilie der 3 1 Einleitung großen GTPasen gehört, besitzt eine vergrößerte G-Domäne mit einer Vielzahl von zusätzlichen Sekundärstrukturelementen sowie einer Coiled-Coil-Domäne am C-Terminus13. Zwei, drei und vier zusätzliche Domänen weisen der Elongationsfaktor Tu, Initiationsfaktor 2 und der Elongationsfaktor G auf13,14. 1.1.3 BIOCHEMIE UND REGULATION DER GTPASEN DER RAS-SUPERFAMILIEN GNBPs übernehmen in der Zelle die Aufgabe eines molekularen Schalters, indem sie in zwei Zuständen vorliegen können: in einem GTP-gebundenen, aktiven und in einem GDP-gebundenen, inaktiven Zustand. Die Hydrolyse von GTP zu GDP und Phosphat ist ein nahezu irreversibler Schritt und der Nukleotidaustausch von GDP zu GTP erfolgt nur langsam. Durch den Austausch des Nukleotids erfährt das Protein besonders in den switch-Regionen eine große Konformationsänderung. Durch NMR-Studien und Röntgenstrukturanalyse konnten diese großen Konformationsänderungen bestätigt werden15,16. Während im GTP-gebundenen Zustand große Ähnlichkeiten bei unterschiedlichen GTPasen vorhanden sind, zeigen die GDP-Formen häufig sehr große strukturelle Unterschiede17. In allen GTPasen liegt GTP als Komplex mit einem Magnesium-Ion vor. Die Koordination des Mg2+ erfolgt über je ĞŝŶ^ĂƵĞƌƐƚŽĨĨĚĞƐɴ- und des ɶ-Phosphats. In Gegenwart von Magnesium liegen die Dissoziationsraten von GTP und GDP im Bereich von 10-4 bis 10-5 s-1 18. In der aktiven Form bilden sich njǁĞŝ tĂƐƐĞƌƐƚŽĨĨďƌƺĐŬĞŶ njǁŝƐĐŚĞŶ ĚĞŶ ^ĂƵĞƌƐƚŽĨĨĞŶ ĚĞƐ ɶ-Phosphats zur Aminogruppe des konservierten Threonins, welches über seine Seitenkette das Magnesium-Ion bindet, und des Glycins (aus DXXG-Element) in den beiden switch-Regionen. Der durch den Austausch des Nukleotids erfolgende Mechanismus wird auch als loaded-spring-Mechanismus11 bezeichnet, bei dem durch die HydrŽůLJƐĞĚĞƐ'dWƐĚĂƐɶ-Phosphat freigesetzt wird und es zur Entspannung des gesamten Systems kommt. So allgemeingültig dieses Prinzip des loaded-spring-Mechanismus ist, so unterscheiden sich die Änderungen der Konformation untereinander sehr stark. Große Konformationsunterschiede lassen sich besonders bei Ran beobachten, wo in switch /ĞŝŶnjƵƐćƚnjůŝĐŚĞƐɴ-Faltblatt entfaltet wird und es eine sehr große Änderungen bei der Lokalisation des Carboxyterminus gibt19. Neben diesem so genannten C-terminalen switch wird bei Arf-Proteinen eine Konformationsänderung beobachtet, die räumlich weit entfernt von der eigentlichen Nukleotidbindung stattfindet. Hier wird durch GTPBindung der N-Terminus von Arf herausgedrückt, wodurch eine Membrananbindung über diesen Terminus möglich wird20. 4 1 Einleitung Abbildung 1.2: Schema des universalen Mechanismus bei der GTP-Hydrolyse von kleinen GTPasen11. In der aktiven, gespannten GTP-gebundenen Form sind die switch I und switch II Regionen über das konservierte Threonin ƵŶĚ'ůLJĐŝŶ ĂŶ ĚĂƐ ɶ-Phosphat gebunden, was sehr gut mit einer gespannten Feder verglŝĐŚĞŶ ǁĞƌĚĞŶ ŬĂŶŶ͘tŝƌĚ ĚĂƐ ɶPhosphat bei der GTP-Hydrolyse freigegeben, so relaxieren die switch-Regionen in eine entspannte Konformation. Bei allen G-Proteinen ist der Hydrolyseprozess von GTP zu GDP sowie der Austausch von GDP zu GTP in vitro sehr langsam, sodass regulierende Faktoren eingesetzt werden müssen. GTPase-aktivierende Proteine (GAPs) beschleunigen die GTP-Hydrolyse um ein Vielfaches und fördern somit den GDPgebundenen Zustand. Die Guaninnukleotid-Austausch-Faktoren (GEF) hingegen fördern den aktiven GTP-gebundenen Zustand, indem sie den Austausch des gebundenen GDPs erleichtern. Abbildung 1.3: GTPase-Zyklus reguliert über GAP- und GEF-Proteine21 Zum Austausch von an das G-Protein (G) gebundenem GDP wird ein spezifischer Austauschfaktor (GEF) benötigt, um die GTPase in einen aktiven Zustand zu überführen, in dem eine Interaktion mit nachgeschalteten Effektoren möglich ist. Das GAP-Protein fördert die Hydrolyse von GTP zu GDP und Pi, was die GTPase wieder in einen inaktiven Zustand zurückführt. 1.1.3.1 GEF-PROTEINE Zum Austausch von GDP gegen GTP werden GEF-Proteine benötigt, welche den sehr langsamen Austauschvorgang um ein Vielfaches beschleunigen. Die GEF-Proteine katalysieren diese Reaktion, indem sie die Nukleotidbindetasche so verändern, dass die Nukleotidaffinität sinkt und somit eine 5 1 Einleitung Freigabe des Nukleotids erfolgen kann. Da die Affinität der GTPasen zu GTP und GDP nahezu identisch ist, kann ein Austausch von GDP hin zu GTP nur durch die erhöhte zelluläre GTPKonzentration erreicht werden. Somit schwächt die GEF- die GDP-Bindung und umgekehrt die Nukleotid-Bindung die GEF-Bindung (Abbildung 1.4)11. 22 Abbildung 1.4: Mechanismus des Nukleotidaustausches, induziert durch GEF Das Nukleotid (gelb; B=Base, P=Phosphat) interagiert mit seiner Base und dem Nukleotid mit der GTPase. Das GEF konkurriert mit dem Nukleotid um die Bindung an der GTPase und fördert auf diese Weise die Freisetzung des Nukleotids. Neben dem Anfangs- und Endzustand existieren auch Zwischenzustände, bei denen eine Bindung (T) oder ein Lösen (L) des Komplexes vorangegangen ist. Das GEF interagiert mit switch I und switch II sowie dem P-Loop und der Magnesium-Ion-Bindestelle, wodurch das Phosphat nicht weiter gebunden werden kann. Das Magnesium kann durch das GNBP selbst (Ala59 bei Ras und Rac) oder durch Reste des GEFs aus seiner Position gedrückt werden. Studien an Ras, Ran und Rho zeigten jedoch eine eher untergeordnete Rolle des Mg2+ bei dem Nukleotidaustausch23–25. Selbst bei Abwesenheit des Magnesiums konnte ein erhöhter Nukleotidaustausch durch Zugabe eines GEFs beobachtet werden23. Daher scheint die Interaktion zwischen P-Loop und ɴ-Phosphat des Nukleotids ausschlaggebend für die enge Nukleotidbindung zu sein. Das positiv geladene Lysin des P-Loops, welches mit den negativen Ladungen des Phosphats interagiert, kann nun mit sauren Resten aus dem GNBP oder mit dem ebenfalls sauren, konservierten Glutamin (ArfGEF) interagieren. Die Phosphatgruppe wird nach Bindung des GEFs freigegeben und die Base des neu eintretenden Nukleotids bindet während der Verdrängung des GEFs (Abbildung 1.4). Studien von Renault26 zeigten anhand von Arf, dass ein Glutamat die Phosphatbindung durch seine negative Ladung abschwächt. Da dieses Glutamat in allen kleinen GTPasen konserviert ist und im binären Komplex von Ras mit seinem GEF eine ionische Bindung ausbildet27, spielt diese Aminosäure offenbar eine zentrale Rolle bei der Verdrängung des Nukleotids aus der Bindungstasche. GEFs einer Subfamilie zeigen strukturelle Gemeinsamkeiten, wohingegen GEFs verschiedener Unterfamilien sehr große Unterschiede aufweisen können. So bestehen die bisher bekannten GEFs der Ras-, Ral- und Rap-Familie aus einer CDC25 (cell division cycle 25 homology)- und oft auch aus einer REM (Ras exchange motif)-Untereinheit. Ein sehr großer Teil der RhoGEFs besitzt neben einer 6 1 Einleitung PH (pleckstrin homology)- auch eine DH (Dbl homology)-Domäne. Die DOCK (Dedicator of cytocinesis)-Familie der GEFs weist eine gänzlich andere Struktur auf. Eine detaillierte Übersicht der bekannten GEFs findet sich in der Literatur28. 1.1.3.2 GAP-PROTEINE Da die GTPase-Reaktion der meisten GNBPs sehr langsam abläuft und somit nicht als regulatorisches Element in der Signaltransduktion eingesetzt werden kann, sind GAP-Proteine notwendig, um eine schnelle Hydrolyse und somit die Inaktivierung des GNBPs zu gewährleisten. GAP-Proteine innerhalb der Ras-Superfamilie unterscheiden sich sehr stark in ihrer Struktur, Sequenz und dem ablaufenden Reaktionsmechanismus. In dem aktiven Zentrum der Ras-verwandten Proteine liegt ein konserviertes Glutamin ŝŶ ĚĞƌ EćŚĞ ĚĞƐ ɶ-Phosphats des Nukleotids. 'ɲ-Proteine weisen zusätzlich noch ein konserviertes Arginin auf. Studien mit GDP-ŐĞďƵŶĚĞŶĞŶ'ɲ-Proteinen in Gegenwart von AlF4- weisen darauf hin, dass sowohl das Glutamin als auch der so genannte Arginin-Finger den Übergangszustand stabilisieren. Das AlFx wird zur Imitation des pentavalenten Übergangszustandes29 Ăŵɶ-Phosphat des Nukleotids eingesetzt. Durch den Arginin-Finger (aus dem GAP) werden positive Reste in der Nähe der Phosphatgruppe des Nukleotids positioniert. Hierdurch und durch die Positionierung des Glutamins des G-Proteins ǁŝƌĚĚĞƌŶƵŬůĞŽƉŚŝůĞŶŐƌŝĨĨĚĞƐtĂƐƐĞƌƐĂƵĨĚĂƐɶ-Phosphat ermöglicht30. Ist dieses Glutamin (Gln61 in Ras) mutiert, ist ein Hydrolysemechanismus nicht mehr möglich. Veränderungen in den Positionen 12 und 13 führen zu einer falschen Positionierung des ArgininFingers und des konservierten Glutamins, wodurch der Übergangszustand nicht mehr eingenommen werden kann31. Eine Inaktivierung durch ein GAP kann somit nicht mehr erfolgen und führt zu einem stets aktivierten Ras-Protein, welches eine häufige Ursache für Krebs darstellt. Bei RhoGAPs läuft der Mechanismus, trotz großer Sequenz- und Strukturunterschiede, sehr ähnlich ab22. Für 'ɲ-Proteine, die eine sehr hohe intrinsische Hydrolyserate aufweisen, konnten ebenfalls GAPs beschrieben werden, die allerdings einen etwas anderen Funktionsmechanismus haben. Hierbei üben sie eine stabilisierende Wirkung auf das G-Protein aus und greifen nicht aktiv in den eigentlichen ,LJĚƌŽůLJƐĞŵĞĐŚĂŶŝƐŵƵƐ ĞŝŶ͘ ƵƌĐŚ ĞŝŶ ŬŽŶƐĞƌǀŝĞƌƚĞƐ ƐƉĂƌĂŐŝŶ ĚĞƌ 'ɲ-GAPs wird das katalytische 'ůƵƚĂŵŝŶĚĞƌ'ɲ-Proteine stabilisiert, wodurch die Hydrolyse beschleunigt wird32. AlFx kann nur den inaktiven, GDP-gebundenen Zustand dieser GTPase binden, wenn das zugehörige GAP anwesend ist. Die meisten Rab-GAPs, welche zuerst in Saccharomyces cerevisiae identifiziert wurden, beinhalten eine konservierte Tre-2/Cdc16/Bub2 (TCB)-Domäne33,34. Neben einem Arginin-Finger, welcher auch in Ras- und Rho-GAPs auftritt, gibt es bei Rab-GAPs einen zusätzlichen Glutamin-Finger, welcher das angreifende Wasser koordiniert. TCBs, welche beide katalytische Aminosäuren nicht aufweisen, fungieren nicht als GAPs. Dies lässt den Schluss zu, dass GABs mit TBC-Domäne ihre Funktion immer 7 1 Einleitung über den beschriebenen Zwei-Finger-Katalysemechanismus ausüben35. Alle bisher bekannten Ran-GAPs weisen eine modulare Architektur mit einer etwa 350 Aminosäuren langen Leucin-reichen Region (LRR), gefolgt von einer kürzeren, etwa 50 Aminosäuren langen, eher sauren Region, auf36. Eine zusätzliche carboxyterminale Domäne, welche Ran-GAPs an den Kernporen-Komplex lokalisiert, tritt in höheren Eukaryoten auf37. Ran-GAPs stabilisieren die switch IIRegion und somit das konservierte Glutamin der GTPase, welches das Wasser koordiniert38. Rap1 besitzt, anders als andere Ras-verwandte GTPasen, anstatt des sonst vorkommenden Glutamins (Gln61 in Ras) ein Threonin, was für die Bindung und für die eigentliche Katalyse wichtig ist39. Der sonst bei der GAP-Katalyse wichtige Arginin-Finger tritt im RapGAP-Komplex nicht auf40. Hingegen findet sich bei GAPs dieser Klasse ein so genannter Asparagin-Daumen, welcher, wie das Gln61 in Ras, die Aufgabe übernimmt, das angreifende Wasser zu positionieren41. Eine große Sequenzähnlichkeit mit der katalytischen Domäne von Rap1GAP zeigt Tuberin (TSC2), welches zusammen mit Harmatin (TSC1) den Tuberous Sclerosis Complex (TSC) formt und ein GAP für Rheb darstellt42. Mutationen in TSC1 oder TSC2 führen zu weit gestreuten Hamartomen. Aufgrund der hohen Sequenzidentität und des Vorhandenseins eines Asparagin-Daumens wird ein ähnlicher GAP-Mechanismus wie im Rap-Rap1GAP-Komplex vermutet42,43. Im Menschen sind bisher zehn verschiedene Unterfamilien der ArfGAPs nachgewiesen worden, welche über das Vorhandensein eines Zink-Fingers charakterisiert werden44. Biochemische Studien45 sowie die Aufklärung der Röntgenstruktur von Arf-ArfGAP im Übergangszustand46 konnten das Vorhandensein eines katalytischen Arginins zeigen. Viele GAPs werden durch posttranslationale Modifikationen, second messenger und/oder ProteinProtein- oder Protein-Ligand-Interaktionen reguliert22. Zum Beispiel erfolgt beim RasGAP Neurofibromin eine Phosphorylierung an mehreren Regionen des Carboxy-Terminus durch Proteinkinase A. Hierdurch kann Neurofibromin mit 14-3-3 Proteinen interagieren, wodurch seine GAP-Aktivität herabgesetzt wird47. Einige GTPasen benötigen keine GAP-Proteine, da sie eine sehr große intrinsische Hydrolyse aufweisen. Dabei wird die Aktivierung der Hydrolyse durch Bildung von Homodimeren in Gang gesetzt48. 1.1.3.3 POSTTRANSLATIONALE MODIFIKATIONEN Viele Proteine benötigen zur Ausübung ihrer Funktion eine (temporäre) Membrananbindung49. Um eine Anbindung an Phospholipide oder Sphingolipide der Membran zu erreichen, können zum einen konservierte Lipidbindemotive aus dem Protein selbst genutzt werden50 oder zum anderen durch 8 1 Einleitung posttranslationale Modifikationen Lipidgruppen kovalent angeknüpft werden51. Parallele Studien an Lamin B52 und einem A-Faktor Peptid aus Saccharomyces cerevisiae53 konnten das so genannte CaaXMotiv am Carboxyterminus als Ankerpunkt für die Lipidgruppe ausmachen. Der Name dieses Motivs leitet sich von den beteiligten Aminosäuren ab: C steht für Cystein, das a für eine aliphatische und das X für jede beliebige Aminosäure. Bei Mutationen des Cysteins ist eine Membrananbindung nicht mehr möglich54. Bei der Isoprenylierung wird ein Farnesyl- (15 Kohlenstoffatome) oder ein Geranylgeranylisoprenoid (20 Kohlenstoffatome) angefügt. Die Reaktion, welche an einem oder zwei Cysteinen am oder nahe des Carboxyterminus durchgeführt werden kann, wird von Protein-PrenylTransferasen (PPTase) katalysiert. Die PPTasen sind Zink-bindende ɲɴ-Heterodimere, die identische ɲ-Untereinheiten aufweisen. Die letzte variable Aminosäure ist entscheidend dafür, welche PPTase angreift und welche Modifikation vorgenommen wird. Die Geranylgeranyltransferase erkennt als letzte Aminosäureposition ein Leucin, wohingegen die Farnesyltransferase hauptsächlich Methionin, Serin, Cystein oder Glutamin erkennt51. Nach dem Anknüpfen des Lipidrestes an der CaaX-Box müssen die dem Cystein folgenden Aminosäuren proteolytisch abgespalten und eine Carboxymethylierung vorgenommen werden55. H-, K- und N-Ras sowie Rheb werden ausschließlich farnesyliert, Rab1 und Rab2 werden geranylgeranylisiert. Wird jedoch zum Beispiel die Farnesyltransferase inhibiert, wird K-Ras4B (CVIM) geranylgeranylisiert. Eine detaillierte Übersicht über mögliche Modifikationen findet sich in der Literatur56. Die Palmitoylierung, bei der eine Anknüpfung einer gesättigten C16Palmitinsäure über eine Thioesterbindung vorgenommen wird, ist im Gegensatz zu den vorher genannten Modifikationen reversibel. Dieser dynamische Prozess erlaubt eine Regulation der Lokalisation der betreffenden Proteine57, reicht jedoch allein für eine feste Membranverankerung nicht aus. Nach der Proteinbiosynthese am Endoplasmatischen Retikulum werden N- und H-Ras farnesyliert und am Golgi-Apparat palmitoyliert, wodurch sie zur Plasmamembran rekrutiert werden können58. Anstatt einer Palmitoylierung weist K-Ras in seiner hypervariablen Region, welche N-terminal der CaaX-Box lokalisiert ist, eine polybasische Sequenz mit einer Vielzahl von Lysinen auf. Über elektrostatische Wechselwirkungen kann sich diese GTPase an die negativ geladenen Phosphatgruppe der Plasmamembran anlagern59. Für palmitoylierte Ras-Proteine liegt eine dynamische Verteilung in zellulären Membranen vor60. Die eigentlich langsame Diffusion Rasverwandter GTPasen von Endomembranen zur Plasmamembran wird durch die erst in neuester Zeit für diese Funktion entdeckte Phosphodiesterase, Typ 6, delta-hŶƚĞƌĞŝŶŚĞŝƚ;WɷͿŐĞƐƚĞƵĞƌƚ61. Wɷ besitzt eine tiefe hydrophobe Tasche, welche den Membrananker vieler prenylierter GTPasen aufnehmen kann62. Eine Bindung von WɷĂŶArl-2 konnte bereits ebenfalls beschrieben werden. 9 1 Einleitung 1.1.4 REGULATION VON MTOR DURCH KLEINE GTPASEN Der Wachstums-kontrollierende Signalweg target of rapamycin (TOR) ist von Eukaryoten bis hin zum Menschen hoch konserviert21. Über diesen Signalweg fließen diverse Stimuli zusammen, welche neben dem Wachstum auch den Metabolismus kontrollieren. Die molekulare Regulation von TOR ist hoch komplex, was bei mTOR durch seine Regulation durch Wachstumsfaktoren, Nährstoffe und Stress deutlich wird. mTOR setzt sich aus den Multiproteinkomplexen mTORC1 und mTORC2 zusammen, welche funktional und strukturell verschieden sind63. Neben der Proteinsynthese werden viele zelluläre Prozesse wie die Autophagie und die Nährstoffaufnahme von mTORC1 reguliert. Die Organisation des Aktin-Zytoskeletts, das Zellüberleben und die Lipidsynthese werden hingegen von mTORC2 reguliert21. Ein großer Unterschied zwischen den beiden mTOR-Einheiten besteht in der Nährstoffsensitivität: mTORC1 weist eine solche Sensitivität im Gegensatz zu mTORC2 auf. Jedoch weisen beide Komplexe eine Sensitivität gegenüber Rapamycin und Wachstumsfaktoren auf. mTORC1 beinhaltet Raptor (regulatory-associated protein of mTOR)64, mLST8 (mammalian lethal with Sec thirteen 8) sowie PRAS 40 (proline-rich Akt substrate 40 kDa)65. Hingegen besteht mTORC2 aus Rictor (rapamycin-intensive companion of mTOR)66, mSin1 (mammalian stress-activated protein kinase interacting protein)67, mLST8 und Protor (protein observed with ricTOR)68. Zu den am besten charakterisierten Substraten von mTORC1 gehören die ribosomale Protein-S6Kinase (S6K) sowie das eukaryotische Translationsinitiationsfaktor-4E-bindende Protein (4E-BP1), welche über Phosphorylierung reguliert werden, wodurch schließlich die Proteinsynthese gesteuert wird. Über mTORC2 ist bekannt, dass es eine Reihe spezifischer Substrate phosphoryliert. Der Mechanismus der mTORC2-Regulation ist bisher weitgehend unbekannt.63 mTORC1 zählt zu den bedeutendsten Regulatoren der Autophagozytose, bei der Zellbestandteile abgebaut werden können. mTORC1 wirk somit als Positiv-Regulator des Zellwachstums, indem er Autophagozytose inhibiert und die Translation aktiviert. Diese Funktion übt mTORC1 auf downstream-Targets über Phosphorylierungsreaktionen aus. Eine Inhibierung von mTORC1 erfolgt hauptsächlich durch osmotischen Stress, Hypoxie und Nährstoffmangel, wohingegen Aminosäuren und Wachstumsfaktoren mTORC1 aktivieren. Von mTORC2 ist bisher nur bekannt, dass es direkt wahrscheinlich nur von Wachstumsfaktoren reguliert wird69. Eine Übersicht über die wichtigsten Regulationsmechanismen des mTORC1-Signalwegs ist in Abbildung 1.5 gezeigt. Auch reguliert mTORC1 die Aktivität verschiedener Transkriptionsfaktoren, welche die Lipidsynthese und den Mitochondrienmetabolismus steuern. In Zellkulturexperimenten konnte gezeigt werden, dass das Vorhandensein von Aminosäuren für eine mTORC1-Regulation notwendig ist und nicht durch andere externe Stimuli ersetzt werden kann. Eine 10 1 Einleitung direkte Verknüpfung zwischen mTORC1 und Aminosäuren lässt sich über die GTPase Rag beschreiben70,71. Diese außergewöhnliche GTPase bildet Heterodimere aus RagA oder RagB mit RagC oder RagD, wobei die Partner dieses Dimers entgegengesetzte Nukleotidbeladung aufweisen. Abbildung 1.5: Regulation des mTORC1-Signalwegs63 Der mTORC1-Signalweg wird über den Wachstumsfaktor-Signalweg aktiviert, welcher die PI3K-PKB/Akt und den ERK-RSKSignalweg über den TSC1/2-Komplex beinhaltet. Rheb, für das der TSC1/2-Komplex als GAP wirkt, interagiert direkt mit mTORC1. Wird PRAS40, ein mTORC1-Inhibitor, von PKB/Akt phosphoryliert, so dissoziiert es von mTORC1 ab. Der RagKomplex rekrutiert als Folge eines Aminosäure-Signals mTORC1 zu den Lysosomen. AMPK kann durch Phosphorylierung TSC2 aktivieren und Raptor inaktivieren, was mTOR inhibiert. Durch Phosphorylierung von S6K und 4E-BP1 wird die Proteinsynthese aktiviert, was zu Zellwachstum führt. Aktivierende Phosphorylierungen sind mit einem blau eingefärbten, inhibierende Phosphorylierungen mit einem rot eingefärbten P gekennzeichnet. Eine Aktivierung, bei der RagA oder RagB im GTP- und RagC oder RagD im GDP-beladenen Zustand sind, wird durch Aminosäuren gesteuert (Abbildung 1.6). Über den genauen Vorgang des Nukleotidaustauschs des Rag-Komplexes ist bisher wenig bekannt. Das Rag-Heterodimer ist im aktiven Zustand an der Oberfläche des Lysosomen angeheftet. Dort kann das aktive Rag-Heterodimer mit Raptor interagieren, wodurch mTORC1 am Lysosomen oder am späten Endosomen clustert70. Ist mTORC1 dort lokalisiert, wird durch Interaktion mit GTP-gebundenem Rheb die Kinaseaktivität von mTORC1 stimuliert72. Der PI3K-Signalweg wird durch die Bindung von Insulin an seinen Rezeptor aktiviert. Dies führt zur Phosphorylierung von Akt/PKB, was zu einer Aktivierung führt. Akt/PKB phosphoryliert seinerseits dann TSC2 an mehreren Stellen, wodurch TSC2 inaktiviert wird. Durch diese Inaktivierung werden wiederum Rheb und mTORC1 aktiviert73. TSC2 ist ein GAP für Rheb und überführt es somit vom aktiven in den inaktiven Zustand74, wodurch Rheb mTORC1 nicht mehr aktivieren kann. Wird TSC2 11 1 Einleitung phosphoryliert, kann es nicht mehr als GAP für Rheb wirken, wodurch mTORC1 aktiviert wird. Die Phosphorylierung von TSC2 hat eine Translokation von der Membran ins Zytosol zur Folge, wodurch es das membrangebundene Rheb nicht mehr inaktivieren kann75. Auch durch Redox-Stress kann mTORC1 reguliert werden: Rheb liegt bei Redox-Stress hauptsächlich im GTP-gebundenen Zustand vor und kann somit mTORC1 aktivieren. Zusätzlich kann Akt/PKB (Proteinkinas B) auch PRas40 phosphorylieren, wodurch es nicht mehr an mTORC1 bindet und somit keine inhibierende Wirkung mehr ausüben kann76. 21 Abbildung 1.6: Aminosäure-abhängige Regulation von mTORC1 durch die GTPase Rag Der Rag-Komplex ist am Lysosomen angebunden. In Abwesenheit von Aminosäuren (links, -AA) ist RagA bzw. RagB mit GDPund RagC bzw. RagD mit GTP beladen, was den inaktiven Zustand des Heterodimers kennzeichnet. mTORC1 kann somit nicht zum Lysosom rekrutiert werden. In Gegenwart von Aminosäuren (rechts, +AA) ändert sich der Nukleotidzustand, sodass RagA bzw. RagB GTP- und RagC bzw. RagD GDP-gebunden vorliegen. Der Komplex ist somit aktiv und kann mTORC1 binden und zum Lysosom rekrutieren, an dem es mit aktivem Rheb interagieren kann. Über Phosphoinositol-3-Kinase (PI3K)-Akt können Wachstumsfaktoren mTORC1 regulieren. TSC2 kann durch extracellular signal-regulated kinase (ERK) phosphoryliert werden, was eine Inhibierung zur Folge hat. Auch der Wnt-Signalweg (wingless Integration 1) nimmt Einfluss auf dem mTORC1Signalweg77. Eine weitere Regulation von mTORC1 durch Rheb konnte kürzlich nachgewiesen werden. Rheb aktiviert GTP-abhängig Phospholipase D (PLD), was wiederum TSC-abhängig ist. Die durch Hydrolyse von Phosphatidylcholin gebildete Phosphatidsäure (PA) kann über Raptor mit mTORC1 interagieren und es aktivieren. mTORC1 wird in einem Rheb-abhängigen Mechanismus über Aminosäuren aktiviert, wobei die Nukleotidbeladung der kleinen GTPase nicht über Aminosäuren reguliert werden kann78. Durch die Aminosäuren wird mTORC1 zum Lysosom rekrutiert, an dem es auf das GTPgebundene Rheb trifft und deshalb aktiviert werden kann. Der genaue Mechanismus der Aminosäure-abhängigen Aktivierung und der zugrundeliegenden Signalwege sind Gegenstand 12 1 Einleitung aktueller Forschung21. Darüber hinaus konnte in neuesten Studien die Regulation von mTORabhängiger T-Zellen-Produktion durch Rheb anhand eines Mausmodells gezeigt werden79. Der Signalweg über PLD und PA wird neben Rheb auch über die GTPase RalA gesteuert, welche auch an einer Aminosäure-abhängigen Regulation von mTORC1 beteiligt ist80. RalA fördert die Assoziation der PLD mit der GTPase Arf6, wodurch die Aktivität der Phospholipase gesteigert wird81. Bekannt ist, dass Arf6 mTORC1 in Abhängigkeit von Glukose reguliert82, indem die GTPase vom Zytosol zur Membran wandert und dort an PLD bindet. Interessanterweise ist RalA für eine Nährstoff-abhängige Regulation von mTORC1 durch Rheb notwendig80, was den Schluss zulässt, dass RalA downstream von Rheb seine regulatorische Funktion ausübt. Auch mTORC2 wird durch GTPasen reguliert. So interagiert das Rac1-GEF phosphatidylinositol-3,4,5trisphosphate-dependent Rac exchange-factor 1 (P-Rex1)83 direkt mit mTORC2, wodurch das GEF aktiviert wird. Somit verbindet P-Rex1 den mTOR-Signalweg mit Rec1 und der Zellmigration84. Rac1 kann jedoch in einer Nukleotid-unabhängigen Reaktion mit mTOR direkt interagieren, wodurch die Lokalisation von mTOR gesteuert wird85. Zur Zeit wird der Einfluss von Rab6 sowie von seinem GAP und GEF auf den mTOR-Signalweg diskutiert86. Wahrscheinlich inhibieren einige Mitglieder der RabFamilie die Phosphorylierung der S6K, interagieren aber selbst nicht mit dem mTOR-Komplex87. Bisher ist noch unklar, wie genau alle Mechanismen zur Regulation des mTOR-Signalwegs zusammenhängen. Die Klärung der Frage, wie mTOR und die aufgeführten GTPasen sowie deren GAPs und GEFs mechanistisch zusammenhängen, wird ein Fokus zukünftiger Forschung sein, da alle Komponenten unabhängig voneinander schon seit geraumer Zeit als therapeutische Targets untersucht werden88. 1.1.5 RHEB- EINE BESONDERE KLEINE GTPASE Die kleine GTPase Rheb weist eine etwa 30-40 %ige Sequenzähnlichkeit zu H-Ras und Rap2 auf89. Das 184 Aminosäuren große Protein besitzt eine Molekularmasse von 20,4 kDa. 1994 wurde das RhebGen zunächst in hippocampalen Granulazellen des Rattenhirns entdeckt, wo es nach Iktus und NMDA-abhängiger Synapsenaktivität hochreguliert wird89. In der gleichen Studie konnte auch eine vermehrte Rheb-Expression nach Zugabe von Wachstumsfaktoren wie des Fibroblastenwachstumsfaktors (FGF) und des Epidermalen Wachstumsfaktors (EGF) beobachtet werden. Beim Menschen ist das zunächst in Ratten gefundene Gen ebenfalls enthalten und auf Chromosom 7 lokalisiert90. In Säugetierzellen konnte neben dem zuerst gefundenen Rheb1-Gen ein weiteres Gen auf Chromosom 12 identifiziert werden, das als Rheb2- oder RhebL1-Gen bezeichnet wird91. Die beiden Proteine Rheb1 und Rheb2 weisen eine 51 %ige Sequenzidentität auf. 13 1 Einleitung Einige Homologe des Rheb-Gens konnten in Saccharomyces cerevisae, in Schizosaccharomyces pombe sowie in Drosophila melanogaster und im Zebrafisch gefunden werden92. N C 93 Abbildung 1.7: NMR-Struktur von RhebͻGDP Rheb weist die typische G-Domänen-Faltung auf. Die variablen switch-Regionen sind in Gelb (switch I) und Rot (switch II) sowie der P-Loop in Orange eingefärbt. Die Termini sind mit N und C gekennzeichnet und aus Gründen der Übersichtlichkeit verkürzt. (PDB: 2L0X) Aufgrund von Sequenzähnlichkeit wird Rheb zur Familie von Ras/Rap-GTPasen gezählt. Anhand der Röntgen94,95- und NMR-Struktur93 konnte das Vorhandensein der typischen G-Domänen-Faltung nachgewiesen werden. Die switch I-Region erstreckt sich von D33 bis N41 und die switch II-Region von G63 bis N79. Der P-Loop ist zwischen G13 und S20 lokalisiert. Durch die Aufklärung der Struktur konnte auch die Nukleotid-abhängige Konformationsänderung in den switch-Regionen nachgewiesen werden. Wie viele GTPasen weist Rheb eine carboxyterminale CaaX-Box (CSVM) auf, die eine Erkennungssequenz für die Farnesyltransferase darstellt96. Das Besondere an Rheb ist, dass es weder eine Palmitoylierungsstelle noch eine polybasische Sequenz N-terminal der CaaX-Box aufweist97. Die Farnesylierung stellt somit die einzige bisher bekannte posttranslationale Modifikation von Rheb dar. Durch die Farnesylierung wird Rheb an Endomembranen angeknüpft und kann dort mit mTORC1 interagieren98. Ohne diese Membrananbindung kann Rheb seine regulatorische Funktion nicht ausüben93. Rheb wird wie viele andere prenylierten GTPasen auch von PDEɷ gebunden. Bei den Ras-verwandten GTPasen wird die Nukleotidhydrolyse über ein konserviertes Glutamin (Gln61 in Ras) und einen Asparagin-Finger aus dem jeweiligen GAP-Protein katalysiert. 1.1.5.1 GAP- UND GEF-PROTEINE VON RHEB Um den Mechanismus und den Signaltransduktionsweg von GTPasen zu erforschen, ist neben der bekannten Struktur auch die Zugänglichkeit zu den zugehörigen GAPs und GEFs von großer 14 1 Einleitung Bedeutung. Für Rheb ist bisher als einziges GAP TSC2 bekannt42,99. Die GAP-Domäne von TSC2 ist homolog zur Rap1GAP-Domäne, sodass ein ähnlicher Reaktionsmechanismus vermutet wird. Rheb besitzt anstatt des konservierten Glycins (Gly12 in Ras) ein Arginin (Arg15), wodurch ein anderer Hydrolysemechanismus als bei vielen anderen kleinen GTPasen wahrscheinlich ist. Rheb weist eine geringere intrinsische GTPase-Aktivität als Ras55 auf. Diese kann jedoch nicht durch eine Mutation von Arg15 hin zu Gly15 gesteigert werden43. Rheb besitzt an Position 64 ein Glutamin, welches an einer ähnlichen Position wie das katalytische Glutamin in Ras lokalisiert ist. Da Gln64 jedoch in einer hydrophoben Tasche sitzt und vom katalytischen Zentrum weg orientiert ist, ist eine Beteiligung am Hydrolysemechanismus sehr unwahrscheinlich. Eine Mutation an dieser Aminosäureposition verhindert jedoch die Interaktion von Rheb mit seinem GAP TSC2. Abbildung 1.8: Mechanismus verschiedener GAPs100. (A) Beim GAP-Mechanismus von TSC2 wird das Asn1643 in die GTPase eingebracht. Gln64 nimmt nicht direkt am Katalysemechanismus teil, sondern stabilisiert die Protein-Protein-Interaktion. (B) Im Rab1-Rab1GAP-Komplex übernimmt ebenfalls ein Asparagin-Daumen eine wichtige Rolle in der Hydrolyse. (C) Beim RasGAP wird ein stabilisierender ArgininFinger zur Katalyse eingebracht, welcher das konservierte Gln61 stabilisiert. Wie im Rap1GAP (Asn290, Asparagin-Daumen) übernimmt auch Asn1643 aus TSC2 eine katalytische Funktion während der Hydrolyse, was durch Mutationsexperimente gezeigt werden konnte100. Durch eine Mutation von Asn1643 ї Asp1643 konnte die essentielle Bedeutung der Amidgruppe der Seitenkette und ihrer Orientierung an der stattfindenden Katalyse bewiesen werden. Auch Mutationen zu Arginin (wie es bei RasGAP der Fall ist) führten zu einer starken Inhibierung des Katalysemechanismus. Ein kompletter Verlust der GAP-Aktivität konnte bei der Mutation Asn1643 ї Gln1643 beobachtet werden. Die Annahme, dass ein ähnlicher Mechanismus der GAP-katalysierten Hydrolyse wie durch Rap1GAPs vorliegt, wird auch durch Mutationen an Position Gln64 unterstützt. 15 1 Einleitung Wird bei Rap1 die analoge Aminosäureposition Thr61 verändert, so kann ein Angriff des GAPs nicht mehr stattfinden. Ein vergleichbares Resultat wurde ebenfalls bei dem TSC2-Rheb-Komplex erzielt. Bekannte Mutationen, welche bei Patienten mit Tuberöser Skleorose auftreten, konnten in Einklang mit dem postulierten GAP-Mechanismus von TSC2 gebracht werden. Neueste Studien zeigen, dass Tyr35 einen autoinhibitorischen Effekt auf die Hydrolyse ausübt, indem es mit dem ɶ-Phosphat des Nukleotids wechselwirkt101. Auch Asp65 aus switch II scheint eine Rolle bei der Hydrolyse zu spielen. Im Gegensatz hierzu wird ein potentielles GEF für Rheb in der Literatur kontrovers diskutiert. Erste Hinweise auf ein GEF wurden in Drosophila-Studien102 gefunden, als das translationally controlled tumor protein (TCTP) in Verbindung mit dem TOR-Signalweg gebracht werden konnte. In zwei zeitgleichen Studien konnte die GEF-Wirkung von TCTP jedoch nicht belegt werden. NMRBindungsstudien103, welche allgemein recht sensitiv sind und somit auch schwache Bindungen detektieren können, konnten keine Interaktion zwischen Rheb und TCTP nachweisen. Auch ein Zusammenhang von TCTP und mTORC1, welcher von aktivem Rheb aktiviert wird, konnte in knockdown-Experimenten nicht beobachtet werden104. Zusätzlich wurde durch Überexpression von TCTP keine Phosphorylierung von S6K, was als Marker der TOR-Signalkaskade gilt, nachgewiesen. In anderen Studien konnte genau der gegenteilige Effekt gezeigt werden105. Diese kontroversen Forschungsergebnisse lassen den Schluss zu, dass TCTP höchsten sehr schwach mit Rheb interagiert und somit wahrscheinlich nicht das physiologisch-relevante GEF für Rheb darstellt. 1.1.5.2 PHYSIOLOGISCHE FUNKTION VON RHEB Das Rheb1-Gen ist für die fetale Entwicklung von Mäusen von größter Wichtigkeit. Durch eine Unterentwicklung des kardiovaskulären Systems sterben Mäuseembryonen ohne dieses Gen etwa in der Mitte der Fetalphase, wohingegen Embryonen ohne das TOR-kodierende Gen deutlich früher sterben106,107. Möglicherweise kann RhebL1 den Wegfall des Rheb kompensieren und/oder TORC1 benötigt für seine Aktivität nicht zwingend Rheb. Eine Funktion von Rheb in der Ausbildung des kardiovaskulären Systems konnte auch in isolierten adulten Zellen gezeigt werden108. Eine Deletion des Rheb-Gens in neuronalen Vorläuferzellen führt zu einer verringerten Myelinisierung im postnatalen Gehirn109. Ebenso ist Rheb ein Aktivator des TORC1 in T-Zellen im Immunsystem79. Rheb scheint auch bei Diabetes eine Rolle zu spielen. In transgenen Mäusen mit einer Überexpression von Rheb in den ɴ-Zellen des Pankreas konnte eine erhöhte Insulinausschüttung nach Glukosezugabe beobachtet werden, was auf eine Vergrößerung des Volumens und der Anzahl der ɴ-Zellen zurückgeführt werden konnte. 16 1 Einleitung Ein weiterer nicht-kanonischer TSC-Rheb-Signalweg wird aktuell diskutiert110: Erste Experimente deuten auf eine Interaktion von Rheb mit dem FK506-binding protein 38 (FKBP38), das Chaperonähnliche Aufgaben übernimmt, hin111. Andere Arbeitsgruppen konnten diese Ergebnisse nicht reproduzieren112, sodass eine direkte Interaktion von Rheb mit FKBP38 wohl nicht stattfindet. Auch eine FKBP38-vermitteltet Regulation von den Apoptoseproteinen BcL-XL und BcL-2 durch Rheb113 erscheint demnach fraglich. Weiterhin wird eine Beteiligung von Rheb und TSC am Notch-Signalweg diskutiert114. 1.1.5.3 LOKALISATION Wie viele GTPasen besitzt auch Rheb eine C-terminale CaaX-Box, an der eine Farnesylierung stattfinden kann. Im Gegensatz zu anderen kleinen GTPasen wird Rheb nicht noch weiter modifiziert und besitzt auch keinen Lysin-Stretch, welcher bei K-Ras4B oder Rac1 zur Membranlokalisiation beiträgt. Bei Ras und Rho ist die an der CaaX-Box stattfindende posttranslationale Modifikation für eine Membranassoziation zwar notwendig, doch allein nicht hinreichend115. Rheb ist im Gegensatz zu vielen anderen kleinen G-Proteinen nicht an der Plasmamembran, sondern am Endoplasmatischen Retikulum und am Golgi-Apparat lokalisiert116. Dies steht in Übereinstimmung mit der Interaktion von Rheb mit dem mTOR-Komplex, da dieser Komplex an Endomembranen aktiv ist. Nach der erfolgten Prenylierung werden die letzten drei Aminosäuren proteolytisch abgespalten und es erfolgt eine Carboxymethylierung. Die Abspaltung der letzten Aminosäuren spielt offenbar für eine korrekte Lokalisation eine untergeordnete Rolle. Fehlt das methylierende Enzym isoprenylcysteine carboxylmethyltransferase (Icmt), ist eine Anbindung an Endomembranen gestört116. Die subzelluläre Lokalisiation und Plasmamembrananbindung von K-Ras4B ist jedoch nicht von der Carboxymethylierung abhängig. Die richtige Memranlokalisiation dieser GTPase wird somit maßgeblich von der polybasischen Lysinregion beeinflusst. Der Guaninnukleotid-Dissoziations-Inhibitor (GDI)-ähnliche Solubilisationsfaktor PDEɷ besitzt eine wichtige Aufgabe bei der Verteilung von vielen GTPasen in der Zelle. Durch seine hydrophobe Tasche bindet er die Prenylgruppe kleiner GTPasen. Studien zeigen, dass K-Ras4B aufgrund seiner vielen basischen Reste deutlich schlechter als Rheb, welches nur wenige basischen Reste nahe des Carboxyterminus aufweist, von PDEɷ solubilisiert werden kann. Dies deutet darauf hin, dass eine Verteilung von Rheb im Zytoplasma vorliegt61. Somit kann eine feste Anbindung von Rheb an die Plasmamembran, wie es bei K-Ras4B der Fall ist, ausgeschlossen werden. Bei anderen GTPasen übernimmt PDEɷ die Funktion, depalmitoylierte Proteinformen wieder zum Golgi-Apparat zu transportieren, damit sie dort (re-)palmitoyliert werden können. Die Nukleotid-unabhängige Bindung 17 1 Einleitung zwischen farnesyliertem Rheb und PDEɷ erfährt durch Arl2 und Arl3 eine GTP-abhängige allosterische Regulation117. 117 Abbildung 1.9: Komplex aus RhebͻPDEɷ . (A) Cartoon-Darstellung aus RhebͻGDP (grün) im Komplex mit PDEɷ (grau). Die switch-Regionen der GTPase sind in magenta und die Farnesylgruppe in blau dargestellt. Der flexible Loop aus PDEɷ ist in braun eingefärbt. (B) Die Aminosäureseitenketten aus PDEɷ, welche die hydrophobe Tasche für den Farnesylrest (blau) formen, sind in grau dargestellt. 1.2 IN-CELL NMR-SPEKTROSKOPIE Die "Flüssig-NMR" stellt bisher die einzige Technik dar, Informationen über biologische Makromoleküle unter physiologischen oder quasi-physiologischen Bedingungen zu erhalten. Im Gegensatz zur Röntgenstrukturanalyse, Elektronenmikroskopie und Festkörper-NMR können mit dieser Technik auch verdünnte und nicht zu kristallisierende Proben in Lösung vermessen werden. Die Technik der in-vivo NMR umfasst neben der Untersuchung von Metaboliten in verschiedenen Zellsuspensionen auch die Technik des Magnetic Resonance Imaging (MRI, auch MRT)118,119, wobei die hochaufgelöste in-cell NMR keine bildgebende Technik darstellt. Das MRI stellt eine in der klinischen Diagnostik standardmäßig eingesetzte Technik dar, welche die Darstellung durchbluteter Organe erlaubt und daher häufig zur Tumordiagnostik eingesetzt wird. Das neuartige Echtzeit-MRIVerfahren erlaubt neben der dauerhaften Beobachtung einzelner Organe auch eine Überwachung der Positionierung von Operationsinstrumenten während eines chirurgischen Eingriffs. Die hochaufgelöste NMR-Spektroskopie weist eine Selektivität für bestimmte in der Natur relativ selten vorkommende Kerne (ausgenommen 1H) wie 13 C (1,11 %) und 15 N (0,37 %) auf. Die häufig vorkommenden Kerne 12C und 14N sind nicht NMR-aktiv und sind in NMR-Messung nicht detektierbar. Somit können gezielt isotopenangereicherte Proteine eingebracht und differenziert von allen 18 1 Einleitung weiteren vorhandenen Makromolekülen untersucht werden. Ändert sich die chemische Umgebung eines NMR-aktiven Kerns, was zum Beispiel bei einer Bindung an einen Interaktionspartner der Fall sein kann, so lässt sich diese Änderung über eine Verschiebung der jeweiligen Resonanzfrequenz nachverfolgen. Somit stellt die NMR-Spektroskopie eine leistungsfähige Technik dar, um die Interaktion von Makromolekülen mit ihren Bindungspartnern wie anderen Makromolekülen, kleinen Liganden oder sogar Medikamenten zu untersuchen120–123. Diese Eigenschaften der NMRSpektroskopie ermöglichen daher erstmals die Erforschung von Makromolekülen in lebenden Zellen124. Die Methode der in-cell NMR-Spektroskopie bietet zwar die Möglichkeit, isotopenangereicherte Proteine in ihrer natürlichen Umgebung zu untersuchen, jedoch lässt sich hiermit kein Bild der Zelle im Ganzen abbilden. Studien an einzelnen Proteinen können nur indirekt Hinweise über molekulare Mechanismen und Verteilungen in der Zelle geben. Die in-cell NMRSpektroskopie stellt bisher die einzige Methode dar, um Makromoleküle in physiologisch-ähnlichen Umgebungen auch strukturell zu untersuchen. Jedoch kann aufgrund der Arbeitsschritte, welche zum Einbringen der isotopenangereicherten Moleküle in die Zelle notwendig sind und der eigentlichen Messung zwar von Messungen in der Zelle, jedoch nicht von in vivo-Messungen im eigentlichen Sinne gesprochen werden125. Die momentane Forschung konzentriert sich nicht auf die Aufklärung von Proteinstrukturen direkt in der Zelle, sondern will über solche in vivo-Messungen Aufschluss über mögliche posttranslationale Modifikationen, Konformationsänderungen und mögliche Interaktionen erhalten120,124,126. Auch unterschiedliche Lokalisationen an Zellorganellen können quantitativ über die Veränderung der Resonanzfrequenz des jeweiligen Kerns detektiert werden. 1.2.1 PROKARYOTISCHE ZELLEN Für Messungen in prokaryotischen Zellsytemen werden bisher ausschließlich E. coli-Zellen eingesetzt. Dieses System bietet sind an, da die meisten mittels NMR-Spektroskopie charakterisierten Makromoleküle aus Bakterienzellen, welche in isotopenangereichertem Medium kultiviert werden, gewonnen werden und somit bestehende Anzuchtprotokolle weiterhin genutzt werden können. Bei der prokatyotischen in-cell NMR-Spektroskopie wird ein starkes Expressionssystem, zum Beispiel durch einen T7-Promotor reguliert, benötigt. Hierdurch kann in kurzer Zeit sehr viel Plasmidcodiertes Protein hergestellt werden. Durch Verwendung eines IPTG-induzierbaren lac-Operators und eines pLys-Systems kann die Expression des Zielproteins derart gesteuert werden, dass sie erst nach Überführung in ein isotopenangereichertes Minimalmedium erfolgt. Somit wird hauptsächlich das Zielprotein isotopenangereichert, die übrigen Proteine, Zellorganellen und Membranen jedoch kaum. 19 1 Einleitung Messungen an E. coli-Extrakten wurden schon vor 20 Jahren durchgeführt, um ohne zeit- und kostenintensive Aufreinigung die Faltung und die ausreichende Überexpression von Proteinproben direkt zu untersuchen127. Erste NMR-spektroskopische Messungen an intakten E. coli-Zellen wurden an einer Metallbindungs-Domäne eines bakteriellen Proteins zur Quecksilberentgiftung durchgeführt128. Abbildung 1.10: Schematische Darstellung des Protokolls zur Herstellung von in-cell Präparaten125. (a) Die unter Kontrolle eines starken Plasmid-Promotors in isotopenangereichertem Medium hergestellten Proteine können direkt in der E. coli-Zelle mittels In-cell NMR vermessen werden (b) Bei der STINT-NMR werden zwei rekombinante Proteine auf zwei verschiedenen Plasmiden mit zwei unterschiedlich induzierbaren Promotoren hergestellt (rot: isotopenangereichert; grau: nicht isotopenangereichert). (c) Für die Nutzung von Xenopus-Oozyten wird das isotopenangereicherte Protein in jede einzelne Zelle injiziert. (d) Durch Anfügen eines CPPs kann ein Protein direkt in die Zellen eindringen. (e) Durch Nutzung von SLO wird die Zelle kurzzeitig permeabel, sodass das isotopenangereicherte Protein 2+ eindringen kann. Durch Zugabe von Ca kann die Membran anschließend wieder verschlossen werden. Die meisten Proteine bedürfen zur Ausübung ihrer physiologischen Funktion einer definierten dreidimensionalen Struktur. Viele in der Literatur als intrinsisch ungefaltet beschriebene Proteine üben jedoch oftmals wichtige Funktionen zum Beispiel bei der Signaltransduktion oder Transkription aus129,130. Durch NMR-Studien in E. coli konnte für das in vitro völlig ungefaltete bakterielle Protein FlgM eine partielle Faltung in vivo nachgewiesen werden131. Die beobachteten Spektren konnten durch Zugabe von Glukose oder größeren Mengen Rinderalbumin (BSA) auch in vitro reproduziert werden. Somit konnten bezüglich FlgM der Schluss gezogen werden, dass für eine Faltung von FlgM eine hohe Konzentration von anderen (Makro-)Molekülen vorliegen muss, was mit konventionellen NMR-Methoden unter isolierten Bedingungen nicht hätte beschrieben werden können. Auch konnten Studien zur Bindung von Proteinen an Metall-Ionen132 und Studien über die Ca2+Bindung von Calmodulin133 erfolgreich mittels in-cell NMR-Spektroskopie in Bakterien-Zellen durchgeführt werden. Neben diesen Untersuchungen bezüglich biochemischer Fragestellungen werden auch Untersuchungen an potentiellen Medikamenten mittels in-cell NMR durchgeführt. Die 20 1 Einleitung Entwicklung potentieller Medikamente scheitert häufig an der schlechten Membrangängigkeit der ausgewählten Substanzen. Für das in die Chemotaxis von Bakterien involvierte Protein CheY konnte über in vitro Studien ein Ligand (BRL-16492PA) identifiziert werden. Durch in-cell NMR-Studien konnte anhand der auftretenden Resonanzverschiebungen die Membrangängigkeit sowie die Bindung von BRL-16492PA an CheY im zellulären Milieu gezeigt werden132. Eine Übersicht über diverse in-cell NMR-Studien an prokaryotischen Systemen findet sich in der Literatur 134,135 . Auch konnten DNA-Protein-Interaktionen136 analysiert und Protein-Dynamiken erfolgreich verfolgt137 werden. Neben Detektion von 13 C- und 15 N-Kernen konnten 19 F-NMR-Studien an bis zu 100 kDa großen Proteinen durchgeführt werden138. Die NMR-Technik wird häufig für Bindungsstudien eingesetzt, in der ein Protein isotopenangereichert vorliegt und ein möglicher Ligand nicht isotopenangereichert hinzu titriert wird. Da der Ligand (ausgenommen seiner Protonen) in der NMR-Spektroskopie nicht detektierbar ist, können Bindungsereignisse am isotopenangereicherten Protein ohne größere Beeinflussung der spektralen Auflösung verfolgt werden. Bei der structural interaction using in-cell NMR (STINT-NMR)139,140 können solche Titrationen auch in E. coli-Zellen durchgeführt werden. Dabei wird die Interaktion zweier sequentiell exprimierter Polypeptide verfolgt. Hierbei werden zwei Plasmide mit unterschiedlich induzierbaren, sehr gut kontrollierten Promotoren genutzt, um eine individuelle Expression der Proteine zu gewährleisten. Die Überexpression des mittels NMR-Spektroskopie zu detektierenden Zielproteins wird in isotopenangereichertem Medium induziert. Das andere Protein wird nichtisotopenangereichert hergestellt, indem die Zellkultur vor Induktion in nicht-isotopenangereichertes Kulturmedium überführt wird. Abbildung 1.11: Schnelle Aquisition der 3D-NMR-Spektren durch Verwendung eines nicht-linearen Aufnahmeschemas 141 (nonlinear sampling) mit reduzierten Datenpunkten . Schema des nonlinear samplings zur Aufnahme von 3D-NMR-Spektren zur Strukturbestimmung (DFT: diskrete FourierTransformation; dim: Dimension; MaxEnt: Maximum-Entropie Prozessierung). 21 1 Einleitung Durch Probenentnahme nach unterschiedlichen Expressionsdauern kann die Interaktion der beiden Proteine mittels hochaufgelöster NMR-Spektroskopie verfolgt werden. Diese Technik eignet sich zur Überprüfung vorangegangener in vitro Titrationsexperimente oder zur Analyse der Wirkung kleiner Moleküle auf das Bindungsereignis. Durch nonlinear (oder auch non-uniform) sampling142,143 und eine Maximum-Entropie- Rekonstruktion144 kann die Aufnahme der notwendigen Daten von einigen Tagen auf wenige Stunden reduziert werden. Durch einen Vergleich mit einer in vitro gelösten Struktur konnte die durch in-cell NMR erhaltene Struktur bestätigt werden. 1.2.2 EUKARYOTISCHE ZELLEN Das prokaryotische System eignet sich aufgrund der einfachen Handhabung für erste NMR-Studien von Proteinen im zellulären System. Jedoch besteht ein großes Interesse der Forschung an der Übertragung solcher Messungen auf eukaryotische Zellen, da zum Beispiel posttranslationale Modifikationen hauptsächlich in Eukaryoten und nicht in Prokaryoten vorkommen. Bei in-cell NMRMessungen in E. coli-Zellen können solche Modifikationen nicht beobachtet werden. Ebenfalls ist die Zusammensetzung der Zellen in Bezug auf die vorhandenen Zellorganellen und Transportwege sehr unterschiedlich134. Bisher ist nicht klar, ob und wie das Vorhandensein von diversen subzellulären Kompartimenten die Struktur und Funktion von Proteinen beeinflusst. Durch diesen hohen Grad an Differenzierung gibt es hoch spezifische Funktion und große Unterschiede in der biochemischen Funktion benachbarter Zellkompartimente145. Im Gegensatz zu den in-cell NMR-Messungen in prokaryotischen Zellen, bei der es nur eine Arbeitstechnik zur Einbringung der zu analysierenden Makromoleküle gibt, werden bei der eukaryotischen in-cell NMR-Spektroskopie verschiedene Methoden eingesetzt. Das Hauptproblem beim eukaryotischen System besteht im Einbringen des Polypeptids, da es bisher keine Möglichkeit zur direkten isotopenangereicherten Expression gibt. 1.2.2.1 XENOPUS LAEVIS OOZYTEN Die Oozyten des Afrikanischen Krallenfrosches (Xenopus laevis) werden aufgrund ihrer guten Zugänglichkeit und enormen Größe (1 mm Durchmesser) schon seit langer Zeit in der Entwicklungsund Zellbiologie verwendet146. Oozyten, welche sich im Entwicklungsstadium VI befinden, werden standardmäßig für Mikroinjektionen eingesetzt, wobei definierte Mengen von exogenem Material (z. B. cRNA) in die Oozyte eingebracht werden147. Für in-cell NMR-Messung an Xenopus-Oozyten müssen die Zielproteine zunächst in E. coli exprimiert und aufgereinigt werden, um später mittels 22 1 Einleitung Mikroinjektor in jede einzelne Oozyte appliziert zu werden148. Für erste Untersuchungen können Extrakte aus Xenopus-Oozyten eingesetzt werden149. Für in-cell NMR-Messungen sind etwa 200-300 intakte Oozyten vonnöten, was einem Volumen von 250 μl bis 450 μl entspricht. Pro Oozyte können bis zu 50 nl Proteinlösung injiziert werden, sodass nur etwa 10-20 μl einer isotopenangereicherten Proteinlösung eingesetzt werden120,134. Entscheidende Voraussetzungen für die Durchführbarkeit solcher in-cell NMR-Messung sind die gute Löslichkeit und eine geringe Viskosität der zu injizierenden Probe. Mittels in-cell NMR-Spektroskopie an Xenopus-Oozyten konnten Phosphorylierungen der eingebrachten Proteine durch endogene Xenopus-Kinasen nachgewiesen werden. Hierbei wurde das humane neuronale Protein Tau untersucht150. Es konnten Hinweise auf eine mögliche Assoziation von Tau mit Mikrotubuli durch einen Vergleich der in vivo Spektren mit in vitro NMR-Spektren von Mirkotubuli-gebundenen Tau nachgewiesen werden. In der gleichen Studie konnten auch eindeutig posttranslationale Modifikationen in Form von Phosphorylierungen bestätigt werden. In Studien an Casein Kinase 2 (CK2) und Cyclin-dependent protein kinase 1 (Cdk1) konnte zeitaufgelöst die Phophorylierung an verschiedenen Stellen mittels NMR-Spektroskopie in Oozyten-Extrakten und intakten Xenopus-Oozyten gezeigt werden151. 134 Abbildung 1.12: Arbeitsschritte zur Probenvorbereitung einer Xenopus-Oozyten in-cell NMR-Messung . (a) Die Oozyten des Xenopus laevis werden bei einer Operation entfernt und sortiert. (b) Übersicht über die verschiedenen Stadien bei der Entwicklung der Oozyte. Die durchschnittliche Zellgröße ist in Klammern angegeben. (c) Injektion der isotopenangereicherten Proteine mit einer Glaskapillare (Durchmesser um 20 μm). (d) Schematische Darstellung des Injektionsvorganges. Das isotopenangereicherte Protein (rot) wird in die nicht-isotopenangereicherte Oozyte injiziert. (e) Abbildung eines mit etwa 200 Oozyten gefüllten ShigemiTM-NMR-Röhrchens. 1.2.2.2 SÄUGERZELLEN Ein großer technischer Fortschritt der in-cell NMR-Spektroskopie stellt die Möglichkeit zur Analyse von Makromolekülen in Säugerzellen dar. Wie bei der Verwendung von Xenopus-Oozyten müssen die 23 1 Einleitung zu untersuchenden Makromoleküle zunächst isotopenangereichert hergestellt und später in die Zellen eingebracht werden. Aufgrund der Größe dieser Zellen ist eine manuelle Injektion nicht möglich. Ebenso müssten in etwa 1000 bis 1500 Zellen Proteinlösung injiziert werden, um eine ausreichende Füllhöhe im NMR-Röhrchen zu erhalten. Momentan stehen zwei Methoden zur Verfügung. Bei der ersten Methode wird an das zu untersuchende Protein ein zellpenetrierendes Peptid (CPP) angeheftet, welches den einfachen Durchtritt durch die Zellmembran erlaubt152,153. In ersten Versuchen wurde die Sequenz des Tat-Proteins von HIV1 N-terminal an das zu untersuchende Ubiquitin-Protein angefügt. Die Expression erfolgte in isotopenangereichertem Medium in E. coliZellen. Die Inkubation mit HeLa-Zellen erfolgte unter Zugabe von Pyrenbutyrat, was die direkte Translokation von CPP-verbundenen Peptiden in das Zytosol vermittelt153. Durch das Vorhandensein einer speziellen endogenen Ubiquitin-spezifischen Protease wurde das CPP in der Zelle abgespalten. Die 1 erhaltenen H-15N-HSQC-Spektren konnten in vitro reproduziert werden. Um die Allgemeingültigkeit dieses Systems zu testen, wurden weitere Proteine und andere Zelllinien (COSZellen) verwendet. Bei der zweiten Methode (Abbildung 1.13) ist keine spezielle Modifikation des zu untersuchenden Proteins erforderlich: Hier wird das bakterielle Poren-formende Toxin Streptolysin O (SLO) eingesetzt154. Poren von 35 nm Durchmesser können durch SLO in der Cholesterin-enthaltenden Plasmamembran entstehen, durch die Proteine bis etwa 150 kDa Größe ein- und austreten können. Um den Austritt der isotopenangereicherten Proteine zu verhindern, können durch Abbildung 1.13: Inkorporierung des Porenbildungen Darstellung durch SLO (1-2), Calcium die Poren wieder der wurden von isotopenangereichertem Thymosin ɴ4 der (Tɴa), was Aktin binden und somit die Polymerisation Inkorporation des Zielproteins (3) und dem Verschließen der Zelle durch Zugabe von Calcium (4). von verschlossen werden155. Hochaufgelöste NMR-Spektren Zielproteins in 293F-Zellen154. Schematische Zugabe verhindern kann, in humanen 293F-Zellen aufgenommen. Mit dieser Technik konnte die N-Acetylierung von Tɴa in vivo eindeutig nachgewiesen werden. Für diese Technik ist eine sehr hohe Löslichkeit des Zielproteins essentiell. Da die Proteine durch Diffusion passiv in die Zellen gelangen, muss ein sehr hoher Proteinkonzentrationsgradient zwischen 24 1 Einleitung Zellinnerem und -äußerem vorliegen. Dies ist nur bei wenigen Proteinen möglich, zumal hohe Proteinkonzentrationen im Medium für gewisse Zellen toxisch sein könnten. Studien von isotopenangereicherten Proteinen in Säugerzellen sind erste Versuche, Proteine in zellulärer Umgebung mittels mehrdimensionaler NMR-Spektroskopie zu charakterisieren. Problematisch bei allen in-cell NMR-Techniken ist der Austritt des Proteins während der Messungen. Dieses Problem ist erst seit kurzer Zeit bekannt und führte zur Rücknahme zweier Veröffentlichungen über die Proteindynamik bei in-cell NMR-Messungen156–158. Die stetige Kontrolle des Puffermediums stellt somit einen wichtigen Bestandteil einer korrekt ausgeführten in-cell NMR-Messung dar. 1.3 VOM SCREENING ZUM VALIDIERTEN HIT Protein-Protein-Interaktionen sind zentrale Elemente in biologischen Prozessen und stehen somit im Fokus der biologischen sowie pharmazeutischen Forschung. Besonders in der pharmazeutischen Chemie werden jedes Jahr mehrere Milliarden Dollar159 hauptsächlich für die Erforschung Antikörperbasierter Antagonisten investiert. Neben einer hohen Spezifität für ihr jeweiliges Target weisen die therapeutischen Antikörper eine sehr hohe Stabilität im menschlichen Plasma auf, was sie zu idealen Medikamenten macht. Hohe Kosten bei der Erforschung und Produktion sowie fehlende Möglichkeiten zur oralen Verabreichung schmälern jedoch die Attraktivität der Antikörper als Medikamente. Antikörper sind außerdem nicht zellpermeabel und wirken nur als Antisense-Therapie, bei der die Expression des Zielproteins blockiert wird160. Es ist nicht trivial, synthetische Komponenten zu entwickeln, die die Protein-Protein-Interaktion inhibieren. Für jede Fragestellung muss individuell eine Anfangsauswahl an synthetischen Molekülen festgelegt werden, was ohne festgelegte Kriterien geschieht. Des Weiteren sind Protein-Protein-Interaktionsflächen häufig flach und bieten so eine geringe Angriffsfläche für Liganden. Neben der Oberflächenkomplexität müssen auch Porengröße und -geometrie sowie die Polarität beachtet werden, wodurch ein hochkomplexes System (bio-)physikalischer Wechselwirkungen entsteht. Eine weitere Herausforderung stellt die Unterscheidung zwischen realen und physiologisch artifiziellen Bindungen dar. Es sind bisher recht wenige natürliche kleine Moleküle, welche direkt an Protein-ProteinInteraktionsflächen binden, bekannt. Die Interaktionsfläche zweier Proteine beträgt in der Regel 7501500 Å2 161 . Anhand von Röntgenstrukturen und gezielten Mutationen konnte gezeigt werden, wie das Prinzip von Protein-Protein-Interaktionen funktioniert162–164. Bei der sogenannten Scanning mutagenesis Methode werden Teile der Proteinoberfläche systematisch mutiert. Durch diese Mutationen konnte gezeigt werden, dass eine Vielzahl von Protein-Protein-Interaktionsflächen eine zentralisierte Region mit wenigen Resten besitzt, die hauptverantwortlich für die Interaktion sind165. Diese sogenannten Hot-Spots sind in der Regel auf beiden Seiten des Protein-Protein-Interfaces 25 1 Einleitung lokalisiert. Die Hot-Spots der verschiedenen Interaktionsflächen sind komplementär zueinander: Negative Ladungen auf der einen Seite passen zu positiven auf der anderen Seite. Gleiches gilt für Salzbrücken sowie hydrophobe oder hydrophile Reste. Besonders an solchen Hot-Spot-Regionen befindet sich ihre funktionale und strukturelle Adaptionsfähigkeit166. Aus diesen Gründen lassen sich mögliche Bindungsregionen nur schwer mit einzelnen Röntgen- oder NMR-Strukturen identifizieren. Auch kann ein Hot-Spot für eine Vielzahl von unterschiedlichen Interaktionen genutzt werden, was die Identifizierung weiter erschwert. Durch Rotation von Seitenketten und Änderungen in flexiblen Regionen des Proteinrückgrats können verschiedene Wechselwirkungsmechanismen ermöglicht werden. In Studien der Fc-Domäne des Immunoglobulins (IgG) konnte ein einziger Hot-Spot für drei verschiedene Bindungspartner ausgemacht werden (Abbildung 1.14). Diese Bindungsregion wird erst durch Bindung der jeweiligen Targets sichtbar, was ein großes Problem bei verschiedenen Screeningverfahren darstellt. Abbildung 1.14: Hot-Spot auf einer Protein-Protein-Interaktionsfläche160. Bei der Analyse von Bindungspartnern der Fc-Domäne von IgG konnte ein Hot-Spot auf der Proteinoberfläche ausgemacht werden. Die verschiedenen Atomtypen sind in unterschiedlichen Farben dargestellt. Die zentrale hydrophobe Bindestelle ist durch weiße Umrandung der beteiligten Aminosäuren hervorgehoben. Wasserstoffbrückenbindungen sind durch schraffierte und Salzbrücken durch gelbe Umrandungen markiert. Ein weiteres großes Problem stellt die Identifikation der genauen Bindestelle sowie des stöchiometrischen Verhältnisses dar. Außerdem muss eine Denaturierung eines Bindungspartners ausgeschlossen werden. Ebenfalls ist es häufig nicht einfach festzustellen, welche Aminosäurereste des Protein-Protein-Komplexes für die Bindung essentiell sind. Liegen mehrere Bindungsstellen vor, wird eine Bewertung der Resultate weiter erschwert. Um den Bindungsmechanismus aufzuklären, können Methoden wie die analytische Ultrazentrifugation (AUC), Oberflächen-Plasmonresonanz (SPR), Röntgenstrukturanalyse und NMR eingesetzt werden167. 26 1 Einleitung Um eine Bibliothek von Verbindungen für biologische Screening-Verfahren zu erstellen, gibt es zwei Verfahren. Bei dem Verfahren des Fragment-basierten Screenings (FBS) wird eine Bibliothek von kleinen Molekülen getestet, um aus Kombinationen der am besten bindenden Moleküle, des so genannten Hit, eine Struktur mit verbesserten Bindungseigenschaften zu entwickeln. Die eingesetzten Bibliotheken sind zwar klein, jedoch lässt sich hieraus eine Vielzahl von größeren Substanzen kreieren, wodurch ein großer Pool von Verbindungen entstehen kann. Abbildung 1.15: Syntheseprinzip von Leitstrukturen bei der Wirkstoffentwicklung168. (a) Bei dem Fragment-basierten Screening (FBS) werden Bibliotheken von vergleichsweise wenigen kleinen Molekülen untersucht. Aus diesen Fragmenten, welche häufig nur sehr schwache Affinitäten zum Zielprotein aufweisen, soll durch Kombination ein Wirkstoff mit einer starken Bindung erzeugt werden. Unterschiedlich eingefärbte Kreise stellen verschiedene chemische Gruppen dar. (b) Bei der Diversität-orientierten Synthese (DOS) werden komplexe Verbindungen, welche Eigenschaften bekannter Wirkstoffe aufweisen, getestet. Die erhaltenen Hits weisen häufig deutlich höhere Affinitäten zum Zielprotein als die durch FBS erhaltenen Leitstrukturen auf. Der große Vorteil des FBS ist es, dass zunächst nur recht wenige Targets getestet werden müssen, da aus den Hits neue, komplexere Strukturen generiert werden, die wiederum getestet werden können. So können schon im ersten Schritt viele Substanzen ausgeschlossen werden, was eine Zeit- und somit auch Kostenersparnis darstellt. Es ist bekannt, dass sich kleine Strukturen häufiger als Wirkstoffe eignen als größere, da ihre physiko-chemischen Eigenschaften eher denen eines Medikamentes entsprechen169. Diese Methode eröffnet somit die Möglichkeit, das kleinste mögliche Target zu finden, welches die besten Bindungseigenschaften an gewählte Zielproteine aufweist. Einer der 27 1 Einleitung größten Nachteile dieser Technik ist jedoch, dass kleine Komponenten oft nur schwache Bindungen an das Zielprotein zeigen und eine Detektion der Interaktion daher sehr sensitiver Techniken bedarf. Somit ist es wahrscheinlich, dass gerade bei schnellen Screeningverfahren viele potentielle Wirkstoffe aufgrund ihrer schwachen Affinität nicht erkannt werden. Bei der Diversität-orientierten Synthese (DOS) wird eine Bibliothek von strukturell unterschiedlichen, Medikament-ähnlichen Komponenten erstellt, die schon bekannte Verbindungen enthalten. Hierbei wird darauf geachtet, dass die möglichen Wirkstoffe passende Molekulargewichte aufweisen. Der Vorteil der DOS-Methode besteht darin, dass bei einem Hit durch die Größe der eingesetzten Verbindung leicht zu detektierende Effekte auftreten und somit der Hit eindeutig identifiziert werden kann. Bei der FBS-Technik werden häufig planare Moleküle als potentielle Wirkstoffe generiert, was die dreidimensionalen Struktur der Proteine außer Acht lässt und zu Hits mit niedriger Affinität führt. Bei DOS hingegen werden gleich zu Beginn komplexe, dreidimensionale Verbindungen auf ihre Bindung an das Target hin untersucht. Die Entwicklung von Wirkstoffen mit FBS setzt häufig den Zugang zur dreidimensionalen Struktur des Zielproteins voraus, da letztendlich die Wirkstoffe hypothesengetrieben anhand von geometrischen, chemischen und physikalischen Eigenschaft für eine optimale Bindung des Hits an das Protein modelliert werden168. 1.3.1 FBS ZUR IDENTIFIZIERUNG VON LIGANDEN VON K-RAS Das vor über 30 Jahren charakterisierte humane Onkogen Ras ist bei über 20 % der menschlichen Tumore mutiert. Von den drei existenten Isoformen K-, H- und N-Ras weist K-Ras die häufigste Mutationsrate auf170. Aktuelle Forschungen konzentrieren sich auf das Design kleiner Liganden für K-Ras, um eine Interaktion mit son of sevenless (SOS), dem Ras-GEF, zu inhibieren. Dabei wird die NMRSpektroskopie zur Analyse der ausgewählten Komponenten eingesetzt171,172, da mit dieser Technik aufgrund ihrer Sensitivität Hits mit einer schwachen Assoziation zum Protein identifiziert werden können. Durch Einsatz dieser Technik sowie das Vorhandensein der gelösten dreidimensionalen Struktur von K-Ras wird der Einsatz des Fragment-basierten Screenings als sehr erfolgsversprechend angesehen. Solche Screenings können auf dem Level eindimensionaler NMR-Spektroskopie durchgeführt werden, indem die Sättigungstransfer-Differenz (STD) NMR-Technik eingesetzt wird173. Das STDExperiment basiert auf dem Kern-Overhauser-Effekt (NOE) und auf der Beobachtung des Resonanzsignals des Liganden. Bei schwach bindenden Liganden existiert ein stetiger Austausch zwischen gebundenem und freiem Liganden. Bei dem STD-Experiment wird die Differenz zwischen einem Spektrum, bei dem das Protein selektiv gesättigt wurde (Isat), und einem Spektrum ohne 28 1 Einleitung Proteinsättigung (I0), auch off-resonance Spektrum genannt, gebildet. Das selektiv vorgesättigte Spektrum wird als on-resonance Spektrum bezeichnet. Das Spektrum wird dadurch erhalten, dass nur auf eine Frequenz im Spektrum eingestrahlt wird, in der ausschließlich Rezeptor-/Protein-Signale (wie z. B. 0 ppm) enthalten sind. Im Differenzsspektrum (ISTD= I0-ISAT) sind nur noch die Signale des Liganden enthalten, welche einen Sättigungstransfer vom Protein erhalten (Abbildung 1.16). Liganden, welche nicht an das Protein binden, erfahren keinen Sättigungstransfer. Deren Signalintensitäten sind somit im off- sowie im on-resonance Spektrum identisch und im Differenzspektrum nicht sichtbar. Für ein an den Rezeptor bindendes Molekül sind daher nur die Wasserstoffatome sichtbar, die im engen Kontakt (ч 5 Å) zum Protein stehen und als Folge eine Übertragung der Magnetisierung erfahren174. Vorteile der STD-Technik sind ihre Robustheit sowie die Tatsache, dass nur der Ligand betrachtet wird. Informationen über den Rezeptor selbst werden nicht benötigt. Da dieses Experiment auf Basis eines konventionellen eindimensionalen Protonenspektrums erfolgt, werden pro Ansatz nur kleinste Mengen an Rezeptor sowie Ligand benötigt. Ebenfalls eignet sich das STD-Experiment aufgrund der Kürze der Aufnahmezeit zum Screening größerer Substanzbibliotheken. Jedoch können durch dieses Experiment keine Informationen über die exakte Bindungsregion auf der Proteinoberfläche gewonnen werden. Abbildung 1.16: Schema des STD-Experiments174. Der Austausch zwischen freiem und gebundenem Liganden erlaubt einen quantitativen intermolekularen Magnetisierungstransfer von dem Rezeptor zu dem gebundenen Liganden. Für die weitere Analyse von Substanzbibliotheken bieten sich zweidimensionale, heteronukleare NMR-Experimente an. Für die Analyse von Protein-Ligand-Interaktionen wird routinemäßig das 1H15 N-HSQC-Experiment eingesetzt, in dem alle H-N-1J-Kopplungen analysiert werden können. Ist die Struktur eines Proteins bekannt und ein Rückgrat-Assignment vorhanden, lassen sich durch dieses 29 1 Einleitung Experiment detailierte Aussagen über die Lokalisation der Interaktionsfläche auf der Proteinoberfläche gewinnen. Ändert sich die chemische Umgebung einer Aminosäure, wie es bei der Bindung eines Liganden der Fall ist, so lässt sich dies durch eine Änderung der detektierten Resonanzfrequenz nachverfolgen. Abbildung 1.17: NMR-basiertes Liganden-Screening: 1H-15N-HSQC-Spektren171 1 H-15N-HSQC-Spektren von 15 N-isotopenangereichertem K-Ras bei Zugabe verschiedener Konzentrationen des Bindungspartners (steigende Konzentration von schwarz nach lila). 1.4 NMR-SPEKTROSKOPIE Seit der Entdeckung des Phänomens der kernmagnetischen Resonanz im Jahr 1945 von Bloch175 und Purcell176 (Nobelpreis für Physik 1952177,178) hat es eine enorme Entwicklung auf dem Gebiet der NMR-Spektroskopie gegeben. Heutzutage zählt sie zu den Routinetechniken der strukturellen Analytik in chemischen Labors. Einen großen Fortschritt stellt die Erweiterung der NMRSpektroskopie auf mehrere Dimensionen dar, wodurch es möglich wurde, die Struktur von Proteinen mittels NMR-Spektroskopie aufzuklären179. Neben der NMR-Spektroskopie werden makromolekulare Strukturen sowie Dynamiken routinemäßig auch mittels Röntgenstrukturanalyse untersucht. Bei der Röntgenstrukturanalyse können im Gegensatz zur NMR-Spektroskopie sehr große Makromolekülkomplexe analysiert werden, wenn geeignete Kristalle vorliegen. Die Entwicklung neuer Techniken der NMR-Spektroskopie für den Einsatz bei größeren Proteinkomplexe ist Gegenstand aktueller Forschungen180. Die NMR-Spektroskopie besitzt gegenüber der Röntgenstrukturanalyse diverse Vorteile. Die NMRSpektroskopie erlaubt neben der Untersuchung von Makromolekülen in Lösung eine Analyse diverser zeitabhängiger Prozesse, wodurch diese Technik nicht nur zur reinen Strukturaufklärung genutzt werden kann, sondern auch für die Untersuchung molekularer Dynamiken, Faltungsereignissen sowie Reaktionskinetiken. Neueste Entwicklungen erlauben es, mittels NMR-Spektroskopie Makromoleküle unter quasi-physiologischen (wie die in- cell NMR-Technik) zu charakterisieren. 30 1 Einleitung An dieser Stelle soll nicht weiter auf die Grundlagen der NMR-Spektroskopie eingegangen werden, da hierzu umfassende, quantenmechanische Beschreibungen notwendig sind. Vereinfachte Darstellungen finden sich in gängigen Lehrbüchern der biomolekularen NMR-Spektroskopie. Einen Einstieg ermöglichen die Bücher „Bioanalytik“ von F. Lottspeich aus dem Spektrum Verlag und „Ein- und zweidimensionale NMR-Spektroskopie“ von H. Friebolin aus dem Wiley-Vch Verlag. Die Bücher „Understanding NMR Spectroscopy“ von J. Keeler und „Spin Dynamics: Basis of Nuclear Magnetic Resonance“ von M. H. Levitt, beide aus dem Wiley-VCh Verlag, empfehlen sich zum vertiefenden Studium der NMR-Spektroskopie. Die hier eingesetzten NMR-spektroskopischen Techniken werden kurz im Kapitel „Material und Methoden“ dieser Arbeit beschrieben. 31 2 Zielsetzung und Gliederung 2 ZIELSETZUNG UND GLIEDERUNG GTPasen übernehmen eine Vielzahl von wichtigen Regulationsprozessen in der Zelle. Die hier untersuchte kleine GTPase Rheb übernimmt beispielsweise eine zentrale Rolle bei der Regulation von TOR und nimmt somit Einfluss auf die Proteinbiosynthese sowie das Zellwachstum. Im Gegensatz zu vielen anderen GTPasen ist über Rheb relativ wenig bekannt. Das für die GTPase-Reaktion essentielle GEF-Protein ist für Rheb bisher noch nicht identifiziert worden. Ebenso besitzt Rheb im Gegensatz zu anderen GTPasen nur eine posttranslationale Modifikation, die essentiell für die Membranverankerung und somit die Lokalisation ist. Im ersten Teil dieser Arbeit soll farnesyliertes Rheb rekombinant hergestellt, aufgereinigt und NMR-spektroskopisch charakterisiert werden. Diese Vorarbeiten sollen zur Nutzung der Nanodisc-Technologie für posttranslational modifizierte Proteine in der NMR-Spektroskopie beitragen. Ebenso soll eine Untersuchung von Rheb in physiologischer Umgebung durch Nutzung der innovativen in-cell NMR-Technik durchgeführt werden. Der zweite Teil dieser Arbeit konzentriert sich auf die Analyse von Rheb als Drug Target. Dabei soll durch ein Fragment-basiertes Screening eine Ligandenbibliothek bezüglich einer Interaktion mit Rheb untersucht werden. Ein weiteres Ziel besteht darin, bisher unbekannte Hot Spots auf der Proteinoberfläche zu identifizieren. Der Komplex aus Protein und Ligand soll mit dem Programm HADDOCK modelliert und weiter charakterisiert werden. Diese Arbeiten dienen zur späteren Entwicklung einer Leitstruktur im Rahmen eines Medikamentendesigns. 32 3 Material und Methoden 3 MATERIAL UND METHODEN 3.1 MATERIAL 3.1.1 CHEMIKALIEN 1-(2-Pyridyl)piperazin Sigma Aldrich 1-(2-Pyrimidinyl)piperazin Sigma Aldrich 1-(3,4-Dichlorophenyl)piperazin Sigma Aldrich 1,3-Diphenylharnstoff Sigma Aldrich 1H-Imidazol-2-carbonsäure Sigma Aldrich 2-(2-Pyridyl)benzimidazol Sigma Aldrich 2-Acetylbenzofuran Sigma Aldrich 2-Acetylpyridin Sigma Aldrich 2-Amino-3-nitropyridin Sigma Aldrich 2-Aminobenzimidazol Sigma Aldrich 2-Aminopyrimidin Sigma Aldrich 2-Hydroxybenzimidazol Sigma Aldrich 2-Naphthoesäure Sigma Aldrich 2-Naphthoxyessigsäure Sigma Aldrich 2-Phenylbenzimidazol Sigma Aldrich 3-(1H-Pyrrol-1-yl)benzoesäure Sigma Aldrich 3-(3-Pyridyl)propionsäure Sigma Aldrich 3-(4-Hydroxyphenyl)propionsäure Sigma Aldrich 33 3 Material und Methoden 3,4-Diaminobenzophenon Sigma Aldrich 3-Acetylindol Sigma Aldrich 3-Benzoylpropionsäure Sigma Aldrich 4-(1-Pyrrolidino)pyridin Sigma Aldrich 4-(4-Hydroxyphenoxy)benzoesäure Sigma Aldrich 4,4´-Biphenol Sigma Aldrich 4,4´-Diacetylbiphenyl Sigma Aldrich 4,4'-Dihydroxybenzophenon Sigma Aldrich 4,4'-Dihydroxydiphenylether Sigma Aldrich 4,4'-Dihydroxydiphenylmethan Sigma Aldrich 4-Amino-1-benzylpiperidin Sigma Aldrich 4-Benzoylpyridin Sigma Aldrich 4-Benzyloxybenzylalkohol Sigma Aldrich 4-Benzyloxyphenol Sigma Aldrich 4-Benzyloxyphenylessigsäure Sigma Aldrich 4-Biphenylcarbonsäure Sigma Aldrich 4-Biphenylessigsäure Sigma Aldrich 4'-Hydroxy-4-biphenylcarbonsäure Sigma Aldrich 4-Phenoxybenzoesäure Sigma Aldrich 4-Phenoxyphenol Sigma Aldrich 4-Phenylbenzylamin Sigma Aldrich 4-Phenylmorpholin Sigma Aldrich 34 3 Material und Methoden 4-Phenylpyridin Sigma Aldrich 5-Amino-2-methylindol Sigma Aldrich 5-Aminouracil Sigma Aldrich 5-Phenyl-2-furansäure Sigma Aldrich 6-Hydroxy-1-tetralon Sigma Aldrich Acrylamid Roth Adenin Sigma Aldrich Ammoniumchlorid (14NH4Cl) J.T. Baker Ammoniumperoxodisulfat (APS) Riedel de Häen Ampicillin Sigma Aldrich Bactotryptone Merck Benzamid Sigma Aldrich Benzamidin Sigma Aldrich Benzidin Sigma Aldrich Benzimidazol-5-carbonsäure Sigma Aldrich Benzimidazol-5-carbonsäure Sigma Aldrich Benzo[b]furan-2-carbonsäure Sigma Aldrich Benzophenon Sigma Aldrich Biotin Sigma Aldrich Bisphenol A Sigma Aldrich Borsäure (H3BO3) Sigma Aldrich Bromphenolblau Merck 35 3 Material und Methoden BSA Roth Calciumchlorid (CaCl2) Sigma Aldrich Carbazol Sigma Aldrich Chinolin-2-carbonsäure Sigma Aldrich Chloramphenicol Fluka Cobalt(II)-Chlorid (CaCl2x6H2O) Sigma Aldrich Coomassie Brillant Blue R-250 Fluka Cumarin Sigma Aldrich Cumarin-3-carbonsäure Sigma Aldrich Deuteriumoxid Isotec Inc. Dikaliumhydrogenphosphat (K2HPO4) Merck Dinatriumhydrogenphosphat (Na2HPO4) Merck Diphenylessigsäure Sigma Aldrich Dithiothreitol (DTT) AppliChem Eisen(III)-chlorid (FeCl3x6H2O) Sigma Aldrich Essigsäure Fluka Ethanol J.T. Baker Ethylendiamintetraessigsäure (EDTA) Sigma Aldrich Farnesylpyrophosphat Sigma Aldrich Glukose AppliChem Glycerin Riedel de Häen Guanidiniumhydrochlorid Acros 36 3 Material und Methoden Hefeextrakt AppliChem Hydrochinon Sigma Aldrich Imidazol J.T. Baker Indazol Sigma Aldrich Indol-2-carbonsäure Sigma Aldrich Indole-6-carbonsäure Sigma Aldrich Isopropanol J.T. Baker Isopropyl-ɴ-D-thiogalactopyranosid (IPTG) Roth Kaliumchlorid (KCl) Riedel de Häen Kaliumdihydrogenphosphat (KH2PO4) Riedel de Häen Kupfer(II)-chlorid (CuCl2x2H2O) Sigma Aldrich L-Pipecolinsäure Sigma Aldrich Magnesiumsulfat (MgSO4) Merck Melamin Sigma Aldrich N,N,N',N'-Tetramethylethylendiamin (TEMED) Biorad N,N'-Diphenylthioharnstoff Sigma Aldrich Natriumacetat J. T. Baker Natriumchlorid (NaCl) J.T. Baker Natriumdihydrogenphosphat (NaH2PO4x2H2O) J.T. Baker Natriumdodecylsulfat (SDS) AppliChem Natriumhydroxid (NaOH) J.T. Baker N-Hydroxybenzamid Sigma Aldrich 37 3 Material und Methoden Nicotinsäure Sigma Aldrich N-Phenyl-o-phenylenediamin Sigma Aldrich Protease Inhibitor Roche, Complete Mini, EDTA free Purin Sigma Aldrich rac-2,3-Diphenylpropionsäure Sigma Aldrich Rotiphorese® Gel 30 Roth ß-Mercaptoethanol Merck Thiamin Sigma Aldrich Thieno[3,2-b]pyridin-7-ol Sigma Aldrich trans-Zimtsäure Sigma Aldrich Triton X-114 AppliChem Trypton Merck Uracil Sigma Aldrich Zinkchlorid (ZnCl2) Sigma Aldrich 3.1.2 VERWENDETE GERÄTE Elektrophorese-Apparatur Mini-PROTEAN® 3 Cell, Bio Rad Extruder LipoFast Liposome Factory Injektor (Nanoliter 2000) WPI Inkubator Minitron AI72, Infors AG Kapillarenziehgerät Shutter Instrument Microfluidizer Microfluidics Corporation Mikroskop Semi 2000-C, Carl Zeiss Mikromanipulator H. Saur Laborbedarf 38 3 Material und Methoden NMR-Spektrometer Probenkopf NMR Röhrchen DRX 600, Bruker, 5mm, TXI 1H/D-13C/15N Z-Gradient WILMAD®NMR tubes, 5 mm, 500 MHz, 7 in L, 528-PP pH-Meter inoLab (WTW) UV/VIS-Spektrometer Specord 200, Analytik Jena AG Sonifizierer Ultrasonic Cleaner, VWR Thermomixer Thermomixer comfort, Eppendorf Vortex Mixer neoLab Zentrifugen Zentrifuge 5810 R, Eppendorf Zentrifuge Evolution RC, Sorvall Zentrifuge 5415 R, Eppendorf Sorvall Discovery M 120 Rotoren F-34-6-38 U (5810 R) A-4-81 (5810 R) SCL-6000 (Evolution RC) S100 AT3-236 3.1.3 VERWENDETE PLASMIDE pQE-30 Qiagen pRSF-DUET Novagen 3.1.4 PROTEINMARKER Als Proteinmarker wird der PageRuler™ Prestaines Protein Ladder 10-170 kDa der Firma Fermentas verwendet. 39 3 Material und Methoden 3.1.5 MEDIEN UND PUFFER Die Angaben für die folgenden Lösungen und Puffer beziehen sich, wenn nicht anders vermerkt, auf ein Zielvolumen von 1 l. Alle Medien bzw. Zusätze werden vor Verwendung autoklaviert oder steril filtriert. LB-Medium 10 g Bactotrypton 10 g NaCl 5g Hefeextrakt 2 ml Ampicillin-Lösung (100 μg/ml) (Stock 50 mg/ml) 1 ml Chloramphenicol-Lösung (34 μg/ml) (Stock 34 mg/ml) auf pH 7,0 einstellen M9-Medium (10x) 75,29 g Na2HPO4 ͻ 2H2O 30 g KH2PO4 5g NaCl Spurenelementlösung (100x) 5g EDTA 0,83 g FeCl3 ͻ 6H2O 84 mg ZnCl2 13 mg CuCl2 ͻ 2H2O 10 mg CoCl2 ͻ 6H2O 10 mg H3BO3 1,35 mg MnCl2 ͻ4H2O auf pH 7,0 einstellen 40 3 Material und Methoden Minimal Medium (zur Herstellung von 15N-isotopenangereicherten Proben) 0,3 ml 1 M CaCl2-Lösung 100 ml M9 Medium (10x) 10 ml Spurenelementlösung (100x) 20 ml 20 % (w/v) Glucose-Lösung 1 ml 1 M MgSO4-Lösung 1 ml Biotin-Lösung (1 mg/ml) 1 ml Thiamin-Lösung (1 mg/ml) 0,5 g 15 2 ml Ampicillin-Lösung (100 μg/ml) (Stock 50 mg/ml) 1 ml Chloramphenicol-Lösung (34 μg/ml) (Stock 34 mg/ml) N-Ammoniumchlorid SOC-Medium 20 g Pepton aus Casein 5g Hefeextrakt 0,5 g NaCl 2,5 mM KCl 10 mM MgCl2 20 mM Glukose Bindepuffer 50 mM TRIS/HCl pH 7,4 50 mM NaCl 10 mM MgCl2 41 3 Material und Methoden Waschpuffer 50 mM TRIS/HCl pH 7,4 50 mM NaCl 10 mM MgCl2 10 mM Imidazol Elutionspuffer 50 mM TRIS/HCl pH 7,4 50 mM NaCl 10 mM MgCl2 250 mM Imidazol Reinigungspuffer 50 mM TRIS/HCl pH 7,4 50 mM NaCl 10 mM MgCl2 500 mM Imidazol Komplexierungspuffer 50 mM TRIS/HCl pH 7,4 50 mM NaCl 50 mM EDTA Regenerationspuffer 0,1 M NiSO4 42 3 Material und Methoden NMR-Puffer 20 mM NaiPO4 5 mM NaCl 5 mM MgCl2 3.1.6 PUFFER UND LÖSUNGEN FÜR SDS-PAGE 15 % SDS-Gel (10 ml) 1,3 ml H2O 1,3 ml TRIS/HCl, pH 8,8 2,25 ml 30 % AA; 0,8 % BisAA 100 μl 10 % SDS 100 μl 10 % APS 10 μl TEMED 4 % Sammelgel (2 ml) 1,18 ml H2 O 500 μl TRIS/HCl, pH 6,8 270 μl 30 % AA; 0,8 % BisAA 20 μl 10 % SDS 20 μl 10 % APS 2 μl TEMED Lämmlipuffer (5x) 20 % (v/v) Glycerin 0,1 % (v/v) ɴ-Mercaptoethanol 0,1 % (w/v) Bromphenolblau 6 % (w/v) SDS 125 mM TRIS/HCl pH 6,8 43 3 Material und Methoden TG-Puffer 30 g TRIS/HCl pH 8,6 144 g Glycin SDS-Laufpuffer 0,1 % (w/v) SDS 100 ml TG (1x) Coomassie-Lösung 0,07 % (w/v) Coomassie Brillant Blue 10 % (v/v) Essigsäure 45 % (v/v) Ethanol Entfärber 10 % (v/v) Essigsäure 10 % (v/v) Isopropanol 3.2 METHODEN 3.2.1 3.2.1.1 MOLEKULARBIOLOGISCHE METHODEN HERSTELLUNG KOMPETENTER E. COLI-ZELLEN Zur Herstellung chemisch kompetenter E. coli BL21 (DE3)-Zellen werden 2 ml LB-Medium mit 100 μl des Glycerin-Stocks angeimpft. Die Bakterienlösung wird im Schüttler für acht Stunden bei 37 °C und 120 rpm inkubiert. Mit der Bakterienlösung werden 200 ml LB Medium angeimpft. Diese Vorkultur wird im Schüttler bei 37 °C und 120 rpm bis zu einer optischen Dichte OD600 von 0,6 inkubiert. Anschließend werden die Zellen für etwa 10 min auf Eis abgekühlt und bei 3000 g und 4 °C innerhalb von 10 min sedimentiert. Das Zellpellet wird in 50 ml kalter 100 mM Calciumchlorid-Lösung resuspendiert und die Lösung 44 3 Material und Methoden erneut zentrifugiert. Zur Resuspendierung der sedimentierten Zellen werden 10 ml eiskalte Calciumchlorid-Lösung verwendet und die Suspension 16 h lang bei 4 °C gelagert, um eine bessere Kompetenz der Bakterien zu erzielen. Daran anschließend wird die Lösung aliquotiert, in flüssigem Stickstoff schockgefroren und bis zur Verwendung bei -80 °C gelagert. 3.2.1.2 TRANSFORMATION VON E. COLI-ZELLEN 50 μl E. coli-Zellen werden auf Eis aufgetaut und mit 2 μl Plasmid-DNA versetzt. Die Zellen werden weitere 30 min auf Eis inkubiert. Im Anschluss daran werden die Zellen 50 s bei 42 °C temperiert und 2 min auf Eis inkubiert. Zu den Zellen werden 800 μl 37 °C warmes SOC-Medium hinzugefügt. Die anschließende Inkubation erfolgt bei 37 °C für 1,5 h. Die Zellen werden innerhalb von 5 min bei 10.000 rpm (Rotor 5415 R) sedimentiert und 700 μl des Überstandes werden verworfen. Die sedimentierten Zellen werden im verbleibenden Überstand resuspendiert, auf eine Agar-Platte mit Zusatz der benötigten Antibiotika ausgestrichen und 18 h im Brutschrank bei 37 °C inkubiert. 3.2.1.3 LAGERUNG TRANSFORMIERTER ZELLEN Zur Lagerung der transformierten Zellen wird eine einzelne Kolonie vom Selektionsmedium extrahiert und in 2 ml LB-Medium (mit entsprechenden Antibiotika) für 16 h bei 37 °C und 180 rpm inkubiert. 1 ml der Kultur wird mit 0,5 ml Glycerin vermischt, in flüssigem Stickstoff schockgefroren und bis zur weiteren Verwendung bei -80 °C gelagert. 3.2.2 3.2.2.1 PROTEINBIOCHEMISCHE METHODEN EXPRESSION VON 14N-PROTEINPROBEN 2 ml LB Medium werden in ein steriles Reagenzglas überführt und mit 50 μl des Glycerin-Stocks angeimpft. Die Bakterienlösung wird im Schüttler für 8 h bei 37 °C und 120 rpm inkubiert. Mit der Bakterienlösung werden 50 ml LB-Medium (Amp und Cl, bei Koexpression zusätzlich Kan) angeimpft. Diese Vorkultur wird im Schüttler bei 37 °C und 120 rpm bis zu einer optischen Dicht OD600 у 0,8 inkubiert. Mit der Vorkultur werden wiederum 2 l LB Medium angeimpft, die im Schüttler bei 37 °C und 100 rpm inkubiert werden. Bei einer OD600 у 0,8 wird mit 1 mM IPTG die Überexpression des Zielproteins für 16 Stunden bei 37 °C (bei der Koexpression 22 °C) induziert. 45 3 Material und Methoden 3.2.2.2 EXPRESSION VON 15N-PROTEINPROBEN 2 ml LB Medium werden in ein steriles Reagenzglas überführt und mit 100 μl des Glycerin-Stocks angeimpft. Die Bakterienlösung wird im Schüttler für 8 h bei 37 °C und 120 rpm inkubiert. Mit der Bakterienlösung werden 50 ml LB-Medium (Amp, Chl) angeimpft. Diese Vorkultur wird im Schüttler bei 37 °C und 120 rpm bis zu einer optischen Dichte OD600 у 0,8 inkubiert. Die Zellen der Hauptkultur werden bei 37 °C und 5000 rpm (Rotor Sorval SLC-6000) innerhalb von 15 min sedimentiert. Die Bakterien werden mit 1x M9-Medium gewaschen und in 1 l Minimalmedium resuspendiert. Nach einstündigem Schütteln bei 37 °C (bei der Koexpression 22 °C) und 100 rpm wird durch Zugabe von 1 mM IPTG die Überexpression des Zielproteins induziert. 3.2.2.3 PROTEINAUFREINIGUNG Die Zellen werden in der Eppendorf Zentrifuge (A-4-81 Rotor) bei 4000 rpm (2900 g) und 4 °C innerhalb von 45 min pelletiert. Von nun an werden die Zellen/Proteine auf Eis gelagert. Der Überstand wird dekantiert und die Zellen in 20 ml kaltem Bindepuffer resuspendiert. Der Zellaufschluss erfolgt mit dem Microfluidizer in mehreren Durchläufen bei 1000 psi. Das Zelllysat wird mit 50 μl Proteaseinhibitor versetzt. Die Aufreinigung erfolgt über den N-terminalen His6-Tag mittels Nickel-Sepharose-High-Performancebeads (Amersham Biosciences, Bindekapazität 10 mg/ml beads). Proteine mit His6-Tag binden spezifisch an Säulenmaterial mit zweiwertigen Kationen (hier: Nickel). Alle anderen Proteine können durch Waschschritte entfernt werden. Durch Zugabe von Imidazol werden die Histidine aus dem mit Nickel gebildeten Chelatkomplex verdrängt. Zu diesem Zweck müssen die beads vorbereitet werden. 4 ml des aufgeschwemmten bead-Materials werden in ein 15 ml Falcon überführt und in der Eppendorf Zentrifuge (34-6-38 Rotor) bei 6800 rpm 10 min zentrifugiert. Der ethanolische Überstand wird verworfen und die beads werden viermal mit je 10 ml Bindepuffer-Puffer gewaschen. Um die löslichen Proteine von unlöslichen Zellwandaggregaten zu trennen, wird das Lysat 45 min in der Eppendorf Zentrifuge (A-4-81 Rotor) bei 4000 rpm (2900 g) und 4 °C zentrifugiert. Der Überstand wird auf die vorbereiteten Nickel-beads gegeben und auf dem Schüttler 1 h bei 20 °C inkubiert. Die beads werden bei 4 °C zentrifugiert und der Überstand verworfen. Anschließend werden die beads mit 10 ml Bindepuffer in ein 15 ml Falcon überführt und zentrifugiert. Die beads werden viermal mit jeweils 10 ml Waschpuffer gewaschen. Die Überstände werden für eine spätere gelektrophoretische Analyse gesammelt. 46 3 Material und Methoden Zur Elution des Zielproteins werden die beads mit 2,5 ml Elutionspuffer versetzt, 5 min bei 20 °C unter leichtem Schütteln inkubiert und der Überstand durch Zentrifugation (4 °C, 2900 g, 10 min) abgetrennt. Der Elutionsvorgang wird insgesamt viermal durchgeführt. Die Eluate werden vereinigt und in Millipore-Konzentratoren (10.000 MWCO) aufkonzentriert. Zur Entfernung des Imidazols erfolgt eine Umpufferung über PD-10-Säulen nach Angaben des Herstellers. Die Proteinkonzentration wird mit Hilfe des Bradford-Tests bestimmt. 3.2.2.4 IN VITRO FARNESYLIERUNG Für die in vitro Farnesylierung werden 10 mg Rheb mit 2,5 mg Farnesyltransferase, 1 μM Farnesylpyrophosphat (FPP) und 20 μM Zinkchlorid in Bindepuffer (Endvolumen 5 ml) für 3 h bei 32 °C inkubiert. Nach 30, 60 und 90 min wird je 1 μM FPP zugegeben. 3.2.2.5 TRITON X-114-EXTRAKTION Die Proteinlösung wird mit 2 ml eiskalter Triton X-114-Lösung (11 %) versetzt und 10 min bei 4 °C inkubiert. Anschließend wird das Gemisch für 10 min bei 35 °C inkubiert, um eine Phasentrennung, die durch beginnendes Eintrüben der Lösung gekennzeichnet ist, zu erreichen. Nach Zentrifugation (A-4-81 Rotor) bei 34 °C für 5 min und 3500 rpm werden die Phasen getrennt und die wässrige Phase noch zweimal mit Triton X-114 extrahiert. Um das Triton für weitere Messungen zu entfernen, wird das Protein über den N-terminalen His6-Tag aufgereinigt (siehe 3.2.2.3). 3.2.2.6 Zur SDS-PAGE Analyse der Proteine wird die diskontinuierliche und denaturierende SDS- Polyacrylamidgelelektrophorese durchgeführt. Dabei werden zwei sich im pH-Wert und in der Porengröße unterscheidende Gele eingesetzt, die aufgrund dieser Eigenschaften eine sehr gute Auftrennung der Proteine und hohe Bandenschärfe garantieren. Durch Behandlung mit SDS und Erhitzen auf 95 °C werden die Proteine vor dem Auftragen auf das SDS-Gel denaturiert. Durch das hinzugefügte SDS erfolgt eine Maskierung der Eigenladung der Proteine, wodurch eine Auftrennung ausschließlich aufgrund ihrer Größe möglich wird. 15 μl Proteinlösung werden mit 5 μl 5x-Laemmli-Puffer versetzt, bei 95 °C für 5 min inkubiert und in die Taschen des Sammelgels aufgetragen. Die Elektrophorese erfolgt so lange bei 100 V, bis die Proben vollständig in das Sammelgel eingelaufen sind. Zur Auftrennung im Trenngel wird die Spannung konstant auf 150 V gehalten. Im Anschluss wird das SDS-Gel für 30 min in der Färbelösung 47 3 Material und Methoden inkubiert, mehrfach mit Wasser gewaschen und bis zur gewünschten Entfärbung in Entfärbelösung inkubiert. 3.2.3 BESTIMMUNG DER PROTEINKONZENTRATION NACH BRADFORD Bei der Bestimmung der Proteinkonzentration nach Bradford wird die Bindung des Farbstoffs Coomassie Brillant Blue-G250 an basische Aminosäuren genutzt. Bei dieser Reaktion verschiebt sich das Absorptionsmaximum des Farbstoffs von 470 nm zu 595 nm. Zur Bestimmung der Proteinkonzentration wird die Absorption bei 595 nm gemessen und dieser Wert mit einer Kalibrierkurve verglichen. Zur Erstellung der Kalibrierkurve wird als Standard Rinder-Serum-Albumin (BSA) verwendet. Für einen Makroansatz (0,2 bis 1 mg/ml Protein) werden 1-20 μl der Proteinlösung mit 1x-Bradfordreagenz auf 1 ml aufgefüllt, 10 min bei 37 °C inkubiert und gegen 1x-Bradfordreagenz als Nullwert bei 595 nm vermessen. 3.2.4 NUKLEOTIDAUSTAUSCH Zur Überführung von Rheb in den aktiven Zustand wird ein Nukleotidaustausch durchgeführt. Dabei wird ausgenutzt, dass G-Proteine für eine affine Bindung des Nukleotids Mg2+ benötigen. Durch EDTA kann das Magnesium komplexiert und auf diese Weise eine Dissoziation des gebundenen GDPs begünstigt werden. Durch Zugabe von Alkaliner Phosphatase wird GDP abgebaut und dem Austauschgleichgewicht entzogen. Für die Austauschreaktion wird 1 mM Rheb in Bindepuffer mit 0,2 mM EDTA und 5 U Alkaline Phosphatase bei 30 °C für 10 min inkubiert. Das EDTA wird über eine PD10-Entsalzungssäule entfernt. Es werden 250 mM GppNHp zugesetzt und eine Inkubation bei 32 °C für 30 min durchgeführt. Es wird MgCl2 in einer finalen Konzentration von 0,2 mM zugegeben und eine Bestimmung der Proteinkonzentration durchgeführt (3.2.3) 3.2.5 PROTEINPRÄPARATION FÜR DIE NMR-SPEKTROSKOPIE Für die hier durchgeführten 1H-15N-HSQC-Experimente werden Proteinkonzentrationen zwischen 0,2 mM und 1 mM eingesetzt. 500 μl Proteinlösung werden mit 50 μl D2O, TSP in einer Finalkonzentration von 1 mM und 1 μl Proteaseinhibitor versetzt und blasenfrei in ein NMR-Röhrchen überführt. 3.2.6 HERSTELLUNG VON E. COLI-LYSAT Die Expression wird wie in 3.2.2.2 beschrieben durchgeführt. Die Zellen werden in der Eppendorf Zentrifuge (A-4-81 Rotor) bei 4000 rpm (2900 g) und 4 °C innerhalb von 45 min pelletiert. Von nun an 48 3 Material und Methoden werden die Zellen/Proteine auf Eis gelagert. Der Überstand wird dekantiert und die Zellen in 20 ml kaltem Bindepuffer resuspendiert. Der Zellaufschluss erfolgt mit dem Microfluidizer in mehreren Durchläufen bei 1000 psi. Das Zelllysat wird mit 50 μl Proteaseinhibitor versetzt. Die Probe kann direkt NMR-spektroskopisch charakterisiert werden. 3.2.7 HERSTELLUNG VON OOZYTEN-EXTRAKT Die Präparierung sowie die Selektion der Xenopus-Oozyten wurde freundlicherweise von den Mitarbeitern der AG Kationenkanäle von Prof. Dr. Guiscard Seebohm (Lehrstuhl Biochemie I der Ruhr-Universität Bochum; jetzt: Universitätsklinikum Münster) durchgeführt. Zur Herstellung von Oozyten-Extrakt wird zunächst das Kulturmedium in einem langsamen Austauschprozess durch NMR-Puffer (+ 10 % D2O) ersetzt. Nach dem erfolgten Austausch wird die verbleibende Flüssigkeit abgenommen. Die Oozyten werden in einem Homogenisator 5 min lang homogenisiert und im Anschluss daran für 20 min in der Eppendorf Zentrifuge (Rotor 5415 R, 10.000 rpm) bei 4 °C zentrifugiert. Hierbei erfolgt die Auftrennung von unlöslichen bzw. löslichen Zellbestandteilen und Lipiden. Die lösliche Proteinfraktion wird abgenommen und der oben dargestellte Zentrifugationsschritt noch einmal durchgeführt. Die restlichen Lipide aus dem ersten Zentrifugationsschritt werden entfernt und die lösliche Proteinfraktion bei - 80 °C schockgefroren. 3.2.8 INJEKTION VON PROTEIN IN XENOPUS-OOZYTEN In die selektierten Oozyten wird mit Hilfe eines Nanoliterinjektors (Nanoliter 2000; WPI, Sarasota, USA) das 15N-isotopenangereicherte Protein injiziert. Glaskapillaren werden mit einem horizontalen Kapillaren-Puller ausgezogen und unter optischer Kontrolle bis zu einem Durchmesser von 20 μm verkürzt. Vor dem Aufnehmen der Proteinlösung wird die so vorbereitete Kapillare mit einem 1:1 (v/v) Gemisch von leichtem und schwerem Mineralöl gefüllt und luftblasenfrei auf den Injektorkolben aufgebracht. Um die Oozyten für den Injektionsvorgang fest zu positionieren, wird eine kleinmaschiges Kunststoffnetz verwendet. Mit Hilfe eines Mikromanipulators wird die vorbereitete Kapillare so positioniert, dass die Spitze durch leichten Druck durch die Zellwand der Oozyte stoßen kann. Für die Mikroinjektion wurden Proteinkonzentrationen von 0,5 mM bis 5 mM eingesetzt. 3.2.9 HERSTELLUNG VON LIPID-VESIKELN Folch fraction I brain Lipids sowie Phosphatidylinositol-4-phosphat (PI(4)P) werden in Chloroform gelöst und bei -80 °C gelagert. Die Lipidlösung wird in einem Gemisch von Chloroform/Methanol (19:1) aufgenommen, im Argonstrom und anschließend im Vakuum getrocknet. 49 3 Material und Methoden Zur Rehydrierung der Lipide wird 1 ml NMR-Puffer hinzugefügt. Das Gemisch wird für 15 min bei 37 °C erwärmt und 15 min im Ultraschallbad behandelt. Anschließend werden fünf Gefrier-AuftauZyklen durchgeführt. Dazu wird die Probe zunächst in flüssigem Stickstoff schockgefroren und anschließend im Wasserbad für 15 min wieder aufgetaut. Bei diesem Vorgang entstehen Liposome unterschiedlicher Größe. Um Liposome gleicher Größe zu erhalten, wird die Mischung unter hohen Druck durch eine Polycarbonat-Membran mit definierter Porengröße (100 nm) gepresst (ExtruderTechnik). Die auf diese Weise hergestellten Liposome können fünf Tage bei 4 °C gelagert werden. 3.2.10 LIPID-BINDUNGSTEST Für die Liposomenbindung werden frisch präparierte Lipide (1 mg/ml) mit 2 μM des aufgereinigten Proteins gemischt. Abhängig vom jeweiligen Experiment werden noch 200 μM Nukleotid und 300 μM AlCl3 mit 10 mM NaF für 15 min auf Eis inkubiert. Die anschließende Ultrazentrifugation bei 100.000 g wird für 10 min bei 4 °C durchgeführt. Der Überstand sowie das Pellet werden mittel SDS-PAGE analysiert. 3.2.11 ESI-MS Die massenspektrometrischen Analysen wurden freundlicherweise von der AG von Dr. Dirk Wolters der Ruhr-Universität Bochum durchgeführt. Das intakte Protein wird in 0,1 % Ameisensäure in 50 % Methanol gelöst (pH 2) und mittels ESI-MS am LTQ Orbitrap Velos von Thermo Fisher Scientific unter Standardbedingungen vermessen. Die Auswertung erfolgt mit der Software XCalibur und Proteome Discoverer 1.2. 3.3 SCREENING DER SUBSTANZBIBLIOTHEKEN Die Substanzbibliotheken werden durch Aufnahme von 1H-15N-HSQC-Spektren oder 1H-15N-SOFASTHMQC-Spektren gescreent. Alle Substanzen liegen als Lösungen in d6-DMSO vor. Um auszuschließen, dass das in der Lösung enthaltene d6-DMSO das Messergebnis beeinflusst, wird vor jeder Messung eine Referenzmessung mit dem entsprechenden Volumen an d6-DMSO durchgeführt. Um bindende von nichtbindenden Substanzen zu unterscheiden, werden 1H-15N-HSQC-Spektren oder 1 H-15N-SOFAST-HMQC-Spektren aufgenommen und die Änderung der chemischen Verschiebung der Rheb-Resonanzen beobachtet. Werden die Resonanzverschiebungen auf der Oberfläche der Proteinstruktur aufgetragen, lässt sich auf diese Weise die jeweilige Bindungsregion identifizieren und daraus lassen sich Informationen für spätere Optimierungen der Ligandenstruktur extrahieren. 50 3 Material und Methoden Für das Screening der Substanzbibliothek wird eine 0,2 mM 15 N-isotopenangereicherte Rheb- Proteinprobe mit 25 μl d6-DMSO vermischt und als Referenz vermessen. Für jede Substanz wird eine 0,2 mM 15N-isotopenangereicherte Rheb-Proteinprobe mit 25 μl Ligand versetzt, was einem molaren Verhältnis von 1:10 entspricht. 3.3.1 BESTIMMUNG DER DISSOZIATIONSKONSTANTEN Es lässt sich nicht nur eine Aussage über die Qualität eines Bindungsereignis machen, sondern es lässt sich anhand der beobachteten Verschiebungen der Resonanzfrequenzen auch die Dissotiationskonstante (KD-Wert) bestimmen. Hierzu wird der Vektor der Verschiebung gegen die Konzentration des zugegebenen Liganden aufgetragen. Dabei werden folgende Annahmen gemacht: Der Reaktion zwischen Protein P und Ligand L (aus der Substanzbibliothek) liegt ein bimolekularer Schritt zweiter Ordnung zugrunde: ࢇ࢙࢙ ࢊ࢙ ࡼ + ࡸ ሱۛሮ ࡼࡸ ሱۛሮ ࡼ + ࡸ (1) kdis beschreibt dabei die Geschwindigkeitskonstante der Dissoziationsreaktion, während kass die Geschwindingkeitskonstante der Assoziation ist. Über das Verhältnis dieser beiden Reaktionsschritte lässt sich der KD-Wert beschreiben. ܭ = ೞ (2) ೌೞೞ Durch Zugabe des jeweiligen Liganden aus der Bibliothek ändert sich bei Bindung an das isotopenangereicherte Protein die chemische Umgebung der an der Bindung beteiligten Aminosäuren. Dies hat eine Änderung der detektierten Resonanzfrequenzen und damit eine chemische Verschiebung des Resonanzsignals (οߜ) in der Protonen- und Stickstoffebene zur Folge. כ Die gewichtete chemische Verschiebung οߜ௦ trägt den unterschiedlichen Frequenzbereichen der beiden Achsen Rechnung. כ οߜ௦ (3) = ඨ(οߜ భு )ଶ + ቆ ಿ οఋ భఱಿ ଶ ହ ቇ Um den KD-Wert aus den beobachteten Veränderungen zu bestimmen, wird die gewichtete chemische Verschiebung gegen das Verhältnis N von Ligand und Protein aufgetragen. Mit ಽೌ ܰ= (4) ುೝ und 51 3 Material und Methoden כ οߜ௦ = (5) כ ൫்ିξ்మ ିସே൯ οఋೌೣ ଶ kann durch individuelles Fitten der resultierenden Bindungskurve an diese Gleichung der KD-Wert bestimmt werden. In Gleichung (5) ist ߜ௫ die maximal gewichtete chemische Verschiebung, somit der (fiktive) Endpunkt der Titration. Für T gilt folgende Gleichung: ܶ = 1+ܰ+ (6) ವ ൫(ேή)ା൯ ή 3.4 COMPUTERGESTÜTZTE ARBEITEN 3.4.1 3.4.1.1 VERWENDETE COMPUTERPROGRAMME NMRPIPE NMRPipe ist ein Programm zur Konvertierung und Prozessierung mehrdimensionaler NMR-Daten. Weiterhin kann dieses Programm zur Analyse und Darstellung von mehrdimensionalen Spektren verwendet werden 181. 3.4.1.2 NMRDRAW NMRPipe beinhaltet das ergänzendes Graphikinterface NMRDraw, welches zur Visualisierung der bearbeiteten NMR-Spektren verwendet werden kann. Diese Visualisierung der Daten kann zur Optimierung der Prozessierungsparameter durch stetige Überprüfung des Prozessierungsfortschritts verwendet werden. 3.4.1.3 NMRVIEW NMRView ist eine Software, die die Visualisierung und Analyse von mehrdimensionalen NMR-Daten erlaubt182. 3.4.1.4 PYMOL PyMol ist ein Computerprogramm zur Darstellung und Erstellung von Animationen von 3D-Strukturen. Weiterhin erlaubt dieses Programm auch die Bearbeitung von PDB-Dateien. 52 3 Material und Methoden 3.4.1.5 HADDOCK Der traditionelle Weg, um Protein-Protein-Komplexe mittels NMR-Spektroskopie zu untersuchen, erfolgt über die Auswertung von intermolekularen Kern-Overhauser-Effekt-(NOE)-Abständen. Dies erfordert die Durchführung und Analyse zahlreicher komplementärer mehrdimensionaler NMRExperimente. Für die Analyse solcher Protein-Protein-Interaktionen ist eine vollständige sequenzspezifische Zuordnung aller NMR-Signale vonnöten, da intermolekulare NOEs häufig Seitenkettenprotonen enthalten. Zwar können über einfache Titrationsexperimente auf Basis von HSQC-Spektren, welche nur eine Zuordnung des Proteinrückgrats voraussetzen, die an der Bindung beteiligten Aminosäuren identifiziert werden. Aus diesen Experimenten können jedoch keine Rückschlüsse auf die genaue räumliche Ausrichtung der Bindungspartner zueinander gemacht werden. Dockingprogramme ermöglichen es, in vergleichsweise kurzer Zeit mithilfe theoretischer Berechnungen Aufschluss über die genaue Orientierung zweier Moleküle zueinander zu erhalten. Das Programm HADDOCK183 (High Ambiguity Driven protein-protein DOCKing) berücksichtigt im Gegensatz zu vielen anderen Dockingprogrammen direkt Ergebnisse aus biochemischen und/oder biophysikalischen Experimenten, wie zum Beispiel Daten aus NMR-Titrationsexperimenten. Hierbei werden diese Informationen als Ambiguous Interaction Restraints (AIR) in die Berechnung eingebracht. Ein AIR ist dabei eine multivariante Abstandsbedingung zwischen Aminosäuren des Proteins und des Liganden und kann somit als Pseudo-NOE angesehen werden. Das Docking mit HADDOCK läuft, vereinfacht beschrieben, in drei Schritten ab: Zuerst werden die beiden Bindungspartner zufällig im Raum zueinander orientiert und es wird eine Minimierung der Rigid Body Energie durchgeführt. Um die intermolekulare Energiefunktion zu minimieren, erfolgt zunächst eine Optimierung der Orientierung der beiden Bindungspartner, wobei Translationen sowie Rotationen erlaubt sind. Im zweiten Schritt wird ein simuliertes Tempern durchgeführt, wobei die Bindungspartner als starre Körper im Raum betrachtet werden; im zweiten Abschnitt dieses Temperns werden die Seitenketten im Bindungsinterface freigegeben, sodass eine gewisse Beweglichkeit der Bindungsregion gewährleistet wird, und im letzten Schritt des simulierten Temperns wird neben den Seitenketten auch das Rückgrat freigegeben. Im dritte Schritt des Dockings erfolgt eine Verfeinerung der Struktur im kartesischen Raum durch Zufügen von Wasser (water refinement). Die Berechnungen werden mit dem Programm HADDOCK 2.0 lokal durchgeführt und alle Parameter des originalen HADDOCK-Setups verwendet. Für die Strukturrechnung greift HADDOCK auf das Programm CNS184 zurück. 53 3 Material und Methoden 3.5 NMR-EXPERIMENTE 3.5.1 DAS 1D 1H-EXPERIMENT Zur Untersuchung von Makromolekülen stehen eine Reihe von NMR-Experimenten zur Verfügung, wobei das klassische eindimensionale 1H-Experiment das einfachste darstellt. Das 1D 1H-Experiment eignet sich aufgrund seiner sehr kurzen Messdauer von einigen Sekunden bis zu wenigen Minuten, seiner Toleranz gegenüber der zu untersuchenden Probe, der schnellen Auswertbarkeit sowie der sehr hohen Empfindlichkeit für erste NMR-spektroskopische Untersuchungen von Makromolekülen. Durch Einsatz dieses Standardexperiments lassen sich Informationen über Konzentration sowie Faltung und daraus folgend der Funktionsfähigkeit des Makromoleküls gewinnen. Ein 1H-NMR-Spektrum, welches eine große Signaldispersion von 0 bis etwa 10 ppm aufweist, ist ein guter Indikator für ein gefaltetes Protein. Dabei besteht eine Korrelation zwischen dem Anteil an ɴ-Faltblattstrukturen und dem Grad der Signaldispersion. Für rein ɲ-helikale Proteine sind Aussagen über den Faltungszustand auf Basis eines 1D 1H-Spektrums schwer zu treffen. Ungefaltete Proteine weisen eine geringe Signaldispersion auf. Die jeweilige detektierte chemische Verschiebung ist stets abhängig von der chemischen Umgebung. Bei ungefalteten Proteinen sind nahezu alle Protonen lösungsmittelexponiert und weisen somit sehr ähnliche chemische Verschiebungen auf. Bei gefalteten Proteinen hingegen sind die meisten Protonen durch ihre individuelle, aus der tertiären Struktur des Proteins resultierenden chemischen Umgebung in distinkte Konformationen gezwungen. Hieraus resultiert eine Vielzahl von verschiedenen Resonanzfrequenzen und somit ein Spektrum mit großer Signaldispersion185. Neben ersten Resultaten betreffend der Proteinfaltung wird das 1D-1H-Spektrum auch zur Optimierung von Puffer- oder pH-Bedingungen eingesetzt. In Abbildung 3.1 ist eine schematische Darstellung des verwendeten Pulsprogramms gezeigt. 1 186 Abbildung 3.1: Schematische Darstellung des Pulsprogramms p3919gp zur Durchführung eines 1D H-Experiments . 54 3 Material und Methoden Bei allen NMR-Messungen von Proteinproben in wässrigen Lösungen ist eine Wasserunterdrückung eine Grundbedingung zum Erhalt individueller Messdaten. Daher folgt nach dem Delay (d1) und dem 90 °-Puls, was dem eigentliche NMR-Experiment entspricht, sogleich die WATERGATE Pulssequenz. Dabei wird ein 3-9-19 Pulsschema verwendet. ZWEIDIMENSIONALE EXPERIMENTE 3.5.2 DAS 2D 1H-15N-HSQC-EXPERIMENT 3.5.2.1 Eine Übertragung der Magnetisierung von einem 1H-Kern auf einen daran gebundenen Heterokern (hier: 15 N) und wieder zurück findet bei dem Heteronuclear Single Quantum Coherence (HSQC)- Experiment statt. Bei einem 1 H-15N-HSQC-Spektrum wird pro stereochemisch unterscheidbarem Stickstoff- gebundenem Wasserstoff ein Signal erwartet. Vereinfacht dargestellt wird pro Aminosäure ein Signal erhalten. Da Prolin im Protein über kein Stickstoff-gebundenes Proton verfügt, werden keine Signale für diese Aminosäure detektiert. Zusätzlich verfügen einige Aminosäuren über Stickstoff-gebundene Wasserstoffatome in ihren Seitenketten, was, abhängig vom gewählten pH-Wert, die Anzahl der detektierten Signale noch vergrößern kann. Bei dem hier gewählten Pulsschema zur Aufnahme des 1H-15N-HSQC-Spektrums wird die Echo/AntiEcho-Technik zur Lösungsmittelunterdrückung eingesetzt. Allgemein setzt sich ein zweidimensionales Spektrum aus Präparation, Evolution (t1), Mischzeit und Detektion (t2) zusammen. Eine schematische Darstellung dieses Pulsprogramms ist in Abbildung 3.2 gezeigt. 1 15 Abbildung 3.2: Schematische Darstellung des Pulsprogramms hsqcetf3gpsi zur Durchführung eines H- N-HSQCExperiments186. 55 3 Material und Methoden 3.5.2.2 DAS 2D 1H-15N-SOFAST-HMQC-EXPERIMENT Die Messdauer, die zur Aufnahme eines NMR-Spektrums notwendig ist, hängt von der Sensitivität des jeweiligen Experiments gegenüber der zu messenden Probe und des verwendeten Spektrometers sowie von der benötigten Auflösung des Spektrums ab. Besonders bei Screeningverfahren bedarf es aufgrund der großen Anzahl von Proben sehr schneller Messmethoden. Eine Weiterentwicklung auf dem Gebiet der zweidimensionalen heteronuklearen 2D-Experimente stellt das SOFAST (Band-Selective Optimized-Flip-Angle Short-Transient)-HMQC-Experiment187 dar. Eine sehr schnelle Datenerfassung wird bei SOFAST-HMQC-Experimenten durch sehr kurze Delays zwischen einzelnen Scans realisiert. Das SOFAST-HMCQ-Experiment kombiniert die Vorteile einer kleinen Anzahl von Radiofrequenzpulsen, Ernst-Winkel-Anregung188 und longitudinale Relaxationsoptimierung189. Das bei SOFAST-Messungen eingesetzte HMQC (Heteronuclear Multiple Quantum Coherence)Spektrum korreliert, wie das HSQC-Spektrum auch, ein Proton mit einem X-Kern. Während der Evolutionszeit eines HMQC-Experiments entwickeln sich die Protonen- und die X-KernMagnetisierungen. Bei einem HSQC-Experiment entwickelt sich nur die X-Kern-Magnetisierung und es gibt keine Protonenmagnetisierung. Als Folge zeigt das HSQC-Experiment eine aufgrund geringerer Linienbreite deutlich bessere Auflösung. Jedoch ist dieses Experiment aufgrund seiner komplexen Pulssequenz sehr anfällig für Störfaktoren. Das HMQC-Experiment ist allgemein unempfindlicher und somit für sehr schnelle Pulsfolgen, wie sie bei SOFAST-Experimenten vorkommen, besser geeignet. Abbildung 3.3: Schematische Darstellung des Pulsprogramms sfhmqcf3gpph zur Durchführung eines 1H-15N-SOFASTHMQC-Experiments186. 56 3 Material und Methoden 3.6 PROZESSIERUNG VON NMR-DATEN Um aufgenommene NMR-Daten analysieren zu können, muss eine Prozessierung dieser Daten mittels Fouriertransformation von der Zeit- in die Frequenzdomäne erfolgen, was mit dem Programm NMRPipe vorgenommen wird. Nach Konvertierung der aufgenommenen binären Daten mittels eines Spektrometer-spezifischen Programms können alle erfassten Dimensionen des Datensatzes nacheinander bearbeitet werden. Eine Korrektur der Basislinie wird zunächst bei der hochaufgelösten direkten Dimension (FID (free induction decay)) durchgeführt. Darauf folgend wird durch die mathematische Operation der Linearvorhersage eine Erweiterung der Daten der Zeitdomäne vorgenommen. Dieser Schritt erfolgt optional. Durch diese Vorgehensweise kann die digitale Auflösung der Spektren verbessert werden, da eine Variation der Linienstärke durch später angewendete Filterfunktionen herbeigeführt wird; eine weitere Optimierung kann durch Einsatz spezieller Sinus- oder Quadratsinusfunktionen erreicht werden. Dem während der Messphase idealerweise auf Null abfallenden FID kann eine definierte Anzahl an Nullen angefügt werden. Dieses direkt vor der folgenden Fouriertransformation durchgeführte zero filling führt zu einer Vergrößerung der digitalen Auflösung. Um die Phase der Signale zu berücksichtigen, wird im Anschluss an die Fouriertransformation eine Phasenkorrektur durchgeführt. Im letzten Schritt der Prozessierung der ersten Dimension wird eine erneute Basislinienkorrektur durchgeführt. Für alle weiteren aufgenommenen Dimensionen wird in gleicher Weise verfahren. Zwischen jedem Prozessierungsschritt können die Daten mit dem Programm NMRDraw visualisiert werden. Die Bestimmung der korrekten Phase erfolgt ebenfalls mit diesem Programm. Nach erfolgter Prozessierung aller Dimensionen wird das Spektrum in einem NMRView-kompatiblen Format gespeichert und kann mit diesem Programm ausgewertet werden. 57 3 Material und Methoden 3.7 PULSPROGRAMME Nachfolgend sind die verwendeten Pulsprogramme mit den jeweiligen Parametern sowie Originalzitaten angegeben. Tabelle 3.1: Verwendete Pulsprogramme für die Durchführung der angegebenen NMR-Experimente. Experiment F1 F2 Referenz NS SW [Hz] TD SW [Hz] TD 1 H 64 12019,2 32768 - - 190,191 1 H-15N-HSQC 64 12019,2 2048 2736,7 128 192–194 1 H-15N-SOFAST-HMQC 64 12019,2 2048 3040,9 96 195 58 4 Ergebnisse-Teil I 4 ERGEBNISSE-TEIL I 4.1 PROTEINPRÄPARATION VON RHEB Die Vorschriften zur rekombinanten Proteinexpression (3.2.2.1 und 3.2.2.2) sowie zur Proteinaufreinigung (3.2.2.3) liefern konstante Proteinausbeuten. Ein Unterschied bezüglich der Proteinausbeute zwischen ausschließlicher Anzucht in LB-Medium und zusätzlicher Anzucht in 15 N- isotopenangereichertem Minimalmedium ist nicht festzustellen. Minimalmedien beeinflussen üblicherweise nur die Wachstumsphase von Bakterienzellen, nicht jedoch deren Expressionsverhalten. Für diese Arbeit wird das Wachstum der Bakterien ausschließlich in LBMedium durchgeführt. Nach erfolgter Wachstumsphase werden die Bakterienzellen in 15 N- isotopenangereichertem Minimalmedium weiter kultiviert, in dem die Überexpression erfolgt. Der His6-Tag, welcher durch Nutzung eines pQE-30-Vektors angefügt wurde, ermöglicht eine unkomplizierte und rasche Proteinaufreinigung in hohen Konzentrationen. Nach Durchführung von 3-4 Waschschritten kann das Protein in für die NMR-Spektroskopie ausreichender Reinheit gewonnen werden (Abbildung 4.1). Aus 2 l Medium können durchschnittlich 10 mg Protein aufgereinigt werden. Das bei der Elution zugegebene Imidazol kann durch die Nutzung einer PD-10-Entsalzungssäule nahezu vollständig entfernt werden. In diesem Schritt ist ein Proteinverlust von etwa 5 % zu beobachten. In Abbildung 4.1 ist ein 15 %iges SDS-Gel nach Färbung mit Coomassie Brillant Blue dargestellt, wie es bei jeder Proteinprobenaufreinigung angefertigt wird. Links ist die Spur des aufgetragenen Proteinmarkers dargestellt. Wichtige Proteingrößen sind daneben in kDa aufgetragen. Neben dem Proteinmarker ist eine Spur des Zelllysats nach IPTGinduzierter Überexpression aufgetragen. In dieser Spur lässt sich das überexpremierte Rheb nicht von den übrigen Haushaltsproteinen unterscheiden. In der Spur der Waschschritte ist eine intensive Bande bei etwa 24 kDa sichtbar. Da es sich hierbei um Waschschritte handelt, bindet das hier sehr gut sichtbare Protein nur sehr schwach an die Nickel-beads. Rechts neben den Spuren der Waschschritte sind die Spuren der Elutionsschritte dargestellt. In Höhe von etwa 21 kDa (siehe Pfeil) ist eine sehr intensive Bande zu erkennen, welche durch das 20,4 kDa große Rheb-Protein hervorgerufen wird. In diesen Spuren sind noch weitere deutlich schwächere Banden vorhanden, welche Haushaltsproteinen zuzuordnen sind, die eine gewisse Affinität zu den Nickel-beads zeigen und in den Waschschritten nicht vollständig entfernt werden können. 59 4 Ergebnisse-Teil I Um auszuschließen, dass die vorhandenen Proteine bei den durchgeführten NMR-spektroskopischen Untersuchen stören, wurde eine weitere Aufreinigung mittels Gelfiltration durchgeführt, wodurch eine höhere Reinheit der resultierenden Proteinprobe erreicht werden kann (Ergebnisse nicht gezeigt). Die auf diese Weise erhaltene Probe zeigt keine nachweisbaren Unterschiede zu der Proteinprobe, welche ausschließlich mittels Affinitätschromatographie aufgereinigt wurde. Da das in dieser Arbeit untersuchte Protein Rheb eine relativ kurze Halbwertszeit aufweist, wird daher bei den folgenden Aufreinigungen auf eine Gelfiltration verzichtet. Marker Lysat Waschen 1 bis 3 Elution 1 und 2 170 70 40 25 15 10 Abbildung 4.1: Ergebnis einer 15 % SDS-PAGE von Proben einer Rheb-Präparation nach Anfärben des SDS-Gels mit Coomassie Brillant Blue. Auf dem 15 %igen SDS-Gel sind von links nach rechts der Marker (Größen in kDa), eine Probe des Proteinlysats nach Induktion mit IPTG, die Waschfraktionen 1 bis 3 sowie die Elutionsfraktionen 1 und 2 aufgetragen. Der Pfeil kennzeichnet die erwartete Höhe der Rheb-Proteinbande bei etwa 21 kDa. 4.2 AUFNAHME VON EIN- UND MEHRDIMENSIONALEN NMR-SPEKTREN Alle NMR-Spektren werden an einem DRX 600 MHz Spektrometer der Firma Bruker bei 298 K (wenn nicht anders angegeben) und pH 7,8 durchgeführt. Als Probenkopf wird ein 5 mm TXI Probenkopf mit Z-Gradient verwendet. 4.2.1 DAS 1D 1H-NMR-EXPERIMENT Das klassische 1D 1H-NMR-Experiment wird eingesetzt, um eine Angabe über den Faltungszustand des zu untersuchenden Proteins machen zu können. Weiter kann dieses Experiment dazu genutzt werden, um die Reinheit, Konzentration und Halbwertszeit der jeweiligen Proteinprobe zu bestimmen. Auch eignet sich das 1D 1H-NMR-Experiment zur Optimierung von Pufferbedingungen. 60 4 Ergebnisse-Teil I Das eindimensionale Protonenspektrum stellt im übertragenen Sinne den Fingerabdruck eines Moleküls dar. Bei diesem Experiment treten alle vorhandenen Protonen bei einer bestimmten Frequenz mit der elektromagnetischen Strahlung in Resonanz, was im Spektrum als Signal sichtbar wird. Aufgrund unterschiedlicher Zusammensetzung und Struktur resultiert für jedes Molekül ein einzigartiges Linienmuster. Signale kleiner Moleküle lassen sich aufgrund der begrenzten Anzahl an Protonen und somit an Signalen oft schon ausschließlich auf Basis eines 1D 1H-NMR-Spektrums zuordnen. Neben der Anzahl der Signale spielt auch die jeweilige Signaldispersion eine entscheidende Rolle. Dies hängt maßgeblich von der übergeordneten Konformation des Moleküls ab. Bei Makromolekülen ist aufgrund der Vielzahl der enthaltenen Protonen und der daraus resultierenden Signale eine Zuordnung allein auf Grundlage dieses Experiments nicht mehr möglich. Aufgrund der Möglichkeit, mit diesem Experiment den Faltungszustand eines aufgereinigten Proteins zu kontrollieren, wird vor jedem komplexeren NMR-Experiment ein eindimensionales Protonenspektrum aufgenommen. Besonders nach der Zugabe von Liganden der Substanzbibliothek ist es somit zweckmäßig, ein 1D 1H-NMR-Spektrum aufzunehmen, da auf diese Weise unmittelbar festgestellt werden kann, ob das Protein durch den zugesetzten Liganden denaturiert wird. Restwasser aliphatische Seitenketten TSP Aromaten Amide [ppm] Abbildung 4.2: 1D 1H-NMR-Spektrum von RhebͻGDP bei 600 MHz, 298 K und pH 7,8. Signale im Bereich von 10,5 bis 7,0 ppm werden durch Amidprotonen, Signale zwischen 7,5 bis 6,0 ppm durch aromatische 1 CH-Gruppen und Signale zwischen 4,5 bis 0 ppm durch aliphatische Seitenketten hervorgerufen. Die Referenzierung des H- Spektrums wird durch TSP (0 ppm nach IUPAC) durchgeführt. 61 4 Ergebnisse-Teil I Ist dies der Fall, muss kein zusätzliches mehrdimensionales Spektrum mehr aufgenommen werden, was eine Zeitersparnis darstellt. Ein 1H-NMR-Spektrum der kleinen GTPase Rheb ist in Abbildung 4.2 dargestellt. Die Referenzierung des Spektrum wird mittels des zugesetzten TSPs (0 ppm nach IUPAC) durchgeführt. Das Restwassersignal ist bei 4,7 ppm lokalisiert. Neben den aliphatischen Seitenkettensignalen (4,5 bis 0 ppm) sind die aromatischen CH-Resonanzen (7,5 bis 6,0 ppm) und die NH-Resonanzen (10,5 bis 7 ppm) im dargestellten Spektrum gekennzeichnet. Die scharfen Signale im Bereich von 8 ppm sind geringen Resten an Imidazol in der aufgereinigten Probe zuzuordnen. 4.2.2 DAS 2D 1H-15N-HSQC-EXPERIMENT Das 1H-15N-HSQC-Experiment gehört zur Gruppe der zweidimensionalen, heteronuklearen NMRExperimente. Hierbei werden die Frequenzen der Stickstoffatome (15N) mit den Frequenzen der daran gebundenen Protonen (1HN) korreliert. Also entspricht die Anzahl der detektierten Signale im 1 H-15N-HSQC-Spektrum ungefähr der Anzahl der im Protein enthaltenen Aminosäuren. Die Aminosäure Prolin ist aufgrund von fehlenden Amidprotonen in der Proteinkette im 1H-15N-HSQCSpektrum nicht detektierbar. Jedoch können zusätzliche Signale von Seitenketten einiger Aminosäuren, die über NH-Gruppen verfügen, hervorgerufen werden. NH-Bindungen aus strukturierten Bereichen weisen meist sehr intensive Signale im 1H-15N-HSQC-Spektrum auf, wohingegen nur wenige Signale aus flexiblen Bereichen wie Loops oder den Termini zu erwarten sind. Aufgrund der Größe des Rheb-Proteins von 20,4 kDa kommt es in einigen Bereichen des 1H-15NHSQC-Spektrum zu stärkeren Signalüberlagerungen, sodass einige Aminosäuren nur unter Verwendung von höher dimensionalen Spektren aufgelöst werden können. Lässt sich eine Aussage über den Faltungszustand des Proteins nicht anhand des eindimensionalen Protonenspektrums machen, kann dies durch Aufnahme eines 1H-15N-HSQC-Spektrums erfolgen: Spektren ungefalteter Proteine zeichnen sich durch eine nur geringe Signaldispersion aus. Bei allen hier dargestellten 1H-15N-HSQC-Spektren wird auf der Ordinate die Frequenz (F1) der Stickstoffatome (15N) und auf der Abszisse die Frequenz (F2) der an diese Stickstoffe gebundenen Protonen (1HN) in ppm-Werten aufgetragen. Um die Bindung von Liganden, posttranslationale Modifikationen, Änderungen der Struktur oder des Nukleotidzustandes nachzuweisen, sind 1H-15N-HSQC-Spektren geeignete NMR-Experimente. Da für die hier untersuchte GTPase Rheb eine Zuordnung der Rückgratresonanzen196,197 (BackboneAssignment) sowie die gelöste dreidimensionale Struktur93,95 vorliegen, kann auf der Grundlage von 1 H-15N-HSQC-Spektren eine Aussage über die Lokalisation des Bindungsereignisses auf der Proteinoberfläche gemacht werden. 62 4 Ergebnisse-Teil I Bei der Titration potentieller Liganden kann somit direkt zwischen bindenden und nicht-bindenden Substanzen unterschieden werden. Bei Liganden, die mit Rheb interagieren, kann über die detektierte Verschiebung einzelner Signale die Bindungsregion auf der Proteinoberfläche identifiziert werden. Rheb liegt wie alle GTPasen in zwei Zuständen vor. Im inaktiven Zustand ist das Nukleotid GDP und im aktiven Zustand das Nukleotid GTP gebunden. Aufgrund der schnellen intrinsischen Hydrolysegeschwindigkeit von Rheb im Vergleich zur NMR-Zeitskala können 1H-15N-HSQC-Spektren, welche den aktiven Zustand von Rheb detektieren sollen, nicht mit GTP durchgeführt werden. Zu diesem Zweck wird daher das nicht-hydrolysierbare Analogon GppNHp eingesetzt. 1 15 Abbildung 4.3: Überlagerung der H- N-HSQC-Spektren von RhebͻGDP (schwarz) und RhebͻGppNHp (rot), aufgenommen bei 600 MHz, 298 K und pH 7,8. Auf der Ordinate ist die Frequenz der Stickstoffatome 15N , auf der Abszisse die Frequenz der Amidprotonen 1HN in ppm aufgetragen. Im Vergleich zum 1H-15N-HSQC-Spektrum des GDP-gebundenen Rhebs finden sich im 1H-15N-HSQCSpektrum von RhebͻGppNHp deutlich weniger Signale (Abbildung 4.3) aufgrund von Austauscheffekten der Amidprotonen mit dem Lösungsmittel. Weitere Unterschiede der 1H-15N63 4 Ergebnisse-Teil I HSQC-Spektren der beiden Formen der GTPase sind das Verschwinden charakteristischer Signale aus dem 1H-15N-HSQC-Spektrum von RhebͻGDP im Bereich von 10,7 bis 9,7 ppm im Spektrum des GppNHp-gebundenen Rhebs und deutliche Signalverschiebungen (Abbildung 4.4). 1 15 Abbildung 4.4: Vergrößerung der Überlagerung der H- N-HSQC-Spektren von GDP- (schwarz) und GppNHpgebundenem (rot) Rheb. Durch Austausch des Nukleotids von GDP zu GppNHp ergeben sich teilweise sehr große Unterschiede in den Resonanzfrequenzen einzelner Aminosäuren. Bei den Aminosäuren S16 und S20 gibt es starke Veränderungen der 1 15 Resonanzfrequenzen im H- N-HSQC-Spektrum. 4.3 IN-CELL NMR-SPEKTROSKOPIE Die NMR-Spektroskopie stellt momentan die einzige Technik dar, mit der Strukturinformationen von Proteinen unter physiologischen oder zumindest physiologisch-ähnlichen Bedingungen gewonnen werden können. Die hochaufgelöste NMR-Spektroskopie weist eine Selektivität für bestimmte, in der Natur relativ selten vorkommende Atomkerne (ausgenommen 1H) wie 13C und 15N auf, während 12C und 14 N nicht NMR-aktiv sind und in der Messung nicht detektiert werden. Somit werden die am häufigsten in der Zelle vorkommenden Atomkerne nicht detektiert. Isotopenangereicherte Proteine können von außen in die Zelle eingebracht und differenziert von allen weiteren vorhandenen 64 4 Ergebnisse-Teil I Makromolekülen untersucht werden. Bei einer Änderung der chemischen Umgebung eines NMRaktiven Kerns, was zum Beispiel bei einer Bindung an einen Bindungspartner möglich sein kann, lässt sich diese Änderung über eine Verschiebung der jeweiligen Resonanzfrequenz nachverfolgen. Dadurch ist es möglich, Änderungen des Nukleotidzustandes und weitere Bindungsereignisse nachzuvollziehen. Hierbei können die isotopenangereicherten Proteine auf unterschiedliche Weise in die Zelle eingebracht werden. Für Messungen in E. coli-Zellen oder in E. coli-Lysat erfolgt nach der Anzucht in Bakterien in unmarkiertem Medium ein Wechsel in isotopenangereichertes Medium vor der Induktion der Überexpression. Für Messungen in Xenopus-Oozyten muss das zuvor aufgereinigte, isotopenangereicherte Protein mechanisch in die Zellen eingebracht werden. Für Messungen in Oozyten-Extrakt wird zu der aufgereinigten, isotopenangereicherten Proteinprobe eine definierte Menge an zuvor gewonnenem Oozyten-Extrakt hinzugefügt. 4.3.1 4.3.1.1 E. COLI-ZELLEN INTAKTE E. COLI-ZELLEN Zur Untersuchung der kleinen GTPase Rheb in physiologischer Umgebung werden NMRspektroskopische Untersuchungen des Proteins in intakten E. coli-Zellen durchgeführt. Die Anzucht der Bakterien erfolgt dabei zunächst in nicht-isotopenangereichertem Vollmedium. Vor Induktion der Expression des Zielproteins erfolgt ein Wechsel in 15N-isotopenangereichertes Minimalmedium. Abbildung 4.5 zeigt das resultierende 1H-15N-HSQC-Spektrum. Auf der Ordinate ist die Frequenz der 15 N-Stickstoffatome und auf der Abszisse die der 1HN-Amidprotonen aufgetragen. Das 1H-15N-HSQC- Referenzspektrum des 15 N-isotopenangereicherten RhebͻGDP in Puffer ist schwarz und das Spektrum des überexprimierten Rhebs in E. coli-Zellen (15N-isotopenangereichertes Minimalmedium) ist grün dargestellt. Im gesamten Spektrum der E. coli-Messung ist nur eine sehr geringe Signaldispersion zu beobachten. Intensive Signale sind in einem schmalen Streifen von 8,5 bis 7,5 ppm sowie in Höhe der Amidseitenkettensignale lokalisiert. Alle dargestellten Signale zeigen eine hohe Intensität und liegen deutlich über dem Rauschlevel. 65 4 Ergebnisse-Teil I 1 15 Abbildung 4.5: H- N-HSQC von RhebͻGDP (schwarz) im Vergleich mit Rheb in intakten E. coli-Zellen (grün). Im in-cell NMR-Spektrum ist nur eine geringe Anzahl von sehr intensiven Signale zu erkennen, welche in Höhe der Seitenketten-Amidprotonen sowie in einem schmalen Streifen zwischen 8,5 und 7,5 ppm lokalisiert sind. Um auszuschließen, dass die detektierten Signale aus den Bakterienzellen in den Messpuffer austreten und deshalb Signale hervorrufen, wird ein 1H-15N-HSQC-Spektrum des Puffers direkt nach der in-cell NMR-Messung durchgeführt. Das aufgenommene 1H-15N-Spekrum weist keinerlei Signale auf (Abbildung nicht gezeigt). Um eine Aussage darüber machen zu können, ob die detektierten Signale im 1H-15N-HSQC-Spektrum von dem enthaltenen Protein oder den E. coli-Zellen selbst stammen, wird ein ebensolches Spektrum von E. coli-Zellen ohne Plasmid, welches das Rheb-Gen trägt, aufgenommen. Das daraus gewonnene Spektrum zeigt das identische Signalmuster wie das 1H15 N-HSQC-Spektrum von Rheb in E. coli-Zellen. Die im in-cell NMR-Spektrum detektierten Signale stammen somit von den E. coli-Zellen und nicht von dem enthaltenen überexprimierten RhebProtein. Um nachzuweisen, dass für eine NMR-Messung ausreichend 15 N-isotopenangereichertes Rheb in den E. coli-Zellen vorliegt, werden die eingesetzten Zellen lysiert und erneut einer NMRMessung unterzogen. 66 4 Ergebnisse-Teil I 4.3.1.2 E. COLI-ZELLLYSAT Die in 4.3.1 verwendeten E. coli-Zellen werden lysiert und nach dem Entfernen unlöslicher Zellbestandteile durch Zentrifugation für das 1H-15N-HSQC-Experiment eingesetzt. In Abbildung 4.6 ist das erhaltene 1 H-15N-HSQC-Spektrum der Messung von 15 N- isotopenangereichertem Rheb in E. coli-Lysat dargestellt (rot). Dieses Spektrum ist einem Referenzspektrum von affinitätschromatographisch aufgereinigtem 15 N-isotopenangereichertem RhebͻGDP (schwarz) überlagert. 1 15 Abbildung 4.6: H- N-HSQC von RhebͻGDP (schwarz) im Vergleich mit Rheb aus E. coli-Lysat (rot). Die E. coli-Zellen, die zuvor für die in-cell NMR-Messung eingesetzt wurden, werden lysiert und ein 1H-15N-HSQC-Spektrum 1 15 des Überstands aufgenommen. Im erhaltenen H- N-HSQC-Spektrum sind fast alle Signale des Rhebͻ'W-Spektrums detektierbar. Im Gegensatz zu der Messung in intakten E. coli-Zellen ist bei diesem 1H-15N-HSQC-Spektrum eine gute Signaldispersion zu erkennen. Durch eine Überlagerung mit einem 1H-15N-HSQC-Spektrum einer Rheb-Referenzprobe lässt sich eine große Ähnlichkeit der beiden Spektren beobachten: Alle Signale 67 4 Ergebnisse-Teil I sind in beiden Spektren vorhanden; auch die Intensität der Signale des 1H-15N-HSCQ-Spektrums der Lysat-Probe entspricht dem der aufgereinigten Probe. 1 15 Abbildung 4.7: Überlagerung der H- N-HSQC-Spektren von RhebͻGDP (schwarz), Rheb in E. coli-Lysat (rot) und Rheb in intakten E. coli-Zellen (grün). Die im 1H-15N-HSQC von Rheb in intakten E. coli-Zellen auftretenden intensiven Signale fehlen im Spektrum von Rheb in E. coli-Lysat. Im Bereich von 8,2 bis 7,8 ppm erscheinen auch im Spektrum von Rheb in E. coli-Lysat einige schlecht aufgelöste Signale. Die Signale, die im Spektrum der intakten E. coli-Zellen zu beobachten sind, treten im E. coli-Lysat-Spektrum nicht auf. 4.3.2 XENOPUS-OOZYTEN Ein weiteres mögliches Zellsystem, das sich zur Durchführung von in-cell NMR-Messungen anbieten, sind die Oozyten des Krallenfrosches Xenopus laevis. In dessen sehr großen Oozyten kann unter optischer Kontrolle 15N-isotopenangereichertes Protein injiziert werden. Auch kann ein Extrakt aus diesen Zellen gewonnen und für NMR-Messungen eingesetzt werden. 68 4 Ergebnisse-Teil I 4.3.2.1 INTAKTE XENOPUS-OOZYTEN Zur Messung von Protein in Xenopus-Oozyten werden je 50 nl des 15 N-isotopenangereicherten, affinitätschromatographisch aufgereinigten Rheb-Proteins (3 mM) unter optischer Kontrolle in 400 Oozyten injiziert. Das daraus resultierende 1H-15N-HSQC-Spektrum zeigt keine Signale. 4.3.2.2 OOZYTEN-EXTRAKT Da in den in-cell NMR-Messungen von 15 N-isotopenangereichertem Rheb, welches in Xenopus- Oozyten injiziert wurde, keine Signale detektiert werden können (4.3.2.1), werden weitere Messungen unter Verwendung von Oozyten-Extrakt durchgeführt. Zur Herstellung von OozytenExtrakt werden die Zellen lysiert und die löslichen Bestandteile von den Lipiden und unlöslichen Bestandteilen durch Zentrifugation getrennt. Zu dem affinitätschromatographisch aufgereinigten, 15 N-isotopenangereicherten Rheb wird ein definiertes Volumen an Zellextrakt (mit und ohne Phosphataseinhibitor) zugefügt und ein 1H-15N-HSQC-Spektrum aufgenommen. In Abbildung 4.8 wird eine Überlagerung des affinitätschromatographisch aufgereinigten, 15 N-isotopenangereicherten Rheb-Proteins (schwarz), des Rheb-Proteins in Oozyten-Extrakt (lila) und des Rheb-Proteins in Oozyten-Extrakt bei Zugabe von Phosphataseinhibitor (grün) gezeigt. Das 1H-15N-HSQC-Spektrum von Rheb in Oozyten-Extrakt weist sehr große Ähnlichkeit mit dem Referenzspektrum von 15 N-isotopenangereichertem Rheb auf. In einzelnen Bereichen sind Verschiebungen der Frequenzen einzelner Aminosäuren, sogenannte shifts, zu erkennen. Bei dem Vergleich der Spektren von Rheb in Oozyten-Extrakt mit und ohne Zugabe von Phosphataseinhibitor fallen große Unterschiede bei den einzelnen Resonanzfrequenzen der detektieren Aminosäuren auf. Signale vieler Aminosäuren (wie die für die GDP-Form charakteristischen Signale zwischen 10,7 und 9,7 ppm) fehlen oder weisen größere Verschiebungen auf. 69 4 Ergebnisse-Teil I 1 15 Abbildung 4.8: Überlagerung der H- N-HSQC-Spektren von RhebͻGDP (schwarz), Rheb in Oozytenextrakt (lila) und Rheb in Oozytenextrakt mit Zugabe von Phosphataseinhibitor (grün). 1 15 Die H- N-HSQC-Spektren von RhebͻGDP und Rheb in Oozyten-Extrakt zeigen bei vielen Signalen eine sehr große 1 15 Übereinstimmung. Die Signalanzahl in dem H- N-HSQC-Spektrum von Rheb in Oozytenextrakt mit Phosphataseinhibitor ist deutlich geringer als in den beiden anderen Spektren. Ebenso tritt eine Reihe neuer Signale auf. 4.4 FARNESYLIERUNG VON RHEB Wie bei vielen anderen kleinen GTPasen ist die posttranslationale Modifikation in Form der Prenylierung bei Rheb essentiell für dessen Funktion. Die am Carboxy-Terminus lokalisierte CaaX-Box dient dabei als Erkennungssequenz für das die Prenylierung durchführende Enzym. Durch die die CaaX-Box bildenden Aminosäuren ist festgelegt, welche Art der Modifikation durchgeführt wird. Rheb wird wie KRas-4B ausschließlich farnesyliert, besitzt im Gegensatz zum Ras-Protein jedoch keine weiteren zur Lokalisierung beitragenden Elemente. KRas-4B besitzt N-terminal der CaaX-Box eine Lysin-reiche Region, welche für die Membranlokalisation dieser kleinen GTPase entscheidend ist. 70 4 Ergebnisse-Teil I 4.4.1 IN VITRO FARNESYLIERUNG Bei einer Überexpression in E. coli-Zellen erfolgt aufgrund des Fehlens der Farnesyltransferase in den Bakterienzellen keine Farnesylierung. Das Protein muss nach der affinitätschromatographischen Aufreinigung durch Zugabe von Farnesylpyrophosphat und Farnesyltransferase in vitro farnesyliert werden. Die Trennung von farnesyliertem und unfarnesyliertem Rheb geschieht durch Triton X-114Extraktion. Der angefügte Farnesylanker ist stark hydrophob und führt zu einer Anreicherung des modifizierten Proteins in der Triton-Phase. Das unfarnesylierte Rheb verbleibt hingegen in der wässrigen Phase und kann abgetrennt werden. Marker 1 2 170 70 40 25 15 10 Abbildung 4.9: Ergebnis einer 15 % SDS-PAGE von affinitätschromatographisch aufgereinigtem Rheb nach in vitro Farnesylierung und Triton X-114-Extraktion nach Anfärben des Gels mit Coomassie Brillant Blue. In der linken Spur ist der Marker aufgetragen. In der mittleren Spur ist die wässrige (1) und in der rechten Spur die TritonPhase (2) aufgetragen. In der wässrigen Phase findet sich bei etwa 20 kDa eine sehr intensive Bande. Im selben Größenbereich wird auch eine Bande in der Spur der Triton-Phase sichtbar, wobei die Intensität deutlich geringer ist, obwohl die Probe sehr stark aufkonzentriert wurde. Abbildung 4.9 zeigt das Ergebnis der SDS-PAGE von der Triton X-114-Extraktion des in vitro farnesylierten Rhebs. In Spur 1 ist die wässrige und in Spur 2 die Triton X-114-Phase aufgetragen. In der wässrigen Phase ist eine sehr intensive Bande bei etwa 20 kDa zu erkennen. Diese Bande wird von unfarnesyliertem Rheb hervorgerufen. Das farnesylierte Rheb reichert sich aufgrund seiner größeren Hydrophobizität in der Triton X-114-Phase an und ist in Spur 2 als schwache Bande bei etwa 20 kDa zu erkennen. 4.4.2 IN VIVO FARNESYLIERUNG In der Zelle wird eine Farnesylierung von Rheb durch das Enzym Farnesyltransferase durchgeführt. Unstrukturierte Termini in Proteinen sind häufiger Angriffspunkt für Proteasen198, weshalb ein gewisser Abbau des Carboxyterminus der GTPase Rheb zu erwarten ist. 71 4 Ergebnisse-Teil I Die Farnesylierung direkt in der Bakterienzelle durch Koexpression der GTPase mit der Farnesyltransferase ermöglicht eine direkte Prenylierung ohne vorherige Proteinaufreinigung, wodurch ein Abbau der Termini und somit auch der für die Farnesylierung entscheidenden CaaX-Box verhindert werden kann. Marker Elution 170 70 40 25 15 10 Abbildung 4.10: Ergebnis einer 15 % SDS-PAGE von Proben einer Rheb/Farnesyltransferase-Koexpressions-Präparation nach Anfärben des SDS-Gels mit Coomassie Brillant Blue. Auf dem 15 %igen SDS-Gel sind links der Proteinmarker in kDa und eine Elutionsfraktion aufgetragen. Die beiden oberen Pfeile kennzeichnen die Untereinheiten der Farnesyltransferase und die beiden unteren Pfeile das farnesylierte und das unfarnesylierte Rheb. Durch eine Koexpression von Rheb und Farnesyltransferase kann eine Farnesylierung direkt in vivo erfolgen. Die Aufreinigung erfolgt affinitätschromatographisch über Ni2+-beads und es können Proteinausbeuten wie in 4.1 beschrieben erzielt werden. Die exprimierte Farnesyltransferase besitzt ebenfalls einen His6-Tag, wodurch unter Nutzung von Ni2+-beads auch das Enzym aufgereinigt wird. Auf dem 15%igen SDS-Gel nach Anfärben mit Coomassie Brillant Blue (Abbildung 4.10) ist neben dem Proteinmarker ein Elutionsschritt der Proteinaufreinigung aufgetragen. Hier sind vier unterschiedliche Banden zu erkennen, wobei die beiden oberen Banden (um 45 kDa) die Untereinheiten der Farnesyltransferase darstellen. Die beiden unteren, eher schlecht getrennten Banden repräsentieren das farnesylierte und unfarnesylierte Rheb (jeweils um 20 kDa). Aufreinigungen von Rheb ohne Koexpression von Farnesyltransferase führen nicht zu den hier beobachteten Doppelbanden bei einer Größe von 20 kDa (siehe Abbildung 4.1). Der angefügte Farnesylrest scheint einen großen Einfluss auf das Verhalten des Proteins im SDS-Gel zu haben, da Unterschiede unter 1 kDa mit dieser Art von SDS-Gel sonst nicht aufgetrennt werden können. 72 4 Ergebnisse-Teil I Zur Trennung von farnesyliertem und unfarnesyliertem Protein wird die Probe, wie in 4.4.1 beschrieben, einer Triton X-114-Extraktion unterworfen. Marker 1 2 170 70 40 25 15 10 Abbildung 4.11: Mit Coomassie Brillant Blue gefärbtes 15 %iges SDS-Gel der Triton X-114-Extraktion der Koexpression von Rheb und Farnesyltransferase. Das 15 %ige SDS-Gel wird mit Coomassie Brillant Blue gefärbt. In der linken Spur ist der Marker aufgetragen (Angaben in kDa). Die mittlere Spur (1) repräsentiert die wässrige Phase der Extraktion. In der rechten Spur (2) ist die Triton X-114-Phase aufgetragen, die vor der Beladung des SDS-Gels nicht aufkonzentriert wurde. In Abbildung 4.11 ist das 15 %ige SDS-Gel nach Anfärben mit Coomassie Brillant Blue der Triton X-114-Extraktion gezeigt. Das farnesylierte Rheb reichert sich aufgrund seiner größeren Hydrophobizität in der Triton X-114-Phase an. Das unfarnesylierte Rheb verbleibt in der wässrigen Phase und kann abgetrennt werden. In der ersten Spur (1) ist die wässrige Phase aufgetragen, welche das unfarnesylierte Rheb enthält. Die rechte Spur (2) repräsentiert die Triton X-114-Phase. In der Spur der wässrigen sowie in der Triton X-114-Phase ist eine Bande in Höhe von 20 kDa lokalisiert, was dem unfarnesylierten bzw. dem farnesyliertem Rheb zuzurechnen ist. Die Bande in Spur 1 ist deutlich intensiver als die Bande aus Spur 2. Im Gegensatz zur Probenvorbereitung für das SDS-Gel der Triton X-114-Extraktion der in vitro-Farnesylierungsprobe wurde die in diesem Experiment erhaltene Triton X-114-Phase nicht aufkonzentriert. 4.4.3 ESI-MS Zum eindeutigen Nachweis, dass eine in vivo Farnesylierung erfolgt ist und es sich bei den aufgereinigten Proben um farnesyliertes Rheb handelt, wird eine massenspektrometrische Analyse mittels ESI-MS am intakten Protein durchgeführt. 73 rel. Häufigkeit 4 Ergebnisse-Teil I m/z Abbildung 4.12: Direktmessung mittels ESI-MS von einer Mischung aus farnesyliertem und unfarnesyliertem Rheb (intaktes Protein). Im ESI-Massenspektrum des Gemischs aus farnesyliertem und unfarnesyliertem Rheb ist eine Differenz von 207 Da der gemessenen Proteine zu beobachten. Dies entspricht der Größe des Farnesylrestes (erwartet: 204 Da). Durch Berechnung der Differenz der effektiven Massen ergibt sich ein Unterschied von 207 Da, was der Größe des Farnesylankers entspricht. Durch Abschätzen der Intensitäten der Signale von farnesyliertem und unfarnesyliertem Rheb kann das Verhältnis der beiden Formen zueinander abgeschätzt werden. Dies ist möglich, da beide untersuchten Proteine sich nur durch den angefügten Farnesylanker unterscheiden und somit ein nahezu identisches Ionisationsverhalten aufweisen. Demnach liegt das farnesylierte Protein etwa zu 65 % und das unfarnesylierte Protein zu 35 % vor. 4.4.4 KOSEDIMENTATIONSANALYSE Um die Lipidbindeeigenschaften von farnesyliertem Rheb zu untersuchen, wird eine Inkubation mit kleinen, unilamellaren Vesikeln (SUV) durchgeführt. Diese Liposome bestehen aus Folch Typ I Lipidextrakt ohne oder mit Zusatz von Phosphatidylinositol-4-phosphat (PI(4)P). Ebenso wird die Abhängigkeit der Lipidbindung von der Nukleotidbeladung charakterisiert. Bei der Kosedimentationsanalyse wird zwischen einer sedimentierten (Pellet) und einer Überstandfraktion unterschieden. Der Nachweis, dass eine Anbindung des Proteins an die Liposome erfolgt ist, wird durch eine SDS-PAGE der sedimentierten (P) und der Überstandfraktion (S) erbracht. Bei ausschließlicher Verwendung von Folch fraction I brain Lipids ist keine Interaktion von farnesyliertem Rheb mit den Liposomen zu beobachten. Werden hingegen zusätzlich 5 % 74 4 Ergebnisse-Teil I Phosphatidylinositol-4-phosphat (PI(4)P) für die Liposombildung verwendet, ist eine Lipidbindung möglich (Abbildung 4.13). Unfarnesyliertes Rheb zeigt keine Bindung an die genutzten Liposome. Um die Nukleotidabhängigkeit der Lipidbindung von farnesyliertem Rheb zu überprüfen, werden für die Kosedimentationsanalyse GDP und GDPͻAlFx zugegeben (Abbildung 4.13). Nur das GDPͻAlFxgebundene Rheb sedimentiert, wohingegen im GDP-gebundenen Zustand die Proteine hauptsächlich im Überstand verbleiben. Abbildung 4.13: Mit Coomassie Brillant Blue gefärbtes 15 %iges SDS-Gel zur Charakterisierung der Nukleotid-abhängigen Bindung von unfarnesyliertem und farnesyliertem Rheb an Folch/PI(4)P-Liposomen. Farnesyliertes Rheb bindet unter Verwendung von GDPͻAlFx an Liposomen, welche aus Folch Typ I Lipidextrakt und 5 % PI(4)P bestehen (S: Überstand, P: Pellet). Unfarnesyliertes Rheb zeigt keine Bindung an Liposome. 4.4.5 NMR-SPEKTROSKOPISCHE CHARAKTERISIERUNG VON FARNESYLIERTEM RHEB Modifikationen von Proteinen beeinflussen in vielen Fällen die Proteinfaltung oder führen zu Veränderungen in der Struktur und der Löslichkeit der Proteine. Um eine mögliche Änderung der Faltung oder eine Interaktion mit dem angefügten C-terminalen Farnesylanker zu charakterisieren, werden 1H-15N-HSQC-Spektren von rekombinant farnesyliertem Rheb aufgenommen. Neben möglichen Änderungen auf struktureller Ebene sind auch veränderte Löslichkeitseigenschaften des modifizierten Proteins einfach zu detektieren. Abbildung 4.14 zeigt eine Überlagerung der 1H-15N-HSQC-Spektren von GDP-gebundenem, 15 N- isotopenangereichertem Rheb (schwarz) und farnesyliertem, 15N-isotopenangereichertem Rheb (rot). Beide Spektren weisen nur sehr geringe Unterschiede bei einzelnen Signalen auf. Das Spektrum des modifizierten Rhebs zeigt die gleiche Anzahl an Signalen wie das nicht-prenylierte Protein. Das 1 H-15N-HSQC-Spektrum des rekombinant farnesylierten RhebͻGDP weist eine große Signaldispersion und eine dem RhebͻGDP-Spektrum ähnliche Intensität der Aminosäuresignale auf. 75 4 Ergebnisse-Teil I Abbildung 4.14: Überlagerung der 1H-15N-HSQC-Spektren von RhebͻGDP (schwarz) mit farnesyliertem RhebͻGDP (rot). 1 15 Im Vergleich der beiden H- N-HSQC-Spektren lassen sich weder Unterschiede bei der Lage der Signale noch bei der Anzahl 1 15 oder Intensität der Signale beobachten. Das H- N-HSQC-Spektrum des farnesylierten Proteins besitzt eine große Signaldispersion. 76 5 Diskussion - Teil I 5 DISKUSSION - TEIL I 5.1 IN-CELL NMR-SPEKTROSKOPIE Protein-Protein-Interaktionen regulieren eine Vielzahl von zellulären Prozessen. Um einen Einblick in diese Vorgänge zu erhalten, ist die Aufklärung und die Veränderung der Proteinstruktur während solcher Prozesse essentiell. Neben der Röntgenstrukturanalyse ist die hochaufgelöste NMRSpektroskopie die einzige Methode, um Proteinstrukturen auf atomarer Ebene aufzuklären. Hierbei wird das zu untersuchende Protein allerdings immer isoliert und nicht innerhalb des physiologischen Systems betrachtet. Neueste Studien zeigen jedoch die Wichtigkeit der physiologischen Umgebung für die Funktion von Proteinen, sodass Charakterisierungen unter physiologisch-ähnlichen Bedingungen an Bedeutung gewinnen. Momentan bietet die hochaufgelöste NMR-Spektroskopie die einzige Möglichkeit, Proteine auf atomarer Ebene in ihrer natürlichen Umgebung zu charakterisieren. Dabei muss eine selektive Isotopenanreicherung der zu untersuchenden Proteine durchgeführt werden, da die in der Natur häufig vorkommenden Kerne 12C und 14N nicht NMR-aktiv sind und somit mit dieser Technik nicht detektiert werden können. Diese Isotopenanreicherung erlaubt schließlich die selektive Detektion des Zielproteins. 5.1.1 E. COLI-ZELLEN Um Proteine zu charakterisieren, bedarf es häufig einer sehr hohen Proteinkonzentration. Standardmäßig werden Proteine in E. coli-Zellen überexprimiert und anschließend affinitätschromatographisch aufgereinigt. Wird ein starkes Expressionssystem genutzt, so wird während der Überexpressionsphase fast ausschließlich das Zielprotein exprimiert. Durch einen Wechsel von nicht-isotopenangereichertem zu isotopenangereicherem Medium unmittelbar vor der Induktion ist es möglich, ausschließlich das Zielprotein isotopenangereichert zu gewinnen. Diese Strategie kann für die Durchführung von in-cell NMR-Experimenten genutzt werden. Dabei entfällt die sonst für proteinbiochemische Untersuchungsmethoden nötige Lyse der Zellen und affinitätschromatographische Abtrennung des Zielproteins von Haushaltsproteinen der Bakterienzellen. Das isotopenangereicherte Zielprotein kann direkt in den E. coli-Zellen NMRspektroskopisch charakterisiert werden. Die in dieser Arbeit zu untersuchende kleine GTPase Rheb erreicht ein hohes Level an Überexpression und führt daher zu sehr hohen Proteinausbeuten bei der 77 5 Diskussion - Teil I affinitätschromatographischen Aufreinigung. Aus diesem Grund scheint es möglich, in-cell NMRMessungen in E. coli-Zellen mit diesem Protein durchzuführen. Zur Charakterisierung des Proteins werden 1H-15N-HSQC-Spektren von Rheb in E. coli-Zellen aufgenommen und mit einer affinitätschromatographisch aufgereinigten, 15 N- isotopenangereicherten RhebͻGDP-Probe in Puffer verglichen. Das 1H-15N-HSQC-Spektrum der E. coli in-cell NMR-Messung zeigt nur wenige Signale und eine sehr geringe Signaldispersion. Intensive Signale sind in einer schmalen Region zwischen 8,2 bis 7,8 ppm und 113 ppm lokalisiert. Eine solche geringe Signaldispersion weist oft auf unstrukturierte Proteine hin. Die hier detektierten Signale stammen in diesem Fall von den eingesetzten E. coli-Zellen und von den darin enthaltenen Metaboliten137,156, was durch 1H-15N-HSQC-Spektren von E. coli-Zellen gezeigt werden kann. 1 15 Abbildung 5.1: Überlagerung der H- N-HSQC-Spektren von RhebͻGDP (schwarz) und Rheb in E .coli-Lysat (rot), aufgenommen bei 600 MHz, 298 K und pH 7,8. 1 15 Neu auftretende Signale im H- N-HSQC-Spektrum von Rheb in E. coli-Lysat sind mit Pfeilen gekennzeichnet. Der Bereich großer Überlagerung ist grün umrandet. 78 5 Diskussion - Teil I Demnach führt das überexprimierte Rheb-Protein in in-cell NMR-Messungen bei der Verwendung von E. coli-Zellen zu nicht-detektierbaren Signalen im Spektrum. Um auszuschließen, dass eine zu geringe Überexpression der Grund für das Fehlen von Signalen ist, werden die für die in-cell NMRMessung eingesetzten Bakterienzellen lysiert und das Zelllysat nach dem Abtrennen unlöslicher Bestandteile für eine erneute NMR-Messung eingesetzt (Abbildung 5.1). Das auf diese Weise erhaltene 1H-15N-HSQC-Spektrum zeigt eine große Signaldispersion. Ebenso sind alle Signale der freien GDP-Form von Rheb vorhanden. Im Bereich zwischen 7,8 und 8,4 ppm kommt es zu starken Überlagerungen im 1H-15N-HSQC-Spektrum von Rheb in E. coli-Lysat (grüne Markierung). Dies könnte auf eine Beteiligung von großen Molekülkomplexen hinweisen. Die im 1H-15N-HSQC-Spektrum der intakten E. coli-Zellen auftretenden einzelnen intensiven Signale sind im Spektrum des E. coli-Lysats nicht detektierbar. Wahrscheinlich werden diese Signale durch unlösliche Bestandteile der Bakterien wie Zellmembranen hervorgerufen, da vor der NMR-Messung eine Abtrennung dieser Bestandteile durch Zentrifugation erfolgt. Die im 1H-15N-HSQC-Spektrum neu auftretenden Signale können von Proteinen oder anderen Zellbestandteilen, welche ebenfalls 15N-isotopenangereichert vorliegen, stammen. Durch den Einsatz eines starken Expressionssystems soll die Expression anderer Proteine während der Überexpression des Zielproteins zwar unterdrückt werden, jedoch ist bei der SDS-PAGE der Aufreinigung von Rheb deutlich zu erkennen, dass weitere Proteine stark hochreguliert sind (siehe Bande in Höhe von 25 kDa, Abbildung 4.1). Dabei kann es sich zum Beispiel um Komponenten der Proteinbiosynthese handeln. Die zusätzlichen Signale könnten auch von Aminosäuren von Rheb herrühren, welche in vorherigen NMR-Experimenten nicht detektierbar waren; hierzu gehören Aminosäuren aus flexiblen Loop-Regionen sowie den Termini. Aufgrund des Protonenaustausches resultiert eine sehr starke Linienverbreiterung des jeweiligen Signals, was dazu führt, dass das Signal in Höhe des Rauschens liegt. Durch das Vorhandensein möglicher Bindungspartner im E. coli-Lysat könnten unter anderem die Termini des Proteins gebunden und somit stabilisiert werden, was zu neuen Signalen im 1H-15NHSQC-Spektrum führen könnte. Um den Ursprung dieser neuen Signale zu klären, bedarf es jedoch weiterer Analysen. Dass bei NMR-Messungen in der E. coli-Zelle kein Spektrum von Rheb beobachtet werden kann, ist somit nicht durch eine zu geringe Überexpression des Proteins zu erklären. Möglicherweise kommt es aufgrund der hohen Proteinkonzentration in der Zelle zur Bildung von sehr großen ProteinProtein-Komplexen (zum Beispiel eine Bindung von Rheb an TOR), was eine Erklärung für das Fehlen von Signalen im 1H-15N-HSQC-Spektrum sein könnte. Auch könnte die erhöhte Viskosität in den E. coli-Zellen dazu führen, dass keine Signale detektierbar sind. Durch eine hohe Viskosität wird das Molekültumbling verlangsamt, wodurch die Signale verbreitert werden und somit nicht mehr 79 5 Diskussion - Teil I detektiert werden können137. Die Stokes-Einstein-Debye-Gleichung199 beschreibt einen Zusammenhang zwischen der Viskosität und der apparenten molekularen Größe eines globulären Proteins. Durch die Annahme, dass die intrazelluläre Viskosität 3-12 Mal größer ist als die von Wasser138 ergibt sich für Rheb eine theoretische Größe von mindestens 60 kDa. Mit der hier eingesetzten NMR-Technik ist es nicht möglich, solch große Proteine ohne Deuterierung zu untersuchen. Da es sich hier jedoch um ein System mit einer großen Anzahl von Komponenten handelt und die Stokes-Einstein-Debye-Gleichung nur für Systeme gilt, bei denen das Testprotein deutlich größer als die Moleküle ist, die die Viskosität bestimmen, kann diese Gleichung nur als Anhaltspunkt bezüglich der tatsächliche Molekülgröße dienen200. Die in der Literatur beschriebenen in-cell NMR-Spektroskopie-Experimente in E. coli-Zellen untersuchen deutlich kleinere Proteine (um 5 kDa139). Bei größeren Proteinen werden ebenfalls keine oder sehr schlecht aufgelöste 1H-15N-HSQCSpektren beobachtet138,201. Auch können unspezifische Protein-Protein-Wechselwirkungen zu einer schlechten Auflösung beitragen. In der Literatur wird zur Zeit diskutiert, ob Rheb in vivo aufgrund seiner geringen intrinsischen Hydrolyse und dem hohen GTP-Gehalt der Zelle hauptsächlich im GTP-gebundenen Zustand vorliegt202. Das 1H-15N-HSQC-Spektrum von Rheb in E. coli-Lysat zeigt ausschließlich die GDPgebundene Form der kleinen GTPase. Wahrscheinlich ist die langwierige Probenvorbereitung und Aufnahmedauer ein möglicher Grund für das ausschließliche Existenz der inaktiven GDP-Form. Durch das Vorhandensein von Phosphatasen im Zelllysat ist es möglich, dass nach kurzer Zeit relativ wenig GTP vorliegt und somit ein Austauschprozess nicht stattfinden kann. Geringe Unterschiede in den Resonanzfrequenzen einzelner Aminosäuresignale können durch einen leicht anderen pH-Wert und eine veränderte Salzkonzentration in der Zelle hervorgerufen werden. Auch kann die veränderte Viskosität der Lysat-Probe zu einem geringfügig veränderten Spektrum führen. Aufgrund einiger weniger Veränderungen im 1H-15N-HSQC-Spektrum von Rheb in E. coli-Lysat kann eine Bindung von Liganden oder anderen Proteinen ausgeschlossen werden. Um Effekte von potentiellen Bindungspartnern zu untersuchen, müssen diese in ausreichenenden Menge vorliegen, um ein ausreichendes stöchiometrisches Verhältnis der beiden Interaktionspartner zu erzeugen. Möglicherweise liegt ein geringer Anteil von Rheb gebunden an Bindungspartner vor, was jedoch aufgrund der niedrigen Konzentration des Liganden nicht zu Veränderungen im 1H-15N-HSQCSpektrum führt. Eine Möglichkeit, um in-cell NMR-Titrationen durchführen zu können, besteht darin, noch ein weiteres Protein in den gleichen Bakterienzellen zu exprimieren, wobei dieses Protein nichtisotopenangereichert vorliegen muss139,140. 80 5 Diskussion - Teil I 5.1.2 XENOPUS-OOZYTEN Neben in-cell NMR-Messungen in E. coli-Zellen und -Lysat werden ebenso Experimente mit XenopusOozyten und Oozyten-Extrakt durchgeführt. Bei den aufgenommenen 1H-15N-HSQC-Spektren von 15N-isotopenangereichertem Rheb in XenopusOozyten können keine Signale detektiert werden. Dieses Ergebnis ist vergleichbar mit den in-cell NMR-Messungen in intakten E. coli-Zellen. Möglicherweise spielt auch hier die große Viskosität im Inneren der Oozyten-Zelle eine große Rolle. Erfolgreiche in-cell NMR-Messungen in Xenopus-Oozyten wurden bisher nur mit verhältnismäßig kleinen Proteinen (7-8 kDa120,134,203) durchgeführt. Bei den Messungen von Rheb in Oozyten-Lysat können hingegen gut aufgelöste 1H-15N-HSQCSpektren erhalten werden. Das resultierende Spektrum zeigt sehr große Übereinstimmungen mit dem in vitro 1H-15N-HSQC-Spektrum von RhebͻGDP. Kleinere Unterschiede können durch eine veränderte Salzkonzentration und einen leicht veränderten pH-Wert erklärt werden. Nach Zugabe von Oozyten-Extrakt, der mit Phosphataseinhibitor behandelt wurde, können jedoch größere Veränderungen im 1H-15N-HSQC-Sepktrum beobachtet werden (Abbildung 5.2). 180 ° Abbildung 5.2: Vergrößerung der Überlagerung der 1H-15N-HSQC-NMR-Spektren von RhebͻGDP (schwarz) und RhebͻGDP in Oozyten-Extrakt (lila) mit Zugabe von Phosphataseinhibitor (links) und Auftragung der fehlenden Aminosäuren (lila) im 1 H-15N-HSQC-Sepktrum auf der Oberfläche von Rheb. 1 15 Ein Ausschnitt aus der Überlagerung der aufgenommenen H- N-HSQC-Spektren zeigt, dass nur einige Aminosäuren im Rheb-Spektrum in Oozyten-Extrakt mit Zusatz von Phosphataseinhibitor fehlen (links). Alle fehlenden Aminosäuren sind auf der Oberfläche des Proteins lila eingefärbt. Das gebundene GDP ist ebenfalls dargestellt. In Abbildung 5.2 ist eine Ausschnittsvergrößerung der Überlagerung der 1H-15N-HSQC-Spektren von RhebͻGDP (schwarz) und RhebͻGDP in Oozyten-Extrakt mit Phosphataseinhibitor (lila) gezeigt. Im Spektrum von Rheb in Oozyten-Extrakt fehlen Signale einiger Aminosäuren. Durch farbliche 81 5 Diskussion - Teil I Hervorhebung der entsprechenden Aminosäuren auf der Proteinoberfläche von Rheb wird deutlich, dass die meisten Veränderungen in der Nähe der Nukleotid-Bindungstasche stattfinden. Eine mögliche Ursache könnte ein Verlust oder auch der Austausch des Nukleotids sein. Es wäre möglich, dass durch die Zugabe des Phosphataseinhibitors das natürlich in den Oozyten vorkommende GTP nicht mehr hydrolyisert werden kann und somit für den Nukleotidaustausch zur Verfügung steht. Ein Verlust des Nukleotids könnte ebenfalls zu Änderungen der Resonanzfrequenzen einzelner Aminosäuren führen. Die Messungen in intakten E. coli-Zellen und Xenopus-Oozyten sind aufgrund der in den Zellen deutlich erhöhten Viskosität und der damit verbundenen langsamen Tumblinggeschwindigkeit des Rheb-Proteins nicht möglich. Experimente an Rheb in E. coli-Lysat und Oozyten-Extrakt liefern sehr gut aufgelöste NMR-Spektren. Interaktionen mit Metaboliten oder anderen Interaktionspartnern können aufgrund der geringen Konzentration dieser nicht nachgewiesen werden. Die Möglichkeit, NMR-Spektren von größeren Proteinen in Zelllysat aufzunehmen, eröffnet die Perspektive zur Charakterisierung von Makromolekülen in physiologischem Umfeld. Alle bisher in der Literatur beschriebenen in-cell NMR-Messungen wurden an kleinen Proteinen vorgenommen, wodurch das Problem der hohen Viskosität im Zellinneren nicht entscheidend war. Bei größeren Proteinen muss, um tatsächliche in-cell NMR-Messungen durchführen zu können, eine Optimierung der Aufnahmetechnik erfolgen. Eine weitere Möglichkeit, um intakte Oozyten für weitere NMRspektroskopische Messungen zu nutzen wäre, den Inhalt der Zelle teilweise oder vollständig gegen Puffer zu ersetzen. Somit wäre zwar eine Interaktion zwischen dem Zielprotein und möglichen Interaktionspartnern nicht mehr gegeben, jedoch könnte die Zellmembran als Ankerpunkt für Proteine mit Membrananker genutzt werden. 5.2 FARNESYLIERUNG VON RHEB Die Farnesylierung stellt die einzig bisher bekannte posttranslationale Modifikation von Rheb zur Membrananbindung dar. Bei KRas-4B, das ebenfalls nur farnesyliert wird, dient eine Lysin-reiche Region als zusätzlicher Membrananker. Im Gegensatz zu vielen anderen kleinen GTPasen ist Rheb am Endoplasmatischen Retikulum und am Golgi-Apparat lokalisiert, an dem es mit TOR interagieren kann116. Bei der Expression von Rheb in E. coli-Zellen wird das Protein unmodifiziert hergestellt, da Bakterien nicht über die Farnesyltransferase, welche die Prenylierungsreaktion durchführt, verfügen. Eine Farnesylierung kann jedoch im Anschluss an die Proteinaufreinigung in vitro durch Zusatz von Farnesyltransferase und Farnesylpyrophosphat erfolgen. Um farnesyliertes von unfarnesyliertem Rheb abzutrennen, wird eine Extraktion mit Triton X-114 durchgeführt. Unterhalb von 4 °C bildet sich 82 5 Diskussion - Teil I hier ein homogenes Gemisch, welches sich bei einer Temperaturerhöhung auf 25 °C völlig in die wässrige und die Detergenzphase trennt. So kann die durch den Farnesylanker erhöhte Hydrophobizität des modifizierten Proteins zur Separation eingesetzt werden. Die in vitro Farnesylierung von Rheb liefert jedoch nur eine geringe Ausbeute an farnesyliertem Protein. Durch Veränderung der Reaktionsbedingungen wie Temperatur, pH-Wert und Konzentration der Reaktanten kann keine Erhöhung der Ausbeute an modifiziertem Rheb erreicht werden. Bei vielen Proteinen sind besonders die flexiblen Termini ein Angriffspunkt für Proteasen. Durch Zugabe von Proteaseinhibitor ist zwar eine Unterdrückung der Proteaseaktivität möglich, jedoch ist eine völlige Inhibierung nicht möglich. Das hier eingesetzte Rheb wird wahrscheinlich vor der in vitroFarnesylierung an seinen Termini von Proteasen angegriffen. Die für die Erkennung der Farnesyltransferase und somit für die Farnesylierung essentielle CaaX-Region kann auf diese Weise leicht hydrolysiert werden. Bei dem Aufreinigungsprozess von H-Ras konnte ebenfalls ein Abbau des C-Terminus beobachtet werde198. Um eine ausreichende Menge von prenyliertem Rheb zu erhalten, wird daher eine Koexpression von Rheb mit den beiden Untereinheiten der humanen Farnesyltransferase in E. coli-Zellen durchgeführt204. Das benötigte Farnesylpyrophosphat wird aus der Ubichinon-10- (bekannt auch als Coenzym Q10) Biosynthese in Bakterienzellen bereitgestellt205. Bei der Koexpression von Rheb mit den Untereinheiten der Farnesyltransferase können sehr gute Proteinausbeuten erzielt werden. Die erfolgreiche Farnesylierung kann mittels ESI und MS/MS nachgewiesen und das am Carboxy-Terminus lokalisierte Cystein eindeutig als Ankerpunkt des Lipidrests ausgemacht werden. Das prenylierte Protein zeigt im SDS-Gel ein minimal anderes Laufverhalten als das nicht-modifizierte Protein. Dieses Ergebnis wird ebenfalls in der Literatur beschrieben104. Zur Trennung von farnesyliertem und unfarnesyliertem Protein wird die Triton X-114-Extraktionsmethode mit Temperatursprung genutzt. Im Vergleich zur in vitro-Farnesylierung liefert die Farnesylierung in der Zelle deutlich höhere Ausbeuten an modifiziertem Protein, was sicherlich auch dem geringeren Abbau des C-Terminus der kleinen GTPase zugeschrieben werden kann. Da Rheb seine Funktion im Membran-gebundenen Zustand wahrnimmt, soll die Bindung von Rheb an Liposome und die Nukleotidabhängigkeit dieser Bindung untersucht werden. Eine Bindung von farnesyliertem Rheb wird an Liposome, welche ausschließlich aus Folch fraction I brain Lipids bestehen, nicht beobachtet. Folch fraction I brain Lipids bestehen aus etwa 10 % Phosphatidylcholin, 15 % Phosphatidylethanolamin, 10 % Phosphatidylserin, 3 % Phosphatidsäure und zu 1,5 % aus Phosphatidylinositol. Der restliche Anteil wird von nicht genau spezifizierten Lipiden gebildet206. Da Rheb in vivo an den PI(4)P-reichen Membranen des Golgi-Apparats lokalisiert ist207, 83 5 Diskussion - Teil I wird ein Anteil PI(4)P für die Herstellung der Liposome verwendet. Es kann eine Bindung von farnesyliertem RhebͻGDPͻAlFx an diese Liposome beobachtet werden. Ohne Nukleotid oder bei Zugabe von ausschließlich GDP erfolgt keine Bindung an die Liposome. Unfarnesyliertes Rheb bindet unabhängig von der Nukleotidbeladung nicht an die hier verwendeten Liposome. Somit kann ein zusätzliches eine Membranbindung begünstigendes Motiv (wie die Lysin-reiche Region bei KRas-4B) bei Rheb ausgeschlossen werden. Bei Ras wird momentan eine Nukleotid-abhängige Orientierung zur Membran diskutiert208. Ebenfalls wird eine Bildung von Dimeren bei membrangebundenem N-Ras diskutiert209. Nach der Farnesylierung erfolgt in vivo eine Abspaltung der letzten drei Aminosäuren sowie eine Carboxymethylierung. Beide Prozessierungsschritte werden in dieser Arbeit nicht durchgeführt. Es ist allerdings bekannt, dass bei fehlender Carboxymethylierung die Anbindung an Endomembranen gestört ist116,210. Um eine mögliche Interaktion des Farnesylankers mit dem Rheb-Protein, wie es etwa bei der kleinen GTPase Ran der Fall ist211, auszuschließen, werden 1 H-15N-HSQC-Spektren von 15 N- isotopenangereichertem, farnesyliertem Rheb aufgenommen. Dabei zeigen sich im Vergleich zum 1H15 N-HSQC von unmodifiziertem RhebͻGDP keine Unterschiede in den Resonanzfrequenzen der Aminosäuren. Ebenso sind keine zusätzlichen Signale zu beobachten, was auf keine feste Fixierung des C-Terminus schließen lässt. Ähnliche Studien an H-Ras zeigten ebenfalls keine strukturellen Änderungen198. Es ist gelungen, farnesyliertes Rheb in einer in vivo-Reaktion herzustellen, aufzureinigen und an Liposome zu binden. Des Weiteren eignet sich farnesyliertes Rheb zur Charakterisierung mittels NMR-Spektroskopie. Für weitere Analysen soll die neue Technik der Nanodiscs dazu genutzt werden, um das Verhalten von modifiziertem Rheb bei der Bindung an eine Membran zu untersuchen. 84 6 Ergebnisse - Teil II 6 ERGEBNISSE - TEIL II 6.1 1 H-15N-SOFAST-HMQC-SPEKTRUM Zur Untersuchung größerer Ligandenbibliotheken bedarf es Untersuchungsmethoden, die schnell und eindeutig eine Entscheidung erlauben, ob ein bindendes Molekül (Hit) oder ein nicht-bindendes Molekül vorliegt. Für das hier eingesetzte Fragment-basierte Screening (FBS) stellt die NMR-Spektroskopie das adäquate Verfahren dar, da durch die hohe Sensitivität dieser Methode auch schwach bindende Liganden, wie sie bei dem FBS zu erwarten sind, sehr gut detektiert werden können. Um ein ausreichendes Signal/Rausch-Verhältnis in einem Spektrum zu erhalten, müssen Faktoren wie die eingesetzte Proteinkonzentration und die Anzahl benötigter Scans aufeinander abgestimmt werden. Aufgrund der großen Anzahl von durchzuführenden Messungen und der damit verbundenen großen Menge an benötigtem Protein ist es zeit- und kostensparend, möglichst geringe Proteinmengen pro NMR-Messung zu verwenden. Die einzusetzende Proteinmenge ist jedoch aufgrund des zur Verfügung stehende Equipments dahingehend beschränkt, dass bei geringeren Proteinmengen deutlich längere Messungen notwendig werden, um ein gleichbleibend gutes Signal/Rausch-Verhältnis zu erhalten. Bei den hier durchgeführten Messungen wird eine Proteinkonzentration von 0,2 mM verwendet. Um ein optimales Signal/Rausch-Verhältnis mit dem zur Verfügung stehenden technischen Equipment zu erhalten, sind 128 Scans in F1 und 64 Inkremente in F2 bei einer Auflösung von 2048 digitalen Punkten in der Akquisition notwendig. Um bei den gewählten Versuchsbedingungen ein ausreichendes Signal/Rausch-Verhältnis bei verringerter Aufnahmedauer zu erhalten, wird das 1H-15N-SOFAST-HMQC-Experiment genutzt, da sich hier aufgrund von kürzeren Relaxations-Delays zwischen den einzelnen FID-Aufnahmen eine verkürzte Messzeit ergibt. Das bei dieser Messmethode zu Grunde liegende 1H-15N-SOFAST-HMQC-Experiment zeigt im Vergleich zu dem sonst eingesetzten 1H-15N-HSQC-Experiment eine deutlich schlechtere Auflösung (Abbildung 6.1). Besonders im Bereich der Seitenkettenamide ist solch eine verschlechterte Auflösung der Signale zu erkennen. Der prinzipielle Unterschied der HMQC- zu HSQC-Spektren, dass sich während der Evolutionszeit das Proton sowie der Heterokern (hier: 15N) entwickeln können. Bei HSQC-Spektren gibt es während der Evolutionszeit keine Protonmagnetisierung, wodurch eine bessere Auflösung der Heterokerndimension resultiert. Durch die spezielle Pulsfolge des SOFASTExperiments ist es jedoch nur möglich, eine Aufnahme auf Basis eines HMQC-Experiments durchzuführen. Das Signal/Rausch-Verhältnis ist bei dem 1H-15N-SOFAST-HMQC-Spektrum jedoch bei 85 6 Ergebnisse - Teil II gleicher Aufnahmedauer bei den gewählten experimentellen Bedingungen deutlich besser als beim 1 H-15N-HSQC-Spektrum (1H-15N-HSQC: 2,5 h; 1H-15N-SOFAST-HMQC: 35 min). Um zwischen bindenden und nicht-bindenden Liganden zu unterscheiden, liefert dieses Experiment Spektren mit ausreichend guter Auflösung. 1 15 1 15 Abbildung 6.1: Überlagerung eines H- N-HSQC-Spektrums (grün) mit einem H- N-SOFAST-HMQC-Spektrum (schwarz) von RhebͻGDP. 1 15 Das H- N-SOFAST-HMQC-Spektrum zeigt bei gleichen Aufnahmebedingungen ein deutlich besseres Signal/Rausch1 15 Verhältnis als das H- N-HSQC-Spektrum. In einigen Bereichen (besonders in Höhe der Seitenkettensignale) kommt es beim 1 15 H- N-SOFAST-HMQC-Spektrum zu starken Überlagerungen aufgrund der größeren Linienbreite. 6.2 SCREENING DER SUBSTANZBIBLIOTHEK Viele GTPasen übernehmen eine zentrale Rolle im zellulären Signalnetzwerk und sind somit ein Ausgangspunkt der Grundlagen- sowie der pharmazeutischen Forschung. Sind natürliche oder sogar synthetische Bindungspartner bekannt, lassen sich oftmals aus diesen Strukturen oder Strukturelementen andere potentiell pharmazeutisch relevante Bindungspartner designen. Fragment-basierte Screeningmethoden eignen sich besonders für das Design einer Leitstruktur bei Makromolekülen, deren dreidimensionale Struktur bekannt ist und ein Zugang zu sehr sensitiven 86 6 Ergebnisse - Teil II Detektionsmethoden besteht, da die Affinität kleiner Moleküle zum Target oftmals nur sehr gering ist. Mit der FBS konnten erste Bindungspartner sogar für „undruggable“ Proteine wie B-cell lymphoma 2 (BCl-2)212 beschrieben werden. Für die GTPase Rheb sind bisher nur wenige Bindungspartner bekannt, obwohl sie im gesamten Organismus eine zentrale Rolle, wie beispielsweise bei der Regulation des TOR-Komplexes, einnimmt. Da natürliche Bindungspartner unbekannt sind, wird in dieser Arbeit eine neue Substanzbibliothek generiert, welche als Grundlage zur Ermittlung von Hot Spots auf der Proteinoberfläche und zum Erstellen einer Leitstruktur für ein späteres Medikamentendesign dienen soll. 6.2.1 AUFBAU EINER SUBSTANZBIBLIOTHEK Ein bekanntes Prinzip, um die Wahrscheinlichkeit der oralen Absorption von Medikamenten zu beschreiben, ist die sogenannte Rule of Five213. Diese Regel besagt, dass mehr als fünf Wasserstoffbrücken-Donoren vorhanden sein sollen, die Molekülmasse unter 500 g/mol liegen soll, der cLogP-Wert (zur Bestimmung der Lipophilie) größer als fünf sein soll und die Summe der Stickstoff- und Sauerstoff-Atome im Molekül größer als zehn sein soll. Diese Regel kann zur Einschätzung beitragen, ob ein potentielles Medikament alle Testphasen bis zur Markteinführung positiv bestehen kann oder nicht. Zur Bewertung einer Leitstruktur ist dieses Prinzip ungeeignet, da Leitstrukturen meistens sehr klein sind und somit z. B. nur über eine geringe Anzahl an Wasserstoffbrücken-Donoren verfügen. Stattdessen wird die Rule of Three herangezogen. Neben dem Screening von kleinen Fragmenten ist zum Beschreiben einer Leitstruktur ebenfalls ein HochDurchsatz-Screening von größeren Komponenten notwendig (Abbildung 6.2). Aufgrund des bestehenden Zugangs zur NMR-Spektroskopie als hochsensitive Testmethode zur Bewertung der Affinität der Bindung sowie der zeitlichen und finanziellen Limitierung des Projekts erfolgt in dieser Arbeit die Bestimmung einer Leitstruktur durch Fragment-basiertes Screening. Die Fragment-basierte Screeningmethode bietet den Vorteil, dass zunächst Messungen mit kommerziell zu erwerbenden Substanzen durchgeführt werden können und somit zunächst keine Synthesearbeiten anfallen. Bei der Auswahl der Substanzen muss neben einem geringen Molekulargewicht (etwa 200 g/mol) auf eine ausreichend hohe Wasserlöslichkeit bei neutralem pH-Wert geachtet werden. Neben dem geringen Molekulargewicht wird die Höchstanzahl von drei Wasserstoffbrücken-Donoren aus der Rule of Three (Molekülmasse < 300 g/mol, cLogP < 3), was als Mindestanforderung für viele Leitstrukturen gilt214, übernommen. Darüber hinaus werden Strukturen aus früheren Arbeiten171,172,215 analysiert und daraus zu testende Komponenten abgeleitet. 87 rel. molecular mass 6 Ergebnisse - Teil II Drug candidate 500 HTS Hit Drug 300 Fragment 100 1 mM 100 ʅM 1 ʅM 10 nM 1 nM Potency/efficacy Abbildung 6.2: Schematische Darstellung der durchschnittlichen Molekülmasse und der Effektivität der Bindung für unterschiedliche Ansätze der Medikamentenentwicklung 216 . Es werden acht ausgewählte Gemische mit jeweils sechs bis elf Substanzen getestet. Jede Substanz wird in einem zehnfach molekularen Überschuss zum 15 N-isotopenangereicherten Protein hinzugegeben und die Veränderungen im 1H-15N-HSQC-Spektrum ausgewertet. Eine Bindung einer oder mehrerer Komponenten kann anhand einer Änderung der Resonanzfrequenz der an der Bindung beteiligten Aminosäuresignale im 1H-15N-HSQC-Spektrum beobachtet werden. Treten solche Veränderungen bei der Zugabe eines Gemisches auf, werden alle Komponenten der Gemische einzeln analysiert, um die jeweiligen Hits eines Gemisches zu identifizieren. Dies erfolgt mittels 1H15 N-SOFAST-HMQC-Experimenten, welche eine geringere Messdauer bei ausreichender Auflösung garantieren. Die eingesetzten Gemische werden nach Gesichtspunkten wie der Art und Anzahl der enthaltenen Heterozyklen und der funktionalen Gruppen unterteilt. 6.3 VERWENDETE SUBSTANZGEMISCHE Aus Gründen der Übersichtlichkeit werden die Substanzgemische in getrennten Unterkapiteln beschrieben. Für jede Messung wird ein zehnfach molarer Überschuss jeder Substanz zur 15 N-isotopenangereicherten 15 N-isotopenangereicherte Proteinprobe mit einem entsprechenden Anteil d6-DMSO verwendet und Proteinprobe gegeben. Als Referenzwert wird eine NMR-spektroskopisch vermessen, da alle Substanzgemische und Einzelsubstanzen in diesem Lösungsmittelgemisch vorliegen. Jede Messung wird bei 298 K und pH 7,8 durchgeführt. 88 6 Ergebnisse - Teil II Zur Bewertung der erzeugten Spektren werden charakteristische Aminosäuresignale bezüglich einer Veränderung ihrer Resonanzfrequenz untersucht. Treten hierbei signifikante Veränderungen auf, wird diese Substanz als Hit eingeordnet. Da für alle eingesetzten Gemische das Vorgehen identisch ist, werden nur für Gemisch 1 alle Arbeitsschritte detailliert aufgeführt. Für alle weiteren Gemische werden nur die Diagramme zur Visualisierung der Veränderungen im 1H-15N-HSQC-Spektrum nach Zugabe des jeweiligen Gemisches dargestellt. Dabei sind nur diejenigen Aminosäuren aufgeführt, die eine Veränderung ihrer Resonanzfrequenz aufweisen. Das Ergebnis der Vermessung der Einzelverbindungen wird nicht in dieser ausführlichen Form dargestellt, sondern nur die Substanzen benannt, die eine signifikante Änderung der chemischen Verschiebung im 1H-15N-SOFAST-HMQC-Spektrum hervorgerufen haben. Die Substanzen, die als erfolgversprechendste Liganden zur Bildung einer Leitstruktur angesehen werden, werden im nächsten Abschnitt diskutiert. GEMISCH 1 6.3.1 In Gemisch 1 wird eine Reihe von aromatischen Carbonsäureverbindungen getestet. Dabei werden Verbindungen bis zu einem Molekulargewicht von etwa 200 g/mol eingesetzt. In Tabelle 6.1 sind die ausgewählten Substanzen mit Strukturformel, der gängigen Benennung und CAS-Nummer aufgeführt. Tabelle 6.1: Verwendete Substanzen für Gemisch 1. Zur besseren Übersichtlichkeit sowie zur eindeutigen Identifikation der jeweiligen Verbindung sind für jede Substanz die Strukturformel, die Benennung und die CAS-Nummer angegeben. Die Verbindung, welche nach Zugabe zu RhebͻGDP zu 1 15 den größten shifts im H- N-SOFAST-HMQC-Spektrum führt, ist grau unterlegt. Strukturformel Name CAS 2-Naphthoxyessigsäure 120-23-0 L-Pipecolinsäure 3105-95-1 O O N H OH OH O O HO N Benzimidazol-5-carbonsäure 15788-16-6 N H 89 6 Ergebnisse - Teil II O OH 3-(3-Pyridyl)propionsäure 3724-19-4 Indol-2-carbonsäure 1477-50-5 4-Phenoxybenzoesäure 2215-77-2 Nicotinsäure 59-67-6 N O N H OH O OH O O OH N O 3-(4-Hydroxyphenyl) 501-97-3 OH propionsäure HO O Indole-6-carbonsäure N H 1670-82-2 OH Um zu prüfen, ob eine im Gemisch enthaltene Substanz die GDP-Form von Rheb bindet, wird für jedes Gemisch jeweils ein 1 H-15N-HSQC-Spektren aufgenommen. Durch Vergleich mit dem Referenzspektrum (Zugabe des äquivalenten Volumenanteils an d6-DMSO zu RhebͻGDP) können Veränderungen der Resonanzfrequenzen einzelner Aminosäuresignale bestimmt werden. In Abbildung 6.3 ist eine solche Überlagerung gezeigt, wobei das Referenzspektrum in schwarz und das 1 H-15N-HSQC-Spektrum nach Zugabe von Gemisch 1 (zehnfachfach molarer Überschuss jeder Substanz) in blau gezeigt ist. In einigen Regionen des 1H-15N-HSQC-Spektrums ist eine Veränderung der Resonanzfrequenz der Aminosäuresignale sichtbar. Eine Veränderung der Aminosäurefrequenz von Aminosäure S75 ist in der Vergrößerung (Abbildung 6.4) gezeigt. Dabei ist der auftretende shift durch einen Pfeil gekennzeichnet. Zur besseren Visualisierung wird die gewichtete chemische Verschiebung jeder Aminosäure in Abbildung 6.5 dargestellt. 90 6 Ergebnisse - Teil II 1 15 Abbildung 6.3: Überlagerung der H- N-HSQC-Spektren der Referenzprobe (Rheb mit d6-DMSO, schwarz) und nach Zugabe von Gemisch 1 (zehnfach molarer Überschuss von jeder Substanz) zu einer 15 N-isotopenangereicherten Rheb- Proteinprobe (blau). Nach Zugabe von Gemisch 1 treten einige Verschiebungen bei den Resonanzfrequenzen einzelner Aminosäuresignale auf (rote Umrandung). Der größte Teil der detektierten Resonanzfrequenzen zeigt nach Zugabe von Gemisch 1 (zehnfacher molarer Überschuss) keine Veränderung im 1 H-15N-HSQC- Spektrum. Verschiebungen treten jedoch im Bereich der Aminosäuren 13-14, 40-41, 69-78 und 114 auf. Durch Einfärben der Aminosäuren, bei denen eine signifikante Verschiebung der Resonanzfrequenz im 1 H-15N-HSQC-Spektrum sichtbar wird, wird auf der Proteinoberfläche ein Cluster sichtbar. Abbildung 6.4: Vergrößerung aus Abbildung 6.3. Zur besseren Orientierung ist das gebundene GDP ebenfalls aufgetragen (Abbildung 6.6). 91 6 Ergebnisse - Teil II Die shiftenden Aminosäuren liegen nahezu auf der gegenüberliegenden Seite der Nukleotidbindetasche. gew. chem. Verschiebung [ppm] 0,25 0,20 0,15 0,10 0,05 8 13 14 16 35 36 38 39 40 41 43 52 55 57 61 62 63 65 66 69 70 74 75 76 78 79 85 103 106 107 109 114 118 136 140 161 0,00 Aminosäure Abbildung 6.5: Auftragung der gewichteten chemischen Verschiebung [ppm] gegen die Aminosäuresequenz. Nach Zugabe von Gemisch 1 (zehnfacher molarer Überschuss jeder Verbindung) kann durch Vergleich der 1 15 Resonanzfrequenzen der Aminosäuresignale im H- N-HSQC-Spektrum mit der Referenzmessung ein Vektor für jede signifikante Veränderung bestimmt werden. Der Bereich von Aminosäure 69-78 umfasst die switch II Region der kleinen GTPase Rheb. Neben diesen Aminosäuren aus der switch II Region zeigen auch benachbarte Aminosäuren Veränderungen in der Resonanzfrequenz. Da in Gemisch 1 eine oder mehrere Substanzen an RhebͻGDP binden, werden alle Verbindungen einzeln vermessen, um die Hits zu identifizieren. Zur Reduzierung der Messdauer werden anstatt der zuvor durchgeführten 1H-15N-HSQC-Experimente nun 1H-15N-SOFAST-HMQC-Experimente genutzt. Des Weiteren werden zur Beschleunigung des Verfahrens nur noch Resonanzfrequenzen ausgewählter Aminosäuresignale auf ihre Veränderung nach Zugabe des Liganden überprüft, in diesem Fall die Signale der Aminosäuren I69, F70 und S75. Die Substanz 4-Phenoxybenzoesäure aus Gemisch 1 führt bei den ausgewählten Aminosäuren zu starken Verschiebungen der Resonanzfrequenzen im 1H-15N-SOFAST-HMSQC-Spektrum. 92 6 Ergebnisse - Teil II 180 ° Abbildung 6.6: Auftragung der Aminosäuren (blau) , welche eine Veränderung der Resonanzfrequenz im 1H-15N-HSQCSpektrum nach Zugabe von Gemisch 1 zeigen. Die shiftenden Aminosäuren sind auf der Proteinoberfläche blau eingefärbt. Alle Aminosäuren liegen in einem zusammenhängenden Cluster auf der Proteinoberfläche. Auf der Rückseite des Proteins sind keine shifts sichtbar. Zur Orientierung ist das gebundene GDP eingezeichnet. 93 6 Ergebnisse - Teil II 6.3.2 GEMISCH 2 Auch in Gemisch 2 wird eine Reihe von aromatischen Säureverbindungen getestet. Dabei werden vorwiegend stickstoffhaltige, zyklische Verbindungen bis zu einem Molekulargewicht von etwa 200 g/mol untersucht. In Tabelle 6.2 sind die ausgewählten Substanzen mit Strukturformel, der gängigen Benennung und CAS-Nummer aufgeführt. Tabelle 6.2: Verwendete Substanzen für Gemisch 2. Zur besseren Übersichtlichkeit sowie zur eindeutigen Identifikation der jeweiligen Verbindung sind für jede Substanz die Strukturformel, die Benennung und die CAS-Nummer angegeben. Die Verbindung, welche nach Zugabe zu RhebͻGDP zu den größten shifts im 1H-15N-SOFAST-HMQC-Spektrum führt, ist grau unterlegt. Strukturformel CH3 N Name CAS 2-Acetylpyridin 1122-62-9 2-Phenylbenzimidazol 716-79-0 2-(2-Pyridyl)benzimidazol 1137-68-4 4-Phenylpyridin 939-23-1 N-Hydroxybenzamid 495-18-1 Cumarin 91-64-5 Uracil 66-22-8 O N N H N N N H N O NH OH O O O NH N H O 94 6 Ergebnisse - Teil II N N Purin N H N 120-73-0 N Thieno[3,2-b]pyridin-7-ol 107818-20-2 S OH S NH N,N'-Diphenylthioharnstoff 102-08-9 NH Um zu prüfen, ob eine im Gemisch enthaltene Substanz die GDP-gebundene Form von Rheb bindet, wird ein 1H-15N-HSQC-Spektren aufgenommen. Durch Überlagerung mit dem Referenzspektrum (Zugabe des äquivalenten Volumenanteils an d6-DMSO) können Veränderungen der Resonanzfrequenzen einzelner Aminosäuresignale bestimmt werden. gew. chem. Verschiebung [ppm] 0,25 0,20 0,15 0,10 0,05 0,00 11 12 13 21 61 62 69 70 75 76 77 78 79 82 103 106 111 Aminosäure Abbildung 6.7: Auftragung der gewichteten chemischen Verschiebung [ppm] gegen die Aminosäuresequenz. Nach Zugabe von Gemisch 2 (zehnfacher molarer Überschuss jeder Verbindung) kann durch Vergleich der Resonanzfrequenzen der Aminosäuresignale im 1H-15N-HSQC-Spektrum mit der Referenzmessung ein Vektor für jede signifikante Veränderung bestimmt werden. 95 6 Ergebnisse - Teil II Da es bei Zugabe von Gemisch 2 zur 15 N-isotopenangereicherten Rheb-Proteinprobe zu Veränderungen einzelner Resonanzsignale im 1 H-15N-HSQC-Spektrum kommt (Abbildung 6.7), werden alle Verbindungen separat charakterisiert. Bei Zugabe von 2-(2-Pyridyl)benzimidazol und 4-Phenylpyridin gibt es signifikante Veränderungen der Resonanzfrequenzen ausgewählter Aminosäuresignale (69, F70, F75) im 1H-15N-SOFAST-HMQCSpektrum. 2-(2-Pyridyl)benzimidazol zeigt im Gegensatz zu 4-Phenylpyridin jedoch nur sehr geringe Veränderungen. Somit stellt 4-Phenylpyridin den besten Hit aus Gemisch 2 dar. 96 6 Ergebnisse - Teil II 6.3.3 GEMISCH 3 In Gemisch 3 wird eine Reihe von Verbindungen getestet, die strukturell an Piperazin sowie an die Purin- und Pyrimidinbasen angelehnt sind. Dabei werden stickstoffhaltige, zyklische Verbindungen bis zu einem Molekulargewicht von etwa 200 g/mol eingesetzt. In Tabelle 6.3 sind die ausgewählten Substanzen mit Strukturformel, der gängigen Benennung und CAS-Nummer aufgeführt. Tabelle 6.3: Verwendete Substanzen für Gemisch 3 Zur besseren Übersichtlichkeit sowie zur eindeutigen Identifikation der jeweiligen Verbindung sind für jede Substanz die Strukturformel, die Benennung und die CAS-Nummer angegeben. Die Verbindung, welche nach Zugabe zu RhebͻGDP zu den größten shifts im 1H-15N-SOFAST-HMQC-Spektrum führt, ist grau unterlegt. Struktur Name CAS 4-Amino-1-benzylpiperidin 50541-93-0 4-(1-Pyrrolidino)pyridin 2456-81-7 1-(2-Pyrimidinyl)piperazin 20980-22-7 Indazol 271-44-3 Melamin 108-78-1 1-(3,4-Dichlorophenyl)piperazin 57260-67-0 NH2 N N N NH N N N N N H NH2 N N H2N N NH2 H N N Cl Cl 97 6 Ergebnisse - Teil II O H2N NH N H 5-Aminouracil 932-52-5 Benzamidin 618-39-3 Adenin 73-24-5 2-Aminobenzimidazol 934-32-7 O NH NH2 NH2 N N N H N N NH2 N H Um zu prüfen, ob eine im Gemisch enthaltene Substanz die GDP-gebundene Form von Rheb bindet, wird ein 1H-15N-HSQC-Spektren aufgenommen. Durch Überlagerung mit dem Referenzspektrum (Zugabe des äquivalenten Volumenanteils an d6-DMSO) können Veränderungen der Resonanzfrequenzen einzelner Aminosäuresignale bestimmt werden (Abbildung 6.8). gew. chem. Verschiebung [ppm] 0,25 0,20 0,15 0,10 0,05 0,00 6 11 13 41 60 61 62 67 69 70 75 76 78 79 80 103 107 110 Aminosäure Abbildung 6.8: Auftragung der gewichteten chemischen Verschiebung [ppm] gegen die Aminosäuresequenz. Nach Zugabe von Gemisch 3 (zehnfacher molarer Überschuss jeder Verbindung) kann durch Vergleich der Resonanzfrequenzen der Aminosäuresignale im 1H-15N-HSQC-Spektrum mit der Referenzmessung ein Vektor für jede signifikante Veränderung bestimmt werden. 98 6 Ergebnisse - Teil II Da es bei Zugabe von Gemisch 3 zur 15 N-isotopenangereicherten Rheb-Proteinprobe zu Veränderungen einzelner Resonanzsignale im 1 H-15N-HSQC-Spektrum kommt (Abbildung 6.8), werden alle Verbindungen separat charakterisiert. Nur bei Zugabe eines zehnfachen molaren Überschusses von 1-(3,4-Dichlorophenyl)piperazin aus Gemisch 3 zur 15 N-istotopenangereicherten RhebͻGDP-Probe können Veränderungen einzelner Aminosäuresignalresonanzen im 1H-15N-SOFAST-HMQC-Spektrum beobachtet werden. 99 6 Ergebnisse - Teil II 6.3.4 GEMISCH 4 In Gemisch 4 wird eine Reihe von aromatischen Carbonsäureverbindungen getestet. Die Verbindungen verfügen über ein oder zwei zum Teil verbrückte aromatische Ringsysteme. Das Molekulargewicht beträgt höchstens 200 g/mol. In Tabelle 6.4 sind die ausgewählten Substanzen mit Strukturformel, der gängigen Benennung und CAS-Nummer aufgeführt. Tabelle 6.4: Verwendete Substanzen für Gemisch 4. Zur besseren Übersichtlichkeit sowie zur eindeutigen Identifikation der jeweiligen Verbindung sind für jede Substanz die Strukturformel, die Benennung und die CAS-Nummer angegeben. Die Verbindung, welche nach Zugabe zu RhebͻGDP zu den größten shifts im 1H-15N-SOFAST-HMQC-Spektrum führt, ist grau unterlegt. Struktur Name CAS 3-Benzoylpropionsäure 2051-95-8 5-Phenyl-2-furansäure 52938-97-3 2-Naphthoesäure 93-09-4 rac-2,3-Diphenylpropionsäure 3333-15-1 Diphenylessigsäure 117-34-0 Benzo[b]furan-2-carbonsäure 496-41-3 O O OH O O OH O OH O CH3 O CH3 O O OH 100 6 Ergebnisse - Teil II O N Chinolin-2-carbonsäure 93-10-7 1H-Imidazol-2-carbonsäure 16042-25-4 4-Biphenylcarbonsäure 92-92-2 CH3 O N OH N H O OH Um zu prüfen, ob eine im Gemisch enthaltene Substanz die GDP-gebundene Form von Rheb bindet, wird ein 1H-15N-HSQC-Spektren aufgenommen. Durch Überlagerung mit dem Referenzspektrum (Zugabe des äquivalenten Volumenanteils an d6-DMSO) können Veränderungen der Resonanzfrequenzen einzelner Aminosäuresignale bestimmt werden (Abbildung 6.9). gew. chem. Verschiebung [ppm] 0,25 0,20 0,15 0,10 0,05 0,00 11 16 35 38 39 40 41 43 48 49 61 62 63 66 69 70 74 75 76 77 78 103106107114 Aminosäure Abbildung 6.9: Auftragung der gewichteten chemischen Verschiebung [ppm] gegen die Aminosäuresequenz. Nach Zugabe von Gemisch 4 (zehnfacher molarer Überschuss jeder Verbindung) kann durch Vergleich der 1 15 Resonanzfrequenzen der Aminosäuresignale im H- N-HSQC-Spektrum mit der Referenzmessung ein Vektor für jede signifikante Veränderung bestimmt werden. 101 6 Ergebnisse - Teil II Da es bei Zugabe von Gemisch 4 zur 15 N-isotopenangereicherten Rheb-Proteinprobe zu Veränderungen einzelner Resonanzsignale im 1 H-15N-HSQC-Spektrum kommt, werden alle Verbindungen separat charakterisiert. Nur bei Zugabe eines zehnfachen molaren Überschusses von 4-Biphenylcarbonsäure zur 15 N-istotopenangereicherten RhebͻGDP-Probe können Veränderungen einzelner Aminosäuresignalresonanzen im 1H-15N-SOFAST-HMQC-Spektrum beobachtet werden 102 6 Ergebnisse - Teil II 6.3.5 GEMISCH 5 In Gemisch 5 werden weitere aromatische Carbonsäureverbindungen getestet. Die Verbindungen verfügen über eine oder zwei aromatische Ringsysteme und Variationen der Säurefunktion. Das Molekulargewicht beträgt höchstens 200 g/mol. In Tabelle 6.5 sind die ausgewählten Substanzen mit Strukturformel, der gängigen Benennung und CAS-Nummer aufgeführt. Tabelle 6.5: Verwendete Substanzen für Gemisch 5. Zur besseren Übersichtlichkeit sowie zur eindeutigen Identifikation der jeweiligen Verbindung sind für jede Substanz die Strukturformel, die Benennung und die CAS-Nummer angegeben. Die Verbindung, welche nach Zugabe zu RhebͻGDP zu den größten shifts im 1H-15N-SOFAST-HMQC-Spektrum führt, ist grau unterlegt. Struktur Name CAS trans-Zimtsäure 140-10-3 Cumarin-3-carbonsäure 531-81-7 3-(1H-Pyrrol-1-yl)benzoesäure 61471-45-2 4-Biphenylessigsäure 5728-52-9 O OH O OH O O O N OH O OH O O OH 4-Benzyloxyphenylessigsäure 6547-53-1 Um zu prüfen, ob eine im Gemisch enthaltene Substanz die GDP-gebundene Form von Rheb bindet, werden 1H-15N-HSQC-Spektren aufgenommen. Durch Überlagerung mit dem Referenzspektrum (Zugabe des äquivalenten Volumenanteils an d6-DMSO) können Veränderungen der Resonanzfrequenzen einzelner Aminosäuresignale bestimmt werden (Abbildung 6.10). 103 6 Ergebnisse - Teil II gew. chem. Verschiebung [ppm] 0,25 0,20 0,15 0,10 0,05 0,00 9 16 38 40 41 42 43 62 67 69 70 74 75 77 79 103 107 114 138 Aminosäure Abbildung 6.10: Auftragung der gewichteten chemischen Verschiebung [ppm] gegen die Aminosäuresequenz. Nach Zugabe von Gemisch 5 (zehnfacher molarer Überschuss jeder Verbindung) kann durch Vergleich der 1 15 Resonanzfrequenzen der Aminosäuresignale im H- N-HSQC-Spektrum mit der Referenzmessung ein Vektor für jede signifikante Veränderung bestimmt werden. Da es bei Zugabe von Gemisch 5 zur 15 N-isotopenangereicherten Rheb-Proteinprobe zu Veränderungen einzelner Resonanzsignale im 1 H-15N-HSQC-Spektrum kommt, werden alle Einzelverbindungen separat charakterisiert. Nur bei Zugabe eines zehnfachen molaren Überschusses von 4-Benzyloxyphenylessigsäure können Veränderungen einzelner Aminosäuresignalresonanzen im 1 H-15N-SOFAST-HMQC-Spektrum beobachtet werden. 104 6 Ergebnisse - Teil II 6.3.6 In GEMISCH 6 Gemisch 6 wird eine Reihe von aromatischen Ketonen sowie heterozyklische Stickstoffverbindungen eingesetzt, die sich von Pyrrol oder Imidazol ableiten. In Tabelle 6.6 sind die ausgewählten Substanzen mit Strukturformel, der gängigen Benennung und CAS-Nummer aufgeführt. Tabelle 6.6: Verwendete Substanzen für Gemisch 6. Zur besseren Übersichtlichkeit sowie zur eindeutigen Identifikation der jeweiligen Verbindung sind für jede Substanz die Strukturformel, die Benennung und die CAS-Nummer angegeben. Die Verbindung, welche nach Zugabe zu RhebͻGDP zu den größten shifts im 1H-15N-SOFAST-HMQC-Spektrum führt, ist grau unterlegt. Struktur Name CAS 2-Acetylbenzofuran 1646-26-0 2-Hydroxybenzimidazol 615-16-7 4,4´-Biphenol 92-88-6 Benzamid 55-21-0 1,3-Diphenylharnstoff 102-07-8 4-Benzyloxybenzylalkohol 836-43-1 4-Benzoylpyridin 14548-46-0 O O CH3 H N CH2 N H OH HO O NH2 O NH NH OH O O N 105 6 Ergebnisse - Teil II O 6-Hydroxy-1-tetralon 3470-50-6 Carbazol 86-74-8 Benzophenon 119-61-9 HO N H O Um zu prüfen, ob eine im Gemisch enthaltene Substanz die GDP-gebundene Form von Rheb bindet, wird ein 1H-15N-HSQC-Spektren aufgenommen. Durch Überlagerung mit dem Referenzspektrum (Zugabe des äquivalenten Volumenanteils an d6-DMSO) können Veränderungen der Resonanzfrequenzen einzelner Aminosäuresignale bestimmt werden (Abbildung 6.11). gew. chem. Verschiebung [ppm] 0,35 0,30 0,25 0,20 0,15 0,10 0,05 0,00 9 12 13 14 40 64 66 69 70 75 77 78 79 80 99 103 106 108 109 114 Aminosäure Abbildung 6.11: Auftragung der gewichteten chemischen Verschiebung [ppm] gegen die Aminosäuresequenz. Nach Zugabe von Gemisch 6 (zehnfacher molarer Überschuss jeder Verbindung) kann durch Vergleich der 1 15 Resonanzfrequenzen der Aminosäuresignale im H- N-HSQC-Spektrum mit der Referenzmessung ein Vektor für jede signifikante Veränderung bestimmt werden. Da es bei Zugabe von Gemisch 6 zur 15 N-isotopenangereicherten Rheb-Proteinprobe zu Veränderungen einzelner Resonanzsignale im 1 H-15N-HSQC-Spektrum kommt, werden alle Einzelverbindungen separat charakterisiert. 106 6 Ergebnisse - Teil II Nur bei Zugabe eines zehnfachen molaren Überschusses von 4,4´-Biphenol können sehr starke Veränderungen einzelner Aminosäuresignalresonanzen im 1 H-15N-SOFAST-HMQC-Spektrum beobachtet werden. 107 6 Ergebnisse - Teil II 6.3.7 GEMISCH 7 In Gemisch 7 wird eine Reihe von heterozyklischen Stickstoffverbindungen eingesetzt. Dabei leiten sich die Verbindungen u. a. von Pyridin ab. In Tabelle 6.7 sind die ausgewählten Substanzen mit Strukturformel, der gängigen Benennung und CAS-Nummer aufgeführt. Tabelle 6.7: Verwendete Substanzen für Gemisch 7. Zur besseren Übersichtlichkeit sowie zur eindeutigen Identifikation der jeweiligen Verbindung sind für jede Substanz die Strukturformel, die Benennung und die CAS-Nummer angegeben. Die Verbindung, welche nach Zugabe zu RhebͻGDP zu den größten shifts im 1H-15N-SOFAST-HMQC-Spektrum führt, ist grau unterlegt. Struktur N N Name CAS 1-(2-Pyridyl)piperazin 34803-66-2 N-Phenyl-o-phenylendiamin 534-85-0 3-Acetylindol 703-80-0 4-Phenylbenzylamin 712-76-5 2-Amino-3-nitropyridin 4214-75-9 2-Aminopyrimidin 109-12-6 3,4-Diaminobenzophenon 39070-63-8 NH NH2 NH O CH3 N H NH2 O + N N O - NH2 N N NH2 O NH2 NH2 108 6 Ergebnisse - Teil II H2N CH3 5-Amino-2-methylindol 7570-49-2 4-Phenylmorpholin 92-53-5 N H O N Um zu prüfen, ob eine im Gemisch enthaltene Substanz die GDP-gebundene Form von Rheb bindet, werden 1H-15N-HSQC-Spektren aufgenommen. Durch Überlagerung mit dem Referenzspektrum (Zugabe des äquivalenten Volumenanteils an d6-DMSO) können Veränderungen der Resonanzfrequenzen einzelner Aminosäuresignale bestimmt werden (Abbildung 6.12). gew. chem. Verschiebung [ppm] 0,25 0,20 0,15 0,10 0,05 0,00 6 9 13 61 62 68 69 70 74 75 77 78 79 80 103 106 107 114 Aminosäure Abbildung 6.12: Auftragung der gewichteten chemischen Verschiebung [ppm] gegen die Aminosäuresequenz. Nach Zugabe von Gemisch 7 (zehnfacher molarer Überschuss jeder Verbindung) kann durch Vergleich der 1 15 Resonanzfrequenzen der Aminosäuresignale im H- N-HSQC-Spektrum mit der Referenzmessung ein Vektor für jede signifikante Veränderung bestimmt werden. Da es bei Zugabe von Gemisch 7 zur 15 N-isotopenangereicherten Rheb-Proteinprobe zu Veränderungen einzelner Resonanzsignale im 1 H-15N-HSQC-Spektrum kommt, werden alle Einzelverbindungen separat charakterisiert. 109 6 Ergebnisse - Teil II Nur bei Zugabe eines zehnfachen molaren Überschusses von N-Phenyl-o-phenylendiamin können Veränderungen einzelner Aminosäuresignalresonanzen im 1 H-15N-SOFAST-HMQC-Spektrum beobachtet werden. 110 6 Ergebnisse - Teil II 6.4 BIPHENYL- UND PHENOLDERIVATE Bei den dargestellten Messungen zeigen stets die Verbindungen shifts in den 1H-15N-SOFAST-HMQCSpektren, welche strukturelle Ähnlichkeiten zu Biphenyl oder Phenol aufweisen. Aus diesem Grund wird gezielt eine Reihe von Biphenyl- und Phenolderivaten auf ihre Interaktion mit RhebͻGDP hin untersucht. In Tabelle 6.8 sind die ausgewählten Substanzen mit Strukturformel, der gängigen Benennung und CAS-Nummer aufgeführt. Da es schlussfolgernd aus den Vormessungen ist, dass eine Vielzahl der neu ausgewählten Substanzen eine Bindung mit RhebͻGDP eingeht, werden alle Verbindungen von vornherein separat charakterisiert. Daher werden 1H-15N-SOFAST-HMQC-Spektren bei Zugabe eines zehnfach molaren Überschusses der jeweiligen Substanz zu 15N-isotopenangereichertem RhebͻGDP aufgenommen. Tabelle 6.8: Verwendete Biphenyl- und Phenolderivate. Zur besseren Übersichtlichkeit sowie zur eindeutigen Identifikation der jeweiligen Verbindung sind für jede Substanz die Strukturformel, die Benennung und die CAS-Nummer angegeben. Struktur Name CAS 4-Benzyloxyphenol 103-16-2 4,4'-Dihydroxydiphenylether 1965-09-9 Benzidin 92-87-5 4-Phenoxyphenol 831-82-3 4-Phenoxybenzoesäure 2215-77-2 OH O O HO OH NH2 H2N HO O O OH O 111 6 Ergebnisse - Teil II O HO OH 4-(4-Hydroxyphenoxy)benzoesäure 500-76-5 4,4´-Diacetylbiphenyl 787-69-9 4'-Hydroxy-4-biphenylcarbonsäure 58574-03-1 Hydrochinon 123-31-9 4,4'-Dihydroxydiphenylmethan 620-92-8 4,4'-Dihydroxybenzophenon 611-99-4 O O CH3 H3C O O OH HO OH OH HO OH O HO OH Abbildung 6.13 zeigt die Überlagerung einiger 1H-15N-SOFAST-HMQC-Spektren der Charakterisierung der Biphenyl- und Phenolderivate. Das Referenzspektrum (15N-isotopenangereichertes RhebͻGDP) ist schwarz und das 1H-15N-SOFAST-HMQC-Spektrum bei Zugabe von Benzidin blau dargestellt. Beim Vergleich der beiden Spektren können keine signifikanten shifts festgestellt werden. Bei Zugabe von 4,4'-Dihydroxydiphenylmethan (lila) und 4,4'-Dihydroxybenzophenon (grün) hingegen treten Veränderungen der Resonanzfrequenzen einiger Aminosäuren auf. In Abbildung 6.14 ist eine Ausschnittsvergrößerung der Überlagerung der 1H-15N-SOFAST-HMQC-Spektren dargestellt. Die Signale der Aminosäuren I9, D60 und L116 zeigen keinerlei shifts. Bei dem Signal von Aminosäure F70 wird eine geringe Verschiebung der detektierten Resonanzfrequenz bei Zugabe von 4,4'-Dihydroxydiphenylmethan sichtbar. Bei Zugabe von 4,4'-Dihydroxybenzophenon tritt eine deutlich stärkere Verschiebungen auf, was auf eine stärkere Bindung hindeuten kann. 112 6 Ergebnisse - Teil II 1 15 Abbildung 6.13: Überlagerung der H- N-SOFAST-HMCQ-Spektren der Referenzprobe (Rheb mit d6-DMSO, schwarz), nach Zugabe von Benzidin (blau), 4,4'-Dihydroxydiphenylmethan (lila) und 4,4'-Dihydroxybenzophenon (grün) (zehnfach 15 molarer Überschuss von jeder Substanz) zu einer N-isotopenangereicherten Rheb-Proteinprobe. Bei einigen Verbindungen treten nach Zugabe der Liganden Verschiebungen bei einzelnen Aminosäureresonanzfrequenzen auf. Verschiebungen bei Aminosäure F70 treten bei Zugabe von 4,4'-Dihydroxydiphenylmethan und 4,4'Dihydroxybenzophenon auf, wobei der shift bei der letztgenannten Substanz deutlich größer ausfällt. Bei Zugabe von Benzidin sind keine shifts zu beobachten. Die Größe des Verschiebungsvektors muss jedoch nicht ursächlich auf eine stärkere Bindung schließen lassen, da starke Veränderungen der Resonanzfrequenz nicht unbedingt mit der Bindungsstärke korrelieren. Die Affinität der Verbindung zum Protein kann nur durch Bestimmung der Dissoziationskonstanten z. B. durch ein NMR-Titrationsexperiment bestimmt werden. Alle anderen Verbindungen führen zu keinen Veränderungen im Spektrum. 113 6 Ergebnisse - Teil II 1 15 Abbildung 6.14: Ausschnittsvergrößerung der Überlagerung der H- N-SOFAST-HMCQ-Spektren aus Abbildung 6.13. Nach Zugabe von Benzidin (blau) wird kein, bei Zugabe von 4,4'-Dihydroxydiphenylmethan (lila) ein geringer und bei Zugabe von 4,4'-Dihydroxybenzophenon (grün) (je zehnfach molarer Überschuss) ein deutlicher shift bei Aminosäure F70 beobachtet. 114 6 Ergebnisse - Teil II 6.5 BISPHENOL A Die in den Gemischen zuvor identifizierten Hits weisen eine große Ähnlichkeit mit Bisphenol A auf, welches seit Jahrzehnten immer wieder in den Fokus der Öffentlichkeit gerät, da ihm gesundheitsschädliche Eigenschaften zugeschrieben werden. Das Bundesinstitut für Risikobewertung stellt in seinem aktuellen Bericht217 dar, dass Bisphenol A in Deutschland momentan nicht als gesundheitsgefährdend angesehen wird. Eine erneute Prüfung und Bewertung soll aber aufgrund neuster Forschungsergebnisse schon im Jahr 2013 erfolgen. Auf der FAO/WHO-Konferenz im Jahr 2010 konnte eine gesundheitsschädliche Wirkung von Bisphenol A weder eindeutig bestätigt noch ausgeschlossen werden218. Bisphenol A wird aufgrund der aktuellen Berichterstattung in den Medien und seiner strukturellen Ähnlichkeit zu den bisher gefundenen Hits ebenfalls auf seine Interaktion mit RhebͻGDP hin untersucht. Die Bindung von Bisphenol A an RhebͻGDP wird in einem 1H-15N-HSQC-Experiment charakterisiert, indem ein zehnfach molarer Überschuss der Verbindungen zum 15N-isotopenangereichertem Protein hinzu gegeben wird. Die Veränderungen der Resonanzfrequenzen der Aminosäuresignale sind in Abbildung 6.15 dargestellt. gew. chem. Verschiebung [ppm] 0,25 0,20 0,15 0,10 0,05 9 10 13 18 22 40 45 52 60 62 63 66 67 69 70 75 77 78 79 80 91 103 106 107 122 165 0,00 Aminosäure Abbildung 6.15: Auftragung der gewichteten chemischen Verschiebung [ppm] gegen die Aminosäuresequenz. Nach Zugabe von Bisphenol A (zehnfacher molarer Überschuss) kann durch Vergleich der Resonanzfrequenzen der 1 15 Aminosäuresignale im H- N-HSQC-Spektrum mit der Referenzmessung ein Vektor für jede signifikante Veränderung bestimmt werden. 115 6 Ergebnisse - Teil II Bei Zugabe von Bisphenol A zeigen sich sehr starke Veränderungen von Resonanzfrequenzen einiger Aminosäuresignale im 1H-15N-HSQC-Spektrum. 6.6 ANALYSE DER BESTEN HITS Durch Analyse der Gemische und Einzelverbindungen können mehrere Substanzen identifiziert werden, die in einer definierten Region an RhebͻGDP binden. Die Größe des auftretenden shifts wird als erster Anhaltspunkt zur Bewertung der Affinität einer Substanz zu Rheb angesehen. Die untersuchten Verbindungen weisen sehr ähnliche Strukturen auf und binden an die gleiche Region von Rheb, sodass eine ähnliche Art der Bindung angenommen werden kann. Als erfolgversprechendste Hits werden Bisphenol A, 4,4´-Biphenol, 1-(3,4-Dichlorophenyl)piperazin und 4,4'-Dihydroxybenzophenon in weiteren Untersuchungen charakterisiert. Diese ausgewählten Substanzen zeigen alle maximale Shiftvektoren von mindestens 0,25 ppm und einen Cluster im Bereich von Aminosäure 75. 1-(3,4-Dichlorophenyl)piperazin wird aufgrund seiner von den anderen Verbindungen verschiedenen funktionalen Gruppen charakterisiert. 6.6.1 MODELLIERUNG MIT HADDOCK Für alle Hits werden Analysen mit dem Programm HADDOCK183 durchgeführt, in welches für die Strukturberechnung CNS implementiert ist. Mit diesem Programm können biomolekulare Komplexe unter Berücksichtigung von experimentellen Daten modelliert werden. Durch die NMR-Titrationen können Aminosäuren des Proteins identifiziert werden, die an der Interaktion beteiligt sind. Die Aminosäuren, welche signifikante Verschiebungen in den aufgenommenen NMR-Spektren zeigen, fließen als „aktive“ Reste in Form der Ambiguous Interaction Restraint (AIR)-Tabelle in die Berechnung ein. Die Aminosäuren 72 und 73 können im 1H-15N-HSQCSpektrum aufgrund von Überlagerungen oder Linienverbreiterung nicht zugeordnet werden. Die Aminosäure 71 ist ein Prolin und somit in diesen Spektren nicht sichtbar. Da allerdings die benachbarten Aminosäuren 69, 70 sowie 74-78 in vielen Fällen deutliche shifts aufweisen, werden die Aminosäuren 71-73 ebenfalls als „aktive“ Reste definiert. Für die HADDOCK-Rechnung wird neben der AIR-Tabelle auch ein PDB-File des Proteins benötigt. Da die hier gewonnenen Daten ausnahmslos aus NMR-Experimenten stammen, wird die gelöste NMR-Struktur und nicht die Kristallstruktur von RhebͻGDP, welche ebenfalls zur Verfügung steht, verwendet (PDB: 2L0X). Die HADDOCK-Rechnung besteht aus drei Schritten: (i) zufällige Anordnung der Orientierung und rigid body Energieminimierung; (ii) semi-rigides simuliertes Annähern (simulated annealing) im Torsionswinkelraum und (iii) finale Verfeinerung im kartesischen Raum unter Berücksichtigung des Lösungsmittels. Während des simulated annealings und der Verfeinerung in Wasser werden die Seitenketten sowie das Proteinrückgrat freigegeben, um das Interface im Kraftfeld zu optimieren. 116 6 Ergebnisse - Teil II Dabei werden nur die Aminosäuren freigegeben, die in den AIRs ±2sequentielle Aminosäuren angegeben sind. Die finalen Strukturen werden geclustert, indem der RMSD im Proteinrückgrat am Interface verwendet wird, wobei sowohl die Interaktionsenergien (Summe Evan-der-Waals, Eelektrostatisch, EAIR, EDesolvatation) und die Interaktionsfläche an sich in die Berechnung des HADDOCK-Scores einfließen. Für die vier potentiell besten Hits werden Modellierungen mit dem Programm HADDOCK durchgeführt. In den folgenden Kapiteln werden die erhaltenen Ergebnisse für jeden Liganden dargestellt. 6.6.2 4,4´-BIPHENOL Abbildung 6.16 zeigt das Ergebnis der HADDOCK-Rechnung von 4,4´-Biphenol an RhebͻGDP. Der berechnete HADDOCK-Score ist gegen den RMSD [A] der jeweiligen Struktur im Vergleich zur Struktur mit dem geringsten HADDOCK-Score aufgetragen. Zur Analyse und weiteren Bewertung der durchgeführten Modellierungen sollen Cluster gebildet werden. Für die Clusterbildung wird eine maximale Abweichung des RMSDs der Strukturen untereinander sowie eine Mindestanzahl von Strukturen, welche in einem jeden Cluster vorliegen sollen, vorgegeben. Für die Berechnung des HADDOCK-Scores nach der Verfeinerung in Wasser werden die verschiedenen Energieterme unterschiedlich gewichtet: Desolvatationsenergie (1,0), intermolekulare elektrostatische Energie (0,2), intermolekulare van-der-Waals-Energie (1,0) und Verletzung der AIRs (0,1). Zur Berechnung des Haddock-Scores wird diese Gewichtung als optimal angesehen183. Da für die Entwicklung einer Leitstruktur sehr kleine Moleküle eingesetzt werden, sind nur wenige verschiedene Konformationen möglich. Durch eine sehr genaue Beschreibung des Interaktionsinterfaces wird die Anzahl an möglichen Lösungen weiter reduziert, sodass ein einzelner Ergebniscluster entsteht. 117 6 Ergebnisse - Teil II 0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0 -5 Haddock-Score -10 -15 -20 -25 -30 -35 -40 -45 RMSD [A] Abbildung 6.16: Visualisierung der Ergebnisse der Modellierung mit HADDOCK. Der berechnete Haddock-Score ist gegen des RMSD [A] der jeweiligen Struktur im Vergleich zur Struktur mit dem geringsten HADDOCK-Score aufgetragen. Dabei wird ein einziger Ergebniscluster erhalten Um eine Aussage darüber machen zu können, welche Strukturen aus dem Ergebnispool die besten möglichen Strukturen sind, wird eine andere Gewichtung der Ergebnisse vorgenommen. Für die hier durchgeführten Haddock-Modellierungen werden nur wenige Daten benötigt. Neben der Struktur des Proteins und des Liganden werden Aminosäuren des Proteins angegeben, die bei Ligandenbindung eine Veränderung ihrer chemischen Umgebung erfahren. Eine genaue Bestimmung der Struktur des Proteins mit dem jeweiligen Liganden wäre durch Ermittlung von Abstandsinformationen durch NOE-NMR-Experimente und anschließender Strukturrechnung möglich. Hierfür ist die Aufnahme einer Reihe von mehrdimensionalen Spektren, welche auch einer 13 C-Isotopenanreicherung bedürfen, notwendig. Diese Experimente sowie die anschließende Strukturrechnung sind aufgrund der erwarteten sehr geringen Affinität des Liganden zum Protein kosten- und, durch die große Anzahl der benötigten Daten, sehr zeitintensiv. Die Modellierung eines solchen Komplexes mit HADDOCK hat sich für vergleichbare Fragestellungen in der Literatur durchgesetzt. Als Maß für die Güte einer modellierten Struktur wird der HADDOCK-Score herangezogen. 118 6 Ergebnisse - Teil II abgelehnt AIR-Energie abgelehnt akzeptiert abgelehnt intermolekulare van-der-Waals-Energie Abbildung 6.17: Prinzip der Auswahl der besten Strukturen219. Intermolekulare van-der-Waals-Energien und experimentelle AIR-Energien werden für jede berechnete Struktur bestimmt und gegeneinander aufgetragen. Strukturen, die niedrige van-der-Waals- und AIR-Energien aufweisen, werden als bestmögliche Ergebnisse angesehen. Strukturen, die dieses Kriterium nicht erfüllen, werden abgelehnt. Eine weitere Möglichkeit zur Bewertung der Ergebnisse aus HADDOCK bietet sich durch den Vergleich mit der gelösten Röntgenstruktur des Protein-Ligand-Komplexes an, wodurch eine Aussage über die Genauigkeit des Komplexmodells gemacht werden kann. In Studien219 konnte darüber hinaus gezeigt werden, dass bei Modellierungen die Strukturen die größte Genauigkeit zeigten, die die geringsten van-der-Waals- sowie AIR-Energien aufweisen (Abbildung 6.17). Um die hier erhaltenen 200 Strukturen zu bewerten, werden die Werte der AIR- gegen die Werte der van-der-Waals-Energien aufgetragen (für 4,4´-Biphenol in Abbildung 6.18 gezeigt). 119 6 Ergebnisse - Teil II 60 2 50 30 20 1 10 3 AIR-Energie [kcal/mol] 40 0 -35 -30 -25 -20 -15 -10 -5 0 intermolekulare van-der-Waals-Energie [kcal/mol] Abbildung 6.18: Auftragung der AIR- gegen die van-der-Waals-Energien der mit HADDOCK modellierten Komplexstrukturen von RhebͻGDP mit 4,4´-Bisphenol. Durch Auftragung der beiden Energien können die modellierten Strukturen in drei Cluster eingeteilt werden. Durch die Auftragung der beiden Energien gegeneinander können drei Cluster unterschieden werden: ein Cluster mit AIR-Energien ग़ 17 kcal/mol (Cluster 2), ein Cluster mit van-der-WaalsEnergien ग़ -17 kcal/mol (Cluster 3) und alle Strukturen, auf die diese beiden Bedingungen nicht zutreffen (Cluster 1). Die Strukturen mit einer AIR-Energie ख़ 17 kcal/mol und einer van-der-WaalsEnergie ख़ -17 kcal/mol werden als bestmögliche Lösungen angesehen. Die so gefundenen Lösungen beinhalten auch die Struktur, die von HADDOCK als beste Struktur gefunden wurde, sowie den Hauptanteil der Strukturen, die den niedrigsten Haddock-Score zeigen (Abbildung 6.19). Die Strukturen, die anhand der AIR/van-der-Waals-Energie-Analyse als beste Strukturen gefunden wurden, sind grün gekennzeichnet. Die Strukturen mit großen AIR-EnergieWerten sind Blau und die mit großen van-der-Waals-Energien in Rot hervorgehoben. Neben der Bewertung der Strukturen anhand der berechneten Energien muss immer auch eine Bewertung anhand der chemisch-physikalischen Plausibilität der Strukturen erfolgen. 120 6 Ergebnisse - Teil II 0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0 Haddock-Score -5 AIR -10 van-der-Waals -15 AIR/vdW -20 -25 -30 -35 -40 -45 RMSD [A] Abbildung 6.19: Vergleich der mit HADDOCK-modellierten Strukturen von 4,4`-Biphenol anhand der van-der-Waals- und AIR-Energien. Die Strukturen mit einer AIR-Energie ग़ 17 kcal/mol sind blau und die Strukturen mit van-der-Waals-Energie ग़ -17 kcal/mol sind rot hervorgehoben. Strukturen, die diese Kriterien nicht erfüllen und somit als die besten Lösungen angesehen werden, sind grün eingezeichnet (Cluster 1: grün; Cluster 2: blau; Cluster 3: rot). Für die weiteren zuvor bestimmten Hits werden ebenfalls Modellierungen mit dem Programm HADDOCK durchgeführt. Für alle durchgeführten Modellierungen ergibt sich ein ähnliches Bild bei der Auftragung des Haddock-Scores gegen den RMSD wie bei der Analyse von 4,4´-Biphenol. Daher wird bei allen Verbindungen eine Bewertung der Ergebnisse anhand der besten Strukturen aus der Auftragung der AIR-Energie gegen die intermolekulare van-der-Waals-Energie vorgenommen. Abbildung 6.20 zeigt beispielhaft eine Struktur aus jedem erstellten Cluster, welcher durch die Bewertung der van-der-Waals- sowie der AIR-Energie ermittelt wurde. Bei den Strukturen aus den Clustern 1 und 2 liegt das 4,4´-Biphenol in den zuvor beobachteten Interaktionsinterphase zwischen Aminosäure 69-78. Bei den Strukturen, die eine hohe intermolekulare van-der-Waals-Energie aufweisen (Cluster 3), liegt das 4,4´-Biphenol nicht in dieser Tasche (Struktur lila eingefärbt). Der Ligand ist seitlich an der GTPase lokalisiert. 121 6 Ergebnisse - Teil II Abbildung 6.20: Überlagerung einiger HADDOCK-Modellierungen von RhebͻGDP mit 4,4´-Biphenol. Aufgetragen sind Strukturen mit einer großen EAir (hellblau), einer großen EvdW (lila) und einer kleinen EAir sowie kleinen EvdW (grün). Links ist die Oberfläche von Rheb (blau: positive Reste, rot: negative Reste; berechnet über Pymol) mit dem gebundenen Liganden dargestellt. Rechts ist das Protein in der Ribbon-Projektion dargestellt. 6.6.3 1-(3,4-DICHLOROPHENYL)PIPERAZIN Um zu überprüfen, ob eine Bewertung anhand der EAIR und EvdW auch bei den anderen mit HADDOCK angefertigten Modellierungen erfolgen kann, wird dies auch für die Haddock-Modellierung von RhebͻGDP mit 1-(3,4-Dichlorophenyl)piperazin untersucht (Abbildung 6.21). Bei dieser Auftragung werden drei voneinander unterscheidbare Cluster generiert. Cluster 3 beinhaltet Strukturen mit einer AIR-Energie ग़ 62 kcal/mo. Strukturen, welche eine AIR-Energie zwischen 20 und 62 kcal/mol zeigen, bilden Cluster 2. Im Cluster 1 befinden sich Strukturen, welche eine AIR-Energie ख़ 20 kcal/mol aufweisen. Bei der Analyse der Orientierung des 1-(3,4-Dichlorophenyl)piperazin an der Rheb-Oberfläche wird exemplarisch jeweils eine Struktur mit geringen EAIR und geringer EvdW, eine Struktur mit großer EvdW und eine Struktur mit großer EAIR untersucht. In Abbildung 6.22 ist die Orientierung von 1-(3,4Dichlorophenyl)piperazin an RhebͻGDP dargestellt. 122 6 Ergebnisse - Teil II 120 3 80 60 2 40 AIR-Energie [kcal/mol] 100 20 1 0 -30 -25 -20 -15 -10 -5 0 van-der-Waals-Energie [kcal/mol] Abbildung 6.21: Auftragung der AIR- gegen die van-der-Waals-Energien der mit HADDOCK modellierten Komplexstrukturen von RhebͻGDP mit 1-(3,4-Dichlorophenyl)piperazin. Durch diese Auftragung können die modellierten Strukturen in drei Cluster eingeteilt werden: der obere Cluster beinhaltet Strukturen, welche eine AIR-Energie ग़ 62 kcal/mol aufweisen (Cluster 3). Die Strukturen aus dem mittleren Cluster zeigen eine AIR-Energie zwischen 20-62 kcal/mol (Cluster 2). Strukturen, die eine EAIR ख़ 20 kcal/mol aufweisen, bilden Cluster 1. Abbildung 6.22: Überlagerung einiger HADDOCK-Modellierungen von RhebͻGDP mit 1-(3,4-Dichlorophenyl)piperazin. Aufgetragen sind eine Struktur mit einer hohen EvdW (lila), eine Struktur mit hohem EAir (hellblau) und eine Struktur mit geringer EvdW und geringer EAIR (grün) auf der Oberfläche (blau: positive Reste, rot: negative Reste; berechnet über Pymol) von RhebͻGDP. Rechts ist das Protein in der Ribbon-Projektion dargestellt. 123 6 Ergebnisse - Teil II Wie bei 4,4´-Biphenol sind die Liganden der Modellierungen mit hohen EvdW nicht in der Bindungstasche, sondern außen an der GTPase lokalisiert. Bei Komplexstrukturen, die einen niedrigen EAIR aufweisen, bindet der Ligand auch in der zuvor beschriebenen Bindetasche. 6.6.4 4,4'-DIHYDROXYBENZOPHENON Die mittels HADDOCK modellierten Komplexe aus RhebͻGDP und 4,4'-Dihydroxybenzophenon werden mit der zuvor beschriebenen Methode charakterisiert. Aus der Auftragung der AIR-Energie gegen die intermolekulare van-der-Waals-Energie können zwei große Cluster bestimmt werden. Der obere Cluster wird von Strukturen mit einer AIR-Energie ग़ 12 kcal/mol gebildet (Cluster 2). Strukturen mit einer EAIR ч 12 kcal/mol bilden den unteren Cluster (Cluster 1). Dabei zeigen beide Cluster eine große Verteilung bei ihren EvdW. Die Komplexstrukturen weisen bei ähnlich niedriger AIR-Energie intermolekulare van-der-Waals-Energien zwischen -12 kcal/mol und -32 kcal/mol auf. 80 2 70 50 40 30 20 1 AIR-Energie [kacl/mol] 60 10 0 -35 -30 -25 -20 -15 -10 -5 0 intermolekulare van-der-Waals-Energie [kcal/mol] Abbildung 6.23: Auftragung der AIR- gegen die van-der-Waals-Energien der mit HADDOCK modellierten Komplexstrukturen von RhebͻGDP mit 4,4'-Dihydroxybenzophenon. Durch diese Auftragung können die modellierten Strukturen in zwei große Cluster eingeteilt werden: der obere Cluster beinhaltet Strukturen, welche eine AIR-Energie ग़ 12 kcal/mol aufweisen (Cluster 2). Die Strukturen aus dem unteren Cluster zeigen eine AIR-Energie ख़ 12 kcal/mol (Cluster 1). 124 6 Ergebnisse - Teil II Abbildung 6.24: Überlagerung einiger HADDOCK-Modellierungen von RhebͻGDP mit 4,4'-Dihydroxybenzophenon. Aufgetragen sind Strukturen mit einer hohen EAir (hellblau), Strukturen mit einer großen EvdW (lila) und Strukturen mit einer kleinen EAir sowie kleinen EvdW (grün). Links ist die Oberfläche von Rheb (blau: positive Reste, rot: negative Reste; berechnet über Pymol) mit dem gebundenen Liganden dargestellt. Rechts ist das Protein in der Ribbon-Projektion dargestellt. Wie bei den zuvor untersuchten Verbindungen sind die Liganden der Modellierungen mit hohen EvdW nicht in der Bindungstasche, sondern außen an der GTPase lokalisiert. Bei Komplexstrukturen, die einen niedrigen EAIR aufweisen, bindet der Ligand auch in der zuvor beschriebenen Bindetasche. Die Orientierung der in der Bindetasche gebundenen Liganden zeigt eine große Ähnlichkeit zu der Orientierung von 4,4´-Biphenol. 6.6.5 BISPHENOL A Ebenso wird eine Bewertung anhand der EAIR und EvdW der mit HADDOCK durchgeführten Modellierungen von Bisphenol A im Komplex mit RhebͻGDP durchgeführt. Hierbei können drei Cluster voneinander abgegrenzt werden. Strukturen in Cluster 3 zeigen eine EvdW ग़ -23 kcal/mol. Strukturen, welche eine EAIR ग़ 15 kcal/mol aufweisen, werden Cluster 2 zugeordnet. Den besten Cluster 1 bilden Komplexstrukturen, die einen niedrigen Wert für beide hier untersuchte Energien aufweisen. 125 6 Ergebnisse - Teil II 60 2 50 30 20 3 1 10 AIR-Energie [kacl/mol] 40 0 -40 -35 -30 -25 -20 -15 -10 -5 0 intermolekulare van-der-Waals-Energie [kcal/mol] Abbildung 6.25: Auftragung der AIR- gegen die van-der-Waals-Energien der mit HADDOCK modellierten Komplexstrukturen von RhebͻGDP mit Bisphenol A. Durch diese Auftragung können die modellierten Strukturen in drei große Cluster eingeteilt werden: der rechte Cluster beinhaltet Strukturen, welche eine intermolekulare van-der-Waals-Energie ग़ -23 kcal/mol aufweisen (Cluster 3). Die Strukturen aus dem oberen Cluster zeigen AIR-Energien ग़ 15 kcal/mol (Cluster 2). Strukturen, welche niedrigere EAIR und niedrigere EvdW aufweisen, werden Cluster 1 zugeordnet. Abbildung 6.26: Überlagerung einiger HADDOCK-Modellierungen von RhebͻGDP mit Bisphenol A. Aufgetragen sind Strukturen mit einer hohen EAir (hellblau), einer großen EvdW (lila) und Strukturen mit einer kleinen EAir sowie kleinen EvdW (grün). Links ist die Oberfläche von Rheb (blau: positive Reste, rot: negative Reste; berechnet über Pymol) mit dem gebundenen Liganden dargestellt. Rechts ist das Protein in der Ribbon-Projektion dargestellt. 126 6 Ergebnisse - Teil II 6.7 BESTIMMUNG DER DISSOZIATIONSKONSTANTEN Neben der Identifikation eines Hot-Spots auf der Proteinoberfläche ist die Bestimmung einer Leitstruktur für die Medikamentenentwicklung von größter Bedeutung. Zur Optimierung der Leistruktur ist es wichtig, stets zu prüfen, wie hoch die Affinität der gefundenen Verbindung zum Target ist, um nur die erfolgversprechendsten Verbindungen für weitere Analysen auszuwählen. Eine erste Bewertung der Affinität wird in dieser Arbeit durch die Größe des auftretenden shifts nach Zugabe eines zehnfach molaren Überschusses jeder Substanz zu RhebͻGDP im 1H-15N-HSQCSpektrum vorgenommen. Der auftretende shift wird durch Änderungen der chemischen Umgebung eines Atomkerns hervorgerufen, wie es bei der Bindung eines Liganden der Fall ist. Die Größe des shifts wird jedoch auch durch die Geometrie des Liganden selbst bestimmt. So können durch aromatische Systeme bedingte Ringstromeffekte zu größeren Veränderungen in der chemischen Umgebung und somit zu einem großen shift führen, obwohl nur eine verhältnismäßig schwache Bindung vorliegt. Eine geeignete Methode, um Affinitäten zwischen einem Protein und einem Liganden zu bestimmen, ist zum Beispiel die isotherme Titrationskalorimetrie (ITC). Ein Vorteil dieser Methode ist ihre sehr hohe Empfindlichkeit und somit Genauigkeit bei der Affinitätsbestimmung. Bei den in dieser Arbeit verwendeten Substanzen besteht das Problem, ein geeignetes Lösungsmittels zu finden. Die getesteten Verbindungen weisen nur eine geringe Löslichkeit in Wasser auf, sodass alle Verbindungen zunächst in reinem d6-DMSO gelöst werden müssen, um einen zehnfach molaren Überschuss über das Protein zu erreichen. Bei der ITC können schon kleinste Änderungen der Pufferbedingungen zu Verfälschungen des Messergebnis führen. Während der Titration des Liganden ändert sich die Pufferzusammensetzung bezüglich der DMSO-Konzentration. Die resultierenden Misch- und Lösungsmittelverhältnisse überlagern die eigentliche Messung, sodass die ITC-Technik in diesem speziellen Fall für die Bestimmung der Affinität ungeeignet ist. Affinitäten können auch mittels NMR-Titration bestimmt werden. Hierbei werden 1H-15N-HSQCSpektren (oder wie hier 1H-15N-SOFAST-HMQC-Spektren) bei jedem Titrationsschritt aufgenommen und die jeweilige Verschiebung bestimmt. Dabei wird als Referenz immer das Spektrum mit dem gleichen Volumenanteils an d6-DMSO ohne Zugabe des Liganden eingesetzt, um Effekte des Lösungsmittels bei der Auswertung zu eliminieren. Unter Verwendung des Programms NMRView können die jeweiligen Verschiebungen gegen die zugegebene Ligandmenge aufgetragen und von Gleichung (6) gefittet werden. So kann eine Dissoziationskonstante von 0,678 ± 0,107 mM für 4,4´-Biphenol und eine Dissoziationskonstante von 1,100 ± 0,213 mM für Bisphenol A an Rheb bestimmt werden. Für die anderen eingesetzten Verbindungen kann auch bei 20fach molarem Überschuss kein Endpunkt der 127 6 Ergebnisse - Teil II Titration ausgemacht werden, sodass an dieser Stelle eine Bestimmung der Dissoziationskonstante nicht möglich ist. Diese Dissoziationskonstanten liegen, wie für einen Hit auf dieser Ebene erwartet, im sehr schwach affinen Bereich. 128 7 Diskussion - Teil II 7 DISKUSSION - TEIL II 7.1 AUFBAU UND SCREENING DER SUBSTANZBIBLIOTHEK Am Angang jedes Fragment-basierten Screenings muss eine Bibliothek von Substanzen zusammengestellt werden, welche potentiell an das gewünschte Protein binden. Dabei wird meistens eine zufällige Vorauswahl getroffen, da es ein sehr großes Angebot an kommerziellen Chemikalien gibt und allein Sigma-Aldrich über 100.000 davon anbietet. Eine adäquate Vorauswahl ist jedoch für das Gelingen eines Projekts entscheidend, da im ungünstigsten Fall nur Substanzen ausgewählt werden, welche keine Bindung zum Target aufweisen. Dadurch, dass durch NMR-Spektroskopie oder Röntgenstrukturanalyse immer mehr dreidimensionale Strukturen von Proteintargets zugänglich werden, werden computerunterstützte Hilfsmittel zum Medikamentendesign immer häufiger eingesetzt. Ebenfalls werden computergestützte Untersuchungen aufgrund der immens hohen Kosten (etwa 880 Millionen Dollar für jedes später zugelassene Medikament) und der langen Phase bis zur Markteinführung (ein Zeitraum von etwa 14 Jahren)220 immer attraktiver. Zur Zeit existiert eine Vielzahl von Computerprogrammen, welche für ein Drug Design in silico eingesetzt werden können. Bisher gibt es allerdings nur eine geringe Anzahl von Medikamenten, die mit Hilfe eines solchen Prozesses entwickelt werden konnten. Ein erfolgreiches Bespiel ist ein Integrase-Hemmer (Isentress®)221 gegen den HI-Virus. Aufgrund der zur Verfügung stehenden Ressourcen wird in dieser Arbeit eine Vorauswahl ohne die Nutzung eines in silico dockings durchgeführt. Alle untersuchten Verbindungen weisen dabei ein Molekulargewicht von unter 200 g/mol auf und können im Handel erworben werden. Für die Generierung der hier getesteten Bibliothek werden Verbindungen herangezogen, welche bei ähnlichen Fragestellungen positiv getestet wurden. Dabei werden Verbindungen eingesetzt, welche über ein konjugiertes Ringsystem verfügen und aufgrund ihrer chemischen Gruppen die Möglichkeit zu weiteren Synthesen eröffnen. Das in vorherigen Studien an K-Ras identifizierte Benzimidazol171 dient dabei als Startstruktur. Die eingesetzten Verbindungen werden in Gruppen eingeordnet, um im Idealfall ganze Gruppen von Verbindungen durch eine einzelne Messung ausschließen zu können. Da in jedem Gemisch mindestens eine Verbindung eine Interaktion mit Rheb zeigt, werden alle Verbindungen über schnell durchzuführende 1H-15N-SOFAST-HMQC-Experimente charakterisiert. Die bei dem Screening identifizierten Hits weisen überwiegend (bis auf Piperazin) zwei aromatische Ringe auf, welche direkt oder als Amin, Ether oder Keton verknüpft sind. Auch scheinen 129 7 Diskussion - Teil II Carbonsäuren sowie Phenolderivate geeignete Hits zur Bildung einer Leitstruktur zu sein. Interessanterweise zeigt Diphenylessigsäure keine und Benzophenon nur eine sehr geringe Interaktion mit RhebͻGDP, obwohl sie große strukturelle Ähnlichkeiten zu den identifizierten Hits aufweisen. Wie in der Literatur für K-RasͻGDP bereits beschrieben171, bindet Benzamidin auch nicht an RhebͻGDP. Um die experimentell ermittelte Daten anhand des dreidimensionalen Strukturmodells von Rheb erklären zu können und Modifikationen der identifizierten Hits zu vereinfachen, werden Modellierungen mit dem Programm HADDOCK durchgeführt. 7.2 MODELLIERUNG MIT HADDOCK Das Programm HADDOCK ist momentan das einzige Programm, welches experimentell ermittelte Interaktionsinformationen mit einer Modellierungssoftware verknüpfen kann. Für eine HADDOCK-Modellierung wird eine feste Interaktionsfläche am Protein vorgegeben (AIRTabelle), wodurch der Lösungsraum begrenzt wird. Diese als „aktiv“ bezeichneten Aminosäuren werden aus mit 1H-15N-SOFAST-HMQC-Spektren verfolgten Titrationen von RhebͻGDP mit den Liganden gewonnen. Dabei werden nur die Liganden weiterhin charakterisiert, die zu einer deutlichen chemischen Verschiebung einiger Resonanzfrequenzen von Aminosäuren führen. Mit dem Programm HADDOCK werden in zwei Iterationsschritten die energieärmsten 200 Komplexstrukturen berechnet. Anschließend erfahren die Komplexstrukturen eine Verfeinerung in Wasser. Um eine Bewertung dieser 200 finalen Strukturen vornehmen zu können, wird der sogenannte HADDOCK-Score herangezogen. Dieser ist ein Pseudoenergiewert, der sich aus unterschiedlichen Beiträgen zusammensetzt (Desolvatationsenergie, intermolekulare elektrostatische Energie, intermolekulare van-der-Waals-Energie, Verletzung der AIRs). Dieser ermittelte HADDOCK-Score wird gegen den RMSD zur Struktur mit dem niedrigsten HADDOCK-Score aufgetragen. Dadurch werden Cluster erhalten, welche anhand ihres RMDS und HADDOCK-Scores geordnet sind. Neben dieser Untersuchungsmethode ist es jedoch von größter Wichtigkeit, die Strukturen auch anhand ihrer Plausibilität zu bewerten. Eine eindeutige Unterteilung in Cluster ist mit dieser Methode bei den hier gewählten ProteinLigand-Komplexen nicht möglich. Bei allen durchgeführten Simulationen ergibt sich ausschließlich ein Cluster, in dem alle Komplexstrukturen enthalten sind. Bei der Analyse der resultierenden Strukturen fällt jedoch auf, dass die Liganden verschiedene Konformationen einnehmen und teilweise nicht direkt an der vorgegebenen Interaktionsfläche lokalisiert sind. Somit scheint eine Bewertung der Komplexstrukturen aus Protein und sehr kleinen Liganden ausschließlich aufgrund des HADDOCKScores nicht ausreichend zu sein. 130 7 Diskussion - Teil II Daher werden die generierten Komplexe zusätzlich anhand ihrer AIR-Energie und der intermolekularen van-der-Waals-Energie bewertet. Strukturen, welche in beiden Energietermen einen niedrigen Wert aufweisen, werden als beste Lösung definiert. Bei Komplexstrukturen, die eine große van-der-Waals-Energie ausweisen, ist der Ligand stets an dem Protein angelagert und befindet sich nicht in der identifizierten Bindetasche. Abbildung 7.1: Überlagerung einiger HADDOCK-Modellierungen von RhebͻGDP mit Bisphenol A. Aufgetragen sind Liganden mit einer hohen EvdW (lila), Liganden mit einer geringen EAIR (hellblau) sowie Liganden mit einer kleinen EAir und geringen EvdW (grün) auf der Oberfläche (blau: positive Reste, rot: negative Reste; berechnet über Pymol) von RhebͻGDP. In Abbildung 7.2 ist ein Vergleich der Orientierung von Bisphenol A im Komplex mit RhebͻGDP von HADDOCK-Modellierungen mit unterschiedlichen EAIR dargestellt. Eine Komplexstruktur mit einem niedrigen EAIR ist in Grün und eine Komplexstruktur mit einem hohen EAIR in Hellblau dargestellt. Die Aminosäure G80 ist in Orange hervorgehoben. Der Abstand der Hydroxylgruppe zum Rückgrat-Glycin an Position 80 beträgt 3,6 Å. Im Gegensatz dazu beträgt dieser Abstand in der Komplexstruktur mit einer hohen AIR-Energie 6,4 Å. Somit wird die experimentelle Randbedingung in der grünen Komplexstruktur besser erfüllt. Allgemein muss bei der Analyse der mit HADDOCK generierten Komplexstrukturen auch immer auf die Plausibilität der erzeugten Ergebnisse geachtet werden und auch eine manuelle Analyse vorgenommen werden. Diese Problematik ist schon lange bekannt183. 131 7 Diskussion - Teil II Abbildung 7.2: Detaillierte Ansicht zweier HADDOCK-Komplexe von RhebͻGDP mit Bisphenol A. Die beiden Strukturen zeigen detailliert die Orientierung von Bisphenol A im Komplex mit RhebͻGDP. Dabei ist in Grün ein modellierter Komplex mit geringer EAIR sowie geringer EvdW (grün) und ein Komplex mit großer EAIR und kleiner EvdW (hellblau) aufgetragen. Die Aminosäure G80 ist in Orange hervorgehoben. Der Abstand zwischen der Hydroxylgruppe und G80 ist in Å aufgetragen. Anscheinend ist es dem Programm HADDOCK in manchen Fällen nicht möglich, die generierten Komplexstrukturen richtig zu bewerten. Die in dieser Arbeit gewählte Bewertung anhand der EAIR sowie der EvdW scheint für die Fragestellung dieser Arbeit jedoch gut geeignet zu sein. 7.2.1 VERGLEICH DER UNTERSCHIEDLICHEN KOMPLEXE Die Analyse und die Bewertung der resultierenden Hits wird durchgeführt, um eine Leitstruktur zum Medikamentendesign generieren zu können. Alle als Hits charakterisierte Liganden zeigen eine Bindung in derselben Bindungsregion von Rheb, sodass ein bisher unbekannter Hot Spot auf der Oberfläche dieser kleinen GTPase lokalisiert werden kann. Alle als Hit untersuchten Verbindungen liegen mit der identischen Orientierung in der Bindetasche. Als Beispiel ist der Vergleich der Orientierung von 4,4´-Biphenol und 4,4'-Dihydroxybenzophenon in Abbildung 7.3 gezeigt. Die Hydroxygruppen weisen auf beiden Seiten der Bindetasche etwa einen Abstand von 3,5 Å zu den Aminosäuren des Proteinrückgrats auf. Somit kann die schlechtere Bindung von Liganden mit para-substituierter Säurefunktion durch den größeren sterischen Anspruch dieser Gruppe erklärt werden. Da eine Bindung von 1-(3,4-Dichlorophenyl)piperazin an RhebͻGDP beobachtet werden kann, scheint eine Möglichkeit für zukünftige Synthesen zu sein, Substitutionen mit Chlor vorzunehmen. 132 7 Diskussion - Teil II Abbildung 7.3: Orientierung von 4,4´-Biphenol (hellblau) und 4,4'-Dihydroxybenzophenon (lila) in der RhebͻGDPBindetasche (grün). Das Proteinrückgrat von RhebͻGDP ist von Aminosäureposition 71 bis 81 in der Stäbchenkonformation abgebildet (grün). Die Orientierung der Liganden stammt aus den HADDOCK-Modellierungen mit den geringsten Werten für EAIR sowie EvdW. Auch sollten weitere Synthesen dahingehend durchgeführt werden, die eher flache Struktur der bisherigen Liganden auf eine komplexere dreidimensionale Struktur zu erweitern. 7.3 VERGLEICH MIT RAS Aktuelle Forschungen befassen sich mit der Erzeugung einer Leitstruktur, um ein potentielles Medikament gegen (H- oder K-) Ras zu generieren. Dabei wird eine Fragment-basierte Screeningmethode eingesetzt, bei der die verschiedenen Liganden mittels NMR-Spektroskopie charakterisiert und bewertet werden. Bei allen in der Literatur diskutierten Beispielen wird ein Hot Spot auf der Oberfläche von Ras beschrieben, an dem alle Hits binden. Alle ermittelten Hits sind kleine zyklische Verbindungen wie Benzimidazol- und Phenolderivate. 133 7 Diskussion - Teil II Abbildung 7.4: Vergleich der Orientierung des gebundenen Liganden an H-RasͻGDP172 (links, PDB: 4EPX) und KRasͻGDP171 (rechts, PDB: 4DST). 215 Für die Arbeiten von Rosnizeck et al. sind keine Strukturen abgelegt. Die switch II Region ist zur besseren Orientierung rot eingefärbt. Der Ligand ist in der Stäbchen- und das Protein in der Ribbon-Darstellung visualisiert. Abbildung 7.4 zeigt einen Vergleich der mittels Röntgenstrukturanalyse bestimmten Komplexstrukturen von RasͻGDP mit einem bindenden Liganden. Die switch II Region ist zur besseren Übersicht rot eingefärbt. Die beiden unterschiedlichen Liganden binden an die nahezu identische Bindungstasche an Ras. Die Ligandenbindung bei der GTPase Rheb erfolgt für die hier untersuchten Liganden auf der anderen Seite der switch II Region (Abbildung 7.5). Durch Vergleich der Orientierung des Liganden auf der Proteinoberfläche wird sichtbar, dass die Tasche bei RhebͻGDP, an welche die in dieser Arbeit getesteten Liganden binden, Abbildung 7.5: RhebͻGDP im Komplex mit 4,4´-Biphenol. weder bei H- noch bei K-RasͻGDP existiert. Die switch II Region rot eingefärbt. 134 7 Diskussion - Teil II Abbildung 7.6 zeigt den Vergleich der Komplexstrukturen von H-RasͻGDP172 und K-RasͻGDP171 mit den publizierten Liganden und das HADDOCK-Modell von RhebͻGDP im Komplex mit 4,4´-Biphenol in der Oberflächendarstellung. Die beiden Ras-Proteine zeigen eine sehr ähnliche Oberflächenstruktur mit einer vergleichbaren Ladungsverteilung. Rheb zeigt eine deutlich andere Oberflächenstruktur und auch eine andere Ladungsverteilung. Auffällig ist jedoch das Fehlen der Bindungstasche, in welche bei RhebͻGDP das 4,4´-Biphenol bindet, in beiden Ras-Strukturen (siehe Pfeil). H-Ras K-Ras Rheb Abbildung 7.6: Vergleich der Komplexe der GTPase mit dem jeweiligen Liganden. Die publizierten Komplexstrukturen von H-RasͻGDP172, K-RasͻGDP171 und das Ergebnis der Haddockmodellierung von RhebͻGDP mit 4,4´-Biphenol sind gezeigt. Die gebundenen Liganden sind in der Stäbchendarstellung mit der zuvor verwendeten Farbkodierung (Abbildung 7.4, Abbildung 7.5) dargestellt. Die Lokalisation der Bindetasche von Rheb ist auf den Ras-Oberflächen mit einem Pfeil gekennzeichnet. 135 7 Diskussion - Teil II Die bei Ras bevorzugte Bindungstasche der Liganden scheint bei Rheb zwar vorhanden, jedoch weniger ausgeprägt zu sein. Die in der Literatur diskutierten Liganden zeigen nicht nur eine Bindung an Ras, sondern können auch die Interaktion von Ras mit seinem GEF-Protein SOS behindern. Da für Rheb zur Zeit noch kein GEFProtein bekannt ist, können solche Experimente nicht durchgeführt werden. Weitere Testes, ob möglicherweise die hier untersuchten Liganden auch in die bekannte RasBindetasche binden und somit keine Selektivität gegenüber verschiedenen kleinen GTPasen vorliegt, könnten Aufschluss über mögliche Unterschiede dieser beiden GTPasen bezüglich des Ablaufs der GEF-Reaktion geben. 136 8 Zusammenfassung 8 ZUSAMMENFASSUNG Kleine GTPasen übernehmen eine Vielzahl von Schlüsselfunktionen in diversen biochemischen Prozessen in der Zelle. Neben der Regulation des Zellwachstums und der -differenzierung regulieren sie unter anderem den intrazellulären Vesikeltransport und die Zellmigration. Der bekannteste Vertreter kleiner GTPasen ist Ras, welches, wenn spezielle Mutationen vorliegen, eine große Anzahl an Krebsformen bedingen kann. Obwohl GTPasen eine große Anzahl von verschiedenen Aufgaben im Organismus erfüllen, binden alle Guaninnukleotide mit einer hohen Affinität und nutzen den Prozess der Hydrolyse von GTP zu GDP und Phosphat als Energiequelle. Neben diesem zentralen Prozess liegen alle bisher bekannten kleinen GTPasen membranassoziiert vor. Dabei werden durch posttranslationale Modifikationen des Carboxy-Terminus eine oder mehrere Fettsäurereste angefügt, welche für die Membranbindung notwendig sind. Dabei gibt es bei den kleinen GTPasen große Unterschiede bei den durchgeführten Modifikationen und somit auch bei der Lokalisation in der Zelle. In dieser Arbeit wurde die kleine GTPase Rheb untersucht. Rheb wurde erfolgreich rekombinant in E. coli-Zellen durch Koexpression mit den beiden Untereinheiten der humanen Farnesyltransferase farnesyliert, 15 N-isotopenangereichert und mittels mehrdimensionaler NMR-Spektroskopie charakterisiert. Es konnte gezeigt werden, dass farnesyliertes Rheb eine gute Löslichkeit bei den gewählten Pufferbedingungen besitzt und somit für weitere NMR-spektroskopische Untersuchungen geeignet ist, die farnesyliertes Rheb im membrangebundenen Zustand charakterisieren können. Für die Herstellung von Nanodiscs musste zunächst eine geeignete Lipidkomposition ermittelt werden, was durch den Einsatz von Folch Brain Lipids mit einem Zusatz von 5 % PI(4)P erfolgreich realisiert werden konnte. Die Charakterisierung von Rheb in einer physiologisch-ähnlichen Umgebung konnte durch NMRMessungen in E. coli-Lysat und durch Zugabe von Oozyten-Extrakt zum aufgereinigten Rheb-Protein realisiert werden. Hingegen konnte eine Charaktersierung von Rheb in intakten Bakterienzellen oder Xenopus-Oozyten aufgrund der hohen Viskosität im Zellinneren nicht durchgeführt werden. GTPasen stehen aufgrund ihrer Wichtigkeit für diverse Prozesse in der Zelle nicht nur im Fokus der Grundlagen-, sondern auch der pharmazeutischen Forschung. Obwohl viele Funktionen der kleinen GTPasen bekannt sind, sind bisher nur wenige natürliche Bindungspartner insbesondere für Rheb identifiziert worden. Somit kann zur Entwicklung eines Medikaments gegen bestimmte GTPasen, deren Fehlregulation häufiger Auslöser diverser Krebserkrankungen ist, nicht auf bekannte Strukturmotive natürlicher etablierter Liganden zurückgegriffen werden. In dieser Arbeit wurde ein Fragment-basiertes Screeningverfahren eingesetzt, mit welchem eine Substanzbibliothek bezüglich 137 8 Zusammenfassung der Interaktion mit RhebͻGDP mittels NMR-Spektroskopie untersucht wurde. Dabei wurden Bisphenol A und 4,4´-Biphenol als besonders vielversprechende Kandidaten zur Entwicklung einer Leitstruktur ermittelt. Außerdem wurde auf der Proteinoberfläche von Rheb ein Hot Spot im Bereich von Aminosäure F70-I78 ermittelt. Ein Vergleich mit dem in der aktuellen Literatur diskutierten Hot Spot auf der Proteinoberfläche von Ras zeigte einen großen Unterschied zu RhebͻGDP. H- sowie KRas weisen an der gleichen Stelle einen Hot Spot für kleine zyklische Liganden auf, wohingegen der in dieser Arbeit entdeckte Hot Spot bei RhebͻGDP auf der anderen Seite der Helix liegt, welche die switch II Region bildet. Die in der aktuellen Literatur diskutierten Liganden für K- und H-Ras zeigen sehr große strukturelle Ähnlichkeiten zu den in dieser Arbeit ermittelten Hits. 138 9 Conclusion 9 CONCLUSION Small GTPases play a key role in different types of biochemical processes in the cell. They regulate the cell growth and differentiation, the intracellular vesicle transport and the migration of cells. The best known representative of small GTPases is Ras. Mutations of the ras gene can lead to several forms of cancer. Although GTPases perform different functions in the organism, they all bind with high affinity to guanine nucleotides. The hydrolysis from GTP to GDP and inorganic phosphate is of fundamental importance for their physiological function. Next to this central process, all GTPases exist in a membrane-associated manner. The carboxy-terminus is prenylated through posttranslational processes. This modification is essential for the membrane association, but different for the various types of small GTPases and therefore determines different localizations in the cell. Here, the small GTPase Rheb was investigated. A recombinant farnesylation in E. coli with the two subunits of human farnesyltransferase was performed. The modified protein was 15 N-isotopically enriched and characterized by multidimensional NMR-spectroscopy. The farnesylated Rheb shows a high solubility for the buffer conditions used and is suitable for further NMR-spectroscopic characterizations of membrane-bound Rheb. For the preparation of nanodiscs, a suitable composition of lipids had to be determined, which was achieved by mixing Folch Brain Lipids with 5 % PI(4)P. The characterization of Rheb under near-physiological conditions was successfully performed by multidimensional NMR-spectroscopy in E.coli-lysate and oocyte-extract. However, the characterization of Rheb inside living Xenopus oocytes failed due to the high viscosity inside of the cytosol. Due to their importance for different cell-processes, GTPases are in the center of fundamental and pharmaceutical research. Although a lot of functions are known, only some natural binding partners have been identified so far and thus only few structures are known. Because of this, the development of drugs against cancer-causing GTPases cannot rely on known structure motifs of natural established ligands. In this study, a fragment-based screening was performed, where the interaction of a potential ligand with Rheb was tested by NMR. Bisphenol A and 4,4´-Biphenol have been found to be the most potential candidates for the design of a lead-component. Moreover, a hot spot between F70 and I78 was identified on Rheb’s surface. The results of this work reveal a big difference between the hot spot of Rheb and the hot spot of Ras, reported recently in the literature. The hot spots for small cyclic ligands on H- and K-Ras are located at the same position, whereas Rheb’s hot spot is located on the other side of the switch II forming helix. Nevertheless, the ligands 139 9 Conclusion for H- and K-Ras discussed in the recent literature and the ligands described in this work for Rheb exhibit a high structural similarity. 140 10 Ausblick 10 AUSBLICK In dieser Arbeit konnte RhebͻGDP erstmals in physiologisch-ähnlicher Umgebung mittels NMRSpektroskopie charakterisiert werden. Die Untersuchung in intakten E. coli-Zellen oder XenopusOozyten ist aufgrund der in der Zelle vorherrschenden hohen Viskosität mit dem zur Verfügung stehenden technischen Equipment nicht möglich. Um Messungen in Xenopus-Oozyten realisieren zu können, wäre es möglich, einen Teil des viskosen Zellinneren durch Pufferlösung zu ersetzten. Die Membran der Oozyte könnte auf diese Weise als natürlicher Ankerpunkt für farnesyliertes Rheb dienen. Darüber hinaus sollte membrangebundenes Rheb NMR-spektroskopisch charakterisiert werden. Dazu müssen Nanodiscs mit mindestens 5 % PI(4)P hergestellt und farnesyliertes, 15 N- isotopenangereichertes Rheb damit assoziiert werden. Ebenso könnte zur Herstellung verschiedener Nanodiscs die Bindung von farnesyliertem Rheb an andere Phospholipide untersucht werden. Die jeweilige Nukleotidbeladung scheint eine wichtige Rolle bei der Membranassoziation zu spielen. Möglicherweise können NMR-spektroskopische Untersuchungen einen Aufschluss über die Orientierung von Rheb relativ zur Membran geben. Auf der Proteinoberfläche von RhebͻGDP wurde ein bisher noch unbekannter Hot Spot entdeckt. Weitere Untersuchungen sollten Rheb im aktiven Zustand mit einschließen, um eine mögliche Nukleotidabhängigkeit der Ligandenbindung zu erforschen. Auch kann durch die in der aktuellen Forschung beschriebenen Liganden für K- und H-Ras eine Überprüfung der Selektivität dieser Liganden erfolgen. Der auf Ras entdeckte Hot Spot liegt in der Interaktionsfläche von Ras-SOS. Binden die für Ras beschriebenen Liganden auch Rheb in der für Rheb gefundenen Bindetasche, können daraus mögliche Unterschiede bei der Bindung des entsprechenden GEF-Proteins, welches bisher noch unbekannt ist, abgeleitet werden. Neben der Entdeckung des Hot Spots auf der Oberfläche von RhebͻGDP konnten Hits bestimmt werden, die zur Synthese einer Leitstruktur genutzt werden können, um die Affinität des Liganden zum Protein zu erhöhen. Es konnten erste Zelltoxizitätstest mit den in dieser Arbeit gefundenen Hits an Zellkulturen durchgeführt werden, wodurch eine Entwicklung einer Leitstruktur auf Basis dieser Hits möglich erscheint. Neben NMR-spektroskopischen Untersuchungen sollten die mit HADDOCK modellierten Komplexe von Protein und Ligand mittels Röntgenstrukturanalyse bestätigt werden. 141 11 Abkürzungsverzeichnis 11 ABKÜRZUNGSVERZEICHNIS 4E-BP1 Translationsinitiationsfaktor-4E-bindendes Protein AIR Ambiguous Interaction Restraint APS Ammoniumperoxodisulfat Arf ADP-ribosyltion factor ATP Adenosintriphosphat AUC Ultrazentrifugation BcL-2 B-cell lymphoma 2 BcL-XL B-cell lymphoma-extra large BSA Rinderalbumin CDC25 cell division cycle 25 homology Cdk1 Cyclin-dependent protein kinase 1 CK2 Casein Kinase 2 CPP zellpenetrierendes Peptid DH Dbl homology DNA Desoxyribonukleinsäure DOCK Dedicator of cytocinesis DOS Diversität-orientierte Synthese E. coli Escherichia Coli EDTA Ethylendiamintetraessigsäure EGF Epidermaler Wachstumsfaktor ERK extracellular signal-regulated kinase 142 11 Abkürzungsverzeichnis FBS Fragment-basiertes Screening FGF Fibroblastenwachstumsfaktor FID free induction decay FKBP38 FK506-binding protein 38 FPP Farnesylpyrophosphat GAP GTPase-aktivierendes Protein GDI Guaninnukleotid-Dissoziations-Inhibitor GDP Guanosinmonophosphat GEF Guaninnukleotid-Austausch-Faktor GNBP Guaninenukleotid-bindendes Protein GTP Guanosintriphosphat HADDOCK High Ambiguity Driven protein-protein DOCKing hGBP1 humanes Guanylat-bindendes Protein 1 HIV Humaner Immundefizienz-Virus HMQC Heteronuclear Multiple Quantum Coherence HSQC Heteronuclear Single Quantum Coherence Icmt isoprenylcysteine carboxylmethyltransferase IgG Immunoglobulin IPTG Isopropyl-ɴ-D-thiogalactopyranosid LB Lysogeny broth LRR Leucin-reiche Region mLST8 mammalian lethal with Sec thirteen 8 MRI Magnetic Resonance Imaging 143 11 Abkürzungsverzeichnis mSin1 mammalian stress-activated protein kinase interacting protein MWCO Molecular Weight Cut Off NMR Nuclear Magnetic Resonance NOE Kern-Overhauser-Effekt PA Phosphatidsäure PAGE polyacrylamide gel electrophoresis PDB Protein-Datenbank Wɷ Phospohidiesterase 6 delta Untereinheit PH pleckstrin homology pH potentia Hydrogenii PI3K Phosphoinositol-3-Kinase PKB Proteinkinase B PLD Phospholipase D PPTase Protein-Prenyl-Transferasen PRAS 40 proline-rich Akt substrate 40 kDa P-Rex1 phosphatidylinositol-3,4,5-triphosphate-dependent Rac exchange-factor 1 Protor protein observed with ricTOR Rab Ras-like proteins in brain Rac Ras-related C3 botulinum toxin substrate Ral Ras-like protein Ran Ras-like nuclear Rap Ras-related protein Raptor regulatory-associated protein of mTOR 144 11 Abkürzungsverzeichnis Ras Rat sarcoma REM Ras exchange motif Rheb Ras homolog enriched in brain Rho Ras homology Rictor rapamycin-intensive companion of mTOR RNA Ribonukleinsäure S6K Protein-S6-Kinase SAR structure-activity relationships SIMIBI signal recognition particle, MInD, BIoD SLO Streptolysin O SOFAST Band-Selective Optimized-Flip-Angle Short-Transient SOS son of sevenless SRP signal recognition particle STD Sättigungstransfer-Differenz STINT-NMR structural interaction using in-cell NMR TCB Tre-2/Cdc16/Bub2 TCTP translationally controlled tumor protein TEMED N,N,N',N'-Tetramethylethylendiamin TG TRIS/Glycin TOR target of rapamycin TRAFAC translation factors TRIS Tris(hydroxymethyl)-aminomethan TSC Tuberous Sclerosis Complex 145 11 Abkürzungsverzeichnis TSP Natriumtrimethylsilylpropionat dɴϰ dŚLJŵŽƐŝŶɴϰ VdW Van der Waals Wnt wingless Integration 1 Formelzeichen °C Grad Celsius Da Dalton ɷ chemische Verschiebung g Gramm h Stunde K Kelvin l Liter M molar rpm Umdrehungen pro Minute U Unit (μmol/min) v/v Volumen pro Volumen w/v Gewicht pro Volumen 146 11 Abkürzungsverzeichnis Kanonische Aminosäuren Name der Aminosäure Drei-Buchstaben-Code Ein-Buchstaben-Code Alanin Ala A Cystein Cys C Aspartat Asp D Glutamat Glu E Phenylalanin Phe F Glycin Gly G Histidin His H Isoleucin Ile I Lysin Lys K Leucin Leu L Methionin Met M Asparagin Asn N Prolin Pro P Glutamin Gln Q Arginin Arg R Serin Ser S Threonin Thr T Valin Val V Tryptophan Try W Tyrosin Tyr Y 147 12 Abbildungsverzeichnis 12 ABBILDUNGSVERZEICHNIS ABBILDUNG 1.1: STRUKTUR VON GUANINNUKLEOTID-BINDENDEN PROTEINEN UND WECHSELWIRKUNGEN ZUM NUKLEOTID............ 3 11 ABBILDUNG 1.2: SCHEMA DES UNIVERSALEN MECHANISMUS BEI DER GTP-HYDROLYSE VON KLEINEN GTPASEN . ....................... 5 ABBILDUNG 1.3: GTPASE-ZYKLUS REGULIERT ÜBER GAP- UND GEF-PROTEINE21 ................................................................. 5 22 ABBILDUNG 1.4: MECHANISMUS DES NUKLEOTIDAUSTAUSCHES, INDUZIERT DURCH GEF ...................................................... 6 ABBILDUNG 1.5: REGULATION DES MTORC1-SIGNALWEGS63 ........................................................................................ 11 ABBILDUNG 1.6: AMINOSÄURE-ABHÄNGIGE REGULATION VON MTORC1 DURCH DIE GTPASE RAG21 ...................................... 12 93 ABBILDUNG 1.7: NMR-STRUKTUR VON RHEBͻ'W ................................................................................................. 14 ABBILDUNG 1.8: MECHANISMUS VERSCHIEDENER GAPS100. .......................................................................................... 15 117 ABBILDUNG 1.9: KOMPLEX AUS RHEBͻWȴ . ........................................................................................................ 18 ABBILDUNG 1.10: SCHEMATISCHE DARSTELLUNG DES PROTOKOLLS ZUR HERSTELLUNG VON IN-CELL PRÄPARATEN125. ................. 20 ABBILDUNG 1.11: SCHNELLE AQUISITION DER 3D-NMR-SPEKTREN DURCH VERWENDUNG EINES NICHT-LINEAREN AUFNAHMESCHEMAS (NONLINEAR SAMPLING) MIT REDUZIERTEN DATENPUNKTEN141. .................................................. 21 ABBILDUNG 1.12: ARBEITSSCHRITTE ZUR PROBENVORBEREITUNG EINER XENOPUS-OOZYTEN IN-CELL NMR-MESSUNG ABBILDUNG 1.13: INKORPORIERUNG DES ZIELPROTEINS IN 293F-ZELLEN 154 134 . .......... 23 . ..................................................................... 24 ABBILDUNG 1.14: HOT-SPOT AUF EINER PROTEIN-PROTEIN-INTERAKTIONSFLÄCHE 160 . ......................................................... 26 ABBILDUNG 1.15: SYNTHESEPRINZIP VON LEITSTRUKTUREN BEI DER WIRKSTOFFENTWICKLUNG168. .......................................... 27 174 ABBILDUNG 1.16: SCHEMA DES STD-EXPERIMENTS . ................................................................................................ 29 ABBILDUNG 1.17: NMR-BASIERTES LIGANDEN-SCREENING: 1H-15N-HSQC-SPEKTREN171 .................................................... 30 ABBILDUNG 3.1: SCHEMATISCHE DARSTELLUNG DES PULSPROGRAMMS P3919GP ZUR DURCHFÜHRUNG EINES 1D 1HEXPERIMENTS186. ........................................................................................................................................ 54 ABBILDUNG 3.2: SCHEMATISCHE DARSTELLUNG DES PULSPROGRAMMS HSQCETF3GPSI ZUR DURCHFÜHRUNG EINES 1H-15N-HSQC186 EXPERIMENTS . ........................................................................................................................................ 55 ABBILDUNG 3.3: SCHEMATISCHE DARSTELLUNG DES PULSPROGRAMMS SFHMQCF3GPPH ZUR DURCHFÜHRUNG EINES 1H-15N186 SOFAST-HMQC-EXPERIMENTS . ................................................................................................................ 56 ABBILDUNG 4.1: ERGEBNIS EINER 15 % SDS-PAGE VON PROBEN EINER RHEB-PRÄPARATION NACH ANFÄRBEN DES SDS-GELS MIT COOMASSIE BRILLANT BLUE. .......................................................................................................................... 60 ABBILDUNG 4.2: 1D 1H-NMR-SPEKTRUM VON RHEBͻ'W BEI 600 MHZ, 298 K UND PH 7,8. .......................................... 61 1 15 ABBILDUNG 4.3: ÜBERLAGERUNG DER H- N-HSQC-SPEKTREN VON RHEBͻ'W (SCHWARZ) UND RHEBͻ'PPNHP (ROT), AUFGENOMMEN BEI 600 MHZ, 298 K UND PH 7,8. ........................................................................................... 63 ABBILDUNG 4.4: VERGRÖßERUNG DER ÜBERLAGERUNG DER 1H-15N-HSQC-SPEKTREN VON GDP- (SCHWARZ) UND GPPNHPGEBUNDENEM (ROT) RHEB. ........................................................................................................................... 64 ABBILDUNG 4.5: 1H-15N-HSQC VON RHEBͻ'W (SCHWARZ) IM VERGLEICH MIT RHEB IN INTAKTEN E. COLI-ZELLEN (GRÜN). ...... 66 1 15 ABBILDUNG 4.6: H- N-HSQC VON RHEBͻ'W (SCHWARZ) IM VERGLEICH MIT RHEB AUS E. COLI-LYSAT (ROT). ..................... 67 ABBILDUNG 4.7: ÜBERLAGERUNG DER 1H-15N-HSQC-SPEKTREN VON RHEBͻ'W (SCHWARZ), RHEB IN E. COLI-LYSAT (ROT) UND RHEB IN INTAKTEN E. COLI-ZELLEN (GRÜN). ....................................................................................................... 68 148 12 Abbildungsverzeichnis 1 15 ABBILDUNG 4.8: ÜBERLAGERUNG DER H- N-HSQC-SPEKTREN VON RHEBͻ'W (SCHWARZ), RHEB IN OOZYTENEXTRAKT (LILA) UND RHEB IN OOZYTENEXTRAKT MIT ZUGABE VON PHOSPHATASEINHIBITOR (GRÜN). ................................................... 70 ABBILDUNG 4.9: ERGEBNIS EINER 15 % SDS-PAGE VON AFFINITÄTSCHROMATOGRAPHISCH AUFGEREINIGTEM RHEB NACH IN VITRO FARNESYLIERUNG UND TRITON X-114-EXTRAKTION NACH ANFÄRBEN DES GELS MIT COOMASSIE BRILLANT BLUE. .............. 71 ABBILDUNG 4.10: ERGEBNIS EINER 15 % SDS-PAGE VON PROBEN EINER RHEB/FARNESYLTRANSFERASE-KOEXPRESSIONSPRÄPARATION NACH ANFÄRBEN DES SDS-GELS MIT COOMASSIE BRILLANT BLUE. ....................................................... 72 ABBILDUNG 4.11: MIT COOMASSIE BRILLANT BLUE GEFÄRBTES 15 %IGES SDS-GEL DER TRITON X-114-EXTRAKTION DER KOEXPRESSION VON RHEB UND FARNESYLTRANSFERASE. ....................................................................................... 73 ABBILDUNG 4.12: DIREKTMESSUNG MITTELS ESI-MS VON EINER MISCHUNG AUS FARNESYLIERTEM UND UNFARNESYLIERTEM RHEB (INTAKTES PROTEIN). ................................................................................................................................... 74 ABBILDUNG 4.13: MIT COOMASSIE BRILLANT BLUE GEFÄRBTES 15 %IGES SDS-GEL ZUR CHARAKTERISIERUNG DER NUKLEOTIDABHÄNGIGEN BINDUNG VON UNFARNESYLIERTEM UND FARNESYLIERTEM RHEB AN FOLCH/PI(4)P-LIPOSOMEN. ................. 75 ABBILDUNG 4.14: ÜBERLAGERUNG DER 1H-15N-HSQC-SPEKTREN VON RHEBͻ'W (SCHWARZ) MIT FARNESYLIERTEM RHEBͻ'W (ROT). ...................................................................................................................................................... 76 ABBILDUNG 5.1: ÜBERLAGERUNG DER 1H-15N-HSQC-SPEKTREN VON RHEBͻ'W (SCHWARZ) UND RHEB IN E .COLI-LYSAT (ROT), AUFGENOMMEN BEI 600 MHZ, 298 K UND PH 7,8. ........................................................................................... 78 ABBILDUNG 5.2: VERGRÖßERUNG DER ÜBERLAGERUNG DER 1H-15N-HSQC-NMR-SPEKTREN VON RHEBͻ'W (SCHWARZ) UND RHEBͻ'W IN OOZYTEN-EXTRAKT (LILA) MIT ZUGABE VON PHOSPHATASEINHIBITOR (LINKS) UND AUFTRAGUNG DER FEHLENDEN AMINOSÄUREN (LILA) IM 1 1 H-15N-HSQC-SEPKTRUM AUF DER OBERFLÄCHE VON RHEB. ................................ 81 15 1 15 ABBILDUNG 6.1: ÜBERLAGERUNG EINES H- N-HSQC-SPEKTRUMS (GRÜN) MIT EINEM H- N-SOFAST-HMQC-SPEKTRUM (SCHWARZ) VON RHEBͻ'W͘ ........................................................................................................................ 86 ABBILDUNG 6.2: SCHEMATISCHE DARSTELLUNG DER DURCHSCHNITTLICHEN MOLEKÜLMASSE UND DER EFFEKTIVITÄT DER BINDUNG FÜR UNTERSCHIEDLICHE ANSÄTZE DER MEDIKAMENTENENTWICKLUNG 1 216 . ................................................................ 88 15 ABBILDUNG 6.3: ÜBERLAGERUNG DER H- N-HSQC-SPEKTREN DER REFERENZPROBE (RHEB MIT D6-DMSO, SCHWARZ) UND NACH ZUGABE VON GEMISCH 1 (ZEHNFACH MOLARER ÜBERSCHUSS VON JEDER SUBSTANZ) ZU EINER 15N-ISOTOPENANGEREICHERTEN RHEB-PROTEINPROBE (BLAU). ........................................................................................................................ 91 ABBILDUNG 6.4: VERGRÖßERUNG AUS ABBILDUNG 6.3. ............................................................................................... 91 ABBILDUNG 6.5: AUFTRAGUNG DER GEWICHTETEN CHEMISCHEN VERSCHIEBUNG [PPM] GEGEN DIE AMINOSÄURESEQUENZ. .......... 92 1 15 ABBILDUNG 6.6: AUFTRAGUNG DER AMINOSÄUREN (BLAU) , WELCHE EINE VERÄNDERUNG DER RESONANZFREQUENZ IM H- NHSQC-SPEKTRUM NACH ZUGABE VON GEMISCH 1 ZEIGEN. ................................................................................... 93 ABBILDUNG 6.7: AUFTRAGUNG DER GEWICHTETEN CHEMISCHEN VERSCHIEBUNG [PPM] GEGEN DIE AMINOSÄURESEQUENZ. .......... 95 ABBILDUNG 6.8: AUFTRAGUNG DER GEWICHTETEN CHEMISCHEN VERSCHIEBUNG [PPM] GEGEN DIE AMINOSÄURESEQUENZ. .......... 98 ABBILDUNG 6.9: AUFTRAGUNG DER GEWICHTETEN CHEMISCHEN VERSCHIEBUNG [PPM] GEGEN DIE AMINOSÄURESEQUENZ. ........ 101 ABBILDUNG 6.10: AUFTRAGUNG DER GEWICHTETEN CHEMISCHEN VERSCHIEBUNG [PPM] GEGEN DIE AMINOSÄURESEQUENZ. ...... 104 ABBILDUNG 6.11: AUFTRAGUNG DER GEWICHTETEN CHEMISCHEN VERSCHIEBUNG [PPM] GEGEN DIE AMINOSÄURESEQUENZ. ...... 106 ABBILDUNG 6.12: AUFTRAGUNG DER GEWICHTETEN CHEMISCHEN VERSCHIEBUNG [PPM] GEGEN DIE AMINOSÄURESEQUENZ. ...... 109 1 15 ABBILDUNG 6.13: ÜBERLAGERUNG DER H- N-SOFAST-HMCQ-SPEKTREN DER REFERENZPROBE (RHEB MIT D6-DMSO, SCHWARZ), NACH ZUGABE VON BENZIDIN (BLAU), 4,4'-DIHYDROXYDIPHENYLMETHAN (LILA) UND 4,4'- 149 12 Abbildungsverzeichnis 15 DIHYDROXYBENZOPHENON (GRÜN) (ZEHNFACH MOLARER ÜBERSCHUSS VON JEDER SUBSTANZ) ZU EINER NISOTOPENANGEREICHERTEN RHEB-PROTEINPROBE. ............................................................................................ 113 1 15 ABBILDUNG 6.14: AUSSCHNITTSVERGRÖßERUNG DER ÜBERLAGERUNG DER H- N-SOFAST-HMCQ-SPEKTREN AUS ABBILDUNG 6.13. ..................................................................................................................................................... 114 ABBILDUNG 6.15: AUFTRAGUNG DER GEWICHTETEN CHEMISCHEN VERSCHIEBUNG [PPM] GEGEN DIE AMINOSÄURESEQUENZ. ...... 115 ABBILDUNG 6.16: VISUALISIERUNG DER ERGEBNISSE DER MODELLIERUNG MIT HADDOCK. ............................................... 118 219 ABBILDUNG 6.17: PRINZIP DER AUSWAHL DER BESTEN STRUKTUREN . ......................................................................... 119 ABBILDUNG 6.18: AUFTRAGUNG DER AIR- GEGEN DIE VAN-DER-WAALS-ENERGIEN DER MIT HADDOCK MODELLIERTEN KOMPLEXSTRUKTUREN VON RHEBͻ'W MIT 4,4´-BISPHENOL. ............................................................................ 120 ABBILDUNG 6.19: VERGLEICH DER MIT HADDOCK-MODELLIERTEN STRUKTUREN VON 4,4`-BIPHENOL ANHAND DER VAN-DERWAALS- UND AIR-ENERGIEN. ...................................................................................................................... 121 ABBILDUNG 6.20: ÜBERLAGERUNG EINIGER HADDOCK-MODELLIERUNGEN VON RHEBͻ'W MIT 4,4´-BIPHENOL. ................. 122 ABBILDUNG 6.21: AUFTRAGUNG DER AIR- GEGEN DIE VAN-DER-WAALS-ENERGIEN DER MIT HADDOCK MODELLIERTEN KOMPLEXSTRUKTUREN VON RHEBͻ'W MIT 1-(3,4-DICHLOROPHENYL) PIPERAZIN. .................................................. 123 ABBILDUNG 6.22: ÜBERLAGERUNG EINIGER HADDOCK-MODELLIERUNGEN VON RHEBͻ'W MIT 1-(3,4DICHLOROPHENYL)PIPERAZIN. ...................................................................................................................... 123 ABBILDUNG 6.23: AUFTRAGUNG DER AIR- GEGEN DIE VAN-DER-WAALS-ENERGIEN DER MIT HADDOCK MODELLIERTEN KOMPLEXSTRUKTUREN VON RHEBͻ'W MIT 4,4'-DIHYDROXYBENZOPHENON. ........................................................ 124 ABBILDUNG 6.24: ÜBERLAGERUNG EINIGER HADDOCK-MODELLIERUNGEN VON RHEBͻ'W MIT 4,4'-DIHYDROXYBENZOPHENON. ............................................................................................................................................................ 125 ABBILDUNG 6.25: AUFTRAGUNG DER AIR- GEGEN DIE VAN-DER-WAALS-ENERGIEN DER MIT HADDOCK MODELLIERTEN KOMPLEXSTRUKTUREN VON RHEBͻ'W MIT BISPHENOL A. ................................................................................ 126 ABBILDUNG 6.26: ÜBERLAGERUNG EINIGER HADDOCK-MODELLIERUNGEN VON RHEBͻ'W MIT BISPHENOL A. .................... 126 ABBILDUNG 7.1: ÜBERLAGERUNG EINIGER HADDOCK-MODELLIERUNGEN VON RHEBͻ'W MIT BISPHENOL A. ...................... 131 ABBILDUNG 7.2: DETAILLIERTE ANSICHT ZWEIER HADDOCK-KOMPLEXE VON RHEBͻ'W MIT BISPHENOL A. ........................ 132 ABBILDUNG 7.3: ORIENTIERUNG VON 4,4´-BIPHENOL (HELLBLAU) UND 4,4'-DIHYDROXYBENZOPHENON (LILA) IN DER RHEBͻ'WBINDETASCHE (GRÜN). ............................................................................................................................... 133 ABBILDUNG 7.4: VERGLEICH DER ORIENTIERUNG DES GEBUNDENEN LIGANDEN AN H-RASͻ'W172 (LINKS, PDB: 4EPX) UND KRASͻ'W 171 (RECHTS, PDB: 4DST). ............................................................................................................ 134 ABBILDUNG 7.5: RHEBͻ'W IM KOMPLEX MIT 4,4´-BIPHENOL. .................................................................................. 134 ABBILDUNG 7.6: VERGLEICH DER KOMPLEXE DER GTPASE MIT DEM JEWEILIGEN LIGANDEN. ................................................ 135 150 13 Tabellenverzeichnis 13 TABELLENVERZEICHNIS TABELLE 3.1: VERWENDETE PULSPROGRAMME FÜR DIE DURCHFÜHRUNG DER ANGEGEBENEN NMR-EXPERIMENTE. .................... 58 TABELLE 6.1: VERWENDETE SUBSTANZEN FÜR GEMISCH 1. ............................................................................................ 89 TABELLE 6.2: VERWENDETE SUBSTANZEN FÜR GEMISCH 2. ............................................................................................ 94 TABELLE 6.3: VERWENDETE SUBSTANZEN FÜR GEMISCH 3............................................................................................. 97 TABELLE 6.4: VERWENDETE SUBSTANZEN FÜR GEMISCH 4. .......................................................................................... 100 TABELLE 6.5: VERWENDETE SUBSTANZEN FÜR GEMISCH 5. .......................................................................................... 103 TABELLE 6.6: VERWENDETE SUBSTANZEN FÜR GEMISCH 6. .......................................................................................... 105 TABELLE 6.7: VERWENDETE SUBSTANZEN FÜR GEMISCH 7. .......................................................................................... 108 TABELLE 6.8: VERWENDETE BIPHENYL- UND PHENOLDERIVATE. .................................................................................... 111 151 14 Literaturverzeichnis 14 LITERATURVERZEICHNIS 1. Scheffzek, K. & Ahmadian, M. R. GTPase activating proteins: structural and functional insights 18 years after discovery. Cell Mol Life Sci 62, 3014–3038 (2005). 2. Leipe, D. D., Wolf, Y. I., Koonin, E. V. & Aravind, L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol 317, 41–72 (2002). 3. Wennerberg, K., Rossman, K. L. & Der, C. J. The Ras superfamily at a glance. J Cell Sci 118, 843–846 (2005). 4. Takai, Y., Sasaki, T. & Matozaki, T. Small GTP-binding proteins. Physiol Rev 81, 153–208 (2001). 5. Slebos, R., Hoppin, J. & Tolbert, P. K-ras and p53 in pancreatic cancer: association with medical history, histopathology, and environmental exposures in a population-based study. Cancer Epidemiol Biomarkers Prev 9, 1223–1232 (2000). 6. Etienne-Manneville, S. & Hall, A. Rho GTPases in cell biology. Nature 420, 629–635 (2002). 7. Pereira-Leal, J. B. & Seabra, M. C. Evolution of the Rab family of small GTP-binding proteins. J Mol Biol 313, 889–901 (2001). 8. Zerial, M. & McBride, H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2, 107–117 (2001). 9. tĞŝƐ͕<͘ZĞŐƵůĂƚŝŶŐĐĐĞƐƐƚŽƚŚĞ'ĞŶŽŵĞථ͗EƵĐůĞŽĐLJƚŽƉůĂƐŵŝĐdƌĂŶƐƉŽƌƚƚŚƌŽƵŐŚŽƵƚƚŚĞĞůůLJĐůĞ͘Cell 112, 441–451 (2003). 10. Memon, A. The role of ADP-ribosylation factor and SAR1 in vesicular trafficking in plants. Biochim Biophys Acta 1664, 9–30 (2004). 11. Vetter, I. R. & Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 294, 1299–1304 (2001). 12. Wittinghofer, A. & Waldmann, H. Ras- Ein molekularer Schalter bei der Tumorentstehung. Angewandte Chemie 112, 4360–4383 (2000). 13. Prakash, B., Praefcke, G. J., Renault, L., Wittinghofer, A. & Herrmann, C. Structure of human guanylatebinding protein 1 representing a unique class of GTP-binding proteins. Nature 403, 567–571 (2000). 14. Roll-Mecak, A., Cao, C., Dever, T. E. & Burley, S. K. X-Ray structures of the universal translation initiation factor IF2/eIF5B: conformational changes on GDP and GTP binding. Cell 103, 781–792 (2000). 152 14 Literaturverzeichnis 15. Farrar, C., Halkides, C. & Singel, D. The frozen solution structure of p21 ras determined by ESEEM spectroscopy reveals weak coordination of Thr35 to the active site metal ion. Structure 5, 1055–1066 (1997). 16. Ito, Y. et al. Regional polysterism in the GTP-bound form of the human c-Ha-Ras protein. Biochemistry 36, 9109–9119 (1997). 17. Sprang, S. R. G protein mechanisms: insights from structural analysis. Annu Rev Biochem 66, 639–78 (1997). 18. Feuerstein, J., Kalbitzer, H., Goody, R. & Wittinghofer, A. Characterisation of the metal-ion–GDP complex at the active sites of transforming and nontransforming p21 proteins by observation of the 17O-Mn superhyperfine coupling and by kinetic methods. Eur J Biochem 55, 49–55 (1987). 19. Vetter, I. R., Nowak, C., Nishimoto, T., Kuhlmann, J. & Wittinghofer, A. Structure of a Ran-binding domain complexed with Ran bound to a GTP analogue: implications for nuclear transport. Nature 398, 39–46 (1999). 20. Pasqualato, S., Renault, L. & Cherfils, J. Arf, Arl, Arp and Sar proteins: a family of GTP-binding proteins with a structural device for “front-back” communication. EMBO Rep 3, 1035–1041 (2002). 21. Durán, R. V. & Hall, M. N. Regulation of TOR by small GTPases. EMBO Rep 13, 121–128 (2012). 22. Bos, J. L., Rehmann, H. & Wittinghofer, A. GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877 (2007). 23. Lenzen, C., Cool, R. & Prinz, H. Kinetic analysis by fluorescence of the interaction between Ras and the catalytic domain of the guanine nucleotide exchange factor Cdc25Mm. Biochemistry 2960, 7420–7430 (1998). 24. Hutchinson, J. P. & Eccleston, J. F. Mechanism of nucleotide release from Rho by the GDP dissociation stimulator protein. Biochemistry 39, 11348–11359 (2000). 25. Klebe, C., Prinz, H., Wittinghofer, A. & Goody, R. S. The kinetic mechanism of Ran--nucleotide exchange catalyzed by RCC1. Biochemistry 34, 12543–12552 (1995). 26. Renault, L., Guibert, B. & Cherfils, J. Structural snapshots of the mechanism and inhibition of a guanine nucleotide exchange factor. Nature 426, 525–530 (2003). 27. Boriack-Sjodin, P. A., Margarit, S. M., Bar-Sagi, D. & Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 394, 337–343 (1998). 153 14 Literaturverzeichnis 28. Vigil, D., Cherfils, J., Rossman, K. L. & Der, C. J. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 10, 842–857 (2010). 29. Chabre, M. Aluminofluoride and beryllofluoride complexes: a new phosphate analogs in enzymology. Trends Biochem Sci 15, 16–10 (1990). 30. Scheffzek, K., Ahmadian, M. R. & Wittinghofer, A. GTPase-activating proteins: helping hands to complement an active site. Trends Biochem Sci 23, 257–262 (1998). 31. Scheffzek, K. The Ras-RasGAP Complex: Structural Basis for GTPase Activation and Its Loss in Oncogenic Ras Mutants. Science 277, 333–338 (1997). 32. Coleman, D. E. & Sprang, S. R. Structure of GIALPHA1.GPPNHP, autoinhibition in a Galpha proteinsubstrate complex. J Biol Chem 274, 16669–16672 (1999). 33. Barr, F. & Lambright, D. G. Rab GEFs and GAPs. Current opinion in cell biology 22, 461–470 (2010). 34. Strom, M. & Vollmer, P. A yeast GTPase-activating protein that interacts specifically with a member of the Ypt/Rab family. Nature 361, 736–739 (1993). 35. Pan, X., Eathiraj, S., Munson, M. & Lambright, D. G. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature 442, 303–306 (2006). 36. Hillig, R. C. et al. The crystal structure of rna1p: a new fold for a GTPase-activating protein. Mol Cell 3, 781–791 (1999). 37. Mahajan, R., Delphin, C., Guan, T., Gerace, L. & Melchior, F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 88, 97–107 (1997). 38. Klebe, C., Bischoff, F. R., Ponstingl, H. & Wittinghofer, A. Interaction of the nuclear GTP-binding protein Ran with its regulatory proteins RCC1 and RanGAP1. Biochemistry 34, 639–647 (1995). 39. Chakrabarti, P. et al. Insight into Catalysis of a Unique GTPase Reaction by a Combined Biochemical and FTIR Approach. J Mol Biol 367, 983–995 (2007). 40. Scrima, A., Thomas, C., Deaconescu, D. & Wittinghofer, A. The Rap-RapGAP complex: GTP hydrolysis without catalytic glutamine and arginine residues. EMBO J 27, 1145–1153 (2008). 41. Daumke, O., Weyand, M., Chakrabarti, P. P., Vetter, I. R. & Wittinghofer, A. The GTPase-activating protein Rap1GAP uses a catalytic asparagine. Nature 429, 197–201 (2004). 154 14 Literaturverzeichnis 42. Inoki, K., Li, Y., Xu, T. & Guan, K. L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 17, 1829–1834 (2003). 43. Li, Y., Inoki, K. & Guan, K. L. Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol Cell Biol 24, 7965–7975 (2004). 44. Kahn, R. a et al. Consensus nomenclature for the human ArfGAP domain-containing proteins. J Cell Biol 182, 1039–1044 (2008). 45. Luo, R. et al. Kinetic analysis of GTP hydrolysis catalysed by the Arf1-GTP-ASAP1 complex. Biochem. J 402, 439–447 (2007). 46. Ismail, S., Vetter, I., Sot, B. & Wittinghofer, A. The structure of an Arf-ArfGAP complex reveals a Ca2+ regulatory mechanism. Cell 141, 812–821 (2010). 47. Oh, J. S., Manzerra, P. & Kennedy, M. B. Regulation of the neuron-specific Ras GTPase-activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. J Biochem 279, 17980–17988 (2004). 48. Praefcke, G., Geyer, M., Schwemmle, M., Kalbitzer, H. & Herrmann, C. Nucleotide-binding characteristics of human guanylate-binding protein 1 (hGBP1) and identification of the third GTPbinding motif1. J Mol Biol 292, 321–332 (1999). 49. Triola, G., Waldmann, H. & Hedberg, C. Chemical biology of lipidated proteins. ACS chemical biology 7, 87–99 (2012). 50. Stahelin, R. V. Lipid binding domains: more than simple lipid effectors. J Lipid Res 50, 299–304 (2009). 51. Casey, P. J. Biochemistry of protein prenylation. J Lipid Res 33, 1731–1740 (1992). 52. Wolda, S. & Glomsets, J. A. Evidence for modification of lamin B by a product of mevalonic acid. J Biol Chem 263, 5997–6000 (1988). 53. Anderegg, R., Betz, R. & Carr, S. Structure of Saccharomyces cerevisiae mating hormone a-factor. Identification of S-farnesyl cysteine as a structural component. J Biol Chem 263, 18236–18240 (1988). 54. Jackson, J. H. et al. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc Natl Acad Sci U S A 87, 3042–3046 (1990). 55. Aspuria, P. J. & Tamanoi, F. The Rheb family of GTP-binding proteins. Cell Signal 16, 1105–1112 (2004). 155 14 Literaturverzeichnis 56. Benetka, W., Koranda, M. & Eisenhaber, F. Protein Prenylation: An (Almost) Comprehensive Overview on Discovery History, Enzymology, and Significance in Physiology and Disease. Monatshefte für Chemie Chemical Monthly 137, 1241–1281 (2006). 57. Hancock, J. & Magee, A. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 57, 1167–1177 (1989). 58. Rocks, O. et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 307, 1746–1752 (2005). 59. Hancock, J. F., Paterson, H. & Marshall, C. J. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63, 133–139 (1990). 60. Rocks, O. et al. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 141, 458–471 (2010). 61. Chandra, A. et al. The GDI-like solubilizing factor PDE sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol 14, 148–158 (2011). 62. Hanzal-Bayer, M., Renault, L., Roversi, P., Wittinghofer, A. & Hillig, R. C. The complex of Arl2-GTP and PDEdelta: from structure to function. EMBO J 21, 2095–2106 (2002). 63. Kim, J. & Guan, K.-L. Amino acid signaling in TOR activation. Annu Rev Biochem 80, 1001–1032 (2011). 64. Hara, K. et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189 (2002). 65. Kim, D.-H. et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell 11, 895–904 (2003). 66. Sarbassov, D., Ali, S., Kim, D. & Guertin, D. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14, 1296–1302 (2004). 67. Yang, Q., Inoki, K., Ikenoue, T. & Guan, K.-L. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev 20, 2820–2832 (2006). 68. Pearce, L. et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J 405, 513–522 (2007). 69. Zoncu, R., Efeyan, A. & Sabatini, D. M. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12, 21–35 (2011). 156 14 Literaturverzeichnis 70. Sancak, Y. et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008). 71. Kim, E., Goraksha-Hicks, P. & Li, L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol 10, 935–945 (2008). 72. Saucedo, L. J. et al. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol 5, 566–571 (2003). 73. Inoki, K., Li, Y., Zhu, T. & Guan, K. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4, 648–657 (2002). 74. Manning, B. D. & Cantley, L. C. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci 28, 573–576 (2003). 75. Cai, S.-L. et al. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol 173, 279–289 (2006). 76. Sancak, Y. et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25, 903–915 (2007). 77. Inoki, K. et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126, 955–68 (2006). 78. Long, X., Ortiz-Vega, S., Lin, Y. & Avruch, J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J Biochem 280, 23433–23436 (2005). 79. Delgoffe, G. et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12, 295–303 (2011). 80. Maehama, T. et al. RalA functions as an indispensable signal mediator for the nutrient-sensing system. J Biol Chem 283, 35053–35059 (2008). 81. Luo, J. Q. et al. Functional association between Arf and RalA in active phospholipase D complex. Proc Natl Acad Sci U S A 95, 3632–3637 (1998). 82. Ma, W., Park, S.-Y. & Han, J.-S. Role of phospholipase D1 in glucose-induced insulin secretion in pancreatic ɴ cells. Exp Mol Med 42, 456–464 (2010). 83. Welch, H., Coadwell, W. & Ellson, C. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guaninenucleotide exchange factor for Rac. Cell 108, 809–821 (2002). 157 14 Literaturverzeichnis 84. Hernández-Negrete, I. et al. P-Rex1 links mammalian target of rapamycin signaling to Rac activation and cell migration. J Biol Chem 282, 23708–23715 (2007). 85. Saci, A., Cantley, L. C. & Carpenter, C. L. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell 42, 50–61 (2011). 86. Tatebe, H. & Shiozaki, K. Rab small GTPase emerges as a regulator of TOR complex 2. Small GTPases 1, 180–182 (2010). 87. Li, L. et al. Regulation of mTORC1 by the Rab and Arf GTPases. J Biol Chem 285, 19705–19709 (2010). 88. Wang, X. & Proud, C. G. mTORC1 signaling: what we still don’t know. J Mol Cell Biol 3, 206–220 (2011). 89. Yamagata, K. et al. rheb, a growth factor- and synaptic activity-regulated gene, encodes a novel Rasrelated protein. J Biol Chem 269, 16333–16339 (1994). 90. Gromov, P. S., Madsen, P., Tomerup, N. & Celis, J. E. A novel approach for expression cloning of small GTPases: identification, tissue distribution and chromosome mapping of the human homolog of rheb. FEBS letters 377, 221–226 (1995). 91. Patel, P. H. et al. Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J Cell Sci 116, 3601–3610 (2003). 92. Urano, J., Tabancay, a P., Yang, W. & Tamanoi, F. The Saccharomyces cerevisiae Rheb G-protein is involved in regulating canavanine resistance and arginine uptake. J Biol Chem 275, 11198–11206 (2000). 93. Karassek, S. et al. Ras homolog enriched in brain (Rheb) enhances apoptotic signaling. J Biol Chem 285, 33979–33991 (2010). 94. Yu, Y. et al. Expression, purification, crystallization and preliminary structural characterization of the GTPase domain of human Rheb. Acta Crystallogr D Biol Cristallogr 60, 1883–1887 (2004). 95. Yu, Y. et al. Structural basis for the unique biological function of small GTPase RHEB. J Biol Chem 280, 17093–17100 (2005). 96. Clark, G. J. et al. The Ras-related protein Rheb is farnesylated and antagonizes Ras signaling and transformation. J Biol Chem 272, 10608–10615 (1997). 97. Choy, E. et al. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 98, 69–80 (1999). 158 14 Literaturverzeichnis 98. Buerger, C., B, D. & Stambolic, V. Localization of Rheb to the endomembrane is critical for its signaling function. Biochem Biophys Res Commun 344, 869–880 (2006). 99. Castro, A. F., Rebhun, J. F., Clark, G. J. & Quilliam, L. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem 278, 32493–32496 (2003). 100. Marshall, C. B. et al. Characterization of the intrinsic and TSC2-GAP-regulated GTPase activity of Rheb by real-time NMR. Sci Signal 2, ra3 (2009). 101. Mazhab-Jafari, M. T. et al. Real-time NMR study of three small GTPases reveals that fluorescent 2’(3')O-(N-methylanthraniloyl)-tagged nucleotides alter hydrolysis and exchange kinetics. The Journal of biological chemistry 285, 5132–5136 (2010). 102. Hsu, Y.-C., Chern, J. J., Cai, Y., Liu, M. & Choi, K.-W. Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature 445, 785–788 (2007). 103. Rehmann, H. et al. Biochemical characterisation of TCTP questions its function as a guanine nucleotide exchange factor for Rheb. FEBS Lett 582, 3005–3010 (2008). 104. Wang, X. et al. Re-evaluating the roles of proposed modulators of mammalian target of rapamycin complex 1 (mTORC1) signaling. The Journal of biological chemistry 283, 30482–30492 (2008). 105. Dong, X., Yang, B., Li, Y., Zhong, C. & Ding, J. Molecular basis of the acceleration of the GDP-GTP exchange of human ras homolog enriched in brain by human translationally controlled tumor protein. J Biol Chem 284, 23754–64 (2009). 106. Gangloff, Y. et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol 24, 9508–9516 (2004). 107. Goorden, S. M. I. et al. Rheb is essential for murine development. Mol Cell Biol 31, 1672–1678 (2011). 108. Wang, Y. et al. Rheb activates protein synthesis and growth in adult rat ventricular cardiomyocytes. J Mol Cell Cardiol 45, 812–820 (2008). 109. Zou, J. et al. Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev Cell 20, 97–108 (2011). 110. Neuman, N. & Henske, E. P. Non-canonical functions of the tuberous sclerosis complex-Rheb signalling axis. EMBO Mol Med 3, 189–200 (2011). 159 14 Literaturverzeichnis 111. Ma, D., Bai, X., Guo, S. & Jiang, Y. The switch I region of Rheb is critical for its interaction with FKBP38. J Biol Chem 283, 25963–25970 (2008). 112. Uhlenbrock, K. et al. Reassessment of the role of FKBP38 in the Rheb/mTORC1 pathway. FEBS letters 583, 965–970 (2009). 113. Ma, D., Bai, X., Zou, H., Lai, Y. & Jiang, Y. Rheb GTPase controls apoptosis by regulating interaction of FKBP38 with Bcl-2 and Bcl-XL. J Biol Chem 285, 8621–8627 (2010). 114. Karbowniczek, M. The evolutionarily conserved TSC/Rheb pathway activates Notch in tuberous sclerosis complex and Drosophila external sensory organ development. J Clin Invest 120, 93–102 (2010). 115. Cox, A. & Der, C. Farnesyltransferase inhibitors: promises and realities. Curr Opin Pharmacol 2, 388–393 (2002). 116. Hanker, A. B. et al. Differential requirement of CAAX-mediated posttranslational processing for Rheb localization and signaling. Oncogene 29, 380–391 (2010). 117. Ismail, S. A. et al. Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat Chem Biol 7, 942–949 (2011). 118. Kanamori, K., Bluml, S. & Ross, B. Magnetic resonance spectroscopy in the study of hyperammonemia and hepatic encephalopathy. Adv Exp Med Biol 420, 185–194 (1997). 119. Kanamori, K. & Ross, B. Localized 15N NMR spectroscopy of rat brain by ISIS. Magn Reson Med 41, 456– 463 (1999). 120. Serber, Z. et al. Investigating macromolecules inside cultured and injected cells by in-cell NMR spectroscopy. Nat Protoc 1, 2701–2709 (2006). 121. Hajduk, P. J., Huth, J. R. & Fesik, S. W. Druggability indices for protein targets derived from NMR-based screening data. J Med 48, 2518–2525 (2005). 122. Sirockin, F. et al. Structure activity relationship by NMR and by computer: a comparative study. J Am Chem Soc 124, 11073–11084 (2002). 123. Shuker, S. B., Hajduk, P. J., Meadows, R. P. & Fesik, S. W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 274, 1531–1534 (1996). 124. Reckel, S., Lohr, F. & Dotsch, V. In-cell NMR spectroscopy. Chembiochem 6, 1601–1606 (2005). 125. Ito, Y. & Selenko, P. Cellular structural biology. Curr Opin Struct Biol 20, 640–648 (2010). 160 14 Literaturverzeichnis 126. Serber, Z. & Dotsch, V. In-cell NMR spectroscopy. Biochemistry 40, 14317–14323 (2001). 127. Gronenborn, A. M. & Clore, G. M. Rapid screening for structural integrity of expressed proteins by heteronuclear NMR spectroscopy. Protein Sci 5, 174–177 (1996). 128. Serber, Z. et al. High-resolution macromolecular NMR spectroscopy inside living cells. J Am Chem Soc 123, 2446–2447 (2001). 129. Iakouchevam, L., Brown, C., Lawson, J., Obradovic, Z. & Dunker, A. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol 323, 573–584 (2002). 130. Dunker, K., Cortese, M., Romero, P., Iakoucheva, L. & Uversky, V. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J 272, 5129–5148 (2005). 131. Dedmon, M. M., Patel, C. N., Young, G. B. & Pielak, G. J. FlgM gains structure in living cells. Proc Natl Acad Sci U S A 99, 12681–12684 (2002). 132. Hubbard, J. A., MacLachlan, L. K., King, G. W., Jones, J. J. & Fosberry, A. P. Nuclear magnetic resonance spectroscopy reveals the functional state of the signalling protein CheY in vivo in Escherichia coli. Mol Microbiol 49, 1191–1200 (2003). 133. Serber, Z., Ledwidge, R., Miller, S. M. & Dotsch, V. Evaluation of parameters critical to observing proteins inside living Escherichia coli by in-cell NMR spectroscopy. J Am Chem Soc 123, 8895–8901 (2001). 134. Selenko, P. & Wagner, G. Looking into live cells with in-cell NMR spectroscopy. J Struct Biol 158, 244– 253 (2007). 135. Pielak, G. J. & Tian, F. Membrane proteins, magic-angle spinning, and in-cell NMR. Proc Natl Acad Sci U S A 109, 4715–6 (2012). 136. Augustus, A., Reardon, P. & Spicer, L. MetJ repressor interactions with DNA probed by in-cell NMR. Proc Natl Acad Sci U S A 106, 5065–5069 (2009). 137. Li, C. et al. Differential dynamical effects of macromolecular crowding on an intrinsically disordered protein and a globular protein: implications for in-cell NMR spectroscopy. J Am Chem Soc 130, 6310– 6311 (2008). 138. Li, C. et al. Protein (19)F NMR in Escherichia coli. J Am Chem Soc 132, 321–327 (2010). 139. Burz, D. S., Dutta, K., Cowburn, D. & Shekhtman, A. Mapping structural interactions using in-cell NMR spectroscopy (STINT-NMR). Nat Methods 3, 91–93 (2006). 161 14 Literaturverzeichnis 140. Burz, D. S., Dutta, K., Cowburn, D. & Shekhtman, A. In-cell NMR for protein-protein interactions (STINTNMR). Nat Protoc 1, 146–152 (2006). 141. Sakakibara, D. et al. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature 458, 102–105 (2009). 142. Rovnyak, D. et al. Accelerated acquisition of high resolution triple-resonance spectra using non-uniform sampling and maximum entropy reconstruction. J Magn Reson 170, 15–21 (2004). 143. Schmieder, P., Stern, A., Wagner, G. & Hoch, J. Improved resolution in triple-resonance spectra by nonlinear sampling in the constant-time domain. J Biomol NMR 4, 483–490 (1994). 144. Laue, E., Mayger, M., Skilling, J. & Staunton, J. Reconstruction of phase sensitive 2D NMR spectra by maximum entropy. J Magn Reson 68, 14–29 (1986). 145. Dyson, H. & Wright, P. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol 6, 197–208 (2005). 146. Peng, H. Xenopus laevis: Practical uses in cell and molecular biology. Solutions and protocols. Methods Cell Biol 36, 657–662 (1991). 147. Colman, A. Transcription of DNAs of Known Sequence after Injection into the Eggs and Oocytes of Xenopus laevis. Eur J Biochem 57, 85–96 (1975). 148. Selenko, P., Serber, Z., Gadea, B., Ruderman, J. & Wagner, G. Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. Proc Natl Acad Sci U S A 103, 11904– 11909 (2006). 149. Hänsel, R., Foldynova-Trantirkova, S., Dötsch, V. & Trantirek, L. Investigation of Quadruplex Structure Under Physiological Conditions Using In-Cell NMR. Topics in Current Chemistry 1–19 (2012).doi:10.1007/128 150. Bodart, J. et al. NMR observation of Tau in Xenopus oocytes. J Magn Reson 192, 252–257 (2008). 151. Selenko, P. et al. In situ observation of protein phosphorylation by high-resolution NMR spectroscopy. Nat Struct Mol Biol 15, 321–329 (2008). 152. Almeida, P. F. & Pokorny, A. Mechanisms of antimicrobial, cytolytic, and cell-penetrating peptides: from kinetics to thermodynamics. Biochemistry 48, 8083–8093 (2009). 153. Takeuchi, T. et al. Direct and rapid cytosolic delivery using cell-penetrating peptides mediated by pyrenebutyrate. ACS Chem Biol 20, 299–303 (2006). 162 14 Literaturverzeichnis 154. Ogino, S. et al. Observation of NMR signals from proteins introduced into living mammalian cells by reversible membrane permeabilization using a pore-forming toxin, streptolysin O. J Am Chem Soc 131, 10834–10835 (2009). 155. Walev, I. et al. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci U S A 98, 3185–3190 (2001). 156. Bryant, J. E., Lecomte, J. T., Lee, A. L., Young, G. B. & Pielak, G. J. Protein dynamics in living cells. Biochemistry 44, 9275–9279 (2005). 157. Bryant, J., Lecomte, J., Lee, A., Young, G. & Pielak, G. Cytosol has a small effect on protein backbone dynamics. Biochemistry 45, 10085–10091 (2006). 158. Pielak, G. Retraction. Biochemistry 46, 8206 (2007). 159. Visiongain dŚĞƌĂƉĞƵƚŝĐDŽŶŽĐůŽŶĂůŶƚŝďŽĚŝĞƐ෴͗tŽƌůĚDĂƌŬĞƚϮϬϭϭ-2021. (2011). 160. Arkin, M. R. & Wells, J. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov 3, 301–317 (2004). 161. Lo Conte, L., Chothia, C. & Janin, J. The atomic structure of protein-protein recognition sites. J Mol Biol 285, 2177–2198 (1999). 162. Tsai, C. & Lin, S. Protein-protein interfaces: architectures and interactions in protein-protein interfaces and in protein cores. Their similarities and differences. Crit Rev Biochem Mol Biol 31, 127–152 (1996). 163. ^ƚŝƚĞƐ͕t͘WƌŽƚĞŝŶоWƌŽƚĞŝŶ/ŶƚĞƌĂĐƚŝŽŶƐ͗/ŶƚĞƌĨĂĐe Structure, Binding Thermodynamics, and Mutational Analysis. Chem Rev 97, 1233–1250 (1997). 164. Arkin, M. R. et al. Binding of small molecules to an adaptive protein-protein interface. Proc Natl Acad Sci U S A 100, 1603–1608 (2003). 165. Bonan, A. & Thorn, K. Anatomy of hot spots in protein interfaces. J Mol Biol 280, 1–9 (1998). 166. DeLano, W. L., Ultsch, M., de Voss, A. & Wells, J. Convergent solutions to binding at a protein-protein interface. Science 287, 1279–1283 (2000). 167. Boehm, H. et al. Novel inhibitors of DNA gyrase: 3D structure based biased needle screening, hit validation by biophysical methods, and 3D guided optimization. A promising alternative to random screening. J Med Chem 43, 2664–2667 (2000). 168. Hajduk, P. J., Galloway, W. R. J. & Spring, D. R. A question of library design. Nature 470, 42–43 (2011). 163 14 Literaturverzeichnis 169. Leeson, P. & Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat rev Drug Discov 6, 881–890 (2007). 170. Schubbert, S., Shannon, K. & Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7, 295–308 (2007). 171. Maurer, T. et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A 109, 5299–5304 (2012). 172. Sun, Q. et al. Discovery of Small Molecules that Bind to K-Ras and Inhibit Sos-Mediated Activation. Angew Chem Int Ed Engl 51, 6140–6143 (2012). 173. Meyer, B. & Peters, T. NMR spectroscopy techniques for screening and identifying ligand binding to protein receptors. Angew Chem Int Ed Engl 42, 864–890 (2003). 174. Viegas, A., Manso, J., Nobrega, F. L. & Cabrita, E. J. Saturation-Transfer Difference (STD) NMR: A Simple and Fast Method for Ligand Screening and Characterization of Protein Binding. J Chem Educ 88, 990– 994 (2011). 175. Bloch, F., Hansen, W. & Packard, M. Nuclear Induction. Phys Rev 69, 127 (1946). 176. Purcell, E., Torrey, H. & Pound, R. Resonance Absorption by Nuclear Magnetic Moments in a Solid. Phys Rev 69, 37–38 (1946). 177. Bloch, F. The Principle of Nuclear Induction. The Nobel Prize in Physics (1952). 178. Purcell, E. Research in Nuclear Magnetism. The Nobel Prize in Physics (1952). 179. Williamson, M., Havel, T. & Wüthrich, K. Solution conformation of proteinase inhibitor IIA from bull seminal plasma by 1H nuclear magnetic resonance and distance geometry. J Mol Biol 182, 295–315 (1985). 180. Tzeng, S., Pai, M. & Kalodimos, C. NMR studies of large protein systems. Meth Mol Biol 831, 133–140 (2012). 181. Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of biomolecular NMR 6, 277–93 (1995). 182. :ŽŚŶƐŽŶĂ͕͘͘ΘůĞǀŝŶƐď͕͘EDZsŝĞǁථ͗ĐŽŵƉƵƚĞƌprogram for the visualization and analysis of NMR data. J Biomol NMR 4, 603–614 (1994). 164 14 Literaturverzeichnis 183. Dominguez, C., Boelens, R. & Bonvin, A. M. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125, 1731–1737 (2003). 184. Simonson, T. & Warren, G. L. Crystallography & NMR System: A New Software Suite for Macromolecular Structure Determination. Acta Cryst. D54, 905–921 (1998). 185. Page, R., Peti, W., Wilson, I., Stevens, R. & Wüthrich, K. NMR screening and crystal quality of bacterially expressed prokaryotic and eukaryotic proteins in a structural genomics pipeline. Proc Natl Acad Sci U S A 102, 1901–1905 (2005). 186. Parelle, T. Bruker BioSpin Puls Program Catalogue. (2006). 187. Schanda, P., Kupce, E. & Brutscher, B. SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J Biomol NMR 33, 199–211 (2005). 188. Ernst, R. Principles of Nuclear Magnetic Resonance in One and Two Dimensions. (1987). 189. Pervushin, K., Vögeli, B. & Eletsky, A. Longitudinal (1)H relaxation optimization in TROSY NMR spectroscopy. J Am Chem Soc 124, 12898–12902 (2002). 190. Piotto, M., Saudek, V. & Sklenar, V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR 2, 661–665 (1992). 191. Sklenar, V., Piotto, M., Leppik, R. & Saudek, V. Gradient-Tailored Water Suppression for 1H-15N HSQC Experiments Optimized to Retain Full Sensitivity. J Magn Reson 102, 241–245 (1993). 192. Palmer, A., Cavanahg, J., Wright, P. & Rance, M. Sensitivity improvement in proton-detected 2dimensional heteronuclear correlation NMR spectroscopy. J Magn Reson 93, 151–170 (1991). 193. Kay, L., Keifer, P. & Saarinen, T. Pure absorption gradient enhanced heteronuclear single quantum correlation spectroscopy with improved sensitivity. J Am Chem Soc 114, 10663–10665 (1992). 194. Schleucher, J. et al. A general enhancement scheme in heteronuclear multidimensional NMR employing pulsed field gradients. J Biomol NMR 4, 301–306 (1994). 195. Schanda, P. & Brutscher, B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J Am Chem Soc 127, 8014–8015 (2005). 196. Schwarten, M., Berghaus, C., Heumann, R. & Stoll, R. Sequence-specific 1H, 13C, and 15N backbone assignment of the activated 21 kDa GTPase rRheb. Biomol NMR Assign 1, 105–108 (2007). 165 14 Literaturverzeichnis 197. Berghaus, C., Schwarten, M., Heumann, R. & Stoll, R. Sequence-specific 1H, 13C, and 15N backbone assignment of the GTPase rRheb in its GDP-bound form. Biomol NMR Assign 1, 45–47 (2007). 198. Thapar, R., Williams, J. G. & Campbell, S. L. NMR characterization of full-length farnesylated and nonfarnesylated H-Ras and its implications for Raf activation. J Mol Biol 343, 1391–1408 (2004). 199. Debye, P. J. Polar Molecules. Angewandte Chemie 42, 995 (1929). 200. Li, C., Wang, Y. & Pielak, G. Translational and rotational diffusion of a small globular protein under crowded conditions. J Phys Chem B 113, 13390–13392 (2009). 201. Banci, L., Barbieri, L., Bertini, I., Cantini, F. & Luchinat, E. In-cell NMR in E. coli to monitor maturation steps of hSOD1. PLoS One 6, e23561 (2011). 202. Im, E. et al. Rheb is in a high activation state and inhibits B-Raf kinase in mammalian cells. Oncogene 21, 6356–6365 (2002). 203. Sakai, T. et al. In-cell NMR spectroscopy of proteins inside Xenopus laevis oocytes. J Biomol NMR 36, 179–188 (2006). 204. Fres, J. M., Muller, S. & Praefcke, G. J. Purification of the CaaX-modified, dynamin-related large GTPase hGBP1 by coexpression with farnesyltransferase. J Lipid Res 51, 2454–2459 (2010). 205. Szkopinska, A. Ubiquinone. Biosynthesis of quinone ring and its isoprenoid side chain. Intracellular localization. Acta Biochim Pol 47, 469–480 (2000). 206. Avanti Polar Lipids Datasheet for 131101P. (2012). 207. Hammond, G. R. V. et al. PI4P and PI(4,5)P2 Are Essential But Independent Lipid Determinants of Membrane Identity. Science 727, 2–6 (2012). 208. Kapoor, S. et al. The role of G-domain orientation and nucleotide state on the Ras isoform-specific membrane interaction. Eur Biophys J Epub ahead, (2012). 209. Güldenhaupt, J. et al. N-Ras Forms Dimers at POPC Membranes. Biophysical journal 103, 1585–1593 (2012). 210. Fres, J. M. Post-translational lipid modification and membrane targeting of the dynaminrelated large GTPase hGBP1. (2011). 211. Nilsson, J., Weis, K. & Kjems, J. The C-terminal extension of the small GTPase Ran is essential for defining the GDP-bound form. J Mol Biol 318, 583–593 (2002). 166 14 Literaturverzeichnis 212. Chessari, G. & Woodhead, A. J. From fragment to clinical candidate—a historical perspective. Drug Discov Today 14, 668–675 (2009). 213. Limpinski, C., Lombardo, F., Dominy, B. & Freeney, P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46, 3–26 (2001). 214. Song, C. M., Lim, S. J. & Tong, J. C. Recent advances in computer-aided drug design. Brief Bioinform 10, 579–591 (2009). 215. Rosnizeck, I. C. et al. Metal-Bis(2-picolyl)amine Complexes as State 1(T) Inhibitors of Activated Ras Protein. Angew Chem Int Ed Engl 1, 1–6 (2012). 216. Rees, D. C., Congreve, M., Murray, C. W. & Carr, R. Fragment-based lead discovery. Nat rev Drug Discov 3, 660–672 (2004). 217. http://www.bfr.bund.de, Bundesinstitut für Risikobewertung. Fragen und Antworten zu Bisphenol A in verbrauchernahen Produkten (2012). 218. FAO/WHO Expert Meeting to Review Toxicological and Health Aspects of Bisphenol A. (2010). 219. Schieborr, U. et al. How much NMR data is required to determine a protein-ligand complex structure? Chembiochem 6, 1891–1898 (2005). 220. Rao, V. & Srinivas, K. Modern drug discovery process: An in silico approach. JBSA 2, 89–94 (2011). 221. Quashie, P. K., Sloan, R. D. & Wainberg, M. a Novel therapeutic strategies targeting HIV integrase. BMC medicine 10, 34 (2012). 167 15 Anhang 15 ANHANG 15.1 AIR-TABELLE FÜR DIE HADDOCK-MODELLIERUNG Bisphenol A: 66, 70, 71, 72, 73, 74, 75, 76, 77, 78, 80, 106 4,4´-Biphenol: 66, 70, 71, 72, 73, 75, 76, 77, 78, 80, 106 1-(3,4-Dichlorophenyl)piperazin: 11, 61, 70, 71, 72, 73, 75, 76, 78, 79, 101, 107 4,4'-Dihydroxybenzophenon: 66, 70, 71, 72, 73, 75, 76, 77, 78, 80, 106 15.2 ÄNDERUNGEN HADDOCK-RUN Für die Modellierungen der Komplexe aus RhebͻGDP mit dem entsprechenden Liganden wird das von A. Bonvin vorgeschlagene Set-Up mit den entsprechenden Änderungen für kleine Liganden verwendet. Zur Übersicht sind folgend die für kleine Liganden zu übernehmenden Änderungen aufgeführt: DOCKING protocol initial temperature for second TAD cooling step with flexible side chain at the interface: 500 K number of MD steps for rigid body high temperature TAD: 0 number of MD steps during first rigid body cooling step: 0 number of MD steps during second cooling stage with flexible side chains at interface: 500 number of MD steps during third cooling stage with fully flexible interface: 500 168