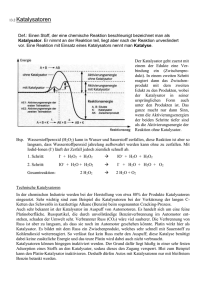
8. Enantioselektive Reaktionen an prochiralen Doppelbindungen
Werbung

8. Enantioselektive Reaktionen an prochiralen Doppelbindungen
8.1 . Enantioselektive Hydroborierungen
Eine der leistungsfähigsten Methoden zur Erzeugung chiraler sekundärer Alkohole ist heute
die enantioselektive Hydroborierung mit nachfolgender Oxidation. Die Hydroborierung läuft
in der Regel als reine syn-Addition an die Doppelbindung mit hoher Regiospezifität für das
sterisch weniger gehinderte C-Atom. Die Regioselektivitäten sind hierbei bis zu 80-100%.
Die syn:anti-Selektivitäten ca. 87:13 bis 97:3.
Eines der gebräuchlichsten Reagentien ist das Diisopinocampheylboran (Ipc)2BH. Es reagiert
sehr gut mit ungehinderten Z-Olefinen. Die E-Olefine reagieren langsamer und geben
schlechtere
Selektivitäten.
Trisubstituierte
Olefine
reagieren
auch
mit
schlechten
Selektivitäten.
B H
B2H6
2
R1 = R2 = Me, 98%ee
2
R1 = R2 = Et, 95%ee
R1 = iPr R2 = Me, 82%ee
1
2
R
R
R1
1) (-)-Ipc2BH
2) H2O2, NaOH
1) (-)-Ipc2BH
2) H2O2, NaOH
R1
OH
R2
R1
OH
Das Monoisopinocampheylboran ist wesentlich reaktiver und natürlich daher auch schlechtern
handzuhaben. Es zeigt auch moderate Selektivitäten bei E-Olefinen. Mit tribubstituierten
Olefinen ergeben sich aus den E-Olefinen die syn Produkte und aus den Z-Olefinen die anti
Produkte
BH2
E-Alkene
trisubstituierte Alkene
70 - 90 % ee
70 - 90 % ee
In dem vorliegenden Beispiel ist das Edukt erneut eine prochirale Substanz, die zwei
enantiotope Halbräume besitzt. Damit existieren, wenn ein chirales Reagenz angreift zwei
diastereomorphe ÜZ. Die Reaktion über den energetisch niedrigsten ÜZ ist wieder die
schnellere.
Ein gutes Hydroborierungsreagenz ist das S,S-2,5-Dimethylborolan
B
+
OH
B
H
Hier liegt im Fall der Hydroborierung erneut eine starke Reagenzkontrolle der
Stereoselektivität vor.
Diese
Hydroborierungen
können
auch
katalytisch
gestaltet
werden.
Als
Hydroborierungsreagenz setzt man dann meistens Catecholboran ein. Katalysiert wird mit RhDiphosphin Komplexen, wie z.B. mit BINAP. Die erreichten ee-Werte liegen bei um die 95%
ee.
PCy2
Josiphos
Cl
O
+
HB
O
Me
Fe PPh2
Cl
Rh, Chiraler Kat.
H2O2 / NaOH
HO
Ph
1) Catecholboran
2) H2O2 / NaOH
Ph
OH OH
Katalysator = Rh-BINAP
8.2.
Enantioselektive Hydrosilylierungen
Hydrosilylierungen können wie Hydroborierungen zur Synthese chiraler Alkohole verwendet
werden. Die Methode ist jedoch längst nicht so gut ausgearbeitet. Als Katalysatoren für
Hydrosilylierungen fungieren Rh, Pt und Pd-Katalysatoren. Grundsätzlich verlaufen
Hydrosilylierungen wie in den unteren Beispielen, in denen diastereoselektiv hydrosilyliert
wird.
OH
Tetramethylsilazan
OH
(Me2HSi)2NH
(Ph3P)3RhCl
OH
Tetramethylsilazan
(Me2HSi)2NH
Ph
Ph
H2PtCl6
HO
HO
OH
Für eine enantioselektive Reaktionsführung müssen erneut chirale Katalysatoren eingesetzt
werden.
Katalysator
H
1) HSiCl3, PdCl2, Katalysator
2) H2O/EtOH, Et3N, H2O2
OH
PPh2
Fe
"Tamao Oxidation"
NMeR
H
CH3
1) HSiCl3, PdCl2, Katalysator
2) H2O/EtOH, Et3N, H2O2
OH
"Tamao Oxidation"
Deratige Reaktionen lassen sich vorteilhaft auch mit Pd-Katalysatoren durchführen
PPh2
HO
O
OMe
PdCl2
O
Interessant die kürzlich ausgearbeiteten intramolekularen Möglichkeiten dieser Reaktion:
(-)-DIOP
RhC2H4Cl/2 (2mol%)
OSiHAr2
H2O2, KF, KHCO3
Rh-Chiraphos
H2O2
H
Si Ph
Ph
homo-Allylsilan
OH
OSiHAr2
Me
OH
OH
66% Ausbeute, 93%ee
Die Regioselektivität bei der Addition von Trichlorsilan an eine Doppelbindung ist
(verzweigt:linear) zwischen 80:20 und 93:7. Bei 1-Aryl-1-alkenen kann die Regioselektivität
auch 99:1 betragen. Die Enantioselektivitäten beragen ca. 71-85% ee.
Si-H
Pd.MOP
Ar
H
Ar
R
*
H2O2
R
H
Ar
*
R
OH
Si
MOP
PPh2
Eine interessante Anwendung der Reaktion:
HSiCl3
Pd-Katalysator
HO
Ph-CHO
DMF
SiCl 3
Ph
H
8.3
Enantioselektive Hydrierungen
Der 1966 von Wilkinson entwickelte Hydrierkatalysator [RhCl(PPh3)3] ermöglicht die
homogene Hydrierung aktivierter Doppelbindungen. Werden die PPh3-Gruppen durch chirale
Gruppen ersetzt so gelingt die enantioselektive homogene Hydrierung. Heute gibt es eine
vielzahl von chiralen Hydrierkatalysatoren mit denen aktivierte Doppelbindungen,
Carbonylgruppen und auch Imin enantioselektiv hydriert werden können.
Eine schnelle Hydrierung gelingt vor allem von α -(Acylaminoacrylsäuren). Diese
Verbindungen besitzen eine Acyl-geschützte Enamin-Substruktur, die für das Gelingen der
Hydrierung von entscheidender Bedeutung ist. Die enantioselektive Hydrierung dieser
Substanzen ermöglich die Synthese enantiomerenreiner α -Aminosäuren, was heute zu einer
Standardmethode geworden ist. Auf dieser Methode beruht z. B. die erste industrielle
assymetrische Synthese von (S)-DOPA.
O
O
COOH
NHAc
H2
L*RhI
O
O
COOH
NHAc
HO
HO
H
COOH
NH2
Ein weiteres Beispiel:
O
Ph
Ph
O
Ph
Ru (S)-BINAP
N
H
H2, 3 bar
COOH
Ph
N
H
COOH
Die enantioselektive Hydrierung von derartigen Doppelbindungen gelingt unter Verwendung
von Katalysatoren mit Bisphosphin Liganden und Rh, Ru als Metall. Die chirale Information
befindet sich hierbei nicht am Phosphor Atom. Bsp. der DIOP-Ligand.
O
PAr2
O
PAr2
(-)-DIOP
Allerdings ist mit dem obigen Katalysator das Substratspektrum der umsetzbaren Substanzen
sehr eng. So sind lediglich die erwähnten Substanzen und Zimtsäure-Derivate problemlos
umsetzbar. Das untenstehende Beispiel verdeutlich die Regioselektivitäten
COOCH3
COOCH3
NHCOCH3
NHCOCH3
Das untenstehende Beispiel erläutert die erreichbaren Selektivitäten mit den Rh-Katalysatoren
R2
CO2H
[Rh]-Katalysator
R2
CO2H
R1
Ph
H2, MeOH
R1
Ph
Fe
PPh2
Me
N
Ph2P Me
N
R1
Ph
CD3
Et
Me
R2
Me
Me
Me
Et
% ee
92
98
97
>94
Für eine brauchbare Synthese braucht man einem Enantiomerenüberschuß von mindestens
80% ee und eine anschließende Kristallisation um eine Steigerung auf > 98% ee zu erreichen.
Das ist mit der Hydrierung in der Regel zu erfüllen. Die Katalysatoren selber sind sehr
effizient. Man ereicht Substrat/Katalysator Verhältnisse von ca. 50.000.
Die Reaktionen lassen sich auch in überkritischem CO2 und in ionischen Flüssigkeiten
durchführen. Die Katalysatoren lassen sich auch an festen Trägern, wie z.B. Polystyrole,
immobilisieren um eine Rückgewinnung zu ermöglichen.
Die untenstehende Abbildung zeigt einige typische heute industriell angewendeten
Katalysatoren.
Der immer noch erfolgreichste Katalysatortyp fusst auf dem BINAP-Liganden, der von
Noyori entwickelt wurde. Die untenstehende Abbildung zeigt drei typische BINAPbasierende Katalysatoren und die Substrate, die mit Ihnen und Wasserstoff zu den
entsprechenden chiralen Produkten umgesetzt wurden. Das im ursprünglichen Wilkinson
Katalysator verwendetet Rh wurde durch Ru ersetzt. Diese Ru-Katalysatoren zählen heute zu
den erfolgreichsten Katalysator Systemen.
1995 wurde entdeckt, dass Ru-BINAP-Diamin Komplexe bevorzugt die Carbonylgruppe in
α , β -ungesättigten Carbonylgruppen reduzieren, was eine Plethora neuer chiraler Bausteine
für die Synthese zugänglich machte.
Verwendet man unterschiedliche Metalle, so kann es passieren, dass man die Konfiguration
des Liganden ändern muss um zu dem einen, gewollten Enantiomeren zu gelangen. Die
Acylaminogruppe ist hierbei für die Koordination an das Metallzentrum entscheidend.
COOR
R1
NHAc
R1
COOR
COOR
(S)-BINAP-Rh
R1
COOR
(S)-BINAP-Ru
R1
NHAc
NHAc
NHAc
R1 = Aryl
Die BINAP Katalysatoren können allerdings Doppelbindungsisomerisierungen einleiten, wie
das untenstehende Beispiel zeigt. So reagieren die Z-Substrate schnell und mit hoher
Enantioselektivität. Die E-Olfine reagieren hingegen langsam und geben significante E/Z
Isomerisierung während der Reaktion.
COOR
R1
(S)-BINAP-Rh+
NHAc
COOR
R1
NHAc
R1
COOR
(R)-BINAP-Rh+
R1
COOR
(S)-BINAP-Rh+
NHAc
NHAc
1
R = Aryl
Ein Katalysator, der sowohl E als auch die Z-Enamide zuverlässig ohne Isomerisierung
reduziert ist DuPhos (auch in alkoholischen Lösungsmittel). Hiermit reagieren auch die
β , β -disubstituierten Substrate glatt zu den β -verzweigten α -Aminosäuren. Wie man oben
sieht, lässt sich die Stereochemie am β -Zentrum durch die Konfiguration der Doppelbindung
im Substrat kontrollieren.
P
DuPhos-Ligand
P
Heute werden sehr viele Ruthenium-Katalysatoren verwendet. Vor allem werden diese
Katalysatoren für die enantioselektive Keton Reduktion eingesetzt. Neue Katalysatoren
verwenden auch Iridium!
Weniger aktivierte Doppelbindungen benötigen sehr viel drastischere Hydrierbedingungen
(S)-BINAP-Ru
H2, 100 atm
O
O
O
O
(S)-BINAP-Ru
H2, 100 atm
O
O
O
O
R3
COOH
R2
R1
(S)-BINAP-Ru
H2, 100 atm
COOH
R3
COOH
R
R1
**
2
(S)-BINAP-Ru
H2, 40 atm
O
COOH
O
(S)-Naproxen
Einfache β, γ - oder α, β -ungesättigte Carbonsäuren sind ebenso wie Allylalkohole, homoAllylalkohole oder nicht-aktivierte Doppelbindungen sehr schwierig zu reduzieren. Hier
fehlen die für die Koordination an das Metall nötigen funktionelle Gruppen. Dieses kann
erneut für regioselektive Hydrierungen ausgenutzt werden.
(S)-BINAP-Ru
COOH
COOH
(S)-BINAP-Ru
OH
OH
*R
R
Zr
O
O
65% ee mit R = H
Noyori synthetisierte später Katalysatoren mit Ruthenium, welche für Reaktionen mit
Carbonylgruppen sehr geeignet sind. Z.B. Ru-BINAP Biscarboxylatkomplex:
Ph Ph
O
P
Ru
P O
Ph Ph
Me
O
O
R
Ru-BINAP
O
HO
COOCH3
O
`R
O
COOCH3
OH OH
R``
`R
R``
Auch Imine lassen sich enantioselektiv hydrieren. Man erhält chirale Amine.
N
R1
NHCOPh
R2
[Rh], H2
iPrOH, 0°C
HN
R1
NHCOPh
NH2
SmI2
R2
R1
R1
Ph
CO2Et
CO2Et
CHex
R2
Me
Me
Ph
Me
% ee
92
89
91
72
R2
Allgemein haben die auf BINAP basierenden chiralen Katalysator bei der enantioselektiven
Hydrierung von Doppelbindungen den Vorteil relativ grosser Substratbreite. Dieser
Katalysator kann zur Hydrierung von Allylalkoholen, α -(Acylamin)-Acrylsäuren oder der
Carbonylgruppe eines β -Ketoesters verwendet werden.
Ph
Ph
P
Ru Cl
P Ph
Ph
Ru-BINAP
Neuerdings werden auch Iridiumkatalysatoren für die enantioselektive Hydrierung vor allem
von nicht-aktivierten Doppelbindungen entwickelt:
Angew. Chem. Int. Ed. 1998, 37, 2897
R1
R2
R1
H2
R2
HetAr
HetAr
O
R2P
N
Ir
8.4.
R
Weitere interessante Reaktionen an aktivierten Doppelbindungen
1. Michael Addition
Die Michael Reaktion ist von unschätzbarem Wert in der organischen Synthese. Besonders
weil das zunächst enstehende Addukt ein Enolat ist, welches weiterreagieren kann. Ist der
erste Schritt enantioselektiv kann auch der zweite diastereoselektiv verlaufen. So werden
gleich zwei neue Chiralitätszentren kreiert. Heute wird die selektive 1,4-Addition so erklärt,
dass Lithium oder Magnesium an den Sauerstoff des Michael-Akzeptors koordinieren. Cu
aber bildete als sehr weiches Metall zunächst einen d, π ∗ -Komplex mit dem π -System aus
und überträgt dann das „Carbanion“ auf die 4-Position. Während Organokupfer Reagentien in
der Regel effizient eine Michael-Addition eingehen, sind Organozink-Verbindungen hierzu
auf Grund der geringeren Reaktivität nicht in der Lage. In Gegenwart kleiner Mengen an Cu
oder Ni, sowie HMPA oder TMSCl findet aber eine Reaktion statt. Angenehm ist, dass diese
Zn-Verbindungen entweder aus Alkenen nach Hydroborierung und Bor Zn Austausch oder
aus den Grignard-Verbindungen leicht zugänglich gemacht werden können.
Ein gut funktionierender Katalysator, der exzellente enantioselektive Michael-Reaktionen
ermöglicht ist das von Feringa entwickelte Phosphoramidit. Es konnte gezeigt werden, dass
der matched-Fall Eintritt wenn (R,R)-Bis(1-phenylethyl)amin und (S)-2,2’-Binaphthol
kombiniert werden. Mit diesem Katalysator und Cyclohexenonen lassen sich ee-Werte von
93-98% realisieren. Addiert werden enantioselektiv Dialkylzink-Verbindungen in Gegenwart
kleiner Mengen an Cu(OTf)2.
Phosphoramidite
O
O
Ph
Katalysator
R2Zn +
Toluol, -30°C, 3-12h
R1
R1 R1
R1
O
P N
O
R
4mol%
Ph
93-98%ee
+ Cu(OTf)2 2mol%
Das intermediär gebildete Zn-Enolat kann durch Aldehyde abgefangen werden, wodurch
dann ein zweites Stereozentrum entsteht.
trans-cis
O
R2Zn +
OZnR
Katalysator
Toluol, -30°C, 3-12h
O
R1-CHO
H
trans-trans
O
OH
H
OH
+
R
R
R
trans-erythro
trans-threo
PCC
O
H
O
>91%ee
R
Die direkte Michael Addition von stabilisierten Kohlenstoffnukleophilen wie Malonate oder
Nitroalkane ist möglich, wenn der Katalysator zwei Eigenschaften besitzt. Zum Einen muss er
eine hohe Brønsted Basizität besitzen, damit das Enolat gebildet werden kann. Gleichzeitig
muss der Katalysator Lewis-sauer sein damit er die Carbonylkomponente aktivieren kann und
damit sich eine fester Komplex bilden kann aus dem heraus das Kohlenstoffnukleophil
enanatioselektiv übertragen werden kann.
Katalysatoren, die diese Anforderungen erfüllen sind die von Shibasaki eingeführten
bimetallischen Substanzen (bifunctional catalysis). Die untenstehende Tabelle gibt einen
Einblick in Reaktionsdauern und erzielte ee-Werte.
Der vorgeschlagene Katalysezyklus ist unten dargestellt.
Die zwei unten aufgeführen Beispiele sollen die Einsatzmöglichkeiten dieser Katalysatoren
verdeutlichen
2. Hydroformylierung:
Me
O
H2 / CO
180 atü
H
z. B. 70% ee
chiraler Rh-Katalysator
Der Mechanismus der Hydroformylierung:
H
H
L2 Rh
L2 Rh
CO
CO
CO
R
CO
R
L2 Rh
CO
CO
CO
L2 Rh
+
O
R
L2 Rh
O
H H
L2 Rh
R
R
L2
H
Rh
CO
O
H
R
3. Die katalytische enantioselektive Cyclopropanierung
Elektronenreiche Alkene und Diene wie z.B. Vinylether lassen sich gut enantioselektiv
cyclopropanieren. Nicht sehr effizient verläuft die Reaktion mit elektronenarmen Alkenen wie
z. B. α , β -ungesättigten Nitrilen oder Ketonen etc. Die Cyclopropanierung leitet sich im
Prinzip von der Simmons-Smith Reaktion her, bei der Alkene mit Methyliodid und Zn/Cu
umgesetzt werden. Hier werden eben sehr elektrophile Carbenspezies eingesetzt und der
Übergangszustand ist früh.
CH3-I
Zn/Cu
R2
R1
R1
R2
In der enantioselektiven Variante fungieren Cu-Bisoxazoline als Katalysatoren und
Verbindungen wie N2=CHCOOMe (Methyldiazoacetat) als Carbenquelle (allgemein:
Diazocarbonylverbindungen wie Diazoketone oder Diazoester). Intermediär scheinen sich
Metallcarbenkomplexe zu bilden, die als metallstabilisierte Carbokationen verstanden werden
können.
LnM C
N2 C
Z
MLn, -N2
Z
R
D
D
D
CZR
R
Diazoverbindung
MLn C
Z
LnM
R
R
Als Katalysatoren fungieren z.B. C2-symmerische Cu-Bisoxazolin Komplexe
O A
R'
R
A
N
O
N
Cu
L
R'
R
A = H oder Me
R' = H oder tBu oder Phenyl
R = Me, Ph, tBu, iPr
L
Betont werden muss, dass in dieser Reaktion die Diastereoselektivität nicht wirklich gut
kontrolliert werden kann. In der Regel liegen die trans:cis Verhältnisse der Produkte
zwischen 60:40 und 85:15. Die Wahl der Substituenten R hat auf dieses Verhältnis nur einen
kleinen Einfluss. Oft erhält man das trans-Produkt mit höherem Enantiomerenüberschuss.
Z
cis
N2=CHCOOMe
Kat*
Ph
trans
H
H
Ph
R
COOMe
Ph
R
H
R
COOMe
H
60%
40% 24% ee
>99% ee
Industrielle Anwendung der Cyclopropanierung
8.5.
Jacobsen-Katsuki Epoxidierung
Asymmetrische Epoxidierungen können prinzipiell mit chiralen Persäuren durchgeführt
werden. Allerdings sind die erzielten Enantiomerenüberschüsse in der Regel < 20% ee. Die
chirale Information ist scheinbar zu weit entfernt um in den diastereomorphen
Übergangszustand signifikante Energieunterschiede erzeugen zu können. Bessere Ergebniss
erzielt man mit chiralen Dioxiranen und Oxaziridinen. Hiermit sind ee-Werte von bis zu 73%
ee realisiert worden.
R2
R4
R1
R3
R1
R2
O
R2
[O]
R4
R1 R3
O
O
R1
2
R
O
NR
Chirale Oxaziridinium-Salze und chirale Dioxirane lassen sich auch vorteilhaft in situ
herstellen.
O
R2
1
R
R2*
[O]
3
O
*
O
O
N
R1 R3
R
O
O
BPh4
+ Oxone = Kaliumhydrogenpersulfat
KOOSO2OH
Sehr interessante Enantioselektivitäten erhält man mit dem aus der Fructose zugänglichen
Reagenz das unten gezeigt ist. Hier konkurriert allerdings die Epoxidierung der
Doppelbindung mit der Bayer-Villiger Oxidation des Reagenzes. Man muss deshalb basisch
arbeiten, um diese konkurrierende Reaktion zu unterdrücken.
O
O
+
Ph
O
O
O
Oxone, pH > 8
H2O, MeCN, -15-0°C
O
Ph
93% Ausbeute, 92% ee
In der Natur werden derartige Epoxidierungen enantioselektiv von Enzymen, sogenannten
Monooxidasen ausgeführt. Hier sind vor allem die Cyctochrom P450 Enzyme zu nennen, die
innerhalb eines Porphyrin-artigen Liganden ein Eisenatom komplexieren. Aus diesem Eisen
wird durch Sausterstoff eine Oxometall-Spezies erzeugt, die hervorragend Doppelbindungen
epoxidiert. Abgeleitet von den Enzymen wurden daher chirale Porphyrinliganden entworfen
und für Epoxidierungen genutzt. Als Metall wurde sowohl mit Fe eines als auch mit Mn und
einer externen Sauerstoffquelle, bei der es sich oft um das Iodosylbenzol oder -mesitylen
handelt gearbeitet. So lassen sich in der Tat enentioselektive Epoxidierungen mit moderaten
ee-Werten von 10-60% erzielen.
R*
O
O
NH
N
*R
R*
N
HN
I O
+
O
O
O
O
Iodosylmesitylen
N Cl
Mn N
ON
N
O
*R
R* wurde und wird verändert!
P450 Modelle!
O
O
Überbrücktes Porphyrin als P450 Modell
von Collman
Sehr effiziente Katalysatoren sind die von Jacobsen und Katsuki entwickelten SalenKomplexe die vor allem als Mn(III)-Komplex effiziente Katalysatoren sind.
R
A
N
1
N
Mn
A
R2
R
O
O
A
Cl
R3
+
B
NaOCl
R
R2
B
O
*
1
R
(0.5 - 8 mol%)
*
R3
bis zu 97% Ausbeute und 98+% ee
Ein sehr guter Katalysator zusammen mit der katalysierten Reaktion ist unten dargestellt.
H
H
* *
N
N
Mn
O
O
Cl
Ph
COOEt
NaOCl
8% Kat.
O
Ph
COOEt
er 98 : 2
JACS 1991, 113, 7063
Dieser Katalysator ist vor allem für die Epoxidierung von cis-di und trisubstituierte Olefine zu
empfehlen. Für tetrasubstituierte Olefine muss der Katalysator auf die vorhandenen Reste
abgestimmt werden. Katalysator
A
mit A = Me, B = Isobutyl, und R = Ph ist hier
empfehlenswert.
Mechanistisch verläuft die Epoxidierung im Fall konjugiert stabilisierter Olefine
möglicherweise radikalisch, was es dem Intermediat erlauben würde, durch eine
Bindungsrotation zu isomerisieren. Tatsächlich beobachtet man das. So enstehen aus den ZOlefinen oft Gemische. Bei niedriger Temperatur lässt sich die Rotation unterdrücken. In
Gegenwart von Ammoniumsalzen hingegen wird die Rotation beschleunigt und es entstehen
ausschliesslich die aus den E-Olefinen erwarteten Produkten. Diese Methode erlaubt daher
den selektiven Zugang zu enantiomerenreinen trans-Stilbenoxiden!
R
Ph
R
Mn(V)=O
O
Mn(IV)
Ph
R
Ph
O
R = Me oder H
R
Ph
R
O
Mn(IV)
Ph O
Beispiele für enantioselektive Epoxidierungen mit diesen Katalysatoren finden sich in der
untenstehenden Tabelle
Wohin geht die Reise bei der Epoxidierung?
Neben dem Erreichen höherer ee-Werte und dem Finden allgemein anwendbarer
Katalysatoren
werden
Verfahren
entwickelt,
in
denen
umweltfreundliche
Oxidationsreagentien wie O2 oder H2O2 eingesetzt werden können. Jacobsen und Que haben
Katalysatoren entwickelt, die nun sogar mit harmlosem Eisen operieren das ja auch von der
Natur verwendet wird. Diese Komplexe sind wahre P450 Modelle. Als Oxidationsmittel
konnte H2O2 eingesetzt werden. Beide Systeme sind aber noch in der Entwicklung. So sind
die ee’s im Fall des Que Katalysators gut, aber die Ausbeuten noch schlecht (35%). Der
gezeigte Jacobsen Katalysator ist noch achiral. (C&EN, 23. Juli 2001, Seite 9).
JACS 2001, 123, 7194
JACS 2001, 123, 6722
H2O2
C8H17
H3C
N
O
C8H17
Fe
C5H11
OH
H2O2
HO
85% Ausbeute
kein ee
CH3
N
N
H3C
H3C
N
N
N
Jacobsen Katalysator
CH3
N
Fe
C5H11
CH3
82% ee
aber nur 38% Ausbeute
N
H3C
CH3
Que Katalysator
8.6.
Enantioselektive cis-Hydroxylierung (B. Sharpless)
Die enantioselektive cis-Dihydroxylierung gehört heute zu den leistungsfähigsten Methoden
zur Funktionalisierung von nicht-aktivierten Doppelbindungen. Zwei Reaktionen, die
Sharpless-cis-Dihydroxylierung und die cis-Aminhydroxylierung erlauben es mit Hilfe
chiraler Hilfsstoffe, Doppelbindungen enantioselektiv zu funktionalisieren. Grundsätzlich
handelt es sich um die schon aus dem Grundstudium bekannte cis-Dihydroxylierung mit
Osmiumtetroxid. Schon Criegee beobachtete, dass die Reaktion in Gegenwart von Aminen
sehr stark beschleunigt wird. Diese Beobachtung nutzt das Sharpless-Verfahren aus. Die cisDihydroxylierung wir in Gegenwart chiraler Amine durchgeführt. Die Reaktion wird mit dem
schon in groben Zügen von Criegee vorgeschlagenen Mechanismus erklärt:
Mechanismus:
[3+2] Cycloaddition
R
O
O
Os
O
O
+ NR3
O
O
R
O
Os
R
O
NR3
O
O Os
O
O
NR3
OH
Hydrolyse
R
NMO oder Kaliumferricyanid
R
R
OH
+ OsO2
Criegee-Mechanismus, Liebigs Ann. Chem. 1936, 522, 75.
Frenking und Mitarbeiter konnten durch Quantenmechanische Berechnungen zeigen, dass ein
konzertierter [3+2] Mechanismus, statt des ebenfalls möglichen [2+2] Mechanismus
wahrscheinlich ist (Veldkamp. Frenking, J. Am. Chem. Soc. 1994, 116, 4937).
Im Liganden NR3 wird die chirale Information untergebracht. Das OsO4 sehr teuer und auch
giftig ist wird die Reaktion heute vorteilhaft katalytisch durchgeführt. Der Katalysator OsO4
wird durch entweder durch Zugabe von NMO (N-Methyl-morpholin-N-oxid) oder von
Kaliumferricyanid regeneriert
Die NR3 Ligandenoptimierung ist eine sehr wichtige Arbeit, die auch im Fall der cisDihydroxylierung eine Reihe von Liganden ergeben hat, die je nach Olefin Anwendung
finden. Alle chiralen Amine lassen sich jedoch im Fall dieser Reaktion auf die
Pseudoenantiomeren Dihydrochinidin (DHQD) und Dihydrochinin (DHQ) zurückführen. Die
Liganden unterscheiden sich durch die Art des Substituenten an der sekundären OH-Gruppe.
Die heute gebräuchlichsten Liganden sind die Dimeren [(DHQD)2-PHAL] und [(DHQ)2PHAL], oder seit neuestem auch die Anthrachinon (AQN)-verbrückten Liganden. Die AQNDimere eignen sich insbesondere für terminale Olefine und einige verzweigte aliphatische
Olefine.
R
O
N
H
O
O
N
R
H
H
O
H
N
N
Dihydrochinidin (DHQD)
quinidine
Dihydrochinin (DHQ)
quinine
N N
R
N N
ODHQD
R
ODHQ
R
ODHQ
PHAL-Liganden
R
ODHQD
O
O
AQN-Liganden O
O
Insgesamt wurden mehr als 250 Derivate der Alkaloide getestet.
Die Reaktion ist sehr leicht durchzuführen. Sie ist nicht sauerstoffenpfindlich und man
benötigt
sogar
etwas
Wasser
um
die
sonst
ratenbestimmende
Hydrolyse
des
Os(VI)monoglycolatesters zu beschleunigen. Tatsächlich scheint bei 1,2-disubstituierten
Olefinen und bei trisubstituierten Olefinen -nicht jedoch bei ungehinderten endständigen
Olefinen- die Hydrolyse des OsO4-Adduktes ratenbestimmend zu werden. In diesen Fällen
ann man 1 Eq. Methansulfonamid zur Reaktion zusetzen, was die Reaktion beschleunigt und
die ee’s verbessert.
Bei Verwendung der oben aufgeführten Katalysatoren müssen die zum Beispiel die folgenden
Reaktionsbedingungen eingehalten werden:
1 mmol Olefin in Aceton/H2O oder in t-Butanol/H2O lösen. 3 mmol Kaliumferricyanid
(Hiermit ist auch eine Elektrokatalyse möglich) oder 2 mmol NMO, sowie 3 mmol K2CO3
zugeben. 0.01 mmol Katalysator (DHQD)2-PHAL oder (DHQ)2-PHAL und Osmiumtetraoxid
als H4K2OsO6 (0.002 mmol) bei 0°C zugeben.
OH
HO
OK
Os
KO
OH
OH
Kaliumosmat-(VI)-dihydrat
Heute sind fertige Reagenzmischung käuflich erhältlich. Sie werden unter dem Namen ADMix- β bzw. AD-Mix- α verkauft.
Mit (DHQD)2-PHAL ≡
AD-mix- β
Der Angriff erfolgt von der β -Seite
Mit (DHQ)2-PHAL ≡
AD-mix- α
Der Angriff erfolgt von der α -Seite
Das Originalrezept für diese Mischungen (1 kg) lautet:
699.96g K3Fe(CN)6, 294g K2CO3, 5.22g (DHQD)2-PHAL oder (DHQ)2-PHAL, 0.52g
K2OsO2(OH)4. Will man etwas reaktivere Mischungen, so kann die Menge an K2OsO2(OH)4
auf 2.6 g um den Faktor 5 hochgesetzt werden.
Entscheidend bei der cis-Dihydroxylierung ist erneut die enorme Ligandenbeschleunigung,
die das Amin verursacht. Die Beschleunigung ist so gross, dass die nicht-enantioselektive
Reaktion des Olefins mit nicht-komplexiertem K2OsO2(OH)4 vernachlässigt werden kann.
Unter den Origonal Reaktionsbedingungen werden von 10.000 Molekülen trans-Stilben mit
Hilfe von 20 Molekülen OsO4 bei 96%ee ca. 9600 vom Aminkomplex cis-dihydroxyliert. Nur
400 werden direkt vom OsO4 angegriffen.
Mit dem unten stehenden Mnemonik ist es meist möglich die Seite von der das Reagenz das
Olefin angreift vorherzusagen. Bei der cis-Dihydroxylierung gilt jedoch, dass das je nach
Substitutionsmuster und nach der aktuellen Raumerfüllung der Reste durchaus auch
abweichende Selektivitäten beobachtet werden. An der exakten Bestimmung der
Produktkonfiguration geht also kein Weg vorbei. Das Olefin wird wie unten angegeben in
dem Schema plaziert.
Et
OR
N
H
O
HO
H
RS
N
RL
OH
H
RM
K2OsO2(OH)4
K3Fe(CN)6, K2CO3
t-BuOH/H2O 1:1 v/v
Et
N
RS
RM
H
HO
OH
RL
OR
H
H
O
N
Die Enantioselektivitäten sind rein empirisch abgeleitet, daher müssen Vorhersagen mit
Vorsicht genossen werden. Hier nur suggestiv.
Cis-Dihydroxylierung von Olefinklassen:
•
Monosubstituierte
Olefine
reagieren
gut
mit
guten
~70-90%
Enantiomerenüberschüssen
O
50-80% ee
O
50-80% ee
•
trans-disubstituierte Olefine, sind die besten Substrate. Es werden sehr gute
Enantiomerenüberschüsse erzielt und die Reaktionen verlaufen schnell.
´R
R``
O
O
COOEt
>90% ee
•
1,1`-Disubstituierte Olefine sind bislang eher sporadisch als Substrate verwendet
worden. Die erreichten Enantiomerenüberschüsse sind ordentlich, wenn sich die
Substituenten räumlich starlk voneinander unterscheiden.
•
Trisubstituierte
Olefine
werden
ebenfalls
zum
Teil
mit
sehr
guten
Enantiomerenüberschüssen Dihydroxyliert
•
cis-disubstituierte Olefine sind mit die am schwierigsten einsetzbaren Substrate. Die
errecihten Enantiomerenüberschüsse sind oftmals nur mässig (60-80% ee)
•
Tetrasubstituierte
Olefine,
z.B.
Silylenolether
geben
sehr
gute
Enantiomerenüberschüsse von bis zu 97% ee. Die Reaktionsgeschwindigkeiten
können niedrig sein, was die Verwendung von Rezepten mit bis zu 1% K2OsO2(OH)4,
5 mol% Ligand und bis zu 3 Eq. Methansulfonamid verlangt.
OTMS
R
OTMS
OH
HO
O
R
HO
R
Interessant sind heute auch die polymergebundenen Liganden, die sich leicht durch Filtartion
abtrennen lassen.
8.6.1 Doppelte Diastereoselektivität
Setzt man die die enantioselektive cis-Dihydroxylierung bereits ein chirales Olefin ein, so
wird aus der enantioselektiven Reaktion eine diastereoselektive Reaktion. Man erhält nun
jenachdem welchen chiralen Liganden man für die Reaktion wählt entweder den matched
Fall, in dem der dirigierende Einfluss des Substrates und der des chiralen Additivs gemeinsam
in die gleiche Richtung wirken und den mismatched Fall in dem entgegensteuernde Effekte
auftreten. Oft, aber nicht immer wird der dirigierende Einfluss des Substrates durch den
chiralen Liganden überschrieben (Reagenz- oder hier Ligandenkontrolle). Allerdings wird im
mismatched Fall die Ligandenkontrolle abgeschwächt, was zu schlechteren Selektivitäten
führt.
Um die Substratkontrolle abschätzen zu können setzt man das Substrat am besten zunächst
ohne die Zugabe des chiralen Amins um.
MeOOC
MeOOC
HO
BnO
BnO
O
BnO
BnO
BnO
O
BnO
MeO
OsO4
DHQD-CLB
DHQ-CLB
MeOOC
HO
OH
10.3
1.3
20.5
OH
O
BnO
BnO
BnO
OMe
:
:
:
OMe
1
1
1
OH
S
O
OH
Ph
OH
2.5
40
1
OsO4
DHQD-CLB
DHQ-CLB
:
:
:
OH
1
1
16
In allen solchen Fällen erhält man im matched Fall sehr viel höhere Diaseteroselektivitäten.
8.6.2 Folgereaktionen
Interessant sind die nach der cis-Dihydroxylierung möglichen Folgereaktionen. So lassen sich
die Diole sehr effizient in cyclische Sulfite umwandeln. Diese können -nach anfänglichen
Schwierigkeiten- heute effizient mit RuO4, oder RuO4/NaIO3 (Periodat) zu den cyclischen
Sulfaten aufoxisiert werden. Die cyclischen Sulfate verhalten sich wie Epoxide, d.h. die
können leicht durch viele Nukleophile unter Inversion der Konfiguration am betroffenen CAtom geöffnet werden
OH
R1
R2
OH
SOCl2
CCl4
O
S
R1
{
RuO4
O
R2
O
Cyclische Sulfite
NaIO3
RuCl3
CH3CN
H2O
O
R1
O
S
O
R2
O
Cyclische Sulfate
O
O
S
R1
O
R2
O
Nu: H-, F-, N3-, PhCOO-
O
Cycl. Sulfat
1
R
SCN-, PhCH2-, RNH2
R C CI-
SO3R2
OH
H2O
R2
R1
Nu
Nu
Interessant ist die folgende Variante:
Bei Umsetzung eines cyclischen Sulfates mit Benzamidin entsteht zunächst unter Inversion
der Konfiguration an beiden C-Atomen ein Imidazolin, welches zum Diamin hydrolysiert
werden kann.
O
O
O S
O
Ph
Ph
H2N
Ph
HN
NH
Ph
Benzamidin
N
NH2
AcOH HBr
Ph
Ph
Imidazolin
Ph
NH2
Ph
Die Synthese chiraler Epoxide aus den Diolen lässt ist auf zwei Wegen möglich. 2,3Dihydroxyester lassen sich selektiv an der 2-Position monosulfonylieren. In Gegenwart von
Base wird dann das Oxiran gebildet.
OH
COOMe
Ph
OH
ClSO2Ar
OH
COOMe
Ph
(OTs)OSO2Ar
K2CO3, MeOH
O
COOMe
Ph
Alternativ können die 1,2-Diole auch mit Trimethylorthoacetat in 1,3-Dioxolan-2yliumkationen überführt werden, aus denen durch einen nukleophilen Angriff mit Chlorid
Acetoxychloride gewonnen werden können. Diese reagieren in Anwesenheit von Base zu den
Epoxiden. Der nukleophile Angriff erfolgt im Fall von Arylolefinen an der benzylischen
Position. Im Fall aliphatischer Diole wird das Halogenatom an der stersich weniger
gehinderten Seite eingeführt.
OMe
OMe
OMe
Ph
OH
HO
+
Me
Cl
Me3SiCl
OAc
Ph
O
K2CO3
MeOH
Ph
Trimethylorthoacetat
K2CO3
MeOH
MeCOBr
Ph
O
Br
Ph
Nu
O
OAc
8.6.3. Sharpless Aminhydroxylierung
Die Aminhydroxylierung ist längst nicht so gut ausgearbeitet wir die cis-Dihydroxylierung.
Neben der Enantioselektivität gilt es nun auch noch die Regioselektivität in den Griff zu
bekommen. Grundsätzlich gilt. Das Amin wird an der β -Position eingeführt an der eine
negative Ladung am besten stabilisiert ist. β zum elektronenziehendsten Substituenten. Unten
ist eine Aminhydroxylierung gezeigt.
Ts
Ph
4% OsO4, 5% L*
TsNClNa, t-BuOCl
CO2Me
Ph
Angew. Chem. 1996, 108, 449
NH
O
OMe
OH
er 94.5 : 5.5
Die Abbildung unten verdeutlicht den Mechanismus.
O
Os
O
O
O
O
+ ClN-X
O
O
Os
L*
N-X
O
O
Os N-X
O
L*
R
R
R
O
O Os
N
O
L* X
R
R
-L*
+ ClN-X
O
O Os
N
O N
X
X
OH
Hydrolyse
R
R
R
OH
1
Die Hydrolyse des Intermediates 1 ist erneut ratenbestimmend, weshalb die Reaktion in
wässrigem Medium durchgeführt wird. Die Selektivitäten werden durch denselben
Sachzusammenhang wie bei der cis-Dihydroxylierung beschrieben. Es kann das gleiche
mnemotechnische Hilfsmittel verwendet werden. Problematisch ist auch die Tatsache, das
Intermediat 1 mit einem Alken zu einem Bisaddukt reagieren kann. Intermediat 1 verfügt aber
nicht mehr über den chiralen Amin-Liganden, weshalb die Weiterreaktion dann ohne
bemerkenswerte Induktion ablaufen würde. Die Hydrolyse von 1 in 50%-Wasser ist aber so
weit beschleunigt, dass dieser Nebenweg nicht beschritten wird. Die Aminhydroxylierung
konkurriert auch mit der cis-Dihydroxylierung. Die Aminquelle wird deshalb in grossem
Überschuss eingesetzt.
Als Quelle für den Stickstoff fungiert meistens ein Nitren aus einem Sulfonamid, Carbamat
oder Amid. Besonders beliebt ist das Chloramin-T oder Chloramin-M.
R
O
S
O
O
BnO
NClNa
R = p-Tol, Chloramin-T
R = Me, Chloramin-M
O
NClNa
H3C
NBrLi
O
TMS
O
NClNa
Eine Anwendung: Die Synthese von cis und trans α, β -Diaminocarbonsäuren (K. Janda, J.
Org. Chem. 1998, 63, 2045).
(DHQ)2PHAL (5mol%)
K2Os2(OH)4 (4mol%)
CbzNHCl (300mol%)
O
O
CbzHN
MeCN, H2O, 90%ee
O
CbzHN
O
O
O
OH
OMs
NaN3
Reduktion
Hydrolyse mit H+
Me3SiN3
t-BuOK, THF
Reduktion
Cbz O
N
Hydrolyse mit H+
H2N
O
O
OH
OH
NH2
cis
Ein weiteres Beispiel:
H2N
O
NH2
trans
1
2
(DHQ)2PHAL (5mol%)
K2Os2(OH)4 (4mol%)
BnOCONHCl (310mol%)
NHCbz
OH
MeCN, H2O, 90%ee
OH
NHCbz
1:1
RuCl3, H5IO6
oder
TEMPO, NaOCl
1 : 2 = 55 : 45, 1: 93%ee
NH2
O
O
NHCbz
OH
8.7.
Die Sharpless Epoxidierung
Die Sharpless Epoxidierung von Allyl- und homo-Allylalkoholen ist eine der verlässlichsten
asymmetrischen Synthesen. Benötigt wird ein Allyl- oder homo-Allylalkohole (die letzteren
geben
schlechtere
(Diethyltartrat
Ergebnisse),
DET
Alkylhydroperoxid
oder
(meist
ein
Ti(IV)-alkoxid,
Diisopropyltartrat
DIPT)
tert-Butylhydroperoxid,
einer
und
selten
chiraler
als
Weinsäureester
Sauerstoffquelle
Cumylhydroperoxid
ein
oder
Tritylhydroperoxid). Setzt man Molekularsieb zu, so kann die Reaktion katalytisch bezüglich
des Ti-Weinsäurekomplexes gefahren werden, was die direkte Weiterfunktionalisierung der
Produkte sehr erleichtert.
In der Reaktion werden ausschliesslich die Allylalkohol-Funktionen epoxidiert. Alle anderen
im Substrat vorhandenen Doppelbindungen incl. Allylether werden in der Reaktion nicht!
umgesetzt.
tert-BuOOH
Ti(OiPr)4, 3Å Molekularsieb
OH
HO
COOEt
HO
COOEt
O
OH
L-(+)-DET (6-12 mol%)
Die Selektivitäten in der Reaktion folgen meistens dem unten stehenden Schema. Ausnahmen
bilden Allylakohole mit chiralen Substituenten an C-1, C-2 und/oder C-3. In diesen Fällen
können unter Umständen andere Seitenpräferenzen auftreten. Wir der Allylalkohol wie im
unteren Schema in die Papierebene gelegt, so erfolgt die Sauerstoffübertragung mit D-(-)DET von oben mit L-(+)-DET von unten.
Die chirale Information, die sich möglicherweise im Substrat befindet, wird in der Sharpless
Epoxidierung durch das DET in der Regel überschrieben. Es gilt eine strenge Additivkontrolle
der Stereoselektivität. Verwendet amn Substrate mit einem weiteren Substituenten an C-1, so
erfolgt die Sauerstoffübertragung wie oben beschrieben. Allerdings befindet sich in einem
Fall (mismatched) der Substituent in Richtung O-Übertrageung, was die Geschwindigkeit der
Reaktion stark beeinträchtigt. Im matched Fall ist der Substituent auf der anderen Seite. In
diesem Fall wird mit der normalen Rate der Sauerstoff übertragen. Statt einer geändertern
Stereoselektivität beobachtet man also sehr stark unterschiedliche Raten.
Dieser Ratenunterschied kann so stark werden, dass das mismatched Substrat quasi gar nicht
umgesetzt werden kann. Dieses ermöglicht hervorragende kinetische Razematspaltungen wie
im unteren Beispiel verdeutlicht wird.
O
OH
OH
R
R
tert-BuOOH
Ti(OiPr)4, 3Å Molekularsieb
L-(+)-DET (6-12 mol%)
OH
OH
R
R
tert-BuOOH
Ti(OiPr)4, 3Å Molekularsieb
OH
R
D-(-)-DET (6-12 mol%)
O
OH
R
Eine erfolgreiche kinetische Razematspaltung hängt demnach in erster Linien von den
Ratenunterschieden ab mit denen die enantiomeren Allylalkohole reagieren. Diese
Ratenunterschiede werden durch die Art des Weinsäureestern massgeblich mitbestimmt. So
steigen die Ratenunterschiede in der Regel stark an wenn man vom Dimethyltartrat über das
Diethyltartrat zum Diisopropyltratrat geht, wie die tabellierten Beispiele zeigen. Das
Verhältnis der Raten kfast/kslow mit denen die beiden Enantiomeren reagieren nennt man auch
die relative Rate krel. Diese relative Rate hängt vom Umsatz und von der
Enantiomerenreinheit des verbliebenen Allylalkohols ab. Diese drei Parameter a) krel, b)
Umsatz und %ee des verbliebenen Allylalkohols hängen mathematisch voneinander ab. Eine
grafische Darstellung des Zusammenhanges gibt das untenstehende Diagramm. Deutlich zu
sehen ist, das relative Raten ab ca. 25 für eine gute kinetische Razematspaltung (50% Umsatz)
ausreichend sind.
S − Faktor = k rel =
k fast
> 25
k slow
relative Raten bei 0°C
DMT
DET
OH
DIPT
%ee
relative Rate bei -20°C
60
83
83
C6H13
OH
15
28
74
>96
104
38
60
96
>96
83
13
82
16
C6H9
OH
CH3
OH
C2H5
Die Sharpless Epoxidierung ist kompatibel mit einer grossen Zahl funktioneller Gruppen, was
ihren Wert als Methode in komplexen Naturstoffsynthesen enorm steigert. Sie kann
durchgeführt werden in Gegenwart von: Acetalen, Ketalen, Acetylenen, entfernt liegenden
Alkoholen und Phenole, Aldehyden, Amiden, Aziden, Estern und Carbonsäuren, Epoxiden,
Ethern, Mercaptanen und Thioether, Hydraziden, Ketonen, Nitrilen, Nitrogruppen, Olefinen,
Silylethern, Sulfonen, Sulfoxiden, Tetrazolen, Harnstoffen, Urethanen, Aminen, Phosphinen.
Allylalkohole selber sind gut zugängliche Ausgangsmaterialien. Sie lassen sich durch z.B.
Carbonylolefinierungen oder auch aus Propargylalkoholen gut aufbauen.
R
(COCl)2, DMSO
NEt3
OH
R
Horner-Emmons
(trans-selektiv)
O
R
R
DIBAL-H
H
COOR'
OH
E-Allylakohol
Still-Gennari
(cis-Selektiv)
R
R
DIBAL-H
COOR'
OH
Z-Allylakohol
R-X
+
Lindlar
H2, Pd/C, Chinolin
CH2OR
R
R
+ H2CO
R
OH
Z-Allylakohol
CH2OH
Birch, e- in NH3
Hydrozirkonierung
Hydroaluminierung
R
OH
E-Allylakohol
Anwendung der Sharpless Epoxidierung. Hier soll exemplarisch nur auf die Synthese von
Zuckern mit ihren vielen stereogenen Zentren aufmerksam gemacht werden. In der Tat
wurden mit dem dargestellten Synthesecyclus alle 8 möglichen L-Hexosen synthetisiert. Die
Synthese beginnt mit Benzyloxyacetaldehyd 1, welcher durch eine Wittig Reaktion und
nachfolgende Reduktion mit DIBAL-H in den Allylakohol 2 überführt wird. Es folgt eine
katalytische Sharpless Epoxidierung. Durch eine basenkatalysierte Payne-Umlagerung wird
ein Gleichgewicht zwischen den Epoxiden 3 und 4 eingestellt. Das primäre Epoxid wird nun
selektiv mit Phenylthiolat aus dem Gleichgewicht zu 5 entfernt. Das Thiophenolat reagiert
hierbei regioselektiv nur mit dem primären C-Atom des primären Epoxids. Das Diol 5 wird
nun nachfolgend als Acetonit zu 6 geschützt. Es folgt die Oxidation des Sulfids zum Sulfoxid
und eine Pummerer Umlagerung zum Acetoxythioacetal 7. Reduktive Hydrolyse zum
Aldehyd 8 ermöglicht die Wiederholung des ganzen Synthesezyclus bis zu den Hexosen.
CH3OP(O)CH2COOCH3
DIBAL-H
OBn
CHO
(+)-DET, Ti(OiPr)4,
tert-BuOOH
OBn
Wittig
1
2
O
Sharpless
OH
S
Abfangen des
primären Epoxides
OBn
S-Ph
O
8.7.
Schützen
S-Ph
6
4
Payne Umlagerung
Pummerer
Umlagerung
O
O
S-Ph
O
Methyl-Sulfoxid
OBn
H
O
O
O
O
HO
OBn
1) mCPBA
2) Ac2O
+
5
OBn
O
O
H
MeO
HO
HO
OH
3
OBn
OBn
OBn
H
S-Ph
OAc
OAc
O
O
OBn
H
O
S-Ph
O
O
O
OAc
1eq. DIBAL-H
H2O
S-Ph
7
OBn
O
O
CHO
8
Desymmetrisierungen
Alle oben genannten Reaktionen und Katalysatoren können auch verwendet werden um
symmetrische Verbindungen so zu desymmetrisieren, dass am Ende eine chirale Verbindung
gebildet wird. Hierbei geht man oft von meso-Verbindungen aus (haben eine Spiegelebene im
Molekül), die dann in das eine oder andere Enantiomere überführt werden.
So kann eine symmetrische Verbindung mit einer chiralen Substanz umgesetzt werden, wobei
zum Teil erhebliche Diastereoselektivitäten beobachtet werden.
OH
OTBS
TBSO
HOOC
+
O
O
O
O
O
15:1 dr, 75% Ausbeute
1) Ar*NH2
O
O
1) LiBH4
2) CH2N2
O
O
O CONHR*
2) H+, H2O
O
O
NH2
NHC5H11
Ar*NH2 =
Auch der Einsatz der oben beschriebenen Katalysatoren kann zum Erfolg führen. Unten sind
zwei Beispiele gezeigt, wie symmetrische Epoxide zu asymmetrsichen Verbindungen
geöffnet werden können.
R
TMS-N3
O
Kat.
R
R
t-Bu-SH
O
Kat.
R
R
N3
HO
N
R
OH
O
R
OH
Cr
O
Cl
O
O
S-tBu
R
OH
N
O
Ga O
Li
Lewis-Säure
Broensted-Base
Eine Anwendung dieser Reaktion:
O
O
HO
Shibasaki
O
S-tBu
S-tBu
O
1) SO3, Pyridin, DMSO
2) NaIO4
TBSO
TBSO
TBSO
TBSO
90% Ausbeute, 98%ee
Weitere Möglichkeiten bieten die Epoxidierung und die cis-Dihydroxylierung von Sharpless
(-)DIPT
Ti(OiPr)4
tert-BuOOH
OH
OH
O
OTES
OTES
K2OsO2(OH)4, K2CO3
K3Fe(CN)6, tBuOH/H2O
OH
OH
Zur Desymmetrisierung haben sich vor allem enzymatische Reaktionen wie z.B.
Veresterungen oder Esterspaltungen sehr bewährt.
HO
Candida antarctica Lipase
OH
HO
OAc
Ac2O, Et3N
COOMe
90% Ausbeute, >99%ee
COOMe
PLE, 93%ee
NHX
NHX
COOMe
COOH
OAc
OH
PLE, 93%ee
R
R
OAc
OAc
pro-S
HO
OH
HO
HO
PLE
OH
AcO
OH
OAc
78% Ausbeute, 100%ee
pro-R
Eine Anwendung der enzymatischen Desymmetrisierung
AcO
OAc
HO
Enzym, PPL
OAc
87% Ausbeute, 92% ee
N
HO
HO
HO
HO
NH2
O
OsO4
N
OH
HO HO
N
N
Aristeromycin
OAc
OH
Die besten Enzyme, sind billig, einsetzbar ohne besonderes biochemisches Equipment und
zeigen eine hohe Substratbbreite bei gleichbleibend hohem ee-Wert der Produkte. Heute
werden die Eigenschaften von Enzymen u.a. durch evolutive Methoden bzg. Der
Substratspezifität und dem erzielbaren ee-Wert optimiert.
Enzyme werden heute als zellfreie Pulver feilgeboten und so auch eingesetzt. Einige Enzyme
können hingegen nur in der Zelle funktionieren. Dann setzt man der Reaktion ganze Zellen
zu. Nun muss das Substrat durch die Zellmembran diffundieren können. Viele Enzyme
benötigen zusätzliche Cofaktoren für die chemische Tranformation. Setzt man der Reaktion
ganze Zellen zu, so sind die Cofaktoren in der Regel vorhanden. Bei Verwendung zellfreier
Enyzme müssen die Cofaktoren oft zugesetzt werden. In diesem Fall bereitet die
Cofaktoregenerierung manchmal Schwierigkeiten. Durch gekoppelte Enzymreaktionen lässt
sich der Cofaktoreinsatz dann vorteilhaft katalytisch gestalten.
Problematisch ist oft auch die mangelnde Löslichkeit der Substrate in Wasser. Hieraus erklärt
sich warum in der Chemie hauptsächlich Lipasen zum Einsatz kommen. Diese arbeiten auch
im Organismus an der Öl/Wasser Grenzschicht und sind daher in Gegenwart organischer
Lösungsmittel recht stabil.
Müssen andere Enzyme eingesetzt werden, so können diese an festen Trägern immobilisiert
werden.
Enantioselektive Hydrolysen lassen sich auch mit organischen Katalysatoren durchführen,
wie sie z.B von G. Fu entwickelt werden.
Katalysator, chirales DMAP
MeN
OH
HO
OH
Ac2O, NEt3, 0°C
AcO
N
Fe
Katalysator
8.8.
Enantioselektive Protonierungen und Deprotonierungen
Mit Hilfe von chiralen Lithiumbasen lassen sich auch prochirale acide H-Atome
stereoselektiv entfernen. So können mit Hilfe chiraler Basen symmetrische Epoxide in
Allylalkohole umgewandelt werden. Das Prinzip ist das Gleiche wie bei der Unterscheidung
von enantiotopen Seiten in einem Molekül. Durch Assoziation mit einem chiralen Reagenz
z.B. einer chiralen Base werden die enantiotopen Gruppen im Übergangszustand zu
diastereotopen Atomen und Gruppen.
H
N
N
H
BuLi
O
OH
97% Auseute ee > 90%
In tetradeuteriertem Cyclopentanon lassen sich selektiv die pro-S D-Atome durch H-Atome
austauschen
NMe2
O
D
D
D
D
O
NH2
D
H
H
D
Auch n-BuLi kann durch Zugabe der chiralen Base Spartein zu einer chiralen Base werden.
H
H
H
N H
Boc
Retention der Konfiguration
n-BuLi
(-)Spartein
E+
Li(Spartein)
N
Boc
n-BuLi
(-)Spartein
N
Boc
Me
Me
N
Boc
E = MeI,
Spartein
MeI
Me
N
Li(Spartein)
N
Boc
Me
Me
N
Boc
N
H
Mit Hilfe dieser chiralen Basen lässt sich selektiv der pro-R oder pro-S Wasserstoff an einer
Methylengruppe abstrahieren.
Cl
n-BuLi
(-)Spartein
O
H H
OH
Cl
H
O
Cl
MeI
OH
Li(Spartein)
O
OH
H Me
Retention
Auch die beiden prochiralen H-Atome von cis-2,6-Dimethylcyclohexanon lassen sich mit
einer chiralen Base differenzieren. Die beiden H-Atome werden also mit unterschiedlicher
Geschwindigkeit abstrahiert. Allerdings kann das Enolat von beiden Seiten durch das
Allylbromid angegriffen werden. Das führt zur Razemisierung.
H
O
Ha
Ph
N
Li
Ha'
O
Ph
65% Ausbeute, 25%ee
Br
H
Ph
N
Li
O
Ph
Br
Man kann das Enolat auch mit Trimethylsilylchorid abfangen. Dann erhält man bessere eeWerte.
N
O
Ph
N
H
N
Li
OSiMe3
Ph
73% Ausbeute, 96%ee
Me3SiCl
Nocheinmal zur Verdeutlichung: Enantiotope und Diastereotope Protonen
H
Enantiotop
H
H
O
Enantiotop
H
Diastereotop