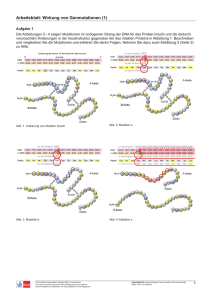
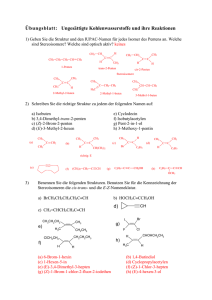
Grundlagen der Organischen Chemie
Werbung

Gr u n dla g e n de r O rg a ni sc he n C he mi e Prof. Martens Vorlesung zu den GRUNDLAGEN DER ORGANISCHEN CHEMIE Wintersemester 2003 / 2004 Universität Oldenburg O OH H H H3C H HH H OH HH H H H H H H CH3 H H O CH3 H C O O H H H Cl H3C H CH3 Mitschrift verfasst von Mika Nashan Letzter Stand: 03. Juli 2004 1. korrigierte Fassung -1- Gr u n dla g e n de r O rg a ni sc he n C he mi e Inhaltsverzeichnis 0. Vorbemerkungen ................................................................................. 2 1. Einführung .......................................................................................... 3 1.1 Historische Entwicklungen ................................................................ 3 1.2 Allgemeine chemische Grundlagen .................................................... 4 1.3 Stereochemie .................................................................................... 6 2. Chemie der organischen Verbindungen .............................................. 10 2.1 Alkane (Parafine) ........................................................................... 10 2.2 Halogenalkane und Alkohole ........................................................... 16 2.3 Alkene ........................................................................................... 23 2.4 Hochmolekulare Stoffe (Makromoleküle ) ......................................... 44 2.5 Alkohole ........................................................................................ 49 2.6 Ether ............................................................................................. 58 2.7 Schwefelanaloga von Alkoholen und Ethern ..................................... 60 2.8 Epoxide (Oxirane) .......................................................................... 62 2.9 Alkine ........................................................................................... 65 2.10 Aromaten ..................................................................................... 68 2.11 Aldehyde & Ketone ....................................................................... 79 2.12 Carbonsäuren und deren Derivate ................................................... 91 2.13 Carbanionen I .............................................................................. 101 2.14 Amine ......................................................................................... 103 2.15 Phenole ....................................................................................... 111 2.16 α – β ungesättigte Carbonylverbindungen ...................................... 113 2.17 Carbanionen II ............................................................................ 116 2.18 Zucker / Kohlenhydrate ................................................................ 120 2.19 Aminosäuren , Peptide und Proteine ............................................... 123 A1 - Index ............................................................................................ 128 -2- Gr u n dla g e n de r O rg a ni sc he n C he mi e 0 . Vo r b e m e r k u n g e n Folgende Mitschrift entstand im Verlauf der Vorlesung „Grundlagen der Organischen Chemie“, vorgetragen von Herrn Prof. Martens an der Universität Oldenburg im Wintersemester 2003 / 2004. Sie umfasst praktisch alle Tafelanschriften sowie einige von mir persönlich hinzugefügte Kommentare, die allerdings selten mehr als mündliche Erläuterungen seitens des Professors enthalten. In einigen Fällen habe ich mich zudem entschlossen, eng verwandte Absätze zusammenzufassen bzw. die Gliederung von der zeitlichen Abfolge in der Vorlesung abzukoppeln. Obwohl ich bei der Digitalisierung auf einige Fehler in meiner handschriftlichen Abschrift aufmerksam wurde, können sich immer wieder einige Fehler eingeschlichen haben bzw. manches habe ich bestimmt auch wieder übersehen. Somit kann ich für die (absolute) Richtigkeit dieser Notizen keine Gewähr geben. In Kooperation mit Herrn Prof. Martens habe ich diese Mitschrift durchgesehen und mehrmals korrigiert. An einigen Stellen wurden zudem kleine Veränderungen vorgenommen, die sich allerdings nicht auf den Inhalt auswirken. Besonderer Dank gilt natürlich in aller erster Linie Herrn Prof. Martens für sein Engagement und der interessanten Gestaltung seiner Vorlesung, sowie für seine Mithilfe bei der Korrektur dieser Mitschrift. Weiterhin möchte ich mich bei meinen Kommilitoninnen und Kommilitonen bedanken, die mich bei dieser Aufgabe unterstützten, indem sie ihrerseits mich auf Fehler aufmerksam machen oder auch meine Abwesenheit bei der Vorlesung durch ihre Mitschriften ermöglicht haben. Bei der Erstellung der Mitschrift benutzte ich unter anderem ISISTM/Draw 2.5 von MDL. Mika Nashan, im Juli 2004 -2- Gr u n dla g e n de r O rg a ni sc he n C he mi e 1. Einführung 1.1 Historische Entwicklungen Im 19. Jahrhundert beschreibt Berzelius die organischen Stoffe als nur durch die Natur erzeugbar, ermöglicht durch die sog. Lebenskraft („vis vitalis“). Die organische Chemie ist somit die Chemie der lebenden Materie. 1827 gelingt Wöhler die Harnstoffsynthese, und damit auch der Beweis, dass organische Stoffe sehr wohl chemisch herzustellen sind: O + NH4 O C N H2N C NH2 Heute versteht man unter der organischen Chemie die Chemie der Kohlenstoffverbindungen, die neben Kohlenstoff auch noch aus weiteren Elementen wie H, O, N, P, S, Halogenen, usw. bestehen (können). Unterschiede zwischen der Anorganischen Chemie (AC) und der Organischen Chemie (OC) Anorganische Chemie Organische Chemie Anorganische Stoffe sind beständig gegenüber hohen Temperaturen Organische Stoffe hingegen zersetzen sich bei Temperaturen über 300°C Meist erfolgen zwischen anorganischen Stoffen Ionenreaktionen, die deshalb recht schnell erfolgen Die Reaktionen sind meist langsamer Natur, und -3- der vorherrschende Bindungstyp ist der kovalente. Gr u n dla g e n de r O rg a ni sc he n C he mi e 1.2 Allgemeine chemische Grundlagen 1.2.2 Hybridisierung des Kohlenstoffs Das Element Kohlenstoff C weist 4 Valenzelektronen auf: 12 6 C : He 2s 2 2p 2 4e- 1. sp 3 - Hybridisierung Das Kohlenstoffatom bildet hierbei mit seinen 4 Substituenten einen Tetraeder, wie er bei den Alkanen z.B. auftritt. H H H 2. sp 2 - Hybridisierung Es verbinden sich zwei p-Orbitale von zwei Kohlenstoffatomen zu einer π-Bindung, die zusammen mit der σ-Bindung eine planare Stellung ergibt. Dies ist z.B. bei Alkenen zu beobachten. 3. sp - Hybridisierung Bei der sp-Hybridisierung binden sich jeweils zwei p-Orbitale pro CAtom zu insgesamt zwei π-Bindungen, die zusammen eine lineare Koordination ergeben. Als Beispiel seien hier die Alkine zu nennen. H H H H H H C C H Beispiel: sp sp2 Alle weiteren Kohlenstoffatome sind sp3-hybridisiert. 1.2.3 Spaltung kovalenter Bindungen Homolyse A B A H3C Cl H3C Heterolyse + + B Cl + A B A H3C Cl H3C + B + + 1.2.4 Bindungspolarität H3C + H3C Br + C O H3C O + Essigsäuremethylester -4- Cl Gr u n dla g e n de r O rg a ni sc he n C he mi e 1.2.5 Isomerie Isomere sind Verbindungen, die zwar dieselbe Summenformel besitzen, aber unterschiedlich aufgebaut sind, also sich in ihrer Strukturen unterscheiden. Bis auf die Ausnahme der Spiegelbildisomerie, sind die physikalischen und chemischen Eigenschaften von isomeren Verbindungen verschieden. 1. Strukturisomerie Beispiel für die Summenformel C2H6O: H3C CH2 OH H3C O CH3 Ethanol Kp = 78 °C Dimethylester Kp = -24 °C 2. Stellungsisomerie CH3 CH3 CH3 CH3 CH3 CH3 CH3 H3C 3. cis / trans – Isomerie (E / Z – Isomerie) E = entgegen / Z = zusammen 1,2 Dibromethen H H H Br Br Br Br H cis (Z) H H H CH3 COOH CH3 trans (E) COOH H cis (Z) trans (E) 4. Spiegelbildisomerie Hierbei gleichen sich alle physikalischen (z.B. Siede- und Schmelzpunkte, NMR-Spektrum uvm.) und chemischen Eigenschaften mit Ausnahme der optischen Aktivität (Drehung des polarisierten Lichtes) und den physiologischen Eigenschaften . (Siehe 1.3.1) 1.2.6 verschieden substituierte Kohlenstoffatome H H C H H H H H H H C C C C H H H H H Primäres C-Atom sekundäres C-Atom -5- H H C C H C H HH tertiäres C-Atom Gr u n dla g e n de r O rg a ni sc he n C he mi e 1. 3 Stereochemie 1.3.1 Enantiomere Bei folgender Reaktion entstehen zwei Produkte, die sich nur in ihrer räumlichen Struktur unterscheiden: HH H Br H C BrH H3C H3C 3 Br2 * * + . h HH CH3 HH CH3 H H CH3 n-Butan * C-Atom mit vier verschiedenen Substituenten (assymetrisches C-Atom, stereogenes Zentrum) Die beiden Produkte entstehen im Verhältnis 1:1, und bilden als 1:1 Gemisch eines Enantiomerenpaares ein Racemat. Enantiomere verhalten sich wie Bild und Spiegelbild, ähnlich wie rechte und linke Hand. Sie haben identische physikalische Eigenschaften mit Ausnahme der optischen Aktivität . D.h., sie drehen den Winkel des polarisierten Lichtes um denselben Betrag, aber in entgegen gesetzter Richtung. Dreht das eine Enantiomer das Licht im Uhrzeigersinn, so bezeichnet man es als rechtsdrehend und wird (+)-Enantiomer genannt. Das andere linksdrehende Enantiomer nennt man dementsprechend (-)-Enantiomer. Auch das Verhalten in chemischen Reaktionen ist dasselbe, allerdings gilt dies nicht bei Reaktionen mit Stoffen, die ihrerseits Enantiomere sind, wodurch auch Unterschiede in den physiologischen Eigenschaften , wie im Geruch und Geschmack, bedingt werden. 1. Beispiele Enantiomer A H * R'' Br R' H3C CH3 Enantiomer B H * R'' Br R' H3C CH3 OH * HO H H CH3 CH3 COOH H3C * COOH * H NH2 H L-Valin (bitter) Naturstoff, Aminosäure * NH2 CH3 D-Valin (süß) -6- Gr u n dla g e n de r O rg a ni sc he n C he mi e 2. R, S – Nomenklatur (CIP-Nomenklatur nach Cahn, Ingold und Prelog) Mit den Substituenten a, b, c und d in absteigender Priorität ergibt sich folgendes Bild: * c * c b d c Blickrichtung c * b d a Blickrichtung a a * a b b links rechts (S)-Konfiguration Die Abfolge verläuft entgegen dem Uhrzeigersinn, also linksherum. (R)-Konfiguration Die Abfolge verläuft mit dem Uhrzeigersinn, also rechtsherum. Sinister (lat.) = links Rectus (lat.) = rechts Festlegung der Prioritäten der Substituenten am stereogenen Zentrum : 1. Regel Hohe Ordnungszahl vor niedriger Ordnungszahl d H c * Cl I Br a b a I * c Cl Br b rechts 2. Regel Bei gleicher Ordnungszahl geht man die Kette entlang, bis ein Unterschied erkennbar wird. CH3 CH2 F CH2 CH3 CH2 CH 2 O CH3 CH3 CH 2 CH3 3. Regel ‚schwere’ Isotope vor leichten: DH 13 C 12 C -7- Gr u n dla g e n de r O rg a ni sc he n C he mi e 1.3.2 Diastereomere Diastereomere weisen neben einem stereogenen Zentrum noch mindestens ein weiteres auf: H H H H Cl 2 * H3C C * C CH3 H3C C * C CH3 h. Br H Br Cl Racemat Aus dem Racemat mit seinen beiden Enantiomeren in der (R)- und der (S)-Konfiguration bilden sich bei Halogenierung also vier Stereoisomere in den Konfigurationen (R)-(R), (R)(S), (S)-(R) und (S)-(S). Diastereomere weisen unterschiedliche chemische und physikalische Eigenschaften auf. Die Anzahl der Stereoisomere einer Verbindung berechnet sich mit der Formel: ΣStereoisomere 2n n Anzahl der stereogenen Zentren 1. Beispiele a) H3C R Cl R Br C C H H CH3 Enantiomere Diastereomere H3C R S H Br C C Cl H CH3 Cl S S CH3 H C C Br H3C H Diastereomere Enantiomere H S R CH3 Cl C C Br H H3C b) Br H Br R H R Enantiomere Cl Cl Br Br Enantiomere S H S Diastereomere R Cl H H Diastereomere S S Cl H H H -8- R Gr u n dla g e n de r O rg a ni sc he n C he mi e c) H Br Br R H S Enantiomere R Br S H H Diastereomere H Diastereomere meso - Form H S Br Br ' = 0° H H R = 0° R Br S Br Br ' = 0° Die beiden unteren Moleküle besitzen eine innere Symmetrie, d.h. eine Spiegelachse. Sie sind daher nach außen optisch innaktiv, haben also einen spezifischen Drehwert [α] = 0. So besitzen zwei gleich substituierte Moleküle mit zwei stereogenen Zentren nur 3 Stereoisomere. 2. Racematspaltung einer Säure R,S -Säure R -Amin Salz aus R -Säure R -Amin 1. Trennung durch fraktionierende Kristallisation 2. Zugabe einer Mineralsäure R -Amin H+ R -Säure Salz aus S -Säure R -Amin S -Säure Beispiel: Ph COOH S S H OH + H2N OH R H Ph COOH COO- Ph R H H3N + R OH Ph H CH3 Ph H CH3 Diastereomere OH H R Ph COO- H3N + R Ph H CH3 Die entstehenden Salze sind Diastereomere und können dementsprechend mit chemischen und physikalischen Methoden getrennt werden. -9- Gr u n dla g e n de r O rg a ni sc he n C he mi e 2 . C h e m i e d e r o r g a n i s c h e n Ve r b i n d u n g e n Kohlenwasserstoffe Aromaten1 Aliphaten Alkane Alkene H Alkine Cycloaliphate H H C C CH3 H3C CH2 CH3 H Propan CH3 Propen Propin Cylopentan Benzol Cn H 2 n 2 , n 2.1 Alkane (Parafine) 2.1.1 Beispiele Summenformel Alkan Anzahl der Strukturisomeren CH4 Methan (Erdgas) 1 CH3 CH3 Ethan 1 CH3 CH 2 CH3 Propan 1 CH3 CH2 2 CH3 Butan 2 CH3 CH2 3 CH3 Pentan 3 CH3 CH2 4 CH3 Hexan 6 CH3 CH2 6 CH3 Oktan 18 CH3 CH2 18 CH3 Eicosan 366.319 1 Aus den Bezeichnungen Aliphaten und Aromaten leitet sich auch der Name eines bekannten Erdöllieferanten ab. - 10 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.1.2 Beispiele zu Strukturisomeren 1. Ethan C 2 H 6 109° H H H H3C CH3 C C H H 109° σ-Bindung (frei drehbar) H Sägebockschreibweise H H H H H H H H H H H H Newman-Projektion H HH HH H HH H verdeckte Konformation (eclipsed) H H H gestaffelte Konformation (stagged) 2. Propan C3H8 H H H CH3 H H 4. Butan C4 H10 H3C CH CH3 CH3 H3C CH2 CH2 CH3 n-Butan (normal) Kp = 0 °C i-Butan (iso) Kp = -12 °C 5. Pentan C5 H12 H3C CH2 CH2 CH2 CH3 n-Pentan CH3 CH3 H3C CH2 CH i-Pentan - 11 - CH3 CH3 C CH3 CH3 neo-Pentan Gr u n dla g e n de r O rg a ni sc he n C he mi e 6. Hexan C6 H14 CH3 H3C CH2 4 CH3 H3C CH H3C CH2 C CH3 n-Hexan H3C CH2 CH2 CH CH2 CH3 CH3 CH3 CH3 CH3 H3C CH2 CH CH CH3 CH3 CH3 2.1.2 Cycloalkane Struktur Name CH2 H2C Cyclopropan CH2 Cyclobutan Cyclopentan Cyclohexan CH3 CH3 1,1-Dimethylcyclobutan Methylcyclopropan CH3 Darstellungen des Cyclohexans Sesselkonformationen H H H H H H H H H Umklappen H H H H H H H H H H H H H H H axiale H-Atome / equatoriale H-Atome Beim Umklappen werden aus axialen (equatorialen) H-Atomen equatoriale (axiale) H-Atome. - 12 - Gr u n dla g e n de r O rg a ni sc he n C he mi e Die equatoriale Stellung wird von (größeren) Substituenten bevorzugt: CH3 Umklappen euquatorial CH3 axial Wannenkonformation (energiereicher) Twistkonformation 2.1.4 Nomenklatur nach UIPAC (International Union of Pure and Applied Chemistry) Alkan Alkylrest Methan Methyl Ethan Ethyl n-Propan n-Propyl Beispiel CH3 Br (Mono-) Brommethan CH3 CH 2 - OH Ethanol CH3 CH2 2 Cl (1-) Chlorpropan i-Propan i-Propyl H3C CH Cl CH3 2-Chlorpropan Butan Butyl CH3 CH2 3 SH (Butanthiol) - 13 - Gr u n dla g e n de r O rg a ni sc he n C he mi e Beispiele zur Nomenklatur: F F Br 3 H3C6 CH 2 CH 5 4 5 CH CH 2 Cl CH 2 CH 2 CH 1 2 3 CH 3 1 Cl 2-Brom-3-chlor-4-fluorohexan CH 2 CH 4 Br 5-Brom-1-chlor-3-fluorohexan 2.1.5 Labormethoden zur Alkansynthese 1. Hydrierung von Alkenen H H H H H2 Pt 2. Hydrolyse von Grignard-Verbindungen R Hal Mg R MgHal R MgHal R H Mg OH Hal H2O 3. Reduktion mit naszierendem Wasserstoff R Hal Zn H + R H Zn 2+ Hal- 4. Reaktion m it Organometallverbindungen + - -+ R Hal R Li R R LiHal 5. Wurtz-Synthese Na 2 R Hal R R 2 NaHal gefährlich 2.1.6 Reaktionen der Alkane Die Alkane sind im Allgemeinen reaktionsträge. 1. Verbrennungsreaktion (wichtigste Reaktion) CH4 CH4 + O2 Licht- O2 + bogen CO2 H C C H Alkin + + H2O CO + H2O 2. Photochlorierung von Methan CH4 H2C + Cl2 h. - HCl Cl Cl2 Cl h Dichlormethan CH 3 6 h. H3C Cl - HCl Monochlormethan Cl HC Cl2 . Cl Cl Cl h. Cl Cl Cl Tetrachlormethan Trichlormethan - 14 - Gr u n dla g e n de r O rg a ni sc he n C he mi e Es folgt der Reaktionsmechanismus der Radikalkettenreaktion : h. Kettenstart Cl2 Cl + Cl Cl + CH4 CH3 + CH3 + Cl2 Cl Cl + Cl Cl HCl CH3 + Cl Cl2 + CH3 Cl CH3 + CH3 Kettenfortpflanzungsreaktionen Kettenabbruchsreaktionen CH3 CH3 CH3 3. Sulfochlorierung mit Cl 2 / SO 2 h. Cl2 Cl + R R + SO2 Cl + Cl H R + HCl R SO2 R SO2 + Cl2 RSO2Cl + Cl Cl + Cl Cl + R Kettenfortpflanzungsreaktionen Kettenabbruchsreaktionen Cl2 SO2 O . R SO2 Cl R S Cl Sulfonsäurechlorid Kettenstart RSO2Cl O - HCl O R S OH - H2O NaOH + R S O Na O Sulfonsäure O O O H3C + S in Shampoo oder Zahnpasta O Na O 2.1.7 Reaktionen der Cycloalkane Die Reaktionen der Cycloalkane ähneln denen der kettenförmigen Alkane. 1. Photohalogenierung Cl2 Cl + h. 2. Besondere Reaktionen des Cyclopropans Ni, H2 Br2 60° HI weitere halogenierte Cyclobutane H H Br Br I I Das Cyclopropan ist deshalb so reaktiv, weil der Bindungswinkel C-C 60° beträgt, und somit stark von dem idealen Bindungswinkel im Tetraeder von 109° abweicht. Daraus resultiert eine recht hohe Ringspannung. - 15 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.2 Halogenalkane und Alkohole H H R R C X R C X R C X H primär R sekundär R tertiär 2.2.1 Beispiele 1. Halogenalkane CH3 CHCl2 CCl4 Dichlormethan Tetrachlormethan 2. Alkohole H3C CH CH3 OH CH3 CH 2 OH CH3 CH2 2 OH Methanol Ethanol n-Propanol (Propan-1-ol) CH3 OH i-Propanol (Propan-2-ol) 2.2.2 Eigenschaften der Alkohole 1. Wasserstoffbrückenbindungen R H O H R O WBB Alkohole weisen aufgrund der sich bildenden Wasserstoffbrückenbindungen (WBB) relativ hohe Siedepunkte auf. 2. Solvatisierung von Ionen R H O R O R H H H R O R O O R O Me+ O H X- H H H O R Anionen Kationen Aufgrund dieser Eigenschaften vermögen die Alkohole Ionen zu solvatisieren. - 16 - R Gr u n dla g e n de r O rg a ni sc he n C he mi e 3. Alkohole als Säuren und Basen R OH R OH + + Na + R O Na + Natriumalkoholat H2 + H2SO4 + R OH2 Oxoniumion HSO4- 2.2.3 Synthese von Halogenalkanen 1. aus Alkoholen R OH HX oder R X + H2O R X + HX P - X3 2. Photohalogenierung X2 R H CH3 H3C C CH3 CH3 h. CH3 Br2 H3C C CH2 h. Br + HBr CH3 3. Addition von H-X an Alkene H C C HX + C C X 4. Addition von Halogenen X C C + X2 C C X 5. Addition von Halogenen an Alkine C C Alkin Br2 - 17 - Br Br C C Br Br Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.2.4 Nucleophile Substitution + R X + Halogenalkan Halogenalkan + Nu- R Nu Nucleophil Substitutionsprodukt Nucleophil XHalogenid Substitutionsprodukt + Abgangsgruppe R X + Nu - R Nu + X- R X + HO - R OH + X- R X + R O - R O R (Ether) + X- R X + + X- R X + + X- R X + N C + X- R X + NH3 + H + + X- + Nebenprodukte R X + HNR2 + H + + X- + P Ph 3 + X- + X- R X R' C (Acetylit) . R' + Li Triphenylphosphan R C R' (Alkin) R R (Alkan) R C N (Nitril) R NH 2 (Amin) R NR 2 R P Ph 3 (Phosphoniumsalz) R X + HS- R SH (Thioalkohol, Mercaptan) R X + SR (Thiolat) R S R' (Sulfid) + X- R X + H Ar 2 AlCl3 R Ar + H + + X- + X- + X- - R X R X 2 + O O + O C R' COOR' + HC COOR'' R O C R' (Ester) Ar = Aromat - 18 - COOR' R CH COOR'' Gr u n dla g e n de r O rg a ni sc he n C he mi e 1. S N 2 – Mechanism us Beispiel: H3C Br -OR + Kinetik: H3C OR Br + (Reaktionskinetik 2. Ordnung) RG K CH3 Br - OR Mechanismus: R O + H3C Br H R O CH3 C2 H13 RO Br - H C2 H13 RO Br Br H3C R O CH3 + Br C2 H13 H RO Br CH3 CH3 (S) - Konformation (R) - Konformation Walden’sche Umkehr (Regenschirmmechanismus) Man spricht hierbei von einer vollständigen Inversion der Konfiguration . Demzufolge verlaufen SN2-Reaktionen stereospezifisch ab, d.h., ein reines Stereoisomer reagiert zu einem reinen Stereoisomer mit der entgegengesetzten Konfiguration. 2. S N 1 – Mechanism us Beispiel: CH3 CH3 H3C C Br -OR + H3C C OR CH3 Kinetik: + Br CH3 (Reaktionskinetik 1. Ordnung) CH3 RG = K . H3C C Br CH3 Mechanismus: CH3 H3C C Br CH3 langsam - Brgeschwindigkeitsbestimmende Teilreaktion CH3 H3C C + CH3 planares Carbeniumion - 19 - schnell -OR CH3 H3C C OR CH3 Gr u n dla g e n de r O rg a ni sc he n C he mi e Die SN1 - Reaktion verläuft nicht stereospezifisch und liefert als Produkte immer Racemate: (Annäherung von links) 50% CH3 R'' + -OR R' Enantiomerenpaar + Br R'' RO CH3 H3C 50% CH3 - Br- R' R'' R' R'' + -OR ebenes R' Carbeniumion (Annäherung von rechts) OR 3. Konkurrenz zwischen S N 1 und S N 2 Es stellt sich nun die Frage, wann welcher Mechanismus auftritt. Einfluss des Halogenalkans Bei tertiären Halogenalkanen wird der SN1- Mechanismus bevorzugt, während bei primären der SN2-Mechanismus auftritt. Bei sekundären Halogenalkanen können sowohl SN1 als auch SN2 vorkommen. Entscheidend ist die Stabilität der zwischenzeitlich entstehenden positiv geladenen Carbeniumionen: abnehmende Stabilität CH3 H3C C + CH3 tertiär > HC 3 + CH CH3 sekundär > HC 3 + CH2 primär Verantwortlich hierfür ist der (+)-I-Effekt (Elektronenspendender, d.h. positiv induktiver Effekt), bei dem die Methylgruppen wie auch andere Alkylgruppen stabilisierend auf das betroffene C-Atom wirken. Darüber hinaus sind manche Carbeniumion zusätzlich mesomeriestabilisiert, die im Vergleich zu anderen noch stabiler sind. Einfluss des Lösungsmittels Aufgrund der starken Dipolwirkung begünstigen polare Lösungsmittel einen Ablauf nach SN1, indem die negativ geladenen Pole der Lösungsmittelmoleküle das positiv geladene Carbeniumion stabilisieren. Unpolare Lösungsmittel hingegen führen zu Reaktionsabläufen nach SN2. - 20 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.2.5 Reaktionen der Halogenalkane 1. Abspaltung von H -X Base C C C C H X Halogenalkan Alken (Eliminierung) 2. Bildung von Grignard-Verbindungen + - - + - Mg R X R Mg X Ersichtlich an den Partialladungen erfolgt eine Umpolung, bei der sich die Vorzeichen der Ladungsverteilung ändern. 3. Umlagerung von Carbeniumionen CH3 a) CH3 Nu- H3C C CH2 Br H3C C CH3 H SN1 - Br- NuCH3 + + H3C C CH2 H3C C CH3 H primär tertiär (stabiler) CH3 CH3 Nu- H3C C CH CH3 H3C C CH CH3 + H3C Br SN 1 Br Nu CH3 b) + Nu CH3 - Br- Nu- CH3 CH3 + + H3C C CH CH3 H3C C CH CH3 tertiär CH 3 (stabiler) CH3 sekundär - 21 - Br Gr u n dla g e n de r O rg a ni sc he n C he mi e 4. Reaktionen zwischen Alkoholen und Halogenalkanen R OH + H X R X + OH H2O Br + H Br H3C CH2 CH2 OH + H Cl R OH + NaCl + H2O H3C CH2 CH2 Cl R Cl + + H2O NaOH nicht direkt möglich, da sich OH-Gruppen nur schlecht substituieren lassen Reaktionsmechanismus: H O H O + H Br H+ + Säure H2O Br Oxoniumion (lässt sich leichter substituieren) 5. Analyse der Halogenalkane t R X verd. alkoholische Lösung AgNO3 AgX warten Die Reaktionsgeschwindigkeit ist vom Halogenalkan und dessen Struktur abhängig: R I R Br R Cl tertiäres sekundäres primäres Halogenalkan Folgende Verbindungen sind besonders reaktiv: H2C CH CH2 X Allylhalogenid Benzylhalogenid CH2 X Der Grund für die erhöhte Reaktivität liegt darin, dass die bei Halogenidabspaltung entstehenden Carbanionen + CH2 + H2C CH CH2 und mesomeriestabilisiert sind damit stabiler sind als z.B. Carbanionen, die sich aus tertiären Halogenalkanen gebildet haben. - 22 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.3 Alkene 2.3.1 Beispiele Strukturformel Name (und weitere Verwendung) H2C CH2 Ethen (Ethylen Polyethylen PE) H2C CH CH3 Propen (Propylen Polypropylen PP) H H cis-2-Buten C C H3C CH3 H CH3 C C H3C trans-2-Buten H 1-Buten H2C CH CH2 CH3 H3C H3C Isobuten C CH2 H2C CH CH Butadien CH2 Isopren 2-Methyl-1,3-butadien ( Terpene, Terpentin, Steroide) CH3 H2C CH C CH2 Cyclopenten Cyclohexen Chlorethen, Vinylchlorid ( Polyvinylchlorid PVC) H2C CH Cl H2C CH CH2 Br Allylbromid H2C CH CH2 OH Allylalkohol - 23 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.3.2 Industrielle Herstellung von Alkenen C C C C thermisch katalytisch H H Alkan H2 + Alken 2.3.3 Laborverfahren zur Synthese von Alk enen 1. Abspaltung von Halogenwasserstoff KOH C C C C H X Halogenalkan + KX + H2O Alken Beispiel H3C CH CH2 CH3 KOH - Cl H3C CH CH CH3 + H2C CH CH2 CH3 Cl 2. Wasserabspaltung aus Alkoholen C C H OH Alkohol Säure + C C (H2SO4) H2O Alken 3. Abspaltung von Halogenen aus vicinalen Dihalogenen C C Zn + C C ZnX2 X X vicinal, d.h. benachbart 4. Reduktion von Alkinen R C C R' Alkin H2 R Kat. H Na o. Li R NH3 R' C C H 5. Wittig-Reaktion später in VL (siehe 2.11.4.8) - 24 - cis-Alken H H C C R' trans-Alken Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.3.4 Eliminierungsreaktionen 1. E 2 – Mechanismus (detaillierte Darstellung) (zu 2. 3.3.1) X C C C C X + + H B H simultaner Prozess B RG K Halogenalkan B- Kinetik zweiter Ordnung: Eliminierungsreaktionen konkurrieren mit Substitutionsreaktionen, da die Base B( B OH - , ) neben dem H-Atom auch natürlich am halogenierten C-Atom angreifen kann. Stereochemie der E 2 – Eliminierung H H Ph C C B- Ph - HBr H Ph Br CH3 Ph C C CH3 4 Stereoisomere In der Sägebockschreibweise ergeben sich die vier Stereoisomere wie folgt: B- H H H3C Ph H Ph Ph Br B- Ph CH3 H Ph H3C H Br H3C B- H Ph Ph H CH3 Ph Br Ph Br H3C C C H B- H Ph C C Ph Ph H trans cis H und Br müssen antiperiplanar vorliegen, d.h. sie stehen sich in einer Ebene gegenüber. 2. E 1 – Mechanismus (detaillierte Darstellung) (zu 2.3 .3.1) X langsam C C H - X- schnell + C C C C H B Kinetik erster Ordnung: RG K Halogenalkan - 25 - + H B Gr u n dla g e n de r O rg a ni sc he n C he mi e 3. Möglichkeiten zur Fallunterscheidung zwischen E 1 und E 2 zunehmende Bevorzugung von E2 H3C H H3C C CH2 H H >HC 3 X C CH2 H > HC 2 X X primär sekundär tertiär CH2 zunehmende Bevorzugung von E1 4. Konkurrenz zwischen Eliminierung und Substitution CH3 3% (tertiär) 99% CH3CH2CH2 Br 91% CH CH2 Br 40% Geometrie der Reste behindert eine Annäherung - 26 - Eliminierung 1% 9% 60% Carbeniumionen nimmt zu CH3CH2 Br H3C 3 Substitution Stabilität der Halogenalkan H3C 97% nimmt zu CH3 Br Sterische Hinderung1 H3C C 80% Carbeniumionen nimmt zu 20% (sekundär) nimmt zu 9% H3C CH CH3 Br Eliminierung 91% Sterische Hinderung3 CH3CH2CH2 Br (primär) Substitution Stabilität der Halogenalkan Gr u n dla g e n de r O rg a ni sc he n C he mi e a) Vergleich E 2 gegen S N 2 Nu SN2 C C SN2 X + X + X H C C H E2 Nu- C C + H Nu E2 Beispiel: H3C CH2 CH2 O C2 H5 Substitutionsprodukt -OC H 2 5 H3C CH2 CH2 Br + H3C CH CH2 + HBr + -OC2H5 Eliminerungsprodukte b) Vergleich E 1 gegen S N 1 SN1 Nu- R X - X- R Nu + R Carbeniumion E1 Alken + H X Eliminerung nimmt zu prim./ sek ./ tert. Substitution nimmt zu c) Abhängigkeit vom Lösungsmittel Während polare Lösungsmittel sowie niedrige Reaktionstemperaturen die Substitution begünstigen, führen unpolare Lösungsmittel und / oder hohe Reaktionstemperaturen zu einem vermehrten Auftreten der Eliminierung. d) Abhängigkeit vom Nucleophil CH3 H3C C O H3C CH2 O CH3 mehr Eliminierung weniger Eliminierung - - SR viel Substitution OR wenig Substitution Sterisch gehinderte Nucleophile gehen bevorzugen Eliminierungsreaktionen ein. - 27 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.3.5 Dehydratisierung von Alkoholen (1,2 – Eliminierung von H 2 O) Säure C C C C Kat. + H2O HO H Die Dehydratiserungsneigung nimmt mit in der Reihenfolge primärer, sekundärer, tertiärer Alkohol ab. Mechanismus + - H2O H C C + C C C C C C H Alken + O H O H H H H 2.3.6 Saytzeff-Regel Bei zwei β-C-Atomen wird bevorzugt das höher substituierte Alken entstehen. CH3 H CH3 B CH3 + - X- X H CH3 + H3C CH2 C CH3 OH H - H2O H3C CH C CH3 CH3 CH3 + H3C Hauptprodukt Nebenprodukt 2.3.7 Reaktionen der Alkene Generell sind Alkene reaktionsfreudiger als die Alkane. C C planar X X Y C C Y -Elektronen stehen senkrecht zur Ebene - 28 - CH2 C CH2 Gr u n dla g e n de r O rg a ni sc he n C he mi e 1. Katalytische Hydri erung (Addition von Wasserstoff) C C H2 + Pd, Pt, Ni C C Alken H H Alkan Details H H H H + H-H Kat. + H2C Kat. - H-H C CH2 Kat. - H2C CH2 H H C C H C C C Kat. Kat. CH3 C C H H H H H CH3 H2 H3C Kat. + C2 H5 C2 H5 C2 H5 H H 50% 50% Heterogene Katalyse cis - spezifisch (syn - spezifisch) H3C C C H CH3 D2 H3C Kat. CH3 H C C H H D D meso H3C H H D2 D S H Kat. C C CH3 H3C H - 29 - D S D R + CH3 H D R CH3 CH3 H Gr u n dla g e n de r O rg a ni sc he n C he mi e CH3 CH3 H H H2 H3C H + Pt H CH3 H H H H3C H H3C H CH3 H CH3 CH3 nicht möglich wegen sterischer Hinderung 2. Halogenierung Br C C Br2 + C C Br Beispiel Br H3C CH2 CH2 CH CH2 Br2 H3C CH2 CH2 CH CH2 (braun) (farblos) Br Br vicinales Halogenalkan Br + 50% Br Br immer trans (anti) 50% Details In Lösungen mit Brom besteht folgendes Gleichgewicht: katalytische Br2 Br + + Br Spaltung + + Br + Br + Br + Br Bromoniumion Br Br Br Br Br immer trans (anti) Br + 50% - 30 - 50% wegen sterischer Hinderung nicht möglich Gr u n dla g e n de r O rg a ni sc he n C he mi e Belege für die Existenz des Bromoniumions: Man fängt das Bromoniumion durch andere Nucleophile ab. Br greift von oben an Cl und Br greifen von unten an Br2, Cl- Cl- H Br + Br + Br -OR H Bromoniumion Cl Br -OH Br Br OR OH rac.* rac.* * rac. = racematisch Das Gemischprodukt ist ein Hinweis auf das Vorhandensein des Bromoniumions. 3. Addition von Halogenwasserstoff X C C + H X C C H Halogenalkan MARKOWNIKOWProdukt H3C CH CH3 HBr H3C CH CH2 HBr Peroxide Br antiH3C CH2 CH2 Br MARKOWNIKOWProdukt Details H3C CH2 CH2 I H3C CH CH2 + HI unsymetrisches Alken I H3C CH CH3 CH3 Br CH3 + HBr H CH3 + H H H - 31 - Br H Gr u n dla g e n de r O rg a ni sc he n C he mi e Radikalische H-Br – Addition Peroxid Rad Rad H Br Rad H Br Br Br + C C C C Br + HBr Br C C C C H + Br Andere Regioselektivität beobachtet man im folgenden Fall: Br H3C CH CH2 Propen H3C CH CH2 instabil Br Br H3C CH CH2 Br H3C CH2 CH2 Br + Anti-Markownikow-Produkt stabiler 4. Addition von Schwefelsäure OSO3 H C C + H2SO4 C C H 5. Addition von Wasser OH C C + H2O C C H 6. Halogenhydrinbildung C C Br2, H2O Br Br C C OH Base C C O Halogenhydrin C C O Peroxide (Oxidane) - 32 - Br Gr u n dla g e n de r O rg a ni sc he n C he mi e 7. Dimerisierung H3C H3C C CH2 + CH2 C CH3 CH3 CH3 H3C C CH C CH3 CH3 CH3 + CH3 CH3 H3C C CH2 C CH2 CH3 8. Alkylierung H3C H3C CH3 CH3 Säure C CH2 + H C CH3 H3C C CH2 CH3 CH3 CH3 C CH3 CH3 9. Addition von Quecksilberacetat (veraltet) C C + O H2O + Hg O C CH3 NaBH4 C C C C O HO Hg HO H O C CH3 Aus Umweltschutzgründen ist dieses Verfahren selbstverständlich nicht mehr gängig. 10. Hydroborierung C C + (BH3 )2 C C H2O2 H BH2 C C H OH Details H3C H2O2 H3C C CH2 H3C B2 H6 H3C HC CH2 H3C CH CH2 OH H3C B 3 H3C H2O CH CH3 H3C - 33 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 11. Addition von Radikalen CCl3 H13C6 CH CH2 + Br Peroxid CCl3 H13C6 CH CH2 Br 12. vicinale Dih ydroxylierung (Glykolbildung) C C H H H2O KMnO4 o. C C OsO4 o. R-COOOH H C C H OH OH OH OH Glykol 13. Halogenierung in Allylstellung Allylstellung Cl2 H2C CH CH2 Cl HCl + p / 600°C H2C CH CH3 NBS* H2C CH CH2 Br *NBS = N-Brom-succinimid 14. Ozonolyse C C + O O3 C Zn C O C H2O O O + Ozonid 1. O3 H3C CH2 CH CH2 H3C H3C C CH2 O H3C CH2 C 2. Zn, H2O 1. O3 H3C 2. Zn, H2O H3C CH3 O C H + H H O C O C H + H O 1. O3 O 2. Zn, H2O CH3 CH2 H3C C (CH)4 C H CH3 O 1. O3 O C H + 2. Zn, H2O H - 34 - C O Gr u n dla g e n de r O rg a ni sc he n C he mi e 15. Carbenübertragung H H C C| H H Triplettcarben (reagiert wie ein Diradikal) Singulettcarben H H H C C H3C H CH3 H H H H H H3C C + H H + CH3 H C C + CH3 H H CH3 + H3C H H H3C H H3C H H H C| H H CH3 H H H3C CH3 Details h. + H2C N N Diazomethan CHCl3 + N2 + CO TriplettCarben h. H2C C O Keten -O-Cl(CH H2C 3)2 (Tertiärbutylat) H2C |CCl3- + H O C(CH3 )3 - Cl2 |CCl2 Dichlorcarben (Singulett-Carben) CH2 I2 Zn (Cu) I CH2 Zn I Diiodmethan C C CH2 + ZnI2 - 35 - C C C C CH2 C I Zn I Carben komplexiert mit ZnI2 (instabil) Gr u n dla g e n de r O rg a ni sc he n C he mi e 16. Reaktion mit Per carbonsäuren O C C + R C C C O Oxiran O OH Details zur Bildung von Oxiranen durch Oxidation von Alkenen mit elektrophilen Oxidationsmitteln C C + C C O+ "+OH" C C O H Alternativ dazu ist folgende Überlegung möglich: C C C C O O H O O C R Percarbonsäure verwendete Carbonsäuren O C O O H CH3 COOOH Cl meta-Chlor-perbenzoesäure Peressigsäure MCPBA (zersetzt sich explosionsartig) Viele Percarbonsäuren zersetzen sich explosionsartig, wobei MCPBA relativ stabil und daher im Labor Verwendung findet. In der chemischen Industrie verwendet man aus Sicherheitsgründen dementsprechend die Peressigsäure, die man zur Risikominimierung direkt nach der Herstellung weiter umsetzt und nie größere Mengen lagert. O D D D D CH3COOOH C C C C H H H H cis-Dideuterioethen D H C C CH3COOOH H D trans-Dideuterioethen D H C C H - 36 - O D Gr u n dla g e n de r O rg a ni sc he n C he mi e Relative Reaktionsgeschwindigkeit der Epoxidation niedrige Elektronendichte, da nur ein (+)-I-Substituent Cl O C H O O H O H Hauptprodukt + höhere Elektronendichte, da zwei (+) - I - Substituenten H O Nebenprodukt Die beiden aus der Reaktion hervorgehenden Oxirane entstehen nicht im Verhältnis 1:1. Die Reaktionsgeschwindigkeiten ergeben sich nach der folgenden Reihenfolge. langsam H2C CH2 H3C C CH2 H3C H3C CH CH2 H3C C C H3C H3C CH CH CH3 CH3 H3C H C C H3C CH3 CH3 schnell H H3C H C C H MCPBA CH3 H3C H C C H CH3 O = 60° gespannter Dreiring (reaktionsfreudig) (giftig, cancerogen) CH3 OH S H2O R OH H3C H H H H C C H3C CH3 MCPBA H H C C H3C CH3 O OH H2O CH3 R R 50% OH CH3 H + H OH R H - 37 - CH3 R OH CH3 50% Gr u n dla g e n de r O rg a ni sc he n C he mi e Details zur Herstellung der cis-Diole a) Man kann das Reaktionsprodukt eindeutig herstellen, und zwar mit KMnO4 oder OsO4, so dass cis-1,2-Diole bzw. durch b) Epoxidation mit Percarbonsäuren und anschließend H2O (mit Säure), wodurch dann trans-1,2-Diole entstehen. O O 1. KMnO4 Mn O H O O OH 2. H2O Mn O O H OH cis-Diol O O 1. OsO4 O O O O OH O Os O 2. H2O OH rac. 2.3.8 Markownikow -Regel (1900) „Addiert man H-X an eine C=C-Doppelbindung, so lagert sich der Wasserstoff an das Kohlenstoffatom, welches am meisten H-Atome besitzt.“ Diese Regel jedoch mehr als Definition des Markownikow-Produktes zu verstehen, da sie in vielen Fällen verletzt wird und dann mehrheitlich oder einzig und allein das antiMarkownikow-Produkt entsteht. Stattdessen ist die Stabilität der zwischenzeitlich entstehenden Carbeniumionen bzw. Radikale von Bedeutung. Durch stabilisierende Einflüsse wie z.B. einem mit einer Phenylgruppe ausgeübten M-Effekt werden hierbei auch anti-Markownikow-Produkte bevorzugt gebildet. - 38 - Gr u n dla g e n de r O rg a ni sc he n C he mi e Vergleich: Markownikow-Produkt – anti-Markownikow -Produkt C CH H3C CH CH2 + H + H + + CH CH C CH2 + + H3C CH CH3 + H3C CH2 CH2 sekundär primär (stabiler) (instabil) + tertiär (instabiler) sekundär, mesomeriestabilisiert I I I CH CH H3C CH CH3 I anti-Markownikow-Produkt Markownikow-Produkt Der Grund für das Auftreten des anti-Markownikow-Produktes ist der +-M-Effekt, der von dem Phenylring ausübt wird: + CH CH CH CH CH CH CH CH C + C + C + Im Allgemeinen gilt, dass geladene Atome wie auch Radikale durch mesomere Effekte stärker stabilisiert werden als solche, die mehrfach substituiert sind. Unter letzteren gilt die Reihenfolge, dass Teilchen mit sekundär substituierten Kohlenstoffatomen eine größere Stabilität aufweisen als primäre, tertiäre stabiler als sekundäre sind, usw. - 39 - Gr u n dla g e n de r O rg a ni sc he n C he mi e Beispiele für Markownikow -Produkte: H H CH3 H H + + H + CH3 H CH3 + tertiär H3C Br sekundär H H H CH3 Br H3C H+ H + H H3C + H3C H Br + H H sekundär Mesomeriestabilisierung H H H Br sekundär (instabiler) Man beachte bei der radikalischen Addition von Halogenwasserstoffen die Bildung von anti-Markownikow-Produkten (2.3.7.3). Zur Wiederholung: CH3 CH2 Br CH3 H3C Br Br h. oder oder Br2 Alkan Hauptprodukt Letzteres Produkt entsteht bei dieser Reaktion am meisten, da das hierbei zwischenzeitlich entstehende Radikal das stabilste ist: CH3 C - 40 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.3.9 Chemie der konjugierten Doppelbindungen kumulierte Doppelbindungen konjugierte Doppelbindungen isolierte Doppelbindungen H2C C CH2 CH2 CH CH CH2 CH2 CH CH2 CH CH2 1,3-Butadien (normale Chemie der Alkene) Allen (nicht besonderes relevante Verbindung) C C CH3 Herstellung konjugierter Diene Laborsynthese: Br alkhol. KOH NBS - HBr Allylstellung Industrielle Herstellung: CH3 CH2 CH2 CH3 CH2 CH CH2 CH3 Cracken CH2 CH CH CH2 , Kat. , Kat. Butadien CH3 CH CH CH3 Reaktionen in Allylstellung CH3 CH CH2 Allylstellung (Doppelbindung nicht an Reaktionen beteiligt) 13 CH3 CH CH2 Additionsreaktionen Allylradikale . . 13 13 CH2 CH CH2 CH2 CH CH H32 niedrige Konz. Br 2 Radikalbildner Br2 Br + Br (+ HBr) Br 13 CH2 CH CH2 50% - 41 - Br + 13 CH2 CH CH2 Br 50% Gr u n dla g e n de r O rg a ni sc he n C he mi e Halogenierungsmittel (Rad ikalbildner) O N Br O N-Brom-succinimid (NBS) Reaktivität konjugierter Diene Br2 CH2 CH CH CH2 CH2 CH CH CH2 + CH2 CH CH CH2 Br HBr Br 1,2-Addition Br Br 1,4-Addition CH2 CH CH CH2 + CH2 CH CH CH2 H Br H Br H2 CH2 CH CH CH2 + CH2 CH CH CH2 H H H H Details entsteht praktisch nicht + + H3C CH CH CH CH CH3 1 2 3 4 5 6 H3C CH CH CH CH CH3 H + H+ + H3C CH CH2 CH CH CH3 kinetisches Produkt H H3C CH CH CH2 CH2 CH3 H + H3C CH CH CH CH H2 CH3 Cl H H3C CH CH2 CH2 CH CH3 H Cl thermodynamisches Produkt + Cl- Das bei der 1,2-Addition entstandene kinetische Produkt , welches bei -80°C fast ausschließlich entsteht, kann durch Umlagerung bei hohen Temperaturen in das thermodynamische Produkt überführt werden, dass bei der 1,4-Addition gebildet wird. - 42 - Gr u n dla g e n de r O rg a ni sc he n C he mi e Bromierung von 1,3,5 -Hexatrien CH2 CH CH CH CH CH2 Br2 dreifache Mesomerie + Br CH2 CH H2 CH CH CH CH2 ++ Br CH2 CH CH C H CH CH2 ++ Br CH2 CH CH CH CH C H2 Br CH2 CH CH2 CH2 CH2 CH3 Br 1,2 - Addition Br Br CH2 CH2 CH2 CH CH2 CH3 CH2 CH2 CH2 CH2 CH2 CH2 Br 1,6 - Additition Br 1,4 - Addition Beispiel der konjugierten Doppe lbindungen in der Natur CH3 CH3 CH3 CH2 OH CH3 CH3 Vitamin A CH3 CH3 H3C CH3 H3C CH3 CH3 CH3 β-Carotin (bestehend aus 2 Vitamin-A-Molekülen) - 43 - CH3 CH3 Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.4 Hochmolekulare Stoffe (Makromoleküle ) 2.4.1 Vorkommen und Verwendung in der Natur a) b) c) d) Polysaccharide: Stärke, Zellulose Proteine (Eiweiß in Pflanzen und Tieren) Nucleinsäuren, z.B. DNS Naturkautschuk industriell erzeugte Produkte Einteilung nach Gebrauchseigenschaften a) Elastomere (dehnbar) b) Thermoplaste (durch Wärme verformbar) c) Duroplaste (irreversibel vernetzt, d.h. nach Vernetzung der Ketten nicht mehr verformbar) d) Kunstfasern (geringe Dehnbarkeit sowie hoher Ordnungsgrad) Einteilung nach Herstellungsmethode a) Polymere (Polymerisation) b) Polykondensate (Polykondensation) c) Polyaddukte (Polyaddition) 2.4.2 Polymere Monomer Polymer n . H2C CH2 O2, Wärme CH2 CH2 CH2 CH2 CH2 CH2 CH2 Ethen Druck Polyethlyen (PE) n . H2C CH Peroxid CH2 CH CH2 CH CH2 CH Cl Cl Cl Cl Polyvinylchlorid (PVC) Vinylchlorid n . H2C CH X CH2 CH X= C N C Acrylnitril N Polyacrylnitril - 44 - n Gr u n dla g e n de r O rg a ni sc he n C he mi e Monomer Polymer X = Ph Styrol CH2 CH Ph O n CH2 CH X= C O CH3 COOCH3 n Acrylsäuremethylester Polyacrylat Regelmäßigkeiten bei Polymeren Struktur Bezeichnung CH2 CH CH2 CH CH2 CH CH2 CH isotaktisch (auf einer Seite) X X X X X X CH2 CH CH2 CH CH2 CH CH2 CH2 X syndiotaktisch X X X X Ataktisch CH2 CH CH2 CH CH2 CH CH2 CH (unregelmäßig) X Regelmäßige Polymere können einen fast kristallinen Charakter besitzen. Monoterpene Monoterpene sind aus zwei Isopren-Einheiten aufgebaut. CH3 CH3 CH3 OH CH H3C CH C CH3 γ-Terpinen H3C CH2 Limonen - 45 - H3C CH3 Menthol Gr u n dla g e n de r O rg a ni sc he n C he mi e Demgegenüber sind Sequiterpene aus drei und Diterpene aus 4 Isopren-Einheiten aufgebaut. CH3 CH3 H3C CH3 CH2 OH CH3 Vitamin A1 Auch die Sexualhormone sind Diterpene. 2.4.3 Polymerisationsreaktionen a) Homopolymerisation Nur ein Monomer reagiert. b) Copolymerisation Man geht von zwei oder mehr verschiedenen Monomeren aus. 1. Radikalische Polymerisation C = Radikal Rad Peroxid Kettenstart Rad C + Wärme H2C CH X Kettenfortpflanzung Rad CH2 CH + HC 2 X CH X C Rad Rad CH2 CH X Rad CH2 CH H2C CH X X usw. Kettenabbruch Rad CH2 CH H2C CH + Rad C X X Rad CH2 CH H2C CH Rad X - 46 - X Gr u n dla g e n de r O rg a ni sc he n C he mi e Beispiele CH2 H2C CH2 H2C CH2 H2C usw. Rad C Rad CH2 CH2 CH2 CH2 CH2 CH2 ... CH3 CH3 CH3 CH2 H2C CH2 Vulkanisation CH2 CH2 S CH3 S n Isopren CH2 natürlicher Kautschuk (durchschnittliche Molmasse = 350.000 Dalton) CH2 CH2 Kautschuk altert, indem die S-Brücken oxidiert werden, und somit der Kautschuk brüchig wird. 2. Ionische Polymerisation Kationische Polymerisation (durch Säuren) CH3 + H CH2 C CH3 Isobuten CH3 CH2 C CH3 CH3 CH2 C CH3 H CH2 C CH3 CH3 CH2 C CH3 CH3 CH2 CH CH3 CH3 Polybutylen Anionische Polymerisation (durch Basen) (z.B. Li+ -NH2 oder K+ -NH2) + K NH2 + CH2 CH H2N CH2 CH Ph Ph K + CH2 CH Ph H2N CH2 CH CH2 CH Ph - 47 - Ph K + usw. Gr u n dla g e n de r O rg a ni sc he n C he mi e Koordinationspolymerisation / Ziegler-Natta-Katalyse CH2 TiCl4 / AlR3 + CH2 CH2 Ti Ti CH2 CH3 CH2 Ti CH2 CH2 CH2 CH3 PE CH2 CH2 2.4.4 Polykondensate O + O C C HO OH + HO CH2 CH2 OH Diol (Dialkohol) Dicarbonsäure O H - H2O O O C O CH2 CH2 O C O C O CH2 CH2 O C Polyester Alternativ: + HO COOH -Hydroxycarbonsäure H - H2O O O O C O C O C O Polyester 2.4.5 Polyaddukte HO OH + O C N Diol N C O Urethan O O O C NH O C NH Polyurethan (Schaumisolator) - 48 - O O C NH Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.5 Alkohole 2.5.1 Einteilung der Alkohole Primäre Alkohole CH3OH C2H5OH Methanol Ethanol Sekundäre Alkohole H3C OH CH OH H3C Isopropanol Cyclohexanol (iso-Propanol / 2-Propanol) Tertiäre Alkohole CH3 OH H3C C OH CH3 CH3 t-Butanol (tertiär-Butanol / 2-Methyl-2-Propanol) 1-Methyl-1-cyclopentanol Die Eigenschaften der Alkohole liegen zwischen denen des Wassers und den Alkanen, wobei kürzere Ketten deutlich den dem Wasser ähnlichen polaren Charakter bewirken, während längere Ketten eher zu einem hydrophoben Verhalten analog zu den Alkanen führen. R-H R-OH H2 O Alkan Alkohol Wasser 2.5.2 Industrielle Herstellung von Alkohol en 1. Erdöl Cracken CH2 CH CH3 H2SO4 CH3 CH CH3 CH3 CH CH3 O SO3H CH4 O2 - 49 - H2O CH3OH OH Gr u n dla g e n de r O rg a ni sc he n C he mi e 2. Oxosynthese CO, H2 C C H2 C C Kat. C C Kat. H C O H Aldehyd H CH2 OH 3. Alkoholische Gärung Zucker Hefe Ethanol Destillation 95%iger Alkohol 2.5.3 Laborsynthesen 1. Hydroxymercurierung C C + Hg(OAc)2 + H2O NaBH4 C C C C OH HgOAc OH 2. Hydroborierung + Oxidation C C B2 H6 H2O2 C C C C OH OH BH2 Vergleich zwischen der Hydroxymercurierung und der Hydroborierung CH3 Hydroxymercurierung Hg(OAc)2 H2O CH3 H3C C CH CH2 CH3 NaBH4 H3C C CH CH2 Hydroborierung H3C C CH CH3 H3C OH HgOAc CH3 B2 H6 CH3 H2O2 H3C C CH CH2 H3C H H3C OH CH3 H3C C CH2 CH2 BH2 H3C 3. Verfahren der nucleophilen Substitution (S N ) R X CH2 Cl CH2 CH2 Cl2, H2O OH R OH NaOH + X CH2 OH C C Cl Cl - 50 - NaCO3 + NaCl C C OH OH OH Gr u n dla g e n de r O rg a ni sc he n C he mi e 4. Hydroxylierung von Alkanen KMnO4 C C C C oder OsO4 OH OH syn OH R-COOOH -OH anti C C C C O OH 5. Grignard-Reaktion (Organomagnesiumverbindungen) R X R Mg X Mg (Umpolung) Ether GRIGNARD-Verbindung Halogenalkan R MgBr C O O MgBr OH H2O C C R H C O H R MgBr R O MgBr H H H H2O C OH C R H primär R Formaldehyd R' C O R' R MgBr O MgBr C H H R' H2O OH C R H sekundär R Aldehyd R' C O R' R MgBr R'' O MgBr H2O C R'' R R' OH C R'' tertiär R Keton MgBr H2C CH2 O Epoxid (Oxiran) R' C O R'' O R MgBr O OH H2C CH2 H2O H2C CH2 primär R 2 R MgBr R' R O MgBr C R R Ester - 51 - H2O R' OH C R R tertiär Gr u n dla g e n de r O rg a ni sc he n C he mi e Statt Grignard-Verbindungen lassen sich auch andere metallorganische Verbindungen wie z.B. Lithiumorganyle einsetzen: R' C O R' 2 R Li O R' O Li H2O C R R' OH C R R tertiär R Ester Zur Wiederholung: R Li H3C C O R = CH3 H H H3C C H H Li + HC 3 O CH3 C CH3 H2O O Li Aldehyd 6. Aldolkondensation 7. Reduktion von Carboyxylverbindungen (siehe 2.13.1.1) 8. Reduktion von Säuren oder Estern - 52 - H3C C CH3 OH Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.5.4 Einführung in Synthesestrategien Nucleophile Substitution S N IF CH2 CH2 CH2 CH2 Br F CH2 CH2 CH2 CH2 I Aceton + Br Dihalogenalkan HBr R OH ? Mg R Br Br2 R H Ether h. ? R MgBr H2O OH H R CH2 CH2 R CH2 CH2 CH2 CH2 OH O H C O CH3 + R CH2 CH2 MgBr + O CH3 C H CH3 R CH2 CH2 C H CH2 CH2 O CH3 R CH2 CH2 CH2 OH + C O CH3 CH3 R CH2 CH2 C OH CH3 OH * H2C O - D2O* R CH2 CH2 D R CH2 CH2 OMgBr GRIGNARDVerbindung R CH2 CH2 OH R CH2 CH2 C H2 C CH2 O = Deuterierung zur Erforschung von Metabolismen 2.5.5 Retrosynthetische Analyse 1. Beispiel H3C CH2 OH H H3C MgBr + C O H - 53 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2. Beispiel O H3C MgBr + C CH2 CH2 CH3 CH2 CH3 OH O H3C C CH2 CH2 CH3 H3C C CH2 CH3 + BrMg CH2 CH2 CH3 CH2 CH3 O BrMg H3C C CH2 CH2 CH3 + CH2 CH3 2.5.6 Reaktionen der Alkohole 1. Spaltung der R -OH-Bindung Reaktion mit Halogenwasserstoff HX H3C CH2 OH H3C CH2 X OH H2O + HBr Br H2O + 2. Reaktion mit PX 3 (X = Halogen) H3C CH2 CH2 OH PBr3 H3C CH2 CH2 Br + H3PO3 3. Abspaltung von H 2 O (Eliminierung) OH C C OH Bei Wärme reicht eine schwache Säure, bei Kälte muss eine stärkere Säure verwendet werden. Säure Kat. C C Alken CH3 C OH CH2 H2SO4 C CH3 CH3 OH Säure Spaltung der O-H-Bindung - 54 - + H2O Gr u n dla g e n de r O rg a ni sc he n C he mi e 4. Reaktion mit Metallen R O H Metall R O- Metall + 12 H 2 schnelle Reaktion langsame Reaktion primär > sekundär > tertiär H3C CH2 OH + Na H3C CH2 O Na + + 1 H2 2 Natriumethylat CH3 CH3 3 . H3C C OH + Al H3C C O Al + 1 H 3 Aluminium-triisopropylat H 1 2 H2 Verwendung der Alkoholate z.B. bei der WILLIAMSON’schen Ethersynthese: R O + Na + Br + CH3 R O CH3 Ether 5. Veresterung O O + H3C C 18 18O O CH2 CH3 Ethylacetat Ester 18O CH CH H3C C OH + H18 2 3 Essigsäure H2O O C O CH3 Methylbenzoat (Duftstoff in unbestäubten Blüten) H3C SO3H + H O R H3C Sulfonsäure SO3 R Methylsulfonat Laborverfahren: O H3C CH2 C + H O R Cl - HCl Base Säurechlorid - 55 - O H3C CH2 C O R Gr u n dla g e n de r O rg a ni sc he n C he mi e 6. Oxidation Primäre Alkohole Pyridin*/ CrO3 / HCl oder DMSO** O KMnO4 R C H O R CH2 OH R C KMnO4 OH O KMnO4 R C Carbonsäure H CH2 OH KMnO4 COOH oder H2O2 Sekundäre Alkohole R R KMnO4 CH OH C O R CH3 R CH3 R CH3 KMnO4 oder H2O2 HO O Cholestanol Tertiäre Alkohole R R C OH KMnO4 keine Reaktion R * Pyridin: ** DMSO = Dimethylsulfoxid H3C N S CH3 O - 56 - CH3 R Gr u n dla g e n de r O rg a ni sc he n C he mi e 7. Ether aus Alkoholen und Mineralsäuren H2SO4 2 . H3C CH2 OH H3C CH2 O CH2 CH3 Diethylether 130°C Mechanismus: H H2SO4 H3C CH2 OH H3C CH2 O 130°C + H O CH2 CH3 H H H3C CH2 - H2O + O CH2 CH3 H3C CH2 O CH2 CH3 8. Periodsäurespaltung von 1,2 -Diolen R CH CH R' OH OH O HIO4 R C H2O O C R' + H H Aldehyde R CH CH2 CH R' OH OH HIO4 H2O keine Reaktion Anhand letzter Reaktion bzw. ‚Nicht-Reaktion’ lassen sich Diole auf das Vorhandensein von vicinalen, d.h. aneinander liegenden OH-Gruppen, analysieren. OH C OH HIO4 C O C OH C O O I C O + O - 57 - C O + HIO4 Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.6 Ether 2.6.1 Einteilung der Ether Symmetrische Ether H3C H5C2 CH3 HC O CH O C2H5 H3C Diethylether CH3 Diisopropylether Unsymmetrische Ether H3C CH2 O CH3 Ethylmethylether 2.6.2 Industrielle Herstellung von Ethern H2SO4 R OH R O R + H2O SN R O + Hal R' OH HO R O H+ + OH O2 + Hal- O - H2O HO O 1,3-Dioxan (Dioxan) Glykol H3C CH2 O CH2 CH3 R' h. H3C CH O CH2 CH3 H3C CH O CH2 CH3 - C2H2-OH O O H CH3 CH3 CH O O CH O O Polymeres Peroxid (hochexplosiv) Hydroperoxid Sicherheitsmaßnahmen zum Diethylether 1. Lagerung nur in (lichtundurchlässigen) brauen Flaschen oder Metallbehältern 2. Behälter nicht über längere Zeiträume offen stehen lassen 3. Analyse des Diethylethers auf Anwesenheit von polymeren Peroxiden mittels Peroxidnachweis: - 58 - n Gr u n dla g e n de r O rg a ni sc he n C he mi e Fe Peroxid 2+ Fe 3+ -SCN -SCN farblos Rhodanit (rot) Das Zerstören von Peroxiden erfolgt durch Ausschütteln mit einer FeSO4-Lösung. 2.6.3 Laborverfahren zu Ethersynthese O C C R OH + + Hg O C CF3 2 Alken NaBH4 C C R O C C O R O H Hg O C CF3 2.6.4 Reaktionen der Ether Die meisten Ether sind relativ reaktionsträge und werden deshalb unter anderem als Lösungsmittel in der organischen Chemie verwendet. Spaltung durch Halogenwasserstoffsäuren H -X H O R Ether + + + X- + H H O R H OH + Alkohol H Oxoniumion R X Halogenalkan Bei unsymmetrischen Ethern stellt sich dann natürlich die Frage, welcher Alkohol bzw. Halogenalkan entsteht. Anwendung von cylischen Ethern: Schutzgruppe für Alkohole (Maskierung) + H O 2,3-Dihydro4-H-pyran HO R Alkohol H+ H+ H C O O R C-Atom mit 2 O-Atomen verbunden (Acetal) (ähnliche Eigenschaften wie C=O) - 59 - H2O HO R Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.7 Schwefelanaloga von Alkoholen und Ethern 2.7.1 Beispiele Struktur Name CH3SH Methanthiol, Methylmercaptan H3C CH2 CH CH2 CH3 3-Pentanthiol SH H3C CH2 S CH3 Ethylmethylsulfid H3C O Methoxylat-Anion H3C Methanthiolat-Anion (sehr starkes Nucleophil) S 2.7.2 Synthesen 1. H3C CH CH3 + NaSH NatriumBr hydrogensulfid H3C CH CH3 + NaBr SH Isopropylmercaptan 1. Na/NH3 (l) 2. R S S R R SH 2. H2O 3. H3C OH H+ / H2S H3C SH + H2O (für Industrie bedeutsamstes Verfahren) 2.7.3 Reaktionen 1. Herstellung von Sulfiden R SH R'O- Na+ + R S Na + R'OH Br R'' R S Sulfid - 60 - R'' + NaBr Gr u n dla g e n de r O rg a ni sc he n C he mi e 2. Oxidation von Mercaptanen a) viel KMnO4 R SH R SO3 H Sulfonsäure oder H2O2 I2 / Br2 b) R SH R S S R + 2 HI oder O2 Natürliches Vorkommen in Haaren, Nägeln und Hörnern in Form von Kreatin 1. Lockenwickler 2. O2 CH2 SH CH2 HS CH2 COOH S Disulfidbrücke S oder anderes CH2 SH CH2 Reduktionsmittel Kreatine (Polymere, Eiweiße) starr, da vernetzt S ("Haube") CH2 S CH2 flexibel neue Vernetzung (neue Starheit, neue Form) 3. Oxidation der Sulfide H2O2 H3C O H2O2 S CH3 H3C - H2O S CH3 H3C O O Dimethylsulfon DMSO H2O2 R' S H2O2, chiraler Katalysator R' R'' R' S R'' O rac. chirales Sulfoxid (unsymmetrisch) gezielt O O S S R'' R'' - 61 - R' S CH3 Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.8 Epoxide (Oxirane) 2.8.1 Industrielle Herstellung O2, Ag H2C CH2 250°C O Ethylenoxid Ethylen (Desinfektionsmittel) 2.8.2 Labormethoden 1. über Halogenhydrine Alken + X2 + H O 2 C C Halogen X OH SN -OH C C C O Oxiran O X H3C CH CH2 C Br2, H2O H3C CH CH2 -OH H3C CH OH Br O Oxiran 2. mit Percarbonsäuren O2, Ag O C C + R C C O O H - 62 - CH2 C O Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.8.3 Reaktionen der Oxirane 1. Säurekatalysierte Ringöffnung Z H2C CH2 + O + H H2C CH2 O OH H H2C CH2 O H3O + H2C CH2 OH OH H O H2C CH2 Z- HO OH H3O + + + H OH HO 50% 50% Enantiomerenpaar Über Oxirane lassen sich demnach trans-Diole als Enantiomerengemisch darstellen. 2. Basenkatalysierte Ringöffnung Z- C C O Z H Z C C - Z- Z C C O OH H H5C2O- Na+ + H2C CH2 Natriumethylat O H5C2 O CH2 CH2 OH + O Na O + H2C CH2 CH2 CH2 O OH Natriumphenolat NH3 + H2C CH2 O H2N CH2 CH2 + Mehrfachalkylierung OH (mono) - 63 - Gr u n dla g e n de r O rg a ni sc he n C he mi e Alkylantien: H2C CH2 O ist ein Alkylanz (Alkylierungsmittel), die in der Regel giftig sind. Diese Stoffgruppe wird wegen folgendem Mechanismus für die Chemotherapie verwendet: NH2 + H2C CH2 O NH CH2 CH2 OH Protein Zusätzlich dazu verwendet man diese Reaktion in der Biotechnologie, um an Makromoleküle Enzyme zu binden, wodurch diese immobilisiert werden: Makromolekül mit Epoxidgruppen in der Seitenkette CH HO CH O CH2 CH2 NH2 NH Enzym (Protein) immobilisiertes Enzym 3. Reaktionen mit Grignard-Verbindungen R MgBr + H2C CH2 O R CH2 CH2 O MgBr + H2C H3O CH2 O - 64 - H3O + R CH2 CH2 OH + CH2 CH2 OH Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.9 Alkine 2.9.1 Aufbau 108pm H C C Ethin / Acetylen (Schweißgas) H Linearer Aufbau 121pm 2.9.2 Industrielle Herstellung 1. veraltetes Verfahr en Kohle H2O CaC2 Calciumcarbid Koks CaO Hydrierung HC CH H2 / Kat. H2C CH2 (Carbidlampen) 2. modernes Verfahren CH4 + O2 2 HC CH + CO + H2 1500°C 2.9.3 Labormethoden 1. Halogenwasserstoffabspaltung H H C C H H H H Cl2 Add. Ethen Cl KOH C C Cl Alkohol H H H Na oder KOH H H Alkohol + R' X Cl C C HC CH 2. Reaktionen von Metallacetyliden R C C R LiNH2 R C C Li Lithiumacetylid - 65 - SN R C C R' Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.9.4 Reaktionen der Alkine 1. Addition von Halogenwasserstoff H Br H Br H Br C C C C H C C Br H Br 2. Addition von Wasserstoff H2 R Pd-Kat. H H Na oder Li R H R cis C C R C C R trans C C NH3(l) H R 3. Addition von Halogenen X X C C + X2 X2 C C X X C C X X 4. Addition von H 2 O C C H2O / H2SO4 H H C C C C H O O Enol (relativ instabiler) H Keton Keto – Enol - Tautomerie - 66 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 5. Metallacetylide LiNH2 R C C H a) R C C Li 1. + R C C Li + H2C CH2 O R C C CH2 CH2 OH 2. H2O primär b) R C C Li 1. R'-Br + R C C R' 2. H2O c) R C C Li O 1. + C R H 2. H2O d) 1. O OH * R C C C R sekundär (Racemat) H + R C C Li OH R C C 2. H2O tertiär e) 1. + O H C H R C C Li 2. H2O - 67 - R C C CH2 OH primär Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.10 Aromaten Der Name der Aromaten leitet sich ab von der Beschreibung der „wohlriechenden Verbindungen.“ 2.10.1 Schreibweisen des Benzols Strukturformel Entdecker (Bezeichnung) korrekte Formel synthetisierte Verbindungen instabil Belege für die Kekulé-Formel: Schreibweise nach Kekulé … DEWAR … LADENBURG (Prisma) (Benzvalen) 1. Es gibt nur ein Monosubstitutionsprodukt (C6H5X) 2. Benzol liefert drei Disubstitutionsprodukte (C6H4XY) 1 X 1 X 1 X 2 Y 2 Y 2 Y 1,2 - substituiert ortho - ~ o-~ - 68 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 1 1 X X 3 3 Y 1 X 1 X 3 Y Y 1,3 - substituiert meta - ~ m-~ 1 1 X Y 4 X Y 4 Y 4 1,4 - substituiert para - ~ p-~ Das Benzolmolekül ist mit π-Elektronen oberhalb und unterhalb der Ringebene planar: 2.10.2 Kriterien des aromatischen Zustandes 1. Gegenbeispiele Cyclobutatien Cyclooctatetraen kein Aromat kein Aromat reagiert wie ein Alken < 20K stabil > 35K: 2 2. Hückel-Regel für Aromaten Ein Aromat muss nach Definition folgende Eigenschaften aufweisen: Das Molekül muss eben und ringförmig aufgebaut sein, und (4n + 2) delokalisierte π – Elektronen aufweisen. ( n ). - 69 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 3. Beispiele für Aromate n mit n = 0 4 ∙ 0 + 2 = 2 delokalisierte π – Elektronen C6 H5 C6 H5 C N C6 H5 C6 H5 C6 H5 + C6 H5 C -C N BF3 + C6 H5 + C6 H5 C C6 H5 Dreiring ist nicht aromatisch C6 H5 CC H 6 5 C6 H5 Cyclopropenyl-Kation (aromatisch) O O C C C6 H5 C6 H5 + C6 H5 Dreiring (formal kein Aromat) C6 H5 Cyclopropenylkation (Aromat) Hierbei ist die Carbonylgruppe nicht experimentell bestimmbar. + C C 6 2 getrennte Aromatensysteme 2 4. Beispiele für Aromate n mit n = 1 4 ∙ 1 + 2 = 6 delokalisierte π – Elektronen C C N N + + C N N N Pyridin N N N N S N N Pyrimidin Pyrazin O Thiophen Furan N H S Pyrrol Thiazol - H+ H H kein Aromat C H Cyclopentadienyl-Anion (aromatisch) - 70 - + Gr u n dla g e n de r O rg a ni sc he n C he mi e 5. Beispiele für Aromaten mit n = 2 10 delokalisierte π – Elektronen Naphtalin N N Isochinolin Chinolin 6. Beispiele für Aromate n mit n = 3 14 delokalisierte π – Elektronen Anthacen Phenanthren 2.10.3 Reaktivitätsvergleich Cyclohexen Benzol + KMnO4 rasche Oxidation keine Reaktion + Br2 Entfärbung durch Addition keine Reaktion + HI Addition keine Reaktion + H2 (Ni-Kat.) rasche Hydrierung bei 25°C / 1 – 1,5bar langsame Hydrierung bei 100-200°C - 71 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.10.4 Nomenklatur von Benzolderivaten und de r Einfluss der Substituenten 1. Nomenklatur Cl Br F Chlorbenzol Fluorbenzol NH2 Brombenzol OH Anilin SO2H Benzoesäure Br Br 1,2 - / o - Dibrombenzol Toluol COOH Phenol Br CH3 Benzosulfonsäure Br Br Br 1,4 - / p - Dibrombenzol 1,3 - / m - Dibrombenzol COOH NO2 Br CH3 Cl m – Chlorbenzoesäure O2N O2N p – Bromnitrobenzol NO2 2,4,6 – Trinitrotoluol (TNT) 2. Elektrophile Substitution am Aromaten H X X - Komplex X H X H C - H+ H + H C + H - 72 - C + X H Gr u n dla g e n de r O rg a ni sc he n C he mi e 3. Substituenteneinfluss CH3 CH3 CH3 CH3 SO3 H H2SO4 / SO3 95°C SO3 H SO3 H 32% 62% Substituent Verhalten bei weiterer Substitution CH3, C2H5, Alkyl, (-) - I – Effekt -OH, -O-CH3, -NH2 (+) – M – Effekt Cl, Br, (+) – M – Effekt CH2Cl (-) - I – Effekt -NO2, -COOC2H5, O O C CH3 C H, , -CF3, -N+(CH3)2 6% Der Substituent dirigiert in ortho- und paraStellung, und erhöht die Reaktionsgeschwindigkeit im Vergleich zum Benzol. Es liegt zwischenzeitlich der Mesomeriestabilisierte σ – Komplex vor. Diese Substituenten dirigieren in o- und para- Stellung, wobei sie die Reaktion im Vergleich zum Benzol verlangsamen, da eine elektrophile Substitution erfolgt. Der elektronenziehende Substituent dirigiert in m-Stellung und verringert die Reaktivität. - 73 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 4. Beispielreaktionen NO2 ? +NO 2 HNO3 Nitriersäure H2SO4 NO2 Br +Br FeBr3 Br2 NO2 ? + Br +Br Br 62% FeBr3 Br2 38% NO2 +NO 2 HNO3, H2SO4 Nitriersäure Br CH3 COOH ? NO2 KMnO4 COOH HNO3, H2SO4 Nitriersäure - 74 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.10.5 Reaktionen des Benzols Benzol geht bevorzugt Substitutionsreaktionen ein. 1. Nitrierung H2SO4 C6 H6 + HO NO2 Benzol C6 H5 NO2 + H2O Nitrobenzol Salpetersäure Mechanismus: H2SO4 + HNO3 H3O + - HSO4 + + + NO2 elektrophil NO2 +NO 2 C + NO2 -H+ H H mesomeriestabilisierter - Komplex 2. Sulfonierung C6H6 + HO SO3H C6H5 Schwefelsäure SO3H + H2O Benzosulfonsäure Mechanismus: 2 H2SO4 H3O SO3 + - HSO4 + SO3 + SO3 H H SO3 C + H 3. Halogenierung C6H6 + Cl2 Fe oder FeCl3 - 75 - C6H5 Cl Chlorbenzol + HCl Gr u n dla g e n de r O rg a ni sc he n C he mi e 4. Friedel-Crafts-Alkylierung C6 H6 AlCl3 R Cl + C6 H5 R Alkylbenzol + HCl Mechanismus: R X AlCl3 R + AlCl3X R R H R+ C + - Komplex 5. Friedel-Crafts-Acylierung Cl C6H6 + C R O Carbonsäurechlorid AlCl3 C6H5 C R + HCl O (Benzolalkylketon) Mechanismus: O O R C + + AlCl3 R C + AlCl4- Cl O O R C O + H C + + C R - Komplex - 76 - C R Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.10.6 Alkylbenzole 1. Darstellung R R Cl AlCl3 O R C Cl AlCl3 O C R CH3 SO2 LiAlH4 OH O R Cl R SO2 CH3 CH2 LiAlH4 R 2. Reaktionen a) Hydrierung R R H2 R H Ni / Pd b) Oxidation C2 H5 H3C COOH KMnO4 CH3 + KMnO4 para-Xylol HOOC CO2 COOH Terephthalsäure - 77 - Gr u n dla g e n de r O rg a ni sc he n C he mi e c) Halogenierung CH3 Kälte Kern Katalysator (KKK) CH3 Sonne Siedehitze Seitenkette (SSS) CH2 Br h. + Br2, 0 - 20°C HBr CH3 CH3 Br2, AlBr3 Br Br Br CH2 CH3 Br CH CH3 h. CH2 CH2 + Br2, C H2 C CH3 CH3 C stabileres Benzylradikal instabiler C + Ph Ph Ph C C Ph Ph Ph C CHOCH2 - 78 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.11 Aldehyde & Ketone 2.11.1 Beispiele Strukturformel O HC H Name Strukturformel Formaldehyd (Methanal, Biozid) H3C C CH3 Acetalaldehyd (Ethanal) H3C CH2 C CH3 O H3C C H Name Aceton O 4 2 3 1 Methylethylketen (2-Butanon) O O Benzaldehyd C Cyclohexanon O H Trioxan4 O O O 5' H3C Acetophenon C CH3 NO2 O 6' 4' 1' 3' O (• = C-Atom mit zwei O-Atomen verbunden) C 2 3 3-Nitro-4’-methylbenzophenon 4 1 2' 6 5 2.11.2 Darstellung der Aldehyde 1. Oxidation primärer Alkohole O Pyridin R CH2 OH R C CrO3 / HCl oder DMSO H 2. Oxidation von Methylbenzolen Ar h. CH2 Cl Ar CHCl2 Cl2 Ar H2O CH3 CrO3 Ac2O O Ar CH O C CH3 Ar = aromatischer Rest 4 Trimeres des Formaldehyds - 79 - H2O 2 O Ar C H Gr u n dla g e n de r O rg a ni sc he n C he mi e 3. Reduktion von Säurechloriden O R C LiAlH(O Bu) Bu 3 Cl (Carbon-) Säurechlorid O R C H 5 4. Reimer-Thiemann-Reaktion OH CHCl2 CHCl3 OH CH OH O NaOH O 70°C O O C C H - H2O O H + H2O OH - -OH 2.11.3 Darstellung der Ketone 1. Oxidation sekundärer Alkohole R CH OH R KMnO4 C O R R Beispiel sekundär OH O KMnO4 CH3 CH3 sekundär HO OH HO CH2 O OH tertiär CH3 O OH C O primär 5 O KMnO4 Bu = Butylrest - 80 - H C O OH Gr u n dla g e n de r O rg a ni sc he n C he mi e 2. Friedel-Crafts-Acylierung Cl AlCl3 C R + C R O O O O + C CH3 O AlCl3 C CH3 C CH3 O 3. Fries’sche Verschiebung O O C R H3C AlCl3 H3C OH Arylether C O R 4. Säurechloride + Organokupferverbindungen CuX R MgBr GrignardVerbindung R BuLi BuLi O O C R' + Cl R 5. Acetessigestersynthese später in VL - 81 - R C R' Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.11.4 Reaktionen der Aldehyde und Ketone O C R Elektrophil E+ Nucleophil Nu- C H sauer 1. Reduktion a) katalytische Hydrierung O H3C CH2 C H2 / Ru H3C CH2 CH2 OH H primärer Alkohol Aldehyd H2 / Pa - Ni H3C C CH2 CH3 H3C CH CH2 CH3 OH O Keton sekundärer Alkohol b) Reduktion zu Kohlenwasserstoffen H Zn / HCl C H CLEMMENSEN-Reduktion C O H2N NH2 H KOH C WOLFF-KISHNER-Reduktion H Reaktionsmechanismus: O + HN 2 - H2O NH2 H N N H Cyclohexanon Hydrazon KOH C N N N N H H H H2O KOH N NH N N H C + N2 - -OH + H2O - 82 - H H H Gr u n dla g e n de r O rg a ni sc he n C he mi e 2. Oxidation Aldehydnachweis a) Fehling-Test O R C NaOH, H2O 2+ Cu + Tatrat H O Cu2O + + R C R C OH Das Kupfer wird also von dem Aldehyd reduziert. b) Tollens-Nachweis (Silberspiegel) + O Ag R C NH3 / H2O H O Ag OH "Silberspiegel" c) Baeyer-Villiger - Oxidation O + Cyclohexanon O O O H O O H O O R C C R Percarbonsäure O - O R C OH O Polyester Siebenringacton (innermulekularer Ester) Caprolacton O HOOC (CH2 )5 OH H+ HOOC - H2O O (CH2 )5 C O (CH2 )5 C O ... 3. Reaktionen mit Wasser und / oder Alkoholen a) Basenkatalysierte Hydratisierung C O -OH O C H H2O OH C OH - -OH Gleichgewicht liegt auf linker Seite b) Säurekatalysierte Hydratisierung C O H+ C OH H2O OH C O H - 83 - OH C + H OH Gr u n dla g e n de r O rg a ni sc he n C he mi e c) Bildung von Halbacetalen C O HO R / H+ OH C O R Halbacetal OH O O H+ OH H3O + Halbacetal d) Acetale, Vollacetale HO R / H+ C O H3O O R C + O H C O R Vollacetal (Diether) (stabil) C O O H Mechanismus H+ + C O R OH + C O H OH C O H C O + H H H+ OH + O H C H - H2O + C O R + O R C O R R O R - H+ O R C O R Vollacetal - 84 - O C O R H R Gr u n dla g e n de r O rg a ni sc he n C he mi e 4. Reaktionen mit Grignard-Verbindungen O HO O H+ R C + H R C HO H O cyclisches Vollacetal H+ / H2O O Br HO CH2 CH2 C + H Mg Br Mg O H+ Br CH2 CH2 CH HO CH2 CH2 CH Grignard-Verbindung O O CH3 H3C C CH3 O Aceton O H3C C CH2 CH2 CH O OH O OH H3O+ CH3 O H+ H3C C CH2 CH2 C OH O H H3C CH3 cyclisches Halbacetal Cyclische Halbacetale bilden sich auch bei Aldosen (Zucker): H O OH C C O OH - 85 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 5. Reaktionen mit primären Aminen O C O + H2N R C + N R H H - H2O OH H+- C Umlagerung C N R NH R Imin (SCHIFF'sche Base) H3C C O + H3C - H2O H2N H3C C N H3C C O + H H2N Benzaldehyd mit Ring in para-Stellung C N Flüssigkristalle (Flüssigkeit mit Ordnungsstruktur) R N OH + C O N NH2 R' Hydrazon Oxim - H2O - H2O H2N NH2 H2N OH O2N O O H2N NH O2N NO2 - H2O - H2O H2N NH C NH2 semi-Carbazid O N NH NO2 N N C NH2 semi-Carbazon 2,4-Dinitrophylhydrazon - 86 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 6. Reaktionen mit sekundären Aminen H3C H2C C O + H3C H2C H N H3C H2C OH C H3C H2C - H2O H3C HC N C N H3C H2C Enamin O + HN - H2O CH3 CH3 N CH3 CH3 Enamin O C + H N - H2O S CH N S H Enamin 7. Addition von Blausäure OH O R C + H C N R CH C N Cyanhydrin H OH O + OH HCN C N Cyanhydrin CH2 NH2 Aminoalkohol NH2 COOH Aminosäure - 87 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 8. Wittig-Reaktion Ph R' P Ph + O C Ph R'' C R' C C R'' Alken Ylid O Ph3P CH2 + CH2 O R C + Ph3P CH CH3 R CH CH CH3 H Mechanismus: R' + Ph3P CH R Ph3P C - H R + R' C O C O R'' R'' H2C R' R' C O C R'' R'' + O CH2 H2C PPh3 PPh3 Zur Herstellung des Ylids: Ph3P + CH2 R X BuLi + SN Ph3P CH R Ph3P CH2 R + X Phosphoniumsalz + Ph3P C- H R Ylid ist ein inneres Salz - 88 - + PPh3 Gr u n dla g e n de r O rg a ni sc he n C he mi e 9. Industrielle Vitamin -A 1 -Synthese H3C CH3 CH3 CH3 PPh3 O C CH3 + CH3 H Verknüpfung durch H3C O O WITTIG-Reaktion CH3 CH3 CH3 O O C CH3 H CH3 1. -OH 2. H2O H3C CH3 CH3 CH3 CH2 OH H CH3 Vitamin A1 10. Pinakol -Kupplung O O H3C C CH3 Mg o. Na + H3C C CH3 + Na Radikal-Anion O O HO OH Dimerisierung Kupplung + H3C C C CH3 + 2 Na H2O H3C CH3 H3C C C CH3 H3C CH3 Pinakol 11. Bildung von Hydraten Aldehyde und Ketone bilden in Wasser geringe Konzentrationen an Diolen. OH C C O Keton / Aldehyd - 89 - OH Diol Gr u n dla g e n de r O rg a ni sc he n C he mi e O R C H Aldehyd + H2O OH H R C OH H H Hydrat des Aldehyds O R C + NH3 R C H H OH H N H2 O R C R' + H2O OH H R C OH H R Hydrat des Ketons Keton O R C R' + NHR2 R C Keton H R' - HCl Cl C H OH H N R2 + H2O H C O Cl + H2O H Cl C H H Cl Normalerweise sind die hydratisierten bzw. mehrfach mit Heteroatomen substituierten Aldehyde nicht so stabil wie der eigentliche Aldehyd, allerdings gibt es Ausnahmen, falls es sich um einen elektronenziehenden Rest handelt: O OH H Cl3C C + H2O Cl3C C H OH H H Aldehyd mit stabileres Hydrat elektrodenziehendem des Aldehyds Rest 2.11.5 Acetale der Mercaptane Mercaptane (R-SH) reagieren wie Alkohole (R-OH): O H+ + R C HS R H BuLi S R S R R' S R R CH S R Dithioacetal SR Br HgCl2 R' R C R C S Carb-Anion (Nucleophil) - 90 - R H2O O R C R' Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.12 Carbonsäuren und deren Derivate O R C O H 2.12.1 Beispiele Strukturformel Name O Ameisensäure H C O (Methansäure) H O Essigsäure H3C C O (Ethansäure) H O Propionsäure H3C CH2 C O H O Buttersäure H3C CH2 CH2 C O H O Benzoesäure C O H O Acrylsäure H2C CH C O H O O R C + R C O H O O H O H O + R C + NH4 O KOH R C O O NH3 R C + H - H2O - 91 - O R C + K O + Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.12.2 Industrielle Herstellung Carbonsäuren werden industriell aus Kohlenwasserstoffen, Aldehyden, Alkoholen sowie Alkoholen mit Kohlenmonoxid hergestellt. 2.12.3 Laborsynthesen 1. Oxidation primärer Alkohole KMnO4 R CH2 OH R COOH 2. Oxidation von Alkylbenzolen O2N K2Cr2O7 H2SO4 CH3 O 2N COOH Parantriotoluol 3. Grignard-Verbindungen + CO 2 R X + Mg R MgX O 13CO O2 13 R C O H+ 13 R C O MgX OH Dies ist eine wichtige Methode zur Markierung, um zum Beispiel Metabolismen in der Natur zu erforschen. K2Cr2O7 13CO Ba13CO3 2 H2SO4 4. Hydrolyse von Nitrilen R C N H3O+ O R C H3O+ NH2 Carbonsäureamid - 92 - O R C OH Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.12.4 Reaktionen der Carbonsäuren 1. Salzbildung NaOH R COOH + Na R COO O- - H2O 2. Ersatz der OH -Gruppe O O R C R C OH Z Säurechloride O O R C + OH SOCl2 oder PCl3 oder POCl3 R C - SO2, -HCl Cl Esterbindung O R C + Et3N HCl R'' O O Amidbindung R C HO R'' NEt3 NH3 OH SOCl2 O R'2CuLi R C R' O R C Cl O R C NH2CH R C H Aldehyd R C NH CH3 O R C N HN CH3 LiAlH O C CH3 CH3 3 O NH2 O Carbonsäureamide 3. Reaktionen mit Estern und Amiden O R C + OH HO R' H+ O R C Esterbindung O + H2O R' Die Reaktionsgeschwindigkeit nimmt hierbei mit zunehmendem Substitutionsgrad ab: primär > sekundär > tertiär. - 93 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 4. Reduktion O O oder R C 1. LiAlH4 R C O H R' O R CH2 O H 2. H2O 5. Substitutionsreaktionen a) Halogenierung in α – Stellung Br2 R CH2 COOH R CH COOH Prot Br b) aromatische Carbonsäuren COOH +NO 2 O 2N COOH 2.12.5 Carbonsäureanhydride 1. Industrielle Herstellung CH3 COOH AlPO4 CH2 C O + H2O Keten O H3C C O H3C C O Ac2O Essigsäureanhydrid (Acetanhydrid) 700°C CH3COOH O COOH - H2O O 200°C COOH O Phthalsäureanhydrid Phthalsäure - 94 - Gr u n dla g e n de r O rg a ni sc he n C he mi e Aspirin ® -Herstellung O H3C C O HOOC HOOC HO H3C C O + H3C C O Salicylsäure O Acetylsalicylsäure ASS / Aspirin 2. Reaktionen a) Esterbildung O O H3C C O + HO H3C C O + CH3COOH H3C C O Essigsäurephenylester O O HO CH3 COOCH3 O C OH O COOCH3 SOCl2 O COOCH3 HO C2H5 NEt3 C COOC2H5 Cl unterschiedlich substituierter Ester b) Reaktion mit Amiden O O H3C C O + H2N H3C C Kochen O - 95 - H3C C NH Gr u n dla g e n de r O rg a ni sc he n C he mi e c) Friedel-Crafts-Acylierung CH3 O CH3 CH3 AlCl3 H3C C O CH3 + CH3 CH3 H3C H3C C O C O O O Benzol O AlCl3 COOH O O O AlCl3 SOCl2 COCl O tricyclisches Diketon 2.12.6 Reaktionen der Carbonsäureamide 1. Hydrolyse O HCl R C OH O R C + NH4Cl + NH2 H2O H+ NaOH - NH3 O R C + + Na O 2. Hofmann-Abbau Die Kohlenstoffkette wird an der funktionellen Gruppe um ein C-Atom verkürzt. O NaOCl oder R C R NH2 + CO2 NaOBr NH2 Amin Amid - 96 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.12.7 Reaktionen der Ester 1. Hydrolyse Verseifung O R C H+ R' OH Na + R' OH + OH O R C O R' NaOH O R C + O Seife O O C CH2 CH3 n O Umesterung O C CH2 CH3 + m H / HO CH3 O O C CH2 CH3 o Triester des Glycerins (Fett) CH2 OH O H3C O C CH2 CH3 n O H3C O C CH2 CH3 m O H3C O C CH2 CH3 Glycerin Biodiesel CH2 OH CH OH + o 12 < n,m,o < 20 n,m,o gerade Interessant hierbei ist, dass infolge der vermehrten Erzeugung von Biodiesel der Preis des ‚Nebenproduktes’ Glycerins von 1 €/kg im Jahre 2003 auf 0,5 €/kg gesunken ist. O O C Ölsäure ungesättigte O Fettsäuren O C Linolsäure O O C Alkansäure Exkurs - Warum man Seife nicht für die Haare benutzt Seife ist zum einen alkalisch (Brennen in den Augen), und bildet außerdem noch wasserunlösliche Calcium- und Magnesiumsalze der Fettsäuren: - 97 - Gr u n dla g e n de r O rg a ni sc he n C he mi e -OOC O O + C O Na Myzel -OOC O Dies wird bei Emulgatoren zur Homogenisierung von organischen Verbindungen in Wasser benutzt. Statt Fettsäuren bzw. deren Salzen verwendet man stattdessen Alkylsufonate (Tenside), deren Ca- / Mg- Salze wasserlöslich sind: - SO3 + Na 2. Aminonolyse O O R C + H N R C R' O N O O R C Ph H2N + R C R' O NH Ph 3. Reaktion mit Grignard-Verbindungen O R C O + BrMg R'' R' OH R C R' R'' tertiärer Alkohol 4. Reduktion mit LiAlH 4 O 1. LiAlH4 O 2. H2O R C R' - 98 - R CH2 O Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.12.8 Kohlensäurederivate HO C OH - H2O CO2 O Kohlensäure 1. Beispiele Cl C Cl H2N C NH2 O O Phosgen (gasförmig, giftig) (wird direkt bei Verwendung hergestellt) H5C2 Harnstoff (Diamid) C C2 H5 CH2 O C Cl O O Diethylcarbonat (Diester) Chlorkohlensäurebenzylester HO C NH2 spontaner NH3 CO2 Zerfall O Carbaminsäure Isocyanat, Isocyansäure NH2 O C N R O C Isocyanat NH3 R NH2 NH2 Cl O C Cl H2O OH O C NH3 HO R O R O C Cl O C NH Isocyansäure Cl HO R O R CO2 + HCl O C O R - 99 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2. Anwendungsbeispiele Phosgen H2N CH2 6 NH2 O C N CH2 6 N C O Polyaddition Diisocyanat O HO O C NH O O CH2 6NH C O O OH HO O C NH CH2 6 NH C O Urethan Polyurethan (Verwendung u.a. als Bauschaum) O O H NH2 H5C2 O C N O C O C CH2 + O C NH2 H5C2 O C N O C H O O Barbitursäure Malonester (Schlafmittel) Warum Barbitur-‚Säure’? Urethan, Polyurethan, Malonester, Barbitursäure O H N O C H O C H N O C O C N O C H H O O + C H N O C H H O Carbanion H N O C O + O C H H N O C H O Das rote C-Atom besitzt zwei Carbonylgruppen als Substituenten, wodurch ein (-)-I-Effekt resultiert. - 100 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.13 Carbanionen I H C C O Mesomerie- B - B-H C C C C stabilisiert O O Enolat Carbanion Carbonylverbindung 2.13.1 Anwendungen 1. Aldolreaktion, Aldolkondensation O O -OH CH3 C CH2 C - H2O H Acetaldehyd O CH3 C H O CH3 C CH2 C H H Carbanion OH O * CH3 CH CH2 C H rac-Aldol (Ende der Aldolreaktion) H2O O H+ O CH3 CH CH C O + H H O CH3 CH CH C H H --ungesättigte Carbonylverbindung, als Produkt der Aldolkondensation H H 2. Knoevenagel -Kondensation Z C O + H2C Z OH -OH C H2O Z CH Z Z C C Z Alken Es entsteht das analoge Reaktionsprodukt des rac-Aldols. Z ist ein elektronenziehender Substituent wie z.B. COOR , CN , C R O C H O , Mechanismus COOR H2C B- COOR C O HC COOR COOR Malonsäureester Carbanion - 101 - COOR 1. + H O 2 CH 2. - H2O O COOR COOR C COOR --ungesättigter Ester Gr u n dla g e n de r O rg a ni sc he n C he mi e 3. Perkin-Kondensation O O C H3C C B- O + CH CH COOH H3C C H Zimtsäure O 4. Claisen-Kondensation O O H3C C O C2 H5 Ethylacetat O H3 C C -OR O C2 H5 H2C COOC2 H5 Carbanion H3C C CH2 COOC2 H5 O 1. NaOH H3C C CH3 2. H+ 3. 5. Dieckmann-Kondensation (innermolekulare Claisen-Kondensation) H NaOR COOC2H5 H5C2OOC C COOC2H5 H5C2OOC C6-Carbonsäureester Letzteres Produkt lässt sich auch folgendermaßen aufschreiben: H5C2OOC O H5C2OOC C C H OC H 2 O C 5 5-Ring--Ketonester HOOC -OH O H O O C C O C Verseifung -Keto-Carbonsäure H O Keto-Enol O Tautomerie cyclisches Keton In der Regel sind OH-Gruppen an C-C-Doppelbindungen instabil. Keto – Enol - Tautomerie Enol - 102 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.14 Amine 2.14.1 Beispiele CH3 4 N+ Cl- CH3 NH2 CH3 2 NH CH3 3 N Methylamin (primär) Diemethylamin (sekundär) Trimethylamin (tertiär) N H N H N Pyrrolidin Pyriol Pyridin NH2 Anilin Tetramethylammoniumchlorid (quartär) Amin +Säure Salz + R-NH 2 + HCl R- NH 3 ClR-NH 2 + HOOC-R R- + NH 3 - OOC-R 2.14.2 Darstellung der Amine 1. Reduktion von Nitroverbindungen Zn / HCl NO2 NH2 2. Alkylhalogenide + NH 3 (ergibt Gemische) R X + NH3 R NH2 primäres Amin R X ... RNH2 + R2NH + R3N Das Stickstoffatom ist hierbei durch den (+)-I-Effekt elektronenreicher. gezielte Herstellung primärer Amine durch Gabriel-Synthese O COOH NH3 COOH NH 300°C Phthalsäure Phthalimid O - 103 - Gr u n dla g e n de r O rg a ni sc he n C he mi e O K2CO3 O N K R X + N R SN O O O N H H2N NH2 + N H H2N R primäres Amin O 3. Reduktive Aminieru ng a) O + NH3 NH H2NR NR H2 NH2 Ni b) O + H2 NHR Ni b’) O + H2N CH2 Ph H2 Ni N CH2 Ph NH CH2 Ph c) O + HNR2 - 104 - NH2 - CH2 Ph H2 Ni H NR2 Gr u n dla g e n de r O rg a ni sc he n C he mi e Modernes Beispiel für b’) H H H CH3 O + H2N N * H3C Ph H H2 H H CH3 H3C * H3C Ph H NH * * Ni H CH3 H NH2 * Ph H3C ca. 80% ee* zwei Diastereomere nicht 1:1 *ee = Enantiomerenüberschuss 4. Reduktion von Oximen 1. LiNH2 N OH NH2 2. H2O Oxim 5. Reduktion von Nitrilen R C N 1. LiNH2 2. H2O R CH2 NH2 6. Reduktion von (Carbonsäure-) Amiden O 1. LiNH2 R C 2. H2O NH2 R CH2 NH2 7. Hofmann-Abbau O NaOBr R C R NH2 + CO2 NH2 Mechanismus O R C O -OBr R C NH2 - -OH -OH Br N - H2O O R N C O Isocyanat H2O R C N Br H O R NH C OH Carbaminsäure (instabil) - 105 - O - Br- R C - CO2 Nitren R NH2 N Gr u n dla g e n de r O rg a ni sc he n C he mi e 8. Curtius-Abbau O NaN3 Cl - NaCl R C O O R C R C + N3 N N N Carbonsäureazid O R C - N2 R NH2 siehe HOFMANN-Abbau N Nitren 2.14.3 Reaktionen der Amine 1. Salzbildung H NH + + HCl N Cl H wasserlöslich wasserunlöslich NH HCl H NH + CH3COOH N O + H3C C H O Diese Salzbildung wird bei Pharmazeutika ausgenutzt, um an sich wasserunlösliche Stoffe durch ihre Salze in Wasser lösen zu können. 2. Alkylierung R' R NH2 + Br R' R NHR' + R N R' + R N R' R' R' Gemisch mehrerer Amine SN + Bei großen Resten können manche der entstehenden Amine aufgrund sterischer Hinderung nicht gebildet werden. 3. Umwandlung zu Amiden O O R NH C R' Amid C R' Cl Ac2O Base R NH2 R' OOC R'' O R NH Ac Amid R' SO2 Cl - R' OH R NH SO2 R' Sulfonamid R NH C R'' - 106 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 4. Hofmann-Eliminierung quartärer Am moniumsalze H / -OH C C C C Alken +NR N 3 Details HO H H AgOH R X C C C C SN NR2 C C E2Mechanismus +NR N 3 Beispiel H ? N NR2 H H HO R-Br N R + (AgOH) R 5. Reaktion mit HNO 2 NH2 N HNO2 + N Cl NaNO2 / HCl Diazoniumsalz Normalerweise sind Diazoniumsalze instabil, allerdings ist dieses spezielle Salz durch den Aromaten mesostabilisiert und ist daher bei Temperaturen unter 0°C stabil. + N N+ Cl O CH3 N N O CH3 Diazofarbstoff Diazofarbstoffe werden zur Färben von Textilien und Leder verwendet, wobei eine breite Vielfalt an Farben durch unterschiedliche Substituenten an den beiden Ringsystemen möglich ist. Allerdings stehen diese Stoffe auch im Verdacht krebserregend zu sein und wurden deshalb, zumindest in Deutschland, verboten. H3C CH2 CH2 NH2 NaNO2 HCl H3C CH CH2 + H3C CH2 CH2 N N Cl (instabil) - N2 + H3C CH2 CH2 OH Gemisch - 107 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 6. Mannich-Reaktion O O C H H HCl H2CO + H2N R + C H CH2 NHR Formaldehyd Amin O + H2CO + HN H CH3 HCl O CH3 CH2 N Dimethylamin CH3 CH3 Mechanismus H2C O + HN O CH3 HCl CH3 H2C N H O Cl CH3 O +CH3 H2 C N CH3 Keto-Enol- +CH3 Cl CH2 N CH3 CH3 Tautomerie 7. Nitrosamine (CH3)2NH NH NaNO2 H + (CH3)2 N NO NaNO2 (CH3)2N NO H + N NO N NO Beide Endprodukte sind krebserregend, und tauchen teilweise in Lebensmitteln wie z.B. gebratenem Fleisch auf. - 108 - Gr u n dla g e n de r O rg a ni sc he n C he mi e O O NO+ - H+ R C NH CH3 Diazomethan verhält sich nach Abspaltung von N2 wie Carben H C H R C N CH3 NO Ether + 40% KOH 0°C + + CH2 N N h . - N2 CH2 N N O Diazomethan oder R C OH H O H R C O CH3 H H Die hier dargestellte Estersynthese ist eine besonders ‚milde’ Variante, da sie ohne starke Säuren und Hitze auskommt. 8. Herstellung von Nylon 66 (Polyamid) COOH (CH2 )4 COOH + Diamin Dicarbonsäure -O OOC (CH2 )4 COO O- + H3N O 270°C (CH2 )6 NH2 H2N + + (CH2 )6 NH3 O O (CH2)4 C NH (CH2)6 NH C NH (CH2)4 C NH -H2O 9. UGI-Reaktion 2 H2N + 1 5 + R N C + + HOOC R C 4 2 3 R R R Isonitril Amin Carbonsäure Keton - 109 - 4 O R R O O 1 R NH C C N C R 5 R 3 Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.14.4 Physiologisch wichtige Amine HN CH3 H NH2 H H3C CH3 N CH3 HO OH Adrenalin Amphetamin Nikotin (Verwendung als Dopingmittel) (ein Alkaloid, reagieren schwach basisch) - 110 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.15 Phenole 2.15.1 Synthesen 1. NaOH (SN) Cl HCl + O Na OH + NaCl 360°C / 300bar Natriumphenolat Chlorbenzol 2. Cumol-Verfahren O2 H3O + O + (radikalisch) H H3C H3C O O H CH3 H3C Isopropylbenzol CH3 CH3 Aceton OH Cumolhydroperoxid Phenol Das hierbei entstehende Kopplungsprodukt Aceton deckt ca. 90% des Bedarfs in Laboratorien. Mechanismus + H - H2O O O H H3C O O H3C O CH3 H2O O H3C H H CH3 C + O CH3 CH3 H+- H + H3C + + O Wanderung H O + OH + O H C H3C CH3 H C H3C CH3 O H3C 3. Hydrolyse von Diazoniumsalzen + N2 Cl H2O / OH + "Verkochen" Diazoniumsalz - 111 - N2 CH3 Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.15.2 Reaktionen der Phenole 1. Salzbildung Ph OH + NaOH - H2O + Ph O Na Br R Ph O R SN 2. Esterbildung H+ R COOH + HO Ph O R C O Ph O R C + HO O Ph R C Cl O Ph 3. Ringsubstitution O OH 125°C + Na + CO2 4-7 bar + COO O- Na Salicylsäure Phenolat O O Ac2O C O CH3 O C H+ + COO O- Na CH3 COOH Ester Acetylsalicylsäure Die OH-Gruppe dirigiert in diesem Fall in ortho- bzw. para-Stellung. Ersteres wird zum Beispiel durch Kaliumionen sterisch verhindert. O K + + CO2 Phenolat OH -OOC O OH H+ HOOC PHB-Säure Die PHB-Säure wird weiter zu PHB-Estern umgewandelt, die als Konservierungsmittel verwendet werden. - 112 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.16 α – β ungesättigte Carbonylverbindungen C C + 4 C C C O 3 C C 2 C O + + C O elektronenziehender Rest 2.16.1 Beispiele O CH2 C C H CH2 C COOH CH2 C COOR Ph CH C COOH Acrylsäure Acrylsäureester Zimtsäure Acrolein O HOOC H H COOH C C C C O H H COOH COOH O Fumarsäure Maleinsäure Maleinsäureanhydrid 2.16.2 Reaktionen von α – β ungesättigten Carbonylverbindungen Konkurrenz zwischen 1,2 - und 1,4- Addition 1,2 – Addition C C + A 1,4 - Addition B C O C C + A B C O C C 2 1 C O B 4 A B (A=H) 2 2 C OA oder C C 3 C C H 4 3 C C 1 Enol C O H 2 C O B B 1. Blausäureaddition O + Ph C CH CH2 O H C N 1,4 - Add. - 113 - Ph C CH2 CH2 C N Gr u n dla g e n de r O rg a ni sc he n C he mi e Mechanismus H O O H+ Ph C CH CH2 + OH OH N + Ph C CH CH2 Ph C CH CH2 O Keto - Enol - C Ph C CH CH2 C N Ph C CH2 CH2 C N Tautomerie Enol 2. Metallorganische Reagenzien 1,2 – Addition O H3C H3C C CH C CH3 1,4 - Addition CH 3 R CH C O C H - + 1. H3C Li 2. H2O OH H3C H3C CH3 C CH C CH3 R CH CH CH3 O 1. (CH3)2CuLi 2. H2O CH3 O C H O 1. (CH3)2CuLi 2. H2O 1,4 - Addition CH CH2 4. Michael-Addition elektronenziehende Gruppe Z Z CH2 + H3C CH C O -OR (als Katalysator) R R O CH CH2 CH C - ungesättigte Carbonylverbindung Beim Z2CH2 kann leicht ein Proton abgespalten werden, wodurch sich leicht ein Carbanion bilden kann. Beispiele O H3C C H3C C CH2 O elektronenziehender Rest -OR + CH2 C C N Acrylnitril - 114 - O H3C C H3C C CH CH2 CH2 C N O Gr u n dla g e n de r O rg a ni sc he n C he mi e Mechanismus O H3C C H3C C HC H CH CH2 CH C N H3C C H3C C HC H3C C O O H3C C + CH2 C O -OR C N O O H2O H3C C CH CH2 CH2 C N H3C C O O 5. Robinson -Anelierung HC C O - + CH2 + H CH3 CH3 -OR + - CH3 C H+ O O CH3 C O CH3 O CH3 O CH3 -OR + + Aldolkondensation O + H+ CH3 H+ C - H2O OH C A B O H H+ A B Gonan Gonan ist Grundgerüst mancher Hormone, unter Anderem von Sexualhormonen (Steroiden). Bei dieser Art von Synthese entstehen Diastereomere, bei gezielter Synthese verwendet man einen optisch aktiven Katalysator. - 115 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.17 Carbanionen II 2.17.1 Reaktionen 1. Decarboxylierung - Br COOR H2C -OR COOR Malonsäureester 1. -OH HOOC 2. H3O+ HOOC CH3 CH2 CH CH3 + COOR HC ROOC COOR CH CH2 CH CH CH2 CH ROOC CH3 CH3 - CO2 HOOC CH2 CH2 CH CH3 CH3 CH3 CH3 - Ketocarbonsäure Es erfolgt also eine Verseifung der Estergruppe COOR mit anschließender Abspaltung von CO2 (Decarboxylierung) von der Malonsäure durch das Erhitzen. Weitere Verbindungen, bei denen sich leicht Protonen substituieren lassen, sind O C CH2 Z H2C H2C COOR Z Acetessigester Mechanismus der Decarboxylierung H HOOC HOOC CH CH2 CH CH3 O O C C HO O CH3 CH2 CH H3C CH3 H O - CO2 O Keto - Enol - C HO C HO Tautomerie CH2 CH2 CH H3C CH3 - 116 - CH H3C CH3 Gr u n dla g e n de r O rg a ni sc he n C he mi e Beispiele COOR H2C 1. -OR 2. C2H5 Br COOR 1. -OR H3C CH 2. CH3 Br COOR H3C COOR 1. -OH 2. H3O+ H5C2 COOR 3. -CO2 COOR H3C CH COOH H5C2 Dialkylmalonsäurederivat O H2C 1. -OR COOR 2. CH3 Br H3C CH C CH3 1. -OH COOR 2. H3O+ H O H3C CH O C CH3 C CH3 COOH O O C C H3C OH O -CO2 C H3C CH3 O Enol Tautomerie HO CH3 2. CH3 Br - Ketonsäureester H O O O COOC2H5 1. -OR O CH3 CH3 2. H3O+ O CH3 -CO2 COOH 1. -OH OH C C Keton CH3 CH3 O COOC2H5 O Keto - Enol - Enol Keto - Enol - CH3 Tautomerie Keton Die Bezeichnung des α – Kohlenstoffatoms richtet sich nach der am höchsten oxidierten Carbonylgruppe der jeweiligen α – β ungesättigten Carbonylverbindung. - 117 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2. α – Alkylierung von Ketonen in α - Stellung über Enamine (Storck-Verfahren ) H3C CH2 C CH2 CH3 + N H O (sek. Amin) + H3C Br N C H3C CH2 CH CH3 CH CH3 H3C CH2 CH CH3 Enamin - N H2O N CH3 N H H3C CH2 C CH CH3 O C CH3 H3C CH2 CH O CH3 H Mechanismus + H3C CH2 C CH2 CH3 O H N (sek. Amin) - H N H3C CH2 C + CH2 CH3 O N H3C CH2 C CH N CH3 HO H - 118 - - H2O C H3C CH2 CH CH3 Gr u n dla g e n de r O rg a ni sc he n C he mi e Beispiele O O R ? H3C N CH3 HN(CH3 )2 1. R-Hal 2. H3O+ 1. H N O C C H3C H H3C 2. C2H5-Hal 3. H3O+ - 119 - H3C H3C O C C H C2H5 Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.18 Zucker / Kohlenhydrate 2.18.1 Allgemeines Die allgemeine Summenformel für Kohlenhydrate bzw. Zucker lautet: Cn n H2O Ein C-6-Zucker wäre demnach C6(H2O)6. Zucker werden in der Natur bei Pflanzen durch Photosynthese hergestellt: Photosynthese CO2 + H2O D - (+) - Glucose Reagieren diese Produkte weiter in Form von Polymerisation, können Stärke oder Cellulose entstehen. 2.18.2 Einteilungsmöglichkeiten a) Nach Anzahl der Kohlenstoffatome Tetrosen, Pentosen, Hexosen, Heptosen, usw. b) Anzahl der Zuckereinheiten Mono-, Di-, Trisaccharide c) Aldosen / Ketosen Glucose (Aldose) Fructose (Ketose) H2C OH H PhNH-NH3 H N C * CH-OH NH - H2O PH 4 CH2 OH O C * CH-OH C O * CH OH CH2 OH CH2 OH H-CN HNO3 COOH * CH-OH COOH 4 3 4 H2 / Ni H-CN CH2 OH * CH-OH 4 OH * H C C N * CH-OH 4 CH2 OH CH2 OH H2C OH * N C C OH * CH OH 3 CH2 OH Es handelt sich hier bei beiden Kohlenhydraten Glucose und Fructose um Monosaccharide mit 6 Kohlenstoffatomen (Hexosen). Die Reaktionen mit den blauen Pfeilen wurden von Emil Fischer untersucht. d) Ringgrößen Anzahl C-Atome 5 6 Name des Rings Furanoid Pyranoid - 120 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.18.3 Stereochemie für Glucose Fischer-Projektion (nach Emil Fischer) Das höchst oxidierte Kohlenstoffatom wird in der der Strukturformel ganz oben geschrieben, das mit dem niedrigsten Oxidationsgrad unten. H CHO HO *C H CHO HO *C H CH2OH CH2OH O C *CH OH * HO CH *CH OH Ta *CH OH Ta Tü Ta CH2OH Glycerinaldehyd D – (+) - Glucose (Triose) 2.18.4 Bildung von Polysacc hariden Bei einem Disaccharid haben sich zwei Monosaccharide unter Wasserabspaltung über eine Sauerstoffbrücke verbunden, in diesem Fall der Saccharose („Zucker“): H CH2OH O CH2OH H O H HO H H HO H HO OH O CH2OH H OH H α - (D) – Glucopyranosil – β - (D) - Fructofuranosid - 121 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.18.5 Spezielle Reaktionen der Zucker Bei Betrachtung der Zucker fällt auf, dass manche typischen Aldehydreaktionen nicht ablaufen Es gibt α – (D)- Glucose und β – (D) – Glucose, d.h. es gibt Diastereomere durch eine innermolekulare Halbacetalbildung Es bilden sich dabei zwei isomere Methyl-D-Glucoside 1 H *C OH 1 HO *C H 2 H * C OH 3 HO *C H 4 H *C OH cyclische Halbacetale 2 H * C OH 3 HO *C H 4 H *C OH 5 6 H *C O 5 6 H *C O H2C OH H CH2OH H2C OH H O Distereomere H HO OH H HO OH O H HO H H HO CH2OH OH OH H H H - (D) - Glucose - (D) - Glucose [] = +19° [] = +112° Durch diesen, als Mutarotation bezeichnete Gleichgewichtsreaktion zwischen α- und βGlucose ist zu sehen, dass beide Zucker in Form von Halbacetalen keine Carbonylfunktion mehr besitzen. - 122 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.19 Aminosäuren, Peptide und Proteine 2.19.1 Einteilung und Beispiele * H3C CH COOH CH2 COOH NH2 NH2 Glycin (Gly) Alanin (Ala) Aminoessigsäure (nicht chiral) 1. essentielle / nichtessentiell Während nichtessentielle Aminosäuren vom Körper selbst hergestellt werden können, ist er auf eine Aufnahme von nicht selber synthetisierbaren essentiellen Aminosäuren angewiesen. 2. neutral / sauer / basisc h * HOOC CH2 CH COOH H2N NH2 CH COOH NH2 Asparaginsäure (Asp) (sauer) Lysin (Lys) (basisch) 3. offenkettige / cyclische N COOH H2N CH2 COOH H Prolin (Pro) + H3N CH2 C O O Zwitterion (Betain) 4. proteinogene / nichtproteinogene Es gibt ca. 20 proteinogene Aminosäuren, aus denen die Proteine in der Natur aufgebaut sind. Ein Beispiel für eine nichtproteinogene Aminosäure ist das Penicillamin COOH NH2 H3C SH H3C - 123 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 2.19.2 Synthesen 1. Strecker-Synthese H3C CH CHO H3C Aldehyd NH3 H3C -H2O H3C CH CH NH Imin + * C N H3O CH CH H3C NH2 HCN H3C H3C * COOH CH CH H3C NH2 rac. Aminonitril rac. D,L - Valin O H3 C C * COOH R CH NH2 CH2 H3 C C * COOH R CH NH C CH3 O O N-Acetyl-Aminsäure (rac.) mit NaOH zu pH 7 H R C L-Acylase HOOC COOH CH R HN + NH2 C CH3 O (R)-N-Acetylaminosäure (S)-Aminosäure 2. Cystein -Synthese O CHO H2C + N NH3 + NaSH S + H3C C CH3 Cl Chloracetaldehyd N C HCN N S CH3 3-Thiazolin COOH CH3 H3O+ S CH3 Aminonitril CH3 NH2 + SH Cystein * O H3C C CH3 Cystein wird insbesondere in der Tierfutterindustrie benötigt, und wurde früher aus Haaren gewonnen. - 124 - Gr u n dla g e n de r O rg a ni sc he n C he mi e 3. Peptidsynthese Gly + Val Gly Gly Val Gly Gly Val Val Val + Gly Val Val Dipeptide viele andere Produkte Tripeptid Zur Unterscheidung bei der Reihenfolge hat man sich auf folgende Nomenklatur geeinigt: (links) N-terminal C-terminal (rechts) Val - Gly Peptidbindung O C-Terminus H2N CH2 C NH CH COOH N-Terminus Gly CH H3C CH3 Val Beispiel - Anwendung der Schutzgruppenchemie zur Peptidsynthese O O Ph H2N CH2 C OH O CH2 O C Cl Ph CH2 O C NH CH2 COOH NaOH Glycin N-geschütztes Glycin O O H2N CH C OH CH H3C CH3 CH3OH SOCl2 H2N CH C O CH3 CH H3C CH3 Valin C-geschütztes Valin H O O Ph CH2 O C NH CH2 COOH + H2N CH C O CH3 N-geschütztes Glycin CH H3C CH3 C-geschütztes Valin - 125 - N C N H (Wasserabfänger) Gr u n dla g e n de r O rg a ni sc he n C he mi e H leicht abspaltbar durch katalytische Hydrierung O O Ph CH2 O C NH CH2 C neue Peptidbindung NH O NH CH C O CH3 + C O NH CH H3C CH3 H O 1. H2 / Pd NH2 CH2 C 2. -OH NH CH COOH CH H3C CH3 Auf diese Art und Weise erreicht man Ketten mit Längen mit bis zu 120 Aminosäuren. Festphasensynthese eine Dipetids nach Merrifield O O C NH CH CH COO O- + Cl P CH2 1 R N-BOC-geschützte Aminosäure O O Polymer CF3 COOH O C NH CH CH C O CH2 R P 25°C 1 O O C NH CH CH COOH O H2N CH CH C O CH2 R R P H 1 O O O C NH CH CH C R CF3 COOH 25°C NH CH CH C O CH2 2 R CH CH C R C N H O P 1 O H2N N 2 O NH CH CH C O CH2 2 R P 1 O HF Abspaltung vom Polymer H2N CH CH C R NH 2 CH CH COOH R 1 Dipeptid Bei der Festphasensynthese erhält man hohe Ausbeuten, bis zu 99,9%, wegen dem vielfachen Abtrennens des CF3COOH durch das Abspülen mit Wasser. - 126 - Gr u n dla g e n de r O rg a ni sc he n C he mi e + H2N CH2 COOH H H CH2 O C Cl CH2 O O C NH CH2 COOH O Das hier verwendete Fmoc-Cl ist besonders leicht wieder abspaltbar, sogar unter basischen Bedingungen. 2.19.3 Anwendungen 1. Süßstoff Phenylanalin Ph CH2 CH COOCH CH3 Methylester NH C O Asparaginsäure CH NH2 CH2 COOH Aspartam / Nutrasweet® - 127 - Gr u n dla g e n de r O rg a ni sc he n C he mi e A1 - Index A F Acetale ....................................... 84, 90 Acetessigestersynthese.....................81 Addition 17, 29, 31, 32, 33, 34, 40, 42, 66, 71, 87, 113, 114 Aldehyde........ 2, 79, 82, 83, 89, 90, 92 Aldehydnachweis .............................83 Aldolkondensation ................... 52, 101 Aldolreaktion .................................101 Aliphate ...........................................10 Alkane 2, 4, 10, 13, 14, 15, 18, 28, 49, 51 Alkene 2, 4, 10, 14, 17, 23, 24, 26, 28, 36, 41, 69 Alkine ........ 2, 4, 10, 17, 18, 24, 65, 66 Alkohole . 2, 16, 17, 22, 24, 28, 49, 54, 56, 57, 60, 79, 80, 83, 90, 92 Alkoholische Gärung .......................50 Alkylanz ..........................................64 Alkylbenzole .............................. 77, 92 Allylstellung .............................. 34, 41 Amide ........................ 93, 95, 105, 106 Amine ...... 2, 18, 86, 87, 103, 106, 110 Aminonolyse ....................................98 Aminosäuren.................. 2, 6, 123, 126 Aromat ... 2, 10, 68, 69, 70, 71, 72, 107 axial .................................................12 Festphasensynthese nach Merrifield126 Fischer-Projektion..........................121 Friedel-Crafts-Acylierung .... 76, 81, 96 Friedel-Crafts-Alkylierung.............. 76 Fries’sche Verschiebung ................. 81 B Bindungspolarität...............................4 Blausäureaddition ..........................113 C Carbaminsäure .................................99 Carbanion ......................................114 Carbanionen ......... 2, 22, 101, 114, 116 Carbonsäureamide ...........................96 Carbonsäureanhydride .....................94 Carbonsäuren 2, 36, 91, 92, 93, 94, 105 Chlorierung ......................................14 CIP-Nomenklatur...............................7 Claisen-Kondensation ....................102 Cumol-Verfahren ...........................111 Curtius-Abbau ...............................106 Cycloalkane ............................... 12, 15 Cystein-Synthese ...........................124 D Decarboxylierung ..........................116 Diastereomere ................ 8, 9, 115, 122 Diazoniumsalze...................... 107, 111 Dieckmann -Kondensation.............102 Diene ......................................... 41, 42 Diethylcarbonat................................99 Diethylether .....................................58 Doppelbindung ............ 38, 41, 43, 102 E elektrophil ........................................36 Eliminierungsreaktion ................ 25, 27 E1 25, 26, 27 E2 25, 26, 27 Enantiomere .......................................6 Epoxidation................................ 37, 38 Epoxide / Oxirane ...... 2, 36, 37, 62, 63 equatorial .........................................12 Ester ....................... 18, 52, 93, 97, 112 Esterbildung ............................. 95, 112 Ether .................... 2, 18, 57, 58, 59, 60 G Gabriel-Synthese............................103 Grignard-Verbindungen 14, 21, 52, 64, 85, 92, 98 H Halbacetale ........................ 84, 85, 122 Halogenalkan 2, 16, 17, 18, 20, 21, 22, 26, 59 Halogenierung...... 8, 30, 34, 75, 78, 94 Harnstoff ......................................... 99 Heterolyse ......................................... 4 Hofmann-Abbau ...................... 96, 105 Hofmann-Eliminierung ..................107 Homolyse .......................................... 4 Hückel-Regel .................................. 69 Hybridisierung .................................. 4 Hydratisierung ................................ 83 basenkatalysiert ......................... 83 säurekatalysiert ......................... 83 Hydrierung ............... 14, 29, 71, 77, 82 Hydrolyse .............. 14, 92, 96, 97, 111 Hydroxymercurierung ..................... 50 I innermolekulare Halbacetalbildung122 Isomerie ............................................ 5 K Keto-Enol-Tautomerie ............. 66, 102 Ketone ............... 2, 79, 80, 82, 89, 118 Knoevenagel-Kondensation ...........101 Kohlenhydrate 2, 50, 85, 120, 121, 122 Kohlensäurederivate ....................... 99 Kriterien des aromatischen Zustandes .................................................. 69 M Makromolekül........................ 2, 44, 64 Mannich-Reaktion .........................108 Markownikow-Regel ...................... 38 MARKOWNIKOW-Regel.............. 38 Mercaptane .......................... 18, 61, 90 Metallacetylide ..........................65, 67 Metallorganische Reagenzien ........114 Methylamin....................................103 Michael-Addition ...........................114 Monomere............................ 44, 45, 46 Monoterpene ................................... 45 Mutarotation ..................................122 N Newman-Projektion ........................ 11 Nitrierung ....................................... 75 Nitrile................................. 18, 92, 105 Nitrosamine ...................................108 Nucleophil ........................... 18, 27, 60 Nucleophile Substitution ............18, 53 SN1 ................................ 19, 20, 27 SN2 ................................ 19, 20, 27 O optische Aktivität ...........................5, 6 Oxidation36, 50, 56, 61, 71, 77, 79, 80, 83, 92 - 128 - Oxime ............................................105 P Peptide .......................................2, 123 Peptidsynthese ...............................125 Percarbonsäuren ................... 36, 38, 62 Perkin-Kondensation .....................102 Phenole ........................ 2, 72, 111, 112 Phosgen........................................... 99 Photohalogenierung ...................15, 17 Pinakol-Kupplung ........................... 89 Polyaddukte ...............................44, 48 Polykondensate ..........................44, 48 Polymere ....................................44, 45 Polymerisation ................... 44, 47, 120 ionische ..................................... 47 radikalische ............................... 46 Proteine ................................ 2, 44, 123 R R, S - Nomenklatur ........................... 7 Racemate ................................. 6, 8, 20 Racematspaltung ............................... 9 Radikalkettenreaktion ..................... 15 Reduktion ... 14, 24, 52, 80, 82, 94, 98, 103, 105 Reduktive Aminierung ...................104 Reimer-Thiemann-Reaktion............ 80 Retrosynthetische Analyse .............. 53 Ringöffnung .................................... 63 Robinson-Anelierung .....................115 S Sägebockschreibweise ...............11, 25 Säurechloride ....................... 80, 81, 93 Saytzeff-Regel ................................ 28 Schutzgruppenchemie ....................125 Sesselkonformation ......................... 12 Stereochemie ................... 2, 6, 25, 121 Storck-Verfahren ...........................118 Strecker-Synthese ..........................124 Sulfide ................................. 18, 60, 61 Sulfochlorierung ............................. 15 Sulfonierung ................................... 75 Synthesen ........................................ 53 Harnstoff ..................................... 3 T Twistkonformation.......................... 13 U UGI-Reaktion ................................109 V Veresterung..................................... 55 W Walden’sche Umkehr...................... 19 Wannenkonformation ..................... 13 Wittig-Reaktion .........................24, 88 Wöhler .............................................. 3 Wurtz-Synthese............................... 14 Z Ziegler-Natta-Katalyse / Koordinationspolymerisation .... 48 Zucker ............ 2, 50, 85, 120, 121, 122 Α α – Alkylierung ..............................118 α – β ungesättigte Carbonylverbindungen .........2, 113
![6.3.1 1-Oxa-spiro[2.5]octan - Institut für Organische Chemie](http://s1.studylibde.com/store/data/001356875_1-96e669e5c88ad586db9f9f199d424d05-300x300.png)