On the Complexity of Fundamental Problems in Pedigree Analysis
Werbung

On the Complexity of Fundamental
Problems in Pedigree Analysis
Antonio Piccolboni, Dan Gusfield 2003
Journal of Computational Biology, Vol. 10, No. 5, Pp. 763-773
Seminar „Aktuelle Themen der Bioinformatik“
SS 2004
Martin Löwer
Johann Wolfgang Goethe Universität Frankfurt a.M.
Fachbereich für Biologie und Informatik
-1-
Inhalt
Motivation
2
Allgemeines über Stammbäume
3
Problemstellungen
4
Einschränkungen des Modells
5
MAXP ist NP-hart
5
Approximierung von MAXP
7
Die Klasse #P
8
MP ist #P-hart
8
Diskussion der Ergebnise
12
Motivation
Die Frage nach dem Warum einer Analyse der Komplexität von Problemen bei
Stammbaumberechnungen lässt sich leicht mit der hohen Bedeutung dieser
Berechnungen beantworten: Sie sind oft der erste Schritt beim positionellen Klonen,
sehr nützlich beim Auffinden von bestimmten, für Krankheiten verantwortlichen
Genen und sind in der Landwirtschaft von ökonmischer Bedeutung. Auch bei der
genetischen Epidemiologie und der Kartierung des menschlichen Genoms sind sie
ein wichtiger Bestandteil.
Bei den meisten Analysen ist das Schlüsselelement die Berechnung von gewissen
Wahrscheinlichkeiten im
Stammbaum, wie
z.B.
die Wahrscheinlichkeit der
beobachteten Daten unter der Bedingung, daß einige Daten im Stammbaum fehlen
oder das Zuweisen von Daten, um die Wahrscheinlichkeit des Stammbaums zu
maximieren. Wenn man die genetische Rekombination berücksichtigt, müssen diese
Berechnungen in einer Schleife des jeweiligen Programms wiederholt ausgeführt
werden.
Zwar exisitieren Programme zur Berechnung der Wahrscheinlichkeiten für bestimmte
Spezialfälle von Stammbäumen, aber es ist keine Methode bekannt, die für alle
Stammbäume auch im worst-case effizient arbeitet und es werden in der Praxis
teilweise monatelange Berechnungen in Kauf genommen [1].
Bei der hohen
Bedeutung von Stammbaumanalysen ist dies sehr überraschend.
Die NP-Härte verschiedener Berechnungen in Stammbäumen bei nur geringer
Abweichung von den Fällen, in denen effiziente Berechnungen möglich sind, wurde
-2-
bereits von den Autoren gezeigt [2], der hier diskutierte Artikel wird diese Ergebnisse
noch bestärken und weiterhin zeigen, daß ein spezielles Problem effizient nur bis zu
einem bestimmten Faktor approximierbar ist.
Allgemeines über Stammbäume
Vor der Diskussion der Probleme werden folgende Definitionen und Konventionen
eingeführt:
Ein Stammbaum ist ein gerichteter, azyklischer Graph, der Grad der eingehenden
Kanten ist bei jedem Knoten 0 oder 2 und der zugehörige Heiratsgraph ist bipartit.
Der Heiratsgraph eines gerichteten Graphen G = (V,E) ist ein ungerichteter Graph H
= (V,E*) mit E* = {(v,w) : v,w V und z V : (v,z) E (w,z) E)}
In einem Stammbaum ist also jeder Knoten ein Inividuum, welches entweder als
Gründer der Population bezeichnet wird und somit keine eingehenden Kanten hat
oder als Nichtgründer bezeichnet wird und für jedes Elternteil eine eingehende Kante
hat. Der Heiratsgraph eines Stammbaums verbindet die Individuuen, die ein
gemeinsames Kind haben. Da dazu immer Mann und Frau gehören, ist er
offensichtlich bipartit, andernfalls wäre der zugrunde liegende Graph kein
Stammbaum.
Im zu einem Stammbaum gehörenden ungerichteten Graphen kann es zu Zyklen
kommen, obwohl der Stammbaum ja azyklisch ist. Das geschieht zum einen durch
Inzest, d.h. wenn sich zwei Individuuen, die einen gemeinsamen Vorfahren haben,
paaren und zum anderen wenn sich sowohl Zwillinge (oder Mehrlinge) als auch ihre
Eltern im selben Stammbaum befinden.
Schon seit längerer Zeit sind zwei Arten von Algorythmen [3] [4] bekannt, die
Stammbaumprobleme lösen, aber sie benötigen von der Anzahl der Individuuen bzw.
der Loci abhänig exponentielle Zeit. Dabei wurde lange Zeit angenommen, daß
gerade die Inzestzyklen eine Quelle für Berechnungsschwierigkeiten sind, da der
erste Algorythmus effizient ist, wenn sowohl Zwillings- als auch Inzestzyklen
ausgeschlossen werden. Es wird noch gezeigt, daß diese Annahme im Allgemeinen
nicht richtig ist.
In einer kompletten Instanz eines Stammbaumproblems, gibt es neben dem Graphen
auch zwei zu den Knoten gehörende Zufallsvariabeln, den Genotyp und den
Phänotyp, wobei der Genotyp durch einen Vektor von Paaren mit Allelen genannten
-3-
Zuständen beschrieben wird und ein Modell für die genetische Information ist. Der
Phänotyp ist die Menge der beobachtbaren Eigenschaften eines Individuums. Oft
geht es darum, fehlende Daten für diese Variablen zu berechnen.
Die Zufallsverteilung des Genotyps für einen Gründer i sei P(gi), für einen
Nichtgründer i sei sie P(gi|gv(i),gm(i)), also abhängig von Genotypen von Vater und
Mutter. Der Phänotyp eine Individuums i sei durch die Zufallsverteilung P(yi|gi)
bestimmt, da der Phänotyp aus Wechselwirkung von Umwelt und Genotyp entsteht.
Die
Gesamtwahrscheinlichkeit
(im
folgenden
auch
Score
genannt)
eines
Stammbaumes ergibt sich aus der Multiplikation aller Einzelwahrscheinlichkeiten:
P(G,Y) = Gründer i P(gi) P(yi|gi) Nichtgründer i P(gi|gv(i),gm(i)) P(yi|gi)
G ist dabei dei Menge der Genotypen und Y ist die Menge der Phänotypen des
Stammbaums. Die Wahrscheinlichkeit für den Genotyp von Nichtgründern muß dabei
die genetische Realität wiederspiegeln und wird wie auch im folgenden mit den
einfachen Regeln der Mendelschen Genetik beschrieben.
Problemstellungen
Folgende Probleme der Stammbaumanalyse werden im weiteren Text behandelt:
MAXP
(Maximale
Wahrscheinlichkeit):
Gegeben
sei
ein
Stammbaum
mit
Wahrscheinlichkeitskeitsverteilungen und einer Teilmenge G* der Genotypen G bzw.
Y* Phänotypen Y. Berechne maxG\G*,Y\Y* P(G,Y).
MP
(Marginale
Wahrscheinlichkeit):
Gegeben
sei
ein
Stammbaum
mit
Wahrscheinlichkeitsverteilungen und einer Teilmenge G* der Genotypen G bzw. Y*
Phänotypen Y. Berechne P(G*,Y*) = G\G*,Y\Y* P(G,Y).
Bei MAXP müssen den fehlenden Werten von G mit Daten belegt werden um den
Score zu maximieren, womit man die wahrscheinlichste Erklärung für die bekannten
Daten bekommt. MP liefert eine Berwertung der allgemeinen Güte der gegebenen
Daten, d.h. wenn es möglich ist, eine gute Ergänzung für die lückenhaften Daten zu
finden, sind sie besser zu erklären.
-4-
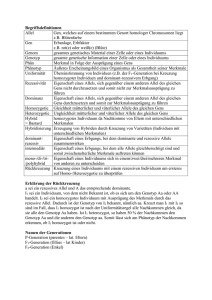
Einschränkungen des Modells
Um die Komplexität der folgenden Beweise zu reduzieren, wird das beschriebene
Modell eingeschränkt. Dies ist unproblematisch, da diese Spezialfälle auch in allen
anderen, komplexeren Szenarien enthalten sind.
So wird nur ein Locus auf einem Chromosom betrachtet. Es gibt nur zwei Alelle, die
mit A und a bezeichnet werden. Der Phänotyp wird vernachlässigt, da er als
deterministische Eins zu Eins Abbbildung des Genotyps bertachtet wird. Zusätzlich
werden Inzestzyklen ausgeschlossen. Der Genotyp eines Individuums kann also nur
die Werte AA, Aa und aa annehmen. Für die Bestimmung der Wahrscheinlichkeiten
bei der Vererbung wird die Mendel’sche Formalgenetik ohne Mutationen benutzt.
MAXP ist NP-hart
Der Beweis wird durch Reduktion vom Problem 3-MIS [5] (maximale unabhängige
Knotenmenge bei maximalen Knotengrad 3) geführt. Eine Instanz von 3-MIS besteht
aus einem Graphen G=(V,E). v sei die Anzahl der Knoten, e die Anzahl der Kanten.
Die Knoten sind mit 1 bis n durchnummeriert und eine Kante ist ein Paar von Knoten.
Aus einem Graphen G wird nun ein Stammbaum MAXP(G) entstehen, der l = 5v + 2e
Individuuen enthält. Dafür wird für jeden Knoten i der Teilbaum in Abbildung 1 in den
neuen Stammbaum eingefügt. Für jede Kante (i, j) in G wird der Teilbaum aus
Abbildung 2 in den neuen Stammbaum eingefügt.
5i, aa
5i+2 5i+4, AA
5i+1, Aa
5i+3
Abbildung 1: Knotengadget für MAXP
5i+2
5j+3
5i+3
{i, j}, Aa
5j+2
{i, j}*, Aa
Abbildung 2: Kantengadget für MAXP
-5-
So sind für jeden Knoten i aus G nur die Genotypen der Individuuen 5i+2 und 5i+3
unbestimmt.
Für
die
komplette
Problembeschreibung
werden
noch
die
Wahrscheinlichkeitsverteilungen für den Genotyp der Gründer 5i, 5i+2 und 5i+4
benötigt. In Übereinstimmung mit dem Hardy-Weinberg-Gesetz wird P(a)=1/3
benutzt. Daraus ergibt sich P(aa)=1/9 und P(Aa)=P(AA)=4/9.
Man kann nun beobachten, daß wenn eine Lösung von MAXP(G) einen Score von
größer als 0 hat, folgendes gilt:
1. i V : Weder Individuum 5i+2 noch 5i+3 hat den Genotyp aa.
Beweis: Individuum 5i+1 muß ein A von 5i+2 erben, 5i+3 erbt auf jeden Fall
ein A von 5i+4.
2. (i, j) E : Die Individuuen 5i+2 und 5j+3 können nicht beide den Genotyp
AA annehmen.
Beweis: Individuum {i, j} muß von einem seiner Elternteile ein a erben.
3. i V : Wenn Individuum 5i+2 den Genotyp AA hat, muß 5i+3 auch AA
haben.
Beweis: 5i+3 erbt dann jeweils ein A von jedem seiner Elternteile.
Diese Beobachtungen werden im weiteren noch gebraucht, um das Verhältnis von G
zu MAXP(G) genauer zu verstehen. Außerdem werden von nun an Stammbäume
ignoriert, die durch ungünstige, in der Natur nicht mögliche Belegung der fehlenden
Daten einen Score von 0 haben.
Zuerst wird der Score von MAXP(G) nach unten abgeschätzt. Dies geschieht indem
man allen Individuuen ohne Genotyp Aa zuweist und dann die einzelnen
Wahrscheinlichkeiten für den Genotyp für jeden Knoten aufmultipliziert:
1/9v 4v/9v 4v/9v 1/2v 1/2v 1/22e =
1
>0
36v 22e-2v
(1)
Der Score kann nur noch größer werden. Wenn s die Größe der Menge M der
Knoten i aus G ist, bei deren zugehörigen Individuuen 5i+2 in MAXP(G) AA
eingesetzt ist, so gilt, daß der Score genau gleich
1
36v 22e-2v-2s
(2)
ist. Beweis: Für jeden Knote i aus G gilt: Wenn wie im obigen Minimalfall (1) Aa für
den Genotyp von 5i+2 eingesetzt wird, hat 5i+3 Aa oder AA als Genotyp, nach
Mendel jeweils mit Wahrscheinlichkeit ½. Würde bei 5i+2 AA eingesetzt, hätte wie
-6-
schon beobachtet auch 5i+3 AA als Genotyp, mit Wahrscheinlichkeit 1. Dann aber
würde in Gleichung (1) ein ½ fehlen, da es durch 1 ersetzt würde. Auch für
Individuum 5i+1 gilt, daß sich seine Genotyp-Wahrscheinlichkeit von ½ auf 1 ändert,
wenn 5i+2 AA statt Aa als Genotyp hat. Daher wird immer, wenn ein Individuum 5i+2
AA trägt, 2 mal der Faktor ½ durch 1 ersetzt.
Grob gesagt werden also so viele Aa wie möglich durch AA ersetzt, ohne daß der
Score 0 wird. Wie oft das geht wird durch die Größe der unabhänigen Knotenmenge
in G beschränkt, wie glecih gezeigt wird.
Die Menge M muss allerdings eine unabhänige Knotenmenge sein. Beweis: Seien i
und j beliebige Knoten aus M, dann hätten durch Definition der Menge M 5i+2 und
5j+2 AA als Genotyp. Dann hätte auch wie beobachtet 5j+3 AA als Genotyp. Aber
5i+2 und 5j+3 können nicht beide AA haben, wie bereits beobachtet.
Wenn M nun maximal ist, wird auch (2) maximiert, denn s, die Größe von M, ist die
einzige Variable in (2). Da aber (2), den Score von MAXP(G), laut Problemstellung
von MAXP maximieren werden soll, muss hierfür die Größe der unabhänigen
Knotenmenge M maximiert werden, was NP-hart ist [5]. Damit ist auch MAXP NPhart.
Approximierung von MAXP
Die NP-Härte von MAXP bedeutet, daß die optimale Lösung des Problems im
schlimmsten Fall nicht effizient berechenbar ist. Die Lösung ist also mit effizienten
Algorythmen nur approximierbar. Bis zu welchem Grad das möglich ist, wird im
folgenden diskutiert. Dazu wird zuerst der negative Logarythmus zur Basis Zwei der
im vorherigen Absatz berechneten Wahrscheinlichkeit betrachtet. Dieser Wert
entspricht
(6 log2 3 – 2)v + 2e – 2s
(3)
Sei s* nun die optimale Lösung von 3-MIS und s die effizient berechenbare. Aufgrund
der Beschränkung auf einen maximalen Knotengrad von Drei ist s* größer als v/4
und e ist höchstens 3v/2. Außerdem ist bekannt daß s*/s größer als 1.0005 ist [6].
Die Approximationsrate von –log(MAXP(G)) ist dann
(6 log2 3 – 2)v + 2e – 2s
1.000071393 = R
(6 log2 3 – 2)v + 2e – 2s*
-7-
(4)
, wobei zu beachten ist, daß das Optimum s* im Nenner liegt, das wir den negativen
Logarythmus betrachten und daher minimieren. Die Richtigkeit dieser Abschätzung
läßt sich leicht überprüfen, indem man obige Schranken für e und v einsetzt. Da auch
das Verhältnis s*/s bekannt ist, kann man R direkt ausrechnen.
Mit Hilfe dieser Konstante und den Beobachtungen, daß ev-1, sv und l8v gilt (l ist
die Größe des Stammbaums MAXP(G), also die Anzahl der Individuuen), kann man
den optimalen Score von MAXP(G) abschätzen.
Score(MAXP(G)) <
1
2(6 log 3 -2)/8(R-1)l-2R+2
(5)
Der optimale Score fällt also exponentiell mit zunehmender Größe l des
Stammbaums, der Rest in (5) ist konstant.
Das bedeutet, solange nicht P=NP gilt, ist wächst das Verhältnis von optimalem
Score zu dem durch effiziente Algorythmen erreichbarem Score exponentiell mit der
Stammbaumgröße.
Die Klasse #P
Ein Problem L gehört zur Komplexitätsklasse #P (Anzahl P), wenn es eine
nichtdeterministische Touringmaschine gibt, die die Anzahl von Lösungen für eine
Instanz von L in polynomieller Zeit berechnen kann.
Die Härt für #P ist analog zur NP-Härte über die polynomielle Reduktion definiert.
MP ist #P-hart
Der Beweis wird durch Reduktion vom Problem #IS [7] (Anzahl von unabhängigen
Knotenmengen in einem Graphen bei maximalen Knotengrad 3) geführt. Eine
Instanz von #IS besteht aus einem Graphen G=(V,E). v sei die Anzahl der Knoten, e
die Anzahl der Kanten. Die Knoten sind mit 1 bis n durchnummeriert und eine Kante
ist ein Paar von Knoten.
Aus einem Graphen G wird nun ein Stammbaum MP(G) entstehen, der l = 3v + 10e
Individuuen enthält. Dafür wird für jeden Knoten i der Teilbaum in Abbildung 3 in den
-8-
neuen Stammbaum eingefügt. Für jede Kante (i, j) in G wird der Teilbaum aus
Abbildung 4 in den neuen Stammbaum eingefügt.
3i, AA
3i+2, Aa
3i+1
Abbildung 3: Knotengadget für MP(G)
({i, j},0), AA 3i+1
({i, j},1)
({i, j},3), Aa ({i, j},6), Aa
({i, j},2)
3j+1
({i, j},7)
({i, j},9), AA
({i, j},8)
({i, j},4), AA
({i, j},5), Aa
Abbildung 4: Kantengadget für MP(G)
Nur die Genotypen der Individuuen 3i+1 und 3i+2 und ihrer direkten Nachkommen
sind unbestimmt.
Die Wahrscheinlichkeitsverteilung für die Genotypen der Gründer wird aus dem
vorherigen Beweis übernommen. Für alle i V ist die Wahrscheinlichkeit für den
Genotyp des Individuums 3i+1 gleich 1/2, was leicht aus Abbildung 3 entnehmbar ist.
Da für alle i V Individuum 3i+1 nicht Genotyp aa haben kann, ohne den Score des
Stammbaums auf Null zu setzen (der triviale und uninteressante Fall), können wir
diese Belegung für die Genotypen der Individuuen 3i+1 und 3j+1 (für die Kante (i,j)
E) bei der Betrachtung der marginalen Wahrscheinlichkeit des Kantengadgets
ignorieren. Diese Wahrscheinlichkeit wird nun als Funktion der eben genannten
Individuuen betrachtet, da sie die einzigen Gründer (bezogen auf das einzelne
-9-
Gadget) ohne belegten Genotyp sind. Tabelle 1 zeigt die entsprechenden
Funktionswerte.
Tabelle 1: Marginale Wahrscheinlichkeit P des Kantengadgets in Abhängigkeit vom
Genotyp von Individuum 3i+1 und 3j+1
3i+1
3j+1
P
AA
AA
0
AA
Aa
3/32
Aa
AA
3/32
Aa
Aa
3/32
Beweis: Zuerst kann man beobachten, daß wenn die Individuuen 3i+1 und 3j+1
beide Genotyp AA tragen, müssen auch die Individuuen ({i,j},1) und ({i,j},8) diesen
Genotyp haben. Da sie aber paaren, kann keiner ein a an ihr Kind ({i,j},5) vererben
und der Score des Gadgets wird Null. Außerdem ist das Kantengadget symmetrisch
und daher resultiern die Genotypbelegungen für die Individuuen 3i+1 und 3j+1 mit
AA und Aa bzw. Aa und AA in gleichen marginalen Wahrscheinlichkeiten für das
Kantengadget.
Nun müssen nur noch die marginalen Wahrscheinlichkeiten für die Vorfahren der
Individuuen ({i,j},4) und ({i,j},5) betrachtet werden. Die Tabellen 2 und 3 decken alle
Fälle für die Belegungen für 3i+1 und 3j+1 außer den eden beschriebenen
Spezialfällen ab. Px ist dabei die Wahrscheinlichkeit für den Genotyp des Individuums
({i,j},x). In der letzten Spalte steht das Produkt der Zeilenwahrscheinlichkeiten und für
jeweils gleiche Zeilen im Bezug auf die Spalten 3i+1 und 3j+1 werden diese
aufsummiert, was jeweils in der Zeile „Summe für ....“ steht.
Tabelle 2: Wahrscheinlichkeiten für das Kantengadget, Vorfahren von ({i,j},5)
3i+1
3j+1
({i,j},1)
P1
({i,j},8)
P8
P5
AA
Aa
AA
1
AA
1/2
0
0
AA
Aa
AA
1
Aa
1/2
1/2
1/4
Summe für Belegung AA, Aa
1/4
Aa
Aa
AA
1/2
AA
1/2
0
0
Aa
Aa
AA
1/2
Aa
1/2
1/2
1/8
Aa
Aa
Aa
1/2
AA
1/2
1/2
1/8
Aa
Aa
Aa
1/2
Aa
1/2
1/2
1/8
Summe für Belegung Aa, Aa
3/8
- 10 -
Tabelle 3: Wahrscheinlichkeiten für das Kantengadget, Vorfahren von ({i,j},4)
3i+1
3j+1
({i,j},2)
P2
({i,j},7)
P7
P4
AA
Aa
AA
1/2
AA
1/4
1
1/8
AA
Aa
AA
1/2
Aa
1/2
1/2
1/8
AA
Aa
Aa
1/2
AA
1/4
1/2
1/16
AA
Aa
Aa
1/2
Aa
1/2
1/4
1/16
Summe für Belegung AA, Aa
3/8
Aa
Aa
AA
1/4
AA
1/4
1
1/16
Aa
Aa
AA
1/4
Aa
1/2
1/2
1/16
Aa
Aa
Aa
1/2
AA
1/4
1/2
1/16
Aa
Aa
Aa
1/2
Aa
1/2
1/4
1/16
Summe für Belegung Aa, Aa
1/4
Für die Wahrscheinlichkeiten aud Tabelle 1 werden nun die Summen für die
einzelnen Belegungen von 3i+1 und 3j+1 jeweils passend multipliziert, wodurch die
Wahrscheinlichkeiten von 3/32 entstehen.
Der Score des Gesamten Stammbaumes MP(G) wird nun mit folgender Gleichung
berechnet, welche durch die genannten Vereinfachungen (Phänotyp fällt weg) und
die Problemstellung (wir betrachten nur die Individuuen ohne bekannten Genotyp)
entsteht. Da aber der Genotyp aller Gründer bekannt ist, fallen diese auch weg.
G\G* Nichtgründer i P(gi | gv(i), gm(i))
(6)
Für alle i V hat Individuum 3i+1 die Wahrscheinlichkeit ½ für seinen Genotyp. Da in
(6) über alle möglichen Belegungen für die unbelegten Genotypen summiert wird,
muß man sich nun überlegen, bei welcher Belegung für die 3i+1 die
Wahrscheinlichkeit des Kantengadgets gleich Null wird, da das Produkt in (6)
entweder (½)v (aufmultiplizierte Wahrscheinlichkeit für alle 3i+1) mal (3/32)e
(aufmultiplizierte Wahrscheinlichkeit für alle Kantengadgets) oder (½) v mal 0 (wenn
ein Kantengadget wahrscheinlichkeit Null hat) lautet. Da natürlich (½)v mal 0 gleich 0
ist, ist (6) gleich (7).
S (½)v (3/32)e
- 11 -
(7)
S ist hierbei die Anzahl der Belegungen für die unbekannten Genotypen in MP(G),
die die marginale Wahrscheinlichkeit eines Kantengadgets nicht gleich Null machen.
Diese Wahrscheinlichkeit eines Kantengadgets für Kante (i,j) ist (wie schon gezeigt)
genau dann Null, wenn 3i+1 und 3j+1 beide AA tragen. Wenn man nun beim
(hypotetischen) Erzeugen der verschiedenen Belegungen für jedes i V und jedes j
V darauf auchtet, den Individuuen 3i+1 und 3j+1 nur dann gleichzeitig AA zu
geben, wenn keine Kante in G zwischen ihnen existiert, wird es kein Kantengadget
für (i,j) geben und es kann auch nicht Wahrscheinlichkeit Null haben. Wenn man aber
nur Individuuen 3i+1 und 3j+1 gleichzeitig AA gibt, wenn sie nicht in einem
Kantengadget liegen, bilden ihre zugehörigen Knoten eine unabhänige Knotenmege,
genauer gesagt ist dann die Menge {i: 3i+1 hat AA als Genotyp} eine unabhänige
Knotenmenge. Wenn man S als mögliche Anzahl für solche Mengen in G nimmt (es
kann ja mehrere verschiedene unabhänige Knotenmengen in einem gegebenen
Graph geben) ist der Score von MP(G) genau gleich (7). Damit ist die Reduktion
komplett und die #P-Härte von MP wurde gezeigt.
Diskussion der Ergebnise
Da die vorgenommenen Einschränkungen des Modells auch in komplexeren
Modellen vorkommen, ist natürlich auch die NP-Härte bzw. #P-Härte übertragbar.
Das bedeutet, daß kein worst-case-effizienter Algorythmus für MAXP und MP
existiert (solange nicht P=NP) bzw. deren Ergebnisse aufgrund der Härte der
Approximation bis zu einem gewissen Grad total unbrauchbar sind. Die Ergebnisse
der obigen Analysen können helfen, Spezialfälle auszuschließen, die eine hohe
Komplexität hervorrufen.
Weiterhin wurde gezeigt, daß auch der Ausschluß von
Inzestzyklen nicht einer hohen Komplexität vorbeugt, womit eine verbreitete
Annahme wiederlegt wurde.
- 12 -
[1] Lin, S. 1996. Mont Carlo methods in genetic analysis, in T. Speed and M.S.
Waterman, eds., Genetic Mapping and DNA Sequencing, Springer, Berlin.
[2] Piccolboni, A., and Gusfield, D. 1999, Sept. On the Complexity of Fundamental
Problems in Pedigree Analysis. Technical Report CSE 99-8, University of California,
Davis.
[3] Lander, E.S. and Green, P. 1987. Construction of multilocus genetic linkage maps
in humans. Proc. Natl. Acad. Sci USA 84, 2363-2367
[4] Elston, R.C. and Stewart, J. 1971. A general model for analysis of pedigree data.
Human Heredity 21, 523-542.
[5] Garey et al., 1976. Some simplified NP-complete graph problems. Theoret.
Comput. Sci., 237-267
[6] Berman, P., and Karpinski, M. 1999. On some tighter inapproximability results
(extended abstract). Proc. ICALP. 200-209.
[7] Dyer, M.E. and Greenhill, C.S. 2000. On Markov chains for independent sets. J.
Algorithms 35(1), 17-49
- 13 -