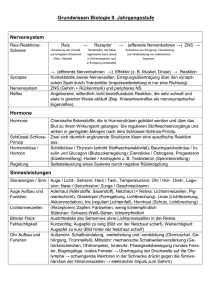
Aus dem Zentrum für Pathologie der Universität zu Köln Allgemeine
Werbung

Aus dem Zentrum für Pathologie der Universität zu Köln Allgemeine Pathologie und Pathologische Anatomie Direktor: Universitätsprofessor Dr. med. H. P. Dienes Immunreaktivität von MUC1 in kolorektalen Adenomen und Karzinomen unter besonderer Berücksichtigung der Glykosylierungsabhängigkeit Inaugural-Dissertation zur Erlangung der Doktorwürde der Hohen Medizinischen Fakultät der Universität zu Köln vorgelegt von Sonja Christiane Zuber aus Donaueschingen promoviert am 13. Oktober 2010 Gedruckt mit Genehmigung der Medizinischen Fakultät der Universität zu Köln 2010 Dekan: Universitätsprofessor Dr. med. J. Klosterkötter 1. Berichterstatterin/Berichterstatter: Universitätsprofessor Dr. med. S. E. Baldus 2. Berichterstatterin/Berichterstatter: Professor Dr. med. S. P. Mönig Erklärung Ich erkläre hiermit, dass ich die vorliegende Dissertationsschrift ohne unzulässige Hilfe Dritter und ohne Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe; die aus fremden Quellen direkt oder indirekt übernommenen Gedanken sind als solche kenntlich gemacht. Bei der Auswahl und Auswertung des Materials sowie bei der Herstellung des Manuskriptes habe ich, außer von Herrn Universitätsprofessor Dr. med. S. E. Baldus, keine Unterstützungsleistungen erhalten. Weitere Personen waren an der geistigen Herstellung der vorliegenden Arbeit nicht beteiligt. Insbesondere habe ich nicht die Hilfe einer Promotionsberaterin/eines Promotionsberaters in Anspruch genommen. Dritte haben von mir weder unmittelbar noch mittelbar geldwerte Leistungen für Arbeiten erhalten, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertationsschrift stehen. Die Dissertationsschrift wurde von mir bisher weder im Inland noch im Ausland in gleicher oder ähnlicher Form einer anderen Prüfungsbehörde vorgelegt. Köln, den 20.01.2010 Die dieser Arbeit zugrunde liegenden Daten wurden von mir persönlich im Archiv des Zentrums für Pathologie der Universität zu Köln (Direktor: Herr Universitätsprofessor Dr. med. H. P. Dienes) erhoben. Die in dieser Arbeit angegebenen immunhistochemischen Färbungen wurden von mir selbst im Labor von Herrn Universitätsprofessor Dr. med. S. E. Baldus durchgeführt. Die Beurteilung und Auswertung der Präparate wurde nach entsprechender Anleitung durch Herrn Universitätsprofessor Dr. med. S. E. Baldus von mir selbstständig durchgeführt. Die Fotografien der immunhistochemischen Färbung wurden mir von Herrn Universitätsprofessor Dr. med. S. E. Baldus zur Verfügung gestellt. Danksagung Für die Möglichkeit, meine Dissertation am Zentrum für Pathologie der Universität zu Köln erstellen zu können, danke ich Herrn Universitätsprofessor Dr. med. H. P. Dienes. Weiterhin bedanke ich mich bei Herrn Universitätsprofessor Dr. med. S. E. Baldus für die Überlassung des Themas und die freundliche und hilfsbereite Unterstützung bei der Durchführung dieser Arbeit. Ebenso bedanke ich mich bei Herrn Prof. Dr. rer. nat. F.-G. Hanisch und Frau Dr. rer. nat. K. Engelmann, Zentrum für Biochemie der Universität zu Köln, für die freundliche Bereitstellung der MUC1-spezifischen monoklonalen Antikörper, sowie die hilfsbereiten und fachkundigen Gespräche bei der Planung dieser Arbeit. Insbesondere bedanke ich mich bei Frau S. Landsberg, biologisch-technische Assistentin, für ihre freundliche und fachkundige Unterstützung bei der praktischen Umsetzung der immunhistochemischen Färbung. Gewidmet meiner Familie Inhaltsverzeichnis Inhaltsverzeichnis ___________________________________________________________________________ 1 Einleitung _____________________________________________________________ 1 1.1 Kolorektale Adenom-Karzinom-Sequenz _________________________________ 1 1.1.1 Definition _______________________________________________________ 1 1.1.2 Epidemiologie ____________________________________________________ 1 1.1.3 Ätiologie ________________________________________________________ 3 1.1.4 Histomorphologische Einteilung ______________________________________ 7 1.1.4.1 Kolorektale Adenome __________________________________________ 7 1.1.4.2 Kolorektale Karzinome _________________________________________ 9 1.1.5 Therapeutische Aspekte ___________________________________________ 13 1.1.5.1 Prognostische Parameter _______________________________________ 13 1.1.5.2 Therapieoptionen _____________________________________________ 15 1.1.6 Molekulare Kanzerogenese _________________________________________ 19 1.2 Muzine___________________________________________________________ 26 1.2.1 Grundstrukturen der Muzine ________________________________________ 26 1.2.1.1 Synthese der Oligosaccharidketten _______________________________ 32 1.2.2 MUC1: Struktur__________________________________________________ 33 1.2.3 MUC1: Biologische Relevanz _______________________________________ 36 1.2.4 Expression von Muzinantigenen im Kolon _____________________________ 39 1.2.4.1 Expression von Muzinantigenen in normalem Kolongewebe___________ 39 1.2.4.1.1 Peptid-Core-Antigene ______________________________________ 39 1.2.4.1.2 Kohlenhydratantigene ______________________________________ 40 1.2.4.2 Expression von Muzinantigenen in kolorektalen Adenomen und Karzinomen _________________________________________________________ 41 1.2.4.2.1 Peptid-Core-Antigene ______________________________________ 42 1.2.4.2.2 Kohlenhydrat-Core-Antigene ________________________________ 43 1.2.4.2.3 Kohlenhydratantigene der Backbone- und peripheren Region _______ 44 1.3 2 Fragestellung der Arbeit _____________________________________________ 46 Material und Methoden ________________________________________________ 47 2.1 Patienten und Gewebe _______________________________________________ 47 2.2 MUC1-spezifische monoklonale Antikörper _____________________________ 47 2.2.1 HMFG2 ________________________________________________________ 47 2.2.2 BCP8 (#133) ____________________________________________________ 48 Inhaltsverzeichnis 2.2.3 115D8 (#148) ___________________________________________________ 48 2.2.4 MA695 (#154) ___________________________________________________ 48 2.3 Immunhistochemische Methode _______________________________________ 48 2.3.1 Ablauf der Färbung _______________________________________________ 48 2.3.1.1 Allgemeines über die Labeled-Streptavidin-Biotin-Methode ___________ 48 2.3.1.2 Coverplate-Methode __________________________________________ 49 2.3.1.3 Entparaffinierung ____________________________________________ 50 2.3.1.4 Partielle Deglykosylierung durch Vorbehandlung mit Neuraminidase ___ 50 2.3.1.5 Partielle Deglykosylierung durch Vorbehandlung mit Perjodat _________ 50 2.3.1.6 Ohne Vorbehandlung _________________________________________ 51 2.3.1.7 Hemmung der endogenen Peroxidase _____________________________ 51 2.3.1.8 Hemmung unspezifischer Proteinbindungen _______________________ 51 2.3.1.9 Die Immunmarkierung ________________________________________ 51 2.3.1.10 Markierung mit dem Sekundärantikörper ________________________ 52 2.3.1.11 Kopplung des Meerrettichperoxidase-konjugierten Streptavidins _____ 52 2.3.1.12 Entwicklung ______________________________________________ 52 2.3.2 Rezepte ________________________________________________________ 53 2.3.2.1 Zitratpuffer _________________________________________________ 53 2.3.2.2 Azetatpuffer _________________________________________________ 53 2.3.2.3 Peroxidase-Blocker ___________________________________________ 53 2.3.2.4 TBS-Puffer (tris buffer solution) _________________________________ 53 2.3.2.5 Entwicklerlösung _____________________________________________ 53 2.3.2.5.1 Lösung I _________________________________________________ 53 2.3.2.5.2 Lösung II ________________________________________________ 53 2.3.2.6 Hämalaun __________________________________________________ 54 2.3.3 Lichtmikroskopische Auswertung____________________________________ 54 3 Ergebnisse ___________________________________________________________ 55 3.1 Immunreaktivität MUC1-spezifischer monoklonaler Antikörper in kolorektalen Adenomen vor und nach partieller Deglykosylierung ____________________________ 57 3.1.1 HMFG2 ________________________________________________________ 59 3.1.2 BCP8 __________________________________________________________ 61 3.1.3 115D8 _________________________________________________________ 63 3.1.4 MA695 ________________________________________________________ 65 3.2 Immunreaktivität MUC1-spezifischer monoklonaler Antikörper in kolorektalen Karzinomen vor und nach partieller Deglykosylierung ___________________________ 67 3.2.1 HMFG2 ________________________________________________________ 68 3.2.2 BCP8 __________________________________________________________ 69 3.2.3 115D8 _________________________________________________________ 70 3.2.4 MA695 ________________________________________________________ 72 4 Diskussion ___________________________________________________________ 74 4.1 Relevanz von MUC1 in der kolorektalen Adenom-Karzinom-Sequenz _________ 74 4.2 Expression von MUC1 in der Adenom-Karzinom-Sequenz __________________ 76 4.3 Methoden der partiellen Deglykosylierung _______________________________ 80 4.3.1 Perjodat-Oxidation _______________________________________________ 80 4.3.2 Neuraminidase-Behandlung ________________________________________ 82 4.4 Fazit _____________________________________________________________ 85 5 Zusammenfassung _____________________________________________________ 86 6 Literaturverzeichnis ___________________________________________________ 88 7 Abbildungsverzeichnis ________________________________________________ 106 8 Tabellenverzeichnis ___________________________________________________ 107 9 Lebenslauf __________________________________________________________ 109 Einleitung 1 1 Einleitung ___________________________________________________________________________ 1.1 Kolorektale Adenom-Karzinom-Sequenz 1.1.1 Definition Die kolorektale Adenom-Karzinom-Sequenz beschreibt die morphologischen und genetischen Ereignisse, die über ein Adenom zu einem kolorektalen Karzinom führen. Adenome sind gutartige, polypös erhabene epitheliale Tumoren der Dickdarmschleimhaut mit drüsiger Differenzierung. Sie können sich im Laufe der Zeit über eine Veränderung ihres Dysplasiegrades in ein kolorektales Karzinom umwandeln [210, 223, 270]. Das kolorektale Karzinom ist ein Sammelbegriff für alle malignen epithelialen Primärtumoren des Kolons, einschließlich des Rektums mit drüsiger Differenzierung. Dabei zählen international alle Tumoren, deren aboraler Rand bei der Messung mit dem starren Rektoskop mehr als 16 cm von der Anokutanlinie entfernt ist, zu den Kolonkarzinomen. Nach der UICC (Union Internationale Contre le Cancer) von 2003 werden die Rektumkarzinome entsprechend ihrem Abstand von der Anokutanlinie in Karzinome des oberen Rektumdrittels (12 bis 16 cm), des mittleren Rektumdrittels (6 bis < 12 cm) und des unteren Rektumdrittels (< 6 cm) eingeteilt. Demgegenüber gelten in den USA als Kolonkarzinome Tumoren, die mehr als 12 cm und als Rektumkarzinome Tumoren, die 12 cm und weniger von der Linea anocutanea entfernt sind [125, 197, 223, 263]. 1.1.2 Epidemiologie Zahlreiche Menschen in westlichen Ländern entwickeln im Laufe ihres Lebens ein oder mehrere kolorektale Adenome. Vor allem bei Menschen mittleren und höheren Alters findet man adenomatöse Polypen. Männer sind dabei häufiger betroffen als Frauen [104, 210, 223, 240]. Es bereitet Schwierigkeiten, genaue Angaben zur Epidemiologie kolorektaler Adenome zu machen, da die meist asymptomatischen Adenome erst bei Autopsien oder bei Früherkennungskoloskopien zufällig entdeckt werden. Die Angaben in Studien variieren erheblich. In koloskopischen Studien unter asymptomatischen Patienten mit einem Durchschnittsrisiko ergab sich eine Prävalenz von mehr als 25%. In sigmoidoskopischen Studien von 10% [104]. Autopsiestudien zeigen eine altersabhängige Prävalenz mit 29% zwischen dem 50. und 60., 33% zwischen dem 60. und 70. und 42% zwischen dem 70. und 2 Einleitung 80. Lebensjahr [44]. Die Prävalenz von kolorektalen Adenomen variiert stark von Land zu Land und korreliert mit der Prävalenz und der Inzidenz von kolorektalen Karzinomen in jedem Land [104, 210]. Das kolorektale Karzinom gehört weltweit zu den dritthäufigsten Tumorerkrankungen, wobei Männer und Frauen etwa gleich häufig betroffen sind. In den entwickelten westlichen Ländern ist es sogar die zweithäufigste Krebserkrankung [3, 20, 33, 207]. Es zeigen sich international erhebliche Unterschiede in der Inzidenz des kolorektalen Karzinoms. Während die Neuerkrankungen in Ländern der 3. Welt sehr gering sind, steigen die Zahlen mit der sozioökonomischen Entwicklung. Die niedrigsten Erkrankungsraten findet man in Afrika und Indien mit etwa 3 bis 4 Neuerkrankungen pro 100.000 Einwohner pro Jahr, die Höchsten in den Wohlstandsländern Nordamerikas und Westeuropas, sowie in Australien und Neuseeland mit über 30 Erkrankungen pro 100.000 Einwohner pro Jahr. In Ländern, die große Veränderungen in ihren Lebensstil- und Ernährungsgewohnheiten erfahren, steigt die Inzidenz für kolorektale Karzinome. Dieser Trend ist deutlicher unter den höheren Einkommensgruppen zu beobachten [2, 125, 127, 279]. Die jährlichen Neuerkrankungen in Deutschland werden auf etwas über 70.000 (etwa 37.000 bei Männern und 36.000 bei Frauen) geschätzt, das sind mehr als 16% aller Neuerkrankungen an Krebs. Damit steht das kolorektale Karzinom an zweiter Stelle nach dem Prostatakarzinom bei Männern und dem Mammakarzinom bei Frauen. Allerdings beruhen diese Zahlen bisher vor allem auf dem Krebsregister Saarland [33]. Innerhalb der EU zeigen epidemiologische Untersuchungen ein signifikant häufigeres Auftreten des kolorektalen Karzinoms in nördlichen und westlichen als in südlichen Ländern. Die niedrigsten Inzidenzraten in Europa findet man in Griechenland mit etwa 20.000 [33, 125]. Während die Inzidenzraten in den USA in der Zeit von 1998 bis 2002 um 1,8% jährlich abnahmen, wird für Deutschland in den letzten 20 Jahren ein eher konstantes Niveau der Inzidenz angegeben. Ein Grund hierfür sind verbesserte Früherkennungsprogramme, da zum Beispiel durch die koloskopische Polypektomie die Progression von Adenomen zu Karzinomen verhindert werden kann [3, 33, 153]. Das Risiko, im Laufe seines Lebens an einem kolorektalen Karzinom zu erkranken, liegt bei etwa 5 bis 6%, daran zu sterben bei etwa 3%. Einleitung 3 Ab einem Alter von 50 Jahren steigt das Erkrankungsrisiko deutlich an. Mehr als 90% der Fälle werden bei Personen, die älter als 50 sind, diagnostiziert, wobei Männer im Mittel mit 69 und Frauen mit 75 Jahren erkranken [3, 33, 240, 270]. Darüber hinaus steht das kolorektale Karzinom sowohl bei Männern als auch bei Frauen an dritter Stelle der Krebstodesursachen der westlichen Welt [114, 159]. In Deutschland ist das kolorektale Karzinom die zweithäufigste Krebstodesursache nach dem Lungenkarzinom bei Männern und dem Mammakarzinom bei Frauen [33]. 2004 starben in Deutschland 28.000 Menschen an einem kolorektalen Karzinom oder dessen Folgen. Etwa 50% der Patienten sterben innerhalb von 5 Jahren an der Erkrankung, meist aufgrund von metastatischen Läsionen. Seit Mitte der 1970er Jahre nimmt die Mortalität für beide Geschlechter stetig ab. In der EU ist die Sterberate bei Frauen in den Jahren von 1959 bis 1998 um 14,9% gesunken, bei Männern um 5,7% [158]. Dies kann auf verbesserte Maßnahmen für Prävention, Screening, Therapie und Nachsorge zurückgeführt werden [3, 125, 158]. 1.1.3 Ätiologie Etwa 80 bis 85% der kolorektalen Karzinome entstehen sporadisch über die AdenomKarzinom-Sequenz. Von den sporadischen Adenomen, die etwa 98% der adenomatösen Polypen ausmachen, ist nur bei etwa 1 bis 10% im Rahmen der Adenom-Karzinom-Sequenz mit einer malignen Entartung zu rechnen. Das Entartungsrisiko ist insbesondere abhängig von der Größe, vom histologischen Typ und dem Grad der Dysplasie bzw. der intraepithelialen Neoplasie (IEN) der Adenome [7, 253]. Die Auslösungsmechanismen für die Entstehung von kolorektalen Adenomen und Karzinomen sind bis heute noch nicht vollständig geklärt und Gegenstand vieler wissenschaftlicher Studien. Viele Faktoren scheinen in der Ätiologie der kolorektalen Adenome und Karzinome eine Rolle zu spielen. Als gesichert gilt, dass sowohl umweltbedingte als auch konstitutionelle Faktoren zusammenspielen, die Entstehung und Verlauf eines kolorektalen Karzinoms beeinflussen [7, 162, 256, 279]. So gibt es einige genetische Alterationen, die im Laufe der Entwicklung von einem Adenom zu einem kolorektalen Karzinom auftauchen. Dabei handelt es sich um Mutationen von ProtoOnkogenen, Tumorsuppressorgenen und DNA-Reparaturgenen. Die großen geografischen Unterschiede in den Inzidenzraten und Ergebnisse von Migrationsstudien deuten auf exogene Faktoren bei der Entstehung des kolorektalen 4 Einleitung Karzinoms hin. Dabei spielen vor allem Ernährungsgewohnheiten, Lebensstilfaktoren, sowie kulturelle und soziale Praktiken eine große Rolle. Migranten, die üblicherweise von ärmeren Ländern in reichere Länder umziehen, haben nach einigen Jahren das gleiche Risiko, an einem kolorektalen Karzinom zu erkranken, wie die einheimische Bevölkerung. So zeigten schon frühe Migrationsstudien, dass das Risiko, an einem kolorektalen Karzinom zu erkranken, für Japaner, die in die USA eingewandert sind, sich nach zehn Jahren dem Erkrankungsrisiko eines Amerikaners angepasst hat. Einen ähnlichen Trend zeigte sich bei polnischen und chinesischen Einwanderern in die USA. Geringer war das Risiko, wenn sich die Migranten weiterhin mit ihrer ursprünglichen Kost ernährten [127, 279]. Epidemiologische Studien der letzten 40 Jahre haben geholfen, die Hauptrisikofaktoren, die mit einem kolorektalen Karzinom assoziiert sind, aufzudecken und protektive Faktoren zu identifizieren. Man schätzt, dass etwa 50 bis 80% der kolorektalen Karzinome mit der Ernährung assoziiert sind. Es gibt jedoch zahlreiche kontroverse Studienergebnisse, so dass eine genaue Risikoeinschätzung nicht möglich ist. Es ist anzunehmen, dass ein „westlicher Ernährungsstil“ mit einem erhöhten Darmkrebsrisiko assoziiert ist. Darunter versteht man eine ballaststoffarme Ernährung mit einem hohen Anteil an rotem Fleisch und Fett und wenig Obst und Gemüse [99, 161, 270, 293]. Auch mangelnde körperliche Bewegung stellt einen Risikofaktor für die Entstehung eines kolorektalen Karzinoms dar. So belegen Studien, dass regelmäßige körperliche Aktivität die Häufigkeit von Adenomen und dass Risiko für das Auftreten eines kolorektalen Karzinoms reduziert [98, 174, 253, 272]. Ebenso scheint Übergewicht bei der Karzinomentstehung eine Rolle zu spielen. Bei Personen mit einem erhöhten BMI findet man häufiger Adenome, das Risiko für ein kolorektales Karzinom ist bis zu zweifach erhöht. Unklar bleibt, ob das Risiko auf die mangelnde Bewegung oder allein auf das Übergewicht zurückzuführen ist [106-107]. Bei postmenopausalen Frauen scheint eine Hormonersatztherapie das Karzinomrisiko zu senken [253, 270]. Nikotinabusus ist ebenfalls mit einem erhöhten Adenom- und Karzinomrisiko im Kolorektum assoziiert [67, 222, 253]. So haben starke Raucher ein etwa zwei- bis dreifach erhöhtes Risiko kolorektale Adenome zu entwickeln. Tabakrauch ist eine bedeutende Quelle für verschiedene Karzinogene, einschließlich Nitrosamine, heterozyklische Amine und polyzyklische Kohlenwasserstoffe. Starker Alkoholkonsum ist vor allem bei unzureichender Folsäurezufuhr mit einem erhöhten Risiko assoziiert. Entscheidend scheint dabei die Menge (≥ 30 g/d) und nicht die Art des Alkohols zu sein [45, 208, 257]. Es konnte gezeigt werden, dass eine Langzeit-Aufnahme von Einleitung 5 Alkohol die Wahrscheinlichkeit für das Auftreten eines Tumors mit Mikrosatelliteninstabilität erhöht [127]. Ein weiterer Risikofaktor für das Auftreten eines kolorektalen Karzinoms ist eine Familienanamnese. Für Verwandte ersten Grades von Patienten mit einem kolorektalen Karzinom ist das mittlere Risiko, ebenfalls zu erkranken zwei- bis dreifach erhöht. Es ist unklar, ob es sich dabei um genetische Faktoren oder um eine ähnliche Lebensweise als Risikofaktor oder um beides handelt. Das Risiko erhöht sich weiterhin auf das drei- bis vierfache, wenn das Karzinom bei dem Indexpatienten vor dem 60. Lebensjahr aufgetreten ist und /oder mehr als ein Verwandter ersten Grades von einem kolorektalen Karzinom betroffen ist. Ebenfalls ein erhöhtes Risiko an einem kolorektalen Karzinom zu erkranken, haben Verwandte ersten Grades von Patienten mit kolorektalen Adenomen (ein- bis zweifach erhöhtes Risiko) und Patienten, die selbst kolorektale Adenome (zwei- bis dreifach erhöhtes Risiko) haben oder hatten. Patienten, die bereits ein kolorektales Karzinom hatten, haben nach 10 Jahren ein Risiko von 2% ein zweites kolorektales Karzinom zu entwickeln [14, 125, 154, 237]. Es gibt hereditäre Syndrome, die mit kolorektalen Adenomen und/oder Karzinomen einhergehen und für die bestimmte Genmutationen gefunden wurden. Man unterteilt sie in zwei Gruppen: 1. Familiäre adenomatöse Polyposis (FAP): Die FAP ist für etwa 1% der kolorektalen Karzinome verantwortlich. Es handelt sich um eine autosomal-dominant vererbte Erkrankung mit einer Mutation des APC-Gens, bei der es zur Ausbildung von mehr als 100 adenomatösen Polypen im gesamten Kolon kommt. Das Risiko für die Entstehung eines kolorektalen Karzinoms liegt nach 35 bis 40 Lebensjahren bei nahezu 100%. Andere Polyposissyndrome mit einer ähnlich hohen Entartungstendenz sind das GardnerSyndrom und das Turcot-Syndrom. Eine niedrigere Karzinominzidenz haben das PeutzJeghers-Syndrom, die generelle juvenile Polyposis und das Cowden-Syndrom [93, 113, 125, 159]. 2. Hereditäres Non-Polyposis-Cancer-Syndrom (HNPCC): Etwa 5% der kolorektalen Karzinome entwickeln sich auf dem Boden eines HNPCCSyndroms. Hierbei kommt es aufgrund einer autosomal-dominant vererbter Mutation von DNA-Reparaturgenen oft zur Ausbildung eines kolorektalen Karzinoms noch vor dem 50. Lebensjahr ohne eine Adenomatose. Auch entstehen spezifische extrakolonische 6 Einleitung Tumoren. Betroffen sind vor allem das Ovar, der Magen, der Dünndarm und der Urogenitaltrakt. Das Lebenszeitrisiko von Genträgern, ein kolorektales Karzinom zu entwickeln, wird auf 80 bis 90% geschätzt. Aufgrund eines fehlenden auffälligen Phänotyps wurden die sogenannten Bethesda- und Amsterdamkriterien definiert, bei deren Zutreffen die Diagnose gestellt werden kann und eine Mutationssuche erfolgen sollte [78, 113, 159]. Weitere 5% der kolorektalen Karzinome entwickeln sich auf dem Boden einer chronischentzündlichen Darmerkrankung. Patienten mit einer Colitis ulcerosa weisen ein erhöhtes Risiko für die Entwicklung eines kolorektalen Karzinoms auf. Das Risiko ist abhängig von Beginn, Dauer und Ausdehnung der Erkrankung, sowie von dem Vorhandensein einer primär sklerosierenden Cholangitis. Bei Befall des gesamten Kolorektums mit schwerer Colitis ulcerosa ist nach 7 bis 10 Jahren mit einem etwa 15% erhöhtem Karzinomrisiko zu rechnen. Die Angaben des kumulativen Risikos variieren von 1,4 bis 34 % [71, 183]. Patienten mit einem Morbus Crohn und einem langwierigen Verlauf haben ebenfalls ein erhöhtes Risiko für die Entstehung eines kolorektalen Karzinoms. Dieses ist im Vergleich zur Colitis ulcerosa möglicherweise geringer. Die Angaben schwanken aufgrund uneinheitlicher und spärlicher Datenlage zwischen keinem und einem 3,5- bis 7-fach erhöhten Karzinomrisiko [35, 90]. Durch primäre Prävention (Krankheitsverhütung) und sekundäre Prävention (Krankheitsfrüherkennung) kann aus epidemiologischer Sicht die Inzidenz, die Morbidität und die Mortalität positiv beeinflusst werden. Da bei kolorektalen Karzinomen die sogenannten „Lebensstilfaktoren“ eine wichtige Rolle spielen, sollte zur primären Prävention die Bevölkerung über eine gesunde Lebensweise informiert werden. Wichtig ist eine ausgewogene vitamin- und ballaststoffreiche Ernährung, die viel Obst und Gemüse und wenig rotes Fleisch enthält. Für eine effiziente Prävention werden fünf Portionen Obst oder Gemüse am Tag empfohlen. Zusammen mit geringem Alkoholkonsum und Nikotinabstinenz, sowie täglicher körperlicher Bewegung kann das Risiko für das Auftreten eines kolorektalen Karzinoms am besten reduziert werden [5, 38, 127, 254]. Für die wirksame Prävention des kolorektalen Karzinoms durch Mikronährstoffe und Medikamente gibt es bisher keine gesicherten Daten. In einigen Studien konnte ein Einleitung 7 präventiver Effekt durch die Einnahme von NSAR bestätigt werden [65, 243]. Wahrscheinlich wirken die NSAR über eine Hemmung der Cyclooxygenase, die bei kolorektalen Karzinomen deutlich erhöht ist [166, 191]. Aufgrund der ungünstigen RisikoNutzen-Relation und widersprüchlicher Studiendaten kann eine protektive Gabe von NSAR derzeit jedoch nicht empfohlen werden. Die Einnahme von Mikronährstoffen, wie die Vitamine A, C, D und E, Kalzium, Karotinoide und Folsäure zeigten in einigen Studien protektiven Charakter bezüglich des Darmkrebsrisikos. Empfehlen kann man aktuell eine folsäurereiche, kalziumreiche Ernährung, da diese mit einem erniedrigten kolorektalen Karzinomrisiko assoziiert ist [5, 167, 287]. Da bei kolorektalen Karzinomen die frühe Diagnosestellung für die Prognose entscheidend ist, ist die sekundäre Prävention von großer Bedeutung. Im Rahmen der sekundären Prävention sollten sich Männer und Frauen mit durchschnittlichem Risiko ab einem Alter von 50 Jahren einem Screening in Form einer Koloskopie unterziehen, die bei negativem Befund alle 10 Jahre wiederholt werden sollte. Die Koloskopie ist das Standardverfahren mit der höchsten Sensitivität und Spezifität [3, 47, 280]. Bei Patienten, die eine Darmspiegelung ablehnen, sollte alle 5 Jahre eine Sigmoidoskopie durchgeführt werden, sowie jährlich ein Guaiak-Test. Die alleinige jährliche Untersuchung auf okkultes Blut im Stuhl (Guaiak-Test) sollte nur bei Patienten durchgeführt werden, die jegliche endoskopische Untersuchung ablehnen [220, 232, 268]. Ein positives Testergebnis macht die endoskopische Untersuchung des gesamten Dickdarms erforderlich. Von Bedeutung für die Prävention ist auch das Erkennen von Risikopatienten, die dann je nach Risikoprofil engmaschiger untersucht werden können. Aufgrund der relativ geringen Teilnahme kann der Einfluss des Koloskopie-Screenings auf die Inzidenz des kolorektalen Karzinoms bisher nicht bewertet werden [33]. 1.1.4 1.1.4.1 Histomorphologische Einteilung Kolorektale Adenome a. Lokalisation Etwa 60% der kolorektalen Adenome entwickeln sich im rektosigmoidalen Bereich. Das Alter des Patienten scheint einen entscheidenden Einfluss auf die Lokalisation von Adenomen zu haben. So entstehen bei jüngeren Patienten Adenome eher im linken Kolon, 8 Einleitung bei älteren Patienten eher im rechten Kolon. Ebenso beobachtet man große Adenome mit schwerer intraepithelialer Neoplasie eher im Rektum bzw. im distalen Kolon [210]. Kolorektale Adenome können solitär, aber auch multipel vorkommen, daher ist stets eine komplette Koloskopie zum Ausschluss von Adenomen angezeigt. b. Makroskopie Nach der Wuchsform werden die Adenome eingeteilt in gestielte, schmalbasige (taillierte) und breitbasige (sessile) Adenome [226]. c. Histologie Histologisch bestehen alle Adenome aus atypischen Drüsenformationen atypischer Epithelzellen. Diese Epitheldysplasie wird je nach Ausdifferenzierung, Kerngröße und form, Chromatinverteilung, Nukleolengröße und Mitosezahl in gering-, mittel- und hochgradige Dysplasie eingeteilt [223, 226]. Nach dem histologischen Feinbau unterscheidet man tubuläre, tubulovillöse und villöse Adenome. Tubuläre Adenome, die etwa 66% der Adenome ausmachen, haben meist eine sessile oder gestielte Form und eine glatte Oberfläche. Typisch sind verzweigte drüsig-tubuläre Wucherungen der Kryptenschläuche. Villöse Adenome sind mit 8 bis 10% eher selten. Sie sind meist breitbasig und durch ausgestülpte, fingerartige Schleimhautzotten mit manchmal exzessiver Schleimbildung charakterisiert. Tubulovillöse Adenome zeigen sowohl tubuläre als auch villöse Merkmale und machen 22% der Adenome aus [7, 226]. In den letzten Jahren wurde ein weiterer Adenomtyp beschrieben, das „serrated adenoma“ (Sägeblatt-Adenom), das Anteile eines hyperplastischen Polypen mit zellulären Charakteristika eines kolorektalen Adenoms in sich kombiniert. Kleine Sägeblattadenome ähneln eher hyperplastischen Polypen, während große den villösen Adenomen zugeordnet werden können. Histomorphologisch findet man gezackte oder sägezahnartige Krypten, eine zytoplasmatische Eosinophilie und ein geringes Kern-/Zytoplasma-Verhältnis. Es ist bisher nicht klar, ob sich die „serrated adenomas“ aus hyperplastischen Polypen oder de novo entwickeln [152, 179]. Einen weiteren histologischen Phänotyp stellt das mikrotubuläre Adenom dar, das ebenfalls erst in den letzten Jahren beschrieben wurde. Es vereint villöse Anteile mit mikrotubulären Strukturen [225]. Einleitung 9 Kolorektale Adenome können sich im Laufe der Zeit in ein Adenokarzinom umwandeln. Dabei steigt das Karzinomrisiko mit der Anzahl, der Größe (Tabelle 1/1), dem histologischen Typ und dem Grad der intraepithelialen Neoplasie der Adenome [152, 253]. Während das Entartungsrisiko bei tubulären Adenomen je nach Größe zwischen 1 bis 10% liegt, steigt es bei tubulovillösen Adenomen auf 20% und bei villösen Adenomen auf 20 bis 40% an. Bei einer mäßigen intraepithelialen Neoplasie besteht ein Karzinomrisiko von 6%, bei einer schweren intraepithelialen Neoplasie steigt es auf 35% an [41, 125, 223]. Es konnte gezeigt werden, dass 37% der Sägeblattadenome Anteile mit hochgradiger intraepithelialer Neoplasie enthielten. Daher nimmt man an, dass diese Adenome einem sehr schnellen Progress folgen [37]. Adenomgröße Karzinomrisiko bis 1 cm < 1% bis 2 cm 6% bis 3 cm 30% bis 4 cm 40% > 4 cm 70% Tabelle 1/1: Entartungsrisiko in Abhängigkeit der Adenomgröße 1.1.4.2 Kolorektale Karzinome a. Lokalisation Die kolorektalen Karzinome entstehen zu 40% im Rektum und zu 60% im Kolon, wovon etwa 20% im Sigma zu finden sind. Nach der UICC 2003 liegt ein Rektumkarzinom vor, wenn der makroskopisch erkennbare aborale Tumorrand bei starrer Rektosigmoidoskopie 16 cm oder weniger von der Linea anocutanea entfernt ist. Die gleiche Lokalisation von Polypen und Karzinomen im Kolon liefert einen weiteren Hinweis dafür, dass sich die sporadischen Karzinome über die Adenom-Karzinom-Sequenz entwickeln [41, 223, 263]. b. Makroskopie Kolorektale Karzinome zeigen unterschiedliche makroskopische Wachstumsformen. Man unterscheidet ulzerierte, polypoide, flache und diffus infiltrierende Tumoren. Die makroskopische Wuchsform erlaubt eine grobe Beurteilung des Tumors, da zum Beispiel 10 Einleitung polypoide Karzinome später und seltener metastasieren als diffus infiltrierende Karzinome [223]. c. Histologie Histologisch werden die kolorektalen Karzinome nach der WHO-Klassifikation (2000) in acht verschiedene Typen eingeteilt: 1. Adenokarzinom 2. Muzinöses Adenokarzinom (Gallertkarzinom) 3. Siegelringzellkarzinom 4. Kleinzelliges (neuroendokrines) Karzinom 5. Plattenepithelkarzinom 6. Adeno-squamöses Karzinom 7. Medulläres Karzinom 8. Undifferenziertes Karzinom Bei über 80% der kolorektalen Karzinome handelt es sich um ein Adenokarzinom. Unter den Adenokarzinomen gibt es tubuläre, papilläre und papillotubuläre Subtypen. Die meisten Adenokarzinome sind mäßig differenziert (G2). Das muzinöse Adenokarzinom macht etwa 10 bis 20% der Fälle aus. Es zeigt eine ausgedehnte extrazelluläre Verschleimung, die mehr als 50% des Tumors einnimmt. Das seltene Siegelringzellkarzinom ist zu mehr als 50% aus Siegelringzellen aufgebaut. Dabei kommt es aufgrund einer Sekretionsstörung zu einer intrazellulären Anhäufung von Schleim. Es hat eine schlechte Prognose mit einem hohen Malignitätsgrad. Die übrigen Karzinome kommen sehr selten vor. Es können vor allem in größeren Tumorarealen oder in Metastasen verschiedene Typen nebeneinander vorkommen. Von grundlegender Bedeutung für die Prognose und Therapie des kolorektalen Karzinoms bleibt die exakte makro- und mikroskopische Untersuchung und Beschreibung des Tumors durch den Pathologen. Die Tumorausbreitung wird international nach der TNM-Klassifizierung der UICC (Union Internationale Contre le Cancer) eingeteilt [263]. Hierin werden die Infiltration des Tumors in die Darmwand, seine Ausdehnung in benachbarte Strukturen, das Ausmaß des Lymphknotenbefalls und das Auftreten von Fernmetastasen erfasst (Tabelle 1/2). Für eine sichere pathologische Stadieneinteilung müssen mindestens 12 Lymphknoten entnommen und histologisch untersucht worden sein. Ein Carcinoma in situ liegt vor, wenn Tumorzellen innerhalb der Basalmembran der Drüsen (intraepithelial) oder in der Lamina propria Einleitung 11 (intramukös) nachweisbar sind, ohne dass eine Ausbreitung durch die Muscularis mucosae in die Submukosa feststellbar ist. Auch andere Klassifikationssysteme, zum Beispiel die Einteilung nach Dukes, werden in einigen Ländern noch angewandt. Die Dukes-Klassifikation von 1932 (Tabelle 1/3) berücksichtigt jedoch nicht die Tumorinfiltration in Nachbarorgane und das Vorliegen von Fernmetastasen. Zahleiche Modifikationen dieser Einteilung erschweren den Vergleich von Literaturdaten. T Primärtumor TX Primärtumor kann nicht beurteilt werden T0 Kein Anhalt für Primärtumor Tis Carcinoma in situ T1 Tumor infiltriert Submukosa T2 Tumor infiltriert Muscularis propria T3 Tumor infiltriert durch die Muscularis propria in Subserosa oder in nicht peritonealisiertes, perikolisches oder perirektales Gewebe T4 Tumor perforiert das viszerale Peritoneum oder infiltriert direkt in andere Organe oder Strukturen N Regionäre Lymphknoten NX Regionäre Lymphknoten können nicht beurteilt werden N0 Keine regionären Lymphknotenmetastasen nach regionärer Lymphadenektomie N1 Metastasen in 1-3 perikolischen bzw. perirektalen Lymphknoten N2 Metastasen in 4 oder mehr perikolischen Lymphknoten M Fernmetastasen MX Das Vorliegen von Fernmetastasen kann nicht beurteilt werden M0 Keine Fernmetastasen M1 Fernmetastasen Tabelle 1/2: pTNM-Klassifikation (UICC 2002) 12 Einleitung Stadium A Tumor reicht maximal in die Muscularis propria; keine Lymphknotenmetastasen Stadium B Tumor breitet sich über die Muscularis propria hinaus aus; keine Lymphknotenmetastasen Stadium C1 Lymphknotenmetastasen liegen vor, aber nicht an der Grenze des Resektates (Grenzlymphknoten negativ) Stadium C2 Lymphknotenmetastasen liegen vor, Grenzlymphknoten positiv Tabelle 1/3: Stadieneinteilung des kolorektalen Karzinoms nach Dukes (1932) Tis N0 M0 T1 N0 M0 T2 N0 M0 Stadium II A T3 N0 M0 Stadium II B T4 N0 M0 Stadium III A T1,T2 N1 M0 Stadium III B T3,T4 N1 M0 Stadium III C Jedes T N2 M0 Stadium IV Jedes T Jedes N M1 Stadium 0 Dukes A Stadium I Dukes B Dukes C Tabelle 1/4: Stadieneinteilung des kolorektalen Karzinoms (UICC 2002) Die Ausbreitung eines kolorektalen Karzinoms erfolgt kontinuierlich lymphogen oder hämatogen in die Leber (Pfortader-Typ), erst sekundär in die Lunge. Bei etwa 15 bis 20% der Tumoren finden sich bereits zum Zeitpunkt der Diagnosestellung sonografisch Lebermetastasen. Entsprechend der Metastasierungswege finden sich beim Kolonkarzinom eher Lymphknoten-, Leber-, Lungen- und Skelettmetastasen, beim Rektumkarzinom vor allem lokoregionäre Metastasen, gefolgt von Leber- und Lungenmetastasen. In Abhängigkeit von der Lokalisation des Rektumkarzinoms verläuft die lymphogene Ausbreitung über drei verschiedene Metastasierungswege. Hochsitzende Karzinome (8 bis 16 cm von der Anokutanlinie) metastasieren in paraaortale Lymphknoten, Tumoren der mittleren Etage (4 bis 8 cm von der Anokutanlinie) betreffen zusätzlich die Beckenwand, tiefsitzende Tumoren (0 bis 4 cm von der Anokutanlinie) befallen außerdem die inguinalen Lymphknoten. Als Ausnahme metastasiert das tiefsitzende Rektumkarzinom primär in die Lunge (Cava-Typ Einleitung 13 der Metastasierung). Daher ist die Lokalisation des Primärtumors ausschlaggebend für die Art der operativen Therapie. 1.1.5 1.1.5.1 Therapeutische Aspekte Prognostische Parameter Um eine individuelle wirkungsvolle Tumortherapie zu erreichen, sind neben der TNMKlassifikation der UICC weitere Prognosefaktoren wichtig, die eine genauere Einteilung und Risikoabschätzung erlauben. Entscheidend für die Prognose und auch für weitere Therapieentscheidungen eines kolorektalen Karzinoms ist die postoperative pathologische Untersuchung mit folgenden Angaben [20, 70, 129, 173]: Lokalisation des Tumors Tumortyp nach WHO-Klassifikation Tumorinvasionstiefe (pT-Stadium) Status der regionären Lymphknoten (pN-Stadium) Anzahl der untersuchten Lymphknoten Anzahl der befallenen Lymphknoten Tumor-Grading (Tabelle 1/7) Abstand von den Resektionsrändern R-Klassifikation (Tabelle 1/6) Lymph-/Blutgefäßinvasion Weitere histopathologische Prognoseparameter sind außerdem das Vorhandensein von Fernmetastasen, eine perineurale Infiltration und eine Dedifferenzierung im Bereich der Invasionsfront [20, 70]. Die 5-Jahres-Überlebensraten nach R0-Resektion des Tumors sind in Abhängigkeit vom TNM-Stadium in Tabelle 1/5 angegeben. Im Falle von unresezierbarem residuellem oder metastatischem Tumorgewebe liegt die mediane Überlebenszeit bei weniger als einem Jahr [20]. Differenzierungsgrade geben Hinweise auf die biologische Qualität von Karzinomen. Dabei zeigen wenig differenzierte Tumoren im Allgemeinen eine höhere Progressionsneigung als gut differenzierte. Ein Nachteil des Tumor-Grading besteht in der Subjektivität der Beurteilung, sowie in der beschränkten Reproduzierbarkeit der Ergebnisse. Trotzdem haben Patienten mit hochdifferenzierten Karzinomen eine signifikant bessere Prognose [20]. 14 Einleitung TNM-Stadium Ungefähres 5-Jahres Überleben Stadium I 85-90% Stadium II 70-75% Stadium III 35-40% Stadium IV < 5% Tabelle 1/5: TNM-Stadium und Überlebensrate bei R0-Resektion des Primärtumors R0 Kein Residualtumor R1 Histologisch Residualtumor R2 Makroskopisch Residualtumor Tabelle 1/6: R-Klassifikation (UICC 2002) GX Differenzierungsgrad kann nicht bestimmt werden G1 Gut differenziert G2 Mäßig differenziert G3 Schlecht differenziert G4 Undifferenziert Tabelle 1/7: Histopathologisches Grading (UICC 2002) Neben den histopathologischen Faktoren, gibt es therapieassoziierte Parameter, die die Prognose eines kolorektalen Karzinoms beeinflussen. So ist zum Beispiel die Erfahrung des Chirurgen von großer Bedeutung, wie auch die Operationsmethode [130, 185]. Eine erweiterte Resektion bewirkt eine höhere Überlebenszeit [42, 149]. Auch kann die Prognose durch eine adjuvante Chemotherapie im Stadium III verbessert werden [188, 283]. Aufgrund der enormen prognostischen Unterschiede zwischen Früh- und Spätstadium lokaler Tumoren, stellt die Suche nach nützlichen prognostischen Markern einen wichtigen Bereich der klinischen Forschung dar. Die Identifikation von genetischen Risikofaktoren für die Entwicklung von Adenomen und assoziierten Karzinomen des Kolon und Rektum ermöglicht eine prädiktive molekulare Diagnose von malignen Erkrankungen. Einleitung 15 Es gibt inzwischen zahlreiche immunhistochemische und molekularbiologische Methoden, die als potenzielle Prognosemarker in Frage kommen. Etabliert hat sich bisher die präoperative Bestimmung des CEA-Wertes (Carcinoembryonales Antigen) als unabhängiger Prognoseparameter. Ein erhöhter Wert geht mit einer signifikant schlechteren Prognose einher [68, 88]. 1.1.5.2 Therapieoptionen Da typischerweise 8 bis 15 Jahre vergehen, bis aus einem Adenom ein kolorektales Karzinom entsteht, ergibt sich ein relativ großes Zeitfenster für Früherkennungsmaßnahmen, insbesondere die Koloskopie. Koloskopisch entdeckte Adenome sollten immer vollständig polypektomiert und histologisch untersucht werden, da dadurch die Entstehung von Karzinomen im Sinne einer Unterbrechung der Adenom-Karzinom-Sequenz verhindert wird [69, 165, 280]. Die Polypen müssen unter Angabe der Lokalisation entsprechend den WHOKriterien histologisch befundet werden. Die Therapie kolorektaler Karzinome sollte grundsätzlich auf der Basis einer histologischen Untersuchung geplant werden. Als Karzinome gelten Veränderungen, bei denen atypische epitheliale Formationen in die Submukosa infiltrieren (pT1 oder mehr). Nicht einbezogen sind sogenannte Mukosakarzinome oder intraepitheliale Karzinome (pTis), bei denen keine Metastasierung erfolgt und die durch lokale Abtragung im Gesunden ausreichend behandelt werden. Prätherapeutisch wird die Stadieneinteilung aufgrund von klinischen, endoskopischen und röntgenologischen Untersuchungsbefunden als Basis für die Therapieplanung vorgenommen. Neben der Anamnese und klinischen Untersuchung mit digitaler rektaler Austastung zählen hierzu eine vollständige Koloskopie mit Stufenbiopsien. Ist eine Koloskopie nicht möglich, kann stattdessen ein Doppelkontrasteinlauf mit Rektoskopie und Biopsie durchgeführt werden. Des Weiteren sind bildgebende Verfahren, wie die Endosonographie des Abdomens und ein Röntgen-Thorax in zwei Ebenen indiziert. Bei Rektumkarzinomen ist zusätzlich eine starre Rektoskopie obligat. Nach radikaler Tumorresektion sind für die weitere Therapieplanung das TNM-Stadium und der R-Status notwendig [68, 84, 88, 125, 232]. a. Operative Behandlung des kolorektalen Karzinoms Die primäre Behandlung eines kolorektalen Karzinoms ist die komplette chirurgische Entfernung des Tumors. Dies stellt bisher die einzige kurative Therapie für den Patienten dar [3, 79]. 16 Einleitung Kolorektale Karzinome wachsen vorwiegend zirkulär und metastasieren weitgehend konstant in die regionären Lymphknoten. Daher ist das Standardverfahren die radikale Resektion des Primärtumors mit partieller oder totaler Entfernung des Mesorektums und damit des Lymphabflussgebietes [42, 84]. Das Ausmaß der Darmresektion wird durch die Resektion der versorgenden Gefäße und das hierdurch definierte Lymphabflussgebiet bestimmt. Ein Großteil aller Rektumkarzinome kann heute sphinktererhaltend reseziert werden. Eine palliative Tumorentfernung beugt Komplikationen wie Blutungen, Perforation oder Ileus vor. Kolorektale Karzinome auf dem Boden einer familiären adenomatösen Polyposis (FAP) sollten mit einer restaurativen Proktokolektomie mit Dünndarm-Pouch behandelt werden. Bei HNPCC-Karzinomen werden zwei Vorgehensweisen diskutiert. Es kommen entweder eine operative Therapie entsprechend dem sporadischen Karzinom in Frage oder die regelhafte Kolektomie bei Kolonkarzinomen bzw. die restaurative Proktokolektomie bei Rektumkarzinomen. Bei Karzinomen auf dem Boden einer Colitis ulcerosa stellt die restaurative Proktokolektomie mit Dünndarm-Pouch die Regeloperation dar. b. Adjuvante Therapie Eine adjuvante Therapieform dient der Vorbeugung eines Tumorrezidivs. Nach der chirurgischen Resektion erleiden etwa 1/3 der Patienten einen Rückfall der Erkrankung. Um die Rezidivrate zu reduzieren, ist eine postoperative Chemotherapie sinnvoll, auf die jedoch nur etwa 26% der Patienten ansprechen [79]. Voraussetzung für jede adjuvante Therapie ist die R0-Resektion des Primärtumors, das heißt es können weder durch klinische noch durch pathologisch-histologische Untersuchungen Tumorreste nachgewiesen werden. Es muss aber davon ausgegangen werden, dass zum Zeitpunkt der Diagnose schon Lymphknoten- und Fernmetastasen vorhanden sein können. Vor allem die Fernmetastasierung bestimmt die Prognose der Patienten erheblich. Durch eine adjuvante Chemotherapie versucht man diese noch nicht nachweisbaren, okkulten Streuherde und Mikrometastasen zu erreichen. Grundlage für die Indikation einer adjuvanten Therapie ist die pathohistologische Stadienbestimmung, insbesondere die Bestimmung des Lymphknotenstatus. Für Patienten mit einem kurativ resezierten Kolonkarzinom im Stadium I ist eine adjuvante Therapie nicht indiziert. Bei Patienten mit einem R0 resezierten Kolonkarzinom im UICC-Stadium III ist eine adjuvante Chemotherapie indiziert. In zahlreichen Studien Einleitung 17 konnte dadurch ein Überlebensvorteil nachgewiesen werden [230]. Die Letalitätsrate konnte um 10 bis 15% reduziert werden [1, 230]. Bei Patienten mit einem kurativ resezierten Kolonkarzinom im Stadium II kann eine adjuvante Therapie in Betracht gezogen werden, da ein Nutzen nicht ausgeschlossen werden kann. In der britischen Quasar-Studie fand sich ein absoluter Überlebensvorteil von 3 % [219]. In vielen Studien fand sich jedoch bisher kein signifikanter Überlebensvorteil [1, 34, 94]. Für die adjuvante Chemotherapie des Kolonkarzinoms im Stadium III sollte eine Oxiplatin-haltige Therapie, wie zum Beispiel das FOLFOX-Schema (5-FU plus Folinsäure plus Oxaliplatin) eingesetzt werden. [1, 202]. In mehreren randomisierten Studien konnte durch eine Kombination von 5-FU und Folinsäure eine signifikante Senkung der Rezidivrate und eine Verbesserung des Gesamtüberlebens gezeigt werden. Durch eine zusätzliche Gabe von Oxaliplatin konnte das Gesamtüberleben weiter erhöht werden [1, 12, 84, 230, 233]. Bei Patienten mit einem Rektumkarzinom im Stadium II und III, bei denen keine neoadjuvante Radiochemotherapie durchgeführt wurde, ist eine postoperative kombinierte Radiochemotherapie indiziert. Es konnte sowohl die Rezidivrate gesenkt als auch das Gesamtüberleben verbessert werden [262, 284]. Bei Patienten mit vorangegangener neoadjuvanter Therapie ist im Stadium III eine adjuvante Therapie wie beim Kolonkarzinom indiziert. Eine Indikation für eine alleinige adjuvante Chemo- oder Radiotherapie besteht beim Rektumkarzinom nicht. c. Neoadjuvante Therapie Eine neoadjuvante Therapie sollte, präoperativ verabreicht, zu einer deutlichen Tumorreduktion führen und den inoperablen Tumor in einen operablen Zustand führen. Das Problem der Indikation zur neoadjuvanten Therapie ist die Selektion der geeigneten Patienten, das heißt die Auswahl der Patienten mit einem hohem Lokalrezidivrisiko und die Vermeidung einer Übertherapie der Patienten mit einem niedrigem Lokalrezidivrisiko [124, 261]. Für das Kolonkarzinom liegen beweisende Untersuchungen für die Wirksamkeit neoadjuvanter Maßnahmen nicht vor. Bei Rektumkarzinomen im Stadium II und III hat sich eine präoperative Radiotherapie bzw. Radiochemotherapie bewährt [157, 231]. Sie ist verglichen zur postoperativen Bestrahlung wirksamer und bringt weniger akute und chronische Morbidität mit sich [56]. Die Rate der sphinktererhaltenden Operationen konnte durch eine neoadjuvante Behandlung verdoppelt werden. Als ein Nachteil der präoperativen Radiochemotherapie 18 Einleitung muss das potenzielle „Overstaging“ und die daraus resultierende Überbehandlung des Patienten gewertet werden. Als Strahlentherapie stehen die Kurzzeitbestrahlung und die konventionell fraktionierte Bestrahlung zur Verfügung. Die Rolle der Chemotherapie im Rahmen der präoperativen Radiochemotherapie ist bislang noch nicht gesichert und wird in Studien untersucht. d. Immuntherapie bei kolorektalen Karzinomen Es werden zahlreiche neue Behandlungsmethoden des kolorektalen Karzinoms in klinischen Studien evaluiert, von denen die Immuntherapie einen großen Nutzen verspricht, die Prognose dieser Erkrankung zu verbessern. Die passive Immunisierung bezieht sich auf die Verwendung monoklonaler Antikörper gegen Tumor-assoziierte Antigene, die auf der Oberfläche von Tumoren überexprimiert werden. Diese Antikörper haben durch Komplementaktivierung oder durch zelluläre Zytotoxizität einen antitumorösen Effekt. Auch können die Antikörper Zelloberflächenrezeptoren hemmen, die für das Überleben und die Proliferation der Tumorzellen wichtig sind. Die Vorteile so einer Therapie liegen in der geringen Zytotoxizität und dem spezifischeren Angriff der Tumorzellen ohne wesentliche Schädigung des gesunden Gewebes. Von großer Bedeutung sind Bevacizumab [110, 229], ein Antikörper gegen den VEGF-Faktor (vascular endothelial growth factor) und Cetuximab [97, 155], sowie Panitumumab [128, 265], Antikörper gegen den EGFR-Rezeptor (epidermal growth factor receptor). Durch die Blockierung dieser Rezeptoren kommt es zu einer Hemmung angiogenetisch wirksamer Faktoren bzw. Zellwachstums-fördernder Faktoren und dadurch zu einer Beeinträchtigung von Tumorinvasion und Metastasierung [3]. In Phase III-Studien konnte bereits in Kombination mit einer Chemotherapie eine signifikante Verbesserung des progressionsfreien Überlebens erreicht werden. Bevacizumab ist für die Erstlinientherapie zugelassen. Die aktive Immuntherapie basiert auf der Hypothese, dass neoplastische Zellen keine effektive Immunantwort auslösen. Durch Vakzinierung können Zellen vom Immunsystem als maligne erkannt werden und von zytotoxischen T-Lymphozyten eliminiert werden. In diesem Fall kommt die Immunantwort vom Patienten selbst. Eine Möglichkeit besteht darin, dem Patienten autologe Vakzine zu geben. Dabei entnimmt man Zytokin-produzierende Zellen aus dem Tumor des Patienten und verändert diese gentechnisch. Bei der allogenen Vakzinierung gewinnt man die Tumorzellen aus Tumoren anderer Patienten. Eine weitere Strategie ist die Immunisierung mit dendritischen Zellen. Diese Zellen sind die leistungsfähigsten Antigen-präsentierenden Zellen. Man isoliert die dendritischen Zellen aus dem peripheren Blut des Patienten und bringt sie mit Einleitung 19 synthetischen Peptid-Epitopen, wie zum Beispiel Her-2/neu, CEA, p53, MUC1 zusammen oder man injiziert diese Peptide direkt dem Patienten. So kann der Tumor dem Immunsystem präsentiert werden [3, 79]. 1.1.6 Molekulare Kanzerogenese Während exogene Umwelteinflüsse eine wichtige Rolle in der Adenom-Karzinom-Sequenz spielen, hat man in den letzten Jahrzehnten typische molekulare Alterationen entdeckt, die während der malignen Transformation zu einem kolorektalen Karzinom auftreten. Kritische genetische Faktoren in der Krebsentwicklung betreffen vor allem Gene der Zellregulation, der Zellproliferation und der Zelldifferenzierung [127]. Das kolorektale Karzinom ist eines der besten Modelle für die Erforschung genetischer Veränderungen, die zu maligner Transformation und Tumorprogression führen. Verschiedene chromosomale Mutationen und Deletionen sind bekannt, die über verschiedene Stadien der Hyperplasie und malignen Transformation zu einem invasiven Karzinom führen. Betroffen sind unter anderem Tumorsuppressorgene, Onkogene und Mismatch-Repair-Gene (Tabellen 1/8 und 1/9). Aktuell gibt es zwei bedeutende genetische „Pathways“, die zu einem kolorektalen Karzinom führen [37, 40, 111, 162, 248, 258]. Etwa 80 bis 90% der kolorektalen Karzinome entstehen über den sogenannten Tumorsuppressor-Pathway. Er ist durch die sequenzielle Inaktivierung einer Reihe von Tumorsuppressorgenen und dem Verlust der Heterozygotie („loss of heterozygosity“) charakterisiert [93, 109, 172]. Tumorsuppressorgene arbeiten in einer rezessiven Weise und fördern die Krebsentwicklung nur, wenn sie in beiden Allelen inaktiviert sind, entweder durch Allelverlust oder durch Mutation beider Allele [18, 113]. Dieses erste molekulare Modell der Kolorektalkarzinogenese wurde von Vogelstein und Fearon für die familiäre adenomatöse Polyposis beschrieben und ist als das klassische Modell der Entstehung eines kolorektalen Karzinoms generell akzeptiert [93, 100, 113, 127]. Entscheidend ist dabei offenbar nicht die Reihenfolge, sondern das Aufeinandertreffen mehrerer dieser genetischen Alterationen (Abbildung 1/1). Für die autosomal dominant vererbte familiäre adenomatöse Polyposis ist eine Keimbahnmutation des Tumorsuppressorgens APC (Adenomatöse-Polyposis-Coli-Gen) auf Chromosom 5q21 verantwortlich [125, 127, 200]. Auch bei 80% der Patienten mit einem sporadischen kolorektalen Karzinom wurde dieser Gendefekt gefunden, wobei es sich in diesem Fall um eine somatische Mutation handelt. Alterationen des APC-Gens werden als initiales Ereignis im Rahmen des Tumorsuppressor-Pathways angesehen. Die Funktionen des vom APC-Gen codierten Proteins beinhalten unter anderem Signaltransduktion über den sogenannten wnt-Pathway, 20 Einleitung Vermittlung interzellulärer Adhäsion und Stabilisierung des Zytoskeletts. Zusammen mit βCatenin reguliert APC über den wnt/β-Catenin-Pathway die Transkription verschiedener Zielgene, wie c-myc und Zyklin D1, die durch ihren Einfluss auf Proliferation, Apoptose und Zell-Zyklus-Progression wichtig in der Tumorentstehung sind. Durch den Funktionsverlust des APC-Gens entsteht aus dem normalen Kolonepithel ein hyperproliferatives Epithel. Durch die Inaktivierung des APC-Gens steigt die Wahrscheinlichkeit für weitere Mutationen [96, 109, 113, 127, 217, 248]. Durch zusätzliche Störungen in der DNA, besonders bei Methylierungsvorgängen, kommt es zur Ausbildung eines sogenannten frühen Adenoms. Die DNA-Methylierung ist ein epigenetisches Ereignis, das eine wichtige Rolle in der Regulierung von Genen spielt. Aus einer Hypomethylierung der DNA könnte eine Überexpression von Onkogenen resultieren, die die Tumorentstehung vorantreibt. In kolorektalen Karzinomen konnte aber auch eine Hypermethylierung der DNA beobachtet werden, die mit einigen Genen, wie APC und hMLH1 assoziiert war. Man vermutet, dass diese Hypermethylierung wichtige Tumorsuppressorgene inaktiviert [111]. 5q Verlust APC 12p Aktivierung k-ras H-ras(?) 18q Verlust DCC DNA HypoMethylierung Normales Epithel Hyperproliferatives Epithel Frühes Adenom Chromosom Änderung Gen 17p Verlust p53 Weitere Änderungen Intermediäres Adenom spätes Adenom Karzinom Metastase Abbildung 1/1: Molekulargenetische Kolorektalkarzinogenese modifiziert nach Fearon und Vogelstein [93] Eine andere genetische Mutation, die relativ früh in der Adenom-Karzinom-Sequenz erscheint, ist eine Aktivierung des Proto-Onkogens K-ras. Sie wird in 30 bis 50% der Adenome und Karzinome gefunden. Es handelt sich dabei um eine Punktmutation im Kodon 12, 13 und 64 auf Chromosom 12. Das mutierte Onkogen aktiviert eine Reihe von Einleitung 21 Effektorwegen, die eine Zunahme der Zellproliferation bewirken [127, 248]. Beim Übergang eines Adenoms in ein invasives Karzinom wird zudem häufig eine Mutation des Tumorsuppressorgens p53 auf Chromosom 17p beobachtet. Das p53-Protein spielt eine wichtige Rolle in vielen Zell-Prozessen, wie DNA-Reparatur, Apoptose und transkriptioneller Aktivierung. Das p53-Protein induziert normalerweise die Apoptose von Zellen und den Verbleib der Zelle in der G1-Phase. Mutationen dieses Gens findet man häufiger in Tumoren des distalen Kolon und Rektum, als in Tumoren des proximalen Kolon [113, 115, 127, 210]. Mit dem Übergang zu Malignität findet man weitere Allelverluste auf dem langen Arm von Chromosom 18. „Loss of heterozygosity“ findet man auf Chromosom 18q21 bei 13% der kleinen Adenome, bei 47% der größeren Adenome und bei 73% der Karzinome. Das Tumorsuppressorgen DCC („deleted in colorectal cancer“), von dem man ursprünglich dachte, dass es eines der wichtigen Zielgene dieser Region sei, codiert für ein großes Zelladhäsionsmolekül und scheint keinen hohen Stellenwert zu haben. Die Proteinprodukte des Smad2- und Smad4-Gens sind in intrazellulären Signalwegen involviert, die mit dem TransformingGrowth-Factor (TGF-β) reagieren, ein Protein, dass viele Zellprozesse, einschließlich Proliferation, Differenzierung, Adhäsion und Apoptose reguliert. Eine Mutation des SMAD4Gens scheint mit der Progression eines Karzinoms assoziiert zu sein [127, 248]. Ein weiteres Gen, das in der Kanzerogenese eine Rolle zu spielen scheint, ist das MCC-Gen (mutated in colorectal cancer). Es liegt nahe dem APC-Gen auf Chromosom 5p21, seine Funktion ist bisher unklar [109]. Zusätzliche genetische Veränderungen ermöglichen es einzelnen Zellen, sich aus dem Tumor zu lösen und in anderen Organen Metastasen zu entwickeln. Der Prozess der Entwicklung eines invasiven Karzinoms aus einem adenomatösen Polypen nimmt etwa 8 bis 15 Jahre in Anspruch. 22 Genetische Läsion Einleitung Chromosom Häufigkeit Zellbiologische Veränderung Vermutetes Stadium innerhalb der AdenomKarzinom-Sequenz Tumorsuppressorgene APC 5q21 30-70% Zellproliferation, Adhäsionsverlust Frühes Adenom (Epitheliale Hyperproliferation) P53 17p12 40-50% Verlust der DNAReparatur-Kontrolle, Verlust der Zellzykluskontrolle Spätes Adenom/Karzinom DCC 18q21 6% Adhäsionsverlust Intermediäres/spätes Adenom/Karzinom MCC 5p21 selten nicht bekannt nicht bekannt SMAD 18q21 16-25% Zellproliferation nicht bekannt Onkogene K-ras 12p12(M) 30-50% Zellproliferation Deregulierung eines Abschaltmechanismus in der Zellaktivierung Intermediäres Adenom c-myc nicht bekannt < 5% Zellproliferation nicht bekannt DNA-Hypomethylierung Gesamtes Genom Überexpression von Onkogenen, DNAFunktionsfehler Frühes Adenom Tabelle 1/8: Genetische Veränderungen in der Adenom-Karzinom-Sequenz Ein zweiter genetischer Pathway, der sogenannte Mutator-Pathway, ist bei der Entwicklung von kolorektalen Karzinomen bei Patienten mit dem „hereditary non-polyposis colorectal cancer-Syndrom“ (HNPCC) involviert, das für etwa 2 bis 5% der kolorektalen Karzinome verantwortlich ist [100, 113, 159]. Patienten mit dieser autosomal-dominant vererbten Erkrankung tragen eine Mutation in den sogenannten Mismatch-Repair-Genen. Aktuell sind fünf dieser DNA-Mismatch-Gene bekannt (Tabelle 1/9). Die durch die Punktmutation inaktivierten MMR-Gene sind nicht mehr in der Lage, spontan auftretende Fehler bei der DNA-Replikation zu korrigieren. Bei Patienten mit einem HNPCC-assoziierten Karzinom ist in über 90% der Fälle das hMLH1- oder das hMSH2-Gen betroffen. In den restlichen 10% der Einleitung 23 Fälle betrifft die Mutation das hPMS2- und das hMSH6-Gen. Defekte in den MismatchRepair-Genen werden auch in 10 bis 15% der sporadischen kolorektalen Karzinome gefunden [100, 196, 255]. Die Mutation der „mismatch-repair-(MMR-)Gene“ führt zu einem weiteren genetischen Phänomen des HNPCC-Syndroms, der Mikrosatelliteninstabilität (MSI). Es handelt sich dabei um eine genetische Instabilität, die molekulargenetisch nachgewiesen werden kann. Man findet Längenpolymorphismen im Bereich einfacher repetitiver DNA-Sequenzen, der sogenannten Mikrosatelliten. Die Mikrosatellitenregionen sind besonders anfällig für Mutationen während der DNA-Replikation, da Fehler nicht effizient repariert werden können. Diese genetische Instabilität führt weiterhin zu Frameshift-Mutationen, die Gene betreffen, die an die Mikrosatellitenregion gebunden sind. Dabei sind bevorzugt Onkogene und Tumorsuppressorgene betroffen, die kurze repetitive Gebiete in ihrer Sequenz besitzen, wie zum Beispiel der TGF-β-TypII-Rezeptor und BAX, die in Zellwachstum und Apoptose involviert sind. Der TGF-β-TypII-Rezeptor ist von besonderem Interesse, da in 90% der kolorektalen Karzinome eine Mutation in diesem Gen über den Mikrosatelliteninstabilitäten-Pathway gefunden wird. Eine hochgradige Satelliteninstabilität (MSI-H, 2 oder mehr Loci betroffen) unterscheidet man von einer niedriggradigen Satelliteninstabilität (MSI-L, ein Locus betroffen). Dabei werden nach internationaler Übereinkunft fünf definierte Loci auf Mikrosatelliteninstabilität untersucht. Man findet sie bei etwa 90% der HNPCC-Tumoren und bei 10 bis 20% der sporadischen kolorektalen Karzinome. Bei den sporadischen Karzinomen wird meist eine hochgradige Mikrosatelliteninstabilität beobachtet [20, 37, 100, 127, 159, 248]. Diese Karzinome weisen teilweise klinische, morphologische und molekulare Gemeinsamkeiten mit HNPCC-Tumoren auf. Histologisch sind MSI-H-Karzinome schlecht, medullär oder muzinös differenziert und zeigen eine starke intratumorale lymphozytäre Infiltration. Trotzdem haben diese Tumoren eine bessere Prognose als mikrosatellitenstabile kolorektale Karzinome und Karzinome mit einer geringgradigen Mikrosatelliteninstabilität. Sie werden häufig bei älteren Frauen im proximalen Kolon gefunden, während p53 und K-ras Mutationen eher im linken Kolon erscheinen [8, 150, 159, 180]. 24 Einleitung hMLH1 Chromosom 3p 60% der Patienten hMSH2 Chromosom 2p 35% der Patienten hPMS1 Chromosom 2q 1% der Patienten hPMS2 Chromosom 7p 1% der Patienten hMSH6 Chromosom 2p 1% der Patienten Tabelle 1/9: Mismatch-Repair-Gene Es wurden noch weitere Wege der molekularen Karzinogenese beschrieben, zum Beispiel der Serrated-Pathway. Sägeblatt-Adenome zeigen oft ein „loss of heterozygosity“ auf Chromosom 18q. Daher vermutet man, dass Sägeblatt-Adenome über einen anderen genetischen Pathway entstehen als andere Adenome im Kolon. Longacre und FenoglioPreiser zeigten, dass 37% dieser gezähnten Adenome Anteile mit hochgradiger intraepithelialer Neoplasie enthielten. Man vermutet, dass die Entwicklung eines „serrated adenoma“ in ein Karzinom sehr schnell verläuft [37, 210, 248, 290]. Durch die genaue Kenntnis der molekularen Alterationen, die in die Entstehung eines kolorektalen Karzinoms involviert sind, eröffnen sich viele neue Anwendungsmöglichkeiten für eine frühe Detektion, Diagnose und Prognose des kolorektalen Karzinoms (Tabelle 1/10). Auch können spezifische molekulare Marker genutzt werden, um die Biologie einer Erkrankung zu erforschen, um neue Therapiemöglichkeiten zu finden und um die Auswahl der richtigen Therapie zu ermöglichen. So könnten Individuen, die endogenen (genetischen) und exogenen (umweltbedingten) Risikofaktoren ausgesetzt sind, identifiziert werden, in ein engmaschiges Überwachungsprogramm aufgenommen werden und/oder eine präventive chirurgische Intervention erhalten. Patienten mit einem hohen Rezidivrisiko und einer schlechten Prognose könnten von einer radikaleren Therapie profitieren, während Patienten mit einem niedrigen Risiko und einer günstigen Prognose eine weniger aggressive Therapie erhalten könnten. Molekulare Marker könnten genutzt werden, um frühe, intermediäre Endpunkte in der Entwicklung kolorektaler Karzinome zu charakterisieren. Da die K-ras-Onkogen-Mutation gewöhnlich in einem frühen Stadium der Adenom-Karzinom-Sequenz auftritt, könnte sie vor der Umwandlung eines kleinen Adenoms in ein Karzinom detektiert werden. So könnte die Entstehung eines Karzinoms verhindert werden [113]. Eine Stuhluntersuchung auf DNAVeränderungen als Screening-Maßnahme des kolorektalen Karzinoms ist inzwischen möglich, jedoch aufgrund unzureichender Daten bisher nur innerhalb von Studien empfohlen. Einleitung 25 Dabei wird die DNA aus Kolonepithelzellen isoliert und auf Mutationen untersucht. Für die APC-Mutation wurde in mehreren Studien eine hohe Sensitivität beobachtet [4, 86, 260]. Jedoch ist für die Mutation des APC-Gens als initiales Ereignis im Rahmen der AdenomKarzinom-Sequenz keine hohe prognostische Relevanz zu erwarten. Eine Mutation des p53Gens ist mit einer schlechteren Prognose assoziiert [20]. Weitere molekulare Marker, die mit einer schlechteren Prognose einhergehen, sind Mutation des Onkogens k-ras und Verlust des Tumorsuppressorgens DCC [139, 186]. Der Nachweis einer Mikrosatelliteninstabilität bei kolorektalen Karzinomen korreliert mit einer besseren Prognose [52, 100]. Anwendung Ziel Detektion Screening der asymptomatischen Bevölkerung Diagnostik Unterscheidung zwischen maligne und benigne Monitoring Einschätzung der Therapiewirksamkeit und prognostische Vorhersage des Tumorverhaltens Klassifikation Therapiewahl und Prognose Staging Definition der Krankheitsausbreitung Lokalisation Nukleäre Methoden mit injizierten radioaktiven Antikörpern Therapie Zytotoxische Agentien, Immuntherapie Tabelle 1/10: Anwendungsmöglichkeiten molekularer Marker Zusätzlich zu den schon beschriebenen Veränderungen von Onkogenen, Tumorsuppressorgenen und der Mikrosatelliteninstabilität stehen auch andere Veränderungen in der Expression und der Aktivität von Genen und ihren Genprodukten im Interesse der Forschung. Dies betrifft vor allem Faktoren, die in Vorgänge der Proteolyse, Adhäsion, Angiogenese und Zellproliferation involviert sind. Hierbei spielen verschiedene an der Zelloberfläche exprimierte Moleküle, wie zum Beispiel β-Catenin und Muzine, sowie ihre assoziierten Antigene als potenzielle prognostische Tumormarker eine große Rolle [20, 93, 248]. 26 Einleitung 1.2 Muzine 1.2.1 Grundstrukturen der Muzine Der Begriff „Muzin“ geht zurück auf De Saussure (1835) und bezog sich anfangs auf die Hauptkomponente des Mukus, der die Oberflächen der glandulären Epithelien mit einem viskoelastischen Gel überzieht. Typisch für die Muzine ist eine dichte O-Glykosylierung, die größtenteils aus einfachen Glykanen mit einem hohen Anteil an sialylierten Bindungen bestanden. Durch die Entwicklung neuer analytischer Methoden wurde offensichtlich, das die Glykane, die an die Muzinpeptid-Cores gebunden sind, viel größer und strukturell komplexer sind, als man erwartet hatte. Seit man entdeckt hat, dass einige blutgruppenverwandte Marker in diese Gruppe fallen, spielen diese komplexen O-gebundenen Glykane eine wichtige Rolle in der Tumordiagnostik. Die Sequenzierung der DNA der Muzine gelang in den späten 1980igern. Als strukturelle Charakterisierung eines „echten“ Muzins besitzen alle Muzine große Peptiddomänen von Tandem-repeats und können so von den Muzin-like-Glykoproteinen klar unterschieden werden [21, 118, 140, 194]. Muzine werden an der duktalen Zelloberfläche glandulärer Epithelien exprimiert und sind die Hauptbestandteile der Glykokalix, die die Schleimhautepithelien bedeckt. Das gewebsspezifische Vorkommen der Muzine wird in Tabelle 1/11 dargestellt. Ihr molekularer Aufbau und ihre Polymerisation gewährleisten die physikochemischen Eigenschaften des Schleims, der ein viskös-elastisches, hochgradig hydriertes Gel darstellt [23]. Dieser Mukus, der die epithelialen Oberflächen des Intestinal-, Bronchial- und Urogenitaltrakts überzieht, spielt eine wichtige Rolle in der Lubrikation und der Protektion der Zellen vor Schäden durch Säuren, proteolytischen Enzymen, pathogenen Organismen und anderen luminalen Noxen [77, 269]. Zu den weiteren Funktionen gehören die Modulation des Wasser- und Elektrolythaushaltes und die Bindung von Proteinen. Zudem sind Muzine Träger von Antigenen, insbesondere Kohlenhydraten, die im Rahmen der Entstehung und Ausbreitung von Neoplasien typische Veränderungen erfahren [23, 259]. Im Kolon fungiert der Mukus als Gleitmittel und sorgt für eine leichtere Darmpassage [9-10]. Sekretorische luminale Muzine können unlösliche Bestandteile, auch Bakterien und Parasiten, ummanteln und dadurch den Abtransport erleichtern. Die Zucker der Oligosaccharidketten sind verantwortlich für Interaktionen mit Parasiten, Viren und Bakterien. Dadurch kann eine Besiedelung und Schädigung der Schleimhaut durch Adhäsion verhindert werden [266]. Einleitung Muzin Hauptsächliches Vorkommen MUC1 Membranassoziiert auf fast allen glandulären Epithelien, vor allem Pankreas und Glandula mammaria MUC2 Kolon (Becherzellen) MUC3 Jejunum, Ileum, Kolon MUC4 Respirationstrakt 27 MUC5AC Magen, Respirationstrakt, Konjunktiva MUC5B Gallenblase, Speicheldrüsen MUC6 Magen MUC7 Speicheldrüsen, Trachea MUC8 Trachea MUC9 Urogenitaltrakt MUC11 Jejunum, Ileum, Kolon, Urogenitaltrakt, Respirationstrakt MUC12 Jejunum, Ileum, Kolon Tabelle 1/11: Gewebsspezifisches Vorkommen der Muzine Muzine sind hochmolekulare Glykoproteine mit einem Molekulargewicht von 300-1000 kDa. Diese Größenvariationen resultieren aus der unterschiedlichen Länge der Kohlenhydratseitenketten. Charakteristisch für alle Muzine ist eine große zentrale Region, die aus Serin- und Threoninreichen (25 bis 70% der Aminosäuren) repetitiven Sequenzen besteht und die man aufgrund ihrer allelabhängig schwankenden Anzahl „variable number of tandem repeats“ (VNTR) nennt. Auch Alanin, Glyzin und Prolin werden in den Tandem-repeats gefunden. Jede dieser Muzineinheiten hat eine spezifische Länge und Sequenz und weist einen genetischen Polymorphismus auf. Diese Eigenschaften sind offenbar organcharakteristisch, jedoch nicht organspezifisch [23, 151]. Jede Muzinuntereinheit besteht aus einer Kette von etwa 1000 Aminosäuren. Diese sind teilweise glykosyliert („T-Domäne“, trypsinresistent) und zum Teil nicht glykosyliert („BDomäne“). Während die T-Domäne hohe Anteile der Hydroxyaminosäuren Serin und Threonin enthält, ist die B-Domäne reich an Asparaginsäure und Cystein. An das Proteingerüst werden in der Länge variierende neutrale, sialylierte oder sulfatierte Oligosaccharidketten durch eine meist O-glykosidische Bindung an Serin bzw. Threonin 28 Einleitung angeknüpft. Jede Muzinuntereinheit enthält etwa 200 Kohlenhydratseitenketten. Die Oligosaccharidketten machen 50 bis 80% der Masse eines Muzins aus. Die Regionen mit zahlreichen Oligosaccharidketten, die mit Regionen niedrigem Kohlenhydratgehaltes abwechseln, bilden eine relativ starre für Proteasen wenig anfällige Struktur [24, 190, 242, 259]. Die Oligosaccharidketten kann man in drei strukturelle Regionen aufteilen (Abbildung 1/2 und Tabelle 1/12). Die innere Kernregion („core region“) ist durch die O-glykosidische Bindung von N-Azetylgalaktosamin an Serin oder Threonin, sowie durch die Zuckerreste, die direkt an N-Azetylgalaktosamin gebunden sind, charakterisiert. Bisher sind acht dieser CoreStrukturen aufgeklärt. Der inneren schließt sich die äußere Kernregion („backbone region“) an. Sie besteht aus linearen oder verzweigten, teilweise repetitiven Galaktose-NAzetylglukosamin-Disacchariden (Gal-GlcNAc), die man aufgrund ihrer Bindung in die Typen 1-Laktosamin (Galβ1-3GlcNAc) und 2-Laktosamin (Galβ1-4GlcNAc) unterscheidet. Die periphere Region zeigt die größte Vielfalt von Kohlenhydratstrukturen und ist die wichtigste Region für die Antigenität der muzinassoziierten Kohlenhydrate. Sie enthält unter anderem ABH- und Lewis-Blutgruppenantigene, sowie eine Reihe anderer Blutgruppenverwandter Strukturen. Einleitung Peptid Innere Kernregion Äußere Kernregion Periphere Region Abbildung 1/2: Schematische Darstellung der Kohlenhydratseitenketten der Muzine Innere Kernregion Core 1 Galβ1-3GalNAc Core 2 Galβ1-3(GlcNAcβ1-6)GalNAc Core 3 GlcNAcβ1-3GalNAc Core 4 GlcNAcβ1-3(GlcNAc1-6)GalNAc Core 5 GalNAcα1-3GalNAc Core 6 GlcNAcβ1-6GalNAc Core 7 GalNAcα1-6GalNAc Core 8 Galα1-3GalNAc Tn-Antigen GalNAcα 29 30 Sialyl-Tn-Antigen Einleitung NeuAcα2-6GalNAc Äußere Kernregion Typ1-Kette Galβ1-3GlcNAc Typ2-Kette Galβ1-4GlcNAc i-Antigen (unverzweigt) (Galβ1-4GlcNAcβ1-3)n I-Antigen (verzweigt) Galβ1-4GlcNAcβ1-3(Galβ1-4GlcNAcβ16)Galβ1 Periphere Region H-Typ 1 Fucα1-2Galβ1-3GlcNAcβ1- Blutgruppe A-Typ 1 GalNAcα1-3(Fucα1-2)Galβ1-3GlcNAcβ1- Blutgruppe B-Typ 1 Galα1-3(Fucα1-2)Galβ1-3GlcNAcβ1- Lewisa Galβ1-3(Fucα1-4)GlcNAcβ1- Lewisb Fucα1-2Galβ1-3(Fucα1-4)GlcNAcβ1- Lewisx Galβ1-4(Fucα1-3)GlcNAcβ1- Lewisy Fucα1-2Galβ1-4(Fucα1-3)GlcNAcβ1- Sialyl-Lewisa NeuAcα2-3Galβ1-3(Fucα1-4)GlcNAcβ1- Sialyl-Lewisx NeuAcα2-3Galβ1-4(Fucα1-3)GlcNAcβ1- Tabelle 1/12: Glykanstrukturen in Oligosacchariden vom Muzin-Typ [21] Bis heute sind 20 Gene identifiziert worden, die für jeweils unterschiedliche Muzin-Typen codieren [49]. Die Muzine werden unterteilt in sekretorische und membrangebundene Muzine, zwischen denen es große Unterschiede gibt (Tabelle 1/13). Die sekretorischen Muzine besitzen in den nicht repetitiven Anteilen cysteinreiche Peptide („vonWillebrandtfactor-like D-Domains“), die über Bildung kovalenter Disulfidbrücken bei der Oligomerisation von Muzin-Monomeren und an der Gestaltung der sekretorischen Vesikel eine Rolle spielen. Im Gegensatz dazu enthalten die membrangebundenen Muzine weniger Cystein und bilden keine oligomerischen Komplexe, besitzen aber hydrophobe Sequenzen („Transmembran-Domäne“), die für die Verankerung in der Lipid-Doppelschicht verantwortlich sind und C-terminale Peptide, die in das Zytosol hineinreichen. Zu den sekretorischen Muzinen zählen MUC2, MUC5AC, MUC5B, MUC6, MUC7, MUC8, MUC9, und MUC19. Zu den Membrangebundenen gehören MUC1, MUC3A, MUC3B, MUC4, Einleitung 31 MUC11-13, MUC15-18 und MUC20, wobei zumindest MUC1 auch in einer sekretorischen Isoform existiert [55, 59, 118, 212]. MuzinSpezies Sekretorisch / membrangebunden ChromosomenLokalisation Anzahl der Tandem-repeats Anzahl der Aminosäuren / Tandemrepeat DDo mä ne MUC1 m, s 1q21 variabel (21-125) 20 - MUC2 s 11p15.5 21/51-115 23/16 + MUC3A m, s 7q22 20/? 17/375 - MUC3B m, s 7q22 33*/? 17/375 - MUC4 m, s 3q29 145-395 16 - MUC5AC s 11p15.5 124,17,34,66** 8/5 + MUC5B s 11p15.5 11,11,17,11,22** 29 + MUC6 s 11p15.5 15-26 169 + MUC7 s 4q13-21 5-6 23 - MUC8 s 12q24.3 6*/3* 13/41 + MUC9 s 1p13 6 15 + MUC11 m 7q22 36* 28 - MUC12 m, s 7q22 22* 28 + MUC13 m 3q13.3 10 15 - MUC15 m 11p14.3 Keine TR 0 - MUC16 m 19p13.3 9 156 - MUC17 m 7q22 5 59 - MUC18 m 11q23.3 Keine TR 0 - MUC19 s 12q12 6*/3/4/2/2/4/3 7/7/15/16/9/8/5 + MUC20 m 3q29 2-6 19 Tabelle 1/13: Muzine [21, 224] *Die vollständige DNA-Sequenz ist noch unbekannt **Die Anzahl der TR ist in spezifischen Regionen von MUC5AC und MUC5B unterschiedlich. ? 32 1.2.1.1 Einleitung Synthese der Oligosaccharidketten Typische post-translationelle Modifikationen von Glykoproteinen sind die N- oder OGlykosylierung. Muzine enthalten meist O-glykosidisch gebundene Oligosaccharide, die in einem schrittweisen Prozess in den Golgikomplexen gebildet werden. Die Synthese beginnt mit der Übertragung von N-Azetyl-D-Glalaktosamin (GalNAc) aus UDP-GalNAc auf die Hydroxyaminosäuren Serin oder Threonin innerhalb der VNTRRegion. Es entsteht das sogenannte Tn-Antigen GalNAc-O-Ser/Thr. Dieser Schritt wird von den Polypeptid-N-Azetylgalaktosaminyl-Transferasen katalysiert. Es liegen verschiedene Isoformen dieser Enzymgruppe vor, die in unterschiedlichen Kompartimenten des Golgiapparates verteilt sind und eine unterschiedliche, teilweise überlappende Spezifität beim Erkennen der Peptid-Sequenz haben. Dadurch, dass die Enzyme in den Organen unterschiedlich aktiv sind, ergeben sich regional unterschiedliche Muster der Kohlenhydratexpression. Bisher sind 30 verschiedene Glykosyltransferasen bekannt, von denen mindestens 12 an der Synthese der O-Glykane der Muzine beteiligt sind [49, 224, 271]. Außer von dem Vorhandensein der Transferasen, hängt die initiale O-Glykosylierung auch von der Aminosäuresequenz ab, die die Aktivität der Transferasen negativ oder positiv beeinflussen kann. Auch die post-translationalen Veränderungen der Peptidsubstrate und die Sekundär- und Tertiärstruktur der Proteine, die potenzielle Glykosylierungspositionen maskieren können, haben einen Einfluss auf die initiale O-Glykosylierung. Als nächster Schritt folgt die Substitution von zwei bis drei weiteren Monosacchariden, meist Galaktose (Gal) und/oder N-Azetylglukosamin (GlcNAc), aber auch Neuraminsäure (NeuAc, NeuGc) oder Fukose (Fuc) mit N-Azetylgalaktosamin an den Positionen C3 und C6. Auf diese Weise entstehen die bereits erwähnten Core-Strukturen, die alle weiter glykosyliert werden können. Core 1 und Core 3 entstehen durch Addition von Gal bzw. GlcNAc an Position C3. Wird zusätzlich an Position C6 GlcNAc addiert, entsteht Core 2 bzw. Core 4. Core 5 entsteht durch Substitution von GalNAc in Position C3. Durch Bindung von GlcNAc bzw. GalNAc in Position C6 entsteht Core 6 bzw. Core 7. Core 8 enthält Gal in Position C3 (Abbildung 1/2). Während Core 1 und Core 2 im menschlichen Organismus weit verbreitet sind und Core 3 und Core 4 im Gastrointestinal- und Bronchialtrakt gefunden werden, kommt Core 5 eher in fetalen Muzinen und in kolorektalen Karzinomen vor. Core 6 findet man in Mekonium und in Magenkarzinomen, Core 7 und Core 8 im menschlichen Bronchialtrakt. Im Kolon kommen vor allem die Core-Strukturen 1 bis 4 vor [49]. An die Core-Region schließt sich die Backbone-Region an durch Substitution des Disaccharids Galβ1-4GlcNAc, entweder über eine β1-3-Bindung, so dass lineare Strukturen entstehen, oder über eine β1-6-Bindung an Galaktose, wodurch verzweigte Ketten entstehen. Einleitung 33 Die Position C6 wird hierbei meistens bevorzugt. Es entstehen Polylaktosaminstrukturen mit über 20 Monosacchariden. Im menschlichen Magen und Kolon sind bis zu 21 verschiedene Backbone-Strukturen bekannt. Diese linearen oder verzweigten Polylaktosamine von Typ1und Typ 2-Ketten repräsentieren die strukturelle Basis der i/I Blutgruppenantigene. Die Ketten werden mit α-Galaktose, α-N-Azetylgalaktosamin oder mit terminierenden Monosacchariden wie α-Neuraminsäure, α-Fukose oder Sulfat substituiert [21, 252]. Die strukturelle Variabilität erhöht sich ferner durch Veränderung der Neuraminsäure, die Nazetyliert, N-glykosyliert oder O-azetyliert sein kann. Die Addition von Neuraminsäure und Sulfat verleihen den Muzinen seine negative Ladung, was den niedrigen pH-Wert erklärt, während Fukose für die Hydrophobizität verantwortlich ist. Mit der negativen Ladung und/oder der Hydrophobizität tragen die terminalen Zucker zu den physikalischen und biologischen Eigenschaften der Muzine bei. Während im Magen hauptsächlich neutrale Muzine und im Dünndarm neutrale und sialinierte Muzine enthalten sind, kommen im Kolon neutrale, sialinierte und sulfatierte Muzine vor. Der hohe Anteil von O-azetylierter Neuraminsäure ist charakteristisch für das menschliche Kolon und wurde in keinem anderen menschlichen Gewebe gefunden [7, 76]. Die poly-o-azetylierte Neuraminsäure führt zu einer Hydrophobizität der Muzine, was im Kolon den Widerstand gegen bakterielle Enzyme erhöht [23]. 1.2.2 MUC1: Struktur MUC1 wurde zuerst in menschlicher Milch als ein hochmolekulares Glykoprotein reich an Serin, Threonin und Prolin mit einem hohen Prozentanteil von O-gebundenen Kohlenhydraten identifiziert [101, 251-252]. MUC1 ist auch bekannt unter den Namen HMFGAntigen, DF2-Antigen, EMA (epithelial membrane antigen), MAM-6, PEM (polymorphic epithelial mucin), PAS-O, PUM und Episialin. Diese vielen Bezeichnungen unterstreichen die polymorphe Struktur von MUC1 und seine verschiedenen Funktionen [59, 269]. Es wurde 1990 parallel von vier Arbeitsgruppen als erstes menschliches Muzin-Gen sequenziert [103, 169, 177, 251, 286]. MUC1 ist das bisher bekannteste Muzin, dessen Gen auf Chromosom 1q21 lokalisiert ist. Es besteht aus sieben Exons, wobei Exon 2 die multiplen Tandem-repeatSequenzen aus Einheiten mit 60 bp enthält. Abhängig von der Zahl der Tandem-repeats, macht diese Region 50 bis 80% des ganzen Gens aus. Die VNTR-Domäne codiert für 20 bis 120 Repeats und weist einen hereditären Polymorphismus auf. Die meisten Menschen Nordeuropas haben zwischen 40 und 80 Repeats [21, 95, 101-102, 236, 244]. Die VNTR-Region enthält einen hohen Anteil (82%) von G/C Basenpaaren. Die hoch konservierte VNTR- 34 Einleitung Region wird flankiert von kurzen nicht repetitiven Peptid-Sequenzen. Die kleine N-terminaleDomäne enthält ein Signalpeptid. Der C-terminale Anteil enthält eine transmembranäre und eine intrazelluläre Domäne, deren Sequenzen hoch konserviert sind (Abbildung 1/3) [24, 103, 169, 177]. Es werden einige MUC1 Isoformen beschrieben, die durch alternatives Splicen generiert wurden. Das MUC1/TM, die vollständige Form von MUC1, bildet ein Heterodimer mit einer extrazellulären, O-glykosylierten Untereinheit, die an eine transmembranäre Untereinheit gebunden ist. Das MUC1/Y ist ein verzweigtes Transmembran-Protein und enthält keine Tandem Repeat-Domäne [102, 298]. Es dient als Bindungspartner für eine andere Isoform, das sekretorische MUC1/sec, ein lösliches Glykoprotein, das keine TransmembranDomäne enthält. Das MUC1/sec kann mit spezifischen Antikörpern in Sekreten von Karzinomzellen und im Serum von Patienten mit einem Mammakarzinom detektiert werden [21, 32, 55, 95, 102]. Sig Tandem-repeat Tandem-repeat Domäne Domäne (VNTR) (VNTR) N TM CP C HGVTSAPDTRPAPGS TAPPA Immundominante Region Abbildung 1/3: Primärstruktur von MUC1, abgeleitet aus der DNA-Sequenz von Chromosom 1q21. Sig=Signalsequenz, TM=Transmembran-Sequenz, CP=cytoplasmatische Domäne MUC1 ist ein membrangebundenes Glykoprotein. Mit einem Molekulargewicht des Apomuzins von 120-300 kDa, in glykosylierter Form von 300-600 kDa, ist es im Vergleich zu den anderen Muzinen ein relativ kleines Muzin. Das reife Glykoprotein bildet einen stabilen heterodimeren Komplex. Es wird von den meisten glandulären Epithelzellen und epithelialen Tumoren gebildet. MUC1 wird in Normalgeweben ausschließlich auf der apikalen Oberfläche der Zellen exprimiert, wo es stabförmig 100 bis 500 nm über die Oberfläche hinausragt. Das ist 100-mal weiter als die meisten anderen Oberflächenmoleküle [95, 163, 218, 252]. Es kommt vor allem in der Mamma, in der Speicheldrüse, im Magen, im Pankreas, in der Einleitung 35 Harnblase, im Uterus, im Dünndarm und im Kolon vor. Auch wurde MUC1 in hämatopoetischen Zellen, wie Granulozyten, Plasmazellen, erythropoetischen Zellen, aktivierten TZellen, Myelomzellen und Lymphomzellen detektiert [269]. In Karzinomen der entsprechenden Organe ist eine unterschiedlich deutliche Überexpression zu beobachten [6, 80, 87, 108, 136, 198]. Vom Aufbau her können die Untereinheiten des Muzins in drei unterschiedliche Regionen aufgeteilt werden: Die zytoplasmatische Domäne mit 69 Aminosäuren, die hydrophobe Transmembran-Domäne mit 31 Aminosäuren und eine große hoch glykosylierte extrazelluläre Domäne, die die große Anzahl an Tandem-repeats enthält. Diese Tandem-repeat-Domäne enthält die bis zu 120-fach wiederholte aus 20 Aminosäuren bestehende Sequenz (PDTRPAPGSTAPPAHGVTSA) mit den Varianten (PD/ET/SRPAPGSTAPP/Q/A/TAHGVTSA). Jede Tandem-repeat-Einheit enthält fünf potenzielle O-Glykosylierungsstellen. Zwei werden von Serin, drei von Threonin repräsentiert Während in normalem Drüsengewebe der Mamma durchschnittlich nur 50% der potenziellen Bindungsstellen besetzt sind, werden in malignen Zellen alle fünf Bindungsstellen zur OGlykosylierung genutzt [118, 192]. Die VNTR-Region ist reich an Serin, Threonin und Prolin. Mit Alanin und Glyzin machen diese fünf Aminosäuren 50% der gesamten Aminosäuren in der VNTR-Region aus [24, 95, 218]. Cystein ist nur in der zytoplasmatischen Domäne vorhanden, so dass eine Polymerisation zwischen einzelnen MUC1-Molekülen unmöglich ist. Zwischen der VNTR-Region und der transmembranären Domäne liegen fünf potenzielle N-Glykosylierungsstellen, die mit komplexen Glykanen substituiert sind. Die Ngebundenen Oligosaccharide machen quantitativ einen kleinen Teil aus, können aber funktionell wichtig sein [55, 177]. Die an Serin, Threonin und Prolin O-glykosidisch gebundenen Kohlenhydratketten machen einen hohen Prozentanteil des Moleküls aus und erlauben dem Muzin, hochgradig hydratisiert zu sein. Die O-gebundenen Glykane in MUC1 variieren in Abhängigkeit des Organs und der Differenzierung des Gewebes. In Epithelien der laktierenden Mamma werden hauptsächlich lange Polylaktosamin-Typ-Ketten exprimiert, die durch Verlängerung des C6-Astes von Core2 gebildet werden [119, 121, 142]. Nur etwa 20% der Glykane enthalten Neuraminsäure. Sie sind stark fukosyliert. In Zellen von Mammakarzinomen findet man demgegenüber kurze Ketten begrenzt auf die Core-Strukturen. Sialylierte Glykane dominieren über Neutrale [120, 178]. Über die Glykosylierung von MUC1 in kolorektalen Karzinomen gibt es weniger Informationen. In rektalen Adenokarzinomen konnte gezeigt werden, dass sich die Glykane auf kurze Core-Typ-Sequenzen beschränken, die aus Core1, 3 und 5 bestehen. Sie enthalten als gemeinsame Struktureinheit das Disaccharid NeuAcα2-6GalNAc (Sialyl-Tn-Antigen) [168]. Enzymatische Studien lassen vermuten, dass es in kolorektalen Karzinomen zu einer generellen Verlagerung der Core- 36 Einleitung Synthese von Core 3, das in normalen Kolongewebe dominiert, zu Core 2 kommt [50, 75, 215-216]. Die Typ 2-Polylaktosamine werden mit Neuraminsäure oder Fukose terminiert [95, 252]. 1.2.3 MUC1: Biologische Relevanz Als membrangebundenes Muzin wird MUC1 in normalen duktalen Epithelien auf der apikalen Oberfläche der Zellen exprimiert. Die exakten Funktionen von MUC1 sind noch nicht vollständig aufgeklärt. Die extrazelluläre Domäne, die stabförmig über die Zelloberfläche hinausragt, ist verantwortlich für die protektive Funktion von MUC1 und spielt eine Rolle in der Immunregulation [170]. Der muköse Schleim bildet eine protektive Barriere zwischen der Epitheloberfläche und dem das Epithel schädigende Milieu im Lumen des gastrointestinalen, respiratorischen und reproduktiven Traktes. MUC1 ist für die Lubrikation der Zelloberfläche und das Aufhalten von pathogenen Partikeln verantwortlich. Eine protektive Funktion gegen bakterielle oder virale Pathogene konnte für MUC1 in menschlicher Milch gezeigt werden [102, 234, 291]. Auch ist MUC1 in Zell-Zell- und ZellMatrix-Interaktionen involviert [189]. Im Rahmen der Kanzerogenese kommt es zu einer deutlichen Überexpression von MUC1, die zum Beispiel in Adenokarzinomen bis auf das 10-fache gesteigert sein kann. MUC1 ist nicht mehr auf die apikale Zellmembran beschränkt, sondern aufgrund des Polarisationsverlustes der Tumorzellen auf der gesamten Zelloberfläche verteilt [189, 206]. Zusätzlich findet man eine Veränderung der Muzin-Glykosylierung, im Sinne einer atypischen verminderten Glykosylierung [49, 74, 95, 136]. Diese typischen Veränderungen in neoplastischen Geweben könnten eine große Aussagekraft sowohl im Hinblick auf Differenzierung und Biologie als auch in Bezug auf Diagnose, Progression und Prognose von Tumoren besitzen. So konnte in Adenokarzinomen der Lunge gezeigt werden, dass eine Membranexpression von glykosylierten MUC1 signifikant mit Lymphknotenmetastasen und fortgeschrittenen pathologischen Stadien assoziiert ist [112]. Cao et al. hat eine Membranexpression von unterglykosyliertem MUC1 in kolorektalen Adenomen mit schwerer Dysplasie und in invasiven kolorektalen Karzinomen nachgewiesen [62]. Hier könnte eine anormale Expression von MUC1 in die Entwicklung von Metastasen und in den Übergang von normalen zu (prä-) malignen Läsionen involviert sein. Auch in der Entwicklung von neuen Therapieansätzen, wie zum Beispiel der Immuntherapie könnte MUC1 wichtig sein [95, 137, 239, 252]. Die Überexpression von MUC1 in Tumoren von Lunge, Magen, Mamma, Pankreas, Kolons, Prostata, Ovarien, Endometrium, Zervix und Einleitung in vielen Myelomen und einigen Lymphomen 37 machen MUC1 zu einem attraktiven therapeutischen Ziel. Man geht inzwischen davon aus, dass MUC1 einen unabhängigen negativen prognostischen Parameter für das kolorektale Karzinom darstellt und dass eine Überexpression von MUC1 in Tumoren mit der Progression und der Metastasierung assoziiert ist [27, 89, 102, 170, 195, 264]. Als Serumtumormarker CA 15.3 hat sich MUC1 bereits bei Patientinnen mit einem Mammakarzinom etabliert [126, 227]. Der Nachweis dient hauptsächlich der Verlaufskontrolle. Zum Nachweis von Primärtumoren ist die diagnostische Empfindlichkeit der Methoden zu gering und der Nachweis zu unspezifisch [238]. Dennoch ist die Aussagekraft und Bedeutung von MUC1, gerade in Bezug auf das Glykosylierungsprofil, trotz zahlreicher Studien noch nicht eindeutig geklärt und wird teilweise kontrovers diskutiert. Eine Theorie für den Zusammenhang einer gesteigerten MUC1-Expression und einer erhöhten Metastasierungsbereitschaft eines Tumors bezieht sich auf die adhäsiven Eigenschaften von MUC1. Die gestörte anormale Glykosylierung von MUC1 im Rahmen der Kanzerogenese führt zu kürzeren und weniger verzweigten Oligosaccharidketten, der Akkumulation von Vorläuferstukturen und der Demaskierung von Regionen des MUC1-Proteincores [214, 228]. Es entstehen neue Kohlenhydrat-Epitope, wie zum Beispiel das T- und das Tn-Antigen, das Sialyl- Le x - und das Sialyl- Le a -Antigen [49]. Diese strukturellen Veränderungen haben einen Einfluss auf die Zellfunktion und somit auf das Potenzial eines Tumors, invasiv zu wachsen und zu metastasieren. So geht ein starker Titeranstieg des muzinassoziierten Sialyl-TnAntigens bei kolorektalen Karzinom-Patienten mit einer schlechteren Prognose einher [145]. Sialyl- Le x und Sialyl- Le a tragen über die Liganden P- und E-Selektin und das intra-zelluläre Adhäsionsmolekül ICAM-1 zur Adhäsion von malignen Zellen an vaskuläre Epithelien bei und könnten so eine entscheidende Rolle in der hämatogenen Metastasierung einnehmen [6, 89, 181-182, 221, 246-247]. In Tumoren findet man eine Steigerung der Sialylierung von MUC1. Die Neuraminsäure, ein terminierendes Monosaccharid der peripheren Region scheint ebenfalls einen Einfluss auf die Metastasierung zu haben. So konnte gezeigt werden, dass ein Entfernen der Neuraminsäure das Metastasierungspotenzial von Karzinomzellen reduzieren kann [48-49]. Auch haben In-vitro-Ergebnisse gezeigt, dass Kolonkarzinomzelllinien mit einer starken Muzinproduktion eine starke Adhärenz an Basalmembranproteine aufweisen. Dieser Effekt konnte durch spezifische Hemmung der O-Glykosylierung aufgehoben werden [235]. 38 Einleitung Neben diesen adhäsiven Eigenschaften besitzt MUC1 aber auch anti-adhäsive Funktionen, die eine entscheidende Rolle in der Progression und Metastasierung eines Tumors spielen. Aufgrund der großen, stabähnlichen Konformation des MUC1-Proteins, das sich 200 bis 500 nm über die Zelloberfläche ausdehnt, können Zell-Zell- und Zell-Matrix-Interaktionen gehemmt werden. So könnten sich Tumorzellen aus dem Zellverband lösen und metastasieren. Da MUC1 in Karzinomen durch den Polarisationsverlust der Zelle auf der ganzen Zelloberfläche verteilt ist und aufgrund seiner Länge und Stabilität, kann MUC1 andere Adhäsionsmoleküle maskieren und so zu einer reduzierten Zell-Zell-Aggregation beitragen [131, 176]. MUC1 kann auch die Interaktion zwischen Glykoproteinen gegenüberliegender Membranen hemmen, wodurch das Lumen der Drüsen und Venolen aufrechterhalten wird [170]. MUC1 interagiert mit einer großen Anzahl an Proteinen, die einen Einfluss auf Karzinogenese und Zell-Adhäsion haben. Vor allem für die über ECadherin vermittelte Zell-Zell-Adhäsion scheint MUC1 ein wichtiger adhäsionshemmender Faktor zu sein [275]. Durch eine MUC1-Überexpression konnte auch die Integrin-vermittelte Adhäsion von Karzinom-Zellen an Bestandteile der Extrazellulärmatrix, wie Laminin, Kalinin und E-Cadherin, wie auch die Bindung zwischen E-Cadherin und β-Catenin gehemmt werden [160, 252, 275-276, 288]. Die intrazelluläre Domäne von MUC1 interagiert außerdem direkt mit β-Catenin, einem nukleären Transkriptionsfaktor, der zur Hemmung von E-Cadherin vermittelter Zell-Zell-Adhäsion führt [144]. Eine Überexpression von MUC1 geht mit einem erhöhtem Level an nukleären β-Catenin einher [274]. Ein Einfluss von β-Catenin auf die Tumorprogression konnte gezeigt werden. So ist eine Koexpression von MUC1 und βCatenin in der Invasionsfront von kolorektalen Tumoren mit einer schlechteren Prognose korreliert [46, 89]. β-Catenin bindet auch an APC. Dies ist ein essenzieller Partner im SignalPathway Wingless/wnt-1 [143, 193]. Eine APC-Überexpression reduziert den Anteil an freiem zytoplasmatischen β-Catenin und somit auch die Komplexbildung von β-Catenin und E-Cadherin. Dies reduziert die intrazelluläre Adhärenz [54, 209]. Die Blockierung der Zell-Zell-Interaktionen durch MUC1 zerstört die epitheliale Struktur und Stabilität, was die Neigung zur Invasivität steigern kann. Dies konnte an Ovarialkarzinomen gezeigt werden [87]. Einleitung 1.2.4 1.2.4.1 39 Expression von Muzinantigenen im Kolon Expression von Muzinantigenen in normalem Kolongewebe 1.2.4.1.1 Peptid-Core-Antigene In Normalgeweben sind die Muzinpeptidantigene infolge der regulär starken Glykosylierung oft maskiert und ihr Nachweis durch Antikörper daher erschwert. Im Gastrointestinaltrakt werden von der Mundhöhle bis zum Kolon Muzine exprimiert. Dabei findet sich in den verschiedenen Geweben ein spezifisches Expressionsmuster der Muzingene. Im Kolon ist es häufig problematisch, die genaue Lokalisation der Muzin produzierenden Zellen zu entdecken. Das liegt auf der einen Seite an der Schnittdicke beim Lichtmikroskop, andererseits an der Schwierigkeit, die vollständig glykosylierten und oligomerisierten Muzine zu detektieren [281]. Daher findet man in der Literatur einige widersprüchliche Angaben in Bezug auf die Expression der Peptidantigene der Muzine. In den vergangenen Jahren wurden in mehreren Workshops viele monoklonale und polyklonale Antikörper entwickelt, die inzwischen gut charakterisierte Epitope auf den Muzin-Molekülen erkennen und binden [64, 213, 218, 251]. Die meisten dieser Peptid-spezifischen Antikörper sind gegen Epitope auf der Repeat-Domäne gerichtet und reagieren mit einem definierten Cluster der Aminosäuren. Dabei stellt das PDTRP-Motiv des MUC1- Muzins eine sehr immundominante Region dar [269]. Im Kolon werden hauptsächlich MUC2 und MUC3 exprimiert [77]. Für MUC2 gibt es kontroverse Ergebnisse. Zum einen soll MUC2 vorwiegend in den Becherzellen und nicht in den Enterozyten exprimiert werden, in denen anti-MUC2-VNTR-Antikörper eine intensive zytoplasmatische Färbung hervorrufen, jedoch nicht das reife Muzin oder das anhängende Mukusgel färben [11, 66, 136, 273]. Nach Entfernung von Oligosacchariden konnte sekretorisches MUC2 nachgewiesen werden [281]. Zum anderen konnte MUC2 sowohl in Becherzellen als auch in Zylinderepithelien nachgewiesen werden. MUC3 wurde sowohl im supra- und perinukleären Zytoplasma der Becherzellen als auch in den absorbierenden Enterozyten detektiert. MUC3 ist eher in den oberflächlichen Epithelien als in der Kryptenbasis vorhanden [55, 66, 136, 273]. Mit Hilfe der In situ Hybridisierung ließen sich im Kolon jedoch nicht nur Muzine vom intestinalen (MUC2 und MUC3), sondern auch vom Mamma- (MUC1) und Tracheobronchialtyp (MUC4, MUC5) nachweisen [17]. In einer Studie wurde neben einer MUC1-, MUC2- und MUC3-mRNA-Expression auch eine MUC4-mRNA-Expression in normalem Kolongewebe gefunden [204]. MUC4 wird sehr stark in den Krypten des normalen Kolons exprimiert, wo die Färbung der Becherzellen am 40 Einleitung auffälligsten war [55, 281]. Obwohl in normalem Kolongewebe MUC1-mRNA detektierbar ist, haben die meisten Studien herausgefunden, dass normale intestinale Epithelzellen eine nur schwache oder gar keine Reaktion mit monoklonalen Antikörpern gegen MUC1-PeptidEpitope zeigen [55, 108, 136, 195, 297]. Das MUC1-Protein scheint in normalem Kolongewebe durch die starke Glykosylierung maskiert zu sein. Andererseits haben einige Autoren berichtet, dass sie mit ihren monoklonalen Antikörpern MUC1 in normalem Kolon detektieren konnten [59, 63]. Durch Perjodat-Oxidation, durch die teilweise Kohlenhydrate entfernt werden, konnte die MUC1-Reaktivität verstärkt werden [6, 59]. Das MUC1-Protein wurde im Zytoplasma von Becher- und Zylinderzellen gefunden. Am stärksten wurde das MUC1-Apomuzin auf der apikalen Membranoberfläche vor allem in der Kryptenbasis exprimiert [6, 37, 281]. Obwohl das MUC5B nur sehr gering in normalem Kolongewebe exprimiert wird [55], konnte in einer Studie eine MUC5B- und MUC2-Koexpression in Becherzellen nachgewiesen werden [267]. MUC5B zeigte eine Reaktivität vor allem in der Kryptenbasis. MUC5AC und MUC6 wurden nur schwach oder überhaupt nicht in der Mukosa des normalen Kolonepithels nachgewiesen [31, 55]. MUC11, MUC12, MUC13 und MUC17 wurden ebenfalls in normalem Kolongewebe detektiert [55, 278]. 1.2.4.1.2 Kohlenhydratantigene Die Synthese der Oligosaccharidketten erfolgt durch die gewebe- und substratspezifischen Glykosyltransferasen, die für ein organspezifisches Verteilungsmuster verantwortlich sind. Sie haben teilweise überlappende Akzeptorspezifitäten. So kann zum Beispiel die α1-2Fukosylierung peripherer Antigene von H- oder von Sekretor (Se) gencodierten Transferasen bewirkt werden. Die Sekretoreigenschaft beinhaltet die Fähigkeit zur Sekretion von ABHBlutgruppen mit dem Mukus. Diese Konstellation weisen etwa 75% der Europäer auf. Da die Glykosyltransferasen jedoch nicht gleichermaßen in allen Anteilen gastrointestinaler Organe aktiv sind, ergeben sich vom Gentyp abhängige, regional unterschiedliche Muster der Kohlenhydratexpression [23]. Es wurden viele spezifische Antikörper generiert, die gegen bestimmte Kohlenhydratantigene gerichtet sind. Ein Problem des Nachweises der Kohlenhydratantigene in normalem Kolongewebe ist die O-azetylierte Neuraminsäure, die viele Antigene maskiert. Im Kolon steht der proximale Organabschnitt bis zur Flexura sinistra unter der Kontrolle des Sekretor-Gens. Hier findet man daher eine starke Immunreaktivität von H-Typ-2 und Le y . Im adulten distalen Kolon findet eine α1-2-Fukosylierung und somit eine Expression von ABHund Le b -/ Le y -Antigenen nicht mehr statt [30, 277, 292]. Das Le a -Antigen wird von Individuen, die das Lewis-Gen nicht exprimieren, im Kolon nicht synthetisiert [91]. Le x ist im gesamten Kolon schwach nachweisbar. Es wird vor allem im apikalen Zytoplasma von Einleitung Zylinderzellen an der Kryptenbasis exprimiert, während man 41 Le y in der Glykokalix beobachten kann [6]. Während Sialyl- Le x und Sialyl- Le a im normalen Kolongewebe nicht oder nur schwach nachweisbar sind, konnte nach De-O-Azetylierung eine starke Expression beobachtet werden [203, 296]. Dies zeigt, dass sie in normalem Kolongewebe exprimiert werden, aber durch O-azetylierte Neuraminsäurereste maskiert vorliegen. Kohlenhydratantigene der inneren Kernregion, sogenannte Kryptantigene wie das Tn-, das TF-Antigen und ihre sialylierten Formen, konnten bisher in normalen Kolongewebe nicht oder nur gering mit Hilfe monoklonaler Antikörper nachgewiesen werden, da sie in der Regel maskiert vorliegen [80]. Allerdings konnte nach Esterspaltung der O-azetylierten Neuraminsäure Sialyl-Tn oder Tn in Becherzellen detektiert werden [205, 241]. Dagegen war jedoch eine Bindung des Dolichos-biflorus-Agglutinins DBA an GalNAcα-Reste, die in Tnund A-Blutgruppenantigenen vorkommen und des Peanut-Agglutinins (PNA) zu beobachten [148, 294]. Für die Expression des Thomsen-Friedenreich-Antigens (TF-Antigen) in normalem Kolongewebe findet man sehr widersprüchliche Daten. In einigen Studien konnte das TF-Antigen mit PNA in Becherzellen und Zylinderepithelien nachgewiesen werden [58, 72, 294], während in anderen Untersuchungen nur eine geringe oder gar keine Färbung zu beobachten war [43, 164]. Mit spezifischen monoklonalen Antikörpern konnte das TF-Antigen nur schwach oder gar nicht detektiert werden [61, 148, 205, 294]. Durch Entfernung der Neuraminsäure- und Fukosereste gelang der Nachweis des TF-Antigens, was ein Hinweis darauf ist, dass es in normalem Kolongewebe maskiert vorliegt [25, 57]. 1.2.4.2 Expression von Muzinantigenen in kolorektalen Adenomen und Karzinomen Im Rahmen der Karzinogenese konnte bisher in vielen Organen eine Reihe charakteristischer Veränderungen der Antigeneigenschaften von Muzinen beschrieben werden. So steigt bei einigen Muzinen die Expression stark an, während bei anderen Muzinen eine verminderte Expression zu beobachten ist. Eine gestörte Glykosylierung führt zu kurzen verzweigten Ketten und zur Synthese von neuen Zuckern. Beschrieben wurde dies für Tumoren der Brust, des Endometriums, der Ovarien, des Pharynx und Larynx, der Lunge, des Magens, der Leber, der Gallenblase, des Pankreas und des Dünn- und Dickdarms. Die spezifische O-Glykosylierung macht Muzine zu sensitiven Indikatoren von differenzierungsabhängigen Veränderungen in der zellulären Glykosylierung [7, 281]. In den letzten Jahren gewannen die Kohlenhydrate als prognostische Marker vieler menschlicher Karzinome an klinischem Interesse. Neben den Kohlenhydrat-Core-Antigenen und den peripheren 42 Einleitung Oligosaccharid-Antigenen könnten auch die Peptid-Core-Antigene prognostische Relevanz besitzen. 1.2.4.2.1 Peptid-Core-Antigene Antigene des Muzinpeptid-Cores sind infolge ihrer starken Glykosylierung in Normalgeweben oft maskiert und durch Antikörper nicht oder nur schwach nachweisbar. In kolorektalen Karzinomen wird der MUC1-Peptid-Core stark exprimiert [136]. In gastrointestinalen Karzinomen konnte man neben den intestinalen MUC2- und MUC3-Epitopen auch das mammatypische MUC1 detektieren [13, 297]. Im Gegensatz zu normalem Kolongewebe ist MUC1 in Adenokarzinomen und Adenomen weniger glykosyliert und daher immunhistochemisch besser zu detektieren [55]. Vor allem in fortgeschrittenen Tumorstadien und in metastasierenden Karzinomen wurde eine starke Expression von MUC1 beobachtet [184, 195]. In Adenokarzinomen kann die Überexpression von MUC1 bis auf das 10-fache gesteigert sein. Eine starke Immunreaktivität zeigen vor allem tubuläre und papilläre Tumoren. Muzinöse Karzinome exprimieren MUC1 demgegenüber gar nicht oder nur schwach [25, 37]. MUC1 konnte auch in Adenomen, vor allem mit schwerer intraepithelialer Neoplasie, detektiert werden. Es scheint, dass die MUC1-Expression mit zunehmenden Grad der intraepithelialen Neoplasie ansteigt. Eine MUC1-Immunreaktivität korrelierte mit einer p53-Expression und mit einem hohen Apoptoseindex in intramukosalen Karzinomen [250]. Demgegenüber zeigen das MUC2- und das MUC3-Peptid mit zunehmendem Grad der intraepithelialen Neoplasie in Adenomen eine Abnahme der Expression. In kolorektalen Adenokarzinomen findet man eine Abnahme der MUC2-Expression, obwohl muzinöse Karzinome MUC2-positiv sind [6, 37, 55, 136, 184]. Für MUC3 wurde sowohl eine Abnahme als auch eine Zunahme der Immunreaktivität in kolorektalen Karzinomen gefunden [136, 204]. Inzwischen weiß man, dass es ein MUC3A und ein MUC3B gibt, was in diesen Studien nicht beachtet wurde [55]. Zusammenfassend ist MUC1 ein Marker der späten Stadien der Adenom-Karzinom-Sequenz, während MUC2, MUC3 und MUC5A Marker der frühen Stadien sind [21]. Es gibt einige Studien über die MUC4-Expression in kolorektalen Karzinomen. Einerseits wurde für MUC4-mRNA in kolorektalen Karzinomen ein Anstieg im Vergleich zu Normalgewebe beobachtet, andererseits eine Abnahme [204]. Es ist dennoch ungewiss, ob eine MUC4-Expression in Beziehung zur Progression des kolorektalen Karzinoms steht. MUC5AC, das in normalem Kolon nicht detektierbar ist, wird in kolorektalen Adenomen und Karzinomen oft de novo exprimiert. Kolorektale Karzinome mit einer hohen Mikrosatelliteninstabilität zeigten in einer Studie eine höhere Positivität für MUC2 und MUC5AC als Mikro- Einleitung 43 satellitenstabile Tumoren [37]. Für MUC6 gibt es widersprüchliche Ergebnisse. In einer Studie konnte MUC6 in kolorektale Karzinomen nicht detektiert werden [245], während eine andere Studie MUC6 in Adenomen immunhistochemisch darstellen konnte [31]. Für die Expression von MUC11 und MUC12 wurde in kolorektalen Karzinomen eine Downregulation im Vergleich zu Normalgewebe beobachtet [278]. 1.2.4.2.2 Kohlenhydrat-Core-Antigene Während in normalem Kolongewebe hauptsächlich Core 3-Srukturen vorhanden sind, findet man in kolorektalen Karzinomen hauptsächlich die Kohlenhydratstrukturen Core 1, Core 2 und Core 4. Core 3 ist nicht mehr vorhanden. Dies ist auf eine veränderte Aktivität einiger Glykosyltransferasen zurückzuführen [289]. Die sogenannten Kryptantigene sind Strukturen der inneren Kernregion, wie zum Beispiel das TF- und das Tn-Antigen, die im Normalgewebe in der Regel maskiert sind. Untersuchungen der Enzymaktivität der für die Synthese der Kryptantigene verantwortlichen Glykosyltransferasen zeigten eine höhere Aktivität der α6-Sialyl-Transferase in muzinreichen, der β3-Gal-Transferase in muzinärmeren Karzinomen. Dabei korrelierte die Höhe der Enzymaktivität nicht mit Tumorlokalisation, Stadium oder histologischem Typ. Daher scheint die fehlende Expression von Sialyl-Tn, TF und Tn in Normalgeweben nicht auf mangelnde Synthese, sondern auf Antigenmaskierung zu beruhen [23, 80]. In kolorektalen Karzinomen kommt es zu einem deutlichen Anstieg der Expression der drei Core-Strukturen Tn-Antigen, TF-Antigen und Sialyl-Tn-Antigen. Das TF-Antigen repräsentiert das am umfangreichsten erforschte Kohlenhydratantigen in kolorektalen Neoplasien. Sowohl mit PNA, als auch mit spezifischen poly- und monoklonalen Antikörpern konnte in kolorektalen Karzinomen eine starke Färbung erreicht werden [25, 43, 117, 148, 164, 294]. In kolorektalen Adenomen korrelierte die Reaktivität mit Zunahme der Polypengröße, dem histologischen Typ und dem Grad der intraepithelialen Neoplasie [25, 62, 294]. Die TF-Positivität war in Lebermetastasen stärker als in primären kolorektalen Karzinomen [61]. Für das Tn-Antigen konnte keine signifikante Veränderung in den verschiedenen Dysplasiegraden von Adenomen beobachtet werden [62, 146]. In kolorektalen Karzinomen zeigte sich jedoch eine starke Tn-Expression [148, 205]. Da Tn das obligate Zwischenprodukt für die Synthese aller Muzin-Oligosaccharide ist, deutet seine Detektierbarkeit in kolorektalen Karzinomen eine Abnahme der weiteren Glykosylierung an, die Tn im normalen Kolon maskiert. Das Sialyl-Tn-Antigen war in kolorektalen Adenomen und Karzinomen sehr reaktiv. Seine Expression korrelierte mit dem Grad der intraepithelialen Neoplasie, der Polypengröße und 44 Einleitung dem histologischen Typ [62, 146, 241, 282]. Diese Core-Struktur wird vor allem in metastasierten kolorektalen Karzinomen stark exprimiert und stellt einen unabhängigen Wert für eine schlechte Prognose dar [48, 145]. 1.2.4.2.3 Kohlenhydratantigene der Backbone- und peripheren Region Die Typ2-Kette der Backbone-Region Galβ1-4GlcNAc zeigte, detektiert mit ErythrinaLektin, eine schwache Reaktivität in weniger als 50% der hyperplastischen Polypen und Adenome. Hingegen waren Primärtumoren und Lebermetastasen stark positiv [24]. Das nichtfukosylierte Blutgruppen-i-Antigen wurde in etwa 65% der Karzinome beobachtet [187]. Die Immunreaktivität von fukosylierten Antigenen, die Typ2-Ketten-abhängig sind, war in kolorektalen Adenokarzinomen generell erhöht. Ihre Bildung ist Folge einer verstärkten Synthese von Typ 2-Ketten. Das Le x -Antigen wurde von etwa 85 bis 95% der Karzinome unabhängig vom Sekretorstatus im proximalen und distalen Kolon exprimiert [147-148], während das difukosylierte Le y -Antigen, normalerweise nur im rechten Darmabschnitt vorkommend, eine stärkere Reaktivität in Karzinomen des distalen Kolon zeigte [30, 51]. Das Le y -Antigen findet man in 91% der Karzinome des distalen Kolons, aber nur in 43% der Karzinome des Zäkums [30]. Hierbei handelt es sich um eine ektope Antigenexpression, da das distale Kolon auch beim Fetus kein Le y exprimiert. Die ABH-Blutgruppenantigene weisen eine Reihe von möglichen Tumor-assoziierten Veränderungen auf. So sind in einigen Tumoren auftretende ABH-Blutgruppenantigene inkompatibel zu der Blutgruppe des Patienten, die im Normalgewebe, zum Beispiel im proximalen Kolon exprimiert wird [73, 82-83, 295]. Dies könnte eine Immunantwort auslösen. Eine andere typische Veränderung ist die Neoexpression von Blutgruppenantigenen. Normalerweise weist das distale Kolon keine Aktivität der Fukosyltransferase auf. In Karzinomen des distalen Kolons kommt es jedoch oft zu einer onkofetalen Synthese von ABH-Antigenen und der α1-2-fukosylierter Strukturen Le b und Le y [30, 91, 295]. Auch kann es zu einem Verlust der ABH-Blutgruppenantigene kommen. Selten kommt es zu einem kompletten Verlust dieser Antigene, in 75% ist der Verlust fokal begrenzt [156, 295]. Dies wurde vor allem für Karzinome des proximalen Kolon beschrieben. In kolorektalen Adenomen nimmt die Expression der ABH-Antigene mit dem Grad der intraepithelialen Neoplasie zu. Ein weiterer typischer Mechanismus der Modifikation Muzin-assoziierter Antigene ist die angestiegene Expression von sialylierten Lewis-Antigenen. Sialyl- Le x zeigt eine Expression in „low-grade“-Adenomen und in „high-grade“-Adenomen [123]. Mit Zunahme des Grades der intraepithelialen Neoplasie nimmt auch die Expression zu [147]. Sialyl- Le a wird in kolo- Einleitung 45 rektalen Karzinomen ebenfalls exprimiert, vor allem in Lymphknotenmetastasen [16, 196]. Der Anstieg der Immunreaktivität von Sialyl- Le x und Sialyl- Le a in Karzinomen und Metastasen lässt sich durch eine reduzierte O-Azetylierung der Neuraminsäure erklären. Der graduelle Verlust der O-Azetylierung wurde als ein frühes Ereignis in der AdenomKarzinom-Sequenz beobachtet [75]. 46 Einleitung 1.3 Fragestellung der Arbeit Das kolorektale Karzinom steht weltweit an dritter Stelle aller Tumorneuerkrankungen und ist die dritthäufigste Krebstodesursache in den westlichen Ländern. Die meisten kolorektalen Karzinome entwickeln sich über die Adenom-Karzinom-Sequenz. Daher ist es für die onkologisch-pathologische Forschung von besonderer Bedeutung, objektive Parameter zu finden, die eine Aussage über Diagnose, Tumorprogression, Prognose und Therapie bestimmter Patientengruppen möglich machen. Neben Tumorsuppressorgenen und Onkogenen stellen die Muzine dabei einen interessanten Bereich in der Tumorforschung dar. Muzine sind wichtige Marker der gastrointestinalen Karzinogenese. Viele Studien haben sich bereits mit der Bedeutung von MUC1 im Rahmen der malignen Transformation und der Metastasierung beschäftigt. Die Ergebnisse von MUC1 als Prognoseparameter werden teilweise widersprüchlich diskutiert. Bisherige Ergebnisse belegen, dass MUC1 einen Marker für Progression und Prognose kolorektaler Karzinome darstellt. Unklar bleibt jedoch bisher, in welchem Maße eine Änderung der Glykosylierung des MUC1-Peptids zu diesem Effekt beiträgt. In dieser Arbeit soll daher die Immunreaktivität von nativen und deglykosylierten MUC1-Protein verglichen werden. Dazu wurden Serienschnitte von kolorektalen Adenomen und Karzinomen mit MUC1spezifischen monoklonalen Antikörpern unterschiedlicher Epitop-Spezifität nach der Streptavidin-Biotin-Peroxidase-Methode immunhistochemisch gefärbt. Die Antikörper waren teils glykosylierungsabhängig, teils –unabhängig. Als weitere Kontrollen zur Charakterisierung der Glykosylierungsabhängigkeit der Immunreaktivität von MUC1 wurden chemische und enzymatische Methoden der Deglykosylierung durchgeführt. Hierzu wurde zum einen die Perjodat-Oxidation, zum anderen Neuraminidase aus Clostridium perfringens verwendet. Des Weiteren wurde die Immunreaktivität von MUC1 in Adenomen mit leichtgradiger intraepithelialer Neoplasie, Adenomen mit hochgradiger intraepithelialer Neoplasie und Karzinomen verglichen. Material und Methoden 47 2 Material und Methoden ___________________________________________________________________________ 2.1 Patienten und Gewebe Das in dieser Arbeit verwendete Kollektiv setzt sich aus zwanzig Patientengeweben zusammen. Es handelt sich dabei um zehn Resektate von Patienten mit einem kolorektalen Karzinom und zehn Resektate von Patienten mit einem kolorektalen Adenom. Von den Adenomen wiesen fünf eine leichte intraepitheliale Neoplasie und fünf eine schwere intraepitheliale Neoplasie auf. Die Gewebe wurden in den Jahren von 1987-2003 reseziert und dem Zentrum für Pathologie der Universität zu Köln (Direktor: Universitätsprofessor H. P. Dienes) zur histopathologischen Untersuchung zugesandt. Hier wurden die Resektate zugeschnitten, zur Konservierung in 5%igem Formalin fixiert und in Paraffin eingebettet. Zur histologischen Untersuchung und Klassifizierung erfolgte an Schnitten dieser Paraffinblöcke eine Hämatoxylin-Eosin-Färbung. Von jedem Paraffinblock wurden mit dem Mikrotom mehrere 5 µm dicke Gewebeschnitte angefertigt, die dann auf Superfrost Plus Objektträger aufgezogen und mit verschiedenen monoklonalen Antikörpern gegen MUC 1 nach der Labeled-Streptavidin-Biotin-Methode angefärbt wurden. 2.2 MUC1-spezifische monoklonale Antikörper In der vorliegenden Arbeit wurden verschiedene MUC1-spezifische Antikörper verwendet, die gegen unterschiedliche Epitope des MUC1-Muzins gerichtet sind. 2.2.1 HMFG2 HMFG2 ist ein monoklonaler Antikörper der Maus vom Isotyp IgG1-k. Er reagiert mit Komponenten des Human Milk Fat Globulin und bindet an die Aminosäuresequenz DTR innerhalb der VNTR des MUC1-Peptid-Cores. Das Epitop detektiert einen glykosylierungsabhängigen Teil des MUC1-Peptid-Cores (DTR) [26, 36, 251]. 48 2.2.2 Material und Methoden BCP8 (#133) BCP8 ist ein monoklonaler Antikörper der Maus vom Isotyp IgG2b und definiert ebenfalls das Epitop PDTRPA auf dem MUC1-Repeat-Peptid (Epitop-Cluster: VNTR (DTR). 2.2.3 115D8 (#148) 115D8 ist ein monoklonaler Antikörper der Maus vom Isotyp IgG2b-k. Er ist gegen ein Glykopeptid-Epitop gerichtet und ist abhängig von Neuraminsäure (Epitop-Cluster: sialylVNTR/Non-VNTR) [189, 218]. 2.2.4 MA695 (#154) Ma695 ist ein monoklonaler Antikörper der Maus vom Isotyp IgG1 und reagiert ebenfalls mit einem sialylierten Kohlenhydrat-Epitop auf MUC1-Repeat-Peptiden (Epitop-Cluster: sialylVNTR/Non-VNTR) [199, 218]. 2.3 Immunhistochemische Methode 2.3.1 2.3.1.1 Ablauf der Färbung Allgemeines über die Labeled-Streptavidin-Biotin-Methode (LSAB-Methode) Das Grundprinzip der immunhistochemischen Methoden beruht auf dem Einsatz von Antikörpern zur Markierung der korrespondierenden Antigene in Geweben. Die Antigenbindungsstelle kann durch Enzym-Kopplung mit farbgebender Reaktion sichtbar gemacht werden (Abbildung 2/1). Die LSAB-Methode macht sich die Affinität von Avidin/Streptavidin zu Biotin zunutze. Avidin ist ein aus Hühnereiweiß gewonnenes Glykoprotein mit einem Molekulargewicht von 68 kDa, das vier Bindungsstellen für Biotin enthält. Das aus dem Bakterium Streptomyces avidinii gentechnisch gewonnene Streptavidin hat gegenüber Avidin den Vorteil, dass es zu weniger unspezifischen Reaktionen kommt. An einen Brückenantikörper wird Biotin gekoppelt. In dieser Studie wurde ein monoklonaler Kaninchen-Anti-Maus-Antikörper verwendet. Biotin ist ein wasserlösliches Vitamin, über das das Streptavidin an den Brückenantikörper gebunden werden kann [141]. Das Streptavidin ist mit einem Enzym (Peroxidase oder alkalische Phosphatase) direkt gekoppelt. Der biotinylierte Brückenantikörper bindet an das Fc-Fragment des Primärantikörpers und markiert so das gesuchte Antigen. Durch die Bindung zwischen Streptavidin und dem Biotin des Brückenantikörpers wird die Peroxidase an den Brückenantikörper gekoppelt. Die Material und Methoden 49 Peroxidase fördert die Freisetzung von Radikalen durch Oxidation von H 2 O 2 , die an farblose Diazoniumsalze (Chromogene) binden und unlösliche Azofarbstoffe bilden. In dieser Studie wurde 3-Amino-9-Ethylcarbazol als Chromogen verwendet, das ein rotes Reaktionsprodukt liefert. Der immunologische Nachweis mit dieser Methode ist sehr sensitiv und derzeit eines der spezifischsten Verfahren. Es erlaubt den Nachweis auch weniger Antigen-Epitope [105]. Streptavidin-Enzymkonjugat Brückenantikörper, biotinyliert Primärantikörper Antigen Abbildung 2/1: Schema der LSAB-Methode mit Darstellung von Primärantikörper, biotinyliertem Brückenantikörper und Streptavidin-Enzym-Komplex 2.3.1.2 Coverplate-Methode Zur Durchführung der Immunmarkierung und anschließender Färbung der Gewebeschnitte bis zur Entwicklung wurde das Coverplate-System der Firma Shandon zu Hilfe genommen. Ein Coverplate ist ein aus Kunststoff bestehender Rahmen, der eine mit Kunstharz beschichtete Fläche trägt. Dieser Fläche wurden die Objektträger feucht angelegt. Anschließend wurden jeweils zehn Coverplates in einen Schlitten verankert. Der Kopfteil des Coverplates bildet eine trichterförmige Öffnung mit dem Objektträger, in die man die Reaktionslösungen pipettieren kann. Bei sorgfältigem und luftblasenfreiem Aufziehen entsteht ein Flüssigkeitsfilm zwischen Objektträger und Coverplate, durch den eine Oberflächenspannung aufgebaut wird. Dies gewährleistet eine gleichmäßige Passage der Flüssigkeiten über den Schnitt. Das Coverplate-System macht eine Standardisierung des Verfahrens möglich. Weitere Vorteile sind die Benötigung minimaler Reagenzvolumina, sowie der physikalische Schutz während des mehrstündigen Färbevorgangs. 50 2.3.1.3 Material und Methoden Entparaffinierung Vor der immunhistochemischen Färbung müssen die Gewebeschnitte entparaffiniert werden. Das Einbettmedium muss vollständig aus dem Gewebeschnitt entfernt werden, um eine Farbreaktion des Gewebes zu ermöglichen und um unspezifische Hintergrundreaktionen zu vermeiden. Dazu wurden die Objektträger für 10 Minuten in einen 80°C heißen Ofen gestellt, so dass das Paraffin ablaufen konnte. Zur Hydrierung wurden die abgekühlten Präparate für 12 Minuten in Xylol gebadet. Anschließend wurde eine absteigende Alkoholreihe durchlaufen. Dabei blieben die Objektträger für jeweils 30 Sekunden zweimal in 100% Ethanol, dann für je 30 Sekunden in 96% und in 75% Ethanol. Abschließend wurden die Objektträger zuerst mit Leitungswasser und dann mit Aqua dest. gewaschen. Die Objektträger mussten vollständig frei von Paraffin- und Alkoholschlieren sein. 2.3.1.4 Partielle Deglykosylierung durch Vorbehandlung mit Neuraminidase In einem Färbedurchgang wurde eine Vorbehandlung mit dem Enzym Neuraminidase (Fa. New England Bio Labs) durchgeführt. Dadurch werden sialinierte Bindungsstellen demaskiert. Die Neuraminidase wurde im Verhältnis 1:5000 mit Zitratpuffer verdünnt. Von dieser Mischung wurde auf jeden Objektträger 100 µl aufgetragen, dies entsprach 1 Unit des Enzyms je Schnitt. Anschließend wurden die Präparate bei 37° C für 30 Minuten inkubiert. Dabei war es wichtig, dass die Präparate nicht austrocknen. Abschließend wurde mit TBS (tris buffered solution) gespült. 2.3.1.5 Partielle Deglykosylierung durch Vorbehandlung mit Perjodat In einem weiteren Färbedurchgang wurde von jedem Fall des Kollektivs je ein Schnitt mit Perjodat (Fa. Merck) vorbehandelt. Perjodat demaskiert verschiedene Muzingen-Produkte und Glykoproteine, die O-glykosidische Bindungen enthalten, ohne Peptid-Epitope zu zerstören. Nach der Deparaffinierung wurden die Schnitte für 1 Stunde im Dunkeln bei Raumtemperatur mit 100 µl einer 10 mM Perjodatlösung inkubiert, die im Verhältnis 1:348 mit 0,05 M Azetatpuffer (pH 4,5) verdünnt wurde. Danach wurden die Präparate mit TBS gewaschen. Anschließend wurden saure reaktive Gruppen mit 100 µl 1%igen Glyzerols (1g Glyzin (Fa. Merck) in 100 ml Aqua dest. aufgelöst) für 30 Minuten neutralisiert. Material und Methoden 2.3.1.6 51 Ohne Vorbehandlung In einem letzten Durchgang wurden die Schnitte mit der Streptavidin-Biotin-Methode gefärbt, ohne eine Vorbehandlung zu erhalten. 2.3.1.7 Hemmung der endogenen Peroxidase Bei der Streptavidin-Biotin-Methode spielen peroxidasekatalysierte Reaktionen eine große Rolle. Um die unerwünschte Reaktion des Chromogens mit der endogenen Peroxidase und damit eine unspezifische Hintergrundfärbung zu verhindern, ist es notwendig, die endogene Peroxidase zu hemmen. Dazu wurde ein Peroxidaseblock aus 30%igem H 2 O 2 und Methanol angesetzt, worin die Objektträger 30 Minuten inkubiert wurden. Daraufhin wurden die Objektträger mit TBS gespült. 2.3.1.8 Hemmung unspezifischer Proteinbindungen Um die unspezifischen Bindungsstellen des Primärantikörpers zu blockieren, wurden die Objektträger mit je 200 µl Schweineserum (X 0901, Fa. DAKO), das mit Antikörperverdünnungspuffer (Fa. Zymed) 1:20 verdünnt wurde, beschickt und für 30 Minuten bei 40°C inkubiert. 2.3.1.9 Die Immunmarkierung Die Schnitte wurden mit jeweils 100 µl des entsprechenden Primärantikörpers, der mit Antikörperverdünnungspuffer in dem angegebenen Verhältnis verdünnt wurde (Tabelle 2/1), beschickt. Das optimale Verdünnungsverhältnis, bei dem es zu einer intensivroten Färbung der Zellen kam und gleichzeitig die unspezifischen Hintergrundreaktionen möglichst gering waren, wurde durch Vorversuche ermittelt. Die Schnitte wurden bei 4°C über Nacht inkubiert. In jedem Färbedurchgang wurden Gewebeschnitte eines Mammakarzinoms als Positivkontrolle zugefügt. Die Negativkontrolle wurde mit 100 µl TBS ohne Primärantikörper beschickt. Vor der Bindung an den Sekundärantikörper wurden die Objektträger mit TBS gespült. 52 Material und Methoden Primärantikörper Verdünnung HMFG 2 1:100 BCP 8 1:100 115 D 8 1:20 MA 695 1:100 Tabelle 2/1: Verdünnung der Primärantikörper 2.3.1.10 Markierung mit dem Sekundärantikörper Als Sekundärantikörper, der an den Primärantikörper bindet, wurde ein biotinylierter, monoklonaler Kaninchen-Anti-Maus-Antikörper verwendet (E 0354, Fa. DAKO). Auf jeden Schnitt wurden 100 µl des Antikörpers aufgetragen, der im Verhältnis 1:400 mit Antikörperverdünnungspuffer gemischt war. Dann wurde bei Raumtemperatur für 30 Minuten inkubiert. Anschließend wurden die Schnitte mit TBS gespült. 2.3.1.11 Kopplung des Meerrettichperoxidase-konjugierten Streptavidins In diesem Schritt wurde auf die Schnitte das mit Meerrettichperoxidase konjugierte Streptavidin aufgetragen (P 0397, Fa. DAKO). Auch hier erwies sich ein Verdünnungsverhältnis von 1:400 als optimal. Für jeden Schnitt wurden 100µl verwendet. Anschließend wurde wiederum bei Raumtemperatur für 30 Minuten inkubiert und danach mit TBS gespült. Dabei wurden die Objektträger aus den Coverplates in Küvetten mit TBS umgesetzt. 2.3.1.12 Entwicklung Der Entwicklerlösung wurde H 2 O 2 als Substrat der Peroxidase und AEC (3-Amino-9-ethylcarbazol) als notwendiges Chromogen zugesetzt. AEC färbt sich bei entsprechender Bindung rot. Die fertige Entwicklerlösung wurde in die Küvetten gegeben. Die optimale Entwicklungszeit, die unter mikroskopischer Kontrolle ermittelt wurde, war für alle verwendeten Antikörper 30 Minuten. Der Vorgang war abgeschlossen, wenn sich sie Zellen lichtmikroskopisch in intensivroter Farbe präsentierten. Durch Einbringen der Objektträger in H 2 O wurde die Farbreaktion gestoppt. Anschließend folgte eine Zellkerngegenfärbung mit Meyers Hämalaun (Fa. Merck) und die Eindeckung der Präparate mit Kaisers Glyceringelatine (Fa. Merck). Material und Methoden 2.3.2 2.3.2.1 53 Rezepte Zitratpuffer 0,52 g Zitronensäure-Monohydrat (Fa. Merck) Diese Komponente wird mit 50 ml Aqua dest. gemischt und mit NAOH auf einen pH von 6 titriert. 2.3.2.2 Azetatpuffer 1 ml Essigsäure, 99,7% (Fa. Merck) Diese mit 347 ml Aqua dest. mischen, dann mit NAOH auf einen pH von 4,5 titrieren. 2.3.2.3 Peroxidase-Blocker 50 ml Methanol, > 99% Methylalkohol (Fa. Roth) 1,5 ml H 2 O 2 , 30% (Fa. Roth) Beide Komponenten werden zusammengegeben und abgedeckt gut gemischt. 2.3.2.4 TBS-Puffer (tris buffer solution) 84 g NaCl (Fa. Merck) 12 g Trishydroxymethylammoniummethan (Fa. Sigma) 10 l Aqua dest. Einstellen des pH mit 25% HCL (Fa. Merck)auf 7,4 2.3.2.5 Entwicklerlösung 2.3.2.5.1 Lösung I 0,05 ml H 2 O 2 , 30% (Fa. Merck) 5 ml Dimethylformamid (Fa. Merck) 1 Tablette AEC (3-Amino-9-ethyl-carbazol) (Fa. Sigma) 2.3.2.5.2 Lösung II 7,4 ml Lösung A: 30 ml 96%-98%ige Essigsäure ad 1000 ml Aqua dest. 17,6 ml Lösung B: 16,4 g Natriumacetat-Trihydrat ad 1000 ml Aqua dest. 54 Material und Methoden Lösung A und B mit Bidest auf 100 ml auffüllen und mit HCl/NaOH (beide 1 Mol/l) auf einen pH von 5 einstellen. Lösung I und II erst kurz vor Gebrauch vermischen. 2.3.2.6 Hämalaun 5 g Hämatoxylin (Fa. Merck) 1 g Natriumtrijodid (Fa. Merck) 250 g Aluminiumkaliumsulfat (Fa. Merck) 250 g Chloralhydrat (Fa. Merck) 5 g Zitronensäure (Fa. Merck) Lösung in 5 l Aqua dest. und Reifung für 3 Wochen 2.3.3 Lichtmikroskopische Auswertung Für die Auswertung der Färbung wurden die Präparate lichtmikroskopisch untersucht und semiquantitativ beurteilt. Zuerst wurden die Präparate in Lupenvergrößerung betrachtet, um einen Eindruck über die Menge und die Verteilung der immun markierten Zellen zu erhalten. Dann wurden sie mäanderförmig mit 250- bzw. 400-facher Vergrößerung durchstreift. Es wurde der prozentuale Anteil der positiv markierten Adenom- bzw. Tumorzellen am Gesamtanteil aller Adenom- bzw. Tumorzellen bestimmt. Außerdem wurden die Präparate nach der Intensität der Färbung eingeteilt in die Stufen negativ (0), gering (1), mäßig (2) und stark (3). Eine geringe Intensität war als eine schwach rosa Zellfärbung definiert, während eine sehr rote Färbung der Stufe 3 (stark) zugeteilt wurde. Ergebnisse 55 3 Ergebnisse ___________________________________________________________________________ In der vorliegenden Arbeit wurden Serienschnitte von fünf „low-grade“-Adenomen, fünf „high-grade“-Adenomen und zehn kolorektalen Karzinomen mit vier verschiedenen monoklonalen MUC1-Antikörpern jeweils unterschiedlicher Epitop-Spezifität nach der LabeledStreptavidin-Biotin-Methode gefärbt. Die Antikörper HMFG2 und BCP8 binden an die immundominante Aminosäuresequenz DTR innerhalb der VNTR-Region des MUC1-PeptidCores. Die Epitope von MA695 und 115D8 sind bisher nicht genau bekannt. Man weiß jedoch, dass es sich um Glykopeptid-Epitope innerhalb der VNTR-Region handelt. 115D8 und MA695 sind gegen Sialidase-sensitive Epitope gerichtet. Als weitere Kontrollen zur Charakterisierung der Glykosylierungsabhängigkeit der Immunreaktivität von MUC1 wurden an den gleichen Gewebeschnitten eine chemische und eine enzymatische partielle Deglykosylierung durchgeführt. Für die chemische Deglykosylierung wurde die Perjodat-Oxidation verwendet. Hierbei werden Verbindungen zwischen Kohlenstoff- und Hydroxylgruppen gespalten, ohne dass die Peptid-Epitope zerstört werden. Die enzymatische Deglykosylierung wurde mit Neuraminidase, einem Enzym, das aus dem Bakterium Clostridium perfringens geklont wurde, durchgeführt. Dieses Enzym katalysiert die Hydrolyse von N-Azetyl-Neuraminsäureresten von Glykoproteinen. Die Farbkomplexablagerungen wurden lichtmikroskopisch semiquantitativ ausgewertet. Der Anteil des Adenom- bzw. Tumorgewebes, der mit dem jeweiligen Antikörper eine positive Reaktion aufwies, wurde in Prozent des Gesamtadenomgewebes bzw. Gesamttumorgewebes angegeben. Die Intensität der Färbung wurde in die Stufen negativ (0), gering (1), mäßig (2) und stark (3) eingeteilt. Eine Farbintensität wurde als gering eingestuft, wenn die Zellen leicht rosa angefärbt waren. Eine starke Intensität wurde als intensiv roter Zellinhalt definiert. Zum Vergleich der Daten wurden die Mittelwerte mit Standardabweichung, sowie die Mediane mit dem Quartilsabstand berechnet. Zusätzlich wurden zur statistischen Auswertung die nicht-parametrischen Testverfahren nach Wilcoxon (Rangtest für Paardifferenzen) und Mann-Whitney-U angewendet. Sie setzen keine Normalverteilung der Daten voraus. Der Grenzwert des Signifikanz-Niveaus wurde mit p<0,05 so gewählt, dass die Irrtumswahrscheinlichkeit unter 5% liegt. 56 Ergebnisse Immunhistochemische Expressionsmuster Abbildung 3/1: Adenokarzinom des Dickdarms, Antikörper BCP8 ohne Vorbehandlung, x200 Abbildung 3/2: Adenokarzinom des Dickdarms, Antikörper BCP8 nach Perjodatbehandlung, x200 Abbildung 3/3: Adenokarzinom des Dickdarms, Antikörper 115D8 ohne Vorbehandlung, x200 Abbildung 3/4: Adenokarzinom des Dickdarms, Antikörper 115D8 nach Neuraminidasebehandlung, x200 Ergebnisse 57 3.1 Immunreaktivität MUC1-spezifischer monoklonaler Antikörper in kolorektalen Adenomen vor und nach partieller Deglykosylierung Insgesamt ließ sich mit den vier verwendeten monoklonalen MUC1-Antikörpern bei den untersuchten kolorektalen Adenomen, von denen fünf eine geringe intraepitheliale Neoplasie und fünf eine hochgradige intraepitheliale Neoplasie aufwiesen, eine durchgehend positive Immunreaktion erzielen. Lediglich der Antikörper MA695 reagierte gering bis gar nicht mit den Geweben. Es waren durchschnittlich 7,7% der Adenomzellen angefärbt. Mit dem Antikörper 115D8 zeigten die Adenomgewebe die stärkste Reaktion. Es wurden durchschnittlich 71% der Adenomzellen angefärbt mit einer durchgehend starken Farbreaktion. Die Antikörper HMFG2 und BCP8, die das gleiche Epitop DTR innerhalb der VNTR-Region von MUC1 erkennen, zeigten mit durchschnittlich 37% bzw. 36% positives Adenomgewebe ein ähnliches Ergebnis bezüglich Quantität und Intensität der Farbreaktion. Nach partieller Deglykosylierung der „low-grade“-Adenomgewebe mit dem Enzym Neuraminidase nahm die Immunreaktivität von MUC1 bei den verwendeten Antikörpern ab. MA695 zeigte als einziger Antikörper eine Zunahme der positiven Zellen. Bei den „highgrade“-Adenomen kam es mit den Antikörpern HMFG2, BCP8 und MA695 zu einem Anstieg der MUC1-Immunreaktivität, bei dem Antikörper 115D8 zu einer Abnahme. Mit der Perjodat-Oxidation konnte ein starker Einfluss auf die Immunreaktivität beobachtet werden. Mit den Antikörpern HMFG2 und BCP8 kam es zu einer starken Zunahme der Immunreaktivität. Der Antikörper 115D8 detektierte weniger MUC1 nach der Vorbehandlung. Eine Ausnahme bildete der Antikörper MA695. Bei den „low-grade“-Adenomen kam es zu einer starken Abnahme der Reaktion mit dem Gewebe, bei den „high-grade“-Adenomen zu einer Zunahme. Statistisch konnte kein signifikanter Unterschied zwischen der Immunreaktivität von MUC1 mit den verwendeten Antikörper in den Adenomen mit leichter intraepithelialer Neoplasie und in den Adenomen mit schwerer intraepithelialer Neoplasie beobachtet werden. 58 Ergebnisse 90 80 70 60 Ohne Vorbehandlung 50 Nach NeuraminidaseBehandlung 40 30 Nach PerjodatOxidation 20 10 0 HMFG2 BCP8 115D8 MA695 Abbildung 3/5: Häufigkeitsverteilung (Mittelwerte) der Antikörper bei den „low-grade“Adenomen 90 80 70 60 Ohne Vorbehandlung 50 Nach NeuraminidaseBehandlung 40 30 Nach PerjodatOxidation 20 10 0 HMFG2 BCP8 115D8 MA695 Abbildung 3/6: Häufigkeitsverteilung (Mittelwerte) der Antikörper bei den „high-grade“Adenomen Ergebnisse 3.1.1 59 HMFG2 Die Ergebnisse der Immunreaktivität von HMFG2 in den Adenomgeweben sind in den Tabellen 3/1 bis 3/3 zu sehen. Dieser Antikörper zeigte eine durchgehend positive Gewebsreaktion, sowohl in den „low-grade“-Adenomen, als auch in den „high-grade“-Adenomen. Der Anteil des MUC1-markierten Gewebes variierte stark von 5% bis 90%. Bei den „lowgrade“-Adenomen konnten im Durchschnitt 39% (STABW 35,07%) des Gewebes angefärbt werden, bei den „high-grade“-Adenomen 35% (STABW 29,15%). Die Intensität der Färbung fiel - bis auf einen gering gefärbten Fall - mäßig aus. Nach Vorbehandlung der Adenomgewebe mit Neuraminidase nahm der Anteil des MUC1markierten Gewebes der „low-grade“-Adenome in zwei Gewebeschnitten zu, in zwei anderen gab es keinen Unterschied zu den unbehandelten Schnitte. Ein Fall zeigte nach partieller Deglykosylierung mit Neuraminidase einen starken Abfall der Immunreaktivität von 90% auf 10%. Im Mittel wurden 26% (STABW 20,74%) des Adenomgewebes angefärbt. Bei den „high-grade“-Adenomen kam es in drei Fällen zu einem Anstieg der immunmarkierten Zellen. In zwei Fällen gab es keinen Unterschied. Im Mittel reagierten nach NeuraminidaseBehandlung 40% (STABW 28,88%) des Adenomgewebes mit dem Antikörper HMFG2. Die Intensität der Färbung bei den Adenomgeweben war mäßig bis stark, dass heißt im Vergleich zu den unbehandelten Fällen nahm die Intensität in vier Fällen um eine Stufe zu. Nach Vorbehandlung der Adenomgewebe mit Perjodat-Oxidation kam es in den Adenomgeweben zu einer deutlichen Zunahme der MUC1-Immunreaktivität mit HMFG2. Lediglich in einem Fall der „low-grade“-Adenome nahm der Anteil des reagierenden Gewebes von 90% auf 70% ab. Im Mittel reagierten 62% (STABW 19,24%) der Adenomzellen mit leichter intraepithelialer Neoplasie und 68% (STABW 21,68%) der Adenomzellen mit starker intraepithelialer Neoplasie mit HMFG2. In fünf Gewebeschnitten kam es zu einer Zunahme der Farbintensität. Nach dem nicht-parametrischen Testverfahren nach Wilcoxon gab es einen statistisch signifikanten Unterschied zwischen dem Ergebnis der Adenome ohne Vorbehandlung und dem nach Perjodat-Oxidation, während der Unterschied nach Neuraminidase-Behandlung nicht signifikant war. Es gab nach dem Mann-Whitney-U-Test keinen signifikanten Unterschied zwischen den Adenomen mit leichter und schwerer Dysplasie. 60 Ergebnisse „low-grade“-Adenome „high-grade“-Adenome 1 2 3 4 5 1 2 3 4 5 Ohne Vorbehandlung 60 20 5 90 20 5 10 30 70 60 Farbintensität 2 2 1 2 2 2 2 2 2 2 Nach Neuraminidase-Behandlung 60 20 10 10 30 10 20 30 70 70 Farbintensität 2 3 2 3 2 2 2 2 2 3 Nach Perjodat-Oxidation 80 30 70 70 60 80 40 50 80 90 Farbintensität 2 3 2 2 3 3 2 2 2 3 Tabelle 3/1: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper HMFG2 in den Adenomgeweben Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 39 26 62 STABW 35,07 20,74 19,24 Median 20 20 70 25%-Quantil 20 10 60 75%-Quantil 60 30 70 Quartilsabstand 40 20 10 Tabelle 3/2: Lage- und Streumaße von HMFG2 in den kolorektalen „low-grade“-Adenomen Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 35 40 68 STABW 29,16 28,28 21,68 Median 30 30 80 25%-Quantil 10 20 50 75%-Quantil 60 70 80 Quartilsabstand 50 50 30 Tabelle 3/3: Lage- und Streumaße von HMFG2 in den kolorektalen „high-grade“-Adenomen Ergebnisse 3.1.2 61 BCP8 Der Antikörper BCP8 zeigte ein ähnliches Reaktionsverhalten wie der Antikörper HMFG2. In den Adenomgeweben ließ sich mit BCP8 eine durchgehend positive Reaktion erzielen. So reagierten 42% (STABW 29,5%) der „low-grade“-Adenomzellen und 30% (STABW 20%) der „high-grade“-Adenomzellen mit dem Antikörper (Tabellen 3/4 bis 3/6). Die Intensität der Färbung war bei den Adenomen mit leichter intraepithelialer Neoplasie in einem Fall gering, in drei Präparaten mäßig und in einem stark. Bei den Adenomen mit schwerer intraepithelialer Neoplasie war die Intensität der Färbung bis auf einen stark angefärbten Schnitt mäßig. Nach Vorbehandlung der Gewebe mit Neuraminidase kam es in sechs Schnitten zu einer Zunahme der mit dem Antikörper reagierenden Adenomzellen. Bei den „low-grade“Adenomen nahm in einem Fall der Anteil des angefärbten Gewebes von 90% auf 20% ab. In den übrigen Schnitten blieb das Ergebnis unverändert. Nach Neuraminidase-Behandlung lag die Menge des angefärbten Gewebes bei den „low-grade“-Adenomen bei 36% (STABW 18,17%) und bei den „high-grade“-Adenomen bei 40% (STABW 20%). Die Intensität der Färbung war insgesamt mäßig bis stark ausgeprägt, dass heißt in drei Schnitten kam es zu einer Zunahme. Durch eine Vorbehandlung mit Perjodat konnte die Immunreaktivität von MUC1 mit dem Antikörper BCP8 sowohl bei den „low-grade“-Adenomen, als auch bei den „high-grade“Adenomen deutlich verstärkt werden. Lediglich in einem Fall der „low-grade“-Adenome sah man keine Veränderung der Quantität, aber eine um eine Reaktionsstufe höhere Intensität der Färbung. Im Durchschnitt wurde bei den „low-grade“-Adenomen 78% (STABW 17,89%) des Gewebes angefärbt. Bei den „high-grade“-Adenomen reagierten ebenfalls 78% (STABW 8,37%) der Adenomzellen mit dem Antikörper. Die Intensität der Färbung war in zwei der Fälle mäßig, in drei Präparaten stark. Im Vergleich zu den Fällen ohne Vorbehandlung kam es in drei Präparaten zu einer Zunahme der Intensität. Die Farbintensität war bei den Adenomen mit leichter intraepithelialer Neoplasie in drei Schnitten stark, bei den Adenomen mit schwerer intraepithelialer Neoplasie durchgehend stark. Statistisch zeigte sich, wie bei dem Antikörper HMFG2 für die partielle Deglykosylierung mit Perjodat-Oxidation ein signifikanter Unterschied im Vergleich zum nativen Gewebe. Demgegenüber konnte für die Vorbehandlung mit Neuraminidase kein signifikanter Unterschied gezeigt werden. Auch konnte mit dem Mann-Withney-U-Test kein signifikanter Unterschied zwischen den „low-grade“- und den „high-grade“-Adenomen gefunden werden. 62 Ergebnisse „low-grade“-Adenome „high-grade“-Adenome 1 2 3 4 5 1 2 3 4 5 Ohne Vorbehandlung 20 50 30 90 20 10 50 10 30 50 Farbintensität 1 2 3 2 2 2 2 3 2 2 Nach Neuraminidase-Behandlung 50 60 30 20 20 10 60 30 50 50 Farbintensität 2 3 3 3 2 2 2 3 2 2 Nach Perjodat-Oxidation 90 70 90 90 50 80 70 90 80 70 Farbintensität 2 3 3 3 2 3 3 3 3 3 Tabelle 3/4: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper BCP8 in den Adenomgeweben Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 42 36 78 STABW 29,5 18,17 17,89 Median 30 30 90 25%-Quantil 20 20 70 75%-Quantil 50 50 90 Quartilsabstand 30 30 20 Tabelle 3/5: Lage- und Streumaße von BCP8 in den kolorektalen „low-grade“-Adenomen Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 30 40 78 STABW 20 20 8,37 Median 30 50 80 25%-Quantil 10 30 70 75%-Quantil 50 50 80 Quartilsabstand 40 20 10 Tabelle 3/6: Lage- und Streumaße von BCP8 in den kolorektalen „high-grade“-Adenomen Ergebnisse 3.1.3 63 115D8 Der Antikörper 115D8, der gegen ein Glykopeptid-Epitop gerichtet ist und dessen genaues Zielepitop noch nicht bekannt ist, zeigte mit den nativen Gewebeschnitten die stärkste Reaktion im Vergleich zu den anderen Antikörpern (Tabellen 3/7 bis 3/9). Bei den „low-grade“-Adenomen reagierten 70% (STABW 18,71%) der Zellen positiv mit dem Antikörper, bei den „high-grade“-Adenomen sogar 72% (STABW 13,04%). Die Farbintensität war durchgehend stark. Nach Vorbehandlung der Schnitte mit Neuraminidase nahm der Anteil der immunmarkierten Adenomzellen in drei Fällen der Adenome mit leichter intraepithelialer Neoplasie und in vier Fällen der Adenome mit schwerer intraepithelialer Neoplasie ab. Die übrigen Präparate zeigten keine quantitative Veränderung. Der Mittelwert betrug 54% (STABW 26,08%) angefärbter „low-grade“-Adenomzellen und 52% (STABW 24,90%) angefärbter „high- grade“-Adenomzellen. Die Intensität der Färbung war in allen Schnitten weiterhin sehr stark. Durch die Vorbehandlung der Schnitte mit Perjodat kam es in allen Adenomgeweben zu einer mehr oder weniger staken Abnahme der immunmarkierten Zellen. Nur in einem Fall der „high-grade“-Adenome blieb es bei 70% positiven Gewebes. Durchschnittlich sank der Anteil des positiv markierten Gewebes bei den Adenomen mit leichter intraepithelialer Neoplasie von 70% auf 34% (STABW 19,49%) und bei den Adenomen mit schwerer intraepithelialer Neoplasie von 72% auf 34% (STABW 20,74%). In der Hälfte der Fälle kam es zu einer Abnahme der Farbintensität um eine Stufe. Insgesamt zeigte sich für die partielle Deglykosylierung sowohl mit Neuraminidase, als auch mit Perjodat-Oxidation ein signifikanter Unterschied im Vergleich zum nativen Gewebe. Jedoch war auch diesmal kein signifikanter Unterschied zwischen den „low-grade“- und den „high-grade“-Adenomen zu beobachten. 64 Ergebnisse „low-grade“-Adenome „high-grade“-Adenome 1 2 3 4 5 1 2 3 4 5 Ohne Vorbehandlung 50 50 80 90 80 90 60 70 60 80 Farbintensität 3 3 3 3 3 3 3 3 3 3 Nach Neuraminidase-Behandlung 40 50 80 80 20 70 20 70 30 70 Farbintensität 3 3 3 3 3 3 3 3 3 3 Nach Perjodat-Oxidation 20 40 60 10 40 10 20 50 30 60 Farbintensität 2 3 2 3 2 2 2 3 3 3 Tabelle 3/7: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper 115D8 in den Adenomgeweben Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 70 54 34 STABW 18,71 26,08 19,49 Median 80 50 40 25%-Quantil 50 40 20 75%-Quantil 80 80 40 Quartilsabstand 30 40 20 Tabelle 3/8: Lage- und Streumaße von 115D8 in den kolorektalen „low-grade“-Adenomen Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 72 52 34 STABW 13,04 24,9 20,74 Median 70 70 30 25%-Quantil 60 30 20 75%-Quantil 80 70 50 Quartilsabstand 20 40 30 Tabelle 3/9: Lage- und Streumaße von 115D8 in den kolorektalen „high-grade“-Adenomen Ergebnisse 3.1.4 65 MA695 Der monoklonale Antikörper MA695, der ähnlich 115D8, ein Glykopeptid-Epitop definiert, zeigte insgesamt die schwächste Reaktion mit den Adenomgeweben (Tabellen 3/10 bis 3/12). In zwei Fällen der „low-grade“-Adenome reagierte er gar nicht mit dem Gewebe. Im Durchschnitt wurden nur 7,4% (STABW 12,8%) der Adenomzellen positiv angefärbt. Die Intensität der Färbung war gering bis mäßig. Bei den „high-grade“-Adenomen reagierten 5% bis 10% der Zellen mit dem Antikörper. Der Mittelwert lag bei 8% (STABW 2,74). Die Farbintensität war mäßig bis stark. Bei den „low-grade“-Adenomen zeigten nach Neuraminidase-Vorbehandlung alle Fälle eine deutliche Reaktionszunahme des Gewebes. Alle Schnitte waren positiv (Mittelwert 26%, STABW 20,74%). Die Intensität der Färbung fiel gering bis mäßig aus. Die Adenome mit schwerer intraepithelialer Neoplasie zeigten nach Desialylierung in drei Fällen eine Zunahme des angefärbten Gewebes, in zwei Fällen blieb der Anteil der positiven Zellen gleich. Im Mittel reagierte der Antikörper mit 23% der schwer dysplastischen Zellen (STABW 17,89%). Die Intensität der Färbung blieb in zwei Fällen im Vergleich zu den Adenomen ohne Vorbehandlung mäßig, in einem Fall stark. In einem Präparat kam es zu einer Abnahme der Intensität, in einem Anderen zu einer Zunahme. Durch Perjodat-Oxidation kam es bei den „low-grade“-Adenomen durchschnittlich zu einer Abnahme des Anteils positiv markierter Zellen (Mittelwert 2%, STABW 2,74%). Drei der Gewebeschnitte waren nun negativ, die zwei anderen zeigten 5 % positiv gefärbtes Gewebe. Die Färbung war negativ bis gering ausgeprägt. Bei den „high-grade“-Adenomen zeigten zwei Präparate nach Perjodat-Behandlung keine Veränderung im Vergleich zu den unbehandelten Schnitten. In zwei Fällen kam es zu einer Zunahme des angefärbten Gewebes. Ein Schnitt, der vor Behandlung positiv war, zeigte nach Perjodat-Oxidation keine Reaktion mit dem Antikörper. Im Mittel wurden 10% (STABW 7,07%) der Adenomzellen mit schwerer Dysplasie angefärbt. Die Intensität der Färbung war gering bis mäßig. Nach dem Testverfahren nach Mann-Whitney-U zeigte sich ein signifikanter Unterschied zwischen den Ergebnissen der „low-grade“-Adenome und der „high-grade“-Adenome. Zwischen den jeweiligen Methoden der partiellen Deglykosylierung und den nativen Schnitten war kein signifikanter Unterschied zu sehen. 66 Ergebnisse „low-grade“-Adenome „high-grade“-Adenome 1 2 3 4 5 1 2 3 4 5 Ohne Vorbehandlung 2 0 30 5 0 10 10 5 5 10 Farbintensität 1 0 2 1 0 2 3 2 3 2 Nach Neuraminidase-Behandlung 60 20 10 30 10 10 50 20 5 30 Farbintensität 2 1 2 1 2 2 1 3 3 2 Nach Perjodat-Oxidation 0 0 5 0 5 10 10 10 0 20 Farbintensität 0 0 1 0 1 2 1 2 0 1 Tabelle 3/10: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper MA695 in den Adenomgeweben Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 7,4 26 2 STABW 12,8 20,74 2,74 Median 2 20 0 25%-Quantil 0 10 0 75%-Quantil 5 30 5 Quartilsabstand 5 20 5 Tabelle 3/11: Lage- und Streumaße von MA695 in den kolorektalen „low-grade“-Adenomen Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 8 23 10 STABW 2,74 17,89 7,07 Median 10 20 10 25%-Quantil 5 10 10 75%-Quantil 10 30 10 Quartilsabstand 5 20 0 Tabelle 3/12: Lage- und Streumaße von MA695 in den kolorektalen „high-grade“-Adenomen Ergebnisse 3.2 Immunreaktivität MUC1-spezifischer 67 monoklonaler Antikörper in kolorektalen Karzinomen vor und nach partieller Deglykosylierung Die Karzinomfälle zeigten ebenfalls eine durchgehend positive Farbreaktion mit den vier MUC1-spezifischen monoklonalen Antikörpern. Die Intensität der Färbung war bis auf einen gering angefärbten Schnitt durchgehend mäßig bis stark. Auffällig war, dass sowohl die MUC1-Immunreaktivität als auch die Farbintensität bei den Antikörpern 115D8 und MA695 stärker ausfiel, als bei den Antikörpern HMFG2 und BCP8. Nach partieller Deglykosylierung nahm die Immunreaktivität der Antikörper HMFG2 und BCP8 zu, während die der beiden anderen Antikörper abnahm. Im Vergleich zu den Adenomen zeigten die Antikörper 115D8 und MA695, die ein Glykopeptid-Epitop definieren, einen deutlichen Anstieg der Immunreaktivität von MUC1 in den kolorektalen Karzinomen. Während mit MA695 in den „low-grade“-Adenomen 7,4% des Adenomgewebes und in den „high-grade“-Adenomen 8% der Adenomzellen angefärbt waren, reagierten in den kolorektalen Karzinomen 38,5% der Tumorzellen mit dem Antikörper. Ein Anstieg der Immunreaktivität von 71% positiver Zellen in den Adenomen auf 79% angefärbter Tumorzellen in den Karzinomen konnte bei dem Antikörper 115D8 beobachtet werden. Demgegenüber reagierten die Antikörper HMFG2 und BCP8, die gegen die DTRSequenz innerhalb der VNTR-Region gerichtet sind, insgesamt schwächer mit dem Karzinomgewebe als mit dem Adenomgewebe. Nach dem nicht-parametrischen Testverfahren Mann-Whitney-U gab es jedoch keinen statistisch signifikanten Unterschied zwischen den Ergebnissen der Adenome und der Karzinome. 68 Ergebnisse 90 80 70 60 Ohne Vorbehandlung 50 Nach NeuraminidaseBehandlung 40 30 Nach PerjodatOxidation 20 10 0 HMFG2 BCP8 115D8 MA695 Abbildung 3/7: Häufigkeitsverteilung (Mittelwerte) der Antikörper bei den Karzinomen 3.2.1 HMFG2 Mit dem Antikörper HMFG2 ließen sich 17% (STABW 15,31%) des Tumorgewebes positiv markieren. Die Farbintensität fiel in einem Fall gering, in acht Fällen mäßig und in einem Fall stark aus (Tabellen 3/13 und 3/14). Nach Vorbehandlung der Karzinomgewebe mit Neuraminidase kam es in allen Schnitten zu einer Zunahme des positiv angefärbten Tumoranteils. Lediglich in einem Fall sah man keine Veränderung. Der Mittelwert lag bei 28% positiver Karzinomzellen (STABW 16,87%). In zwei Präparaten nahm die Intensität der Färbung um eine Stufe zu, ansonsten fiel sie mäßig aus. Mit einer Vorbehandlung der Gewebe mit Perjodat konnte die Reaktivität von HMFG2 in den Karzinomen deutlich verstärkt werden. Es färbten sich 65% (STABW 21,21%) der Tumorzellen positiv an. Die Intensität der Färbung war durchgehend stark, dass heißt man sah eine Zunahme der Farbintensität in allen zehn Präparaten. Es gab statistisch einen signifikanten Unterschied zwischen den Ergebnissen der Gewebe ohne Vorbehandlung und den Geweben nach Perjodat-Oxidation bzw. NeuraminidaseBehandlung. Ergebnisse 69 1 2 3 4 5 6 7 8 Ohne Vorbehandlung 5 5 10 5 10 5 30 30 50 20 Farbintensität 2 2 2 2 2 2 3 2 9 2 10 1 Nach Neuraminidase-Behandlung 10 10 30 20 30 10 50 30 60 30 Farbintensität 2 2 Nach Perjodat-Oxidation 80 40 70 90 70 30 70 70 90 40 Farbintensität 3 3 2 3 2 3 2 3 3 3 3 3 2 3 2 3 2 3 Tabelle 3/13: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper HMFG2 in den kolorektalen Karzinomen Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 17 28 65 STABW 15,31 16,87 21,21 Median 10 30 70 25%-Quantil 5 12,5 47,5 75%-Quantil 27,5 30 77,5 Quartilsabstand 22,5 17,5 30 Tabelle 3/14: Lage- und Streumaße von HMFG2 in den kolorektalen Karzinomen 3.2.2 BCP8 Mit dem Antikörper BCP8 konnten 29,5% (STABW 20,06%) des Karzinomgewebes positiv angefärbt werden. Die Intensität der Färbung fiel bei allen Präparaten mäßig aus Tabellen 3/15 und 3/16). Durch die Vorbehandlung der Präparate mit Neuraminidase konnte die Reaktion des Gewebes mit dem Antikörper in sechs Fällen verstärkt werden. Die übrigen vier zeigten keinen Unterschied zu den nativen Schnitten. Es reagierten durchschnittlich 37% (STABW 17,67%) der Karzinomzellen positiv. Die Intensität der Färbung blieb auch mit der NeuraminidaseVorbehandlung durchgehend mäßig. Nach Perjodat-Oxidation zeigte sich, ähnlich wie mit dem Antikörper HMFG2, eine deutliche Zunahme der MUC1-Reaktivität des Karzinomgewebes mit dem Antikörper BCP8 in allen 70 Ergebnisse zehn Präparaten. Es färbten sich 63% (STABW 21,11%) der Karzinomzellen an. Die Intensität der Färbung war unverändert mäßig. In einem Präparat kam es zu einer starken Färbung. Statistisch zeigte sich sowohl für die partielle Deglykosylierung mit Neuraminidase, als auch mit Perjodat-Oxidation ein signifikanter Unterschied im Vergleich zum nativen Gewebe. 1 2 3 4 Ohne Vorbehandlung 10 5 50 20 40 10 30 30 70 30 Farbintensität 2 2 2 2 5 2 6 2 7 2 8 2 9 2 10 2 Nach Neuraminidase-Behandlung 10 20 50 30 40 20 40 50 70 40 Farbintensität 2 2 Nach Perjodat-Oxidation 80 50 70 70 80 20 70 60 90 40 Farbintensität 2 2 2 2 2 2 2 2 2 2 2 3 2 2 2 2 2 2 Tabelle 3/15: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper BCP8 in den kolorektalen Karzinomen Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 29,5 37 63 STABW 20,06 17,67 21,11 Median 30 40 70 25%-Quantil 12,5 22,5 52,5 75%-Quantil 37,5 47,5 77,5 Quartilsabstand 25 25 25 Tabelle 3/16: Lage- und Streumaße von BCP8 in den kolorektalen Karzinomen 3.2.3 115D8 Der Antikörper 115D8 zeigte die stärkste MUC1-Immunreaktivität mit den Karzinomgeweben. Es konnte ein Tumoranteil von 79% (STABW 13,7%) angefärbt werden. Die Werte lagen zwischen 50% bis 90% positiv markierter Tumorzellen. Die Farbintensität war durchgehend stark (Tabellen 3/17 und 3/18). Ergebnisse 71 Nach partieller Deglykosylierung mit Neuraminidase nahm der Anteil gefärbter Karzinomzellen leicht ab. In zwei Präparaten sah man keine Veränderung. Es konnten nur noch 67% (STABW 15,67%) der Karzinomzellen angefärbt werden. In zwei Schnitten sank auch die Farbintensität um eine Stufe, ansonsten blieb die Intensität stark. Mit der Perjodat-Vorbehandlung war die Abnahme der Immunreaktivität noch deutlich stärker ausgeprägt. Durchschnittlich reagierte nur noch 45% (STABW 22,24%) des Tumorgewebes mit dem Antikörper. In einem Fall kam es nach Perjodat-Behandlung sogar zu einem negativen Ergebnis. Die Intensität der Färbung nahm um ein bis zwei Stufen ab, in drei Schnitten blieb sie unverändert. Auch mit dem Antikörper 115D8 zeigte sich ein statistisch signifikanter Unterschied zwischen den Ergebnissen vor und nach partieller Deglykosylierung von MUC1 in den Karzinomen. 1 2 3 4 5 6 7 8 9 10 Ohne Vorbehandlung 90 60 90 80 80 50 80 90 90 80 Farbintensität 3 3 3 3 3 3 3 3 3 3 Nach Neuraminidase-Behandlung 90 40 80 50 70 50 70 80 70 70 Farbintensität 3 2 3 3 3 Nach Perjodat-Oxidation 70 40 40 30 70 30 70 50 0 50 Farbintensität 3 0 2 2 3 2 2 1 3 3 3 3 3 2 2 Tabelle 3/17: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper 115D8 in den kolorektalen Karzinomen Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 79 67 45 STABW 13,7 15,67 22,24 Median 80 70 45 25%-Quantil 80 55 32,5 75%-Quantil 90 77,5 65 Quartilsabstand 10 22,5 32,5 Tabelle 3/18: Lage- und Streumaße von 115D8 in den kolorektalen Karzinomen 72 3.2.4 Ergebnisse MA695 Der Antikörper MA695, der in den Adenomgeweben gar keine oder nur eine schwache Reaktion zeigte, reagierte mit den Karzinomgeweben deutlich stärker. Während bei den „lowgrade“-Adenomen einige Fälle negativ blieben, färbten sich in den kolorektalen Karzinomen 38,5% (STABW 21,09%) der Tumorzellen an. Die Intensität der Färbung fiel mäßig bis stark aus (Tabellen 3/19 und 3/20). Ähnlich wie die Reaktion des Antikörpers 115D8 nahm die Immunreaktivität von MUC1 mit MA695 nach partieller Deglykosylierung ab. Nach einer Vorbehandlung der kolorektalen Karzinomgewebe mit dem Enzym Neuraminidase reagierten noch 29,5% (STABW 22.54%) mit MA695. Während in sechs Schnitten der positiv gefärbte Tumoranteil abnahm, beobachtete man in zwei Fällen eine Zunahme der Reaktivität, in einem Fall sah man keine Veränderung. Die Intensität der Färbung war in vier Fällen stark, in vier Fällen mäßig und in einem Fall gering ausgeprägt. Damit kam es bei vier der Gewebeschnitte zu einer Intensitätsabnahme mit der Neuraminidase-Vorbehandlung, während sie in den übrigen sechs Fällen gleich blieb. Die Intensität der Färbung nahm in vier Fällen ab und blieb in den übrigen Präparaten gleich. Nach Perjodat-Oxidation sank die Immunreaktivität von MA695 stark ab. Es konnten nur noch 6% (STABW 4,6%) der Karzinomzellen positiv markiert werden. Drei der Gewebeschnitte waren nach Perjodat-Behandlung negativ. Auch die Intensität der Färbung nahm in allen Präparaten stark ab. In drei Fällen war sie mäßig, die übrigen Schnitte zeigten gar keine bis eine geringe Färbung der Karzinomzellen. Mit dem Wilcoxon-Test konnte ein statistisch signifikanter Unterschied für die Ergebnisse der unbehandelten und die mit Perjodat behandelten Karzinome festgestellt werden. Die Neuraminidase-Behandlung war im Vergleich zu den unbehandelten Karzinomen nicht signifikant. Ergebnisse 1 2 3 73 4 5 6 7 8 9 10 Ohne Vorbehandlung 80 40 30 50 40 5 60 30 20 20 Farbintensität 2 2 3 3 Nach Neuraminidase-Behandlung 5 30 5 40 40 5 40 70 10 50 Farbintensität 1 2 2 2 2 2 3 3 Nach Perjodat-Oxidation 0 10 5 10 10 0 0 5 10 10 Farbintensität 0 1 1 1 0 2 1 3 3 2 2 0 3 3 3 3 3 2 Tabelle 3/19: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper MA695 in den kolorektalen Karzinomen Ohne Vorbehandlung Nach NeuraminidaseBehandlung Nach PerjodatOxidation Mittelwert 38,5 29,5 6 STABW 21,09 22,54 4,59 Median 35 35 7,5 25%-Quantil 30 6,25 1,25 75%-Quantil 47,5 40 10 Quartilsabstand 17,5 33,75 8,75 Tabelle 3/20: Lage- und Streumaße von MA695 in den kolorektalen Karzinomen 74 Diskussion 4 Diskussion ___________________________________________________________________________ 4.1 Relevanz von MUC1 in der kolorektalen Adenom-KarzinomSequenz Das kolorektale Karzinom stellt in den westlichen Industrieländern die zweithäufigste Tumorerkrankung sowohl bei Männern als auch bei Frauen dar. Etwa 80 bis 85 % der kolorektalen Karzinome entstehen sporadisch über die Adenom-Karzinom-Sequenz. Kolorektale Adenome gelten heute als fakultative Präkanzerosen. Das Risiko der malignen Entartung nimmt vor allem mit der Größe und dem Grad der intraepithelialen Neoplasie zu. Entscheidender Prädiktor für die Prognose des primären kolorektalen Karzinoms ist nach wie vor das pathologische Tumorstadium. Dies gilt sowohl bei Verwendung der TNMKlassifikation der UICC als auch bei Anwendung anderer Klassifikationssysteme, zum Beispiel nach Dukes oder Astler-Coller. In den vergangenen Jahren wurden verschiedene Aspekte der Progression und Prognose des kolorektalen Karzinoms erforscht, um neue unabhängige Parameter für dieses Krankheitsbild zu definieren. Dabei rückten zunehmend genetische Veränderungen sowie die entsprechenden Genprodukte in den Mittelpunkt des pathologisch-wissenschaftlichen Interesses. Durch Anwendung neuer immunhistochemischer und molekularbiologischer Methoden konnten verschiedene Faktoren der molekularen Pathogenese kolorektaler Karzinome beschrieben und hinsichtlich ihrer Bedeutung für den Krankheitsverlauf und die Prognose evaluiert werden. Als wichtiger Marker der gastrointestinalen Karzinogenese entwickelte sich ein besonderes Interesse für die Gruppe der Muzine. Sie stellen eine heterogene Gruppe hochmolekularer, ausgeprägt glykosylierter Proteine dar, deren Peptidcore durch das Auftreten von Tandemrepeat-Sequenzen charakterisiert ist. Die komplexen Oligosaccharid-Seitenketten sind durch O-glykosidische Bindungen an das Apomuzin angeheftet. Muzine findet man in dem mukösen Gel, das die epithelialen Oberflächen in den Hohlorganen des Körpers schützt. Außerdem werden sie als transmembranäre Proteine an der apikalen Oberfläche glandulärer und duktaler Epithelien verschiedener Organe exprimiert. In Normalgeweben zeigt die Expression ein Organ- und Zell-spezifisches Muster. Während der Karzinogenese kommt es zu typischen Veränderungen der Muzin-Expression. Durch die Einführung monoklonaler Antikörper in die Immunhistochemie konnten die histologische Verteilung und die Diskussion 75 Veränderungen der Muzine im Rahmen der Zelldifferenzierung und während der Karzinogenese untersucht werden. Die O-glykosidisch gebundenen Kohlenhydratseitenketten erfahren im Rahmen der neoplastischen Transformation Veränderungen, die für bestimmte Zell- oder Tumorarten typisch sind. Einige dieser Veränderungen konnten in Zusammenhang mit Zelladhäsion, Metastasierung und klinischer Prognose gebracht werden. Als Marker der Zelldifferenzierung könnten muzinassoziierte Antigene eine Relevanz als funktionelle tumorbiologische Marker und Prognosemarker haben. MUC1 repräsentiert als Produkt des MUC1-Gens das am besten charakterisierte membranassoziierte Muzin. Die Glykoproteinstruktur besteht aus einer extrazellulären Region, einer transmembranären Domäne und einem zytoplasmatischen Teil. MUC1 wird an der apikalen Oberfläche der meisten epithelialen Zellen, vor allem im Gastrointestinal-, respiratorischem und genitourethralem Trakt, sowie im Mammagewebe, exprimiert. Es spielt eine wichtige Rolle in der Modulation der Zell-Adhäsion, in der Erhaltung der Zellpolarität, in Signaltransduktionswegen und zellulärer Immunologie [37, 281]. In der Karzinomimmunologie und -therapie besitzt MUC1 inzwischen eine große klinische Bedeutung, da es im Rahmen der Karzinogenese zu einer generellen Überexpression und zu meist typischen, karzinom-bedingten Veränderungen der Kohlenhydratstrukturen kommt. Bisherige Ergebnisse belegen, dass MUC1 ein Marker für Progression und Prognose kolorektaler Karzinome darstellt. Unklar ist jedoch, in welchem Maße eine Änderung der Glykosylierung des MUC1-Peptids zu diesem Effekt beiträgt. Bisher sind die immunhistochemischen und biochemischen Daten über die Glykosylierung normaler oder neoplastischer kolorektaler Muzine sehr dürftig. In den letzten Jahren wurde eine große Anzahl monoklonaler Antikörper gegen MUC1 entwickelt. Ihre Zielepitope können entsprechend ihrer Position in der Tandem-repeat-Region des Muzins und in Hinsicht auf Effekte, die durch die ortsspezifische Glykosylierung ausgeübt werden, klassifiziert werden [22]. Die meisten dieser Antikörper binden an die immundominante hydrophile Sequenz der VNTR-Region, die PDTRPAP-Sequenz. Die Tandem-repeat-Region enthält fünf O-Glykosylierungsstellen, die die Spezifität der monoklonalen Antikörper gegen MUC1 modulieren [189]. Eine Reihe von MUC1spezifischen monoklonalen Antikörpern erkennen oft Epitope, die aus Peptid- und Kohlenhydratanteilen bestehen. Von einigen Antikörpern weiß man, dass sie von dem Glykosylierungsprofil des MUC1-Moleküls beeinflusst werden [116, 249]. Die Immun- 76 Diskussion reaktivität des MUC1-Peptids wird stark von seiner O-Glykosylierung beeinflusst. Veränderungen der MUC1-Glykosylierung und Mechanismen einer anormalen Glykosylierung wurden gründlich in Mamma-Karzinomzellen erforscht. Die biosynthetischen Wege werden von der Substratspezifität der verschiedenen Glykosyltransferasen bestimmt. Daraus resultieren eine starke Reduktion der O-gebundenen Kohlenhydratseitenketten und der O-azetylierten Neuraminsäure. Diese Aspekte haben direkte Auswirkungen auf die Antigenität von MUC1 und auf das Bindungsmuster spezifischer monoklonaler MUC1-Antikörper. In diesem Kontext spielt die Feinspezifität MUC1-spezifischer AK und ihre Epitopkonformation eine wichtige Rolle. Aus Ergebnissen einiger Studien wurde deutlich, dass dies nicht nur von der unterschiedlichen Peptid-Epitopspezifität abhängen kann, sondern auch von der MUC1-Glykosylierung und seiner Veränderung während der Karzinogenese. Daraus kann eine Reduzierung der Länge der O-Glykane resultieren, was zu einem stärkeren immunologischen Erkennen einiger Peptidsequenzen führt. Mit mehreren solcher Antikörper könnte man zeigen, dass bestimmte Veränderungen der Peptid- oder Kohlenhydrat-Epitope mit der Tumorentwicklung assoziiert sind und es wäre möglich, Tumore früh zu detektieren, indem man diese Epitope als spezifische Tumormarker nutzt. In dieser Arbeit wurden vier monoklonale Antikörper verwendet. Zwei von ihnen, HMFG2 und BCP8 haben ihr Epitop innerhalb der VNTR-Region. Das exakte Zielepitop von 115D8 und MA695 ist bisher nicht bekannt. Bei der Auswertung der Expressionsmuster der Antikörper zeigten sich in den kolorektalen Geweben teilweise deutliche Quantitäts- und Intensitätsunterschiede. 4.2 Expression von MUC1 in der Adenom-Karzinom-Sequenz Bisher konnte gezeigt werden, dass die Expression von MUC1 in einigen Karzinomen sehr stark ansteigt [19, 85, 126]. Diese Überexpression von MUC1 wird in Verbindung mit Tumorprogression und Metastasierung gebracht [37, 102, 184, 195, 201]. Mehrfach wurde eine Assoziation zwischen der MUC1-Expression und dem Risiko der malignen Transformation bzw. einer schlechten Prognose kolorektaler Karzinome beschrieben. Dabei stellte sich MUC1 als ein unabhängiger Prognosemarker für eine ungünstige Prognose bei Patienten mit einem kolorektalen Karzinom dar [20, 27, 89, 133]. Diskussion 77 Auch die klinische Verwendung von MUC1 als Tumormarker bei Mamma-Karzinomen wurde bereits in mehreren Studien demonstriert [92, 126, 227]. In kolorektalen Geweben wird das Vorkommen von MUC1 bis heute kontrovers diskutiert. In den meisten Studien wurde angedeutet, dass normale intestinale Epithelzellen nicht oder nur schwach mit monoklonalen Antikörpern gegen MUC1 reagieren [59, 108, 136, 151]. Andere Autoren berichteten, dass normales adultes intestinales Gewebe mit ihren MUC1-spezifischen monoklonalen Antikörpern angefärbt wurde [63, 132]. Auch MUC1-mRNA konnte mit Hilfe der in-situ-Hybridisierung nachgewiesen werden [59]. Man nimmt an, dass MUC1 in normaler kolorektaler Mukosa exprimiert wird, jedoch aufgrund der starken Glykosylierung des MUC1-Proteins nicht durch anti-MUC1-Peptid erkennende monoklonale Antikörper detektiert werden kann, da die Zielepitope maskiert sind [23, 136, 195, 204]. Im Rahmen der Entwicklung eines Kolonkarzinoms über die Adenom-Karzinom-Sequenz wurde in vielen Studien demonstriert, dass im Vergleich zu normalem und hyperplastischem Gewebe, die Immunreaktivität von MUC1 in kolorektalen Adenomen mit steigender villöser Histologie, Größe und Dysplasie signifikant ansteigt [59, 135, 152]. So wurde in kolorektalen Adenomen mit leichter und moderater Dysplasie kein oder nur wenig MUC1 detektiert [59]. In kolorektalen Adenomen mit schwerer Dysplasie und in Karzinomen wurde eine MUC1-Überexpression beschrieben. In einer Studie von Li et al. [175], in der 200 kolorektale Adenome und 58 Karzinome auf MUC1 untersucht wurden, wurde MUC1 in kolorektalen Karzinomen in 24% der Zellen und in den Adenomen mit schwerer Dysplasie in 4% der Zellen exprimiert, während die Adenome mit leichter und mäßiger Dysplasie MUC1-negativ waren. In den Karzinomen neigten die MUC1-positiven Fälle in den fortgeschrittenen Stadien signifikant anzusteigen. Ein ähnliches Ergebnis erhielt Nakamori et al. [195], der eine Korrelation zwischen der MUC1-Expression und der Progression kolorektaler Karzinome untersuchte. Es stellte sich heraus, dass die MUC1-Expression in Karzinomgewebe merklich höher als in Normalgewebe und in Primärtumoren von Patienten mit Metastasen oder metastasierten Tumoren höher als in Primärtumoren von Patienten ohne Metastasen war. Dies könnte ein Hinweis sein, dass das MUC1-Muzin in kolorektalen Karzinomen fortgeschrittener Metastasenstadien ektopisch exprimiert wird und ein nützlicher Marker für das fortgeschrittene kolorektale Karzinom darstellen könnte. Auch Perçınel et al. [211] zeigten, dass die Expression von MUC1 in enger Korrelation mit dem neoplastischen Prozess signifikant zunahm. MUC1, detektiert mit dem monoklonalen Antikörper MA695, konnte in dieser Studie sowohl in normaler Mukosa, als auch in 78 Diskussion hyperplastischen Polypen und tubulären Adenomen nicht nachgewiesen werden, während 24,4% der tubulo-villösen, 19,3% der villösen, 20% der „serrated adenomas“, 37,5% der Adenome mit karzinomatösen Anteil und 89,3% der Adenokarzinome MUC1 positiv waren. Zusammenfassend beweisen diese Daten, dass eine zunehmende MUC1-Expression mit der malignen Transformation kolorektaler Neoplasien zusammenhängt. In dieser Arbeit konnte ebenfalls eine Überexpression von MUC-1 in den untersuchten Adenom- und Karzinomgeweben demonstriert werden. Mit den monoklonalen Antikörpern HMFG2, BCP8 und 115D8 zeigten alle Präparate eine durchgehend positive Gewebereaktion. Lediglich der Antikörper MA695 reagierte in drei Fällen der „low-grade“-Adenome negativ. Insgesamt zeigte sich kein signifikanter Unterschied zwischen der MUC1-Positivität in den „low-grade“- und den „high-grade“-Adenomen. Viele Studien zeigten, dass der Antikörper HMFG2 der Beobachtung von Veränderungen des Glykosylierungsstatus von MUC1 im Rahmen der Kanzerogenese dient, da die Bindungsaffinität durch lange, regulär glykosylierte Kohlenhydratseitenketten normaler Muzine inhibiert wird. Die Überexpression in Karzinomen wurde mit dem Fehlen normal glykosylierter MUC1-Seitenketten erklärt [28, 53]. Im Gegensatz zu diesen Beobachtungen, fiel die Immunreaktivität von MUC1 mit den Antikörpern HMFG2 und BCP8 in den Karzinomen schwächer aus als in den Adenomen. In den „low-grade“-Adenomen waren mit dem Antikörper HMFG2 39%, in den „high-grade“Adenomen 35% der dysplastischen Zellen positiv markiert, während in den Karzinomgeweben nur 17% der Tumorzellen MUC1-positiv waren. Das spricht gegen die allgemeine Annahme, dass das Zielepitop von HMFG2 vornehmlich von Tumorzellen exprimiert wird. Demgegenüber reagierte der Antikörper MA695 gar nicht oder kaum mit den „low-grade“Adenomen (7,7%), während 38,5% des Karzinomgewebes stark positiv war. In den meisten Untersuchungen konnte nachgewiesen werden, dass im Kolon nur Tumorgewebe und kein Normalgewebe durch MA695 angefärbt wird. Dieses Ergebnis geht konform mit der Annahme, dass der Antikörper MA695 eine erhöhte Bindungsaffinität zu Karzinomzellen mit dichter, kurzer O-Glykosylierung hat. Auch für den Antikörper 115D8 zeigte sich eine Zunahme der Immunreaktivität mit steigendem Grad der intraepithelialen Neoplasie. Der Antikörper 115D8 reagierte mit 70 bis 80% immunmarkierter Zellen am stärksten mit den kolorektalen Geweben. Diskussion 79 Diese Ergebnisse reflektieren deutlich die Unterschiede der Feinspezifität der monoklonalen Antikörper und der damit zusammenhängenden Immunreaktivität. Das heißt, dass in Bezug auf immunhistologische Studien, die die Korrelation zwischen Antigen-Expression und histopathologischen Subtyp, Tumorprogression und Prognose untersuchen, die Auswahl der MUC1-spezifischen monoklonalen Antikörper das Ergebnis erheblich beeinflussen kann. Jedoch scheinen diese Beobachtungen nicht nur durch eine unterschiedliche Peptid-Epitopspezifität, sondern auch durch die MUC1-Glykosylierung und ihre Veränderung während der Karzinogenese beeinflusst zu sein. Ein Grund für die zunehmende MUC1-Expression in kolorektalen Adenomen und Karzinomen könnte sein, dass MUC1 in Karzinomen tatsächlich unkontrolliert überexprimiert wird. In Tumorzellen wird das Glykoprotein nicht mehr nur an der apikalen Zelloberfläche exprimiert, sondern ist durch Polaritätsverlust auf der gesamten Zelloberfläche vorhanden. Andererseits wird die Immunreaktivität von MUC1-Peptid-Epitopen stark durch die OGlykosylierung beeinflusst. Die biosynthetischen Wege sind durch die Substratspezifität der verschiedenen Glykosyltransferasen determiniert [22]. Die Glykosylierungsprozesse sind bei krankhaften Prozessen, wie Karzinomen und Entzündungen häufig gestört im Sinne einer abnormalen Glykosylierung und Sialylierung. Es dominieren statt langer Kohlenhydratseitenketten kürzere Core-Strukturen [81, 138, 204, 281, 289]. Studien haben einen Wechsel von Core 3 (GalNAcβ1-3GalNAc) zu Core 2 (Galβ1-3[GlcNAcβ1-6]GalNAc) in kolorektalen Karzinomen beschrieben [15, 21, 39, 122]. Diese Aspekte haben direkte Auswirkungen auf die Antigenität von MUC1 und auf das Bindungsmuster MUC1-spezifischer monoklonaler Antikörper und führen wohl zu einer Demaskierung von MUC1-Peptid-Antigenen, was die immunhistochemische Detektion möglich macht [27, 55]. Außerdem wurde vermutet, dass die Bindung einiger monoklonaler AK an Karzinomassoziiertem MUC1 mit ihrer höheren Affinität für weniger glykosyliertes und sialyliertes MUC1 zu tun hat. Andererseits brauchen einige Antikörper Neuraminsäurereste, um an MUC1 binden zu können [81]. Insgesamt zeigte der Grad an MUC1-Positivität in den kolorektalen Adenomen und Karzinomen erhebliche Unterschiede. Diese Beobachtungen reflektieren die Präsenz 80 Diskussion verschiedener Epitope in oder außerhalb der VNTR-Region, die glykosylierungsabhängig oder -unabhängig sein können. 4.3 Methoden der partiellen Deglykosylierung Hochmolekulare Glykoproteine wie die Muzine enthalten zahlreiche Epitope, die mit Kohlenhydrat- und Peptidketten verbunden sind. Die Kohlenhydrate machen bis zu 80% des Gewichtes dieser Moleküle aus, der Peptid-Core die übrigen 20%. Einige Peptid-Epitope können durch diese Kohlenhydratketten in Gewebeschnitten maskiert vorliegen und somit für Antikörper schwer erreichbar sein [29, 138]. Bisher wurden verschiedene Methoden entwickelt, um solche maskierten Antigene in paraffinierten Gewebeschnitten zu demaskieren, damit diese versteckten Epitope den Antikörpern zugänglich gemacht werden. Da die Tandem-repeat-Einheiten aus verschiedenen Aminosäuresequenzen bestehen, die für jedes Muzingenprodukt spezifisch sind, basieren immunhistochemische Studien des Apomuzins hauptsächlich auf Antikörpern, die mit der Peptid-Determinanten innerhalb der Tandem-repeat-Domäne reagieren. Eine Schwierigkeit dieses Ansatzes ist, dass die starke Glykosylierung das Erkennen des Peptid-Epitops durch die Antikörper verhindern kann und dies falsch negative Ergebnisse liefert. Der Glykosylierungsstatus von MUC1, der von Zellen exprimiert wird, ist ein interessantes Thema der Tumorimmunologie, da die Glykosylierung von MUC1 seine Immunogenität sowie seine biologische Funktion zu determinieren scheint. MUC1 spezifische monoklonale Antikörper, die spezifische Glykosylierungsmuster erkennen, würden in Forschung und klinischer Anwendung von großem Nutzen sein. In dieser Arbeit wurden zur partiellen Deglykosylierung des MUC1-Glykoproteins eine enzymatische Methode mit Neuraminidase, sowie eine chemische Methode mit PerjodatOxidation angewendet. 4.3.1 Perjodat-Oxidation Die Perjodat-Oxidation von formalinfixierten Gewebeschnitten, die partiell Kohlenhydratreste spaltet, um die Darstellung von Muzin-Antigenen zu verbessern, wurde erstmals von Diskussion 81 Woodward et al. 1985 [285] beschrieben und wurde erfolgreich immunelektronenmikroskopisch angewendet ohne ernsthaft die Gewebemorphologie zu beeinträchtigen. Eine milde Perjodat-Oxidation bei saurem pH spaltet die den Kohlenhydraten benachbarten Hydroxylgruppen ohne die Struktur der Peptidketten zu verändern. Durch diese Prozedur wird die rigide Ringstruktur der Saccharide zerstört. So werden Kohlenhydrat-Epitope, die Galaktose, N-Azetylgalaktosamine oder Fukose enthalten, fast vollständig zerstört. Dies könnte zu einer größeren Flexibilität des MuzinMoleküls führen. Maskierte Peptid-Epitope können dadurch enthüllt und für Antikörper zugänglich gemacht werden [29, 59, 103]. Obwohl der Effekt der Perjodat-Oxidation noch nicht vollständig aufgeklärt ist, ist er deutlich und spezifisch. In der vorliegenden Arbeit nahm die MUC1-Immunreaktivität mit den monoklonalen Antikörpern HMFG2 und BCP8 nach Vorbehandlung der Gewebe mit Perjodat signifikant zu. Mit dem Antikörper HMFG2 vervierfachte sich das immunmarkierte Karzinomgewebe sogar. Die starke Zunahme der Immunreaktivität dieser beiden Antikörper deutet darauf hin, dass das Zielepitop bei nativen MUC1-Molekülen durch Glykane maskiert ist. Auch bestätigt dieses Ergebnis die Annahme, dass die Antikörper HMFG2 und BCP8 eine Affinität für kurze Kohlenhydratketten haben. Auf diese Weise eignen sich die Antikörper HMFG2 und BCP8 als Marker für Veränderungen der MUC1-Glykosylierung im Rahmen pathologischer Prozesse während der Karzinogenese. Ähnlich den Ergebnissen anderer Studien [29, 114, 116] zeigte sich in dieser Arbeit, dass die Immunreaktivität von MUC1 mit dem Antikörper MA695 nach einer Perjodat-Oxidation der Gewebeschnitte vor allem in den Kolon-Karzinomgeweben signifikant abnahm, nämlich von 38.5% auf 6% markierter Tumorzellen. Bei den „low-grade“-Adenomen kam es prozentual ebenfalls zu einer Abnahme von 7,4% auf 2%, jedoch zeigte ein Fall eine Zunahme positiv markierter Zellen, ein anderer zeigte keinen Unterschied. Bei den „high-grade“-Adenomen kam es überraschenderweise zu einer prozentualen Zunahme von 8% auf 10% angefärbter Zellen. Mit dem Antikörper 115D8 kam es in allen Geweben zu einer deutlichen Abnahme der MUC1-Immunreaktivität nach Perjodat-Behandlung. Dies ist ein Hinweis dafür, dass diese Antikörper wahrscheinlich pure Kohlenhydratantigene repräsentieren, die durch die partielle Deglykosylierung komplett zerstört werden. 82 Diskussion Auch Bara et al. [29] unterstützt dieses Ergebnis. So nahm die Immunreaktivität von monoklonalen Antikörpern gegen Histoblutgruppen, die gegen Kohlenhydrat-Epitope gerichtet sind, wie zum Beispiel A, Le a , Le x , Sialyl- Le a , H, I, T, TN und Sialyl-Tn nach PerjodatBehandlung drastisch ab. Im Gegensatz dazu blieb die Immunreaktivität von monoklonalen Antikörpern gegen Peptid-Epitope meistens unbeeinflusst. Nach einer Perjodat-Behandlung der Gewebeschnitte konnte MUC1 mit monoklonalen Antikörpern auch in Normalgewebe detektiert werden. In Adenomen und Karzinomen wurde die Immunreaktivität nach Perjodat-Oxidation verstärkt. Dieser Effekt wurde auch in mehreren anderen Studien bestätigt [6, 59, 138, 281]. Cao et al. [59] beschrieb einen schwächer verstärkenden Effekt durch Perjodat-Oxidation in malignen Läsionen im Vergleich zu weniger dysplastischen Läsionen. Er deutete dies damit, dass MUC1 in hoch dysplastischen Geweben und Karzinomen weniger glykosyliert vorliegt, was dazu führt, dass die Epitope den monoklonalen Antikörpern auch ohne PerjodatBehandlung besser zugänglich sind. Dieses Ergebnis konnte in dieser Arbeit nicht bestätigt werden. In einer anderen Studie von Cao et al. [60] wurden 51 Antikörper gegen MUC1 an Warthin Tumoren untersucht. Nach milder Kohlenhydrat-spezifischer Perjodat-Behandlung wurden die Gewebe in den meisten Fällen mit einer stärkeren Intensität angefärbt als ohne Vorbehandlung. Cao erklärte diese scheinbare Heterogenität der Bindungsspezifität durch die Glykosylierung und nicht durch Unterschiede in der Aminosäuresequenz der Epitope. Die Abwesenheit oder Schwäche einer immunhistologischen Färbung eines Antikörpers ist oft durch die Maskierung des Epitops durch die Glykosylierung bedingt, die je nach Gewebetyp differiert. Die Perjodat-Oxidation kann somit empfohlen werden als Methode, um die Kohlenhydratbedingte Epitop-Maskierung zu bewältigen und um die immunhistochemische Muzinfärbung zu verbessern. Dabei sollte man sich nicht auf einen Antikörper verlassen. 4.3.2 Neuraminidase-Behandlung Das MUC1-Glykoprotein enthält Neuraminsäurereste als terminierende Monosaccharide [81]. Die Neuraminsäure verleiht den Muzinen eine negative Ladung. Dadurch trägt sie zu den physikalischen und biologischen Eigenschaften der Muzine bei. Der hohe Anteil von O- Diskussion 83 azetylierter Neuraminsäure ist charakteristisch für das menschliche Kolon und wurde in keinem anderen menschlichen Gewebe gefunden [7, 76]. Wahrscheinlich erhöht die poly-oazetylierte Neuraminsäure im Kolon den Widerstand gegen bakterielle Enzyme [21, 152]. In kolorektalen Adenomen und Karzinomen enthält MUC1 weniger Kohlenhydratseitenketten. Sie sind kürzer und enthalten weniger Neuraminsäurereste. Dieser Verlust der Seitenketten-O-Azetylierung erscheint als ein frühes Ereignis in der kolorektalen AdenomKarzinom-Sequenz und hat zwei Effekte: eine steigende Sensitivität der Neuraminsäure für eine Neuraminidaseverdauung sowie eine veränderte Antigenität [55, 76, 152]. Es wird angenommen, dass die Neuraminsäure in MUC1 zur Maskierung der Peptid-Epitope beiträgt, so dass ein Entfernen der Neuraminsäurereste durch eine Behandlung der Gewebe mit dem Enzym Neuraminidase die Immunreaktivität positiv beeinflussen könnte. Durch eine Vorbehandlung der Gewebeschnitte mit Neuraminidase werden endständige Neuraminsäurereste entfernt, wodurch Kryptantigene, wie zum Beispiel T oder Le a enthüllt werden können [29, 171]. Larena et al. [171] konnte zum Beispiel in papillären Karzinomen der Schilddrüse zeigen, dass durch Demaskierung der sialinierten Bindungsstellen mit Neuraminidase die Färbeergebnisse des Le a - und Le x -Antikörper deutlich verstärkt wurden. Es konnte in einigen Studien gezeigt werden, dass die Desialylierung von MUC1 durch Neuraminidase die Bindung einiger monoklonaler Antikörper, wie 115D8 und MA695 vermindert, da diese Antikörper von Neuraminsäure abhängig sind. Die größte Abnahme der Bindungskapazität wurde für den Antikörper 115D8 beobachtet [114, 249]. Demgegenüber konnte für die Antikörper HMFG2 und BCP8 nach Desialylierung ein Anstieg der Antikörperbindung gefunden werden [81, 218]. Ho et al. [134] zeigte in einer Studie, dass die Reaktivität von HMFG2, spezifisch für das Tripeptid DTR innerhalb der Tandem-repeat-Region von MUC1, durch eine Desialylierung der Zelloberfläche mit dem Enzym Neuraminidase deutlich verstärkt wurde. Damit sah Ho einen Beweis, dass Neuraminsäurereste an benachbarten Oligosaccharidenden den Zugang zu Peptidregionen, die von HMFG2 erkannt werden, merklich beschränken. In einer anderen Studie war eine Behandlung mit dem Enzym Neuraminidase ohne wesentlichen Effekt bezüglich der Immunreaktivität von MUC1 [59]. Dies wurde als ein 84 Diskussion Hinweis gedeutet, dass die Neuraminsäurereste allein nicht die Hauptrolle im Maskieren der Peptid-Epitope in gastrointestinalen Geweben spielen kann. In dieser Arbeit zeigte sich bei den kolorektalen Adenomen und Karzinomen nach Neuraminidasebehandlung ein sehr unterschiedliches Ergebnis. Bei den „low-grade“Adenomen kam es mit den Antikörpern HMFG2 und BCP8 zu einer Zunahme, Abnahme oder zu keiner Änderung der Immunreaktivität. Bei den „high-grade“-Adenomen und Karzinomen verhielt es sich einheitlicher. Die Immunreaktivität nahm von 17% angefärbter Karzinomzellen auf 28% zu. Ein ähnliches Ergebnis erhielt man mit dem Antikörper BCP8. In den Karzinomgeweben nahmen die positiv gefärbten Tumorzellen von 29,5% auf 37% zu, wobei vier von den zehn Fällen keinen Unterschied zeigten. Nach Desialylierung nahm die Bindungsaffinität des Antikörpers 115D8 in allen Geweben deutlich ab. Dieses Ergebnis geht konform mit dem anderer Studien [249] und beweist, dass die Bindungsaffinität des Antikörpers 115D8 an das Zielepitop von Neuraminsäureresten abhängig ist. Diese Beobachtungen indizieren, dass dieser Antikörper sehr nützlich sein könnte, um MUC1 zu detektieren, das α2-3-gebundene Neuraminsäurereste enthält. Mit dem Antikörper MA695 kam es bei den „low-grade“-Adenomen nach Neuraminidasebehandlung zu einer starken Zunahme des MUC1-markierten Gewebes von 7,4% auf 26%. Ebenso stieg die Immunreaktivität bei den „high-grade“-Adenomen von 8% auf 23%. Dieses Ergebnis spricht gegen die These, dass auch das Epitop von MA695 von Neuraminsäure abhängig ist. Bei den Karzinomen zeigte sich eine Abnahme des mit dem Antikörper MA695 markierten Tumorgewebes von 38,5% auf 29,5%. Daj et al. [81] untersuchte den Effekt einer Desialylierung bei 56 monoklonale Antikörper gegen MUC1. Sein Ergebnis zeigte, dass eine Desialylierung verschiedene Effekte, wie einen Anstieg, eine Abnahme oder keine Veränderung der Bindungsaffinität hatte. Alle monoklonalen Antikörper, die an Epitope innerhalb der Aminosäuresequenz VTSAPDTRPA von MUC1 binden, wiesen nach Desialylierung einen Anstieg der Bindungskapazität auf. Für die Antikörper, deren Bindung sich durch eine Desialylierung veränderte, könnten laut Daj einige Mechanismen in Frage kommen. Terminale Neuraminsäurereste könnten die Diskussion 85 Erreichbarkeit der Epitope durch elektrostatische Abstoßung positiv oder negativ beeinflussen. Umgekehrt könnte das Entfernen der Neuraminsäure entweder zu sterischer Behinderung oder zu einem besseren Zugang der Antikörper-Bindung zu den Peptid-Epitopen führen. Zusammenfassend wird die Bindung von monoklonalen Antikörpern an MUC1 stark von dem Sialylierungsstatus beeinflusst. Der Vergleich von nativen und desialylierten MUC1 könnte ein nützliches Instrument zur Selektion von Antikörpern sein, die weniger sialyliertes MUC1 favorisieren. Antikörper wie BCP8 oder HMFG2 könnten in der Entwicklung von neuen Assays zur Detektion von Tumor-assoziiertem MUC1-Muzin genutzt werden. 4.4 Fazit Die Ergebnisse in dieser Arbeit, wie auch in anderen Studien demonstrieren die Überexpression von MUC1 innerhalb der Adenom-Karzinom-Sequenz. Auch konnte der Einfluss der Glykosylierung von MUC1 auf die Immunreaktivität bestätigt werden. Nach Deglykosylierung der kolorektalen Gewebe konnte die Immunreaktivität der PeptidAntikörper HMFG2 und BCP8 verstärkt werden. Demgegenüber nahm die Immunreaktivität der Antikörper 115D8 und MA695 nach Perjodat-Behandlung stark ab. Ein ähnliches Ergebnis zeigte sich nach Entfernen der Neuraminsäurereste mit Neuraminidase. Dies verdeutlicht, dass neben der unterschiedlichen Feinspezifität der monoklonalen Antikörper auch das Glykosylierungsprofil von MUC1 einen großen Einfluss auf die Antigenität des MUC1-Moleküls hat. In Bezug auf immunhistologische Studien, die ihren Fokus auf die Korrelation zwischen Antigen-Expression und histopathologischen Subtypen, Tumorprogression und Prognose haben, kann die Auswahl der MUC1 spezifischen monoklonalen Antikörper somit das Ergebnis entscheidend beeinflussen. Die Ergebnisse verdeutlichen weiterhin die diagnostische Aussagekraft spezifischer MUC1Antikörper und weisen auf ihre mögliche Rolle bezüglich immuntherapeutischer Überlegungen hin. Um die mit der malignen Transformation von Zellen assoziierten Veränderungen des MuzinMoleküls für Prävention, Therapie und Nachsorge eines Karzinoms anwenden zu können, sollte die Expression dieser Moleküle in unterschiedlichen Stadien der Kanzerogenese weiter untersucht werden. 86 Zusammenfassung 5 Zusammenfassung ___________________________________________________________________________ Muzine stellen wichtige Marker der gastrointestinalen Karzinogenese dar. Von besonderer Bedeutung für das kolorektale Karzinom ist vor allem das MUC1-Muzin. Im Rahmen der Karzinogenese kommt es zu einer generellen Überexpression und zu meist typischen, Karzinom-bedingten Veränderungen der O-glykosidisch gebundenen Kohlenhydratseitenketten des MUC1-Moleküls. Bisherige Ergebnisse belegen, dass MUC1 ein Marker für Progression und Prognose kolorektaler Karzinome darstellt. Unklar ist jedoch, in welchem Maße eine Änderung der Glykosylierung des MUC1-Peptids zu diesem Effekt beiträgt. In der vorliegenden vergleichenden Studie wurde an Paraffin-eingebetteten Serienschnitten von zehn kolorektalen Adenomen (je fünf mit gering- und hochgradiger intraepithelialer Neoplasie (IEN)) und zehn Karzinomen die Immunreaktivität von MUC1 in Hinblick auf das Glykosylierungsprofil untersucht und bewertet. Dazu wurden die Gewebe mit vier MUC1spezifischen monoklonalen Antikörpern unterschiedlicher Epitop-Spezifität nach der Streptavidin-Biotin-Peroxidase-Methode immunhistochemisch gefärbt. Als weitere Kontrollen zur Charakterisierung der Glykosylierungsabhängigkeit wurden chemische und enzymatische Methoden der partiellen Deglykosylierung durchgeführt. Bei der PerjodatOxidation werden Verbindungen zwischen Kohlenstoff- und Hydroxylgruppen gespalten, ohne dass die Peptid-Epitope zerstört werden. Das Enzym Neuraminidase katalysiert die Hydrolyse von N-Azetyl-Neuraminsäureresten von Glykoproteinen. Bei der Auswertung der Expressionsmuster der Antikörper zeigten sich in den kolorektalen Geweben deutliche Quantitäts- und Intensitätsunterschiede. Diese Beobachtungen reflektieren die Präsenz verschiedener Epitope innerhalb des MUC1-Muzins, die glykosylierungsabhängig oder –unabhängig sein können. Eine Überexpression von MUC-1 konnte in den untersuchten kolorektalen Adenom- und Karzinomgeweben bestätigt werden. Die Immunreaktivität von MUC1 fiel mit den Antikörpern HMFG2 und BCP8 in den Karzinomen schwächer aus als in den Adenomen. Das spricht gegen die allgemeine Annahme, dass das Zielepitop von HMFG2 und BCP8 vornehmlich von Tumorzellen exprimiert wird. Mit den Antikörpern Ma695 und 115D8 zeigte sich eine Zunahme der Immunreaktivität von MUC1 mit zunehmender IEN. Der Antikörper MA695 reagierte hauptsächlich mit dem Karzinomgewebe. Er scheint eine erhöhte Bindungsaffinität zu Karzinomzellen mit dichter, kurzer O-Glykosylierung zu haben. Zusammenfassung 87 Nach Vorbehandlung der kolorektalen Gewebe mit Perjodat konnte die Immunreaktivität von MUC1 mit den Antikörpern HMFG2 und BCP8 deutlich verstärkt werden. Dies ist ein Hinweis darauf, dass die Zielepitope durch die normale Glykosylierung des MUC1-Moleküls maskiert sind. Demgegenüber nahm die Immunreaktivität der Antikörper MA695 und 115D8 stark ab. Dies beweist, dass ihr Zielepitop ein Glykopeptid darstellt, das durch die Deglykosylierung zerstört wurde. Insgesamt kann die Perjodat-Oxidation als Methode empfohlen werden, um die Kohlenhydrat-bedingte Epitop-Maskierung zu bewältigen und die immunhistochemische Muzinfärbung zu verbessern. Nach Deglykosylierung der kolorektalen Gewebe mit Neuraminidase kam es mit den Antikörpern HMFG2, BCP8 und Ma695 zu einer Abnahme, einer Zunahme oder zu keinem Unterschied der Immunaktivität. Die Bindungsaffinität des Antikörpers 115D8 nahm nach Desialylierung in den kolorektalen Geweben deutlich ab, ein Hinweis, dass das Epitop des Antikörpers 115D8 Neuraminsäureabhängig ist. Mit dem Antikörper MA695 kam es bei den kolorektalen Adenomen nach Desialylierung zu einer starken Zunahme des MUC1-markierten Gewebes. Dieses Ergebnis spricht gegen die These, dass auch das Epitop von MA695 Neuraminsäure-abhängig ist. Zusammenfassend verdeutlichen die Ergebnisse, dass neben der unterschiedlichen Feinspezifität der monoklonalen Antikörper auch das Glykosylierungsprofil von MUC1 einen großen Einfluss auf die Antigenität des MUC1-Moleküls hat. In Bezug auf immunhistologische Studien, die ihren Fokus auf die Korrelation zwischen Antigen-Expression und histopathologischen Subtypen, Tumorprogression und Prognose haben, kann die Auswahl der MUC1 spezifischen monoklonalen Antikörper das Ergebnis entscheidend beeinflussen. 88 Literaturverzeichnis 6 Literaturverzeichnis ___________________________________________________________________________ 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. Efficacy of adjuvant fluorouracil and folinic acid in colon cancer. International Multicentre Pooled Analysis of Colon Cancer Trials (IMPACT) investigators. Lancet, 1995. 345: 939-944. Cancer incidence in five continents. Volume IX. IARC Sci Publ, 2008: 1-837. Cancer, Facts and Figures 2009. Herausgeber: American Cancer Society, 2009: 1213. Ahlquist, D.A., Skoletsky, J.E., Boynton, K.A., Harrington, J.J., Mahoney, D.W., Pierceall, W.E., Thibodeau, S.N., Shuber, A.P., Colorectal cancer screening by detection of altered human DNA in stool: feasibility of a multitarget assay panel. Gastroenterology, 2000. 119: 1219-1227. Ahmed, F.E., Effect of diet, life style, and other environmental/chemopreventive factors on colorectal cancer development, and assessment of the risks. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev, 2004. 22: 91-147. Ajioka, Y., Allison, L.J., Jass, J.R., Significance of MUC1 and MUC2 mucin expression in colorectal cancer. J Clin Pathol, 1996. 49: 560-564. Aksoy, N., Akinci, O.F., Mucin macromolecules in normal, adenomatous, and carcinomatous colon: evidence for the neotransformation. Macromol Biosci, 2004. 4: 483-496. Alexander, J., Watanabe, T., Wu, T.T., Rashid, A., Li, S., Hamilton, S.R., Histopathological identification of colon cancer with microsatellite instability. Am J Pathol, 2001. 158: 527-535. Allen, A., Flemstrom, G., Garner, A., Kivilaakso, E., Gastroduodenal mucosal protection. Physiol Rev, 1993. 73: 823-857. Allen, A., Hutton, D.A., Leonard, A.J., Pearson, J.P., Sellers, L.A., The role of mucus in the protection of the gastroduodenal mucosa. Scand J Gastroenterol Suppl, 1986. 125: 71-78. Allen, A., Hutton, D.A., Pearson, J.P., The MUC2 gene product: a human intestinal mucin. Int J Biochem Cell Biol, 1998. 30: 797-801. Andre, T., Boni, C., Mounedji-Boudiaf, L., Navarro, M., Tabernero, J., Hickish, T., Topham, C., Zaninelli, M., Clingan, P., Bridgewater, J., Tabah-Fisch, I., de Gramont, A., Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med, 2004. 350: 2343-2351. Andrews, C.W., Jr., Jessup, J.M., Goldman, H., Hayes, D.F., Kufe, D.W., O'Hara, C.J., Steele, G.D., Jr., Localization of tumor-associated glycoprotein DF3 in normal, inflammatory, and neoplastic lesions of the colon. Cancer, 1993. 72: 3185-3190. Andrieu, N., Launoy, G., Guillois, R., Ory-Paoletti, C., Gignoux, M., Familial relative risk of colorectal cancer: a population-based study. Eur J Cancer, 2003. 39: 19041911. Ashwell, G., Morell, A.G., The role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteins. Adv Enzymol Relat Areas Mol Biol, 1974. 41: 99-128. Atkinson, B.F., Ernst, C.S., Herlyn, M., Steplewski, Z., Sears, H.F., Koprowski, H., Gastrointestinal cancer-associated antigen in immunoperoxidase assay. Cancer Res, 1982. 42: 4820-4823. Audie, J.P., Janin, A., Porchet, N., Copin, M.C., Gosselin, B., Aubert, J.P., Expression of human mucin genes in respiratory, digestive, and reproductive tracts ascertained by in situ hybridization. J Histochem Cytochem, 1993. 41: 1479-1485. Literaturverzeichnis 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 89 Baba, S., Recent advances in molecular genetics of colorectal cancer. World J Surg, 1997. 21: 678-687. Baeckstrom, D., Nilsson, O., Price, M.R., Lindholm, L., Hansson, G.C., Discrimination of MUC1 mucins from other sialyl-Le(a)-carrying glycoproteins produced by colon carcinoma cells using a novel monoclonal antibody. Cancer Res, 1993. 53: 755-761. Baldus, S.E., Klinisch-pathologische und molekulare Prognosefaktoren kolorektaler Karzinome. Pathologe, 2003. 24: 49-60. Baldus, S.E., Engelmann, K., Hanisch, F.G., MUC1 and the MUCs: a family of human mucins with impact in cancer biology. Crit Rev Clin Lab Sci, 2004. 41: 189-231. Baldus, S.E., Goergen, D., Hanisch, F.G., Dienes, H.P., Epitope-dependent differential immunoreactivities of anti-MUC1 monoclonal antibodies in human carcinomas. Int J Oncol, 2001. 18: 507-512. Baldus, S.E., Hanisch, F.G., Muzinassoziierte Antigene als Marker gastrointestinaler Differenzierung und Karzinogenese. Pathologe, 1995. 16: 94-105. Baldus, S.E., Hanisch, F.G., Biochemistry and pathological importance of mucinassociated antigens in gastrointestinal neoplasia. Adv Cancer Res, 2000. 79: 201-248. Baldus, S.E., Hanisch, F.G., Kotlarek, G.M., Zirbes, T.K., Thiele, J., Isenberg, J., Karsten, U.R., Devine, P.L., Dienes, H.P., Coexpression of MUC1 mucin peptide core and the Thomsen-Friedenreich antigen in colorectal neoplasms. Cancer, 1998. 82: 1019-1027. Baldus, S.E., Hanisch, F.G., Putz, C., Flucke, U., Monig, S.P., Schneider, P.M., Thiele, J., Holscher, A.H., Dienes, H.P., Immunoreactivity of Lewis blood group and mucin peptide core antigens: correlations with grade of dysplasia and malignant transformation in the colorectal adenoma-carcinoma sequence. Histol Histopathol, 2002. 17: 191-198. Baldus, S.E., Monig, S.P., Hanisch, F.G., Zirbes, T.K., Flucke, U., Oelert, S., Zilkens, G., Madejczik, B., Thiele, J., Schneider, P.M., Holscher, A.H., Dienes, H.P., Comparative evaluation of the prognostic value of MUC1, MUC2, sialyl-Lewis(a) and sialyl-Lewis(x) antigens in colorectal adenocarcinoma. Histopathology, 2002. 40: 440-449. Baldus, S.E., Wienand, J.R., Werner, J.P., Landsberg, S., Drebber, U., Hanisch, F.G., Dienes, H.P., Expression of MUC1, MUC2 and oligosaccharide epitopes in breast cancer: prognostic significance of a sialylated MUC1 epitope. Int J Oncol, 2005. 27: 1289-1297. Bara, J., Decaens, C., Loridon-Rosa, B., Oriol, R., Immunohistological characterization of mucin epitopes by pre-treatment of gastro-intestinal sections with periodic acid. J Immunol Methods, 1992. 149: 105-113. Bara, J., Mollicone, R., Herrero-Zabaleta, E., Gautier, R., Daher, N., Oriol, R., Ectopic expression of the Y (Ley) antigen defined by monoclonal antibody 12-4LE in distal colonic adenocarcinomas. Int J Cancer, 1988. 41: 683-689. Bartman, A.E., Sanderson, S.J., Ewing, S.L., Niehans, G.A., Wiehr, C.L., Evans, M.K., Ho, S.B., Aberrant expression of MUC5AC and MUC6 gastric mucin genes in colorectal polyps. Int J Cancer, 1999. 80: 210-218. Baruch, A., Hartmann, M., Yoeli, M., Adereth, Y., Greenstein, S., Stadler, Y., Skornik, Y., Zaretsky, J., Smorodinsky, N.I., Keydar, I., Wreschner, D.H., The breast cancer-associated MUC1 gene generates both a receptor and its cognate binding protein. Cancer Res, 1999. 59: 1552-1561. Batzler, W.U., Giersiepen, K., Hentschel, S., Husmann, G., Kaatsch, P., Katalinic, A., Kieschke, J., Kraywinkel, K., Meyer, M., Stabenow, R., Stegmaier, C., Bertz, J., Haberland, J., Wolf, U., Krebs in Deutschland 2003-2004-Häufigkeiten und Trends. 90 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. Literaturverzeichnis Herausgeber: Robert Koch-Institut in Zusammenarbeit mit der Gesellschaft der epidemiologischen Krebsregister in Deutschland e. V., 2008. 6: 34-37. Benson, A.B., 3rd, Schrag, D., Somerfield, M.R., Cohen, A.M., Figueredo, A.T., Flynn, P.J., Krzyzanowska, M.K., Maroun, J., McAllister, P., Van Cutsem, E., Brouwers, M., Charette, M., Haller, D.G., American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J Clin Oncol, 2004. 22: 3408-3419. Bernstein, C.N., Blanchard, J.F., Kliewer, E., Wajda, A., Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer, 2001. 91: 854-862. Berry, N., Jones, D.B., Smallwood, J., Taylor, I., Kirkham, N., Taylor-Papadimitriou, J., The prognostic value of the monoclonal antibodies HMFG1 and HMFG2 in breast cancer. Br J Cancer, 1985. 51: 179-186. Biemer-Hüttmann, A.-E., Mucin Core Protein Expression in Colorectal Cancers with High Levels of Microsatellite Instability Indicates a Novel Pathway of Morphogenesis. Clinical Cancer Research, 2000. Vol. 6: 1909-1916. Bingham, S.A., Day, N.E., Luben, R., Ferrari, P., Slimani, N., Norat, T., ClavelChapelon, F., Kesse, E., Nieters, A., Boeing, H., Tjonneland, A., Overvad, K., Martinez, C., Dorronsoro, M., Gonzalez, C.A., Key, T.J., Trichopoulou, A., Naska, A., Vineis, P., Tumino, R., Krogh, V., Bueno-de-Mesquita, H.B., Peeters, P.H., Berglund, G., Hallmans, G., Lund, E., Skeie, G., Kaaks, R., Riboli, E., Dietary fibre in food and protection against colorectal cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC): an observational study. Lancet, 2003. 361: 1496-1501. Blank, M., Klussmann, E., Kruger-Krasagakes, S., Schmitt-Graff, A., Stolte, M., Bornhoeft, G., Stein, H., Xing, P.X., McKenzie, I.F., Verstijnen, C.P., Expression of MUC2-mucin in colorectal adenomas and carcinomas of different histological types. Int J Cancer, 1994. 59: 301-306. Bodmer, W.F., Cancer genetics: colorectal cancer as a model. J Hum Genet, 2006. 51: 391-396. Boese-Landgraf, J., Epidemiologie, Vorstufen und Pathogenese des kolorektalen Karzinoms. Der Onkologe, 1998. 4: 2. Bokey, E.L., Ojerskog, B., Chapuis, P.H., Dent, O.F., Newland, R.C., Sinclair, G., Local recurrence after curative excision of the rectum for cancer without adjuvant therapy: role of total anatomical dissection. Br J Surg, 1999. 86: 1164-1170. Boland, C.R., Montgomery, C.K., Kim, Y.S., Alterations in human colonic mucin occurring with cellular differentiation and malignant transformation. Proc Natl Acad Sci U S A, 1982. 79: 2051-2055. Bombi, J.A., Polyps of the colon in Barcelona, Spain. An autopsy study. Cancer, 1988. 61: 1472-1476. Bongaerts, B.W., de Goeij, A.F., de Vogel, S., van den Brandt, P.A., Goldbohm, R.A., Weijenberg, M.P., Alcohol consumption and distinct molecular pathways to colorectal cancer. Br J Nutr, 2007. 97: 430-434. Brabletz, T., Herrmann, K., Jung, A., Faller, G., Kirchner, T., Expression of nuclear beta-catenin and c-myc is correlated with tumor size but not with proliferative activity of colorectal adenomas. Am J Pathol, 2000. 156: 865-870. Brenner, H., Arndt, V., Sturmer, T., Stegmaier, C., Ziegler, H., Dhom, G., Longlasting reduction of risk of colorectal cancer following screening endoscopy. Br J Cancer, 2001. 85: 972-976. Bresalier, R.S., Ho, S.B., Schoeppner, H.L., Kim, Y.S., Sleisenger, M.H., Brodt, P., Byrd, J.C., Enhanced sialylation of mucin-associated carbohydrate structures in human colon cancer metastasis. Gastroenterology, 1996. 110: 1354-1367. Literaturverzeichnis 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. 64. 91 Brockhausen, I., Mucin-type O-glycans in human colon and breast cancer: glycodynamics and functions. EMBO Rep, 2006. 7: 599-604. Brockhausen, I., Romero, P.A., Herscovics, A., Glycosyltransferase changes upon differentiation of CaCo-2 human colonic adenocarcinoma cells. Cancer Res, 1991. 51: 3136-3142. Brown, A., Ellis, I.O., Embleton, M.J., Baldwin, R.W., Turner, D.R., Hardcastle, J.D., Immunohistochemical localization of Y hapten and the structurally related H type-2 blood-group antigen on large-bowel tumours and normal adult tissues. Int J Cancer, 1984. 33: 727-736. Bubb, V.J., Curtis, L.J., Cunningham, C., Dunlop, M.G., Carothers, A.D., Morris, R.G., White, S., Bird, C.C., Wyllie, A.H., Microsatellite instability and the role of hMSH2 in sporadic colorectalcancer. Oncogene, 1996. 12: 2641-2649. Burchell, J., Taylor-Papadimitriou, J., Effect of modification of carbohydrate side chains on the reactivity of antibodies with core-protein epitopes of the MUC1 gene product. Epithelial Cell Biol, 1993. 2: 155-162. Burchill, S.A., The tumour suppressor APC gene product is associated with cell adhesion. Bioessays, 1994. 16: 225-227. Byrd, J.C., Bresalier, R.S., Mucins and mucin binding proteins in colorectal cancer. Cancer Metastasis Rev, 2004. 23: 77-99. Camma, C., Giunta, M., Fiorica, F., Pagliaro, L., Craxi, A., Cottone, M., Preoperative radiotherapy for resectable rectal cancer: A meta-analysis. Jama, 2000. 284: 10081015. Campbell, B.J., Finnie, I.A., Hounsell, E.F., Rhodes, J.M., Direct demonstration of increased expression of Thomsen-Friedenreich (TF) antigen in colonic adenocarcinoma and ulcerative colitis mucin and its concealment in normal mucin. J Clin Invest, 1995. 95: 571-576. Campo, E., Condom, E., Palacin, A., Quesada, E., Cardesa, A., Lectin binding patterns in normal and neoplastic colonic mucosa. A study of Dolichos biflorus agglutinin, peanut agglutinin, and wheat germ agglutinin. Dis Colon Rectum, 1988. 31: 892-899. Cao, Y., Blohm, D., Ghadimi, B.M., Stosiek, P., Xing, P.X., Karsten, U., Mucins (MUC1 and MUC3) of gastrointestinal and breast epithelia reveal different and heterogeneous tumor-associated aberrations in glycosylation. J Histochem Cytochem, 1997. 45: 1547-1557. Cao, Y., Karsten, U., Binding patterns of 51 monoclonal antibodies to peptide and carbohydrate epitopes of the epithelial mucin (MUC1) on tissue sections of adenolymphomas of the parotid (Warthin's tumours): role of epitope masking by glycans. Histochem Cell Biol, 2001. 115: 349-356. Cao, Y., Karsten, U.R., Liebrich, W., Haensch, W., Springer, G.F., Schlag, P.M., Expression of Thomsen-Friedenreich-related antigens in primary and metastatic colorectal carcinomas. A reevaluation. Cancer, 1995. 76: 1700-1708. Cao, Y., Schlag, P.M., Karsten, U., Immunodetection of epithelial mucin (MUC1, MUC3) and mucin-associated glycotopes (TF, Tn, and sialosyl-Tn) in benign and malignant lesions of colonic epithelium: apolar localization corresponds to malignant transformation. Virchows Arch, 1997. 431: 159-166. Carrato, C., Balague, C., de Bolos, C., Gonzalez, E., Gambus, G., Planas, J., Perini, J.M., Andreu, D., Real, F.X., Differential apomucin expression in normal and neoplastic human gastrointestinal tissues. Gastroenterology, 1994. 107: 160-172. Ceriani, R.L., Geradts, J., Peterson, J.A., Second international workshop on monoclonal antibodies and breast cancer. San Francisco, November 20-21, 1986. Breast Cancer Res Treat, 1987. 10: 55-63. 92 65. 66. 67. 68. 69. 70. 71. 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. Literaturverzeichnis Chan, A.T., Giovannucci, E.L., Schernhammer, E.S., Colditz, G.A., Hunter, D.J., Willett, W.C., Fuchs, C.S., A prospective study of aspirin use and the risk for colorectal adenoma. Ann Intern Med, 2004. 140: 157-166. Chang, S.K., Dohrman, A.F., Basbaum, C.B., Ho, S.B., Tsuda, T., Toribara, N.W., Gum, J.R., Kim, Y.S., Localization of mucin (MUC2 and MUC3) messenger RNA and peptide expression in human normal intestine and colon cancer. Gastroenterology, 1994. 107: 28-36. Chao, A., Thun, M.J., Jacobs, E.J., Henley, S.J., Rodriguez, C., Calle, E.E., Cigarette smoking and colorectal cancer mortality in the cancer prevention study II. J Natl Cancer Inst, 2000. 92: 1888-1896. Chapman, M.A., Buckley, D., Henson, D.B., Armitage, N.C., Preoperative carcinoembryonic antigen is related to tumour stage and long-term survival in colorectal cancer. Br J Cancer, 1998. 78: 1346-1349. Citarda, F., Tomaselli, G., Capocaccia, R., Barcherini, S., Crespi, M., Efficacy in standard clinical practice of colonoscopic polypectomy in reducing colorectal cancer incidence. Gut, 2001. 48: 812-815. Compton, C.C., Pathology report in colon cancer: what is prognostically important? Dig Dis, 1999. 17: 67-79. Connell, W.R., Talbot, I.C., Harpaz, N., Britto, N., Wilkinson, K.H., Kamm, M.A., Lennard-Jones, J.E., Clinicopathological characteristics of colorectal carcinoma complicating ulcerative colitis. Gut, 1994. 35: 1419-1423. Cooper, H.S., Peanut lectin-binding sites in large bowel carcinoma. Lab Invest, 1982. 47: 383-390. Cooper, H.S., Cox, J., Patchefsky, A.S., Immunohistologic study of blood group substances in polyps of the distal colon. Expression of a fetal antigen. Am J Clin Pathol, 1980. 73: 345-350. Corfield, A.P., Carroll, D., Myerscough, N., Probert, C.S., Mucins in the gastrointestinal tract in health and disease. Front Biosci, 2001. 6: 1321-1357. Corfield, A.P., Myerscough, N., Gough, M., Brockhausen, I., Schauer, R., Paraskeva, C., Glycosylation patterns of mucins in colonic disease. Biochem Soc Trans, 1995. 23: 840-845. Corfield, A.P., Myerscough, N., Warren, B.F., Durdey, P., Paraskeva, C., Schauer, R., Reduction of sialic acid O-acetylation in human colonic mucins in the adenomacarcinoma sequence. Glycoconj J, 1999. 16: 307-317. Cornberg, M., Differenzielle Muzinexpression im Gastrointestinaltrakt. 2000. Cruz-Correra, M., Giradiello, FM., Diagnosis and management of hereditary colon cancer. Gastroenterol Clin North Am, 2002. 31: 537-549. Cubas, R., Li, M., Chen, C., Yao, Q., Colorectal cancer: new advances in immunotherapy. Cancer Biol Ther, 2007. 6: 11-17. Dahiya, R., Itzkowitz, S.H., Byrd, J.C., Kim, Y.S., Mucin oligosaccharide biosynthesis in human colonic cancerous tissues and cell lines. Cancer, 1992. 70: 1467-1476. Dai, J., Allard, W.J., Davis, G., Yeung, K.K., Effect of desialylation on binding, affinity, and specificity of 56 monoclonal antibodies against MUC1 mucin. Tumour Biol, 1998. 19 Suppl 1: 100-110. David, L., Leitao, D., Sobrinho-Simoes, M., Bennett, E.P., White, T., Mandel, U., Dabelsteen, E., Clausen, H., Biosynthetic basis of incompatible histo-blood group A antigen expression: anti-A transferase antibodies reactive with gastric cancer tissue of type O individuals. Cancer Res, 1993. 53: 5494-5500. Literaturverzeichnis 83. 84. 85. 86. 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. 98. 99. 100. 93 Denk, H., Tappeiner, G., Davidovits, A., Eckerstorfer, R., Holzner, J.H., Carcinoembryonic antigen and blood group substances in carcinomas of the stomach and colon. J Natl Cancer Inst, 1974. 53: 933-942. Deutsche-Krebsgesellschaft, Kolonkarzinom-Interdisziplinäre Leitlinien. Coloproctology, 2000. 22: 145. Devine, P.L., McGuckin, M.A., Ward, B.G., Circulating mucins as tumor markers in ovarian cancer (review). Anticancer Res, 1992. 12: 709-717. Dong, S.M., Traverso, G., Johnson, C., Geng, L., Favis, R., Boynton, K., Hibi, K., Goodman, S.N., D'Allessio, M., Paty, P., Hamilton, S.R., Sidransky, D., Barany, F., Levin, B., Shuber, A., Kinzler, K.W., Vogelstein, B., Jen, J., Detecting colorectal cancer in stool with the use of multiple genetic targets. J Natl Cancer Inst, 2001. 93: 858-865. Dong, Y., Walsh, M.D., Cummings, M.C., Wright, R.G., Khoo, S.K., Parsons, P.G., McGuckin, M.A., Expression of MUC1 and MUC2 mucins in epithelial ovarian tumours. J Pathol, 1997. 183: 311-317. Duffy, M.J., Carcinoembryonic antigen as a marker for colorectal cancer: is it clinically useful? Clin Chem, 2001. 47: 624-630. Duncan, T.J., Watson, N.F., Al-Attar, A.H., Scholefield, J.H., Durrant, L.G., The role of MUC1 and MUC3 in the biology and prognosis of colorectal cancer. World J Surg Oncol, 2007. 5: 31. Ekbom, A., Helmick, C., Zack, M., Adami, H.O., Increased risk of large-bowel cancer in Crohn's disease with colonic involvement. Lancet, 1990. 336: 357-359. Ernst, C., Atkinson, B., Wysocka, M., Blaszczyk, M., Herlyn, M., Sears, H., Steplewski, Z., Koprowski, H., Monoclonal antibody localization of Lewis antigens in fixed tissue. Lab Invest, 1984. 50: 394-400. Eskelinen, M., Tikanoja, S., Valkamo, E., Loikkanen, M., Collan, Y., Cancerassociated antigen CA 15-3 in the diagnostics of breast tumours. Scand J Clin Lab Invest, 1988. 48: 653-658. Fearon, E.R., Vogelstein, B., A genetic model for colorectal tumorigenesis. Cell, 1990. 61: 759-767. Figueredo, A., Germond, C., Maroun, J., Browman, G., Walker-Dilks, C., Wong, S., Adjuvant therapy for stage II colon cancer after complete resection. Provincial Gastrointestinal Disease Site Group. Cancer Prev Control, 1997. 1: 379-392. Finn, O.J., Jerome, K.R., Henderson, R.A., Pecher, G., Domenech, N., MagarianBlander, J., Barratt-Boyes, S.M., MUC-1 epithelial tumor mucin-based immunity and cancer vaccines. Immunol Rev, 1995. 145: 61-89. Fodde, R., The APC gene in colorectal cancer. Eur J Cancer, 2002. 38: 867-871. Folprecht, G., Lutz, M.P., Schoffski, P., Seufferlein, T., Nolting, A., Pollert, P., Kohne, C.H., Cetuximab and irinotecan/5-fluorouracil/folinic acid is a safe combination for the first-line treatment of patients with epidermal growth factor receptor expressing metastatic colorectal carcinoma. Ann Oncol, 2006. 17: 450-6. Friedenreich, C.M., Orenstein, M.R., Physical activity and cancer prevention: etiologic evidence and biological mechanisms. J Nutr, 2002. 132: 3456-3464. Fung, T., Hu, F.B., Fuchs, C., Giovannucci, E., Hunter, D.J., Stampfer, M.J., Colditz, G.A., Willett, W.C., Major dietary patterns and the risk of colorectal cancer in women. Arch Intern Med, 2003. 163: 309-314. Gafa, R., Maestri, I., Matteuzzi, M., Santini, A., Ferretti, S., Cavazzini, L., Lanza, G., Sporadic colorectal adenocarcinomas with high-frequency microsatellite instability. Cancer, 2000. 89: 2025-2037. 94 101. 102. 103. 104. 105. 106. 107. 108. 109. 110. 111. 112. 113. 114. 115. 116. 117. 118. Literaturverzeichnis Gendler, S., Taylor-Papadimitriou, J., Duhig, T., Rothbard, J., Burchell, J., A highly immunogenic region of a human polymorphic epithelial mucin expressed by carcinomas is made up of tandem repeats. J Biol Chem, 1988. 263: 12820-12823. Gendler, S.J., MUC1, the renaissance molecule. J Mammary Gland Biol Neoplasia, 2001. 6: 339-353. Gendler, S.J., Lancaster, C.A., Taylor-Papadimitriou, J., Duhig, T., Peat, N., Burchell, J., Pemberton, L., Lalani, E.N., Wilson, D., Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem, 1990. 265: 15286-15293. Giacosa, A., Frascio, F., Munizzi, F., Epidemiology of colorectal polyps. Tech Coloproctol, 2004. 8: 243-247. Giorno, R., A comparison of two immunoperoxidase staining methods based on the avidin-biotin interaction. Diagn Immunol, 1984. 2: 161-166. Giovannucci, E., Modifiable risk factors for colon cancer. Gastroenterol Clin North Am, 2002. 31: 925-943. Giovannucci, E., Diet, body weight, and colorectal cancer: a summary of the epidemiologic evidence. J Womens Health (Larchmt), 2003. 12: 173-182. Girling, A., Bartkova, J., Burchell, J., Gendler, S., Gillett, C., Taylor-Papadimitriou, J., A core protein epitope of the polymorphic epithelial mucin detected by the monoclonal antibody SM-3 is selectively exposed in a range of primary carcinomas. Int J Cancer, 1989. 43: 1072-1076. Grizzle, W.E., Manne, U., Jhala, N.C., Weiss, H.L., Molecular characterization of colorectal neoplasia in translational research. Arch Pathol Lab Med, 2001. 125: 9198. Gruenberger, B., Tamandl, D., Schueller, J., Scheithauer, W., Zielinski, C., Herbst, F., Gruenberger, T., Bevacizumab, capecitabine, and oxaliplatin as neoadjuvant therapy for patients with potentially curable metastatic colorectal cancer. J Clin Oncol, 2008. 26: 1830-5. Gryfe, R., Swallow, C., Bapat, B., Redston, M., Gallinger, S., Couture, J., Molecular biology of colorectal cancer. Curr Probl Cancer, 1997. 21: 233-300. Guddo, F., Giatromanolaki, A., Patriarca, C., Hilkens, J., Reina, C., Alfano, R.M., Vignola, A.M., Koukourakis, M.I., Gambacorta, M., Pruneri, G., Coggi, G., Bonsignore, G., Depolarized expression of episialin (EMA, MUC1) in lung adenocarcinoma is associated with tumor progression. Anticancer Res, 1998. 18: 1915-1920. Hahn, M., Koufaki, O.N., Schackert, H.K., Molecular biology of colorectal cancer and clinical consequences for colorectal cancer syndromes. Langenbecks Arch Surg, 1998. 383: 389-396. Haier, J., Nasralla, M., Nicolson, G.L., Cell surface molecules and their prognostic values in assessing colorectal carcinomas. Ann Surg, 2000. 231: 11-24. Hamelin, R., Laurent-Puig, P., Olschwang, S., Jego, N., Asselain, B., Remvikos, Y., Girodet, J., Salmon, R.J., Thomas, G., Association of p53 mutations with short survival in colorectal cancer. Gastroenterology, 1994. 106: 42-48. Hanisch, F.G., Specificity clusters of MUC1-reactive mouse monoclonal antibodies. Tumour Biol, 1998. 19 111-117. Hanisch, F.G., Baldus, S.E., The Thomsen-Friedenreich (TF) antigen: a critical review on the structural, biosynthetic and histochemical aspects of a pancarcinomaassociated antigen. Histol Histopathol, 1997. 12: 263-281. Hanisch, F.G., Muller, S., MUC1: the polymorphic appearance of a human mucin. Glycobiology, 2000. 10: 439-449. Literaturverzeichnis 119. 120. 121. 122. 123. 124. 125. 126. 127. 128. 129. 130. 131. 132. 133. 134. 135. 136. 137. 95 Hanisch, F.G., Peter-Katalinic, J., Egge, H., Dabrowski, U., Uhlenbruck, G., Structures of acidic O-linked polylactosaminoglycans on human skim milk mucins. Glycoconj J, 1990. 7: 525-543. Hanisch, F.G., Stadie, T.R., Deutzmann, F., Peter-Katalinic, J., MUC1 glycoforms in breast cancer-cell line T47D as a model for carcinoma-associated alterations of 0glycosylation. Eur J Biochem, 1996. 236: 318-327. Hanisch, F.G., Uhlenbruck, G., Peter-Katalinic, J., Egge, H., Dabrowski, J., Dabrowski, U., Structures of neutral O-linked polylactosaminoglycans on human skim milk mucins. A novel type of linearly extended poly-N-acetyllactosamine backbones with Gal beta(1-4)GlcNAc beta(1-6) repeating units. J Biol Chem, 1989. 264: 872883. Hanski, C., Is mucinous carcinoma of the colorectum a distinct genetic entity? Br J Cancer, 1995. 72: 1350-1356. Hanski, C., Bornhoeft, G., Topf, N., Hermann, U., Stein, H., Riecken, E.O., Detection of a mucin marker for the adenoma-carcinoma sequence inhuman colonic mucosa by monoclonal antibody AM-3. J Clin Pathol, 1990. 43: 379-384. Hatfield, P., Sebag-Montefiore, D., The use of radiotherapy in rectal cancer. Scand J Surg, 2003. 92: 65-73. Hauser, H., Das kolorektale Karzinom-Epidemiologie, Präkanzerosen, Primär- und Sekundärprävention. J Gastroenterol 2004. 2: 6. Hayes, D.F., Zurawski, V.R., Jr., Kufe, D.W., Comparison of circulating CA15-3 and carcinoembryonic antigen levels in patients with breast cancer. J Clin Oncol, 1986. 4: 1542-1550. Heavey, P.M., McKenna, D., Rowland, I.R., Colorectal cancer and the relationship between genes and the environment. Nutr Cancer, 2004. 48: 124-141. Hecht, J.R., Patnaik, A., Berlin, J., Venook, A., Malik, I., Tchekmedyian, S., Navale, L., Amado, R.G., Meropol, N.J., Panitumumab monotherapy in patients with previously treated metastatic colorectal cancer. Cancer, 2007. 110: 980-8. Hermanek, P., Methodik der histopathologischen Untersuchung von Resektionen kolorektaler Karzinome. Chir Gastroenterol, 2000. 16: 255-259. Hermanek, P., Mansmann, U., Staimmer, D.S., Riedl, S., Hermanek, P., The German experience: the surgeon as a prognostic factor in colon and rectal cancer surgery. Surg Oncol Clin N Am, 2000. 9: 33-49. Hilkens, J., Ligtenberg, M.J., Vos, H.L., Litvinov, S.V., Cell membrane-associated mucins and their adhesion-modulating property. Trends Biochem Sci, 1992. 17: 359363. Hilkens, J., Wesseling, J., Vos, H.L., Storm, J., Boer, B., van der Valk, S.W., Maas, M.C., Involvement of the cell surface-bound mucin, episialin/MUC1, in progression of human carcinomas. Biochem Soc Trans, 1995. 23: 822-826. Hiraga, Y., Tanaka, S., Haruma, K., Yoshihara, M., Sumii, K., Kajiyama, G., Shimamoto, F., Kohno, N., Immunoreactive MUC1 expression at the deepest invasive portion correlates with prognosis of colorectal cancer. Oncology, 1998. 55: 307-319. Ho, J.J., Cheng, S., Kim, Y.S., Access to peptide regions of a surface mucin (MUC1) is reduced by sialic acids. Biochem Biophys Res Commun, 1995. 210: 866-873. Ho, S.B., Ewing, S.L., Montgomery, C.K., Kim, Y.S., Altered mucin core peptide immunoreactivity in the colon polyp-carcinoma sequence. Oncol Res, 1996. 8: 53-61. Ho, S.B., Niehans, G.A., Lyftogt, C., Yan, P.S., Cherwitz, D.L., Gum, E.T., Dahiya, R., Kim, Y.S., Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res, 1993. 53: 641-651. Hollingsworth, M.A., Swanson, B.J., Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer, 2004. 4: 45-60. 96 138. 139. 140. 141. 142. 143. 144. 145. 146. 147. 148. 149. 150. 151. 152. 153. 154. 155. Literaturverzeichnis Hong, J.C., Kim, Y.S., Alkali-catalyzed beta-elimination of periodate-oxidized glycans: a novel method of chemical deglycosylation of mucin gene products in paraffin embedded sections. Glycoconj J, 2000. 17: 691-703. Houlston, R.S., What we could do now: molecular pathology of colorectal cancer. Mol Pathol, 2001. 54: 206-214. Hounsell, E.F., Lawson, A.M., Stoll, M.S., Kane, D.P., Cashmore, G.C., Carruthers, R.A., Feeney, J., Feizi, T., Characterisation by mass spectrometry and 500-MHz proton nuclear magnetic resonance spectroscopy of penta- and hexasaccharide chains of human foetal gastrointestinal mucins (meconium glycoproteins). Eur J Biochem, 1989. 186: 597-610. Hsu, S.M., Raine, L., Fanger, H., A comparative study of the peroxidaseantiperoxidase method and an avidin-biotin complex method for studying polypeptide hormones with radioimmunoassay antibodies. Am J Clin Pathol, 1981. 75: 734-738. Hull, S.R., Bright, A., Carraway, K.L., Abe, M., Hayes, D.F., Kufe, D.W., Oligosaccharide differences in the DF3 sialomucin antigen from normal human milk and the BT-20 human breast carcinoma cell line. Cancer Commun, 1989. 1: 261-267. Hulsken, J., Behrens, J., Birchmeier, W., Tumor-suppressor gene products in cell contacts: the cadherin-APC-armadillo connection. Curr Opin Cell Biol, 1994. 6: 711716. Hulsken, J., Birchmeier, W., Behrens, J., E-cadherin and APC compete for the interaction with beta-catenin and the cytoskeleton. J Cell Biol, 1994. 127: 2061-2069. Itzkowitz, S.H., Bloom, E.J., Kokal, W.A., Modin, G., Hakomori, S., Kim, Y.S., Sialosyl-Tn. A novel mucin antigen associated with prognosis in colorectal cancer patients. Cancer, 1990. 66: 1960-1966. Itzkowitz, S.H., Bloom, E.J., Lau, T.S., Kim, Y.S., Mucin associated Tn and sialosylTn antigen expression in colorectal polyps. Gut, 1992. 33: 518-523. Itzkowitz, S.H., Yuan, M., Fukushi, Y., Palekar, A., Phelps, P.C., Shamsuddin, A.M., Trump, B.F., Hakomori, S., Kim, Y.S., Lewisx- and sialylated Lewisx-related antigen expression in human malignant and nonmalignant colonic tissues. Cancer Res, 1986. 46: 2627-2632. Itzkowitz, S.H., Yuan, M., Montgomery, C.K., Kjeldsen, T., Takahashi, H.K., Bigbee, W.L., Kim, Y.S., Expression of Tn, sialosyl-Tn, and T antigens in human colon cancer. Cancer Res, 1989. 49: 197-204. Izbicki, J.R., Hosch, S.B., Knoefel, W.T., Passlick, B., Bloechle, C., Broelsch, C.E., Extended resections are beneficial for patients with locally advanced colorectal cancer. Dis Colon Rectum, 1995. 38: 1251-1256. Jass, J.R., Do, K.A., Simms, L.A., Iino, H., Wynter, C., Pillay, S.P., Searle, J., Radford-Smith, G., Young, J., Leggett, B., Morphology of sporadic colorectal cancer with DNA replication errors. Gut, 1998. 42: 673-679. Jass, J.R., Roberton, A.M., Colorectal mucin histochemistry in health and disease: a critical review. Pathol Int, 1994. 44: 487-504. Jass, J.R., Walsh, M.D., Altered mucin expression in the gastrointestinal tract: a review. J Cell Mol Med, 2001. 5: 327-351. Jemal, A., Siegel, R., Ward, E., Murray, T., Xu, J., Thun, M.J., Cancer statistics, 2007. CA Cancer J Clin, 2007. 57: 43-66. Johns, L.E., Houlston, R.S., A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol, 2001. 96: 2992-3003. Jonker, D.J., O'Callaghan, C.J., Karapetis, C.S., Zalcberg, J.R., Tu, D., Au, H.J., Berry, S.R., Krahn, M., Price, T., Simes, R.J., Tebbutt, N.C., van Hazel, G., Wierzbicki, R., Langer, C., Moore, M.J., Cetuximab for the treatment of colorectal cancer. N Engl J Med, 2007. 357: 2040-8. Literaturverzeichnis 156. 157. 158. 159. 160. 161. 162. 163. 164. 165. 166. 167. 168. 169. 170. 171. 97 Kapadia, A., Feizi, T., Jewell, D., Keeling, J., Slavin, G., Immunocytochemical studies of blood group A, H, I, and i antigens in gastric mucosae of infants with normal gastric histology and of patients with gastric carcinoma and chronic benign peptic ulceration. J Clin Pathol, 1981. 34: 320-337. Kapiteijn, E., Marijnen, C.A., Nagtegaal, I.D., Putter, H., Steup, W.H., Wiggers, T., Rutten, H.J., Pahlman, L., Glimelius, B., van Krieken, J.H., Leer, J.W., van de Velde, C.J., Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer. N Engl J Med, 2001. 345: 638-646. Karner-Hanusch, J., Marian, B., Genderspezifische Aspekte bei kolorektalen Tumoren. Wien Med Wochenschr, 2006. 156: 541-544. Kemp, Z., Thirlwell, C., Sieber, O., Silver, A., Tomlinson, I., An update on the genetics of colorectal cancer. Hum Mol Genet, 2004. 13: 177-185. Kemperman, H., Wijnands, Y., Wesseling, J., Niessen, C.M., Sonnenberg, A., Roos, E., The mucin epiglycanin on TA3/Ha carcinoma cells prevents alpha 6 beta 4mediated adhesion to laminin and kalinin and E-cadherin-mediated cell-cell interaction. J Cell Biol, 1994. 127: 2071-2080. Kesse, E., Clavel-Chapelon, F., Boutron-Ruault, M.C., Dietary patterns and risk of colorectal tumors: a cohort of French women of the National Education System (E3N). Am J Epidemiol, 2006. 164: 1085-1093. Kinzler, K.W., Vogelstein, B., Lessons from hereditary colorectal cancer. Cell, 1996. 87: 159-170. Kitamura, H., Yonezawa, S., Tanaka, S., Kim, Y.S., Sato, E., Expression of mucin carbohydrates and core proteins in carcinomas of the ampulla of Vater: their relationship to prognosis. Jpn J Cancer Res, 1996. 87: 631-640. Klein, P.J., Osmers, R., Vierbuchen, M., Ortmann, M., Kania, J., Uhlenbruck, G., The importance of lectin binding sites and carcinoembryonic antigen with regard to normal, hyperplastic, adenomatous, and carcinomatous colonic mucosa. Recent Results Cancer Res, 1981. 79: 1-9. Klimpfinger, M., Hauser, H., Hermanek, P., Pathologie kolorektaler Präkanzerosen. Chirurgische Gastroenterologie, 1992. 8: 16-19. Kokawa, A., Kondo, H., Gotoda, T., Ono, H., Saito, D., Nakadaira, S., Kosuge, T., Yoshida, S., Increased expression of cyclooxygenase-2 in human pancreatic neoplasms and potential for chemoprevention by cyclooxygenase inhibitors. Cancer, 2001. 91: 333-338. Konings, E.J., Goldbohm, R.A., Brants, H.A., Saris, W.H., van den Brandt, P.A., Intake of dietary folate vitamers and risk of colorectal carcinoma: results from The Netherlands Cohort Study. Cancer, 2002. 95: 1421-1433. Kurosaka, A., Nakajima, H., Funakoshi, I., Matsuyama, M., Nagayo, T., Yamashina, I., Structures of the major oligosaccharides from a human rectal adenocarcinoma glycoprotein. J Biol Chem, 1983. 258: 11594-11598. Lan, M.S., Batra, S.K., Qi, W.N., Metzgar, R.S., Hollingsworth, M.A., Cloning and sequencing of a human pancreatic tumor mucin cDNA. J Biol Chem, 1990. 265: 15294-15299. Langner, C., Ratschek, M., Rehak, P., Schips, L., Zigeuner, R., Expression of MUC1 (EMA) and E-cadherin in renal cell carcinoma: a systematic immunohistochemical analysis of 188 cases. Mod Pathol, 2004. 17: 180-188. Larena, A., Vierbuchen, M., Schroder, S., Larena-Avellaneda, A., Hadshiew, I., Fischer, R., Blutgruppenantigenexpression in papillaren Karzinomen der Schilddruse. Eine immunhistochemische und klinische Studie uber das Vorkommen von Lewis-, ABO- und verwandten Antigenen. Langenbecks Arch Chir, 1996. 381: 102-113. 98 172. 173. 174. 175. 176. 177. 178. 179. 180. 181. 182. 183. 184. 185. 186. 187. 188. Literaturverzeichnis Laurent-Puig, P., Blons, H., Cugnenc, P.H., Sequence of molecular genetic events in colorectal tumorigenesis. Eur J Cancer Prev, 1999. 8: 39-47. Le Voyer, T.E., Sigurdson, E.R., Hanlon, A.L., Mayer, R.J., Macdonald, J.S., Catalano, P.J., Haller, D.G., Colon cancer survival is associated with increasing number of lymph nodes analyzed: a secondary survey of intergroup trial INT-0089. J Clin Oncol, 2003. 21: 2912-2919. Lee, I.M., Physical activity and cancer prevention-data from epidemiologic studies. Med Sci Sports Exerc, 2003. 35: 1823-1827. Li, A., Goto, M., Horinouchi, M., Tanaka, S., Imai, K., Kim, Y.S., Sato, E., Yonezawa, S., Expression of MUC1 and MUC2 mucins and relationship with cell proliferative activity in human colorectal neoplasia. Pathol Int, 2001. 51: 853-860. Ligtenberg, M.J., Buijs, F., Vos, H.L., Hilkens, J., Suppression of cellular aggregation by high levels of episialin. Cancer Res, 1992. 52: 2318-2324. Ligtenberg, M.J., Vos, H.L., Gennissen, A.M., Hilkens, J., Episialin, a carcinomaassociated mucin, is generated by a polymorphic gene encoding splice variants with alternative amino termini. J Biol Chem, 1990. 265: 5573-5578. Lloyd, K.O., Burchell, J., Kudryashov, V., Yin, B.W., Taylor-Papadimitriou, J., Comparison of O-linked carbohydrate chains in MUC-1 mucin from normal breast epithelial cell lines and breast carcinoma cell lines. Demonstration of simpler and fewer glycan chains in tumor cells. J Biol Chem, 1996. 271: 33325-33334. Longacre, T.A., Fenoglio-Preiser, C.M., Mixed hyperplastic adenomatous polyps/serrated adenomas. A distinct form of colorectal neoplasia. Am J Surg Pathol, 1990. 14: 524-537. Lothe, R.A., Peltomaki, P., Meling, G.I., Aaltonen, L.A., Nystrom-Lahti, M., Pylkkanen, L., Heimdal, K., Andersen, T.I., Moller, P., Rognum, T.O., Genomic instability in colorectal cancer: relationship to clinicopathological variables and family history. Cancer Res, 1993. 53: 5849-5852. Lowe, J.B., Stoolman, L.M., Nair, R.P., Larsen, R.D., Berhend, T.L., Marks, R.M., ELAM-1-dependent cell adhesion to vascular endothelium determined by a transfected human fucosyltransferase cDNA. Cell, 1990. 63: 475-484. Majuri, M.L., Mattila, P., Renkonen, R., Recombinant E-selectin-protein mediates tumor cell adhesion via sialyl-Le(a) and sialyl-Le(x). Biochem Biophys Res Commun, 1992. 182: 1376-1382. Marchesa, P., Lashner, B.A., Lavery, I.C., Milsom, J., Hull, T.L., Strong, S.A., Church, J.M., Navarro, G., Fazio, V.W., The risk of cancer and dysplasia among ulcerative colitis patients with primary sclerosing cholangitis. Am J Gastroenterol, 1997. 92: 1285-1288. Matsuda, K., Masaki, T., Watanabe, T., Kitayama, J., Nagawa, H., Muto, T., Ajioka, Y., Clinical significance of MUC1 and MUC2 mucin and p53 protein expression in colorectal carcinoma. Jpn J Clin Oncol, 2000. 30: 89-94. McArdle, C.S., Hole, D., Impact of variability among surgeons on postoperative morbidity and mortality and ultimate survival. Bmj, 1991. 302: 1501-1505. McLeod, H.L., Murray, G.I., Tumour markers of prognosis in colorectal cancer. Br J Cancer, 1999. 79: 191-203. Miyake, M., Kohno, N., Nudelman, E.D., Hakomori, S., Human IgG3 monoclonal antibody directed to an unbranched repeating type 2 chain (Gal beta 1-4GlcNAc beta 1-3Gal beta 1-4GlcNAc beta 1-3Gal beta 1-R) which is highly expressed in colonic and hepatocellular carcinoma. Cancer Res, 1989. 49: 5689-5695. Moertel, C.G., Fleming, T.R., Macdonald, J.S., Haller, D.G., Laurie, J.A., Tangen, C.M., Ungerleider, J.S., Emerson, W.A., Tormey, D.C., Glick, J.H., Veeder, M.H., Mailliard, J.A., Fluorouracil plus levamisole as effective adjuvant therapy after Literaturverzeichnis 189. 190. 191. 192. 193. 194. 195. 196. 197. 198. 199. 200. 201. 202. 203. 99 resection of stage III colon carcinoma: a final report. Ann Intern Med, 1995. 122: 321-326. Mommers, E.C., Leonhart, A.M., von Mensdorff-Pouilly, S., Schol, D.J., Hilgers, J., Meijer, C.J., Baak, J.P., van Diest, P.J., Aberrant expression of MUC1 mucin in ductal hyperplasia and ductal carcinoma In situ of the breast. Int J Cancer, 1999. 84: 466469. Moniaux, N., Escande, F., Porchet, N., Aubert, J.P., Batra, S.K., Structural organization and classification of the human mucin genes. Front Biosci, 2001. 6: 1192-1206. Morris, C.D., Armstrong, G.R., Bigley, G., Green, H., Attwood, S.E., Cyclooxygenase-2 expression in the Barrett's metaplasia-dysplasia-adenocarcinoma sequence. Am J Gastroenterol, 2001. 96: 990-996. Muller, S., Goletz, S., Packer, N., Gooley, A., Lawson, A.M., Hanisch, F.G., Localization of O-glycosylation sites on glycopeptide fragments from lactationassociated MUC1. All putative sites within the tandem repeat are glycosylation targets in vivo. J Biol Chem, 1997. 272: 24780-24793. Munemitsu, S., Albert, I., Souza, B., Rubinfeld, B., Polakis, P., Regulation of intracellular beta-catenin levels by the adenomatous polyposis coli (APC) tumorsuppressor protein. Proc Natl Acad Sci U S A, 1995. 92: 3046-3050. Mutsaers, J.H., van Halbeek, H., Vliegenthart, J.F., Wu, A.M., Kabat, E.A., Typing of core and backbone domains of mucin-type oligosaccharides from human ovarian-cyst glycoproteins by 500-MHz 1H-NMR spectroscopy. Eur J Biochem, 1986. 157: 139146. Nakamori, S., Ota, D.M., Cleary, K.R., Shirotani, K., Irimura, T., MUC1 mucin expression as a marker of progression and metastasis of human colorectal carcinoma. Gastroenterology, 1994. 106: 353-361. Nakayama, T., Watanabe, M., Katsumata, T., Teramoto, T., Kitajima, M., Expression of sialyl Lewis(a) as a new prognostic factor for patients with advanced colorectal carcinoma. Cancer, 1995. 75: 2051-2056. Nelson, H., Petrelli, N., Carlin, A., Couture, J., Fleshman, J., Guillem, J., Miedema, B., Ota, D., Sargent, D., Guidelines 2000 for colon and rectal cancer surgery. J Natl Cancer Inst, 2001. 93: 583-596. Nguyen, P.L., Niehans, G.A., Cherwitz, D.L., Kim, Y.S., Ho, S.B., Membrane-bound (MUC1) and secretory (MUC2, MUC3, and MUC4) mucin gene expression in human lung cancer. Tumour Biol, 1996. 17: 176-192. Nilsson, O.B., Karlsson, A., Petterson, L., Lindholm, L., Development of a new serological assay for determination of the MUC1 breast cancer associated antigen. Current tumor diagnosis: applications, clinical relevance, research trends, ed. Klapdor, R. 1994: W. Zuckerschwerdt Verlag, München, Bern, Wien, New York. Nishisho, I., Nakamura, Y., Miyoshi, Y., Miki, Y., Ando, H., Horii, A., Koyama, K., Utsunomiya, J., Baba, S., Hedge, P., Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science, 1991. 253: 665-669. Niv, Y., MUC1 and colorectal cancer pathophysiology considerations. World J Gastroenterol, 2008. 14: 2139-2141. O'Connell, M.J., Mailliard, J.A., Kahn, M.J., Macdonald, J.S., Haller, D.G., Mayer, R.J., Wieand, H.S., Controlled trial of fluorouracil and low-dose leucovorin given for 6 months as postoperative adjuvant therapy for colon cancer. J Clin Oncol, 1997. 15: 246-250. Ogata, S., Ho, I., Chen, A., Dubois, D., Maklansky, J., Singhal, A., Hakomori, S., Itzkowitz, S.H., Tumor-associated sialylated antigens are constitutively expressed in normal human colonic mucosa. Cancer Res, 1995. 55: 1869-1874. 100 204. 205. 206. 207. 208. 209. 210. 211. 212. 213. 214. 215. 216. 217. 218. 219. 220. 221. Literaturverzeichnis Ogata, S., Uehara, H., Chen, A., Itzkowitz, S.H., Mucin gene expression in colonic tissues and cell lines. Cancer Res, 1992. 52: 5971-5978. Orntoft, T.F., Harving, N., Langkilde, N.C., O-linked mucin-type glycoproteins in normal and malignant colon mucosa: lack of T-antigen expression and accumulation of Tn and sialosyl-Tn antigens in carcinomas. Int J Cancer, 1990. 45: 666-672. Park, S.Y., Lee, H.S., Choe, G., Chung, J.H., Kim, W.H., Clinicopathological characteristics, microsatellite instability, and expression of mucin core proteins and p53 in colorectal mucinous adenocarcinomas in relation to location. Virchows Arch, 2006. 449: 40-47. Parkin, D.M., Global cancer statistics in the year 2000. Lancet Oncol, 2001. 2: 533543. Pedersen, A., Johansen, C., Gronbaek, M., Relations between amount and type of alcohol and colon and rectal cancer in a Danish population based cohort study. Gut, 2003. 52: 861-867. Peifer, M., Cancer, catenins, and cuticle pattern: a complex connection. Science, 1993. 262: 1667-1668. Peipins, L.A., Sandler, R.S., Epidemiology of colorectal adenomas. Epidemiol Rev, 1994. 16: 273-297. Perçınel S, S.B., Ensarı A, Kuzu I, Kuzu MA, Bektaş M, Cetınkaya H, Kurşun N. , Mucins in the colorectal neoplastic spectrum with reference to conventional and serrated adenomas. Turk J Gastroenterol., 2007. 18: 230-238. Perez-Vilar, J., Hill, R.L., The structure and assembly of secreted mucins. J Biol Chem, 1999. 274: 31751-31754. Peterson, J.A., Ceriani, R.L., International workshop on monoclonal antibodies and breast cancer. San Francisco, California; November 8 and 9, 1984. Breast Cancer Res Treat, 1985. 5: 207-214. Petrakou, E., Murray, A., Price, M.R., Epitope mapping of anti-MUC1 mucin protein core monoclonal antibodies. Tumour Biol, 1998. 19: 21-29. Podolsky, D.K., Oligosaccharide structures of human colonic mucin. J Biol Chem, 1985. 260: 8262-8271. Podolsky, D.K., Oligosaccharide structures of isolated human colonic mucin species. J Biol Chem, 1985. 260: 15510-15515. Powell, S.M., Zilz, N., Beazer-Barclay, Y., Bryan, T.M., Hamilton, S.R., Thibodeau, S.N., Vogelstein, B., Kinzler, K.W., APC mutations occur early during colorectal tumorigenesis. Nature, 1992. 359: 235-237. Price, M.R., Rye, P.D., Petrakou, E., Murray, A., Brady, K., Imai, S., Haga, S., Kiyozuka, Y., Schol, D., Meulenbroek, M.F., Snijdewint, F.G., von MensdorffPouilly, S., Verstraeten, R.A., Kenemans, P., Blockzjil, A., Nilsson, K., Nilsson, O., Reddish, M., Suresh, M.R., Koganty, R.R., Fortier, S., Baronic, L., Berg, A., Longenecker, M.B., Hilgers, J., Summary report on the ISOBM TD-4 Workshop: analysis of 56 monoclonal antibodies against the MUC1 mucin. San Diego, Calif., November 17-23, 1996. Tumour Biol, 1998. 19: 1-20. Quasar Collaborative, G., Gray, R., Barnwell, J., McConkey, C., Hills, R.K., Williams, N.S., Kerr, D.J., Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet, 2007. 370: 2020-2029. Rasmussen, M., Kronborg, O., Fenger, C., Jorgensen, O.D., Possible advantages and drawbacks of adding flexible sigmoidoscopy to hemoccult-II in screening for colorectal cancer. A randomized study. Scand J Gastroenterol, 1999. 34: 73-78. Regimbald, L.H., Pilarski, L.M., Longenecker, B.M., Reddish, M.A., Zimmermann, G., Hugh, J.C., The breast mucin MUCI as a novel adhesion ligand for endothelial intercellular adhesion molecule 1 in breast cancer. Cancer Res, 1996. 56: 4244-4249. Literaturverzeichnis 222. 223. 224. 225. 226. 227. 228. 229. 230. 231. 232. 233. 234. 235. 236. 237. 101 Reid, M.E., Marshall, J.R., Roe, D., Lebowitz, M., Alberts, D., Battacharyya, A.K., Martinez, M.E., Smoking exposure as a risk factor for prevalent and recurrent colorectal adenomas. Cancer Epidemiol Biomarkers Prev, 2003. 12: 1006-1011. Riede, U.N., Werner, M., Schäfer H.E., Allgemeine und spezielle Pathologie. Thieme, 2004. 5: 719. Rose, M.C., Voynow, J.A., Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev, 2006. 86: 245-278. Rubio, C.A., Colorectal adenomas: time for reappraisal. Pathol Res Pract, 2002. 198: 615-620. Rubio, C.A., Jaramillo, E., Lindblom, A., Fogt, F., Classification of colorectal polyps: guidelines for the endoscopist. Endoscopy, 2002. 34: 226-236. Safi, F., Kohler, I., Rottinger, E., Beger, H., The value of the tumor marker CA 15-3 in diagnosing and monitoring breast cancer. A comparative study with carcinoembryonic antigen. Cancer, 1991. 68: 574-582. Sagara, M., Yonezawa, S., Nagata, K., Tezuka, Y., Natsugoe, S., Xing, P.X., McKenzie, I.F., Aikou, T., Sato, E., Expression of mucin 1 (MUC1) in esophageal squamous-cell carcinoma: its relationship with prognosis. Int J Cancer, 1999. 84: 251257. Saltz, L.B., Clarke, S., Diaz-Rubio, E., Scheithauer, W., Figer, A., Wong, R., Koski, S., Lichinitser, M., Yang, T.S., Rivera, F., Couture, F., Sirzen, F., Cassidy, J., Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol, 2008. 26: 2013-9. Sargent, D.J., Goldberg, R.M., Jacobson, S.D., Macdonald, J.S., Labianca, R., Haller, D.G., Shepherd, L.E., Seitz, J.F., Francini, G., A pooled analysis of adjuvant chemotherapy for resected colon cancer in elderly patients. N Engl J Med, 2001. 345: 1091-1097. Sauer, R., Fietkau, R., Wittekind, C., Rodel, C., Martus, P., Hohenberger, W., Tschmelitsch, J., Sabitzer, H., Karstens, J.H., Becker, H., Hess, C., Raab, R., Adjuvant vs. neoadjuvant radiochemotherapy for locally advanced rectal cancer: the German trial CAO/ARO/AIO-94. Colorectal Dis, 2003. 5: 406-415. Schmiegel, W., Pox, C., Adler, G., Fleig, W., Folsch, U.R., Fruhmorgen, P., Graeven, U., Hohenberger, W., Holstege, A., Junginger, T., Kuhlbacher, T., Porschen, R., Propping, P., Riemann, J.F., Sauer, R., Sauerbruch, T., Schmoll, H.J., Zeitz, M., Selbmann, H.K., S3-Leitlinienkonferenz "Kolorektales Karzinom" 2004. Z Gastroenterol, 2004. 42: 1129-1177. Schmoll, H.J., Neoadjuvante und adjuvante Therapiekonzepte beim Kolorektalkarzinom. Onkologie, 2000. 23: 35. Schroten, H., Hanisch, F.G., Plogmann, R., Hacker, J., Uhlenbruck, G., Nobis-Bosch, R., Wahn, V., Inhibition of adhesion of S-fimbriated Escherichia coli to buccal epithelial cells by human milk fat globule membrane components: a novel aspect of the protective function of mucins in the nonimmunoglobulin fraction. Infect Immun, 1992. 60: 2893-2899. Schwartz, B., Bresalier, R.S., Kim, Y.S., The role of mucin in colon-cancer metastasis. Int J Cancer, 1992. 52: 60-65. Siddiqui, J., Abe, M., Hayes, D., Shani, E., Yunis, E., Kufe, D., Isolation and sequencing of a cDNA coding for the human DF3 breast carcinoma-associated antigen. Proc Natl Acad Sci U S A, 1988. 85: 2320-2323. Slattery, M.L., Levin, T.R., Ma, K., Goldgar, D., Holubkov, R., Edwards, S., Family history and colorectal cancer: predictors of risk. Cancer Causes Control, 2003. 14: 879-887. 102 238. 239. 240. 241. 242. 243. 244. 245. 246. 247. 248. 249. 250. 251. 252. 253. 254. 255. Literaturverzeichnis Stenman, U.H., Heikkinen, R., Serum markers for breast cancer. Scand J Clin Lab Invest Suppl, 1991. 206: 52-59. Stepensky, D., Tzehoval, E., Vadai, E., Eisenbach, L., O-glycosylated versus nonglycosylated MUC1-derived peptides as potential targets for cytotoxic immunotherapy of carcinoma. Clin Exp Immunol, 2006. 143: 139-149. Stevens, T., Burke, C.A., Colonoscopy screening in the elderly: when to stop? Am J Gastroenterol, 2003. 98: 1881-1885. Stramignoni, D., Bowen, R., Atkinson, B.F., Schlom, J., Differential reactivity of monoclonal antibodies with human colon adenocarcinomas and adenomas. Int J Cancer, 1983. 31: 543-552. Strous, G.J., Dekker, J., Mucin-type glycoproteins. Crit Rev Biochem Mol Biol, 1992. 27: 57-92. Sturmer, T., Glynn, R.J., Lee, I.M., Manson, J.E., Buring, J.E., Hennekens, C.H., Aspirin use and colorectal cancer: post-trial follow-up data from the Physicians' Health Study. Ann Intern Med, 1998. 128: 713-720. Swallow, D.M., Gendler, S., Griffiths, B., Corney, G., Taylor-Papadimitriou, J., Bramwell, M.E., The human tumour-associated epithelial mucins are coded by an expressed hypervariable gene locus PUM. Nature, 1987. 328: 82-84. Sylvester, P.A., Myerscough, N., Warren, B.F., Carlstedt, I., Corfield, A.P., Durdey, P., Thomas, M.G., Differential expression of the chromosome 11 mucin genes in colorectal cancer. J Pathol, 2001. 195: 327-335. Takada, A., Ohmori, K., Takahashi, N., Tsuyuoka, K., Yago, A., Zenita, K., Hasegawa, A., Kannagi, R., Adhesion of human cancer cells to vascular endothelium mediated by a carbohydrate antigen, sialyl Lewis A. Biochem Biophys Res Commun, 1991. 179: 713-719. Takada, A., Ohmori, K., Yoneda, T., Tsuyuoka, K., Hasegawa, A., Kiso, M., Kannagi, R., Contribution of carbohydrate antigens sialyl Lewis A and sialyl Lewis X to adhesion of human cancer cells to vascular endothelium. Cancer Res, 1993. 53: 354361. Takayama, T., Miyanishi, K., Hayashi, T., Sato, Y., Niitsu, Y., Colorectal cancer: genetics of development and metastasis. J Gastroenterol, 2006. 41: 185-192. Takeuchi, H., Kato, K., Denda-Nagai, K., Hanisch, F.G., Clausen, H., Irimura, T., The epitope recognized by the unique anti-MUC1 monoclonal antibody MY.1E12 involves sialyl alpha 2-3galactosyl beta 1-3N-acetylgalactosaminide linked to a distinct threonine residue in the MUC1 tandem repeat. J Immunol Methods, 2002. 270: 199209. Tanimoto, T., Tanaka, S., Haruma, K., Yoshihara, M., Sumii, K., Kajiyama, G., Shimamoto, F., Kohno, N., MUC1 expression in intramucosal colorectal neoplasms. Possible involvement in histogenesis and progression. Oncology, 1999. 56: 223-231. Taylor-Papadimitriou, J., Report on the first international workshop on carcinomaassociated mucins. Int J Cancer, 1991. 49: 1-5. Taylor-Papadimitriou, J., Burchell, J., Miles, D.W., Dalziel, M., MUC1 and cancer. Biochim Biophys Acta, 1999. 1455: 301-313. Terry, M.B., Neugut, A.I., Bostick, R.M., Sandler, R.S., Haile, R.W., Jacobson, J.S., Fenoglio-Preiser, C.M., Potter, J.D., Risk factors for advanced colorectal adenomas: a pooled analysis. Cancer Epidemiol Biomarkers Prev, 2002. 11: 622-629. Terry, P., Giovannucci, E., Michels, K.B., Bergkvist, L., Hansen, H., Holmberg, L., Wolk, A., Fruit, vegetables, dietary fiber, and risk of colorectal cancer. J Natl Cancer Inst, 2001. 93: 525-533. Thibodeau, S.N., Bren, G., Schaid, D., Microsatellite instability in cancer of the proximal colon. Science, 1993. 260: 816-819. Literaturverzeichnis 256. 257. 258. 259. 260. 261. 262. 263. 264. 265. 266. 267. 268. 269. 270. 271. 103 Thomas, D.B., Karagas, M.R., Cancer in first and second generation Americans. Cancer Res, 1987. 47: 5771-5776. Tiemersma, E.W., Wark, P.A., Ocke, M.C., Bunschoten, A., Otten, M.H., Kok, F.J., Kampman, E., Alcohol consumption, alcohol dehydrogenase 3 polymorphism, and colorectal adenomas. Cancer Epidemiol Biomarkers Prev, 2003. 12: 419-45. Toft, N.J., Arends, M.J., DNA mismatch repair and colorectal cancer. J Pathol, 1998. 185: 123-129. Toribara, N.W., Roberton, A.M., Ho, S.B., Kuo, W.L., Gum, E., Hicks, J.W., Gum, J.R., Jr., Byrd, J.C., Siddiki, B., Kim, Y.S., Human gastric mucin. Identification of a unique species by expression cloning. J Biol Chem, 1993. 268: 5879-5885. Traverso, G., Shuber, A., Levin, B., Johnson, C., Olsson, L., Schoetz, D.J., Jr., Hamilton, S.R., Boynton, K., Kinzler, K.W., Vogelstein, B., Detection of APC mutations in fecal DNA from patients with colorectal tumors. N Engl J Med, 2002. 346: 311-320. Tveit, K.M., Radiotherapy in rectal cancer. Acta Oncol, 1999. 38: 5-6. Tveit, K.M., Guldvog, I., Hagen, S., Trondsen, E., Harbitz, T., Nygaard, K., Nilsen, J.B., Wist, E., Hannisdal, E., Randomized controlled trial of postoperative radiotherapy and short-term time-scheduled 5-fluorouracil against surgery alone in the treatment of Dukes B and C rectal cancer. Norwegian Adjuvant Rectal Cancer Project Group. Br J Surg, 1997. 84: 1130-1135. UICC, TNM Classification of malignant tumours. New York: John Wiley & Sons, 2002. Utsunomiya, T., Yonezawa, S., Sakamoto, H., Kitamura, H., Hokita, S., Aiko, T., Tanaka, S., Irimura, T., Kim, Y.S., Sato, E., Expression of MUC1 and MUC2 mucins in gastric carcinomas: its relationship with the prognosis of the patients. Clin Cancer Res, 1998. 4: 2605-2614. Van Cutsem, E., Peeters, M., Siena, S., Humblet, Y., Hendlisz, A., Neyns, B., Canon, J.L., Van Laethem, J.L., Maurel, J., Richardson, G., Wolf, M., Amado, R.G., Openlabel phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol, 2007. 25: 1658-64. Van Klinken, B.J., Dekker, J., Buller, H.A., Einerhand, A.W., Mucin gene structure and expression: protection vs. adhesion. Am J Physiol, 1995. 269: 613-627. van Klinken, B.J., Dekker, J., van Gool, S.A., van Marle, J., Buller, H.A., Einerhand, A.W., MUC5B is the prominent mucin in human gallbladder and is also expressed in a subset of colonic goblet cells. Am J Physiol, 1998. 274: 871-878. Verne, J.E., Aubrey, R., Love, S.B., Talbot, I.C., Northover, J.M., Population based randomized study of uptake and yield of screening by flexible sigmoidoscopy compared with screening by faecal occult blood testing. Bmj, 1998. 317: 182-185. von Mensdorff-Pouilly, S., Snijdewint, F.G., Verstraeten, A.A., Verheijen, R.H., Kenemans, P., Human MUC1 mucin: a multifaceted glycoprotein. Int J Biol Markers, 2000. 15: 343-356. Wagner, M., Adler, G., Seufferlein, T., Kolorektale Karzinome: Neue Entwicklungen in der Tumorprävention und in der Diagnostik der Tumorausbreitung. Chir Gastroenterol, 2005. 21: 109. Wandall, H.H., Hassan, H., Mirgorodskaya, E., Kristensen, A.K., Roepstorff, P., Bennett, E.P., Nielsen, P.A., Hollingsworth, M.A., Burchell, J., Taylor-Papadimitriou, J., Clausen, H., Substrate specificities of three members of the human UDP-N-acetylalpha-D-galactosamine:Polypeptide N-acetylgalactosaminyltransferase family, GalNAc-T1, -T2, and -T3. J Biol Chem, 1997. 272: 23503-23514. 104 272. 273. 274. 275. 276. 277. 278. 279. 280. 281. 282. 283. 284. 285. 286. Literaturverzeichnis Wei, E.K., Giovannucci, E., Wu, K., Rosner, B., Fuchs, C.S., Willett, W.C., Colditz, G.A., Comparison of risk factors for colon and rectal cancer. Int J Cancer, 2004. 108: 433-442. Weiss, A.A., Babyatsky, M.W., Ogata, S., Chen, A., Itzkowitz, S.H., Expression of MUC2 and MUC3 mRNA in human normal, malignant, and inflammatory intestinal tissues. J Histochem Cytochem, 1996. 44: 1161-1166. Wen, Y., Caffrey, T.C., Wheelock, M.J., Johnson, K.R., Hollingsworth, M.A., Nuclear association of the cytoplasmic tail of MUC1 and beta-catenin. J Biol Chem, 2003. 278: 38029-38039. Wesseling, J., van der Valk, S.W., Hilkens, J., A mechanism for inhibition of Ecadherin-mediated cell-cell adhesion by the membrane-associated mucin episialin/MUC1. Mol Biol Cell, 1996. 7: 565-577. Wesseling, J., van der Valk, S.W., Vos, H.L., Sonnenberg, A., Hilkens, J., Episialin (MUC1) overexpression inhibits integrin-mediated cell adhesion to extracellular matrix components. J Cell Biol, 1995. 129: 255-265. Wiley, E.L., Murphy, P., Mendelsohn, G., Eggleston, J.C., Distribution of blood group substances in normal human colon. Use of the unlabeled antibody (PAP) immunoperoxidase technic to identify A and B blood group substances. Am J Clin Pathol, 1981. 76: 806-809. Williams, S.J., McGuckin, M.A., Gotley, D.C., Eyre, H.J., Sutherland, G.R., Antalis, T.M., Two novel mucin genes down-regulated in colorectal cancer identified by differential display. Cancer Res, 1999. 59: 4083-4089. Wilmink, A.B., Overview of the epidemiology of colorectal cancer. Dis Colon Rectum, 1997. 40: 483-493. Winawer, S.J., Zauber, A.G., Ho, M.N., O'Brien, M.J., Gottlieb, L.S., Sternberg, S.S., Waye, J.D., Schapiro, M., Bond, J.H., Panish, J.F., et al., Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Workgroup. N Engl J Med, 1993. 329: 1977-1981. Winterford, C.M., Walsh, M.D., Leggett, B.A., Jass, J.R., Ultrastructural localization of epithelial mucin core proteins in colorectal tissues. J Histochem Cytochem, 1999. 47: 1063-1074. Wolf, B.C., D'Emilia, J.C., Salem, R.R., DeCoste, D., Sears, H.F., Gottlieb, L.S., Steele, G.D., Jr., Detection of the tumor-associated glycoprotein antigen (TAG-72) in premalignant lesions of the colon. J Natl Cancer Inst, 1989. 81: 1913-1917. Wolmark, N., Rockette, H., Fisher, B., Wickerham, D.L., Redmond, C., Fisher, E.R., Jones, J., Mamounas, E.P., Ore, L., Petrelli, N.J., et al., The benefit of leucovorinmodulated fluorouracil as postoperative adjuvant therapy for primary colon cancer: results from National Surgical Adjuvant Breast and Bowel Project protocol C-03. J Clin Oncol, 1993. 11: 1879-1887. Wolmark, N., Wieand, H.S., Hyams, D.M., Colangelo, L., Dimitrov, N.V., Romond, E.H., Wexler, M., Prager, D., Cruz, A.B., Jr., Gordon, P.H., Petrelli, N.J., Deutsch, M., Mamounas, E., Wickerham, D.L., Fisher, E.R., Rockette, H., Fisher, B., Randomized trial of postoperative adjuvant chemotherapy with or without radiotherapy for carcinoma of the rectum: National Surgical Adjuvant Breast and Bowel Project Protocol R-02. J Natl Cancer Inst, 2000. 92: 388-396. Woodward, M.P., Young, W.W., Jr., Bloodgood, R.A., Detection of monoclonal antibodies specific for carbohydrate epitopes using periodate oxidation. J Immunol Methods, 1985. 78: 143-153. Wreschner, D.H., Hareuveni, M., Tsarfaty, I., Smorodinsky, N., Horev, J., Zaretsky, J., Kotkes, P., Weiss, M., Lathe, R., Dion, A., et al., Human epithelial tumor antigen Literaturverzeichnis 287. 288. 289. 290. 291. 292. 293. 294. 295. 296. 297. 298. 105 cDNA sequences. Differential splicing may generate multiple protein forms. Eur J Biochem, 1990. 189: 463-473. Wu, K., Willett, W.C., Fuchs, C.S., Colditz, G.A., Giovannucci, E.L., Calcium intake and risk of colon cancer in women and men. J Natl Cancer Inst, 2002. 94: 437-446. Yamamoto, M., Bharti, A., Li, Y., Kufe, D., Interaction of the DF3/MUC1 breast carcinoma-associated antigen and beta-catenin in cell adhesion. J Biol Chem, 1997. 272: 12492-12494. Yang, J.M., Byrd, J.C., Siddiki, B.B., Chung, Y.S., Okuno, M., Sowa, M., Kim, Y.S., Matta, K.L., Brockhausen, I., Alterations of O-glycan biosynthesis in human colon cancer tissues. Glycobiology, 1994. 4: 873-884. Yashiro, M., Carethers, J.M., Laghi, L., Saito, K., Slezak, P., Jaramillo, E., Rubio, C., Koizumi, K., Hirakawa, K., Boland, C.R., Genetic pathways in the evolution of morphologically distinct colorectal neoplasms. Cancer Res, 2001. 61: 2676-2683. Yolken, R.H., Peterson, J.A., Vonderfecht, S.L., Fouts, E.T., Midthun, K., Newburg, D.S., Human milk mucin inhibits rotavirus replication and prevents experimental gastroenteritis. J Clin Invest, 1992. 90: 1984-1991. Yonezawa, S., Nakamura, T., Tanaka, S., Sato, E., Glycoconjugate with Ulex europaeus agglutinin-I-binding sites in normal mucosa, adenoma, and carcinoma of the human large bowel. J Natl Cancer Inst, 1982. 69: 777-785. Yoon, H., Benamouzig, R., Little, J., Francois-Collange, M., Tome, D., Systematic review of epidemiological studies on meat, dairy products and egg consumption and risk of colorectal adenomas. Eur J Cancer Prev, 2000. 9: 151-164. Yuan, M., Itzkowitz, S.H., Boland, C.R., Kim, Y.D., Tomita, J.T., Palekar, A., Bennington, J.L., Trump, B.F., Kim, Y.S., Comparison of T-antigen expression in normal, premalignant, and malignant human colonic tissue using lectin and antibody immunohistochemistry. Cancer Res, 1986. 46: 4841-4847. Yuan, M., Itzkowitz, S.H., Palekar, A., Shamsuddin, A.M., Phelps, P.C., Trump, B.F., Kim, Y.S., Distribution of blood group antigens A, B, H, Lewisa, and Lewisb in human normal, fetal, and malignant colonic tissue. Cancer Res, 1985. 45: 4499-4511. Zhang, K., Baeckstrom, D., Brevinge, H., Hansson, G.C., Secreted MUC1 mucins lacking their cytoplasmic part and carrying sialyl-Lewis a and x epitopes from a tumor cell line and sera of colon carcinoma patients can inhibit HL-60 leukocyte adhesion to E-selectin-expressing endothelial cells. J Cell Biochem, 1996. 60: 538549. Zotter, S., Lossnitzer, A., Hageman, P.C., Delemarre, J.F., Hilkens, J., Hilgers, J., Immunohistochemical localization of the epithelial marker MAM-6 in invasive malignancies and highly dysplastic adenomas of the large intestine. Lab Invest, 1987. 57: 193-199. Zrihan-Licht, S., Vos, H.L., Baruch, A., Elroy-Stein, O., Sagiv, D., Keydar, I., Hilkens, J., Wreschner, D.H., Characterization and molecular cloning of a novel MUC1 protein, devoid of tandem repeats, expressed in human breast cancer tissue. Eur J Biochem, 1994. 224: 787-795. 106 Abbildungsverzeichnis 7 Abbildungsverzeichnis ___________________________________________________________________________ Abbildung 1/1: Molekulargenetische Kolorektalkarzinogenese ______________________ 20 Abbildung 1/2: Schematische Darstellung der Kohlenhydratseitenketten der Muzine _____ 29 Abbildung 1/3: Primärstruktur von MUC1 ______________________________________ 34 Abbildung 2/1: Schema der LSAB-Methode _____________________________________ 49 Abbildung 3/1: Adenokarzinom des Dickdarms, Antikörper BCP8 ohne Vorbehandlung, x200 _____________________________________________________________________ 56 Abbildung 3/2: Adenokarzinom des Dickdarms, Antikörper BCP8 nach Perjodatbehandlung, x200 _____________________________________________________________________ 56 Abbildung 3/3: Adenokarzinom des Dickdarms, Antikörper 115D8 ohne Vorbehandlung, x200 _____________________________________________________________________ 56 Abbildung 3/4: Adenokarzinom des Dickdarms, Antikörper 115D8 nach Neuraminidasebehandlung, x200 __________________________________________________________ 56 Abbildung 3/5: Häufigkeitsverteilung (Mittelwerte) der Antikörper bei den „low-grade“Adenomen ________________________________________________________________ 58 Abbildung 3/6: Häufigkeitsverteilung (Mittelwerte) der Antikörper bei den „high-grade“Adenomen ________________________________________________________________ 58 Abbildung 3/7: Häufigkeitsverteilung (Mittelwerte) der Antikörper bei den Karzinomen _ 68 Tabellenverzeichnis 107 8 Tabellenverzeichnis ___________________________________________________________________________ Tabelle 1/1: Entartungsrisiko in Abhängigkeit der Adenomgröße _____________________ 9 Tabelle 1/2: pTNM-Klassifikation (UICC 2002) __________________________________ 11 Tabelle 1/3: Stadieneinteilung des kolorektalen Karzinoms nach Dukes (1932) _________ 12 Tabelle 1/4: Stadieneinteilung des kolorektalen Karzinoms (UICC 2002) ______________ 12 Tabelle 1/5: TNM-Stadium und Überlebensrate bei R0-Resektion des Primärtumors _____ 14 Tabelle 1/6: R-Klassifikation (UICC 2002) ______________________________________ 14 Tabelle 1/7: Histopathologisches Grading (UICC 2002) ____________________________ 14 Tabelle 1/8: Genetische Veränderungen in der Adenom-Karzinom-Sequenz ____________ 22 Tabelle 1/9: Mismatch-Repair-Gene ___________________________________________ 24 Tabelle 1/10: Anwendungsmöglichkeiten molekularer Marker _______________________ 25 Tabelle 1/11: Gewebsspezifisches Vorkommen der Muzine _________________________ 27 Tabelle 1/12: Glykanstrukturen in Oligosacchariden vom Muzin-Typ _________________ 30 Tabelle 1/13: Muzine _______________________________________________________ 31 Tabelle 2/1: Verdünnung der Primärantikörper ___________________________________ 52 Tabelle 3/1: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper HMFG2 in den Adenomgeweben ___________________________________________________________ 60 Tabelle 3/2: Lage- und Streumaße von HMFG2 in den kolorektalen „low-grade“-Adenomen _________________________________________________________________________ 60 Tabelle 3/3: Lage- und Streumaße von HMFG2 in den kolorektalen „high-grade“-Adenomen _________________________________________________________________________ 60 Tabelle 3/4: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper BCP8 in den Adenomgeweben ___________________________________________________________ 62 Tabelle 3/5: Lage- und Streumaße von BCP8 in den kolorektalen „low-grade“-Adenomen _________________________________________________________________________ 62 Tabelle 3/6: Lage- und Streumaße von BCP8 in den kolorektalen „high-grade“-Adenomen _________________________________________________________________________ 62 Tabelle 3/7: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper 115D8 in den Adenomgeweben ___________________________________________________________ 64 Tabelle 3/8: Lage- und Streumaße von 115D8 in den kolorektalen „low-grade“-Adenomen _________________________________________________________________________ 64 Tabelle 3/9: Lage- und Streumaße von 115D8 in den kolorektalen „high-grade“-Adenomen _________________________________________________________________________ 64 108 Tabellenverzeichnis Tabelle 3/10: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper MA695 in den Adenomgeweben ___________________________________________________________ 66 Tabelle 3/11: Lage- und Streumaße von MA695 in den kolorektalen „low-grade“-Adenomen _________________________________________________________________________ 66 Tabelle 3/12: Lage- und Streumaße von MA695 in den kolorektalen „high-grade“-Adenomen _________________________________________________________________________ 66 Tabelle 3/13: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper HMFG2 in den kolorektalen Karzinomen _________________________________________________ 69 Tabelle 3/14: Lage- und Streumaße von HMFG2 in den kolorektalen Karzinomen _______ 69 Tabelle 3/15: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper BCP8 in den kolorektalen Karzinomen ____________________________________________________ 70 Tabelle 3/16: Lage- und Streumaße von BCP8 in den kolorektalen Karzinomen _________ 70 Tabelle 3/17: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper 115D8 in den kolorektalen Karzinomen ____________________________________________________ 71 Tabelle 3/18: Lage- und Streumaße von 115D8 in den kolorektalen Karzinomen ________ 71 Tabelle 3/19: MUC1-Immunreaktivität und Farbintensität mit dem Antikörper MA695 in den kolorektalen Karzinomen ____________________________________________________ 73 Tabelle 3/20: Lage- und Streumaße von MA695 in den kolorektalen Karzinomen _______ 73 Lebenslauf 109 9 Lebenslauf ___________________________________________________________________________ Mein Lebenslauf wird aus Gründen des Datenschutzes in der elektronischen Fassung meiner Arbeit nicht veröffentlicht.