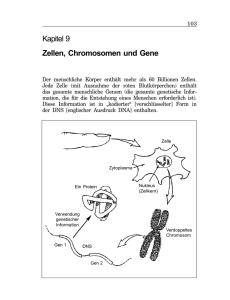
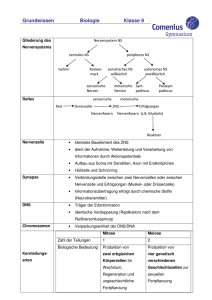
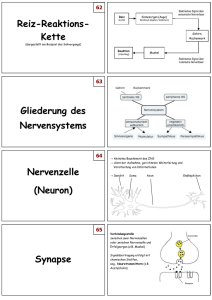
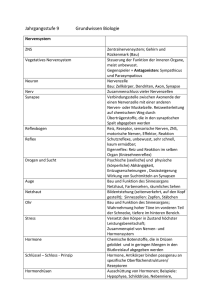
E.coli
Werbung

Kolloquiumsunterlagen Seite 1 Molekularbiologische Übungen I Kolloquiumsunterlagen Beispiel: E.coli a.o. Prof. Mag. Dr. Herta Steinkellner Dr. Rudolf Mitterbauer Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 2 Einführung: Molekularbiologische Verfahren unterliegen einer raschen Weiterentwicklung. Viele Methoden konnten sich deshalb so rasch entwickeln, da sie dem großen Erfahrungsschatz entstammen, der im Umgang mit Bakterien und Bakteriophagen gewonnen wurden. Escherichia coli ist mit Abstand der wichtigste Organismus der Molekularbiologie. Fast jede gentechnische Modifikation wird zuerst in E.coli realisiert und das eigentliche Klonieren, d.h. das Vereinzeln und die Vervielfältigung von einem bestimmten DNS-Abschnitt, findet nahezu ausnahmslos in E. coli statt. Die Basis des Klonierens ist die Isolierung, die Modifikation und die Verknüpfung spezifischer DNS Moleküle zu neuen Funktionseinheiten. (Der Begriff wird heute allerdings viel weiter gefasst und wird als Überbegriff für gentechnisches Arbeiten im allgemeinen verwendet). Damit die neu kombinierte DNS schließlich in E. coli erhalten bleibt braucht man geeignete Vektorsysteme (Plasmide) und Verfahren, mit deren Hilfe sich die neu kombinierte DNS effizient in die E.coli Zellen einschleusen lässt (Transformation). Ziel dieses Beispiels ist (i) das Kennenlernen einiger einfacher Klonierungsstrategien und der Umgang mit den wichtigsten DNS-modifizierenden Enzymen, (ii) die Herstellung von Bakterienzellen zur Transformation, (iii) sowie eine einfache Methode zur Isolierung und Analyse von Plasmid DNS. Diverse Eigenschaften von Klonierungsplasmiden sollen näher gebracht, sowie ein prinzipielles Verständnis oftmals kompliziert erscheinender bakterieller Genotypen ermöglicht werden. Im wesentlichen werden zwei verschiedende Klonierungen durchgeführt: Einerseits wird ein für eine Antibiotikaresistenz codierender Abschnitt eines Klonierungsplasmids (Plasmid pCR4-TOPO) dermaßen verändert, dass die Resistenz verloren geht und somit auf entsprechenden Selektionsplatten kein Wachstum der Klone mehr möglich ist. Andererseits wird ein Insert aus einem Pool unterschiedlich großer Fragmente von λ-DNS in die „multiple cloning site“ eines gebräuchlich verwendeten Klonierungsvektors (pUC18) gesetzt. Von einigen ausgewählten Transformanten wird die Plasmid DNS isoliert und diese auf die Größe und Orientierung der erhaltenen Fragmente überprüft. Tipp: dieses Skriptum ersetzt auf keinen Fall ein Lehrbuch. Hier werden nur Minimalinformationen gegeben die sich auf bestimmte Themen konzentrieren. Nehmen Sie eines der empfohlenen Bücher (Kapitel 10) zur Hand um genügend Hintergrund Informationen zu erhalten um die Experimente auch tatsächlich zu verstehen! Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 3 INHALTSVERZEICHNIS 1. Plasmid-Vektoren 1.1. Replikation von Plasmiden 1.2. Inkompatibilität von Plasmiden 1.3. Plasmid-codierte Selektionsmarker 1.4. Andere Eigenschaften von Plasmid Vektoren 1.5. Typische Plasmid Vektoren: Plasmide der pUC Serie (Beispiel pUC18) 2. Alpha Komplementation 3. E. coli: Phänotyp, Genotyp 4. Bakterielle Modifikations- und Restriktionssysteme 5. Restriktionsenzyme 6. Sonstige DNS-modifizierende Enzyme 7. Transformation von E.coli 7.1. Herstellung kompetenter Zellen 8. Anzucht der Bakterienkulturen und Plasmid-Präparation 8.1. Aufschluß der Bakterienkultur und Reinigung von Plasmiden 9. Gelelektrophorese von DNS 9.1. Agarosegele 10. Weiterführende Lehrbücher 11. Auswahl einiger Übungsbeispiele und Fragen Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 4 1. Transformation von E. coli Einer der wichtigsten Schritte bei der Etablierung eines rekombinanten Organismus ist das Einbringen der zuvor in vitro neu kombinierten DNS in die Wirtszelle. Dieser Prozess wird Transformation genannt, wenn die DNS passiv, d.h. auf physikalischem Weg in die Wirtszelle eingeschleust wird. Als Infektion bezeichnet man hingegen, wenn die DNS mit Hilfe von Phagen, also auf biologischem Weg, in die E. coli Zelle gebracht wird. Für die Transformation werden in der Regel Derivate des Escherichia coli-Stammes K12 verwendet. Dieser Stamm ist ein sogen. "Sicherheitsstamm", das bedeutet, dass ihm die für die Pathogenizität essentiellen Gene fehlen. Der Stamm XL1-Blue besitzt z. B. den großen Vorteil, - recombinationsdefizient (recA ) zu sein (unerwünschte DNS-Rekombinationen kommen nicht vor). Transformieren lassen sich sogenannte kompetente Zellen. Deren Membranen wurden durch gezielte Behandlung (chemisch, physikalisch) durchlässig gemacht. Grundsätzlich können zwei Arten von kompetenten E. coli Zellen unterschieden werden: chemisch-kompetente und elektrokompetente Zellen. Sowohl das Protokoll zur Präparation als auch zur Transformation der kompetenten Zellen unterscheidet sich in wesentlichen Aspekten. • Chemische Transformation mittes CaCl2: Für diese Art von Transformation werden die Membranen von E. coli Zellen durch Behandlung mit CaCl2 vorübergehend für DNS durchlässig gemacht. Präpariert werden die Zellen durch Herstellen einer "log-phase" Bakterien Zellensuspension in eiskaltem 50 mM Calciumchlorid. Für die eigentliche Transformation inkubiert man die kompetenten Zellen zusammen mit der zu transformierenden DNS in einer CaCl2 Lösung für 30 min bei 0oC. Durch die Ca2+ Ionen in der Zellsuspension wird die DNS als Polyanion mikrokristallin auf der Oberfläche der kompetenten Zellen präzipitiert. Danach erfolgen rasche Temperaturwechsel: ein sogenannter Hitzeschock (90 sec bei 42oC) mit darauffolgender kurzer Inkubation bei 0oC. Durch diese Wechsel wird die DNS über nicht genau geklärte Mechanismen passiv in der Zelle aufgenommen. Zur Regeneration wird der Transformationsansatz danach in Nährmedium (LB, SOC) bei 37oC inkubiert. Während dieser Inkubation können neue phänotypische Eigenschaften die vom Plasmid in die Zelle eingebracht werden exprimiert werden (beispielsweise eine Antibiotikaresistenz). Zum Abschluss werden die Zellen auf dem geeigneten Selektivmedium ausplattiert. Die Transformationseffizienzen liegen bei diesem Verfahren bei ca 105 – 107 Transformanten pro µg DNS. • Elekrische Transformation (Elektroporation): Eine weitere Methode, um DNS in mikrobielle (also auch E. coli), aber auch pflanzliche oder tierische Zellen einzubringen, ist die Elektroporation. Diese Methode beruht auf der Beobachtung, dass Strompulse mit hoher Spannung die Fusion von zellulären Plasmamembranen induzieren können. Zusätzlich können Zellen nach einem "elektrischen Schock" exogene DNS-Fragmente aus der Umgebungslösung aufnehmen. Auch hier gilt, dass neben vielen exogenen/technischen Faktoren (z. B. Temperatur, Spannung, Leitfähigkeit, DNS-Topologie) auch genetische, Bakterienstammabhängige Faktoren die Effizienz der Transformation beeinflußen. Die Transformationseffizienz bei diesem Verfahren kann bei zu 109 Transformanten pro µg DNS (109cfu: colony forming units) sein, Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 5 was deutlich höher ist als bei der chemischen Methode. Ein Vorteil der Elektroporation gegenüber der Transformation mit chemisch-kompetenten Zellen ist weiters, dass hierbei auch sehr große DNSFragmente erfolgreich transformiert werden können (Größenlimit bei chemisch kompetenten Zellen ist um die 50 kb). Plasmid CaCl2 30 min OoC E.coli Zelle Elektroporation Feldstärke 10-14kV/cm 90 sec 42oC 1-2 min, 0oC + Medium, 37oC + Medium, 37oC Ausplattieren auf Selektionsagar Abbildung 1: Die bakterielle Transformation: Um Bakterien zu transformieren (linke Hälfte der Abbildung), müssen ihre Membranen durch eine Behandlung von CaCl2 vorgeschädigt sein. Die Bakterien werden „kompetent“ gemacht. Die Transformation erfolgt durch Co-Inkubation des Plasmides mit Zellen in Anwesenheit von CaCl2 bei nierdrigen Temperaturen. Danach erfolgt ein sog. Hitzeschock bei 42oC. Bei der Elektroporation (rechte Hälfte der Abbildung) werden Zellen vor der Transformation salzfrei gewaschen und dann zusammen mit DNS in eine Küvette gegeben, die an zwei gegenüberliegenden Seiten mit Plattenelektroden ausgekleidet sind. Nach anlegen einer Feldstärke wird die DNS bei der Entladung in die Bakterienzelle aufgenommen. Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 6 2. Vektoren für die E.coli transformation: 2.1. Plasmid-Vektoren Bakterielle Plasmide sind extrachromosomale DNS Moleküle, meist doppelsträngig zirkulär und in der Regel zwischen 2 und 200 kilobasen (1kb=1000 basen) groß. Ihre Replikation erfolgt unabhängig vom Bakterien-Chromosom, ist aber trotzdem von Enzymen der Wirtszelle abhängig. In den vergangenen Jahren wurde eine Vielzahl, zum Teil sehr spezieller, Plasmid Vektoren entwickelt. All diesen Vektoren ist gemeinsam, dass sie eine Sequenz besitzen, die man als Replicon bezeichnet (siehe Anhang und Abbildung 4). Dies ist eine genetische Einheit, welche neben einem Replikationsursprung („origin of replication“ ori) zur Initiierung der Replikation die dazugehörigen (cisacting) Kontrollelemente besitzt. Die Art eines Plasmid-Replicons bestimmt letztendlich die Kopienzahl (plasmid copynumber, PCN) eines Plasmids. Die ori Sequenz wird von der DNS Polymerase von E. coli als Initiationsstartpunkt für die Replikation erkannt und stellt sicher, dass das Plasmid unabhängig von der genomischen DNS repliziert werden kann und neben der genomischen DNS in der Zelle persistieren. Mehr als 30 verschiedene Replicons wurden bereits in Plasmiden identifiziert. Die meisten, gegenwärtig verwendeten Plasmid-Vektoren tragen jedoch dasselbe Replicon, abgeleitet vom Plasmid pMB1 (nahe verwandt zum ebenso häufigen colE1 Replicon). Plasmide mit diesem Replicon liegen mit einer Kopienzahl von zumindest 15 bis 20 pro Zelle vor (multicopy plasmid). Plasmid Plasmidgröße Kopienzahl Ertrag/ml Kultur pGEM 2700 bp 300-700 1,8 – 4,1 µg pUC 2700 bp 500-700 2,9 – 4,1 µg pBR322 4400 bp >25 >0,23 µg ColE1 4500 bp >15 >0,15 µg pACYC 4000 bp ~10 ~0,09µg pSC101 9000bp ~6 ~0,12µg Tabelle 1: Kopienzahl häufig verwendeter Plasmide Zusätzlich zum pMB1/Col E1 origin gibt es auch weitere origins wie den f1-origin, welcher die Replikation des Plasmids als Einzelstrang ermöglicht. T3 und T7 Regionen sind Promotoren, die von den RNA-Polymerasen der T3- und T7-Phagen spezifisch erkannt werden und so zur in vitro Synthese von RNA-Transkripten des klonierten Fragments benutzt werden können. 2.2. Inkompatibilität von Plasmiden Zwei Plasmide, die den gleichen Replicon-Typ (Regulation der Replikation) aufweisen, können ohne entsprechende Selektion in einer bakteriellen Kultur nicht koexistieren. Grund ist die Kompetition der Plasmide während der Replikation und der Weitergabe (partitioning) in die Tochterzellen. Schon Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 7 innerhalb weniger Generationen kann so eines der Plasmide eliminiert werden. Dieses Phänomen ist als "Inkompatibilität" bekannt. Inkompatibilitätsgruppe negatives Kontrollelement colE1, pMB1 RNAI (Kontrolle des pre-RNAII processing) IncFII, pT181 RNA (Kontrolle der RepA Synthese) P1, F, R6K, pSC101, p15A Iterons (Kontrolle der RepA Konz.) Tabelle 2: Inkompatibilitätsgruppen 2.3. Plasmid-codierte Selektionsmarker Weiters enthalten alle Vektoren einen oder mehrere Selektionsmarker. Dabei handelt es sich um Gene, deren Produkte der E. coli Zelle besondere Eigenschaften verleihen, sodass sie leicht von nicht transformierten Zellen zu unterscheiden sind. Meistens handelt es sich bei einem Selektionsmarker um ein Resistenzgen, das ein Enzym für die Inaktivierung eines spezifischen Antibiotikums codiert. So codiert beispielsweise das Ampicillinresistenzgen (bla, ampr) das Enzym ß-Lactamase, welches das Antibiotikum Ampicillin inaktiviert. Auf Ampicillin-hältigem Medium können somit die Zellen, die während der Transformation nun tatsächlich ein Plasmid aufgenommen haben, aus dem großen Überschuss an nicht transformierten Zellen selektiert werden. Weit verbreitet sind auch andere Antibiotikaresistenzen wie: Tetracyclin. Kanamycin, Chloramphenicol, Streptomycin 2.4. Andere Eigenschaften von Plasmid Vektoren Ein weiterer wichtiger Bestandteil von Klonierungsvektoren ist ein speziell ausgewiesener Bereich, der die zu klonierenden DNS Fragmente aufnehmen soll. Dieser Bereich enthält eine Vielzahl von Erkennungssequenzen für verschiedene Restriktionsendonukleasen (= DNS schneidende Enzyme), die jeweils nur einmal in dem Plasmid vorhanden sind. Diese Sequenz bezeichnet man „multiple cloning site (MCS)“. Durch diese „unique“ vorkommenden Restriktionsschnittstellen ist es möglich ein Plasmid gezielt an einer Stelle zu schneiden (zu linearisieren) und eine fremde Sequenz zu insertieren. Die MCS liegt oft innerhalb des codierten Teils des lacZ-Gens welches eine weitere wichtige Eigenschaft zur Unterscheidung von rekombinanten Plasmiden von nicht rekombinanten Plasmiden erlaubt. Der Einbau der Insert-DNS in die MCS zerstört den Leseraster dieses lacZ-GenFragments und die betroffenen Bakterien-Kolonien bleiben in Gegenwart von X-Gal farblos (AlphaKomplementierung). 3. Das lacZ Gen („Alpha-Komplementierung") Mithilfe der sogenannten "Alpha-Komplementation" können beim Klonieren positive Klone (BakterienZellen, die ein rekombinantes Plasmid enthalten) sehr einfach identifiziert werden. Das System beruht Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 8 darauf, dass das Gen für die β-Galactosidase in zwei Fragmente zerlegt wird: Der N-Terminale Teil (α-Fragment) ist auf dem Plasmidvektor codiert, der C-terminale Teil (ω-Fragment) liegt auf dem Bakterienchromosom verankert. Durch die gleichzeitige Expression beider Fragmente kann die Aktivität der β-Galactosidase hergestellt werden. Die entsprechenden Zellen/Kolonien verfärben sich dann in der Gegenwart des β-Galactosidase-Substrates X-Gal blau. X-Gal (5-Brom-4-chlor-3-indolyl-βD-galactosid) ist normalerweise farblos, wird es jedoch durch β-Galactosidase gespalten wird, entsteht das tiefblaue 5-Brom-4-Chlor-Indigo. Falls die Expression des α-Fragments unter der Kontrolle des lac-Repressors (Hintergrundinformation dazu in jedem Standardlehrbuch „lac-operon“) steht (z.B. pUC18) muss natürlich neben dem Substrat (X-Gal ist kein Inductor der Lactose Gene!) auch ein Induktor wie das IPTG (Isopropyl- β-D-thiogalactosid) zugegeben werden. Befindet sich nun die MCS (multiple cloning site) innerhalb des Plasmid-codierten N-Terminus des lacZ Gens, wird durch die Insertion eines DNS-Fragments in die MCS der Leserahmen des αFragments unterbrochen bzw. dessen Funktion zerstört. Es kommt zu keiner α-Komplementation, die entsprechende Kolonie erscheint weiß. lacZ alpha MCS a (ampR) pUC18 2686 bp ori (pMB1 derived) Abbildung 4: Das Klonierungsplasmid pUC 18 enthält den kodierenden Bereich für das α-Komplement der β-Galactosidase als Reporter Gen (lacZ alpha) Die Transkription des Gens wird durch den regulierbaren lac Promotor/Operator kontrolliert. Im „vorderen“ Bereich des β-Galactosidase Gens befindet sich die MCS. Diese enthält eine Vielzahl von Erkennungssequenzen für unterschiedliche Restriktionsendonukleasen, die das Plasmid jeweils nur einmal schneiden. Fügt man in die MCS ein DNS Frafment ein, so wird dadurch das β-Galactosidase Gen zerstört. Das ist die Basis für ein blau/weiss screening mit Xgal. Wie fast alle gängigen Vetktoren enthält pUC18 einen Replikationsursprung (ori), den die Polymerasae aus E. coli erkennt und damit das Plasmid autonom, d.h. unabhängig von bakteriellen Genom replizieren kann. Weiters enthält pUC19 das AntibiotikaResistenzgen ampr (bla), das Ampicillin Resistenz vermittelt. Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 9 Abbildung 5a: Funktion der β-Galactosidase und die Verwendung von Xgal pUC18 (2,7 kb) Recomb. pUC18 MCS Transforamtion in E.coli und Selektion auf X-Gal hälttigen Agarplatten Expression von β-Galactosidase MCS (+ Insert) β-Galactosidasegen zerstört Abbildung 5b: Blau/Weiss Selektion durch das β-Galctosidase Reporter-Gen. Zellen, in denen Teilbereiche des β-Galctosidase Gens deletiert sind, können keine β-Galctosidase synthetisieren. Dieser Defekt kann in Plasmiden der pUC Serie korrigiert (komplementiert) werden, da auf diesen Plasmiden der fehlende Genbereich (α-Fragment) zusammen mit den Kontrollelementen vorhanden ist. Solche komplementierten Zellen können leicht dadurch identifiziert werden, dass dem Medium der Indikator X-Gal dazugegeben wird. Dieses farblose Substrat der β-Galctosidase wird von dem Enzym an der glycosididischen Bindung hydrolysiert und dann an der Luft zu einem blauen Indigo-Farbstoff oxydiert. Die Kulturen färben sich also blau. Enthält die MCS in den Klonierungsvektoren ein FremdDNS Fragment, so wird dadurch das Gen für das β-Galctosidase-Teilprotein zerstört. Zellen die ein derartiges rekombinantes Plasmid aufgenommen haben, können keine funktionsfähige β-Galctosidase synthetisieren und bleiben daher auch in Gegenward des Indikators farblos (weiss). Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 10 3. E. coli: Phänotyp, Genotyp Phänotyp: sichtbare oder sonstwie messbare Eigenschaften eines Organismus Der Phänotyp wird in Normalschrift angegeben, der erste Buchstabe wird groß geschrieben wird und gefolgt von einem Superscript + oder – (manchmal auch r, resistent; s, sensitiv). Genotyp: die für die Ausprägung des Phänotyps verantwortlichen genetischen Faktoren • Gene in "unverändertem Zustand" (Wildtyp-Allele) werden bei der Beschreibung des Genotyps nicht explizit angeführt. • Gene werden mit drei kursiv geschriebenen Kleinbuchstaben (üblicherweise ein Mnemonic für die Funktion des jeweiligen Gens) benannt. Manchmal wird zur Spezifikation einer bestimmten Mutation (eines best. Alleles) noch eine Nummer, oder ein Großbuchstabe angehängt. Die Superscripts + und – sind zur korrekten Angabe des Genotyps nicht notwendig (redundant!). • Die Kennzeichnung von Deletionsmutanten erfolgt durch ein ∆ (manchmal auch "del" oder "d"), gefolgt von der in Klammern gesetzten Bezeichnung des deletierten Gens. • Manchmal folgt, in Klammern gestellt, der Bezeichnung des Genotyps eine Bezeichnung des Phänotyps (wenn dieser nicht klar aus ersterem ersichtlich). z.B. Genotyp des E. coli Stammes DH10B: F- mcrA ∆(mrr-hsdRMS-mcrBC) Φ80dlacZ∆M15 ∆lacX74 endA1 recA1 deoR ∆(ara, leu)7697 araD139 galU galK nupG rpsL Marker - Anmerkung F Stamm enthält kein F Episom, keine Konjugation möglich mcrA Mutation verhindert die McrA-Restriktion methylierter DNS ∆(mrr-hsdRMS-mcrBC) Deletion eines ganzen Genclusters von 6 Restriktionsenzymen (mrr-hsdR-hsdM-hsdSmrcB-mrcC) Φ80dlacZ∆M15 Stamm trägt den defekten Lambda-Prophagen Φ80, dieser trägt das dlacZ∆M15-Allel, dlacZ∆M15codiert das ω-Fragment der β-Galactosidase ∆lacX74 Deletion des kompletten lac Operons endA1 Mutation der unspezifischen Endonuclease I (ermöglicht stabilere DNS Präparationen) recA1 Mutation verhindert homologe Rekombination deoR Mutation in einem Repressor des deoCABD Operons ⇒ konstitutive Expression (Aufnahme großer Plasmide möglich) ∆(ara, leu)7697 Deletion eines Genclusters (reicht vom ara bis ins leu-Operon) araD139 Mutation der L-ribulose 5-phosphate 4-epimerase ⇒ L-Arabinose kann nicht metabolisiert werden galU Mutation der UDP-Glucose Pyrophosphorylase ⇒ Galaktose kann nicht metabolisiert werden galK Mutation der Galaktokinase ⇒ Galaktose kann nicht metabolisiert werden nupG Mutation eines Nucleoside-Transporters rpsL Mutation im Protein S12 der 30S Untereinheit ⇒ Steptomycin-Resistenz Tabelle 3: Genotyp des E. coli Stammes DH10B Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 11 5. Bakterielle Modifikations-und Restriktionssysteme Sowohl Pro- als auch Eukaryoten enthalten Enzyme, die DNS methylieren, obwohl die Funktionen der Methylierung verschieden sind. Bei Eukaryoten dient die Methylierung der Unterscheidung von Genen in verschiedenen funktionellen Zuständen. Bei Prokaryoten dient das Methylierungsmuster zum Erkennen "fremder" DNS. Es scheint zu keinem der bakteriellen Methylierungssysteme ein eukaryotisches Gegenstück zu geben. E.coli DNS selbst enthält kleine Mengen an 6-Methyladenin und 5-Methylcytosin. Das Methylierungsmuster der E. coli DNS wird durch die Aktivität dreier Methylasen erzeugt, diese werden von den Genen dam und dcm sowie vom Gencluster hsdRMS codiert (hsdRMS codiert ein Typ I Methylierungs- und Restriktionssystem). Die vom dam-Gen codierte Methylase transferiert eine Methylgruppe vom S-Adenosinmethionin zur N6-Position eines Adeninrestes in der Sequenz GATC. Die dcm codierte Methylase modifiziert einen Cytosinrest innerhalb der Sequenz CCAGG und CCTGG an der C5-Position. In DNS mit einem GC-Gehalt von 50% trifft man statistisch alle 44 = 256 bp bzw. 45 = 512 bp auf solch eine dam bzw dcm-Modifizierung. Die Erkennungssequenz für die im hsdRMS Gencluster codierte EcoKI Methylase (5´-AACNNNNNGTGC) wird viel seltener angetroffen, statistisch einmal in 8 kb. dam G m C dcm C A T m C G G dcm C m C G G hsdRMS A T m A T m C A G A G T m G C C T G G A C T G m C C N N N N N N N N N N G C m T G C A C G Das dam-System unterscheidet überdies die Stränge gerade replizierter DNS, indem es Adenin methyliert. Es ist auch an der Kontrolle der Replikation sowie an der Markierung von DNS-Strängen für die Reparatur beteiligt. E. coli Stämme mit Mutationen in allen drei Systemen haben keine methylierten Basen. Sie sind aber lebensfähig, weshalb die Methylierung kein lebenswichtiges Ereignis darstellen kann. Ein unterschiedliches Methylierungsmuster macht fremde DNS dem Angriff durch Restriktionsenzyme zugänglich, die das Fehlen von Methylgruppen an den entsprechenden Stellen erkennen. E. coli hat zumindest vier Restriktionssysteme, um fremde DNS zu erkennen und zu zerstören: Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 12 hsdRMS-System Die hsdRMS Gene codieren das sogenannte EcoKI Restriktionssystem. Der EcoKI Enzymkomplex, attackiert eine spezifische DNS Sequenz (5´-AACNNNNNGTGC) und führt in die DNS (in variablem Abstand von der Erkennungssequenz) eine Doppelstrang-Schnitt ein. Wenn diese Sequenz nicht vorhanden, bzw. die darin vorkommenden Adenine methyliert sind, wird die DNS nicht angegriffen. Sollte eine der drei Untereinheiten defekt sein, kommt es ebenso zu keiner Restriktion. Der selbe Enzymkomplex methyliert jedoch auch sein eigenes Substrat (doppelsträngige DNS die 5´AACNNNNNGTGC enthält, siehe oben), allerdings sehr langsam, wodurch nicht methylierte "FremdDNS" selten "überlebt". mcrA, mcrBC, mrr-System Im Gegensatz zum EcoKI Restriktionsystem wird von diesen Restriktionssystemen DNS attackiert, die an spezifischen Positionen methyliert ist, z. B. schneiden sowohl McrA, als auch McrB und Mrrr mCG (Produkt der CG Methylase Sss I). Genauere Informationen zur Spezifität dieser Restriktionssysteme finden sich z.B. in den Katalogen der diversen Anbieter für Restriktions- und Modifikationsenzyme (z.B. http://www.fermentas.com/). Bedeutung für das molekularbiologische Arbeiten • DNS von Säugern, höheren Pflanzen und auch vielen Prokaryonten (nicht Drosophila melanogaster und Saccharomyces cerevisiae) enthält Methyl-Cytosin (mCG, m CNG). Zur Konstruktion genomischer „Libraries“ müssen somit unbedingt mcrA, mcrB, mrr-defiziente (restriktions-defiziente) E. coli Stämme verwendet werden! • Durch die CG-Methylation können bestimmte Restriktionenzyme (nämlich solche, deren Erkennungssequenz das CG-Motiv enthält oder auch nur überlappt) blockiert werden. Informationen zur Sensitivität von Restriktionsenzymen hinsichtlich einer CG-Methylierung finden sich in den Katalogen der diversen Anbieter (z. B. http://www.fermentas.com/). • DNS, die aus einem Dcm+- oder Dam+- E. coli Stamm präpariert wurde, kann von manchen Restriktionsenzymen deren Erkennungssequenz das Dam- bzw Dcm- Motiv enthält (bzw. auch nur überlappt) nicht mehr geschnitten werden. So kann zum Beispiel DNS aus einem Dam+- E. coli Stamm nicht mehr vom Restriktionsenzym MboI geschnitten werden, sehr wohl aber von Sau3AI - obwohl beide die gleiche Erkennungssequenz GATC aufweisen. Der Grund ist, dass MboI (im Gegensatz zu seinem Isoschizomer Sau3AI) methylierungssensitiv ist und die dammethylierte GATC Restriktionsschnittstelle nicht mehr geschnitten werden kann. Informationen zur Sensitivität von Restriktionsenzymen hinsichtlich einer dam- bzw dcm-Methylierung finden sich in den Katalogen der diversen Anbieter (z. B. http://www.fermentas.com/). Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen • Seite 13 Der Methylierungsstatus einer Plasmid DNS kann auch die Transformationseffizienz beeinflussen. So kann zum Beispiel eine dam-methylierte Plasmid-DNS nur mit weit geringer Effizienz in einen Dam- E.coli Stamm transformiert werden. 6. Restriktionsenzyme Restriktionsenzyme sind Endonucleasen bakteriellen Ursprungs. Sie sind in der Lage doppelsträngige DNS-Moleküle an spezifischen Erkennungsstellen zu binden und zu schneiden. Die entstehenden Restriktionsfragmente besitzen eine je nach Lage der Schnittstellen definierte Länge und können durch ein anschließende Auftrennungsverfahren (Gelelektrophorese) entsprechend ihrer Größe geortet werden. Die ungefähre Bestimmung der Größe (in Basenpaaren, bp) erfolgt durch Vergleich mit einem parallel aufgetragenen Größenstandard wo man die Länge der einzelnen Fragmente genau kennt. Die biologische Funktion der Restriktionsenzyme ist die Zerkleinerung und somit Inaktivierung eingedrungener Fremd-DNS, beispielsweise von Phagen-DNS. Eigene DNS wird durch Modifizierung wie zum Beispiel Methylierung vor dem Eingriff der eigenen Restriktionsenzyme geschützt. Restriktionenzyme spalten hydrolytisch die Phosphodiesterbindungen beider Stränge eines DNSMoleküls und unterscheiden sich in hinsichtlich ihrer Erkennungssequenz, ihrer Spaltstelle und ihres Ursprungsorganismus. Man unterscheidet mindestens 3 Typen: Typ I, bzw. Typ III Restriktionsenzyme besitzen sowohl Endonuclease als auch Methylase-Funktion, benötigen ATP und kommen aufgrund ihrer unspezifischeren Spaltstelle nicht in der DNS-Analytik zum Einsatz. Typ II-Restriktionsenzyme besitzen im Gegensatz dazu nur eine Restriktionsaktivität und haben den großen Vorteil, DNS innerhalb einer definierten Erkennungssequenz zu spalten. Die Erkennungssequenzen der Restriktionsenzyme sind typischerweise 4-8 Nukleotide lang und meist palindromisch, d.h. . Die Spaltstelle liegt in der Regel innerhalb der Erkennungssequenz, wodurch die entstehenden Fragmente definierte Enden erhalten, welches für eine nachfolgende Klonierung von großer Bedeutung ist. Man untescheidet zwischen sogenannten sticky und blunt ends . 5´ 3´ 3´ 5´ b a 5´ 3´ c Abbildung 6: a: Blunt end DNS, z.B. generiert durch SmaI, b und c: sticky ends DNS mit 5´-overhang (z.B. generiert durch HamHI) bzw. mit 3´-overhang (z.B. generiert durch NcoI). Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 14 Restriktionsenzym Isoliert aus Erkennungsseq. Ende Isoschizomere G/GATCC sticky (5´-overhang) BstI Bacillus amyloliquefacines BamHI Stamm H erstes isoliertes Enzym (I) SmaI Serratia marcecescens Sb CCC/GGG blunt XmaI SacI Streptomyces achromogenes GAGC/TC sticky (3´-overhang) SstI Tabelle 4: Beispiele der Namensgebung einiger gebräuchlicher Restriktionsenzyme Nachfolgend sei am Bsp. dreier Restriktionsenzymen ihre Herkunft, die Namensgebung, die Art der entstehenden Enden und Kompatibilität zu anderen Restriktionsenzymen veranschaulicht: Detaillierte Listen aller für das Labor verfügbaren Restriktionsenzyme können in Laborkatalogen der jeweiligen Anbieter nachgesehen werden (siehe Anhang). Außerdem findet man dort detaillierte Information zu • Inkubationstemperaturen (meistens 37°C; manche Enzyme benötigen jedoch 50°C oder 65°C zur Enfaltung der optimalen Aktivität) • Puffersystemen • Hitzeinaktivierung • Isoschizomere (2 Enzyme haben eine gemeinsame Erkennungssequenz und Schnittstelle) und Neoschizomere (2 Enzyme haben eine gemeinsame Erkennungssequenz, aber unterschiedliche Schnittstellen) • Listen kompatibler Enden (je nach 5`bzw. 3`Überhang) • Methylierungssensitivität 7. Sonstige DNS-modifizierende Enzyme Zusätzlich zu den Restriktionsenzymen sind mittlerweile eine große Anzahl an modifizierenden Enzymen erhältlich, welche in der Molekularbiologie Anwendung finden. An dieser Stelle sei nur auf jene verwiesen, die hauptsächlich für die Klonierung zum Einsatz kommen. Taq DNS Polymerase (siehe Beispiel „Genotyp“) Dieses Enzym aus Thermus aquaticus ist eine thermostabile DNS-abhängige DNS-Polymerase (Temperaturoptimum: 75 – 80°C). Native Taq DNS Polymerase hat keine 3´→5´ Exonuclease-Aktivität (proof reading-activity) und eine schwache 5´→3´ Exonuclease-Aktivität. Anwendungen Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen • Seite 15 PCR (Polymerase Chain Reaction): zur Amplifikation von definierten DNS Fragmenten (genaue Beschreibung: siehe Beispiel A) • Sequenzierung (genaue Beschreibung: siehe Beispiel A) • DNS-Markierung mit Radionucleotiden, oder chemischen Farbstoffen (Digoxigenin, Biotin,.... genaue Beschreibung: siehe Beispiel A) Will man ein PCR-Produkt ohne weiteren Verdau klonieren, kann man sich die Terminale TransferaseNebenaktivität der Taq DNS Polymerase zunutze machen: diese hängt an das 3`-OH Ende mit hoher Effizienz ein zusätzliches Deoxyadenosin an. Das PCR Produkt erhält somit ein "sticky end" (AÜberhang) und kann in Klonierungsvektoren mit einem 3`-Thymidin Überhang ligiert werden (diese Vektoren sind käuflich erwerblich). Durch einen andernt Typ von Taq-Polymerase, zum Bsp. die Pfu Polymerase können aber auch „blunt end“ Fragmente generiert werden (genaue Beschreibung: siehe Beispiel A). T4 DNS Ligase Die Ligase des Bakteriophagen T4 katalysiert die Verknüpfung von 5´-Phosphat und 3´Hydroxylgruppen zweier DNS Moleküle. Das Enzym kann somit zur Ligation von kompatiblen „sticky“oder „blunt-end“- Fragmenten doppelsträngiger DNS eingesetzt werden. Dieses Enzym wird daher zur Wiedervereinigung von manipulierten DNS Fragmenten (z.B. Konstruktion von rekombinanten Plasmiden) verwendet. .Die T4 DNS Ligase benötigt Mg2+, bzw. Mn 2+ und ATP (befindet sich üblicherweise im Puffer). Die optimale Temperatur einer Ligation (4-15°C) stellt eine Kompromiss zwischen der optimalen Temperatur für die Aktivität des Enzyms (37°C) und der optimalen Temperatur zur Assoziation der DNS-Enden dar. Eine hohe DNS-Konzentrationen (z. B. durch Zugabe von Polyethylenglycol), die Dephosphorylierung des Vektors und ein Überschuss an Insert DNS sollten selbst bei blunt-end Ligationen eine vernünftige Ausbeute an rekombinanten Plasmide ergeben. DNS Polymerase I Large Fragment (Klenow) Das auch oft kurz "Klenow" bezeichnete Enzym besteht aus den C-terminalen-Teil der E. coli DNS Polymerase I (eine DNS-abhängige DNS Polymerase). Es wurde früher proteolytisch hergestellt, heute wird es rekombinant exprimiert. Das Klenow Fragment besitzt eine starke 5´→3´ PolymeraseAktivität und eine relativ schwache 3´→5´ Exonuclease-Aktivität (proof reading-activity) der DNS Polymerase I, hat aber keine 5´→3´ Exonuclease-Aktivität mehr. Klenow ist allerdings auch bereits ohne 3´→5´ Exonuclease-Aktivität erhältlich. Anwendungen • Reparatur von 5´-Überhängen (⇒ generieren von blunt ends) Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 16 Die Reparatur von 5´-Überhängen (fill-in) wird von der die 5´→3´-Polymerase-Aktivität bewirkt, gelegentlich auch für die Reparatur von 3´-Überhängen durch die 3´→5´ Exonuclease-Aktivität (T4 DNS Polymerase ist hierbei allerdings die bessere Wahl) • Markieren des 3´-Endes von DNS (z.B. wenn eine Detektionssonde gebraucht wird wie in Beispiel „Genotyp“ beschrieben) Zur Aktivität benötigt das Klenow-Fragment Mg2+ und es wird nach Abschluss der Reaktion hitzeinaktiviert (für 10 min bei 75°C). T4 DNS Polymerase Die T4 DNS Polymerase stammt vom Bakteriophagen T4 und wird entweder aus infiziertem E.coli oder rekombinant gewonnen. DieT4 DNS Polymerase ist eine DNS-abhängige DNS Polymerase mit einer sehr aktiven 3´→5´ Exonuclease-Aktivität, besitzt aber keine 5´→3´ Exonuclease-Aktivität mehr. Daher ist sie das Enzym der Wahl wenn man eine DNS mit 3´ Überhängen vorliegen hat blunt ends generieren will. Im Gegensatz zum Klenow Fragment erfolgt hier aber eine „cut-back“ Reaktion. 8. Extraktion und Reinigung von Plasmid DNS: Nach erfolgter Manipulation von DNS Fragmenten und Transfer von vermeintlich rekombinanten Plasmiden in E.coli müssen diese nun hinsichtlich der gewünschten Eigenschaften überprüft werden. Dazu ist es notwendig die Plasmide von Bakterien die auf Selektonsagar gewachsen sind zu isolieren und analysieren. Es wurden viele Methoden zur Reinigung von Plasmiden aus Bakterien entwickelt wobei alle drei Schritte gemeinsam haben: (i) Anzucht der Bakterien (ii) Ernte und Lyse der Bakterien (iii) Reinigung der Plasmid DNS • Anzucht der Bakterien Plasmide werden fast ausschließlich aus Flüssigkulturen (mit geeignetem Antibiotikum) gereinigt, die zuvor mit Einzelkolonien von Agarplatten inokuliert wurden. Viele der heute verwendeten Plasmide (e.g. aus der pUC Serie) replizieren in einer derart hoher „copy number“, dass sie in ausreichender Menge bei einfachem Wachstum bis zur log-Phase in einem Standard LB Medium geerntet werden können. Bei sehr großen Plasmiden oder „low-copy number“ Plasmiden kann man die Erträge durch selektive Vermehrung von Plasmiden durch Zugabe von Chloramphenicol steigern. Chloramphenicol inhibiert die Proteinsynthese des Wirtes und verhindert so eine Replikation der chromosomalen DNS. Die Plasmid DNS wird aber weiterhin repliziert wodurch es zu einer selektiven Anreicherung kommt. Durch Verwendung von besonders „nahrhaften“ Medien wie „Terrific Broth“ kann es ebenfalls zur Vermehrten Ausbeutung von Plasmid DNS kommen. Nach dem Volumen, der für die PlasmidPräparation eingesetzten Bakterienkultur, unterscheidet man Miniprep (2 – 10ml), Midiprep (25 – 100ml) und Maxiprep (> 100ml). Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen • Seite 17 Ernte und Lyse der Bakterien Für die Lyse der Bakterien zur Gewinnung niedermolekularer DNS stehen mehrere Methoden zur Verfügung. Die am häufigsten verwendete Methode ist die alkalische Lyse. Dabei wird die Bakterienkultur zentrifungiert, der Kultur-Überstand verworfen und das Pellet in einem EDTA-haltigen Puffer resuspendiert. EDTA komplexiert zweiwertige Kationen (Mg2+, Ca2+), welche für die Stabilität der bakteriellen Zellwände wichtig sind. Dem Resuspensions-Puffer wird bei manchen Protokollen auch RNaseA (degradiert bakterielle RNA) und Lysozym (greift die bakterielle Zellwand an) zugesetzt. • Reinigung der Plasmid DNS Grundsätzlich erfolgt die Reinigung von Plasmiden über eine „alkalische-Lyse“ oder über eine „Koch-Lyse“. Beide Methoden nützen die relativ gute Stabilität der niedermolekularen zirkulären Natur der Plasmide gegenüber der leichter zu denaturierenden hochmolekularen linearen chromosomalen DNS. alkalische-Lyse Die lysierte Bakteriensuspension wird einer alkalischen Lösung (NaOH) in Kombination mit einem starken Detergens (SDS) ausgesetzt. Das Einwirken von einem anionischem Detergens bei hohem pH löst die Phospholipide und Proteinkomponenten der Zellwände, denaturiert chromosomale DNS und Proteine und entläßt Plasmid DNS in den Überstand. Obwohl durch Zugabe einer alkalischen Lösung die Basebpaarbildung der chromosomalen DNS aufgelöst wird, kommt diese Eigenschaft bei zirkulären Molekülen wie der Plasmid DNS nicht zum tragen, da diese durch ihre topologisch enge Ausrichtung schwer angreifbar sind. Solange die Dauer und die Intensität der Einwirkung nicht zu lange ist, kann Plasmid DNS drurch anschießende Neutralisation ohne weiteres wieder in ihre Ursprungsform gebracht werden. Im Gegensatz zur chromosomalen DNS kann die Plasmid DNS renaturiert werden. Dieses Lysat wird dann anschließend mit saurem Kaliumacetat-Puffer neutralisiert. Denaturierte Proteine, hochmolekulare RNA, denaturierte chromosomale DNS (chromosomale DNS haftet an der Zellmembran) und bakterielle Zell-Abbauprodukte bilden in Anwesenheit von Kaliumdodecylsulfat unlösliche Komplexe und werden gemeinsam mit dem Salz präzipitiert (Kaliumdodecylsulfat ist wesentlich schlechter in Wasser löslich als Natriumdodecylsulfat). Die unlöslichen Komponenten werden dann abzentrifugiert und die Plasmid DNS kann mit Ethanol oder Isopropanol gefällt und gewaschen werden. „Koch-Lyse“ Eine andere Aufschlußmethoden stellt z. B. die sogenannte „Koch-Lyse“ dar. Dabei werden die bakteriellen Zellwände durch Zugabe von Lysozym zerstört und die lysierten Bakterien für kurze Zeit aufgekocht. Die bakteriellen Abbauprodukte werden abzentrifugiert und die Plasmid DNS kann anschließend präzipitiert werden. Anschließend werden durch Zugabe von Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 18 Phenol/Chloroform/Isoamylalkohol bakterielle Proteine und Membranen denaturiert und die niedermolekulare Plasmid-DNS bleibt in der wässrigen Phase gelöst. Nach dem Zentrifugationsschritt kann aus dem Überstand die Plasmid-DNS isoliert werden. Weiters können große Plasmide über CsCl-Dichtegradientenzentrifugation weiter aufgereinigt werden. 9.Analyse von DNS durch Agarose-Gelelektrophorese Agarose ist das wichtigste Trägermaterial für die Elektrophorese von Nukleinsäuren. Es handelt sich dabei um ein Polymer, das aus verknüpften Galactoseeinheiten besteht. Die Wanderungsgeschwindigkeit der DNS-Moleküle wird dabei von mehreren Faktoren beeinflusst: der Form der DNS (superhelikal, offen, linear doppelsträngig, einzelsträngig), den Laufbedingungen, der Agarosekonzentration, der angelegten Spannung, der Wahl des Laufpuffers oder auch die Präsenz eines interkalierenden Farbstoffs, wie zum Beispiel Ethidiumbromid. In der Regel wandert die superhelikale (SC für „supercoiled“) Form der DNS schneller im Gel als die lineare (L). Die offene Form (OC für „open circular“) wandert im Agarosegel wesentlich langsamer als die superhelikale oder lineare DNS (siehe Abbildung 7). Generell ist die Wanderungsgeschwindigkeit von DNS Fragmenten in Agarosegelen proportional zur angelegten Spannung. Große DNS Fragmente wandern mit zunehmender Spannung jedoch zunehmend langsamer im Gel, so dass sich hohe Spannungen für die Auftrennung großer Fragmente nicht eignen. Sehr kleine Fragmente trennt man am besten durch Erhöhung der Agarosekonzentration in 2-3%igen Agarosegelen auf. Für die Auftrennung verwendet man Tris-Acetat-(TAE) oder Tris-Borat-(TBE)Laufpuffer. Die Konzentration der Ionen im Laufpuffer ist von großer Bedeutung. Sind zuwenige Ionen im Laufpuffer vorhanden, so ist die elektrische Leitfähigkeit minimal und die Wanderungsgeschwindigkeit der DNS zu gering. Ist die Ionenkonzentration zu hoch, wird der Laufpuffer durch die sehr hohe Leitfähigkeit zu stark erhitzt, die DNS möglicherweise denaturiert und die Agarose geschmolzen. Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 19 A B . Abbildung 7: DNA-Elektrophorese in Agarose Gelen. Die Wanderung der DNS erfolgt in Pfleilrichtung. Die DNS wurde durch Inkubation des Geles in EtBr- unter UV Licht sichtbar gemacht (EtBr- interkaliert zwischen den Basenpaaren). A: OC entspricht der „open circle“ und SC der „super coiled“ Form des Plasmides. Die SC Form läuft somit schneller im Gel als die OC Form. B: L= linearisiertes Plasmid. Das in Spur B aufgetragen Plasmid wurde mir dem „single-cutter“ EcoRI geschnitten. Weitere Informationen zu Agarose-Gelelektrophorese entnehmen Sie bitte den Praktikumsunterlagen zum Beispiel „Genotyp“. Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 20 Flussdiagramm eines Klonierungvorganges: 1 2 Linearisieren des Vektors Herstellen des Inserts Die Enden der DNS müssen kompatibel sein 3 Ligation von Vektor und Insert 4 5 Herstellen von kompetenten E.coli Zellen 6 Transformation Selektion auf geeignetem Medium (blau/weiss; Antibiotikum) Anzucht der interessanten Klone 7 Plasmid (Mini-) Präparation 8 Analyse der Plasmide (Ararosegelelektrophorese) Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 21 10. Empfohlene Literatur (in der Fachbibliothek Muthgasse zu finden) • Der Experimentator: Molekularbiologie, Genomics (Spektrum Verlag) C. Mühlhardt (für Einsteiger). Kapitel: 1., 2.5., 3.1., 3.2., 2.1., 2.2., 6.1. (nicht 6.1.2.)., 6.2.1., 6.3., 6.4., 6.5., • Gentechnische Methoden (Spektrum Verlag) G. Schrimpf (zur Vertiefung) Kapitel: 1.10., 1.11., 3.2., (pp: 100-107). Weiterführende Literatur: • Principles of Gene Manipulation. Primrose, S.B., Twyman, R.M., and Old, R.W. 2001. Blackwell Science Ltd. (Lesesaal, 53.02; Kapitel 1,2,3,4,6) • Gentechnologie für Einsteiger. T.A. Brown 2001 (Lesesaal, 53.02) • Gene Cloning & DNS Analysis. T.A. Brown 2001 (Lesesaal, 53.02), Part 1: pp 3-196 • Molekulare Biotechnologie Glick & Pasternack 1995 (Lesesaal 53.02) Kapitel 2: 19-59. • Molecular Biotechnology Glick & Pasternack 1995 (Lesesaal 53.02) Kapitel 2. • Molekulare Genetik. Knippers, R. 2001. Georg Thieme Verlag Stuttgart (7.Auflage, 8. neubearbeitete Auflage erscheint demnächst). (Lehrbuchsammlung, 53.10; pp 23-28, LacZ: 112-125. Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 22 Anhang: Typische Plasmid Vektoren: Plasmide der pUC Serie (Beispiel pUC18) lacZ alpha MCS a (ampR) pUC18 2686 bp ori (pMB1 derived) pUC18 und pUC19 sind kleine (2686 bp), high copy number E. coli Plasmide. Bis auf die entgegengesetzt orientierte MCS sind sie ident. pUC Plasmide besitzen • ein pMB1 Replicon, liegen aber in 500-700 Kopien pro Zelle vor. Grund dafür ist eine PunktMutation im RNAII Primer, welche (abhängig von der Temperatur) die Interaktion mit dem Repressor RNAI beeinträchtigt, außerdem fehlt pUC Plasmiden das rop Gen • ein bla Gen (ampr), welches das Enzym β-Lactamase kodiert. Dieses vermittelt Resistenz gegen Ampicillin • den Promotor (incl. CAP-Protein- und lac-Repressor-Bindungsstelle) und das 5´- Ende des lacZ Gens, welches das N-terminale Fragment der β-Galaktosidase codiert. Dieses ist somit IPTG induzierbar und zur sogenannten α-Komplementation fähig Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 23 Antibiotikaresistenzen und ihre Gene Ampicillin Abbildung 2: Ampicillin Das Ampicillin ist ein Aminopenicillin, interferiert mit der bakteriellen Zellwandsynthese (u.a. wird eine Transpeptidase und somit die Quervernetzung des Mureins blockiert) und zerstört so wachsende Zellen (Zellwände können dem wachsenden Turgordruck nicht mehr standhalten). Das Resistenzgen ampr (bla) codiert ein periplasmatisches Enzym, eine ß-Lactamase, welche den cyclischen ßLactamring des Antibiotikums spaltet. Gebräuchlich verwendete Konzentrationen von Ampicillin sind 50-125 µg/ml. Chloramphenicol Chloramphenicol bindet an die 50S Untereinheit der bakteriellen Ribosomen (70S), verhindert die Ausbildung von Peptidbrücken und greift so in die bakterielle Proteinsynthese ein. Das entsprechende Resistenzgen Cmr (cat) codiert eine Acetyltransferase welche das Antibiotikum acetyliert und inaktiviert. Arbeitskonzentrationen: 20-170 µg/ml Abbildung 3: Chloramphenicol Kanamycin Kanamycin (ein Aminoglycosid) bindet an 70S Ribosomen und inhibiert so u. a. auch die ProteinBiosynthese. Das Resistenzgen kanr codiert eine Aminoglycosid-Phosphotransferase, die das Antibiotikum phosphoryliert und so vermutlich den aktiven Transport in die Zelle und die Interaktion mit den Ribosomen verhindert. Arbeitskonzentration: 2550 µg/ml. Abbildung 4: Kanamycin Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 24 Streptomycin Streptomycin (ein Aminoglycosid) bindet an die 30S Untereinheit prokaryotischer Ribosomen und verursacht ebenfalls Fehler beim Lesen der mRNA (stört die Bildung des Initiationskomplexes). Resistenzgene (str) codieren Enzyme welche das Antibiotikum modifizieren (Phosphorylierung, Adenylierung) und so die Ribosomenbindung verhindert. Arbeitskonzentrationen sind ebenfalls 30µg/ml. Tetracycline Tetracycline binden an die 30S Untereinheit der Ribosomen, verhindern den Transfer der aktivierten Aminosäuren zum Ribosom und stoppen so die bakterielle Proteinsynthese. Effluxpumpen, Tetracyclin-Resistenzgene die Tertacyclin aktiv in codieren Form von Metallkomplexen aus der Zelle ausschleusen. Arbeitskonz.: 10 µg/ml in Flüssigkultur, 12,5 µg/ml auf Platten. Abbildung 5: Tetracyclin Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen Seite 25 11. Auswahl einiger Übungsbeispiele/Fragen (teilweise mit Lösungen) (Die Anzahl der Punkte 1 oder 2, stehen zu Beginn jeder Frage) • 2 Was versteht man beim Klonieren unter „kompetente Zellen“ ? • 2 Erklären sie das Prinzip einer E. coli Transformation ? • 2 Welche 3 Minimalanforderungen muss ein Plasmid besitzen um es als Klonierungsvektor verwenden zu können? • 2 Was ist der Unterschied zwischen physikalischer und chemischer Transformation? • 2 Welche Selektionsmarker (Antibiotikaresistenzen) sind häufig auf Labor-Plasmiden zu finden? • 2 Was bezeichnet man „Kopienzahl“ bei Plasmiden und wovon hängt diese hauptsächlich ab? • 1 Was versteht man unter „Inkompatibiliät“ von Plasmiden? • 1 Was ist eine „multiple cloning site“ (MCS) in einem Plasmid? • 2 Welche Eigenschaften besitzt eine multiple cloning site (MCS) und wofür wird sie beim Klonieren verwendet? • 2 Was ist das lacZ Gen ? • 2 Wie funktioniert die blau weiss Selektion durch das b-Galactosidase Gen? • 2 Was sind Restriktionsendonukleasen, und wofür werden sie beim Klonieren verwendet? • 2 Wie entstehen blunt und sticky ends einer DNS? • 1 Kann aus einer blunt end DNS durch eine in vitro Modifikation ein sticky end gemacht werden? • 1 Auf welchem Prinzip beruht die Namensgebung der Restriktionsendonukleasen (z.B. BamHI, SmaI,.....) • 2 Was ist ein Isoschizomer, bzw. Neoschizomer? • 1 Wie kann man DNS Produkte die mittels Taq-Polymerase (PCR) erzeugt werden klonieren? • 1 Wie sehen die DNS Enden aus die mittels Taq-Polymerase generiert werden? • 1 Welche Eigenschaften besitzt das Enzym alkalische Phosphatase und wozu verwendet man es beim Klonieren? • 2 Wie kann man bei einer Ligation verhindern, dass sich ein linearisierter Vektor alleine religiert? • 1 Welche Eigenschaften besitzt das Enzym T4 DNS Ligase und wozu verwendet man es beim Klonieren ? • 1 Beschreiben Sie die Eigenshaften des DNS Polymerase I Large Fragment (Klenow- Fragment) ? • 2 Wozu wird das Klenow-Fragment in der Molekularbiologie werwendet? • 1 Wie kann eine Lyse von E.coli Zellen für eine darauffolgende Plasmid Reinigung erfolgen? • 2 Auf welchem Prinzip beruht die Plasmid Reinigung mittels „alkaliascher Lyse“ ? • 1 Was ist Agarose und wozu wird es in der Molekularbiologie verwendet? • 1 Was ist eine Gelelektrophorese? • 1 Wie kann man DNS nach einer Gel-Elektrophorese sichtbar machen? Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen • Seite 26 Sie habe einen Vektor mit 3´ Überhängen und ein Insert mit 5´ Überhängen. Wie können Sie ein rekombinantes Plasmid daraus machen? • Sie habe einen Vektor mit blunt ends und ein Insert mit 5´ Überhängen. Wie können Sie ein rekombinantes Plasmid daraus machen? • Sie habe einen SmaI geschnittenen Vektor ein SmaI geschnittenes Insert. Wie können Sie effizient ein rekombinantes Plasmid daraus machen? Welchem Problem stehen Sie gegenüber? • 1 Wieviel ng einer Insert-DNS von 600 bp Länge müssen in einer Ligationsreaktion zu 50 ng eines 3 kb großen Vektors zugegeben werden, wenn das Verhältnis Insert:Vektor 3:1 sein soll? Lösung: 30 ng • 1 Schreiben Sie die DNS Sequenz an (5´→3´), die ensteht, wenn eine NcoI Site (5´-C/CATGG- 3´) mit NcoI geschnitten, anschließend mit Klenow aufgefüllt und religiert wird? Lösung: 5´-CCATGCATGG-3´ • 1 Schreiben Sie die DNS Sequenz an (5´→3´), die ensteht, wenn eine XhoI Site (5´-C/TCGAG- 3´) mit XhoI geschnitten, anschließend mit Klenow aufgefüllt und religiert wird? • 1 Die Definition der Aktivität für HindIII lautet wie folgt: 1 Unit (U) HindIII ist jene Menge Enzym, die - in einem 50 µl Ansatz - 1 µg lambda DNS (48502 bp, enthält 7 HindIII Schnittstellen) innerhalb einer Stunde bei 37°C völlig verdaut. Wieviele Units HindIII sind demnach notwendig, um 1µg pUC18 (2686 bp, 1 HindIII Schnittstelle) vollständig zu schneiden? Lösung: 2.6 Units • 1 Die Definition der Aktivität für EcoRI lautet wie folgt: 1 Unit (U) EcoRI ist jene Menge Enzym, die - in einem 50 µl Ansatz - 1 µg lambda DNS (48502 bp, enthält 5 EcoRI Schnittstellen) innerhalb einer Stunde bei 37°C völlig verdaut. Wieviele Units EcoRI sind demnach notwendig, um 1µg pCR4-TOPO (3957 bp, 2 EcoRI Schnittstellen) vollständig zu schneiden? Lösung: 4.9 Units • 2 Wie funktioniert die Präparation von Plasmid-DNS nach dem Prinzip der alkalischen Lyse. Warum findet sich am Ende kaum genomische DNS in der Präparation? • 1 In einen HindIII verdauten Klonierungsvektor pUC18 werden verschieden große Lambda DNS Inserts kloniert (ebenfalls HindIII geschnitten, s.o.). Können diese in den entstehenden Klonen in verschiedenen Orientierungen vorhanden sein und wenn ja wie überprüfen Sie die Orientierung? • 2 Welche Eigenschaften (Genotyp) des E. coli Stamms DH10B sind für das Funktionieren der Alpha-Komplementation notwendig ? • 1 Im Zuge einer Klonierung soll in die MCS eines pUC Plasmids ein Insert ligiert werden. Welche Kolonien sind nach dem Ausplattieren auf Xgal/IPTG Platten bevorzugt zur Restriktionsanalyse (Screening nach Rekombinanten) heranzuziehen (Begründung)? • 2 Welche Eigenschaften (Genotyp) des E. coli Stamms DH10B machen ihn für die Klonierung genomischer DNS (aus Pflanzen, tierischen Zellen, ...) geeignet? • 2 DNS wurde aus einem Dam+- E.coli Stamm präpariert, kann jedoch nicht mehr vom Restriktionsenzym MboI geschnitten werden, obwohl die Schnittstelle vorhanden ist. Was ist die Ursache und wie lösen Sie dieses Problem? • 1 Sie erhalten im Zuge einer Klonierung eine große Anzahl an Klonen mit Plasmid ohne Insert ("leerer" Klonierungsvektor). Welche Gründe kann es dafür geben? Wie können Sie in diesem Fall die Klonierungseffizienz verbessern? Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08 Kolloquiumsunterlagen • 1 Seite 27 Sie tragen geschnittene doppelsträngige versus ungeschnittene Plasmid-DNS auf einem Agarosegel parallel nebeneinander auf. Wieviele Banden erwarten Sie in beiden Fällen und wie wird das Laufverhalten ungefähr sein? Machen Sie eine Skizze. • 1 Sie wollen in einen blunt end geschnittenen Klonierungsvektor ein Insert setzen, welches NcoI verdaut ist. Wie lösen Sie dieses Klonierungsproblem? • 2 Suchen Sie die Erkennungssequenz der Restriktionsenzyme BamHI, BglII, XbaI, BclI, EcoRV, SacI, XhoI, SmaI, SalI und PstI. Enthält (überlappt) die jeweilige Erkennungssequenz ein damdcm-, bzw. GC-Methylierungsmotiv? Wird die Aktivität des jeweiligen Enzyms dadurch inhibiert? • 1 Wie bestimmt man die Transformationseffizienz kompetenter Zellen? • 1 Wie hoch ist die Transformationseffizienz von kompetenten DH10B Zellen wenn nach einer Transformation ein Zehntel des Gemisches - transformiert mit 2 µl einer pUC18 DNS (1 ng/ml) auf eine LB Amp100 Platten ausplattiert und am nächsten Tag darauf 26 Kolonien gezählt wurden? Lösung: 1.3 x 108 cfu/µg pUC18 • Was ist λ (lamda) DNS und wozu wird sie im E. coli Beispier eingesetzt? • Welche Komponenten müssen in einem Ligationsansatz sein? • Welche Komponenten müssen Sie zusammengeben, damit sie bei einem mit blunt-end geschnittenen Vektor die Phosphatreste weg bekommne? Molekularbiologie Übungen I (LV-Nr. 954.101): Plasmidvektoren - Klonieren in Escherichia coli (Steinkellner + Mitterbauer) 11.03.08