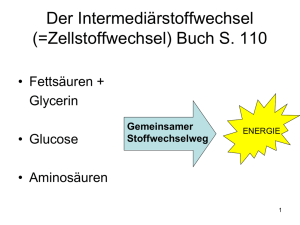
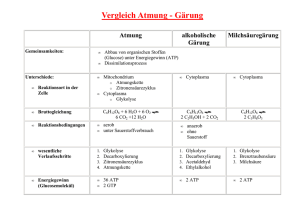
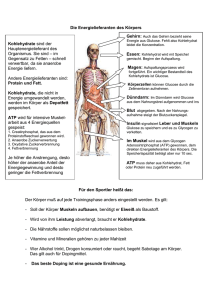
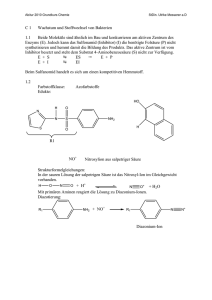
OH - BioKemika
Werbung

Zusammenfassung Biochemie Vordiplom (Stoffwechselseminar, Biochemie I, Biochemie I Praktikum) Gliederung 1.1 1.2 1.3 1.4 1.5 1.6 1.7 1.8 1.9 1.10 1.11 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 2.9 2.10 2.11 2.12 2.13 2.14 2.15 2.16 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 Teil 1: Biomoleküle Lipide Biologische Membranen, deren Stoffwechsel sowie Funktionen deren Lipide Kohlenhydrate Nukleinsäuren Aminosäuren Peptide und Proteine Myoglobin und Hämoglobin Enzyme Proteasen Membranproteine Coenzyme, prosthetische Gruppen und Cofaktoren Teil 2: Metabolismus Metabolismus, ATP und Gruppenübertragungspotentiale Glykolyse Gluconeogenese Gärung Pentosephosphatweg Glykogenmetabolismus (Schwerpunkt:Regukation) Citratzyklus oxidative Phosphorylierung Fettsäuremetabolismus Ketonkörperbiosynthese Cholesterinbiosynthese Aminosäuredegradation Harnstoffzyklus Vernetzung des Stoffwechsels Der Muskel Die Leber Teil 3: Praktikum Diagonaltechnik DNA Immunologische Grundlagen ELISA Immunologie Glyoxisomen Bromelain Anionentransport 1 TEIL 1: BIOMOLEKÜLE Hierarchische Organisation biologischer Strukturen Glucose und einige andere wenige Moleküle (z.B. Ammoniak) sind Grundsubstanzen des Lebens, mit Hilfe derer man andere Substanzen (AS, Nukleotidbasen, Monosaccharide, Fettsäuren, Glycerin, Cholin) im Stoffwechsel generieren kann; Diese Substanzen stellen Grundbaustein für höhere Strukturen (Nukleotide, Phospholipide, Triacylglycerine) und Makromoleküle (Proteine, Nukleinsäuren, Polysaccharide) dar; Proteine, Nukleinsäuren, Polysaccharide und Phospholipide können sich zu größeren Strukturen zusammenlagern (Multienzymkomplexe, Ribosomen, Chromosomen, membranen, Mikrotubuli), welche wiederum Zellorganellen bilden (Kerne, Mitochondrien, Peroxisomen, ER, Golgi, Lysosomen); Zellorganelle bauen Zellen auf, diese lagern sich zu Geweben zusammen; ein Organ besteht aus mehreren Geweben, während ein Organismus aus mehreren Organen besteht; Dimensionen: Teilungsrate einer Säugetierzelle: 12-24h Kalottenmodell zeigt Van-der-Waals Radius Proteinfunktionen Enzymkatalyse, Transport und Speicherung (Aufrechterhaltung eines Energiegradienten), koordinierte Bewegung (molekulare Motoren, Bewegung von Schleimpilzen in Richtung einer cAMP Quelle), Materialeigenschaft (Stabilität!), Immunabwehr, Erzeugung und Weiterleitung von Nervenimpulsen (Signaltransduktion), Kontrolle von Zellwachstum und Differenzierung (koordinierter Zelltod) 2 AS unterscheiden sich in Größe, Form (Erkennung), Wasserstoff-Donor-Akzeptor Wirkung (Ladung) und chemischer Reaktivität voneinander; Van-der-Waals Kontaktradien: zwischen 1 und 2 Å ist die attraktive WW maximal; Zusammensetzung aus abstoßender WW, die mit zunehmender Nähe r sehr stark zunimmt (proportional zu r -12) und anziehender WW, die mit zunehmender Nähe r schwächer zunimmt (proportional zu r -6); Die Summe ergibt folgendes Potential Metalle und ihre biologische Funktion: Funktion Na+ K+ Ladungstransport, Aufbau eines Ladungsgradienten Beweglichkeit Liganden Komplexbildung Austausch hoch O schwach Sehr schnell Mg2+ Ca2+ Stabilisierung von Strukturen, Informationsträger (second messenger, Signalweiterleitung, Kanalsteuerung) mittel O mittel mittel Zn2+ Lewis-SäureKatalysatoren (polarisiert Wasser) Fe, Cu, Co, Mn RedoxKatalysatoren keine S, N Sehr stabil keine Keine S, N Sehr stabil keine Fluor ist Wachstumsfaktor bei Ratten und Bestandteil von Zähnen und Knochen; Silicium bildet Struktureinheit der Kieselalgen und ist essentiell bei Hühnern; Vanadium ist essentiell für höhere Tiere; Chrom ist essentiell für höhere Tiere; Mangan ist essentiell für die Aktivität einiger Enzyme; Eisen ist wichtiges Übergangselement; Kobalt ist essentiell für Vitamin B12; Kupfer ist wesentlicher Bestandteil von Redoxenzymen; Selen ist essentiell für Gluthationperoxidase; Molybdän ist essentiell für die Aktivität einiger Enzyme; Zinn ist essentiell für Ratten; 3 Säuren und Basen katalysieren wichtige Reaktionen O OH R O + R C OH O - H CH2 H2C OH H H H BH Veresterung B - H O R Aldoladdition 1 H O R R 1 O 1 + H3C CH2 H3C C OH OH H H O R R BH O OH O B - O CH2 H3C C H O - H Säure-Base-Definition: Brönstedt Säure = Protonen-Donor Brönstedt Base = Protonen-Akzeptor Lewis-Säure = Elektronenpaarakzeptor (Elektrophil) Lewis-Base = Elektronenpaardonor (Nukleophil) Ionisierung von Wasser: H H2O + + OH - [H ]⋅ [OH ] , [H O] = 55,5M , Dissoziationsgleichgewicht: K = [H O] Ionenprodukt des Wassers: K = [H ]⋅ [OH ] = 10 M + − eq 2 2 + − −14 W Definition von pH und pK Wert: Für schwache Säuren und Basen gilt: H HA + + - A [ ] pH = − log H + pK S = − log K S Dissoziationsgleichgewicht: K S = [A ] − [HA]⋅ K S = [H ]⋅ [A ] + [HA] − [ ] 1 A− (log) pK + = pH Hendersen-Hasselbalch Gleichung log S [HA] H+ [ ] Pufferkapazität ist im Bereich des pKS Wertes am größten: HA + OH - H2O + - A (Basizität von OH- wird kompensiert, indem Puffersubstanz Proton abgibt) Puffer kann auch Acidität von H+ kompensieren, indem es das Proton aufnimmt! 4 Wenn der pH Wert < pKS Wert der Gruppe liegt die Gruppe protoniert vor (je kleiner pKs Wert desto acider die Verbindung) Wenn der pH Wert > pKS Wert der Gruppe liegt die Gruppe deprotoniert vor; (je größer pKs Wert desto basischer die Verbindung) Wenn der pH Wert = pKS Wert der Gruppe gilt protonierter Anteil der Gruppe = deprotonierter Anteil der Gruppe (im GG) Ladung der Aminosäuren bei verschiedenen pH-Werten: Proteine als Ampholyte COOH + H3N +H Cα H H2C COOH pH=1 Nettoladung: +1 +H COO + + H3N - H H2C COOH pH=3 Nettoladung: 0 Cα - H R dominante Form pH=11 +H Cα COO + H2N H Cα R dominante Form pH=7 +H Cα + - pKs: 8,0 COOH H3N pKs: 3,1 COO H3N H R dominante Form pH=1 + + COO + + H3N - +H Cα H H2C COO COO + H2N Cα - H H2C - pH=6,6 Nettoladung: -1 COO - pH=11 Nettoladung: -2 Isoelektrischer Punkt ist der pH Wert, bei dem Nettoladung = 0 ist; IEP ist für jede AS spezifisch; 5 Lipide Gliederung in versteifbare (enthalten Esterbindungen) und nicht versteifbare (ohne Esterbindungen) Lipide; Einteilung in Lipidklassen: 1.) Fettsäuren - - - - Fettsäuren (aliphatische Monocarbonsäuren): bestehen aus unverzweigter Kohlenwasserstoffkette und einer Carboxylgruppe (nicht versteifbar, amphiphil: hydrophober Schwanz, hydrophiler Kopf); meist geradzahlige Anzahl an C Atomen (aus C2 Körper synthetisiert) Löslichkeit in Wasser nimmt mit Kettenlänge und Sättigungsgrad ab Kohlenstoffkette: gesättigt beweglicher, oder ungesättigt bzw. mehrfach ungesättigt; gesättigte FS sind bei Raumtemperatur fest; Z DB erniedrigen den Schmelzpunkt; polyungesättigte FS werden im Körper schnell oxidiert; manche ungesättigte FS sind für Menschen essentiell: Omega-n-FS; Omega-3-FS hat DB an dritter Stelle von hinten gezählt; Die Doppelbindungen in ungesättigten Fettsäuren sind meist Z konfiguriert (es resultiert ein 30° Knick in der Kette, wodurch die Van-der-Waals WW zu anderen Molekülen abgeschwächt werden und der Schmelzpunkt verringert ist); bei Schmelzpunkt findet Übergang von Wachs nach Flüssigkeit statt; 6 - selten sind konjugierte und E-Fettsäuren; E DB haben höheren Schmelzpunkt als Z DB; je kürzer die Kette, desto hydrophiler ist die FS und somit auch das Fett C12 C14 CH3 -OOC C16 -OOC C18 -OOC C16 ∆ C18 ∆ C18 ∆ C18 ∆ - CH3 CH3 9 CH3 -OOC 9 -OOC 9,12 Laurinsäure Myristinsäure Palmitinsäure Stearinsäure Palmitoleinsäure CH3 -OOC -OOC 9,12,15 CH3 Ölsäure CH3 Linolsäure α-Linolensäure CH3 -OOC T SCHMELZ steigt T SCHMELZ sinkt Funktion von FS: Bausteine von Phospho- und Glykolipiden (=amphipathische Moleküle); Proteine können durch kovalente Bindung an FS modifiziert werden (Dirigierung an bestimmte Membranorte); Brennstoffmoleküle (werde oxidiert, um Energiebedarf zu decken); Derivate der FS dienen als Hormone und intrazelluläre Botenstoffe 2.) Triacylglycerine dreifach-Ester des dreiwertigen Alkohols Glycerin (versteifbar, apolar: absolut wasserunlöslich); Hauptenergiespeicherform; hydrophob, unlöslich in Wasser; kommen in Fetttröpfchen in den Adipocyten vor; dienen dem Langzeitspeicher O H2C OH H2C O HC O H2C OH Glycerin + 3 HO R 2 O R Fettsäure 1 O O CH OH R H2C 3 H2O O R 3 Triacylglycerin 3.) Triglyceride zweifach-Ester des dreiwertigen Alkohols Glycerin, wobei die dritte Position mit einer nichtFS X verbunden ist; meist amphipatisch (X ist teilweise hydrophil) 7 OH O H2C O R 1 O HC H R O X= - P O Triglycerid H OH CH3 O O O OH HO HPO 3 Phosphatidsäure 2 X H2C Diacylglycerin (DAG) - CH2 O + CH2 N - H H CH3 H H CH3 OH Galactolipid Phosphatidylcholin A.) Phosphoglyceride: bestehen aus zwei mit Glycerin veresterten Fettsäuren und einer Phosphorsäure, die mit einer weiterer Alkoholverbindung verestert ist (Phosphorsäurediester) (Versteifbar und amphiphil); Hauptbestandteil zellulärer Membranen; häufigstes Phosphoglycerid ist Phosphatidylcholin; gefolgt von Phosphatidylethanolamin (halb soviel in Membran enthalten); Das einfachste Phosphoglycerid ist Phosphatidsäure; O OH O H2C R 1 O R O P O CH2 CH2 N 2 Cholin O H2C + HO O HC CH3 O X - Phosphoglycerid X= HO OH OH CH3 CH3 OH Inosit + NH3 Ethanolamin OH COO + H3N Serin - OH CH2OH HOHC OH CH2OH Glycerin - - R1 ist eine gesättigte C16 bis C18 FS; R2 eine ungesättigte C18 bis C20 FS; Verknüpfung erfolgt durch nukleophilen Angriff der Hydroxylgruppe von X an die Phosphorgruppe im Phosphoglycerid In der Regel sind Phospholipide neutral (Phosphatidyl-ethanolamin und -cholin), einfach negativ (Phosphatidyl-serin und –glycerin, sowie Phosphatidsäure) oder zweifach negativ geladen (Cardiolipin) wenn Phosphatidylglycerol als Rest X fungiert spricht man von Cardiolipin 8 O H2C HC 1 O C R O O C R 2 Cardiolipin O H2C P O O CH2 O - CH2OH O O H2C P O O O 4 R C O CH O R C O CH2 3 CH2 B.) Glykoglyceride bestehen aus zwei mit Glycerin veresterten Fettsäuren und einem Zuckerrest; In Chloroplastenmembran dominieren Galactolipide; OH O H2C O HC O R 1 O R O O HO OH 2 (MGDG) Monogalactosyl-Diglycerid X Triglycerid O HO OH H2C OH R O X= O - OH O O O OH HO S R OH O O OH OH O HO R (DGDG) Digalactosyl-Diglycerid OH Sulfochinovosyl-Diglycerid 4.) Sphingolipide bestehen aus einer mit Sphingosin (C18 Aminoalkohol) veresterten, ungesättigten Fettsäure ( Ceramide; Bildung eines Säureamids); Zusätzlich können als Rest X fungieren: A.) Phospho-Cholin oder seltener Phospho-ethanolamin ( Sphingomyelin), kommt in Plasmamembranen von Schwannschen Zellen (Myelinschicht) vor B.) Monosaccharide: Glucose oder Galactose ( Glucosylcerebrosid oder Galactosylcerebrosid = Cerebroside), Gal-Cerebroside in Plasmamembran von Neuronen und Glc-Cerebroside in Plasmamembran von nicht-neuronalen Zellen C.) Neutrale di-, tri- oder tetrasaccharide (Globoside; z.B. Lactosylceramide, wenn Disaccharid Lactose dranhängt) 9 D.) eine komplexe Zuckerstruktur, negativ geladen ( Ganglioside; entstehen aus Cerebrosiden) (versteifbar und amphiphil); Bestandteil von Membranen; Rückgrat ist Dihydroxylamin OH H3C OH C18 Sphingosin OH H2N OH CH3 O R OH Ceramide R NH O NH O R O 1 O R 1 O P CH2 + CH2 N - CH3 CH3 Spingomyelin A.) Ganglioside Bei den Gangliosiden ist eine Oligosaccharidkette an das Ceramid gebunden, die mindestens einen sauren Zucker (=Salinsäure) enthält; Bestandteil von Nervenzellen (störungen des Gangliosidabbaus enrste klinische Konsequenzen; führt bis zum Tod) CH3 O HN Salinsäure HO HO O COO OH OH OH NeuAc N-Acetyl-Neuraminidat 10 5.) Plasmalogene Plasmalogene sind Ether-verknüpfte Phospholipide im Herzmuskel und im zentralen Nervensystem (Gehirn); kommen in halophilen Bakterien vor; Etherbindung ist beständiger gegen Hydrolyse als Esterbindung; Außerdem: Resistenz gegen Verdau durch Phospholipasen, die spezifisch Ester-verknüpfte Phospholipide verdauen; Plasmalogen ist das namensgebende Lipid dieser Klasse (das C1 trägt eine über Etherbindung verknüpfte C16 FS mit DB zwischen den ersten beiden C Atomen; das C2 trägt ebenso eine C16 FS, ohne DB, verestert) Platelet-aktivierender Faktor (PFE) ist ein Etherlipid mit Signalfunktion bei Entzündungen; er wird durch Leukozyten (Basophile) freigesetzt und löst die Vasokonstriktion (Regulierung der Organdurchblutung durch Gefäßverengung, wodurch der Strömungswiderstand des arteriellen Systems und damit der Blutdruck erhöht wird) und Thrombozyten-Aggregation (Thrombocyten = platelets = Blutblättchen) aus; durch die Acetyl-gruppe wird dieses Lipid wasserlöslich; es enthält am C1 eine Ether-verknüpfte C16 FS ohne DB O H2C CH CH O CH O R 2 R 1 O O + CH2 CH2 N - 1 PFE O CH CH3 CH3 P O R O Plasmalogen O H2C O H2C CH3 O CH3 O H2C CH3 P O O + CH2 CH2 N - CH3 CH3 6.) Isoprenoide (vor allem Steroide) Isopren (C5) Grundgerüst; Polymerisierung und Cyclisierung möglich; Prenyle (Farnesyl, geranyl etc.), Gallensäuren und Steroide gehören in diese Klasse; Steroide bestehen aus einem Sterangerüst, einer verzweigten aliphatischen Seitenkette, mehreren Methylgruppen und Alkohol oder Säuregruppen; (nicht versteifbar, Ausnahme: Cholesterinester; amphiphil); Bestandteil von Membranen (Cholesterin), Hormone (Steroidhormone), Emulgatoren (Gallensäuren) 24 H3C 21 H3C 19 11 1 CH3 9 C A B D 2 10 8 3 4 Steran H3C 29 5 6 CH3 28 13 24 1 24 22 18 12 CH3 17 Sterol HO 20 2 23 16 14 26 CH3 H3C 25 CH3 27 CH3 CH3 CH3 CH3 H 15 CH3 7 30 H H Cholesterin HO Steroid-Gerüst Cholesterin wird mittels Lipoproteine im Körper transportiert; 70% liegt dort in Form von Cholesterinester vor; 11 7.) andere Lipidklassen, Vergleich der Lipide Phospholipide = Phosphoglyceride + Phosphosphingolipide + Plasmalogen Glykolipide = Glykoglyceride + Glykosphingolipide Sphingolipide und Glycerphospholipide sind strukturell sehr ähnlich: Biologische Membranen, deren Stoffwechsel, sowie Funktionen deren Lipide flächenförmige Struktur (5-6 nm Dicke eine Phospholipiddoppelschicht; Membrandicke: 6 10 nm); Barriere für polare Moleküle; Nichtkovalente Zusammenlagerung von Lipiden, Proteinen und Kohlenhydraten; asymmetrische, flüssige Struktur, ermöglicht elektrische Polarisierung und Kompartimentierung!! Komponenten der biologischen Membran: Membranlipide: Phospholipide (glycerophospholipide und Sphingomyelin), Glykolipide (Ganglioside), Cholesterol Membranproteine: integrale Proteine innerhalb der Membran und periphere Proteine an der Peripherie; Transmembranproteine durchspannen die gesamte Membran mit einer oder mehreren hydrophoben AS-Sequenzen oder sind über hydrophobe Anker mit der Membran verbunden; 12 Innerhalb der Zelle ist das Cytoskellet mit der Membran verbunden; außerhalb der Zelle ist die Glykokalix befestigt; Die Glykokalix besteht aus Oligosachariden, die kovalent an die Membranproteine und – Lipide gebunden sind; sind sie an Membranproteine gebunden, heißt der Membranbaustein Glykoprotein; sind sie an Membranlipide gebunden, heißt er Glykolipid; Spektrin ist ein Makromolekül und wesentlicher Bestnadteil des Zytoskeletts der Erythrozyten, kommt aber auch in anderen Zellen vor; es stabilisiert zusammen mit anderen Proteinen wie z.B. Aktin die Innenseite der Zellmembran und somit die gesamte Zelle; Abbildung: Innenseite der Erythrozyten-Membran mit Spektringerüst 1.) Struktur und Dynamik der biologischen Membran Detergentien sind kegelförmig und bilden Micellen (große Micellen können im zentrum auch Wasser beinhalten); Phospholipide sind zylindrisch und bilden Membrandoppelschichten und Vesikel; Detergenz Phospholipid Liposome sind kleine, kugelförmige Vesikel aus konzentrisch angeordneten Lipiddoppelschichten (Bilayer), welche sowohl hydrophile Stoffe in der wässrigen 13 Innenphase als auch lipophile Stoffe innerhalb der Bilayer transportierten können; als Hauptkomponente tritt meist Phosphatidylcholin (Lecithin) auf; sie entstehen automatisch durch Selbstassoziation von Amphiphilen im wässrigen milieu; Liposomen werden durch ihre Lamellarität und Größe unterschieden: A.) kleine unilamelare Vesikel (SUV; 20 bis 100 nm Durchmesser) B.) multilamelare Vesikel (MLV) C.) große unilamelaren Vesikel (LUV) D.) multivesikuläre Vesikel (MVV) MLV, LUV, MVV haben einen Durchmesser von mehreren hundert nm bis mehreren Mikrometern; Phasenumwandlungen: - Kristalline Membranstruktur bei geringer Temperatur, da nicht genug thermische Energie vorhanden, um die energetisch günstigen trans Konformation zu verlassen Fluide Membranstruktur bei Überschreitung des Schmelzpunktes der Membran, da vereinzelnd trans Konformationen zu gauche Konformationen umgewandelt werden; Faktoren der Phasenumwandlung: Saturierung und Länge der Fettsäure, sowie Hydrophilie der Kopfgruppe sind entscheidend für den Schmelzpunkt der Lipiddoppelschicht; PE hat einen höheren Schmelzpunkt als PC bei selber Kettenlänge und Saturierung; 14 Cholesterol ist Mediator der Fluidität: Cholesterol verringert die Bewegungsfreiheit von C1 bis C9 der Fettsäuren innerhalb der Membran und erhöht die Bewegungsfreiheit der terminalen Methylengruppen; Unterhalb der Schmelztemperatur der Membran nimmt daher die Fluidität zu, während sie oberhalb der Schmelztemperatur abnimmt (Cholesterol beeinflusst das Schmelzverhalten allosterisch); Ohne Cholesterin wäre die Membran entweder kristallin oder fluid-ungeordnet; mit Cholesterol ist sie fluid-geordnet; Wenn die Transition-Temperatur unterschritten wird, die Membran also im parakristallinen Zustand vorliegt, ist ihre Permeabilität erhöht, die Selektivität für den Stofftransport jedoch vermindert. Außerdem steigt die Aktivierungsenergie für membrangebundene Enzyme. Es gibt zwei Arten der Lipid-Diffusion: laterale Diffusion mit Geschwindigkeit von 2 μm/s; ein flip flop (transversale Diffusion) findet sehr selten statt (unkatalysiert: einmal in Stunden bis Tage / Flippase-katalysiert: Sekunden); Untersuchung der lateralen Diffusion: Laser-Bleaching of fluorescent marker fluorescence recovery after photo-bleching (FRAP) 15 2.) Permeabilität der Membran Geringe Permeabilität für Ionen und polare Moleküle; permeabel für apoalare Substanzen; 3.) Membranzusammensetzung und Assymetrie der Membran Die Zusammensetzung der Membran ist vom zell- und Organelltyp abhängig 16 Die Asymmetrie zwischen innerer und äußerer Lage der Doppelschicht bewirkt, dass die cytosolische Seite negativ geladen und die exoplasmatische Seite positiv geladen ist; 4.) Ankerlipide und „lipid rafts“ - - Viele intra- und extrazelluläre Proteine werden posttranslational durch lipophile Gruppen (Anker Gruppen) modifiziert; in erster Linie vermitteln lipohile Anker die Bindung des Proteins an Membranen und spezifische Membrankompartimente; verändern aber auch physikochemische Eigenschaften des Proteins (nehmen so Einfluss auf Signaltransduktion) Überblick: Der Glucosyl-Phosphatidyl-Inositol-Anker (GPI-Anker) befestigt Proteine, wie die alkalische Phosphatase an der extrazellulären Membran; Als Membrananker fungieren auch Cholesterol, sowie verschiedene Fettsäuren; bekannte Fettsäureanker sind die Acylgruppen Myristoyl und Palmitoyl und die Prenylgruppen Geranylgeranyl und Farnesyl. 17 GPI Anker: vermitteln Verankerung von Proteinen auf Zelloberfläche; dazu gehören Rezeptoren, Adhesionsproteine, Enzyme und Zelloberflächenmarker; hängt am C terminus; Cholesterol dient als Lipidanker für Proteine der Hedgehog-Familie; diese sind an Zelloberflächen lokalisiert und wirken als morphogenetische Faktoren bei Embryonalentwicklung; Prenylierte Proteine sind involviert in Signaltransduktion (G-Proteine), in der Organisation des Cytoskeletts, den vesikulären Transport und die Ausbildung der Kernstruktur; Prenylierung erfolgt über die Ausbildung eines Thioethers zwischen Cys nahe des C terminus und den Isoprenoiden Farnesyl (C15) oder Geranylgeranyl (C20) (mittels Farnseyltransferase bzw. Geranylgeranyl-Transferase I und II) Acylierte Proteine; die Acylierung erfolgt durch die Fettsäuren Myristat und Palmitat; Die Myristoylgruppe wird an einem N-terminalen Glycin über eine stabile Säureamidbindung angehängt (N-Myristoyl-Transferase); Protein kann nicht mehr abgespalten werden (irreversible Verknüpfung) Die Palmitoylgruppe wird in der Nähe von Transmembrandomänen über eine Thioesterbindung mit einem Cys verbunden (S-Palmitoylierung) oder über ein Säureamid mit einer AS gekoppelt (N-Palmitoylierung); Palmitoyl-Thioesterase kann jederzeit das Protein abspalten (reversible Verknüpfung) - - - Lipophil modifizierte Proteine werden nicht zufällig in einer Membran verteilt, sondern zum Teil in bestimmten Membranregionen (Micordomänen), den lipid rafts oder Caveolae verankert; lipid rafts sind reich an Sphingolipiden, Cholesterol, PC und langen, gesättigten FS, die sehr eng gepackt sind; daher werden diese Membranbereiche als geordnet und die übrigen als ungeordnet bezeichnet; diese Anordung macht sie resistent gegen Detergentien (Isolation mittels Detergenz Triton X-100 möglich); Caveolae entstehen durch Polymerisation von Caveoline (Proteinklasse; spielt bei Signaltransduktionen Apoptose, Zellwachstum, Membrantransport u.a. wichtige 18 - Rollen) aus lipid rafts und formen Einstülpungen in der Zelloberfläche (sind ebenso resistent gegen Detergentien); In geordneten Membrandomänen sind vor allem extrazelluläre Proteine mit GPI Ankern und intrazelluläre Proteine mit Myristoylierung und Palmitoylierung oder zweifacher Palmitoylierung angereichert; Proteine, die große, verzweigte Prenylgruppen tragen sind eher gering; in lipid rafts reichern sich spezielle Transmembranproteine an; 5.) Funktion von Glycerophospholipiden in der Membran Lipidabbau: Der Abbau der Lipide erfolgt in Lysosomen durch spezifische Phospholipasen: Typ A: Entfernung einer Fettsäure Typ C: Hydrolyse der Phosphodiesterbindung zur Kopfgruppe (Entstehung von DAG) Typ D: Hydrolyse der Phosphodiesterbindung zur Kopfgruppe (aus PC: Phosphatidylcholin entsteht PA: Phosphatidsäure) A.) Phosphatidyl-Inosit und Phospholipase C Phosphatidyl-Inosit kann innerhalb der Plasmamembran durch ATP Verbrauch zweimalig phosphoryliert werden (mehrere Enzyme beteiligt), wodurch Phosphatidyl-Inositol-4,5bisphosphat entsteht (bei pH=7 ist es 4fach negativ geladen); Durch Hydrolyse mittels 19 Hormon-sensitiver Phospholipase C (Hydrolase) innerhalb der Plasmamemrban entsteht ein Diacylglycerin (DAG) und Inositol-1,4,5-trisphosphat (IP3); Phopspholipase C wird durch das G-Protein Gq/11 und Rezeptor-Tyrosin-Kinasen kontrolliert; DAG und IP3 sind die sekundären Botenstoffe (second messenger) des Gq/11-IP3 Signalweges (neben dem G-Protein gekoppelten Adenylatcyclase Weg einer der häufigsten Signalwege, die ein extrazelluläres in ein intrazelluläres Signal übersetzen); DAG aktiviert Protein Kinase C; IP3 bewirkt die Fresetzung von intrazellulärem Ca2+, das ebenso die Protein Kinase C und weitere Enzyme aktiviert; Die protein Kinase C kontrolliert weitere Enzyme durch reversible Phosphorylierung; O Phosphatidyl-Inositol-4,5-bisphosphat OH 2- O 3 PO OPO 3 2- O O OH OH O HC O O O CH2 O HC O O R R R R P O H2C O H2C 1 H2C H2O 1 2 OH Diacylglycerin (DAG) + 2 OH 2- O 3 PO OPO 3 - 2- OPO 3 OH 2- OH Inositol-1,4,5-trisphosphat (IP3) B.) Archidonsäure und Phospholipase A2 Die Phospholipase A2 kann Archidonsäure (20:4) aus Phospholipiden freisetzen; Archidonsäure ist die wichtigste Quelle für Eikosanoide (hormonähnliche Substanzen, dienen als Immunmodulatoren und Neurotransmitter); Eikosanoide lassen sich in vier Substanzklassen unterteilen: Prostaglandine, Prostacycin, Thromboxane und Leukotriene; Prostaglandine sind Gewebshormone (wirken nur im Gewebe, wo sie ausgeschüttet werden; Grund: sehr instabil, nicht speicherbar, keine Zeit Arachidonsäure-Derivate durchs Blut zu transportieren); Sie werden mittels Cyclooxygenase aus der Archidonsäure gebildet; man unterteilt nach Oxidationskgrad in verschiedene Gruppen, die jeweils andere Wirkungen auf verschiedene Rezeptoren haben; 20 6.) Ganglioside an der Zelloberfläche dienen der Erkennung - - Sphingolipide spielen eine wichtige Rolle in der Zell-Zell Erkennung und Wechselwirkung, in der zellulären Differentierung und in der Zellwachstumskontrolle; sie treten als Blutgruppenantigene und als Tumor-assoziierte Antigene auf; sie sind Rezeptoren für Glykoproteinhormone der Hirnanhangdrüße und Rezeptoren bakterieller Proteingifte (Cholera Toxin GM1); Die Biosynthese der Glycosphingolipide findet im ER und im Golgi statt; Genetische Defekte im Abbau der Glykosphingolipide führen zu schwerwiegenden Erkrankungen, da es zu abnormen Anreicherungen von Gangliosiden innerhalb der Zellmembran kommt; Gangliosid GM1 Abbau-Defekt bei Tay-Sachs Krankheit; Symptome: motorische Retardierung, Demenz, Spastik, Krämpfe, Erblindung, kurze Extremitäten 21 7.) Transport durch Membranen A.) Die drei Haupttypen von Transportproteinen Pumpen = ATPasen (1 bis 103 Ionen/s) nutzen die Energie, die bei der Hydrolyse von ATP frei wird, um bestimmte Ionen oder kleine Moleküle gegen den elektrochemischen Gradienten zu transportieren (primärer aktiver Transport); Pumpen sind für die Aufrechterhaltung der Ca2+ und Na+ Konzentration innerhalb nahezu aller tierischen Zellen verantwortlich; Außerdem dienen sie der Erzeugung eines niedrigen pH Wertes in den tierischen Lysosomen, den pflanzlichen Vakuolen und im Magenlumen; Kanäle (107 bis 108 Ionen/s) erleichtern den Transport bestimmter Ionen oder von Wasser entlang ihres elektrochemischen Gradienten (passiver Transport); Kanalproteine kleiden einen Kanal in der Membran aus, durch den mehrere Wassermoleküle oder Ionen gleichzeitig transportiert werden können; alle Plasmamembranen tierischer Zellen enthalten stets geöffnete K+ Kanäle, wodurch ein konstantes elektrisches Potential an der Plasmamembran erzeugt wird; viele weitere Kanäle sind in der Regel geschlossen und öffnen sich nur auf bestimmte Signale hin; Kanäle interagieren nur schwach mit ihrem Substrat, sind dennoch hochspezifisch; Die Spezifität basiert auf der Ladung und der Hydrathülle des zu transportierenden Ions; der Transport durch Kanäle ist erheblich schneller als der Transport durch Carrier; 22 Transportproteine (102 bis 104 Moleküle/s) transportieren Ionen und kleinere Moleküle; man differenziert Uniporter (transportieren ein Molekül entlang des elektrochemsciehn Gradienten: passiver Transport) und Cotransporter (katalysieren die Bewegung eines Moleküls gegen seinen Konzentrationsgradienten, indem sie gleichzeitig ein oder mehrere Ionen entlang eines elektrochemischen Gradienten transportieren: meist also sekundär aktiver Transport; Antiporter transportieren Molekül und Ion in unterschiedliche Richtung; Symporter in dieselbe) Transporter binden immer nur ein oder wenige Substratmoleküle; nach der Bindung des Substrats erfolgt eine Konformationsveränderung, so dass ausschließlich die gebundenen Moleküle die Membran passieren können; Carrier ist die allgemeine Bezeichnung für ein Transportmolekül, das mit dem zu transportierenden Stoff wechselwirkt (u.a. Transportproteine) B.) Die erleichterte Diffusion durch Carrier Erleichterte Diffusion, Carrier-Diffusion: Diffusionsgeschwindigkeit der Substanz durch die Membran ist höher, als sie spontan durch ein Loch vergleichbarer Größe innerhalb der Membran wäre; Nachweis mittels zwei wichtiger Kriterien: System muss absättigbar sein (Diffusionsgeschwindigkeit darf sich ab einer bestimmten Konzentration mit zunehmender Konzentration nicht mehr erhöhen) und es sollten Inhibitoren existieren, die selektiv den Transport einer bestimmten Molekülsorte hemmen, während andere Transportfunktionen derselben Membran in takt bleiben (siehe Versuch: Anionentransport) Die erleichterte Diffusion wird durch Trägermoleküle (Carrier = Permeasen), die die Membran durchspannen und spezifische Bindestellen für ihr Substrat aufweisen, ermöglicht; häufig sind solche Carrier für den Kombinationstransport eingerichtet (Durchtritt durch Membran erfolgt erst dann, wenn alle Substrate gebunden haben); entweder folgt Bindung auf der gleichen Seite (Symport) oder auf unterschiedlichen Seiten der Membran (Antiport durch Wechsel-Carrier); ATP-ADP Shuttle ist ein Wechselcarrier; C.) Der gruppen-Transfer Gruppen-Transfer: hier findet der Transport nur unter gleichzeitiger chemischer Modifizierung (z.B. Phosphorylierung oder Acylierung) des Substrats statt; solche Transporteinrichtungen sind sehr komplex gebaut; Beispiel ist der Transport von Glucose durch die Muskelmembran (gleichzeitige Umsetzung zu Glc-6-p durch ATP Hydrolyse mittels Hexokinase); das modifizierte Agens kann auch wieder nach dem Transport abgespalten werden (z.B. beim Transport von einigen AS unter gleichzeitiger Spaltung von Glutathion; An der Innenseite der Membran werden die AS wieder frei gesetzt) D.) ATPasen Man unterscheidet vier Klassen von ATPasen: P, V und F ATPasen können nur Ionen transportieren, während ATPasen der ABC-Transportfamilie auch kleine Moleküle pumpen können; zur P-Klasse gehören z.B. Ca2+ ATPasen und die Na+/K+ ATPase; ATPasen der F und V Klassen transportieren ausschließlich Protonen; 23 Die Natrium Kalium ATPase: Die Calcium-ATPase: E.) Carrier als Antibiotika - - Der Carrier Valinomycin (Abbildung): es handelt sich um ein Makrolid-Antibiotika mit zyklischer Stuktur bestehend aus der dreifach repetetiven Einheit L-Lac-L-Val-DHiv-D-Val; Es wirkt als Ionophor, das selektiv Kalium Ionen transportiert, wobei Kalium in einer käfigartigen Struktur komplexiert und so durch die Zellmembran transportiert wird; durch den Transportvorgang bricht das Membranpotential zusammen, weshalb die Zelle abstirbt; Der Carrier Monensin: In Zellen blockiert dieses Antibiotikum die Sekretion von Glykoproteinen und fungiert als Na+ / Protonen Antiporter; Der Carrier Gramicidin A: es handelt sich um ein Peptid-Antibiotika, dass sich in die Zellmembran einlagert; zwei Moleküle Gramicidin bilden einen Ionenkanal zwischen Cytoplasma und Zelläußerem; dieser Kanal ist spezifisch für monovalente Kationen wie Kalium; der unregulierte Ionenfluss entlang des jeweiligen Konzentrationsgradienten führt zum Zelltod; 24 F.) Transportproteine: Uniporter - - Die Plasmamembran der meisten Zellen enthällt mehrere Uniporter; sie transportortieren beispielsweise KH, AS, Nukleoside und weitere kleine Moleküle durch die Zellmembran entlang ihres Gradienten (in die Zelle oder aus der Zelle); der Transport entlang eines Konzentrationsgradienten wird als erleichterte Diffusion bezeichnet, da der ΔG Wert immer denselben negativen Wert hat – ob mit oder ohne Transporter; die Aktivierungsenergie für den Transport durch die Membran ohne Transporter wäre nur zur hoch; Unterschiede zwischen Uniport und passiver Diffusion: die Geschwindigkeit der erleichterten Diffusion ist sehr viel höher als die der passiven durch die hydrophobe Membran; der Uniport ist spezifisch; Die Geschwindigkeit der erleichterten Diffusion folgt einer Michaelis Menten Kinetik, während die Geschwindigkeit der passiven Diffusion linear (sehr geringe Steigung) mit dem Konzentrationsgradienten zunimmt; G.) Die GLUT Transporter sind Uniporter - - - Alle GLUTs ermöglichen die erleichterte Diffusion (ohne Energieverbrauch aufgrund des chemischen Gradienten für Glucose); folgende GLUTs gehören zu Typ1; GLUT1 sorgt in fast allen Säugerzellen für eine kontinuierliche Glucose Aufnahme; insulinUnabhängig; hohe Affinität zu Glucose GLUT2 in Leberzellen = Hepatozyten, β-Zellen des Pankreas, Darm, Niere; insulinUnabhängiger Transporter; geringe Glucose-Affinität (hoher Km Wert schleust bei hohem Blutzuckerspiegel mehr Glucose ein; aktiviert dadurch Insulinsynthese in den β-Zellen und hemmt den Glykogenabbau in der Leber) GLUT3 in Nervenzellen des Gehirns; insulinUnabhängig; geringerer KM als GLUT1 (gewährleistet Glucoseaufnahme bei niedrigem Blutzuckerspiegel, da höhere Affinität) GLUT4 in Fetzellen und Muskelzellen; insulinAbhängig; hohe Affinität zu Glucose; Speicherung intrazellulär in Vesikeln (wird vermehrt in die Membran eingeschleust, wenn Insulin im Blut vorhanden Signalkaskade) H.) Transportproteine: Symporter und Antiporter Wie auch Pumpen koppeln Antiporter und Symporter eine energetisch ungünstige (Transport eines Moleküls gegen seines Gradienten) an eine energetisch günstige (Transport eines Ions entlang seines Gradienten) Reaktion; der Cotransport von Na+ (in die Zelle) ist häufig, da er sowohl vom Na+-Konzentrationsgradienten, als auch durch das elektrische Membranpotential angetrieben wird; dieser Transport kann mit z.B. dem Transport von Glucose gegen seinen Gradienten gekoppelt werden; Antiporter und Symporter sind sekündär aktive Transporter, da 25 der Gradient von Natrium durch eine Na-K-ATPase (3 Natrium aus der Zelle, 2 Kalium in die Zelle) aufrecht erhalten werden muss, wodurch ATP verbraucht wird; I.) Natrium-Glucose-Symport im Dünndarmepithel Die Dünndarmepithelzellen und die Nierentubuliepithelzellen müssen Glucose gegen eines steilen Konzentrationsgradienten aufnehmen; dieser Transport wird durch den ZweiNatrium-Ein-Glucose-Symporter katalyisert; dadurch werden 2 Natrium-Ionen zusammen mit einem Glucose Molekül in die Zelle aufgenommen; nach der gleichzeitigen Besetzung aller Natrium und Glucosebindestellen auf der extrazellulären Oberfläche des Transporters wird eine Konformationsveränderung ausgelöst, durch die sich eine Pore in der Membran bildet; hierdurch können die drei Teilchen auf entsprechende Bindestellen auf der cytosolischen Seite des Proteins gelangen, von denen sie ins Cytosol übergehen; nach dem Transport erlangt das Protein seine ursprüngliche Konformation zurück; Alle inneren und äußeren Körperoberflächen sind mit einer Schicht aus Epithelzellen, dem Epithel, ausgekleidet; Epithelzellen des Magens transportieren Protonen ins Lumen, wodurch ein niedriger pH=1 erreicht wird, während Epithelzellen des Dünndarms Verdauungsprodukte wie Glucose und AS ins Blut transportieren; Das Darmepithel ist stark polarisiert, weil sich die zwei Seiten dieser Zellen sowohl in Struktur als auch in Funktion stark unterscheiden; das Blut enthält viel Natrium und wenig Kalium, mittels Na+ K+ATPase in der basolateralen Membran werden 2 Kalium-Ionen vom Blut in die Epithelzelle und 3 Natrium Ionen von den Epithelzellen ins Blut transportiert, wodurch die Epithelzellen negativ aufgeladen werden und ein Mangel an Natrium aufweisen; Sowohl der Na-Konzentrationsgradient, als auch das Membranpotential, werden dazu genutzt den Glucosetransport mittels Zwei-Natrium-Ein-GlucoseSymporter innerhalb der Mikrovilli der apikalen Membran vom Darmlumen in die Epithelzellen zu transportieren; die Glucose verlässt die Zelle über GLUT2 (erleichterte Diffusion) innerhalb der basolateralen Membran; für den Transport der AS gilt das gleiche Prinzip! J.) Anionentransport im Erythrozyten Obere Abbildung: im Muskel Untere Abbildung: in der Lunge Die Umgebung ist elektroneutral! 26 Kohlenhydrate 1.) Monosaccharide - - Kohlenhydrate sind Ketone oder Aldehyde mit mehreren Hydroxylgruppen: Cn(H2O)n Die einfachsten Kohlenhydrate sind Triosen: Dihydroxaceton (Ketose) und D oder L Glycerinaldehyd (enantiomere Aldosen). D (Rechts) oder L (links) beschreibt die absolute konfiguration an dem Chiralitätszentrum (assymetrisches C Atom), das am weitesten von der Aldehydgruppe entfernt ist. Ketosen haben ein Chiralitätszentrum weniger als Aldosen Epimere unterscheiden sich nur in der Konfiguration an einem C Atom Keto-Enol Tautomerie ermöglicht die Isomerisierung zwischen Ketose und Aldose D- Struktufromeln merken: eine Aldose mit n C Atomen hat n-2 Chiralitätszentren. Eine Ketose mit n C Atomen hat n-3 Chiralitätszentren. Für L Konfiguration einfach spiegeln! o Für das erste Chiralitätszentrum (Alodse: C2 Atom, Ketose: C3 Atom) schreibt man die Hydroxylgruppe abwechselnd rechts links. o Für das zweite Chiralitätszentrum (C3 bzw. C4) schreibt man die hydroxylgruppe abwechselnd 2 mal rechts, 2 mal links o Für das dritte Chiralitätszentrum schreibt man die Hydroxylgruppe abwechselnd 4 mal rechts, 4 mal links o 8 mal rechts, 8 mal links etc. D-Aldosen: O O O H H OH OH H OH OH OH OH HO H OH H OH HO H OH H OH H OH H OH D-Threose D-Ribose OH OH HO H H OH H HO OH HO H H OH HO H H OH H HO H OH H OH H OH HO H OH H OH H OH H OH H OH H H H HO OH H OH OH D-Xylose D-Lyxose Pentosen H OH OH OH HO H H H OH HO H OH OH O O O O OH OH OH HO OH D-Arabinose O H OH H OH O H O O H Tetrosen O O H H H D-Erythrose Triose H HO OH D-Glycerinaldehyd O O HO H OH HO H H HO H H HO H OH OH H OH OH D-Allose D-Altrose D-Glucose D-Mannose D-Gulose D-Idose D-Galactose D-Talose Alle Alten Glucken Möchten Gerne Im Garten Tanzen Hexosen 27 D-Ketosen: OH OH O O OH OH O O H OH OH H OH HO H OH H H OH OH Dihydroxyaceton OH OH D-Erythrulose D-Ribulose D-Xylulose Triose Tetrose OH OH O O HO H OH H OH HO H OH H OH H OH D-Psicose H OH D-Fructose OH O OH H H Pentosen OH O OH HO H H HO H OH OH D-Sorbose H OH OH D-Tagatose Hexosen - - - Pentosen und Hexosen zyklisieren in Lösung unter Bildung eines Halbacetals bzw. Halbketals. Aldohexosen zyklisieren zu Pyranosen (sechsgliedriger Ring). Ketohexosen zyklisieren zu Furanosen (fünfgliedriger Ring) oder Pyranosen. Pyranoseform wird bevorzugt, da weniger Ringspannung. Geradkettige Form: Fischerprojektion / Ringform: Haworth Projektion Durch Bildung des zyklischen Halbacetals entsteht zusätzliches Chiralitätszentrum: das C Atom der ehemaligen Carbonylgruppe. Es wird als anomeres C Atom beschrieben. In Ringschreibweise unterscheidet man daher nochmals die diastereomeren α und β Formen (= Anomere Formen) voneinander. Bei der α Form befindet sich die Hydroxylgruppe am anomeren C Atom unterhalb der Ringebene; bei der β Form oberhalb Mutarotation: α und β Anomere können ineinander übergehen. Nach bestimmter Zeit herrscht Gleichgewicht: 1/3 α, 2/3 β und 1% Kettenform 28 D-Fructose O O CH2OH OH O H OH O HO OH HO OH α-D-Fructo-Pyranose OH HO CH2OH H OH H OH H OH β -D-Fructo-Pyranose OH HOH2C HOH2C CH2OH O HO O OH OH α-D-Fructo-Furanose HO H H OH H OH HO H O O OH CH2OH OH H β -D-Fructo-Furanose Endiol OH CH2OH O OH HO OH OH α-D-Gluco-Pyranose O H HO OH H H OH H OH OH D-Glucose - - CH2OH O OH OH HO OH β -D-Gluco-Pyranose O Furan O Pyran Monosaccharide bilden mit Alkoholen und Aminen Addukte (=Glykoside). Die entsprechenden Bindungen heißen O- und N-glykosidisch Addukte, bei denen das anomere C-Atom an der Bindung beteiligt ist, können nicht mehr in die Kettenform übergehen und reagieren daher nicht mehr mit Oxidationsmitteln. Polysaccharide weisen in diesem Falle ein nicht reduzierendes Ende auf. Wenn das anomere C Atom nicht beteiligt ist, liegt ein reduzierendes Ende vor. Reduzierende Zucker werden durch Oxidationsmittel oxidiert (Cyrbonylgruppe wird nach Freilegung zur Carboxylgruppe). 29 2.) Disaccharide α Bindung liegt unterhalb der Ringebene, β Bindung oberhalb - 6 CH2OH 5 O 4 OH 2 HO 3 HOH2C 5 1 + OH CH2OH O OH 2 OH O HO 4 OH CH2OH 1 3 OH β -D-Fructo-Furanose α-D-Gluco-Pyranose CH2OH 5 O 4 OH 2 HO HOH2C CH2OH HO HO OH O OH Cellobiose -1,4-O-glykosidische Bindung β - - - - CH2OH O OH OH OH OH 1 4 3 5 4 HO OH CH2OH O OH OH 2 3 1 OH β -D-Galacto-Pyranose D-Gluco-Pyranose HO OH 6 CH2OH HO 5 O OH 1 OH 2 OH CH2OH O OH OH D-Gluco-Pyranose 6 D-Gluco-Pyranose β -D-Gluco-Pyranose CH2OH O OH 3 1 Maltose α-1,4-O-glykosidische Bindung 6 OH 3 OH CH2OH 5 O OH 4 2 OH 2 HO HO Saccharose α-1,2-O-glykosidische Bindung 3 4 OH OH CH2OH O OH O OH CH2OH 5 O OH 4 OH 2 1 1 CH2OH O OH HO OH 5 α-D-Gluco-Pyranose O HO HO 3 O 6 6 6 6 CH2OH O OH OH O CH2OH O OH OH OH Laktose -1,4-O-glykosidische Bindung β Saccharose (Rohrzucker): α-D-Glucopyranosyl-1,2-β-D-Fructofuranose (Spaltung durch Saccharase) o Hat nicht reduzierende OH Gruppe (Trehalose Typ) Maltose (Malzzucker): α-D-Glucopyranosyl-1,4-α-DGlucopyranose (Spaltung durch Maltase) o Hat reduzierende OH Gruppe (Maltose Typ) Lactose (Milchzucker): β-D-Galactopyanosyl-1,4-α-D-Glucopyranose (Spaltung durch Lactase, bzw. β Galactosidase in Bakterien) o Hat reduzierende OH Gruppe (Maltose Typ) Cellobiose 30 3.) Polysaccharide - Glykogen (Speicherform der Glucose tierischer Zellen): verzweigtes Homopolymer, das durch α-1,4 glyk. Bindungen (Kette) und α-1,6 glyk. Bindungen (Zweige) verknüpft ist. Zweige alle 10 Monomere Stärke (Speicherform der Glucose pflanzlicher Zellen): Amylose (unverzweigter Teil aus α-1,4 glyk verknüpften Glucoseeinheiten) und Amylopektin (verzweigter Teil wie Glykogen, Zweige alle 30 Monomere). Stärke wird durch die α Amylase abgebaut Cellulose (Zellwand von Pflanzen) ist ein unverzweigtes, langkettiges Polymer mit β1,4 glyk Bindungen. Parallele Ketten bilden Fibrillen, die untereinander H-Brücken bilden. Säuger haben keine Cellulasen Stärke und Glykogen liegen als Helixform vor; α glykosidische Bindungen erzeugen offene helikale Polymere. β glykosidisce Bindungen erzeugen gerade Stränge - - - Nukleinsäuren - Nukleinsäuremonomere bestehen aus einem Kohlenhydrat, einem Phosphat und einer Base Basen: 3 5 N O 1 Pyrimidin 1 NH HO O - P OH - 5 4 O Monohydrogenphosphat 4 N 3 Indol O 8 N 2 NH 9 Purin N H O Cytosin Thymin O N N N HN N Adenin NH H2N NH N Guanin OH O 1 3 OH N H O HN NH2 7 5 N 6 N H Uracil NH2 CH3 HN HN 6 N 2 N Pyridin O O 4 2 2-Desoxy- β -D-Ribo-Furanose 31 NH2 Nukleosid HO NH2 5'-Nukleotid N N Nukleosid-5'-Mono-Phosphat N N O N N O O CH - O P O N CH2 O - N CH dAMP OH OH 2-Desoxy-Adenosin-Monophosphat 2-Desoxy-Adenosin Ohne Phosphat: Adenosin bzw. Desoxyadenosin Guanosin Cytidin Uridin Thymidin Mit Phosphat: Adenylat bzw. Desoxyadenylat Guanylat Cytidylat Thymidylat NH2 N O O - O O P O - O O P O - O P O N ATP N CH2 O - N CH Adenosin-Triphosphat OH OH β-1,9-N-glykosidische Bindung Phosphorsäure-Ester-Bindung Phosphorsäure-Anhydrid-Bindung DNA und RNA: - - Nukleotide sind Monomere der DNA bzw. RNA; sie sind verbunden über Phosphodiesterbrücken Die DNA besteht aus komplementär gepaarten Basen und einem Rückgrat aus Desoxyribose und Phosphatgruppen; Zwei gepaarte Stränge bilden eine Doppelhelix, die im Zentrum durch H-Brücken zwischen den gepaarten Basen zusammengehalten werden; Thymin und Adenin paaren über 2 H Brücken; Cytosin und Guanin paaren über 3 H Brücken Die eukaryotische DNA liegt als Chromosomen im Zellkern und von Histonen (basische Proteine) umwunden vor; Die prokaryotische ringförmige DNA liegt dagegen frei in der Zelle vor, wobei diese zusätzlich noch einen DNA-Plasmidring auf dem z.B. Antibiotikaresistenzen liegen aufweisen; 32 O O - O P O OH O - CH O O P O O OH O - O O O P O O OH - OH - - DNA weist 2-Desoxyribose auf, RNA hat Ribose als Zucker (kann also sowohl 3’ 5’ Verbindung, als auch 2’ 5’ Verbindung eingehen) Man schreibt Basensequenzen von 5’ (Phosphatgruppe) zum 3’ Ende (unverknüpfte OH Gruppe) Uracil ersetzt in der RNA die Base Thymin Negative Ladung der Phosphatgruppen macht die Nukleinsäuren resistent gegenüber Hydrolyse durch nukleophile Reagenzien (Stabilität höher). Das Fehlen der 2’ OH Gruppe erhöht die Resistenz gegenüber Hydrolyse weiter (DNA in Natur bevorzugt gegenüber RNA) Das Abbauprodukt des Nukleinsäurestoffwechsels ist die Harnsäure. Sie wird wie Harnstoff über die Nieren ausgeschieden O H N O HN NH O N H Harnsäure Aminosäuren pKs: 3,1 COO + H3N pKs: 8,0 - - Cα - H R α Aminosäuren sind optisch aktiv, da α C Atom chiral L Aminosäuren (R Konfiguration ) sind proteinogen (D Aminosäuren sind Enantiomere und weisen S Konfiguration auf) Aminosäuren in Lösung: Zwitterionen (Aminogruppe protoniert, Carboxylgruppe dissoziiert) Jede Aminosäure hat einen spezifischen isoelektrischen Punkt pI (derjenige pH Wert, bei dem ein Ampholyt (können als Base und als Säure reagieren) im elektrischen Feld weder zur Anode noch zur Katode wandert). Aminosäuren sind Puffer (da sie Protonen aufnehmen und abgeben können). PI berechnet sich aus dem arithmetischen Mittel aller pKs Werte. Wenn der pKs Wert der funktionellen Gruppe kleiner (größer) ist als der pH Wert des umgebenden Mediums ist die Seitenkette deprotoniert (protoniert) 33 - Der pH Wert berechnet sich mittels Hendersen Hasselbachgleichung (es wird der pI Wert eingesetzt) Proteinogene Aminosäuren (die 20 kanonischen AS): G | Gly | Glycin + - COO H H NH3 L | Leu | Leucin I | Ile | Isoleucin A | Ala | Alanin V | Val | Valin - + NH3 - - COO H CH3 + NH3 H3C COO H H CH3 + NH3 H H3C aliphatisch, unpolar + NH3 H3C H COO H C* H H CH3 Isoleucin hat am β C Atom ein Chiralitätszentrum Glycin ist die einzig nicht optisch aktive Aminosäure, da das α C Atom nicht chiral ist. Je größer die aliphatische Gruppe, desto stärker der Hydrophobe Effekt (die Seitenketten im Protein lagern sich mehr zusammen, wodurch 3D Struktur stabilisiert wird) - D | Asp | Asparaginsäure K | Lys | Lysin R | Arg | Arginin - E | Glu | Glutaminsäure - - + - COO H H H CH3 COO NH3 H H H O HO + NH3 H H HO + COO H H H O NH3 H H H H pKs: 4,0 COO H H H H H NH2 H2N + C - COO NH3 H H H H H H H N H H2N NH2 + NH2 NH2 Guanidinium pKs: 4,3 saure pKs: 10,8 pKs: 12,5 basische - Lysin: epsilon-Aminogruppe Arginin: Guanidiniumgruppe (positive Ladung am C delokalisiert) Arginin ist Metabolit im Harnstoffzyklus; wird zu Ornithin und Harnstoff gespalten C | Cys | Cystein M | Met | Methionin - - + NH3 H COO H H S H pKs: 8,3 + NH3 H H COO H H H S CH3 schwefelhaltig - H O + NH3 R H O H N N H H R HS COO H O - + NH3 kovalente Dimerisierung R R H H N N - H H Cystin SH O OC H O N O H reduziert R -H2 + O H NH3 N N H - S H O OC COO H R S R H - H N O H R + NH3 O H oxidiert Methionin: Thioether (Methylgruppenübertragung möglich) Cystein: Thiol (Disulfidbrückenbildung zum Cystin möglich, was Struktur des Proteins stabilisiert) 34 S | Ser | Serin T | Thr | Threonin N | Asn | Asparagin Q | Gln | Glutamin - + - COO H H O H NH3 H + NH3 H3C + NH3 H neutral, polar - - - COO H C* H O H COO H H O NH2 + NH3 H H COO H H H O NH2 Asparagin und Glutamin: Carboxamid (keine Aminogruppe, da nicht protonierbar) Glutamin ist universeller NH2 Donor im Stoffwechsel Serin (und Threonin) kann im Protein phosphoryliert werden und befindet sich häufig im aktiven Zentrum von Proteasen. Threonin weist am β C Atom ein Chiralitätszentrum auf. P | Pro | Prolin COO - zyklische HN - es handelt sich um eine Iminosäure mit Pyrrolidinring die proteinstruktur wird durch prolin maßgeblich beeinflusst, da es in seiner Konformation stark eingeschränkt ist (α Helices und β Faltblätter werden unterbrochen, um eigene Motive zu initiieren) F | Phe | Phenylalanin Y | Tyr | Tyrosin W | Trp | Tryptophan H | His | Histidin - - + NH3 H COO H H + NH3 H COO H H - + NH3 H COO H H - + NH3 H COO H H OH pKs: 10,1 - - aromatische Indol H N HN NH NH N pKs: 6,0 N Imidazol Tryptophan: Indolring Histidin: Imidazolring His befindet sich oft im aktiven Zentrum, wo der Ring je nach Bedarf Protonen binden oder abgeben kann: Säure-Base katalyse (N hat geringe Affinität zu H+) Tyr und Trp absorbieren besonders gut ultraviolettes Licht (bei 280nm), wodurch Proteinkonzentrationen bei bekannter Anzahl dieser Aminosäuren abgeschätzt werden kann Tyrosin kann im Protein phosphoryliert werden Selenocystein und Pyrrolysin sind ebenso proteinogen 35 Aminosäure-Modifikationen AS können u.a. lipidiert und acetyliert werden etwa 100 post-translationale AS Modifikationen sind bekannt; nur 24 AS werden ribosomal ins protein eingebaut; Hypovitaminose = Vitaminmangel 1.) Skorbut = Ascorbat-Hypovitaminose Ascorbat = Vitamin C Vitamin C ist wichtiger Cofaktor bei der Modifizierung der Aminosäuren Prolin und Lysin zu Hydroxyprolin und Hydroxylysin (Hydroxylierung); bei fehlender Hydroxylierung werden nur schadhafte Kollagenmoleküle (Bestandteil von Haut und Bindegewebe) gebildet, die ihrer Funktion als Strukturprotein nicht nachkommen können; Synthese von Hydroxyprolin: Prolin + α-Ketoglutarat + O2 Hydroxylprolin + Succinat + CO2; Prolyl-Hydroxylase (FeIII FeII; Cofaktor: Ascorbat) Durch Skorbut ausgelöste Depression kommt durch Mangel an Adrenalin und Noradrenalin zustande (Synthese mittels Dopamin-β-Hydroxylase ist Vitamin C abhängig) 2.) Vitamin K-Hypovitaminose Vitamin K dient der Synthese von Prothrombin und anderen Gerinnungsfaktoren; bei Vitamin K Mangel wird abnormales Prothrombin gebildet, das keine Ca2+ Ionen binden kann, da γCarboxyglutamat fehlt (es kommt zu starken Blutungen); normalerweise würde eine Vitamin-K abhängige Carboxylierung aus dem schwachen Ca2+ Gelator Glutamat den starken Ca2+ Gelator Carboxyglutamat synthetisieren; COO + H3N C - Stabilisierung und Verknotung der Kollagenfibrillen H (CH2)2 H COO + CH2 OH C C N CH2 CH + NH3 CH Hydroxy-Lysin COO - CH + H2N CH2 CH2 HC Kommt nur in Actin und Myosin vor H C H3N - N CH3 3-Methyl-Histidin Erhöht Löslichkeit und stabilisiert Kollagenfibrillen COO + H3N C - Prothrombin, CalciumKomplexierung; H CH2 CH OH Hydroxy-Prolin - O OC COO - Carboxy-Glutamat 36 COO + C H3N - Serinphosphat, Threoninphosphat, Tyrosylsulfat H Signaltransduktion (Phosphatasen und Kinasen), Regulation von Proteinen, reversible Schalter, Regulation von zellulären Prozessen; Enzymkaskadeneffekt; HORMONE; ONCOGENE; CH2 OPO3 2- Tyrosyl-phosphat Aminosäurederivate: - + NH3 H CH3 - COO H H + NH3 H NH2 COO H H H H H H OH OH OH - L Tyrosin DOPA NH2 H HO HN H H OH H HO OH OH OH Dopamin Noradrenalin H H OH OH Adrenalin Peptide und Proteine 1.) Peptide 3 + NH3 pKs: 2,5 H O H O H COO Kondensationsreaktion + NH3 N N COO H -2H2O R pKs: 10,5 R H R H R N-terminus H O + NH3 H O N N H G - Peptid C-terminus H COO - H P F α Carboxylgruppe reagiert mit α Aminogruppe einer anderen Aminosäure unter Wasserabspaltung zu einer Peptidbindung. Polypeptide bestehen aus Hauptkette, Rückgrat (Carbonyl und NH Gruppen) und den einzelnen Seitenketten. Wasserstoffbrücken innerhalb des Rückgrats führen zur Sekundärstruktur. Bindungen zwischen Rückgrat und Seitenketten sowie innerhalb der Seitenketten führen zur Tertiärstruktur. 37 Peptidbindungen sind immer trans-konfiguriert Glutathion (Antioxidanzmittel) Aspartam (Süßstoff) SH O N O + O H3N N H - C CH3 O O OC D O H F Methylester + H3N O O O - O C N H C - H G E γ-Carboxylgruppe von Glutaminsäure bildet mit Aminogruppe des Cystein Isopeptidbindung Große Polypeptide ab ca. 100 AS sind Proteine und weisen bis zu 2000 Aminosäuren auf. 2.) Primärstruktur: genetisch codierte Aminosäuresequenz 38 - - Die Länge der Bindung zwischen C und N innerhalb der Peptidbindung liegt zwischen der Länge einer C-N und einer C=N Bindung; der partielle Doppelbindungscharakter lässt sich durch den Mesomerie Effekt erklären; Durch die Mesomerie innerhalb der Peptidbindung wird die Peptid-Einheit planar; lediglich die Bindungen zwischen C und Cα sowie zwischen N und Cα sind drehbar; Deshalb ist bei der Strukturaufklärung eines Protein-backbones die Position von C α mit seinem Diederwinkelpaar φ und Ψ das einzige was man benötigt; o φ = entlang der Cα – N Bindung schauen; der Winkel wird definiert durch CO am N und CO am Cα; o Ψ = entlang der Cα – C Bindung schauen; der Winkel wird definiert durch NH am Cα und NH am C; - Es sind nicht viele Diederwinkelpaare sterisch erlaubt; jedes Diederwinkelpaar hat eine bestimmte (auf Statistik und Entropie beruhende) Zustandswahrscheinlichkeit, die im Ramachandran-plot visualisiert wird; jedes Diederwinkelpaar begünstigt darüber hinaus eine bestimmte Sekundärstruktur (wenn eine Reihe von aufeinander folgenden Cα Atome etwa das gleiche Diederwinkelpaar aufweisen); dadurch lassen sich die Häufigkeit bestimmter Sekundärstrukturen innerhalb eines Moleküls abschätzen; - bei kleinem Rest am C α sind mehr Winkelpositionen möglich, als bei einem großen Rest (Glycin kann am meisten Diederwinkelpaare einnehmen); grüne Inseln stellen den Bereich dar, der erlaubt ist; blaue Inseln sind die wahrscheinlichsten Bereiche; Im rechten Bild symbolisiert ein punkt ein Diederwinkelpaar; Punkte die nahe beieinander liegen, haben fast dieselbe Winkelwahrscheinlichkeit und bilden daher - 39 - - - - meist ein gemeinsames Sekundärstrukturelement (deren Positionen im linken Bild durch die orangene Punkte angedeutet werden) Beispiel: wenn φ = Ψ = 180° gilt, liegt die Polypeptidkette gerade gestreckt vor; dies ist entropisch aufgrund des hydrophoben Effekts sehr ungünstig und ist somit nicht erlaubt (es käme zur Bildung eines Zufallsknäuels; unten rechts im Plot); wenn φ = Ψ = 0° gilt, dann stoßen die Reste der planaren Peptidbindungen direkt aneinander, was sterisch absolut ungünstig ist (in der Mitte des Plots) Prolin schränkt die Struktur eines Proteins erheblich ein, da es φ = -65° vorgibt; Da sich zwischen den einzelnen Sekundärstrukturelementen loops und turns befinden, welche die Tertiärstruktur definieren, gibt es auch einzelne Punkte außerhalb des erlaubten Bereichs; Folgende Sekundärstrukturen findet man im Ramachandran-plot: o Paralleles Faltblatt und antiparalleles Faltblatt befinden sich im blauen Bereich oben links; das antiparallele etwas über dem parallelen; o Links-drehende α Helix befindet sich im grünen Bereich rechts o Rechts-drehende α Helix (wesentlich häufiger) befindet sich unten links im blauen bereich o Die Pi und die 310 Helix befinden sich direkt unter- und oberhalb der α Helix 3.) Sekundärstruktur Sekundärstruktur meint die räumliche Anordnung des Rückgrats, aufgrund der DiederWinkel. Der Zusammenhalt der Sekundärstrukturen erfolgt lediglich aufgrund der H-brückenBindungen und der delokalisierten Pi Elektrktronensysteme in der Peptidbindung; Man unterscheidet bestimmte Motive, die häufigsten sind: A.) α Helix indem zwischen jeder vierten Peptidbindung eine intramolekulare Wasserstoffbrücke (jede Peptidbindungen geht 2 H-Brücken ein) ausgebildet wird (C=O Gruppe der Peptidbindung i mit N-H Gruppe der Peptidbindung i+4), entsteht eine starre Helix (Seitenketten ragen nach außen heraus) mit 3,6 AS pro 360° Windung (100° Drehung und 0,15 nm Verschiebung zwischen jeder AS); daraus ergibt sich eine Ganghöhe von 3,6 AS * 0,15nm = 0,54 nm; die AS i liegt exakt unterhalb der AS i+18 (18*0,15 = 2,7nm und 2,7/0,54 = 5 Ganghöhen darüber); Näherungsweise kann man sagen, dass bereits AS i unterhalb von AS i+7 liegt (7*0,15=1,05 ≈ 1nm und 1,05 /0,54 = 1,94 ≈ 2 Ganghöhen darüber); man spricht in diesem Fall von einer Heptade, die 1nm hoch ist und 2 Windungen hat; die Membrandomänen von Membranproteinen sind meist α-Helices: 21AS entsprechen 3 Heptaden, die benötigt werden um eine 3 nm dicke Membran zu durchspannen; Prolin bricht die Helixstruktur! So genannte helical wheels (Heptadenmuster) eignen sich zur Aufsichts-Darstellung einer Helix; die 7 AS werden um 100° versetzt in einen Kreis gemalt; so lässt sich erkennen, ob 40 z.B. alle geladenen AS auf einer Seite der Helix sind; solche Helices, in denen die eine Seite überwiegend hydrophobe und die andere Seite überwiegend hydrophile Elemente enthält heißen amphipathisch; in der Abbildung liegt der achte Rest (näherungsweise) genau unter Rest a; B.) seltenere Helices - - 310 Helix: H-Brücke bildet sich zwischen AS i und AS i+3, wodurch nur 3 AS eine 360° Windung der Helix ermöglichen; dadurch ist diese Helix enger gepackt und länger gestreckt als die α Helix; П Helix: H-Brücke zwischen i und i+5; 4 AS pro 360° Windung; breiter und weniger gestreckt als die α-Helix C.) β Faltblatt Die Polypeptidketten liegen nahezu ausgestreckt vor (β Strang); ein Faltblatt entsteht durch die Verknüpfung von β Strängen über H Brücken. Der Knick (109,5°), welcher die Faltoptik bewirkt, kann lediglich am Cα Atom stattfinden; der Abstand zwischen der AS i und der AS i+2 beträgt 0,7nm; Die Stabilisierung erfolgt durch H-brücken der gegenüberliegenden Peptidgruppen; 41 Die Reste aufeinander folgender Aminosäuren ragen senkrecht in unterschiedliche Richtungen nach oben (wenn Knick oben) bzw. nach unten (wenn Knick unten) von der Faltblatt-Ebene weg (Die β Faltblatt-Stuktur wird daher durch ein alternierendes auftreten von hydrophoben und hydrophilen Resten stabilisiert); Das antiparallele Faltblatt ist stabiler als das parallele! Parallele β Stränge: ausschließlich in globulären Proteinen Antiparallele β Stränge: häufig in fibrillären Proteinen D.) Turns und loops Die meisten Proteine haben eine kompakte, globuläre Gestalt, was häufige Richtungsänderungen im Verlauf ihrer Polypeptidkette, immer an der Oberfläche des Proteins voraussetzt; Turns und Loops sind nichtrepetitive Sekundärstrukturen, die die Richtung einer Polypeptidkette umkehren; ein Turn (eine Kehre) ist charakterisiert durch eine H-Brücke zwischen Akzeptor C=O und dem n Einheiten entfernten Donor N-H; 42 Der β-Turn tritt am häufigsten auf, da ein Turn mit einer H-Brücke von AS i zur AS i+3 am stabilsten ist; dieser Turn wird am besten durch die AS Glycin und Prolin ermöglicht; Außerdem: loops (Schleifen) (flexiblere Strukturen) 1.) Omega Schleife (omega loop): Richtung wird gewechselt, keine periodische Struktur; es sind 6 bis 16 AS Reste beteiligt; 2.) Ausgedehnte Schleife (extended loop): mehr als 16 AS Reste beteiligt Antikörper (Immonuglobuline) haben Domänen, welche die Kontaktstellen an den Loops aufweisen; diese sind hypervariabel! 4.) Suprasekundärstrukturen: A.) Superhelices: Kollagentripelhelix Es handelt sich um keine α-Helix; Kollagen ist aus Tropokollagen aufgebaut; dabei handelt es sich um eine Tripelhelix zu je 1000 AS pro Helix; Die AS Zusammensetzung ist charakteristisch: 35% Glycin, 21% Prolin und Hydroxyprolin; jeder dritte Rest einer Helix liegt im Zentrum der Tripelhelix; hier hat allerdings nur Glycin Platz, weshalb jede dritte AS Glycin sein muss, um diese Struktur zu ermöglichen; Prolin und Hydroxyprolin versteift die Anordnung, was die starke Zugfestigkeit des Kollagens bewirkt; die Verdillung der Einzelhelices ineinander verhindert, dass Windungen unter Spannung auseinander gezogen werden; Der Zusammenhalt erfolgt nicht über H-Brücken, sondern ausschließlich über die Verdrillung und Van-der-Waals WW; 43 Die Tripel Helix ist 300 nm lang, hat aber nur 1,5 nm Durchmesser ( längste Persistenzlänge in der Biologie überhaupt); es existieren keine H-Brücken innerhalb der Helix! B.) Superhelices: Coiled coil Stuktur in Myosin, Tropomyosin (Muskel), Fibrin (Blutgerinsel), Keratin (Haar) Es handelt sich um eine Helix, die ihrerseits wiederum zu einer Helix mit größerem Radius gewunden ist (sehr stabile Einheit; Funktion: Abstandhalter?); sie besteht aus mindestens zwei Einzelhelices; die coiled coil Struktur basiert auf die WW zwischen den Aminosäureresten a und d’ bzw. a’ und d (leicht erkennbar im helical wheel); in Position 1 und 4 befinden sich hydrophobe AS Reste, weshalb die coiled coil Struktur durch Van-der-Waals WW dieser AS stabilisiert wird; zusätzlich kann eine Stabilisierung erfolgen, indem die AS der Positionen 2, 3, 5, 7 Coulomb-WW aufweisen (lange Reichweite) und die AS der Position 6 hydrophile Eigenschaften hat (WW mit Wasser); 1XXX5 XX1XXX5XX 1 LXXXLXXLXXXLXXL LXXXLXXLXXXLXXL Einer der häufigsten Coiled coil Strukturen ist die bZIP Domäne (= basic leucin zipper domain: Leucin-Zipper); Diese Proteindomäne findet man in vielen eukaryotischen DNABindeproteinen (Bsp. CREB) C.) β barrel Struktur Anordnung von antiparallelen β Faltblättern, sodass eine geschlossene Struktur entsteht, in der der erste β Strang mit dem letzten β Strang eine H Brücke eingeht; β Strukturen findet man häufig in Porinen oder anderen Membrandurchspannenden Proteinen; hier findet man Stränge mit abwechselnd polaren und hydrophoben AS, sodass die hydrophoben Reste nach außen ragen (WW mit Lipiden in membran); Im Zentrum bildet sich ein hydrophiler Kern (genau entgegengesetzt zum allgemeinen Prinzip bei globulären Proteinen: hydrophober Kern) 44 4.) Tertiärstruktur Tertiärstruktur meint die räumliche Beziehung der Reste, die in der Sequenz weiter voneinander entfernt liegen. Im Allgemeinen ordnen sich die hydrophoben Reste zum Zentrum der Struktur (hydrophober Kern: WW untereinander), während die hydrophilen Reste nach außen ragen (WW mit dem Wasser); so stellen Proteine eine ideale Verpackungsform für prosthetische Gruppen dar, die sich in die hydrophobe Tasche einlagern; Modifizierte AS-Reste können die Tertiärstruktur maßgeblich beeinflussen; Im groben unterscheidet man globuläre lösliche Proteine, Membranproteine mit hydrophober Membrandomäne und fibriläre Proteine; Die dreidimensionale Anordnung der Sekundärstruktur folgt keiner Symmetrie, sie ist durch die Aminosäuresequenz determiniert, da sie auf die WW zwischen den Resten basiert; Enthalpische Beiträge: - Ionenbindungen (80kJ/mol, geringen Beitrag, da Enthalpiegewinn nicht ausreicht, um Entropieverlust auszugleichen), - Van-der-Waals Bindungen (0,3kJ/mol Bindungsenergie, aber Entropiegewinn des Wassers enorm groß), - Disulfidbindungen (200kJ/mol, aber oft weniger wichtig für Faltung, da auch mit derivatisiertem Cystein aktive Konformationen vorliegen können) - H-Brücken (20 kJ/mol, haben geringen Einfluss; in denaturierten proteinen bilden AS H Brücken mit Wasser) Entropische Beiträge: - Der hydrophobe Effekt als Triebkraft: Unpolare Moleküle haben in Wasser das Bestreben sich zusammenzulagern, um die Größe der geordneten, entropisch ungünstigen Hydrathülle zu minimieren; dadurch erfährt das polare Medium ein Entropiegewinn; der hydrophobe Effekt spielt bei Proteinfaltung (Entropie im Protein nimmt nämlich nach Faltung ab, aber Entropiegewinn durch Freisetzung des Wassers 45 - aus Hydrathülle überwiegt, wodurch Faltung ohne Energieaufwand möglich ist) und beim Aufbau von Membranen eine entscheidende Rolle; die Entropieerniedrigung durch Einschränkung der Bewegungsfreiheitsgrade der Polypeptidkette, ist nicht so stark wie die Entropieerhöhung durch den hydrophoben Effekt; 5.) Proteinfaltung Experiment von Christian Anfinsen H-Brücken der nativen Ribonuklease A werden mittels Harnstoff gebrochen; β-Mercaptoethanol wird als Reduktionsmittel eingesetzt, um Disulfidbrücken zu spalten; Die denaturierte und reduzierte Ribunuklease A (zufällige Knäuelkonformation: random-coil) wird durch Dialyse von Harnstoff und β Mercaptoethanol getrennt, an der Luft oxidiert und in Puffer gegeben; es ist eine Strukturrückbildung zu beobachten; Schlussfolgerung: In der Primärstruktur liegt die Information für die Tertiärstruktur (Proteinfaltung ist unter physiologischen Bedingungen ein spontaner Prozess) Theoretisch gibt es bei vier gespaltenen Disulfidbrücken 105 Möglichkeiten der S-S Paarung, wobei nur eine enzymatisch aktiv ist; das Protein „sucht“ automatisch nach der stabilsten Konformation (die native Form ist also immer die thermodynamisch stabilste Form) Chaperone (Klasse: Hitzeschockproteine Hsp) Auch in vivo kommt es zu Ausbeute minimierender Aggregation der Proteine während der Faltung; Als Hilfe für Proteinfaltung setzt die Zelle Faltungshelferproteine (Chaperone) ein; Sie interagieren selektiv mit nichtnativen Proteinen über exponierte, hydrophobe Oberflächen, wodurch sie nicht legitime WW zwischen den faltenden Polypeptidketten unterbinden und somit der Aggregation als unspezifischer, dominierender Nebenreaktion entgegenwirken; sie bilden einen schützenden Käfig um das faltende Protein, um die Aggregation mit anderen sich faltenden Proteinen zu vermeiden; Chaperone sind Enzyme, die während des Faltungsprozesses helfen; sie setzen die Aktivierungsenergie für den Faltungsprozess herab, beschleunigen ihn damit, verändern aber nicht das Gleichgewicht der Faltung; Beispiel: Protein-Disulfid-Isomerasen (PDI) Intramolekulare Disulfidbrücken werden aufgebrochen, um dem faltenden Protein neue Chance zugeben, die richtige Kombination zu finden; Die Reaktion (intra- und intermolekulare Sulfhydryl-Disulfid-Austausch Reaktionen) verläuft solange, bis die 46 thermodynamisch günstigste Form erreicht ist; dieses Enzym beschleunigt die Faltung zum nativen Protein um kcat x6000! Beispiel: Protein-Prolyl-cis-trans-Isomerasen (PPI) Manchmal wird eine cis und manchmal eine trans Konfiguration im Prolin benötigt, damit das Protein weiter falten kann; dieses Enzym beschleunigt die Faltung zum nativen Protein um kcat x600! Das Proteinfaltungs-Problem: Levinthal’sches Paradoxon Für die Faltung von 1nem Protein mit 100 AS, von denen jeder Rest 3 Konformationen annehmen kann resultieren 3100=5*1034 mögliche Strukturen; für einen Konformationswechsel werden 10-13s (Ratenkonstante der Isomerisierung) benötigt, weshalb es 1,6*1027 Jahre dauern würde, um alle möglichen Strukturen auszuprobieren; Die Proteinfaltung erfolgt jedoch im ms bis Sekunden Bereich; Die Differenz zwischen der berechneten und der tatsächlichen Faltungszeit wird als Levinthal’sches Paradoxon bezeichnet; Wie lange braucht ein Affe um „methinks its like a weasel“ auf die schreibmaschine zu tippen? Ca. 1040 Anschläge notwendig; wenn allerdings immer die korrekten Buchstaben bewahrt werden würden, benötigt man nur noch wenige tausend Anschläge; das erste Modell geht von zufälliger Suche aus, das zweite geht davon aus, dass korrekte Zwischenprodukte bewahrt werden; letzteres ist ein Grundprinzip der Proteinfaltung; Für protein mit 100 AS gilt: ΔG Faltung = 10 kcal/mol = 42 kJ/mol jeder Rest trägt nur ΔG = 0,1 kcal/mol = 0,42 kJ/mol zur Erhaltung der gefalteten Struktur bei; Dieser Betrag ist geringer als der der thermisches Energie bei Raumtemperatur (ΔG= 0,6 kcal/mol = 2,52 kJ/mol); Diese schwache Stabilisierung bedeutet: korrekte Zwischenprodukte können verloren gehen; Es müssen sich mindestens ca. 7 AS gleichzeitig in einer korrekten Struktur zusammenfinden, damit die gefaltete Struktur nicht durch das thermische Rauschen gebrochen werden kann; ein zu bewahrendes korrektes Faltungszwischenprodukt muss also mind. 7 Reste aufweisen; Proteinfaltungsmodelle Proteinfaltung erfolgt in vivo meist erst, wenn vollständiges Protein das Ribosom verlassen hat (Bsp: Ribonuklease A, hier ist C terminus für native Struktur essentiell); kann aber auch Domänenweise, beginnend am N terminus stattfinden, während ein Teil des Polypeptids noch am Ribosom translatiert wird; Die Proteinfaltung erfolgt über mehrere Wege hin zum Energieminimum Die Faltung und Denaturierung bzw. Renaturierung ist ein hoch kooperativer Prozess! Die Faltung von Proteinen kann als hierarichischer Prozess angesehen werden (initiiert durch lokale, nichtkovalente WW zwischen benachbarten AS Resten); es kommt so zur Ausbildung von Sekundärstrukturelementen, welche über weitere Interaktion die Tertiärstruktur bilden; 47 Der molten globule state = Übergangszustand (transition state) nativ molten globule denaturiert - - Wenn Denaturierung durch Hitze oder Säure erfolgt, bilden sich häufig molten globules (hydrophobe AS Reste aus wässriger LÖMI Umgebung werden ins Proteininnere verlagert); es entsteht ein kompakte denaturierte Konformation mit hydrophoben Kontakten zwischen benachbarten AS Resten, die denen im nativen Protein entsprechen können; molten globules weisen Sekundärstrukturelemente auf; außerdem charakteristisch sind eine fluktuierende Tertiärstruktur, lösungsmittelexponierte hydrophobe Oberflächen und eine unkooperative Entfaltung; dieser Zustand hat im Vergleich zum nativen protein nur eine geringe Stabilität, weshalb sich der molten globule Zustand in einem schnellen GG zum vollständig denaturierten Protein befindet; ausgehend vom molten globule kann die Faltung zum nativen Protein erfolgen (Ausbildung von H Brücken, elektrostatischen Interaktionen und Tertiärkontakte); es entstehen Subdomänen, die eine geringere Packungsdichte als die des nativen Proteins aufweisen; außerdem existieren mobile loops an der Oberfläche; Faltungsphasen dieser Art treten teilweise unabhängig voneinander in verschiedenen Domänen desselben Proteins auf (Protein kann also auf mehreren unterschiedlichen Faltungswegen zum nativen Zustand gelangen); in den Modellen A und B verläuft der erste Schritt über einen pre-molten globule state zu einem molten globule state; der molten globule state wird im Fall A durch die Formation von Sekundärstrukturen und im Fall B durch den Hydrophoben Kollaps stabilisiert; - Framework model: konformationeller zustand mit nativ-ähnlichen, aber instabilen Sekundärstrukturelementen Kompaktierung molten globule Hydrophobic collapse model: Faltungszustand mit unspezifischen, nicht nativen WW Ausbildung einer nativ-ähnlichen Struktur molten globule A.) Das framework model Zu Beginn der Faltung werden Teile der Sekundärstruktur gebildet, die in Größe und Position fluktuieren; durch Ausbildung von unspezifischen hydrophoben WW innerhalb dieser Sekundärstrukturelemente werden sie anschließend stabilisiert; die gebildeten festen Sekundär- und Suprasekundärstrukturen legen den Faltungsweg fest (Voraussetzung für Assembierung der Tertiärstruktur, die mit Verdrängung von Wasser aus dem Proteininneren und der Entstehung des hydrophoben Kerns einhergeht) B.) hydrophobic collapse model Zu Beginn der Faltung wird ein unspezifischer Kollaps der Polypeptidkette angenommen; durch Zusammenlagerung von hydrophoben AS wird so der Kontakt mit dem wässrigen LÖMI vermieden; der Kollaps bewirkt die Ausbildung von Sekundärstrukturelementen wie Helices und β loops; die entstehende Sekundärstruktur bewirkt die Neuordnung der hydrophoben Seitenketten zu einer nativen Struktur; C.) Nukleation condensation model 48 Es bildet sich ein Übergangszustand der nativ ähnliche hydrophobe WW aufweist und die schwachen Sekundärstrukturen stabilisiert; Synthese aus Modellen A und B Methoden zur Beobachtung der Faltung 1.) enzymatische Aktivität 2.) Absorption 3.) Fluoreszenz (Tryptophan hat im gefalteten Protein ein Maximum bei Wellenlänge von 330nm und im ungefalteten Protein bei 350nm, wobei hier die Emissionslinie wesentlich breiter ist) 4.) Viskosität (ungefaltetes Protein wandert langsamer) 5.) Circular dichroismus 6.) Deuterium Austausch (denaturiertes protein in D2O für kurze zeit falten lassen; danach in H2O bei pH=9 für 10ms und dann bei pH=4 geben; das teilweise gefaltete Protein hat nun zum Teil Deuterium eingebaut; Der Deuteriumeinbau ist proportional zum Faltungsfortschritt) 7.) Proteaseverdau 8.) Molecular dynamic simulation Krankheiten, die durch Protein-Faltungsfehler ausgelöst werden: Punktmutationen führen zu Polymerisationsreaktionen der Proteine: - Alzheimer - Kreuzfeld-Jakob Syndrom - BSE falsch gefaltete proteine polymerisieren z.B. zu Fibrillen. Wenn diese extrazellulär sind, können sie nicht mehr in die Zelle aufgenommen und degradiert werden! 6.) Quartiärstruktur Quartiärstrukturen weisen Proteine auf, die aus mehreren Polypeptidketten (Untereinheiten) bestehen. Sie beschreibt die räumliche Anordnung der Untereinheiten und die WW untereinander. Bsp.1 Hämoglobin mit 4 Untereinheiten zu je einer prosthetischen Gruppe (Zusammenhalt über schwache, nichtkovalente Bindungen) Bsp.2 Poliovirus: Capsid ist Verpackung für kondensierte RNA und besteht aus 60 mal 4 Untereinheiten; 49 o Dimer: Homodimer (zwei identische Untereinheiten) / Heterodimer (zwei unterschiedliche Untereinheiten) o α2β2 Tetramer: vier Untereinheiten, von denen jeweils zwei identisch sind (Bsp: Hämoglobin) Apoprotein: Proteinanteil eines Komplexes (inaktiv) Holoprotein: Apoprotein + Kofaktor/Koenzym/prosthetische Gruppe (aktiv) Kofaktor (anorganisch): Metalle über transiente Bindung an Protein gebunden Koenzym (organisch): organisches Molekül über transiente Bindung an Protein gebunden Prosthetische Gruppe: nicht peptidisches organisches Molekül über dauerhafte (kovalente oder nichtkovalente) Bindung an Protein gebunden Myoglobin und Hämoglobin 1.) Allgemeines Struktur: Myoglobin (4,5 x 3,5 x 2,5 nm) ist ein Monomer mit 153 AS (16kD); 75% des Proteins hat αHelix Struktur; es existieren 8 Helices von A bis H, wobei zwischen Helix E und Helix F die prosthetische Gruppe sitzt; Die Helices haben 5 nicht-helikale Übergangselemente AB, BC, CD, DE, EF; die unpoalren Reste ragen nach innen, während die polaren Reste nach außen ragen; Hämoglobin (6,4 x 5,5 x 5 nm) ist ein Modell für ein allosterisches Protein; es handelt sich um ein α2β2 Tetramer (α-Ketten haben 141 AS, β Ketten haben 146 AS; 18% Sequenzidentität zwischen den Untereinheiten); 50 Häm = Mesomerie stabilisiertes, planares System aus 4 Pi konjugierten Pyrrolringen mit Propionsäure- und Methylgruppen sowie Acetylengruppe und einem Eisen (= Sauerstoffbindestelle) im Zentrum; das Eisen ist komplexiert durch die 4 Stickstoffatome der Pyrrolringe, sowie dem proximalen HisF8 des Proteins; Sauerstoff kann reversibel durch FeII koordiniert werden Funktion: Hämoglobin stellt das Transportmolekül für Sauerstoff im Blut (Kreislaufsystem für Sauerstoff) dar (98% im oxy-Hb, nur 1-2% im Blut direkt gelöst, da Sauerstoff schlecht wasserlöslich); 5l Blutplasma binden 14 ml Sauerstoff (1mM); 5l Blut binden 1l Sauerstoff (Hb erhöht die Bindungsfähigkeit um das 70-fache); Sauerstoffverbrauch: 300 bis 3500 ml pro Minute; 32 pg Hb pro Erythrozyt; der Mensch hat 25 * 1012 Erythrozyten (Lebensdauer: 120 Tage) 800 g Hb hat ein Mensch im Blut (1l Blut hat ca. 100 bis 150g Hb) in Gliederfüßlern fungiert Hämocyanin mit seinen zwei Kupfer-Ionen als Sauerstofftransporter; die Kupfer-Ionen sind über Histidin direkt am Protein gebunden; deshalb hat das Blut eine blaue Farbe; 2.) Die Sauerstoffbindestelle – was kann binden? Man unterscheidet mehrere Zustände der Sauerstoffbindestelle, wobei nur der Ferro Zustand Sauerstoff binden kann; die Dyshämoglobine, welche im Ferrozustand vorliegen, sind ebenfalls nicht zum Sauerstofftransport befähigt; 51 Desoxymyoglobin (Ferro) Oxymyoglobin (Ferro) Ferrimyoglobin = Met-Hb Sulfhämoglobin = SHb (Ferro) CO-Hb (Ferro) Fe Fe2+ Fe3+ Fe2+ Bindestelle Leer Sauerstoff Wasser H2S (irreversibel) Farbe Blau/violett Hellrot Gelb-braun grün Fe2+ CO (reversibel) kirschrot 2+ Kein O2 Transport möglich: Dyshämoglobine CO-Hb: Kohlenmonoxid bindet 25.000 mal stärker an isoliertes Häm als Sauerstoff, aber nur 250 mal stärker an Hämoglobin und Myoglobin (durch hohen Sauerstoffpartialdruck nahe der Lunge wird CO aus Hb tweilweise verdrängt; Bindung ist daher nicht vollständig irreversibel); Ursache: distales HisE7 (verhindert die lineare Koordination von CO, und schwächt somit die Bindung); wegen des endogenen Häm-Abbaus mittels Häm-Oxygenase (baut Häm zu Biliverdin, CO und Eisen ab; CO entsteht im Körper; immer geringe CO Konzentration im Blut) sind im Durchschnitt 1% der Bindestellen mit CO besetzt; Wozu Proteinumgebung für Häm? das Protein ist für den Sauerstofftransport wichtig: Häm wird vom Wasser isoliert, und den Ferri Zustand zu vermeiden; außerdem wird so die Bindung an andere Moleküle vermieden; Hinzu kommt die Möglichkeit der Regulation des Sauerstofftransports durch die Proteinumgebung; Met-Hb = Methämoglobin; einziger Unterschied zu Hämoglobin: FeII wurde zu FeIII oxidiert ( keine Sauerstoffbindung mehr möglich), Met-Hb entsteht ständig; die Konzentration wird allerdings durch das NADH-Methämoglobin-Reduktase System in den Erythrozyten gering gehalten (normaler Anteil von Met-Hb im Blut liegt bei 1%); Trick: Myoglobin ist weniger oxidationsempfindlich als Hämoglobin, weshalb es kein bis kaum Met-Mb gibt; 52 3.) Polypeptidkette determiniert Funktion der prosthetischen Gruppe Vorkommen der prosthetischen Gruppe Häm je nach polypeptidkette hat die prosthetische Gruppe eine andere Funktion Prosthetische Gruppe Häm (Fe2+) Hämin (Fe3+) Häm / Hämin Vorkommen Funktion Hb O2-Transport Mb O2-Speicher Oxygenasen O2-Einführung Katalase 2 H2O2 2 H2O + O2 Peroxydase Met-Hb, Met-Mb Redoxkomponenten: Transport, Cytochrome, OH Einfuhr bei Atmungskette, Hormonen oder Hydroxylierung Fremdstoffen Andere Liganden CO CN 4.) Vergleich Hb und Mb - - Tertiärstruktur ist sehr ähnlich; jedoch nur 20% Sequenzidentität; Exon-Intron Organisation: Hb / Mb besteht aus 3 funktionellen Einheiten unterschiedlicher Bereiche: N-terminale Domäne, Häm-bindende Domäne (Exon; wird auch für Atmungskette verwendet: AS 31-105), C-terminale Domäne Myoglobin = Mb: Speicherung und Transport von Sauerstoff im Muskelgewebe (Pottwal-Muskel: 80g/kg / Mensch-Muskel: 8g/kg) Hämoglobin = Hb: Transport von Sauerstoff im Blut; in den Erythrocyten hochkonzentriert vorhanden: 20mM (Sauerstofflöslichkeit 0,1mM in Wasser) 53 5.) Sauerstoffbindungskurven: Das Prinzip der Allosterie - - - - - P50 ist der Sauerstoffpartialdruck, bei dem 50% der Hb/Mb mit O2 gesättigt sind; P50(Mb) = 2,8 Torr (schnelle Sauerstoffaufnahme; Abgabe nur bei sehr niedriger O2 Konzentration; Bindung des Liganden ist einstufige, bimolekulare Reaktion); P50(Hb) = 26 Torr (jedoch: kooperatives Verhalten) Allosterie ist ein Maß für die Kooperativität; Sie wird in relativen Einheiten n (Hill Koeffizient) angegeben; je größer n, desto allosterischer ist ein System; n=1 bedeutet keine Kooperativität; n kann maximal den Wert der Anzahl aller miteinander wechselwirkenden Untereinheiten annehmen; je größer n ist, desto steiler wird die Sigmoidalität der Kurve; für n=1 handelt es sich um einen hyperbolen Kurvenverlauf; Für Hb gilt n= 2,8 (max. möglich n=4), weswegen Hb kein perfektes allosterisches Protein ist; Allosterie wird dadurch ermöglicht, dass die Bindung eines Substrates die Bindung weiterer Substrate aufgrund der Kommunikation zwischen den Untereinheiten über Gleitkontakte ermöglicht; Vorraussetzung für Allosterie: Oligomer, Zahl der Liganden > 1; Monomere zeigen nur hyperbolisches Bindungsverhalten; Den Hill-Koeffizienten erhält man aus der Steigung des Hill-Plots kooperative Sauerstoffbindung erleichtert den Transport: in den Alveolen der Lunge herrscht ein Sauerstoffpartialdruck von pO2= 100 Torr ( Y(Hb) = 0,98), während in den Kapillaren aktiver Muskeln pO2 = 20 Torr ( Y(Hb) = 0,32) herrscht; da P50(Hb) = 26 Torr wird 98% der Hb in der Lunge mit Sauerstoff beladen und 32% 54 kommen nur beladen zurück; für n=2,8 wird 1,83 mal mehr Sauerstoff transportiert las für n=1; 6.) Konformationsänderungen: Das Prinzip der Allosterie - - - - - Pack-Kontakte: α1β1 / α2β2 sind feste Bindungen (verändern kaum ihre Position wenn Untereinheit von Desoxy in die Oxy Form übergeht), an denen jeweils 35 AS beteiligt sind; Gleit-Kontakte: α1β2 / α2β1 sind flexible Kontakte, an denen 19 AS beteiligt sind; Schwache WW: α1α2 / β1β2 Die Bindung von Sauerstoff einer Untereinheit bewirkt den Übergang von der Desoxyform (tense = T Form) in die Oxyform (relaxed = R Form); Die T Form wird durch Salzbrücken, H-Brücken und BPG stabilisiert; Diese Kontakte brechen, wenn Sauerstoff bindet; es folgt eine Verdrehung der α1β1 gegen α2β2 um 15° (Atomverschiebung um 0,6 nm); Die Untereinheiten rücken näher zusammen, wodurch das zentrale Loch kleiner wird und 2,3BPG abdissoziiert; die Abstände der Fe Zentren zwischen α1 und α2 sowie β1 und β2 werden kleiner (39,9 A 33,4 A); Außerdem gehen die Salzbrücken zwischen und innerhalb der Untereinheiten verloren; Desoxy-Hb ist fest verklammert; ist dieser Zustand erreicht, nimmt Hämoglobin leichter Sauerstoff auf; Nach Sauerstoffbindung erfolgt die Verschiebung des zentralen Fe2+; es steht im desoxy-Hb 55 pm aus der Porphyrinebene, im oxy-Hb nur noch 22pm heraus (im desoxy-Hb hat Eisen 5 Liganden, im Oxy-Hb hat es 6 Liganden, wodurch der Spinzustand verändert und der Ionenradius reduziert wird; so passt es erst jetzt in die Hämebene hinein); dies bewirkt die Kippung und Drehung des proximalen His F8 und somit eine Verschiebung des Proteingerüsts; ein Val FG5 Rest kommt dadurch mit einer Ecke des Häm in sterische Probleme; diese Spannung wird gelöst, indem eine HBrücke zwischen Val FG5 und Tyr HC2 gelöst wird; als Resultat ergeben sich erhebliche Bewegungen der FG Ecke und des HC Bereiches; dadurch werden Interaktionen aufgehoben bzw. verändert; Veränderung der Gleitkontakte: β2FG Asp – α1C Tyr = FG Ecke der β2 Untereinheit kontaktiert die C Helix der α1 Untereinheit; hier findet die entscheidene Geometire Veränderung statt (switch transition); α1FG - β2C = Die FG Ecke der α1 Untereinheit kontaktiert die C Helix der β2 Untereinheit; diese Interaktion ist für Desoxy und Oxy Zustand gleich; das selbe gilt für den Kontakt α2β1; im Desoxy Zustand existieren 19 Salzbrücken; im Oxy-Zustand werden diese verändert; α1β1 und α2β2 gleiten aneinander vorbei, wodurch ein neuer stabiler OxyZustand entsteht, der weniger Salzbrücken aufweist; 55 - In den β Ketten ist der Zugang des Eisens schwiereiger als in den α Ketten, da das Häm in der T Form relativ schwer zugänglich hinter dem distalen His-E7 und Val-E11 (nur bei β Kette vorhanden) liegt; wenn Sauerstoff an eine α Untereinheit bindet, löst es die genannten Konformationsveränderungen aus, wodurch die β Ketten in die R Form übergehen, in der ValE11 und HisE7 genug abstand von Eisen haben, damit Sauerstoff binden kann; Bewegung des Fe2+ in die Hämebene Bewegung der Peptidkette, initiiert durch Fe2+ Bewegung Gleitkontakte werden umorientiert 56 - Sauerstoff bindet an Desoxy-Hb im T state mit niedriger Affinität; je mehr Sauerstoff gebunden ist, desto höher ist die Bindungsenergie zwischen Sauerstoff und Eisen im T state, weshalb er in den R state springt; der R state hat eine hohe Affinität zu Sauerstoff; die sigmoidale Kurve kommt durch den Überlapp der linearen (T state) und der hyperbolen (R state) bindungskurve zustande 7.) mathematische Modelle zur Beschreibung der Kooperativität nach Hill („Alles oder Nichts“) im Jahr 1910 gab es weitere Modelle zur Beschreibung der Kooperativität (man spricht von homotropen WW; heterotrope WW werden durch andere Liganden induziert: Inhibitoren und Aktivatoren; siehe unten) A.) Das Sequenzielle Modell nach Gilbert Adair (1924) „kommunizierende Intermediate“ Gutes und einfaches mathematisches Modell; Strukturbiologisch jedoch nicht korrekt; es gibt einen T Zustand, der keinen Sauerstoff gebunden hat und einen R Zustand, der voll mit Sauerstoff besetzt ist; Grundgedanke: Bindung eines Liganden an eine Bindestelle beeinflusst die Bindestellen in der Umgebung so, dass ihre Affinität steigt, ohne dass es zu einem Übergang kommt, der sich auf das gesamte Enzym auswirkt; je nach geometrischer Anordnung unterscheidet sich die Anzahl der direkt beeinflussten Nachbarn; Der Grad der Allosterie hängt von der Kopplung ab; Ki ist die apparente GG Konstante; ki ist die mikroskopische intrinsische Konstante [ES i −1 ]⋅ [S ] [ES i ] n − i +1 ki = ⋅ Ki i Das sequentielle Modell kann im Gegensatz zum konzertierten Modell auch negative Kooperativität erklären; Ki = 57 B.) Das konzertierte Modell nach Monod, Wymann und Changeux (1965) In diesem Modell wechseln alle Untereinheiten konzertiert von der einen in die andere Konformation; Die beiden Konformationen werden mit T und R bezeichnet; innerhalb einer Konformation sind die Bindungsstellen unabhängig, weshalb sich das Bindungsverhalten innerhalb einer Konformation durch das nicht-kooperative Modell mit einer intrinsischen Gleichgewichtskonstanten kR bzw. kT beschreiben lässt; Die beiden Konformationen stehen im Gleichgewicht, das durch die Gleichgewichtskonstante L0 zwischen den unbeladenen Zuständen R0 und T0 (bzw. durch die GGK Lj zwischen den beladenen Zuständen Tj und Rj, wobei j der Anzahl der Sauerstoffatome entspricht, mit der das Molekül beladen ist) beschrieben wird; Positiv kooperatives Verhalten tritt auf, wenn GG bei niedriger Ligandenkonzentration beim T Zustand liegt (T0 > R0 bzw. L0 >> 1) und die Affinität des R Zustands höher ist (kR > kT bzw. c > 1); L0 = [T0 ] [R0 ] c= kR kT L j = j ⋅ c ⋅ L0 = [T ] [R ] j j α= [S ] kR n ist Anzahl der Untereinheiten; S ist Ligandenkonzentration (hier: pO2) L ist relative Stabilität T/R (wenn L groß ist, folgt T stabiler als R bessere Kooperativität) α ist normierte Ligandenkonzentration wenn c groß ist, folgt große Affinität zum Liganden YS = [Ligand an R] + [Ligand an T ] = α ⋅ (1 + α ) + L ⋅ c ⋅ α ⋅ (1 + c ⋅ α ) (1 + α ) + L ⋅ (1 + c ⋅ α ) n ⋅ [R Zustände ] + [T Zustände ] n −1 n n −1 n 58 mit zunehmenden L (T immer stabilerer als R) sinkt die Sättigung des allosterischen Proteins bei gleicher normierter Ligandenkonzentration α mit zunehmendem c steigt die Sättigung des allosterischen Proteins, allerdings unter Verlust des sigmoidalen Verhaltens; 8.) andere Transportmechanismen Erythrocyten transportieren neben Sauerstoff durch Hb auch andere Verbindungen durchs Blut; CO2 und H+ Ionen (vom Muskel produziert) werden vom Gewebe in die Lunge transportiert; Außerdem dienen die roten Blutkörperchen der Pufferung des Intra- und Extrazellularraumes; A.) HCO3- / Cl- Antiporter spielt wichtige Rolle beim Ausatmen! Im Gewebe diffundiert CO2 in die Erythrozyten; es folgt die Umsetzung mittels Carboanhydrase zu H2CO3, das in H+ und HCO3- dissoziiert; Die Protonen werden von Desoxy-Hb gebunden, Hydrogencarbonat wird vom Antiporter ins Blut transportiert; In der Lunge wird das Desoxy-Hb zu Oxy-Hb umgewandelt, wodurch Protonen abdissoziieren und mit Hydrogencarbonat zu Kohlensäure reagieren; Kohlensäure dissoziiert in Wasser und CO2; CO2 wird ausgeatmet, während HCO3- durch den Antiport in die Zelle gelangt; B.) Transport von CO2 62% des im Blut gelösten CO2 befindet sich im Plasma (6% in gelöster Form und 56% in der Form von HCO3-) und 38% in den Erythrocyten (26% als Carbamat an Hb und 12% in Form von HCO3-) Die α-Aminogruppen aller vier Ketten können im Desoxy-Zustand Kohlenstoffdioxid binden, wodurch ein Carbamat gebildet wird; es entsteht eine negative Ladung pro gebundenem CO2, statt NH3+; es werden im Schnitt 3,6 Moleküle CO2 pro Desoxy-Hb transportiert; C.) Der Bohr Effekt und Transport von H+ / ClDer Bohr Effekt (Christian Bohr, 1904) beschreibt den Anstieg der Affinität von Hb zum Sauerstoff, mit steigendem CO2 Partialdruck und sinkendem pH Wert (steigende ProtonenKonzentration) der Umgebung; 59 da der stark kontrahierende Muskel sowohl CO2 freisetzt (Citratcyclus), als auch das Medium stark ansäuert (Lactat-Bildung) wird es durch den Bohr Effekt möglich besonders bedürftige Organe bevorzugt mit Sauerstoff zu versorgen; pro abgegebenen O2 werden 0,5 H+ aufgenommen (Protonen werden von Desoxy-Hb transportiert); Protonen stabilisieren den T Zustand des Hb, je saurer des Medium, desto stärker verschiebt sich die Sauerstoffbindungskurve nach rechts (Abgabe von Sauerstoff wird erleichtert) Im Desoxy-Hb existiert eine Salzbrücke zwischen Asp94 und His146 der β Kette; H+ und Clwerden durch diese Salzbrücke fest gebunden; wenn Oxy-Hb entsteht dissoziiert H+ leichter von His146 ab, da die Salzbrücke nicht mehr existiert; pro Desoxy-Hb werden ca. 2 Protonen und 2 Chlorid-Ionen (da Salzbrücke nur in β Kette vorhanden) auf diese Weise transportiert; Protonen und Chlorid-Ionen können auch alternativ durch His-122 (25%) oder dem Aminoterminus der α Kette (25%) transportiert werden; an His-146 sind sie zu 50%; D.) Transport von 2,3-BPG Pro Desoxy-Hb wird 1 Molekül 2,3-BPG transportiert; erst dadurch, dass dieses kleine, fünffach negativ geladene Molekül in das zentrale Loch (positiv geadene Bindestelle; zwei β Ketten: Lys82, His143, His2) von Hämpglobin bindet, wird die Sauerstoff-Affinität um den Faktor 26 vermindert und das kooperative verhalten ermöglicht (BPG ist Vaurausetzung für 60 die Allosterie des Hb); 2,3-BPG stabilisiert die T Form, wird allderings bei der Bindung von Sauerstoff aus der Bindetasche verdrängt (Platzmangel), wodurch Sauerstoff besser binden kann (R Form ist stabiler); Die Konzentration von BPG in den Erythrozyten ist gleich der HbKonzentration; In Sauerstoff-unterversorgten Gewebe wird die BPG Konzentration erhöht, um die Sauerstoffabgabe aus Hb zu erhöhen; Physiologische Funktion von BPG (Pathobiochemie): 1.) durch Blutkonserven fällt der BPG Wert, weshalb Stabilisatoren (D-Glucose) notwendig sind 2.) hypoxische Zustände (z.B. Asthma) bewirken eine Erhöhung des BPG Spiegels, weshalb mehr Sauerstoff entladen wird und länger im Gewebe zugänglich ist 3.) durch die Höhenadaption werden kurzfristig mehr BPG und langfristig mehr Erythrozyten im Blut sein 4.) Fötales Hämoglobin (HbF) bindet BPG weniger stark als das mütterliche Hämoglobin (HbA), weshalb HbF eine höhere Sauerstoffaffinität aufweist; E.) Heterotrope WW WW eines allosterischen Proteins mit Inhibitoren oder Aktivatoren, die nicht dem Liganden entsprechen werden als heterotrop bezeichnet; Beispiele sind die genannten Stoffe: Kohlenstoffdioxid, Protonen, Chlorid und BPG (alles Inhibitoren, wobei BPG den mit Abstand größten Einfluss hat, da es mit allen 4 Untereinheiten wechselwirkt); Das Bindungsverhalten wird also beeinflusst: 61 9.) Funktion der verschiedenen Polypeptidketten des Hb Es gibt 6 verschiedene Polypeptidketten mit unterschiedlichen Funktionen und Transportfähigkeiten für Sauerstoff, Kohlenstoffdioxid und Protonen; Alternativen für die β Kette im adulten Tier: Die ε Polypeptidkette ist Teil des embryonalen Hb (hat höchste Sauerstoff-Affinität); Die γ Polypeptidkette ist Teil des fötalen Hb (hat hohe Sauerstoff-Affinität); 2% der β Polypeptidkette im adulten Organismus ist durch die δ Polypeptidkette ersetzt; Alternativen für die α Kette im adulten Tier: Die ξ Polypeptidkette ist Teil des embryonalen Hb; 62 Bei der Geburt wird HbF abgebaut; Häm wird zu Bilirubin abgebaut; 10.) Pathobiochemie des Hämoglobin 1.) Met-Hb kann mit Nitrite und Nitrate zu unlöslichen Heinz’schen Innenkörper aggregieren 2.) Eine CN Vergiftung führt zur Blockierung der Atmungskette (Cytochrom Oxidase), wodurch der ATP Spiegel sinkt; Gegenmaßnahme: Thiosulfat + Cyanid Rodanit SCN Hämoglobinopathien sind angeborene Störungen der Hämoglobinbildung infolge unterschiedlicher genetischer Defekte bei der Bildung der α- bzw. β-Kette; es sind etwa 500 Varianten bekannt (95% Punktmutationen); 5% der Weltpopulation tragen Hb Varianten; Hämpglobinopathien können u.a. die Bildung von Dyshämoglobine begünstigen; 63 - - Eine abweichende Primärstruktur kann mehrere Folgen haben: am häufigsten tritt eine erhöhte Sauerstoff-Affinität auf; erniedrigte Sauerstoff-Affinität; Met-Hb Bildung; Instabilität des Hb und Verlust der Häm-Gruppe; Veränderte Löslichkeit und Aggregation auf molekularer Ebene werden diese Symptome erreicht durch: Änderung der Oberflächenreste (führt zu HbS: Sichelzellenbildung); Änderung interner Reste (Degradation; Heinz-Körper); Stabilisierung von Met-Hb (durch anionisches OxyAtom am Fe: Punktutation bewirkt Austausch von HisF8 mit TyrF8); Änderung des α1 β1 Gleitkontakts (wenn so T Zustand stabilisiert wird, wird Sauerstoff-Affinität erniedrigt) Sichelzellenhämoglobin - - - Die häufigste Hämoglobinopathie ist die homozygote Sichelzellenanämie (Anämie = Verminderung der Zahl der Erythrozyten und des Hämoglobins im Blut; es handelt sich um einen hämolytischen Defekt: wenn viele Sichelzellen vorhanden sind, wird Hämoglobin durch Zerstörung der Erythrozyten freigesetzt); Sie entsteht durch eine Punkt-Mutation des β-Globingens, wodurch an Position 6 der β Kette ein Austausch von Glutaminsäure zu Valin stattfindet (Glu6 Val6); so entstandenes HbS bildet bevorzugt unter Sauerstoffentzug Aggregate, die zu einer Sichelform der Erythrozyten führen; Solche Erythrozyten werden von der Milz sequestriert (und durch Hämolyse abgebaut), können aber Gefäßverschlüsse auslösen. Außerdem haben sie eine wesentlich kürzere Lebensdauer; Erkrankung endet meist vor dem 30. lebensjahr tödlich (Infektion, Nieren- oder Herzversagen, Thrombosen, Schock); 0,4% der schwarzen Bevölkerung betroffen; Patienten, welche Sichelzellenanämie-Symptome aufweisen sind homozygot erkrankt (50% Sichelzellen); heterozygote Individuen zeigen keine Symptome (nur 1% Sichelzellen); darüber hinaus haben heterozygote Individuen einen Selektionsvorteil, da sie gegen Malaria resistent sind (Malaria Viren bilden in Erythrozyten eigene Vakuolen und ernähren sich von Hb, bis sie ein bestimmtes Entwicklungsstadium erreicht haben); deshalb ist diese krankheit in malaria Gebieten (Mittelafrika) weit verbreitet; Sichelzellenanämie findet man nur in Sauerstoffarmen Blut (Die ärztliche Verordnung „Ruhe“ ist daher sinnvoll, da kaum Sauerstoff veratmet wird und das Blut sauerstoffreich bleibt), weil die Desoxy Form am meisten betroffen ist; 64 - HbS läuft im elektrischen Feld langsamer als HbA, weshalb man so die Hb Sorten voneinander unterscheiden kann; Dies liegt an der Veränderung des Isoelektrischen Punktes: Oxy-HbA hat IEP = 6,87, während Oxy-HBS IEP=7,09 aufweist; DesoxyHbA hat IEP = 6,68 und Desoxy-HbS hat IEP=6,91; in beiden Fällen also resultiert ein Anstieg des IEP von ca. 0,22; Der IEP befindet sich bei dem pH Wert, bei dem das Protein keine Ladung aufweist; - Außerdem enthält der Abschnitt des β-Globulin-Gens eine Schnittstelle für das Restriktionsenzym MstII, die durch Punktmutation verloren geht; nach der DNA Isolierung, einer PCR und Zugabe von MstII lässt sich eine Fragmentanalyse mittels Gelelektrophorese machen; mutierte DNA läuft nicht so weit wie gesunde, da sie länger ist; Fingerprintanalyse: Anfänge der Molekulardiagnostik tryptischer Verdau des Hb und anschließende 2D-Trennung der Fragmente; so konnte auf die Punktmutation geschlossen werden; - Die Sichelbildung wird bei Sauerstoffmangel eingeleitet; es handelt sich um eine Polymerisation von Desoxy-HbS mit Verzögerungskinetik bei der Keimbildung; kleine HbS Aggregate liegen im GG mit einzelnen HbS Molekülen, können also wieder dissoziieren; erst ab einer kritischen Keimgröße von 10 folgt die weitere irreversible Aggragation; 10 HbS bilden zusammen einen Keim und aggregieren weiter unter Bildung einer Fibrille; diese bilden bei Sichelzellen das Cytoskelett und bewirken eine Bewegungseinschränkung der Erythrozyten; Dadurch ist weniger Sauerstoff-Transport möglich, wodurch mehr Desoxy-Hb vorhanden ist und somit mehr Fibrillen gebildet werden; Eine Fibrille ist eine 14strängige Helix, wobei 8 Desoxy-Hbs in Kontakt miteinander stehen 65 Thalassämie: Erkrankung der Erythrozyten, bei denen durch ein Gendefekt das Hb nicht ausreichend gebildet bzw. gesteigert abgebaut wird; Gendefekte auf Chromosom 11 (β Thalassämie) oder 16 (α-Thalassämie) führen zur vermiderten β- bzw. α-Globinkettenbildung Enzyme 1.) Enzymklassen Klassifizierung Reaktionstyp Oxidoreduktasen Redoxreaktion Transfer funktioneller Gruppen Transferasen Hydrolasen Bindungsspaltung durch Hydrolyse Lyasen Spalten oder knüpfen Bindungen ohne Beteiligung oxidativer oder hydrolytischer Schritte Isomerisierung Isomerasen Ligasen Verknüpfen zwei Moleküle unter Hydrolyse einer energiereichen bindung Beispiele Alkohol-Dehydrogenase, Katalase Hexokinase, Phosphoglyceratkinase, Transketolase Asparaginase, Peptidasen, Amylasen, Phosphatasen Pyruvat-Decarboxylase, AdenylatCyclase, Carboanhydrase Phosphoglycerat-Mutase, DNATopoisomerase Pyruvat-Carboxylase 2.) Regulationsprinzipien A.) Feedback Mechanismus z.B. durch allosterische Inhibition des Endprodukts B.) Regulatorproteine (z.B. Calmodulin) C.) Kovalente Modifikation (z.B. reversible Kopplung von Phosphorylgruppen durch Kinasen und Phosphatasen; Serin, Threonin und Tyrosin können phosphoryliert werden) D.) Enzym-Kaskaden – proteolytische Aktivierung (z.B. blutgerinnung, Trypsinogen, Pankreas) 66 3.) Thermodynamische Grundlagen Die thermodynamik fragt nicht nach dem Weg, sondern betrachtet nur Anfangs- und Endzustand; Erster HS der Thermodynamik: Energie kann weder erzeugt, noch vernichtet werden Änderung der inneren Energie: Änderung der Enthalpie: ΔU = Ufinal – Uinitial ΔH = ΔU + PΔV Energie kann nur im Vakuum gemessen werden; Enthalpie in Lösung Exothermer Prozess, endothermer Prozess Zweiter HS der Thermodynamik: spontane Prozesse bewirken immer eine Entropieerhöhung des universums ΔSSystem + ΔSUmgebung = ΔSUniversum > 0 S = kblnW ΔS = q/T Gibbs Enthalpie (Gibbs Energie): ΔG = ΔH – TΔS Exergonischer Prozess, endergonischer prozess Entropie oder enthalpiegetrieben GG, Massenwirkungsgesetz ∆G = ∆G0 + RT ln [C ]⋅ [D] für A+B <-> C + D [A]⋅ [B] ∆G0 = − RT ln K eq Gekoppelte Reaktionen: ΔG1 + ΔG2 = ΔG3 ; wenn ΔG3 negativ, dann laufen beide Reaktionen ab 4.) Wirkungsweise von Enzymen Enzyme können Reaktionsgleichgewichte nicht verschieben, sie beschleunigen lediglich die Einstellung des Gleichgewichts; dies geschieht durch eine Herabsetzung der Aktivierungsenergie 67 Entweder Enzyme funktionieren nach dem Schlüssel-Schloss Prinzip (Substrat passt perfekt in Bindetasche des Enzyms) oder nach dem induced-fit (Enzymtasche bildet sich erst aus, wenn Ligand in die Nähe kommt; dynamisches Modell); beide Modelle bewirken die Ausbildung eines Enzym-Substrat-Komplexes; Substratspezifität: Enzyme weisen unterschiedliche geometrische Selektivitäten in Bezug auf ihre Substrate auf; die geometrische Selektivität variiert im Ausmaß von Enzym zu Enzym; ein geometrisch unsepzifisches Enzym ist z.B. die Alkohol-Dehydrogenase; sie setzt als Hauptedukt Ethanol zu Ethanal um; kann jedoch auch Methanol und Isopropanol umsetzen; Enzyme mit hoher Substratspezifität setzen nur ein Substrat um; Die Maltase z.B. spaltet nur Maltose, nicht aber die Diastereomere Cellobiose; Enzyme haben besonders eine hohe Spezifität in Bindung und Reaktionskatalyse chiraler Substrate: sie sind aufgrund der Geometrie des aktiven Zentrums dazu in der Lage prochirale Gruppen zu differenzieren; Die Hydridübertragung von NADH ist ein beispiel für eine stereospezifische Reaktion; 68 Wirkungsspezifität: Die Fähigkeit von verschiedenen Möglichkeiten der chemischen Umsetzung eine ganz bestimmte auszuwählen Innerhalb eines aktiven Zentrums differenziert man den bindenden Bereich (binding site) von dem katalysierenden Bereich (active site), wobei beide Bereiche auch zusammenfallen können; die binding site ist verantwortlich für die Substratspezifität; die active site, sowie die Coenzyme sind verantwortlich für die Wirkungsspezifität; 5.) Quantifizierung von Enzymen: Enzymkinetik Enzyme werden durch ihre Aktivität quantifiziert; Die Messung der Aktivität erfolgt durch spektroskopische Methoden; Die Aktivität einer NADH-abhängigen Dehydrogenase kann gemessen werden durch die Abnahme der spezifischen Extinktion bei 340nm (Absorptionsmaximum von NADH und NADPH); das Absorptionsmaximum von NAD+ und NADP+ liegt bei 260nm, wobei diese bei 340nm keine Absorption mehr zeigen; Die katalysierte Reaktion erfolgt schneller, weil durch die Herabsetzung der Aktivierungsenergie die Wahrscheinlichkeit einer Reaktion sehr viel höher ist; findet man einen hohen Aktivierungsenergie Berg (so wie bei einer unkatalysierten Reaktion), so handelt es sich um eine statistisch unwahrscheinliche Reaktion (Kinetik); die Thermodynamik, also das Gleichgewicht zwischen Produkt und Edukt jedoch wird nicht beeinflusst (katalysierte Rückreaktion ist um den selben Betrag begünstigt) Herabsetzung der AE erfolgt durch Stabilisierung des Übergangszustandes (äquivalent zum Enzym-Substrat-Komplex) im katalytischen Zentrum des Enzyms durch verschiedene Enzym-Substrat-WW; Michaelis Menten Kinetik: Enzymkinetik im quasi stationären Bereich dc ES =0 dt 69 (Annahmen: kleine Enzymkonzentration, sodass sehr viel mehr Substrat vorliegt als Enzyme; Lebensdauer des Enzym-Produkt-Komplexes geht gegen 0; Enzym und Produkt reagieren nicht zurück zum ES Komplex, da Produkt aus dem GG entzogen wird biochemische Weiterreaktion) KS = k1 [ES ] = k −1 [E ] ⋅ [S ] KS ist Bildungskonstante des Enzym-SubstratKomplexes KI = l −1 [E ] ⋅ [I ] = [EJ ] l1 KI ist Zerfallskonstante des Enzym-InhibitorKomplexes cS cS v = v max ⋅ k −1 + k 2 K M + cS + cS k1 KM ist diejenige Substratkonzentration, für die die Halbmaximale Reaktionsgeschwindigkeit erreicht ist, es handelt sich um die Zerfallskonstante des Enzymsubstratkomplexes; v = k 2 ⋅ c ES = k 2 ⋅ c E0 ⋅ KM = c ES k + k2 = −1 cE ⋅ cS k1 Die Gleichgewichtslage zwischen E+S und ES kennzeichnet die Affinität zum Substrat; Wenn die Substratkonzentration oder die Affinität zum Substrat sehr groß ist, wird die Umsetzung nur noch von ES E+P bestimmt; Grenzfälle: c S << K M c S >> K M cS = K M v max ⋅ cS KM v = v max v v = max 2 v= 70 v vmax vmax / 2 Michaelis-Menten Plot KM cs Die Leistungsfähigkeit des Enzyms wird als Wechselzahl (Turnover) k2 angegeben max imalerUmsatz k 2 = k cat = c E0 k2 ist Reaktionsgeschwindigkeitskonstante und bestimmt den Zerfall des ES Komplexes; Die Einheit ist sek-1 (Substratumsatz pro Sekunde) 5.1) lineare Auftragungen 1/v Lineweaver-Burk Plot Doppel-reziproke Darstellung: 1 KM 1 1 = ⋅ + v v max c S v max KM / vmax -1 / KM 1 / vmax 1/cs v vmax Eady-Hofstee Plot Halb-reziproke Darstellung: v v = − K M ⋅ + v max cS -KM vmax / KM v/cs 71 5.2) Enzyminhibition k1 E+S k -1 + ES k2 E+P v = v max ⋅ I l-1 kompetetive Inhibition l1 cS c c S + K M ⋅ 1 + I KI EI Kompetetive inhibitoren konkurieren mit dem Substrat um dieselbe Bindestelle v v0-v v0 KM Inhibition = KM (1+cI / KI) cs v0 − v sinkt mit steigender Substratkonzentration v0 1/v -1 / KM - 1 KM (1 + cI / KI) 1/cs 72 E+S + k1 k -1 I v = v max ⋅ k1 nicht-kompetetive l1 l-1 l1 EI + S E+P + I l-1 k2 ES (c S + K M ) ⋅ 1 + cI KI v max cS v= ⋅ c cS + K M 1+ I KI Inhibition ESI k -1 cS Nicht-kompetetive inhibitoren binden außerhalb des aktiven zentrums an das Enzym und stabilsiert es in seiner Konformation in einer Weise, dass das substrat nicht umgesetzt werden kann v vmax v0-v vmax 1 + c I / KI v0 cs Inhibition = v0 − v ist unabhängig von der Substratkonzentration v0 1/v 1 + c I / KI vmax 1 / vmax 1/cs 73 k1 E+S ES k -1 k2 v = v max ⋅ E+P + I unkompetetive l1 l-1 Inhibition ESI cS c c S ⋅ 1 + I + K M KI v cS v = max ⋅ c KM 1 + I cS + c KI 1+ I KI vmax v v0-v vmax 1 + c I / KI v0 KM cs KM 1 + c I / KI v −v steigt mit steigender Substratkonzentration Inhibition = 0 v0 1/v 1 + c I / KI vmax 1 / vmax - 1 + c I / KI -1 / KM 1/cs KM 74 Experimentelle Ermittlung des KM Wertes eines Enzyms: 1.) man setzt eine Pufferlösung mit einer bestimmten Konzentration eines Substrats an; nach Zugabe der Enzymsupsension startet die Reaktion, welche im UV VIS Spektrometer aufgrund von Absorptionsverschiebungen beobachtet wird; dabei wird die Zunahme oder Abnahme der spezifischen Absorption bei einer bestimmten Wellenlänge (z.B. Zunahme bei 340nm, wenn NADH gebildet wird) gemessen; 2.) die Anfangssteigung des so erhaltenen „spezifische Absorption bei xxx nm vs. Zeit [min]“ Diagramms (exponentielle Produktbildung gegen Sättigungsgrad; keine Hyperbole) entspricht der Umsetzungs-Geschwindigkeit des Enzyms bei der entsprechenden Anfangssubstratkonzentration; 3.) mehrere Versuche bei unterschiedlichen Substratkonzentrationen ergeben mehrere Geschwindigkeit-Substratkonzentration Paare; diese können im Michaelis-Menten Plot aufgetragen werden, der hyperbolen Verlauf zeigt 4.) nun folgt die Linearisierung z.B. mittels Lineweaver-Burk Plot; der Schnittpunkt mit der 1/S Achse liefert den KM Wert; Katalytische Effizienz: KM Werte sollten idealerweise im Bereich der Substratkonzentration liegen; Je niedriger der KM Wert desto höher ist die Affinität des Enzyms zum Substrat; die katalytische Effizienz steigt mit sinkendem KM und steigendem kcat; das kinetische Optimum (kcat / KM Kriterium) liegt bei max. 108 bis 109 M-1s-1. je größer dieser Quotient desto besser arbeitet das Enzym; die Umsetzungsgeschwindigkeit von einem kinetisch perfekten Enzym wird lediglich durch die Diffusionsgeschwindigkeit von Enzym und Substrat eingeschränkt; 5.3) allosterische Enzyme Allosterische Enzyme gehorchen nicht der Michaelis Menten Kinetik (keine Hyperbole Kinetik! Alles sigmoidal); allosterische Effektoren lagern sich außerhalb des aktiven Zentrums an das Enzym an und verändern die Konformation des Enzyms so, dass eine Substratumsetzung verlangsamt (ES-Komplex wird durch allosterische Inhibitoren destabilisiert; T state wird stabilisiert) oder beschleunigt (ES-Komplex wird durch allosterische Aktivatoren stabilisiert; R state wird stabilisiert) wird; auf diese Enzyme können die selben Modelle wie bei der Beschreibung von Hämoglobin angewendet werden: 75 Die Besetzung der ersten Untereinheit mit einem Substrat beeinflusst die Affinität der nächsten Untereinheit zum Substrat (homotroper kooperativer Effekt); je höher die Substratkonzentration, desto eher wird eine Bindestelle besetzt, desto leichter können weitere Bindestellen besetzt werden; es resultiert eine sigmoidale Beziehung zwischn der geschwindigkeit und der Substratkonzentration; allosterische Inhibitoren und Aktiviatoren bewirken das gleiche (heterotroper kooperativer Effekt) Man unterscheidet zwei typen von Effektoren: solche vom K Typ wirken sich auf den KM Wert aus und solche vom V-Typ beeinflussen den vmax Wert; A: ohne Effektor / B: allosterischer Aktivator / C: allosterischer Inhibitor 6.) Wirkung von Inhibitoren Übergangsanaloga sind potente Inhibitoren; Die Prolin-Racemase katalysiert die Isomerisierung zwischen dem tetraedrischen L- und D-Prolin, wobei ein planarer 76 Übergangszustand entsteht; als Inhibitor für die Prolin-Racemase eignet sich daher das planare Molekül Pyrrol-2-carboxylat; Katalytische Antikörper: Abzyme = Antikörper, die an einen Übergangszustand einer bestimmten Reaktion binden und als Enzym fungieren; es wäre eine Massenerzeugung von künstlichen Enzymen möglich; Wirkung von Penicillin: bewirkt die Inhibition der Bakterienzellwandsynthese (Glykopeptid-Transpeptidase); gram positive Bakterien haben eine dicke PeptidoglykanSchicht in der Zellwand; Das Peptidoglycan-Gerüst besteht aus einem Rückgrat aus abwechselnd auftretenden Molekülen N-Acetyl-Glukosamin und N-Acetyl-Muraminsäure; an jedem N-Acetyl-Muraminsäure-Molekül befindet sich ein Tetrapeptid aus bestimmten AS; dieses Tetrapeptid vermittelt die Quervernetzung zwischen den einzelnen Rückgrats mittels Pentaglycin-Resten; Pentaglyin ist auf der einen Seite mit der ε-Aminogruppe von Lysin und auf der anderen Seite mit dem C terminus von D-Ala verknüpft; Die Glykopeptid-Transpeptidase (Protease) katalysiert die Knüpfung der D-Ala-Glycin Bindung; Ein anderes Enzym spaltet die Quervernetzungen im Bereich der Z Scheibe des Bakteriums (Zellmittelpunkt) wieder; Dieser Umbau der Zellwand ist im Zuge des Zellwachstums notwendig; Penicillin wirkt als Inhibitor der Glykopeptid-Transpeptidase; es besetzt das aktive Zentrum des Enzyms, weil es in seiner Struktur analog zum Substrat (der terminalen D-Ala-D-Ala Einheit) ist; das Enzym wird durch die Spaltung des β Lactam-Ringes von Penicillin unter der Bildung eines Penicilloyl-Enzym-Komplexes irreversibel blockiert, wodurch keine 77 weitere Quervernetzung der Zellwand geschieht; da aber das andere Enzym weiter die Zellwand auflockert, kommt es zur Lyse; Enzym-Mechanismus: das terminale Glycin der Pentaglycin-Brücke und die terminale DAla-D-Ala Einheit des Pentapeptids sind Erkennungsstrukturen des Enzyms; zunächst erfolgt die Abspaltung der terminalen D-Ala Einheit (Zwischenprodukt: Acyl-Enzym Intermediat), woraufhin die neue Peptidbindung zum terminalen Glycin geknüpft wird; 7.) katalytische Mechanismen A.) Säure-Base Katalyse Enzym stellt stereoselektiv Protonen zur Verfügung oder zieht sie vom Substrat ab) B.) Kovalente Katalyse Enzym ist kurzzeitig mit AS über ein Intermediat kovalent verbunden; das Intermediat ist kein ÜZ, sondern isolierbar, jedoch schwer nachzuweisen; wichtige Rolle: Serin, OH Gruppe 78 C.) Metallionen Katalyse (z.B. Zn katalysiert) D.) Elektrostatische Katalyse Die Bindung eines Substrats an das aktive Zentrum findet in Abwesenheit von Wasser statt E.) Nachbarschafts- und Orientierungskräfte Durch eine richtige räumliche Anordnung der Reaktanden (Nähe und Orientierung) kann eine Reaktion begünstigt werden F.) Bindung des Übergangskomplexes Durch die Stabilisierung des Übergangszustandes wird die Aktivierungsenergie herabgesetzt; tatsächlich findet meist eine Kombination aus mehreren dieser Mechanismen statt; A.) Säure-Base Katalyse Ein partieller Protonentransfer erniedrigt die freie Enthalpie des Übergangszustandes; Es handelt sich um die in der Biochemie häufigst vorzufindende Katalyse; Die Seitenketten der AS Asp, Glu, His, Cys, Tyr und Lys können sowohl als Säure- als auch als Basenkatalysatoren fungieren; Enzyme haben oft mehrere ionisierbare Seitengruppen so um das aktive Zentrum angeordnet, dass sie eine konzertierte Säure-Base-Katalyse durchführen können; Beispiele: Hydrolyse von Peptiden, Estern; Reaktionen von Phosphatgruppen; Tautomerisierungen, Isomerisierungen; Addition an Carbonylgruppen; Redoxreaktionen Reaktive Gruppen: Imidazolgruppen von Histidin, Thiolgruppen von Cys, Hydroxylgruppen von Tyr, Aminogruppen von Lys, Carboxylatanionen als Base; 79 Ribonuklease A Mechanismus: Ribonuklease A (124AS, in vitro Faltung) spaltet RNA; es handelt sich um eine Endonuklease; das Enzym erkennt die Pyrimidine U und C (Purine wären zu groß) und spaltet die Phosphodiesterbindung an der 5’-Position der Ribose des in der Kette folgenden Nukleotids; als Intermediat entsteht ein cyclisches Phosphat; einzelsträngige DNA bindet ebenso an RNAse A, wird aber nicht umgesetzt; RNAse A ist häufig extrazellulär vorkommende Umweltnuklease, da sie mit dem Schweis sekretiert wird, um RNA Viren abzubauen; R2 OCH2 Pyrimidin O H H O O - R2 O O Protein H H H His12 N H O NH P + H2O O O - R1OH H + NH3 + NH3 Protein + OCH2 H NH H O O H His12 N His119 NH NH - + NH3 Protein N OH P O Protein H H H Protein + N Protein O Protein + N Pyrimidin O H O H O Lys41 His119 Lys41 P Protein NH Protein - O H N R2 Pyrimidin O H His12 H R1 O H His119 NH Lys41 Im ersten Schritt abstrahiert His12 aus der 3’ OH Gruppe der Ribose ein Proton, wodurch das entstandene Alkoholat nukleophil an das 5’ Phosphat angreifen kann; es folgt die Bildung eines pentavalenten cyclischen Phosphorübergangszustand (welcher von Lys41 stabilisiert wird) und die Eliminierung eines Alkoholats (beinhaltet Rest-RNA), das konzertiert mit seiner Eliminierung ein Proton von His119 abstrahiert; Im zweiten Schritt wird die Aktivität des Enzyms regeneriert: His119 abstrahiert ein Proton vom Wasser, wodurch ein Hydroxidion 80 entsteht; dieses greift nukleophil an das cyclische Phosphat an, wodurch es hydrolysiert wird und zugleich das Proton von His12 zurück zur 3’ OH Gruppe der Ribose gelangt; Ein Hinweis auf eine Säure-Base Katalyse unter Beteiligung von Histidin liefert eine Auftragung von log(vmax / KM) gegen den pH Wert; für das Enzym Ribonuklease A resultiert eine umgekehrte Parabel, das Maximum liegt bei pH = 6 (zwischen KS = 5,4 und KS 6,4); oberhalb und unterhalb dieses pH Wertes sinkt die Kurve fast linear ab; auch möglich ist der Austausch von AS im Protein durch Punkt-Mutation, um für die Aktivität spezifische AS zu finden (es ist aber auch möglich, dass der N-terminus katalytische Aktivität aufweist); = Potente Inhibitoren dieses Enzyms wären folgende 3 Moleküle; sie bewirken eine kovalente Modifikation aktiven Zentrum: am Lactat-Dehydrogenase Mechanismus: Die Lactat-Dehydrogenase weist eine Nukleotidbindedomäne mit zwei Hälften (Nikotinamidbindehälfte und Adeninbindehälfte) auf; die Hydridübertragung von Lactat auf NAD+ würde erheblich länger dauern, wenn His119 nicht das Proton aus der Hydroxylgruppe von Lactat abstrahieren würde; Arg171 stabilisiert Lactat in der Bindetasche; O R1 + N NAD O + R1 NH2 N NAD + NH2 H Protein CH3 H C O O - C His119 + N O NH O His119 + NH2 H2N C C NH NH Arg171 Arg171 Protein + C O NH2 H2N N NH C O H Protein H CH3 H Protein 81 Alkoholdehydrogenase Mechanismus: Die Alkoholdehydrogenase ist ein Beispiel für eine hoch-stereospezifische Reaktion; sie katalysiert die Umsetzung der prochiralen Edukte Ethanol und NAD+ zu Acetaldehyd (Ethanal) und NADH+H+ Westheimer und Vennesland: Nachweis einer Stereospezifischen Reaktion (ADH katalysiert 100% stereospezifisch) 1.) Hinreaktion von Deuterium-markierten Ethanol mit dem prochiralen NAD+ mittels YADH (Hefe-Alokohol-Dehydrogenase); es entsteht chirales NADD+H+ (pro-R Transfer hat stattgefunden; kein racemisches Gemisch) 2.) Rückreaktion von NADD+H+ mit Deuterium-freien Ethanal mittels YADH; es entsteht ausschließlich chirales Deuterium-markiertes Ethanol; dies bedeutet das selektiv nur Deuterium vom NADD+H+ auf Ethanal übertragen wurde 3.) Achirales Deuterium-markiertes Ethanal mit NADH+H+ reagiert zu NAD+ und chiralem Ethanol; kein plan xD 4.) Kein plan xD 82 Lysozym: Lysozym (in Speichel, Tränen, Schmalz, Schleimhäuten etc. enthalten; 129AS, 14,4kD) spaltet die β-1,4-glykosidischen Bindungen zwischen N-Acetyl-Muraminsäure und N-AcetylGlucosamin und wirkt somit bakteriolytisch; dient der Abwehr bakterieller Infektionen; Die Wechselzahl von Lysozym ist sehr gering und wird nochmals reduziert, wenn NAG auf NAG folgt, anstelle von NAM; mit zunehmender Länge der Kette steigt die Wechselzahl; es sind 6 Glieder notwendig, damit es zur effizientesten Spaltung kommt; das Peptidoglykangerüst zieht sich wie ein Faden durch das Enzym; Wenn man ein Bakterium in isotonische Lösung gibt und diese anschließend mit Lysozym versetzt, bleibt ein überlebensfähiger Sphäroblast zurück (dieser besteht nur aus der Membran, ist aber auch ohne Zellwand stabil); „Phillips-Mechanismus“ (Lysozym) Zunächst wird der Ring D in eine Halbsessel Konformation verdreht, woraufhin ein Glutaminsäurerest des Proteins ein Proton auf die glykosidische Bindung überträgt, wodurch sie gespalten wird; Die Spaltung bringt ein Carbokation (im GG zum Oxoniumion) hervor, das von einem Aspartatrest stabilisiert wird; der Ring E wird im zweiten Schritt aus dem aktiven Zentrum entlassen, es gelangt H2O in die Bindetasche, das nukleophil an das Carbokation angreifen kann und zugleich ein Proton zur Regeneration von Glutaminsäure liefert; so wird Ring D zum neuen reduzierten Ende der Peptidoglykankette; Nach neuesten Erkenntnissen tritt darüber hinaus ein kurzzeitiges kovalentes Intermediat zwischen Carbokation und Protein auf; 83 Protein O Asp52 C O O H3C - C C O O O 1 O D OH - O CH2OH CH2OH R O H E O +H2O O R O NH H C O 2 2 -R EOH NH O C O C H3C Glu35 Asp52 Protein Protein C CH2OH R O O O O C O O - O 1 + D OH R Asp52 C CH2OH - O 1 CH3 Protein OH D OH H H NH O C H3C O H O H O - O C Glu35 NH H C O H3C O C Glu35 Protein Protein Beweise des Phillips-Mechanismus: - Spaltmuster und Substratspezifität: Zugabe von NAG6 und H2O; Spaltung in NAG4 und NAG2; keine weitere Spaltung, da Affinität des Enzyms zu gering; eine HPLC des Produktgemischs ist der Beweis dafür, dass nur bis zum Tetrasaccharid gespalten wird und jeweils nur Disaccharide abgespalten werden - Übergangsanaloga: das Lacton-Analoga zum Übergangszustand ist kompetetetiver Inhibitor des Enzyms; dies beweist, dass es sich um einen planaren ÜZ handelt (gegeben durch Mesomeres GG zwischen Oxoniumion und Carbokation) - pH-Abhängigkeit der Aktivität: das pH Optimum liegt bei ca. pH=5, was beweist, dass Aspartat und / oder Glutamat am Mechanismus beteiligt sind; - Substrat-Enzym-Cokristalle: Während der Substrat-Umsetzung kann die Struktur des Komplexes „eingefroren“ und somit analysiert werden; 84 B.) Kovalente Katalyse Bei der kovalenten Katalyse wird die Reaktionsgeschwindigkeit durch die vorübergehende Bildung eines kovalent gebundenen, häufig tetraedrischen Zwischnprodukts erhöht; In den meisten Fällen entsteht diese Bindung durch die nukleophile Reaktion zwischen Katalysator und Substrat; Die kovalente Katalyse lässt sich in drei Schritte gliedern: Erstens: zwischen Katalysator und Substrat findet eine nukleophile Reaktion statt, die zur Ausbildung einer kovalenten Bindung führt; Zweitens: Entfernen der Elektronen aus dem Reaktionszentrum durch einen elektrophilen Katalysator; Drittens: Durch eine Spaltung der kovalenten Bindung wird der Katalysator wieder frei gesetzt (Eliminierung); durch kovalentem Katalysator (z.B. PLP oder TPP in Assoziation mit ihren Apoenzymen) wird die Reaktion erheblich erleichtert und somit beschleunigt Das kovalente Intermediat kann mittels Reduktion durch NaBH4 bewiesen werden; so wird das Imin zum Amin reduziert, wodurch die Katalyse zum stoppen kommt; zu funktionellen Gruppen mit hoher Nukleophilie gehören in Proteinen die Aminogruppe von Lys, die Imidazolgruppe von His, die Thiolgruppe von Cys und die Hydroxylgruppe des Ser; Histidin-Decarboxylase Mechanismus: Pyridin sorgt für den Elektronenabfluss, wodurch die Einfachbindung zur Carboxylgruppe destabilisiert und die Decarboxylierung begünstigt wird; das Intermediat kann durch transiente Vergiftung nachgewiesen werden; Das Produkt dieser Reaktion ist Histamin; es wirkt im Organismus als Gewebshormon und Neurotransmitter, spielt eine zentrale Rolle bei allergischen Reaktionen und ist am Immunsystem beteiligt; 85 O + O O - OH P O - O O Iminbildung C + PLP NH HC CH3 N H + CH -H2O / 2H CH N H O N + C O - NH O N O O - C O H CH N H +H2O / 2H Histamin + O CH2 O H O - OH P - O H2C + C NH HC + CH3 CH3 H3N O N H -CO 2 + Retro-Iminbildung O - Schiff-Base OH + O C O H N H OH P - CH CH N H O - C H2C CH P + NH C H2C H2C + CH CH + O Histidin C H3N H - N H CH3 + N H CH Fructose-1,6-bisphosphat-Aldolase (Aldolase A) Mechanismus: Dieses Enzym bildet ein kovalentes Intermediat mit einem Lysinrest, um das Substrat zu fixieren und macht anschließend eine Säure-Base Katalyse mittels Tyrosin; 86 Fructose-1,6-bp Protein H + N HC O (CH2)4 C H2C OPO 3 C C H H C OH H C OH Lys H2C O + OPO 3 (CH2)4 NH O C O H C OH H2C Protein OPO 3 H H O C O H C OH O OPO 3 O + - 2- Protein protonierte Schiff-Base H2C (CH2)4 C + NH HO OPO 3 2- C CH2 O - Protein Protein 2- H2C NH2 OPO 3 N Protein H O H 2- HC CH C CH H Protein Tyr N + 2- C HO Protein Protein (CH2)4 OPO 3 2- H2C -H C + GA-3p + NH (CH2)4 C HO HC H2C - H2C C H N HC O O Protein + H2O H - H2O N HC +H Enamin Protein H protonierte Schiff-Base 2- O NH2 HO Protein Protein OPO 3 DHAP C H2C O 2- OH - Protein Protein C.) Metallionen-Katalyse Metalle mit katalytischer Funktion: Fe2+, Fe3+, Cu2+, Zn2+, Mn2+, Co3+ Metalle mit Aktivierungsfunktion: Na+, K+, Mg2+, Ca2+ Funktionen: Substratbindung zur optimalen Orientierung, Redox-Katalyse, elektrostatische Stabilisierung oder Abschirmung von negativer Ladung; Wasserionisierung (nukleophile Katalyse) Carboanhydrase Mechanismus: Das Zn2+ Ion im katalytischen Zentrum wird von drei Histidinen tetraedrisch koordiniert, während die vierte Koordinationsstelle in Lösung von Wasser besetzt ist; Die Histidine und das Molekül Wasser werden in ihrer Position durch H-Brücken stabilisiert; Durch die Koordination von Wasser an Zink wird es stark polarisiert, weshalb es als OH- Ion sehr nukleophil ist und somit das Elektrophil CO2 angreifen kann; Die Wechselzahl der Carboanhydrase beträgt 106 s-1; es handelt sich um eine sehr schnelle Reaktion, die mittels Enzym noch sehr viel schneller gemacht wird! 87 O C H HN O His96 N Hydrogencarbonat O + H2O - N Protein N HN His94 O 2+ Zn Protein Protein - HO O H - + 2+ Zn Protein Protein N N NH C H HN O HN N NH His119 Protein Siehe auch Carboxypeptidase als Metallionenkatalyse bei Proteasen D.) weitere Beispiele Glutathion-Reduktase Mechanismus Gluthation ist ein Tripeptid und kann in der oxidierten Form GSSG und der reduzierten Form GSH vorliegen; Die Gluthation-Reduktase katalysiert die oxidative Bildung der Disulfidbrücke zwischen zwei GSH Molekülen unter NADPH+H+ Verbrauch; GSH dient als universelles Reduktionsmittel im Stoffwechsel; Funktionen von GSH: Intrazelluläres Reduktionsmittel (Erythrozyten, Cytosol), Transport von Aminosäuren (Neurophysiologie), Antioxidanz (Schutz vor Oxidativem Stress, Entgiftung von Wasserstoffperoxid, Neurobiologie, CFTR), Coenzym verschiedener Enzymkatalysierter Reduktionen, Detoxifizierung (Glutathion bindet an toxische Moleküle und organischen Müll, wodurch dieser zum Abbau markiert wird); 88 GSH / GSSG = 100 innerhalb der Zelle Die Glutathion-Reduktase gehört zur Familie der Disulfid-Oxidoreduktasen; zu dieser Familie zählen auch 1.) Dihydrolipoyl-Dehydrogenase (Pyruvatdehydrogenase) 2.) Quecksilber-Reduktaste (reduziert Quecksilber Ionen zu weniger giftigem metallischem Hg) 3.) Trypanothion-Reduktase (Malaria-Erreger hat kein Gluthation; Trypanothion ist ein ähnliches Molekül, das dieselben Funktionen hat) 4.) NADH Peroxidase Sie weist zwei identische Untereinheiten auf: Enzymmechanismus: Im ersten Teil werden die Elektronen von NADPH+H+ auf das Enzym übertragen; im zweiten Schritt werden die Elektronen vom Enzym mittels Säure-BaseKatalyse auf GSSG übertragen, das über ein Cys kovalent gebunden ist; 89 1.) zunächst überträgt NADPH+H+ seine Elektronen auf FAD 2.) von dort aus werden sie an zwei Cys-Seitenketten weitergegeben, wodurch die Disulfidbrücke im Protein zwischen Cys58 und Cys63 reduziert wird; 3.) His467 stabilisiert die Thiolgruppe des Cys58, während S- des Cys63 von FAD stabilisiert wird; 4.) Erst jetzt verlässt NADP+ die Bindetasche und GSSG gelangt ins aktive Zentrum 5.) ein Schwefelatom der Disulfidbrücke von GSSG wird nun nukleophil von der Thiolgruppe des Cys58 angegriffen, wodurch eine gemischte Disulfidbrücke zwischen Protein und einem Teil von GSSG entsteht und das frei werdende Proton der Thiolgruppe von His467 abstrahiert wird; 6.) der andere Teil von GSSG liegt kurzzeitig als GS- vor und abstrahiert das Proton von His467, wodurch Histidin wieder deprotoniert und GSH frei wird; die noch existierende gemischte Disulfidbrücke wird unter Bildung von GS- gespalten, indem S- von Cys63 nukleophil an das Schwefelatom von Cys53 angreift und erneut das intramolekulare Disulfid bildet; 7.) das so frei werdende GS- abstrahiert ein Proton aus dem Medium; 90 8.) Regulatorische Mechanismen Enzym-Regulation: Aspartat-Transcarbamoylase (ATCase) Dieses Enzym katalysiert bei der Biosynthese der Pyrimidine den ersten Schritt (Kondensation von Aspartat und Carbamoylphosphat zu N-Carbamoylaspartat und Orthophosphat); Am Ende der Biosynthese stehen die Pyrimidinnukleotide (Cytidintriphosphat, CTP); CTP ist das Endprodukt und zugleich allosterischer Inhibitor der ATCase; dies ist ein Beispiel für eine Rückkopplungs- und Endprodukthemmung (feedback inhibition); CTP ist ein allosterischer Inhibitor (passt nicht ins aktive Zentrum; bindet an allosterisches Zentrum); ATP dagegen ist ein allosterischer Aktivator; Die ATCase ist ein C6R6 Oligomer (6 regulatorische und 6 katalytische Untereinheiten auf getrennten Polypeptidketten) mit 300kD; Die Quartiärstruktur der ATCase kann zwei unterschiedliche Formen annehmen; die T Form dominiert, wenn kein Substrat, kein ATP und viel CTP vorhanden ist , die R Form dominiert, wenn viel Substrat, viel ATP und kein CTP vorhanden ist; Die T-Form bewirkt die Erniedrigung der Aktivität, Die R-Form bewirkt die Erhöhung der Aktivität; Es herrscht also Konkurrenz zwischen ATP und CTP – die allosterische Bindung wirkt sich immer auf alle Untereinheiten aus (konzertierter Mechanismus); die regulatorischen Domänen kontrollieren somit die katalytischen Domänen; O NH2 O - ATCase C + C OPO 3 O + H3N 2- Carbamoylphosphat H2PO4 C O O - C 6 enzymatische NH CH CH2 Reaktionen C - O O N-CarbamoylAspartat O O CH2 - - H P O O - P O O - O P O - N O O O O NH2 N CTP - Aspartat Cytidintriphosphat C O CH CH2 O O H2N H H H OH OH 91 Proteasen Proteasen = Peptidasen: man unterscheidet Exopeptidasen (Aminopeptidasen spalten am Nterminus und Carboxypeptidasen spalten am C terminus) von Endopeptidasen (spalten im znetrum der Kette); Peptidasen wirken als nano-Rasierapparate: sie greifen nur freie Peptide oder „Überhänge“ an, aber keine gefalteten Proteine, da spezifischer Terminus verborgen liegt C terminale Sequenzanalyse: im Massenspektrometer wird ein Anstieg der ASKonzentrationen in bestimmter Reihenfolge detektiert; Rückschluss auf AS-Sequenz möglich; Es gibt 5 große Proteasefamilien: Familie Serin Proteasen Threonin Proteasen Zink Proteasen Beispiele Chymotrypsin, Trypsin, Elastase Proteasome, β-Lactamase Cyrboxypeptidase A Cystein Proteasen Aspartat Proteasen Papain, Cathepsine, Bromelaine Pepsin, Renin, Chymosin, HIV-1-Protease Inhibitoren DFP = Diisopropyl-Fluorophosphat Lactacystin EDTA = EthylendiaminTetraessigsäure Jodacetat Pepstatin Bei den letzten drei Protease klassen besitzt das active zentrum merkmale, welche die Aktivierung von einer nukleophilen Gruppe sowie die Polarisierung der Peptidcarbonylgruppe und die anschließende Stabilisierung eines tetraedrischen zwischenprodukts ermöglichen 1.) Zink Proteasen Inhibitor: Alle Puffer enthalten EDTA; EDTA hat eine hohe Komplexbildungskonstante und komplexiert somit sehr effizient zweiwertige Metallionen; so wird die Aktivität von Metalloproteasen verhindert; bei der Koordination umhüllt das Molekül das Metallion und komplexiert es; OH HO M O O N O O O O - N HO OH 2+ N - O O O - O N O O - EDTA 92 Carboxypeptidase A Carboxypeptidase A ist eine Zink-Protease; sie ist ein Beispiel für den induced fit Mechanismus: Glu270, Tyr248 und Arg145 verschließen den C terminus im aktiven Zentrum; Zink-gebundenes Wasser verhält sich wie OH-; es greift nukleophil an die Carbonylgruppe an, woraufhin die Peptidbindung zum Stickstoff hin gespalten wird; das Stickstoffatom abstrahiert ein Proton von Glu270; Der Übergangszustand ist tetraedrisch, und wird durch Arg127 stabilisiert; die Bindungsumlagerungen verlaufen alle konzertiert; nach der Reaktion kann das Substrat das katalytische Zentrum nur über den induced fit verlassen; es sind weitere AS beteiligt, welche stabilisierend auf einzelne Bereiche des Peptids wirken; Glu270 wird durch das nächste Molekül Wasser regeneriert; Protein NH Arg127 C His196 NH2 + R H2N O Glu72 C CH2 N 2+ Zn H O His69 H COO - C terminus H H R O C + OH - O CH2 N COO - Glycin H neuer C-terminus O Protein Glu270 2.) Serin Proteasen Zu den wichtigen Serin-Proteasen zählen Chymotrypsin, Trypsin und Elastase; sie haben alle etwa 240 AS und 40% Identität der Primärstruktur; die 3D Struktur ist sehr ähnlich; sie weisen alle denselben katalytischen Mechanismus auf; 93 Die katalytische Triade Ser-His-Asp Das aktive Zentrum von Chymotrypsin liegt in einer Spalte an der Oberfläche des Enzyms und enthält Ser195; Die Seitenkette von Ser195 ist über eine H-Brücke mit dem Imidazolring von His57 verbunden; Die NH Gruppe des Imidazolrings ist wiederum über eine H-Brücke mit der Carboxylatgruppe von Asp102 verbunden; Diese Anordnung der AS Reste wird als katalytische Triade bezeichnet; durch sie wird eine stark erhöhte Reaktivität (Nukleophilie) von Ser195 ermöglicht; Histidin polarisiert die OH Gruppe des Ser195, wodurch das Gleichgewicht zur Deprotonierung hin verschoben wird; das entstehende Alkoholat ist wesentlich nukleophiler als das Alkohol; der Aspartatrest stützt die Orientierung des Histidins und macht es zu einem besseren Protonenakzeptor; Mechanismus: Säure-Base Katalyse (am Besipiel von Chymotrypsin) Zunächst erfolgt die Substratbindung, worauf der nukleophile Angriff der Alkoholatgruppe von Ser195 an das Carbonyl C des Substrats folgt; der nukleophile Angriff verändert die Umgebung des Carbonyl-C von trigonal planarer zu tetraedrischer Struktur; instabiles, tetraedrisches ZP enthält negative Ladung am Carbonyl-Sauerstoff; diese Ladung wird durch NH-Gruppen eines Proteinbereichs stabilisiert, der als Oxyaniontasche bezeichnet wird; sie bewirkt die Herabseknung der Aktivierungsenergie und somit die Beschleunigung der Reaktion; Nun zerfällt das ZP, woraufhin das Acyl-Enzym entsteht; dieser Schritt erfolgt durch die Übertragung eines Protons vom positiv geladenen Histidin Rest auf die Aminogruppe, die bei der Spaltung der Peptidbindung entsteht; Die Aminogruppe kann sich vom Enzym lösen; Nun gelangt Wasser in die Bindetasche: Histidin abstrahiert ein Proton, wodurch OHnukleophil an den Ester des Acyl-Enzyms angreifen kann; es folgt die Hydrolyse des Esters, aus der eine Carbonsäure und der Alkoholat-Rest von Ser195 hervorgehen; Die Bevorzugung von großen hydrophoben Resten wird durch die S1 Tasche erklärt: es handelt sich um eine tiefe, hydrophobe Tasche, in die die ungeladenen Seitenketten von Phe und Trp hineinpassen; Die Bindung einer geeigneten Seitenkette in S1 positioniert die anschließende Peptidbindung im aktiven Zentrum, sodass die Spaltung erfolgen kann; Diese Tasche ist für die Substratspezifität verantwortlich; 94 His57 Asp102 Protein O C Protein Protein N H N H - Ser195 O + H N O Protein R 1 R N Protein 2 1 R tetraedrischer ÜZ C Protein H R Michaelis Komplex O N Peptid N 2 C H O - O H Protein N H N H O N R 1 H Protein R N + H2O O H O Acyl-Enzym-Intermediat R O Protein Protein H N + O N R H N N Protein H O 2 C H Amin H Protein - NR HH C H O N 1 2 2 C O H - O R Carbonsäure O H Protein 2 C O tetraedrischer ÜZ Der erste nukleophile Angriff und die Eliminierung des Amins verlaufen schnell; der nukleophile Angriff von Wasser, um das Acyl-Enzym Intermediat zu spalten verläuft langsam; 95 Abbildung: Trypsin (Links markiert: katalytische Triade / Rechts markiert: Bereich für Substratspezifität) Substratspezifität und S1 Taschen von Chymotrypsin, Trypsin und Elastase Serin Proteasen binden bevorzugt den Übergangszustand; linke Abbildung zeigt trigonal planare Anordnung; rechte Abbildung zeigt tetraedrischen ÜZ; oben links: Oxyaniontasche; unten links: S1 Tasche; 96 Trypsin und Elastase sind zu Chymotrypsin homolog, unterscheiden sich aber in der Substratspezifität; Die Ursache der Spezifität liegt in der Struktur der S1 Taschen Trypsin spaltet bei Peptidbindungen nach AS mit langen, positiv geladenen Seitenketten (Arg und Lys); am Grund der S1 Tasche liegt der negativ geladene Asp189 Rest; (Spaltung der Peptidbindung nach jedem Arginin und Lysin tryptischer Verdau liefert Peptidfingerprint); Elastase spaltet bei Peptidbindungen nach AS mit kleinen Seitenketten (Ala, Ser, Gly etc.); am oberen Ende der Tasche befinden sich Val190 und Val216 (alternativ auch Thr bei Enzymen mit dieser Substratspezifität), welche die Öffnung der Tasche verschließen; Andere Proteasen (neben Chymotrypsin mit der großen, hydrophoben S1 Tasche, die ein Serinrest am Grund aufweist) zeigen komplexere Spezifitätsmuster, da sie für die Erkennung weitererer AS Reste im Substrat an ihren Oberflächen zusätzliche Taschen aufweisen; S2 (S3) Tasche stabilisiert zweite (dritte) AS im Peptid (gezählt wird ab der zu spaltenden Peptidbindung); je nach Beschaffenheit der unterschiedlichen Taschen resultiert andere Spezifität; Konvergente Evolution: Der den Ser-Proteasen zugrunde liegende katalytische Mechanismus wurde mindestens dreimal unabhängig voneinander erfunden; Chymotrypsin, Trypsin und Elastase haben eine sehr ähnliche Struktur, weshalb sie durch Genduplikation eines Vorläufers entstanden sind; Protease A aus einem Prokaryont ist auch sehr ähnlich, weshalb das Ur-Trypsin schon vor der Trennung von Eukaryonten und Prokaryonten entstanden ist; Aber: Das Vorhandensein von sehr ähnlicher aktiver Zentren in verschiedenen Proteinfamilien ist Resultat der konvergenten Evolution; es ist ein ineinander Überführen der Enzyme durch Mutation / Evolution nicht möglich; sie sind divergent! Beispiel: Die thermostabile Protease Subtilisin (Vorkommen: Bakterien); das aktive Zentrum dieses Enzyms enthält die katalytische Triade und die Oxyaniontasche; eine der NH Gruppen der Oxyaniontasche gehört jedoch zur Seitenkette von Asp und nicht zum Peptidrückgrat! Beispiel: Die Carboxypeptidase II (Vorkommen: Weizen); keine Strukturähnlichkeit zu Chymotrypsin oder Subtilisin; 97 Proenzym Konzept: Inaktive Pro-Form: Alle Proteasen werden in einer inaktiven pro-Form synthetisiert! Trypsin ist ein Verdauungsenzym, das im Dünndarm Peptide spaltet; Trypsin wird als inaktives Trypsinogen vom Pankreas sekretiert; das Darmenzym Enteropeptidase, das an das Darmepithel gebunden ist, steuert die Umwandlung der Vorstufe Trypsinogen zu Trypsin, indem es das Hexapeptid Val-Asp4-Lys abspaltet; Trypsin aktiviert sich selbst (Autokatalyse; positive Rückkopplung) und wandelt Chymotrypsinogen, Proelastase und weitere Proenzyme in deren aktive Formen um; Chymotrypsin und Elastase werden auch im Pankreas in ihrer pro-Form gebildet; Pro-Formen nennt man allgemein Cymogene; Irreversibler Inhibitor: DFP H3C CH3 F Ser195 Protein H3C O H H3C O O P O O CH3 Protein CH3 Diisopropyl-Fluorophosphat DFP P O O + HF O CH3 H3C 3.) Cystein Proteasen Die nukleophile Gruppe ist ein Cysteinrest, der von einem Histidin aktiviert wird und in einer sehr ähnlichen Weise wie der Serinrest bei den Serinproteasen die Peptidbindung nukleophil angreift; dieser Mechanismus ist zwei mal während der Evolution unabhängig voneinander erfunden worden; es handelt sich um dieselbe katalytische Triade, nur mit Cys statt Ser! Irreversibler Inhibitor: Jodacetat modifiziert SH Gruppe des Enzyms, was durch Mercaptane aufgehoben werden kann; 98 4.) Aspartat Proteasen Das Merkmal dieses Protease-Typs ist ein Paar von Asparaginsäure-Resten im aktiven Zentrum, die zusammenwirken und es so einem Wassermolekül ermöglichen eine Peptidbindung nukleophil anzugreifen; der eine deprotonierte Asparaginsäurerest aktiviert H2O, indem er dessen Gleichgewicht zur Deprotonierung verschiebt; der andere protonierte Asparaginsäurerest aktiviert die Peptidcarbonylgruppe und erhöht so deren Empfindlichkeit für den nukleophilen Angriff; Beispiel: HIV1 Protease Die HIV Protease (Homodimer, je 99AS) ist in der viralen RNA codiert (bereits im Capsid enthalten) und zeigt zweizählige Symmetrie; sie spielt bei der Bildung neuer Viren eine entscheidende Rolle, da sie das Zerschneiden von Vorläufer-Multidomänen-Proteinen katalysiert; erst die Bruchstücke dieser Vorläufer-Proteine werden benötigt, damit neue Viruspartikel zusammen gesetzt werden können; dieses Enzym spaltet vor allem bei hydrophoben und aromatischen AS, aber auch Proline und Asparagine; jedes Monomer der HIV Protease besitzt ein Aspartet des aktiven Zentrums; andere AspartatProteasen (z.B.Pepsin) bestehen aus einer Polypeptidkette ( die Dimere sind im Verlauf der Evolution fusioniert); HIV-Protease Dimer Pepsin Monomer HIV-Protease-Inhibitoren: Nach Blockierung der HIV-Protease können die Multidomänen-Proteine nicht mehr in ihre aktiven Formen gespalten werden, weshalb HIV nicht mehr infektiös ist; es muss jedoch darauf geachtet werden, dass der Inhibitor spezifisch für das zu hemmende Enzym ist, da keine weiteren Enzyme im Körper gehemmt werden dürfen; Crixivan ist ein Inhibitor der HIV-Protease; er wurde auf Grundlage eines Alkoholmoleküls entwickelt, welches das tetraedrische Zwischenprodukt nachbildet; Röntgenstrukturanalyse zeigt, dass Crixivan im Komplex mit dem Enzym eine Konformation annimmt, die ebenso wie das Enzym eine zweizählige Symmetrie zeigt; Das aktive Zentrum der HIV Protease wird von zwei Laschen bedeckt, die sich über dem gebundenen Inhibitor zusammenfalten; Die Hydroxylgruppe des zentralen Alkohols tritt mit den beiden Asparaginsäure-Resten des aktiven Zentrums in wechselwirkung; 99 Renin / Pepsin Inhibitor: Pepstatin Wechselwirkung, aber keine kovalente Bindung, zwischen katalytischen Aspartat-Resten und Pepstatin; oben: Abbildung HIV-1-Protease mit Pepstatin Inhibitor (unten) 5.) Threonin Protease Proteasom: Proteasom ist ein 1700kD schwerer proteinkomplex, der im Cytoplasma und im Zellkern lokalisiert ist; eine 20S und zwei 19S Untereinheiten, die jeweils aus mehreren Proteinen zusammengesetzt sind; 20S Untereinheit ist ein hohler Zylinder und wirkt als multikatalytische protease (proteolytische Aktivität an der Innenwand); Die 19S Komplexe sitzen als Deckel auf den beiden Öffnungen des 20S Komplexes; sie erkennen und entfalten durch Ubiquitin für den Abbau markierte Proteine 100 Inhibitor: Lactacystin bildet ein Ester Addukt mit dem N-terminalem Threonin des Proteasomes β-Lactamase: es handelt sich um ein Enzym, das von Bakterien gebildet wird, um den β-Lactam Ring ( Resistenz gegen β-Lactam Antibiotika, u.a. Penicillin) zu hydrolysieren; es sind über 340 verschiedene Varianten bekannt; z.B. Klasse B: Metallo-β-Lactamase (Mechanismus abgebildet) 101 Membranproteine 1.) Porine Porine sind porenformende Transmembranproteine in der äußeren membranen von gramnegativen Bakterien und Mitochondrien; sie dienen dem Stoffaustausch durch die Membran und ermöglichen die freie Diffusion von Molekülen bis 600 Da; Porine werden auch VDAC (voltage dependend anion channel) genannt, da sie den geregelten Durchtritt von anionischen Molekülen (Chlorid, Phosphat, Nukleotide etc.) erlaubt; sie bestehen aus einer Kette von 300 bis 420 AS, die zu einem 16 oder 18 strängigem β Fass gefaltet sind (ein Strang ist ein β-Faltblatt); Die Wandung der Pore besitzt die Stärke einer AS (sehr dünn); Im inneren des Porins befindet sich eine Engstelle mit einigen ionisierbaren AS, an der die Durchlasseigenschaften der Pore festgelegt sind; 2.) Struktur von membranproteinen Die Transmembrandomäne weist immer eine stark hydrophobe Sequenz auf; diese kann ermittelt werden, indem die freie Enthalpie für die Solvatation in Wasser gemessen wird; das größte ΔG kennzeichnet den hydrophobsten bereich, also die Transmembrandomäne; 102 3.) Analyse von Membranproteinen Man untersucht die Funktion eines spezifischen Membranproteins, indem man das entsprechende Protein extrahiert, reinigt und anschließend wieder in eine reine Phospholipiddoppelschicht (von Liposomen) integriert; Zweite Möglichkeit: das für ein Transportprotein codierende Gen sehr stark in Zellen zu exprimieren, die dieses Protein normalerweise nicht herstellen; alle unterschiede bezüglich des Transports bestimmter Substanzen zwischen transfizierter und unbehandelter Zelle beruhen dann auf diesen Transporter Gefrierbruchätzung 103 Coenzyme, prosthetische Gruppen und Cofaktoren - - - - - Coenzyme sind organische Moleküle, die eine transiente Assoziation mit dem Enzym eingehen; Cofaktoren sind z.B. Metallionen; eine prosthetische Gruppe ist ein permanent assoziiertes Coenzym Coenzyme, Cofaktoren und prosthetische Gruppen werden im Verlaufe einer enzymkatalysierten Reaktion chemisch verändert und müssen regeneriert werden; für prosthetische gruppen in einer separaten Phase der enzymatischen Reaktion; für Coenzyme ist eine Regeneration durch ein anderes Enzym möglich; Viele Organismen können Vorläufer für essentielle Coenzyme nicht synthetisieren; deshalb ist die Aufnahme der Vitamine über die Nahrung notwendig Fettlösliche Vitamine sind nicht Bestandteile von Coenzymen, obwohl sie für höhere Organismen essentiell sind (Vitamin A fürs Auge und Vitamin D für die Knochenmineralisierung) Unsere Vorfahren konnten möglicherweise verschiedene Vitamine synthetisieren; da viele Vitamine im Überfluss in Nahrung vorhanden sind oder durch Bakterien im Verdauungstrakt synthetisiert werden, konnte durch Weglassen der Vitaminsynthese Energie gespart werden, weshalb sie in der Evolution verloren gegangen ist; Probiotische nahrungsmittel: enthalten modifizierte Bakterien, die vermehrt Vitamine oder andere Substanzen produzieren und sich im Darm aufhalten (funktionelle Lebensmittel, isotonische Getränke) A.) Reduktionspotentiale und redoxreaktive Coenzyme Elektronenübertragungspotentiale ΔE können in bestimmten Stoffwechselwegen in Phosphorylgruppenübertragungspotentiale ΔG umgewandelt werden. 1.) NADH / NADPH - - NADH und NADPH sind die wichtigsten Elektronencarrier. Diese Coenzyme übertragen die Elektronen in Form von Hydrid Ionen (H-). Die aktive Einheit dieser Coenzyme ist der Nikotinamidring. Sie wirken nur in Zusammenarbeit mit Enzymen. NAD+ bindende Proteine gehören in die Klasse der Dehydrogenasen. NAD+ dient vorwiegend als Elektronenakzeptor bei oxidativen katabolen prozessen. NADPH+H+ ist vorwiegend Elektronendonor bei reduktiven Biosynthesen O O NAD NH2 reduzierte Form + NH2 NADH+H+ Nikotinamidadenindinukleotid + N NH2 N N HO N OH O O P O - oxidierte Form O O O P N O R N - O O O R O P O OH OH NADP O - + - bzw. + NADPH+H Nikotinamidadenindinukleotidphosphat 104 O NH2 N Nikotinamid Vitamin B3 Enzyme: 1.) Bestandteil von Oxidoreduktasen 2.) Flavine - - Flavine enthalten als aktive Einheit einen Alloxazinring, der 2 Wasserstoffatome überträgt (es ist möglich nur ein Elektron oder 2 auf einmal zu übertragen; NAD+ kann nur immer 2 gleichzeitig aufnehmen). Die wichtigsten Flavine sind FAD und FMN. Beide sind kovalent am Enzym gebunden (prosthetische Gruppe). Flavin-bindende Proteine weisen eine gelbe Farbe auf und werden als Flavoproteine bezeichnet. Es handelt sich NICHT um Nukleotide, da der Alloxazinring nicht an einem Zuckerrest, sondern an einem Alkohol (Ribitol) hängt. Oxidierte Form: Chinon / reduzierte Form: Dihydrochinon oder Chinol Flavine können sowohl ein, als auch zwei Elektronen aufnehmen; FADH ist die radikalische Semichinon Form CH2OH H OH H OH H OH CH2OH D-Ribitol HOH2C H N O N NH OH OH HO Riboflavin Vitamin B2 CH2 H3C N H3C N O N N Isoalloxazin O NH O 105 NH2 N Flavinadenindinukleotid O O - - P O O H H CH2 N - O H H OH OH H OH OH H O N H NH H3C O O OH H N - P H OH H3C O P H FMN OH HO N O Flavinmononukleotid O O FAD O N H3C N H3C N O N N O NH O oxidierte Form H3C H3C H N N H O N NH ° O radikalische Semichinonform H3C H N H N O NH H3C N H O reduzierte Form Enzyme: FMN ist Bestandteil von Komplex I der Atmungskette FAD ist Cofaktor von Flavoproteinen, die folgende Reaktionen katalysieren 1.) Succinat-Dehydrogenase im Citratcyclus bzw. Koplex II der Atmungskette 2.) Acyl-CoA-Dehydrogenase (erster Schritt der β-Oxidation) 3.) Transhydrogenierungen mittels Dihydrolipoyl-Dehydrogenase der Dehydrogenase Komplexe (Pyruvat-Dehyd / α-Ketoglutarat-Dehyd) 4.) Cholesterin-Biosynthese: 3-iPPP-Isomerase und Squalen-Epoxidase 5.) Oxidative Desaminierung aromatischer proteinogener L-AS und ihrer Derivate (Serotonin, Histamin) durch Monooxidasen 106 3.) Ubichinone (speziell CoQ) - Ubichinone: z.B. Coenzym Q (wirkt an Redoxreaktionen in Mitochondrien [Atmungskette] und Chloroplasten [Photosynthese] mit; Ist überträger zwischen KI bzw. KII und KIII der Atmungskette; ist primärer Elektronenakzeptor im PSII) CoQ ist Chinonderivat mit lipohpiler Isoprenoid-Seitenkette ( Verankerung in Membran); die Anzahl der Isoprenoid-Einheiten ist artspezifisch (Säuger haben meist Q10) Überträgt 2H (Shuttle für 2 Elektronen: 2 Elektronentransporter); kann in drei Oxidationszuständen vorliegen: voll oxidiert (Q; Ubichinon); Semichinonform (QH; kann leicht Proton abgeben, so dass Semichinonradikalanion Q- entsteht); voll reduziert (QH2; Ubichinol) - - O H3C O OH Coenzym Q O CH3 H H3C O R2 R4 R3 10 O O p-Benzochinon R1 OH H3C oxidierte Form Ubichinon reduzierte Form Ubichinol CoQH2 CoQ Ähnliche Strukturen: CH3 O CH3 HO CH3 R O Vitamin K H3C CH3 CH3 O CH3 CH3 CH3 Vitamin E α-Tocopherol Kein Ubichinon! (strukturell aber ähnlich zu CoQ) 4.) Häm - - - Die Hämbiosynthese findet im Knochenmark statt; sie verläuft über zahlreiche Porphyrinvorstufen; Häm besteht aus vier pyrrolringen, die über 4 Methingruppen kreisförmig verlnüpft sind; Cytochrome: elektronenübertragende Proteine, die Häm als prosthetische Gruppe tragen (über Cys mittels R gebunden); dienen zum Schutz der Häm-Gruppe und sorgen mittels Bindungsstellen dafür, dass Elektronen nur gezielt aufgenommen und abgegeben werden können (gerichteter Transport) Während des Elektronentransportes wechselt das Eisenatom im Häm zwischen dem reduzierten Ferro (Fe2+) und dem oxidierten Ferri (Fe3+) Zustand; Häm ist also Einelektronenüberträger 107 H3C HC C CH N H C C CH C HC HC N H Pyrrol HC C CH C C H N CH CH C CH C 2+ Fe N - - C C NH CH C C O OC CH3 N C CH C CH Porphyrin CH C O OC C C NH C CH N C C HC R3 C N R1 C C R2 Häm CH3 Enzyme: 1.) Hämoglobin, Myoglobin (beide Häm b) 2.) Katalase (Häm b) 3.) Atmungskette: Succinat-Dehydrogenase (Häm b) und Cytochrom-c-Oxidase (2Häm a) Häm a Häm b Häm a im Cytochrom 5.) Ascorbinsäure Überträgt 2H 6.) Eisen-Schwefel-Cluster - Eisenatome in diesen Komplexen wechseln zwischen den zuständen Fe3+ (oxidiert) und Fe2+ (reduziert); Einelektronenüberträger; die häufigsten Fe-S-Zentren sind 2Fe2S pder 4Fe-4S konfiguriert, wobei die Eisenatome an die Thiolgruppe von Cys oder an die Stickstoffatome von His gebunden sind; 108 Fe-S Zentrum im Rieske Protein Enzyme: 1.) Aconitase (Citratcyclus) 2.) ETF-Q-Oxidoreduktase 3.) 7.) Liponamid siehe Gruppenübertragende Coenzyme B.) Gruppenübertragende Coenzyme 1.) Nukleosidphosphate - in Phosphotransferasen, Nukleotidyltransferasen und Ligasen Bereits oben!! 2.) Coenzym A - - Coenzym A ist ein Acylgruppenüberträger in Acyltransferasen oder CoA-Transferasen (Acetyl-CoA und Acyl-CoA), wobei die aktive Einheit die Thiolgruppe ist, welche eine energiereiche Thioesterbindung knüpft. β Alanin und Pantoinsäure bilden Pantothensäure. Pantotensäure und Cysteamin bilden Pantethein. Phosphopantethein kommt ebenso im Enzym Fettsäure-Synthase vor. 109 NH2 HS NH2 H H OH N O O N N N O O O S P O HS R Cysteamin N H3C CH3 Coenzym A O O - P O O NH2 HO N - O O β -Alanin CH3 CoA O CH3 O P O Acetyl-CoA OH O - OH HO - OH O CH3 Pantoinsäure 3.) Biotin - Biotin in Carboxylasen; katalysiert eine Carbxylgruppen-Übertragung (bindet CO2 und überträgt es auf Zielmolekül) und ist über einen Lysinrest am Apoenzym gebunden O Biotin NH HN O O Vitamin B7 O OH S HO N R1 NH R2 Carboxy-BiotinC C Enzyme: 1.) Acetyl-CoA-Carboxylase (Fettsäuresynthese: Acetyl-CoA zu Malonyl-CoA) 2.) Pyruvat-Carboxylase (Gluconeogenese: Pyruvat zu Oxalacetat) 3.) Propionyl-CoA-Carboxylase (Fettsäure-Abbau, ungeradzahlig) 4.) Thiaminpyrophosphat - - Thiaminpyrophosphat: TPP in Decarboxylasen oder Transketolasen; katalysiert den Transfer von Aldehyden (genauer: dehydrierende Decarboxylierung von αKetosäuren) CH Gruppe im Thiazolring hat pKs Wert von 10, weshalb diese unter Bildung eines Carbanions ionisiert. Das Carbanion kann nun mit seinem freien Elektronenpaar nukleophil an Carbonyle (z.B. pyruvat Carbonylgruppe) angreifen (es kommt zur Addition des Carbonyls unter Bildung eines Alkohols) 110 NH2 N S Thiazol H3C CH3 + N N N S OH Thiamin Vitamin B1 R1 NH2 R2 + N HO - C S CH3 + N N O O R3 H3C CH3 N TPP Hydroxyethyl-TPP O S P O Thiaminpyrophosphat O - P O O - - Enzyme: 1.) Pyruvat-Dehydrogenase (Pyruvat zu Acetyl-CoA) 2.) α-Ketoglutarat-Dehydrogenase (Citratcyclus) 3.) Transketolase (Pentosephosphatweg) 5.) Liponamid 1. kann sowohl Elektronenüberträger (2H) als auch Acylgruppenüberträger sein S Liponsäure S S OH Liponamid S HS CH3 H S N O Lys O O Acetyl-Liponamid R Enzyme: 1.) Pyruvat-Dehydrogenase 6.) Pyridoxalphosphat 1. Pyridoxalphosphat PLP in Transaminasen (=Aminotransferasen) katalysiert den Transfer von Aminogruppen; 2. Enthält einen leicht basischen Pyridinring und eine leicht saure phenolische Hydroxylgruppe, weshalb Pyridoxalphosphatderivate in stabilen tautomeren Formen vorliegen: N protoniert und OH deprotoniert; 3. Die Aldehydgruppe ermöglicht die Bildung von intermediär kovalenten Schiff Basen mit Aminosäuresubstraten (externes Aldimin); innerhalb des Enzyms ist sie (wenn kein Substrat vorliegt) an die ε Aminogruppe eines Lysins gebunden (internes Aldimin); bei der Reaktion mit einem Substrat ersetzt die α Aminogruppe des Substrats die ε 111 Aminogruppe des Lys Restes im aktiven Zentrum; das so gebildete externe Aldimin bleibt jedoch durch nicht kovalente Kräfte am Enzym gebunden; 4. Die Reaktion verläuft über ein chinoides ZP, einem Ketimin und nach Hydrolyse zum Pyridoxaminphosphat (PMP) und einer α-Ketosäure; PLP wird regeneriert, indem eine α-Ketosäure als Edukt fungiert und dieselbe Reaktion rückwärts beschreitet, wodurch eine α-AS entsteht; H HO Gruppe: Pyridoxin Vitamin B6 H O OH HO OH HO N Pyridoxol H H2N CH3 OH HO N CH3 CH3 N Pyridoxamin Pyridoxal O O P O - O O H OH O PLP N Pyridoxalphosphat R1 HO - CH3 N H Pyridoxaminphosphat R Schiff'sche Base (Imin) Enzyme: 1.) Transaminasen (Alanin-, Aspartat- u.v.m.) 2.) Decarboxylierungen (L-DOPA Dopamin; Histidin Histamin; Phosphatidylserin Phosphatidylethanolamin u.v.m.) 3.) Erster Schritt der Häm-Biosynthese 4.) Glykogen-Phosphorylase (hier funktioniert PLP als Säure-Base-Katalysator durch die Phosphatgruppe) 7.) Tetrahydrofolat 1. Tetrahydrofolat: THF in C1 Transferasen katalysiert den Methylgruppen-Transfer; besonders wichtig im Aminosäure und Purin Stoffwechsel; Sie wird zur Entgiftung von Ameisensäure (gebildet aus Methanol) benötigt; 112 O - O O O - N H OH O HN N N H2N Vitamin B9 (B11) N Folsäure N O - O O O - N OH H N N H2N N H O HN THF Tetrahydrofolat N H 8.) Cobalamin Cobalamin in Mutasen und Methyltransferasen katalysiert eine Alkylierung Enzyme: 1.) Homocystein Methionen durch Methylierung 2.) Methylmalonyl-CoA-Mutase (Fettsäureabbau) Teil 2: METABOLISMUS Metabolismus, ATP und Gruppenübertragungspotentiale Was ist Metabolismus? Zellen sind auf laufende Energiezufuhr angewiesen (mechanische Arbeit, aktiver Transport von Teilchen, Synthese von Makromolekülen aus einfachen Vorstufen). Die verwendete freie Enthalpie wird der Umgebung entzogen; Phototrophe Organismen (Pflanzen) nutzen das Sonnenlicht, um einfache Moleküle in komplexere umzuwandeln (Lichtenergie chemische Energie). Chemotrophe Organismen (Tiere, menschen) erhalten ihre chemische Energie durch Oxidation von Nährstoffen, die durch Phototrophe erzeugt wurden; Die Umwandlung der Energie innerhalb eines Organismus wird als Metabolsimus bezeichnet. Er wird gewährleistet durch eine bestimmte Anzahl an Stoffwechselwegen (Umwandlung eines Moleküls in ein anderes unter Energieaufwand oder Energiefreisetzung). Alle Stoffwechselwege eines Organismus sind miteinander über Kommunikationsmittel verknüpft; 113 Katabole Stoffwechselwege ( Katabolismus; exergone Reaktionen) wandeln Brennstoffe mittels oxidativer Prozesse in zelluläre Energie um (KH, Lipide Wasser, CO2 und Energie) Anabole Stoffwechselwege ( Anabolismus; endergone Reaktionen) benötigen Energie zum Aufbau von größeren Molekülen (Glucose, Lipide, DNA) mittels reduktiver Prozesse aus einfachen Vorstufen. Amphibolische Stoffwechselwege können anabolisch oder katabolisch sein (abhängig von Energiebedingungen in der Zelle) ΔG eines Stoffwechselweges entspricht der Summe der ΔG Werte der einzelnen Reaktionen. Eine thermodynamisch ungünstige Reaktion kann demnach durch eine begünstigte Reaktion, die mit ihr gekoppelt ist, ermöglicht werden: die Gesamtänderung der freien Enthalpie ist aufgrund von Kopplungen verschiedener enzymatisch katalysierter Reaktionen bei jedem Stoffwechselweg negativ; Thermodynamik der Phosphatverbindungen: ATP tritt als Donor freier Enthalpie auf. Gewonnene Energie wird in Form von ATP gespeichert. Der ATP-ADP Zyklus ist der fundamentale Mechanismus des Energieaustauschs in biologischen Systemen. Die reaktive Form ist ein Komplex aus ATP und Mg2+ bzw. Mn2+. Die Energie ist in den zwei Phosphorsäureanhydridbindungen gespeichert und wird durch Hydrolyse frei. ATP ADP + Orthophosphat (ΔG=-30,5kJ/mol) ATP AMP + Pyrophosphat (ΔG=-45,6kJ/mol) Thermodynamisch ungünstige Reaktionen (anabole Stoffwechselwege) werden mit der ATP Hydrolyse gekoppelt (ATP = Energiekoppler). ATP verschiebt das Gleichgewichtsverhältnis gekoppelter Reaktionen (Edukt zu Produkt, Konformationsänderung im Protein, IonenKonzentrationsverhältnis innerhalb und außerhalb der Zelle) um den Faktor 108. nATP Moleküle: 108n. Alle Reaktionen können thermodynamisch günstig werden, wenn sie mit der Hydrolyse genügend Moleküle ATP gekoppelt werden; Phosphorylgruppenübertragung ist allgemeines Mittel zur Energiekopplung. ATP ist ein besonders effizienter Phosphorylgruppendonor, da die terminale Phosphorylgruppe eine hohe Tendenz hat an ein Wassermolekül übertragen zu werden (ΔG ist stark negativ). Daher spricht man von einem hohen Phosphorylgruppenübertragungs-Potenital des ATP (ein positives Potential ergibt immer eine negative Enthalpie). Aufgrund der hohen Aktivierungsenergie AE der Hydrolyse wird ATP nur mittels Enzyme gespalten, wodurch die freie Enthalpie nur gezielt freigesetzt wird. 3 Ursachen für das hohe Phosphorylgruppenübertragungspotenital: Resonanzstabilisierung: ADP und Orthophosphat werden stärker resonanzstabilisiert als ATP (Orthophosphat hat wesentlich mehr mesomerer Grenzstrukturen als die γ-Phosphorylgruppe im ATP) Elektrostatische Abstoßung: ist bei ATP (4 negaitve Ladungen dicht konzentriert) wesentlich höher als bei den Hydrolyseprodukten Stabilisierung aufgrund Hydratation: Wasser koordiniert effizienter an ADP und Orthophosphat als an ATP. Stabilisierung der Hydrolyseprodukte durch Hydratation Die Konzentrationen innerhalb einer Zelle entsprechen ca. [ATP]=2mM [ADP]=0,2mM [AMP]=0,02mM 114 Andere Moleküle mit hohem PGÜP: Phosphoenolpyruvat, 1,3-Bisphosphoglycerat, Kreatinphosphat. Das PGÜP von ATP liegt zwischen den Potentialen anderer biologisch wichtiger Überträger-Moleküle. Diese Zwischenstellung ermöglicht ATP die Funktion als effektiven Phosphorylgruppenüberträger. O OH O O - OPO 3 2O O - O O PEP 1,3-bpG OH OH OH OH Glycerin O OH - OPO 3 2- CH3 Glycerat OPO 3 Pyruvat 2- CH2 Phosphoenolpyruvat 1,3-Bisphospho-glycerat - O O H H N CH3 O OPO 3 2- O N H H H3C NH + NH2 HN CH3 N Kreatinin + NH3 Kreatin HN + NH3 Kreatinphosphat Glykolyse Glykolyse verläuft anaerob, kommt in allen Zellen vor, findet im Cytosol statt; Glykolyse unterteilt sich in drei Stufen: 1. Glucose Fructose-1,6-bisphosphat unter Verbrauch von 2 ATP (Phosphorylierung, Isomerisierung, Phosphorylierung) 2. Fructose-1,6-bisphosphat GAP + DHAP 3. 2 GAP 2 Pyruvat (Oxidation) unter Gewinn von 4ATP 115 Eintritt von Glucose mittels Transportproteine; Hexokinase: überträgt Phosphorylgruppen an Hexosen; ist nur aktiv, wenn zweiwertige Metallionen vorliegen (Komplexbildung mit ATP); besteht aus zwei Lappen, die sich bei Bindung der Glucose aufeinander zu bewegen, bis die Glucose bis auf die C6 OH Gruppe umhüllt ist (gutes Beispiel für induced fit bei enzymatischer Katalyse); unpolarere Umgebung der Glucose im aktiven Zentrum begünstigt die Übergabe der terminalen Phosphorylgruppe des ATP; durch die Konformationsveränderung ist sichergestellt, dass nicht Wasser die γ-Phosphorylgruppe des ATP aufnimmt, sondern eben Glucose. Wenn Wasser Akzeptor wäre, würde es sich um eine ATPase handeln, die ATP hydrolysiert ( Substratinduzierte Schließung einer Spalte ist daher wichtige allgemeine Eigenschaft einer Kinase); Gegenteil Kinase = Phosphatase (hydrolysiert mit Wasser Phosphatgruppen) 116 Glc-6-p ist negativ geladen und kann nicht mehr zurück durch die Membran diffundieren (Hexokinase Reaktion dient der Fixierung der Glucose in der Zelle; Glucose wird durch die Reaktion zu Glc-6-p aus dem Transport-GG entozogen, weshalb Glucose in die Zelle nachdiffundieren kann). Durch Addition der Phosphorylgruppe wurde Glucose destabilisiert und somit für den weiteren Stoffwechsel aktiviert. Die Reaktion verläuft exergonisch, da ein Molekül ATP ein höheres PGÜP hat als ein Molekül Glc-6-p Phosphofructokinase 1 ist ein allosterisches Enzym. Es ist für die Glykolyse geschwindigkeitsbestimmend (comitted step) Aldolase A: gehört zur Klasse der Lyasen (Bindungsspaltung) und enthält ein Zink im aktiven Zentrum; TIM: Im Gleichgewicht liegen 96% der Isomere als DHAP vor (GAP wird schnell aus dem GG entzogen, daher Weiterreaktion); TIM katalysiert eine intramolekulare Redoxreaktion, da es Wasserstoffatome vom C1 zum C2 transferiert; TIM katalysiert die Isomerisierung von einer Ketose (DHAP) zu einer Aldose (GAP) über ein Endiol Zwischenprodukt; TIM ist katalytisch perfektes Enzym (beschleunigt Isomerisierung um Faktor 1010, katalysiert schneller als Substrate ins aktive Zentrum diffundieren!); TIM unterdrückt unerwünschte Nebenreaktion (Zerfall von Endiol zu Methylglyoxal und Orthophosphat), indem eine Schleife aus 10 AS einen Deckel über das aktive Zentrum bildet, sodass das instabile Endiol nicht freigesetzt werden kann. H H OH O O spontaner Zerfall OH O H H OPO 3 2- Endiol Zwischenprodukt + CH3 O - OH P O - Methylglyoxal 2-Oxo-propanal GA-3-P-Dehydrogenase: Enzym, welches für den Nettoenergiegewinn in der Glykolyse verantwortlich ist; es ermöglicht die Addition des niedrig energetischen Moleküls Orthophosphat an GA-3-P unter Bildung einer hochenergetischen Phosphorsäure-esterbindung, die in der darauffolgenden Reaktion (Phosphoglycerat-Kinase) unter Bildung eines Moleküls ATP erneut gespalten wird. Außerdem entsteht NADH+H+, das in der oxidativen phosphorylierung 2,5 ATP liefert; katalysiert zwei Reaktionen: Oxidation (ΔG=-50kJ/mol) und Phosphorylierung (ΔG=+50kJ/mol); diese Reaktionen sind über ein Thioester Zwischenprodukt im aktiven Zentrum gekoppelt (nur so laufen sie nacheinander ab) Enolase: bildet aus einem Alkoholphosphat ein Enolphosphat, das ein wesentlich höheres PGÜP besitzt (Enol Keton Umwandlung ist stark begünstigt, geht aber nur, wenn Phosphat abgespalten wird) und somit den nächsten Schritt der Glykolyse: die zweite Substratkettenphosphorylierung ermöglicht; enthält ein Mg im aktiven Zentrum; Nettogleichung: Glc + 2Pi + 2ADP + 2NAD+ 2ATP + 2NADH+ 2H+ + 2Pyr + 2H2O ΔG=-84kJ/mol Regulation! regulatorisch wirkende Enzyme sind solche, die irreversible Reaktionen katalysieren. Hexokinase: allosterische Inhibition durch Glc-6-p (Endproduktrepression; feedback inhibition) Phosphofructokinase I: Schlüsselrolle in der Glykolyse-Regulation; Inhibition durch ATP (Energieproduktion überflüssig, da genug vorhanden), Säure (verhindert Abfall des Blut-pH Wertes) und Citrat (es sind genug Biosynthesevorstufen vorhanden). Bei Inhibition, wird gleichzeitig die Hexokinase inhibiert, da Glc-6-p nicht weiter verarbeitet wird (Rückkopplung); Aktivierung durch AMP (wenn zuwenig Glykolyse betrieben wird, liegt zu 117 wenig ATP vor; Adenylatkinase katalysiert die Reaktion von 2 Molekülen ADP zu ATP und AMP) und Fructose-2,6-bisphosphat (feed forward stimulation) Pyruvatkinase: Inhibition durch ATP und Alanin; Aktivierung durch Fructose-1,6bisphosphat Die Transkription der drei Schlüsselenzyme wird durch Glucose und Insulin gefördert und durch cAMP inhibiert; Gluconeogenese Aufbau von Glucose aus nicht KH (Pyruvat Malat im Mitochondrium; Malat Glc-6-p im Cytosol; Glc-6-p zu Glucose im ER); wichtig, da Gehirn in hohem Maße Glucoseabhängig und Erythrozyten ausschließlich Glucose als Brennstoff verwenden; Glucosebedarf: Gehirn/Tag: 120g; Gesamtbedarf/Tag: 160g / Glucosereserven: Glykogen: 190g; Körperflüssigkeiten: 20g; Bei längeren Hungerperioden wird Glucose in Leber und Niere aufgebaut, damit Homöostase des Blutzuckerspiegels erhalten bleibt, sodass Gehirn und Muskel genug Glucose zur Verfügung haben Die Gluconeogenese kann bei unterschiedlichen Molekülen beginnen: Lactat zu Pyruvat durch Lactat-Dehydrogenase; Skelettmuskelprotein wird in Hungerperioden zu Aminosäuren abgebaut. Glucogene AS reagieren zu Pyruvat oder Oxalacetat; In Fettzellen werden Triacylglycerine zu Glycerin und Fettsäure abgebaut. Glycerin wird mithilfe der Glycerin-Kinase unter ATP Verbrauch und der GlycerinphosphatDehydrogenase unter NAD+ Verbrauch zu DHAP umgewandelt; Wenn ein Überschuss an Akzeptormolekülen im Citratcyclus vorhanden ist, können auch die über Oxalacetat zu Glucose aufgebaut werden; dies ist allerdings selten der Fall! Die Gluconeogenese aus Acetyl-CoA ist nicht möglich (FS, Ketonkörper und ketogene AS können nich Glucose aufbauen) Die Gluconeogenese ist identisch mit Glykolyse (rückwärts), bis auf drei Reaktionen, die in der Glykolyse irreversibel und zugleich die regulatorischen Schritte sind: Hexokinase (ΔG=34kJ/mol); Phosphofructokinase (ΔG=-22kJ/mol); Pyruvat-Kinase (ΔG=-17kJ/mol) Umgehung der Pyruvat-Kinase: Pyruvat-Carboxylase hat Coenzym Biotin für Carboxylierung. Carboxylierung findet im Mitochondirum statt und ist eine anaplerotische Reaktion des Citratzyklus; Oxalacetat wird dann zu Malat und in dieser Form ins Cytosol transportiert, wo es zu Oxalacetat reoxidiert wird; Die Decarboxylierung ist die treibende Kraft für die Phosphorylierung durch die PEP-Carboxy-Kinase. 118 Umgehung der Phosphofructokinase: OPO 3 2- OPO 3 2- H2O O H OPO3 H HO OH H OH OH OH O HO H 2- Pi H OH Fructose-1,6-bisphosphat Hydrolyse H Fructose-6-phosphat Fructose-1,6-bisphosphatase Umgehung der Hexokinase: meist findet diese Reaktion nicht statt, da Glucose-6-phosphat in Glykogen eingebaut werden kann und verhindert werden muss, dass Glucose aus der Zelle diffundiert. In Leber und Niere findet diese Reaktion jedoch statt (Ort: Lumen des endoplasmatischen Retikulums an einem membrangebundenen Enzym) OPO 3 2- OH O H H H2O H H OH HO OH H OH H O H H Pi HO OH OH Glucose-6-phosphatase H Glucose-6-phosphat Hydrolyse H OH Glucose Nettogleichung: 2Pyr + 4ATP + 2GTP + 2NADH + 2H+ + 6H2O Glc + 4ADP + 2GDP + 6Pi + 2NAD+ 119 4 NTP mehr, um aus reversibler Glykolyse (ΔG=+84kJ/mol) eine exergonische Reaktion wie die Gluconeogenese (ΔG=-38kJ/mol) zu machen; bei Glykolyse entstehehn pro Glucose 2 NTP, bei Gluconeogenese werden 6 NTP verbraucht; Regulation! reziproke Regulation von Glykolyse und Gluconeogenese Pyruvat-Carboxylase: Inhibition durch ADP (Zeichen für zu wenig Energie; im Mitochondrium existiert kein AMP, weshalb hier ADP inhibiert; Glykolyse muss stattfinden); Aktivierung durch Acetyl-CoA (Zeichen dafür, dass in der Zelle mehr als genug Biosynthesevorstufen und Energie vorhanden ist, sodass keine Glykolyse mehr ablaufen muss, sondern im Gegenteil: Glucose aufgebaut und in Glykogen eingebaut werden kann) PEP-Carboxykinase: Inhibition durch ADP Fructose-1,6-bisphosphatase: Inhibition durch AMP und Fructose-2,6-bisphosphat; Aktivierung durch Citrat Das Fructose-2,6-bisphosphat-System: Ist die Konzentration an F-6-p und Orthophosphat hoch, so wird F-2,6-bp gebildet. Dadurch sinkt die Konzentration an F-6-p, während die Konzentration an Triosephosphat steigt (Glykolyse bevorzugt). Ist die Konzentration an Triosephosphaten hoch wird die Bildung von F-2,6bp gestoppt, während die vorhandenen Moleküle F-2,6bp zugleich abgebaut werden. So kann niemals Glykolyse und Gluconeogenese gleichzeitig in einer Zelle stattfinden 120 Gärung Die Chemie des Stoffwechsels entstand durch die Entdeckung der Gärung außerhalb der lebenden Zelle durch die Buchners im Jahre 1897; Die Gärung verläuft anaerob (ist Sauerstoff vorhanden, findet die Atmungskette statt, in der Pyruvat vollständig zu CO2 und Wasser oxidiert wird) es gibt viele Gärungstypen; sie geht vom Endprodukt der Glykolyse (Pyruvat) aus und endet bei verschiedenen Molekülen (wichtig: alkoholische Gärung liefert Ethanol; Milchsäure Gärung liefert Lactat); Obligate Anaerobier: leben nur ohne Sauerstoff; Fakultative Anaerobier: können mit und ohne Sauerstoff leben; Bei plötzlich auftretender Muskelarbeit steigt der ATP Bedarf schneller an, als Sauerstoff zum Muskel transportiert werden kann. Der Muskel arbeitet deshalb solange anaerob bis die Lactat-Konzentration den pH Wert soweit angesäuert hat, dass der anaerobe Weg gehemmt ist (keine Muskelarbeit mehr möglich); einziger Zweck der Gärung ist die Regenerierung des NAD+ aus NADH+H+, um die Glykolyse unter anaeroben Bedingungen aufrecht zu erhalten. Unter aeroben Bedingungen wird NAD+ in der Atmungskette regeneriert, wobei hier zugleich 2,5 ATP pro NADH+H+ enststehen. 1.) Die alkoholische Gärung Pyruvat Decarboxylase: Coenzym: Thiaminpyrophosphat Alkohol-Dehydrogenase: aktives Zentrum enthält Zn2+ Ion, welches die Übertragung des Hydridions vom NADH begünstigt; 121 2.) Die Milchsäuregärung Tritt in Zellen auf, wenn der Sauerstoffgehalt limitierender Faktor ist (Bsp. Muskelzellen der Skelettmuskeln bei intensiver Beanspruchung); Nach Herzinfarkt befindet sich im Blut eine hohen Konzentration der Lactat-Dehydrogenase (zeichen für Überanstrengung); Erythrozyten und Krebszellen (bzw. Tumore) haben keine Mitochondrien, weshalb sie ausschließlich von Glykolyse und Milchsäuregärung am Leben bleiben. (Tumore können aufgrund von Lactatproduktionsraten im Körper ausfindig gemacht werden) Pentosephosphatweg Andere Bezeichnungen: Hexosemonophosphatweg, Phosphogluconat-Zyklus; findet im Cytosol statt; Zweck: Bildung des NADPH+H+ und Bildung von Ribose-5-p für reduktive Biosynthesen (in allen Organismen); 2 Phasen: oxidative Erzeugung von NADPH+H+ und Nichtoxidative Umwandlung von KH; Beginn mit Glc-6-p (10% der Gesamtkonzentration wird im PPW abgebaut) und endet mit unterschiedlichen Produkten, je nachdem ob C5 KH für Biosynthesen oder ob Zwischenprodukte der Glykolyse zur Energiegewinnung benötigt werden. Die Oxidative Phase: Der erste Schritt ist geschwindigkeitsbestimmend für den PPW; Im letzten Schritt entsteht eine β-Ketosäure, die in der Regel instabil sind, und daher spontan unter Decarboxylierung zerfallen; die abgebildete α-D-Glucose-6-p muss zunächst zu β-D-Glucose-6-p isomierisiert werden (Glucose-6-p-Isomerase), damit es von der Glc-6-Dehydrogenase als Substrat erkannt wird; Nettogleichung: Glc + H2O + 2 NADP+ Ribulose-5-p + CO2 + 2NAPH + 3H+ 122 NADPH wird insbesondere in den Erythrozyten zur Regeneration von Glutathion benötigt, da dieses mit seiner reduzierten Thiolgruppe Hämoglobin und andere Proteine vor der Oxidation schützt Die Nichtoxidative Phase: Verwertung (normal): PPW dient der Substratlieferung für reduktive Biosynthesen (Hauptsächlich Ribose-Einbau in Nukleotide unter NADPH+H+ Verbrauch). Im Vergleich zum NADPH+H+ Verbrauch für diesen Einbau herrscht jedoch ein Überschuss an Ribose-5-p, weshalb ein Teil anders verwertet werden kann. Dieser Teil wird im nichtoxidativen Teil des PPW mittels reversiblen Transferase Reaktionen in biokompatible Hexosen und Triosen umgewandelt, um in der Glykolyse der Energiegewinnung zu dienen (vollständige Verwertung aller C Atome); Transketolase hat hohe Substratspezifität (erkennt nur Substrate mit der C3 Konfiguration der Xylulose, weshalb die Epimerase Aktivität notwendig ist; Transketolase und Transaldolase gehören zu den Transferasen; Transketolase enthält als Cofaktor Thiaminpyrophosphat (TPP); Nettogleichung: 2X-5-p + R-5-p GAP + 2F-6-p 123 Ribulose-5-phosphat PhosphopentoseIsomerase OH H OH H OH OH OH O Isomerisierung (Konstitution) OPO3 PhosphopentoseEpimerase O H OH H OH 2- OPO 3 O HO H H Epimerisierung (Konfiguration) OH 2- OPO3 2- Xylulose-5-phosphat Ribose-5-phosphat OH (+) O Transketolase H C2 Aldehyd-Übertragung Sedoheptulose-7-p HO OH X-5-p HO H O H OH OPO3 H OH H OH H OH 2- O + OPO 3 H OPO 3 2- Transaldolase OH OPO3 (+) O + 2- H C3 Keton-Übertragung OH C2 Aldehyd-Übertragung HO H H 2- (+) Transketolase O H OH OH GA-3-P OH OH OPO3 2- O H OH H OH OPO 3 O HO + H OH H OH F-6-p Glykolyse 2- OPO3 Erythrose-4-p GA-3-P H 2- Fructose-6-p Glykolyse Regulation! cytosolischer NADP+ Spiegel reguliert oxidative Phase: Schrittmacherenzym und Regulatorische Einheit ist die Glucose-6-p-Dehydrogenase: NADP+ aktiviert sie und NADPH+H+ inhibiert sie; Verfügbarkeit der Substrate reguliert die nichtoxidative Phase (alles reversible Umwandlungen); je nachdem wie viele Glykolyse Zwischenprodukte (GAP, Frc-6-p) und wie viel Ribose-5-p vorhanden ist läuft dieser Teil vorwärts oder rückwärts; Die Glc-6-p Verwertung muss zwischen Glykolyse und PPW koordiniert sein; Bedarf an NADPH, ATP und Ribose reguliert die Verwertung; Ribose / NADPH benötigt: nur oxidativer Teil des PPW: Glc-6-p + 2 NADP+ + H2O R-5-p + 2NADPH + 2H+ + CO2 Ribose benötigt (DNA Synthese; Nachschub an Nukleotiden gering): Glykolyse bis zu F-6-p und GAP (Glc-6-p + ATP 2GAP + ADP und Glc-6-p F-6-p); diese gehen in nichtoxidativen Teil des PPW und bilden Ribose-5-p (Umkehrung der Transferase-Aktivität im PPW: GAP + 2F-6-p 3R-5-p); somit gilt insgesamt: 5Glc-6-p + ATP 2GAP + 4F-6-p + ADP 6R-5-p + ADP 124 NADPH benötigt (Fettsäuresynthese im Fettgewebe; Nachschub an NADPH gering): Glc-6-p bildet im oxidativen Teil des PPW 2NADPH und 1R-5-p; R-5-p wird im nichtoxidativen Teil des PPW zu GAP und F-6-p; Diese Zwischenprodukte gehen in die Gluconeogenese ein und resynthetisieren Glc-6-p; es resultiert vollständige Oxidation von Glc-6-p zu CO2; 1.) 6 Glc-6-p + 12 NADP+ + 6 H2O 6 R-5-p + 12 NADPH + 12 H+ + 6 CO2 2.) 6 R-5-p 4 F-6-p + 2 GAP 3.) 4 F-6-p + 2GAP + H2O 5 Glc-6-p + Pi Summe: Glc-6-p + 12 NADP+ + 7 H2O 6 CO2 + 12 NADPH + 12 H+ + Pi NADPH / ATP benötigt: Glc-6-p reagiert im oxidativen Teil zu NADPH und R-5-p; R-5-p geht in die nichtoxidative Phase über; deren Produkte gehen in die Glykolyse ein, um ATP zu bilden 1.) 3 Glc-6-p + 6 NADP+ + 3 H2O 3 R-5-p + 6 NADPH + 6 H+ + 3 CO2 2.) 3 R-5-p 2 F-6-p + GAP 3.) 2 F-6-p + 2 ATP + 8 ADP + 4 Pi + 4 NAD+ 4 Pyr + 4 H2O + 2 ADP + 8 ATP + 4 NADH + 4H+ 4.) GAP + 2 ADP + Pi + NAD+ Pyr + 2 ATP + NADH + H+ + H2O Summe: 3 Glc-6-p + 6 NADP+ + 5 NAD+ + 8 ADP + 5 Pi 5 Pyr + 8 ATP + 3 CO2 + 2 H2O + 5 NADH + 5 H+ + 6 NADPH + 6 H+ Glykogenmetabolismus [Regulation] Glykogen ist eine leicht mobilisierbare Speicherform der Glucose (dient als Puffer zur Konstanthaltung des Blutzuckers zwischen den Mahlzeiten und bei plötzlich auftretender intensiver körperlicher Arbeit); ist weniger reduziert als Glucose (hat geringere Energie); kann Energie auch anaerob bereitstellen (im Gegensatz zu Fettsäuren, da Reduktionsäquivalente die im FS-Abbau entstehen nicht ohne oxid. Phosphorylierung regeneriert werden können); Die wichtigsten Glykogenspeicher sind in der Leber (Synthese und Abbau werden so reguliert, dass der Blutzuckerspiegel entsprechend den jeweiligen Bedürfnissen des Gesamtorganismus erhalten bleibt; schwankt zwischen 1g und 150g) und im Skelettmuskel (reguliert nach eigenen Bedürfnissen selbst; Glucosereservoir dient für plötzliche Muskelaktivität; schwankt zwischen 300g und 200g); Der größte Teil befindet sich in der Muskulatur (Grund: mehr Muskelmasse als Lebermasse im Körper); in Leberzelle: [Glucose] = 400mM / [Glykogen] = 10nM Blut enthält 4-6g Glucose pro Liter Glykogen befindet sich im Cytosol in Form von Granula (10 bis 40 nm Durchmesser), welche alle Enzyme zum Glykogen Metabolismus und dessen Regulation enthalten; Im Verdauungstrackt wird Glykogen durch die α-Amylase zu Dextrin gespalten; 1.) Glykogenkatabolismus Phosphorolytische Spaltung: Glykogen-Phosphorylase ist keine Kinase (kein ATP Verbrauch); die Reaktion ist in vitro reversibel, da ΔG klein ist (Bindungen identischen Übertragungspotentials wurden vertauscht: glykosidische Bindung und Phosphorsäureesterbindung), in vivo jedoch zum Glykogenabbau verschoben, da [Pi] >> [Glc-1-p]; Prosthetische Gruppe ist PLP (an Lys680 des Enzyms gebunden, ist Säure-Base-Katalysator) 125 6 5 4 1 OH 2 3 OH nichtreduzierendes Ende 4' OH Gruppe Pi OH H H OH + OPO 3 O H OH OH O O HO Glucose-1-phosphat OH OH OH 2- Glykogen-Phosphorylase OH Phosphorolyse OH O O HO OH OH OH O 3 reduzierendes Ende 1' OH Gruppe Glykogen n Monomere OH 2 O OH OH 1 OH O O HO O 4 OH OH OH 5 O O O 6 OH OH OH H OH H OH Glykogen n-1 Monomere Mechanismus (rechts) es entsteht zunächst ein Enzym-PLPPhosphat-Glykogen Komplex und dann erst ein Carbokation, das nun nukleophil von Orthophosphat angegriffen werden kann; PLP übernimmt die Funktion der Säure-BaseKatalyse Hydrolytische Spaltung: würde Glucose liefern; Glc verbraucht ein ATP zusätzlich in der Glykolyse (Hexokinasereaktion; energetisch unvorteilhaft) und kann durch die Membran diffundieren, geht also der Zelle verloren (logistisch unvorteilhaft); Tatsächlich wird 90% des Glykogens phosphorolytisch und 10% hydrolytisch gespalten. Umbau des Polysaccharids für den weiteren Abbau: 4 Monomere vor einer α1,6-glykosidischen Bindung kann die Glykogen-Phosphorylase nicht mehr katalysieren; Transferase überträgt einen Block aus 3 Glucoseeinheiten von einem äußeren Zweig auf einen anderen, wodurch eine Glucoseeinheit freigelegt wurde, die jeweils einmal 1,4-glyk. und 1,6-glyk. verknüpft ist; α-1,6-Glucosidase (debranching enzyme) hydrolysiert die α-1,6-glyk. Bindung (Glucose wird abgespalten); beide Enzyme bauen das verzweigte Glykogen in eine lineare Struktur um, sodass die Phosphorylase weiter arbeiten kann. 126 Glucosephosphat-Mutase: im aktiven Zentrum der aktiven Mutase befindet sich ein phosphorylierter Serinrest; es folgt die Übertragung der Phosphorylgruppe auf das C6 der Glucose (Zwischenprodukt: Glc-1,6-bp), woraufhin die Phosphorylgruppe am C1 auf den Serinrest übertragen wird. Glc-6-p kann in die Glykolyse eingehen; In der Leber wird Glc-1-p direkt mittels Glucose-1-phosphatase in Glucose umgewandelt (auch Glc-6-p mittels Glc-6-phosphatase; dieses Enzym ist essentiell für die Gluconeogenese und kommt in Niere und Darm, nicht aber in Gehirn und Muskulatur [Muskel setzt keine Glucose frei egoistisches Organ] vor) 2.) Glykogenanabolismus Glucosedonor ist UDP-G (Uridindiphosphatglucose), UDP-G ist die aktivierte Form der Glucose (wie: ATP ist aktivierte Form von Orthophosphat); die UDP-G- Synthese ist gekoppelt mit der darauf folgenden irreversiblen Hydrolyse von Pyrophosphat zu 2 Orthophosphat mittels Pyrophosphatase, was die Synthese thermodynamisch antreibt. O OH O O OH HO - O P O + - O β α O O O O P O O P O O O CH2 N O - O Glc-1-p HN P - - - OH γ CH O UTP OH OH OH H2O UDP-G-Pyrophosphorylase O OH O HO O - O P - O OH O O P O P O O - P O HN UDP-G O O O O O CH2 N O - CH - - O OH OH Nucleosiddiphosphat Kinase: regeneriert UTP aus UDP unter ATP-Verbrauch Die Glykogenkette wird durch die Glykogen-Synthase am nichtreduzierenden Ende verlängert; Zuvor müssen allerdings vier Glucose Moleküle in einer Kette vorliegen; Als Primer fungiert das Glykoprotein Glykogenin (2 identische 37kd Untereinheiten); jede Untereinheit katalysiert mit einem enzymatisch-aktiven Bereich (Tyrosin-GlykosylTransferase) die Addition von Glucose an ihren Partner im Dimer (Autoglykosylierung; reduzierendes Ende ist an Glykogenin gebunden): Die erste UDP-G wird an Tyr194 Rest des aktiven Zentrums gebunden (Glykogenin und UDP-G im Komplex mit Mn2+). Danach werden durch Glykogenin fünf bis acht weitere Glucosereste angehängt. Der Glykogenin-Glc5 Primer bindet nun an die Glykogen-Synthase (übernimmt die weitere Elongation des Glykogenpolymers); 127 Die Glykogen-Verzweigungen sind wichtig, da sie die Löslichkeit und die Anzahl der nichtreduzierenden Enden (mehr Angriffstellen für Phosphorylase und Synthase = schnellerer Auf- und Abbau) erhöhen; Verzweigungsenzym (branching enzyme): überträgt einen Block aus 7 Glucoseeinheiten (Aufbrechen einer α-1,4-glykosidischen Bindung und Bildung einer α1,6-glykosidischen Bindung bzw. Transfer einer Kette von 4’ OH Gruppe an eine 6’OH Gruppe); die zu übertragene Kette muss das nichtreduzierende Ende enthalten, von einer Kette mit mindestens 11 Monomeren stammen und der neue Verzweigungspunkt muss mindestens 4 Einheiten entfernt sein. Nutzeffekt: pro Glucose-6-p, das in Glykogen eingebaut wird, wird ein ATP verbraucht (zur UTP-Regenerierung); Durch vollständige Oxidation von Glc-6-p entstehen 31 ATP; Wirkungsgrad der Speicherung liegt demnach fast bei 97% (10% Hydrolytische Spaltung vernachlässigt, bei der 1ATP mehr benötigt wird) Regulation! Die reziproke Regulation des Glykogenstoffwechsels umfasst drei funktionelle Ebenen: 1.) Ebene der Rezeptoren und Sensoren (Empfang intra- und extrazellulärer Signale) 2.) Ebene der Signalverstärkung und Signalintegration (zyklische Enzymkaskaden) 3.) Ebene der regulierten Enzyme (durch Phosphorylierung oder Bindung von allosterischen Effektoren): Glykogen-Phosphorylase und Glykogen-Synthase Zentrum der Kontrolle: Glykogen-Phosphorylase; Regulation über verschiedene allosterische Effektoren, die den Energiestatus der Zelle signalisieren und durch Kaskaden- gesteuerte, reversible Phosphorylierung, die letztlich durch die Hormone Insulin, Adrenalin und Glucagon initiiert wird; je nach Gewebe (Muskel oder Leber) ist das Enzym anders reguliert; 1. Hormone und deren Wirkung Insulin: steigert die Fähigkeit Glykogen zu synthetisieren, wodurch der Blutzuckerspiegel reduziert wird; Insulin ist ein Polypeptid-Hormon (A und B Kette: 21 und 30 AS), das von βZellen des endokrinen Pankreas bei zu hohem Blutzuckerspiegel ausgeschüttet wird (exokriner Pankreas produziert hydrolytische Enzyme der Verdauung: Proteasen, Lipasen, Glykosidasen). Ist der Insulinspiegel niedrig befindet man sich in einem Hungerzustand (bei hohem Insulinspiegel, ist man gesättigt) Glucagon: stimuliert den Abbau von Glykogen in der Leber, wodurch der Blutzuckerspiegel erhöht wird; Glucagon ist ein Polypeptid-Hormon (29AS), das von α-Zellen des endokrinen Pankreas bei niedrigem Blutzuckerspiegel sezerniert wird. Freisetzung von Glucagon = Hungergefühl. Insulin und Glucagon haben eine antagonistische Wirkung auf die Regulation von KH und Lipid Stoffwechselwege Adrenalin: stimuliert den Abbau von Glykogen im Muskel und teilweise auch in der Leber; es wird im Nebennierenmark aus Tyrosin gebildet, während der Muskelkontraktion (nach nervöser Stimulation) in die Blutbahn freigesetzt und im Körper verteilt; Adrenalinspiegel wird vom Nervensystem kontrolliert. Adrenalin aktiviert darüber hinaus die Glucagon Ausschüttung und Inhibiert die Insulin Ausschüttung des Pankreas 128 Wo wirken welche Hormone? Adrenalin wirkt auf alle Zellen (auf Muskulatur vorwiegend); es existieren α und β Rezeptoren: dockt Adrenalin an den β Rezeptor, resultiert eine Kaskade, welche identisch ist mit der Glucagon Kaskade; dockt Adrenalin an den α Rezeptor, so werden Ca2+ Ionen freigesetzt, die an Calmodulin binden; Der Calmodulin-Ca2+ Komplex bindet an die δ Untereinheit der Phosphorylase-Kinase (siehe unten) Glucagon wirkt ausschließlich auf die Leber! Insulin wirkt auf alle Zellen; die Glucoseaufnahme in der Leber ist jedoch Insulinunabhängig und hängt direkt von der Glucose-Konzentration im Blut (Blutzuckerspiegel) ab (siehe GLUT). 2. Rezeptoren Intrazelluläre Sensoren: Enzyme und einzelne Komponenten der zyklischen Enzymkaskade, welche durch intrazelluläre Faktoren (Edukte, Produkte) allosterisch reguliert werden. Wichtiger intrazellulärer Botenstoff: cAMP Extrazelluläre Sensoren (Membranrezeptoren): erfassen Konzentration der extrazellulären Faktoren: Adrenalin und Glucagon (stimulieren Abbau) und Insulin (stimuliert Aufbau) Der Adrenalin und Glucagon Rezeptor gehört zur Superfamilie der „7TM-G-Protein gekoppelte Rezeptoren“. Diese Rezeptoren durchspannen die Membran siebenmal. Im Liganden gebundenen Zustand aktivieren sie heterotrimere GTP bindendende Proteine (GProteine). Die aktivierten G-Proteine stimulieren ihrerseits die Adenylat-Cyclase (katalysiert cAMP Synthese aus ATP). cAMP ist das universelle, intrazelluläre Hungersignal (auch in Bakterien) und dient als second messenger. cAMP aktiviert nun seinerseits die cAMP abhängige Proteinkinase, die erste Komponente der zyklischen Enzymkaskade. H2N N N N O CH2 Adenylat-Cyclase H ATP N O H O PPi O H O P - cAMP-Phosphodiesterase H AMP OH cAMP 129 Der Insulinrezeptor gehört zu der Superfamilie der „Rezeptor-Tyrosinkinasen“. Diese haben eine extrazelluläre Ligand (Hormon) bindende Domäne, eine einzige transmembrane Domäne und eine oder mehrere intrazelluläre Tyrosinkinase-Domänen. Wenn Insulin an die α UE bindet, folgt Konformationsveränderung (Ligandeninduzierte Dimerisierung) und eine damit verbundene Aktivierung der Tyrosinkinase der β UE (intrazelluläre Tyrosinkinasedomänen kommen in direkten Kontakt und können sich gegenseitig an spezifischen Tyr- Resten unter ATP Verbrauch phosphorylieren Autophosphorylierung); durch Autophosphorylierung kommt es zur erneuten Konformationsveränderung, wodurch Bindestellen für intrazelluläre Adapterproteine frei werden, die verschiedene Signalwege in Gang setzen (Die phosphorylierten Tyr Reste binden und aktivieren verschiedene cytoplasmatische Ser-Kinasen, die ersten Komponenten von zyklischen Enzymkaskaden); 3. Die regulatorischen Enzyme im Glykogenmetabolismus Glykogen-Phosphorylase (Homodimer) kann in zwei Zuständen vorliegen: im T Zustand (tight) ist sie inaktiv und im R Zustand (relaxed) ist sie aktiv. Durch Phosphorylierung an einem Serinrest wird der Übergang von der inaktiven T in die aktive R Form erleichtert (Gleichgewicht wird zur aktiven R Form verschoben) und die Empfindlichkeit für intrazelluläre, allosterische Effektoren moduliert. Die nicht phosphorylierte B Form bindet vorwiegend AMP und ATP (kompetieren für dieselbe Bindungsstelle; AMP stabilisiert die aktive R Form; ATP hemmt die Bindung von AMP kompetetiv und stabilisiert damit die T Form indirekt); Die phosphorylierte A Form bindet vorwiegend Glukose (stabilisiert die inaktive T Form); A-R ist „aktivste“ Form / B-T ist „inaktivste“ Form 130 Glykogen-Synthase: hat mehrere phosphorylierbare Serinreste (mindestens 9), welche von unterschiedlichen Proteinkinasen phosphoryliert (inaktiviert) werden. 4. Kaskadenproteine der Adrenalin / Glucagon Kaskade (ausführlich) Basiselement jeder zyklischen Enzymkaskade: Kinase und Phosphatase (Phosphatase macht Wirkung der Kinase rückgängig; Aktivitäten müssen zeitlich verzögert erfolgen); Alle am Glykogenstoffwechsel beteiligten Enzyme sind an die Oberfläche des unlöslichen Glykogens gebunden. cAMP abhängige Proteinkinase (PKA): ist ein Heterotetramer (R2C2), bestehend aus zwei Katalystischen C und zwei regulatorischen R Untereinheiten. cAMP bindet an die RUntereinheit und bewirkt die Dissoziation des R2C2 Komplexes. Dabei werden die C Untereinheiten frei, die nun Serin-Kinase Aktivität haben. Die freie C Untereinheit phosphoryliert nun die Glykogen-Synthase (wird inhibiert), die Phosphorylase-Kinase (SPK; wird aktiviert) und den Phosphatase-Inhibitor I (wird aktiviert). Phosphorylase-Kinase (SPK): ist ein Heterotetramer (αβγδ), wobei γ Untereinheit Kinaseaktivität besitzt (α, β, δ UE sind regulatorisch); wird von cAMP abhängige Proteinkinase phosphoryliert und somit aktiviert; nun kann sie die Glykogen-Synthase (an anderen Serinen als die cAMP abhängige Proteinkinase) phosphorylieren und diese noch mehr inhibieren; gleichzeitig kann sie die Glykogen-Phosphorylase phosphorylieren und somit aktivieren (Gleichgewicht wird zum R Zustand verschoben, indem b Form in die a Form übergeht). Glykogenabbau findet statt. Reziproke Regulation, da GlykogenPhosphorylase durch Phosphorylierung aktiviert, Glykogen-Synthase allerdings durch Phosphorylierung inaktiviert wird. Phosphatase-Inhibitor I: wird von cAMP abhängige Proteinkinase phosphoryliert und somit aktiviert; nun kann er an die Phosphoproteinphosphatase I binden und diese inhibieren. Dadurch wird die Dephosphorylierung von Glykogen-Synthase, Glykogen-Phosphorylase und Phosphorylasekinase gehemmt (solange wie Adrenalin-cAMP Stimulus anhält) 131 Protein-Phosphatase I (PP1): wird vom phosphorylierten, aktiven Phosphatase-Inhibitor I komplexiert und somit inhibiert; im aktiven zustand würde PP1 Glykogen-Synthase, Glykogen-Phosphorylase und Phosphorylase-Kinase dephosphorylieren (also die Wirkung der PKA rückgängig machen); die Inaktivierung stellt sicher, dass nicht gleichzeitig zwei gegensätzliche Signale wirken (PP1 ist Antagonist zu PKA); Zeitverzögerungsmechanismen: PP1 bindet nicht direkt an Glykogen, sondern über ein Bindeprotein G, dessen Affinität für PP1 durch Phosphorylierung moduliert wird. Somit wird die Aktivität von PP1 indirekt auch über die Phosphorylierung des Bindeprotein G gesteuert. Das Bindeprotein G wird durch die Insulin abhängige Kaskade, letztlich durch die Protein-Phosphatase 2B dephosphoryliert und somit aktiviert, wodurch es PP1 rekrutiert und in Kontakt zum Glykogenkorn bringt (Glykogenaufbau wird ermöglicht); Phosphorylierung von G durch die PKA hemmt G, wodurch PP1 das Glykogenkorn verläst; d.h. solange PKA aktiv ist, kann PP1 nicht aktiv sein. PP1 wird von der Glykogen-Phosphorylase A (aktiv, phosphoryliert) in der R Form (aktiv) gebunden und solange sequestriert (gefangen gehalten) bis die Relaxation in die B - T Form stattfindet. A-R Form ist im Gleichgewicht mit A-T Form (GG liegt jedoch stark auf Seiten der A-R Form); in der A-T Form ist PP1 nur noch locker gebunden, sodass sie die Dephosphorylierung der A-T Form zur B-T Form katalysieren kann. Erst jetzt kann PP1 von der Glykogen-Phosphorylase dissoziieren und zur Glykogen-Synthase diffundieren um sie zu aktivieren; damit ist zusätzlich dafür gesorgt, dass die Glykogen-Synthase erst dann aktiviert werden kann, wenn die Glykogen-Phosphorylase inaktiv ist. Kaskade (Zusammenfassung): 1.) Adrenalin Rezeptorkomplex aktiviert via G-Protein die Adenylat-Cyclase (1. Signalverstärkung) 2.) Adenylat-Cyclase bewirkt Anstieg der intrazellulären cAMP Konzentration (Hungersignal, 2. Signalverstärkung) a. cAMP kann durch Insulin-Kaskade gespalten werden 3.) cAMP aktiviert die cAMP abhängige Proteinkinase (PKA) 4.) PKA phosphoryliert Glykogen Synthase (inaktivierung), Phosphorylase-Kinase (aktivierung) und Phosphatase Inhibitor I (aktivierung) (Signalausbreitung, 3. Signalverstärkung) weniger Glykogenaufbau 5.) Phosphorylase-Kinase phosphoryliert Glykogen Synthase (weitere inaktivierung) und Glykogen-Phosphorylase (aktivierung) Glykogenabbau, kein Glykogenaufbau 6.) Phosphatase Inhibitor I bindet und inhibiert somit die Proteinphosphatase I (PP1) 7.) Proteinphosphatase I kann nicht PKA Aktivität rückgängig machen a. erst wenn cAMP Spiegel sinkt (durch Insulin Kaskade) wird PP1 aktiv und macht PKA Aktivität rückgängig Glykogenaufbau b. PP1 kann auch aktiviert werden durch Insulin Kaskade 5. Kaskadeproteine der Insulin Kaskade (stark vereinfacht) Das Insulin-Signal wird über die Adapterproteine an verschiedene intrazelluläre Signalkaskaden gekoppelt. Es gibt 3 Haupt-Kaskaden, die von Insulin ausgelöst werden. Alle drei Wege (PLC/IP3 Weg, MAP Kinasekaskade, PI3K Weg) bewirken ein sinken des Blutzuckerspiegels durch 132 1.) PI3K Weg: Förderung der Glucose-Aufnahme (GLUT-4-Translokation mittels Vesikel zur Oberfläche; dadurch steigt die Glucose-Konzentration in der Zelle an; [Glc] und [Glc-6-p] regulieren allosterisch die Aktivität der Glykogensynthase und Phosphorylase. 2.) MAP Kinasekaskade: Förderung der Glucosespeicherung (Glykogensynthese) in Muskel und Leber 3.) PLC/IP3 Weg: Förderung des Glucoseverbrauchs (Aktivierung der Phosphofructokinase durch Erhöhung des pH-Wertes der Zelle) Besonders wichtig ist Punkt 2: Glykogensynthese Im Fall des Insulins wird eine Rezeptor-Tyrosinkinase (RTK) aktiviert. Auf dem Wege einer komplexen Signaltransduktion wird hier über die Schlüssel-Kinase raf (ras-activated factor) die Aktivierung der MAP-Kinase-Kaskade (MAP, Mitogen-aktivierte Protein Kinase) initiiert. An deren Ende steht die Insulin-stimulierte Proteinkinase (ISPK), die im phosphorylierten Zustand eine Protein-Phosphatase weitere Wirkungen: Insulin aktiviert über Kaskade Phosphodiesterase, diese spaltet cAMP in AMP Insulin aktiviert über Kaskade Protein-Phosphatase-2B, diese aktiviert das Bindeprotein G; dieses rekrutiert PP1. Citratcyclus abschließender, gemeinsamer, amphibolischer (aufbauend: Zwischenprodukt-Synthese oder abbauend: „Energiegewinnung“) Stoffwechselweg bei der Oxidation von Brennstoffen (AS, FS, KH); die meisten dieser Moleküle treten als Acetyl-CoA in den Zyklus ein; erster Teil der Atmungskette (aorobe Bedingungen notwendig: Sauerstoff dient als Elektronenakzeptor am Ende der oxidatven Phosphorylierung und sorgt somit für die Regeneration von NAD+ und FAD); findet im Mitochondirum statt; Zweck: produziert Reduktionsäquivalente (8 Elektronen werden von Acetyl-CoA übertragen 3 NADH+H+ und 1FADH2 entstehen) , die in der oxidativen Phosphorylierung ATP generieren (95% der vom Menschen benötigten Energie entsteht durch Atmungskette); es können auch Zwischenprodukte abgezogen werden und für die Synthese unterschiedlichster Moleküle (AS, Nukleotidbasen, Cholesterin, Porphyrin) verwendet werden; 1.) Der Pyruvat-Dehydrogenase Komplex Pyr-Transport: unter aeroben Bedingungen wird Pyruvat (Endprodukt der Glykolyse) in die Mitochondrien transportiert: Durch Porine (Filter: nur Moleküle mit weniger als 1kD können passieren) in der Außenmembran gelangt Pyr in den Intermembranraum; In Innenmembran fungiert Antiporter als Pyruvat-Carrier (Membranproteinkomplex; pumpt OH- gegen Pyruvat aus der Matrix). Es folgt die oxidative Carboxylierung von Pyruvat zu Acetyl-CoA im Pyruvat-Dehydrogenase Komplex (irreversible Reaktion; Verbindung zwischen Glykolyse und Citratcyclus) Pyr-Dehyd-Komplex: Multienzymkomplex (3 Enzyme mit ihren prosthetischen Gruppen): E1 ist Pyruvat-Dehydrogenase Komponente ( oxidative Dcarboxylierung mit Cofaktor Thiaminpyrophosphat); E2 ist Dihydrolipoyl-Transacetylase ( Acetylgruppentransfer aus 133 CoA mit Cofaktor Liponamid); E3 ist Dihydrolipoyl-Dehydrogenase ( ox.-LiponamidRegeneration mit Cofaktor FAD); zusätzlich: stöchiometische Cofaktoren CoA und NAD+ notwendig; mindestens zwei zusätzliche Enzyme regulieren Aktivität des Komplexes; Alle drei Schritte verlaufen thermodynamisch gekoppelt: freie Enthalpie der Decarboxylierung wird zur Erzeugung von NADH+H+ und Acetyl-CoA verwendet Netto: Pyruvat + CoA + NAD+ Acetyl-CoA + CO2 + NADH + H+ Gehört zur Familie der 2-Oxoazid-Dehydrogenase-Multienzymkomplexe (homologe Komplexe mit molekularen Massen zwischen 4 und 10 MD): Vorteile: schnelle Umsetzung des Substrats, hohe Reaktionsgeschwindigkeit (wegen hoher Anzahl an Enzyme im Komplex und räumlicher Nähe dieser Enzyme, sodass die Wanderung des Substrats von einem aktiven Zentrum ins andere ermöglicht wird); keine Nebenreaktionen (es werden keine Zwischenprodukte frei); leichtere Regulation und leicht zu kontrollierender Metabolismus (es muss nur ein Enzym im Komplex aktiv oder inaktiv gehalten werden); Metabolon: Bezeichnug für Multienzymkomplexe, bei denen es zu keinem Substratstau kommt, da die hinteren Enzyme schneller katalysieren, als die vorderen; Pyr-Dehy-Komplex ist ein Metabolon 134 E1 Pyruvat-Dehydrogenase Komponente NH2 + + O H3C H N R2 CH3 H3C S R1 OH O + - R2 S O + + C S Liponamid S + H O Redoxreaktion oxidiertes Disulfid E2 Thioester S H3C - Lys R1 O Pyruvat O N S + R H O - C H3C R1 - Acyl-Gruppen-Transfer CH3 N S H Hydroxy-Ethyl- TPP R2 C - O S O O - O O HO Decarboxylierung Addition + N R1 - R1 CH3 + H3C C S Carbanion CH3 O CO2 H3C - O - C - N N C P O O H O P O S Thiaminpyrophosphat R2 CH3 + N N N H3C R2 CH3 H Acetyl- Hydro-Liponamid Dihydrolipoyl-Transacetylase O + CoA + O - S S H3C HS Acyl-Gruppen-Transfer CoA CoA S H + S H3C HS R H R reduziertes Thiol Dihydro-Liponamid -II HS O CoA H3C S Acetyl- CoA + HS Dihydrolipoyl-Dehydrogenase -I S oxid -II + R FAD Redoxreaktion S -I oxid red + NADH+H + FADH2 R NAD red E3 red + oxid Redoxreaktion Zusammenfassung: a.) Decarboxylierung durch Bindung an TPP (Additionsreaktion mit darauf folgender Eliminierung) b.) Oxidation durch Transfer auf Liponamid (Hydroxyethylgruppe wird während des Transfers zur Acetylgruppe oxidiert Acetylliponamid ist energiereich, da 1. reduzierte Form und 2. energiereiche Thioesterbindung) c.) Transfer auf CoA (Substitutionsreaktion: Thioester bleibt erhalten) d.) Regeneration von Liponamid (E3 ist Flavoprotein, die 2 Elektronen werden von Dihydroliponamid auf FAD und von dort weiter auf NAD+ übertragen, das in Form von NADH+H+ den Komplex verlässt) 135 2.) Der Citratcyclus Die zwei in den Citratcyclus eingebundenen C-Atome werden nicht nach einem Durchlauf des Zyklus carboxyliert (Beweis mit radioaktiv markiertem Pyruvat); es werden zunächst „ältere“ C-Atome des Oxalacetats carboxyliert; Oxalacetat (α-Ketosäure) ist Acetylgruppen-Akzeptor! Dieses Molekül kann auch zur ASSynthese verwendet werden und dient auch als Zwischenprodukt in der Gluconeogenese; Die Isomerisierung des Citrats zu Isocitrat ist notwendig, da die tertiäre Hydroxylgruppe im Citrat nicht oxidiert werden kann; Aconitase hat als Cofaktor ein Eisen-Schwefel Cluster; Spontane Decarboxylierung zu α-Ketoglutarat verläuft noch am Enzym gebunden; αKetoglutarat kann auch für die AS-Synthese verwendet werden; α-KetoglutaratDehydrogenase Komplex funktioniert wie Pyruvat-Dehydrogenase Komplex Succinat-Dehydrogenase Reaktion liefert nicht genug freie Enthalpie, um Elektronen auf NAD+ zu übertragen, weshalb die Natur auf FAD zurückgreift; Succinat-Dehydrogenase ist in der inneren Mitochondriemembran verankertsie ist eine Domäne des Komplex II der oxidativen Phosphorylierung; die Elektronen werden von FADH2 also direkt übertragen; Fumarase: stereospezifische trans-Addition von Wasser (H und OH), wodurch L-Isomer entsteht; Die Reaktion Malat Oxalacetat hat positive Änderung der freien Enthalpie (ΔG=+29,7kJ/mol), weshalb GG auf Seiten von Malat liegt. Angetrieben wird die Reaktion durch Weiterverarbeitung der Produkte (Oxalacetat im Citratcyclus und NADH in Atmungskette); Citrat-Synthase verbraucht kein NTP; Succinyl-CoA Synthetase verbraucht NTP (in der betrachteten Richtung wird GTP gebildet; Energiekonservierung: Thioesterbindung des CoA wird zur Phosphorsäureanhydrid Bindung im GTP); ATP ist GTP gleichzusetzen, sie können mittels Nukleosiddiphosphat-Kinase ohne Energieaufwand ineinander umgewandelt werden Ein Acetyl-CoA liefert in der oxidativen Phosphorylierung und im Citratcyclus 10 ATP! es kommen hinzu 1 ATP aus der Glykolyse; 2 NADH = 5 ATP aus Glykolyse und Pyr-DH Komplex; insgesamt 16 ATP je Acetyl-CoA bzw. 32 ATP je Glucose; Regulation! hauptsächlich durch Substratfluss und Produkthemmung; auch allosterische Regulation und kompetetive Rückkopplungsinhibition durch andere Zwischenprodukte; Interkonvertierung (Kontrolle der Aktivität durch reversible Phosphorylierung) bedeutend bei Pyr-Dehyd-Komplex Pyruvat-Dehyd Reaktion ist irreversibel; daher erster Kontrollpunkt E1: Inhibition durch Phosphorylierung mittels Protein Kinase; Aktivierung der Protein Kinase durch NADH+H+, Acetyl-CoA und ATP; Inaktivierung der Protein Kinase durch ADP, NAD+, Pyruvat, CoA; Aktivierung durch Dephosphorylierung mittels Phosphatase; Aktivierung der Phosphatase durch Ca2+ (werden bei Muskelkontraktion freigesetzt, weshalb Energie benötigt wird; Atmungskette muss laufen) und Insulin; E2: Inhibition durch Acetyl-CoA; Aktivierung durch CoA E3: Inhibition durch NADH+H+ und Aktivierung durch NAD+ im Citratzyklus fungieren 3 Enzyme (Reaktionen nicht frei reversibel) als Regulatoren Citrat-Synthase: Inhibition durch NADH+H+, Citrat, Succinyl-CoA; Aktivierung durch Acetyl-CoA, Oxalacetat, NAD+ Isocitrat-Dehydrogenase: Inhibition durch ATP und NADH+H+; Aktivierung durch Isocitrat, NAD+, ADP und Ca2+ α-Ketoglutarat-Dehydrogenase-Komplex: Inhibition durch Succinyl-CoA, ATP, NADH+H+; Aktivierung durch Ca2+, α-Ketoglutarat, NAD+, ADP 136 137 Anabolische Betrachtung: viele Zwischenprodukte des Citatzyklus können dem Zyklus entzogen werden, um andere Stoffwechselprodukte zu erzeugen; Acetyl-CoA: kann im Cytoplasma zur Fettsäure-Synthese und zur Herstellung von Isoprenoiden wie Cholesterin herangezogen werden; muss allerdings erst von Matrix ins Cytosol transportiert werden; dies geschieht indirekt (siehe Citrat-Malat-Antiport): Oxalacetat reagiert mit Acetyl-CoA zu Citrat; Citrat wird aus der Matrix gepumpt, während Malat hineingepumpt wird (Citrat-Malat-Antiporter); Im Cytoplasma katalysiert die ATP-abhängige Citrat-Lyase die Reaktion von Citrat zu Acetyl-CoA und Oxalacetat. Oxalacetat wird über cytosolische Malat-Dehydrogenase in Malat umgewandellt und kann über denselben Antiporter zurück in die Matrix gelangen. α-Ketoglutarat (α-Ketosäure): dient als Grundbaustein für die Aminosäuren der Glutamat Familie (Glutamat, Glutamin, Arginin und Prolin) Succinyl-CoA: dient als Ausgangsstoff für Porphyrine (z.B. Häm); Synthese des Häms geschieht hauptsächlich im Knochenmark (aber auch in der Leber), da hier auch Erythrozyten gebildet werden, die Hämoglobin benötigen; Malat und Oxalacetat: Oxalacetat wird zu Malat reduziert (Malat-Dehydrogenase), um mittels Malat-Shuttle ins Cytosol zu gelangen; dort erfolgt die Reoxidation; Oxalacetat (αKetosäure) kann für die Biosynthese der Aminosäuren der Aspartat Familie genutzt werden; Oxalacetat kann auch in die Gluconeogenese eingehen und Glucose aufbauen: PEPCarboxykinase katalysiert die Reaktion zu PEP (befindet sich hauptsächlich in Leber und Niere); Anaplerotische Reaktionen: Wenn Zwischenprodukte des Citratzyklus abgezogen werden, müssen diese auch rasch wieder aufgefüllt werden, um den Zyklus aufrecht zu erhalten; Prozesse, die einen Stoffwechselweg wieder vervollständigen heißen anaplerotisch (gr. auffüllen) Wird z.B. Oxalacetat für Biosynthese von AS abgezogen, so kann Pyruvat zur Neusynthese von Oxalacetat mitteels Pyruvat-Carboxylase herangezogen werden; dieses Enzym wird von Acetyl-CoA und Pyruvat aktiviert (Zeichen für einen zu langsam ablaufenden Citratcyclus); so erhöht sich die Oxalacetat-Konzentration und der Zyklus wird beschleunigt (Acetyl-CoA Konzentration nimmt wieder ab); Auch Fumarat wirkt anaplerotisch, da es als Abfallprodukt im Harnstoffzyklus entsteht und in den Citratzyklus eingebaut werden kann oxidative Phosphorylierung oxidative Phosphorylierung (letzter Schritt in der Zellatmung; findet in innerer Mitochondrien-Membran statt; liefert den Großteil ATP bei der vollständigen Oxidation von Glucose): Die Elektronen mit hohem Übertragungspotential aus NADH+H+ und FADH2 werden zur Reduktion von molekularem Sauerstoff zu Wasser benutzt, wobei diese Reaktion mit der ATP-Synthese gekoppelt ist. Prinzip: Elektronenmotorische Kraft (Elektronen wandern über 4 Proteinkomplexe) Protonenmotorische Kraft (3 dieser Proteinkomplexe nutzen Energie der Elektronen um Protonengradient über die Membran, also elektrisches Transmembranpotential aufzubauen, wodurch eine protonenmotorische Kraft erzeugt wird) Rotationskraft (diese wird „verbraucht“, indem sie die Untereinheiten der ATP-Synthase zum rotieren bringt) PGÜP (dadurch wird ATP synthetisiert) Mitochondrium: Innenmembran undurchlässig für Ionen und polare Moleküle; Außenmembran ist wegen Mitochondriunporin (VDAC = voltage depended anionchannel) 138 durchlässig für die meisten kleinen Moleküle und steuert den geregelten Durchtritt von Metaboliten; zwei Seiten der Innenmembran: Matrixseite (innen: N-Seite: Membranpotential ist negativ) und Cytosolseite (Intermembranraum: P-Seite: Membranpotential ist positiv); Einstülpungen der Innenmambran = Cristae; Komplexe der Atmungskette: Komplexe I, III und IV fungieren als Protonenpumpen: sie pumpen Protonen von der Matrix in den Intermembranraum! Zusammenfassung: Elektronen von NADH+H+ gelangen von der NADH-QOxidaoreduktase (Komplex I) auf CoQ; Elektronen von FADH2 gelangen von der SuccinatQ-Reduktase (Komplex II) ebenso auf CoQ; CoQ diffundiert schnell innerhalb der inneren Mitochondrien Membran und überträgt die Elektronen auf die Cytochrom-C-Oxidoreduktase (Komplex III); Cyt-c überträgt nun die Elektronen auf die Cytochrom-c-Oxidase (Komplex IV), welche die Reaktion von Sauerstoff zu Wasser katalysiert Komplex I: L-förmiger Komplex mit Bindestelle für NADH+H+; überträgt schrittweise (Besonderheit!) 2 Elektronen auf seine prosthetische Gruppe FMN, wodurch 2 Protonen aus der Matrix aufgenommen werden; FMNH2 überträgt nun die Elektronen auf eine Reihe von Eisen-Schwefel-Clustern (Redoxreaktionen hier leider nicht mir Protonentransfer gekoppelt); Die Elektronen aus den Eisen-Schwefel Clustern werden letztlich auf das CoQ übertragen, wodurch erneut 2 Protonen aus der Matrix aufgenommen werden; CoQ diffundiert innerhalb der Membran Bilanz: NADH + H+ + 4H+MATRIX + Q NAD+ + 4H+INTER + QH2 ΔG = -69,5kJ/mol Komplex II: Im Citratcyclus katalysiert die Succinat-Dehydrogenase (Komponente des Komplex II) die Reaktion Succinat zu Fumarat, wodurch FADH2 entsteht; FADH2 gibt die Elektronen direkt an Eisen-Schwefel-Zentren des Komplex II weiter, von wo aus sie über ein Cytochrom b ebenfalls auf CoQ übertragen werden (FAD wird so direkt regeneriert); da die Energie der Elektronen im FADH2 nicht ausreicht, um im Komplex 2 Protonen zu pumpen, wird aus FADH2 - Elektronen auch weniger ATP gewonnen als aus NADH- Elektronen Bilanz: FADH2 + Q FAD + QH2 139 Komplex III: Dieser Komplex (auch Q-Cytochrom-C-Oxidoreduktase) transportiert die Elektronen vom Zweielektronentransporter QH2 auf den Einelektronentransporter Cytochrom c; Cyt-c- ist ein lösliches Protein und diffundiert im Intermembranraum zu Komplex IV. Q-Zyklus: es werden 2 Elektronen von CoQ (Bindestelle Q0 im Rieske-Protein) auf FeS Zentren übertragen; von hier aus kommt je ein Elektron zum CytbL und zum Cytc1; Das Elektron vom Cytc1 wird auf das Einelektronenshuttle Cytc übertragen und wird zu Komplex IV transportiert; Das Elektron vom CytbL wird weiter auf CytbH übertragen und gelangt von dort aus über eine andere Q-Bindestelle zum CoQ (Ubichinon-Pool); somit wandert jedes Elektron zweimal durch den Komplex III (= jedes Elektron verlässt zweimal QH2), um zu Komplex IV zu gelangen, wodurch für jedes Elektron zwei mal H+ (aus QH2) in den Intermembranraum gelangen. Bilanz: QH2 + 2Cytox + 2H+MATRIX Q + 2Cytred + 4H+INTER ΔG=-36,7kJ/mol Komplex IV: Die Oxidation von Cytochrom c ist mit der Reduktion von Sauerstoff zu Wasser in diesem Komplex gekoppelt; Cyt-c gibt sein Elektron an Kupfer A ab; von hier aus gelangt es zu Häm a, weiter über Häm a3 zu Kupfer B; Kupfer B ist nun von der oxidierten Cupri Form (Cu2+) in die reduzierte Cupro Form (Cu+) übergegangen; Häm a und Häm a3 unterscheiden sich nicht in Struktur, sondern nur in Position und somit Funktion im Komplex; 140 Mechanismus der Sauerstoffreduktion: Wasserbildung erfordert 4 Elektronen, 4 Protonen und somit das viermalige Andocken von Cytc; zu Beginn sind alle prosthetischen Gruppen oxidiert; erstes Cyt-c reduziert CuB; zweites Cyt-c reduziert Häm a3, wodurch ein Sauerstoffmolekül an Fe2+ des Häm a3 gebunden wird; auf Sauerstoff werden 2 Elektronen übertragen, wodurch Peroxidbrücke zwischen Fe und CuB entsteht; drittes Cyt-c führt Elektron ein, das Peroxidbrüücke spaltet und zur Aufnahme eines Protons aus der Matrix führt (CuB trägt Hydroxyylgruppe, Fe trägt Ketogruppe); das letzte Cyt-c führt Elektron ein, das zur Aufnahme dreier weiterer Protonen aus der Matrix führt, wodurch 2 Wassermoleküle abgespalten werden; durch diesen Mechanismus wird verhindert, das Sauerstoffradikale (ROS: reactive oxygene species) entstehen, die dem Mitochondrium und der Zelle erheblichen Schaden zufügen können (Folge: Seneszenz) Bilanz: 2Cytred + 4H+MATRIX + ½O2 2Cytox + H2O + 2H+INTER Für zwei Wasser: ΔG=-112kJ/mol Häm-a3 Häm-a3 O O + CuB 2e - O O CuB - + e H Häm-a3 Häm-a3 O OH HO CuB - + e H HO CuB 2H + Häm-a3 + 2 H2O CuB Komplex V: Dieser Komplex (f0f1-ATPase) funktioniert in beide Richtungen (ATP-Synthese durch Verbrauch des Protonengradienten; in Lysosomen ATP Verbrauch durch Aufrechterhaltung des sauren Milieus im Verdauungssaft; Wirkungsgrad der protonenmotorischen Kraft: 100%) 141 In Membran liegt die f0 Untereinheit, welche Protonen aus dem Intermembranraum aufnimmt und in der Matrix abgibt; In der Matrix befindet sich die f1 Einheit, welche durch die protonenmotorische Kraft erzeugte Rotation ATP synthetisiert; Ein Proton wird über die A Untereinheit (Stator; Halbkanal mit einem Eingang und einem Ausgang) aufgenommen und an die C Untereinheit (Rotor) weitergegeben; C besteht aus mehreren kreisförmig parallel ausgerichteten Doppel-Helices, die im Zentrum Aspartat enthalten. Zu Beginn des Zyklus sind alle Asp Reste protoniert, bis auf einen. Dieser Asp Rest mit negativer Ladung befindet sich in direkter Nähe zum Stator A, dessen positiv geladene Arginin Gruppe den Asp Rest in seiner energetisch ungünstigen Konformation stabilisiert. Nun wird von Asp- ein Proton aus dem Intermembranraum aufgenommen, wodurch eine energetisch günstigere Konformation entsteht; dadurch wird mechanische Energie frei, der Rotor dreht sich um 30° (Das Aspartat ist nun neutralisiert worden, sodass das „Proton“ in diesem Zustand in die hydrophobe Membran gelangen kann); Durch die Drehung gelangt eine andere Doppel-Helix zum Stator; sie gibt ihr Proton durch den Ausgang der A Untereinheit in die Matrix ab; es kommt zur gerichteten Rotation aufgrund des elektrochemischen Gradienten. An der sich nicht bewegenden A Untereinheit hängen zwei B Untereinheiten, welche an einer δ Untereinheit befestigt sind; δ ist an der oberen Peripherie von f1 befestigt; dadurch wird die f1 Einheit unbeweglich An der rotierenden C Untereinheit hängt u.a. eine γ Untereinheit, die mit C rotiert und im Zentrum der f1 Einheit endet. Durch die Rotation innerhalb des f1 Komplexes bewirkt es ständig Konformationsveränderungen (Bindungstaschen öffnen und schließen sich sequentiell) welche die ATP Synthese steuern. Die 3 β Untereinheiten vom f1 Komplex (α3β3 Hexamer) liegen immer in unterschiedlichen Konformationen vor. Eine in der O Form (offen, enthält kein Substrat); die zweite in der L Form (bindet Substrate ADP und PI locker); die dritte in der T Form (enthält ATP fest gebunden); während der Katalyse greift ein terminales Sauerstoffatom des ADP das Phosphoratom des PI unter Bildung einer pentavalenten Zwischenstufe an, die dann zu ATP und Wasser dissoziiert; pro 120° Drehung wird 1 ATP synthetisiert; Die C Untereinheit beim Menschen besteht aus 12 Helices (es werden also 4 Protonen pro synthetisiertes ATP benötigt); Die Anzahl der Helices bei allen Organismen schwankt zwischen 10 und 14. 142 Bilanz: je NADH+H+ werden 2,5 ATP synthetisiert (10 Protonen werden gepumpt, wobei 4 Protonen 1 ATP bauen); je FADH2 werden 1,5 ATP synthetisiert (6 Protonen werden gepumpt) KI pumpt 4 Protonen, KIII pumpt 4 Protonen, KIV pumpt 2 Protonen; die Protonen, welche in der Matrix verbraucht werden (Wasserbildung) verstärken den Gradienten, tragen aber nicht zur ATP-Synthese bei; Shuttle-Systeme: Werden benötigt, um NADH+H+ (entsteht im Cytosol durch Glykolyse u.a.; Reduktionsäquivalente der β-Oxidation und des Citratcyclus sind bereits in Matrix) in die Matrix zu pumpen, sodass es der oxidativen Phosphorylierung zur Verfügung steht; der NADH+H+ Transport verläuft immer indirekt! Außerdem muss ATP zwischen Matrix und Cytosol verteilt werden! 1.) Das Glycerin-3-phosphat-Shuttle Elektronen werden zunächst im Cytosol von NADH+H+ auf DHAP mittels cytosolischer Glycerin-3-phoshpat-Dehydrogenase übertragen; es entsteht Glycerin-3-p. An der Oberfläche der inneren Mitochondrienmembran wird G3P durch ein membrangebundenes Isoenzym der Glycerin-3-phosphat-Dehydrogenase zu DHAP reoxidiert; G3P überträgt Elektronen auf FAD des Isoenzyms, von wo aus sie in den CoQ Pool innerhalb der Membran gelangen (Eintritt in Atmungskette); Da die Elektronen über FAD wandern, werden hier für jedes NADH+H+ nur 1,5ATP, anstatt 2,5 ATP synthetisiert; Vorteil: es wird ein Transport gegen einen NADH+H+ Konzentrationsgradienten ermöglicht (die Kosten sind also 1ATP pro Elektronenpaar für eine sehr hohe Geschwindigkeit der oxidativen Phosphorylierung; Anwendung vor allem in Muskulatur) 143 2.) Malat-Aspartat-Shuttle Dieses Shuttle befindet sich in Herz und Leber; Es besteht aus zwei Membran-Carriern und vier Enzyen; Die Elektronen des cytosolischen NADH+H+ werden auf Oxalacetat übertragen; es entsteht Malat, das durch die innere Mitochondrienmembran in die Matrix (im Antiport gegen α-Ketoglutarat) passieren kann und dort zu Oxalacetat reoxidiert wird (indirekter NADH+H+ Transport); beide Reaktionen werden von der Malat-Dehydrogenase katalysiert. Oxalacetat wird nun mit Glutamat zu Aspartat und α-Ketoglutarat transaminiert; Aspartat kann nun ins Cytosol (im Antiport gegen Glutamat) transportiert werden, wo die gegensätzliche Transaminierung zu Oxalacetat stattfindet. Dieses Shuttle ist leicht reversibel, weshalb es stark vom Konzentrationsgradienten von NADH+H+ abhängig ist, jedoch werden keine Verluste beim Transport erlitten. Abbildung falsch (fehlender Antiport) 144 3.) ATP Shuttle / Verlust des Protonengradienten ATP entsteht innerhalb des Mitochondriums, während es im Cytosol benötigt wird; Der Transport von ATP ins Cytosol wird von einer ATP-ADP Translokase (ANT = AdeninNukleotid-Translokase; häufigste Translokase der inneren Mitochondrienmembren) in der inneren Mitochondrienmembran katalysiert; Sie wirkt als Antiporter (ADP Einstrom und ATP Ausstrom sind miteinander gekoppelt) OH- / Orthophosphat Antiporter: OH- ins Cytosol und Orthophosphat in die Matrix; notwendig, um aus ADP innerhalb der Matrix ATP synthetisieren zu können! Membranpotential wird beim Austausch von ATP gegen ADP abgeschwächt, da die positive Nettoladung im Cytosol 3e- vom ADP verliert, aber 4e- vom ATP hinzubekommt; Wegen des OH- - Orthophosphat Antiports gelangt OH- ins Cytosol, das ein H+ aufnimmt; dies macht einen Nettoverlust von einem Proton je synthetisiertem ATP aus; Außerdem: Pyruvat-Symport mit Protonen in die Matrix, um die Atmungskette am laufen zu halten; ebenso Nettoverlust von einem Proton pro transportiertem Pyruvat; Insgesamt werden also etwas über ein viertel der Energieausbeute aus dem Elektronentransfer für den Transport der Edukte und Produkte verwendet; Die Inhibition dieser Transport-Vorgänge käme mit einer Inhibition der Atmungskette gleich 145 Entkopplungsreaktionen: Oxidation von Brennstoffen und Phosphorylierung von ADP sind mit Protonengradient an der inneren Mitochondrienmembran gekoppelt; Diese Kopplung kann mittels Entkoppler Molekülen aufgehoben werden, wodurch die ATP Synthese stoppt. Leicht protonierbare, lipophile Substanzen nehmen im Intermembranraum Protonen auf und diffundieren durch die Membran; auf der Matrixseite wird das Proton, aufgrund des Gradienten abgegeben; Dinitrophenol (früher: Schlankmacher; toxisch!) fungiert als Entkoppler Fettsäuremetabolismus Vorteile der Triglyceride im Vergleich zu Glykogen: hochkonzentrierter Speicher: Energieausbeute der Triglyceride beträgt mehr als das Doppelte als die der Proteine und KH, da Triglyceride reduzierter; Glykogen ist polarer (1g Glykogen bindet 2g Wasser in Form einer Hydrathülle; 1g wasserfreies Fett speichert 6mal soviel wie 1g hydratisiertes Glykogen Fettspeicher reicht für mehrere Wochen, Glykogenreservoir nur 24h (Fettspeicher hat sich in Evolution durchgesetzt); Speicherung erfolgt im Cytoplasma der Adipocyten (Fettzellen); Tröpfchen vereinigen sich zu großen Kugeln, die fast das gesamte Zellvolumen ausmachen; Diese Zellen sind auf Synthese und Speicherung von Fetten sowie Mobilisierung zu Brennstoffmolekülen spezialisiert 1.) Fettsäureanabolismus Die Fettsäuresynthese (im Cytosol) geht von einer aktivierten Acylgruppe (zu Beginn AcetylCoA) und einer aktivierten Malonylgruppe aus; Kondensation der Monomere zu einer C4 Ketosäure; Reduktion Hydroxylgruppe; Dehydratisierung DB; Reduktion Butyryl-Einheit; Es wird so oft Malonyl-CoA angelagert, bis C16 FS vorliegt. Acetyl-CoA Carboxylase (Cofaktor: Biotin) katalysiert den comitted step des Fettsäureanabolismus; Malonyl-CoA ist aktivierter C2 Donor (hat bessere Abgangsgruppe als Acetyl-CoA: CO2); dieses Enzym ist die wichitgste regulatorische Einheit im Fettsäurestoffwechsel; Wenn der Ausgangspunkt eine freie Fettsäure ist, wird diese zunächst durch die Acyl-CoASynthetase unter Verbrauch von ATP (wird zu PPi) aktiviert; Zwischenprodukt ist ein Acyladenylat, das sofort mit CoA-SH zu Acyl-CoA und AMP zerfällt; 146 zunächst Austausch zwischen CoA und ACP: Acyl-Malonyl-ACP-kondensierendes Enzym: (Definition von Kondensation: Addition zweier Moleküle, unter Abspaltung eines kleinen Moleküls, hier: CO2); Decarboxylierung liefert freie Enthalpie für die Reaktion (daher vorher ATP-Verbrauch für Carboxylierung), sodass sie theromodynamisch günstig wird; Cl Enoyl-ACP-Reduktase: wird inhibiert von Triclosan (Bakterienhemmstoff; in „Colgate total“ Zahncreme enthalten) Cl OH O Cl Triclosan 147 Nächste Verlängerungsrunde: Butyryl ACP + Malonyl-ACP C6-β-Ketoacyl-ACP C6Acyl-ACP; Reaktionssequenz läuft ab, bis C16-Acyl-ACP synthetisiert ist; Jenes wird von Thioesterase zu Palmitat und ACP hydrolysiert; Thioesterase wirkt regulierend auf Fettsäurelänge; Im Mitochondrium und im glatten ER werden Fettsäuren mit Sonderlängen synthetisiert (andere Enzyme); im ER können Doppelbindungen eingefügt werden; Bilanz: 7 Acetyl-CoA + 7 CO2 + 7 ATP 7 Malonyl-CoA + 7 ADP + 7 Pi + 14H+ Acetyl-ACP + 7 Malonyl-ACP + 14NADPH + 21H+ Palmitat + 7 CO2 + 14 NADP+ + 8 ACP + 7 H2O Gesamt: 8 Acetyl-CoA + 7 ATP + 14NADPH +7H+ Palmitat + 7 ADP + 7 Pi + 14 NADP+ + 8 CoA + 7 H2O CoA ist ACP gleichzusetzen 1.) 14 H+ spalten sich vom protonierten ADP + Pi ab 148 2.) 21 H+ = 14 H+ gehen vom NADPH+H+ verloren; 7H+ benötigt das Enolat, um zum Keton zu werden; Der Fettsäure-Synthase Komplex: Alle beschriebenen Enzyme befinden sich in Eukaryonten von Vertebraten innerhalb einer großen Polypeptidkette (Fettsäure-Synthase; bei Hefe findet man sieben Enzym-Aktivitäten in 2 seperaten Polypeptidketten; Bei Bakterien sieben Enzym-Aktivitäten in sieben separaten Polypeptidketten); Es handelt sich beim Fettsäure-Synthase-Komplex um ein Homodimer aus 260kd schweren Untereinheiten zu je 3 Domänen; jede Domäne weist eine bis mehrere katalytische Zentren auf; Domäne I Acetyl-Transacetylase Malonyl-Transacetylase Acyl-Malonyl-ACP kondensierendes Enzym Domäne II Acyl-Carrier Protein (ACP) β-Ketoacyl-ACP-Reduktase Dehydratase Enoyl-ACP-Reduktase Domäne III Thioesterase Wie arbeiten die Domänen zusammen? Vorteile von Multienzymkomplexe: a. multifunktionelles Protein weist mehrere Enzyme in einer Polypeptidkette auf; Somit ist Synthese der einzelnen Enzyme einfacher zu koordinieren b. Zwischenstufen, können effizient von einem aktiven Zentrum zum nächsten gereicht werden, ohne das Aggregat zu verlassen; Reaktion verläuft schneller; c. Die Reaktion verläuft gerichtet, da keine Diffusion der Zwischenprodukte möglich ist und somit Nebenreaktionen verhindert werden d. Regulation eines Multienzymkomplexes ist einfacher, aufgrund einer koordinierten Struktur (Inhibition eines einzelnen Enzyms = Inhibition des Komplexes) aus diesen Vorteilen ergibt sich auch ein erhöhter Wirkungsgrad Multienzymkomplex aus kovalent verknüpften Enzymen ist stabiler, als ein Komplex, der über nicht kovalente Kräfte zusammengehalten wird; ACP: Acyl-Carrier-Protein: die Zwischenstufen der Fettsäuresynthese sind an ACP gebunden; die reaktive Einheit von ACP (wie auch von CoA) ist Phosphopantethein; es ist auf der einen Seite über eine Thioesterbindung mit dem entsprechenden Zwischenprodukt, auf der anderen Seite über ein Serinrest mit dem Protein verknüpft; H H N N OH O O HS O O H3C CH3 Ser P O O - Phosphopantethein Wo kommen die Edukte her? Die Synthese von FS mit 2n C Atomen erfordert n Acetyl-CoA, 2n-2 NADPH und n-1 ATP 149 Acetyl-CoA Transport: Citrat-Malat Antiporter Acetyl-CoA wird im Mitochondrium durch den Fettsäurekatabolismus erzeugt; Zusätzlich entsteht Acetyl-CoA ausschließlich im Mitochondrium aus Pyruvat (Pyruvat-Dehyd Komplex) und kann dort dem Citratcyclus entzogen werden; Es muss ein Transportsystem für Acetyl-CoA existieren, um dem Cytosol den Acetyl-CoA Pool für den FS-Anabolismus zur Verfügung zu stellen; Es gibt zwei Möglichkeiten für den Transport: 1.) Citrat+H+-Malat Antiport (Citrat wird ins Cytosol transportiert, wobei gleichzeitig Malat ins Mitochondrium gelangt, damit die Akzeptormoleküle für den Citratzyklus nicht ausgehen) 2.) Besser: Citrat+2H+-Pyruvat Antiport; Malat wird hier über das NADP+ abhängige Malat Enzym zu Pyruvat oxidiert: Dabei wird gleichzeitig NADPH+H+ (Palmitatsynthese: Transport von 8 Acetyl-CoA + 8 NADPH+H+ ins Cytosol es werden 14 NADPH+H+ benötigt) indirekt mit transportiert, was für den Fettsäureanabolismus notwendig ist; die restlichen 6 Moleküle NADPH+H+ stammen aus dem PPW; das benötigte ATP entspringt der Glykolyse und der oxidativen Phosphorylierung; 150 2.) Fettsäurekatabolismus Der Hauptteil des Fettes wird in der Leber abgebaut; Das meiste der frei werdenden Energie kommt dem Muskel zugute; Periphere Gewebe erhalten in drei Verarbeitungsschritten Zugang zu den als Fett gespeicherten Energiereserven: 1. Mobilisierung der Lipide (Triglyceride FS + Glycerin; FS Transport von Adipocyt zum peripheren Gewebe), 2. Aktivierung der FS (FS Acyl-CoA; Transport ins Mitochondrium), 3. Abbau der FS (β Oxidation zu Acetyl-CoA; weiter im Citratcyclus) A.) Mobilisierung Lipolyse in Adipocyten: Die Lipasen im Fettgewebe werden durch eine Kaskade aktiviert; Hormone (Adrenalin, Noradrenalin, Glucagon und ACTH) stimulieren in den Fettzellen 7TM-Rezeptoren; Diese aktivieren die Adenylat-Cyclase, die unter GTP Verbrauch aus ATP cAMP synthetisiert; ein erhöhter Spiegel an cAMP stimuliert die Protein-Kinase A, welche durch Phosphorylierung die Triacylglycerin-Lipase aktiviert. Es folgt die Spaltung in Diacylglyceride; andere Lipasen spalten weiter, sodass FS und Glycerin entstehen; Transport durchs Blut; Glycerin wird von der Leber absorbiert und zu GA3-p umgesetzt; GAP kann in Glykolyse oder Gluconeogenese eingebaut werden; Diese Reaktion ist leicht reversibel, weshalb Zwischenprodukte der Glykolyse leicht in Glycerin überführt werden können B.) Aktivierung und Transport ins Mitochondrium Aktivierung (muss erfolgen, da reine FS sehr reaktionsträge) erfolgt vor dem Transport in die mitochondriale Matrix; Acyl-Adenylat ist ein gemischtes Anhydrid aus einem Carbonsäureester und einem Phosphorsäureester; Acyl-CoA Synthetase befindet sich an der äußeren Mitochondrienmembran; die Reaktionen sind frei reversibel (ATP-Spaltung setzt Energie zur Bildung des Thioesters frei; Reaktionen gekoppelt) Sie wird erst irreversibel durch die ebenso gekoppelte, anschließende Hydrolyse von Pyrophosphat mittels Pyrophosphatase; AMP reagiert mit ATP zu 2 ADP (Adenylat-Kinase); 151 Transport der aktivierten FS durch innere Mitochondrienmembran mittels Carnitin (zwitterionischer Alkohol): C.) Die β Oxidation In der β-Oxidation (β C-Atom wird oxidiert) wird eine aliphatische Verbindung in eine Reihe aktivierter Acetyl-Einheiten gespalten, die dann in den Citratcyclus eingehen; Die Oxidation verläuft in vier Reaktionen, die in einer immer wiederkehrenden Sequenz (jedes mal wird eine C2 Einheit abgespalten) ein gesättigtes, geradzahliges Acyl-CoA vollständig abbaut. Oxidation einer aktivierten FS mittels FAD DB wird eingeführt; Hydratisierung OH Gruppe; Oxidation mittels NAD+ Ketogruppe; Spaltung des Acyl-CoA durch CoA Acetyl-CoA; Acyl-CoA Dehydrogenase ist geschwindigkeitsbestimmend; FAD ist prosthetische Gruppe des Enzyms, nimmt Elektronen auf und überträgt sie auf das Elektronen-Transferierende Flavoprotein (ETF); Die ETF-Q-Oxidoreduktase (enthält FeS Zentren) katalysiert die Weitergabe der Elektronen an den Ubichinon-Pool der inneren Mitochondrienmembran; Enoyl-CoA-Hydratase katalysiert stereospezifische Wasseranlagerung (Aus trans DB entsteht nur L-Isomer; cis DB würde D-Isomer hervorbringen) 152 Bilanz: vollständiger Abbau einer FS mit 2n C Atomen liefert n-1 FADH2, n-1 NADH+H+ und n Acetyl-CoA; Palmitinsäure (C16) liefert somit 7*1,5+7*2,5+8*10 = 108ATP; Nettoertrag ist 106 ATP, da 2 ATP (ATP AMP und AMP + ATP 2ADP) wegen der Aktivierung der FS verbraucht wurden. 3.) Abbau von speziellen Fettsäuren (ungeradzahlig, ungesättigt) A.) Ungesättigte Fettsäuren einfach ungesättigte Fettsäuren: wenn sich beim Abbau ein cis Δ3 Enoyl-CoA (kein Substrat der Acyl-CoA-Dehydrogenase) bilden sollte, katalysiert die cis Δ3 Enoyl-CoA Isomerase die Isomerisierung der Doppelbindung, sodass die Enoyl-CoA-Hydratase weiter katalysieren kann; 153 Palmitoleoyl-CoA CH H3C CH (CH2)5 O O C (CH2)7 CH H3C CoA C CH CH2 (CH2)5 S CoA S 3 3 cis-∆ -Enoyl-CoA Isomerase O C CH (CH2)5 H3C cis-∆ -Enoyl-CoA CH2 CH CoA S 2 trans-∆ -Enoyl-CoA Mehrfach ungesättigte Fettsäuren: Linoleat ist eine mehrfach ungesättigte Fettsäure mit einer cis Δ9 und einer cis Δ12 Doppelbindung; zunächst erfolgt der Abbau bis zur cis-Δ3 Doppelbindung, die wie oben beschrieben durch die cis Δ3-Enoyl-CoA Isomerase zur trans Δ2 Doppelbindung umgeformt wird; nach einem weiteren Zyklus entsteht eine cis Δ4 Doppelbindung; Diese wird durch die Acyl-CoA Dehydrogenase wie üblich oxidiert, wodurch ein 2,4-Dienoyl-Zwischenprodukt entsteht, das nicht als Substrat für die Enoyl-CoA Hydratase dient; hier katalyisert die 2,4-Dienoyl-CoA-Reduktase, wodurch die zwei Doppelbindungen zu einer Doppelbindung werden, die genau dazwischen (Δ3) liegt; es folgt der Angriff derselben Isomerase, wie zu Beginn, worauf der normale Abbau fortgesetzt werden kann; 154 DB an ungeradzahligen C Atomen werden durch Isomerase behandelt; solche an geradzahligen C Atomen durch die Sequenz Isomerase, Abbauzyklus, Dehydrogenase, Reduktase, Isomerase B.) ungeradzahlige Fettsäuren bei solchen, besonders seltenen Fettsäuren entstehen in der letzten Runde anstelle zweier Moleküle Acetyl-CoA ein Molekül Acetyl-CoA und ein molekül Propionyl-CoA; PropionylCoA wird in Succinyl-CoA umgewandelt, damit es in den Citratcyclus eingehen kann; Die Propionyl-CoA Carboxylase hat Biotin als Cofaktor und ist der Pyruvat-Carboxylase homolog; Das entstehende D-Isomer wird spontan zum L-Isomer racemisiert, das als Edukt für die methyl-Malonyl-CoA-Mutase dient; Dieses Enzym katalysiert die intramolekulare Umlagerung in Succinyl-CoA und enthält als Cofaktor Cobalamin; 4.) Regulation! des Fettsäuremetabolismus Die Schrittmacherreaktion der FS-Biosynthese ist die irreversible Acetyl-CoA-Carboxylase Reaktion; sie wird sowohl allosterisch, als auch kovalent reguliert; Die Fettsäure-Synthase wird über ihre Transkription reguliert (Insulin und Glucose induzieren die Genexpression über Transkriptionsfaktor SREBP; Glucagon hemmt die Expression über cAMP); Aufgrund der Enzymkaskaden folgt eine reziproke Regulation von Anabolismus und Katabolismus: im Hungerzustand werden Adrenalin und Glucagon frei, welche Lipasen der Fettzellen aktivieren, wodurch Fettsäure-Spiegel steigt; Insulin hemmt die Lipolyse Acetyl-CoA-Carboxylase: Aktivierung durch Insulin und Citrat; Inhibition durch Adrenalin, Glucagon, Palmitoyl-CoA und AMP übergeordnete Regulation: mittels reversibler Phosphorylierung wird Acetyl-CoA-Carboxylase deaktiviert; Dephosphorylierung aktiviert sie wieder; AMP aktiviert und ATP inaktiviert die AMPaktivierte-Proteinkinase (AMPK), welche die Acetyl-CoA-Carboxylase phosphoryliert, also deaktiviert; wenig Energie, also keine FS-Synthese; Adrenalin und Glucagon aktivieren die Proteinkinase A, welche die ProteinPhosphatase 2A (PP2A) phosphoryliert, also inaktiviert; PP2A im aktiven Zustand dephosphoryliert, also aktiviert die Acetyl-CoA-Carboxylase; Die Hormone schalten FSSynthese ab, indem sie dafür sorgen, dass Acetyl-CoA-Carboxylase bei geringem Energiegehalt in der Zelle inaktiv bleibt! 155 lokale Regulation: Citrat (Hinweis auf viel Acetyl-CoA, Hinweis auf viel ATP) bewirkt eine allosterische Aktivierung, wodurch die Inhibition durch Phosphorylierung teilweise aufgehoben wird; Palmitoyl-CoA (Hinweis auf FS-Überschuss) bewirkt allosterische Inhibierung; PalmitoylCoA inhibiert ebenso die Citrat-Translokase, welche Citrat aus den Mitochondrien ins Cytosol transportiert (Netto Acetyl-CoA Transport) und die Glc-6-p-Dehydrogenase, welche im PPW NADPH erzeugt; Synthese Cytosol ACP NADPH Multienzymkomplex Verlängerung nur bis C16 D-3-Hydroxybutyryl-ACP Aktivierter C2 Donor ist Malonyl-ACP Abbau Matrix CoA NAD+ und FAD Keine Assoziation der Enzyme Keine Einschränkung L-3-Hydroxybutyryl-CoA / Ketonkörperbiosynthese Nur wenn Fett und KH Abbau in einem ausgewogenen Verhältnis zueinander stehen, tritt das Acetyl-CoA aus dem Fettabbau vollständig in den Citratcyclus ein; ansonsten (z.B. Hungerzustand) dient es der Synthese von Ketonkörpern (Acetacetat); Wenn weniger KH abgebaut werden als Fette, so ist auch weniger Oxalacetat (entsteht aus Pyruvat vom KH Abbau) als Acetyl-CoA (entsteht aus Fettsäureabbau) vorhanden; somit fehlt vielen Acetyl-CoA Molekülen ihr Akzeptor, um in den Citratcyclus eintreten zu können; Dieser Acetyl-CoA Überschuss wird für die Ketonkörpersynthese verwendet; sie findet ebenso im Mitochondrium statt; Synthese von Acetacetat und D-3-Hydroxybutyrat findet nur in Leber (dort im Mitochondrium) statt: 3-Ketothiolase katalysiert die Umkehrung der Thiolyse im Fettsäureabbau; es handelt sich um eine reversible Reaktion, die auf Seiten des Edukts liegt; Die anschließende Synthese von HMG-CoA (auch Edukt der Steroidbiosynthese) geht mit der Hydrolyse einer Thioesterbindung einher, wodurch das Gleichgewicht beim Produkt HMG-CoA liegt; dieses günstig liegende GG kompensiert das ungünstig liegende GG der vorangehenden Reaktion; HMG-CoA wird zu Acetacetat, das weiter reagieren kann zu D-3Hydroxybutyrat und Aceton; diese 3 Ketonkörper werden ins Blut freigesetzt; Ketonkörper haben hohen physiologischen Wert: Acetacetat und D-3-Hydroxybutyrat sind Brennstoffe der Zellatmung und als Energiequellen quantitativ bedeutsam; Herzmuskel und Nierenrinde bevorzugen Acetacetat gegenüber Glucose; Herzmuskel ist vollständig vom Glucose-Metabolismus abgekoppelt, um nicht abhängig von Konzentrationsschwankungen des Glucosespiegels zu sein; bei andauerndem Hunger werden 75% des Brennstoffbedarfs des Gehirns mit Ketonkörpern gedeckt, da von Fettreserven ( Acetyl-CoA Ketonkörper) gelebt wird; auch Muskel verwertet Ketonkörper; In den peripheren Geweben findet die Reaktivierung der 3 Ketonkörper zu AcetacetylCoA statt (CoA- Donor ist Succinyl-CoA), das mittels 3-Ketothiolase zu Acetyl-CoA reagiert (kann in den Citratcyclus eingehen); Die Oxidation von D-3-Hydroxy-butyrat zu Acetacetat in der Peripherie liefert zusätzlich ein Molekül NADH+H+, das in die oxidative Phosphorylierung eingeht; Das Enzym zur Reaktivierung (3-Ketoacyl-CoA-Transferase) befindet sich nicht in der Leber, um sicherzustellen, dass die Leber ihre synthetisierten Ketonkörper nicht selbst verwertet! 156 Diabetes: Insulinmangel führt dazu, dass Aufbau von Depotfett nicht mehr stimuliert wird!! Fettzellen geben viele FS ins Blut ab, Ursache: keine Regulation der Synthese durch Insulin; FS können nicht vollständig abgebaut werden, da ein Überschuss an Acetyl-CoA resultiert; Dies alles geschieht, obwohl genug Glucose vorhanden ist; Resultat: Glucose wird nicht in der Glykolyse abgebaut, sondern in der Gluconeogenese aufgebaut, da Acetyl-CoA Pool viel zu hoch; auch Oxalacetat wird zum Teil zu Glucose umgebaut; dadurch gelangt vermehrt Glucose ins Blut (Glucose geht durch Niere und erscheint im Harn, da wenig Glucose in Leber- und Muskelzellen gepumpt wird kein Bedarf); außerdem ist der Acetyl-CoA Pool im Vergleich zum Oxalacetat Pool sehr viel größer, wodurch die Ketonkörperbiosynthese startet (Das Blut von Diabetikern hat hohen Ketonkörper-Spiegel, am häufigsten: Aceton, weshalb auch der Atom von Diabetikern einen Acetongeruch aufweist; Ketonkörper wandern auch in den Urin kein Bedarf) Die HMG-CoA Synthase wird durch ihre Transkription reguliert (SREBP aktiviert sie) 157 Cholesterinbiosynthese Die Synthese findet ausschließlich in der Leber statt; Alle C Atome des Steroids stammen aus Acetyl-CoA; Synthese verläuft in drei Schritten: Aktivierung der Isopreneinheit zu iPPP; Kondensation von 6 Molekülen iPPP zu Squalen; Zyklisierung von Squalen und Umwandlung des tatracyclischen Produkts in Cholesterin; iPPP wird aus 3 Molekülen AcetylCoA gebildet; Reaktion von Acetyl-CoA im Cytosol zu HMG-CoA mittels cytosolischer β-Ketothiolase und cytosolischer HMG-CoA-Synthase; im Cytosol erfolgt anschließend der comitted step: die Reduktion von HMG-CoA zu Mevalonat mittels HMG-CoA Reduktase (integrales Membranprotein im ER und wichtiger Kontrollpunkt für die Regulation der Cholesterinbiosynthese), die nicht im Mitochondrium zufinden ist; im Mitochondrium hingegen erfolgt die Weiterverarbeitung zu Ketonkörper; Isopentenyl-pyrophosphat-Isomerase ist ein Flavoprotein (enthält FMN oder FAD) und enthält zusätzlich zweiwertige Ionen als Cofaktor; Die Kinasen in dieser Reaktionsfolge befinden sich im Peroxisom; 158 Squalen entsteht durch eine Reihe von Kondensationsreaktionen des gleichen Mechanismus; Die Squalen-Synthase (enthält zweiwertige Ionen) befindet sich im ER; Squalen wird anschließend mithilfe von Sauerstoff und NADPH+H+ aktiviert, wodurch ein Epoxid entsteht (Squalen-Epoxidase, enthält FAD); es folgt die Protonierung des Epoxids mittels SqualenOxidocyclase und die anschließende spontane Umlagerung des entstandenen Carbokations; durch Zyklisierung und Wagner-Meerwein-Umlagerung entsteht Lanosterol; In weiteren 19 Syntheseschritten wird aus diesem Cholesterin gebildet (Entfernung von 3 Methylgruppen durch Abspaltung von 2 CO2 und einer Methansäure, Reduktion einer DB mittels NADPH+H+, Isomerisierung von DB) 159 160 Regulation! Regulation erfolgt durch Kontrolle der Enzymmenge und Enzymaktivität der HMG-CoA Reduktase, also auf mehreren Ebenen; alle Ebenen werden durch die Messung des BlutCholesterinspiegels mittels LDL-Rezeptoren gesteuert; Anzahl der LDL Rezeptoren kann somit auch die Regulation beeinflussen; Steuerung der Transkriptionsrate mittels Bindeprotein SREBP (aktiv bei niedrigem Cholesterinspiegel); Dieser Transkriptionsfaktor bindet im aktiven Zustand an die DNA Sequenz des Reduktasegens und erhöht die Transkriptionsrate; im inaktiven Zustand ist SREBP an der Kernmembran verankert; Vom Mevalonat abgeleitete Metaboliten und mit der Nahrung aufgenommenes Cholesterin verringern die Translationsrate der Reduktase-mRNA Der Abbau der Reduktase wird erleichtert, wenn eine hohe Sterinkonzentration vorherrscht; Sterin kann die Membrandomäne der Reduktase so verändern, dass sie empfindlicher für eine Proteolyse wird; Reversible Phosphorylierung durch eine AMP aktivierte Proteinkinase verringert die Aktivität der Reduktase; wenig ATP = viel AMP = keine Cholesterinsynthese Cholesterin-Derivate: 1.) Glucocorticoide sind Steroidhormone aus der Nebennierenrinde und Derivate des Progesterons; zu diesen zählen auch Cortisol; sie haben verschiedene physiologische Wirkungen (beeinflussen Stoffwechsel, Wasser- und Elektrolythaushalt, Herz-Kreislaufsystem und Nervensystem; wirken entzündungshemmend und immunsuppressiv) 2.) Aldosteron ist ein Steroidhormon, das aus Cholesterin gebildet wird; es wird bei Flüssigkeitsmangel vermeert ausgeschüttet und führt zur Rückresorption von Natrium und Chlridionen in die Niere, sowie zur Ausschüttung von Kalium-, Wasserstoff und Ammoniumionen 3.) Androgene und Östrogene sind meist steroide Sexualhormone 161 4.) Vitamin D3 (Calciol) wird im Körper mit Hilfe von UV-B Licht gebildet und hat die Funktion eines Prohormons; Das endokrine Vitamin-D-System spielt eine wesentliche Rolle für die Regulierung des Calcium Spiegels im Blut und für den Aufbau der Knochen; Vitamin-D-Mangel führt zu den Krankheiten Rachitis bei Kindern und Osteomalazie bei Erwachsenen; 5.) Phytosterine: ähnlich wie Cholesterin in tierischen Geweben sind Phytosterine wichtige Bestandteile von pflanzlichen Zellmembranen; sie haben eine Cholesterin-senkende Wirkung (es wird vermindert Cholesterin von Darm resorbiert, wenn auch Phytosterine vorliegen) a. Stigmasterol b. β-Sitosterol 6.) Mycosterine: Analogon zu den tierischen Sterinen in Pilzen und Hefen; kommen dort in der Zellmembran vor; Ergosterin ist eine Vorstufe (Provitamin) des Vitamin D2 (Ergocaldiferol) Transport von Cholesterin im Blut 1.) Einteilung der Lipoproteinpartikel Die Lipide (Cholesterin, Cholesterinester, Triglyzeride und Phospholipide), mit Ausnahme der freien Fettsäuren sind Bestandteile der Lipoproteine. Die Lipoproteinpartikel enthalten im Kern die hydrophoben Lipide (Cholsterinester und Triglyzeride), welche von spezifischen Apolipoproteinen und polaren Lipiden (Phospholipiden und freiem Cholesterin) umhüllt sind. Die Lipoproteine stellen die wasserlösliche Transportform der hydrophoben Lipide im wässrigen Milieu des Blutes dar. Sie unterscheiden sich in ihren physikochemischen Eigenschaften Dichte, Größe, Ladung und Löslichkeit. Zwischen den Lipid- und Proteinbestandteilen der Lipoproteine findet ein ständiger Austausch statt (metabolic remodeling). Mit zunehmendem Protein- und abnehmendem Lipidgehalt steigt die Dichte der Lipoproteinpartikel. Die Einteilung der Lipoproteine basiert auf der hydratisierten Dichte. Die Ultrazentrifugation liefert 5 Hauptfraktionen nach zunehmender Dichte: 1.) 2.) 3.) 4.) 5.) Chylomikronen Lipoproteine sehr geringer Dichte (very low density lipoproteins, VLDL) Lipoproteine mittlerer Dichte (Intermediate density lipoproteins, IDL) Lipoproteine geringer Dichte (low density lipoproteins, LDL) Lipoproteine hoher Dichte (high density lipoproteins, HDL) 2.) Apolipoproteine Apolipoproteine sind funktioneller Bestandteil der Lipoproteinpartikel; Lipoproteinpartikel können verschiedene Apolipoproteine enthalten; sie wirken stabilisierend auf Partikelstruktur und ermöglichen den gerichteten Transport der Lipide (binden an Membranrezeptoren der Zielzellen, steuern Aufnahme der Lipide in Zielzelle und wirken durch Beeinflussung von Enzymaktivitäten auf den Lipoproteinstoffwechsel); im Folgenden sind einige wichtige Apolipoproteine mit ihrer Funktion genannt: - - Apo B 100 ist die Hauptproteinkomponente und das wichtigste Strukturprotein des VLDL, IDL und LDL. Es wird von LDL-Rezeptoren erkannt und ist für den LDLKatabolismus verantwortlich; Apo C II ist Cofaktor für die endothelständige Lipoproteinlipase und sorgt dafür, dass Triglyceride hydrolysiert und von der Zielzelle aufgenommen werden können; 162 - - Apo E kommt hauptsächlich in den VLDL und den Chylomikronen vor und hat wichtige Funktionen im Stoffwechsel triglyceridreicher Lipoproteine. Es leitet den Abbau der Chylomikronen-Remnants und z.T. auch der IDL ein und wird von den LDL-Rezeptoren der Leber erkannt. Die Apo E-Konzentration ist ein Maß für die Apo E vermittelte Aufnahme dieser Lipoproteine durch die Leber. Apo A-I und Apo A-II sind Strukturproteine der HDL. Apo A-I aktiviert die LecitinCholesterin-Acyltransferase, die freies Cholesterin im Serum verestert. Struktur und Syntheserate der Apoproteine sind genetisch determiniert. Im Gegensatz dazu ist die Lipidzusammensetzung des Serums und der Lipoproteine abhängig von der jeweiligen Ernährungs- und Stoffwechsellage. 3.) Funktion der einzelnen Lipoproteinpartikel A.) Chylomikronen Im Darm werden die Nahrungstriglyceride mit Gallensäuren emulgiert und durch die Pankreas-Lipase zu freien FS und Monoacylglycerinen hydrolysiert, wodurch sich Micellen bilden (Micellen sind amphipatisch, ermöglichen optimalen Kontakt mit Mukosaoberfläche und verbessern Resorption); Während der Aufnahme der Lipide in die Micelle werden ihre Esterbindungen zur Wasseroberfläche hin orientiert, wodurch sie für die Verdauungsenzyme zugänglich werden; Pankreas-Lipase wird durch Gallensäuren aktiviert; O H2C O R 1 R O 2 O H2C O H2C OH O O O HC 2 H2O HC O R 2 + 2 HO R Pankreas-Lipase R 3 Triacylglycerin Hydrolyse / Lipolyse H2C OH FS Monoacylglycerin Die Abbauprodukte werden mittels Micellen innerhalb des Darmlumens zum Dünndarmepithel transportiert; Dort werden sie über die Plasmamembran von Schleimhautzellen aufgenommen; In der Dünndarmschleimhaut werden FS und Monoacylglycerine wieder zu Triglyceride vereint; in Enterocyten erfolgt die Veresterung von Cholesterin und Retinol mit FS zu Cholesterinestern und Retinolestern; langkettige FS werden in Triglyceride eingebaut und mit Apolipoproteinen (ausschließlich essentielle Strukturproteine), Cholesterinestern, Retinylestern, Phospholipiden und Cholesterin zu Chylomikronen (180 bis 500 nm Durchmesser, enthalten nur exogene Lipide aus der Nahrung) zusammengefügt; diese bestehen zu 85 bis 90% aus Triglyceriden; sie werden ins Lymphsystem und anschließend ins Blut freigesetzt; Chylomikronen dienen dem Transport von Nahrungslipiden aus dem Darm zu peripheren Geweben (In den Adipocyten werden die Trigylceride synthetisiert und gespeichert; Im Muskel können sie zur Energiegewinnung oxidiert werden); Erst nach der Sekretion ins Blut nehmen die Chylomikronen Aporpteine aus HDL auf, von denen Apo C II als Cofaktor für die endothelständige, membranassoziierte Lipoprotein-Lipase (LPL; durch Insulin aktiviert; durch Alkohol inhibiert; spaltet FS aus Triglyceride von Chylomikronen und VLDL ab) benötigt wird, um Triglyceride zu hydrolysieren und die entstehenden freien FS in die Zielzelle zu transportieren; 163 Während des Transports der freien FS in die Zielzelle geben die Chylomikronen zahlreiche Struktur-Apolipoproteine und anschließend auch Apo CII (das nicht mehr benötigt wird, da kaum mehr Triglyceride vorhanden sind) an HDL ab, wodurch sie an Größe verlieren; sie nehmen Apo E (welche die Lipoproteinpartikel für den Abbau in Leber zugänglich machen) aus HDL auf; es bleiben Chylomikronen-Remnants (enthalten hauptsächlich Cholesterinester) zurück, die in der Leber abgebaut werden; Leberzellen haben Hepatische Lipase (spaltet FS von Triglyceriden der Chylomikronen-Remnants, der VLDL und der IDL ab) B.) VLDL Chylomikronen Remnants wurden von der Leber abgebaut; VLDL werden in der Leber aus überschüssigen, endogenen Triglycyeriden synthetisiert und über die Zirkulation zu Muskelund Fettgewebe transportiert; VLDL sind Triglyceridreich, enthalten Phospholipide, freies Cholesterin und auch Apolipoproteine (geringer Gehalt an Apo CII, das die LPL aktiviert und Apo E, das den Abbau in der Leber ermöglicht; entahlten ein Molekül Apo B 100, welches das Andocken an LDL-Rezeptoren ermöglicht); Abbau der VLDL erfolgt durch endothelständige LPL (befinden sich an Gefäßwänden der Blutkapillaren; durch Hydrolyse entstandene freie FS werden von den umliegenden Zellen aufgenommen), welche 90% der Triglyceride in den VLDL hydrolysiert; auf dem Weg durch den Organismus erhöht sich durch fortlaufende Absapltung von Triglyceriden auch der Cholesterinanteil; es entstehen cholesterinreiche VLDL und IDL (cholesterinreich); C.) IDL IDL entstehen durch intravasale Lipolyse (= in einem Blut- oder Lymphgefäß befindliche Lipolyse) von Chylomikronen-Remnants und VLDL; enthalten variable Mengen Apo E (verantwortlich für Abbau in der Leber), kaum mehr Triglyceride, dafür viele Cholesterinester; enthalten auch ein Molekül Apo B 100; IDL wird weiter durch hepatische Lipase (es werden weitere Triglyceride entfernt) zu LDL abgebaut; D.) LDL LDL sind die wichtigsten Cholesterin-Carrier im Blut (enthalten ca. 1500 veresterte Cholesterin Moleküle, wobei häufigste FS Kette Linolsäure ist); der hydrophobe Kern ist von Phospholipiden und unverestertem Cholesterin umgeben; Funktion der LDL besteht darin, Cholesterin zu peripheren Geweben zu transportieren und die Cholesterinsynthese in der Leber zu regulieren; in LDL werden 70% des gesamten Plasmacholesterins befördert; dort wird es von den Zellen aufgenommen und für den Aufbau von Zellmembranen, die Synthese von Steroidhormonen und weitere Stoffwechselprozesse verwendet; Regulation der LDL Aufnahme erfolgt über LDL Rezeptor; Apo B 100 ist Ligand des LDL Rezeptors (LDL Rezeptormangel führt zu extrem hohen Cholesterinspiegel im Blut und hoher Sterblichkeit an Gefäßkrankheiten); LDL können auch von Scavenger Rezeptoren in Makrophagen aufgenommen werden; E.) HDL HDL haben hohen Proteinanteil (v.a. Strukturproteine Apo AII und Apo AI = Kofaktor der Lecithin-Cholesterin-Acyl-Transferase: LCAT: katalyisiert Bildung von Cholesterinestern aus Phospholipiden, und Apo E); entstehen in Leber und Darm und reifen im Blut durch Aufnahme von Lipiden und Apoproteinen zu sphärischen Molekülen an; HDL nehmen Cholesterin auf, das von absterbenden Zellen und abgebauten Membranen an das Blutplasma 164 abgegeben wird; Dieses aufgenommene Cholesterin wird von LCAT der HDL Partikel verestert; das veresterte Cholesterin wird entweder an VLDL oder LDL weitergegeben oder durch HDL zurück in die Leber bzw. zu Geweben, die Cholesterin für die Steroid-Synthese benötigen, transportiert; Gutes und schlechtes Cholesterin: mehr HDL (gutes Cholesterin transportiert Cholesterin aus dem Serum in die Leber) verringert das Risiko einer Gefäßerkrankung und Herzerkrankung; LDL ist schlechtes Cholesterin, da es sich (wenn im Überschuss vorhanden) in den Gefäßwänden ablagert; hohes Verhältnis LDL/HDL bzw. Apo B / Apo AI dieses Risiko erhöht; das Verhältnis LDL/HDL liegt bei einem gesunden Menschen bei 3,5; freie FS im Blut liegen an Serum Albumin gebunden vor; Albumin transportiert endogene FS; es handelt sich um ein Plasmaprotein mit 11 Binde-Domänen, sodass sechs FS binden können; es erhöht Löslichkeit von FS im Blut von 10-6M auf 2mM Aminosäuredegradation Synthese neuer Proteine benötigt AS-Pool, der durch Verdauung von Proteinen im Dünndarm und den Proteinabbau innerhalb der Zelle aufrecht erhalten wird; viele Proteine werden ständig abgebaut und neu synthetisiert (Recyclingprozess): Defekte oder nicht benötigte Proteine werden durch kovalente Bindung mehrerer Ubiquitin Moleküle für den Abbau markiert; diese werden durch einen großen ATP-abhängigen Komplex (Proteasom) abgebaut; AS dienen als Bausteine der Proteinbiosynthese, aber auch als Stickstoffquellen zur Synthese anderer AS und Nukleotidbasen; Überschüssige AS lassen sich nicht speichern, werden daher zu Zwischenprodukten des Stoffwechsels abgebaut; Dieser Abbau findet bei Mammalia hauptsächlich in der Leber statt: (1) Entfernung der α Aminogruppe (Desaminierung, da in Energieübertragungswegen kein N vorkommt) und Weiterverarbeitung der Aminogruppe im Harnstoffzyklus; (2) entstandene αKetosäure wird so umgebaut, dass sie in den Hauptstoffwechselstrom eingehen kann (CGerüste werden in Acetyl-CoA Einheiten, Acetacetyl-CoA, Pyruvat oder Zwischenprodukte des Citratcyclus überführt); AS können daher FS, Glucose und Ketonkörper aufbauen; 1.) Transaminierung Die α-Aminogruppen vieler α-AS werden durch Transaminierungsreaktion auf α-Ketoglutarat übertragen, wodurch Glutamat entsteht; die katalysierende Enzymklasse ist eine Aminotransferase, abgekürzt auch Transaminase; die Reaktionen sind reversibel und können somit zum Aufbau von α-AS aus α-Ketosäuren dienen; als Beispiel sei die Transaminierung von Aspartat zu Oxalacetat gezeigt (genauso funktioniert: Alanin Pyruvat): O - O O C + H3N O O C H C O + O O C CH2 C Aspartat AspartatAminotransferase CH2 - O O - O O C C CH2 O - - α-Ketoglutarat O C C + + H3N CH2 C O - O C H CH2 CH2 - Oxalacetat C O O - Glutamat 165 Die Reaktion verläuft in zwei Schritten: zunächst wird die α-Aminogruppe einer AS auf Pyridoxylphosphat (ist prosthetische Gruppe aller Aminotransferasen) übertragen, wodurch als Zwischenprodukt ein externes Aldimin (bleibt durch nicht kovalente Kräfte am Enzym gebunden) und als Produkt nach Hydrolyse Pyridoxaminphosphat (enthält die Aminogruppe der AS) und die α-Ketosäure (enthält sein Sauerstoff vom Wasser) entsteht; nun kann in einem zweiten Schritt eine α-Ketoglutarat exakt dieselbe Reaktion rückwärts durchlaufen, wobei die α-Aminogruppe von Pyridoxaminphosphat auf die α-Ketoglutarat übertragen wird und der Sauerstoff vom α-Ketoglutarat durch Dehydratisierung im Wasser verschwindet; es entsteht Glutamat! H Protein (H2C)4 NH ε H O O + + H3N 2- C H O N H C COO H NH α + + (CH2)4 H3N - OPO 3 2- O + H H N H C R + + H3C PLP Protein - + R α-Aminosäure + H3C O C + OPO 3 - R internes Aldimin - NH - OPO 3 externes Aldimin Pyridoxalphosphat COO Aminotransferase H3C N H 2- Chinon Pyridoxaminphosphat + O H2C - O O C O C R PMP H NH3 C - OPO 3 + H3C α-Ketosäure + N H 2- H + COO - R C O H + H2C O H O - OPO 3 + - H2C H N H COO NH + N H3C Reaktionsteil (1): Reaktionsteil (2) H R O - OPO 3 2- H3C + N H 2- Ketimin α-ASA + E-PLP α-KSA + PMP + E α-KSB + PMP + E α-ASB + E-PLP 2.) Desaminierung Nach dem Abbau einer beliebigen AS zu Glutamat folgt die oxidative Desaminierung und anschließende Hydrolyse, durch die Ammonium-Ionen frei werden, die in den Harnstoffzyklus eintreten können; Das GG der Glutamat-Dehydrogenase Reaktion liegt auf Seiten des Glutamats; thermodynamisch angetrieben wird die Reaktion durch Weiterreaktion von Ammonium; Glutamat Dehydrogenase (6 identische Untereinheiten) wird durch ATP und GTP allosterisch inhibiert und durch ADP und GDP aktiviert (weniger Energie beschleunigung der AS-Oxidation); Das Enzym kann sowohl NAD+ als auch NADP+ als Cofaktor verwerten; Durch Desaminierung von Glutamat wird α-Ketoglutarat regeneriert, das nun wieder PMP zu PLP in der Transaminierungsreaktion regenerieren kann; 166 Die Aminogruppen von Threonin und Serin können direkt zu Ammoniumionen abgebaut werden (keine Transaminierung auf Glutamat nötig); diese direkten Desaminierungen werden von Serin-Dehydratase (zu Pyruvat) und Threonin-Dehydratase (zu α-Ketobutyrat) katalysiert, die ebenfalls PLP als prosthetische gruppe haben; zunächst erfolgt die Spaltung des Hydroxidions, woraufhin das Proton am α-C Atom folgt und Wasser freigesetzt wird; es entsteht das instabile Aminoacrylat, das sofort Hydratisiert und somit desaminiert wird; Serin-Dehydratase Desaminierung Dehydratation O - O O C + H3N C - O C + H CH2 OH Serin H3N H2O H2O O Aminoacrylat O C C CH2 - O NH4 + C CH3 Pyruvat 3.) Kohlenstoff-Gerüste der AS werden dem Hauptenergiestoffwechsel zugeführt Das freie Ammonium wird nun im Harnstoffzyklus zu Harnstoff verstoffwechselt, was ausgeschieden wird; es verbleibt nun das Kohlenstoffgerüst der Aminosäure zu entsorgen, indem es zu wichtigen Stoffwechselzwischenprodukten recycelt wird; alle 20 Aminosäuren werden in 7 verschiedene Moleküle des Hauptenergiestoffwechsels überführt; ketogene Aminosäuren werden zu Acetyl-CoA oder Acetacetyl-CoA abgebaut; aus ihnen gehen Ketonkörper oder Fettsäuren hervor; glucogene Aminosäuren werden zu Pyruvat, α-Ketoglutarat, Succinyl-CoA, Fumarat oder Oxalacetat abgebaut; aus ihnen kann Glucose aufgebaut werden; 1.) Leucin und Lysin: vollständig ketogen 2.) Isoleucin, Phenylalanin, Tryptophan, Tyrosin: ketogen und glucogen 3.) Der Rest: vollständig glucogen 167 A.) Pyruvat als Eintrittsstelle Alanin, Serin und Cystein Alanin + α-Ketoglutarat Pyruvat + Glutamat (Alanin-Aminotransferase) Glutamat regeneriert α-Ketoglutarat mittels Desaminierung (Glutamat-Dehydrogenase) Serin Pyruvat + Ammonium (Serin-Dehydratase) Cystein Pyruvat + H2S oder SCN- oder SO3 Glycin, Threonin, Tryptophan Glycin kann durch enzymatische Addition einer Hydroxymethylgruppe in Serin umgewandelt oder zu CO2, NH4+ und einer aktivierten C1 Einheit gespalten werden (Cofaktor: Tetrahydrofolat: THF: überträgt C1 Einheiten) Threonin kann über Aminoaceton zu Pyruvat reagieren Drei C Atome des Thryptophans können im Alanin auftauchen; dadurch wird Tryptophan zu Pyruvat B.) Oxalacetat als Eintrittstelle Aspartat und Asparagin Aspartat + α-Ketoglutarat Oxalacetat + Glutamat (Aspartat-Aminotransferase) Oder Umwandlung von Aspartat in Fumarat mittels Harnstoffzyklus Asparagin wird von Asparaginase zu Ammonium und Aspartat hydrolysiert C.) α-Ketoglutarat als Eintrittstelle Glutamin, Histidin, Arginin und Prolin C5 AS werden zunächst in Glutamat überführt, das dann zu α-Ketoglutarat oxidativ desaminiert wird; Histidin wird in 4-Inidazolon-5-propionat umgewandelt; nach hydrolyse der RingAmidbindung entsteht Formimino-Derivat des Glutamats, das durch THF in Glutamat überführt wird; Glutamin wird von Glutaminase zu Glutamat und Ammonium hydrolysiert Prolin und Arginin werden in Glutamat-γ-semialdehyd umgewandelt, der anschließend zu Glutamat oxidiert wird; 168 D.) Succinyl-CoA als Eintrittstelle Valin, Methionin, Isoleucin Als Zwischenprodukte des Abbaus dieser AS treten Propionyl-CoA und dann MethylmalonylCoA, wodurch letztlich Succinyl-CoA gebildet wird (selber Mechanismus wie Oxidation von ungeradzahligen FS) Methionin erfordert 9 Schritte im Abbau, wobei zunächst die Adenylierung zu S-AdenosylMethionin (SAM = verbreiteter Methylgruppen-Donor in Zellen) erfolgt; Zwischenprodukte des Abbaus sind Homcystein und α-Ketobutyrat, wonach die Umwandlung zu Propionyl-CoA stattfindet; E.) AS mit verzweigten Seitenketten Valin, Isoleucin, Leucin AS mit verzweigten Seitenketten Leucin α-Ketoisocapronat (entsprechende Ketosäure) Isovaleryl-CoA (durch oxidative Decarboxylierung mittels verzweigtketten-α-Ketosäure-Dehydrogenase: homolog der Pyruvat-Carboxylase) durch Oxidation mittels FAD, Carboxylierung mittels ATP Verbrauch und anschließender hydratisierung entsteht ein ZP (β-hydroxy-β-methylglutarylCoA), das durch Spaltung Acetyl-CoA und Acetacetat bildet; Abbau von Valin und Isoleucin ähnlich: Isoleucin Propionyl-CoA + Acetyl-CoA Valin Propionyl-CoA + CO2 F.) Aromatische AS Phenylalanin, Tryptophan, Tyrosin Aromatische AS Sehr viel komplizierter; Es wird molekularer Sauerstoff zum Öffnen eines aromatischen Rings eingesetzt; Phenylalanin Tyrosin (durch Hydroxylierung: Phenylalanin-Hydroxylase: Monooxygenase, da ein Atom vom molekularem Sauerstoff im produkt das andere im Wasser auftaucht; Reduktionsmittel ist Tetrahydrobiopterin: Elektronen Carrier, der sich vom Cofaktor Bipterin ableitet;) die weitere Umwandlung erfordert die Funktion weiterer Enzyme, darunter zwei Dioxygenasen (beide Sauerstoffatome landen im produkt) und ein Enzym mit dem Cofaktor Gluthation zur Isomerisierung; eine Hydrolyse liefert letztlich Fumarat und Acetacetat Tryptophan erfordert mehrere Oxygenasen; es wird gespalten in Alanin und Acetacetyl-CoA 169 Harnstoffzyklus Harnstoffbiosynthese findet hauptsächlich in der Leber statt; Die Funktion ist die Entgiftung von Ammoniak unter Bicarbonat-Verbrauch zu ungiftigem Harnstoff; Harnstoff wird anschließend über die Niere ausgeschieden; ein Teil der bisher aus der α-Aminogruppe entstandenen Ammoniumionen wird zur Biosynthese stickstoffhaltiger Verbindungen verbraucht; der überschüssige Rest geht bei den meisten terrestrischen Vertebraten (solche heißen ureotelisch) in den Harnstoffzyklus ein und wird in Form von Harnstoff ausgeschieden; ein N Atom im Harnstoff stammt aus Aspartat, das andere vom freien Ammonium; das C stammt aus der Hydratisierung von Kohlenstoffdioxid zu Kohlensäure und ihrer anschließenden Dissoziation zum Hydrogencarbonation HCO3In der mitochondrialen Matirx findet die Synthese des Carbamoyl-phosphat statt, in der das freie Ammonium gebunden wird; die Carbamoyl-phosphat-Synthetase Reaktion ist durch den Verbauch von 2 ATP praktisch irreversibel; N-Acetyl-Glutamat ist allosterischer Aktivator der Carbamoyl-phosphat-Synthetase; Glutamat + Acetyl-CoA mittels N-Acetyl-Glutamat-Synthase (wird durch Glutamat aktviert); bei AS Abbau entsteht Glutamat zuerst, Harnstoffzyklus muss also bei hohem GlutamatGehalt aktiviert werden; es gibt auch eine Carbamoyl-phosphat-Synthetase II, ein Enzym der Pyrimidin Nukleotid Biosynthese; ebenso in der Matrix: Die Carbamoylgruppe kann durch ein hohen Übertragungspotential auf Ornithin übertragen werden; dadurch entsteht Citrulin; beides sind nicht proteinogene Aminosäuren; Die nächsten drei Reaktionen finden im Cytosol statt: Citrullin wird daher durch einen Citrullin Transporter ins Cytosol transportiert; Die Hydrolyse von PPI treibt die Argininosuccinat Synthetase an; Die Arginase enthält ein Mn als Cofaktor; am Ende wird Ornithin über den mitochondrialen Ornithin-Transporter 1 zurück ins Mitochondrium transportiert und beginnt einen neuen Zyklus; Harnstoff wird ausgeschieden; Fumarat verbindet Harnstoffzyklus mit Citratcyclus; es reagiert im Citratcyclus zu Oxalacetat, das (wenn nicht genug Aspartat vorhanden) zu Aspartat transaminiert wird und erneut in den Harnstoffzyklus eingeht; Oxalacetat kann aber auch Glucose aufbauen, Pyruvat bilden oder im Citratcyclus mit Acetyl-CoA kondensieren; Andere Organismen: Die meisten aquatischen Vertebraten und Invertebraten (solche heißen ammoniotelisch) scheiden den überschüssigen Stickstoff direkt in Form von Ammonium aus; Solche Organismen sind jedoch bei der Ausscheidung auf Wasser angewiesen; Organismen, die den Stickstoff in Form von Harnsäure ausscheiden heißen uricotelisch und benötigen nur wenig Wasser (Vorteil für viele Tiere und besonders Vögel) 170 Bilanz: CO2 + NH4+ + 3ATP + Aspartat + 2H2O Harnstoff + 2ADP + 2Pi + AMP + PPi + Fumarat (da AMP + ATP 2 ADP warden 4 äquivalente ATP verbraucht; PPi wird direct hydrolysiert) 171 Vernetzung des Stoffwechsels Prinzipien zur Integration des Stoffwechsels 1.) Prinzipien des Stoffwechsels Erzeugung von ATP, Reduktionsäquivalenten und Bausteinen für Biosynthesen - - - ATP ist universelle Energiewährung; die Hydrolyse von ATP wird an anabole, reduktive Stoffwechselreaktionen gekoppelt, um sie thermodynamisch zu ermöglichen ATP wird durch Oxidation von Brennstoffmolekülen (Glucose, FS, AS) erzeugt; das gemeinsame Zwischenprodukt bei den meisten dieser Reaktionen ist Acetyl-CoA; es wird vollständig oxidiert, wodurch die Atmungskette 30 ATP liefert; NADPH ist wichtigster Elektronendonor bei reduktiven Biosynthesen; ATP genügt nicht, weil die Produkte meist reduzierter sind als die Edukte, weshalb Elektronen hohen Potentials benötigt werden; NAD+ wird als Elektronen-Akzeptor verwendet! Zentrale Stoffwechselwege haben sowohl anabole als auch katabole Funktionen (Zuckerstoffwechsel, Fett-Stoffwechsel; Acetyl-CoA ist Mittelpunkt), wobei Biosynthese und Abbauwege fast immer verschieden sind (effektivere Kontrolle; beide Wege können jederzeit thermodynamisch begünstigt werden) 2. ) Prinzipien der Regulation metabolische Netzwerke empfangen und beantworten Informationen zum Status ihrer einzelnen Komponenten; Empfang von infos und Stoffwechselkontrolle erfolgt auf mehreren Wegen; Die Intensität mit der eine Reaktion stattfindet, kann durch die Verfügbarkeit und durch die Aktivität des Enzyms kontrolliert werden! - - - - Allosterische WW: Durchsatz von Molekülen in einem Stoffwechselweg wird durch Aktivität der Enzyme kontrolliert; Enzyme, die irreversible Reaktionen katalysieren sind gute Kontrollpunkte; die erste irreversible Reaktion ist der comitted step einer Reaktion und fast immer streng allosterisch reguliert; Bsp: Phosphofructokinase in Glykolyse und Acetyl-CoA Carboxylase in FS-Synthese Kovalente Modifikation: neben allosterischen WW ist zusätzliche Kontrolle mittels kovalenter Modifikation möglich; katalysiert durch spezifische Enzyme; Bsp. GlykogenPhosphorylase (Aktivität erhöht sich durch Phosphorylierung), Glykogen-Synthase (wird dagegen inhibiert durch Phosphorylierung); kovalente Modifikationen von Schlüsselenzymen sind oft die letzte Stufe einer Verstärkungskaskade (ermöglichen schnelles An und Abschalten von Stoffwechselwegen durch geringe Konzentrationen an Signal); hält länger an (sekunden bis minuten) als reversible allosterische WW (ms bis sekunden) Enzymmenge: Synthese- und Abbaugeschwindigkeit (Degradationsrate) von Regulationsenzymen sind der hormonellen kontrolle unterworfen; Substratmenge: Kompartimentierung: Schicksal der Moleküle ist je nach Kompartiment anders, weshalb der Transport zwischen Kompartimenten (meist Mitochondrium und Cytosol) häufig reguliert wird; FS werden nur dann ins Mitochondrium Transportiert, wenn Energie benötigt wird, wobei parallel im Cytosol die Lipid-Synthese (Veresterung und Transport ins Blut) stattfindet; Stoffwechselspezialisierungen von Organen: Regulationsvorgänge werden durch die Existenz von organen mit unterschiedlichen Funktionen grundlegend beeinflusst; Stoffwechselspezialisierung ist Folge unterschiedlicher Genexpression; 172 3.) wichtige Kontrollstellen A.) Glykolyse Es entstehen 2 ATP und 2 NADH; Im Muskel (hochaktiv; anaerobe Bedingungen) Regeneration von NAD+ durch Reaktion Pyruvat Lactat; unter aeroben Bedingungen Regeneration durch Übertragung der Elektronen auf O2 in der oxidativen phosphorylierung; Glykolyse hat zwei Funktionen: Abbau der Glucose zur ATP Erzeugung und Bereitstellung von C-gerüsten für Biosynthesen; Comitted step: Phosphofructokinase-Reaktion ATP ist Substrat der PGÜ als auch regulatroisches Molekül; hoher ATP Spiegel hemmt (Regulationsstelle ist nicht Substratbindestelle und hat niedrige Affinität zu ATP); Hemmeffekt wird von Citrat verstärkt und von AMP aufgehoben; Geschwindigkeit der Reaktion und somit der Glykolyse hängt also vom Energiebedarf der Zelle (ATP/AMP Verhältnis) und vom Bedarf an Bausteinen (Citratspiegel) ab; In der Leber ist der wichtigste Regulator der Phposphofructokinaseaktivität das F-2,6-bp System; Die F-2,6bp Konzentrtation wird durch eine Kinase- und eine Phosphatase-Aktivität gesteuert (Synthese aus F-6-p); Bei niedrigem Blutglucosespiegel führt eine vom Glucagon ausgelöste Kaskade zur Aktivierung der Phosphatase und zur Hemmung der Kinase in der Leber, wodurch F-2,6-bp Konzentration und somit auch Phosphofructokinase Aktivität sinkt; Glykolysegeschwindigkeit nimmt ab; Glucose wird für Nutzung anderer Organe ins Blut abgegeben; B.) Atmungskette Die meisten Nahrungsstoffe treten als Acetyl-CoA in den Cyclus ein; Die vollständige Oxidation einer Acetyl-Einheit erzeugt ein GTP, 3 NADH+H+ und ein FADH2. Diese Elektronen werden auf Sauerstoff übertragen, wodurch in der vorangehenden Elektronentransportkette ein Protonengradient erzeugt wird, der die Synthese von 9 ATP antreibt; Die Elektronendonoren werden nur dann oxidiert (Regeneration von Elektronenakzeptoren für Citratzyklus) wenn gleichzeitig ATP aus ADP generiert wird; dadurch wird die Geschwindigkeit des Citratzyklus mit dem ATP-Bedarf gekoppelt (= Atmungskontrolle); ATP Überschuss senkt auch die Aktivität zweier Enzyme des Citratcyclus (Isocitrat-Dehydrogenase und α-Ketoglutarat-Dehydrogenase); Citratcyclus hat auch anabole Funktionen; liefert zusammen mit Pyruvat-Carboxylase ZP für Biosynthesen (Bsp: SuccinylCoA für Porphyrinsynthese; Citrat für FS Synthese) C.) Pentosephosphatweg Die oxidative Decarboxylierung von Glc-6-p liefert 2 NADPH für reduktive Biosynthesen und Ribose-5-p für die Nukleotid-Biosynthese; Comitted step: Dehydrogenierung von Glc-6-p wird kontrolliert von der NADP+ (Elektronenakzeptor) Konzentration; der zweite Teil des PPW ist die nichtoxidative, reversible Umwandlung von phosphorylierten KH in GlykolyseZP (Verbindung zwischen Ribose-5-p für Nukleotid-Biosynthese und Glykolyse zur Energiegewinnung) D.) Gluconeogenese Glucose kann in leber und Niere aus Nicht-KH (Lactat, Glycerin, AS) synthetisiert werden; wichtigste verbindung ist Pyruvat, das im Mitochondrium zu Oxalacetat reagiert (Pyruvat173 Carboxylase); Oxalacetat wird im Cytosol zu PEP umgesetzt; Die Gluconeogenese wird des weiteren charakterisiert durch zwei hydrolytische Schritte, welche irreversible Schritte der Glykolyse umgehen; Gluconeogenese und Glykolyse werden reziprok reguliert (ein Weg liegt fast still, während der andere hochaktiv ist): Fructose-1,6-bisphophatase (Schlüsselenzym der Gluconeogenese) wird von AMP gehemmt und von Citrat aktiviert; bei Phosphofructokinase (Schlüsselenzym der Glykolyse) ist dies genau umgekehrt! Auch F-2,6bp koordiniert die reziproke Regulation durch Hemmung der F-1,6-bisphosphatase und Aktivierung der Phosphofructokinase; E.) Glykogenmetabolismus Beim Abbau katalysiert eine Phosphorylase den Abbau des Glykogens durch Orthophosphat, wodurch Glc-1-p entsteht, dass rasch in Glc-6-p umgewandelt wird; Bei Glykogen-Synthese ist das aktivierte ZP UDP-Glc, die aus Glc-1-p und UTP entsteht; Glykogen-Synthase katalysiert den Glc-Transfer von UDP-Glc auf die endständige Hydroxylgruppe einer wachsenden Kette; Abbau und Synthese unterliegen einer kontrollierten Kontrolle durch hormoninduzierte Verstärkungskaskade, sodass Phosphorylase aktiv, wenn Synthase inaktiv und umgekehrt; Diese Enzyme werden durch Phosphorylierung und allosterische WW kontrolliert; F.) Fettsäuremetabolismus FS werden im Cytosol durch Addition von C2 Einheiten an eine wachsende Kette synthetisiert, die an ACP gebunden ist; das aktivierte ZP Malonyl-CoA entsteht durch Carboxylierung aus Acetyl-CoA; Die Acetylgruppen gelangen durch das Citrat-Malat-Shuttle von den Mitochondrien ins Cytosol (dort wird Citrat zu Acetyl-CoA gespalten); zusätzlich zum Transport von Acetyl-CoA stimuliert Citrat im Cytosol die Acetyl-CoA-Carboxylase (comitted step); viel ATP und viel Acetyl-CoA heißt auch viel Citrat, wodurch Geschwindigkeit der FS-Synthese steigt; Der FS Abbau erfolgt auf einen anderen Weg in einem anderen Kompartiment; Carnitin transportiert FS in Mitochondrien, wo sie mittels β-Oxidation zu Acetyl-CoA abgebaut werden; wenn genug Oxalacetat, kann Acetyl-CoA in Citratzyklus eintreten; wenn nicht, umwandlung in Ketonkörper; Die bei β-Oxidation entstandenen FADH2 und NADH gehen in oxidative Phosphorylierung; deshalb kann β-Oxidation wie auch der Citratzyklus nur dann ablaufen, wenn NAD+ und FAD regeneriert werden; Geschwindigkeit des Fettsäureabbaus ist auch mit ATP Bedarf gekoppelt! Malonyl-CoA (Vorstufe für FS Synthese) hemmt FS Abbau, indem es die Bildung von Acylcarnitin durch Carnitin-Acyltransferase I hemmt; Transport der FS in die Mitochondrien wird verhindert; 4.) Wichtige Knotenpunkte A.) Glc-6-p Wenn Glucose in die Zelle gelangt, wird sie sofort zu Glc-6-p phosphoryliert, das als Glykogen gespeichert, über Pyruvat abgebaut oder in Ribose-5-p umgewandelt werden kann; Glykogen entsteht, wenn Glc-6-p und ATP im Überschuss vorhanden sind; wenn ATP und C Gerüste für Biosynthesen benötigt werden geht es in die Glykolyse ein (Abbau von Glc-6-p zu Pyruvat kann anabol und katabol sein); der Eintritt in den PPW findet statt, wenn NADPH und / oder Ribose-5-p für Nukleotid-Biosynthese benötigt wird; Glucose kann über 174 Gluconeogenese aus Pyruvat, über die Mobilisierung von Glykogen und aus glucogenen AS synthetisiert werden; B.) Pyruvat Pyruvat entsteht in erster Linie aus Glc-6-p, Alanin und Lactat; Pyruvat kann von der LactatDehydrogenase zu Lactat reduziert werden, um NAD+ zu regenerieren (ermöglicht den Fortgang der Glykolyse unter anaeroben Bedingungen in aktiven geweben); Lactat, das so in aktivem gewebe entsteht wird vornehmlich in anderen geweben zu Pyruvat reoxidiert (dieser Mechanismus spart Zeit und überträgt einen Teil der Stoffwechsellast auf inaktivere Gewebe); leicht reversibel ist auch die Transaminierung des Pyruvats (α-Ketosäure) zu Alanin; umgekehrt können mehrere AS in pyruvat umgewandelt werden (Transaminierung ist Verbindungsglied zwischen KH und AS Stoffwechsel); weiter kann Pyruvat im Mitochondrium zu Oxalacetat carboxyliert werden (erster Schritt der Gluconeogenese), wonach unter Umgehung der irreversiblen Schritte der Glykolyse Glucose gebildet werden kann; Die carboxylierung von pyruvat ist auch wichtig zur Auffüllung des Citratcyclus mit ZP; Aktivierung der pyruvatCarboxylase durch Überschuss an Acetyl-CoA sorgt für Oxalacetat-Synthese und somit für beschleunigung des Citratcyclus; letztlich kann Pyruvat auch zu Acetyl-CoA oxidativ decarboxyliert werden; es handelt sich um eine irreversible Reaktion im Mitochondrium, welche die KH- und AS- C Atome der Oxidation im Citratzyklus und der FS-Synthese zur Verfügung stellt (umgekehrt ist dies nicht möglich); daher wird der pyruvat-Dehyd Komplex durch allosterische WW und kovalente Modifkation streng reguliert; Pyruvat wird nur dann in Acetyl-CoA umgewandelt, wenn ein Bedarf an ATP oder C2 Einheiten für die lipidsynthese besteht; C.) Acetyl-CoA Die β Oxidation der FS und die oxidative Decarboxylierung von pyruvat liefern Acetyl-CoA; es entsteht außerdem aus ketogenen AS; Im Stoffwechsel steht Acetyl-CoA nur eine beschränkte Anzahl an Wegen offen: vollständige oxidation im Citratcyclus, Aufbau von FS (Transport über Citrat ins Mitochondrium) oder Bildung von HMG-CoA; letzteres ist eine Vorstufe des Cholesterins und der ketonkörper (Transportform für Acetyl-Einheiten von leber zu einigen peripheren geweben); 175 5.) Organe haben einzigartige Stoffwechselprofile A.) Gehirn Glucose ist praktisch der einzige Brennstoff, den das menschliche Gehirn verwerten kann (Ausnahme: lange Hungerperioden); Gehirn besitzt keine Brennstoffvorräte und ist auf ständige Versorgung mit Glucose angewiesen; täglicher Verbrauch 120g Glc = 1760kJ (60% des Gesamtglucoseumsatzes des ruhenden Organismus); Ein Großteil der Energie wird für Transportmechanismen aufgewandt, die das Na-KMembranpotential aufrecht erhalten, das für Übertragung von Nervenimpulsen notwendig ist; Das Gehirn muss auch Neurotransmitter für Weiterleitung von Nervenimpulsen synthetisieren; Glucosestoffwechsel während Gehirnaktivität bleibt unverändert (dennoch lokaler Anstieg möglich); GLUT3 (hat niedrigen KM Wert und ist daher unter den meisten Bedingungen gesättigt) transportiert Glucose in die Gehirnzellen; FS können dem Gehirn nicht als Brennstoff dienen, weil sie im Plasma an Albumin gebunden sind und damit nicht die Blut-Hirn Schranke passieren können; Im Hungerzustand können von der Leber erzeugte Ketonkörper teilweise Glucose als Energielieferant des Gehirns ersetzen; B.) Muskulatur Die wichtigsten Brennstoffe für den Muskel sind Glucose, Fettsäuren und Ketonkörper; Muskeln besitzen im Gegensatz zum Gehirn einen hohen Glykogenvorrat (5000 kJ, Gehirn: 30 kJ); 75% des gesamten Glykogens sind im Muskel gespeichert; es wird zur Weiterverwertung im Muskel rasch zu Glc-6-p abgebaut; der Muskel besitzt wie das Gehirn keine Glc-6-phosphatase (Glucose wird nicht abgegeben egoistische Organe); Im aktiv kontrahierenden Skelettmuskel ist die Geschwindigkeit der Glykolyse weit höher als die des Citratzyklus; ein Großteil des gebildeten Pyruvats wird daher zu Lactat reduziert, das zur Leber transportiert und dort zu Glucose umgewandelt wird (Cori-Zyklus verlagert Stoffwechsellast von Muskel auf Leber); Zusätzlich entsteht im Muskel durch Transaminierung von Pyruvat viel Alanin, das wie Lactat in der Leber zu Glucose umgewandelt werden kann (Alanin-Zyklus); der Muskel setzt Alanin frei, weil er keinen Harnstoff bilden kann (Abbau von AS nicht möglich); er kann lediglich AS aufnehmen und transaminieren; Im ruhenden Muskel sind FS Hauptbrennstoff, die dann 85% des Energiebedarfs decken; Der Herzmuskel arbeitet im Gegensatz zum Skelettmuskel fast ausschließlich aerob (Herzmuskel hat hohe Mitochondriendichte); Herz hat praktisch keine Glykogenreserven; FS sind Hauptenergielieferant des Herzmuskels (Ketonkörper und Lactat können auch als Brennstoffe dienen); Hermuskel setzt mehr Acetacetat als Glucose um; C.) Fettgewebe Fettgewebe weist einen sehr hohen Vorrat an Triacylglycerine auf (70kg Mensch: 565.000kJ); es ist auf die Veresterung von FS und auf ihre Freisetzung aus Triacylglycerinen spezialisiert; Beim Menschen stellt Leber Hauptort der FS-Synthese dar; von dort aus werden sie zu Triacylglycerinen verestert und in VLDL zum Fettgewebe transportiert; Triacylglycerine 176 werden vom Fettgewebe nicht aufgenommen (zuvor Hydrolyse durch extrazelluläre Lipoprotein-Lipase; Aktivierung der Lipase durch Insulin-Kaskade); nach dem Eintritt der FS in die Zelle müssen sie aktiviert werden; die gebildeten Acyl-CoA Einheiten werden auf Glycerin-3-phosphat übertragen; Glycerin-3-phosphat stammt von der Reduktion von DHAP (ZP der Glykolyse), weshalb das Fettgewebe zur Synthese von Lipiden Glucose benötigt; Intrazelluläre Lipasen hydrolysieren Triacylglycerine zu FS und Glycerin; Freisetzung der ersten FS ist comitted step und wird von hormonsensitiver Lipase katalysiert, die über reversible Phosphorylierung kontrolliert wird (Adrenalin stimuliert cAMP Bildung, die letztlich Protein-Kinase aktiviert); Triacylglycerine in Fettzellen werden ständig hydrolysiert und resynthetisiert; das bei der Hydrolyse anfallende Glycerin wird zur Leber transportiert, wobei die entstehenden FS (wenn genug Glycerin-3-p vorhanden) sofort wieder zu Triacylglycerinen verarbeitet werden; Bei Mangel an Glycerin-3-p werden die FS ins Plasma abgegeben; Glucosespiegel im inneren der Fettzellen kontrolliert also die Abgabe der FS ins Blut; D.) Niere Die Hauptaufgabe der Niere ist die Erzeugung von Harn, der als Transportmittel zur Ausscheidung von Stoffwechselabfällen und zur Aufrechterhaltung der Osmolarität der Körperflüssigkeiten dient; In den Nierentubuli wird das Blutplasma fast 60mal am Tag filtriert; Der größte Teil der aus dem Blut filtrierten Substanzen wird wieder resorbiert (so werden nur ein bis zwei Liter Harn erzeugt); wasserlösliche Bestandteile des Plasmas (Glucose und Wasser selbst) werden wieder resorbiert, um Verschwendung zu vermeiden; für die Rückresorption benötigt die Niere viel Energie; Obwohl sie nur 0,5% der Körpermasse ausmachen, verbrauchen sie 10% des bei der Zellatmung verwendeten Sauerstoffs; der größte Teil der rückresorbierten Glucose wird vom Natrium-Glucose Cotransporter in die Nierenzellen transportiert (Transporter bekommt Energie vom Na-K Gradienten, der von der Na-K-ATPase erzeugt wird); genau nach dem selben Mechanismus läuft der Transport von Glucose aus dem Darm ins Blut! Im Hungerzustand wird Niere zu einem wichtigen Ort der Gluconeogenese und kann bis zur Hälfte der Blutglucose bereitstellen; E.) Leber Die Stoffwechselaktivitäten der Leber sind von entscheidender Bedeutung für die Versorgung des Gehirns, der Muskulatur und anderer peripherer Organe mit Brennstoffen; Leber macht 24% der Körpermasse aus und stellt den Stoffwechselmittelpunkt eines Organismus dar; Die meisten im Verdauungstrakt resorbierten Stoffe passieren zunächst die Leber, die auf diese Weise die Konzentration zahlreicher Metabolite im Blut kontrollieren kann; Die Leber entfernt 2/3 der Glucose und alle anderen Monosaccharide aus dem Blut (ein Teil der Glucose bleibt für andere Organe); Hexokinase und leberspezifische Glucokinase setzen aufgenommene Glucose in Glc-6-p um; ein großer Teil des Glc-6-p wird in Glykogen umgewandelt (1650kJ Speicher in Leber); ein Überschuss an Glc-6-p wird zu Acetyl-CoA abgebaut, aus dem FS, Cholesterin und Gallensäuren (Endprodukt des Cholesterinstoffwechsels, dienen der Fettverdauung und Fettabsorption) gebildet werden können; der PPW (weitere Möglichkeit Glc-6-p umzusetzen) liefert das NADPH für diese reduktiven Biosynthesen; 177 Die Leber kann Glucose durch Mobilisierung des Glykogenvorrats und Gluconeogenese ans Blut abgeben; Die Vorstufen für die Gluconeogenese sind Lactat und Alanin aus dem Muskel, Glycerin aus dem Fettgewebe und glucogene AS aus der Nahrung; Wenn viele Brennstoffe vorhanden sind, synthetisiert die Leber FS (oder verwendet solche aus der Nahrung), verestert sie und gibt sie in Form von VLDL ans Blut ab; Im Hungerzustand wandelt die Leber FS in Ketonkörper um; wie wird die Verwertung zwischen diesen beiden Möglichkeiten reziprok reguliert? Wenn viele FS vorhanden sind werden Lipide synthetisiert: es ist viel Malonyl-CoA (Edukt der FS Synthese) vorhanden; die Carnitin-Acyl-Transferase wird dadurch gehemmt und somit der Transport von FS ins Mitochondrium verhindert ( da nur in der Matrix die Ketonkörpersynthese stattfindet, wird diese verhindert; auch die β-Oxidation wird so verhindert); stattdessen findet die Synthese von Triacylglycerinen im Cytosol statt; Bei Brennstoffmangel ist Malonyl-CoA Spiegel niedrig (daher gelangen die vom Fettgewebe freigesetztem FS in die Matrix und dienen der Ketonkörpersynthese) Die Leber nimmt einen Großteil der aus der Nahrung stammenden und im Blut umherschwirrenden Aminosäuren auf (einige bleiben für periphere Gewebe im Blut); AS werden hauptsächlich für Proteinbiosynthese verwendet und nicht abgebaut; wie wird die Verwertung der AS in diese Richtung gelenkt? Die KM Werte der Aminoacyl-tRNA-Synthetasen liegen niedriger als die der Enzyme für den AS-Abbau, weshalb die AS bevorzugt zur Synthese von Aminoacyl-tRNAs eingesetzt werden, anstatt abgebaut zu werden; wenn der Abbau stattfindet, wird zunächst der Stickstoff entfernt, der dann in Harnstoff umgewandelt wird; Die Leber synthetisiert so 20 bis 30g Harnstoff am Tag; die α-Ketosäuren werden in Gluconeogenese oder Fettsäure-Synthese verwendet; Leber kann von verzweigtkettigen AS (Isoleucin, Leucin, Valin) die Aminogruppe nicht entfernen (das übernimmt der Muskel) Die Leber deckt ihren eigenen Energiebedarf, indem sie α-Ketosäuren, die aus dem Abbau von AS stammen, der Glucose als Brennstoff vorzieht; Glykolyse dient in der Leber hauptsächlich zur Gewinnung von Bausteinen für die Biosynthese; Acetacetat kann die Leber nicht als Brennstoff verwendet werden, da sie nur wenig von der für die Aktivierung zu Acetyl-CoA notwendigen 3-Ketoacyl-CoA-Transferase besitzt; Die Leber verzichtet auf die Brennstoffe, die sie an Muskel und Gehirn weitergibt; 6.) Änderungen des Stoffwechsels Biochemische Antwort auf eine Reihe physiologischer Zustände drei Phasen des nächtlichen Hunger-Sättigungs-Zyklus A.) postresorptive Phase nach einer mahlzeit B.) frühe Hungerphase während der Nacht C.) erneute Sättigungsphase nach dem Frühstück Hauptziel der biochemischen Veränderungen in diesem Zeitrum ist die Erhaltung der Glucosehomöostase (Konstanthaltung des Blutzuckerspiegels) A.) Die postresorptive Phase - Aufnahme und Verdauung einer Abendmahlzeit; Transport von Glc und AS von Darm in Blut; Nahrungslipide werden in Chlomikronen verpackt und über das lymphathische System ins Blut transportiert 178 - - - - - - Sättigungszustand führt zur Ausschüttung von insulin durch die β-Zellen des Pankreas (Stimulierung durch Glucose und durch das parasympathische Nervensystem); Insulin signalisiert Zustand der Sättigung und stimuliert auf verschiedenen Wegen die brennstoffspeicherung und die proteinsynthese; Insulin setzt proteinkinase-Kaskaden in gang (Stimulation der Glykogensynthese in Muskel und Leber, Inhibition der Gluconeogenese in Leber; Beschleunigung der Glykolyse in der Leber, wodurch FSSynthese gefördert wird); Leber trägt zur Begrenzung der Glucosemenge im Blut in Zeiten des Überflusses bei (speichert Glucose in Glykogen); Die Glucoseaufnahme in der Leber wird über GLUT2 durch hohen Blutzucker erhöht; Durch Hexokinasereaktion steigt Glc-6-p Konzentration in der Leber an; die leberspezifische Glucokinase hat zwar eine 50fach geringe Affinität zu Glucose, wird aber nicht von Glc-6-p inhibiert, was den Verbrauch von Glucose in der Leber auch bei sehr hohem Blutzucker sicherstellt (katalysiert selbe Reaktion wie Hexokinase); so wird sichergestellt, dass die Leber schneller Glc-6-p produziert, wenn der Blutzuckerspiegel hoch ist; so werden auch schneller Glykogenspeicher der leber aufgefüllt (zusätzlich aktiviert Insulin die Glykogensynthese, siehe unten; die Beschleunigung der Glc-6-p Produktion hängt allerdings lediglich direkt mit dem hohen Blutzuckerspiegel und dem dadurch verbundenen verstärkten Transport mittels GLUT2 zusammen, nicht aber mit Insulin, da keine Insulinabhängigen GLUTs in der Leber vorhanden) Die Hormoneffekte auf die Glykogensynthese und speicherung werden durch eine direkte Wirkung der Glucose selbst verstärkt; Phosphorylase a baut Glykogen ab und ist zugleich Glucose-Sensor; ein hoher Glc-Spiegel macht sie zugänglicher für eine Phosphatase, die sie in die inaktive Phosphorylase b umwandelt; durch die allosterischen Effekte der Glucose wird das Glykogensystem vom Abbau zur synthese verlagert; Der hohe insulinspiegel im Sättigungszustand fördert den Einstrom von Glucose in Muskel und Fettgewebe (GLUT4); Die Glykogen-Synthese des Muskels und der Leber wird von insulin stimuliert; Die Aufnahme von Glucose in das Fettgewebe liefert Glycerin-3-p für die Synthese von Triacylglycerinen Die Insulin-Wirkung erstreckt sich auch auf den AS- und Proteinstoffwechsel; Insulin fördert die Aufnahme von verzweigtkettigen AS in den Muskel; Insulin hat einen allgemein stimulierenden Effekt auf die Proteinsynthese, was den Aufbau von Muskelprotein begünstigt, zusätzlich hemmt es den intrazellulären Proteinabbau; B.) Der frühe Hungerzustand und die nächtliche Nahrungskarenz - - - Blutzuckerspiegel sinkt einige Stunden nach einer Mahlzeit, wodurch weniger Insulin und mehr Glucagon ausgeschüttet wird; Glucagon wird von den α-Zellen des Pankreas bei niedrigem Blutzuckerspiegel (Hungerzustand) ausgeschüttet; es mobilisiert die Glykogenspeicher, wenn über die Nahrung keine Glucose aufgenommen wird; Das hauptsächliche Zielorgan von Glucagon ist die Leber; es stimuliert den Glykogenabbau und hemmt die Glykogensynthese über eine cAMP Kaskade (Phosphorylierung und Aktivierung der Phosphorylase und Inhibition der Glykogen.Synthase); Glucagon hemmt auch die FS Synthese durch Verminderung der Pyruvatproduktion und durch Inhibition der Acetyl-CoA-Carboxylase (wird so in einem nicht-phosphorylierten Zustand gehalten); Glucagon stimuliert außerdem die Gluconeogenese der Leber und hemmt die Glykolyse durch Senkung des F-2,6-bp Spiegels; Alle Wirkungen des Glucagons werden von Proteinkinasen vermittelt, die durch cAMP aktiviert werden; Die Aktivierung der cAMP Kaskade führt zur höheren Phosphorylase a Aktivität und zur niedrigeren Glykogen-Synthase Aktivität; Die Wirkung von Glucagon 179 - - - wird durch eine verminderte Bindung von Glucose an die Phosphorylase a verstärkt (dadurch wird Phosphorylase a unempfindlicher gegen Hydrolyse durch Phosphatase: Phosphorylase a wird also nicht zur inaktiven Phosphorylase b; Die Phosphatase bleibt dennoch an Phosphorylase a gebunden, sodass die Synthase in der inaktiven phosphorylierten Form bleibt); es kommt zu einer raschen Glykogenmobilisierung; Die große Menge Glucose, die durch Hydrolyse des aus Glykogen entstandenen Glc-6-p entsteht wird nun ins Blut abgegeben; Die verminderte Glucoseverwertung in Muskel und Fettgewebe trägt ebenso zur Aufrechterhaltung des Blutglucosespiegels bei; das Resultat der Glucagon-Ausschüttung ist also ein deutlicher Anstieg der Glucose-Freisetzung durch die Leber Während des Absinkens des Blutzuckerspiegels nutzen sowohl Muskulatur, als auch Leber Fettsäuren als Brennstoff; Blutzuckerspiegel wird durch drei Faktoren bei 80mg/dl gehalten: 1. Mobilisierung von Glykogen und Freisetzung von Glucose durch die Leber; 2. Freisetzung von FS durch das Fetgewebe; 3. Verschiebung der von Muskel und Leber verwendeten Brennstoffe von Glucose zu Fettsäuren Glykogenvorräte der Leber sind erschöpft: Gluconeogenese aus Lactat und Alanin geht weiter (ersetzt allderings nur bereits von der Peripherie verwertete Glucose); Da Gehirn Glucose vollständig oxidiert ist für Nettosynthese von Glucose eine C Quelle erforderlich; Ein Teil stammt aus dem Glycerin der Fettzellen, ein anderer Teil aus der Hydrolye von Muskelproteinen C.) Der erneute Sättigungszustand - nach einem Frühstück wird Fett genauso umgesetzt wie im normalen Sättigungszustand; Bei Glucose ist dies nicht der Fall; Die Leber nimmt zunächst keine Glucose aus dem Blut auf, sondern belässt sie dort für üeriphere gewebe; Leber betreibt weiter Gluconeogenese, wobei die neusynthetisierte Glucose zum Aufbau der erschöpften glykogenvorräte der leber dient; wenn der Blutzucker weiter steigt vollendet die leber das Auffüllen des Glykogen-Speichers und beginnt anschließend mit der Fettsäure-Synthese D.) was passiert bei längerem Fasten? - - - - 70kg Mensch hat Brennstoffreserven von 680000kJ; Energiebedarf/Tag liegt zwischen 6700kJ im Ruhezustand und 25000 kJ, je nach Ausmaß der Aktivität; gespeicherte Brennstoffe genügen also für ein bis dreimonatige Hungerperioden; Die KH Reserven sind schon nach einem Tag erschöpft; Selbst unter Hungerperioden muss der Blutzuckerspiegel über 40mg/dl gehalten werden; während einer Hungerperiode ist es höchste Priorität genügend Glucose für Gehirn und Erythrocyten zur Verfügung zu stellen; Vorstufen der Glucose fehlen jedoch; Die meiste Energie ist in den Fettsäuren der Triacylglycerine gespeichert; FS können jedoch nicht in Glucose umgewandelt werden (Acetyl-CoA kann kein Pyruvat bilden); Glycerinanteil der Triacylglycerine ist nicht ausreichend; Die einzige andere mögliche Glucose-Quelle sind AS aus dem Proteinabbau; da Proteine nicht gespeichert werden bewirkt jeder Abbau eine Einschränkung von Funktionen (zweite Priorität während einer Hungerperiode hat also die Erhaltung der Proteine); Proteinabbau wird verhindert, indem die verwendeten Energielieferanten von Glucose auf FS und Ketonkörper verlagert werden; Erster Fastentag: niedriger Blutzuckerspiegel führt zu vermindeter Insulin- und gesteigerter Glucagonsektretion; es folgt die Mobilisierung von Triacylglycerinen im Fettgewebe und die Gluconeogenese in der Leber (deckt ihren eigenen Energiebedarf 180 - - - - - - durch Oxidation von FS, die vom Fettgewebe stammen); dadurch steigt die Acetyl-CoA und Citratkonzentration an, wodurch die Glykolyse stoppt; niedriger Insulinspiegel bewirkt, dass Glucoseaufnahme im muskel verrringert wird; FS können jedoch frei passieren, weshalb auch der Muskel von Glucose auf FS als Brennstoff umschaltet; FS-Oxidation im Muskel stoppt die Umwandlung von pyruvat zu Acetyl-CoA (Acetyl-CoA bewirkt phosphorylierung und somit Inaktivierung des PyruvatDehydrogenase Komplexes); Pyruvat, Lactat und Alanin werden daher zur Leber exportiert und dort in Glucose umgewandelt; Glycerin aus Triacylglycerinen ist weiteres Edukt der Glucose-Synthese in der Leber; Proteolyse liefert auch C-gerüste für Gluconeogenese; während des Fastens werden abgebaute proteeine nicht ersetzt und dienen als C Quele für Glucose-synthese; zunächst werden proteine mit raschem Umsatz verwertet (Proteine des Darmepithels, Sekretionsproteine des pankreas); Die Proteolyse des Muskelproteins liefert einige C3 Vorstufen der Glucose; Muskelschwund muss jedoch gering gehalten werden, da das Überleben von vielen tierischen lebewesen davon abhängt, schnell laufen zu können xD Einschränkung des Muskelschwunds: nach etwa drei hungertagen bildet Leber große Mengen an Ketonkörper; ihre Synthese aus Acetyl-CoA nimmt deutlich zu, da der Citratzyklus nicht alle Einheiten oxidieren kann (wegen der Gluconeogenese kommt es zum Oxalacetat-Mangel, dem Akzeptor der Acetyl-Einheit), die durch FS Abbau entstehen; die ketonkörper werden ins Blut abgegeben, weshalb das gehirn nun zu einem Drittel Acetacetat anstelle von Glucose verwertet; auch der herzmuskel stellt sich auf ketonkörper um; Nach mehreren Wochen sind ketonkörper Hauptenergiequelle des Gehirns; sie liefern dort Acetyl-CoA, das vor ort in den Citratzyklus eingehen kann; Ketonkörper stellen Äquivalente der FS dar, welche die Blut-Hirn Schranke passieren können; pro Tag benötigt das gehirn dann nur noch 40 g Glucose (vgl. 120g am ersten Fastentag); die effiktive Umwandlung von FS in ketonkörper durch die Leber und ihre Verwertung durch das gehirn setzen den Glucosebedarf deutlich herab; es wird folglich weniger Muskelprotein (nur noch 20g, statt 75g am ersten Fastentag) abgebaut als in den ersten Fastentagen, was für das überleben entscheidend ist; Wenn die Fettdepots erschöpft sind beginnt der Proteinabbau in großem Stil; die Folge ist Muskelschwung und Tod durch Herz-, Leber- oder Nierenversagen; 181 Der Muskel - Kreatin-Kinase bildet aus Kreatin unter ATP Verbrauch Kreatinphosphat, wenn im Muskel ATP Überschuss herrscht. Dieses zerfällt spontan unter Bildung von Phosphat und Kreatinin, das mit dem Harn ausgeschieden wird. Die Kreatin-Konzentration steigt im Muskel, wenn mechanische Arbeit verrichtet wird, da Kreatinphosphat in der Rückreaktion aus ADP und orthophosphat ATP aufbaut. Kreatinphosphat stellt demnach einen Phsophorylpuffer dar (PGÜP ist höher, als das PGÜP von ATP). Kreatinphosphat stellt in den ersten 4 Sekunden Sprint die Hauptquelle an Energie dar. Muskelaufbau - Die Skelettmuskelfaser (7) mit ihrem Zellkern (8) ist ein langes Synzytium (Mehrkernige zelle). Sie wird von Bindegewebe (Endomysium; 6) umgeben. Das Bindegewebe ist für die Reisfestigkeit der Muskeln verantwortlich. Viele Muskelfasern bilden einen Primärbündel (5), der ebenfalls von Bindegewebe (3; Perimysium) umgeben ist. Mehrere Primärbündel bilden einen Sekundärbündel (4; makroskopisch sichtbare Faserung des Muskels), der vom Epimysium (2) umgeben ist. Zusammenhalt bekommen mehrere Sekundärbündel durch eine Faszie aus straffem Bindegewebe (1), die dem Epimysium folgt. - Die Skelettmuskelfasern enthalten mehrere myofibrillen (9), die aus einem Bündel von myofilamenten (10) bestehen. Myofilamente sind Aktin und Myosin (14). Es lässt sich die MLinie (11), und die Z Scheibe (12) erkennen. Zwei Z-Scheiben grenzen das Sakromer (13) ein. Muskelkontraktion - Der kontraktile Apparat der Muskelzelle bilden die Myosfilmante Aktin (7nm Durchmesser) und Myosin (15 nm Durchmesser). Sie sind so regelmäßig angeordnet, sodass sich eine Querstreifung ergibt - Die Kontraktion findet im Sakromer statt, wobei Myosin als Motorprotein der aktive Teil darstellt. Das Myosin Monomer besteht aus einem Schwanz, einem beweglichen Hals und einem Kopfteil, der mit dem Aktinfilament eine Querbrücke bildet und sie wieder löst. So bewegen sich die Aktinfilamente entlang der Myosinköpfchen in Richtung M Linie, wodurch das Sakromer verkleinert wird (I und H Bande werden kleiner; A Bande bleibt gleich). Volle Kontraktion = H Bande ist nicht vorhanden. 182 - Im entspannten Zustand hat der Muskel wenig Ca2+ Ionen im Sakroplasma. Dies hat zur Folge, dass die Myosinbindestelle am Aktinfilament für Myosin nicht zugänglich ist. Bei hoher Ca2+ Ionenkonzentration binden die Ionen an Troponin C, wodurch eine Konformationsveränderung bewirkt wird. Das aktive Troponin C kann Tropomyosin von der Myosinbindestelle „wegschieben“, wodurch Myosin andocken kann. Das Myosinmolekül durchläuft nach Bindung an das Aktinfilament folgende Veränderungen: Bindung an das Aktinfilament in Form von Myosin-ADP (Winkel 90°) Bewegung des Kopfes in Richtung M Linie (Winkel 50°). Diese Bewegung zieht das Aktinfilament um 10nm mit sich. Bindung eines Moleküls ATP (Freisetzung von ADP) bewirkt weitere Konformationsveränderung. Die Affinität zu Aktin wird verringert, wodurch Myosin von der Aktinbindestelle abdissoziiert. Die Abdissoziation bewirkt eine weitere Konformationsveränderung, die zur Spaltung von ATP zu ADP und Orthophosphat führt. Die Hydrolyseprodukte bleiben an Myosin gebunden. Die Spaltung von ATP bewirkt die Rückkehr in die ursprüngliche Konformation (Myosinkopf wandert um etwa 10 nm am Aktinfilament in entgegengesetzte Richtung entlang, wodurch der Winkel sich von 50° wieder auf 90° vergrößert). Dadurch wird eine neue Bindung an das Aktinfilament ermöglicht. Während der zweiten Bindung dissoziiert Orthophosphat vom Myosin ab, wodurch Myosin-ADP entsteht, das im 90° Winkel bindet. Myosin II hat zwei Köpfchen: ist an coiled coil Struktur gebunden, an die ein Protein hängt, das in einer Membran eines Vesikels gebunden ist; Myosin wandert entlang eines Aktinfilaments innerhalb der Zelle und schleppt das Vesikel von A nach B Die Leber 1.) Der Cori Zyklus (= indirekter ATP Transport von Leber zu Muskel) Der kontrahierende Skelettmuskel erhält von der Leber über das Blut Glucose, wodurch er durch die glykolytische Umwandlung der Glucose zu Lactat ATP gewinnt. Lactat wird zurück über das Blut zur Leber transportiert, welche unter ATP Verbrauch Glucose synthetisiert. Die Leber (altruistisches Organ) trägt die gesamte Stoffwechsellast des Muskelgewebes. Der Zyklus geht allerdings mit einem Verlust von 2 ATP pro transportierte Glucose einher, da in Leber 6 ATP verbraucht, im Muskel nur 4 ATP gewonnen werden. Das Koma stellt einen Schutzmechanismus vor einer weiteren Ansäuerung des Blutes durch Lactat während harter körperlicher Belastung dar. 183 2.) Der Alanin-Zyklus (indirekter Ammonium-Transport von Muskel zu Leber) Der AS-Abbau findet größtenteils in anderen Geweben als der Leber statt, da AS als Brennstoffquelle in Fastenperioden und bei längerer Anstrengung im Muskel verbrannt werden; Da dem Muskel die Enzyme des Harnstoffzyklus fehlen, kann der Stickstoff nicht in Form der Ammoniumgruppe freigesetzt werden; deshalb wird im Muskel zunächst wie in der Leber auch durch Transaminierung Glutamat aufgebaut, das (nicht durch Desaminierung wie in der Leber) durch weitere Transaminierung mit Pyruvat α-Ketoglutarat regeneriert; dabei entsteht Alanin, das durchs Blut zur Leber transportiert und dort abgebaut wird; Es ist auch möglich Glutamin durchs Blut zu transportieren: Glutamin-Synthetase katalysiert die ATP abhängige Synthese von Glutamin aus Glutamat und einem Ammonium-Ion; beide Stickstoffatome des Glutamin können in der Leber zu Harnstoff verwertet werden; Alanin-Zyklus Leber Muskel Pyruvat Pyruvat Glutamat Glutamat α-Ketosäure Energiegewinnung Abbau Desaminierung Blut α-Ketoglutarat α-Ketoglutarat Alanin α-Aminosäure Alanin 184 Teil 3: PRAKTIKUM Diagonaltechnik - - - - - - - - - es soll die Quartiärstruktur des Hb durch diagonale Elektrophorese aufgeklärt werden; um die einzelnen Untereinheiten zu identifizieren, muss Hb zerlegt und mit Hilfe der Sodiumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-PAGE) aufgetrennt werden; SDS-PAGE ist ein allgemeines Verfahren zu Bestimmung der Molekülmasse von Proteinen und zur Untersuchung von Nachbarschaftsbeziehungen zwischen Proteinen und zur Reinheitskontrolle; Die diagonale Elektrophorese findet in zwei senkrecht zueinander stehenden Geldimensionen statt: zunächst sorgt ein Cross-linker (hier: Diepoxybutan: DEB; verbindet Lys-Reste miteinander) für die Quervernetzung der einzelnen Proteinuntereinheiten; DEB weist zwei reaktive Zentren auf, wodurch eine kovalente Bindung zwischen zwei Proteinuntereinheiten über DEB ermöglicht wird; für eine erfolgreiche Trennung im Gel nach der Masse ist eine Denaturierung notwendig: Das anionische Detergenz SDS löst die nicht-kovalenten WW im Protein auf und bildet einen Komplex mit dem Protein (negativ geladenes SDS umhüllt das Protein micellenartig), β-Mercaptoethanol bricht die Disulfidbrücken; die quervernetzten und durch SDS komplexierten negativ geladenen Proteinuntereinheiten liegen nun in einer Kette vor; Die Eigenladung der proteine ist zu vernachlässigen, da die negaitve Ladung von SDS überwiegt; Die Auftrennung erfolgt nach der Masse; Grund: Größe ist proportional zur Masse, wenn alle Proteine in einer Kette vorliegen; Die größte Kette wandert am wenigsten Die elektrophoretische Auftrennung in der ersten Dimension erfolgt über zweigeteiltes Acrylamid-Gel (diskontinuierliche Elektrophorese): Das Sammelgel enthält 5% Acrylamid/Methylenbisacrylamid bei pH=6,8; Das darauf folgende Trenngel enthält 7 bis 20 % Acrylamid/Methylenbisacrylamid bei pH=8,8; durch Polymerisation von Acrylamid und Quervernetzung mit Methylenbisacrylamid bildet sich eine der Konzentration entsprechend dichte Matrix aus; In den Proben, die später in die Geltaschen pipetiert werden und im Gel selbst befinden sich Chlorid Ionen; im Laufpuffer ist Glycin enthalten; Die Chlorid-Ionen laufen sehr schnell (klein und negativ geladen), wohingegen Glycin sehr langsam läuft (groß, bei pH=6,8 neutral); zwischen diesen beiden Fronten bildet sich ein Spannungsgradient, in dem sich die Proteine unabhängig von ihrer Größe sammeln; so wird es ermöglicht, dass die Proteine als geschlossene Front das Trenngel erreichen und erst hier die Auftrennung stattfindet Im Trenngel läuft Chlorid weiter; Glycin wird negativ geladen und läuft schneller als die proteine, weshalb der Spannungsgradient zusammenbricht und die Proteine mit einer Geschwindigkeit entsprechend ihrer Größe durch das Gel laufen; Das Ergebnis bei quervernetzten Proteinen ist abhängig von der Anzahl der Untereinheiten des nativen Moleküls; handelt es sich z.B. um einen Trimer so erhält man drei Banden: die erste Bande ist die Monomer-bande, die zweite die Dimer-bande und die am wenigsten gewanderte Bande die Trimer-Bande; In der ersten Dimension werden sechs identische Hb Proben mit Puffer, aber unterschiedlichen DEB-Konzentrationen (eine enthält kein DEB; je mehr DEB, desto mehr Tetramere und weniger Monomere resultieren) versetzt und für 1h inkubiert; Die Zugabe von Saccharose dient der Verdichtung der Probe (erleichtert das pipettieren in die Gel-Taschen); Die Zugabe von Bromphenolblau ermöglicht später eine visuelle 185 Identifizierung der Banden; nach Denaturierung und zusätzlicher Hitzedenaturierung (5 minuten bei 85°C) werden die sechs Proben und der Marker (ermöglicht spätere Zuordnung der Banden zu bestimmten Massen) in die 7 Taschen pipettiert; - - - In der zweiten Dimension wird eine der erlangten Gelspalten herausgeschnitten und in Spaltpuffer, der das Oxidationsmittel Natrium-meta-Periodat (spaltet vicinales Diol) enthält, inkubiert; Dieser greift die durch den Cross-Linker entstandenen Verbindungen an und spaltet sie; der Gelstreifen wird nun senkrecht zur Richtung der ersten Elektrophorese in ein zweites Gel einpolymerisiert; da die Beweglichkeit der Proteine im zweiten Gel identisch ist mit der Beweglichkeit im ersten Gel und außerdem in allen Taschen Monomere vorhanden sind, sollte eine Diagonale resultieren; Nach der zweiten Dimension stellen wir fest, dass die Tetramerbande nach der ersten Dimension in vier gleich weit entfernte Banden aufgeteilt wurde; das Ergebnis: Hb besteht aus vier gleich schweren Untereinheiten reaktive Gruppen der Proteine: Aminogruppe von Lysin, Thiolgruppe von Cystein, Imidazolring von Histidin und Guanidinium Gruppe von Arginin (in bestimmten Fällen auch aromatische Seitenketten von Tyr, Trp und Phe); Die reaktivsten zentren der Nukleinsäuren sind 7N-Stellung des Guanins, 1N-Stellung der Adenins und 3N-Stellung des Cytosins; solche lassen sich am einfachsten Alkylieren oder Acylieren; Die Struktur eines Quervernetzungsreagenzes wird durch diese Reaktionszentren vorgegeben, um eine möglichst hohe Spezifität zu erreichen; 186 - Die Verknüpfung zweier Lysinreste mittels DEB verläuft unter Bildung eines vicinalen Diols; Nach der ersten Dimension wurde bewiesen, dass Hb aus 4 untereinheiten besteht; nach der zweiten Dimension wurde bewiesen, dass diese gleich groß sind; DNA - In diesem Versuch soll eine Plasmid-DNA durch Restriktion und Darstellung einer Gelelektrophorese untersucht werden; als Ausgangsmaterial dient ein Plasmid-Vektor, der aus E.coli isoliert wurde; zusätzlich soll eine Schmelzpunktbestimmung von der natürlichen DNA aus Kalbs-Thymus gemacht werden; TEIL 1 - Um die DNA aus E.coli zu extrahieren, haben wir zwei verschiedene Methoden verwendet; beide Methoden ermöglichen Lyse und Abtrennen der Zelltrümmer, sowie Aufreinigung der DNA; - Erste Methode (herkömmlich): Chloroform wird zur Reinigung der DNA und zum beseitigen der Zelltrümmer während der Zentrifugation verwendet; Zweite Methode (kommerzielles kit): DNA bindet fest an eine Membran, ein Waschpuffer sorgt während der Zentrifugation zum Reinigen und abtrennen der Zelltrümmer, nach dem Waschvorgang wird die DNA aus der Membran gelöst; - über eine Phasentrennung und mehrmahliges abzentrifugieren wird die DNA von den restlichen Zellbestandteilen getrennt; - Danach wird sie mit verschiedenen Restriktionsenzymen an spezifischen Basensequenzen geschnitten und in der Gelelektrophorese nach Größe aufgetrennt; - die DNA wandert im elektrischen Feld bei pH=7 im Agarose-Gel entsprechend ihrer Größe aufgrund der negativ geladenen Phosphatgruppen zum positiven Pol; kleine DNA Stücke wandern schneller, als große, da sie besser durch die Poren des Gels passen - Abschalten der Gleichspannung beendet die Elektrophorese; Die DNA Abschnitte werden mit Hilfe von Färbemittel sichtbar gemacht; mit Hilfe eines Markers lässt sich die Größe und damit auch Masse der DNA Abschnitte bestimmen; Ergebnisse - Tasche 1: Marker; - Tasche 2 (DNA nach Reinigung): 3 Banden (eine oberhalb 5700bp: stabförmige supercoiled Plasmid-DNA; sehr kompakt, wandert weit; ca. 10.000bp: die weniger gewanderte Bande enthält coiled DNA, weniger kompakt als supercoiled; die am wenigsten gewanderte Bande enthält entwundene und somit diffuse Plasmid.DNA) 187 - Tasche 3 + 4 (DNA + BAM HI bzw. DNA + HIND III): jeweils eine dicke Bande an der selben Stelle, da beide Restriktionsenzyme den Ring an nur einer Stelle zerschneiden Tasche 5 (DNA + beide Restriktionsenzyme): zwei Banden, die weniger gewnaderte ist der Vektor, die weiter gewnaderte das kleine insert-DNA-Fragment Tasche 6: wie Tasche 5 nur weniger DNA, identisch nur dünnere Banden Tasche 7 (wie Tasche 3, nur mit kommerzieller Methode): Im kommerziellen Kit konnten RNA nicht von DNA getrennt werden (keine RNAse vorhanden; im herkömmlichen Puffer schon), weshalb die kleinen RNA Abschnitte eine Wolke am Ende des Gels bildeten, sodass die kleinen DNA Abschnitte verdeckt wurden; TEIL 2 - - - - - - Die Denaturierung erfolgt durch Hitze, wobei die H-Brücken im Doppelstrang gebrochen werden, wodurch er in zwei Einzelstränge dissoziiert; um die Verhältnisse zwischen A-T und G-C- Basenpaarungen in der analysierten DNA einschätzen zu können, bedarf es einer Schmelzpunkt-Bestimmung; Der Schmelzpunkt ist die Temperatur, bei der die Hälfte aller H-Brücken gebrochen wurden; da G-C Paare drei H-Brücken ausbilden und somit stabiler sind, liegt der Schmelzpunkt bei einem hohen GC Gehalt umso höher Außerdem: Van-der-Waals WW sind bei G und C größer, als bei A und T; Stapelenergie ist bei GC größer; mehr Energie notwendig um G und C zu trennen; für die Schmelzpunktbestimmung nutzen wir ein Poly-dA-dT Strang (enthält nur AT Basenpaare) mit Mg, einen Poly-dA-dT Strang ohne Mg und die natürliche Kalbs-Thymus DNA; beide werden erhitzt, wonach eine Messung der Extinktion bei 260nm folgt; Die Extinktion bei 260nm ist dann gegeben, wenn die Doppelhelixstruktur gebrochen ist; so erhalten wird charakteristische Kurven, aus, deren Wendepunkte (Punkt, an dem die hälfte der H-Brücken gespalten sind) den Schmelztemperaturen entsprechen; Poly-dA-dT ohne Mg schmilzt bei 50°C als erstes; Poly-dA-dT mit Mg bei 75°C und zuletzt die natürliche DNA; das positiv geladene Mg hat also eine stabilisierende Wirkung auf die H-Brücken In der Zelle: Bei einem GC Gehalt von 50% resultiert eine Schmelztemperatur (Definition: Temperatur, bei der die DNA denaturiert ist, also die hälfte der H-Brücken gebrochen sind) von 90°C; für reine AT DNA ergibt sich 70°C; Lösungsmittel und Ionen beeinflussen die Stärke der H-Brücken und somit den Schmelzpunkt Immunologische Grundlagen - - Immunologie = alle Prozesse und Mechanismen, die zwischen Selbst (angeborene, körpereigene Strukturen) und Nicht-Selbst (alles körperfremde) unterscheiden; zum Nicht-Selbst gehören u.a. infektiöse Mikroorganismen (Protozoen, Pilze, bakterien, Mycoplasmen, Viren), Parasiten, Toxine, Tumoren, Transplantate oder Moleküle von genetisch nicht identischen Lebewesen; Die Immunantwort: Reaktion auf Körperfremde Substanzen mit Abwehr; Die Mechanismen der Immunantwort und ihre Entwicklung sind Untersuchungsgebiet der Immunologie; Immunanworten können angeboren sein (treten ohne vorherigen Kontakt mit dem fremden Stoff auf) oder sie sind erworben (erfordern zunächst Kontakt mit der körperfremden Substanz) 188 - Zur Abwehr von Antigenen existieren verschiedene Mechanismen, die miteinander wechselwirken; die unspezifische Abwehr (nicht adaptiv) beinhaltet Lysozym, Interferone (Proteine, die immunstimulierende Wirkung haben, werden von Leukozyten und TLymphozyten gebildet) und das Komplementsystem; die spezifische Abwehr (adaptiv) nimmt bei wiederholten Infektionen zu; Verantwortlich sind Zellen des Blut- und Lymphsystems: T und B Lymphozyten, sowie Fresszellen; zelluläre Abwehr wird direkt durch diese Zellen vermittelt, während humorale Abwehr durch die Antikörper und durch das Komplementsystem vermittelt wird; - Angeborene Immunität: beruht hauptsächlich auf T-Lymphocyten und Makrophagen und dient der Bekämpfung infizierter Zellen und der Erkennung neuer Antigene (Viren, Toxine, Bakterien); Barrieren für Fremdkörper: Die Haut und schleimigen Sekrete wirken als Barrieren, proteolytische Enzyme in den Körperflüssigkeiten können einige der eindringenden Organismen zerstören; Zellen mit spezifischer, angeborener Immunfunktion (Granulozyten und Monozyten) reagieren rasch auf eindringende Organismen, endocytieren und zerstören sie; sie produzieren darüber hinaus Substanzen, die gegen Infektionen schützen und die Entwicklung der Immunantwort fördern (Zytokine sind Botenstoffe über die das Immunsystem kommuniziert; dadurch wird das adaptive Immunsystem aktiviert) Das Komplementsystem: besteht aus vielen verschiedenen Faktoren, die im Blut als inaktive Vorstufen vorliegen; Die Aktivierung erfolgt entweder über den klassischen Weg durch Antigen-Antikörper Komplexe auf der Fremdzelle oder über den alternativen Weg durch Fremdpolysaccharide; es wird nach der Aktivierung eine Kaskade von Reaktionen ausgelöst, wobei u.a. durch proteolytische Spaltung eines Komplementfaktors eine aktive protease entsteht, die wiederum einen weiteren Faktor aktivieren kann; es resultiert letztlich die Bildung eines lytischen Komplexes, der die Membran von Fremdzellen zerstört; weiterhin kommt es durch das Komplementsytem zu einer Aktivierung von Makrophagen, was zur Phagocytose der Fremdzelle führt; Außerdem entstehen bei der Komplement-Reaktionskaskade Faktoren (Anaphylotoxine), die über die Freisetzung von Histamin aus Mastzellen und die Aktivierung neutrophiler Granulozyten die Bildung von Entzündungsherden fördern; Granulozyten (Fresszellen im Blut) zirkulieren im Blut, können aber schnell in die Gewebe wechseln, sobald Fremdkörper bestimmte Reize verursachen; zirkulierende Monozyten bewegen sich auch von Blut ins Gewebe, wo sie sich zu Makrophagen (Fresszellen im Gewebe) verwandeln; beide werden vom Komplementsystem aktiviert; Angeborene Immunität ist relativ unspezifisch, unterscheidet jedoch eindeutig zwischen Selbst und Nicht-Selbst; sie reagiert rasch und stellt eine schnelle erste Abwehr gegen unerwünschtes eindringen von Fremdkörpern zur Verfügung; - - - - Wie kommen die Antigen-Antikörper-Komplexe auf der Fremdzelle zustande, sodass das Komplementsystem und die Fresszellen aktiviert werden können? Antikörper mit vielen verschiedenen variablen Regionen schwirren im Blut herum und bewirken die Bildung der Antigen-Antikörper-Komplexe - - Erworbene Immunität: Das adaptive Immunsystem basiert auf die Antikörper, die von B-Lymphocyten hergestellt werden; Sie befinden sich entweder frei im Blut (humorele Immunität) oder als Rezeptoren an Lymphozyten (zelluläre Immunität) gebunden Antigene sind körperfremde Stoffe, die eine Gefahr für den Körper darstellen; kommt eine hoch entwickelter Organismus (erworbene Immunität bei Säugern und Vögeln am 189 höchsten entwickelt) mit ihnen in Kontakt, kann er die Fähigkeit erwerben, spezifisch auf sie zu reagieren; - - - Spezifische humorale Immunität: entsteht mittels großer, löslicher, globulärer Proteine (Immunglobuline = Ig oder Antikörper), die im Blut vorhanden und von B-lymphozyten produziert werden Säuger produzieren fünf Klassen von Immunglobulinen, die als Immunglobulin G, M, A, D und E bezeichnet werden; sie bestehen aus zwei identischen leichten (212 Reste) und zwei identischen schweren (450 Reste) Untereinheiten, die über Disulfidbrücken verknüpft sind; der variable Teil (Fab = F antigen bindend) des Antikröpers bindet an das Antigen; der kostante Teil (Fc = F cristallizing) besteht aus Effektoren für Komplementsystem und Maktrophagen; Antikörper weisen eine hohe Variabilität auf und können spezifisch an zwei Stellen Antigene binden (ermöglichen so Agglutination Y Form); Das Agglutinat = AntigenAntikörper-Komplex (hat häufig bereits die unerwünschten Eigenschaften des Antigens verloren) wird wie beschrieben von einer Fresszelle zerstört; - Das erworbene Immunsystem kann weit über eine Millionen verschiedener Antikörper produzieren; Die zahlreichen Unterschiede in den variablen Domänen in Ig entstehen durch die Neuanordnung mehrerer Gene: somatische Rekombination des genetischen Materials während der B-Zellen Reifung; dadurch sind immer sehr viele Ig mit unterschiedlicher variabler Regionen im Blut, die auf unterschiedliche Antigene reagieren; es kommt zur Bildung von Antigen-Antikörper-Komplexen; - IgM werden zu Beginn der Immunreaktion nach Kontakt mit Antigenen im Blut gebildet; sie sind Zeichen einer frischen Infektion; Diese Antikörper besitzen 10 Bindestellen für Antigene und führen zu einer starken Agglutination (effizient, aber unspezifischer) IgE bewirkt bei Antigenkontakt anaphylaktische (Histamin freisetzende) und allergische Reaktionen (Freisetzung von Granulozyten) und dient zum Schutz gegen Parasiten; es handelt sich um ein Membrangebundenes Ig, das praktisch nicht im Blut vorkommt; IgG ist am häufigsten vorhanden; wird in einer verzögerten Abwehrphase (3 Wochen) gebildet und bleibt lange erhalten; sie sind Zeichen einer Impfung oder frühen infektion; IgG wird aktiv über das Blut und in die Plazenta in den Fötus transportiert und sorgt dort nachgeburtlich für einen ersten Schutz gegen Infektionen; über zwei antigen-gebundene IgG wird das Komplementsystem aktiviert; Der Fc-Rezeptor vermittelt Phagocytose; - - 190 - IgA wird auf Schleimhäuten der Atemwege, der Augen, des Magen-Darmtrakts etc. gebildet und schützt dort vor Pathogene; IgD Funktion unbekannt Wie können nun die Makrophagen signalisieren, dass sie etwas zu fressen gefunden haben, und die B-Lymphozyten somit mehr spezifische Antikörper produzieren sollen? Die Makrophagen enthalten MHC Proteine auf der Oberfläche und signalisieren, was der richtige Antikörper war; das Signal wird von T-Zellen aufgenommen und durch Botenstoffe an B Zellen weitergegeben - - Die meisten Zellen tierischer Lebewesen (außer die Erythrocyten) exprimieren auf ihrer Oberfläche Moleküle, die von anderen, genetisch nicht identischen Lebewesen als nichtSelbst erkannt werden; Es handelt sich um MHC I Proteine; Killerzellen sind in der Lage zwischen körpereigenen und körperfremden Molekülen dieser Klasse zu unterscheiden; Außerdem erkennen sie, ob die MHC Proteine fremde Peptide präsentieren und eliminieren diese; diese Peptide stammen aus zytoplasmatischen Proteinverdau und dienen dazu Mikroorganismen die intrazellulär lokalisiert sind ausfindig zu machen; Antigen-präsentierende Zellen (Makrophagen und B-Lymphocyten) verarbeiten Antigene, die durch Endocytose aufgenommen wurden; Die durch Spaltung erhaltenen Antigen-Peptide werden den entsprechenden T-Helferzellen mit-Hilfe der MHC II Proteine gezeigt; - Spezifische zelluläre Immunität: Auslösung durch T-Lymphozyten; Die Präsentation von Antigenen durch MHC der Makrophagen löst die Teilung und Differenzierung der unspezifischen T-Zelle zur antigenspezifischen T-Helferzelle aus; diese produziert spezifische Botenstoffe; alle nicht spezifischen T-Lymphozyten nehmen diese Botenstoffe auf und spezialisieren sich zu antigenspezifischen T-Killerzillen; - Antigenspezifische Killerzellen vernichten Mikroorganismen und infizierte Zellen (Erkennung über MHC), T-Helferzellen und T-Supressorzellen beeinflussen die Immunreaktion, indem sie Zytokine u.a. produzieren, welche die Aktivierung und Reifung der B-Lymphocyten verstärken oder hemmen (somit wird das humorale Immunsystem aktiviert); Unterscheidung zwischen Selbst und Nicht-Selbst - Bricht die Immuntoleranz gegenüber dem Selbst zusammen entwickeln sich Autoimmunkrankheiten; gesunde Zellen und substanzen werden zerstört; Eine Allergie ist die Überreaktion des Immunsystems auf fremde Stoffe; nach einer Organtransplantation muss das Immunsystem mit Medikamenten unterdrückt werden, damit es nicht die fremden Zellen schädigt; 191 Über die an den Immunantworten beteiligten Zellen: - Monocyten werden im Knochenmark gebildet, circulieren etwa drei tage im Blut, um in gewebe im ganzen Körper zu migrieren (besonders in Leber, Milz und Bindegewebe); dort differenzieren sie sich zu Makrophagen; im nicht infizierten organismus sind diese passiv; ihre Hauptaufgabe ist die phagocytose alternder Zellen; im infizierten organismus vernichten sie Krankheitserreger an den Infektionsherden (Endocytose, Fusion mit Lysosom; Verdauung) über die nicht-adaptive Immunantwort; nach der Verdauung entstehen Peptide, die von MHC II Molekülen auf der Zelloberfläche präsentiert werden können; Diese können von B und T Lymphocyten mit passendem Rezeptor erkannt werden; sie leiten die adaptive Immunantwort ein; - Lymphozyten zirkulieren zwischen Blut und lymphatischen Geweben und sind für die adaptive Immunantwort verantwortlich; sie haben gelernt was Selbst und was NichtSelbst ist (durch MHC-Präsentation) und können auf Fremdkörper reagieren; sie werden nach dem ort ihrer Ausdiffernzierung in B-Lymphocyten (Knochenmark) und TLymphozyten (Thymus) unterteilt; - Alle T-Lymphozyten tragen auf ihrer Oberfläche einen T-Zell Rezeptor (TCR) und Marker Proteine; Sie werden aufgrund dieser Marker Proteine und ihrer Funktion in Unterklassen eingeteilt: man unterscheidet T-Helferzellen (CD4+), T-Supressor-Zellen und zytotoxische T-Zellen = T-Killerzellen (CD8+); eine Killerzelle erkennt spezifisch Antigene in verbindung mit MHC Proteinen der Klasse I (auf fast allen Säugerzellen) und soll infizierte Zellen abtöten; eine Helferzelle erkennt Antigene in verbindung mit MHC Klasse II (nur auf Zellen des Immunsystems), wodurch sie selektiv zur klonaren Vermehrung angeregt wird; Helferzellen sollen aktivierte B-Zellen zur weiteren vermehrung stimulieren; - B-Zellen produzieren Antikörper mit verschiedenen variablen Regionen und differenzieren nach ihrer Aktivierung (durch Bindung von Antigenen an die IgM und IgD Rezeptoren ihrer Oberfläche) zu Plasmazellen, welche große Mengen spezifischer Antikörper freisetzen; Die spezifische B-Zelle trägt den produzierenden Antikörper zusätzlich als Rezeptor auf der Zelloberfläche; nach der Aktivierung schalten die B-Zellen von der IgM auf die IgG Produktion um; - alle aktivierten Lymphocyten-Typen kommen auch als Gedächtniszellen vor; diese sorgen bei einem erneuten Antigenkontakt für eine raschere Immunantwort (Sekundärantwort) - Granulozyten gehören mit den Monocyten zu den Leukocyten; man unterscheidet drei Granulozyt-Typen: Neutrophile phagozytieren und zerstören Bakterien ; Basophile sezernieren Histamin und sind an allergischen Reaktionen beteiligt; Eosinophile spielen eine Rolle beim Angriff auf Parasiten 192 ELISA es sollen Immunologische Methoden näher gebracht werden: Antigen-Antikörper-Reaktionen werden in RIA und ELISA zur Quantifizierung bestimmter substanzen genutzt RIA / Radioimmunoassay: - empfindlichste methode (Antigenkonzentrationen von 0,5 pg/ml können bestimmt werden); deshalb können auch plasmabestandteile mit extrem niedriger Konzentration (Proteohormone, Steroidhormone, Drogen) bestimmt werden; Nachteil ist hier, dass ein Antikörper gegen das Antigen bekannt und verfügbar sein muss; außerdem erfordert die radioaktive Strahlung Sicherheitsmaßnahmen - Beispiel: Bestimmung von Prolactin im Serum; Im Versuch werden in Teströhrchen gleiche bekannte Mengen radioaktiv markiertes Prolactin (131Jod) mit gleichen Mengen Anti-Prolactin (Antikörper für Prolactin) zugegeben; außerdem wird eine bestimmte Menge an Serum mit unbekannter Prolactinkonzentration hinzupipettiert; Die zugrende liegende GG-Reaktion ist folgende (Ag und Ag* konkurieren um die selbe Bindestelle) 193 Ak + Ag ⇔ AkAg + Ag* ↓↑ AkAg* - - - Die markierten Komponenten müssen nach Erreichen des GG voneinander getrennt werden; Die Radioaktivität des Gemisches (ohne Ag*) ist ein maß für die Antigenkonzentration (Prolactin-Konzentration) des Serums; Da der Komplex aus Ak und Ag meist sehr viel schwerer ist als Ag, lassen sich als Trennmethoden alle Methoden, die auf das Molekulargewicht basieren auswählen; Häufig wird angewendet: Immunpräzipitation mit einem zweiten Antikörper; Hinzugabe einer Menge Anti-Anti-Prolactin (Antikörper gegen Anti-Prolactin) sorgt für die Bildung von Doppelantikörperkomplexe, die so schwer sind, dass sie abzentrifugiert werden können; Das Präzipitat wird auf seine Radioaktivität vermessen; Auftragung der Radioaktivität (Counts per minutes) gegen die Konzentration des Serums ergibt eine Verdrängungskurve ELISA / Enzyme-linked Immunoasorbant Assay: - Bestimmung der Antikörperkonzentration: Das Antigen ist hier immobile Phase und wird an einer Plastikoberfläche meist kovalent gebunden; in einem zweiten Schritt wird der entsprechende Antikörper in verschiedenen Konzentrationen zugegeben; überschüssige Antikörper werden durch Waschen entfernt; nach einer gewissen Inkubationszeit gibt man den zweiten Antikörper (der an einer alkalischen phosphatase gekoppelt ist) hinzu; der zweite Antikörper erkennt den ersten Antikörper; überschüssiger zweiter Antikörper wird ebenso durch Waschen entfernt - Die alkalische Phosphatase katalysiert eine Chromogenumwandlung; Das farblose Chromogen wird zu einem Farbstoff umgesetzt und vermessen; Die Menge an freigesetztem Farbstoff koreliert mit der Menge des ersten Antikörpers; - kompetetiver Test zur Bestimmung der Antigenkonzentration: Antikörper (immobile Phase) an eine feste Phase gebunden; Enzym markierte und nicht markierte Antigene konkurieren um die Bindung an die Antikörper; ist die Konzentration an unbekannten nicht markierten Antigenen hoch, so binden weniger Enzym markierte und mehr nicht markierte Antigene; durch die messung der Enzymaktivität, kann anschließend indirekt die Konzentration festgestellt werden; - - Im versuch soll die Antikörper-Konzentration im Serum vom Kaninchen ermittelt werden, die zuvor das Antigen BSA gespritzt bekommen haben; es werden nacheinander die Küvetten mit BSA (Antigen), mit dem ersten Antikörper (gegen BSA = Anti-BSA = α-BSA) und dem zweiten Antikörper (Anti-α-BSA), der mit einer alkalischen phosphatase gekoppelt ist, beschichtet; zwischen den beschichtungen werden die Küvetten einer 24-stündigen Inkubationszeit ausgesetzt, wonach sie immer gewaschen werden; zuletzt wird das Substrat (p-Nitrophenyl-phosphat) hinzugegeben; die alkalische Phosphatase spaltet die phosphatgruppe vom Substrat ab, wodurch eine Extinktionszunahme bei 405nm messbar ist; nach einer Inkubation wird im Photometer gemessen; Auftragung: -log(Verdünnung) gegen spezifische Extinktion bei 405 nm; es handelt sich um eine abfallende Kurve (kooperativ); der Titer ist diejenige Verdünnung, bei dem kein Anti-BSA mehr nachweisbar ist (Schnittpunkt mit der –log(Verdünnung) = x Achse); Der 194 Schnittpunkt ist bei 7, also einer Verdünnung von 10^-7; wir gehen davon aus, dass bei dem Titer kein Molekül Anti-BSA in der Lösung vorliegt, da die Extinktion gegen 0 geht; Berechnung der Antikörpermenge, ausgehend von der höchsten verdünnung, die noch Extinktion zeigt; Lamber Beersches Gesetz: Extinktion: E = c ⋅ d ⋅ ε c = Konzentration d = Dicke der Küvette ε = Extinktionskoeffizient für p-Nitrophenol c= E mmol mmol 0,7345 = = 0 , 039 = 0 , 039 2 3 cm d ⋅ ε 1cm ⋅18,5 mmol ml cm Das ist die Konzentration an Farbstoff in der Küvette Nummer 6 und bezeichnet damit den Substratumsatz. Substratumsatz Zeit ⋅ Enzymmenge Substratumsatz 0,039mmol Enzymmenge = = = 1,86 ⋅10 −6 mg mmol U ⋅ Zeit 300 min ⋅ 70 min ⋅mg spezifische Aktivität: U = Mit dieser bestimmten Enzymmenge (mE) kann nun die Stoffmenge (nE) an Enzym ermittelt werden, die sich in der Küvette Nummer 6 befindet. nE = mE 1,86 ⋅10 −7 mg = = 1,33 ⋅10 −11 mmol mg M 140000 mmol Da es sich hier um eine Verdünnung von 10-6 handelt, entspricht die Stoffmenge an Antikörper im Ausgangsserum n AK = 1,33 ⋅10 −5 mmol Daraus ergibt sich eine ml . Antikörperkonzentration von 1,995 mg ml im Serum. mg m AK = n AK ⋅ M IgG = 1,33 ⋅10 −5 mmol ⋅150000 mmol = 1,995 mg ml Immunologie 1.) Toxin-Neutralisation - - - Streptolysin O wird von A-Typ Streptokokken gebildet und zerstört u.a. Erythrozyten; Ist der Körper mit diesem bakterium infiziert, bildet er Antikörper (Antistreptolyin O); in diesem Versuch soll die Antikörperkonzentration von Antistreptolysin O in einem Patientenserum bestimmt werden, um dann zu entscheiden, ob der Patient akut mit Streptokokken infiziert ist; Es werden zwei Versuchsreihen angesetzt: eine mit dem zu untersuchenden Patientenserum und eine Positiv-Kontrolle (enthält Antikörper bekannter Konzentration); Die Seren werden mit dem Reduktionsmittel DTT verdünnt, das Antistreptolysin O aktiviert; nun wird die Verdünnung auf verschieden konzentrierte Streptolysin O Lösungen pipettiert, worauf sich Antigen-Antikörper Komplexe bilden; 195 - - Um freies Streptolysin O nachzuweisen werden gewaschene Erythrozyten aus Kaninchenblut hinzugegeben; In den Ansätzen mit noch freiem Streptolysin O werden die Erythrozyten zerstört; An den Überständen wird nach einer Zentrifugation mit dem Elisa Reader die Absorption bei 585nm (Häm) gemessen; freies Häm von zerstörten Erys befindet sich im überstand; Häm in noch intakten Erys befinden sich im Niederschlag; ERGEBNISSE - Die Serumkontrolle der Positiv-Kontrolle weist kein Strptolysin O auf; es resultiert wie zu erwarten ein farbloser Überstand und ein roter Niederschlag - Die Röhrchen mit geringer Antigenkonzentration sind ebenso farblos; alle Streptolysin O Moleküle sind mit den Antikörpern agglutiniert, weshalb keine Erythrozyten zerstört werden konnten; somit ist der Überstand farblos, weil das Häm vollständig innerhalb der Erythrozyten, die einen dunkelroten niederschlag bilden, vorliegt; In farblosen Überständen wurde also das Toxin durch den Antikörper vollständig neutralisiert;; Ab einer Antigenkonzentration von blubb sind die Lösungen rötlich, wobei die Farbintensität mit steigender Antigenkonzentration steigt und die Intensität des Niedrschlags abnimmt; hieraus ergibt sich der Titer (letzter farbloser Überstand) - Die Serumkontrolle des Patienten-Serums weist kein Streptolysin O auf und ist daher wie zu erwarten farblos; Die weiteren Röhrchen mit geringer Antikörperkonzentration sind ebenso farblos, jedoch ist der Titer ein anderer als bei der Positiv-Kontrolle mit bekannter Antikörperkonzentration; - - vergleicht man den Titer der Positiv Kontrolle mit dem Titer des Petientenserums, so lassen sich Titer-Grenzen festlegen, ab denen ein Patient als erkrankt gilt; diese Grenze ist in diesem Fall 200 IU/ml; erreicht wurde ein Titer von 75IU/ml (bestimmt durch die Abschätzung ab welcher Konzentration der Überstand farbig wird) bzw. 130 IU/ml (aus dem angenäherten Graphen abgelesen; der so genannte EC50 Wert), weshalb keine akute Infektion vorliegt; dennoch existierte eine frühere Infektion, da Antikörper im Patientenserum nachgewiesen werden konnten; Die im Niederschlag befindlichen weißen Erythrozyten bei Röhrchen mit farbigem Überstand, sind durch Hämolyse entstandene Erythrozyten-ghosts ohne Inhalt (leere Hülle); das frei gewordnene Hb befindet sich in Lösung 2.) Immunelektrophorese - In diesem Versuch werden Humanserumalbumin, IgG und IgA durch Antikörper nachgewiesen; hierzu wird ein Antigengemisch nach Ladung und Größe aufgetrennt; 196 - - - Als Antigengemisch dient ein Humanserum, das auf drei Agaroseeplatten aufgetragen wird; es folgt die Auftrennung durch Elektrophorese, wobei bei leicht alkalischem pH Wert Die Ig in entgegengesetzte Richtung zum Serumalbumin laufen; Im Anschluss werden auf jeder der drei Platten ein spezifischer Antikörper längs der Diffusionsrichtung aufgetragen; er diffundiert während einer Inkubationszeit durch das Gel und bildet eine Präzipitationslinie durch Agglutination, wenn er auf eine äquimolare Menge gewanderter Antigene trifft; In den Bereichen, wo ein Antikörperoder Antigenüberschuss existiert bilden sich auch Antigen-Antikörper-Komplexe; diese können aber in Lösung gehalten werden Mit dieser Methode konnten alle drei Proteine im Gemisch qualitativ nachgewiesen werden; Die Dicke der Präzipationslinie ermöglicht eine grobe Einschätzung über die Menge; Wenn die Präzipationslinien nahe am Humanserum-Auftragsloch liegen, bedeutet dies, dass die Proteine kaum geladen sind (kaum Wanderung im elektrischen Feld; pH Wert der Pufferlösung am isoelektrischen Punkt); HSA ist das kleinste Serumprotein und am häufigsten vorhanden (weit gelaufen und dicke Linie); IgA ist selten vorhanden (dünne Linie) 3.) Ouchterlony-Diffusionsmethode - - - - In diesem Versuch soll nachgewiesen werden, ob ein mit BSA immunisiertes Kaninchen Antikörper gebildet hat und in welcher Konzentration diese vorliegen Es wird eine BSA-Verdünnungsreihe hergestellt; Die einzelnen Verdünnungen werden kreisförmig auf das Agarosegel aufgetragen; In der Mitte des Kreises wurde das Kaninchenserum, das auf Anti-BSA getestet werden soll pipettiert; Durch Diffusion innerhalb der Inkubationszeit ergibt sich nebenstehende Präzipationsspirale; diese entsteht immer beim aufeinandertreffen äquimolarer Mengen Antikörper und Antigen; Wenn davon ausgegangen wird, dass Antikörper und Antigen gleich schnell diffundieren (gleiche größe), entspricht die Antikörperkonzentration im Serum der aufgetragenen Antigenkonzentration beim unteren Loch, da Antigen und Antikörper sich in der Mitte getroffen haben; Durch den Vergleich der molaren Konzentration ergibt sich die Konzentration an gebildeten Antikörper; 197 Glyoxisomen - Glyoxisomen sind Zellorganellen der Pflanzen, die hauptsächlich in keimenden Samen vorkommen; sie verwenden als Energiespeicher Fettsäuren und enthalten spezielle Enzyme der Fettsäureoxidation; Ziel des Versuchs ist die Isolierung der Glyoxisomen; Um diese nachzuweisen, benötigen sie ein Merkmal, das sie charakterisiert: Das Leitenzym; Im Fall der Glyoxisomen gibt es zwei Leitenzyme: Malat-Synthase und Isocitrat-Lyase; letztere wird zur eindeutigen Identifizierung der Glyoxisomen verwendet; Peroxisomen: - alle eukaryotischen zellen enthalten peroxysomen (kleine, mit einer membran abgegrenzte Vesikel; Durchmesser 0,2-1,5 μm); Die membran besitzt keine Investigationen (Einstülpungen) und ist ohne Ribosomenbesatz; Peroxysomen enthalten Enzyme, welche die bei verschiedenen Stoffwechselvorgängen entstehenden freien Radikale unschädlich machen; - Glyoxisomen sind spezialisierte Peroxisomen, die nur in Pflanzen und hier vor allem in keimenden Samen vorkommen; In ihnen sind Enzyme der Fettsäureoxidation und des Glyoxylatzyklus lokalisiert; Sie entstehen mit Beginn der Keimung in fettspeichernden Zellen des Endosperms oder der keimblätter durch Abschnürung vom ER; Enzyme werden erst später eingelagert; Funktion der Glyoxisomen: - da der junge Keimling nicht zur Photosynthese fähig ist, aber dennoch KH als Baustoff notwendig sind (Zellulose-Zellwand) enthalten Glyoxysomen einen Teil der Enzyme für den Fettsäureabbau und für die Gluconeogenese; - der Glyoxylat-Zyklus beschreibt die Überführung von Acetyl-CoA (Endprodukt des Fettsäureabbaus) zu Succinat - Glyoxylat-Zyklus kann auch von bakterien durchgeführt werden (hier sind Enzyme an/in Cytoplasmamembran und im Cytoplasma lokalisiert) - Der Glyoxylat-Zyklus in Pflanzenzellen ist in drei Kompartimente aufgeteilt, zwischen denen ein reger Stoffaustausch herrscht; 1.) Abbau der Fettsäuren im Glyoxysom zu Acetyl-CoA über β-Oxidation 2.) Acetyl-CoA + Oxalacetat Citrat Isocitrat; Isocitrat wird von Isocitrat-Lyase (erstes Leitenzym) zu Succinat und Glyoxylat gespalten 3.) Succinat wird in die Mitochondrien transportiert und dort im Citratzyklus zu Oxalacetat umgesetzt; Durch Transaminierung entsteht Aspartat, das wieder in die Glyoxysomen gelangt; erneute Transaminierung ergibt Oxalacetat, dass wieder mit Acetyl-CoA kondensieren kann 4.) Glyoxylat wird durch Malat-synthase (zweites leitenzym) mit Acetyl-CoA zu Malat; Malat wird ins Cytoplasma transportiert und steht hier zur Synthese von Glucose zur verfügung - wenn der Glyoxylat-Zyklus ohne andere Kompartimente verläuft, wird lediglich wie im Citratzyklus auch, Acetyl-CoA verwendet um NADH+H+ zu generieren; jedoch wird Succinat aus dem zyklus augeschleust; 198 Ausgangsstoff waren vorgekeimte Sonnenblumenkerne (enthalten Fettspeicher, weshalb Glyoxylatzyklus wichtig ist und somit Glyoxisomen vorkommen) Isolierung und Aufreinigung: 1.) mörsern in Puffer (zerstören der Zellwände); 2.) Filtration durch Gaze (Glyoxisomen werden von grpßen Zelltrümmern getrennt) 3.) Zentrifugationsschritte (Glyoxisomen letztlich im Pellet; resuspension in Puffer) 4.) Saccharosegradient pipettieren; Dichtegradienten-Zentrifugation; Auftrennung der restlichen zellorganellen nach ihrer Dichte (in höher konzentrierten Saccharose-Schicht befinden sich die schwersten Organellen, z.B. Zellkern; in nieder konzentrierten die leichtenm z.B. ER; Organellen befinden sich zwischen den Sachharosephasen, da Dichte nicht identisch mit Saccharosephase) 5.) Absaugen aller Fraktionen in getrennte Behälter, wobei sie durch eine Photozelle fließen (Menge der aromatischen AS wird bestimmt; Rückschlüsse auf Proteinzusammensetzung) 6.) Ergebnisse visualisiert ein Schreiber (grßer Peak viele aromatische AS) 7.) Aufgrund dieser Ergebnisse wird Fraktion 1,2,3 für eine nähere Untersuchung mittels Brad-Ford Methode ausgewählt Proteinbestimmung nach Bradford: Die einzelnen Proteinfraktionen werden verdünnt und mit Bradford-Reagenz versetzt; durch die Bindung des Frabstoffes im Bradford Reagenz an das Protein kommt es zur Verschiebung der Absorption, die im Photometer messbar ist; die Fraktion mit der höchsten 199 Proteinkonzentration ist die Fraktion, wo sich zur höchsten Wahrscheinlichkeit die Glyoxysomen mit ihrem leitenzym aufhält Bestimmung der Enzymkinetik: Nun muss dum Nachweis der Glyoxisomen die katalytische Aktivität des Leitenzyms Isocitrat-Lyase gemessenen werden; Zugabe von Puffer, Phenylhydrazin und zum Starten der Reaktion Isocitrat; Reaktion von Glyoxylat (entsteht durch Isocitrat-Lyase Reaktion aus Isocitrat) mit Phenylhydrazin zu Glyoxylsäure-Phenylhydrazon mit spezifischer Extinktion; Die Extinktionszunahme nach der Zeit ergibt die Produktbildung des Enzyms; So lässt sich auch die spezifische Aktivität des Enzyms berechnen (siehe ELISA); Aufgrund der Enzymaktivität konnten Glyoxisomen eindeutig in allen drei Fraktionen nachgewiesen werden (Glyoxisomen haben hohe Volumen-, Masse- bzw. Dichte-Diversität gehabt) Bromelain - - In diesem Versuch sollen Bromelaine (Proteasen der Ananasfrucht) isoliert, aufgereinigt und enzymkinetisch untersucht werden Bromelaine sind Cystein-Endopeptidasen; man unterscheidet u.a. Ananain und Comosain (beide nur im Stamm), Papain, das basische Stammbromelain (Hauptkomponente des Stammes; enthält viel Arginin und Lysin; IEP = 9,5) und das saure Fruchtbromelain (Hauptkomponente der Frucht; IEP = 4,6) Stammbromelain und Fruchtbromelain sind mit den gleichen Zuckerresten glykosyliert Bromelaine haben medizinische verwendung: nach oraler Einnahme erscheinen sie in aktiver Form im Blut; helfen beim Auflösen von Proteinkapseln abgestorbener Zellen und zellfragmente, die durch Ödeme verursacht werden (Entzündete gelenke reagieren gut auf Bromelain-Therapie) Methoden zur Trennung von Proteinen - - - SDS-PAGE: siehe Diagonaltechnik Säulenchromatographie: stationäre Phase sind poräse Gelperlen (Molekularsieb); Probe wird zusammen mit Elutionspuffer (Schutz vor Austrocknung der Säule und mobile Phase) auf die Säule gegeben; kleine Moleküle dringen in die Gelporen ein, große wandern außen herum, da sie nicht durch die Poren passen; Proteine passen nicht durch die Poren und wandern somit schneller; mit diesem verfahren können Ionen und andere kleine Moleküle von proteinen getrennt werden; Dialyse: Probe wird in einen Schlauch mit semipermeabler Membran gegeben, durch die nur die kleinen Moleküle austreten können; Der Schlauch endet in einem Puffer, in dem die Proteine landen; Die kleinen Moleküle hingegen wandern durch die membran bis zu einem Konzentrationsausgleich; Wiederholung dieses Vorgangs reduziert die Verunreinigung der Proteinprobe mit kleinen Molekülen erheblich Affinitätschromatographie: Affinität der Proteine zu bestimmten chemischen Gruppen wird zur Auftrennung genutzt; Säule wird z.B. mit Antikörpern gegen das abzutrennende Protein bestückt und mit dem Proteingemisch durchlaufen; das protein kann nach Zugabe von gelösten antikörpern wieder eluiert und als gereinigte Probe aufgefangen werden; nach dem gleichen Prinzip kann auch nach hydrophilen und hydrophoben Proteinen getrennt werden; ebenso möglich: Säule mit Metallionen bestücken, an die ein bestimmtes Protein komplexieren soll; 200 - - Aussalzung: bestimmte Proteine fallen ab einer bestimmten Salzkonzentration aus, weshalb eine Fraktionierung möglich ist Isoelektrische Fokussierung: Auftrennung aufgrund der unterschiedlichen isoelektrischen Punkte; Proteine werden ohne SDS Zusatz in einer Elektrophorese durch einen pH Gradienten aufgetrennt; das Protein wandert solange, bis der pH seines isoelektrischen Punktes erreicht ist; Anionenaustauschchromatographie (verwendet): Proteingemisch wird auf die DEAE-Cellulose-Säule (DEAE = Diethylaminoethyl) gegeben; DEAE-Cellulose ist positiv geladen und bindet Anionen; Vor der Probenzugabe sind daran Citrat-Ionen gebunden; Die basischen Proteine haben jedoch eine größere Affinität zu DEAECellulose und verdrängen Citrat, das in Lösung übergeht; so können die basischen Stammbromelaine entfernt werden; Die aufgefangene Lösung enthält nur saure Proteine; Vorgehensweise: 1.) 2.) 3.) 4.) 5.) 6.) Zerstörung der Zellwände durch Entsafter Zentrifugation des rückständigen zellsaftkonzentrats Filtration des Überstands durch eine Gaze Fällung des Flitrats durch Ammoniumsulfatlösung Zentrifugation und Homogenisierung des Rückstands mit Natriumacetatpuffer Erneute Zentrifugation und Eluierung des Überstands in DEAE-Cellulose mit Natriumcitratpuffer 7.) Durchführung der Anionenaustauschchromatographie Die DEAE-Cellulose Säule ist an einem Photometer angeschlossen, dass die Absorption bei 280nm (hier absorbieren aromatische AS) misst; so kann festgestellt werden, wann die Proteine aufgefangen werden; der Schreiber gibt das Signal der Absorption wieder; so wissen wir in welcher Fraktion Proteine aufgefangen wurden; da die basischen Proteine an die DEAE Säule binden (erst bei erhöhter Citrat-Konzentration in der mobilen phase, werden sie wieder eluiert), kommen die sauren Proteine zu erst! Um die Proteasen auf der Säule zu schützen, wurden sie mit Hg inaktiviert; 8.) Aktivitätsmessung Die aufgereinigten Proteine werden in einer wässrigen Lösung von BAPNA, DMSO, Cystein und Testpuffer hinzu gegeben; Cys dient zur Reaktivierung der proteasen, indem es die durch Hg entstandene Disulfidbindung spaltet; BAPNA (α-N-benzoyl-arginin-p-nitroanilid) wird durch die Cysteinpeptidase abgebaut; Das Produkt (p-Nitro-Aminobenzol) weist ein Absorptionsspektrum bon 405nm auf; Die Verfolgung der Zunahme an spezifischer Extinktion bei dieser Wellenlänge lässt rückschlüsse auf die Enzymaktivität zu (Bestimmung von KM mittels Lineweaver-Burk; es wurden sechs Ansätze mit verschiedenen BAPNA Konzentrationen gemacht) 201 Anionentransport - - - - - - im vorliegenden Fall wird eine erleichterte Diffussion gemessen; Erythrozyten lassen sch sehr vorsichtig auflockern, sodass die membran größere Poren bekommt, durch die das Hb und die wenigen übrigen, zellständigen Proteine ausfließen; Durch Inkubation in Pufferlösung bei 37°C wird die membran „unverletzt wieder geschlossen; Die Erythrozyten sind blass-rosa (erythrozyten-ghosts), weisen aber noch typische Transportfunktionen auf; In der porösen Phase kann die Zelle mit dem gewünschten Transportsubstrat beladen werden; anschließend kann der Transport von innen nach außen verfolgt werden; Ein Transportsystem hoher Kapazität ist der Anionentransporter in der Erythrozytenmembran, das unter physiologischen Bedingungen dem Durchtritt von Clund HCO3- dient; Das System ist notwendig, weil während der Sauerstoffbindung des Hb in der Lunge und der Sauerstoffabgabe des Hb in der Peripherie auch ein kräftiger Cl- und HCO3- Ein- und Austransport ablaufen muss; Mit geringeren Geschwindigkeiten transportiert das System auch andere Anionen wie Sulfat (größer als Chlorid); Zur Messung werden die Erythrozyten während der porösen Phase mit radioaktivem 35S-Sulfat beladen; gemessen wird dann der Austausch zwischen radioaktiven Sulfat (innen) und Sulfat (außen) Die Messung der radioaktiven Strahlung der aus der Zelle diffundierenden Isotope über einen längeren zeitraum mittels LSC (liquid szintillation counter) lässt rückschlüsse auf die Transportleistung des Carriers zu. Eine zweite Erythrozytenprobe wird mit Persantin (Inhibitor des Antiporteers) versetzt; hier ist die Zunahme der cpm (counts per minutes) über die zeit (Steigung der Kurve) wesentlich geringer; Es handelt sich um eine Sättigungskurve; Der Transport verläuft solange, bis die Konzentrationen außen und innen gleich sind; 202