Eigenschaften von Gasen
Werbung

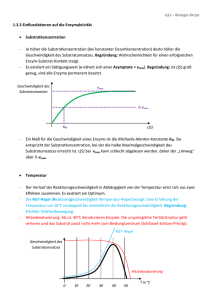
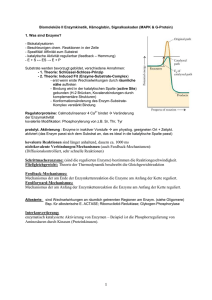
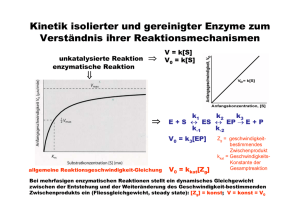
Eigenschaften von Gasen Die Zusammensetzung von Luft Bestandteil Prozentuale Zusammensetzung Volumen Masse Stickstoff, N2 78.09 75.52 Sauerstoff, O2 20.95 23.14 Argon, Ar 0.93 1.29 Kohlendioxid, CO2 0.03 0.05 Die Gasgesetze •Ein Gas füllte jedes Volumen, das ihm zur Verfügung steht, vollständig aus •Verschiedene Gase sind in jedem Verhältnis homogen mischbar •Ein Gas ist eine Ansammlung weit voneinander getrennter Moleküle in chaotischer Bewegung •Gase sind hochkompressibel aufgrund der grossen molekularen Abstände Reaktion auf äussere `Reize`? Druck Def.: † Kraft F p= = Fläche A Einheit: 1Pa = 1kg/(ms2) = 1N/m2 (SI) 1bar = 100 kPa 1atm = 101.3 kPa 1atm = 760 Torr Das Gasgesetze Boyle´sches Gesetz: Volumen, V 1 Vµ p T = const. +Hg V 1/p Charles´ Gesetz : p = const. T V V µT T Experimentell: p µT Zusammengefasst: pV = const. T Avogadro: Das Volumen eines Gases ist bei gegebenem Druck und Temperatur proportional zur Menge der Gasmoleküle, die in der Probe enthalten sind: V µn V Molares Volumen Vm einer Substanz: Vm = n Bei Standardtemperatur und Druck (STP: 273.15K, 1atm), beträgt das Molvolumen der meisten Gase ca: Vm = 22.4 L Ideales Gasgesetz: pV = nRT R = 8.314 J/(mol K) allgemeine Gaskonstante! Beispiel: Airbag gefüllt mit 120 g NaN3 2 NaN3(s) 2Na(l) + 3 N2(g) Reaktion erfolgt bei elektrischer Zündung in ca. 30 Millisekunden. (N2 im Airbag hat einen Druck von 1.25*105 Pa bei 25°C) m(NaN 3 ) 120g n(NaN 3 ) = = = 1.85mol M(NaN 3 ) 65gmol-1 3 n(N 2 ) = n(NaN 3 ) = 2.77mol 2 † n(N 2 )RT 2.77mol * 8.314Jmol-1K -1 * 298K V (N 2 ) = = = 54.9L 5 p 1.25 *10 Pa † Partialdruck In Gasgemischen setzt sich der Gesamtdruck aus den Partialdrucken der einzelnen Komponenten zusammen p = p(A) + p(B) + .... Keine Wechselwirkung zwischen den Teilchen! Bsp: Mischen von 1L des Gases A bei 20 kPa und 1L des Gases B bei 40 kPa in einem Gefäss mit dem Volumen 1L Gesamtdruck des Gasgemisches: 60 kPa! Gemisch besteht aus n(A) Teilchen a und n(B) Teilchen B n(A) Stoffmengenanteil (Molenbruch): x(A) = n(A) + n(B) Partialdruck des Gases A: p(A) = x(A) * p Die kinetische Gastheorie Postulate: • Gase bestehen aus Teilchen mit vernachlässigbarem Volumen (Vteilchen<< Vgesamt) • Die Teilchen sind in ständiger, schneller und geradliniger Bewegung und stossen miteinander und mit der Gefässwand zusammen. Kein Verlust kinetischer Energie. • Die mittlere kinetische Energie hängt von der Temperatur ab • Anziehungskräfte zwischen den Teilchen sind vernachlässigbar) Kinetische Gastheorie und ideales Gasgesetz • N Gasteilchen in einem Würfel der Kantenlänge a • 1/3 der Teilchen bewegt sich in x-Richtung Aufschläge der Gasteilchen gegen die Gefässwände verursachen den Druck des Gases! Kraft die die Teilchen gegen die Wand ausüben? • Kraft F = Impulsänderung pro Zeit ( = 2mv) • Zahl der Stösse eines Teilchens pro Zeiteinheit: 2a/v v mv 2 2mv * = 2a a † N mv 2 F= 3 a F N mv 2 Druck: p = 2 = a 3 a3 † Mit a3 = V und Ekin =mv2/2: pV = 13 Nmv 2 bzw. pV = n 23 N A E kin † 2 3 † † N A E kin = RT Mittlere Geschwindigkeit von Gasteilchen: Ê 3RT ˆ v =Á ˜ Ë M ¯ 1 2 Bei 25°C † [m/s] Reale Gase Zwischenmolekulare Anziehungskräfte (Verflüssigung von Gasen!) pV/RT pV <1 RT Eigenvolumen der Gasmoleküle † † pV >1 RT Van der Waals Gleichung Ê n 2a ˆ Á p + 2 ˜ * (V - b) = nRT V ¯ Ë † Die Geschwindigkeit chemischer Reaktionen ´Kinetik´ Reaktionsgeschwindigkeit rate = † Konzentrationsänderung Zeit D[ X ] rate = Dt Die Geschwindigkeit der meisten chemischen Reaktionen hängt von der Konzentration der Reaktanten ab! (ändert sich mit fortschreitender Reaktion) 2N2O5(g) 4NO2(g) + O2(g) [N2O5] (mol/L) momentane Geschwindigkeit! Tangente Zeit (s) Anfangsgeschwindigkeit = momentane Geschwindigkeit bei t = 0! (keine Produkte, die Sekundärreaktionen durchlaufen können) Anfangsgeschwindikeit Die Anfangsgeschwindigkeiten der meisten Reaktionen hängen von der Anfangskonzentration der Reaktanten ab! Anfangskonzentration an N2O5 vAnfangl= k*[N2O5] k: Geschwindigkeitskonstante Geschwindigkeitsgesetze (momentane Reaktionsgeschwindigkeit in Abhängigkeit der momentanen Konzentrationen der Reaktionsteilnehmer) v = k*[Reaktand]a a = 1: Reaktion erster Ordnung a = 2: Reaktion zweiter Ordnung Das Geschwindigkeitsgesetz und damit auch die Reaktionsordnung muss experimentell bestimmt werden! Sie kann nicht aus der Reaktionsgleichung abgeleitet werden! Häufig findet man komplexere Geschwindigkeitsgesetze: S2O82-(aq) + 3I-(aq) 2SO42-(aq) + I3-(aq) v = k[S2O82-][I-] 1.Ordnung bzgl. S2O82-und 1.Ordnung bzgl. IGesamtreaktionsordnung ist 2! Allg.: v = k[A]a[B]b... Reaktionen 0. Ordnung 2NH3(g) (Pt) N2(g) + 3H2(g) rate Konzentration v = [NH3]0 = konstant! Zeit Zeit Reaktionen 1. Ordnung 2N2O5(g) 4NO2(g) + O2(g) v = k[A] [A]o = Konzentration zur Zeit 0 (bekannt!) [A] = Konzentration zur Zeit t ??? d[ A ] = k [ A] dt d[ A] = -kdt [ A] [A] d[ A ] Ú [ A] = -k Ú dt [A] 0 0 [ A] ln = -kt [ A ]0 t or [ A] = [ A] 0 e -kt ln[A] t Die Halbwertszeit t1/2 einer Substanz ist die Zeit in der ihre Konzentration auf die Hälfte der Anfangskonzentration abgefallen ist. [ A] 0 ln 1 = kt 2 [ A ]0 1 ln 2 t1 2 = k 2 Reaktionen 2. Ordnung 2HI(g) H2(g) + I2(g) r = k[A]2 1 1 = + kt [ A] [ A] 0 1/[A] 1 t1 2 = k [ A] 0 1/[A]0 time Die Temperaturabhängigkeit der Reaktionsgeschwindigkeit Die meisten Reaktionsgeschwindigkeiten nehmen mit steigender Temperatur zu! Arrheniusverhalten Temperaturabhängigkeit der Geschwindigkeitskonstante Ea ln k = ln A RT Frequenzfaktor (Svante Arrhenius, 1889) Aktivierungsenergie lnk Arrhenius Plot Ea/R T-1 (K-1) Potentielle Energie Die Aktivierungsenergie Ea Edukte DH Produkte Reaktionsverlauf Katalyse Potentielle Energie Katalysatoren sind Substanzen, die die Reaktionsgeschwindigkeit erhöhen, ohne bei der Reaktion verbraucht zu werden. Ursprünglicher Pfad, ohne Kat. Reaktionspfad in Gegenwart eines Kat. Reaktionsverlauf Das chemische Gleichgewicht Haber-Bosch Verfahren N2(g) + 3H2(g) 2NH3(g) Reaktionsgeschwindigkeit nimmt mit abnehmender Konzentration an Stickstoff und Wasserstoff ab! Die umgekehrte Reaktion 2NH3(g) N2(g) + 3H2(g) Wird mit zunehmender Ammoniakkonzentration schneller! Dynamisches Gleichgewicht N2(g) + 3H2(g) 2NH3(g) Geschwindigkeiten der Hin- und Rückreaktion sind identisch! Die Gleichgewichtskonstante (Guldberg, Waage, 1864!) 2SO2(g) + O2(g) 2SO2(g) [ SO3 ] 2 Qc = 2 [SO2 ] [O2 ] Reaktionsquotient Qc: Im Gleichgewicht: Qc = const = Kc (Gleichgewichtskonstante) Massenwirkungsgesetz aA + bB cC + dD c d [C] [ D] Qc = a b = Kc [A ] [ B] (molare Gelichgewichtskonzentrationen) Die Grössenordnung von Kc Kc > 103 10-3 < Kc < 103 Kc < 10-3 - Gleichgewicht stark auf Seiten der Produkte - Edukte und Produkte sind im Gleichgewicht in vergleichbaren Konzentrationen vorhanden. - Gleichgewicht stark auf Seiten der Edukte Die Beziehung zwischen Qc und Kc Qc < K c Qc = K c Qc > K c - Reaktion läuft in Richtung Produkte - Reaktion ist im Gleichgewicht - Reaktion läuft (nach links) in Richtung Edukte Gleichgewichtszusammensetzung und Reaktionsgeschwindigkeit vorwärts: H2(g) + I2(g) rückwärts: 2HI(g) 2HI(g) H2(g) + I2(g) Im Gleichgewicht: rforward = kf[H2][I2] rreverse = kr[HI]2 kf[H2][I2] = kr[HI]2 2 kf [HI ] Kc = = kr [H2 ][I 2 ] Gleichgewicht und Reaktionsbedingungen Das Prinzip von Le Chatelier Ein dynamisches Gleichgewicht hat die Tendenz, einer Aenderung der Umgebungsbedingungen entgegenzuwirken. Beispiel N2(g) + 3H2(g) 2NH3(g) Zugabe von überschüssigem Wasserstoff System versucht den Effekt des zusätzlichen Wasserstoffs zu minimieren Reaktion produziert zusätzlichen Ammoniak! Auswirkung einer Druckänderung?? Auswirkung einer Temperaturänderung?? Säuren und Basen Die Arrhenius`sche Definition Arrheniussäure: Arrheniusbase: enthält Wasserstoff und bildet H+ Ionen in Wasser bildet Hydroxid (OH-) Ionen in Wasser Beispiele: HCl NH3 CH3COOH NaOH Entsprechende Reaktionen in Wasser?! NH3 Neutralisation HCl(aq) + NaOH(aq) NaCl(aq) + H2O(l) Allgemeine Form einer Arrhenius Neutralisation Säure + Base Salz + Wasser ‚Netto‘-Gleichung H+ + OH- H2O Die Brønsted-Definition Eine Brønsted-Säure ist ein Protonendonator Eine Brønsted-Base ist ein Protonenakzeptor Eine starke Säure ist normalerweise in Wasser vollständig Ionisiert: HCl(g) + H2O(l) H3O+(aq) + Cl-(aq) Eine schwache Säure ist normalerweise in Wasser nur teilweise Ionisiert: CH3COOH(l) + H2O(l) H3O+(aq) + CH3COO-(aq) Brønsted Säure-Base Gleichgewichte CH3COOH(l) + H2O(l) Säure1 Base2 H3O+(aq) + CH3COO-(aq) Säure2 Base1 Allgemein Säure1 + Base2 Säure2 + Base1!! Konjugate Säure2 ][ Base1 ] [ Qc = [Säure1 ][ Base2 ] Im Gleichgewicht: Kc Autoionisation H2O kann sowohl als Säure als auch als Base reagieren: ‚amphiprotisch‘ H2O(l) + H2O(l) Säure1 Base2 H3O+(aq) + OH-(aq) Säure2 Base1 Autoionisation H O ][OH ] [ = + Qc - 3 2 [H 2O] molare Konzentration des Wassers ª1 [H 2O] = molare Konzentration reinen Wassers Die Autoionisationskonstante (Ionenprodukt) des Wassers Kw = [H3O+][OH-] = 10-14 Die Wasserstoffionenkonzentration und pH pH = -log[H3O+] Allgemein: pX = -logX z.B. pKw = -logKw = 14 pH + pOH = pKw = 14 1 sauer 2 3 Zitronensaft Essig Cola 4 Korrodierend Wein Bier saurer Regen neutral 5 6 7 Milch Blut Speichel Urin Trinkwasser basisch 8 9 Seifen 10 11 ‚Haushalts‘-Ammoniak 12 13 14 Korrodierend Säure-Base-Gleichgewichte In Lösungen von schwachen Säuren und Basen liegen nur ein geringer Bruchteil der Moleküle in ionisierter Form vor. dynamisches Gleichgewicht zwischen ionisierter und nicht-ionisierter Form Die Säuredissoziationskonstante HA(aq) + H2O(l) H3O+(aq) + A-(aq) [H O ][ A ] = K + Im Gleichgewicht: - 3 [ HA] a (acid ionization constant) Die Bedeutung von Ka Je grösser Ka desto stärker die Säure! zunehmende Säurestärke Säuredissoziatiationskonstanten werden i.a. als pKa = -logKa tabelliert Säure pKa Cl3CCOOH 0.52 HIO3 0.77 H3PO4 2.12 HNO2 3.37 HF 3.45 CH3COOH 4.74 H2CO3 6.37 B(OH)3 9.14 HCN 9.31 HIO 10.64 Die Basenkonstante H2O(l) + B(aq) zunehmende Basenstärke Kb = HB+(aq) + OH-(aq) + HB OH [ ][ ] [B ] Base Harnstoff Pyridin Nicotin Morphin Ammoniak Triethylamin pKb 13.9 8.75 5.98 5.79 4.74 2.99 Die Beziehung zwischen pKa, pKb und pKw pKa + pKb = pKw Der pH schwacher Säuren und Basen HA(aq) + H2O(l) H3O+(aq) + A-(aq) [H O ][ A ] = K + - 3 [ HA] pH = pK a + log a A [ ] [ HA] (Henderson-Hasselbalch Gleichung) Pufferlösungen und Indikatoren Puffer: Mischung aus einer schwachen Säure und ihrer konjugierten Base, die den pH einer Lösung stabilisiert, indem sie sowohl Protonen liefern als auch abfangen kann. Bsp: Mischung Essigsäure/Acetat pH Henderson-Hasselbalch-Gleichung! Pufferlösungen vertragen i.a. nur den Zusatz einer begrenzten Menge an Säure und Base! Faustregel: Pufferlösung aus x mol Säure und x mol ihrer konjugierten Base hält den pH-Wert im Bereich pH = pKa ±0.1 stabil, wenn max. 0.115 * x mol Säure oder Base zugesetzt wird. Indikatoren: Schwache Säuren, deren Farbe in Lösung vom pH-Wert abhängt! Indikator Farbe bei niedrigerem pH pH-Umschlagsbereich Farbe bei höherem pH Thymolblau Methylorange Methylrot Lackmus Bromthymolblau Thymolblau Phenophthalein Alizaringelb Rot Rot Rot Rot Gelb Gelb Farblos Gelb 1.2-2.8 3.1-4.5 4.2-6.3 5.0-8.0 6.0-7.6 8.0-9.6 8.3-10.0 10.0-12.1 Gelb gelb Gelb Blau Blau Blau Rot Blauviolett Löslichkeitsgleichgewichte Löslichkeitsgleichgewichte Die Löslichkeitskonstante AgCl(s) Ag+(aq) + Cl-(aq) Kc = + Ag Cl [ ][ ] [ AgCl] [AgCl] = 1: [Ag+][Cl-] = KL Al(OH)3 CaCO3 Bi2S3 AgCl KL = 1*10-33 KL = 8.7*10-9 KL =1*10-97 KL= 1.6*10-10 Ein Beispiel für eine Anwendung (Kinetik/Gleichgewichte): Enzymkatalysierte Reaktionen Ein Katalysator setzt Aktivierungsenergie einer Reaktion herab, indem er einen anderen Reaktionsweg ermöglicht, so dass der geschwindigkeitsbestimmende Schritt der nicht-katalysierten Reaktion vermieden wird. höhere Reaktionsgeschwindigkeit bei derselben Temperatur! Achtung: Gleichgewicht der chemischen Reaktion wird nicht verschoben!! Hin- und Rückreaktion werden um den gleichen Faktor beschleunigt! Homogener Katalysator befindet sich in der gleichen Phase wie die Reaktionsmischung Heterogener Katalysator befindet sich in einer anderen Phase wie die Reaktionsmischung (z.B.: fester Katalysator in Gasphasenreaktion) Spezialfall: Enzyme Enzyme = biochemische Katalysatoren • mit hoher katalytischer Aktivität z.B.: Rohrzucker-Hydrolyse Ea = 107 kJ/mol in Gegenwart von Saccharase Ea = 36 kJ/mol d.h. Reaktionsbeschleunigung um einen Faktor 1012 bei Körpertemperatur • mit hoher Spezifität Enzyme katalysieren nur eine einzige oder eine Serie eng verknüpfter Reaktionen kaum Nebenprodukte! • mit regulierbarer Aktivität Stichworte: - Feedback-Hemmung - Regulatorproteine - kovalente Modifikation - proteolytische Aktivierung Spezialfall: Enzyme Katalytische Aktivität Bildung von Enzym-Substrat (ES) -Komplexen Substrat wird hochselektiv an das aktive Zentrum des Enzyms gebunden! - nicht-kovalente Wechselwirkungen - Schlüssel-Schloss-Prinzip (E.Fischer, 1890) Adsorption des Substrates charakteristische Kinetik (Reaktion mit vorgelagertem Gleichgewicht) Michaelis-Menten-Modell Hypothese: Enzym E und Substrat S stehen mit Enzym-Substrat(ES) Komplex im Gleichgewicht E + S k1 k-1 ES k2 E + P Annahme: Produkt P wird nicht in ursprüngliches Substrat zurückverwandelt Gesucht: Ausdruck der Katalysegeschwindigkeit mit Enzym- und Substratkonzentration, sowie den Geschwindigkeiten der Einzelschritte verknüpft v = k2[ES] [ES]?? Bildung des ES-Komplexes: Zerfall des ES-Komplexes: vf = k1[E][S] vd = k-1[ES] + k2[ES] Stationärer Zustand (Fliessgleichgewicht, ´steady state´) Bildungs- und Zerfallsgeschwindigkeit des ES-Komplexes sind gleich vf = vd d.h. k1[E][S] = (k-1 + k2)[ES] [ES]= [E][S] (k -1 + k 2 )/ k1 Def.: Michaelis-Menten-Konstante KM: K M = [ ES] k -1 + k 2 k1 E ][ S ] [ = KM Konzentration an freiem Enzym: † Konzentration an freiem Substrat: [E] = [E]0 - [ES] [S] ª [S]0 (Annahme: [E]0<<[S]0 !) [ E ] - [ ES])[ S] ( [ ES] = 0 KM oder: † [ E ] 0 [ S] [ ES] = [ S] + K M Reaktionsgeschwindigkeit: Für [E] << [S]: S] [ v = k2 [ ES ] = k 2 [ E ] 0 [S ] + K M - gesamtes Enzym im ES-Komplex gebunden † Bindungsstellen mit Substrat gesättigt - alle maximale Reaktionsgeschwindigkeit : Vmax = k2[E]0 (d.h. Michaelis-Menten-Gleichung: [ S] v = Vmax [S]+ K M (bei [S] = KM v = Vmax/2!) [S] [S]+ K M Æ1 ) Bestimmung von Vmax und KM: Linearisierung! v = Vmax - K M v [S] 1 1 KM 1 = + v Vmax Vmax [S] (Eadie-Hofstee) (Lineweaver-Burk) Bsp: Lineweaver-Burk Diagramm 1 v 1 Vmax - 1 KM KM Vmax 1 [S] Bedeutung von KM und Vmax: • KM = [S] • k2 << k-1: Substratkonzentration, bei der die Hälfte der aktiven Zentren besetzt ist KM = Gleichgewichtskonstante der Dissoziation des ES-Komplex typische Werte für KM: 10-1 - 10-7 M • Vmax = k2 [E]0 kcat Wechselzahl (turnover number) typische Werte für kcat: 1 - 105 s-1 (Zahl der Katalysevorgänge pro sec) Beispiel: Carboanhydrase CO2 + H2O HCO3- + H+ KM = 8 mM kcat = 6*105 s-1 Hemmung von Enzymen - Kontrollmechanismus in biologischen Systemen - Wirkung von Medikamenten und Toxinen irreversible Hemmung Inhibtor ist sehr fest an Enzym gebunden z.B. Wirkung von Nervengasen auf AChE reversible Hemmung schnelle Dissoziation des Enzym-Inhibitor Komplexes a. kompetitive Hemmung: Substrat und Inhibitor werden vom aktiven Zentrum des Enzyms gebunden Enzym kann Substrat oder Inhibitor binden, aber nicht beide [ES] verringert! Kompetitive Hemmung Substrat kompetitiver Inhibitor Enzym Enzym Kinetik: 1 v 1 Vmax mit Inhibitor ohne Inhibitor 1 1 K = + M v Vmax Vmax Ki = 1 [S] [E][I] [EI] Ê [I] ˆ 1 ÁÁ1 + ˜˜ Ë K i ¯ [S] b. nicht-kompetitive Hemmung: Inhibitor und Substrat können gleichzeitig vom selben Enzymmolekül gebunden werden verschiedene Bindungsstellen Substrat Enzym nicht-kompetitiver Inhibitor Nicht-kompetitive Hemmung Kinetik: 1 v mit Inhibitor ohne Inhibitor - 1 KM 1 [S] I Vmax = Vmax [I] 1+ Ki