ENZYME
Werbung

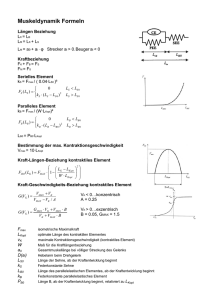
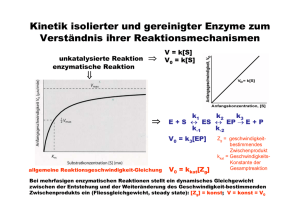
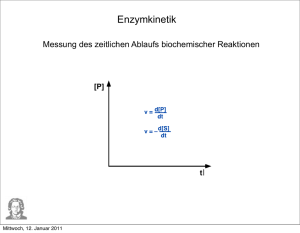
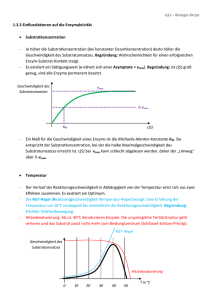
ENZYME Teil 2: Enzymkinetik & Enzymaktivitäts- und Proteinbestimmung Kinetik unkatalysierte Reaktion Wenn [S] >> [P] () S P () z.B. Hydrolyse von Saccharose in saurer Lösung v = -d[S] dt-1 = k x [S] k....Geschwindigkeitskonstante 4 v [S] 3 2 1 0 0 1 2 3 t 4 5 Kinetik unkatalysierte Reaktion Umformung der Grafik: Anfangsgeschwindigkeit der Reaktion gegen die Substratkonzentration linearer Zusammenhang !! 4 v 3 2 1 0 0 1 2 [S] 3 4 Kinetik unkatalysierte Reaktion bimolekulare Reaktion () () A + B C + D () () v = -d[A] dt-1 = -d[B] dt-1 = k x [A] [B] Enzymkinetik enzymkatalysierte Reaktion Anfangsgeschwindigkeit in Abhängigkeit der Substratkonzentration Sättigungsbereich linearer Bereich v prop. [S] Ursache der Sättigungskinetik ?? Ursachen der Sättigung ?? E + S ES EP E + P Wenn [S] >> [E] sind alle Enzymmoleküle mit Substrat besetzt und eine höhere Umsetzungsrate v ist nicht möglich # SÄTTIGUNGSKINETIK # Michaelis-Menten-Gleichung vmax . [S] v = K + [S] M Sonderfall 1 Hohe Substratkonzentration [S] >> KM KM + [S] ~ [S] die Gleichung geht über in v = vmax Sättigung - v unabhängig von [S] Sonderfall 2 [S] << KM Niedrige Substratkonzentration KM + [S] ~ KM v= vmax . [S] vmax = KM KM .[S] Linearität - linearer Anstieg von v mit Zunahme von [S] Ermittlung des Zahlenwertes KM [S] = KM vmax . [S] vmax . [S] v = [S] + [S] = 2 [S] vmax = 2 bei vmax/2 ist die Substratkonzentration gleich KM großer Zahlenwert für KM heißt lockere Bindung von Substrat an das Enzym: geringe Affinität kleiner Zahlenwert für KM heißt feste Bindung von Substrat an das Enzym hohe Affinität Berechnung des KM-Wertes eines Enzyms Lineweaver-Burke-Plot doppelt reziproke Auftragung von v und [S] vmax 5 ausschlaggebend für die maximale Geschwindigkeit ist die Menge an Enzym 4 1/v 3 KM 2 Eigenschaft des Enzyms, die auch ohne Enzymreinigung bestimmt werden kann 1 1/vmax 0 -1 0 1/KM 1 1/[S] 2 3 Verhalten verschiedener Isoenzyme gleicher vmax bei verschiedenen Substrataffinitäten Enzymaktivitätsbestimmung Enzymextraktion Welche Faktoren beeinflussen bzw. beeinträchtigen die Enzymaktivität? Regulative Einflüsse wie Inhibitoren oder Aktivatoren Destruktive Einflüsse Schwermetalle Sauerstoff Phenole und Gerbstoffe Temperatur pH-Wert Proteasen Abhilfen gegen diese Interferenzen ?? Schwermetalle >> Ethylendiamintetraessigsäure (EDTA, Na+) • Komplexierungsreagens (Chelator) mit hoher Affinität zu Schwermetallen, Calcium, Magnesium • nicht für alle Enzyme einsetzbar (Metallkofaktoren) Oxidierendes Milieu >> Dithiothreit (DTT) • Problem der Disulfidbrückenbildung im Enzymprotein • Zusatz von Stoffen mit frei verfügbaren SH-Gruppen (Sulfhydryl-Gruppen), die leicht oxidierbar sind • -SH HS- + 1/2 O2 -> -S-S- + H2O Phenole/Gerbstoffe >> Polyvinylpolypyrrolidon (PVPP) • Phenole bilden mit Proteinen sehr starke H-Brücken aus • Phenoloxidasen oxidieren Phenole zu Quinonen, die mit Proteinen polymerisieren • PVPP bildet ebenfalls starke H-Brücken mit Phenolen aus • zusätzlich Schaffung reduzierender Bedingungen (DTT, Ascorbinsäure) gegen Phenoloxidasen Temperatur >> Arbeiten bei +4°C • Enzymaktivität ist von Erhalt der aktiven Proteinkonformation abhängig • basiert auf hydrophoben Wechselwirkungen im Inneren, die bei höheren Temperaturen reduziert werden • Proteasen pH-Wert >> Pufferung der Medien • Verwendung (an)organischer Puffer (Acetat, Phosphat) • enzymkompatibler, organischer Puffer (sg. Good`sche Puffer) wie Tris, HEPES etc. gegen die Säuredenaturierung Proteasen >> Proteaseinhibitoren (Cocktails) • Metalloproteasen • Serinproteasen • Saure Proteasen • Thiolproteasen EDTA, EGTA Phenylmethylsulfonylfluorid (PMSF) Benzamidin Pepstatin A Leupeptin, Antipain Enzymaktivitätsbestimmung Weitere Aufreinigung # Filtration (Cheesecloth, Miracloth) # Abzentrifugieren # Fällungen (Ammoniumsulfat, Protaminsulfat) # Gelfiltration • Abtrennung störender niedermolekularer Verbindungen • Pufferaustausch # Ultrafiltration (Extraktkonzentrierung) Proteinbestimmung UV-Absorptionsmethoden >> Proteine absorbieren im UV mit 2 Maxima, bei 200 und 280 nm. >> Die Extinktion einer 0.1% Proteinlösung ist 0.4-0.6 bei 280 nm, bzw. 45 bei 200 nm. Probleme Verfälschung durch DNA/ RNA in diesem Bereich >> 2-Wellenlängenmessung Interferenzen durch UV-absorbierende Substanzen sowie Additiva und pflanzliche Inhaltsstoffe nicht denaturierend Proteinbestimmung nach Lowry et al. (1951) >> Unter alkalinen Bedingungen komplexieren Cu2+-Ionen mit Peptidbindungen und werden zu Cu+ reduziert. >> Das Folin-Reagens enthält Phosphomolybdat-Wolframat Mischsäuren, die durch Protein-Cu+-Komplexe [sowie phenolische Seitengruppen von Tyr/Trp, und Cys] zu blauen Mischoxiden reduziert werden z.B. 3H2O . P2O5 . 13WO3 . 5MoO3 . 10H2O 3H2O . P2O5 . 14WO3 . 4MoO3 . 10H2O >> Vermessung bei 750 nm. Proteinbestimmung nach Lowry et al. (1951) # Interferenzen durch Gerbstoffe und Additiva, durch Vorfällung mit Trichloressigsäure/Deoxycholat behebbar # hohe Sensitivität und Linearität für verschiedenste Proteine # höherer Zeit- und Arbeitsaufwand Proteinbestimmung Bradford-Assay >> Protein-Farbstoffkomplex mit Coomassie Brilliant Blue G250 (Schafwollfärberei) >> CBB G250 ist ein synthetischer, amphoterer Farbstoff, der in saurer Lösung in der kationischen braunroten Leukoform vorliegt (Emax 465 nm) >> im Basischen: anionische blaue Form (Emax 595 nm) >> durch WW mit basischen und aromatischen Aminosäuren der Proteine auch im Sauren stabilisiert wird. Proteinbestimmung Bradford-Assay # variable Farbausbeute für verschiedene Proteine # einfach, rasch, sehr empfindlich # nur geringe Interferenzen mit Chemikalien Auf baldiges Wiedersehen bei Sinn und Unsinn von Isotopen stabiler und instabiler Natur







![E + S [ES] E + P Enzyme](http://s1.studylibde.com/store/data/011452180_1-cbdca6d5ea6d6e898aef4e3165d8acb4-300x300.png)