ENZYME
Werbung

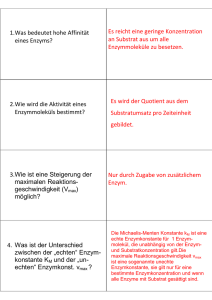
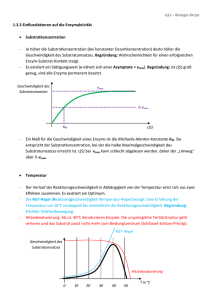
ENZYME Teil 1: Grundlagen und Substratbestimmungen Metastabiler Zustand Beispiel: Glucose-6-Phosphat + H2O • Glucose + Pi [Glc6P] [H20] K= = 1.135 x 10-3 [Glc] [Pi] Gleichgewicht stark auf Seite von Glc + Pi • DGo‘ = - 4.02 kcal mol-1 (Exergonische Reaktion) Reaktion läuft freiwillig ab! ABER: Beim Lösen von Glc6P in Wasser passiert aber scheinbar nichts! Vorrausetzungen für den Ablauf einer Reaktion Zusammenstoß der Moleküle: • • Räumlich richtig ! Energiereich genug ! 25 °C 75 °C Energiezufuhr (Temperaturerhöhung) steigert die Reaktionsgeschwindigkeit ! Aktivierungsenergie notwendige Energie die Energie, die man einem System zuführen muß, damit die Reaktion messbar schnell abläuft Energieprofil (unkatalysierte Reaktion) DGH DGR A+ B DG C+ D Aktivierungsenergie Energieprofil (enzymkatalysierte Reaktion) EAB = Enzym-Substrat Komplex DGH DGR ECD = Enzym-Produkt Komplex A E+A+B E B C E+C+D DG E D A+B+E EAB ECD C+D+E Enzymkatalysierte Reaktionen benötigen geringere Aktivierungsenergie, weil: • • • Näherungseffekt räumlich richtige Ausrichtung der Substrate durch Bindung an Enzym Milieueffekt optimale Bedingungen für Reaktion am Enzym (pH, Solvatisierung, etc) Konformationsstreckungseffekt durch Substratbindung Gestaltänderung des Enzyms (Übergangszustand) Enzymkatalysierte Reaktionen • Gleichgewicht Ein Enzym verändert die Geschwindigkeit einer Reaktion in Richtung des Gleichgewichts. Das Gleichgewicht selbst kann aber nicht verändert werden! Wenn das Gleichgewicht einer Reaktion stark auf einer Seite liegt spricht man von einer nicht umkehrbaren Reaktion. • Hin- und Rückreaktion Ein Enzym kann eine Reaktion in beide Richtungen beschleunigen. Die Reaktion läuft aber immer in Richtung Gleichgewicht ab. Was sind Enzyme Enzyme sind Biokatalysatoren, die die Aktivierungsenergie einer Reaktion herabsetzen können! Unterschiede zu anderen Katalysatoren sind: • • • Spezifität für die Substrate (Gruppen) und Reaktionen Kapazität sehr hohe Effizienz (Beschleunigung bis zu 1014-fach) Regulation Aktivität kann an Umweltbedingungen angepasst werden. Regulation von Enzymen • • Proteinsynthese und Proteinabbau Neusynthese des gewünschten Enzyms; Aktivierung oder Deaktivierung von Genen Konformationsänderung (a) Allosterische Effekte Effektoren (Inhibitoren oder Aktivatoren) binden an regulatorisches Zentrum des Proteins Allosterische Effekte Inhibitor Aktivator Enzym inaktiv Enzym aktiv Regulation von Enzymen • • • Proteinsynthese und Proteinabbau Neusynthese des gewünschten Enzyms; Aktivierung oder Deaktivierung von Genen Konformationsänderung (a) Allosterische Effekte Effektoren (Inhibitoren oder Aktivatoren) binden an regulatorisches Zentrum des Proteins (b) Kompetitive Hemmung „Konkurrenz“ um das aktive Zentrum Chemische Veränderung Phosphorylierungen (Protein-Kinasen) oder Dephosphorylierungen (Phosphatasen) Einteilung von Enzymen 1. Oxidoreduktasen Transfer von H, O oder e- von einem Substrat auf ein anderes 2. Transferasen Transfer von chem. Gruppen zwischen Substraten 3. Hydrolasen hydrolytischer Abbau 4. Lyasen nicht-hydrolytische Spaltung von Bindungen 5. Isomerasen Intramolekulare Umlagerungen 6. Ligasen Knüpfung von kovalenten Bindungen unter ATP-Verbrauch Benennung von Enzymen Beispiel: Fructose + NADP+ 5-Anhydro-D-Fruktose + NADPH + H+ Bezeichnung Fructose 5-Dehydrogenase Fructose:NADP 5-Oxidoreduktase Einteilung EC-Nummer, hier EC 1.1.1.124 (Oxidoreduktase, wirkt an CH-OH Donoren, NAD oder NADP als Akzeptor, Enzymnummer) Analytisches Arbeiten mit Enzymen 1. Enzymbestimmung Bestimmung der Biologische Aktivität Bestimmung der Enzymaktivität Daneben kann auch der Proteingehalt bestimmt werden (z.B. mit Immunologischen Methoden) 2. Substratbestimmung Bestimmung eines Analyten (eines Substrates) mit Hilfe eines Enzyms Das Enzym muß dazu aber käuflich erhältlich sein, oder selbst gereinigt werden. KSubstra onzetraion od.Prukt Enzymreaktionen Z e i t KSubstra onzetraion od. Produkt Enzymreaktionen Z e i t Enzymatische Substratbestimmung Aufbau eines Testsystems 1. Spezifisches Enzym Enzym sollte nur das Substrat umsetzen 2. Co-Substrate wo notwendig muß man Co-Substrate (Co-Enzyme) einsetzen 3. Reaktionsmilieu pH-Wert, Pufferung, Aktivatoren, etc 4. Detektionsmöglichkeit (a) Direkte Messung mittels Extinktion Produkt absorbiert, Substrat nicht (oder umgekehrt); Co-Substrat oder Co-Produkt absorbiert (b) Produktnachweis chemisch 5. Abfangreaktionen sind notwendig wenn das Gleichgewicht ungünstig liegt. Das NAD(P)H-System Die reduzierte Form des Co-Substrats NADH oder NADPH absorbiert Licht im UV-Bereich bei 340 nm. Die oxidierte Form NAD+ oder NADP+ aber nicht. Universell mit Oxidoreduktasen anwendbar! Bespiel: Sorbitbestimmung D-Sorbit + NAD+ SDH D-Fruktose + NADH + H+ Extinko(340nm) DE (NADH) SDH Probe + NAD+ Z e i t Extinko(340nm) Allgemeine Überlegungen 1 A+B C+D Vorraussetzungen • Gleichgewicht der Reaktion liegt auf Seite von C +D • [Cosubstrat] > [Analyt] sonst kann nicht alles an Probe verbraucht werden Z e i t Extinko(340nm) Allgemeine Überlegungen 2 A+B C+D Regeln • v nimmt ab, wenn [c] kleiner wird. Steigung = Geschwindigkeit • Wenn A+B mit C+D im Gleichgewicht, dann v = 0 (kein Netto-Umsatz) • Konzentration (Aktivität) des Enzyms ändert v, hat aber keinen Einfluß auf den Endpunkt (DE bleibt gleich) Z e i t Bestimmung von Glu & Frc 1. Glukose + ATP HK Glukose-6-P + ADP Reaktion nicht umkehrbar (vollständig) Auch Fruktose wird umgesetzt 2. Glu-6-P + NADP+ G6PDH Glukonat-6P + NADPH + H+ Indikatorreaktion 3. Fructose-6-P Zusatzreaktion PGI Glucose-6-P PGI E GDH HK Frc Glu t ENZYME Teil 2: Enzymkinetik & Enzymaktivitäts- und Proteinbestimmung Kinetik unkatalysierte Reaktion Wenn [S] >> [P] () S P () z.B. Hydrolyse von Saccharose in saurer Lösung 4 v [S] 3 2 1 0 0 1 2 3 t 4 5 Kinetik unkatalysierte Reaktion Umformung der Grafik: Anfangsgeschwindigkeit der Reaktion gegen die Substratkonzentration linearer Zusammenhang !! 4 v = -d[S] dt-1 = k x [S] k....Geschwindigkeitskonstante v 3 2 1 0 0 1 2 [S] 3 4 Kinetik unkatalysierte Reaktion bimolekulare Reaktion () () A + B C + D () () v = -d[A] dt-1 = -d[B] dt-1 = k x [A] [B] Enzymkinetik enzymkatalysierte Reaktion Anfangsgeschwindigkeit in Abhängigkeit der Substratkonzentration Sättigungsbereich linearer Bereich v prop. [S] Ursache der Sättigungskinetik ?? Ursachen der Sättigung bei enzymkatalysierten Reaktionen ?? E + S ES EP E + P Wenn [S] >> [E] sind alle Enzymmoleküle mit Substrat besetzt und eine höhere Umsetzungsrate v ist nicht möglich # SÄTTIGUNGSKINETIK # Michaelis-Menten-Gleichung vmax . [S] v = K + [S] M Sonderfall 1 Hohe Substratkonzentration [S] >> KM KM + [S] ~ [S] die Gleichung geht über in v = vmax Sättigung - v unabhängig von [S] Sonderfall 2 [S] << KM Niedrige Substratkonzentration KM + [S] ~ KM v= vmax . [S] vmax = .[S] KM KM Linearität - linearer Anstieg von v mit Zunahme von [S] Ermittlung des Zahlenwertes KM [S] = KM vmax . [S] vmax . [S] v = [S] + [S] = 2 [S] vmax = 2 bei vmax/2 ist die Substratkonzentration gleich KM großer Zahlenwert für KM heißt lockere Bindung von Substrat an das Enzym: geringe Affinität kleiner Zahlenwert für KM heißt feste Bindung von Substrat an das Enzym hohe Affinität Berechnung des KM-Wertes eines Enzyms Vmax Km Lineweaver-Burke-Plot doppelt reziproke Auftragung von v und [S] vmax 5 ausschlaggebend für die maximale Geschwindigkeit ist die Menge an Enzym 4 1/v 3 2 KM 1 1/vmax 0 -1 0 1/KM 1 1/[S] 2 3 Eigenschaft des Enzyms, die auch ohne Enzymreinigung bestimmt werden kann Verhalten verschiedener Isoenzyme gleicher vmax bei verschiedenen Substrataffinitäten Enzymaktivitätsbestimmung Enzymextraktion Welche Faktoren beeinflussen bzw. beeinträchtigen die Enzymaktivität? Regulative Einflüsse > wie Inhibitoren oder Aktivatoren Destruktive Einflüsse > > > > > > Schwermetalle Sauerstoff Phenole und Gerbstoffe Temperatur pH-Wert Proteasen Abhilfen gegen diese Interferenzen ?? Schwermetalle >> Ethylendiamintetraessigsäure (EDTA, Na+ Salz) • Komplexierungsreagens (Chelator) mit hoher Affinität zu Schwermetallen, Calcium, Magnesium • nicht für alle Enzyme einsetzbar (Metallkofaktoren) Oxidierendes Milieu >> Dithiothreit (DTT) • Problem der Disulfidbrückenbildung im Enzymprotein • Zusatz von Stoffen mit frei verfügbaren SH-Gruppen (Sulfhydryl-Gruppen), die leicht oxidierbar sind • -SH HS- + 1/2 O2 -> -S-S- + H2O Phenole/Gerbstoffe >> Polyvinylpolypyrrolidon (PVPP) • Phenole bilden mit Proteinen sehr starke H-Brücken aus • Phenoloxidasen oxidieren Phenole zu Quinonen, die mit Proteinen polymerisieren • PVPP bildet ebenfalls starke H-Brücken mit Phenolen aus • zusätzlich Schaffung reduzierender Bedingungen (DTT, Ascorbinsäure) gegen Phenoloxidasen Temperatur >> Arbeiten bei +4°C • Enzymaktivität ist von Erhalt der aktiven Proteinkonformation abhängig • basiert auf hydrophoben Wechselwirkungen im Inneren, die bei höheren Temperaturen reduziert werden • Proteasen pH-Wert >> Pufferung der Medien • Verwendung (an)organischer Puffer (Acetat, Phosphat) • enzymkompatibler, organischer Puffer (sg. Good`sche Puffer) wie Tris, HEPES etc. gegen die Säuredenaturierung Proteasen >> Proteaseinhibitoren (Cocktails) • Metalloproteasen • Serinproteasen • Saure Proteasen • Thiolproteasen EDTA, EGTA Phenylmethylsulfonylfluorid (PMSF) Benzamidin Pepstatin A Leupeptin, Antipain Enzymaktivitätsbestimmung Weitere Aufreinigung # Filtration (Cheesecloth, Miracloth) # Abzentrifugieren # reversible Fällungen (Ammoniumsulfat, Protaminsulfat) # Gelfiltration • Abtrennung störender niedermolekularer Verbindungen • Pufferaustausch # Ultrafiltration (Extraktkonzentrierung) Proteinbestimmung UV-Absorptionsmethoden >> Proteine absorbieren im UV mit 2 Maxima, bei 200 und 280 nm. >> Die Extinktion einer 0.1% Proteinlösung ist 0.4-0.6 bei 280 nm, bzw. 45 bei 200 nm. Probleme Verfälschung durch DNA/ RNA in diesem Bereich >> 2-Wellenlängenmessung Interferenzen durch UV-absorbierende Substanzen sowie Additiva und pflanzliche Inhaltsstoffe nicht denaturierend Proteinbestimmung nach Lowry et al. (1951) >> Unter alkalinen Bedingungen komplexieren Cu2+-Ionen mit Peptidbindungen und werden zu Cu+ reduziert. >> Das Folin-Reagens enthält Phosphomolybdat-Wolframat Mischsäuren, die durch Protein-Cu+-Komplexe [sowie phenolische Seitengruppen von Tyr/Trp, und Cys] zu blauen Mischoxiden reduziert werden z.B. 3H2O . P2O5 . 13WO3 . 5MoO3 . 10H2O 3H2O . P2O5 . 14WO3 . 4MoO3 . 10H2O >> Vermessung bei 750 nm. Proteinbestimmung nach Lowry et al. (1951) # Interferenzen durch Gerbstoffe und Additiva, durch Vorfällung mit Trichloressigsäure/Deoxycholat behebbar # hohe Sensitivität und Linearität für verschiedenste Proteine # höherer Zeit- und Arbeitsaufwand Proteinbestimmung Bradford-Assay >> Protein-Farbstoffkomplex mit Coomassie Brilliant Blue G250 (Schafwollfärberei) >> CBB G250 ist ein synthetischer, amphoterer Farbstoff, der in saurer Lösung in der kationischen braunroten Leukoform vorliegt (Emax 465 nm) >> im Basischen: anionische blaue Form (Emax 595 nm) >> durch WW mit basischen und aromatischen Aminosäuren der Proteine auch im Sauren stabilisiert wird. Proteinbestimmung Bradford-Assay # variable Farbausbeute für verschiedene Proteine # einfach, rasch, sehr empfindlich # nur geringe Interferenzen mit Chemikalien