1b. Monogenetische Krankheiten
Werbung

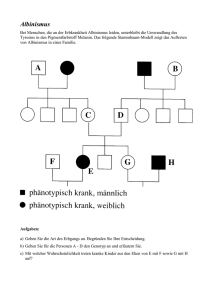
1b. Monogenetische Krankheiten- II Molekularbiologie der Krankheiten Monogenetische Krankheiten Thalassämien DIA 15 Als Thalassämien (griechisch für Mittelmeeranämie) werden Erkrankungen der roten Blutkörperchen bezeichnet, bei denen durch einen Gendefekt das Hämoglobin nicht ausreichend gebildet bzw. gesteigert abgebaut wird. Gendefekte auf Chromosom 11 (bei β-Thalassämie) oder 16 (bei α-Thalassämie), die zu einer verminderten Globinkettenbildung führen, sind für die Entstehung der Thalassämie verantwortlich. Die verschiedenen Thalassämievarianten werden nach den Globinen benannt, die in nicht ausreichender Menge gebildet werden: αund β-Thalassämien. Die meisten Mutationen werden autosomal rezessiv vererbt und treten vor allem in einstigen Malariagebieten im Mittelmeerraum und in Africa. Die β-Thalassämie ist die häufigste Form der Thalassämie. Von ihr sind über 4000 Mutationen bekannt, die in der Regel kleinere Raster-, oder Punktmutationen am β-Globin-Locus und nur selten längere Deletionen ausmachen. Die meisten Mutationen der β-Thalassämie werden autosomal-rezessiv vererbt. Die β-Thalassämie wird in zwei Formen eingeteilt, die Thalassaemia minor und die Thalassaemia major Galaktosämie DIA 16. Galaktosämie (gr. Äma: Blut) ist eine seltene angeborene Stoffwechselstörung bei der sich zu viel Galaktose (ein Zucker) im Blut befindet. Es handelt sich um eine Mutation des sogenannten GALT-Gens die autosomalrezessiv vererbt wird. Das GALT-Gen liegt auf dem Chromosom 9. Galaktosämie tritt weltweit etwa bei einem von 40.000 Neugeborenen auf und wurde zuerst 1917 durch Friedrich Goppert (1870-1927) beschrieben. Die klassische Galaktosämie ist durch das teilweise oder vollständige Fehlen des Enzyms Galactose-1-phosphatUridyltransferase (=Galaktosetransferase, GALT), welches eine wichtige Rolle im Galaktosestoffwechsel hat, gekennzeichnet. Durch den Enzym-Mangel erfolgt eine Anreicherung von Galaktose und Galaktose-1-Phosphat in den Zellen Hämophilie DIA 17 . Hämophilie ist eine klassische X-chromosomal-rezessive Erkrankung. Bei etwa 85 % der Hämophilienfamilien befindet sich einen Mangel an antihämophilem Globulin A, Faktor VIII (Hämophilie A), bei etwa 15 % einen Mangel an antihämophilem Globulin B, Faktor IX (Hämophilie B). Die Gerinnungsstörung führt zu bedrohlichen Blutungen bei Verletzungen. Es treten schmerzhafte tiefe Hämatome der Muskulatur auf. Das Faktor VIII Gen ist 200 kb gross und kodiert für 2351 Aminosäuren. Bei der schwer betroffenen Patienten tritt eine micro-Inversion innerhalb der Promotorregion und im Intron 22. Der Erbgang ist typisch X-Chromosomgekoppelt: Die heterozygotischen Frauen sind Konduktorinnen und weitergeben das Mutation-trägende X Chromosom an Hälfte ihrer Söhne, die hemizygot für X-Chromosom sind EXTRA ANFORDERUNG Boldogkői Zsolt ©Seite 1 1b. Monogenetische Krankheiten- II Marfan Syndrom DIA 18 Bindegewebeserkrankung. Unerkannt kann sie zum plötzlichen Tode führen. Leider bleibt sie in vielen Fällen unerkannt. Bis heute ist das Syndrom unheilbar und nur begrenzt behandelbar. Mögliche Merkmale der Betroffenen: überlange Gliedmaßen und oft schmaler Körperbau Kurzsichtigkeit Netzhautablösung Aortenaneurysmen (Herz-, Gefäßveränderungen) Erkrankungen der Herzklappen unerklärliche Müdigkeit überdehnbare Gelenke schmaler Kiefer mit schief stehenden Zähnen Trichter- oder Kielbrust Veränderungen an der Wirbelsäule (z.B. Skoliose) Das Syndrom wurde erstmals unter von dem französischen Kinderarzt Antoine Marfan beschrieben. In den 1930er Jahren erkannte man, dass die Besonderheit nicht geschlechtsgebunden (gonosomal) vererbt wird, sondern dass Veränderungen auf dem Chromosom 15 ursächlich sind (autosomal dominanter Erbgang). Das Gen für das Marfan-Syndrom liegt auf dem langen Arm des Chromosom 15 (15q21) und ist inzwischen sequenziert. Eine Mutation des Fibrillin-1-Genes (FBN1) führt zu verkürztem Genprodukt oder einer MissenseMutation, Mutation des TGF-β II Rezeptors. Wenn eine Mutation verschiedene, voneinander unabhängige phänotypische Merkmale verursacht, wird dies als Pleiotropie bezeichnet. Marfan-Sybdrom ein klassisches Beispiel für Pleiotropie ist, als dies viele Organe und Gewebe betrifft Achondroplasie DIA 19 Die Achondroplasie ist eine bei vielen Säugetieren – so auch beim Menschen – häufige Mutation, welche das Wachstum des Skelettsystems betrifft. Sie wird in geringerem Teil autosomal-dominant vererbt (ca. 15 %), entsteht zu weit größerem Teil aber durch Neumutation, wobei die Wahrscheinlichkeit des Auftretens insbesondere mit dem Alter des biologischen Vaters ansteigt. Die verkürzten Beine einiger Hunderassen wie Dackel und Basset Hound sind das Resultat einer gezielten Selektion auf Achondroplasie. Die Achondroplasie ist das Resultat einer Punktmutation im Fibroblasten Wachstumsfaktor-Rezeptor Gen (FGFR-3). In 98% der Fällen in der 1138. Position eine Substitution erfolgt: G mutiert auf A, welche zu Gly -> Arg Aminosäueraustauschung in der Proteinkette führt. Diese autosomal dominante Mutation führt zu einer Störung der Knorpelbildung; die Knochenwachstumszone (Epiphysenfuge) wird verfrüht verknöchert, was zur EXTRA ANFORDERUNG Boldogkői Zsolt ©Seite 2 1b. Monogenetische Krankheiten- II Einschränkung des Längenwachstums vor allem der Arme und Beine (Extremitäten) führt (enchondrale Ossifikation) Albinismus DIA 20 Albinismus ist eine Sammelbezeichnung für angeborene Störungen in der Biosynthese der Melanine und der daraus resultierenden helleren Haut-, Haar- und Augenfarbe. Betroffene Tiere nennt man Albinos, betroffene Menschen ziehen meist die neutralere Form „Menschen mit Albinismus“ vor. Menschen mit Albinismus bekommen leichter Sonnenbrand und deshalb auch leichter Hautkrebs. Außerdem sind bei vollständigem Albinismus Sehschärfe und ihr räumliches Sehen eingeschränkt. /Albinismus folgt meist einem rezessiven Erbgang und kommt beim Menschen weltweit mit einer Häufigkeit (Prävalenz) von 1:20.000 vor. Häufungen finden sich vor allem in Afrika mit einer Prävalenz von 1:10.000 und höher./ Bei Säugetieren einschließlich des Menschen tritt der Albinismus mit aufgehellter Augen-, Haut- bzw. Fellfarbe aus denselben Gründen auf, da bei ihnen die Farbstoffsynthese sehr ähnlich ist. Mutationen mehrerer Gene können zum Albinismus führen. Das Tyrosinase Gen (TYR) Albinismus bezeichnet ein Enzymdefekt. Das Enzym synthetisiert dunkles Pigment (Melanin) aus dem Tyrosin Aminosäuer. Das P Gen Albinismus bezeichnet den Mangel eines des Tyrosinase hilfenden P Proteins, es benötigt für das Transport der Tyrosinase aus dem Endoplasmatischen Reticulum heraus, verantwortlich für blondes Haar und blaue Augen bei Europäern. Die helle Hautfarbe der Menschen wurde durch MCR1 Gen Mutationen geschafft. Bei den Neanderthalen war dieses Gen auch mutatnt, aber in einer anderen Position. Das MCR1 Genprodukt spielt eine Rolle in Pigmentsynthese. Seine Mutation in den europischen und asiatischen Menschen ist keine Funktionslose Mutation, sondern eine Allelvariante. DIA 20b Früher wurde gedacht, dass dunkeler Haut gegen UV Strahlung verursachende Krebs schützt. Heute, der wissenschaftliche Überzeugung lieber ist, dass die Farbe der Haut gegen UV Licht als Ergebnis von zwei gegenüberliegenden Prozessen tritt auf: die UV-Strahlung fördert die Bildung von Vitamin D aus Provitamin A, aber erniedrigende Wirkung auf die Folsäure hat, die in der Synthese der Purin-und Pyrimidinbasen eine Rolle spielt . Tay-Sachs Krankheit (TSD) DIA 21 Seltene rezessive Erkrankungen können entstehen unter Isolatbedingungen in erstaunlicher hoher Häufigkeit. Die Entstehung der Isolate kann geographische, historische, ethnische, oder religiöse Ursachen haben. Ein Beispiel für die Zunahme genetischer Erkrankungen unter Isolatbedingungen ist hohe Frequenz von drei Lipidspeichererkrankungen unter den Aschkenazi-Juden. Die Krankheiten beruhen auf Defekten lysosomaler Hydrolasen: Tay-Sachs-Erkrankung (Gangliosidose). Eine Mutation im HEXA Gen (auf dem 15. Chromosom) zu viel Gangliosid Anhäufung in der Nervenzellen verursacht. Es führt schon im 6. Lebensmonat zur Demenz und im 4. Lebensjahr zum Tod. Fettleibigkeit – monogenetischer Hintergrund DIA 21 Die Fettleibigkeit ist eine typisch poligenetische Krankheit, aber leichter sie zu einem Gen binden, deshalb oft kann man solche Nachrichten lesen z.B. eine Forschung stellte es fest, dass das klf14 Gen als ein Hauptschalter andere Gene in der Fettzellen kontrolliert. Das KLF14 Genprodukt (ein Transkriptionsfaktor) wirkt auf die Gene, EXTRA ANFORDERUNG Boldogkői Zsolt ©Seite 3 1b. Monogenetische Krankheiten- II die Cholesterin-, Glükose- und Insulinspiegel im Blut regulieren. Über der genetischen und hormonalen Faktoren der Fettleibigkeit siehe das andere Material. . Die Störungen des Farbsehen: Farbenblindheit und Farbensehschwäche DIA 23 Die Farbenblindheit kann mehrere Ursachen haben, genetische Mutationen, Sehnerven-, Gehirn chädigungren, usw. Die menschliche Netzhaut besteht aus zwei Typen der Lichtempfindingsrezeptoren: aus Stäbchen (die bei niedrigen Lichtintensität aktiv sind) und aus Zäpfchen (die bei dem Tageslicht aktiv sind). Drei verschiedene Zäpfchen können wir unterschieden, jeder andere Photopigment enthält, die unterschiedliche Wellenlänge empfinden: die rote Zäpfchen (L Zäpfchen: long) empfangen den langen, die grüne (M: medium) den mittleren, die blaue Zäpfchen (S: short) den kürzeren Wellenlängen des Lichtes. Eine korrekte Aufteilung der Zäpfchen würde aber so aussehen: gelbe, gelb-grüne, blaue lichtemfindende Zäpfchen. Weiterhin die Zäpfchen können außerdem der Farben die Bewegungen, Farbenmodalität, Farbentief auch empfangen. Von der Genen des Farbsehens die grüne und rote Opsin Gene liegen auf dem X Chromosom, deshalb die Männer sind am meistens betroffen. Es gibt 4 Arten der vererbenden Störungen des Farbsehens: (1) Achromatie (totale Farbenblindeit): jede Typen der Zäpfchen fehlen in der Netzhaut. Die Patienten können nur undeutliche schwarz-weisse Bilder sehen. Darum sind die Bilder undeutlich, denn die Stäbchen nur die Lichtstarkkeit empfangen können. (2) Monochromatie nur ein Typ der Zäpfchen (in der Regel die blaue) ist in der Netzhaut vorhanden. Die Patienten können wiedermal nur schwarz-weisse Bilder sehen, weil mehrere und verschiedene Farben zur Vergleichung der Farbenempfindung des Gehirns notwendig sind. Wenn keine Vergleichung ist, kein Farbsehen stattfindet. Diese Patienten können im Gegensatz der Achromatie scharf sehen. (3) Dichromatie zwei Typ der Zäpfchen sind in der Netzhaut vorhanden. Eine aus der 3 Lichtempfindungssystemen fehlt oder nicht Funktionsfähig ist (Farbfehlsichtigkeit). Wir unterschieden 3 Arten voneinander: (a) Protanopie: die rotempfindliche Rezeptoren fehlen (Rotblindheit), man sieht rote Farbe als graue. Die Häufigkeit ist 1% in den Männern. (b) Deuteranopie: die grünempfindliche Rezeptoren fehlen (Grünblindheit), sie verursacht Störungen in der rotgrün Farbunterscheidungsfähigkeit. (c) Tritanopie: Sehr selten vorkommend die blaue lichtempfindliche Rezeptoren fehlen (Blaublindheit). Da diese Rezeptorgene nicht auf dem X chromosom liegen (so manifestieren sich nur in dem homozygoten rezessiven Zustand) sind die Kranken sehr selten. (4) Anomalien der Trichromatie die verschiedene Zapfchen bleiben funktionsfähig, aber ihre photospektrale Empfindlichkeit wird verändert (das Maximum der Lichtempfindlichkeit in der Spektrumkurve wird verschoben): so sprechen wir über Protanomalie, Deuteranomalie und Tritanomalie. EXTRA ANFORDERUNG Boldogkői Zsolt ©Seite 4 1b. Monogenetische Krankheiten- II Evolutionelle Aspekte des Farbenfehlsehens Die mehr als 5% auftretende rezessive Merkmale stellen eine Frage auf, ob diese bestimmte Allele neben der Nachteilen auch Vorteilen bieten. Die homozygote Patienten im Fall der Zystische Fibrose oder Sichelzellenanämie schwierig krank sind, trotzdem die Heterozygoten resistenter sind gegen einiger Infektionen. Die Heterozygotie ist im Fall eines mit X Chromosom gebundene Gen in Frauen vorhanden. Spekulationen werden unternommen um die in Männer Anomalia Trichromatie verursachende Mutationen, bei Frauen eine bessere Sehensfähigkeit auszulösen, weil sie (in Heterozygoten Zustand) vier verschiedene Photopigmentallele trägen (sie sind also Tetrachromat). Die Deuteranomale Männer haben wirklich Problemen mit Unterscheidung der Rot-Grün Farben, aber bei den Soldaten wurde es erstmal bemerkt, dass sie die kakhi Farbenschattierung (Tarnung) besser erkennen können. Genetik Für die Farbsehstörungen sind in der Regel die Mutationen der auf dem X Chromosom liegenden grünen und roten Opsingene verantwortlich. Die Farbsehenproblemen können selten andere Mutationen verursachen: auf 19 anderen Chromosomen wurde 56 verschiedene Gene bestimmt, die Anomalien im Farbsehen erfolgen. Zirka 8% der Männer, 0,5 % der Frauen leiden von Farbsehenproblemen. Aus den Proportionen können wir feststellen, dass die mit dem X Chromosom verbundene rote und grüne Lichtempfindlichkeiten sind die grösste Problemen. Rote Pigmenten kodieren die OPN1-LW, die grüne Pigmentendie OPN1-MW Gene. Die 75% der Rotgrünfarbenblindner können wir mit molekularen Techniken diagnostisieren (100% der Protanopen und 65 % der Deuteranopen). DIA 24 Eine Variation in der Anzahl der Grünpigmentgene können wir in dem normalen Farbsehen beobachten. Das rote Pigmentgen liegt immer in der 5’ Richtung in einer Kopie. Das Grünpigmentgen kann unter den kaukasien Menschen 1 bis 6 Kopien vorhanden. Auf dem Bild representieren die Quadraten die 6 Exons. Die Länge des Rotpigmentgens 15kb, des Grünpigmentgens 13kb sind. Der Abstand zwischen der Opsin Gene zirka 25kb sind. Das rote und das folgende grün Gen sind nur aktiv, die Reste Grünopsingene bleiben inaktiv (die sind Pseudogene). DIA 25 Die intergenische Rekombinationen verursachen unterschiedliche Genkopien und Fusionsgenen. DIA 26 Die rot-grün Farbfehlsichtigkeit wird durch bestimmte Genvariante erfolgt, welche können Deletionen (B6), Hybridgene/Fusionsgene (A1, A2, A3, B4, B5) sein. Rot-Grün-Sehschwäche oder Rot-Grün-Blindheit (Dyschromatopsie) sind die wissenschaftlichen Fachtermini für über 99 % der Farbfehlsichtigkeiten, die umgangssprachlich als Farbenblindheit bezeichnet werden. Nach ihrem Entdecker John Dalton nennt man sie auch Daltonismus. Die Betroffenen können hierbei die Farben Rot und Grün schlechter als Normalsichtige unterscheiden. Ursache dieser genetisch bedingten Behinderung ist die Veränderungen der Aminosäuresequenz in den Sehpigment-Proteinen (Opsin) der entsprechenden Zapfen der Netzhaut, die aus der Veränderung der Gensequenz des entsprechenden Opsins resultiert. Es existiert bei jedem Menschen jeweils ein Gen für das rotempfindliche Opsin und drei identische Gene für das grünempfindliche Opsin. Alle liegen nahe beieinander auf dem X-Chromosom. Durch Fehler beim Crossingover/assymmetrisches Crossover kommt es zu falschen Genkombinationen. Vor allem zu Kombinationen, die sich phänotypisch durch verschobene Absorptions-Empfindlichkeitsmaxima in den entsprechenden ZapfenTypen äußern, meist bei den Grün-Rezeptoren, da sich diese direkt an einer Crossing over-Stelle des XChromosoms befinden. Fehlt das Gen für eines dieser Opsine komplett, spricht man über einer Rot- oder Grünblindheit (Protanopie oder Deuteranopie). DIA 27 Photopigment Gentherapie: siehe in der Therapien Vorlesung EXTRA ANFORDERUNG Boldogkői Zsolt ©Seite 5