Skript zur Einführung in die Chemie (Experimentalchemie)
Werbung
")
1
Experimentalvorlesung Grundlagen der Chemie
Vorlesungsskriptum
Wintersemester 2006/2007
Prof. Dr. Dieter Rehder
Institut für Anorganische und Angewandte Chemie
Die Vorlesung richtet sich primär an Studenten und Studentinnen der Studiengänge Chemie
(Bachelor of Science), Lebensmittelchemie, sowie Lehramt an der Oberstufe
Allgemeinbildende Schulen mit Fach Chemie, sowie Gewerbelehrer der Fachrichtung
Chemotechnik.
Vorausgesetzt werden schulische Kenntnisse in den Fächern Chemie, Physik und
Mathematik.
Vorbemerkungen
Ein Vorlesungsskriptum ist naturgemäß knapp gefasst. Erläuternde Sätze und detaillierte
Darlegungen werden oft fehlen, ebenso Anmerkungen, welche die Sachverhalte hinterfragen
und in einen breiteren Zusammenhang bringen. Ein Skriptum ist daher kein Ersatz für ein
Lehrbuch. Die im Skriptum knapp dargelegten Sachverhalte sind oft nur im Zusammenhang
mit einem Lehrbuch und/oder der Vorlesung selbst verständlich. Ebenso wenig ersetzt das
Skriptum daher den Besuch der Vorlesung. Eine Vorlesung lebt auch von der Spontaneität des
Augenblicks, von Rückkopplungen zwischen dem Dozenten und den Studenten/Studentinnen.
Es kann – und wird – daher immer wieder Diskrepanzen zwischen dem Duktus (und dem
Inhalt) dieses Skriptums und dem aktuellen Ablauf der Vorlesung geben.
Ein wichtiger methodischer Bestandteil der Wissensvermittlung durch diese spezielle
Vorlesung sind Experimente, die begleitend und erläuternd zum gesprochenen Wort und
formelhaften Darstellungen wo möglich den Zugang zu den zugrunde liegenden Chemismen
veranschaulichen und damit erleichtern sollen. Dieser Bereich fehlt natürlich im Skriptum;
Verweise auf Versuche sind im Skriptum aber vermerkt.
Lesen Sie das Skriptum also kritisch: Ich erhebe keinen Anspruch darauf, das Skriptum völlig
fehlerfrei vorgelegt zu haben. Die eine oder andere Darlegung kann sich zudem unterscheiden
von dem was Sie im Lehrbuch finden. Das kann auf unterschiedlicher Sichtweise beruhen,
oder auch darauf, dass auch Lehrbücher nicht fehlerfrei sind und, vor allem, nicht auf dem
aktuellsten Stand.
Ausgewählte Gefährdungshinweise sind in Rot gekennzeichnet. Das entbindet bei Umgang
mit den in diesem Skriptum angesprochenen Stoffen und Reaktionen nicht von der
Informationspflicht über Gefährdungspotenziale.
2
Buchempfehlungen
Für Studenten/Studentinnen der Chemie:
E. Riedel: Anorganische Chemie, Verlag Walter de Gruyter (6. Auflage, 2004). Behandelt
Konzepte der Anorganischen und Allgemeinen Chemie sowie stoffliche Chemie.
Für Studenten/Studentinnen des Lehramtes:
C.E. Mortimer, U. Müller: Chemie, Thieme Verlag (8. Auflage, 2003). Behandelt primär
Konzepte der Chemie, sowie überblickartig stoffliche Chemie einschließlich der organischen
Chemie.
Für Wissenshungrige aller Studiengänge:
Hollemann · Wiberg, Lehrbuch der Anorganischen Chemie, Verlag Walter de Gruyter (101.
Auflage).
Inhaltsverzeichnis
Maßeinheiten
Seite 4
1. Die Begriffe “Chemie” und “Stoff”
2. Geschichtliches
3. Einteilung und Trennung der Stoffe
4. Die chemische Reaktionsgleichung
5. Der Aufbau der Atome I: Rutherfordsches Atommodell
und Elementarteilchen
6. Radioaktivität und Kernreaktionen
7. Wasserstoff
8. Der Aufbau der Atome II: Das Bohr-Sommerfeldsche Atommodell
9. Aufbau des Periodensystems
9.1. Relativistische Effekte
10. Sauerstoff; die kovalente Bindung I
10.1 Sauerstoff
10.2 Die kovalente Bindung I: die Lewis- oder Valenzstrichformel;
formale Ladungen
11. Der Aufbau der Atome III: Das Orbitalmodell
12. Die kovalente Bindung II: Molekülorbitale
13. Wasserstoffverbindungen des Sauerstoffs
13.1. Wasserstoffperoxid
13.2. Wasser
13.3. Wasser als Lösungsmittel
13.4. Die Struktur des Wassers
14. Die Ionenbindung
15. Die Halogene
15.1. Fluor
15.2. Chlor
15.3. Brom und Iod
5
5
6
9
10
11
15
17
19
22
23
23
25
27
29
30
31
31
33
36
36
37
38
39
40
3
16. Oxidation und Reduktion (Redoxprozesse)
17. Die elektrochemische Spannungsreihe
18. Elektrochemische Reaktionen
19. Elektrochemische Stromquellen
19.1. Batterien und Akkus
19.2. Brennstoffzellen
20. Schwefel, Selen und Tellur
20.1. Schwefel
20.2. Selen und Tellur
20.3. Polonium
20.4. Wasserstoffverbindungen der Chalcogene
20.5. Sauerstoffverbindungen des Schwefels
21. Das Massenwirkungsgesetz
22. Säure-Base-Konzepte
22.1. Brønstedt-Säuren/Basen
22.2. Lewis-Säuren/Basen
23. Die Stickstoff-Phosphorgruppe
23.1. Die Elemente
23.2. Wasserstoffverbindungen
23.3. Sauerstoffverbindungen des Stickstoffs
23.4. Sauerstoffverbindungen des Phosphors
24. Die Elemente der Gruppe 14
24.1. Kohlenstoff
24.2. Silizium
24.3. Blei
25. Bor
26. Aluminium, Erdalkali und Alkalimetalle
26.1. Die metallische Bindung
26.2. Aluminium
26.3. Die Erdalkalimetall
26.4. Die Alkalimetalle
42
44
47
48
48
49
50
51
52
53
53
55
57
57
57
58
58
58
62
65
66
67
68
71
74
74
75
75
76
77
78
4
Maßeinheiten in der Chemie (Auswahl) im SI-System (Systeme Internationale)
Messgröße
Symbol Einheit
Symbol
Länge
l
Meter
m
Masse
m
Gramm (Kilogramm)
g (kg)
Volumen
V
Liter
l
Dichte
Gramm pro Milliliter
g/ml
a)
Stoffmenge
n
Mol
mol
molare Masseb)
M
Masse pro Mol; M = m/n
g/mol
(Stoffmengen-)Konzentration c
Stoffmenge pro Volumen; c mol/l
= Molarität
= n/V
Kraft, Gewicht
K
Newton
N = kg·m/s2
Energie
E
Joule
J = kg·m2/s2
Reaktions-Enthalpie
Kilojoule pro Mol
kJ/mol
Hc)
(absolute) Temperatur
Kelvin
K
Reaktions-Entropie
Enthalpie pro Temperatur;
kJ/mol·K
S
S = H/T
Druck
p
Pascal
Pa = N/m2
a)
23
Die Stoffmenge 1 Mol entspricht 6·10 elementarer Einheiten (Moleküle, Atome,
Elektronen, Photonen, …)
b)
Die molare Masse wird bezogen auf 1/12(12C) 1 g/mol
c)
Die Reaktions-Enthalpie ist die bei konstantem Druck bei einer chemischen Reaktion
umgesetzte Energiemenge.
Weitere gebräuchliche Einheiten und ihre Relation zum Systeme International (SI):
Länge (im atomaren Bereich): Ångström, Å; 1Å = 10-11 m = 1nm
Temperatur: Grad Celsius, °C. 0 °C (Celsius) entsprechen 273,15 K
Druck: Bar, bar. 1 bar = 105 Pa
Druck: Torr (mm Hg). 1 Torr 133.3 Pa.
Energie: Kalorie, cal. 1 cal = 1.484 J
Energie (im atomaren Bereich): Elektronenvolt, eV. 1 eV 1.6·10-19 J [entspricht 96 kJ/mol]
Molalität: Stoffmenge pro Masse, mit der Einheit mol/kg
Ladung: Coulomb (C).
Dipolmoment: Debye (D). 1D = 3.338 C·m
Bruchteile und Vielfache der Grundeinheiten
10-3 Milli (m)
103
Kilo (k)
-6
10
Micro (µ)
106
Mega (M)
-9
9
10
Pico (p)
10
Giga (G)
10-12 Nano (n)
1012 Tera (T)
10-15 Femto (f)
1015 Peta (P)
-18
10
Atto (a)
1018 Exa (E)
5
1. Die Begriffe “Chemie” und “Stoff”
Chemie, die „Misch- und Scheidekunst“, ist die Lehre von den Veränderungen und
Umwandlungen der Stoffe. Sie steht damit der Physik gegenüber, welche
Zustandsänderungen von Stoffen beschreibt. Ein Stoff ist ein System, das durch eine Reihe
spezifischer, d.h. Masse- und Gestalts-unabhängiger Eigenschaften charakterisiert ist.
Beispiel: Eisen, zu dessen charakteristischen physikalischen Eigenschaften seine gute
Leitfähigkeit für Wärme und den elektrischen Strom, der charakteristische metallische
Oberflächenglanz, seine Dichte von 7.8 g/cm3, und Verformbarkeit (Schmiedbarkeit) gehören.
Eine charakteristische chemische Eigenschaft des Eisens ist seine Fähigkeit, sich an feuchter
Luft in Rost umzuwandeln. Dieser Vorgang entspricht einer stofflichen Umwandlung. Rost,
ein Eisenoxidhydrat, ist nicht mehr schmiedbar; seine Farbe ist braunrot; Rost leitet den
Strom nicht; seine Dichte ist von der des Eisens verschieden. Ein weiteres Beispiel: Erhitzt
man einen Platindraht, so erglüht er gelbrot; nach dem Abkühlen liegt keine Veränderung vor.
Die Emission von Licht (das Erglühen) beim Erhitzen ist ein physikalischer Vorgang. Erhitzt
man hingegen ein Stück Magnesiumband, so verbrennt es mit Leuchterscheinung. Es entsteht
Magnesiumoxid, ein weißes Pulver. Hier liegt ein chemischer Vorgang zugrunde. (Versuch).
Und noch ein Beispiel: Durch starkes Abkühlen kann Luft verflüssigt werden. Durch
vorsichtige, farktionierte Destillation gelingt es, die beiden Hauptbestandteile der Luft,
Stickstoff und Sauerstoff, voneinander zu trennen. Dabei wird die stoffliche Natur dieser
beiden Elemente nicht verändert (Verflüssigung, Verdampfen, Destillation sind physikalische
Vorgänge). Eingeatmeter Sauerstoff wird im Körper zum „Verbrennen“ u.a. von Glucose
verwendet. Hierbei wird Energie erzeugt, und Sauerstoff und Glucose in Wasser und
Kohlendioxid überführt. Dies sind chemische Vorgänge.
2. Geschichtliches
Die Herkunft des Namens „Chemie“ liegt im Arabischen, wobei zwei Deutungen favorisiert
werden, nämlich (1) al-kimiyá (die Beschäftigung mit gießbaren Stoffen, der Metallurgie und
der Färbetechnik), und (2) ch’mi (schwarz [schwarze Erde der fruchtbaren Ebenen des Nils]).
Der bewusste Umgang mit Chemie im Sinne einer zielgerichteten Umwandlung von Stoffen
setzte in unserem Kulturraum spätestens im frühen Mittelalter ein. Solange dieser Umgang
eher unwissenschaftlich geschah und mit mythischen Elementen verbrämt wurde, spricht man
von „Alchimie“ und den „Alchimisten“ als ihren Betreibern. Ein wesentliches Ziel der
mittelalterlichen Alchimisten bestand in der Suche nach der „Quinta Essentia“ (dem fünften
Element), dem „Stein der Weisen“, die es gestatten sollten, unedle Materialen in Gold zu
verwandeln. Bei diesen Versuchen wurde natürlich kein Gold gefunden, wohl aber eine Reihe
von Entdeckungen gemacht, darunter
- Salmiak (NH4Cl) und Zinnober (HgS) von Geber (= Abu Musa Jabir ibn Hayyan) in
Arabien im 8./9. Jh.
- Alkohol (Ethanol C2H5OH) in Süditalien um 1100 aus Wein hergestellt
- Schwarzpulver (KNO3 + Schwefel + Holkohle) von Berthold Schwarz in Konstanz um
1300
- Heilmittel auf Quecksilber-, Arsen- und Antimonbasis von Paracelsus Anfang 16. Jh.
- Spiritus fumans Libavii (Zinnchlorid SnCl4) von Andreas Libavius Ende 16. Jh.
- Phosphor von Henning Brandt in Hamburg 1669
- Porzellan von Johann Friedrich Böttger 1707/08
6
Dass die eher bewusste Auseinandersetzung mit chemischen Prozessen in anderen
Kulturkreisen schon länger etabliert war zeigt die folgende chinesische Rezeptur zum Erwerb
der Unsterblichkeit aus dem 6. Jh. vor Christi: „Man thue Goldstaub in Eßig ein, setze
Salpeter und Eisenvitriol bei und bewahre das Gantze 30 Thage in einem Gefäße aus Bambus.
Diese Lösung trinke man und erfreue sich Ewigen Lebens.“ Der in der Rezeptur genannte
Salpeter (NaNO3) enthielt als Verunreinigung Kaliumiodat (KIO3), das von den Eisenionen
(Fe2+) des Eisenvitriols (FeSO4) zu Iod (I2) reduziert wird. Durch das organische Material des
Gefäßes (Bambus) erfolgt die Weiterreduktion zu Iodid (I-). In Gegenwart von Iodid erfolgt
dann die Oxidation von Gold (Au) durch den Luftsauerstoff, wobei Diiodoaurat ([AuI2]-)
gebildet wird. Dies ist der eigentliche Wirkstoff, der zwar keine Unsterblichkeit verleiht, aber
erfolgreich bei der Behandlung rheumatischer Arthritis eingesetzt werden kann.
Die wissenschaftliche Chemie, wie wir sie heute verstehen, ist verknüpft u.a. mit den Namen
Boyle (und dessen Werk „The Sceptical Chymist“, 1661) sowie Priestley, Scheele und
Lavoisier (Arbeiten zum Sauerstoffs als Bestandteil der Luft 1774/77). Erst zu Zeiten Justus
von Liebigs wurden - ca. 1840 - Professuren für Chemie an den Universitäten eingerichtet.
3. Einteilung und Trennung der Stoffe
Die Kategorisierung der stofflichen Vielfalt durch Rückführung auf Grund-„Elemente“ ist in
vielen Kulturen frühzeitig vorgenommen worden und weist überraschende Ähnlichkeiten auf;
s. die Tabelle 1.
Tabelle 1. Der Elementbegriff im Altertum
Griechenland Buddhismus
China (unter Dao)
Feuer (heiß)
Feuer
Feuer
Wasser (nass)
Wasser
Wasser
Erde (trocken) Erde
Erde
Luft (kalt)
Wind
Geist/Äther*)
Raum
Holz
Metall
*)
Erst unter Aristoteles eingeführt; im Mittelalter als quinta essentia uminterpretiert
Eine Einteilungsebene, die die Herkunft der Stoffe widerspiegelt, ist die folgende:
Geogen: z.B. Granit, Pyrit, Marmor
Biogen: z.B. Holz
Biogeogen: z.B. Kohle, Erdöl
Anthropogen: z.B. Stahl, Porzellan, PVC
Eine Einteilung der Stoffe nach Eigenschaften (und damit letztendlich nach den Bindungen,
die die Einzelbausteine zusammenhalten) zeigt Tabelle 2.
Eine hierarchische Einteilung der Stoffe nach Komplexität der Zusammensetzung zeigt
Abbildung 1. Hier sind zusätzlich die Trennverfahren eingetragen, mit deren Hilfe man von
einer Stufe höherer zu einer solchen niederer Komplexität kommt.
7
Tabelle 2. Einteilung der Stoffe nach Eigenschaften
___________________________________________________________________________
Salzartige
Metallische
Molekulare
Stoffe mit
Stoffe
Stoffe
Stoffe
Gerüststruktur
___________________________________________________________________________
Mechanische
Eigenschaften
hart, spröde
hohe Fp./Sp.
duktil
oft hohe Sp.
weich
niedrige Fp./Sp.
sehr hart
sehr hohe Fp
Elektrische
Eigenschaften
Ionenleiter
Elektronenleiter
Nichtleiter
Nichtleiter
Löslichkeit
Wasser
unlöslich
organische
Lösungsm.
unlöslich
Bindungsmodus
Ionenmetallische
kovalente
kovalente
bindung
Bindung
Bindung
Bindung
----------------------------------------------------------------------------------------------------------------Beispiele
NaCl, MgO
Metalle
N2, H2O, S8
Korund, Quarz,
Ether, Wachs
Diamant
___________________________________________________________________________
Fp. = Schmelzpunkt, Sp. = Siedepunkt
Abbildung 1
Dieses Klassifizierungsschema geht aus von heterogen Stoffen, d.h. solchen, die aus zwei
oder mehr Komponenten (= Phasen) zusammengesetzt sind und an den Phasengrenzen
sprunghafte Änderungen der physikalischen und chemischen Eigenschaften aufweisen. Man
unterscheidet folgende heterogenen Systeme (in Klammern: Beispiele):
(1) fest/fest
Gemenge
(Granit, zusammengesetzt aus Feldspat, Quarz und
Glimmer)
8
(2) fest/flüssig
(3) flüssig/flüssig
(4) fest/gasförmig
(5) flüssig/gasf.
Suspension (Blut = Blutkörperchen im Blutplasma)
Emulsion
(Milch = Fetttröpfchen in wässriger Lösung)
Rauch; Schaum
(Zigarettenrauch, Styropor)
Nebel, Schaum
(Nebel, Schlagsahne)
Gase sind stets vollständig mischbar und Mischungen aus Gasen daher stets homogene
Systeme. (4) und (5) werden auch unter dem Begriff Aerosole zusammengefasst, wenn die
dispergierte Phase fest oder flüssig, die dispergierende Phase gasförmig ist, wie im Falle von
Rauch und Nebel.
Durch einfache, physikalische Operationen lassen sich heterogene in homogene – d.h. in sich
einheitliche – Systeme zerlegen. Die Eigenschaften einer homogenen Phase sind im
Regelfalle durchgehend identisch, d.h. unabhängig vom Ort der Probennahme; Phasengrenzen
fehlen. Homogene Systeme können reine Stoffe sein oder Mehrkomponentensysteme
(Lösungen). Folgende Lösungen werden unterschieden:
(1) fest/fest
(Legierungen, z.B. Messing)
(2) fest/flüssig
(Zucker gelöst in Wasser)
(3) flüssig/flüssig
(Alkohol + Wasser)
(4) fest/gasförmig
(eine Lösung von Wasserstoff in Platin)
(5) flüssig/gasförmig
(Luft gelöst in Wasser)
(6) gasförmig/gasförmig
(Luft)
Lösungen können, wiederum durch physikalische Operationen, in einheitliche Stoffe zerlegt
werden.
Wichtige Trennverfahren zur Zerlegung von heterogenen in homogene Systeme, und zur
Auftrennung von Lösungen in reine Stoffe sind:
- Dekantieren („Abgießen“): Trennung auf Grund unterschiedlichen Gewichtes.
- Filtration: Trennung aufgrund unterschiedlicher Teilchengröße (unterschiedlichen
Dispersionsgrades) unter Nutzung der Schwerkraft. Versuch
- Zentrifugieren: Trennung aufgrund unterschiedlichen Teilchengewichtes unter
Vervielfachung der Schwerkraft.
- Flotation: Trennung aufgrund unterschiedlicher Dichte. Versuch
- Destillation: Trennung aufgrund unterschiedlicher Flüchtigkeit. Versuch
- Extraktion: Trennung aufgrund unterschiedlicher Löslichkeit in zwei nicht-mischbaren
Lösungsmitteln (Versuch: Ausschütteln von Jod aus einer wässrigen Lösung durch
Ether)
- Sublimation: Trennung von Feststoffen aufgrund unterschiedlicher Flüchtigkeit
(Versuch: Sulimation von Jod aus einem Jod/Sand-Gemisch).
- Adsorptionschromatografie: Trennung aufgrund unterschiedlicher Adsorption an
einem Trägermaterial.
Homogene Phasen können Verbindungen oder Elemente sein; Elemente wiederum können
molekular (z.B. N2, P4, S8) oder atomar sein (z.B. Ne, Hg-Dampf). Die Zerlegung einer
Verbindung in Elemente erfolgt gegebenenfalls durch Zersetzung mittels Wärme
(Thermolyse; Versuch: Zerlegung von HgO in Hg und O2), Licht (Fotolyse, z.B. Zerlegung
von AgBr in Ag und Br2) oder den elektrischen Strom (Elektrolyse, z.B. Zerlegung von H2O
in H2 und O2), oder durch eine chemische Reaktion, bei der ein Bestandteil „umgebunden“,
der andere frei gesetzt wird (z.B. 2 Mg + CO2 2 MgO + C).
Elemente können polynuklid (trifft für die meisten Elemente zu) oder mononuklid (Fluor)
vorkommen. Zum Begriff „Nuklid“ s. Abschnitt 6.
9
4. Die chemische Reaktionsgleichung
Geschichtliche Vorbemerkung:
Zu den ältesten von Menschen (unbewusst) durchgeführten chemischen Prozessen gehört das
Verbrennen organischer Materialien wie Holz. Der Verbrennung liegt die Vereinigung eines
brennbaren Substrats mit Sauerstoff zugrunde. Hierbei bildet sich ein neuer Stoff, ein Oxid.
(Versuch: Verbrennen von Mg zu MgO). Erst Lavoisier hat diesen Vorgang korrekt gedeutet
(1777). Zuvor glaubte man, dass bei der Verbrennung ein Stoff („Phlogiston“) entweiche, was
bei ungenauem Experimentieren dem Augenschein entspricht (Versuch: Gewichtsabnahme
einer brennenden Kerze auf einer Waage). Tatsächlich nimmt, wenn man die Verbrennung so
vornimmt, dass die Rauchgase nicht entweichen können, das Gewicht zu (Versuch). Nachdem
Lavoisier dies zeigen konnte, versuchten die Anhänger der Phlogistontheorie, ihre Theorie
dadurch zu retten, dass sie dem Phlogiston eine negative Masse zugedachten, eine absurde
Sichtweise, die alsbald aufgegeben werden musste.
Im Gesamtsystem treten weder bei einer Verbrennung noch bei anderen chemischen
Reaktionen wägbare Masseveränderungen auf. Für die Verbrennung von Magnesium heißt
das, dass die Summe der Massen aus Magnesium und Sauerstoff vor der Reaktion gleich der
Masse des entstandenen Magnesiumoxids nach der Reaktion ist. In diesem Sinne ist eine
chemische Reaktionsgleichung auch eine Gleichung hinsichtlich der Massenbilanz, und bei
Aufstellen der korrekten Reaktionsgleichung muss das beachtet werden. Beispiel:
Knallgasreaktion (Verbrennen von Wasserstoff mit Sauerstoff zu Wasser). Die
Reaktionsgleichung einschließlich der Energiebilanz lautet:
2H2 + O2 2 H2O, H = -286 kJ/mol
Die Reaktionsgleichung gibt Auskunft über:
- Die Reaktionsrichtung, gekennzeichnet durch den Reaktionspfeil
- Die Art der Stoffe: Die Edukte (Wasserstoff und Sauerstoff) und Produkte (Wasser)
- Die Stoffmengenbilanz oder Stöchiometrie der Reaktion: 2 mol H2 reagieren mit 1
mol O2 unter Bildung von 2 mol Wasser. Die numerischen Faktoren vor den Edukten
und Produkten heißen „stöchiometrische Faktoren“. Diese Faktoren sind so zu wählen,
dass Edukt- und Produktseite ausgeglichen sind.
- Die Massenbilanz: 4g H2 (= 2 mol) reagieren mit 32g O2 (= 1 mol) zu 36g H2O (= 2
mol). Entsprechendes gilt für Vielfache oder Bruchteile der Stoffmengen bzw.
Massen.
- Den Energieumsatz. Ein negatives Vorzeichen entspricht freiwerdender Energie
(exotherme Reaktion), ein positives Vorzeichen einem Energieverbrauch (endotherme
Reaktion.
Bei Reaktionsgleichungen, in denen auch Ionen (also geladene Teilchen) eine Rolle spielen,
ist neben einer ausgeglichenen Stoffmengenbilanz auch die ausgeglichene Ladungsbilanz zu
beachten.
Viele chemische Reaktionen laufen nicht eindeutig in eine Richtung. Es stellen sich
Gleichgewichtssituationen ein, die mehr auf der linken oder mehr auf der rechten Seite liegen
können. In diesem Falle bedient man sich des Gleichgewichtspfeiles . Beispiel:
H2 + I2 2HI
10
Die Reaktionsgleichung gibt keine Auskunft darüber, ob eine Reaktion tatsächlich abläuft.
Der Reaktionsablauf kann gehemmt sein; z.B. ist ein Gemisch aus Wasserstoff und Sauerstoff
metastabil. Viele chemische Reaktionen, wie auch die Knallgasreaktion, sind „kinetisch
gehemmt“; es bedarf der Überwindung einer Aktivierungsbarriere, vergl. Abb. 2. Die
Aktivierungsenergie kann durch Wärmezufuhr bereitgestellt werden (H2 und O2 reagieren bei
Zündung explosionsartig). Eine Alternative ist die Herabsetzung der Aktivierungsschwelle
durch einen Katalysator. So reagieren H2 und O2 auch bei Raumtemperatur im Kontakt mit
Platin.
Energie
aktivierter
Zustand
4H + O2
Edukte
Aktivierungsenergie
2H2 + O 2
H
Produkt(e)
2H2O
Reaktionskoordinate
Abbildung 2
5. Der Aufbau der Atome I:
Rutherfordsches Atommodell und Elementarteilchen
Rutherfords Streuversuche:
Atome sind die kleinsten, mit gängigen Mitteln nicht weiter teilbaren Grundeinheiten aller
Materie. Erste detaillierte Vorstellungen zum Aufbau der Atome entwickelte Rutherford
durch seine Streuversuche im Jahre 1911 (Abb. 3). Er beschoss eine äußerst dünne Goldfolie
mit -Teilchen (zweifach positiv geladene Heliumkerne) und stellte fest, dass der weitaus
größte Teil ungehindert die Goldfolie passierte. Einige wenige -Teichen wurden abgelenkt,
noch weniger zurückgeworfen. Der Versuch führte zu den folgenden Schlussfolgerungen:
1. Atome sind weitgehend leer
2. Die Masse des Atoms konzentriert sich im positiv geladenen Kern
3. Das Volumen des Atoms wird durch eine negativ geladene Hülle repräsentiert
Das Verhältnis der Durchmesser von Kern und Hülle liegt bei etwa 10-5, der Durchmesser
eines Atoms in der Größenordnung 10-10 m (= 100 pm = 1Å).
-Teilchen
Ra
Radiumquelle im
Bleiblock
Blende
Gold-Folie
.. ..
.. ..
.. ..
11
Abbildung 3. Rutherfordscher Streuversuch. Links: Versuchsaufbau; rechts: Ausschnitt aus
der Goldfolie. Die Pfeile geben die Flugrichtung der -Teichen an.
Die positiven Ladungen des Atomkernes wurden mit Protonen, die negativen Ladungen der
Atomhülle mit Elektronen assoziiert, deren Existenz man bereits aus anderen Versuchen
(Kanalstrahlen, Kathodenstrahlen; s. Lehrbücher der Physik) und Beobachtungen kannte
(radioaktiver Zerfall; s. Abschnitt 6). Hiermit ergab sich ein Atommodell, in dem negativ
geladene Elektronen nur geringer Masse den aus positiv geladenen Protonen
zusammengesetzten Massenschwerpunkt, den Atomkern, in einem großen Abstand
umkreisen, die Atome im Übrigen aber „leer“ sind. [Neutronen als zusätzliche Kernbausteine
wurden erst 1932 durch Chadwick entdeckt]. Grenzen dieses Modells:
- Bewegte Ladungen strahlen Energie ab; die Elektronen müssten also in den Kern
stürzen.
- Die Atomspektren zeigen diskrete Linien statt des im Rutherfordschen Modell
erwarteten Kontinuums.
- Die Leere der Atome ist schwer mit der Alltagserfahrung massiver Materialen in
Einklang zu bringen.
Elementarteilchen:
Die für den Chemiker wichtigsten Elementarteilchen (Bausteine der Atome) sind in Tabelle 3
zusammengestellt. Neben den dort genannten Eigenschaften besitzen die Elementarteilchen
einen „Eigendrehimpuls“ (Spin).
Tabelle 3. Elementarteilchen
________________________________________________________________________
Name
Symbol
Ort/
Ruhemasse (u*)) bzw.
Ladung
Entstehung
Molmasse (g mol-1)
________________________________________________________________________
Proton
p, H+
Atomkern
1,00728
+1
Neutron
n
Atomkern
1,00867
0
Elektron
e-, -
Atomhülle/
Zerfall eines n
0,00055
-1
Positron
e+, +
Zerfall eines p
0,00055
+1
Neutrino und
Zerfall eines p
0
0
Antineutrino
oder n
________________________________________________________________________
*)
Die atomare Masseneinheit (atomic unit), u, ist die Masse eines Zwölftels der Masse des
Isotops 12C (1 u 1,66·10-24 g). Zahlenmäßig ist u gleich der molaren Masse (Einheit: g/mol).
6. Radioaktivität und Kernreaktionen
Der Atomkern enthält (außer beim Wasserstoffisotop 1H) neben Protonen die etwa
massegleichen, neutralen Neutronen. Ein durch Protonenzahl (= Kernladungszahl =
Ordnungszahl) und Massenzahl (Summe aus Protonen und Neutronen) gekennzeichneter Kern
wird als Nuklid bezeichnet. Die Massenzahl steht links oben, die Kernladungszahl links unten
am Elementsymbol. Nuklide mit derselben Kernladungszahl aber unterschiedlicher
12
Massenzahl (und damit unterschiedlicher Neutronenzahl) sind Isotope (von iso topos; an der
selben Stelle des Periodensystems stehend). Beispiele:
1
1H
2
1H
3
1H
12
C
6
13
C
6
14
235
U
92
6
C
238
U
92
Die Wasserstoffisotope Protium, Deuterium und Tritium
Die drei wichtigsten Kohlenstoffisotope
Zwei der gebräuchlichsten Uranisotope
Wird die Neutronenzahl überproportional groß, so wird der Kern instabil; er zerfällt unter
Emission charakteristischer Strahlung. Diesen Vorgang bezeichnet man als radioaktiven
Zerfall. Die wichtigsten Zerfallsarten sind:
- der -Zerfall (Emission zweifach positiv geladener Heliumkerne der Masse 4)
- der --Zerfall (Emission von Elektronen)
- der +-Zerfall (Emission von Positronen)
Begleitend tritt fast immer auf
- die -Strahlung (extrem kurzwellige = energiereiche elektromagnetische Strahlung);
wird beim Übergang des Tochternuklids aus dem angeregten Zustand (symbolisiert
durch {}*) in den Grundzustand emittiert.
Beispiele:
-Zerfall:
-
-Zerfall:
223
88Ra
219
4
86Rn + 2He
14
6C
14
7N
137
55Cs
137
56Ba
+ -1e
+ -1e
137
56Ba
+
-Zerfall:
40
19K
40
18Ar
+
+ 1e
Der dem -Zerfall zugrunde liegende Elementarprozess ist der Zerfall eines Neutrons (Zerfall) bzw. eines Protons (+-Zerfall) unter gleichzeitiger Bildung eines Antineutrinos bzw.
eines Neutrinos:
-
1
0n
1
1p
+ -10e + 00
+
1
1p
1
0n
+ -1e + 0
-Zerfall:
-Zerfall:
0
0
Beim -Zerfall (Erniedrigung der Kernladungszahl um zwei Einheiten) rückt das neu
entstandene Element (das Tochterelement) im Periodensystem um zwei Positionen nach links,
beim --Zerfall (Erhöhung der Kernladungszahl um eine Einheit) um eine Position nach
rechts, beim+-Zerfall (Erniedrigung der Kernladung um eine Einheit) um eine Position nach
links.
Der radioaktive Zerfall wird durch die folgende Gesetzmäßigkeit beschrieben:
-dN/dt = N
13
N ist die Zahl der zum Zeitpunkt t (noch) vorhandenen Kerne, die Zerfallskonstante, eine
für jedes radioaktive Nuklid charakteristische Konstante. Demnach ist die in einem
infinitesimalen Zeitraum dt zerfallende Anzahl von Kernen proportional der jeweils
vorhandenen Zahl von Kernen. Variablentrennung und Integration in den Grenzen N0 und N
bzw. t0 = 0 und t (N0 = Zahl der Kerne zum Zeitpunkt t = 0) ergibt
ln(N/N0) = -t, oder: N = N0·e-t
, die Zerfallskonstante, ist ein Maß für die Aktivität eines radioaktiven Nuklids.
Anschaulicher ist die Halbwertszeit t1/2, d.h. diejenige Zeit, die angibt, wann die Hälfte einer
Ausgangsmenge von Kernen (N = ½N0) zerfallen ist:
t1/2 = ln2/
Je kürzer die Halbwertzeit, um so „aktiver“ ist ein Nuklid. Beispiele für Halbwertszeiten. (a =
Jahr [annum], h = Stunde [hora], s = Sekunde):
232
90Th
238
92U
235
92U
1.3 1010 a
4.5 109 a
7 108 a
99m
43Tc
6h
14
6C
5570 a
3
1H
12.35 a
266
109Mt
3.4 s
Thorium (Th) und Uran werden in Kernkraftwerken, Uran auch waffentechnisch
(Atombombe) verwendet. Technetium-99m (99mTc), ein metastabiler (m) -Strahler, der mit
einer Halbwertszeit von 6 Stunden in 99Tc (-Strahler, Halbwertszeit 21000 Jahre) zerfällt,
spielt in der medizinischen Radiodiagnostik eine wichtige Rolle.
Kohlenstoff-14 und Tritium (beide Isotope sind --Strahler) werden bei der Altersbestimmung
verwendet, Tritium z.B. bei der Alterskontrolle von Wein oder der Datierung von
unterirdischen Wasserreservoirs, 14C bei der Datierung archäologischer Funde, sofern diese
organische Materialien enthalten (Carbon-14-Methode): Solange ein Organismus, z.B. ein
Baum, lebt, tauscht er seine Biomasse und damit auch seinen Kohlenstoff ständig mit der
umgebenden Atmosphärenluft aus, die im Kohlendioxidgehalt stets einen weitgehend
konstanten Prozentsatz an 14CO2 aufweist. Nach dem Fällen des Baumes, z.B. im Rahmen der
Verarbeitung zu Schiffsplanken, erfolgt dieser Austausch nicht mehr. Der Gehalt an 14C
nimmt stetig ab; über die Restaktivität lässt sich der Zeitpunkt bestimmen, zu dem der Baum
gefällt worden war. Gebildet wird 14C in der Stratosphäre durch die folgende Reaktion:
14
N
7
14
6C
+ 10 n
1
+ 1H
Gemessen wird die Aktivität eines Radionuklids in Bequerel (Bq). 1 Bq = 1 Zerfall pro
Sekunde. (Mitunter wird auch noch die Einheit Curie, Ci, verwendet. 1 Ci = 3.7·1010 Bq; dies
entspricht der Aktivität von eines Gramms Radium). Zur Messung dient der Geigerzähler;
Abb. 4 (Versuch).
Ar
Ar+e-
Abbildung 4: Schemazeichnung eines Geigerzählers.
Die radioaktiven Teilchen (im wesentlichen und -)
ionisieren die unter vermindertem Druck mit Argon
gefüllte Kammer. Die Ar+ und e- fliegen zur Kathode
bzw. Anode und lösen so einen Kurzschluss aus, der z.B.
über ein Zählwerk registriert und über einen
Lautsprecher in ein akustisches Signal umgesetzt wird.
14
Die Beurteilung der biologischen Wirkung radioaktiver Strahlung erfolgt durch die
Energiedosis; sie wird in Gray (Gy) angegeben; 1 Gy = 1J/1kg (d.h. absorbierte
Strahlungsenergie pro Masse; J = Joule ist die Einheit für die Energie). Die Äquivalentdosis
berücksichtigt auch die Strahlungsart: Äquivalentdosis = Energiedosis × Q, wo Q ein von der
Strahlungsart abhängiger Faktor ist (Q = 20 für ; 1 für , und Röntgenstrahlen; 3-10 für
Neutronen [je nach Geschwindigkeit der Neutronen]). Angegeben wird die Äquivalentdosis in
Sievert (Sv), Einheit J/kg. 1 Sv entspricht 100 rem (röntgen equivalent man). 1 rem = 10 mSv.
Kernchemische Prozesse spielen weiterhin eine Rolle bei der Kernfusion und bei der
Kernspaltung. Die Kernfusion ist z.B. der energieliefernde Prozess in unserer Sonne (und
vielen anderen Fixsternen; die für die Fusion erforderliche Temperatur liegt bei 1 Million
Grad): Hier verschmelzen 4 Protonen (= Wasserstoffkerne) zu Helium. Dabei werden
Positronen und Neutrinos freigesetzt:
1
0
4
0
He
+
2
+2 0
2
1
4 H
1
Vergleicht man die Masse des Heliumkernes mit der Summe der Massen der den Heliumkern
aufbauenden zwei Protonen und zwei Neutronen, so kommt man zu einem Defizit von
0,036608 g/mol. Die Umrechnung dieses Massendefektes in Energie mit Hilfe der
Einsteinschen Energie-Masse-Beziehung E = mc2 (E = Energie, m = Masse, c =
Lichtgeschwindigkeit = 3·108 m/s) ergibt eine Energiemenge von 3,3·109 kJ/mol; vergl. die
bei der Knallgasreaktion (2H2 + O2 2H2O) frei werdende chemische Energie von 286
kJ/mol).
Künstliche Fusion (Fusionsreaktor, Wasserstoffbombe): Fusion von Tritium mit Deuterium
unter Bildung von Helium und einem Neutron. Das Tritium wird aus 6Li mit Neutronen
„erbrütet“ (dabei entsteht neben Tritium Helium). In der Praxis setzt man Lithiumdeuterid ein:
6
2
3Li + 1H
4
2 2He
In sehr heißen Sternen spielen auch die folgenden, für die Entstehung schwererer Elemente
verantwortlichen Fusionsprozesse eine Rolle:
Helium-Fusion:
4
8
4 Be
2 2 He
12
6C
16
8O
20
10Ne
CNO- oder Bethe-Weizsäcker-Zyklus:
1
12
+ 1H
6C
13
7N
-
1
1H
13
C
6
+
1
1H
14
7N
15
8O
-
+
1
1H
15
N
7
12
6C
4
+ 2 He
Fusionen spielen auch bei der Erzeugung der schwersten, künstlichen Elemente eine Rolle.
Als Beispiel sei die Generierung von Meitnerium genannt:
209
83 Bi
58
+ 26 Fe
266
1
Mt + 0 n
109
Die in Kernkraftwerken (s. die Schemazeichnung Abb. 5) durchgeführte kontrollierte
Kernspaltung z.B. von angereichertem Uran (Anreicherung des Isotops U-235) liefert
Nutzenergie. Die Spaltung wird mit langsamen, so genannten thermischen Neutronen
durchgeführt, z.B. gemäß der Gleichung:
235
U + 1n
92
0
92
Kr
36
+
142
56
Ba + 2 1n
0
15
Abbildung 5.
Schematische
Darstellung eines
Kernkraftwerkes
(DruckwasserReaktors).
Da hierbei mehr Neutronen frei als eingesetzt werden, bedarf es einer diffizilen Steuerung, um
einerseits das Erlöschen, andererseits das Durchbrennen des Reaktors zu verhindern. 1 g Uran
liefert 8·1010 J (entsprechend der Umwandlung von 0.1% der Masse des Urans in Energie);
das entspricht dem Brennwert von 2.4·103 kg Kohle. Auf dem Prinzip der Kernspaltung des
beruhen auch die Atombomben (235U, Hiroshima).
Brutreaktoren produzieren aus 238U Plutonium, das seinerseits als Reaktorbrennstoff und als
Spaltmaterial in Bomben (Nagasaki) Verwendung finden kann:
238
92U
+n
239
92U
-
-e
239
93Np
-
239
94Pu
-e
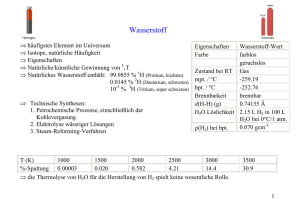
7. Wasserstoff
Wasserstoff ist mit 91 Atom-% das kosmisch häufigste Element und kommt dort als H+ (HIIRegionen), H (HI-Regionen), H2, H2+ und H3+ in interstellaren Wolken sowie in Form von
Protonen in der kondensierten Masse (also in Sternen) vor. Die Fusion von Wasserstoff zu
Helium (s.o.) ist in Sternen wie unserer Sonne der wichtigste Energie liefernde Prozess.
Interstellare Wolken enthalten außerdem eine Vielzahl anorganischer und organischer
Wasserstoffverbindungen. Der Nachweis des Wasserstoffs in den HI-Regionen erfolgt
radioastronomisch mittels der 21cm-Linie, die einem Umklappen des Elektronenspins relativ
zum Kernspin entspricht. [Spin = Eigendrehimpuls]
Die irdische Atmosphäre enthält nur 5·10-5 % freien Wasserstoff. Der Gehalt von Wasserstoff
in Form von Verbindungen in der Erdkruste einschl. Aqua- und Troposphäre liegt bei 0,74
Gew.-%. Vorkommen in Form von Verbindungen: freies Wasser (in allen drei
Aggregatzuständen), H2S, NH3, Hydrate wie CaSO4·2H2O (Gips) und KMgCl3·6H2O
(Carnallit), Hydroxide wie AlO(OH) (Bauxit), organische Verbindungen in Form von
Biomasse und fossilen „Brennstoffen“ (Erdgas = CH4, Methanhydrate, Erdöl, Kohle).
16
Elementarer Wasserstoff H2 existiert in zwei Formen, dem Orthowasserstoff (Kernspins der
beiden Atome parallel ausgerichtet) und dem Parawasserstoff (Kerspins antiparallel). Beide
Formen stehen miteinander in einem temperaturabhängigen Gleichgewicht, das bei
Raumtemperatur zu 75% auf Seiten des Orthowasserstoffs liegt. Die drei Nuklide 1H, 2H
(„schwerer Wasserstoff“, Deuterium) und 3H (Tritium; radioaktiv) wurden bereits erwähnt.
Wasserstoff hat unter allen Elementen die geringste Dichte: = 0,0089·10-3 g/ml (vergl. Luft:
1,29·10-3, CO2: 1,98·10-3 g/ml). Versuch: H2 und CO2 auf der Waage. H2 hat das am stärksten
ausgeprägte Diffusionsvermögen. Die Diffusionsgeschwindigkeiten v1 und v2 zweier Gase
verhalten sich umgekehrt proportional zur Wurzel aus ihren Molmassen: v1/v2 = √M2/M1
(Versuch: Diffusion von Wasserstoff bzw. Luft durch einen Tonzylinder).
Die Gewinnung von Wasserstoff erfolgt heute im Wesentlichen durch das Steamreforming
aus Erdgas und Wasserdampf (1), Dehydrierung von Erdöl (2), Elektrolyse oder Fotolyse von
Wasser (3), im Labor auch aus Zink und Säuren (4). Historisch von Interesse ist die
Gewinnung aus Kohle und Wasserdampf (5).
- CH4 + H2O CO + 3H2
(1)
- CnH2n+2 CnH2n + H2
(2)
- H2O H2 + ½O2
(3) Versuch: Elektrolyse von Wasser
+
2+
- Zn + 2H Zn + H2
(4)
- H2O + C H2 + CO (“Wassergas”)
(5)
Verwendung:
- Ammoniaksynthese: N2 + 3H2 2NH3 (s. hierzu das Kapitel „Stickstoff“)
- Hydrocracking: C12H26 + H2 C4H10 + C8H18 (Dodecan zu Butan und Octan)
- Hydrotreating: C8H16 C8H18 (Octen zu Octan)
- Härtung von Pflanzenölen (z. B. in der Margarineherstellung):
Z-CH3(CH2)7CH=CH-(CH2)7CO2H + H2 CH3(CH2)16CO2H (Ölsäure zu Stearinsäure)
- Hydrodesulfurierung von Erdöl: S-haltige Verbindung + H2 H2S
- Fischer-Tropsch-Synthesen, z.B. CO + 2H2 CH3OH (Methanol)
- Als Reduktionsmittel: Fe2O3 + 3H2 2Fe + 3H2O
- Kohlehydrierung: n C + (n+2) H2 CnH2n+2
- Kernfusion (s.o.)
Reaktionen (s.a.: Verwendung):
- Mit Sauerstoff bei Zünden oder am Pt-Kontakt heftige Reaktion (Knallgasreaktion; die dabei
entstehende Hitze wird auch zum Schweißen genutzt.):
H2 + ½O2 H2O, H = -286 kJ/mol
Wasserstoff-Luft-Gemische sind explosiv!
Diese Reaktion kann in Brennstoffzellen auch „sanft“ und unter Erzeugung elektrischen
Stromes geführt werden (s. Kap. Elektrochemie)
- Mit Chlor bei Initiierung Chlorknallgasreaktion. Einzelschritte dieser Kettenreaktion:
Start:
Cl2 2Cl·
H = +243 kJ/mol
Reaktionskette:
Cl· + H2 HCl + H·
H = +4 kJ/mol
H· + Cl2 HCl + Cl·
H = -188 kJ/mol
Abbruch:
H· + Cl· HCl
Gesamtreaktion:
H2 + Cl2 2HCl
H = -184 kJ/mol
17
Die Überwindung der Aktivierungsschwelle (Startreaktion) kann z.B. durch Einstrahlen
hinreichend energiereichen Lichtes erfolgen (s. Tabelle 4) Hierzu reicht rotes Licht nicht aus,
wohl aber blaues Licht (Versuch).
Tabelle 4. Energieinhalte sichtbaren Lichts
____________________________________________________
Farbe
Wellenlänge (nm)
Energie (kJ/mol Photonen)
____________________________________________________
Rot
700
171
Gelb
600
199
Grün
550
217
Blau
450
266
Violett
400
299
____________________________________________________
- Mit Nichtmetallen: Bildung leicht flüchtiger Nichtmetall-Wasserstoffverbindungen: HCl,
H2S, NH3, CH4 usw.
- Mit Alkali- und Erdalkalimetallen: Bildung salzartig gebauter Hydride (außer bei Mg und
Be), z. B. Na + ½H2 Na+H- Mit Bor bilden sich Diboran B2H6 und höherkernige Borane
- Einige Metall, z.B. Pt, Ti, Mg-Ni, vermögen Wasserstoff legierungsartig zu lösen.
- In interstellaren Gaswolken laufen komplexe Reaktionen ab, die zu einfachen und
komplexen Molekülen führen. Beispiel: Die Bildung von Cyanwasserstoff und
Isocyanwasserstoff .
+
N + H2
+
NH + H
+ H2
+
NH2 + H
+ H2
+
CH3 + N
+
NH3 + H
+ Mg
+
NH3 + Mg
+
+C
+
H2CN + H
-
+e
-H
HCN und HNC
In einer Entladungsröhre, die Wasserstoff unter vermindertem Druck enthält, beobachtet man
bei Anlegen einer Spannung von etwa 10 kV ein intensives und charakteristisches rotes
Leuchten (Versuch): Durch die elektrische Spannung (Energiezufuhr) werden angeregte
Wasserstoffatome (H*) erzeugt; beim Rückfall in den Grundzustand wird die
Energiedifferenz als Licht (h ; h = Plancksches Wirkungsquantum, = Frequenz) emittiert:
H2 2H; H H*; H* H * + h. Zur Erläuterung s. den folgenden Abschnitt.
8. Der Aufbau der Atome II:
Das Bohr-Sommerfeldsche Atommodell
Bei Zerlegung des von angeregtem Wasserstoff ausgesandten Lichtes mittels eines Prismas
und Abbildung durch einen Spalt erhält man ein charakteristisches Linienspektrum. Die
18
Deutung solcher Spektren führte Bohr zur Aufstellung seines Modells von der Strukturierung
der Elektronenhülle (Bohrsches Atommodell, 1913). Beim Wasserstoff umfasst das
Gesamtspektrum 5 Serien, eine davon, die Balmerserie, liegt im sichtbaren Bereich; vergl.
Abb. 6.
Elektronensprünge von angeregten
Zuständen auf den Zustand n = 2
6
5
Blende
4
Licht
3
2
Aufzeichnung
656 nm
1
Prisma
Wasserstoffatom mit
Elektronenbahnen (n = 1, 2, 3, 4, 5, 6)
(n = 1 ist der Grundzustand)
486 nm
Spektrallinien im Vis
(Balmer-Serie)
434 nm
410 nm
für Sprünge, die auf n = 2 enden:
Balmer-Serie (sichtbares Licht, Vis)
Abbildung 6. Entstehung diskreter Lichtemissionen angeregter Atome.
Zur Erklärung stellte Bohr die folgenden Postulate auf:
1. Das Elektron des Wasserstoffatoms umkreist den Atomkern auf einer Bahn (Schale).
Dies geschieht ohne Energieverlust. (nicht vereinbar mit klassischen Vorstellungen
der Physik)
2. Das Elektron kann unterschiedliche Bahnen mit unterschiedlichem Abstand vom Kern
einnehmen. Erlaubt sind aber nur Bahnen, die der folgenden Bedingung genügen:
Bahnumfang × Impuls = ganzzahliges Vielfaches des Plankschen Wirkungsquantums,
2r × mv = nh
r = Bahnradius, m = Masse, v = Geschwindigkeit, n = 1,2,3..., h = Planck-Konstante =
6,626·10-34 Js. In anderen Worten: Der Bahndrehimpuls ist gequantelt.
3. Bei der Aufnahme von Energie springt das Elektron vom Grundzustand (n = 1,
entsprechend der kernnächsten Bahn oder K-Schale) auf einen der höher liegenden
Zustände (n = 2: L-Schale, n = 3: M-Schale, n = 4: N-Schale, usw.). Dieser Übergang
vom Grundzustand zu angeregten Zuständen erfolgt diskontinuierlich.
4. Bei der Rückkehr in tiefer liegende Zustände und in den Grundzustand (auch wieder
diskontinuierlich) werden Energiebeträge emittiert, die den Energiedifferenzen
zwischen den einzelnen Zuständen (Bahnen) entsprechen.
Beim Wasserstoff werden fünf Serien unterschieden (vergl. Abb. 6):
Lyman-Serie (UV)
Sprünge auf die K-Schale (n = 1)
Balmer-Serie (Vis)
Sprünge auf die L-Schale (n = 2)
Paschen-Serie (IR)
Sprünge auf die M-Schale (n = 3)
Brackett-Serie (IR)
Sprünge auf die N-Schale (n = 4)
Pfund-Serie (MW)
Sprünge auf die O-Schale (n = 5)
Die Spektren werden so zum Abbild der verschiedenen möglichen Energiezustände des
Atoms und damit zu einem Abbild der Struktur der Atomhülle.
19
Die Zahl „n“ wird als Hauptquantenzahl bezeichnet. Sie gibt die Hauptenergieniveaus an. Die
Verfeinerung von Sommerfeld trägt der Tatsache Rechnung, dass Bahnen mit n > 1 eine
Substruktur aufweisen. Diese „Unterbahnen“ (Energie-Unterniveaus), charakterisiert durch
den Buchstaben „l“ (Nebenquantenzahl), wurden ursprünglich als Ellipsenbahnen
unterschiedlicher Exzentrizität interpretiert. Die Nebenquantenzahl l kann Werte von l = 0
(Kreisbahn, s-Elektronen), 1 (p-), 2 (d-), 3 (f-Elektronen) bis maximal n-1 annehmen.
9. Aufbau des Periodensystems
Die p (l = 1), d (l = 2) und f-Niveaus (l = 3) spalten im Magnetfeld weiter auf in magnetische
Unterniveaus, gekennzeichnet durch die Magnetquantenzahl m, die Werte von +l über 0 bis –l
annehmen kann:
n = 1: l = 0 m = 0
n = 2: l = 0 m = 0
l = 1 m = +1, 0, -1
n = 3: l = 0 m = 0
l = 1 m = +1, 0, -1
l = 2 m = +2, +1, 0, -1, -2
n = 4: l = 0 m = 0
l = 1 m = +1, 0, -1
l = 2 m = +2, +1, 0, -1, -2
l = 3 m = + 3, +2, +1, 0, -1, -2, -3
Im ungestörten Atom sind die zu einem bestimmten n und l gehörenden magnetischen
Unterniveaus energiegleich = entartet: s-Niveaus sind einfach, p-Niveaus dreifach, d-Niveaus
fünffach und f-Niveaus siebenfach entartet. Jedes der durch n, l und m gekennzeichneten
Niveaus kann mit zwei Elektronen besetzt werden, wobei die Elektronen sich dann durch die
Spinquantenzahl s unterscheiden. Die Spinquantenzahl s kann den Wert +½ () oder -½ ()
annehmen.
Der Aufbau der Atome vollzieht sich nun so, dass, beginnend mit dem Wasserstoff, Elektron
für Elektron in die Energieniveaus eingefüllt wird. Hierbei sind die folgenden drei Regeln zu
beachten:
1. Aufbauprinzip: Die Elektronen gehen stets in die energetisch niedrigsten verfügbaren
Niveaus.
2. Hundsche Regel: Entartete Zustände werden zunächst einzeln besetzt, wobei alle
Elektronen denselben Spinzustand haben (größt mögliche Multiplizität). Erst wenn
jedes entartete Niveau ein Elektron aufgenommen hat, tritt das darauf folgende mit
entgegen gesetztem Spin (Spinpaarung) ein.
3. Pauli-Prinzip: Zwei Elektronen müssen sich mindestens in einer der vier
Quantenzahlen unterscheiden. Jedes Elektron hat also eine eigene „Individualität“.
Die maximal mögliche Zahl der Elektronen in einem neutralen Molekül ist 2n2.
Als Beispiel ist in Abbildung 7 die Elektronenverteilung des Eisens gezeigt. Die
Elektronenverteilung (Elektronenkonfiguration) in der äußersten Schale (bei den d-BlockElementen in den äußeren Schalen), den Valenzschalen, wird als ValenzelektronenKonfiguration bezeichnet.
Die Valenzelektronen-Konfiguration wiederholt sich periodisch. Da sie eng mit dem
chemischen Verhalten der Elemente verknüpft ist, kommt man so zu einer Anordnung der
Elemente in vertikalen Gruppen (Familien), in denen die zur jeweiligen Gruppe gehörenden
Elemente neben derselben Valenzelektronen-Konfiguration auch chemische Ähnlichkeiten
20
aufweisen. Die Gruppen werden von 1 bis 18 durchnummeriert. Horizontal ordnen sich die
Elemente in Perioden an. Eine neue Periode beginnt immer dann, wenn ein HauptenergieNiveau vollständig mit Elektronen gefüllt ist. Die Nummerierung der Perioden entspricht der
Hauptquantenzahl n:
Energie
4f
4d
n=4
4p
3d
4s
ValenzelektronenKonfiguration
des Fe: 4s23d6
3p
n=3
3s
2p
n=2
2s
n=1
Elektronenkonfiguration
für Eisen (Fe)
1s
Kern
Abbildung 7. Energieniveauschema und Elektronenverteilung im Eisenatom.
Perioden:
n=1
n=2
n=3
n=4
n=5
n=6
n=7
Wasserstoff und Helium
Lithium bis Neon
Natrium bis Argon;
Kalium bis Krypton; Scandium bis Zink
Rubidium bis Xenon; Yttrium bis Cadmium;
Cäsium bis Radon; Lanthan bis Quecksilber; Cer bis Luthetium
Francium und Radium; Actinium bis Lawrencium; Rutherfordium bis
Röntgenium (bzw. bis Element 118 [Stand Nov. 2006])
Die Elemente, mit der Valenzelektronenkonfiguration ns1-2p0-6 sind die
Hauptgruppenelemente. Beginnend mit der dritten Periode (n = 3) kommt es zu einer
Besonderheit in der energetischen Abfolge der l-Unterniveaus: Hier liegt das 4s-Niveau
energetisch günstiger als das 3d-Niveau, wird also zuerst gefüllt (Elemente Kalium und
Calcium). Es folgt die Besetzung der 3d-Niveaus (Scandium bis Zink; s.a. Eisen in Abb. 7).
Diese Besonderheiten wiederholen sich in den folgenden Gruppen. Die Elemente der Gruppen
3 bis 12, die so generiert werden, fasst man unter dem Begriff d-Block-Elemente (auch:
Nebengruppenelemente, Übergangsmetalle) zusammen. In der 6. und 7. Periode werden auch
f-Niveaus besetzt (4f für n = 6: Lanthaniden; 5f für n = 7: Actiniden).
21
Gruppen:
1
2
3
ns1
Alkalimetalle (Li bis Fr)
2
ns
Erdalkalimetalle (Be bis Ra)
ns2(n-1)d1
Scandiumgruppe (Sc, Y, La, Ac; n = 4, 5, 6, 7)
2
1
1-14
ns (n-1)d (n-2)f
n = 6: Lanthanide; n = 7: Actinide
4
ns2(n-1)d2
Titangruppe (Ti, Zr, Nb, Hf, Rf)
2
3
5
ns (n-1)d
Vanadiumgruppe (V, Nb, Ta, Db)
6
ns1(n-1)d5
Chromgruppe (Cr, Mo, W, Sg)
7
ns2(n-1)d5
Mangangruppe (Mn, Tc, Re, Bh)
2
6
8
ns (n-1)d
Eisengruppe (Fe, Ru, Os, Hs)
9
ns2(n-1)d7
Cobaltgruppe (Co, Rh, Ir, Mt)
2
8
10
ns (n-1)d
Nickelgruppe (Ni, Pd, Pt, Rg)
11
ns1(n-1)d10
Kupfergruppe (Münzmetalle; Cu, Ag, Au)
2
10
12
ns (n-1)d
Zinkgruppe (Zn, Cd, Hg)
13
ns2p1
Bor-Aluminiumgruppe (B bis Tl)
2 2
14
ns p
Kohlenstoff-Siliziumgruppe (C bis Pb)
15
ns2p3
Stickstoff-Phosphorgruppe (Pnictogene) (N bis Bi)
16
ns2p4
Chalcogene (O bis Po)
2 5
17
ns p
Halogene (F bis At)
18
ns2p8
Edelgase (He bis Rn)
Voll gefüllte Schalen sind besonders stabil: Edelgase (Gruppe 18). Man beachte aber auch die
besondere Stabilität der halb und ganz gefüllten d-Schale (Gruppen 6 und 11).
Die Anordnung der Elemente im Periodensystem reflektiert neben der periodisch
wiederkehrenden Elektronenkonfiguration auch eine Reihe anderer periodisch auftretender
Eigenschaften innerhalb der Gruppen bzw. Trends innerhalb der Perioden. So nimmt der
Atomradius grundsätzlich von links nach rechts ab (Zunahme der Coulomb-Wechselwirkung
zwischen Kern und Hülle), von oben nach unten zu (Zunahme der Anzahl der Schalen); Abb.
8. Ab der Gruppe 13 und der 4. Periode ist dieser Trend wegen der vorgeschalteten
Nebengruppen nur noch schwach ausgeprägt. In der 6. Periode führen die zwischen Lanthan
und Hafnium eingeschalteten Lanthaniden zu einer Kontraktion der Atome, die dazu führt,
dass die Radien der zur selben Familie gehörenden Elemente der 5. und 6. Periode nahezu
identisch sind: Lanthaniden-Kontraktion. Dadurch werden z.B. die Paare Zr/Hf und Nb/Ta
chemisch so ähnlich, dass sie nur schwer trennbar sind.
Periodizität der Atomradien
1
2
13
14
15
16
17 18
n = 2 Li
Ne
n = 3 Na
Ar
Abbildung 8
n=4
K
Kr
Nebengruppen
Die sich aus der Periodizität der Atomradien herleitenden Trends im Periodensystem sind in
Abb. 9 zusammengestellt.
22
In den Paaren Li/Mg, Be/Al und B/Si liegen ähnliche Atomradien vor, was zu chemischer
Verwandtschaft dieser Paare führt (Schrägbeziehung). Durch Abgabe bzw. Aufnahme von
Elektronen erhält man positiv geladene Kationen bzw. negativ geladene Anionen, häufig mit
der stabilen Konfiguration des benachbarten Edelgases. Die Kationen sind deutlich kleiner,
Anionen deutlich größer als die neutralen Atome. Innerhalb der Kationen und Anionen gelten
aber, gleiche Ladung vorausgesetzt,
nimmt ab
vergleichbare Trends wie bei den Atomen.
Atomradius
Die maximal mögliche Zahl an Elektronen,
nimmt zu
die abgegeben werden können, entspricht
bei den Gruppe 1-8 der Gruppennummer,
nimmt zu
bei den Gruppen 13-17 der zweiten Ziffer
Ionisierungsenergie
der Gruppennummer. Die höchst mögliche
nimmt ab
Zahl von Elektronen, die von den
Elementen der Gruppen 14-17
aufgenommen werden können, ist 18 minus
nimmt ab
Metallcharakter
der Gruppennummer.
nimmt zu
Elektronenaffinität
Elektronegativität
nehmen zu
nehmen ab
-
Abbildung 9. Einige periodische Trends
von Eigenschaften der
(Hauptgruppen-)Elemente im
Periodensystem.
Ionisierungsenergie: Die für die Ablösung eines Elektrons erforderliche Energie
Elektronenaffinität: Bei Aufnahme eines Elektrons (meist) frei werdende Energie
Metallcharakter: typische metallische Eigenschaften wie hohe Leitfähigkeit für
Wärme und Strom, hohe Lichtreflexion, Duktilität
Elektronegativität: Befähigung eines Atoms in einem Molekül, Elektronen zu sich
heranzuziehen.
Schrägbeziehungen: In den Gruppen 1, 2 und 13 sind die Radien der Elemente für n = 2 (Li,
Be, B) sehr ähnlich denen der Elemente der Gruppen 2, 13 und 14 für n = 3 (Mg, Al, Si). Das
führt zu einer chemischen Ähnlichkeit der Elementpaare Li/Mg, Be/Al und B/Si.
9.1 Relativistische Effekte
Bei den jeweils schwersten Elementen innerhalb einer Gruppe treten Besonderheiten auf, die
auf relativistische Effekte zurückzuführen sind:
- In den Gruppen 12-15 ist die stabilste Oxidationsstufe der Elemente der 3., 4. und 5.
Periode die Stufe +2, +3, +4 bzw. +5, dagegen in der 6. Periode die um zwei Stufen
verminderte, also Hg0, Tl1+, Pb2+, Bi3+. Dieser Zustand entspricht jeweils der 6s2Valenzelektronen-Konfiguration, die vergleichsweise stabil („inert“) ist. Dadurch wird
z.B. das Hg „edelgasähnlich“, d.h. flüssig und reaktionsträge (edel).
- Gold hat eine relativ hohe Elektronenaffinität und bildet wie die Halogene Anionen,
Au- (Auride); auch hier liegt die 6s2-Konfiguration vor.
Elektronenaffinitäten der Elemente der Gruppe 11: Cu (-1,23), Ag (-1,30), Au (-2,31
eV)
- Auch die Tatsache, dass die Elemente Cäsium und Gold „goldfarben“ sind (entgegen
den „silbrigen“ übrigen Metallen) ist eine Folge des relativistischen Effekts.
23
-
Die Ionenradien nehmen entgegen den allgemeinen Trends bei den schwersten
Elementen wieder ab: Cu (127,8), Ag (144,5), Au (144,2 pm)
Die Ionisierungsenergien nehmen, den allgemeinen Trends entgegenlaufend, bei den
schwersten Elementen wieder zu: Zn (9,39), Cd (8,99), Hg (10,44 eV).
Die experimentelle Ionisierungsenergie für Gold beträgt 11.2 eV, berechnet mit
relativistischer Korrektur 11.7, ohne relativistische Korrektur 14.2 eV.
Erklärung: Für die Masse und Geschwindigkeit der Elektronen eines Atoms gilt die
Einsteinsche Beziehung mv = m0/√(1-v2/c2). mv ist die Masse des Elektrons bei der
Geschwindigkeit v, m0 die Ruhemasse, c die Lichtgeschwindigkeit. Bei den schweren Atomen
der Gruppen ist das 1s-Elektron dem Kern besonders nahe; seine Umlaufgeschwindigkeit
kommt in die Nähe der Lichtgeschwindigkeit, seine Masse nimmt also zu, was die Attraktion
zwischen Kern und Elektron erhöht: die 1s-Bahn kontrahiert (und damit auch alle höheren
s-Niveaus Stabilisierung des 6s-Niveaus). Wegen der größeren Kernabschirmung
expandieren dann die d-Niveaus; der Abstand zwischen 5d und 6s, der u.a. für die optischen
Eigenschaften verantwortlich ist, wird geringer, sodass Au und Cs im Sichtbaren absorbieren.
10. Sauerstoff; die kovalente Bindung I
10.1. Sauerstoff
21 Vol.-% der (trockenen) atmosphärischen Luft bestehen aus Sauerstoff O2. Das entspricht
einer Gesamtmenge von 1015 t. Der jährliche Umsatz (Verbrauch durch Atmung,
Verbrennung; Nachbildung durch Assimilation) liegt bei 1011 t. Daneben kommt freier
Sauerstoff noch in geringen Mengen als Ozon O3 vor, sowie in gebundener Form als CO2
(0.03 %) und H2O (2.3% bei 100% relativer Luftfeuchtigkeit). Die Hydrosphäre enthält 8589% Sauerstoff in Form von H2O, die Lithosphäre 45% gebunden in Mineralien und
Gesteinen, z.B. SiO2, CaCO3, Fe2O3, Ca3(PO4)2.
Der Sauerstoff wurde durch Scheele (1771; nicht veröffentlicht) und Priestley (1774) als
Bestandteil der Luft entdeckt. 1777 ermittelte Lavoisier die Menge des in der Luft enthaltenen
Sauerstoffs durch Überführen in Quecksilberoxid; vergl Abb. 10. Versuch: Überführen des
Sauerstoffs in Phosphorpentoxid.
Abbildung 10. Lavoisiers Versuch zur
Bestimmung des Volumenanteils von
Sauerstoff in der Luft. Die Retorte enthält
Hg. Der Retortenhals taucht in eine
luftgefüllte Glasglocke, deren Öffnung
durch ein Wasserbad abgesperrt ist. Beim
Erhitzen des Quecksilbers verbindet sich
dies mit dem Sauerstoff der Luft zu
Quecksilberoxid; der Wasserspiegel in
der Glasglocke steigt um das verbrauchte
Sauerstoffvolumen (ca. 1/5) an.
Isotopenzusammensetzung: 16O 99.7587%, 17O 0.0374, 18O 0.2039%.
24
O2 kommt in zwei Modifikationen vor, als stabiler Triplettsauerstoff (paramagnetisch, zwei
ungepaarte Elektronen), und als energiereicher, instabiler Singulettsauerstoff (diamagnetisch);
s. hierzu weiter unten. Flüssiger Sauerstoff (Siedepunkt 90.2 K) und fester Sauerstoff
(Schmelzpunkt 54.4 K) sind hellblau gefärbt. Bei Drucken zwischen 8 und 96 GPa liegt eine
dunkelrote, diamagnetische Modifikation der Zusammensetzung (O2)4 vor (-Sauerstoff), in
der vier Moleküle O2 über schwache Bindungen (2.25 Å bei 13.2 GPa) zu einem Rhomboeder
miteinander verknüpft sind.
Herstellung/Gewinnung:
- Verflüssigung von Luft und anschließende fraktionierte Destillation. Der Siedepunkt
von O2 beträgt -183 °C (90 K), der von N2 -196 °C (77 K). In offene, mit flüssigem
Stickstoff gefüllte Dewargefäße, kondensiert Sauerstoff hinein. Da der Stickstoff
zuerst absiedet, kommt es zu einer Anreicherung von Sauerstoff Brandgefahr
- Mittels Bariumoxid aus Luft über Bariumperoxid:
500°
700°
BaO + ½O2 BaO2 BaO + ½O
- Thermolyse von HgO, NaNO3 (Salpeter), KMnO4, KClO3, z.B.
NaNO3 NaNO2 + ½O2
150°
KClO3 (+ 10% MnO2) KCl + 1½O2 (Versuch)
Vorsicht bei Versuchen mit Kaliumchlorat: Explosionsgefahr bei unsachgemäßer
Handhabung und Verwendung verunreinigten Kaliumchlorats.
-
Elektrolyse von Wasser: H2O H2 + ½O2
Darstellung von Ozon durch elektrische Entladung aus O2 im Siemensschen
Ozonisator (Versuch): 3O2 2O3, H = +142 kJ/mol
Verwendung:
- Schweißtechnik unter Verwendung von Wasserstoff oder Acetylen:
H2 + ½O2 H2O H = -286 kJ/mol
C2H2 + 2½O2 2CO2 + H2O
H = -1298 kJ/mol
- Stahlerzeugung: Herausbrennen von Verunreinigungen im Roheisen durch Einblasen
von Sauerstoff (7-10 bar) in das verflüssigte Eisen („Windfrisch-Verfahren“).
- Brennstoffzellen: „Stille“ Verbrennung von Wasserstoff oder Methanol zur direkten
Stromerzeugung
2CH3OH + 2½O2 2CO2 + 3H2O
Sauerstoff fördert generell die Verbrennung, wobei Oxide, Hyperoxide (engl.:
superoxide)oder Peroxide entstehen:
- C + O2 CO2; bei hohen Temperaturen: C + ½O2 CO
- S + O2 SO2
- Mg + ½O2 MgO (Magnesiumoxid; enthält das O2- Ion)
- 2Na + O2 Na2O2 (Natriumperoxid; enthält das O22- Ion)
- K + O2 KO2 (Kaliumhyperoxid; enthält das O2- Ion)
Ausgeprägter noch ist die Oxidationskraft von Ozon (Versuche)
Biologische Bedeutung:
25
-
Sauerstoff „fördert die Atmung“: O2 wird vom O2-Transportprotein Hämoglobin (dem
roten Farbstoff der roten Blutkörperchen mit einem zentralen Eisenion als O2Akzeptor) in der Lunge aufgenommen, über die Blutbahn zum Gewebe transportiert,
dort vom Myoglobin (dem roten Farbstoff im Muskelgewebe) aufgenommen und zu
der mit Energiegewinn verknüpften Oxidation organischen Substrats (z.B. Glucose)
verwendet. Hierbei entstehen CO2 und H2O. Der komplementäre Vorgang, die
Assimilation, findet in den Chlorophyll tragenden Chloroplasten grüner Pflanzen,
Algen und Bakterien unter Lichteinwirkung statt: CO2 und Wasser werden umgesetzt
zu Glucose und Sauerstoff (Fotosynthese).
-
In der Stratosphäre wird molekularer Sauerstoff durch kurzwelliges UV (Wellenlänge
< 242 nm) in Sauerstoffatome gespalten (1). Durch Reaktion von O mit O2 entsteht
Ozon (2), das mit längerwelligem UV ( < 310 nm) zu O2 und O zurückreagiert (UVFilterwirkung des Ozons; (3)). O3 ist in Gegenwart von Sauerstoffatomen metastabil,
d.h. die Reaktion (4) läuft normalerweise nicht ab, obwohl sie exotherm ist. Radikale
R· können die Reaktion jedoch katalysieren; (5a) und (5b). Solche Radikale, häufig
anthropogenen Ursprungs (z.B. Cl·, das durch Fotolyse von Fluor-ChlorKohlenwasserstoffen FCKWs entsteht, oder ·NO in den Abgasen des Kfz-Verkehrs)
bewirken einen Abbau der stratosphärischen Ozonschicht.
O2 2O
(1)
O + O2 O3
(2)
O3 O2 + O
(3)
O3 + O 2O2, H = -391 (4)
O3 + R· O2 + RO
(5a)
RO + O R· + O2
(5b)
-
In der Troposphäre kann es hingegen bei starker NO-Emission insbesondere bei
Sonnenlichteinfall (h) zum Aufbau des toxischen Ozons kommen. Das folgende,
vereinfachte Formelschema gibt den Reaktionsverlauf wider:
NO + ½O2 NO2
NO2 + h NO + O
O + O2 O3, H = -106 kJ/mol
10.2. Die kovalente Bindung I: die Lewisformel (Valenzstrichformel); formale Ladungen
Während die Summenformel (oder Bruttoformel) lediglich die Zusammensetzung eines
Moleküls angibt, beschreibt die Lewisformel (oder Valenzstrichformel) dessen Aufbau, sofern
es sich um kovalent aufgebaute Moleküle handelt. Von kovalenter Bindung oder
Atombindung spricht man dann, wenn die Bindung durch ein oder mehrere gemeinsame
bindende Elektronenpaare zustande kommt.
In den Lewisformeln werden Einzel-Elektronen durch Punkte, Elektronenpaare durch Striche
symbolisiert. Elektronenpaare, die zwei Atome miteinander verknüpfen, sind bindende,
solche, die an der Bindung nicht teilhaben, nicht-bindende oder freie Elektronenpaare.
Bindungen können durch ein, zwei oder drei Elektronenpaare bewirkt werden. Entsprechend
spricht man von Einfach-, Doppel- und Dreifachbindungen. Die oben angesprochenen
Sauerstoffmodifikationen und Sauerstoffanionen haben die folgenden Valenzstrichformeln:
26
O O
TriplettSauerstoff
O
2
Oxid
O O
SingulettSauerstoff
O O
Hyperoxid
O
O
O
O
O
O
Ozon
O O
Peroxid
Valenzstrichformeln werden so aufgestellt, dass alle Atome die Edelgaskonfiguration
(Elektronen-Achterschale; Wasserstoff: Zweierschale) erhalten. Von dieser Regel kann
abgewichen werden, wenn
- eine ungerade Elektronenzahl vorliegt (z.B. NO, Hyperoxid) und bei gerader
Elektronenzahl in Sonderfällen, wie dem Triplett-O2 (s. hierzu Kap. 12): Siebener-Schale
- eine Elektronenmangelverbindung vorliegt, (z.B. bei einigen Borverbindungen):
Sechserschale
- Elemente ab der 3. Perioden vorliegen, also solche Elemente, die über virtuelle dNiveaus verfügen: Schalenerweiterung auf 10 oder 12 ist zulässig.
CH3
N O
N O
H3C
B
O
S
CH3
O
C O
+
-
Die in den Formeln auftretenden Ladungen sind nur formaler Natur und heißen deswegen
formale Ladungen. Man erhält sie, wenn man unter paritätischer Aufteilung der
Bindungselektronen auf die beiden Bindungspartner die Elektronen für jedes Atom abzählt
und mit der Valenzelektronen-Konfiguration vergleicht. Bei Unterschuss werden positive, bei
Überschuss negative formale Ladungen vergeben. Formale Ladungen sind nicht zu
verwechseln mit Partialladungen (+ und -; s. Kohlenmonoxid CO, oben rechts) aufgrund
unterschiedlicher Elektronegativität (der Atome in Dipolen), oder mit realen Ladungen wie
den Ionenladungen.
Bei Molekülen, die aus mehr als zwei Atomen aufgebaut sind, führt ein freies
Elektronenpaare an einem der inneren Atome zu einer Abwinkelung (s. z. B. O3 und SO2).
Kann die Elektronenverteilung in einem Molekül durch mehr als eine Valenzstrichformel
beschrieben werden (z.B. O3, NO), so spricht von mesomeren Grenzformeln (auch:
Resonanzformeln) bzw. von Mesomerie. Hier verwendet man den Mesomeriepfeil , nicht
zu verwechseln mit dem Gleichgewichtspfeil . Der tatsächliche Zustand des Moleküls liegt
zwischen den durch die Grenzformeln repräsentierten Grenzstrukturen. Die Beiträge der
Grenzstrukturen können gleich sein (wie beim Ozon) oder ungleich (wie beim NO).
Ein Sonderfall der kovalenten Bindung ist die dative Bindung (auch: Donorbindung,
koordinative Kovalenz). Während in der „normalen“ kovalenten Bindung beide
Bindungspartner zum Bindungselektronenpaar beitragen, stammt bei der dativen Bindung das
Bindungselektronenpaar von nur einem Bindungspartner; s. das Addukt aus
Ammoniak und Bortrifluorid als Beispiel. Man kann das BindungsF
H
Elektronenpaar hier zur Verdeutlichung mit einer Pfeilspitze versehen, die
F
N H
die Herkunft des Elektronenpaars anzeigt. Auf diese Art der Bindung wird
F
H
im Zusammenhang mit den Begriffen Lewis-Säure/Lewis-Base und
Koordinationsverbindungen wieder zurückgegriffen werden.
27
11. Der Aufbau der Atome III:
Das Orbitalmodell
Bis zum Beginn des vorletzten Jahrhunderts wurde die Erscheinung „Licht“ im Sinne einer
elektromagnetischen Welle gedeutet (Maxwell, Lorentz), charakterisiert durch die
Wellenlänge bzw. die Frequenz (mit = c, c = Lichtgeschwindigkeit) und die Amplitude
a. Als Welle zeigt Licht bei der Beugung am Doppelspalt ein typisches Interferenzmuster; s.
Abb. 11, links. Zu Beginn des 20. Jahrhunderts entwickelten Planck und Einstein, basierend
auf Experimenten zum Fotoeffekt, die Vorstellung von der Teilchenstruktur (Lichtquanten,
Photonen) des Lichtes mit dem Energieinhalt h je Lichtquant. Der Fotoeffekt (auch:
lichtelektrischer Effekt) besagt, dass aus den Oberflächen geeigneter Metalle (z.B. Cäsium)
mit Licht Elektronen dann herausgelöst werden können, wenn die Energie h des Lichtes die
Ionisierungsenergie EI des Metalls übersteigt. Die kinetische Energie der Fotoelektronen, Ekin
= h - EI, hängt allein von ab und nicht von der Intensität (der Amplitude) des Lichtes. Es
gibt demnach Experimente, in denen sich das Licht wie ein Teilchen-Phänomen verhält
(Fotoeffekt), and andere, in denen der Wellencharakter des Lichtes zu Tage tritt (Beugung am
Doppelspalt).
Dieser Welle-Teilchen-Dualismus wurde 1924 von de Broglie auch auf Elementarteilchen,
also auch auf Elektronen übertragen. Durch Kombination der Beziehungen E = m·c2 (Masse
m als Teilcheneigenschaft) und E = h( = c/ mit als Welleneigenschaft) erhält man
m·c = h/ bzw. = h/mc = h/p
p, das Produkt aus Masse und Geschwindigkeit, ist der Impuls, in diesem Kontext die „de
Broglie-Wellenlänge“ des Teilchens. Sie beträgt für Elektronen von ca. 100 keV ca. 10-1 pm,
entsprechend der Wellenlänge harter -Strahlung.
Abbildung 11. Doppelspaltversuch. Die Intensitätsverteilung jenseits des Doppelspaltes
entspricht der für Wellen (links) bzw. Teilchen (rechts).
Auf einer Bohrschen Bahn kann die Elektronenwelle nur dann stabil sein, wenn sich eine
stehende Welle bildet (andernfalls käme es zur Auslöschung). Das ist nur dann möglich, wenn
der Bahnumfang ein ganzzahliges Vielfaches der Wellenlänge ist:
aus der Bohrschen Bedingung mv·2r = nh wird mit der de Broglie Beziehung (mv =
h/): 2r = n·
(Hiermit wird das Bohrsche Postulat, wonach nur bestimmte Bahnen möglich sind, rational).
28
Versuch: zweidimensionale Stehende Wellen.
Heisenberg postulierte 1927 mit seinen Unbestimmtheitsrelationen, dass (1) Ort x und Impuls
p eines Teilchens, und (2) Energie E und Zeit t (Beobachtungszeitraum; Lebensdauer eines
Zustandes) nicht zugleich exakt bestimmbar sind. Das Produkt der Unschärfen dieser Größen
ist gleich dem Plankschen Wirkungsquantum:
(1) p × x h/2
(2) E ×t h/2
Mit diesen Forderungen werden die Bohrschen Bahnen der Elektronen unscharf; aus den
Elektronenbahnen (orbits) werden Orbitale.
z
Heisenbergs Vorstellungen, zusammen mit der wellenmechanische
Beschreibung der Elektronenzustände durch Schrödinger ab 1926,
lieferte das heute gebräuchliche Bild der Aufenthaltsorte der
Elektronen, das Orbitalmodell. Der ortsabhängige Teil der
Wellenfunktion
(r, , )
r
y
x
(r, und sind Polarkoordinaten, die mit den karthesischen Koordinaten wie rechts gezeigt
zusammenhängen) wird üblicherweise separiert in einen Radialanteil R(r) und einen
Winkelanteil Y(, ):
(r, , ) = R(r) × Y(, )
Die Winkelfunktion hat für alle s-Orbitale die Form Yn00 = 1/2√, ist also kugelförmig; Abb.
12. Die Indizes n,0,0 beziehen sich auf die Haupt-, Neben- und Magnetquantenzahl. Die
Radialfunktionen für das 1s (R100) und das 2s Elektron (R200) sind in Abb. 12 dargestellt. Für
n>1 haben diese Funktionen Nullstellen (Knoten):
R100 = b·exp(-r/a)
R200 = b(1-r/2a)exp(-r/2a)
b ist ein Koeffizient, r ist die Variable (der “Radius”), a ist eine Konstante, der so gen.
Bohrsche Radius (Radius des Wasserstoffatoms) = 0.529 Å (52.9 pm).
Abbildung 12. Darstellung der Radial- (oben)
und Winkelanteile (unten) von s-Orbitalen. Für
die Radialanteile ist 4r2R2 (radiale
Aufenthaltswahrscheinlichkeit) gegen r
aufgetragen.
Bei der gängigen Darstellung von Orbitalen wählt man die Winkelfunktion oder deren
Quadrat [genauer: Integral aus dem Produkt der Funktion und der dazu komplex konjugierten
Funktion, integriert wird über den Raum]. Die Quadrate können im Sinne von
Aufenthaltswahrscheinlichkeits-Räumen für das Elektron interpretiert werden. Die
Information über die Vorzeichen geht dabei verloren.
Die analytische Behandlung von Winkelfunktionen sei am Beispiel desjenigen 2p-Elektrons
exemplifiziert, das durch die Magnetquantenzahl m = 0 charakterisiert ist. Der
winkelabhängige Teil der zugehörigen Wellenfunktion lautet
29
Y210 = ½√(3/ · cos
Mit Hilfe der folgenden Wertetabelle lassen sich für ausgewählte Winkel die zugehörigen
Funktionswerte ermitteln und in ein Koordinatensystem übertragen:
_________________________________________________________
(°) 0
45
90
135 180 225 270 315 360
cos +1
+½√2 0
-½√2 -1
-½√2 0
+½√2 +1
_________________________________________________________
z
r
y
Im Orbitalmodell erhalten die Quantenzahlen teils neue Bedeutungen:
x
- Hauptquantenzahl n: Sie gibt auch im Orbitalmodell die
Hauptenergieniveaus an
- Nebenquantenzahl l Orbitalquantenzahl (Form der Orbitale)
- Magnetquantenzahl m Richtungsquantenzahl (Ausrichtung der Orbitale im
Koordinatensystem, gekennzeichnet durch tief gestellte Indices; vergl. Abb. 13)
Man beachte, dass s- und d-Orbitale bezügl. Punktspiegelung (Inversion) gerade (g) sind, pund f-Orbitale hingegen ungerade (u).
Abbildung 13. p- und d-Orbitale
(winkelabhängiger Teil Y2). Die Indices x, y, z
bzw. xy, xz, yz, x2-y2 und z2 kennzeichnen die Ausrichtung der Orbitale im karthesischen
Koordinatensystem.
12. Die kovalente Bindung II:
Molekülorbitale
Im Orbitalmodell wird eine Bindung in einem Molekül mathematisch durch eine
Linearkombination der Atomwellenfunktionen, anschaulich: durch Überlappung der
Atomorbitale zu Molekülorbitalen beschrieben (Molekülorbital-Theorie). Diese
Kombinationen führen zu bindenden, energetisch stabilisierten, und antibindenden,
energetisch destabilisierten Molekülorbitalen (MO). Letztere werden durch * gekennzeichnet.
Siehe das Beispiel H2 in Abb. 14. Antibindende MO haben eine Knotenebene (ElektronenAufenthaltswahrscheinlichkeit zwischen den Bindungspartner ist Null); bei bindenden MO
erreicht die Elektronendichte zwischen den Bindungspartnern ein Maximum. Liegt dieses
Maximum auf der Kernverbindungsline, handelt es sich um -Orbitale (-Bindungen). Bindungen sind rotationssymmetrisch. Sie werden von Atomorbitalen von Typ s, pz und d(z2)
gebildet (Bindungsrichtung ist die z-Richtung). Liegt das Maximum der Elektronendichte
ober- und unterhalb der Kernverbindungslinie, so liegen -Orbitale (-Bindungen) vor. Orbitale ergeben sich beim Überlappen von px, py und allen d-Orbitalen außer dem d(z2); s.
30
Abb. 15. Elektronenpaare, die nicht für Bindungen verwendet werden, besetzen nichtbindende MO. Eine Einfachbindung ist stets eine -Bindungen. Zweifach- und
Dreifachbindungen setzen sich aus einer - und ein bzw. zwei -Bindungen zusammen. Bei
einer Doppelbindung ist die Rotation um die Bindungsachse blockiert
Abbildung 14. Bindende (links, grün) und antibindende MO (Mitte, rot) vom Wasserstoff.
Oben jeweils der radialabhängige Teil R(r), unten der winkelabhängige Y(, ). Rechts:
Darstellung der Bindungswechselwirkungen im Energieniveau-Schema (MO-Schema).
Abbildung 15. (a)-(c): Bindungen; (d) bindende, (e)
antibindende -Bindung zwischen
p-Orbitalen; (f) -Bindung
zwischen d-Orbitalen.
Der unter Normalbedingungen stabile Triplett-Sauerstoff (s. S. 24) wird im MO-Modell
durch das in Abb. 16 links dargestellte MO-Schema beschrieben. Die Besetzung der einzelnen
Molekülorbitale folgt den bereits angesprochenen Besetzungsregeln (Aufbau-Prinzip,
Hundsche Regel, Pauli-Prinzip; s. S. 18). Das höchste besetzte MO ist ein antibindendes Orbital (*); es ist zweifach entartet und wird gemäß der Hundschen Regel nur jeweils
einfach besetzt. Hieraus resultiert der Biradikalcharakter. Zu den beiden weniger stabilen
Formen des Singulett-Sauerstoffs s. Abb. 16 rechts. Die Bindungsordnung = ½(bindende e- –
antibindende e-) ergibt sich in allen drei Fällen zu 2, entspricht also einer Doppelbindung.
31
E (kJ/mol)
2p
2p
155
92
2s
2s
0
}
SingulettO2
Triplett-O 2
Abbildung 16. MO-Schema des Triplett-Sauerstoffs (links) und relative Energien der drei O2Sorten.
Energiereicher Singulett-Sauerstoff kann z.B. gemäß der folgenden Reaktionsgleichung
hergestellt werden:
H2O2 + Cl2 + 2OH- Singulett-O2 + 2Cl- + 2H2O
Er geht spontan in den stabilen Triplett-O2 über, wobei die Energiedifferenz in Form roten
Lichtes abgestrahlt wird (Versuch).
13. Wasserstoffverbindungen des Sauerstoffs
Prolog
In Kapitel 10 wurden bereits im Zusammenhang mit Lewisformeln die Sauerstoffionen Oxid,
Hyperoxid und Peroxid vorgestellt (s. auch die Gegenüberstellung von „O2-Spezies“ in
Tabelle 5). Von diesen Ionen leiten sich kovalente Oxide (in Verbindungen des Sauerstoffs
mit Nichtmetallen) und – mit Metall-Kationen als Gegenionen – ionische Oxide, Hyperoxide
und Peroxide her. Zu den kovalenten Oxiden gehören die Wasserstoff-SauerstoffVerbindungen Wasserstoffperoxid (H2O2) und Wasser (H2O).
Tabelle 5. Einige Eigenschaften der Spezies O2q.
Formel
Bezeichnung
+
O2
Dioxigenyl(ium)
O2
Dioxigen
O2
Hyperoxid*)
2O2
Peroxid
)
* Im Englischen: superoxide
Bindungsordnung
2,5
2,0
1,5
1,0
O-O Abstand (pm)
112
121
133
149
Beispiele
O2[PtF6]
K2O
Na2O2, H2O2
13.1. Wasserstoffperoxid
Darstellung (Versuch):
BaO2 + H2SO4 BaSO4 + H2O2
Technisch wird H2O2 durch anodische Oxidation von Hydrogensulfat und nachfolgende
Hydrolyse des dabei entstehenden Peroxodisulfats dargestellt:
32
O
O
2 HO
S
O
O
O
-2e
-
HO
S
O
O
S
OH
H2O
H2O2 + 2H2SO4
O
O
Nachweis/Eigenschaften (Versuche):
- Mit Titanylsulfat ensteht in sauerer Lösung unter intensiver Orangefärbung
Peroxotitanylhydrogensulfat:
TiO(SO4) + H2O2 + H2SO4 Ti(O2)(HSO4)2 + H2O
O O
O Cr O(C2H5)2
O O
-
Mit Dichromat entsteht in sauerer Lösung blaues Chromperoxid
CrO(O2)2, das mit Ether stabilisiert wird.
-
Durch die Oxidation von Iodid zu Iod (Verfärbung von farblos nach rotbraun bzw.
blau in Gegenwart von Stärke): I- + 2H+ + H2O2 ½I2 + 2H2O
-
Selbstzersetzung in Gegenwart von Katalysatoren (Pt; MnO2; Katalase aus Blut):
H2O2 H2O + ½O2
Wasserstoffperoxid (umgangssprachlich auch: Wasserstoffsuperoxid) wirkt stark bleichend
und ätzend.
94°
Molekülbau: s. nebenstehendes Bild
H
O O
111.5°
H
13.2. Wasser
Wasser kommt frei in allen drei Aggregatzuständen sowie gebunden an
= 1.84 D
Minerale und Gesteine in Form von Kristallwasser vor. Der
H
Sättigungsdampfdruck in Luft bei Normaldruck und 20 °C beträgt 17.5
- O
104,5°
Torr. Das entspricht einem Gehalt von 2.3% absolut (= 100% relative
H
Luftfeuchtigkeit). Wasser ist gewinkelt. Wegen der unterschiedlich stark
ausgeprägten Tendenz von H und O, die Elektronendichte in den
Bindungen zu delokalisieren, resultiert ein Dipolmoment. Die Wasserdipole haben eine
ausgeprägte Tendenz zu größeren Aggregaten zu assoziieren. Viele der Eigenschaften des
Wassers, z.B. die Tatsache, dass es bei Zimmertemperatur flüssig ist, sind darauf
zurückzuführen. In Tabelle 6 sind einige Eigenschaften des Wassers zusammen- und denen
von Wasserstoffperoxid gegenübergestellt.
Tabelle 6: Eigenschaften von H2O und H2O2
H2O
H2O2
Schmelzpunkt(°C) bei 1 bar
0
-0.43
Siedepunkt (°C) bei 1 bar
100
150.2
Dichte (kg/l) bei 20 °C
0.9982
1.448
1)
-16
Dissoziationskonstante bei 25 °C 1,8·10
2,4·10-12
2)
Dipolmoment (D )
1.84
1)
Definiert als Gleichgewichtskonstante für die folgenden Dissoziationsgleichgewichte:
H2O H+ + OH- bzw. H2O2 H+ + HO22)
Debey (Einheit für das Dipolmoment)
33
Von großer biologischer und geologischer Bedeutung ist der Dichteverlauf flüssigen Wassers.
Das Dichtemaximum liegt bei 4 °C:
0° fest
= 0.9168 kg/l
0° flüssig
0.9999
4°
1,0000
10°
0.9997
20°
0.9982
100° flüssig
0.9584
Dieser Dichteverlauf hat zur Folge, dass Gewässer auch bei länger anhaltendem Frost nur
schwer bis zum Boden durchfrieren. Da Wasser sich beim Gefrieren um ca. 9% ausdehnt,
entfaltet in Gesteinsritzen eingedrungenes Wasser beim Gefrieren eine beträchtliche
Sprengwirkung ( Erosion; Versuch mit einer wassergefüllten, gusseisernen Kugel). Das
spezifisch leichtere Eis schwimmt auf dem Wasser, wobei etwa 9% der Eismasse oberhalb der
Wasseroberfläche liegt.
Die Bewegung der Moleküle von Flüssigkeiten ähnelt der von Gasen, d.h. sie ist ungeregelt;
zugleich sind aber die Attraktionskräfte zwischen den Molekülen in einer Flüssigkeit sehr viel
ausgeprägter als in einem Gas. Die kinetische Energie der Moleküle unterliegt einer
Temperaturverteilung. Moleküle hinreichend hoher kinetischer Energie können durch die
Flüssigkeitsoberfläche hindurch treten und so in den Gasraum gelangen. In einem
abgeschlossenen System stellt sich ein Gleichgewicht zwischen der in der Gasphase
befindlichen Menge des Stoffes und der flüssigen Phase ein Dampfdruck. Der Dampfdruck
der Flüssigkeit ist temperaturabhängig; er nimmt mit zunehmender Temperatur zu.
Vergleichbares gilt für die feste Phase.
Schmelzpunkt, Siedepunkt und Sublimationspunkt von Wasser sind druck- und
temparaturabhängig (Versuch zur Druckabhängigkeit des Schmelzpunktes: Ein über einem
Eisblock gehängter, mit Gewichten beschwerter Draht sinkt langsam hindurch). Trägt man
den Sättigungsdampfdruck von festem
bzw. flüssigem Wasser gegen die
Temperatur auf, so erhält man eine
Sublimations- bzw. Dampfdruckkurve.
Entsprechend erhält man eine
Schmelzdruckkurve für die
Abhängigkeit des Schmelzpunktes vom
Druck. Alle drei Kurven zusammen
bilden das Zustandsdiagramm des
Wassers (Abb. 17), dem man folgende
Informationen entnehmen kann:
Abbildung 17. Zustandsdiagramm von
Wasser. Erläuterungen s. Text.
-
-
Jenseits der Kurven kann Wasser in recht variablen Bereichen von Druck (p) und
Temperatur (T) in jeweils einem Aggregatzustand existieren: gasförmig (g), flüssig (l
[für liquid]) oder fest (s [für solid]). Die drei Kurven werden wie folgt bezeichnet: l/g:
Dampfdruckkurve; s/g: Sublimationskurve; s/l: Schmelzdruckkurve.
Auf den Kurven sind zwei Phasen koexistent. p und T sind für jeden Punkt auf der
Kurve festgelegt.
34
-
Im Schnittpunkt der drei Kurven, dem Tripelpunkt (TP: 0.0099°; 0.006 bar = 4,579
Torr), sind alle drei Phasen koexistent.
Siedepunkt und Schmelzpunkt unter Normaldruck ergeben sich als Schnittpunkte der
1 bar (760 Torr; 105 Pa) Geraden mit der Schmelzdruck- bzw. Dampfdruckkurve.
Mit zunehmendem Druck nimmt der Schmelzpunkt ab (negative Steigung der
Schmelzdruckkurve) Gleiten von Gletschern, Schlittschuhläufern.
Oberhalb von 374° und 220.5 bar (kritischer Punkt, KP) sind flüssige und Gasphase
nicht mehr unterscheidbar: kritischer Zustand. Die Dichte des überkritischen Wassers
beträgt 0,324 g/ml.
13.3. Wasser als Lösungsmittel
Viele Gase (z.B. O2, N2), Flüssigkeiten (z.B. Alkohol) kovalente Stoffe (z.B. Zucker) und
ionogene Stoffe (z.B. NaCl) lösen sich in Wasser. Bei 0°C lösen sich z.B. in je 1 l Wasser
49.1 ml O2 und 23.2 ml N2. Versuch: Nachweis von O2 in Leitungswasser mit Mn2+/OH-, und
negativer Befund in ausgekochtem Wasser. Unser Trinkwasser enthält neben N2 und O2 auch
gelöstes CO2 (neben Kohlensäure) und kleinere Mengen an Chloriden und Sulfaten (Versuch:
Nachweis von Cl- und SO42-). Der Salzgehalt (vorwiegend NaCl) in den Weltmeeren liegt bei
3,5%. Stoffe, die sich in Wasser leicht lösen, werden hydrophil (= wasserfreundlich) genannt,
solche, die in Wasser weitgehend unlöslich sind (z.B. Fette) hydrophob (= wasserfeindlich).
Den Salzgehalt von Wasser kann man über eine Messung der elektrischen Leitfähigkeit
ermitteln. Reines Wasser leitet den Strom fast nicht, da in reinem Wasser wegen dessen
minimaler Dissoziation (vergl. Tabelle 6) kaum Ladungsträger vorliegen. Bei Zugabe
ionogener Stoffe (Salze) nimmt die Leitfähigkeit durch deren Dissoziation in hydratisierte
Ionen zu, z.B.
NaCl + aq [Na·aq]+ + [Cl·aq](aq = Wasser [aqua])
Versuch: Zwei in Wasser tauchende, an das Netz angeschlossene und über eine Glühlampe
und Klingel miteinander verbundene Elektroden lassen Stromfluss erst dann zu, wenn Salz im
Wasser gelöst wird. Die Reinigung von Wasser erfolgt durch Destillation ( destilliertes
Wasser) oder Ionenaustausch ( deionisiertes Wasser).
In Lösungen treten pro Zeiteinheit weniger Wassermoleküle aus der Oberfläche aus, sodass
bei gegebener Temperatur der Dampfdruck der Lösung geringer ist als der des reinen
Wassers. Diese Dampfdruckerniedrigung führt zu einer Gefrierpunkterniedrigung (GPE) und
Siedepunkterhöhung (SPE); vergl. Abb. 18; was man sich zur Molmassebestimmung des
gelösten Stoffes mittels der Kryoskopie (Gefrierpunkterniedrigung, GPE) oder Ebullioskopie
(Siedepunkterhöhung, SPE) zunutze machen kann. Versuche zur GPE und SPE.
Abbildung 18: Siedepunkterhöhung (links) und Gefrierpunkterniedrigung (rechts).
35
Es gilt das Raoultsche Gesetz:
T = E·b
T = GPE oder SPE; b = Molalität, Einheit: mol/kg; E ist eine Lösungsmittel-spezifische
Konstante.
Ist i ein beliebiger gelöster Stoff und LM ein Lösungsmittel so wird mit
b = ni/mLM
und ni = mi/Mi
( n = Stoffmenge, m = Masse, M = Molmasse; vergl. Tabelle auf S. 3)
Mi = E·mi/T·mLM
Eine weitere charakteristische Erscheinung von Lösungen, die man immer dann beobachtet,
wenn zwei Flüssigkeiten durch eine semipermeable (= halbdurchlässige) Membran getrennt
sind, ist die Osmose. Die Bewegung der Moleküle oder Ionen in einer Lösung kann man in
Analogie zur Bewegung von Gasteilchen durch eine „allgemeine Gasgleichung“ beschreiben
(Versuch zur ungeregelten Bewegung: Aluminiumflitter im Lichtkegel), in der der
osmotische Druck ist und die anderen Größen dieselbe Bedeutung haben wie in der
allgemeinen Gasgleichung (R = Gaskonstante, n = Molzahl, T = Temperatur, V = Volumen):
= nRT/V = cRT mit R = 8,31 kPa·l·mol-1·K-1
Versuch zur Osmose: „Chemischer Garten“ (verschiedene Salzkristalle werden in Wasserglas
gelegt). Den osmotischen Druck kann man mit nebenstehender Versuchsanordnung messen:
In ein mit Wasser (kleine Kreise) gefülltes Gefäß taucht ein mit der
Lösung (z.B. Zuckerlösung) gefüllter Behälter aus einem Material
ein, das semipermeabel ist, z.B. Ton (rot gestrichelt). Dieser Behälter
ist zusätzlich mit einem Steigrohr aus Glas versehen.
Wassermoleküle vermögen durch die Poren der Membran hindurch
zu diffundieren, nicht aber die großen Zuckermoleküle (große rote
Kreise). Im zeitlichen Mittel gelangen mehr Wassermoleküle von
außen nach innen (Pfeile) als umgekehrt; bezüglich des Wassers ist
die Lösung „verdünnt“. Die Wassersäule im Glasrohr steigt an, und
zwar solange, bis der hydrostatische Druck der Wassersäule ebenso
groß ist wie der entgegenwirkende osmotische Druck.
Osmotische Erscheinungen spielen für Gleichgewichtssituationen
zwischen Zellen und umgebenden Flüssigkeiten sowie für den
Transport von Wasser in Pflanzen eine lebenswichtige Rolle. Der
osmotische Druck in den roten Blutkörperchen von Säugern (T =
310 K) beträgt 7,9 bar. Das entspricht einer 0,9 %igen NaCl-Lösung
( physiologische oder isotonische Kochsalzlösung). Das
nebenstehende Bild zeigt die Auswirkungen auf rote Blutkörperchen
von hypotonischen Medien (geringerer osmotischer Druck =
geringere Salzkonzentration, links unten) und hypertonischen
Medien (größerer osmotischer Druck = höherer Salzkonzentration;
rechts unten). Die roten Blutkörperchen oben im oberen Teil des Bildes befinden sich in
isotonischer Lösung.
36
13.4. Die Struktur des Wassers
Wie weiter oben schon erwähnt, hat Wasser aufgrund seiner
Winkelung ein Dipolmoment. Die Winkelung kann man auf der
Grundlage folgender Überlegung verstehen: Die
Valenzelektronen-Konfiguration des Sauerstoffs ist
s2px2py1pz1.Diese vier Orbitale vermischen (hybridisieren) zu vier
äquivalenten, in die Ecken eines Tetraeders weisenden
Hybridorbitalen vom Typ sp3, von denen zwei mit einem
Elektronenpaar, und zwei mit einem einzelnen Elektron besetzt
sind. Die einzeln besetzten Hybridorbitale überlappen mit den
ebenfalls einzeln besetzten s-Orbitalen zweier Wasserstoffatome zu bindenden Orbitalen, die
beiden anderen sp3-Hybridorbitale bleiben nichtbindend. Der etwas größere Raumanspruch
letzterer bewirkt eine Verengung des Tetraederwinkels (109°28’) auf 104,5°. Die DipolDipol-Attraktion führt zu Nahstrukturen in flüssigem Wasser, bis zur Ausbildung kurzlebiger
Cluster wie in Abb. 19 links gezeigt. Das Aufbrechen solcher Cluster erfordert zusätzliche
Energie, sodass der Siedepunkt des Wassers mit 100 °C „anomal“ hoch liegt. Im Eis liegt eine
hoch geordnete Struktur vor Molekülgitter); Abb. 19 rechts. Bei tieferen Temperaturen und
hohem Druck können die Wassercluster Methan einschließen; hierauf beruht das Vorkommen
so gen. Methanhydrate in tieferen Regionen der Ozeane.
Abbildung 19: Strukturen von Wasser. Rot: O, weiß: H, schwarz: kovalente Bindungen; grau
bzw. gestrichelt: Wasserstoffbrücken-Bindungen. Der linke Teil der Abbildung zeigt
verschiedene Aggregations-Zustände („Wassercluster“) in flüssigem Wasser, der rechte Teil
die Struktur von Eis.
14. Die Ionenbindung
Metalle und Nichtmetalle (typischerweise die Metalle der Gruppen 1 und 2 und Nichtmetalle
der Gruppen 17 und 16) gehen Ionenbindungen (salzartige Bindungen) ein. Treibende Kraft
ist auch hier die Erlangung der stabilen Edelgaskonfiguration, die hier durch
Elektronenübertragung vom Metall auf das Nichtmetall erreicht wird:
Na
+
Cl
+
Na
Cl
Die so gebildeten Kationen (positiv geladene Ionen) und Anionen (negativ geladene Ionen)
unterliegen starken Coulomb-Wechselwirkungen, die – anders als in der kovalenten Bindung
– nicht nur zu einem benachbarten Bindungspartner sondern in alle drei Raumrichtungen
weisen. Dadurch kommt es zum Aufbau geordneter, dreidimensionaler Ionengitter (Salze) mit
den Kationen und Anionen auf definierten Gitterplätzen. Je nach dem Größenverhältnis der
beiden im Gitter vertretenen Ionenarten werden bestimmte Gittertypen ausgebildet. Die für
37
AB-Systeme (z.B. NaCl, CsCl, ZnS) wichtigsten Gittertypen sind in Abb. 20 gezeigt. Die
Zahl der nächsten Nachbarn eines Kations bzw. Anions wird als Koordinationszahl
bezeichnet. Die Koordinationszahl beträgt 6 im Natriumchlorid-Typ, 8 im Cäsiumchlorid-Typ
and 4 im Zinkblende-Typ. Entsprechend befinden sich die Ionen in oktaedrischer (NaCl),
kubischer (CsCl) bzw. tetraedrischer (ZnS) Umgebung. Im NaCl-Typ wird die dichtest
mögliche Packung realisiert (kubisch flächenzentriert); der CsCl-Typ ist kubisch primitiv.
Abbildung 20. Von links nach rechts: NaCl, NaCl (raumfüllende Darstellung), CsCl, ZnS.
Die Radienverhältnisse r(Kation)/r(Anion) betragen 0,414 (NaCl), 0,732 (CsCl) und 0,225
(ZnS).
Die Bildung von Ionengittern ist dann begünstigt, wenn die Reaktionsenthalpie für den
Gesamtvorgang negativ ist. Hierzu müssen aufzuwendende Energiebeiträge durch frei
werdende überkompensiert werden:
Na (fest) Na (gasförmig)
H = +108 kJ/mol Verdampfungsenthalpie
½(Cl2 2Cl)
H = +122 kJ/mol Dissoziationsenergie
+
Na Na + e
H = +496 kJ/mol Ionisierungsenergie
Cl + e Cl
H = -349 kJ/mol
Elektronenaffinität
n(Na+ + Cl-) (NaCl)n (fest)
H = -788 kJ/mol
Gitterenergie
Hieraus ergibt sich eine Bildungsentalpie von -411 kJ/mol.
Beim Auflösen eines Salzes in Wasser, ein Vorgang, der mit dem Zusammenbruch des
Ionengitters einhergeht, muss in der Regel mindestens die Gitterenergie aufgewendet werden.
Dies geschieht durch Hydratation der Ionen. Als Hydratation bezeichnet man die Umhüllung
der Ionen mit Wassermolekülen aufgrund elektrostatischer Wechselwirkungen zwischen den
Ionen und den Wasserdipolen; s.a. Abb. 21.
Na+ + xH2O [Na(H2O)x]+
H = -406 kJ/mol
Cl + yH2O [Cl(H2O)y]
H = -381 kJ/mol
Die Summe der Hydratationsenergien für die beiden Ionen ist -787 kJ/mol.
Abbildung 21. CoulombWechselwirkung zwischen
Ionen und Wassermolekülen
(Hydratation).
38
Neben der Enthalpiebilanz spielen für den Lösungsvorgang (und generell für alle Reaktionen)
auch die Entropieverhältnisse eine Rolle. Der Gesamtvorgang wird durch die GibbsHelmholzsche Gleichung beschrieben:
G = H – TS
Hierin ist G die Freie Reaktionsenthalpie (das ist derjenige Anteil der Enthalpie, die in
Nutzarbeit überführt werden kann), T die Temperatur (in K), und S die Entropieänderung,
anschaulich die Änderung des Ordnungszustandes. Mit zunehmender Unordnung nimmt die
Entropie S und damit S zu. Eine Reaktion läuft dann ab, wenn G negativ ist. Im Falle des
Auflösens eines Salzes und damit des Zusammenbruches des geordneten Ionengitters nimmt
die Entropie zu und liefert somit einen Beitrag in Richtung auf den Lösungsvorgang. Damit
können sich gegebenenfalls auch solche Salze auflösen, für die der Enthalpiebeitrag nicht
ausreicht, um die Gitterenergie zu überwinden. Ein Beispiel ist Natriumthiosulfat (Fixiersalz).
Beim Lösen in Wasser (Versuch) kühlt sich die Lösung ab: Die Summe der
Hydratationsenthalpien ist hier kleiner als die Gitterenergie; der fehlende Betrag wird dem
Wärmeinhalt des Systems entnommen.
15. Die Halogene
Zu den Halogenen (= Salzbildner) gehören die Elemente Fluor (schwach grünlich gefärbtes
Gas), Chlor (gelbgrünes Gas), Brom (rotbraune Flüssigkeit), Iod (schwarz-glänzender
Feststoff; Ioddampf ist violett) und Astatium (radioaktiv). At ist ein Halbmetall, die anderen
Vertreter der Gruppe 17 sind typische Nichtmetalle. Alle Halogene haben molekularen
Aufbau (X2). Zur Gegenüberstellung einiger physikalischer Eigenschaften s. Tabelle 7.
Tabelle 7. Physikalische Daten für Halogene
Fluor
Schmelzpunkt/°C
-223
Siedepunkt/°C
-187
Dissoziationsenergie (X2
2X)/kJmol-1
Atomradius/pm
72
Elektronegativität
3,98
Elektronenaffinität/kJmol-1 -377
Elektroenaffinität/eV
3,91
Ionisierungsenergie/eV
17,42
Chlor
-102
-34,5
Brom
-7,3
+58,8
Iod
+114
+183
99
3,16
-387
4,01
13,01
114
2,96
-364
3,77
11,84
133
2,66
-331
3,43
10,44
Vorsicht beim Umgang mit freien Halogenen (kein Hautkontakt [Ekzeme, Chlorakne]; kein
Einatmen [Gefahr von Lungenödemen])!
15.1 Fluor
Vorkommen: Flussspat CaF2, Kryolith Na3[AlF6], Fluorapatit Ca5(PO4)3F. Knochen und
Zähne enthalten fluorhaltigen (< 0,1%)Hydroxylapatit Ca5(PO4)3(OH,F).
Darstellung:
- Durch Elektolyse von Kaliumhydrogenfluorid KHF2 (aus KF und HF; HF wird aus
Flussspat und Schwefelsäure gewonnen): HF2- → ½F2 + HF + e- Aus K2[MnF6] (KMnO4 + HF) und SbF5 (SbCl3 + HF) gemäß
K2[MnF6] + 2SbF5 2K[SbF6] + MnF4; MnF4 MnF3 + ½F2
Eigenschaften und Verwendung
39
-
-
-
-
-
Fluor setzt sich mit nahezu allen Elementen (einschließlich des Edelgases Xenon)
lebhaft um. Einige Metalle, z.B. Mg, Cu und Monel-Metall (eine Cu-Ni-Legierung)
werden oberflächlich durch dichte und fest haftende Fluoridschichten passiviert.
Fluor hat eine hohe Affinität zu Wasserstoff. So reagiert es mit Wasser unter
Freisetzung von Sauerstoff: H2O + F2 2HF + ½O2
Fluor stabilisiert hohe Oxidationsstufen: SbF6, IF7, XeF6, AgF2
Fluorwasserstoff wirkt auf Glas ätzend: SiO2 + 4HF SiF4 + 2H2O (Versuch)
Etwa ¾ der Gesamtaufkommens an Fluor geht in die Produktion von UF6, eine
flüchtige Verbindung, die zur Anreicherung von 235U im natürlich vorkommenden
238 235
U/ U verwendet wird.
UO2 + 6HF → UF6 + 2H2O + H2
Weitere Anwendungsbereiche sind die Aluminiumherstellung (KHF2 als Flussmittel)
und Fluorierungen mit XeF2, SbF5, SF6, z.B. die Herstellung von
Fluorkohlenwasserstoffen wie Teflon, Hostaflon, Frigen, Freon:
(SbF5)
CHCl3 + 2HF CHClF2 (-F2C-CF2-)n (+ HCl)
Fluorwasserstoff (sehr stark ätzend) assoziiert wegen seines
hohen Dipolmomentes unter Ausbildung von
Wasserstoffbrücken zu höheren Aggregaten (HF)n; s. das
nebenstehende Bild; s.a. den im Vergleich zu anderen
Halogenwasserstoffen hohen Siepunkt (Tabelle 8).
15.2 Chlor
Vorkommen: NaCl (als Steinsalz in Salzlagerstätten oder gelöst im Meerwasser), KCl
(Sylvin) KCl·MgCl2 (Carnallit), AgCl (Hornsilber).
Darstellung:
- Oxidation von HCl (aus NaCl und Schwefelsäure) mit Braunstein (MnO2)
NaCl + H2SO4 NaHSO4 + HCl; 2HCl + MnO2 + 2H+ Cl2 + Mn2+ + 2H2O
- technisch durch Luftoxidation von HCl nach dem Deacon-Verfahren (Versuch):
2CuO + 4HCl 2CuCl2 + 2H2O
2CuCl2 + O2 2CuO + 2Cl2
_________________________________________
-
4HCl + O2 2Cl2 + 2H2O
großtechnisch durch die Chloralkali-Elektrolyse z.B. nach dem Amalgamverfahren
(vergl. Abb. 22) gemäß der folgenden Gesamtgleichung:
Na+ + Cl- + H2O Na+ + OH- + ½H2 + ½Cl2
Der Wasserstoff entsteht hierbei in einem nachgeschalteten Verfahrenen durch
Zersetzung des primär gebildeten Natriumamalgams mit Wasser.
Abbildung 23. Schematische
Darstellung der
Chloralkalielektrolyse nach
dem Amalgam-Verfahren.
40
Weltweit werden jährlich etwa 45 Millionen Tonnen Chlor produziert (zur Verwendung s.u.).
In Deutschland betrug die Chlorproduktion im Jahre 2004 vier Millionen Tonnen.
Nachweis: Der Nachweis von freiem Chlor erfolgt durch dessen Oxidationswirkung
gegenüber Iodid (I- I2 IO3-) und die Bleichwirkung z.B. gegenüber Indigo oder Lackmus
[Indigo bzw. Lackmus werden entfärbt]; Versuche.
Chlorwasserstoff: HCl, bei Raumtemperatur ein aggressives Gas (vergl. Tabelle 8), kann mit
nicht-flüchtigen Säuren wie Schwefelsäure aus Chloriden, z.B. NaCl, ausgetrieben (s.o.), oder
direkt aus H2 und Cl2 dargestellt werden. Eine Mischung dieser beiden Gase ist metastabil, da
die initiierende Reaktion, die Spaltung von Cl2 in zwei Chloratome, eines Energiebetrages
von 243 kJ/mol bedarf. Die erforderliche Energie kann z.B. in Form blauen Lichtes zugeführt
werden; vergl. Tabelle 4 auf S.17; die Reaktion verläuft dann explosionsartig (ChloknallgasReaktion; Versuch); zum Reaktionsablauf s. S. 16.
Einige Eigenschaften von HCl sind denen der anderen Halogenwasserstoffe in Tabelle 9
gegenübergestellt. HCl hat eine hohe Affinität zu Wasser (Versuch: Springbrunnen). Die
Lösung von HCl in Wasser heißt Salzsäure (oder Chlorwasserstoffsäure). Die „Magensäure“
enthält etwa 0.5 % Salzsäure.
Tabelle 8. Einige Eigenschaften der Halogenwasserstoffe
HF
HCl
Siedepunkt (°C)
+19,5
-85,1
Bildungsenthalpie (kJ/mol)
-271
-92
Thermischer Dissoziationsgrad (%) bei
300 °C 3·10-7
1000 °C 0,014
Dipolmoment (D)
1,91
1,07
Ionischer Charakter der Bindung (%)
45
17
HBr
-66,7
-52
HI
-35,4
-4,7
0,003
0,5
0,83
12
19
33
0,45
5
Chlorsauerstoff-Verbindungen:
Chlor und Sauerstoff bilden eine Reihe von Oxiden (darunter Cl2O, Cl2O3, ClO2, Cl2O7), die
Anhydride von Chlorsäuren sind. Einige wichtige Chlorsäuren und ihre Anionen sind in
Tabelle 10, Valenzsztrichformeln von Chloroxiden und Anionen der Chlorsäuren weiter unten
zusammengestellt.
Tabelle 10. Sauerstoffsäuren des Chlors und deren Anionen
Oxidationszahla) Säure
Anion
+I
HClO Hypochlorige
ClO- Hypochloritb); Chlorat(I)
Säure
+III
HClO2 Chlorige Säure
ClO2- Chlorit; Chlorat(III)
+V
HClO3 Chlorsäure
ClO3- Chlorat; Chlorat(V)
+VII
HClO4 Perchlorsäure
ClO4- Perchlorat; Chlorat(VII)
a)
Zum Begriff „Oxidationszahl“ s. das folgende Kapitel
b)
Phonetisch unterscheide man zwischen Chlorit (mit kurzem „i“ und stimmlosem [hartem]
„t“) und Chlorid (mit langem „i“ und stimmhaftem [weichem] „d“)
Valenzstrichformeln einiger Chloroxide and Chlorsäureanionen:
41
2
O Cl
Cl O
Cl
O
Dichloroxid
Cl O
Cl O
O
O
Chlordioxid
O
2
Cl O
Cl O
Hypochlorit
O
Chlorit
Cl
O
O
Chlorat
O
Cl
O
O
3
O
Perchlorat
Alle Sauerstoffverbindungen des Chlors haben eine ausgeprägte Tendenz, Sauerstoff
abzugeben oder Oxido-Ionen zu übertragen; sie sind starke Oxidationsmittel. Insbesondere die
Chlorate und Perchlorate bilden mit oxidierbaren Stoffen – gegebenenfalls schon mit
Bestandteilen aus dem Staub der Luft – hoch explosive Gemische. Auf dieser
Oxidationswirkung beruht auch die Bleichwirkung und bakterizide Wirkung (Desinfektion)
von Chlor und Chlorverbindungen wie den Hypochloriten:
ClO- + H2O HClO + OHHClO HCl + O (atomar in statu nascendi)
Cl2 + H2O HCl + HClO
Bildung und Reaktionen weiterer Chlorsäuren bzw. Chlorate werden im Kapitel 16
(Reduktion und Oxidation) behandelt.
Verwendung:
- Chlor und seine Verbindungen (insbesondere Hypochlorite und Chlorate) finden als
Desinfektionsmittel (Schwimmbäder, Trinkwasser) und Bleichmittel Verwendung.
Diese Eigenschaften beruhen auf der Bildung atomaren Sauerstoffs (s.o.) in statu
nascendi (im Augenblick des Entstehens), dem eine besonders hohe Oxidationskraft
und damit „zerstörerische“ Wirkung zukommt. Mit Chlor oder Chlorverbindungen wie
Chlorkalk CaCl(ClO) desinfiziertes Wasser wird als gechlortes Wasser (nicht
chloriertes Wasser) bezeichnet.). Chlorkalk ist ein gemischtes Chlorid-Hypochlorit,
das aus Chlor und Ca(OH)2 hergestellt wird.
- Große Mengen an Chlor und Chlorwasserstoff werden in der chemischen Industrie zur
Synthese chlorierter Kohlenwasserstoffe (chloriert impliziert die Bindung von Chlor
an den Kohlenstoff einer organischgen Verbindung) benötigt. Beispiel:
Polyvinylchlorid (PVC):
C2H4 + Cl2 CH2Cl-CH2Cl;
n CH2Cl-CH2Cl (-CHCl-CHCl-)n +2n HCl
15.3 Brom und Iod
Vorkommen: Als Bromid und Iodid vergesellschaftet mit Chloriden. Iod außerdem als
Calciumiodat Ca(IO3)2 als Beimengung (ca. 5%) im Chilesalpeter (NaNO3). Iod ist ferner
angereichert in Meeresalgen (Tangen). Iod ist als Bestandteil mehrerer Schilddrüsenhormone
(z.B. Tyroxin) ein essentielles Spurenelement; bei Iodmangel kommt es u.a. zu einer
krankhaften Vergrößerung der Schilddrüse.
Gewinnung:
2X- + Cl2 2Cl- + X2
(X = Br, I)
(Versuch)
42
Bei der technischen Gewinnung von Brom wird Meerwasser bei pH 3,5 mit Cl2 gechlort und
das so entstandene Brom mit Luft ausgetrieben und kondensiert.
Oxidation von Iodid durch Permanganat
Reduktion von Iodat mit Sulfit
Symproportinierung von Iodid und Iodat
Zur Formulierung der drei zuletzt genannten Reaktionen s. das folgende Kapitel 16.
Nachweis: Iod bildet mit Stärke (einem aus
Glucosemolekülen aufgebauten, spiralig
aufgewundenem Polymer) eine intensiv blau bis
blauviolett gefärbte Einschlussverbindung.
Diese Reaktion wird in pflanzlichen Geweben
zum Nachweis von Stärke mittels Lugolscher
Lösung (KI3; s.u.) genutzt (Versuch).
Eigenschaften:
- Brom ist gegenüber Metallen und Nichtmetallen sehr reaktiv. Versuche: Mit Kalium
wird unter heftiger Reaktion KBr gebildet, mit weißem Phosphor PBr3.
- Im festen Zustand ist Iod dunkel metallisch glänzend
und bildet ein Molekülgitter mit Schichtstruktur (s. rechts), in
dem Iodmoleküle (Bindungsabstand 267 pm) innerhalb der
Schichten (357 pm) und zwischen den Schichten (430 pm)
schwach miteinander wechselwirken. Im gasförmigem Zustand
ist Iod violett gefärbt und liegt wie die anderen Halogene in
Form von Molekülen (I2) vor. Lösungen in organischen
Lösungsmitteln (Versuche) enthalten ebenfalls isolierte
Iodmoleküle (z.B. CHCl3; violett) oder Additionsverbindungen
des I2 mit dem Lösungsmittel (z.B. Toluol: rot, organische
Lösungsmittel mit einer Sauerstofffunktion wie Ether: rotbraun).
- In Wasser löst sich Iod nur sehr geringfügig mit schwach rotbrauner Farbe. Die
Löslichkeit kann beträchtlich erhöht werden, wenn man Iodid zusetzt, z.B. in Form von
Kaliumiodid. Eine solche Iod-Iodkalium-Lösung („Lugolsche Lösung“) enthält das linear
gebaute Ion I3- = Triodid (Versuch).
- Untereinander verbinden sich die Halogene zu Interhalogenverbindungen. Dies
macht man sich in der qualitativen Analytik zum Nachweis der Halogene nebeneinander
zunutze. Versetzt man z.B. eine wässrige, mit CCl4 unterschichtete Lösung, die Bromid und
Iodid enthält, portionsweise mit Chlorwasser und zieht die entstehende Verbindung durch
leichtes Schütteln in die organische Phase (Versuch), so beobachtet man folgende
Farbabfolge:
violett (I2) rubinrot (ICl) rot (ICl3) farblos (IO3-) rotbraun (Br2 +
BrCl) farblos (BrO3-)
- Chlorid, Bromid und Iodid bilden mit Ag+ schwerlösliche Halogenide; AgF ist
hingegen gut löslich. Andererseits bildet Fluorid mit Ca2+ schwerlösliches CaF2, während die
anderen Calciumhalogenide leicht löslich sind (Versuche):
Ag+ + X- AgX X = Cl- weiß, Br- blassgelb, I- gelb
Ca2+ + 2F- CaF2 (weiß)
Die Silberhalogenide sind lichtempfindlich und dunkeln infolge Fotolyse:
AgX + h Ag + ½X2.
43
Die Verfärbung nach schwarz bei längerer Lichteinwirkung ist auf die Bildung fein verteilten
metallischen Silbers zurückzuführen. Vergl. den klassischen Fotografischen Prozess.
16. Reduktion und Oxidation (Redoxprozesse)
Begriffe:
- Oxidation ist verknüpft mit der Abgabe von Elektronen bzw. der Erhöhung der
Oxidationszahl (s.u.): M Mn+ + n e- Reduktion ist verknüpft mit der Aufnahme von Elektronen bzw. der Erniedrigung der
Oxidationszahl: X + n e- XnDie beiden Prozesse laufen stets gekoppelt ab: Redoxvorgang: M + X Mn+Xn- (z. B. die
Bildung von Natriumchlorid aus Natrium und Chlor oder von Calciumoxid aus Calcium und
Sauerstoff). X wird reduziert (M ist das Reduktionsmittel), M wird oxidiert (X ist das
Oxidationsmittel).
Mit dem Begriff Oxidationszahl sind die Teilschritte von Redoxvorgängen wie folgt definiert:
- Oxidation entspricht einer Erhöhung der Oxidationszahl
- Reduktion entspricht einer Erniedrigung der Oxidationszahl
Oxidationszahl (OZ)
Sie ist eine formale Zahl, die 0, positiv oder negativ sein kann, ganzzahlige oder
gebrochene Werte annehmen kann. OZ werden mit römischen Buchstaben über das
Elementsymbol platziert. Zur Ermittlung der OZ gelten die folgenden Regeln (hierarchisch
geordnet):
1. Elemente haben die OZ = 0
2. Für einfache (d.h. nicht zusammengesetzte Ionen) gilt, dass die OZ gleich der Ladung
des Ions ist. z.B. +I für Na+, +II für Mg2+, -I für Cl-, -II für S2-.
3. Für Moleküle und zusammengesetzte Ionen wird die OZ wie folgt ermittelt:
(i) Man teilt die Bindungselektronen dem elektronegativeren Element zu [zu
unterscheiden von der Zuteilung formaler Ladungen; dort verteilt man die Bindungselektronen
paritätisch! S. z.B. die Valenzstrichformeln für die Chlor-Sauerstoffverbindungen, S. 40.] Das
elektronegativere Element erhält somit eine negative, das weniger elektronegative
eine positive OZ. Hierbei ist das „Achter-Elektronenschalen-Limit“ zu beachten.
(ii) Die Summe aus positiven und negativen OZ muss gleich Null sein (neutrale
Moleküle) oder, bei zusammengesetzten Ionen, der Ladung des Ions entsprechen.
Folgende Unterregeln erleichtern das Auffinden von OZ
a. Fluor, als elektronegativstes Element, hat stets die OZ -I (es sei denn es
gilt Regel 1.)
b. Metalle haben regelhaft positive, Nichtmetalle negative OZ.
(i) Alkalimetalle: +I
(ii) Erdalkalimetalle: +II
(iii) Aluminium: +III
c. Wasserstoff hat in seinen Verbindungen die OZ +I (es sei denn, es gilt
Regel b, z.B. in Metallhydriden wie LiH; hier kommt dem Wasserstoff die
OZ –I zu).
d. Sauerstoff hat meist die OZ –II. Ausnahmen: Peroxide wie H2O2 (-I),
Hyperoxide wie K2O (-½), Sauerstofffluoride wie OF2 (+II). Bei all diesen
Ausnahmen gelten höher-rangige Regeln.
Beispiel:
44
+I +I -II
+I +V -II
+I +VII -II
NaClO
KIO3
KMnO4
+I -IV -II
HSO3-
+III -I
-III +I
NF3
NH3 CO
+II -II
+IV -II
+II +I -II
+II -II
0
CO2 CH3OH = CH4O S2O32- Cl2
Aufstellen von Redoxgleichungen
Das systematische Vorgehen ist im Folgenden am Beispiel der Oxidation von Eisen(II)chlorid
mit Kaliumpermanganat in saurem Milieu erläutert.
(1) Gesamtreaktion: Kaliumpermanganat (KMnO4) + Eisen(II)chlorid (FeCl2) + Säure
(H+) ergibt Mangan(II)sulfat (MnSO4) + Eisen(III)chlorid (FeCl3) + Wasser (H2O).
(2) Zurückgeführt auf die an der Reaktion beteiligten Ionen:
MnO4- + Fe2+ reagieren in saurem Milieu zu Mn2+ + Fe3+ (K+, und Cl- sind am
Redoxgeschehen nicht beteiligt)
(3) (a) Teilschritt für die Oxidation (Erhöhung der OZ):
+II
+III
Fe2+
-
Fe3+ + e
(b) Teilschritt für die Reduktion (Erniedrigung der OZ)
+VII
+II
MnO4 + 8H + 5e Mn2+ + 4H2O
Bei der Formulierung der Teilschritte ist zu beachten, dass links und rechts des
Reaktionspfeiles die Anzahl der Ladungen ebenso ausgeglichen ist, wie die Anzahl der H+
und O2- (zu ergänzen durch H+ bzw. H2O).
(4) In den beiden Teilschritten wird nun die Elektronenbilanz durch Multiplikation
abgeglichen. Sodann werden die Teilschritte addiert, was die Gesamtreaktion ergibt:
5(Fe2+ Fe3+ + e-)
MnO4- + 8H+ + 5e- Mn2+ + 4H2O
-
+
-
_______________________________________________________
MnO4- + 8H+ + 5Fe2+ Mn2+ + 4H2O + 5Fe3+
Weitere Beispiele für Redoxreaktionen:
- Oxidation von Iodid durch Permanganat: 5I- + MnO4- + 8H+ 2½I2 + Mn2+ + 4H2O
- Reduktion von Iodat mit Sulfit: 2IO3- + 5HSO3- I2 + 5SO42- + H2O + 3H+
Redoxreaktionen, bei denen im Rahmen der Elektronenübertragung eine Oxidationsstufe
erreicht wird, die zwischen denen der Ausgangssituation liegt, nennt man Sym- oder
Komproportionierung:
5I- + IO3- + 6H+ 3I2 + 3H2O (-I und +V 0)
Entsprechend wird eine Redoxreaktion als Disproportionierung bezeichnet, wenn in den
Reaktionsprodukten das am Redoxvorgang beteiligte Element in einer höheren und
niedrigeren Oxidationsstufe vorliegt:
Cl2 + NaOH NaCl + NaClO (0 -I und +I)
3NaClO 2NaCl + NaClO3
(+I -I und +V)
Die folgende gekoppelte Reaktion (a)-(d), die dem qualitativen Nachweis von Chlorat dient,
enthält mehrere Redoxvorgänge:
(a) KClO3 + H2SO4 KHSO4 + HClO3 (keine Redoxreaktion)
(b) 2HClO3 HClO2 + HClO4 (Disproportionierung)
(c) HClO3 + HClO2 2ClO2 + H2O (Symproportionierung)
(d) ClO2 ½Cl2 + O2 (Redoxreaktion) Der Zerfall erfolgt bei Erwärmen
explosionsartig
45
Ein gekoppelter Redoxvorgang ist auch die Landoltsche Zeitreaktion (Versuch):
(a) IO3- + 3HSO3- I- + 3SO42- + 3H+
(b) IO3- + 5I- + 6H+ 3I2 + 3H2O
(c) I2 + HSO3- + H2O 2I- + SO42- + 3H+
Iodat wird von Hydrogensulfit zu Iod reduziert; Reaktionen (a); gefolgt von einer
Symproportionierung; Reaktion (b). Das gemäß (b) entstehende Iod wird durch
überschüssiges Hydrogensulfit wieder zu Iodid zurück reduziert (Reaktion (c)). Erst nach
vollständigem Verbrauch des Sulfits entsteht somit Iod, angezeigt durch die Iodstärkereaktion
(Blaufärbung, s.o.). Je nach Konzentration und Temperatur verläuft die Gesamtreaktion
unterschiedlich schnell.
17. Die elektrochemische Spannungsreihe
Ob eine Spezies, die an einer Redoxreaktion beteiligt ist, oxidierend wirkt oder selbst oxidiert
wird, hängt von ihrer Oxidationskraft in Relation zu der des Redoxpartners ab. Versuchsreihe:
- Kupferblech wird in eine Silbernitratlösung getaucht: es scheidet sich Silber ab;
Kupfer geht in Lösung: Cu + 2Ag+ Cu2+ + 2Ag
- Cu-Blech wird in Zinkchloridlösung getaucht: keine Reaktion
- Zn-Blech wird in Kupfersulfatlösung getaucht: es scheidet sich Cu ab; Zn geht in
Lösung: Zn + Cu2+ Cu + Zn2+
Cu steht danach bezüglich seiner Tendenz, Elektronen abzugeben (reduzierend zu wirken)
zwischen Ag (schwächeres Reduktionsmittel als Cu) und Zn (stärkeres Reduktionsmittel als
Cu). Bzw.: Cu2+ steht danach bezüglich seiner Tendenz, Elektronen aufzunehmen (oxidierend
zu wirken) zwischen Ag+ (stärkeres Oxidationsmittel als Cu2+) und Zn2+ (schwächeres
Oxidationsmittel als Cu2+).
Entsprechend werden Iodid und Bromid von Chlor zu Iod und Brom oxidiert (Chlor dabei zu
Chlorid redzuziert), sowie Iodid von Brom zu Iod, nicht aber umgekehrt.
Quantitativ werden diese Sachverhalte durch die elektrochemischen Standardpotenziale E0 für
die jeweiligen Redoxpaare erfasst. Beispiele für Redoxpaare in den oben angeführten
Reaktionen sind:
Ag Ag+ + eZn Zn2+ + 2e2Cl- Cl2 + 2e2H2O O2 + 4H+ + 4eStandardbedingungen sind: Temperatur = 298 K (25 °C), Druck = 105 Pa (1 bar),
Konzentration c = 1 mol·l-1. Als Bezugssystem wird das Redoxpaar
Wasserstoff/Hydroniumionen verwendet, dessen Standardpotenzial = 0 gesetzt wird:
H2 + 2H2O 2H3O+ + 2e-
E0 = 0 V (25 °C, 1 bar, c(H3O+) = 1 mol·l-1 (pH = 0))
Da Elektronenübertragungen gekoppelt ablaufen, kann ein Redoxpotenzial eines isolierten
Redoxpaares nicht gemessen werden. Messtechnisch zugänglich sind nur Potenzialdifferenzen. Als Messvorrichtung dient z.B. die folgende Anordnung (Daniell-Element):
46
Stromfluss
Cu-Zn Element
Spannungsmessung: E = 1.10 V
Cu-Stab
Zn-Stab
(Cu scheidet sich ab)
(Zn geht in Lösung)
wässrige CuSO4-Lösung
(c = 1 mol/l)
Cu2+
wässrige ZnSO4-Lösung
(c = 1 mol/l)
Zn2+
Membran (nur Stromtransport)
Die Anordnung der Redoxpaare nach steigendem Redoxpotenzial wird als elektrochemische
Spannungsreihe bezeichnet. Ausschnitt:
Li Li+ + eNa Na+ + eZn Zn2+ + 2eFe Fe2+ + 2eH2 + 2H2O 2H3O+ + 2eCu Cu2+ + 2eAg Ag+ + e2H2O O2 + 4H+ + 4e2Cl- Cl2 + 2e2F- F2+ 2e-
E0 = -3,04 V
E0 = -2,71 V
E0 = -0,76 V
E0 = -0,40 V
E0 = 0 V
E0 = +0,34 V
E0 = +0,80 V
E0 = +1,23 V
E0 = +1,63 V
E0 = +2,87 V
Je positiver das Redoxpotenzial, umso stärker ist die Oxidationskraft der oxidierten Form im
Redoxpaar und vice versa. So ist Fluor ein besonders starkes Oxidationsmittel, Natrium ein
besonders starkes Reduktionsmittel. Die reduzierte Form eines Redoxpaares mit positivem
Redoxpotenzial wird auch als „edler“ als Wasserstoff bezeichnet. Hierzu gehören die
„Edelmetalle“, z.B. Gold, dessen reduzierte (also metallische) Form beständig ist.
Sind zwei verschiedene Metalle über ein leitendes Medium (z. B. saures Wasser
[Regenwasser] oder alkalisches Wasser [Speichel], oder eine Salzlösung) miteinander im
Kontakt, so bilden die beiden eine dem Daniell-Element vergleichbares Lokalelement: das
weniger edle Metall geht in Lösung (löst sich anodisch auf): Korrosion; z.B. verchromter
Stahl bei Verletzung der Chromschicht (Fe → Fe2+ + 2e-), Goldkrone plus Amalgamfüllung
im Mund (Hg → Hg2+ + 2e-). Entsprechend bilden auch Metalle, die in der Spannungsreihe
über dem Wasserstoff stehen (also unedler sind als Wasserstoff), mit dem System H2/H3O+
ein Lokalelement: sie lösen sich auf, z.B. Zink, Eisen (das dabei unter gleichzeitiger
Einwirkung des Luftsauerstoffs entstehende Fe3+ bildet schwerlösliches Eisenoxid-Hydrat =
Rost). Einige unedle Metalle, wie Chrom und Aluminium, überziehen sich an der Luft mit
einer dichten Oxidhaut (Passivierung), die sie vor dem Angriff durch die Protonen des
Wassers schützt. Die Oxidschicht kann künstlich verstärkt werden (Eloxieren von Aluminium
= oberflächlich elektrische Oxidation zu Aluminiumoxid). Wasserleitungen aus Blei in
Altbauten überziehen sich mit einer dichten Schicht aus schwerlöslichen basischen
Bleiverbindungen, die das Eintreten gesundheitsschädlicher Mengen freier Bleiionen in das
Wasser hintanhalten.
Die Normalpotenziale sind auf c = 1 mol l-1 normiert. Die Abhängigkeit der Potenziale von
der Konzentration wird durch die Nernstsche Gleichung beschrieben:
0
Ec = E +
[Ox.]
0,059
z log
[Red.]
47
Hierin ist Ec das Potenzial bei einer beliebigen Konzentration c, z ist die Zahl der
übertragenen Elektronen, [Ox.] und [Red.] sind die Gleichgewichtskonzentrationen der
oxidierten und reduzierten Formen.
Beispiel: H2 + 2H2O 2H3O+ + 2e-
+
[H3O ] 2
0,059
0
c
E = E +
log
2
[H2]
Mit E0 = 0 und [H2] = 1 (reine Phase) erhält man
Ec = 0.059·log[H3O+], bzw. Ec = -0.059·pH
pH 7
Für pH = 7 ergibt sich damit E
= -0,413 V.
18. Elektrochemische Reaktionen
Dies sind Redoxvorgänge, deren einer Redoxpartner eine Elektrode ist. Die Elektrode, an der
die Elektronenaufnahme erfolgt (also eine Oxidation) ist die Anode, die Elektrode, an der
Elektronenabgabe erfolgt (also eine Reduktion) die Kathode. Beispiele:
- Elektrolyse von Wasser (Versuch): Durch Autoprotolyse liegen OH- und H3O+ vor.
Anode:
OH- e- + ∙OH ( ½H2O + ½O2)
Kathode:
H3O+ + e- H2O + ½H2
Gesamtvorgang: H2O H2 + ½O2
(aus praktischen Gründen setzt man dem Wasser zur Erhöhung der Leitfähigkeit etwas
Säure zu)
- Schmelzflusselektrolyse von Natriumchlorid (in geschmolzenen ionischen Verbindungen
liegen die Ionen frei beweglich vor)
Anode:
Cl- ½Cl2 + eKathode:
Na+ + e- Na
- Elektrolyse wässriger NaCl-Lösung. Hier liegen die folgenden Ionen vor: Na+, H3O+, Cl-,
OHAnode:
Cl- ½Cl2 + eKathode:
H3O+ + e- H2O + ½H2; da Wasserstoff „edler“ ist als Natrium (s.
Spannungsreihe) wird das Hydroniumion (und nicht das Na+) reduziert. Nimmt man als
Kathodenmaterial Quecksilber, so kommt es zur „Überspannung“ des Wasserstoffs, und statt
Wasserstoff wird kathodisch Natrium abgeschieden und als Natriumamalgam in das
Kathodenmaterial aufgenommen; vergl. Abb. 23 auf S. 39.
- Elektrolyse einer wässrigen Bleinitratlösung (Versuch): Folgende Ionen liegen vor: H3O+,
Pb2+, OH-, NO3-. Anodisch wird OH- oxidiert, kathodisch Pb2+ reduziert (E0Pb = -0,13 V;
wegen der niedrigen [H3O+] wird hier das Blei „edler“ als der Wasserstoff; vergl. Nernstsche
Gleichung).
Anode:
2OH- 2e- + H2O + ½O2
Kathode:
Pb2+ + 2e- Pb
- Technische Bedeutung hat die Schmelzflusselektrolyse von Aluminiumoxid (versetzt mit
Kryolith oder KHF2 zur Schmelzpunktdepression):
Anodenvorgang:
O2- ½O2 + 2eKathodenvorgang: Al3+ + 3e- Al
48
- Ein weiterer bedeutender elektrochemischer Prozess ist die Kupferraffination, wie sie z.B. in
der Norddeutschen Affinerie betrieben wird. Hier wird eine Kathode aus Reinstkupfer gegen
eine Anode aus Rohkupfer geschaltet. Die Spannungsdifferenz muss hier (da es sich um
dieselben Redoxpaare handelt) nur wenige zehntel Volt betragen. Als Elektrolyt dient
Schwefelsäure. Rohkupfer enthält eine Reihe von „Verunreinigungen“, darunter solche, die
edler sind als Kupfer (Silber, Gold, Platinmetalle) und solche die unedler sind als Kupfer
(Nickel, Cobalt, Zink, Eisen, Selen, Tellur). An der Anode gehen Kupfer und die unedleren
Verunreinigungen in Lösung; die edleren Verunreinigungen fallen ab und sammeln sich im
Anodenschlamm, aus dem sie isoliert werden. Auch Tellur geht, in Form von Telluriden (z.B.
Cu2Te, Ag2Te) in den Anodenschlamm. An der Kathode scheidet sich Reinkupfer ab:
Anodenvorgang:
Cu Cu2+ + 2eKathodenvorgang: Cu2+ + 2e- Cu
19. Elektrochemische Stromquellen
Hier werden elektrochemische Prozesse genutzt, um Spannungsgefälle und somit einen
Stromfluss zu erzeugen. Man unterscheidet: Batterien (irreversible Stromerzeuger),
Akkumulatoren („Akkus“; wieder aufladbare Stromerzeuger) und Brennstoffzellen
(Stromerzeugung durch chemische Prozesse, die unkontrolliert Verbrennungen gleichen, und
deren Funktion durch Nachlieferung der Brennstoffe aufrechterhalten wird).
19.1 Batterien und Akkus
Das auf S. 45 oben abgebildete Daniell-Element ist gewissermaßen der Prototyp einer
Batterie. Für die praktische Verwendung sind Trockenbatterien in Gebrauch. Akkus sind im
Unterschied zu konventionellen Batterien wieder aufladbar.
Beispiele:
Taschenlampenbatterie (Lechlaché-Element). Abb. 24 links zeigt einen Schnitt durch die
Batterie, deren Kathode ein mit Mangandioxid ummantelter Graphitstab, und deren Anode ein
Becher aus Zinkblech ist. Als leitendes Medium (und Lieferant für Protonen) dient eine
gelatinöse Paste, die Ammoniumchlorid enthält. Redoxvorgänge:
- Anode:
Zn Zn2+ + 2e- (Die Zinkionen werden durch NH3 komplexiert)
- Kathode:
2MnO2 + 2e- + 2H+ 2MnO(OH)
gekoppelt mit: NH4+ NH3 + H+
In transportablen Geräten häufig eingesetzte Batterien basieren oft auf Lithium. In
Lithiumbatterien wird als Kathode Lihiumcobaltat LiCoO2 (neuerdings auch LithiumEisenphosphat LiFePO4) verwendet. Als Anode dient Graphit mit interkaliertem
Lithiummetall (Bruttozusammensetzung LiC6). Beim Ladevorgang wird ein Bruchteil x der
Lithiumionen des LiCoO2 in die Anode (LiC6) transportiert. Bei Stromentnahme (Entladung)
kehrt sich dieser Vorgang um; die Elektronen fließen dabei über einen externen Leiter. Der
Elektrolyt ist z.B. Dimethylcarbonat:
LiCoO2 + LiC6 + xe- Li1-xCoO2 + Li1+xC6
Knopfbatterien sind meist Silberoxidbatterien (vergl. Abb. 24, rechts); sie basieren auf einem
Elektronenaustausch zwischen Zink und Silberoxid. Leitendes Medium ist KOH oder NaOH:
- Anode: Zn + H2O ZnO + 2e- + 2H+
- Kathode: Ag2O + 2e- + 2H+ 2Ag + H2O
49
Der in Autos verwendete Bleiakkumulator besteht aus Bleiplatten und mit Bleidioxid
beschichteten Platten, die in ca. 25%-ige Schwefelsäure eintauchen (daher Vorsicht im
Umgang mit Bleiakkus). Entladevorgang (liefert eine Spannung von ca. 2V je Zelle):
- Anode:
Pb + H2SO4 PbSO4 + 2e- + 2H+
- Kathode:
PbO2 + H2SO4 + 2H+ + 2e- PbSO4 + 2H2O
Das – schwerlösliche – Bleisulfat haftet fest auf der Bleioberfläche. Beim Ladevorgang wird
durch eine ausreichend hohe Gegenspannung der Ausgangszustand wieder hergestellt.
(Ag2O)
(Zn)
Abbildung 24.
Lechlanché-Element (konventionelle Taschenlampenbatterie, links), eine SilberoxidKnopfbatterie (rechts), und ein Nickel-Cadmium-Akku (Mitte).
Dem z.B. in Laptops gebräuchlichen Nickel-Cadmium-Akku (Abb. 24 Mitte) liegt folgende
Redoxreaktion zugrunde:
Entladung
2NiO(OH) + 2H 2O + Cd
2Ni(OH) 2 + Cd(OH) 2
Ladung
Anodenvorgang: Cd + 2H2O Cd(OH)2 + 2e- + 2H+
Kathodenvorgang: NiO(OH) + H+ + e- Ni(OH)2
Der Elektrolyt ist eine 20%ige KOH-Lösung.
19.2. Brennstoffzellen
Verbrennungsreaktionen sind grundsätzlich Redoxreaktionen. Bei der üblichen Verbrennung
z.B. fossiler Brennstoffe wird die dabei erzeugte Wärme benutzt, um Wasserdampf zu
erzeugen, der dann eine Turbine antreibt und über einen hiermit gekoppelten Dynamo Strom
erzeugt. Der Wirkungsgrad ist nicht optimal, und das Verbrennungsgas CO2 als Treibhausgas
umweltproblematisch. Die direkte Erzeugung elektrischer Energie durch „stille“ Verbrennung
gelingt mit Brennstoffzellen, die allerdings bislang nur in begrenzten Einsatzbereichen
breitere Anwendung finden (z.B. Raumfahrt, Beleuchtung von Seezeichen, versuchsweise in
Kraftfahrzeugen; künftig wahrscheinlich in Laptops). Die wichtigsten derartigen
Verbrennungsreaktionen sind
H2 + ½ O2 H2O
CH4 + 2O2 CO2 + 2H2O
CH3OH + 1½O2 CO2 + 2H2O
Anode: CH3OH + H2O CO2 + 6H+ + 6e-
50
Kathode: ½ O2 + 2H+ + 2e- H2O
Der Sauerstoff wird (in der Regel als Luft) über eine poröse Kathode zugeführt und dort zu
Oxoanionen bzw. unter Aufnahme von zwei Protonen zu Wasser reduziert. Der Brennstoff
wird an der Anode oxidiert. Als Leitmedium findet eine Säure oder Base (zum Binden des
CO2 Bildung von NaHCO3), in modernen Brennstoffzellen meist eine Polymermembran
(für die Protonenleitung) Verwendung. Beispiele sind in Abbildung 25 zu sehen (s. 49).
Abbildung 25. Links: Brennstoffzelle für die Knallgasreaktion. Rechts: Brennstoffzelle mit
Methanol als Brennstoff. Der Elektronenfluss von der Anode (an der Methanol oxidiert wird)
zur Kathode (an der Sauerstoff reduziert wird) über „LOAD“ liefert nutzbare elektrische
Energie. Die Protonen, die für die Überführung der an der Kathode gebildeten Oxoanionen in
Wasser erforderlich sind, wandern über eine Membran, z.B. ein auf Phenylsulfonsäure
basierendes Polymer. PEM = Proton Electrolyte Membrane.
20. Schwefel, Selen, Tellur und Polonium
Zusammen mit dem leichteren Homologen Sauerstoff fasst man diese Elemente in der Gruppe
der Chalcogene zusammen (Chalcogen = „Kalkbildner“; benannt nach dem Oxid des
Calciums = CaO = [gebrannter] Kalk). Die Chalcogene (Gruppe 16) haben die
Elektronenkonfiguration ns2p4. Die höchst mögliche Oxidationszahl ist damit +VI, die
niedrigste –II. Zum Sauerstoff vergl. Kapitel 10.1. Tabelle 11 gibt einen Überblick über einige
wichtige Eigenschaften:
Tabelle 11. Ausgewählte Eigenschaften der Chalcogene
O
-219
hellblau
66
-141,4
Schmelzpunkt (°C)
Farbe
Atomradius (pm)
Elektronenaffinität
(kJ/mol)
Dissoziationsenergie 498,7
von X2 (kJ/mol)
Metallcharakter2)
NM
Elektronegativität
3,50
S
113/119
gelb
104
-200,4
Se
180/220
rot1)
121
-195
Te
450
braun1)
128
-190
Po
254
silbrig
(167)
-180
429,5
308
255
-
NM
2,44
NM und M
2,48
NM und M
2,01
M
1,76
51
1)
2)
Nicht-metallische Modifikation
NM = Nichtmetall, M = Metall
20.1. Schwefel
Vorkommen: elementar; in Form von Sulfaten (z.B. Gips CaSO4∙2H2O und Anhydrit CaSO4),
Kiesen (Pyrit FeS2 [enthält Fe2+ und Disulfid S22-; kristallisiert im NaCl-Gitter], Kupferkies
CuFeS2), Blenden (Zinkblende ZnS) und Glanzen (Bleiglanz PbS); Als S3--Radikal in
Ultramarin und Lapislazuli (verantwortlich für deren Blaufärbung); als H2S (Fäulnisgas), SO2
uns SO3 (in Rauchgasen); in organischen Verbindungen und fossilen „Brennstoffen“; H2S und
Schwefeloxide auch in vulkanischen Exhalationen.
Modifikationen (Abb. 26): Elementarer Schwefel, wie er in Schwefelfundstätten vorkommt,
bildet S8-Ringe; die Ausbildung der Kristalle kann rhombisch sein (thermodynamisch stabile
Modifikation) oder monoklin. Daneben gibt es eine große Zahl von Erscheinungsformen
(Modifikationen) des Schwefels, die sich durch die Ringgröße unterscheiden, darunter den Schwefel (Sechsring, z. B. in den Schwefeltröpfchen von Schwefelbakterien und auf dem
Jupitermond Io), und den -Schwefel, eine instabile und plastische Modifikation, die beim
Abschrecken von Schwefelschmelzen erhalten wird und eine Mischung aus großen Ringen
und Ketten darstellt.
S
S
S
S
S S S
S S
S
S
S S
S
rhombischer und
monokliner Schwefel -Schwefel
S
S
S
S
S
(S)n
S
S
S S
S
S
S
S
S
S
S
S
(S)n
S
-Schwefel
Abbildung 26. Modifikationen des Schwefels. Oben links: monokliner (-S), oben rechts:
rhombischer Schwefel (-S). Beiden Modifikationen liegt der kronenförmige S8-Ring
zugrunde. Neben dem S8 sind weniger stabile Modifikationen abgebildet.
Beim Erhitzen von Schwefel spielen sich folgende Vorgänge ab (Versuch):
- rhombischer Schwefel (-S) wandelt sich bei 95.6 °C in monoklinen Schwefel (-S)
um
- Bei 113 °C (-S) bzw. 119 °C (-S) schmilzt der Schwefel zu einer hellgelben, leicht
beweglichen Flüssigkeit auf. Mit zunehmender Temperatur und zunehmender Dauer
der Erwärmung bildet sich zunehmend der höhermolekulare µ-Schwefel, was mit
einer Farbvertiefung in Richtung orange rot rotbraun einhergeht.
52
-
-
-
Bei 159 °C erfolgt eine plötzliche Zunahme der Viskosität durch sprunghaften Anstieg
der Konzentration an µ-S.
Ab Temperaturen > 200 °C nimmt die Viskosität wieder ab (entsprechend dem
üblichen Verlauf von Viskositäts/Temperaturkurven). Die Schmelze ist jetzt tief
rotbraun gefärbt.
Durch Eingießen in kaltes Wasser (Abschrecken) wird das in der Schmelze
vorliegende Gemisch aus S8 (etwa 45%), µ-S (etwa 55%), und S6 (etwa 1%) als
rotbraune, plastische Masse erhalten, die sich innerhalb weniger Tage vollständig in
-S zurück verwandelt.
Bei 445 °C siedet der Schwefel. In der Dampfphase befindet sich hauptsächlich S8.
Mit weiter zunehmender Temperatur liegen in der Gasphase auch S7, S6, S5, S4, S3 und
(ab 700 °C fast ausschließlich) S2 vor.
Gewinnung:
- Durch Tagebau oder, in tieferen, Schwefel führenden Gesteinsschichten, durch
Ausschmelzen mit überhitztem Wasserdampf und Hochpumpen (Frasch-Verfahren).
- Erhitzen (1200 °C) von Pyrit unter Luftausschluss: FeS2 FeS + S
- Nach dem Claus-Prozess: (a)
H2S + 1½O2 SO2 + H2O
(b)
2H2S + SO2 3S + H2O
__________________________________________
(c)
3H2S + 1½O2 3S + 3H2O
Die Symproportionierungs-Reaktion (b) verläuft nur in Gegenwart geeigneter
Katalysatoren. Technisch kommt hier z.B. Bauxit zum Einsatz. Auch Wasser
katalysiert die Reaktion (Versuch). H2S fällt z.B. bei der Entschwefelung von Erdgas
und Erdöl (durch Hydrodesulfurierung) in großen Mengen an.
Verwendung: Die Hauptmenge geht in die Produktion von Schwefelsäure (und deren
Weiterverwertung in der chemischen, metallurgischen und Düngemittel-Industrie). Weitere
Verwendungsbereiche: Vulkanisierung von Kautschuk (S), Schädlingsbekämpfung (SO2),
Zündholzindustrie (Sb2S3), Feuerwerkerei (S).
20.2. Selen und Tellur
Selen kommt vergesellschaftet mit Schwefel in sulfidischen Erzen vor und reichert sich bei
derem Abrösten (= Überführen der Metallsulfide in Metalloxide und SO2) im Flugstaub an
(SO2 ist gasförmig, SeO2 ist fest). Unter Laborbedingungen kann Selen durch Reduktion
Seleniger Säure (eine Lösung von SeO2 in Wasser = H2SeO3) mit Schwefliger Säure (H2SO3)
- Versuch - oder Hydrazin (N2H4) gewonnen werden. Das Selen fällt dabei in der roten
Modifikation an, der Se8-Ringe zugrunde liegen:
H2SeO3 + 2H2SO3 1/8Se8 + 2H2SO4 + H2O
Durch Erhitzen der wässrigen Suspension roten Selens erhält man schwarzes Selen, das dem
µ-S vergleichbar ist. Erhitzt man über 100 °C, so erhält man graues Selen, eine
halbmetallische Modifikation, die aus spiralig aufgewundenen Selenketten aufgebaut ist und
Halbleitereigenschaften besitzt ( photoelektrischer Belichtungsmesser).
Selen ist ein biologisch essentielles Element und kommt im Selenocystein vor, einem
Homologen der schwefelhaltigen Aminosäure Cystein. Größere Mengen Selen sind aber
toxisch.
53
Tellur hat bereits weitgehend metallische Eigenschaften. Es tritt gleichfalls als Begleiter
sulfidischer Erze auf und wird z.B. aus dem Anodenschlamm der Kupferraffination
gewonnen; vergl. S. 48.
20.3. Polonium
Das radioaktive Polonium wurde 1898 von Pierre und Marie Curie (geb. Sklodowska) aus der
Pechblende (UO2) isoliert. 1000 t Pechblende enthalten ca. 0.03 g Polonium. Das stabilste
Isotop, 210Po, wird heute in Reaktoren u.a. durch Beschuss von 209Bi mit Neutronen
hergestellt. 210Po hat eine Halbwertszeit von 138 Tagen; es zerfällt unter -Emission in 206Pb.
209
83Bi
1
+ 0n
210
210
83Bi
84Po
+
-
206
82Pb
4
+ 2
Die Toxizität ist erheblich: Der LD50-Wert liegt bei 8 ng/kg, die biologische Halbwertszeit bei
50 Tagen. Bekannt geworden ist Polonium-210 in jüngster Zeit durch die Vergiftung des
russischen Exspions Litwinenko. In seinem chemischen Verhalten ähnelt Polonium dem
Tellur und dem Wismut.
20.4. Wasserstoffverbindungen der Chalcogene
Tabelle 12 gibt eine Übersicht über einige Eigenschaften. Zum Wasser s. a. Kap. 13.2.
Tabelle 12
Siedepunkt (°C)
Bildungsenthalpie (kJ/mol)
MAK-Wert (mg/m3)
H2O
+100
-286
-
H2S
-60,8
-20,1
15
H2Se
-41,5
+77,4
0,2
H2Te
-1,8
+143
0,1
Während der Verlauf der Siedepunkte von H2S, H2Se und H2Te insofern den Erwartungen
entspricht, als sie entsprechend der Zunahme der Molmasse in dieser Richtung ansteigen,
zeigt sich beim Wasser eine „Anomalie“: Der „zu hohe“ Siedepunkt ist eine Folge der
Assoziation der Wasser-Dipole; vergl. z.B. Abb. 19. Die Bildungsenthalpien von H2Se und
H2Te sind positiv, d.h. dass diese Verbindungen thermodynamisch instabil sind. Einmal
gebildet, ist der Zerfall jedoch kinetisch gehemmt. H2S, H2Se und H2Te sind hoch-giftige,
übel riechende Gase (H2S riecht nach faulen Eiern, H2Se nach faulem Rettich). Die MAKWerte können zur ersten Orientierung für das Ausmaß der Giftigkeit dienen. MAK = maximal
[zulässige] Arbeitsplatzkonzentration (bezogen auf 40 Wochenstunden Arbeitszeit). Zum
Vergleich: Blausäure (HCN): 11, Kohlenmonoxid (CO) 55 mg/m3 Luft. Die Toxizität von
H2S beruht auf der Blockierung des Eisens im Hämoglobin durch Fällung von Eisensulfid.
Schwefelwasserstoff fällt u.a. bei der Hydrodesulfurierung von Rohöl an (Entschwefelung
durch Umsetzung mit Wasserstoff; der Schwefel liegt im Rohöl in Form organischer
Schwefelverbindungen vor). Zur Darstellung eignen sich die folgenden Reaktionen:
S + H2 H2S
FeS + H2SO4 H2S + FeSO4
Zur Verwendung von H2S als Ausgangsverbindung für die Darstellung elementaren
Schwefels s. S. 52.
54
Bedeutung von H2S in der Analytik:
Im Folgenden werden Begriffe verwendet, die erst in späteren Kapiteln im Einzelnen erläutert
werden: Säure-Base-Gleichgewichte: Kap. 22, Massenwirkungsgesetz, GleichgewichtsKonzentrationen, Löslichkeitsprodukt: Kap. 21.
H2S ist eine sehr schwache Säure, die mit Wasser wie folgt in einem Säure-BaseGleichgewicht steht:
1. Stufe: H2S + H2O HS- + H3O+ KS1 = 10-7
2. Stufe: HS- + H2O S2- + H3O+ KS2 = 10-13
Für die Sulfidionen-Konzentration im Gleichgewicht, [S2-], erhält man hieraus (bei
Anwendung des Massenwirkungsgesetzes auf die beiden Stufen):
[S2-] = 10-7∙10-13∙[H2S]∙[H3O+]-2
Bei einem pH = 0, d.h. [H3O+] = 1, und einer Konzentration [H2S] c(H2S) = 10-1 (das ist die
maximal erreichbare Konzentration an H2S) wird
[S2-] = 10-21
Entsprechend fallen nur solche Metallsulfide quantitativ aus, die hinreichend schwerlöslich
sind, deren Löslichkeitsprodukte also ausreichend klein ist:
Bleisulfid:
LPbS = [Pb2+][S2-] = 10-28
PbS fällt in stärker saurem Melieu
2+
2-23
Zinksulfid: LZnS = [Zn ][S ] = 10
ZnS fällt in sehr schwach saurem Milieu
Mangansulfid: LMnS = [Mn2+][S2-] = 10-15 MnS fällt nur im alkalischen Milieu
Die Sulfide der H2S-Gruppe sind, mit Ausnahme des extrem schwerlöslichen
Quecksilbersulfids, in stärker konzentrierten Säuren löslich, da hier ihr Löslichkeitsprodukt
durch Bildung von Hydrogensulfid (S2- + H+ HS -) unterschritten wird. HgS (L = 10-52)
kann nur noch von oxidierenden Säuren wie Salpetersäure gelöst werden: Halbkonzentrierte
Salpetersäure (HNO3) oxidiert die Sulfidionen zu Sulfat. Auch einige spezialisierte Bakterien
vermögen schwerlösliches HgS in Sedimenten auf diesem Wege oxidativ zu remobilisieren.
Die (weniger schwerlöslichen) Sulfide der Ammonsulfidgruppe werden im alkalischen Milieu
gefällt. Zur Abtrennung von Nickel und Cobalt lässt man die Sulfidfällung im Kontakt mit
Luftsauerstoff „altern“. Nickel- und Cobaltsulfid werden so säureunlöslich und können von
den anderen Sulfiden der Ammonsulfidgruppe abgetrennt werden. Die säureunlöslichen
Sulfide haben die Bruttoformel M2S3 (M = Ni, Co). Der „Alterung“ liegt eine partielle
Oxidation des Sulfids zum Disulfid zugrunde (die Oxidationsstufe von M bleibt +II); die
Zusammensetzung entspricht also z.B. NiII2S-II(S-I2).
Alkalimetallsulfide sind leicht löslich in Wasser. Sie dissoziieren in Alkalimetallionen und
Sulfidionen, die mit Wasser gemäß
S2- + H2O HS- + OHunter Bildung von Hydrogensulfid reagieren. Alkailimetallsalz-Lösungen sind mithin
alkalisch. Mit elementarem Schwefel, oder durch Oxidation, bilden Sulfide Disulfide und
Polysulfide:
S2- + S S22- ( Sn2-)
2S2- S22- + 2eDurch Ansäuern kann aus Disulfid- und Polysulfidlösungen Schwefel ausgefällt werden:
Sn+12- + 2H3O+ H2S + nS + 2H2O
Mit Polysulfid werden in der Arsengruppe Arsen, Antimon und Zinn vom Wismut abgetrennt.
Nur die Sulfide des Arsens und Antimons, As2S3 und Sb2S3, sowie des Zinns, SnS, werden
von Polysulfid zu den löslichen Thioarsenaten, -antimonaten und -stannaten oxidiert:
55
As2S3 + 2HS2- + HS- + 3OH- 2[AsS4]3- + 3H2O
SnS + HS2- + OH- [SnS3]2- + H2O
Die Verwendung von Polysulfid (und damit eines oxidierenden Agens) ist erforderlich, um
auch das Zinn(II)sulfid in Lösung zu bringen: nur in der Oxidationsstufe +IV ist Zinn
hinreichend nichtmetallisch, um ein Thiostannat zu bilden. Arsen bzw. Antimon bilden bereits
in ihren Oxidationsstufen +III lösliche Thioanionen (Thioarsenit bzw. Thioantimonit).
20.5. Sauerstoffverbindungen des Schwefels
Die wichtigsten Oxide des Schwefels sind Schwefeldioxid SO2 und Schwefeltrioxid SO3, die
Anhydride der Schwefligen Säure bzw. der Schwefelsäure. SO2 erhält man durch Verbrennen
von Schwefel oder Schwefelwasserstoff:
S + O2 SO2
H2S + 1½O2 SO2 + H2O
oder durch Abrösten sulfidischer Erze wie Pyrit:
2FeS2 + 5½O2 Fe2O3 + 4SO2
SO3 wird nach dem Kontaktverfahren aus SO2 und Sauerstoff mit Vanadiumpentoxid als
Katalysator gewonnen:
SO2 + V2O5 SO3 + V2O4
V2O4 + ½O2 V2O5
_________________________________
SO2 + ½ O2 SO3
SO2 und SO3 entstehen auch bei der Verbrennung von Restbeständen organischer
Schwefelverbindungen in Benzin, Kerosin, Heizöl und Stadtgas (CH4) und gelangen so in die
Atmosphäre, wo sie mit der Luftfeuchtigkeit Schwefelsäure bilden ( Saurer Regen). Bei der
technischen Schwefelsäure-Gewinnung wird SO3 mit Schwefelsäure zu Dischwefelsäure
(„Pyroschwefelsäure“) umgesetzt, aus der durch Hydrolyse Schwefelsäure entsteht (die
direkte Bildung von Schwefelsäure aus SO3 und Wasser ist nicht begünstigt):
H2SO4 + SO3 H2S2O7;
H2S2O7 + H2O 2H2SO4
Schwefelsäure ist eine starke Säure (KS1 = 103, KS2 = 10-1,9), von der sich zwei Reihen von
Salzen, die Hydrogensulfate und die Sulfate herleiten, z.B. NaHSO4 und Na2SO4
(Glaubersalz). Schweflige Säure ist eine schwache Säure (KS1 = 10-2, KS2 = 10-7,2); ihre Salze
sind die Hydrogensulfite (z. B. NaHSO3) und Sulfite (z.B. Na2SO3). Das Hydrogensulfit-Ion
liegt in zwei tautomeren Formen vor (vergl. die folgende Zusammenstellung der
Valenzstrichformeln), von denen die so gen. Sulfhydrilform – mit dem Proton am Schwefel –
allerdings eine untergeordnete Rolle spielt.
H
O
S
O
O
O
O
S
S
S
O
O
O
S
O
O
O
H
O
O H
S
O
O
O
O
O
O
S
S
O
O
H
H
O
O
O
eisartiges SO 3
S
O
O
O
O
n
asbestartiges SO 3
S
O
O H
56
SO3 ist nur in der Gasphase monomer. Bei Raumtemperatur ist SO3 fest und liegt als
„eisförmige“ Modifikation (SO3)3 oder als „asbestartige“ Modifikation (SO3)n vor.
Schwefeldioxid, Schweflige Säure und Sulfite sind starke Reduktionsmittel:
H2SeO3 + 2H2SO3 Se + 2H2SO4 + H2O (Versuch)
I2 + H2SO3 + H2O 2I- + H2SO4 + 2H+
(Versuch)
Dagegen ist (konzentrierte) Schwefelsäure ein mildes Oxidationsmittel. Während verdünnte
Schwefelsäure Kupfer nicht löst (vergl. die Stellung von Cu und H2 in der Spannungsreihe),
wird Cu von heißer conc. H2SO4 unter Reduktion des Schwefels oxidativ gelöst:
Cu + H2SO4 + 2H+ Cu2+ + SO2 + 2H2O (Versuch)
Schwefelsäure wird u.a. in der Düngemittelindustrie (1) sowie bei der Herstellung von
Pigmenten auf Rutil-Basis verwendet (2):
(1) Phosphatdüngung: Schwerstlöslicher Phosphorit (= tertiäres Calciumphosphat) ist für
Pflanzen nicht zugänglich. Der Aufschluss mit H2SO4 führt zu dem von Pflanzen
verwertbaren Superphosphat, einer Mischung aus primärem Calciumphosphat (CalciumDihydrogenphosphat) und Calciumsulfat:
Ca3(PO4)2 + 2H2SO4 2CaSO4 + Ca(H2PO4)2
(2) Titanerze wie Ilmenit (FeTiO3) werden mit H2SO4 aufgeschlossen. Die so erhaltene
wässrig-sauere Lösung aus Eisensulfat und Titanylsulfat wird eingeengt, wobei das
Eisensulfat auskristallisiert und abgetrennt wird. Das verbleibende Titanylsulfat wird durch
Einleiten heißen Wasserdampfes hydrolysiert, der dabei anfallende Rutil (TiO2) abgetrennt,
und die Schwefelsäure („Dünnsäure“) wieder aufgearbeitet oder verklappt:
FeTiO3 + 2H2SO4 FeSO4 + TiOSO4 + 2H2O
TiOSO4 + H2O TiO2 + H2SO4
Rutil findet Verwendung als weißes Farbpigment in Antrichfarben, sowie in FarbstoffSolarzellen.
Durch anodische Oxidation von Hydrogensulfat erhält man Peroxodischwefelsäure, deren
Hydrolyse Wasserstoffperoxid liefert:
H
O
O
S
O
O
-
-e
O
H
H
O
S
O
x2
O
O
H
O
O
S
S
O
O
O
O
+
+ H2O + 2H
O
- 2H2SO4
H2O2
Eine weitere Sauerstoffverbindung des Schwefels ist die (frei nicht beständige)
Thioschwefelsäure, von der sich Thiosulfate wie Natriumthiosulfat Na2S2O3 (Fixiersalz)
herleiten. Thiosulfate erhält man durch Symproportionierung von Sulfiten und Schwefel:
SO32- + S S2O32Formal entspricht das einem Austausch eines Oxo- durch einen Sulfido-Substituenten im
Sulfat. Die mittlere Oxidationsstufe des Schwefels im Thiosulfat ist +II. Der Trivialname von
Natriumthiosulfat – Fixiersalz– spiegelt die Befähigung von Thiosulfat wider, Silberbromid
„aufzulösen“ und damit eine konventionelle Fotografie zu „fixieren“ (d.h. ein Nachdunkeln
eines Negativs und Positivs am Licht dadurch zu verhindern, dass überschüssiges AgBr aus
57
der lichtempfindlichen Beschichtung entfernt wird). Die Trivialbezeichnung „Antichlor“
kennzeichnet die Befähigung von Thiosulfat, Chlor zu Chlorid zu reduzieren:
„Fixiersalz“: AgBr + 2S2O32- [Ag(S2O3)2]3- + Br„Antichlor“: 4Cl2 + S2O32- + 5H2O 8Cl- + 2SO42- + 10H+
Säuert man eine Thiosulfatlösung an, so entsteht primär Thioschwefelsäure, die sogleich in
Schwefel und Schwefeldioxid zerfällt. Der Schwefel fällt dabei als -Schwefel (S6; s.o.) an
und bleibt zunächst kolloidal in Lösung, aggregiert schließlich und trübt die Lösung
(Versuch):
H2S2O3 1/6S6 + SO2 + H2O
Versetzt man eine wässrige Silbernitratlösung mit Thiosulfat, so bildet sich primär
Silberthiosulfat (weißer Niederschlag), das rasch durch Zersetzung SO3 und eine schwarze
Fällung von Silbersulfid (Ag2S) ergibt. Dabei werden die Stufen Gelb, Orange, Rot und Braun
durchlaufen: „Anorganischer Sonnenuntergang“ (Versuch):
2Ag+ + S2O32- Ag2S2O3 Ag2S + SO3
weiß
schwarz
Thiosulfat spielt eine wichtige Rolle in der Iodometrie zur quantitativen Bestimmung von
Reduktions- und Oxidationsmitteln. Thiosulfat (Oxidationsstufe des Schwefels +II) wird
dabei zu Tetrathionat oxidiert (Oxidationsstufe + 2,5). Die Grundgleichungen der Iodometrie
lauten:
O
2 O S
O
O
S
O S
O
S
O
-
I2 + 2e
S
S O + 2eO
-
2I
I2 + 2S2O32- 2I- + S4O82Beispiele:
1. Bestimmung von Oxidationsmitteln, z.B. Cu2+: Die zu bestimmende Lösung wird
mit überschüssigem Iodid versetzt; es fällt Kupfer(I)iodid aus und eine äquivalente Menge
Iodid wird zu Iod oxidiert.
Cu2+ + 2I- CuI + ½I2
Das Iod wird sodann mit Thiosulfat titriert.
2. Bestimmung von Reduktionsmitteln, z.B. Arsenit: Die Arsenitlösung wird mit einer
bekannten Menge überschüssiger Iodlösung (in Form von Iod-Iodkalium KI3) versetzt Arsenit
reduziert das Iod zu Iodid:
AsO33- + I3- + H2O AsO43- + 3I- + 2H+
Der nicht verbrauchte Anteil an Iod wird mit Thiosulfat rücktitriert
21. Das Massenwirkungsgesetz
Die Gleichgewichtslage dynamischer chemischer Gleichgewichte wird quantitativ durch das
Massenwirkungsgesetz (MWG) beschrieben. Ein Beispiel für eine Gleichgewichtsreaktion ist
die Bildung von Iodwasserstoff aus Iod und Wasserstoff (Hinreaktion) sowie der Zerfall von
Iodwasserstoff zu Iod und Wasserstoff (Rückreaktion).
H2 + I2 2HI
Geschwindigkeit der Hinreaktion: v = kc(H2)∙c(I2)
58
Geschwindigkeit der Rückreaktion: v = kc(HI)2
mit v = dci/dt; k und k sind die Geschwindigkeitskonstanten für die Hin- bzw.
Rückreaktion; ci sind die Konzentrationen der beteiligten Stoffe. Bei
Gleichgewichtseinstellung gilt v = v und damit
[HI]2/[H2][I2] = k/k = K
K ist die von den Konzentrationen unabhängige Gleichgewichtskonstante. K ist aber
temperaturabhängig. Die eckigen Klammern kennzeichnen die Konzentrationen im
Gleichgewicht (Gleichgewichtskonzentrationen).
Allgemein gilt für ein Gleichgewicht, an dem die Stoffe A und B als Edukte und B und C als
Produkte beteiligt sind ( und sind die stöchiometrischen Faktoren)
A + B C + D
K = [C][D]/[A][B]
Ausgewählte Anwendungen:
(1) Autoprotolyse des Wassers:
2H2O H3O+ + OH[H3O+][OH-]/[H2O]2 = 1,8∙10-16
Unter Einbeziehung der als konstant zu erachtenden Konzentration undissoziierten Wassers
([H2O] = 55,56 mol/l) in die Gleichgewichtskonstante erhält man für das
Ionenprodukt des Wassers: [H3O+][OH-] = KW = 10-14 mol2/l2 (bei 25 °C)
Und damit [H3O+] (= [OH-]) = 10-7 mol/l in reinstem Wasser.
(2) Säure-Gleichgewichte in wässriger Lösung (Säure = HX):
HX + H2O X- + H3O+
[X-][H3O+]/[HX][H2O] = K; bzw. mit K[H2O] = KS
Säurekonstante KS = [X-][H3O+]/[HX] mol/l
KS ist ein Maß für die Stärke einer Säure, d.h. die im Gleichgewicht befindliche
Konzentration an Hydroniumionen. Statt des KS-Wertes wird häufig dessen negativer
dekadischer Logarithmus, pKS, angegeben. Beispiele: X = Cl, pKs = -6 (Salzsäure, sehr
starke Säure); X = F, pKs = 3,17 (Flusssäure, schwache Säure); X = CN, pKs = 9,4
(Blausäure, sehr schwache Säure).
(3) Gleichgewichte zwischen gelöstem und ungelöstem Anteil eines schwer-löslichen Stoffes,
z.B. Silberchlorid AgCl:
AgCl(ungelöst) Ag+ + Cl[Ag+][Cl-]/[AgCl(ungelöst)] = K
Die reine Phase AgCl(ungelöst) wird = 1 gesetzt. Damit ergibt sich als Maß für die
Löslichkeit das Löslichkeitsprodukt zu
L = [Ag+][Cl-] = 10-10 mol2/l2
Je kleiner das Löslichkeitsprodukt, umso schwerer löslich ist eine Verbindung. Weitere
Beispiele für Löslichkeitsprodukte s. z.B. die Sulfide (Kap. 20.3., S. 53).
Aus dem Löslichkeitsprodukt erhält man die Löslichkeit l (in gesättigter, mit dem ungelösten
AgCl im Gleichgewicht stehender Lösung) zu: l = [Ag+] = [Cl-] = 10-5 mol/l.
22. Säure-Base Konzepte
22.1. Brønstedt-Säure/Basen
Im Brønstedtschen Begriffssystem ist eine Säure ein Protonendonator, eine Base ein
Protonenakzeptor. Säure-Base-Reaktionen laufen stets gekoppelt ab. HCl (Chlorwasserstoff)
allein ist noch keine Säure; erst die Reaktion mit Wasser (also eine wässrige HCl-Lösung =
Salzsäure) verleiht den Säurecharakter. Allgemein kann ein Säure-Base-Gleichgewicht wie
folgt formuliert werden:
HX + H2O X- + H3O+
59
Den von links nach rechts verlaufenden Vorgang nennt man Protolyse. Wasser fungiert hier
als Brønstedt-Base. Das bei Protonenaufnahme entstehende Hydroniumion ist seinerseits eine
Säure (die zu Wasser konjugierte Säure). Entsprechend ist das Anion X- die zu HCl
konjugierte Base. Im Falle starker Säuren liegt das Gleichgewicht auf der rechten, im Falle
schwacher Säuren auf der linken Seite.
Beispiele für Brønstedtsche Säure-Base-Reaktionen:
(1) Saure Reaktion wässriger Ammoniumsalz-Lösungen, z.B. Ammoniumchlorid
(Salmiak). Durch Dissoziation liegen neben Cl- Anionen Ammonium-Kationen vor,
die den Charakter einer Brønstedt-Säure haben:
NH4+ + H2O NH3 + H3O+
(2) Saure Reaktion einer wässrigen Alaun-Lösung. Alaun ist KAl(SO4)2∙12H2O. In einer
wässrigen Lösung liegen vor: SO42-, K+ und komplexe Hexaquaaluminium-Kationen
[Al(H2O)6]3+. Letztere wirken als Kationsäuren (das komplex gebundene Wasser hat
eine größere Tendenz, Protonen anzugeben, als das Lösungswasser):
[Al(H2O)6]3+ + H2O [Al(H2O)5(OH)]2+ + H3O+
(Die eckigen Klammern geben hier - und generell bei der Formulierung von Komplexen - nicht die
Gleichgewichtskonzentrationen an, sondern kennzeichnen die Integrität des komplexen Teilchens)
(3) Basische Reaktion wässriger Carbonatlösungen, z.B. Natriumcarbonat (Soda). Die
durch Dissoziation entstehenden Carbonationen wirken als Base. Sie gehen dabei über
in Hydrogencarbonat:
CO32- + H2O HCO3- + OHWasser ist hier die Säure. Wasser kann also, wie unter (1) und (2), als Base fungieren,
sowie, wie unter (3) als Säure: Wasser ist ein Ampholyt. S. auch die Eigendissoziation
von Wasser: 2H2O H3O+ + OHTräger des sauren Charakters ist das Hydroniumion H3O+ (verkürzt: H+). Liegt dieses
gegenüber den Hydroxidionen OH- im Überschuss vor, so reagiert die Lösung sauer. Da das
Produkt aus [H3O+] und [OH-] eine Konstante ist, nämlich 10-14 (s.o.), liegt eine saure
Reaktion dann vor, wenn [H3O+] > 10-7. Entsprechend ist ein Medium basisch (alkalisch),
wenn H3O+ gegenüber OH- im Unterschuss ist, also [H3O+] < 10-7. Für [H3O+] = [OH-] = 10-7
ist eine Lösung neutral. Mit der Einführung des pH-Wertes, definiert als
pH = -log10[H3O+]
gilt: pH < 7: sauer; pH = 7: neutral; pH > 7: basisch (alkalisch)
Für starke Säuren ist [H3O+] c(Säure).
22.2. Lewis-Säure/Base
Eine Lewis-Säure ist ein System mit einer Elektronenlücke, eine Lewis-Base ein solches mit
einem freien Elektronenpaar. Bei einer Lewis-Neutralisation treten Säure und Base unter
Ausbildung einer koordinativ-kovalenten Bindung (dative Bindung; Donorbindung)
zusammen. Das kann man dadurch verdeutlichen, dass in der Valenzstrichformel der
resultierenden Verbindung das gemeinsame bindende Elektronenpaar durch einen Pfeil (statt
eines Valenzstriches) symbolisiert wird, der vom Donor- zum Akzeptoratom gerichtet ist.
Beispiele:
F3B + N H3
Fe2+ + O H2
F3B
Fe
NH3
OH2
2+
60
Fe2+ verfügt über 6 Valenzelektronen (in den 3d-Orbitalen), hat gegenüber dem nächst
folgenden Edelgas (Krypton) also ein Elektronendefizit von 12 Elektronen. Diese
Elektronenlücke kann Fe2+ durch Aufnahme von insgesamt 6 Wassermolekülen (jedes
Wasser stellt ein freies Elektronenpaar am Sauerstoff für die Bindung zur Verfügung)
ausgleichen. Man erhält so ein stabiles, komplexes Kation [Fe(H2O)6]3+. Die LewisSäure/Lewis-Base Wechselbeziehung spielt ganz generell in der Komplexchemie
(Koordinationschemie) eine zentrale Rolle.
23. Die Stickstoff-Phosphorgruppe
23.1. Die Elemente
In diese Gruppe (Gruppe 15, auch „Pnictogene“ genannt) gehören die Elemente Stickstoff,
Phosphor, Arsen, Antimon und Bismut, und damit reine Nichtmetalle (N), reine Metalle (Bi),
sowie Elemente, für die es neben nichtmetallischen auch halbmetallische bzw. metallische
Modifikationen gibt (P, As, Sb). Der Halbmetallcharakter von As und Sb spiegelt sich auch in
deren Vorkommen wider (kationisch bzw. anionisch; s.u.). Bi ist das letzte stabile Element im
Periodensystem. Entsprechend der Valenzelektronen-Konfiguration ns2p3 können die
Elemente in den Oxidationsstufen –III bis +V vorliegen.
Vorkommen
Stickstoff ist
Bestandteil der Luft (78%
Strickstoffkreislauf
N2) sowie anorganischer
Verbindungen (nitrose Gase
N2
N2O
NOx, Nitrat, Ammoniak,
NichtDenitriBiogene
Ammoniumsalze) und
biogene
NO fizierung
StickstoffStickstofforganischer Verbindungen
Fixierung
Fixierung
(z.B. Aminosäuren, Amine,
Nitrifizierung
Nukleotide). Mineralisch
NH3
Nitrat
Nitrit
kommt Stickstoff als Salpeter
Ammonifizierung
(Natriumnitrat NaNO3) vor.
Abbau
Stickstoff unterliegt einem
Assimilation
Kreislauf, vergl. Abb. 27, der
Abbildung 27
biologische Prozesse
{C-N}
(biogene Stickstofffixierung,
nitrifzierende, ammonifizierende und denitrifizierende Mikroorganismen) und nicht-biogene
Prozesse (Blitzschlag, Höhenstrahlung, anthropogene Einflüsse) umfasst.
Phosphor bildet die Minerale Phosphorit Ca3(PO4)2 und Apatit Ca5(PO4)3(OH,F)
(Hydroxyl- und Fluorapatit). Hydroxylapatit ist auch Bestandteil der Knochen, des Zahnbeins
und Zahnschmelzes. Phosphat spielt weiterhin eine wichtige Rolle im Energiemetabolismus
der Organismen sowie beim Aufbau von RNA (Ribonucleinsäüre) und DNA
(Desoxyribonucleinsäure).
Arsen und Antimon liegen mineralisch entweder gediegen vor (z.B. Scherbenkobalt =
As), oder kationisch (Realgar As4S4, Auripigment As2S3, Grauspießglanz Sb2S3, Arsenik
As2O3) oder anionisch (Arsenkies Fe2S2As2, Breithauptit NiSb). Von Bismut gibt es nur
kationische Vorkommen (Bismutglanz Bi2S3, Bismutocker Bi2O3).
Gewinnung
61
- N2 durch Luftverflüssigung und fraktionierte Destillation (Siedepunkte: O2 = -183.0, N2 =
-195.8 °C), durch Überleiten von Luft über glühenden Koks:
{O2 + 4N2} + C CO2 + 4N2 (das CO2 wird sodann ausgewaschen)
- Im Labor z.B. durch Erhitzen einer wässrigen Lösung von Ammoniumnitrit:
NH4NO2 N2 + 2H2O (Versuch)
- Phosphor: Ca3(PO4)2 + 5C + 3SiO2 2P + 3CaSiO3 + 5CO
- Arsen: Erhitzen von Arsenkies unter Luftausschluss: FeAsS FeS + As
- Antimon: Sb2S3 + 3Fe 3FeS + 2Sb
- Wismut: 2Bi2O3 + 3C 4Bi + 3CO2
Erscheinungsformen (Modifikationen):
Stickstoff, das einzige Gas in dieser Gruppe, liegt molekular als N2 vor. Die N-NDreifachbindung (Bindungsenergie = 945 kJ/mol) macht dieses Element sehr inert. Vom P,
As und Sb gibt es jeweils mehrere Modifikationen, wobei hier nur auf die des Phosphors
näher eingegangen werden soll; vergl. hierzu auch Abb. 28.
- Weißer (oder gelber) Phosphor, P4, ist tetraedrisch gebaut. Die Bindungswinkel von
60° bedingen eine hohe Spannung und damit hohe Reaktionsfähigkeit. Weißer
Phosphor ist giftig und an der Luft selbstentzündlich. Nur unter Wasser aufbewahren
und schneiden!
- Roter Phosphor entsteht durch Erhitzen aus weißem Phosphor. Roter Phosphor ist
mehr oder weniger amorph. Er findet z.B. in den Reibflächen von Zündholzschachteln
Verwendung.
- Violetter Phosphor entsteht aus rotem Phosphor durch längeres Tempern bei ca. 550
°C. In der Schichtstruktur treten Käfige aus P8- und P9-Einheiten auf.
- Schwarzen Phosphor erhält man aus den anderen Modifikationen durch Erhitzen unter
hohem Druck in Gegenwart von Hg als Katalysator. Er ist aus übereinanderliegenden,
stark gewellten Schichten aufgebaut. Diese Modifikation leitet den elektrischen Strom,
ist also metallisch.
Abbildung 28.
Phosphormodifikationen. Links:
weißer, rechts oben: violetter
(vereinfacht), rechts unten: schwarzer
Phosphor.
Bismut wiederum kommt nur in einer, nämlich metallischen Modifikation vor. Bi bildet mit
anderen Metallen Legierungen, die leicht schmelzen, z.B. Woodsches Metall, eine Legierung
aus Bi, Pb, Sn und Cd, mit einem Schmelzpunkt von 70 °C.
62
23.2. Wasserstoffverbindungen
Die wichtigsten binären (homoleptischen) Wasserstoffverbindungen des Stickstoffs sind
Ammoniak (Azan) NH3, Hydrazin (Diazan) N2H4 und Stickstoffwasserstoffsäure HN3.
Im Labor wird Ammoniak zweckmäßig aus Ammoniumsalzen wie Salmiak
(Ammoniumchlorid) durch „Austreiben“ mit starken Basen dargestellt:
NH4Cl + NaOH NH3 + NaCl + H2O
Versuch
Ammoniak nicht einatmen. Leicht ätzend und giftig!
Wässrige Ammoniaklösungen (Ammoniakwasser; Salmiakgeist) reagieren infolge des SäureBase-Gleichgewichtes NH3 + H2O NH4+ + OH- alkalisch; Ammoniumsalze in Wasser
gelöst reagieren sauer:
NH4Cl + aq. NH4+·aq + Cl-·aq
NH4+ + H2O NH3 + H3O+
2+
Mit Cu bildet NH3 den tief blau gefärbten Komplex „Tetramminkupfer“, dessen Bildung als
Nachweisreaktion für Cu2+ oder NH3 Verwendung findet:
[Cu(H2O)6]2+ + 4NH3 [Cu(NH3)4(H2O)2]2+ + 4H2O
Versuch
Spuren Ammoniak z.B. in Trinkwasser werden mit Neßlers Reagenz K2[HgI4] nachgewiesen
und auch quantitativ bestimmt (colorimetrisch). Nesslers Reagenz reagiert mit Ammoniak zu
orangefarbenen Lösungen, bei höherer Ammoniak-Konzentration zu braunen Fällungen von
[Hg2N]I (Versuch).
Vom Ammoniak leiten sich viele wichtige organische Verbindungen her, z.B. die Amine und
Aminosäuren. Verschiedene Bakterien, darunter frei lebende (Azotobacter), symbiotisch mit
Schmetterlingsblütlern vergesellschaftete (Rhizobium) sowie Blaugrünalgen (Cyanophyceae)
vermögen N2 zu NH3 zu „fixieren“ (vergl. auch den Stickstoffkreislauf, Abb. 27). Beim
biologischen Abbau von Stickstoffverbindungen wie Harnstoff entsteht Ammoniak.
Historisch gesehen kommt dem Ammoniak auch in sofern eine Bedeutung zu, als Wöhler
(1828) nachweisen konnte, dass aus der anorganischen Verbindung Ammoniumcyanat
NH4+OCN- eine organische Verbindung der selben Bruttoformel, nämlich Harnstoff
O=C(NH2)2 (= Kohlensäurediamid) entsteht.
Großtechnisch wird NH3 nach dem Haber-Bosch-Verfahren hergestellt (Abb. 29):
N2 + 3H2 2NH3
H = -92,4 kJ/mol
Da die Bildung des Ammoniaks unter Volumenverminderung erfolgt, kann das Gleichgewicht
durch erhöhten Druck nach rechts verschoben werden. Drucke von 200 bar sind üblich. Die
Reaktion ist exotherm, sodass auch eine Reaktionsführung bei niedriger Temperatur das
Gleichgewicht in Richtung auf die Erhöhung der Ausbeute an Ammoniak beeinflussen sollte.
Tatsächlich ist die Reaktion aber kinetisch gehemmt; bei Raumtemperatur liefe sie unendlich
langsam ab. Man arbeitet daher bei ca. 500 °C, nimmt also Ausbeuteverluste in Kauf. Auch
bei 500 °C bedarf es noch eines Katalysators; hier wird üblicherweise -Eisen dotiert mit
Al2O3 und Alkalimetalloxiden verwendet. Die Ausbeuten liegen bei 17%; nicht umgesetzte
Ausgangsgase werden in den Prozess zurückgeführt. Die chemische Realisierung dieses
Verfahrens in wirtschaftlichem Rahmen gelang Haber, die technische Realisierung ist das
Verdienst Boschs: Wasserstoff diffundiert in den Stahlmantel des Reaktions-Autoklaven ein
und reagiert dort mit dem Kohlenstoff zu Methan, was dazu führt, dass die Autoklaven nach
wenigen Durchläufen bersten. Um dieses Problem zu umgehen, kleidete Bosch den
Stahlmantel mit Weicheisen aus (Weicheisen ist frei von Kohlenstoff), und versah die
Stahlummantelung mit feinen Bohrungen, die eindiffundierenden Wasserstoff entweichen
lassen; vergl. auch Abb. 29 rechts.
63
Abbildung 29. Technische Haber-Bosch Anlage (links) und Details des Autoklaven-Mantels
(Kontaktofens) (rechts).
Gewinnung der Synthesegase:
Stickstoff aus Luft: 4N2 + O2 + 2C (Koks) 4N2 + 2CO
(Generatorgas)
(+ O2 CO2)
H2 aus Methan (Steam-Reforming): CH4 + H2O 3H2 + CO
Historisch gesehen hat auch die Gewinnung von H2 aus Koks und Wasser Bedeutung:
H2O + C H2 + CO
(Wassergas)
Zur Entfernung des Kohlenmonoxids wird dieses zunächst in CO2 überführt:
CO + H2O CO2 + H2 (Konvertierung)
CO2 wird sodann durch Druckwäsche mit Wasser ausgewaschen
Hydrazin wird nach dem Raschig-Verfahren durch Oxidation von Ammoniak mit Hypochlorit
hergestellt:
NH3 + ClO- NH2Cl + OHNH2Cl + OH- „NH“ + H2O + Cl„NH“ + NH3 N2H4
__________________________________________
2NH3 + ClO- N2H4 + Cl- + H2O
(NH2Cl = Chloramin; „NH“ = Nitren)
Von den drei möglichen Orientierungen der beiden NH2-Einheiten zueinander, cis (oder
eclipsed), trans (oder staggered) und gauche, ist die gauche-Konformationen realisiert, bei
der die beiden freien Elektronenpaare unter einem Winkel von 90° stehen. Vergl. hierzu die
Newman-Projektionen, d.i. die Projektion in Blickrichtung entlang der N-N-Achse (vorne
liegende Atome und Orbitale in fett):
H
H
N N
H
H
Hydrazin
H
H
H
H
gauche
H
H
H
H
H
H
H
H
cis (eclipsed) trans (staggered)
N N N
H
StickstoffwasserstoffSäure
64
Stickstoffwasserstoffsäure, eine schwache Säure (KS = 1,2∙10-5), kann z.B. durch
Symproportionierung von Salpetriger Säure und Hydrazin dargestellt werden:
N2H4 + HNO2 HN3 + 2H2O
Die ionisch aufgebauten Alkali- und Erdalkalisalze, Azide, sind unzersetzt schmelzbar. Erst
bei höheren Temperaturen erfolgt Zerfall in das Metall und Stickstoff:
NaN3 Na + 1½N2
Natriumazid findet als Fungizid und Bakterizid (außerhalb des Nahrungsmittelbereiches)
Verwendung. Natriumazid wird ferner in vielen Airbags verwendet: Bei Aufprall werden
Natriumazid-Pellets elektrisch gezündet, die sodann spontan unter Freisetzung von Stickstoff
zerfallen. 65 g NaN3 (1 mol) liefern dabei ca. 33 l N2. Die kovalenten Schwermetallazide, z.B.
Bleiazid PbN3, zersetzen sich beim Erhitzen oder auf Schlag explosionsartig und werden
daher als Initialzünder eingesetzt.
In ammoniakalischen Silbersalzlösungen, die in frisch bereitetem Zustand das komplexe
Kation Diamminsilber [Ag(NH3)2]+enthalten, bildet sich bei längerem Stehen Silbernitrid
Ag3N, das bei Erschütterung detoniert. Ammoniakalische Silbersalzlösungen, wie sie z.B. im
Rahmen der Halogenidnachweise und bei Verspiegelungen anfallen, müssen daher umgehend
durch Ansäuern und Ausfällen des Silbers entschärft werden.
Mit Iod reagiert Ammoniak zu Iodstickstoff, der als Ammoniakat NI3∙NH3 anfällt, und in
feuchtem Zustand gut handhabbar ist. Im trockenen Zustand zerfällt Iodstickstoff dagegen auf
Schlag explosionsartig (Versuch) unter Bildung von Iod, Stickstoff und Ammoniumiodid:
Bildung von Iodstickstoff: 2NH3 + 3I2 NI3∙NH3 + 3HI
Phosphor, Arsen und Antimon bilden dem Ammoniak vergleichbare Phosphane, Arsane und
Stibane, die aber von oben nach unten in der Gruppe zunehmend weniger beständig sind. PH3
ist ein nach Knoblauch riechendes, hochgiftiges Gas, das bei der Hydrolyse von
Calciumphosphid frei gesetzt wird:
Ca3P2 + 6H2O 2PH3 + 3Ca(OH)3
Handelsübliches Carbid CaC2 enthält Verunreinigungen an Calciumphosphiden. Bei der
Zersetzung von Carbid mit Wasser wird daher nicht nur (geruchloses) Acetylen (= Ethin) frei,
sondern auch PH3 und P2H4, die für den „carbidartigen“ Geruch verantwortlich sind. P2H4 ist
im Übrigen selbstentzündlich.
AsH3 und SbH3 spielen beim Nachweis und der Unterscheidung von Arsen und Antimon in
der Analytik eine Rolle (Arsenik-Probe nach Marsh; Arsenik = As2O3): Die zu untersuchende
Probe wird mit Schwefelsäure + Zink versetzt (→ H2). Eventuell vorhandenes Arsen und
Antimon werden dabei in flüchtiges AsH3 bzw. SbH3 überführt und gemeinsam mit H2
ausgetrieben. Die Thermolyse liefert einen schwarzen Arsen- bzw. Antimonspiegel. Im
Unterschied zum Arsenspiegel, der von Hypochlorit durch Oxidation zu Arsenat aufgelöst
wird, ist der Antimospiegel resistent gegen Hypochloritlösung.
As2O3 + 6H2 2AsH3 + 3H2O
AsH3 As + 1½H2
2As + 5ClO- + 3H2O 2AsO43- + 5Cl- + 6H+
65
23.3. Sauerstoffverbindungen des Stickstoffs
(a)
N N O
(b)
N O
N O
O
(c)
O
N
O
N
O
O
O
O
O
N
(d)
O
N
O
O
O
O
(f)
N
O H
(h)
H
N O
O
N
(e)
N
(g) O N
O H
O
H
N
Die wichtigsten Oxide sind Lachgas
(Distickstoffmonoxid N2O (a)), Stickstoffmonoxid NO
(b), Stickstoffdioxid NO2 (c), Distickstofftetroxid N2O4
((d), steht mit NO2 im Gleichgewicht) und
Distickstofftrioxid N2O3 (e). NO und NO2 sind
Radikale; über die Bedeutung dieser beiden Gase im
Zusammenhang mit der Ozonproblematik s. Kap. 10.1.
NO2 ist rotbraun, N2O3 blau gefärbt (Versuch); die
anderen Oxide sind farblos. Stickstoffoxide wirken
generell toxisch und ätzend. NO z.B. ist ein Atemgift.
Geringe Mengen unter physiologischen Bedingungen
gebildetes NO ist allerdings auch ein essentieller
Botenstoff und Regulator verschiedener
physiologischer Abläufe.
NO2 ist das gemischte Anhydrid der Salpetrigen Säure
(f) und Salpetersäure (g):
2NO2 + 2H2O HNO2 + HNO3
H
Lachgas (der Name stammt daher, dass Lachgas euphorisierende und narkotische Wirkung
hat) erhält man durch Erhitzen von Ammoniumnitrat (Versuch):
NH4NO3 N2O + 2H2O
Ammoniumnitrat ist ein wichtiger Stickstoffdünger und wird daher in großen Mengen
produziert. Der unter bestimmten Bedingungen explosionsartige Zerfall in N2O und Wasser
führt gelegentlich zu schweren Unglücken (1921 auf dem Industriegelände in Oppau;
Güterzugunglück 2004 in Nordkorea). N2O unterstützt die Verbrennung (Versuch).
Bei Energieeinwirkung (hohe Temperaturen z.B. im Verbrennungsmotor, elektrische
Entladung bei Gewitter, Höhenstrahlung) auf ein Stickstoff-Sauerstoff-Gemisch – also z.B.
Luft – entstehen „Nitrose Gase“ NOx mit x zwischen 1 und 2. Die Bildung von NO im
Lichtbogen war früher Ausgangsbasis für die Salpetersäure-Herstellung. Heute wird
Salpetersäure nach dem Ostwald-Verfahren durch Verbrennen von Ammoniak gewonnen:
2NH3 + 2½O2 2NO + 3H2O
Die Bildung von NO erfolgt bei 900 °C am Platinnetz-Kontakt. Die Kontaktzeit darf dabei
nur ca. 1/1000 Sekunde betragen, da das bezüglich der Bildung aus den Elementen
endotherme NO metastabil ist und im Kontakt mit dem Katalysator wieder zerfällt:
2NO N2 + O2
H = -180,6 kJ/mol
Salpetersäure erhält man dann durch weitere Oxidation des NO zu NO2 und Einleiten des NO2
unter Luftzufuhr in Wasser:
NO + ½O2 NO2
2NO2 + H2O + ½ O2 2HNO3
Salpetersäure ist eine sehr starke Säure (pKS = -1,44). Sie hat außerdem stark oxidierende
Wirkung, sodass auch solche Metalle von ihr aufgelöst werden, die in der Spannungsreihe auf
der Seite positiver Redoxpotenziale stehen. So wird Silber (nicht aber Gold; daher der
Trivialname „Scheidewasser“ für Salpetersäure) durch HNO3 oxidiert:
HNO3 + Ag + H+ Ag+ + NO2 + H2O
66
Mit Königswasser (aqua regia), einer Mischung aus einem Teil HNO3 und 3 Teilen HCl, wird
auch Gold, „der König der Metalle“ gelöst. Die hohe Oxidationskraft des Königswassers
beruht auf der intermediären Bildung von Nitrosylchlorid NOCl und atomarem Chlor:
HNO3 + 3HCl NOCl + 2Cl + 2H2O
Au + 3Cl + NOCl [AuCl4]- + NO+
Auch Nichtmetalle werden von konzentrierter („rauchender“) Salpetersäure oxidiert.
Holzkohle z.B. verbrennt unter Feuererscheinung zu CO2 (Versuch). Die sich von der
Salpetersäure herleitenden Nitrate sind ebenfalls effiziente Oxidationsmittel. So wird Salpeter
(Natriumnitrat NaNO3) in der Feuerwerkerei und als Bestandteil des Schwarzpulvers (eine
Mischung aus Schwefelpulver, Holzkohlenpulver und Salpeter) eingesetzt. Die Verwendung
von Nitraten als Düngemittel wurde schon erwähnt. Ihre Ausschwämmung aus den Böden
und der Eintrag in Gewässer trägt zu deren Eutrophierung bei. Blattgemüse wie Salat und
Spinat können besonders viel Nitrat aufnehmen, das damit in die Nahrung gelangt. Unter
physiologischen Bedingungen wird Nitrat zu Nitrit reduziert. Nitrit ist toxisch. Zu seinen
toxischen Wirkungen gehört die Blockierung von Hämoglobin und die Bildung kanzerogener
Nitrosamine durch Reaktion mit organischen Aminen:
NO3- + 2e- + 2H+ NO2- + H2O
(Reduktion von Nitrat)
+
R2NH + NO2 + H R2N-NO + H2O
(Bildung von Nitrosaminen)
Dem Nachweis von Nitrat durch die Ringprobe (Versetzen einer wässrigen Lösung der zu
untersuchenden Probe mit Eisen(II)sulfat und Unterschichten dieser Lösung mit
konzentrierter Schwefelsäure) liegt der folgende Reaktionsablauf zugrunde:
(a) H2SO4 + NaNO3 NaHSO4 + HNO3
(b) HNO3 + 3Fe2+ + 3H+ NO + 3Fe3+ + 2H2O
(c) [Fe(H2O)6]2+ + NO H2O + [Fe(H2O)5NO]2+ (mit Fe in der Oxidationsstufe +I
und dem Nitrosylkation NO+ als Ligand)
Die Salze der (schwachen) Salpetrigen Säure HNO2 (KS = 103,3) heißen Nitrite. Mit
Schwefliger Säure bildet Salpetrige Säure Hydroxylamin (NH2OH; (h) im Schema auf der
Vorseite):
HNO2 + 2H2SO3 + H2O NH2OH + 2H2SO4
23.4. Sauerstoffverbindungen des Phosphors
Bei der Oxidation von weißem Phosphor P4 an der Luft erhält man über die Stufe des
Diphosphorhexoxids das Diphosphordecaoxid (meist verkürzt als Phosphorpentoxid P2O5
bezeichnet). Die Lichtemission (das Leuchten) bei Exposition von Phosphor gegenüber Luft
erfolgt beim Übergang von P4O6 zum P4O10. Auf dem Leuchten beruht die Phosphorprobe
nach Mitscherlich (Versuch), die schon in historischen Zeiten gerichtsmedizinische
Bedeutung hatte (weißer Phosphor ist hoch giftig; er ist deswegen, aber auch wegen seiner
Selbstentzündlichkeit an der Luft, mit größter Vorsicht zu behandeln).
O2
O2
+ Licht
67
Phosphorpentoxid ist das Anhydrid der Phosphorsäure H3PO4 (Orthophosphorsäure) bzw.
(HPO3)n (Metaphosphorsäure; nur in Form ihrer Salze bekannt; s.u.). Da Phosphorpentoxid
sehr hygroskopisch ist (und daher auch als Trockenmittel Verwendung findet), bildet sich
Phosphorsäure bereits, wenn das Oxid mit feuchter Luft in Kontakt kommt.
Orthophosphorsäure ist eine dreiprotonige Säure mit den folgenden pKS-Werten: pKs1 = 2,16;
pKs2 = 7,2; pKs3 = 12,3. Entsprechend bildet Phosphorsäure drei Reihen von Salzen:
Dihydrogenphosphate (primäre Phosphate), z.B. NaH2PO4
Monohydrogenphosphate (sekundäre Phosphate), z.B. Na2HPO4
Orthophosphate (tertiäre Phosphate), z.B. Na3PO4 und Phosphorit Ca3(PO4)2.
Beim Erwärmen gehen Monohydrogenphosphate über in Diphosphate, die Salze der
Diphosphorsäure oder Pyrophosphorsäure, H4P2O7:
2Na2HPO4 Na4P2O7 + H2O
Dihydrogenphosphate bilden beim Erhitzen über verschiedene Oligophosphate schließlich
Metaphosphate:
n NaH2PO4 (NaPO3)n + n H2O
Eine isolierbare Zwischenstufe auf dem Weg zu den Metaphosphaten ist das
Pentanatriumtriphosphat Na5P3O10, das wegen seiner guten Komplexierungseigenschaften für
Ca2+ und Mg2+ als Wasserenthärter Verwendung findet und früher dem Waschpulver wegen
eben dieser Eigenschaft zugesetzt wurde. Wegen der Umweltproblematik solcher
Phosphatzusätze (Gewässer-Eutrophierung) werden den Waschmitteln heute nur noch geringe
Mengen Phosphat zugesetzt, die nach wie vor zur Stabilisierung der Micell-Strukturen
(kolloide waschaktive Teilchen mit amphiphilen Eigenschaften) in der Waschflotte
erforderlich sind.
O
O
P
O
O
O
O
P
O
Diphosphat
O
O
P
O
O
O
P
O
O
O
O
P
O
O
O
O
P
O
O
Triphosphat
P
O
P O
O
O
O
O
P
n
O
Metaphosphate
Zum Nachweis von Phosphat spielt die Umsetzung mit Molybdat zu gelbem
Phosphomolybdat PMo6O2412- und dessen Reduktion z.B. mit Hydrazin zu Molybdänblau
(einem Molybdän-Mischoxid mit MoV und MoVI) eine Rolle.
Phosphat nimmt eine zentrale Stellung im Energiestoffwechsel in der belebten Natur ein. Es
dient z.B. der Aktivierung organischer Substrate wie der Glucose. In Form von
Adenosintriphosphat (ATP) wird Energie gespeichert. Bei dessen Hydrolyse zu
Adenosindiphosphat (ADP) und Hydrogenphosphat wird Energie freigesetzt (G ca. -35
kJ/mol). ATP und ADP sind Derivate des Triphosphats: an einen der endständigen
Sauerstoffsubstituenten ist ein Adenosylrest geknüpft.
ATP4- + H2O ADP3- + H2PO4- (in Gegenwart von Mg2+)
24. Die Elemente der Gruppe 14
Diese Gruppe der Valenzelektronenkonfiguration ns2p2 umfasst das Nichtmetall Kohlenstoff,
die Halbmetalle Silicium und Germanium, und die Metalle Zinn und Blei. Gängige positive
Oxidationsstufen sind +IV und +II, wobei die stabilere Stufe bei C, Si, Ge und Sn +IV, bei
Blei +II ist (relativistischer Effekt; s. Kap. 9.1). Während die Stufe +II auch noch bei Sn und
Ge geläufig ist, kommen Silicium(+II)-Verbindungen nur selten vor. Kohlenstoff nimmt eine
Sonderstellung ein.
68
24.1. Kohlenstoff
Vorkommen: Als CO2 in der Luft (0.035 Vol-%, entspricht ca. 1012 t) und gelöst im
Meerwasser (0.005%, entspricht 3∙1013 t), in Form von Carbonaten, z.B. CaCO3 (Kalkstein,
Marmor, Kreide, Schalen von Muscheln und Schnecken, Korallen), CaCO3∙MgCO3
(Dolomit), ZnCO3 (Zinkspat), in elementarer Form (Graphit, Diamant), sowie in Form
organischer Verbindungen in den fossilen „Brennstoffen“ (Erdöl, Erdgas, Stein- und
Braunkohle) und in der von Lebewesen gebundenen Biomasse (ca. 3∙1011 t).
Es gibt drei natürlich vorkommende Isotope: 12C (98,892 %), 13C (1,108 %) und das
radioaktive Nuklid 14C (s. a. S. 12).
Modifikationen:
Die natürlich vorkommenden Modifikationen Graphit und Diamant (s. Abb. 30)
unterscheiden sich u.a. in Dichte, Härte, Farbe und elektrischem Verhalten. Diese
Unterschiede in den Eigenschaften sind eine Folge der unterschiedlichen
Bindungsverhältnisse:
Im Graphit sind die Kohlenstoffatome sp2-hybridisiert; sie bilden ebene Sichten aus
Sechsringen mit kovalenten Bindungen. Die C-C-Abstände innerhalb der Schichten betragen
142 pm, zwischen den Schichten 335 pm. Die Schichten sind, gegeneinander versetzt,
übereinander gestapelt und werden durch van der Waals-Wechselwirkungen (schwache
elektrostatische Anziehungskräfte) zusammengehalten, sind aber entlang der Schichten
verschiebbar. Hierauf beruht die Verwendung von Graphit als Gleitmittel. Die vierten
Elektronen der Kohlenstoffatome sind über die Schicht delokalisiert; hierauf beruhen die
optischen Eigenschaften (metallisch-graue Farbe) und die elektrische Leitfähigkeit des
Graphits entlang der Schichten.
Abbildung 30. Modifikationen des Kohlenstoffs. Links: Graphit; Mitte: Diamant; rechts:
Fullerene und Kohlenstoff-Nanoröhren (von links oben nach rechts unten: C60, C70, C82 mit
eingeschlossenem La-Atom, SWNT = single wall nano tube, MWNT = multiwall nano tube).
Im Diamant sind die C-Atome sp3-hybridisiert (C-C-Abstände 154 pm) und damit ergibt sich
eine tetraedrische Anordung. Das Ergebnis sind gewellte Sechsringschichten mit gleichen
Abständen innerhalb und zwischen den Schichten. Diamant ist extrem hart, nicht mehr
elektrisch leitend und farblos. Durch hohen Druck (105 bar) und Temperatur (1800 °C) geht
Graphit, katalysiert z.B. durch Fe, in Diamant über ( Industriediamanten).
Weitere Modifikation des Kohlenstoffs sind die Fullerene, z.B. C60; vergl. Abb. 30 [die
Benennung erfolgte nach dem Architekten Buckminster Fuller, der in den zwanziger und dreißiger
69
Jahren des vergangenen Jahrhunderts durch seine geodätischen Kuppelbauten mit Fünf- und
Sechsringkonstruktionen berühmt wurde]. Vom sphärischen C60 leiten sich weitere ellipsoide
käfigartige Modifikationen her, sowie Kohlenstoff-Nanoröhren mit einer oder mehreren
Wandungen (single/multi layer carbon nano tubes), die aus Graphit durch Laser-Ablation oder
im Lichtbogen hergestellt werden und u.a. wegen ihrer besonders hohen Zugfestigkeit
potentielle technische Anwendungen haben.
Anorganische Kohlenstoffverbindungen
- Carbide: Viele Metalle reagieren mit Kohlenstoff zu Carbiden. Die Carbide der Alkali- und
Erdalkalimetalle sind ionisch gebaut. Durch Wasser erfolgt Hydrolyse. Je nach dem dabei
gebildeten Kohlenwasserstoff unterscheidet man u.a. Methanide, Acetylide und Propinide
(Allylenide):
Be2C + 4H2O 2Be(OH)2 + CH4 (Methan)
CaC2 + 2H2O Ca(OH)2 + C2H2 (Acetylen = Ethin)
Mg2C3 + 4H2O 2Mg(OH)2 + HCCCH3 (Propin)
Neben diesen ionischen gibt es kovalente Carbide (z.B. SiC, B4C3) sowie legierungsartige
Carbide (z.B. Wolframcarbid, Eisencarbid im Stahl).
- Dicyan und Cyanwasserstoff:
N C C N
a
H C N
b
R C N
c
H N C
d
R N C
e
Dicyan (CN)2 (a), ein Pseudohalogen (d.h. eine Molekül, das den Halogenen vergleichbare
Reaktionen eingeht), erhält man durch direkte Umsetzung von Kohlenstoff mit Stickstoff bei
hohen Temperaturen. Es entsteht auch bei der Reduktion von Cu2+ mit Cyanid gemäß:
5CN- + Cu2+ [Cu(CN)4]3- + ½(CN)2
(vergl. Hierzu die „Maskierung“ von Kupfer bei der Cu/Cd-Trennung in der Analyse)
Cyanwasserstoff HCN (b) entsteht bei der Umsetzung von Dicyan mit Wasserstoff, sowie bei
der Freisetzung aus Cyaniden, z.B. aus Kaliumcyanid (Zyankali):
(CN)2 + H2 2HCN
KCN + HCl KCl + HCN
Die gasförmigen Verbindungen (CN)2 und HCN, sowie die Cyanide sind äußerst toxisch
(Blockierung der Atmungskette). HCN ist eine sehr schwache Säure (Blausäure [zum Namen
s.u.]; pKS = 9,4). Ihre organischen Derivate heißen Nitrile (c). Isocyanwasserstoff (d) ist nicht
beständig; beständig hingegen sind dessen organische Derivate, die Isonitrile (e). Wie die
Halogenide, so bildet auch Cyanid mit Silberionen einen schwerlöslichen Niederschlag, der
sich hier allerdings in überschüssigem Cyanid wieder löst unter Bildung von Dicyanoargentat:
Ag+ + CN- AgCN; AgCN + CN- [Ag(CN)2]Der Name „Blausäure“ für (eine wässrige Lösung von) HCN rührt daher, dass Cyanid mit
Fe2+ plus Fe3+ die intensiv blaue, komplexe Verbindung Berliner Blau bildet, z.B. gemäß
6KCN + Fe2+ K4[Fe(CN)6] + 2K+
K4[Fe(CN)6] + Fe3+ 3K+ + K[FeIIIFeII(CN)6] („Berliner Blau“)
Kaliumhexacyanoferrat(II) (Gelbes Blutlaugensalz) wird dem Kochsalz in geringen Mengen
zugesetzt, um dessen Rieselfähigkeit zu bewahren.
- Cyanat und Thiocyanat
O C N
f
S C N
g
70
Wie Chlor mit Laugen Chlorid und Hypochlorit bildet, so disproportiniert auch Dicyan in
wässrigem alkalischem Milieu, wobei Cyanid und Cyanat (f) gebildet werden:
(CN)2 + 2OH- CN- + NCO- + H2O
In Gegenwart von Ammoniak entsteht dabei Ammoniumcyanat, das durch Erhitzen (in
Gegenwart von Hg2+) in Harnstoff (s.u.) übergeht: Wöhlersche Harnstoffsynthese 1828:
NH4OCN CO(NH2)2
Mit Schwefel setzt sich Cyanid um zu Thiocyanat (Rhodanid (g)):
CN- + S NCSThiocyanat bildet mit Fe3+ einen intensiv rot gefärbten Komplex der Zusammensetzung fac[Fe(SCN)3(H2O)3] (Versuch), eine Reaktion, die zum Nachweis von Fe3+ oder NCSherangezogenen werden kann.
Kohlenmonoxid
Kohlenmonoxid CO ist isoelektronisch zum Cyanid. Es entsteht bei der Verbrennung von
Kohlenstoff und kohlenstoffhaltigen Verbindungen neben Kohlendioxid CO2, sowie im Sinne
einer Symproportionierung aus CO2 und C:
Boudouard-Gleichgewicht: CO2 + C 2CO
H = +172 kJ/mol (für )
Die Bildung von CO aus CO2 und C ist endotherm, die Bildung von CO daher bei h o h e n
Temperaturen begünstigt, während bei niedrigeren Temperaturen Kohlendioxid gebildet wird.
Im Labor stellt man CO aus Ameisensäure und Schwefelsäure her. Der Nachweis erfolgt
durch Weiterverbrennung zu CO2 (Versuch):
HCO2H CO + H2O;
CO + ½O2 CO2
Industrielle Verwendung findet CO u.a. in der Hydroformylierung von Olefinen, in der
Fischer-Tropsch-Synthese und bei der Gewinnung von reinem Nickel (Mond-Verfahren):
Hydroformylierung: R-CH=CH2 + H2 + CO R-CH2-CH2-CHO
Fischer-Tropsch, z.B.: CO + 2H2 CH3OH
Mond-Verfahren: Ni + 4CO Ni(CO)4;
Zersetzung bei erhöhter Temperatur: Ni(CO)4 Ni + 4CO
CO hat eine um das ca. Zweihundertfache höhere Affinität zum Eisen des Hämoglobins als
Sauerstoff und ist damit hochgiftig. 0.025 Vol-% CO in der Atemluft blockieren bereits 25%
des Hämoglobins, 0.5 Vol-% blockieren 90% des Hämoglobins.
Kohlendioxid und Kohlensäure
CO2 hat beträchtliche biologische und geologische Bedeutung. Als eines der wesentlichen
Treibhausgase (neben Methan CH4) kommt dem CO2 im Übrigen erhebliche globale
Bedeutung für die Klimaentwicklung zu.
In der Fotosynthese dient es als Kohlenstoffquelle für die Generierung von Biomasse, z. B.
Glucose:
CO2 + 2H2O* 1/6(C6H12O6) + O*2 + H2O
(* markiert die Herkunft des bei der Fotosynthese freigesetzten Sauerstoffs). Viele
Organismengruppen verwenden Ca2+ und CO2 zum Aufbau von Calciumcarbonat (CaCO3:
Calcit, Aragonit oder Vaterit) in ihren Exo-Skeletten (Muschelschalen, Schneckenhäuser,
Korallen). Gesteinsbildende Mineralien wie Kalkstein (CaCO3) und Dolomit
(CaCO3∙MgCO3) haben geologische Bedeutung. CO2 fällt als Verbrennungsprodukt von
Kohlenstoff und organischen Materialien an (s.o.), bei der Konvertierung, sowie beim
71
Kalkbrennen und beim Versetzen von Carbonaten wie Kalkstein, Soda (Na2CO3) oder
Pottasche (K2CO3) mit Säuren:
Konvertierung: CO + H2O CO2 + H2 H = -41.2 (für )
Kalkbrennen: CaCO3 CaO + CO2
(zur Bedeutung in der Baustoffindustrie s. Kap. 26.3.)
Austreiben mit Säuren: CaCO3 + H2SO4 CaSO4 + CO2 + H2O
CO2 (M = 44 g/mol) ist schwerer als Luft (ca. 29 g/mol) und sammelt sich daher dort, wo es
entsteht, in Bodennähe an (Gärkeller, Grotten; Versuch: Kerzenkasten). Luft mit CO2Gehalten > 5% führt zur Erstickung. Mit Alkali- und Erdalkalimetallen reagiert CO2 zu
Oxiden, Carbonaten und Kohlenstoff:
2Mg + 2CO2 MgO + MgCO3 + C (Versuch: Verbrennen von Magnesium in
Trockeneis. Trockeneis ist festes Kohlendioxid; Festpunkt -78 °C). Man darf somit Brände,
an denen Alkali- oder Erdalkalimetalle beteiligt sind, nicht mit „Kohlensäure“-Löschern (und
auch nicht mit Sand = SiO2, der vergleichbar reagiert)) löschen. Metallbrände werden mit
Salz gelöscht.
CO2 löst sich in Wasser, wobei aber nur 0,2 % zu Kohlensäure umgesetzt werden (CO2 + H2O
H2CO3; die restlichen 99,8 % liegen in Form hydratisierten Kohlendioxids CO2∙aq vor).
Der pKS Wert für das Gleichgewicht H2CO3 + H2O HCO3- + H3O+) beträgt 3,88; der
effektive pKS für das Gleichgewicht {H2CO3 + CO2∙aq} + H2O HCO3- + H3O+ beträgt
6,35. Der Puffer {H2CO3 + CO2∙aq}/HCO3- hat physiologische Bedeutung; er trägt wesentlich
zum konstanten pH-Wert 7,35 des Blutes bei. Freie Kohlensäure ist nur in der Gasphase
existent; sie entsteht durch thermische Zersetzung von Ammoniumhydrogencarbonat:
NH4HCO3 NH3 + H2CO3
Als zweiprotonige Säure bildet Kohlensäure zwei Reihen von Salzen, die Carbonate (wie
Na2CO3) und die Hydrogen- oder Bicarbonate (wie NaHCO3). Wässrige Lösungen von
Carbonaten und Bicarbonaten reagieren alkalisch.
Derivate der Kohlensäure sind die Carbaminsäure (Kohlensäuremonoamid), Harnstoff
(Kohlensäurediamid) und Phosgen (Kohlensäuredichlorid, hoch giftig), ein wichtiger
Grundstoff für die chemische Industrie. Das Ammoniumsalz der Carbaminsäure,
Ammoniumcarbaminat, wird unter dem Handelsnamen Hirschhornsalz als Backpulver
verwendet: Die beim Erhitzen entstehenden Gase NH3 und CO2 treiben den Teig auf:
NH4CO2NH2 2NH3 + CO2
O
O
O
C
C
C
O
OH
Hydrogencarbonat
O
NH2
Carbaminat
H2N
O
NH2
Harnstoff
Cl
C
Cl
Phosgen
24.2. Silizium
Silizium ist ein Halbmetall von zentraler Bedeutung u.a. in der Halbleitertechnik (und damit
auch im Hardwarebereich der EDV), sowie in der Solartechnik. Anorganische
Siliziumverbindungen (z.B. mesoporöse Silikagele) und organische Siliziumverbindungen
(z.B. Silikone) sind Massenprodukte in unterschiedlichsten Anwendungsbereichen, etwa der
Nanotechnologie, Heterogenkatalyse, chemisch widerstandsfähiger Kunststoffe. Elementares
Silizium gewinnt man durch Reduktion von Quarzsand mit Kohlenstoff oder Metallen wie
Aluminium und Magnesium:
SiO2 + 2C Si + 2CO
SiO2 + 2Mg Si + 2MgO
72
Besonders reines Si wird durch Thermolyse von Chlorsilanen wie HSiCl3 (in Gegenwart von
Wasserstoff) oder Silan SiH4 erhalten:
HSiCl3 + H2 Si + 3HCl
SiH4 Si + 2H2
Silan entsteht bei der Hydrolyse von Magnesiumsilicid (Mg2Si; Darstellung durch
Zusammenschmelzen von Mg und Si), Chlorsilan aus SiH4 und HCl:
Mg2Si + 4H2O SiH4 + 2Mg(OH)2
SiH4 + 3HCl SiHCl3 + 3H2
Organische Derivate der Silane, z.B. Alkylchlorsilane, die Ausgangsverbindungen für die
Produktion von Silikonen sind, werden durch die Allred-Rochow-Direktsynthese aus Silizium
und Chloralkanen in Gegenwart von Kupfer hergestellt. Die Hydrolyse liefert die Vorstufen
für die polymeren Silikone (formal Analoga der Ketone), z.B:
Si + 2CH3Cl SiCl2(CH3)2
SiCl2(CH3)2 + H2O 2HCl + 1/n{-Si(CH3)2-O-}n (Silikon)
Siliziumdioxid und Silikate
Mit 27.5 Gew.-% ist Silizium das häufigste Element der Erdrinde. Außer als SiO2 (Quarz und
Quarzvarianten wie Amethyst und Achat, Sand) kommt es in Form verschiedenster Silikate
sowie Alumosilikate vor, die als gesteinsbildende Mineralien und Hauptkomponenten von
Tonen eine umfassende Bedeutung haben. Den Silikaten liegen die – frei nicht existenten –
Orthokieselsäure H4SiO4, Metakieselsäure H2SiO3 und kondensierte Oligo-Kieselsäuren
zugrunde. Die Silikate bilden Insel-, Ketten-, Band- oder Blattstrukturen (vergl. Abb. 31), in
denen SiO4-Tetraeder miteinander eckenverknüft sind. Ein Beispiel für ein Orthosilikat ist der
Olivin MgFeSiO4, der wesentlich am Aufbau des Erdmantels beteiligt ist.
Das Anhydrid der Kieselsäuren, SiO2, weist eine Raumnetzstruktur aus
miteinander verknüpften SiO4-Tetraedern auf (s. die Abb. links), ist also im
Unterschied zum CO2 nicht molekular sondern polymer aufgebaut. Silizium
weicht der Bildung von Doppelbindungen aus. Doppelbindungen sind für
die Elemente ab der 3. Periode grundsätzlich ungünstig
(„Doppelbindungsregel“): Die zweite Bindung einer Doppelbindung ist
eine -Bindung zwischen p-Orbitalen, die senkrecht zur Kern-KernVerbindungslinie
Doppelbindung
C O
C O
(C O)
stehen. Wegen der
geringeren Überlappung
zwischen Si und O (größerer Abstand,
zusätzlicher Knoten im p(Si)-Orbital) ist
keine Doppelbindung
Si
O
der Energiegewinn gering:
73
Inselsilikate
Bandsilikate
Kettensilikate
Blattsilikate
Abbildung 31. Aufbau von Silikaten
Zu den Alumosilikaten gehören die Feldspäte und Zeolithe. Sie leiten sich formal vom (SiO2)n
durch Austausch von Si durch Al und Ladungskompensation mit Alkalimetallionen her: Setzt
man für n = 4 und substituiert eines der Si durch Al, so erhält man einen Feldspat, z.B.
Kalifeldspat K[AlSi3O8]; setzt man für n = 6 und substituiert zwei Si durch Al, so erhält man
einen Zeolith, z.B. K2[Al2Si4O8]. Zeolithe finden wegen ihrer Hohlraumstruktur Verwendung
als Ionenaustauscher und in einer Reihe katalytisch geführter Reaktionen.
Technische Silikate
- Glas: Der Begriff „Glas“ wird ganz allgemein für unterkühlte, ohne Kristallisation erstarrte
(und damit amorphe) Schmelzen verwendet. Im engeren Sinne versteht man hierunter
Gebrauchsgläser wie Fensterglas, Flaschen oder Laborglas. Normales Fensterglas ist ein
Natrium-Calciumsilikat der Bruttozusammensetzung Na2CaSi6O14, das durch
Zusammenschmelzen von Kalk (CaO), Soda (Na2CO3) und Quarzsand (SiO2) hergestellt
wird. Im Duranglas, aus dem widerstandsfähige Laborgeräte hergestellt werden, ist SiO2
teilweise durch Al2O3 und B2O3 ersetzt.
- Tonwaren (Keramiken): Sie bestehen aus Ton, einem schichtförmig aufgebauten
gemischten Aluminium-Siliciumoxid (oder Alumosilikat) der ungefähren Zusammensetzung
AlSiO3(OH)∙nH2O (mit zwischen den Schichten eingelagertem Wasser) und
Magerungsmitteln zur Vermeidung des Schwundes beim Brennen, wie z.B. Sand und fein
gemahlener Feldspat. Ein besonders reiner, weißer Ton, Kaolin, wird zusammen mit Quarz
und Kalifeldspat bei der Porzellan-Herstellung eingesetzt.
- Zement: Zemente wie den Portlandzement erhält man durch durch „Brennen“ von
Gemischen aus gemahlenem Kalkstein (CaCO3) und Tonen bei 1450 °C in Drehrohröfen.
Hierbei bilden sich Calciumsilikate (z. B. Ca2SiO4) und –alumosilikate, sowie
Calciumaluminate (z.B. Ca3Al2O6). Zemente enthalten außerdem zwischen 2 und 5 % Fe2O3.
Das Abbinden von Zement mit Wasser beruht auf der Bildung miteinander verfilzter
Kristallite von Hydraten der Calciumaluminate und -alumosilikate. Durch Einmischen von
Kies erhält man Beton, durch Einbetten von Stahlgeflechten Stahlbeton.
74
24.3. Blei
Blei kommt in der Natur gediegen vor sowie – meist – in Form von Verbindungen als Sulfid
(PbS, Bleiglanz), Sulfat (PbSO4, Bleivitriol), Carbonat (PbCO3, Weißbleierz) und Chromat
(PbCrO4, Rotbleierz). Die Darstellung metallischen Bleis aus Bleiglanz erfolgt nach dem
Röstreduktionsverfahren (1) oder dem Röstreaktionsverfahren (2):
(1)
PbS + 1½ O2 PbO + SO2
PbO + C Pb + CO
(2)
PbS + 1½ O2 PbO + SO2
2PbO + PbS 3Pb + SO2
Die (stabilere) Oxidationsstufe +II liegt z.B. im PbNO3 und den schwerlöslichen
Verbindungen PbCl2, PbSO4 und PbO vor. In der Oxidationsstufe +IV (z.B. PbCl4, PbO2) ist
Blei ein starkes Oxidationsmittel. Z.B. wird Mn2+ von PbO2 zu violettem Permanganat
oxidiert (Versuch):
5PbO2 + 2Mn2+ + 4H+ 2MnO4- + 5Pb2+ + 2H2O
In Menninge, einem Blei(II)-Orthoplumbat(IV) der Zusammensetzung Pb2[PbO4] („Pb3O4“)
liegen beide Oxidationsstufen des Bleis nebeneinander vor. Menninge wurde früher in
Anstrichfarben für den Rostschutz verwendet. Blei(II)-chromat und –molybdat finden
Verwendung als gelbe, organgefarbene und rote Pigmente. Das Weißpigment Bleiweiß (ein
basisches Bleicarbonat) findet wegen Nachdunkelns (Bildung von schwarzem Bleisulfid)
keine Verwendung mehr. Blei und Bleiverbindungen sind hoch-toxisch. Zur Verwendung von
Blei im Bleiakkumulator s. Kap. 19.1.
25. Bor
Bor, das leichteste Element der Gruppe 13, ist das einzige Nichtmetall dieser Gruppe. Mit
anderen Nichtmetallen, z.B. den Halogenen, werden kovalente Verbindungen BX3 gebildet, in
denen Bor sp2-hybridisiert ist, die also trigonal-eben gebaut sind, (a) und (b). In der
mesomeren Grenzform (a) – mit einem Elektronensextett am Bor – ist Bor eine Lewissäure.
Aus dieser Form heraus reagieren trivalente Borverbindungen mit Lewisbasen.
F
F
F
B
F
F
F
F
B
H
B
H
H
F
O
H3C
B
OH
(d)
H
H
B
B
H
H
H
H
H
B
H
(c)
OH
HO
F
B
H
F
(b)
(a)
HO
F
B
F
O
B
(e)
OH
CH3
O
CH3
HO
O B O
O
B OH
B
O B O
HO
2-
(f)
Mit Metallen bildet Bor Boride unterschiedlichster Zusammensetzung. Die Boride z.B. der
Alkalimetalle werden durch Wasser hydrolysiert, wobei Diboran B2H6 ensteht. Im Diboran –
zwei kantenverknüpfte Tetraeder (d.h. Bor ist hier sp3-hybridisiert) – liegen zwei DreizentrenZweielektronen-Bindungen vor (c).
2Na3B + 6H2O B2H6 + 6NaOH
Mit Lithiumhydrid reagiert Boran zu Lithiumboranat (Lithiumborhydrid), einem vielfältig
eingesetzten Hydrierungsmittel:
75
B2H6 + 2LiH 2Li[BH4]
Boroxid B2O3 reagiert mit Wasser zu Borsäure B(OH)3 = H3BO3, deren saurer Charakter
darauf beruht, dass mit weiterem Wasser Tetrahydroxoboranat (d) gebildet wird:
B(OH)3 + 2H2O [B(OH)4]- + H3O+
Von der Borsäure leiten sich die flüchtigen Borsäureester ab, z.B. B(OCH3)3 (e), die mit
grüner Flamme brennen (Versuch).
Borax, Na2B4O5(OH)4∙8H2O, enthält im Anion (vergl. (f)) sp2- und sp3-hybridisiertes B.
Ersatz eines Teils des Kristallwassers durch H2O2 ergibt Perborat, das vielen Waschmitteln als
Bleichkomponente zugesetzt wird.
26. Aluminium, die Erdalkali- und die Alkalimetalle
Diese zu den Gruppen 13 (Al), 2 (Be, Ca, Mg, Sr, Ba, Ra) und 1 (Li, Na, K, Rb, Cs, Fr)
gehörenden Elemente sind typische Metalle, die in den Oxidationsstufen +III (Al), +II
(Erdalkalimetalle) bzw. +I (Alkalimetalle) auftreten. Bedingt durch die vergeleichsweise
geringen Ionisierungsenergien und damit der leichten Überführbarkeit in Kationen haben
diese Elemente die für Metalle charakteristischen Eigenschaften wie metallischen Glanz, gute
thermische und elektrische Leitfähigkeit, die mit zunehmender Temperatur abnimmt, und
Duktilität (Verformbarkeit).
26.1. Die metallische Bindung
In einem einfachen Modell lassen sich die metallischen Eigenschaften im Rahmen des
Elektronengasmodells der metallischen Bindung beschreiben: Durch Abgabe von
Valenzelektronen gehen die Metallatome in kugelige, positiv geladene Ionen (Kationen) über,
die sich in dichter Packung zu einem geordneten, kristallinen Metallionengitter
agglomerieren. Den Zusammenhalt dieses Gitters aus Kationen gewährleistet das aus den
Valenzelektronen gebildete Elektronengas, das im Kationengitter frei vagabundiert. Die
wichtigsten Kristalltypen dieser Gitter sind (vergl. Abb. 32)
- die kubisch dichteste Kugelpackung mit der Schichtenfolge A-B-C-A-B-C(Koordinationszahl 12, Kupfer-Typ)
- die hexagonal dichteste Kugelpackung mit der Schichtenfolge A-B-A-B(Koordinationszahl 12, Magnesium-Typ)
- die kubisch innenzentrierte Packung (Koordinationszahl 8; Wolfram-Typ)
Bei den kubisch bzw. hexagonal dichtesten Packungen beträgt die Raumerfüllung 74%, beim
kubisch-innenzentrierten Gitter nur 68%.
Abbildung 32. Hexagonal dichteste (links), kubisch dichteste (Mitte) und kubisch
innenzentrierte (rechts) Packungen.
76
[Die dichteseten Packungen finden sich auch als „Untergitter“ für Packungen von Anionen in u.a.
ionisch gebauten Halogeniden und Oxiden wieder. Die Kationen besetzen hier dann oktaedrische
oder/und tetraedrische Lücken im Anionengitter]
26.2. Aluminium
Neben den Alumosilikaten (s. Kap. 25) findet sich Aluminium u.a. als Bauxit AlO(OH), dem
Ausgangsprodukt für die Aluminium-Gewinnung. Bauxit enthält Beimengungen an
Eisen(III)oxid-Hydraten (daher die rote Farbe) und SiO2. Die Gewinnung von Al aus Bauxit
erfolgt wie folgt:
1. Abtrennung vom Eisenoxid-Hydrat und SiO2 mittels Natronlauge: Während sich
AlO(OH) in der Lauge löst, verbleiben Eisenhydroxid und SiO2 als unlösliche
Komponenten und kann durch Filtration abgetrennt werden ( Rotschlamm);
vergl. die Eisen-Aluminium-Trennung in der Analyse:
AlO(OH) + NaOH + H2O Na[Al(OH)4]
2. Durch Neutralisation der alkalischen Natriumaluminat-Lösung mit CO2 und
Impfen mit Hydrargillit (Al2O3∙3H2O) wird Aluminiumhydroxid gefällt:
Na[Al(OH)4] + CO2 Al(OH)3+ NaHCO3
3. Überführen des Aluminiumhydroxids in Aluminiumoxid durch Brennen:
2Al(OH)3 Al2O3 + 3H2O
4. Schmelzflusselektrolyse des Aluminiumoxids (Abb. 33) unter Zusatz von
Kaliumhydrogenfluorid KHF2 als Flussmittel, d.h. zur Schmelzpunktsdepression:
Kathodenvorgang: Al3+ + 3 e- Al
Anodenvorgang: O2- ½O2 + 2eDie Kathode – aus Graphit – brennt dabei ab und muss nachgeführt werden:
O2 + C CO2
Daneben wird auch etwas Fluorid oxidiert, das entweichen und mit der
Luftfeuchtigkeit – toxischen und aggressiven (korrosivem) – Fluorwasserstoff
bildet:
F- ½F2 ;
F2 + H2O 2HF + ½O2
Abbildung 33. Schmelzflusselektrolyse von
Aluminiumoxid zur Gewinnung von
Aluminium.
Das Leichtmetall Aluminium findet z.B. im
Flugzeugbau, für Stromleitungen und als
Verpackungsmaterial Verwendung. Da reines
Aluminium sehr oxidationsanfällig ist,
werden entweder Legierungen verwendet,
oder das Aluminium wird elektrisch mit einer
sehr dünnen aber dichten und fest haftenden
Aluminiumoxidhaut überzogen
(„Eloxieren“), die es korrosionsfest macht.
Da Al eine hohe Affinität zu Sauerstoff hat (2Al + 1 ½ O2 Al2O3; H = -1675 kJ/mol), ist
es ein geeignetes Reduktionsmittel zur Gewinnung vieler Metalle und Halbmetalle aus deren
Oxiden: Alumothermische Verfahren (Goldschmidt-Verfahren; Versuch):
3SiO2 + 4Al
3Si + 2Al2O3
Aluminiumchlorid AlCl3 wird in der organischen Synthese als Katalysator bei Friedel-CraftsAlkylierungen und –Acylierungen von Aromaten eingesetzt. In Wasser löst es sich unter stark
77
exothermer Reaktion und Bildung von Hexaquaaluminium-Kationen [Al(H2O)6]3+. Diese
oktaedrisch gebauten Kationen sind Brønstedt-Säuren, die in drei Stufen bis zum
Aluminiumhydroxid reagieren:
[Al(H2O)6]3+ + H2O [Al(H2O)5(OH)]2+ + H3O+
[Al(H2O)5(OH)]2+ + H2O [Al(H2O)4(OH)2]+ + H3O+
[Al(H2O)4(OH)2]+ + H2O [Al(H2O)3(OH)3] (= Al(OH)3) + H3O+
Eine vielfältig Verwendung findende Aluminiumverbindung ist der Alaun, ein Doppelsalz der
Zusammensetzung K[Al(H2O)6](SO4)2∙6H2O.
26.3. Erdalkalimetalle
Der erste Vertreter dieser Gruppe, Beryllium, hat als Kation Be2+ einen ähnlichen
Durchmesser wie Al3+ und ist damit dem Aluminium ähnlich (Schrägbeziehungen im
Periodensystem). So lösen sich die in Wasser schwerlöslichen Hydroxide beider Metalle in
alkalischer Lösung unter Bildung komplexer, tetraedrisch gebauter Anionen auf:
Be(OH)2 + 2OH- [Be(OH)4]2- bzw. Al(OH)3 + OH- [Al(OH)4]Auch Mg(OH)2 ist noch schwerlöslich, bildet aber im Alkalischen keine löslichen Komplexe
mehr. Die Hydroxyde der folgenden Elemente sind zunehmend leichter löslich in Wasser und
zeigen alkalische Reaktion. Das radioaktive Radium, im Jahre 1898 vom Ehepaar Curie aus
der Pechblende UO2 isoliert, zerfällt mit einer Halbwertszeit von 1622 Jahren unter Emission in Radon. 1 t Pechblende enthält etwa 0.14 g Ra. Ra entsteht als Zerfallsprodukt des
Uran-238 in einer Kaskade.
Vorkommen:
Be: Beryll Be3Al2Si6O18
Mg: Dolomit MgCO3∙CaCO3, Carnallit MgCl2∙KCl∙6H2O, Bittersalz MgSO4,
Magnesiumsilikate. Mg ist außerdem Bestandteil des Chlorophylls und spielt eine Rolle bei
der Umwandlung von ATP in ADP.
Ca: Dolomit MgCO3∙CaCO3, Gips CaSO4∙2H2O, Anhydrit CaSO4, Flussspat CaF2,
Kalkstein/Kreide/Marmor/Kalkspat CaCO3, Calciumphosphate (s. beim Phosphor).
Hydroxylapatit ist Bestandteil der Endoskelettstrukturen von Wirbeltieren, Calciumcarbonate
bauen die Exoskelette von Muscheln, Schnecken und Korallen auf.
Sr und Ba: als Sulfate, Carbonate und Fluoride. Beispiele: BaSO4 (Schwerspat), SrSO4
(Coelestin).
Ra: in der Pechblende (s.o.)
Verwendung:
Beryllium, Strontium und Barium sind auf begrenzte Anwendungsbereiche beschränkt (Be
z.B. in Kernreaktoren als Moderator für schnelle Neutronen, Sr und Ba in der Feuerwerkerei
zur Erzeugung roter bzw. grüner Farben). Mg ist Legierungsbestandteil in LeichtmetallLegierungen.
Wirtschaftliche Bedeutung hat vor allem Calciumcarbonat (Kalkstein). Durch Brennen geht
Kalk über in gebrannten Kalk CaO, der mit Wasser zu gelöschtem Kalk Ca(OH)2 umgesetzt
wird. Die Mischung aus gelöschtem Kalk und Sand, Mörtel, finder als Bindemittel in der
Bauindustrie Verwendung. Das Abbinden von Mörtel erfolgt durch Reaktion von Ca(OH)2
mit dem Kohlendioxid der Luft zu Calciumcarbonat:
CaCO3 CaO + CO2
Kalkbrennen
CaO + H2O Ca(OH)2
Kalklöschen
Ca(OH)2 + CO2 CaCO3 + H2O Abbinden
78
Zur Rolle von CaCO3 bzw. CaO im Zement s. das Kapitel „Silizium“.
Die Verwendung von Gips in der Bauindustrie, Medizin und Kunst (Gips-Skulpturen) beruht
auf der Fähigkeit des Halbhydrats (das durch vorsichtiges Erhitzen aus dem Dihydrat erhalten
wird) unter Rückbildung des Dihydrats auszuhärten:
CaSO4∙½H2O + 1½H2O CaSO4∙2H2O
Wasserhärte
Ca2+ und Mg2+ sind Verursacher der Wasserhärte. Diese beiden Ionen bilden mit den
klassischen Seifen – das sind die Natrium- und Kaliumsalze langkettiger Fettsäuren, z.B. der
Stearinsäure C17H35CO2H – schwerlösliche Mg- bzw. Calciumseifen, sodass diese Seifen
„krümeln“ und ihre waschaktiven Eigenschaften verlieren. Je nach Gegenion für Mg2+ und
Ca2+ unterscheidet man zwischen temporärer Härte (Gegenion ist Hydrogencarbonat HCO3-)
und permanenter Härte (Cl-, SO42-, NO3- als Gegenion). Die permanente Härte kann durch
Ionenaustauscher oder Phosphat entfernt werden, die temporäre Härte durch Aufkochen:
Ca(HCO3)2 CaCO3 + CO2 + H2O
Die Bildung von Kesselstein in Wasserkesseln ist hierauf zurückzuführen. Der Vorgang ist
reversibel, d.h. bei Normaltemperatur löst sich Calciumcarbonat in Gegenwart von Wasser
und Kohlendioxid auf. Das führt zu besonders hartem Wasser in bergigen Regionen mit
Kalkformationen. Die Eliminierung der Wasserhärte ist z. B. für industrielle Brauchwässer
(Kühlsysteme) unerlässlich, da es andernfalls durch Abscheidung schwerlöslichen Kalkes zur
Beeinträchtigung der Kühlkreisläufe kommt. Den Waschpulvern sind Zeolithe beigemischt,
die auf Grund ihrer Ionenaustauscheigenschaften das Wasser enthärten und so die Funktion
der waschaktiven Substanzen (Tenside) gewährleisten.
Gemessen wird die Härte in Härtegraden. 1° deutscher Härte (dH) entspricht 1 mg CaO (bzw.
die äquivalente Menge MgO) pro 100 ml Wasser. In Hamburg liegt die Wasserhärte des
Trinkwassers mit 10° dH im mittleren Bereich.
26.4. Alkalimetalle
Lithium (gesprochen Litium [nicht Lizium]) nimmt in dieser Gruppe eine Sonderstellung
insofern ein, als viele seiner Eigenschaften denen des Magnesiums ähnlicher sind
(Schrägbeziehung) als denen der anderen Alkalimetalle. So ist Li2CO3 wie MgCO3 (und im
Unterschied zu den anderen Alkalimetallcarbonaten) schwerlöslich. Mit Sauerstoff bildet Li
ein „normales“ Oxid, nämlich Li2O – wie Mg, aber anders als die schwereren Homologen
(s.u.). Das schwerste Element dieser Gruppe, Francium (entdeckt 1939 durch die Französin
M. Perey) ist radioaktiv. Das längstlebige Isotop, 223Fr, hat eine Halbwertszeit von 22 Min.; es
zerfällt unter-Emission in Radium und unter -Emission in Astatium.
Vorkommen
Li: In Form von Phosphaten und Alumosilikaten, z.B. Spodumen Li[AlSi2O6].
Na: Steinsalz NaCl (als NaCl auch gelöst im Meerwasser), Glaubersalz Na2SO4, Soda
Na2CO3, Salpeter NaNO3, Alumosilikate. Die gewaltigen Salzlagerstätten z.B. der
Nordeutschen Tiefebene und Polens sind durch Eindunsten abgeschlossener Meeresteile und
späterer Überlagerung mit wasserundurchlässigen Sedimenten entstanden.
K: Sylvin KCl, Carnallit MgCl2∙KCl∙6H2O, Alumosilikate.
Rb und Cs: Als Begleiter von Na und K.
Gewinnung und Allgemeines
79
Die Gewinnung erfolgt durch Schmelzflusselektrolyse der Chloride oder Hydroxide oder
auch, im Falle des Natriums, durch Elektrolyse wässriger NaCl-Lösungen nach dem
Amalgamverfahren (s. Abb. 23 in Kap. 15.2.) und Abtrennung des Quecksilbers durch
Abdestillieren.
Die Verbindungen der Alkalimetalle zeigen charakteristische Flammenfärbungen bzw.
Spektrallinien (Li: rot, Na: gelb, K: violett, Rb: rot, Cs: blau) die zum Nachweis
herangezogen werden können und im Falle des Rubidiums und Cäsiums namengebend waren.
Die Reaktivität der Metalle nimmt von oben nach unten zu. Mit Wasser reagieren sie unter
Entwicklung von H2 zu Hydroxiden, die, mit Ausnahme des LiOH (Ähnlichkeit zu Mg(OH)2),
leicht löslich sind:
Na + H2O Na+ + OH- + ½H2
Während diese Reaktion mit Natrium noch vergleichsweise moderat verläuft, setzt sich K
bereits sehr heftig um (Versuche). Rb und Cs reagieren mit Wasser explosionsartig. An der
Luft bilden die Metalle mit dem Luftsauerstoff Oxide (die unter Aufnahme von
Luftfeuchtigkeit weiter zu den Hydroxiden reagieren). Mit Li erhält man herkömmliches
Oxid, mit Na Peroxid, und mit K, Rb und Cs Hyperoxide:
2Li + ½O2 Li2O Oxid
2Na + O2 Na2O2 Peroxid
K + O2 KO2
Hyperoxid
Wegen der beträchtlichen Reaktivität gegenüber Sauerstoff und Wasser werden Natrium und
Kalium unter Petrolether (einem benzinähnlichen Kohlenwasserstoffgemisch) aufbewahrt und
für chemische Versuche in passgerechte Formen unter Petrolether geschnitten (alle
Alkalimetalle sind weich). Beim Umgang mit Na und K ist wegen deren Reaktivität an
feuchter Luft (Entzündungsgefahr) Vorsicht geboten. Zur Entsorgung löst man Metallreste in
Isopropanol und hydrolysiert anschließend mit Wasser:
Na + iC3H7OH iC3H7ONa + ½H2
iC3H7ONa + H2O iC3H7OH + NaOH
Mit Wasserstoff erhält man Hydride (z.B. Li + ½H2 LiH), die im NaCl-Ionengitter
kristallisieren und mit Wasser im Sinne einer Symproportionierung Wasserstoff freisetzen:
LiH + H2O LiOH + H2
Metallisches Natrium wird in bestimmten Kernreaktortypen als Kühlmittel eingesetzt.
Natriumverbindungen wie Na2O2, NaCN, NaNH2 (Natriumamid) finden industrielle
Verwendung in organischen Synthesen, NaOH bei der Seifenfabrikation (Hydrolyse von
Fetten) und zum Aufschluss von Rohzelluloseprodukten, Na2SO4 in der Glas- und
Papierindustrie, Na2CO3 gleichfalls in der Papier- und Glasherstellung, NaNO3 als
Düngemittel und zur Herstellung von Sprengstoff. Kaliumsalze spielen vornehmlich als
Düngemittel eine Rolle.