Herstellung einer Null-Mutation durch homologe Rekombination
Werbung

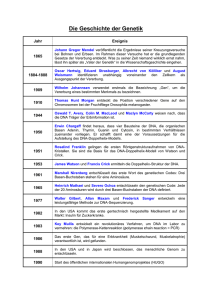
Herstellung einer Null-Mutation durch homologe Rekombination (2006) Jörg Kämper (Tel: 178630), Björn Sandrock (Tel: 2827080) Andrea Hlubek, Kai Heimel, Ramon Wahl Hintergrund Der phytopathogene Basidiomycet Ustilago maydis ist der Erreger des Maisbeulenbrands, einer weltweit verbreiteten Pflanzenkrankheit. Ustilago maydis hat sich in den letzten Jahren zu einem der wenigen Modellorganismen für phytopathogene Pilze entwickelt. Der Pilz kann, im Gegensatz zu vielen anderen phytopathogenen Pilzen, problemlos auf artifiziellen Medien kultiviert werden und ist dabei, ähnlich wie die Bäckerhefe Saccharomyces cerevisiae, für eine Vielzahl von molekularen Techniken zugänglich. So lässt sich U. maydis leicht transformieren, und über das Einbringen von modifizierten Kopien lassen sich endogene Gene durch homologe Rekombination spezifisch ausschalten oder gezielt verändern. Ähnlich wie andere pflanzen- oder humanpathogene Pilze durchläuft U. maydis während seines Lebenszyklus verschiedene morphologisch unterscheidbare Phasen. Seine haploiden, Sporidien genannten Zellen vermehren sich hefeartig durch Sprossung und wachsen ausschließlich saprophytisch, d.h. sie können die Wirtspflanze nicht infizieren. Nach der Fusion zweier Sporidien kommt es zur Bildung eines Dikaryons, das nun filamentös wächst. Im Gegensatz zu den Sporidien ist das Dikaryon in der Lage, Maispflanzen zu infizieren; mehr noch, für die Vermehrung ist es sogar auf die Wirtspflanze angewiesen. Die Fusion der Sporidien, die zur Bildung des infektiösen Dikaryons führt, wird genetisch durch zwei verschiedene Kreuzungstyp-Loci, die a und b genannt werden, kontrolliert. Zur Etablierung eines stabilen Dikaryons müssen die Sporidien sowohl verschiedene Allele des biallelischen aLocus als auch verschiedene Allele des multiallelischen b-Locus tragen. Deshalb ist eine a1b1 Sporidie kompatibel mit einer a2b2 Sporidie, nicht aber mit a1b2 oder a2b1 Sporidien. Der a-Locus kontrolliert die Zellerkennung und Zellfusion über ein Pheromon-Rezeptor System. Nach der Zellfusion wird über ein vom b-Locus kodiertes intrazelluläres Selbst-Nichtselbst Erkennungssystem entschieden, ob es zu den beschriebenen morphologischen Veränderungen, d.h. dem Wechsel von hefeartigem Wachstum zum typischen Spitzenwachstum des Filaments kommt und ob die weiteren Schritte für die sexuelle und pathogene Entwicklung initiiert werden. Da diese Prozesse erwiesenermaßen unabhängig vom a-Locus ablaufen, kommt dem b-Locus die Rolle eines zentralen molekularen Schalters für die Pathogenese zu. Der b-Locus kodiert für zwei unterschiedliche Homeodomänenproteine, bE und bW. Die beiden Proteine zeigen, mit Ausnahme der Homeodomänen, einem konservierten DNA-Binde-Motiv, 1 keine Ähnlichkeiten zueinander auf. Vergleicht man die bE Proteine verschiedener b-Allele, zeigt sich, dass der C-terminale Bereich der Proteine hoch konserviert ist, während sich die allelischen Unterschiede in den N-terminalen Bereichen häufen. Diese Unterscheidung in eine N-terminale, variable Domäne und eine C-terminale konstante Domäne findet man in ähnlicher Form auch bei den bW-Proteinen. rb2.1n rb2.2n Abb.1: oben: Aufbau des b-Locus in U. maydis. unten: Homologie der rb-Flanke (b2) mit dem entsprechenden Bereich in b1. Der Translationsstart ist jeweils durch Pfeile angegeben. 2 Die molekulare Grundlage für die Selbst-Nichtselbst-Erkennung der verschieden Allele liegt auf der Ebene der Protein-Dimerisierung. Die bE-und bW-Proteine können über die variablen Domänen ein Heterodimer bilden, allerdings nur, wenn sie von verschiedenen Allelen stammen. Gene können in U. maydis über homologe Rekombinationsereignisse ausgetauscht werden. In der Regel wird dazu ein Resistenzgen verwendet, das auf beiden Seiten von zum Intergrationsort homologen Bereichen flankiert wird. Diese Bereiche sind so gewählt, dass das auszuschaltende Gen durch das Resistenzgen ersetzt wird. In den meisten Fällen werden Flanken verwendet, die ca. 1000 bp lang sind. In diesem Praktikumsversuch werden wir das bW Gen in U. maydis deletieren. Wir verwenden ein Hygromycin-Resistenzgen, das von Sequenzen des b2-Locus flankiert wird (Abb. 1a). Die lb-Flanke (left border) liegt 3´ vom bW2-Gen, die rb-Flanke umfasst den 5´ Bereich des bW2 Gens und den 5´Bereich des bE2 Gens (Abb. 1a). Da bW2 und bE2 divergent angeordnet sind, wird im Falle einer homologen Rekombination in den b2 Locus des Stammes FB2 (a2b2) ein Großteil des bW2-Gens deletiert, während das bE2 Gen funktionell erhalten bleiben sollte. Bei Transformation in den Stamm FB1 (a1b1), der statt des b2 Locus den b1 Locus trägt, sollte es ebenfalls zu homologen Integrations-Ereignissen kommen. Dabei ist die lb-Flanke 100% homolog zu dem Bereich 3´ des bW1-Gens; außerhalb der bE und bW Gene unterschieden sich b1 und b2 nur in wenigen Nukleotiden. Die rb-Flanke liegt hingegen im variablen Bereich des bE Gens; in Folge ist die rb-Flanke nur zu 73% identisch zum entsprechenden Bereich in b1 (Abb. 1b). Dabei verwenden wir zwei verschiedene Konstrukte, die sich geringfügig in der Länge der rb-Flanke unterscheiden: in einem Fall sind die letzten Nukleotide am Ende des linearen Konstrukts homolog zum Rekombinationsort in bE1, im anderen Fall fehlt dieser kurze homologe Bereich (Abb. 1b). Die Spezifität der bE Gene kann durch die Rekombiantion verändert werden. Speziell bei Rekombination des bE2 Konstrukts in den bE1 Hintergrund können chimäre Allele entstehen. Abhängig davon, an welcher Stelle die Rekombination erfolgt, könnte die Spezifität der bE Gene verändert werden. 3 Fragestellungen: 1: Ist die homologe Rekombinationsfrequenz des b2-KO-Konstrukts im b1 und b2 Hintergrund unterschiedlich? 2: Hat das "homologe" Ende der rb-Flanke einen Einfluss auf die homologe Rekombinationsfrequenz im b1 Hintergrund? 3: Wie (und wo) findet die Rekombination im "heterologen" bE Bereich im b1 Hintergrund statt? An den Enden? Innerhalb des Fragments? 4: Kommt es zur Bildung von chimären bE Allelen? Was ist die Spezifität solcher chimären Allele? GENERELL: Die mit * markierten Puffer/Lösungen erhalten Sie von den Betreuern. ALLE ANDEREN LÖSUNGEN MÜSSEN SIE SELBST ANSETZEN! PLANEN SIE DESHALB 12 TAGE VORAUS! Die von Ihnen anzusetzenden Lösungen/Medien sind als Fettdruck und unterstrichen hervorgehoben! Jede Gruppe hat Puffer und Lösungen für den Eigengebrauch selbst anzusetzen; sprechen Sie sich NICHT ab, um Lösungen/Pufffer für den gesamten Kurs herzustellen (Ausnahme: Puffer für die Gelelektrophorese). Bitte beschriften Sie Ihre Medienplatten und Reaktionsgefäße EINDEUTIG (Grupppennummer, Ansatz)! Reaktionsgefäße, die aufbewahrt werden, sind mit einem Datum zu versehen. 1. Praktikumstag 1. PCR-Amplifikation des KO-Konstrukts 283 Eco RI • 597 Pst I • Abb. 2: Plasmidkarte pCR-b-ko-neu(+) lb 129 6 Sfi I • 158 4 Pst I • 159 4 Not I • pCR-b-ko-neu(+) hph • Nco I 5 785 7837 bp 227 1 Nco I • 229 0 Pst I • 238 0 Eco RI • • Pst I 5 402 262 5 Nco I • 4 318 0 Sfi I • • Not I 4 256 • Pst I 4 235 • Eco RI 4 230 • Sna BI 4 121 rb 376 7 Eco RI • Das Plasmid pCR-b-ko-neu(+) (Abb. 2) enthält das bakterielle Hygromycin-PhosphotransferaseGen (hph) unter Kontrolle des U. maydis hsp70-Promotors und terminiert durch den Agrobakterium Nos-Terminator. Flankiert wird dieses Gen von zwei ca. 1 kb DNA Fragmenten, die eine Integration der DNA über ein homologes Rekombinationsereignis ermöglichen sollen. Wir verwenden für die Amplifikation des KO-Konstrukts zwei verschiedene PrimerKombinationen. Der Primer auf der Seite der lb-Flanke (lb1) wird konstant gehalten, auf der Seite der rb-Flanke verwenden wir die Primer rb2.1n und, alternativ, rb2.2n. Die Amplifikationsprodukte unterscheiden sich, wie in Abb. 1b dargestellt, durch einen kurzen Bereich homologer Nukleotide am Ende der rb-Flanke. Praktischer Teil: PCR-Amplifikation mit Phusion-DNA-Polymerase Primer-Kombinationen: A) lb1, b-rb2.2n B) lb1, b-rb2.1n 1 l 20 l 1 l 1 l 1 l 3 l 0.5 l ad 100 l Plasmid-Template (10 ng/l) 5 x Phusion-Puffer* 10 mM dNTP* Primer 1 (50 pmol/l) Primer 2 (50 pmol/l) DMSO* Phusion Polymerase (2U/l) ddH2O Cycling: 1x 30x 1x 30 sec 98C 10 sec 98C 15 sec 65C 75 sec 72 10 min 72C b-rb2.1n b-rb2.2n lb1 GAAGCATCGGCTTTCTTCAAGGCT CGACACCCTACAACAGGACTTGAA GAAGCTAGCGGAAAAGTGGGTGAG 5 2. Praktikumstag 1. EtOH Präzipitation, Test der PCR-Amplifikation auf Agarosegel 2. Herstellen der Protoplasten von Ustilago maydis 3. Transformation von Ustilago maydis Aufgabe ist es, die zwei verschiedenen PCR Produkte jeweils in Protoplasten der Stämme FB1 und FB2 zu transformieren (4 Transformationen). Bei diesen Ansätzen plattieren Sie bitte den Transformationsansatz komplett aus. Als zusätzliche Kontrolle für die Transformationskompetenz der Protoplasten transformieren Sie 0,1g des autonom replizierenden Plasmids pNEBUH (2 Transformationen). Da bei dieser Kontrolle erwartungsgemäß eine hohe Transformanterate zu erwarten ist, plattieren Sie bitte 250 l des Transformationsansatzes unverdünnt, und zusätzlich 250 l einer 1/10 Verdünnung (in STC-PEG). Alle Transformanden werden auf Hygromycinhaltigen Gradientenplatten selektioniert. Praktischer Teil: EtOH Präzipitation der PCR-Produkte vom Vortag (siehe Material und Methoden) Aufnehmen des Pellets in 25 l TE Puffer Testgel 1 l der DNA werden auf einem 0.8% Agarose-Gel (0.5x TBE oder 1x TAE) aufgetragen. Schätzen Sie die DNA-Menge durch Vergleich mit dem Größenmarker ab! Herstellung von Protoplasten von U. maydis 50 ml Kultur in YEPS Medium bei 28°C bis zu einer Zelldichte von ca. 5x107/ml (OD 0.6 bis 1.0) angezogen (stationäre Vorkultur 1:100, 1:300, 1:1000 verdünnen, über Nacht bei 28°C, 200 rpm in Schikanekolben inkubieren) 7 min. bei 2500g (Hereaus, 3500 rpm) in 50 ml Falkonröhrchen abzentrifugieren Zellpellet wird in 25 ml SCS-Puffer resuspendiert erneut 7 min. bei 2500 g (3500 rpm) zentrifugieren Pellet in 2 ml SCS mit 5 mg/ml Novozym 234 (steril filtriert durch 0.2 m Filter) resuspendieren Protoplastierung erfolgt bei Raumtemperatur und wird mikroskopisch alle 5 min. verfolgt Protoplasten werden mit 10 ml SCS-Puffer gemischt und bei 1100 g (2300 rpm) 10 min. zentrifugiert, der Überstand wird verworfen 6 Pellet wird vorsichtig in 10 ml SCS Puffer resuspendiert (auf und ab-pipettieren mit 10 ml Pipette) und erneut zentrifugiert Waschvorgang mit SCS-Puffer 2 X wiederholen Pellet in 10 ml STC-Puffer vorsichtig resuspendieren, 10 min. zentrifugieren, der Überstand wird verworfen Pellet in 500 µl kaltem STC-Puffer resuspendieren und auf Eis gehalten Aliquots können mehrere Monate bei -80°C gelagert werden (Vorsichtig auf Eis auftauen). SCS-Puffer* 20 mM NaCitrat pH 5.8 1.0 M Sorbit (20 mM NaCitrat/ 1.0 M Sorbit und 20 mM Zitronensäure/ 1.0 M Sorbit mischen und mit pH-Meter auf pH 5.8 einstellen) STC-Puffer* 10 mM Tris/HCL pH 7.5 1.0 M Sorbit 100 mM CaCl2 Transformation von U. maydis Protoplasten Max 10 µl linearisierte DNA (optimal 3-5 µg) wird in ein 2 ml Eppendorfröhrchen übertragen 1 µl Heparin (15 µg/µl) werden zugesetzt (SIGMA H3125) 50 µl Protoplasten werden zugesetzt, 10 min. auf Eis lassen 500 µl STC-PEG* (40% (w/w) PEG3350 (SIGMA P3640) in STC, steril filtriert, wird gestellt) werden zugesetzt, und VORSICHTIG mit der Protoplastensuspension gemischt 15 min. Inkubation auf Eis Ausplattieren auf Gradienten-Platten: 1 ml Glaspipette wird zum Verteilen benutzt. Unterer Agar: 10 ml YEPS-1,5% Agar-1M Sorbitol mit Hygromycin (400 µg/ml). Kurz vor dem Ausplattieren wird diese Agarschicht mit 10 ml YEPS- 1,5% Agar-1M Sorbitol* überschichtet. Medium für Gradientenplatten wird gestellt! Die Platten werden 3-4 Tage bei 28°C inkubiert Stammlösung Hygromycin 50 mg/ml Flüssigmedium 200 µg/ml 7 Festmedium Bottom-Agar 200 µg/ml 400 µg/ml 3. Praktikumstag 1. Auswertung der Transformation, Isolierung von Transformanden 2. Kreuzung mit kompatiblen Stämmen Die Transformanden werden phänotypisch auf homologe Rekombinationsereignisse im b-Locus untersucht. Dazu werden die FB1 Transformanden mit dem Stamm RK1661 (a2bE2) und die FB2 Transformanden mit dem Stamm RK1607 (a1bE1) gekreuzt, um zu untersuchen, ob das bW1-Gen (in FB1) bzw. bW2-Gen (in FB2) inaktiviert wurde. Die Funktionalität der bE Gene wird durch Kreuzung der FB1 Transformanden mit den Stämmen RK1724 (a2bW1) und RK1722 (a2bW2), und der FB2 Transformanden mit RK1725 (a1bW1) und RK1723 (a1bW2) untersucht. Die Spezifität der bE Gene kann durch die Rekombination verändert werden. Speziell bei Rekombination des bE2 Konstrukts in den bE1 Hintergrund können chimäre Allele entstehen. Abhängig davon, an welcher Stelle die Rekombination erfolgt, könnte die Spezifität der bE Gene verändert sein (b1, b2, bNull, oder b1+b2 oder ????) Von interessanten Transformanden soll in der Folge DNA präpariert werden, um die Rekombinationsereignisse genauer zu charakterisieren. Praktischer Teil: Die Anzahl der Transformanden auf den verschiedenen Transformationsplatten wird bestimmt, und die Transformationsfrequenz mit den PCR Produkten in Relation zur Transformationsfrequenz des Kontrollplasmids gesetzt. Von den Transformationsplatten werden pro Transformation 10 Transformanden mit flachen Zahnstochern in 100 l YEPS Medium transferiert und resuspendiert. Von den resuspendierten Transformanden werden ZUERST je 10 µl Zellsuspension auf eine PDPlatte mit 200 µg/ml Hygromycin getropft (MÖGLICHST STERIL ARBEITEN!). Danach werden von jeder Transformande und, als Kontrolle, von den wt-Stämmen FB1 und FB2 jeweils 15 l mit 15 l der entsprechenden Kreuzungspartner in einer Mikrotiterplatte gemischt. Von der Mischung werden 2 l auf der Oberfläche einer PD-Aktivkohleplatte getropft. Die Oberfläche der Platte sollte dabei möglichst nicht beschädigt werden. Die PD-hyg Platten werden bei 28°C, die PD-Aktivkohle Platten bei Raumtemperatur inkubiert. 8 9 4. Praktikumstag 1. Auswertung der Kreuzungen 2. Ansetzen von 3 ml Kulturen 3. PCR Amplifikation der Sonde (siehe auch 5. Praktikumstag) Praktischer Teil: Auswertung der Kreuzungen: Die Kreuzungsplatten werden (u.U. mit Hilfe einer Stereolupe) auf filamentöses Wachstum untersucht. Ansetzen von 3 ml Kulturen: Von insgesamt 10 Transformanden, die einen interessanten Phänotyp zeigen (siehe Seite 4) werden 3 ml Kulturen in YEPS-Medium mit 100 µg/ml Hygromycin von den getropften PD-hyg Platten angesetzt. Als Kontrollen werden FB1 und FB2 in YEPS-Medium ohne Hygromycin angezogen. PCR-Amplifikation der Sonde: 10 l 1 l 1l 1 l 0.01 g 2U 10x PCR-Puffer (100 mM Tris/HCl pH 8.3, 500 mM KCl, 20 mM MgCl2) Primer 1 (50 pmol/l): b-rb2.2n Primer 2 (50 pmol/l): b-rb1n dNTP (10 mM) pCR-b-ko-neu(+) Taq-Polymerase ad 100 l mit dH2O 1x 30 x 1x 10 min 94°C 1 min 94°C 1 min 60°C 1.5 min 72°C 10 min 72°C b-rb2.2n b-rb1n1 CGACACCCTACAACAGGACTTGAA ACTGCTTCGGAGTCGTGTCCACGA 10 5. Praktikumstag 1. Präparation genomischer DNA aus Ustilago maydis Transformanden 2. Restriktion und Gelelektrophorese der genomischen DNA mit SalI 3. PCR-Amplifikation der Integrationsstellen 4. Aufreinigung der Sonde mit PCR-Purification Columns 5. Markierung der Sonde 6. Übertragung der DNA auf einen Nylonfilter (Southern blot) Ziel ist es, die Transformanden näher zu charakterisieren, um die auf S. 3 gestellten Fragen beantworten zu könnnen. Die homologe Integration kann zum einen durch einen Southern-Blot verifiziert werden. Die DNA wird dazu mit SalI geschnitten, gelelektrophoretisch aufgetrennt, und auf eine Nylonmembran übertragen. Durch Hybridisierung mit einer markierten Sonde (der rbFlanke) kann ein definiertes Fragment im Wildtyp detektiert werden, das sich durch die homologe Rekombination verändert. Das KO-Konstrukt hat keine SalI Schnittstelle. Sie können die erwarteten Banden aus der genomischen Karte und der Plasmidkarte errechnen; Orientierungspunkte sind die PstI und SnaBI Schnittstellen. 103 7 Sal I • 339 2 Sal I • 244 8 Pst I • 375 2 Sal I • b1 total.strider 103 7 Sal I • b2 total.strider 674 3 Sal I • 655 0 Sal I • 7969 bp 244 8 Pst I • 766 2 Eco RI • Mode: Global 655 3 Sal I • 579 3 Sna BI • 544 0 Eco RI • 339 2 Sal I • 375 5 Sal I • 7968 bp 766 1 Eco RI • Mode: Global bW1-Gen: 5360 - 3155 bW2-Gen: 5362 - 3155 bE1- Gen: 5564 - 7059 bE2- Gen: 5561 - 7059 Sonde (rb): 4866 - 5895 lb: 2147 - 3132 11 Eine alternative Methodik, homologe Integrationsereignisse zu charakterisieren (NICHT GLEICHBEDEUTEND MIT SOUTHERN-BLOT! WARUM?) ist es, eine diagnostische PCR Reaktion durchzuführen. Dabei wird ein Primer aus der KO-Kassette gewählt, der andere Primer in einem Bereich der genomischen DNA, der kurz ausserhalb des für die Rekombination verwendeten Bereichs liegt. Eine Amplifikation ist demnach nur möglich, wenn das KO-Konstrukt homolog integriert ist. Wir verwenden die Primer hhn5n (liegt am 5´-Ende der HygromycinKassette, gerichtet auf die rb-Flanke; Position 2985 - 3006 in pCR-b-KO-neu(+), und bE_const1, Position 5951 - 5975 in der Sequenz b2 total.strider. Die Primerkombinationen können sowohl für eine PCR im b1 als auch im b2 Hintergrund verwendet werden, bE_const1 liegt im konstanten Bereich der bE-Gene. Durch die beiden Primer wird der variable Bereich der bE-Gene amplifiziert. Dieser Bereich kann durch charakteristische HaeIII und TaqI Restriktionsmuster typisiert werden. Aus den Abweichungen des bE1 bzw bE2 typischen Restriktionsmusters kann auf Rekombinationsereignisse innerhalb dieses Bereichs geschlossen werden. Am Vortag haben Sie bereits eine PCR durchgeführt, um die Sonde für die Hybridisierung zu generieren. Die PCR Reaktion wird über eine PCR-Purification Column (Quiagen) aufgereinigt, durch die die Primermoleküle entfernt werden und die DNA generell aufreinigt wird. Die gereinigte DNA (1 l ) wird auf einem Gel überprüft. Die vier besten DNA-Präparationen des Kurses werden nach Vorschrift (Dig-DNA labelling kit) über Nacht mit Digoxygenin markiert. 12 31 Hae III • 22 Hae III • 490 Hae III • 385 Hae III • b1-PCRn2 149 Taq I • 127 Taq I • 691 Hae III • 1143 bp Mode: Global 366 Taq I • 340 Taq I • 329 Taq I • 787 Taq I • b1-PCRn2 1143 bp 112 0 Taq I • 106 1 Taq I • 967 Taq I • Mode: Global Ende b-rb2.1n entspricht Position 1044 Ende b-rb2.2n entspricht Position 1064 Digested Sequence: b1-PCRn2 Enzymes selected ----------------------------------1 HaeIII gg/cc Sites ----5 Digestion generates 6 fragments: Upstream cut site ------------------------------------------Site Enzyme Site orientation position name Recognition sequence | -------- ---------- -------------------- 691 31 490 385 1 22 HaeIII HaeIII HaeIII HaeIII = HaeIII gg/cc gg/cc gg/cc gg/cc 1 TaqI | | | | | | gg/cc Enzymes selected ----------------------------------t/cga DNA fragment ----------------------------------------Upstream True DS Downstream overhang Order length overhang ------------ --- -------- ---------------------------------------------------- 6 3 5 4 1 2 451 354 201 105 23 9 ----------------------------------------- | | | | | | Downstream cut site ------------------------------------------Site orientation Enzyme Site | Recognition sequence name position - -------------------- ---------- -------gg/cc gg/cc gg/cc gg/cc gg/cc = HaeIII HaeIII HaeIII HaeIII HaeIII 1143 385 691 490 22 31 Sites ----9 Digestion generates 10 fragments: Upstream cut site ------------------------------------------Site Enzyme Site orientation position name Recognition sequence | -------- ---------- -------------------- 366 149 787 1 967 1061 340 1120 127 329 TaqI TaqI TaqI = TaqI TaqI TaqI TaqI TaqI TaqI t/cga t/cga t/cga t/cga t/cga t/cga t/cga t/cga t/cga DNA fragment ----------------------------------------Upstream True DS Downstream overhang Order length overhang ------------ --- -------- -----------5’ 5’ 5’ | 5’ 5’ 5’ 5’ 5’ 5’ -------CG -------CG -------CG -------CG -------CG -------CG -------CG -------CG -------CG 6 3 7 1 8 9 5 10 2 4 419 178 178 127 92 57 24 21 20 9 13 gc------gc------gc------gc------gc------gc------gc------- 5’ 5’ 5’ 5’ 5’ 5’ 5’ | gc------- 5’ gc------- 5’ Downstream cut site ------------------------------------------Site orientation Enzyme Site | Recognition sequence name position - -------------------- ---------- -------t/cga t/cga t/cga t/cga t/cga t/cga t/cga t/cga t/cga TaqI TaqI TaqI TaqI TaqI TaqI TaqI = TaqI TaqI 787 329 967 127 1061 1120 366 1143 149 340 31 Hae III • 22 Hae III • 363 Hae III • 605 Hae III • b2-PCRn2 955 Hae III • 1145 bp Mode: Global 789 Taq I • b2-PCRn2 1145 bp 969 Taq I • 112 2 Taq I • Mode: Global Ende b-rb2.1n entspricht Position 1044 Ende b-rb2.2n entspricht Position 1064 Digested Sequence: b2-PCRn2 Enzymes selected ----------------------------------1 HaeIII gg/cc Sites ----5 Digestion generates 6 fragments: Upstream cut site ------------------------------------------Site Enzyme Site orientation position name Recognition sequence | -------- ---------- -------------------- 605 31 363 955 1 22 HaeIII HaeIII HaeIII HaeIII = HaeIII gg/cc gg/cc gg/cc gg/cc DNA fragment ----------------------------------------Upstream True DS Downstream overhang Order length overhang ------------ --- -------- -----------| | | | | | gg/cc ----------------------------------------- 5 3 4 6 1 2 350 332 242 189 23 9 --------- | --------- | --------- | | --------- | --------- | Downstream cut site ------------------------------------------Site orientation Enzyme Site | Recognition sequence name position - -------------------- ---------- -------gg/cc gg/cc gg/cc gg/cc gg/cc HaeIII HaeIII HaeIII = HaeIII HaeIII 955 363 605 1145 22 31 Digested Sequence: b2-PCRn2 Enzymes selected ----------------------------------1 TaqI t/cga Sites ----3 Digestion generates 4 fragments: Upstream cut site ------------------------------------------Site Enzyme Site orientation position name Recognition sequence | -------- ---------- -------------------- 1 789 969 1122 = TaqI TaqI TaqI t/cga t/cga t/cga DNA fragment ----------------------------------------Upstream True DS Downstream overhang Order length overhang ------------ --- -------- -----------| 5’ -------CG 5’ -------CG 5’ -------CG 1 2 3 4 789 178 151 21 Praktischer Teil: 14 gc------- 5’ gc------- 5’ gc------- 5’ | Downstream cut site ------------------------------------------Site orientation Enzyme Site | Recognition sequence name position - -------------------- ---------- -------t/cga t/cga t/cga TaqI TaqI TaqI = 789 969 1122 1145 DNA-Präparation aus U. maydis 2 ml Eppendorf-Reaktionsgefäße werden mit 0.3 g Glasperlen (425-600 Micron, mit Säure gewaschen) versetzt; 1,8 ml der Kultur zugeben 2 min bei 13000 U/m abzentrifugieren; Überstand verwerfen. 400 µl Usti-Lysis-Puffer zugeben 500 µl Phenol/Chloroform (1:1) zugeben (Handschuhe, Schutzbrille, Abzug!) 6 - 10 min auf Vibrax schütteln (Handschuhe, Schutzbrille, Abzug!) 15 min bei 13000 U/m abzentrifugieren. 400 µl Überstand in neues Eppendorfgefäß überführen (Handschuhe, Schutzbrille, Abzug!), 1 ml 100% EtOH zugeben , gut schütteln! 5 min bei Raumtemperatur stehen lassen. 5 min bei 13000 U/m abzentrifugieren, Überstand verwerfen 1 ml 70% EtOH zugeben, ohne das Pellet von der Wand zu lösen 5 min bei 13000 U/m abzentrifugieren, darauf achten, das das Pellet aussen zu liegen kommt. Überstand verwerfen, erneut anzentrifugieren, EtOH-Reste entfernen, evtl, erneut anzentrifugieren, um EtOH Reste vollständig zu entfernen. Bei RT 5-10 min trocken lassen. NICHT ÜBERTROCKNEN! Pellet in 50 µl TE/RNAse aufnehmen und mind. 30 min bei 60° C lösen. TE/RNAse: 10 mM Tris/HCl pH 8.0 1 mM EDTA 20 µg/ml RNAseA Lysis-Puffer: 2% Triton X-100 1% SDS 100 mM NaCl 10 mM Tris-HCl pH 8.0 1 mM EDTA 15 Restriktion der genomischen DNA: 10 µl der Gesamt-DNA werden mit SalI geschnitten, als Kontrolle dient DNA der Stämme FB1 und FB2. Der Restriktionsansatz besteht in einem Gesamtvolumen von 20 µl aus 10 µl DNA 2.0 µl 10 x SalI Puffer 1 µl BSA (1mg/ml) 1 µl RNaseA (1mg/ml) 1 µl SalI (20 U/µl) Die Inkubation erfolgt für 2 Stunden bei 37°C. Die Fragmente werden auf einem 0.8%igen Agarosegel aufgetrennt (siehe Material und Methoden). Als Standard wird 10 µl eines 1 kb-LeiterGrößenmarkers aufgetragen. Alkalischer Transfer von DNA auf Nylonmembranen - Das Agarosegel wird für 15 Min. in 0,25 N HCl auf dem Schüttler inkubiert. Dies dient dazu, einen Teil der Purine abzuspalten, um den Transfer großer DNA-Moleküle zu erleichtern. - Anschließend wird das Gel für 15 Min. in DENAT (Denaturierungslösung: 1,5 M NaCl, 0,4 M NaOH) inkubiert. - Der Transfer erfolgt mit 0,4 M NaOH durch Kapillarblot. Auf einer Glasplatte wird das Gel auf Blottingpapier, das auf beiden Seiten in 0,4 M NaOH eintaucht, gelegt. Um das Gel herum werden Streifen von Parafilm gelegt. - Auf das Gel wird ein passendes Stück Nylonfilter gelegt und anschließend drei Lagen zugeschnittenes Blottingpapier. Darüber kommt ein Stapel (ca. 10 cm hoch) Einmalhandtücher. Auf diesen Stapel wird noch eine Glasplatte zur Beschwerung gelegt. PCR-Amplifikation der Insertionsstelle: 10 l 1 l 1l 1 l 0.1 g 2U 10x PCR-Puffer (100 mM Tris/HCl pH 8.3, 500 mM KCl, 20 mM MgCl2) Primer 1 (50 pmol/l): hhn3neu2 Primer 2 (50 pmol/l): bE_const1 dNTP (10 mM) genomische DNA Taq-Polymerase ad 100 l mit dH2O 16 Cycling: 1x 30 x 2 min 95°C 45 sec 95°C 60 sec 60°C 60 sec 72°C 10 min 72°C 1x Primer: hhn3neu2 bE_const1 TCGCTGGTAGTTACCACGTTCGGC CTTGAGTCGGATAAGGGTTGTCGAG Aufreinigung von PCR-Produkten: siehe Qiagen-Manual Gelelektrophorese: siehe Material und Methoden Digoxygenin-Markierung der Sonde: siehe Roche-Digoxygenin-Manual 6. Praktikumstag 1. Überprüfung der DIG-Sonde 2. RFLP Analyse der PCR-Amplifikate vom Vortag mit TaqI und HaeIII 3. Auftrennung der Amplifikate auf Agarosegel 4. Hybridisierung des Nylonfilters mit der DNA-Sonde Praktischer Teil: Überprüfung der Sonde: Siehe Original-Vorschrift von Roche. Hybridisierung von Nylonfiltern - Nach dem Blot wird der Filter kurz an der Luft getrocknet und anschließend wird die DNA durch UV-crosslinking an die Membran gebunden (Stratalinker Auto-crosslink). - Die Hybridisierung erfolgt bei 35°C in Dig-Easy-Hyb-Puffer (Roche) in einem Hybridisierungsofen. Nach einer Vorhybridisierung (siehe Vorschrift) wird die denaturierte (siehe Vorschrift) Digoxigenin-markierte Probe direkt in die Hybridisierungslösung gegeben. 17 RFLP-Analyse der PCR-Amplifikate 17 l PCR-Produkt 2 l 10 x HaeIII-Extra -Puffer bzw. 10 x TaqI-Extra-Puffer 1 l HaeIII bzw. TaqI Restriktionsenzym Inkubation für 1.5 h bei 37°C (HaeIII) oder 65°C (TaqI) Auftrennung auf einem 3% 0,5x TBE Agarosegel (Material und Methoden) Marker: 100 bp Leiter (NEB) 10 x HaeIII-Extra* 10 mM Tris/HCl pH 8.3 50 mM KCl 82 mM MgCl2 10 mM DTT 1 mg/ml BSA 10 x TaqI-Extra* 10 mM Tris/HCl pH 8.3 550 mM KCl 82 mM MgCl2 1 mg/ml BSA 7. Praktikumstag 1. Nachweis der markierten DNA mit Hilfe spezifischer Antikörper und Chemilumineszenz 2. Auswertung der Hybridisierung 3. Gesamtauswertung Die Hybridisierungslösung (ca. 30 ml) wird aus der Hybridisierungsröhre in ein Sarstedt– Röhrchen abgekippt und aufbewahrt, da sie mehrmals verwendet werden kann. Die Filter werden gewaschen (siehe Vorschrift, Waschtemperatur: 55°C) und die gebundene Digoxigenin-markierte DNA wird durch monoklonale Digoxigenin-spezifische Antikörper, die mit alkalischer Phosphatase gekoppelt sind, nachgewiesen. Der Nachweis erfolgt durch ein Leuchtreagenz, das durch die alkalische Phosphatase umgesetzt wird. Während der katalysierten Reaktion wird Licht erzeugt, das auf einem Röntgenfilm eine Schwärzung hervorruft. Mit dieser Methode können noch etwa 0,1 pg homologer DNA nachgewiesen werden. Praktischer Teil: Waschen des Nylon-Filters nach dem Southern-Blot Lumineszenz Nachweis von Digoxigenin-markierten Nukleinsäuren auf Nylonmembranen: Siehe Roche- Protokoll (ACHTUNG! LÖSUNGEN MÜSSEN VORBEREITET WERDEN) 18 19 Material und Methoden Medien dYT 18 g Bacto Tryptone 10 g Bacto Yeast Extract 5 g NaCl 1 l H2O dest. YEPS 10 g Yeast Extract (Difco) 4 g Bacto Peptone (Difco) 20 g Saccharose (für Mikrobiologie, Merck) ad 1 l für Platten werden 1.5% Agar vor dem Autoklavieren zugegeben. PD-Aktivkohle-Platten 24 g Potato Dextrose Broth (Difco) 20 g Bacto Agar (Difco) 10 g Aktivkohle (Sigma Nr. 9157) ad 1 l dH2O, autoklavieren bei 117°C Häufig verwendete Puffer und Lösungen TE-Puffer 10 mM Tris-HCl pH 7,9 1 mM Na2EDTA Puffer für Restriktionsenzyme 10 x NEB 1 100 mM Bis Tris Propane/HCl pH 7.0 100 mM MgCl2 10 x NEB 2 100 mM Tris/HCl pH 7.9 100 mM MgCl2 10 mM DTT 500 mMNaCl 10 mM DTE 10 x NEB 3 500 mM Tris/HCl pH 7.9 100 mM MgCl2 1 M NaCl 10 mM DTE 10 x NEB 4 200 mM Tris/Acetate pH 7.9 100 mM MgCl2 500 mM K-Acetate 10 mM DTT 20 10 x SalI-Puffer 100 mM Tris/Hcl pH 7.9 100 mM MgCl22 1.5 M NaCl 10 mM DTT Methoden Auftrennung von DNA auf Agarosegelen Die Seiten der Gelform werden mit Klebeband verschlossen, der Kamm in die dafür vorgesehenen Aussparungen eingesetzt und das Gel mit Agarose in TAE-Puffer gegossen (0.4% bis 4.0% Agarose in TAE, durch Kochen in einer Mikrowelle gelöst und auf ca. 60 °C temperiert) (enthält 0.5 µg/ml Ethidiumbromid) Achtung! Handschuhe und Schutzbrille tragen! EtBr ist ein starkes Cancerogen! Nach dem Erkalten der Agarose wird das Klebeband vorsichtig entfernt und das Gel mit der Gelform in die Gelapparatur eigebracht und mit 1 x TAE-Puffer versehen, so daß das Gel ca. 1-2 mm bedeckt ist. Die DNA wird mit 1/5 Vol. Auftragspuffer versetzt und auf das Gel aufgetragen. Nach dem Beladen des Gels wird der Deckel geschlossen und die Spannung angelegt. Pluspol (rot) an der den Taschen abgewandten Seite!!! Sicherheitshalber wird das Einwandern der blauen Front (Bromphenolblau) nach 2 Min kontrolliert. Nach Beendigung des Laufes wird das Gel auf der Form zu einem Transilluminator mit Videosystem gebracht und unter UV-Licht (302 nm) photographiert. Schutzbrille und Schutzschirm benutzen!!! 1 x TAE-Puffer (Agarosegele): 40 mM Tris-Acetat pH 7,9 2 mM Na2EDTA Auftragspuffer (6x) 0.25 % Bromphenolblau 40 % (w/v) Saccharose in H2O 0.5 x TBE-Puffer (Agarosegele) 50 mM Tris/Borat , pH 7.9 1 mM EDTA Phenolextraktion und Fällung von DNA Phenolextraktion Die DNA sollte in einem Mindestvolumen von 100 µl H2O oder TE vorliegen. Zur DNA wird ein gleiches Volumen Phenol/Chloroform gegeben (Unter dem Abzug arbeiten! Schutzbrille und Handschuhe Tragen! Phenol führt zu starken Verbrennungen auf der Haut! Beim Einatmen 21 kann es zu irreversiblen Schädigungen kommen!). Die beiden Phasen werden auf dem Vortex gründlich (ca. 20 sec) vermischt und anschließend für 2 bis 5 min bei 13000 Upm zentrifugiert. Die obere, wässerige Phase mit der DNA wird vorsichtig, ohne die Interphase oder die Phenolphase mitzunehmen (EXTREM WICHTIG!!!), mit einer Gilson-Pipette in ein neues Eppendorfgefäß überführt. Phenolextrahierte DNA wird in der Regel mit EtOH gefällt. Fällung von DNA Zu einer DNA Lösung wird 1/10 Vol 3 M Na Acetat gegeben. Dann werden 2 Volumina 100 % Ethanol zugegeben und gemischt. Nach 10 min Inkubation bei Raumtemperatur wird die DNA durch 5 - 15 min. Zentrifugation bei 13000 Upm pelletiert; bei sehr geringen DNA-Mengen sollte die Zentrifugation länger erfolgen. Die Gefäße werden so in der Zentrifuge angeordnet, daß das "Scharnier" des Gefäßes nach außen zeigt. Dies erleichtert es, das Pellet wiederzufinden. Der Überstand wird vorsichtig entfernt (z.B. mit einer Pipette oder durch vorsichtiges ausgießen), bitte darauf achten, daß das Pellet nicht verlorengeht. Das Pellet wird mit ca 1 ml 70% Ethanol gewaschen, um überschüssige Salze zu entfernen: der Alkohol wird auf das Pellet pipettiert, nach Mischen wird erneut für 2 - 5 Min. bei 13000 Upm zentrifugiert. Der Überstand wird wie oben entfernt; letzte Alkoholreste können vorsichtig nach erneutem Anzentrifugieren (wenige Sekunden) mit einer gelben Spitze an der dem Pellet abgewandten Seite entfernt werden. Dieser Schritt sollte wiederholt werden, bis kein Alkohol mehr sichtbar ist. Das Pellet wird dann an der Luft getrocknet. Danach wird das Pellet in Wasser oder TE gelöst; für kleine DNAs wie Plasmide geht dies innerhalb weniger Minuten, bei chromosomaler DNA braucht dieser Prozeß in der Regel mehrere Stunden. Die DNA kann dabei auf 60 - 65° erwärmt werden. Restriktion von Plasmid-DNA Dazu werden pro Eppendorfgefäß ca. 1 µg DNA, 2 µl 10 x Restriktionspuffer 1 µl BSA (1 mg/ml) 1 µl RNaseA (1 mg/ml) 2 - 10 U Restriktionsenzym (meist ca 10 U/µl) in einem Gesamtvolumen von 20 µl gemischt und 60 Min. bei (meist) 37°C inkubiert. 22