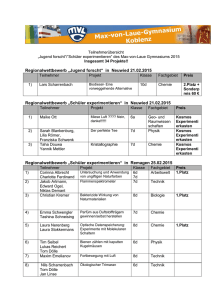
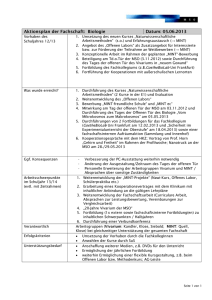
vts_8790_13108 - oparu
Werbung

Universitätsklinik Ulm Klinik für Innere Medizin I Endokrinologie, Diabetes und Stoffwechsel Sektionsleiter Endokrinologie: Prof. Dr. med. B. Böhm Ärztlicher Direktor: Prof. Dr. med. T. Seufferlein Proinsulin 74-90 basierende proteaseresistente Peptidliganden erhöhen die TGF-ß1 Sekretion in humanen peripheren mononukleären Zellen von Typ I Diabetes mellitus Patienten Dissertation zur Erlangung des Doktorgrades der Medizin der Medizinischen Fakultät der Universität Ulm Denise van Aalst Ulm 2013 Amtierender Dekan: Prof. Dr. Thomas Wirth 1. Berichterstatter: PD Dr. Timo Burster 2. Berichterstatter: Prof. Dr. Uwe Knippschild Tag der Promotion: 25.10.2013 I Inhaltsverzeichnis 1. Einleitung ................................................................................................. 1 1.1. Autoimmunität ............................................................................................ 1 1.2. Diabetes mellitus Formen .......................................................................... 1 1.2.1. Typ 1 Diabetes mellitus (T1DM) ................................................................ 2 1.2.2. Typ 2 Diabetes mellitus (T2DM) ................................................................ 3 1.2.3. Spezielle Diabetesformen .......................................................................... 3 1.2.4. Gestationsdiabetes .................................................................................... 3 1.3. Typ 1 Diabetes mellitus (T1DM) ................................................................ 4 1.3.1. Klinik .......................................................................................................... 4 1.3.2. Ätiologie ..................................................................................................... 5 1.3.3. Genetik und Vererbung .............................................................................. 7 1.3.4. Pathogenese des T1DM ............................................................................ 8 1.4. Das Immunsystem ..................................................................................... 9 1.4.1. Die angeborene und die adaptive Immunabwehr ...................................... 9 1.4.2. Regulatorische T-Zellen (Treg-Zellen) ....................................................... 13 1.4.3. Das angeborene Immunsystem: Antigenpräsentierende Zellen, Makrophagen und NK-Zellen ................................................................... 15 1.5. Major Histocompatibility Complex (MHC) ................................................ 16 1.5.1. MHC Klasse I Moleküle ........................................................................... 17 1.5.2. MHC Klasse II Moleküle .......................................................................... 17 1.5.3. MHC-Klasse III Moleküle ......................................................................... 18 1.5.4. T1DM-assozierte HLA-Moleküle .............................................................. 18 1.5.5. MHC Klasse II-Peptid-Komplex und T-Zell-Interaktion ............................ 19 1.5.6. MHC Klasse II Antigenprozessierung ...................................................... 19 1.6. Proinsulin ................................................................................................. 20 1.6.1. Proinsulinpeptid P17 ................................................................................ 21 1.6.2. Proinsulinprozessierung durch Cathepsine ............................................. 21 1.7. Modifizierte Peptidliganden zur Inhibierung von diabetogenen T-Zellen.. 23 1.7.1. Proteaseresistente Peptidliganden .......................................................... 24 1.8. Fragestellung ........................................................................................... 25 2. Material und Methoden .......................................................................... 26 2.1. Material .................................................................................................... 26 2.1.1. Materialien für ELISA ............................................................................... 26 II 2.1.2. Materialien für den T-Zell Assay .............................................................. 28 2.1.3. Fluorescence activated cell sorting (FACS)-Färbung und Analyse .......... 30 2.1.4. Verwendete Geräte.................................................................................. 31 2.2. Methoden................................................................................................. 32 2.2.1. Zellkultur/T-Zell Assay ............................................................................. 32 2.2.2. Lymphozyten-Aktivierung ........................................................................ 32 2.2.3. Cytokin-Detektion mittels ELISA .............................................................. 33 2.2.4. FACS-Färbung und Analyse .................................................................... 35 2.2.5. Signifikanzanalyse ................................................................................... 36 3. Ergebnisse ............................................................................................. 37 3.1. Vorarbeiten: Herstellung von P17-abgeleiteten proteaseresistenten Peptiden durch cleavage-site-directed amino-acid substitution (CS-DAS) ................................................................................................................. 37 3.2. Cytokin-Detektion mittels ELISA .............................................................. 39 3.3. Ergebnisse ............................................................................................... 40 3.4. Intrazelluläre Cytokinbestimmung durch FACS-Analyse ......................... 48 4. Diskussion ............................................................................................. 61 4.1. Vorarbeiten: Herstellung von P17 proteaseresistenten Peptiden durch cleavage-site-directed amino-acid substitution (CS-DAS) ....................... 62 4.2. T-Zell Assay und Cytokin-Detektion mittels ELISA .................................. 63 4.3. Intrazelluläre Cytokinbestimmung durch FACS-Analyse ........................ 64 4.4. Regulatorische T-Zellen (Treg-Zellen) ....................................................... 65 4.5. Perspektive .............................................................................................. 67 5. Zusammenfassung ................................................................................ 68 6. Literaturverzeichnis............................................................................... 69 7. Abbildungsverzeichnis ......................................................................... 75 8. Tabellenverzeichnis............................................................................... 77 9. Danksagung ........................................................................................... 78 10. Curriculum Vitae .................................................................................... 79 III Abkürzungsverzeichnis A AEP Asparagin-Endoprotease Anti-IA-2-AK Anti-Thyrosinphosphatase-2-Antikörper APCs Antigenpräsentierende Zellen APLs engl. altered peptide ligands (Veränderte Peptidliganden) B B-Zelle Bursa Fabrici Zelle BFA Brefeldin A BSA Bovines Serumalbumin C Cat Cathepsin CD engl. cluster of differentiation CLIP Klasse II-assoziiertes-invariante-Ketten-Peptid CS-DAS Cleavage site-directed amino acid substitution (Schnittstellen-spezifische Aminosäuren Substitution) CTLs Cytotoxische T-Lymphozyten CTLA-4 Cytotoxisches T-Lymphozyten-Antigen 4 D DMSO Dimethylsulfoxid E EAD engl. experimental autoimmune diabetes ELISA engl. enzyme linked immunosorbent assay (Enzymgekoppelter Immunadsorptionstest) ER Endoplasmatisches Retikulum F FACS engl. fluorescence activated cell sorting (Durchflusszytometrie) FoxP3 Forkhead Box P3 G GAD Glutamatdecarboxylase GADA Anti-Glutamatdecarboxylase-Antikörper GITR engl.glucocorticoid-induced TNFR-related protein (Glukokortikoid-induziertes TNFR-gebundenes Protein) IV H HBA1c Glykosyliertes Hämoglobin A1 HLA Humanes Leukozyten Antigen HPLC engl. high performance liquid chromatography (Hochleistungsflüssigkeitschromatographie) HRP engl. horseradish peroxidase (Meerrettichperoxidase) HWZ Halbwertzeit I IAA Insulin-Autoantikörper IA2 Inselzellantigen 2 ICA Cytoplasmatische Inselzellantikörper ICOS engl. inducible costimulator IFN-γ Interferon-γ IL Interleukin K KHK Koronare Herzkrankheit L LADA engl. latent autoimmune diabetes with onset in adults LC engl. liquid chromatography (Flüssigkeitschromatographie) li invariante Kette M M Mol m Milli (10-3) µ Mikro (10-6) MHC engl. major histocompatibility complex (Haupthistokompatibilitätskomplex) Min Minute MODY engl. maturity onset diabetes of the young N NK-Zelle Natürliche Killerzelle NOD engl. non-obese-diabetic P P Piko (10-12) P17 Proinsulin Peptid 17 V pAVK Periphere arterielle Verschlusskrankheit PBMC engl. peripheral blood mononuclear cells (mononukleäre Zellen des peripheren Blutes) PBS engl. phosphate buffer saline (Isotonischer Phosphatpuffer) PD-1 engl. programmed cell death-1 PKC Proteinkinase C R RT Raumtemperatur T T1DM Typ 1 Diabetes mellitus TCR T-Zellrezeptor TGF-ß engl. transforming growth factor ß (Transformierender Wachstumsfaktor ß) TH T-Helferzelle TNF-α Tumornekrosefaktor α Treg-Zelle Regulatorische T-Zelle T-Zelle T-Lymphozyt U U/min Umdrehungen pro Minute UV Ultraviolett W WT Wildtyp 1. Einleitung 1.1. Autoimmunität Bei Autoimmunerkrankungen wie dem Typ 1 Diabetes mellitus (T1DM) handelt es sich um Immunreaktionen gegen körpereigene Proteine durch sogenannte autoreaktive T-Zellen. Allerdings können autoreaktive T-Zellen nicht nur bei Autoimmunerkrankten sondern auch bei Gesunden auftreten. Man unterscheidet zwei Formen: Die lokale Gewebsschädigung, bei der sich die Immunreaktion gegen ein spezielles Organ richtet und die Multisystemerkrankung, bei der verschiedene Autoantikörper und Reaktionen gegen verschiedene Organe des Körpers gerichtet sind [90]. Gemeinsam ist ihnen der Verlust der Selbsttoleranz [5]. Für die Aufrechterhaltung der Selbsttoleranz ist immer ein ausgeglichenes Verhältnis an antiinflammatorischen bzw. regulierenden Mechanismen gegenüber einem proinflammatorischen Milieu notwendig. Eine Erklärung für die Entwicklung von Autoimmunität könnte ein Ungleichgewicht zwischen beiden Mechanismen sein, die eine autoreaktiver Abnahme und der regulatorischen proinflammatorischer Mechanismen Mechanismen zugunsten beinhaltet [4]. Das Erkrankungsbild des T1DM erscheint zwar abrupt, allerdings resultiert das Erkrankungsbild aus einer jahrelangen Autoimmunattacke gegen ß-Zellen [90]. Somit ist die Autoimmunität und die autoimmunvermittelte Zerstörung der ß-Zellen eine Ursache des T1DM. 1.2. Diabetes mellitus Formen Diabetes mellitus bezeichnet eine Gruppe von chronischen Stoffwechselerkrankungen, die mit einer Fehlregulation der Kohlenhydrate und Proteine einhergehen. Ihre Gemeinsamkeit besteht in der Hyperglykämie, basierend auf einem absoluten oder relativen Insulinmangel. Eine Einteilung der verschiedenen Diabetesformen erfolgt anhand der Klassifikation der American Diabetes Association nach ihrer Ätiologie [33]. Man unterscheidet folgende vier Diabetesformen: 1.2.1. Typ 1 Diabetes mellitus (T1DM) Beim T1DM handelt es sich um eine chronische Stoffwechselerkrankung, bei der die pankreatischen insulinproduzierenden ß-Zellen selektiv eliminiert werden [52]. Dabei spielen autoreaktive T-Lymphozyten (T-Zellen), welche ßZellen direkt oder indirekt zerstören eine wichtige Rolle in der Entwicklung und im Fortschreiten der Erkrankung [11]. Die Konsequenz der ß-Zell-Destruktion ist ein absoluter Insulinmangel. Der T1DM wird somit auch als insulinabhängiger Diabetes bezeichnet. 5-10% aller Diabetiker sind T1DMDiabetiker. Individuen mit T1DM entwickeln eine Hyperglykämie und können aufgrund des Mangels an Insulin Diabetes-assoziierte Komplikationen, wie Mikro-und Makroangiopathien der Gefäße, Nieren, Augen und Nerven entwickeln [90]. Man unterscheidet 2 Formen des T1DM: IA: Immunologisch bedingter T1DM (T1DM IA) Hierbei handelt es sich um eine chronische Autoimmunreaktion durch Infiltration von autoreaktiven Zellen in den Pankreas. Dort können autoreaktive T-Zellen entzündliche Infiltrat insulinproduzierende besteht aus ß-Zellen autoreaktiven zerstören. CD4+ Das T-Zellen, Makrophagen und CD8+ T-Zellen. Zu einer Manifestation des T1DM kommt es erst, wenn mehr als 90% der ß-Zellen zerstört sind. Der Nachweis von Autoantikörpern gegen Inselzellantigene zeigt ein erhöhtes Risiko für T1DM auf. Sie können bei 80% der T1DM Patienten mit Neumanifestation nachgewiesen werden. Es handelt sich um folgende Autoantikörper (AutoAK): Zytoplasmatische Inselzellantikörper (ICA) gegen Ganglioside, AntiGlutmatdecarboxylase-AK (GADA), Anti-Thyrosinphosphatase-2-AK (AntiIA-2-AK) und Insulin-Auto-AK (IAA) gegen Proinsulin [6, 22]. Man unterscheidet auch zwischen einer schweren insulinpflichtigen Form im Kindes- und Jugendalter und einer milderen, initial nicht-insulinpflichtigen Form im Erwachsenenalter, dem latent autoimmune diabetes with onset in adults (LADA). Bei diesem tritt der Insulinmangel erst verzögert auf. Das Manifestationsalter dieser Form liegt erst im Erwachsenalter (25.40.Lebensjahr) [85]. IB: Idiopathisch bedingter T1DM (T1DM IB) Darunter versteht man ebenfalls eine Zerstörung der ß-Zellen des Pankreas, jedoch ist hier der auslösende Faktor für die Zerstörung der Insulin produzierenden ß-Zellen nicht bekannt [33]. 1.2.2. Typ 2 Diabetes mellitus (T2DM) Im Gegensatz zum T1DM ist der T2DM charakterisiert durch eine gestörte Insulinfreisetzung der ß-Zellen und eine Insulinresistenz des peripheren Gewebes. Letztere scheint ein Hauptfaktor in der Entwicklung des T2DM zu sein. Die Insulinresistenz ist häufig gebunden an eine Adipositas, die in den ß-Zellen Stress verursacht und einen Hyperinsulinismus zur Folge hat. Des Weiteren sind genetische Faktoren für die Entwicklung des T2DM mitverantwortlich. Bei der Entwicklung des T2DM handelt es sich um einen multifaktoriellen Vorgang [33]. 1.2.3. Spezielle Diabetesformen Andere seltenere Diabetesformen können einen genetischen Defekt in der ßZellfunktion aufweisen. Diese Form wird maturity-onset diabetes of the young (MODY) bezeichnet und tritt vor dem 25.Lebensjahr in Erscheinung. Auch ein genetischer Defekt in der Insulinwirkung kann eine seltene Ursache für die Entstehung eines Diabetes sein. Ferner kann ein Diabetes im Rahmen einer chronischen Pankreatitis erscheinen, medikamentös induziert werden, und mit Endokrinopathien, wie z.B. dem Cushing-Syndrom, dem Phäochromozytom und der Akromegalie vergesellschaftet sein [33]. 1.2.4. Gestationsdiabetes Diese Form des Schwangerschaft Diabetes und ist manifestiert durch einen sich erstmals Insulinmangel mit im Laufe der postprandialer Hyperglykämie gekennzeichnet. Das Kind einer an Diabetes erkrankten Mutter kann eine diabetogene Fetopathie entwickeln, die eine Makrosomie und eine Unreife des Kindes bei der Geburt mit sich zieht. Diese Kinder neigen zu einer postpartalen Hypoglykämie aufgrund einer Hypertrophie der pankreatischen Inselzellen als Reaktion auf die mütterliche Hyperglykämie. Die Folge ist eine erhöhte Sekretion von Insulin. Häufig leiden diese Kinder an einem Atemnotsyndrom bei der Geburt, die durch die Insulin basierende Hemmung der Surfactantbildung begründet ist [33]. Im Gegensatz zu den anderen Diabetesformen kann sich der Gestationsdiabetes nach der Entbindung wieder zurückbilden, in einigen Fällen wird aber allerdings auch eine Persistenz des Diabetes beobachtet [33]. 1.3. Typ 1 Diabetes mellitus (T1DM) 1.3.1. Klinik Das Immunsystem ist für die Abwehr von eindringenden Fremdpathogenen verantwortlich, hat aber gleichzeitig die Aufgabe die Selbsttoleranz gegenüber Eigenproteinen zu erhalten. Eine Fehlregulation der Mechanismen, die die Selbsttoleranz normalerweise gewähren, kann in einer Autoimmunkrankheit wie dem T1DM resultieren. Der T1DM ist eine T-Zell-gesteuerte Autoimmunkrankheit, die durch die Zerstörung von endokrinen insulinproduzierenden ß-Zellen der Langerhansinseln des Pankreas charakterisiert ist. Dies resultiert in einer Fehlregulationen der Plasmaglukose, in einer persistierenden Hyperglykämie und in Langzeitkomplikationen [80]. Der T1DM manifestiert sich häufig zwischen dem 12. bis 24. Lebensjahr. Zur klinischen Manifestation des T1DM kommt es erst nach Zerstörung von mehr als 90% der pankreatischen ß-Zellen. Klinisch zeigen sich unspezifische Symptome wie Müdigkeit, Leistungsminderung sowie Symptome infolge der Hyperglykämie und Glucosurie wie Polyurie, Durst, Polydipsie und Gewichtsverlust. Die Patienten neigen zur Ketoazidose aufgrund der vermehrten Ketonkörperbildung. Die Autoimmunkrankheit histologisches resultiert Korrelat findet in einem man eine absoluten Insulinmangel. Autoimmuninsulitis mit Als einem lymphozytenreichen entzündlichen Infiltrat v.a. aus zytotoxischen CD8+-T-Zellen, sowie aus CD4+-T-Zellen und Makrophagen [33]. In der prädiabetischen Phase, vor dem Erscheinen der ersten Symptome, ist bereits eine Insulinsekretionsstörung erkennbar, allerdings liegen die Blutzuckerwerte und der Hämoglobin A1c –Wert (HbA1c), der eine Aussage über die Blutzuckereinstellung über einen längeren Zeitraum trifft, bis zur Manifestation des T1DM noch im Normbereich. Spätkomplikationen des T1DM können aufgrund der sich entwickelnden Makro- und Mikroangiopathien entstehen. Bei T1DM Patienten ist das Risiko für die Entstehung einer koronaren Herzkrankheit (KHK), einer peripheren arteriellen Verschlusskrankheit (pAVK), einer arteriellen Verschlusskrankheit der Hirnarterien oder eines ischämischen Hirninfarkts aufgrund der Makroangiopathie erhöht. Ebenso ist das Risiko für die Entwicklung einer diabetischen Nephropathie, einer diabetischen Neuropathie und einer diabetischen Retinopathie aufgrund der Mikroangiopathien bei diesen Patienten erhöht [33]. 1.3.2. Ätiologie Es wird spekuliert, dass eine Kombination aus genetischen Faktoren und Umweltfaktoren für die Entwicklung des T1DM verantwortlich ist. Als Hauptlokalisationsorte der genetischen Suszeptibilität wurden die HumanenLeukozyten-Antigen (HLA)-Gene genannt. Ferner Umweltfaktoren, die die Entstehung des T1DM begünstigen wie z.B. Viren oder Diätetische Faktoren [43]. Die Auslösung eines T1DM durch Umweltfaktoren wird basierend auf der bestehenden genetischen Suszeptibilität angenommen [52]. Die Rolle der genetischen Faktoren wird deutlich, wenn die familiäre Häufung des T1DM betrachtet wird. Für Verwandte ersten Grades von T1DM Patienten besteht ein Lebenszeitrisiko, d.h. ein Risiko im Laufe des Lebens ebenfalls an T1DM zu erkranken von 5-6% im Vergleich zur kaukasischen Bevölkerung mit einem Lebenszeitrisiko von ca. 0,4% [73]. Einige Studien vermuten eine genetische Assoziation zwischen Melanoma Differentiation-Associated protein 5 (MDA5) Allelvarianten und der Anfälligkeit für T1DM [63]. In einer Zwillingsstudie, die die Konkordanzrate, d.h. die Übereinstimmungrate des Auftretens von T1DM zwischen eineiigen Zwillingen maß, konnte festgestellt werden, dass die Rate der T1DM entwickelnden Zwillinge in den Jahren nach Diagnosestellung bei den Indexzwillingen abfällt. Dies lässt vermuten, dass der initiale Prozess, der zu T1DM führt, innerhalb einer begrenzten Zeit und nicht prolongiert verläuft [64]. Eineiige Zwillinge, die in verschiedenen Ländern lebten, zeigten eine ähnliche Diabetes-Progression. Ferner konnten auch altersbezogene Unterschiede festgestellt werden, wie ein schnelleres Fortschreiten bei Zwillingen von Patienten, bei denen in einem jungen Alter T1DM diagnostiziert wurde [71]. Aufgrund der niedrigen Konkordanzrate zwischen den eineiigen Zwillingen (ca. 30-40%), kann man folgern, dass neben den genetischen Faktoren, nicht-genetische Faktoren wie Umweltfaktoren eine wichtige Rolle in der Pathogenese des T1DM spielen [64]. Die Hypothese der Umweltfaktoren, die die Potenzierung der Krankheit auslösen, wurde durch Studien mit Viren belegt. Folgende Mechanismen, welche einen T1DM auslösen könnten diskutiert werden: Erstens ein molekulares Mimikry zwischen viralen Antigenen und eigenen Antigenen, zweitens die Induktion der Schädigung der ß-Zellen durch Nebensignaleffekte, wie z.B. zwischen Makrophagen und T-Zellen, die die ß-Zell-Zerstörung fördern [52]. Drittens Veränderungen im Gleichgewicht zwischen der Anzahl beziehungsweise Funktion der regulatorischen T-Zellen (Treg-Zellen) und der Anzahl der Effektor-T-Zellen (Teff-Zellen) und viertens eine direkte Schädigung der pankreatischen ß-Zellen durch Viren. Allerdings scheint es meistens einen Bedarf an Immunzellen im ßZell-Zerstörungsprozess zu geben. Bestimmten Viren wird eine Induktion der T1DM Entstehung durch die Auslösung einer Entzündungsantwort, die direkt nach Infektion der ß-Zellen entsteht, zugeschrieben [52]. Während dieser Entzündungsantwort wird unter anderem IFN-γ produziert. Die pankreatische Expression von IFN-γ resultiert in einem Toleranzverlust gegenüber normalen Inselzellen. IFN-γ ist ein proinflammatorisches Cytokin und ist wichtig für die Lymphozytenaktivierung während einer Immunantwort, welches einen ß- Zellverlust hervorrufen kann [77]. Es wurde gezeigt, dass IFN-γ keinen Effekt auf ß-Zellen hat. Allerdings können die Cytokine IFN-γ und TNF-α auch eine Apoptose und somit eine ß-Zellschädigung durch Expression von reaktiven 02 Radikalen induzieren [52]. Nicht-spezifische virale Infektionen, wie z.B. grippale Infekte scheinen der T1DM Manifestation vorauszugehen. Dies wurde anhand einer Studie, bei der zwei Personen eines gleichen Haushalts gleichzeitig an T1DM erkrankten, deutlich. Die erhöhte Insulinresistenz während der Infektion könnte den Insulinmangel in Personen mit unterschiedlicher ß-Zell-Aktivität auslösen [92]. Bei Analyse der Kreuzreaktion von bestimmten viralen Antigenen und Autoantigenen des T1DM konnte eine Kreuzreaktion der viralen Antigene des Rötelnvirus und der des Autoantigens GAD65 festgestellt werden. Die in dieser Studie verwendeten T-Zell Klone erkannten das kreuzreaktive Antigen des Rötelnvirus und des Autoantigens GAD65 und es wurde festgestellt, dass diese auch durch T-Zellen von T1DM Patienten erkannt wurden [65]. Weitere Umweltfaktoren, die ebenfalls eine Rolle zu spielen scheinen, sind Impfungen, die Ernährung im Kindesalter, Toxine, der klimatische Einfluss und Stress [45]. Eine Ursache für die Zunahme der Inzidenz des T1DM in den vergangenen Jahren [47, 70] wird in den verbesserten Hygienestandards, den verbesserten Lebensbedingungen und den Impfstrategien gesehen, die eine verminderte Exposition gegenüber Pathogenen zur Folge haben [23]. Eine Studie, die sich mit den perinatalen und neonatalen Risiken der T1DM Entwicklung beschäftigte, konnte heurausfinden, dass ein signifikant erhöhtes Risiko für die T1DM Entstehung durch Erkrankungen in der Neonatalperiode wie z.B. respiratorische Infekte, besteht. Weitere Faktoren, die die T1DM Entstehung des Kindes begünstigen, sind ein erhöhtes Alter der Mutter bei Feststellung der Schwangerschaft, eine schon bestehende T1DM Erkrankung der Mutter und die Entwicklung einer Präeklampsie der Mutter während der Schwangerschaft. Im Gegensatz hierzu, scheint das Stillen einen protektiven Effekt auf die Entwicklung des T1DM zu haben [58]. Allgemein ist festzuhalten, dass Infektionen die Entstehung des T1DM begünstigen können, die Exposition gegenüber manchen Infektionen allerdings auch paradoxerweise einen protektiven Effekt gegenüber der T1DM Entwicklung hat. 1.3.3. Genetik und Vererbung Die Suzeptibilität für Autoimmunerkrankungen wie dem T1DM ist mit bestimmten HLA-Genen assoziiert, die sich im MHC-Genloci befinden. Es sind auch andere Nicht-MHC-Genloci bekannt, die mit der Entstehung des T1DM in Verbindung gebracht werden. Allerdings ist deren Auswirkung nur relevant, wenn eine zusätzliche MHC II Prädisposition besteht [90]. Insgesamt werden etwa 20 Genloci mit T1DM assoziiert. Die MHC II Gene sind auf Chromosom 6p21 lokalisiert [34]. MHC II beeinflussen den Grad der Autoimmunantwort gegenüber einem ß-ZellAutoantigen oder ein ß-Zell-Autoantigen wird in einer Weise präsentiert, die eine abnorme Immunantwort zur Folge hat [90]. Die Allele des MHC-Klasse II Komplexes, die spezifische Autoantigene binden, sind HLA-DR3 und HLA-DR4. Die T1DM Patienten sind zu 40-50% DR3/DR4 heterozygot, während dies nur bei 5% der Normalbevölkerung der Fall ist. Die kaukasischen T1DM Patienten besitzen zu 90-95% DR3, DR4 oder beide Allele, während dies in der Gesamtbevölkerung nur 5% sind [18]. Der T1DM tritt am häufigsten bei den Nordeuropäern in Erscheinung. Dunkelhäutige, amerikanische Ureinwohner, Südamerikaner und Asiaten sind viel seltener von dieser Autoimmunerkrankung betroffen. Des Weiteren wird ein familiär gehäuftes T1DM Auftreten beobachtet. Bei Erkrankung des Vaters, liegt das Risiko für das Kind einen T1DM zu entwickeln bei 5%, ist die Mutter erkrankt bei 2,5 %, und sind beide erkrankt bei 20% [37]. Allerdings entwickeln 80% ohne familiären Hintergrund einen T1DM, was darauf schließen lässt, dass beide, genetische als auch Umwelteinflüsse bei der Entwicklung des T1DM von Relevanz sind [90]. 1.3.4. Pathogenese des T1DM Wie bereits erläutert, wird der Pathogenese des T1DM eine autoimmune Zerstörung der ß-Zellen des Pankreas zu Grunde gelegt. Diese kann spontan entstehen oder ausgelöst werden. Die Erkrankung resultiert aus einer chronischen Autoimmunantwort gegen ß-Zellen, die meist schon Jahre vor der Erstmanifestation einsetzt und zunächst klinisch inapparent verläuft. Jedoch können während dieser Entwicklung bereits Veränderungen in der humoralen und zellulären Immunreaktion nachgewiesen werden [24] . Bei der Entstehung des T1DM handelt es sich vor allem um eine T-Zell-gesteuerte Erkrankung. Autoreaktive T-Zellen werden bei Donoren, die an einer Autoimmunkrankheit erkrankt sind, aber auch in gesunden Donoren gefunden [3]. Bei der Entwicklung des T1DM ist zusätzlich das angeborene Immunsystem (Dendritische Zellen (DCs), Natürliche Killerzellen (NK-Zellen) und Makrophagen) involviert, die diese autoreaktiven T-Zellen über die Produktion bestimmter proinflammatorsicher Cytokine aktivieren oder durch ihr zytotoxisches Potenzial direkt ß-Zellen zerstören [52]. 1.4. Das Immunsystem 1.4.1. Die angeborene und die adaptive Immunabwehr Zur angeborenen Immunabwehr gehören die physikalischen und chemischen Barrieren, die vor der Invasion von Erregern wie Bakterien und Viren, einen ersten Schutz bieten. Zu den physikalischen Barrieren zählt man die Haut und die Schleimhäute des Respirations-, Gastrointestinal- und Urogenitaltraktes. Chemische Barrieren werden durch Substanzen mit mikrozider Wirkung, wie z.B. Milchsäure, Enzyme und diverse Peptide hergestellt. Auch das Komplementsystem ist durch seine Opsonierung, der Beladung von bakteriellen Oberflächen mit Proteinen zur Erleichterung der Phagozytose, an der chemischen Abwehr beteiligt [35]. Wichtige Komponenten der zellulären Abwehr sind somit die Makrophagen, Granulozyten, dendritischen Zellen (DCs) sowie die Natürlichen Killer T-Zellen (NK T-Zellen), die durch die Sekretion von Cytokinen, Chemokinen und mit Hilfe des Komplemtsystems als induzierbare Effektorsysteme die Abwehr von Erregern erzielen können [7, 41, 52]. Die adaptive Immunabwehr wird dann eingeschaltet, wenn der angeborenen Immunabwehr eine Elimination der Erreger nicht gelungen ist. Der Übergang von angeborener zur adaptiven Immunabwehr ist jedoch fließend und nicht streng voneinander zu trennen. Im Unterschied zur angeborenen Immunabwehr wird die adaptive Immunabwehr erst durch ein Pathogen induziert und zeichnet sich durch ihre Spezifität gegen bestimmte Antigene aus. Man unterscheidet ferner die zelluläre von der humoralen Immunabwehr. Die zelluläre Immunabwehr wird durch die T-Zellen gesteuert, den T-Helferzellen, die eine wichtige Rolle bei der antigenspezifischen Aktivierung von B-Lymphozyten spielen, zu Plasmazellen differenzieren und Antikörper sezernieren. Eine Einteilung der CD4+-T-Zellen erfolgt in TH1-, TH2-, TH17-und regulatorische T-Zellen (Tregs) die sich vor allem durch ihre Cytokinproduktion unterscheiden [48]. TH1 bewirken durch die Ausschüttung von IFN-γ, IL-2 und TNF-α eher eine pathogene, inflammatorische Immunantwort [48, 60] in Autoimmunkrankheiten, während durch die Sekretion von TGF-ß, IL-4, IL-5, IL-10 und IL-13 durch die TH2 Zellen bzw. regulatorische TZellen (Tregs) eine antiinflammatorische Antwort, und somit eine Protektion vor Autoimmunkrankheiten wie T1DM, Multipler Sklerose und Rheumatoider Arthritis erreicht wird. Allerdings wurde festgestellt, dass diese TH2 Zellen auch pathologisch reagieren können und nicht nur antiinflammatorisch wirken [48]. Eine weitere Untereinheit der CD4+-T-Zellen sind die TH17 Zellen. Sie sezernieren hauptsächlich IL-17 [36] und sind somit für eine proinflammatorische Immunantwort verantwortlich. TH17 Zellen spielen eine wichtige Rolle in der Abwehr gegen spezifische Pathogene, aber können auch Autoimmunkrankheiten und Gewebsentzündungen auslösen bzw. diese begünstigen [10]. Im Unterschied zu CD4+-T-Zellen besitzen CD8+-T-Zellen zytotoxische Aktivität. Diese können durch die Sekretion von Perforinen und Granzymen eine direkte Apoptose der Zielzellen, wie z.B. von virusbefallenen Zellen oder Tumorzellen bewirken [36, 90]. CD8+-T-Zellen können die ß-Zellen des Pankreas bereits in einer frühen Phase zerstören [91]. DCs besitzen eine zentrale Rolle in der Immunabwehr, da sie als antigenpräsentierende Zellen (APCs) eine Verbindung zwischen angeborener und adaptiver Immunabwehr herstellen. Ihre Aufgabe ist es als antigenpräsentierende Zellen die Immunität und Toleranz zu gewähren [87]. Eine Einteilung der DCs erfolgt in interstitielle DCs, konventionelle DCs (cDCs), plasmozytoide DCs (pDCs) inklusive der speziellen Form der Langerhanszellen und follikuläre DCs [90]. Die interstitiellen DCs aktivieren bevorzugt die humorale Immunabwehr während die Langerhanszellen bevorzugt die zelluläre Immunabwehr induzieren [87]. DCs sind in der Lage periphere Antigene aufzunehmen, zu verarbeiten, kostimulatorische Moleküle für Lymphozyten zu exprimieren, zu den sekundär lymphatischen Organen zu migrieren und Cytokine auszuschütten, um den Immunprozess zu initieren [7]. Eine weitere wichtige Aufgabe der DCs besteht in der Aktivierung der Differenzierung der naiven CD4+-CD25+-T-Zellen in Foxp3 regulatorische T-Zellen, welche TGF-ß1 und IL-10 sezernieren. Diese haben eine hemmende Wirkung auf inflammatorische Prozesse und sind von großer Relevanz bei der Kontrolle und Entstehung von Autoimmunkrankheiten [55]. Neben dem TCR-MHCII-Peptid-Komplex (siehe 1.6.2.) ist auch die Kostimulation der T-Zelle über Zelloberflächenmoleküle wie cluster of differentiation (CD28), cytotoxic T-Lymphocyte-Antigen 4 (CTLA-4), inducible costimulator (ICOS) und programmed cell death 1 (PD1) durch Liganden der B7 Familie auf APCs oder den ß-Zellen eine wesentliche Rolle bei der Aktivierung der T-Zellen beizumessen. Immunzellen, wie DCs, NK-Zellen und Makrophagen können aber auch durch die Sekretion von Cytokinen wie IL-1ß, IL-12, IFN-γ und TNF-α eine direkte Zerstörung durch Apoptose bewirken [11] (Abb. 1 [52]). Obwohl der T1DM vor allem eine T-Zellgesteuerte Erkrankung ist, sind auch BLymphozyten im pankreatischen Infiltrat zu finden. Ferner sind neben DCs auch NK-Zellen zu sehen, die neben ihrer schädigenden Wirkung auch eine schützende Wirkung auf die ß-Zellen ausüben können. Somit ist eine komplexe Interaktion zwischen pankreatischen ß-Zellen und Zellen des angeborenen und adaptiven Immunsystems an der Pathogenese des T1DM beteiligt [52]. Lehuen A. et al., Nature Reviews Immunology 2010 Abb.1: Interaktionen zwischen der ß-Zelle und den Zellen des angeborenen und adaptiven Immunsystems Zellen des angeborenen Immunsystems wie dendritische Zellen (DCs), natürliche Killerzellen (NK+ + Zellen) und Makrophagen bzw. Zellen des adaptiven Immunsystems wie CD4 - und CD8 -T-Zellen bewirken über verschiedene Rezeptor-Liganden-Interaktionen die Destruktion der ß-Zellen des Pankreas. Abkürzungserläuterung: CXCL=CXC-chemokine ligand; CCL= CC-chemokine-ligand; CD=cluster of differentiation; DC=dendritic cell; FASL= FAS-Ligand; IDO= Indoleamine 2,3-dioxygenase; IFN= Interferon; IL= Interleukin; NK cell= natural killer cell; NKG2D= natural killer group 2 member D; NKp46= natural killer protein 46; MHC= major histocompatibility complex; PD1= programmed cell death 1; PDL1= programmed cell death ligand 1; RAE= retinoic acid early transcript 1; TCR= T-cell receptor; TNF= tumor necrosis factor; TLR= toll like receptor; WE14= chromogranin-derived peptid 1.4.2. Regulatorische T-Zellen (Treg-Zellen) Das Immunsystem ist für die Erkennung, Destruktion und Eliminierung von Pathogenen von entscheidender Bedeutung. Dabei existieren auch Mechanismen, die eine Destruktion von körpereigenem Gewebe durch den Aufbau einer Selbsttoleranz verhindern. Man unterscheidet zwei verschiedene Toleranzmechanismen, die zentrale Toleranz, die durch die negative Selektion der T-Zellen im Thymus gekennzeichnet ist, bei der autoreaktive T-Zellen normalerweise eliminiert werden, von der peripheren Toleranz. Treg-Zellen haben einen großen Stellenwert bei der Aufrechterhaltung dieser peripheren Toleranz im peripheren nicht-thymalen Gewebe [5]. Eine Unterteilung kann in verschiedene Kategorien erfolgen: CD4+-CD25+-TregZellen, FoxP3+-Zellen, IL-10 sekretierende Treg-Zellen (Tr1), TGF-ß1 produzierende T-Helferzellen Typ3 (TH3) [88]. CD4+-CD25+-Treg-Zellen schützen vor Autoimmunkrankheiten, in dem sie die autoreaktiven T-Zellen hemmen und eine Bildung weiterer Treg-Zellen fördern [68]. Peng et al konnten zeigen, dass das Cytokin TGF-ß1 in den Pankreasinselzellen in der Lage ist die Diabetes Entstehung in der Anfangsphase durch CD4+-CD25+-T-Zellen zu hemmen. TGFß1 wie auch IL-10 gehören zu den antiinflammatorischen und immunsuppressiven Cytokinen und werden u.a. von Treg-Zellen sezerniert. Treg-Zellen inhibieren eine Immunantwort, dadurch besitzen sie eine protektive Wirkung vor Autoimmunität in dem sie die Aktivierung und Proliferation autoreaktiver T-Zellen supprimieren [82]. Etwa 40-50% der CD4+-T-Zellen in den Inseln exprimieren das CD25Oberflächenmolekül und üben typische Treg-Zellfunktionen aus. FoxP3 stellt keinen ausreichenden Marker für menschliche T reg-Zellen dar, da einige CD4+-T-Zellen keine regulatorische Funktion zeigen [83]. Allerdings sind diese FoxP3+-CD4+-Treg Zellen wichtig in der Immunhomöostase. Treg-Zellen übernehmen eine wesentliche Rolle bei der Kontrolle der Autoimmunität, bei entzündlichen Erkrankungen wie Asthma bronchiale, chronisch entzündlichen Darmerkrankungen, bei Immunantworten gegenüber Gewebstransplantaten und Tumoren. Treg-Zellen besitzen eine stärkere hemmende Wirkung an Entzündungsorten als in lymphoiden Organen. Sie können sowohl antigenspezifisch als auch antigenunspezifisch wirken. Allgemein zeigen sie eine höhere Sensitivität gegenüber Antigenen als ihre T-Effektorzellen. Diese Eigenschaft erlaubt ihnen die Überwachung der lymphoiden Organe, um eine Aktivierung von autoreaktiven T-Zellen zu verhindern [82]. Naive CD4+-T-Zellen können durch APCs in der Peripherie zu Tr1 Tregs differenzieren und zwei Hauptcytokine TGF-ß1 und IL-10 ausschütten, um T-Effektorzellen zu supprimieren [8, 32, 74, 82]. Auch TGF-ß1 produzierende T-Zellen werden sehr wahrscheinlich durch Tr1 (Tregs) aktiviert [88]. Treg-Zellen besitzen eine wichtige Funktion bei der Kontrolle von Immunantworten gegenüber Eigen-und Fremdantigenen. Dies konnte gezeigt werden, als eine Depletion von CD4+-Treg Zellen in NOD Mäusen in der Induktion von Autoimmunkrankheiten wie z.B. T1DM und in einer erhöhten Immunität gegenüber Tumoren, Transplantaten und Pathogenen resultierte [76, 80]. Ihre Hemmfunktion ist zum Teil von der Expression des FoxP3 Protein abhängig, da T-Zellen, denen das FoxP3 Protein fehlt ihre regulatorische Aktivität verlieren [80]. In der Studie von Luo et al wurde gezeigt, dass DCs von NOD Mäusen durch TGF-ß1 Stimulierung effektiv CD4+-CD25+-FOXP3+-T-Zellen generieren konnten. Somit ist eine periphere Differenzierung zu Treg-Zellen möglich. DCs erfüllen eine wichtige Aufgabe in der Induktion von antigenspezifischen CD4 +-CD25+-FoxP3+T-Zellen, welche man für eine Immuntherapie nutzen könnte, z.B. könnte man spezifische Antigene oder modifizierte Peptidliganden (APLs) einsetzen, die TregZellen aktivieren um autoagressive T-Zellen hemmen [55]. Mitunter vermutet man, dass durch Abweichungen in der DC Reifung bzw. in ihrer Funktion, indirekt die Treg-Zell-Homöostase gestört wird, sodass das Gleichgewicht in Richtung Autoimmunität verschoben wird. Deshalb könnte eine Immunmodulation von TregZellen die Aktivierung von autoreaktiven T-Zellen verhindern und somit die Selbsttoleranz fördern [80]. Das Zelloberflächenmolekül cytotoxisches-T-Lymphozyten-Antigen-4 (CTLA-4) ist auf T-Zellen und Treg-Zellen exprimiert, welche eine unkontrollierte Aktivierung von T-Zellen verhindert. Es wurde festgestellt, dass Mutationen des CTLA-4 Oberflächenmoleküls zu autoimmunen Gewebsschädigungen führen können [86]. Treg-Zellen sind vom IL-2 Milieu abhängig, denn sie benötigen ein IL-2 Signal für ihre Entwicklung, Funktion, und Homöostase [21, 28, 56]. Ferner scheint das Cytokin TNF-α in die Treg-Zellexpansion durch Stimulation der Teff-Zellen mitinvolviert zu sein [31]. Zusammengefasst sind Treg-Zellen wahre Regulationsmeister der Immunhomöostase [82]. Ihre Hauptfunktion besteht darin, die Immunhomöostase in den lymphatischen Organen aufrechtzuerhalten und Entzündungen sowie Gewebsschädigungen zu beheben [82]. 1.4.3. Das angeborene Immunsystem: Antigenpräsentierende Zellen, Makrophagen und NK-Zellen Zu den Zellen des angeborenen Immmunsystems gehören die Dendritischen Zellen (DCs), die in plasmazytoide dentritische Zellen (pDCs) und konventionelle dendritische Zellen (cDCs) unterteilt werden können, sowie die Makrophagen, denen allen gemeinsam die Fähigkeit zur Antigenpräsentation gegeben ist. Sie werden als professionelle APCs bezeichnet. Eine Besonderheit stellen die DCs dar, welche eine Brücke zwischen angeborenem und adaptivem Immunsystem bilden. Weitere Zellen des angeborenen Immunsystems sind die NK-Zellen und die NKT-Zellen. Es gibt Hinweise, dass das angeborene Immunsystem eine wesentliche Rolle in der T1DM Pathogenese z.B. durch die Produktion proinflammatorischer Cytokine spielt. Jedoch sind einige dieser Immunzellen des angeborenen Immunsystems (DCs und NK T-Zellen) in der Lage vor dieser Krankheit zu schützen [52]. Ob autoreaktive T-Zellen aktiviert oder gehemmt werden, hängt u.a. vom Cytokinprofil der Umgebung ab, ob es sich um proinflammatorische Cytokine wie IFN-γ, TNF-α, IL-2, IL-4, IL-5, IL-6 oder um antiinflammatorische Cytokine wie TGF-ß und IL-10 handelt. Die DCs sind in die Kontrolle der B- und T-Zellen integriert. DCs nehmen Proinsulin von ß-Zellen auf, prozessieren diese um sie T-Zellen zu präsentieren. Im Falle von autoreaktiven TZellen, können diese durch DCs aktiviert werden und sind folglich für die Autoimmunität verantwortlich [54, 59, 69] (Abb.2). Weitere Aufgaben der DCs beinhalten die Induktion von naiven T-Zellen zu CD4+-oder CD8+-T-Effektor-Zellen und die Differenzierung von naiven CD4+-CD25--T-Zellen in Treg-Zellen, die an der Aufrechterhaltung der Selbsttoleranz beteiligt sind [55, 93]. Die DCs gehören zu den APCs und sind neben der Präsentation von Fremdantigenen auch an der Präsentation von Autoantigenen beteiligt. Autoantigene, solche wie Proinsulin, GAD65 und IA-2 werden durch DCs präsentiert [2, 72] (Abb2). Die pDCs sezernieren Typ I Interferone, IL-12 und proinflammatorische Cytokine und sind daher wichtig in der Pathogenese des T1DM [50, 53]. Allerdings ist ihre Fähigkeit zur Aufnahme, Prozessierung und Präsentation von Antigenen im Vergleich zu cDCs vermindert [50]. Das Gleichgewicht zwischen pDCs und cDCs ist bei Neudiagnose des T1DM in Richtung pDCs mit einem erhöhten Anteil an pDCs und einem verminderten Anteil an cDCs verlagert. Paradoxerweise können DCs auch eine protektive Funktion einnehmen, indem sie die Differenzierung und Expansion von Treg-Zellen induzieren, welche hemmend auf autoreaktive T-Zellen wirken und dadurch eine ß-Zellschädigung verhindern [87]. Eine therapeutische Induktion von CD4+-CD25+- Treg-Zellen ist möglich. Z.B. konnte gezeigt werden, dass mit einer Behandlung mit G-CSF die Anzahl von pDCs und cDCs stieg. Diese Zellen induzieren CD4+-CD25+-Treg-Zellen, welche TGF-ß1 sezernieren und einen Schutz vor Diabetes bilden [42] . Abb.2: Präsentation von Autoantigenen Dendritische Zellen (DCs) präsentieren als antigenpräsentierende Zellen (APLs) das Autoantigen + über major histocompatibility complex (MHC) Klasse II den CD4 -T-Zellen. Solche autoreaktiven TZellen (Th1 und Th17-Zellen) sezernieren proinflammatorische Cytokine. 1.5. Major Histocompatibility Complex (MHC) Die MHC Moleküle sind für die Immunerkennung, die immunologische Individualiät und die Gewebeverträglichkeit bei Transplantationen von Relevanz. Sie sind von großer Bedeutung für eine T-Zell-vermittelte Immunantwort und befinden sich auf fast allen kernhaltigen Körperzellen. Beim Menschen wird der MHC-Komplex als Humanes Leukozyten Antigen (HLA) bezeichnet und ist auf Chromosom 6 lokalisiert [18]. Unterschiedliche MHC-Moleküle befähigen zur Bindung unterschiedlicher Antigene [90]. Viele Krankheiten stehen im Zusammenhang mit den HLA Molekülen: Autoimmunkrankheiten, wie z.B. autoimmune Endokrinopathien (T1DM), und Entzündungskrankheiten wie postinfektiöse Arthropathien. Man kann MHC Moleküle in 3 Untergruppen unterteilen, Klasse I, Klasse II und Klasse III [90]. 1.5.1. MHC Klasse I Moleküle MHC Klasse I Moleküle befinden sich auf fast allen kernhaltigen Zellen. Viral infizierte Zellen besitzen meist keine MHC Moleküle, da Viren eine Strategie entwickelt haben, indem sie MHC-Moleküle an der Zelloberfläche reduzieren. Human MHC Klasse I können in A, B und C (HLA-A, HLA-B und HLA-C) unterteilt werden [90]. Das MHC Klasse I Molekül besteht aus einem ß2 Mikroglobulin und einer schweren α-Kette aus 3 Untereinheiten α1, α2, α3, die die peptidbindende Grube, an der die Antigene binden können, bilden. Die Funktion des MHC Klasse I Peptid-Komplexes besteht in der Aktivierung von CD8+-T-Zellen [26, 52]. 1.5.2. MHC Klasse II Moleküle Die MHC Klasse II Moleküle werden in die Genloci HLA-DP, HLA-DQ und HLADR unterteilt, die auf APCs exprimiert und von CD4+-T-Helferzellen erkannt werden. Somit befinden sich die MHC Klasse II Moleküle auf Dendritischen Zellen, Makrophagen und B-Lymphozyten, die zu den professionellen APCs gehören. MHC Klasse II Moleküle setzen sich aus der α- und der ß-Kette zusammen, so dass sich die peptidbindende Grube aus beiden Ketten zusammensetzt, aus der α1 und der ß1 Kette. An MHC Klasse II Moleküle können längere Peptide als an MHC Klasse I Moleküle binden. Diese Antigene werden von CD4+-T-Zellen mittels ihres T-Zellrezeptors (TCR) erkannt, woraus eine Aktivierung der T-Zellen erfolgt. Bestimmte Zellen wie Gefäßendothelzellen, Fibroblasten und renale Tubulizellen können durch die Sekretion des Cytokins IFN-γ zur MHC Klasse II Expression stimuliert werden und werden daher fakultativ MHC II exprimierende Zellen genannt [90]. 1.5.3. MHC-Klasse III Moleküle Die Gene der MHC-Klasse III Moleküle werden nicht mit der Peptidpräsentation in Verbindung gebracht. Sie kodieren für Komplementfaktoren wie C2, C4, bestimmte Cytokine wie TNF-α und das Hitzeschockprotein 70 (Hsp 70). 1.5.4. T1DM-assozierte HLA-Moleküle HLA-Moleküle HLA-DQ und HLA–DR sind die wesentlichen genetischen Determinanten bei der Entwicklung des T1DM [20]. Die MHC Moleküle sind auf mehreren Genen kodiert, diese Eigenschaft bezeichnet man als Polygenie. Außerdem weisen sie einen Polymorphismus auf, das heißt es kommt innerhalb des MHC Moleküls zur Entwicklung einer Vielzahl an Varianten, die sich v.a. in der Antigen-bindenden Region unterscheiden. Die Struktur der Region entscheidet darüber, ob das HLA Molekül mit dem Antigen (Peptid) und dem TCR in Verbindung tritt und somit T-Zellen aktiviert [35]. Die Hochrisikogendeterminanten für T1DM sind auf den MHC Klasse II Genen HLA-DQB1 und HLA-DRB1 lokalisiert. Diese Gene sind nicht alleine für die Assoziation mit T1DM und der MHC Region verantwortlich [62]. Eine erhöhte Empfänglichkeit für T1DM wird mit den HLA-Molekülen HLA-DQ2 (HLADQB1*0201) und HLA-DQ8 (HLA-DQB1*0302) in Verbindung gebracht [15, 51], aber auch mit HLA-DR3 (HLA-DRB1*0301) und HLA-DR4 (HLA-DRB1*0401). Vor allem die Kombination HLA-DR3/4 oder HLA DQ2/8 birgt das größte Risiko für die T1DM Entstehung [43]. Nicht unbeachtet sollten auch die protektiven HLAMoleküle bleiben, zu denen HLA-DQ2/HLA-DQ6 (HLA-DQA1*0102/DQB1*0602) gehören, die nicht bei Patienten mit T1DM auftreten [46]. Diesen Schutz vor T1DM könnte man sich durch die Bildung eines stabileren MHC II Komplexes mit Selbstantigenen im Thymus erklären, der eine bessere Peptidpräsentation und eine Eliminierung autoreaktiver T-Zellen nach sich zieht. Damit könnte man bei den T1DM assoziierten HLA-Molekülen einen instabileren Komplex vorstellen, der eine mangelhafte Selektion autoreaktiver T-Zellen im Thymus bewirkt und somit autoreaktive T-Zellen in die Peripherie entkommen [39, 81]. 1.5.5. MHC Klasse II-Peptid-Komplex und T-Zell-Interaktion Die Entscheidung über die Interaktion des T-Zell-Rezeptors mit dem MHC Klasse II-Peptid-Komplex wird im Thymus während der Reifung der Thymozyten getroffen. Dabei ist der Ko-Rezeptor, das CD4+-Molekül von entscheidender Bedeutung. Dieser sorgt für die Stabilität der TCR-MHC Bindung und kann 100fach niedrigere Antigenmengen erkennen als die Bindung ohne Ko-Rezeptor [35]. Ob eine T-Zelle positiv oder negativ selektiert wird, hängt von der Interaktion mit MHC und dem präsentierten Peptid ab. Die spezielle Abfolge der Aminosäuren an den nichtidentischen Bindungsstellen des TCR und auch außerhalb der Kernregion bestimmt, ob T-Zellen durch das präsentierte Peptid angesprochen werden [15]. 1.5.6. MHC Klasse II Antigenprozessierung Durch die rezeptorvermittelte Endozytose werden exogene Antigene und Autoantigene in die APCs aufgenommen. Die Prozessierung dieser Antigene erfolgt anschließend durch Proteasen, den sogenannten Cathepsinen, die im sauren Milieu der Endosomen und Lysosomen zu finden sind. Die Cathepsine tragen unterschiedliche Aminosäuren (AS) in ihrem aktiven Zentrum und werden daher in 3 verschiedene Klassen eingeteilt: Aspartatproteasen (CatD, E), Cysteinproteasen (CatB, C, F, H, L, S, X, AEP) und Serinproteasen (CatA, G). Eine weitere Unterteilung kann in Endo- und Exoproteasen erfolgen. Endoproteasen werden durch die Cathepsine CatG, S, D, E, F und AsparaginEndoprotease (AEP) repräsentiert. Zu den Exoproteasen gehören Cat X, C und A. Cathepsin B und H besitzt beide Eigenschaften, Endoproteasenaktivität als auch Exoproteasenaktivität [17, 75]. Die MHC Moleküle werden im Endoplasmatischen Retikulum (ER) translatiert und die invariante Kette (li) assoziiert an MHC II. li erfüllt unterschiedliche Aufgaben: Sie dient dem Schutz vor Bindung zelleigener Peptide im ER, ist für die richtige Faltung der MHC-Moleküle verantwortlich und unterstützt den Transport des MHC Klasse II Moleküls zum endozytischen Kompartiment. In diesem Kompartiment erfolgt der Abbau der li durch Cathepsine, bis das Klasse II-assozierte-invariante-Ketten-Peptid (CLIP) übrig bleibt. Die Beladung mit hochaffinen antigenen Peptiden erfolgt mit Hilfe des HLA-DM Moleküls, welches den Austausch von CLIP mit den prozessierten Antigenen (Peptide) editiert [17, 75, 89, 96]. Nach Beladung mit Peptid, wird der MHC-Klasse II- Peptid-Komplex an die Zelloberfläche transportiert. 1.6. Proinsulin Die Synthese des Insulins findet in den ß-Zellen der Langerhans Inseln des Pankreas statt. Der Vorläufer des Insulins ist das Präproinsulin und besteht aus einem Signalpeptid, der ß-Kette, dem C-Peptid und der α-Kette (Abb.3). Es befindet sich in gefalteter Form durch die Ausbildung von drei Disulfidbrücken, die eine Stabilisierung des Komplexes gewähren (Abb.3). Nach Abspaltung der Signalsequenz entsteht das Proinsulin, das aus 84 Aminosäuren besteht. Das physiologische aktive Insulin entsteht erst nach Abspaltung des C-Peptids durch spezifische Peptidasen. Es besteht aus zwei Peptidketten, der α-Kette mit 21 Aminosäuren und der ß-Kette mit 30 Aminosäuren, die über zwei intermolare Disulfidbrücken verbunden sind und aus einer dritten intramolekularen Disulfidbrücke innerhalb der a-Kette [66]. Proinsulin 74-90 (P17) ist ein T-ZellEpitop, das heißt dies ist der Bereich eines Antigens, an das der T-Zell-Rezeptor spezifisch bindet und eine T-Zell-Antwort induziert. P17 beinhaltet das Cterminale-Ende des C-Peptids und die enzymatische Schnittstelle der Insulin-αKette [19]. Proinsulin ist ein Schlüsselautoantigen in der Pathogenese des T1DM [29, 38, 61, 95]. In mehreren Studien konnte seine wesentliche Rolle als krankheitsbezogenes Autoantigen gezeigt werden. Nakayama et al haben Experimente mit einem Mausmodell durchgeführt, bei dem modifiziertes Insulin, bei dem Thyrosin gegen Alanin ausgetauscht wurde, exprimiert wurde. Sie konnten zeigen, dass Insulin eine wichtige Rolle als krankheitsbezogenes Autoantigen spielt, da Mäuse, die das modifizierte Insulin exprimierten keinen T1DM entwickelten [15, 61]. Ein weiterer Beleg für die wichtige Rolle des Autoantigens Proinsulin in der Entwicklung eines T1DM, war dessen Prävention durch Inhibieren einer Insulin-Immunantwort in NOD Mäusen. Außer Proinsulin existieren weitere pankreatische Autoantigene, die an der T1DM Entwicklung mitbeteiligt sind. Hierzu gehören die Autoantigene Glutamat Decarboxylase 65 (GAD65) und die Inselspezifische Tyrosinphosphatase (IA-2) [6]. Diese drei Autoantigene stellen somit die drei wichtigsten pankreatischen Autoantigene in der Pathogenese des T1DM dar, die zur Aktiverung von autoreaktiven Zellen führen [6]. Allerdings ist Proinsulin das einzige ßzellspezifische Autoantigen und trägt somit zur Aktivierung von autoreaktiven TZellen, welche ß-Zellen zerstören, bei [44, 78, 97]. Deshalb wird Proinsulin als Schlüsselautoantigen in der T1DM Pathogenese angesehen [38, 61, 95]. Abb.3 : Präproinsulin 1.6.1. Proinsulinpeptid P17 Das Proinsulinpeptid P17 ist ein T-Zell-Epitop und stellt einen Abschnitt des Proinsulins dar. Es entspricht dem Proinsulinabschnitt 74-90 (C18-A1) und überspannt C-Peptid und α-Kette. In einer Studie von Endl et al konnte festgestellt werden, dass P17 von T-Zell Hybridoma, die mit humanen Proinsulin immunisiert wurden und aus HLA-DRB1*0401 transgenen Mäusen stammen, erkannt wurde. Dieses Modell ist auch übertragbar auf den Menschen, da hier eine Erkennung von P17 durch humane T-Zellen beobachtet werden konnte [27]. Bei Messungen der Bindungsaffinität gegenüber P17 wurde die höchste Bindungsaffinität für DRB1*0401 festgestellt [30]. 1.6.2. Proinsulinprozessierung durch Cathepsine Die Proteasen des endosomalen/lysosomalen Kompartiments sind in die Antigenverarbeitung involviert. Die Cathepsine machen den Großteil der Proteasen aus und haben eine wichtige Rolle in der Antigenprozessierung und in der Reifung der MHC Klasse II Moleküle durch die Prozessierung der invarianten Kette, welche die MHC-Klasse II Bindungsgrube benutzt, um ein frühzeitiges Beladen von Peptiden im ER zu verhindern [96]. Das MHC-Klasse II Molekül kann über zwei verschiedene Mechanismen mit Antigenen beladen werden. Das aufgenommene Antigen wird durch die Cathepsine in kleine Fragmente gespalten und anschließend in die Bindungsgrube der MHC-Klasse II Moleküle beladen. Eine andere Möglichkeit zur Beladung besteht darin, dass größere Peptide auf MHC Klasse II beladen werden und anschließend durch Exoproteasen gekürzt werden. Die Kernregion des antigenen Peptids ist allerdings durch die Bindungstasche des MHC Klasse II Moleküls vor der Degradation geschützt. Hierbei spricht man von der MHC-gesteuerten Antigenprozessierung [79]. Es wurden zwei wesentliche humane immundominante T-Zell-Epitope des Proinsulins beschrieben, das P17 (C18-A1) und P21 (A5-A21) [22, 25, 27]. Ein weiteres TZell-Epitop ist das Peptid C19-A3. Es wurde von einer Proinsulin-gepulsten BZelllinie eluiert und stimulierte T-Zellen von T1DM Donoren (HLA- DRB1*0401/0401). Diese T-Zell-Epitope sind innerhalb der Proinsulinsequenz zu finden. Daraus lässt sich schließen, dass Proinsulin das Hauptautoantigen in der T1DM Pathogenese ist [4]. Bevor ein antigenes T-Zell-Epitop generiert werden kann, muss sich das Antigen Proinsulin zunächst einer Prozessierung unterziehen, da eine direkte Bindung von Insulin an MHC Klasse II Moleküle, durch die Disulfidbrücken im Insulin, verhindert wird [15]. CatG, welches nur in primären B-Zellen, DCs, aber nicht in B-Zelllinien oder in vitro differenzierten DCs zu finden ist, dominiert die Prozessierung von Antigenen und Autoantigenen [14] und wurde auch an der Zelloberfläche von primären humanen B-Zellen, T-Zellen und NK-Zellen entdeckt. Man vermutet deshalb, dass die Prozessierung von Proinsulin schon vor bzw. kurz nach der Aufnahme auftritt [15]. 1.7. Modifizierte Peptidliganden zur Inhibierung von diabetogenen TZellen Ziel der Behandlung des T1DM ist es spezifische Therapien zu entwickeln, die selektiv gegen autoreaktive bzw. diabetogene T-Zellen gerichtet sind. Die bisherigen Therapieansätze gegen Autoimmunerkrankungen führen häufig zu einer nicht selektiven Immunsuppression, die mit einer erhöhten Infektanfälligkeit und einem erhöhten Risiko der Kanzerogenese assoziiert sind. Ein viel versprechender Therapieansatz ist der Einsatz von modifizierten Peptidliganden (APLs), die mit dem MHC-Peptid-TCR-Komplex interferieren, um eine autoaggressive T-Zell-Aktivierung zu verhindern [1]. Diese APLs könnten eine neue Strategie in der Prävention des T1DM darstellen. Falcioni et al untersuchten die Bedingungen, unter denen sich ein Peptid an ein bestimmtes MHC Molekül bindet. Somit konnten Peptide hergestellt werden, die spezifisch krankheitsassoziierte MHC Moleküle blockieren. In den 80er Jahren wurden, basierend auf diesem Ansatz, APLs hergestellt, die mit hoher Affinität an MHC Klasse II Moleküle binden [1]. Um in der Immuntherapie Verwendung zu finden, müssen APLs bestimmte Voraussetzungen erfüllen: Sie sollten sich der proteolytischen Aktivität im Serum und gleichzeitig der Antigenprozessierung widersetzen können, damit keine wiederholten Injektionen von hochdosierten Peptid-basierten Immuntherapeutika notwendig werden [88]. Ein weiterer wichtiger Punkt sind die Ankerpositionen der Peptide, welche an MHC II binden. Das Peptid bindet mit den Seitenketten bestimmter Aminosäuren, die Ankerpositionen darstellen. Während andere Seitenketten bestimmter Aminosäuren für die Erkennung durch den TCR der T-Zellen notwendig sind. Das Prinzip dieser Immuntherapie mittels modifizierter Peptidliganden (APLs) beruht darauf, dass ein antigenes Peptid in der MHC Klasse II Bindungstasche durch ein modifiziertes hochaffines Peptid ersetzt wird, das die gleichen Ankerpositionen wie das antigene Peptid besitzt. Allerdings sind die Nicht-Ankerpositionen durch andere Aminosäuren austauschbar, womit eine Generierung von proteaseresistenten Peptidliganden möglich wird. 1.7.1. Ein Proteaseresistente Peptidliganden weiteres wichtiges Kriterium für die Herstellung von modifizierten Peptidliganden zur Immuntherapie ist die Proteasestabilität. Ein Schwachpunkt der bisherigen therapeutisch eingesetzten Peptidliganden ist die Proteasesensitiviät, die in einer geringen Bioverfügbarkeit und in einer Abschwächung der pharmakologischen Wirkung resultiert. Ferner führen hohe Dosen der therapeutisch eingesetzten APLs zu unerwünschten Nebenwirkungen, wie dem anaphylaktischen Schock [67]. Um dieser Proteasesensitivität durch erhöhten Abbau durch Serumproteasen entgegenzuwirken, wurden bestimmte Methoden angewandt, um die Halbwertszeit in der proteolytischen Umgebung zu erhöhen: Eine Strategie, um die Proteasestabilität innerhalb eines Peptids zu verbessern, ist der Einsatz von zyklischen Peptiden [16]. Eine andere Strategie ist die Inkorperation Aminosäuren, von methylierten womit die D-Aminosäuren Hauptangriffspunkte anstelle von von natürlichen Cathepsinen oder Serumproteasen eliminiert werden [16, 88]. Bei der letzten Strategie spricht man von der Schnittstellenspezifischen Aminosäuren Substitution (engl. Cleavage Site Directed Amino Acid Substitution) [16]. Die Herstellung von proteaseresistenten Peptidliganden ist durch dieses neu etablierte Verfahren möglich. Proteaseresistente APLs könnten als Immunmodulatoren fungieren und als möglicher Therapieansatz in Autoimmunerkrankungen wie dem T1DM Anwendung finden (Abb.4). Abb.4: Proteaseresistente Peptidliganden (prAPLs) als Immunmodulatoren Der proteaseresistente Peptidligand (prAPL) bindet über major histocompatibility complex (MHC) Klasse II an eine antigenpräsentierende Zelle (APC), z.B. eine dendritische Zelle (DC). Es erfolgt + die Erkennnung des MHC Klasse II-Peptid-Komplexes durch CD4 -T-Zellen bzw. eine mögliche Aktiverung von regulatorischen T-Zellen (Treg-Zellen), die durch die Ausschüttung des antiinflammatorischen Cytokins TGF-ß1 eine Entzündungsantwort bzw. Immunantwort verhindern können. APL= altered peptide ligands; TGF-β= transforming growth factor β 1.8. Fragestellung Neue antigenspezifische Therapiemöglichkeiten Immunmodulatoren. Proinsulin ist das sind Autoantigen basierende Hauptautoantigen in der T1DM Pathogenese, welches autoreaktive bzw. diabetogene T-Zellen aktivieren kann. Basierend auf der P17 Sequenz wurden proteaseresistente APLs (prAPLs/MuPeptide) generiert. Diese prAPLs wurden in der vorliegenden Arbeit in einem funktionellen T-Zell-Assay getestet, um zu prüfen ob 1. Eine Aktivierung von autoreaktiven T-Zellen verhindert werden kann 2. Eine Aktivierung von Treg-Zellen möglich ist Im Experiment soll mit Hilfe des T-Zell Assays gezeigt werden, ob eine ausreichende Suppression der autoaggressiven T-Zellen erfolgt. Hierbei werden PBMCs von T1DM Patienten mit P17 und Mu-Peptiden/prAPL kokultiviert. Die Bestimmung der T-Zellaktivierung erfolgt durch Detektion der Cytokinsekretion mit Hilfe von ELISA und der Durchflusszytometrie (FACS). Untersucht werden PBMCs von mindestens 17 Patienten, um eine statistische Aussage über einen möglichen Erfolg dieses immunmodulatorischen Therapieansatzes zu machen. 2. Material und Methoden 2.1. Material 2.1.1. Materialien für ELISA Antikörper („coating“).................. Maus Anti-human TNF-α, IFN-γ, IL-6, IL-17, TGF-β1 (R&D Systems, Minneapolis, MN, USA) Antikörper (Detektion)................ Biotinylierter Ziegen Anti-human TNF-α, IFN-γ, IL-6, IL-17, TGF-β1 Antikörper (R&D Systems, Minneapolis, MN, USA) Blockierungspuffer………………. 1% BSA (Albumin bovine Fraktion V (SERON, Minooka, IL, USA) in PBS ELISA Mikrotiterplatte…………… ELISA Platte, Half Area, 96 well, F- Form (Greiner Bio-One, Frickenhausen, Deutschland) PBS……………………………….. 137 mM NaCl; 2,7 mM KCl; 8,1 mM Na2HPO4; 1,5 mM KH2PO4; pH 7,2; 0,2 μm filtriert (Invitrogen, Carlsbad, CA, USA) Reagenzverdünnungsmittel A….. 0,1% BSA; 0,05% Tween20 (SERVA, Heidelberg, Deutschland) in Tris gepuffertem Salz (20 mM Trizma Base; 150 mM NaCl); pH 7,4; 0,2 μm filtriert Reagenzverdünnungsmittel B….. 1% BSA in PBS Reagenzverdünnungsmittel C…..0,05% Tween20 in PBS; 1,4% entlipidiertes bovines Serum (R&D Systems, Minneapolis, MN, USA) Stopplösung……………………… 1 M H2SO4 (Sigma, Taufkirchen, Deutschland) Substratlösung………………....... 1:1 Mischung der Farbreagenzien A (H2O2) und B (Tetramethylbenzidin) (R&D Systems, Minneapolis, MN, USA) Streptavidin-HRP………………… R&D Systems, Minneapolis, MN, USA TGF-β1- Aktivierungslösung…….1 N HCl (Riedel de Haen, Seelze, Deutschland) TGF-β1- Neutralisierungslösung..1 N NaOH; 0,5 M HEPES (Sigma, Taufkirchen, Deutschland) Waschpuffer………………………0,05 % Tween20 in PBS; pH 7,4 (R&D Systems, Minneapolis, MN, USA) Tabelle 1: Liste der beim ELISA verwendeten Lösungskonzentrationen abhängig vom Cytokin Cytokin Coating- Detektions- Standard- Streptavidin- Reagenz- Antikörper- Antikörper- Konzentration HRP Konzentration Konzentration [μg/ml] in PBS [ng/ml] [pg/ml] TNF-α 4 250 IFN-γ 4 175 IL-6 4 IL-17 4 200 1000-30 1/200 B TGF-β1 2 300 1000-30 1/200 C verdünnungs- Verdünnung mittel 1000-30 1/200 B 1000-30 1/200 A 1/200 HRP= horseradish peroxidase; IL= Interleukin; IFN-γ= Interferon-γ; PBS= phosphate buffer saline; TGF-β1= transforming growth factor β1; TNF-α= tumor necrosis factor α 2.1.2. Materialien für den T-Zell Assay T1DM wurde auf Basis der klinischen Diagnose definiert. PBMCs von T1DM Patienten waren im Stickstofftank mit 10% Dimethylsulfoxid (DMSO, Sigma, Taufkirchen Deutschland) und fetal bovine serum (FCS, Invitrogen, Carlsbad, CA, USA) gelagert. Tabelle 2: Genotypen der untersuchten T1DM Patienten Donor Humanes Leukozyten Antigen-Typ 211A DRB1*0401, DRB1*0301 DQB1*02XX, DQB1*0302 597A DRB1*0401, DRB1*1302 DQB1*0302, DQB1*0604 614A DRB1*0401, DRB1**0701 DQB1*02XX, DQB1*0302 643A DRB1*0401, DRB1*0301 DQB1*02XX, DQB1*0302 686A DRB1*0401, DRB1*0101 DQB1*0301, DQB1*0501 849A DRB1*0401, DRB1*0701 DQB1*0202, DQB1*0302 871A DRB1*0401, DRB1*0404 DQB1*0302, DQB1*0302 875A DRB1*0401, DRB1*1102 DQB1*0301, DQB1*0301 911A DRB1*0404, DRB1*0301 DQB1*0301, DQB1*0202 912A DRB1*0401, DRB1*1302 DQB1*0604, DQB1*0302 913A DRB1*0401, DRB1*0401 DQB1*0302, DQB1*0301 914A DRB1*0401, DRB1*0301 DQB1*0302, DQB1*0201 918A DRB1*0401, DRB1*0101 DQB1*0501, DQB1*0302 941A DRB1*0401, DRB1*0701 DQB1*0302, DQB1*0202 945A DRB1*0401, DRB1*1302 DQB1*0604, DQB1*0302 949A DRB1*0101, DRB1*0701 DQB1*0501, DQB1*0202 952A DRB1*0401, DRB1*1303 DQB1*0302, DQB1*0301 Die Proinsulin 74-90 (P17) basierenden Peptidmutanten wurden durch einen Multipeptid-Synthesizer Syro II (MultiSynTech, Witten, Deutschland) generiert und über HPLC (C18 Säule 125x8, Grom, Herrenberg, Deutschland) aufgereinigt (im Labor Dr. Kalbacher, Tübingen, von Dr. Burster). Proinsulin 74-90 (P17) Aminosäurensequenz: AGSLQPLALEGSLQKRG Proinsulin 74-90 (P17) Medium: 10% FCS (Invitrogen, Carlsbad, CA, USA) 1% Penicillin/Streptomycin (Invitrogen, Carlsbad, CA, USA) 0,2% Normocin (Amaxa Biosytems, Köln, Deutschland) in RPMI 1640 (Invitrogen, Carlsbad, CA, USA) Zellkulturplatte: Tissue Culture Plate, 48 Vertiefungen, Polystyrol (BD, Franklin Lakes, NJ, USA) 2.1.3. Fluorescence activated cell sorting (FACS)-Färbung und Analyse Tabelle 3: Verwendete Antikörper für die FACS-Analyse CDAntigene Cytokine Antikörper Farbstoff Firma CD4 Fluorescein (FITC) BD CD4 Peridinin Chlorophyll Protein (PerCP) BD CD25 Allophycocyanin (APC) BD CD45RA Fluorescein (FITC) BD CD134 Fluorescein (FITC) BD TNF-α Allophycocyanin (APC) BD Phycoerythrin (PE) BD TGF-β1 Phycoerythrin (PE) R&D Systems IFN-γ Allophycocyanin (APC) BD Phycoerythrin (PE) BD IL-10 Allophycocyanin (APC) BD IL-17 Phycoerythrin (PE) R&D Systems IL= Interleukin; IFN-γ= Interferon-γ; TGF-β1= transforming growth factor β1; TNF-α= tumor necrosis factor α Brefeldin A (BFA).........................(Sigma-Aldrich, St. Louis, MO, USA) Blockierungspuffer………………. 10% humanes Serum AB in Perm/Wash FACS-Platte................................. MICROTEST U-Bottom, Polystyrol (BD, Franklin Lakes, NJ, USA) FACS-Puffer……………………… 1% BSA in PBS, filtriert Fixierungslösung/ Permeabilisierungslösung………. Cytofix/Cytoperm (BD, Franklin Lakes, NJ, USA) Gefäße (15 ml, 50 ml)…………… (Greiner Bio-One, Frickenhausen, Deutschland) Ionomycin.................................... (Sigma-Aldrich, St. Louis, MO, USA) Medium........................................ RPMI 1640 + GlutaMax (Invitrogen, Carlsbad, CA, USA) Phorbol-12-Myristat-13-Acetat….(Sigma-Aldrich, St. Louis, MO, USA) (PMA) Waschpuffer…………………....... Perm/Wash (BD, Franklin Lakes, NJ, USA) 2.1.4. Verwendete Geräte Brutschrank………………………. BBD 6220 (Heraeus, Hanau, Deutschland) ELISA Analysegerät…………….. EL808 (BioTek, Winooski, Vermont, USA) FACS-Gerät……………………… FACSCalibur 4CA (BD, Franklin Lakes, NJ, USA) Gefrierschrank…………………… (Liebherr, Bulle, Schweiz) Kühlschrank……………………… (Bosch, Gerlingen, Deutschland) Netzgerät…………………………. EPS 500/400 (General Electric, Schenectady, NY, USA) Pipet-Boy…………………………. Pipet boy acu (INTEGRA Biosciences, Fernwald, Deutschland) Pipetten, Einkanal……………….. VWR (VWR, Darmstadt, Deutschland) Pipette, Einkanal………………… Eppendorf Reference (Eppendorf, Hamburg, Deutschland) Pipette, Multikanal………………. Eppendorf Research (Eppendorf, Hamburg, Deutschland) Pipetten, serologisch................... Costar Stripette (Corning Inc. Corning, NY, USA) Sterilbank…………………………. Lamin Air HA2448 (Heraeus, Hanau, Deutschland) Stickstofftank…………………….. Chronos 400 (Messer, Sulzbach, Deutschland) Wasserbad……………………….. 3043 (Köttermann, Hänigsen, Deutschland) Zentrifuge………………………… Multifuge 3S-R (Heraeus, Hanau, Deutschland) 2.2. Methoden 2.2.1. Zellkultur/T-Zell Assay Zunächst erfolgte die Entnahme des Kryoröhrchens mit den in Einfriermedium (90% FCS, 10% DMSO) gelösten PBMCs aus dem Stickstofftank (- 196˚C). Zum Auftauen wurde das Gefäß für zwei Minuten in ein 37° Wasserbad gestellt. Nach dem Auftauprozess wurde der Gefäßinhalt zu 10 ml Medium hinzugegeben. Dieser wie auch die folgenden Arbeitsschritte erfolgten unter der Sterilbank. Die Überstände wurden nach Zentrifugation (1700 U/min., 5min.) verworfen. Die Resuspendierung der Zellpellets erfolgte in 10 ml Medium. Um das DMSO auszuwaschen, wurde der Zentrifugationsprozess wiederholt. Danach wurden die Zellen in 8 ml Medium resuspendiert. Die Übertragung der Zellen auf die Zellkulturplatte erfolgte in Ansätzen zu je 500 μl. In Doppelbestimmung wurde P17 zu diesen Ansätzen (Endkonzentration 10 μg/ml) hinzugegeben und die verschiedenen Mu-Peptide (Mu1-Mu5) pipettiert. Als Referenzwert dienten die beiden Ansätze, die nur P17 enthielten. Danach wurde die Zellkulturplatte bei 37˚C, 6%igem CO2-Gehalt und 94%iger relativer Luftfeuchtigkeit im Brutschrank für 5 Tage inkubiert. 2.2.2. Lymphozyten-Aktivierung Alle folgenden Versuche erfolgten unter sterilen Bedingungen. Zunächst erfolgte die Aktivierung der Lymphozyten mit BFA, PMA und Ionomycin, um die intrazelluläre Cytokinkonzentration via FACS bestimmen zu können. Die Aktivatoren wurden zu 8 ml Medium hinzugegeben. Es wurden folgende Endkonzentrationen hergestellt: BFA [10 μg/ml], PMA [25 ng/ml], Ionomycin [1 μg/ml]. BFA besitzt die Eigenschaft den intrazellulären Transport und die Sekretion, der während der Aktivierung poduzierten Cytokine zu inhibieren. Als Analogon des natürlichen Proteinkinase C (PKC)-Aktivators Diacylglycerin dient PMA als Aktivator der zellulären PKC. PMA und das Ionophor Ionomycin erhöhen beide die intrazelluläre Kalzium-Konzentration und stimulieren folglich die Produktion von Cytokinen. Nach 5 Tagen, konnten die Überstände vorsichtig mit einer Pipette abgenommen werden, da sich die PBMCs am Plattenboden der Zellkulturplatte abgesetzt hatten. Dabei wurde darauf geachtet, dass der Boden der Zellkulturplatte nicht berührt wird. Die Zellüberstände wurden bei -20°C für eine spätere Cytokin-Detektion mittels ELISA eingefroren. Von diesen wurden jeweils zu jedem Ansatz 500 μl pipettiert und eine anschließende Inkubation der Zellkulturplatte erfolgte für 3 Stunden im Brutschrank bei 37°C (6% CO2, 94% relative Luftfeuchtigkeit). Anmerkung: Bei der intrazellulären FACS-Färbung des Transkriptionsfaktors FoxP3 und der vorangegangenen Anfärbung mit Oberflächenmolekülen fand keine Lymphozytenaktivierung statt. 2.2.3. Cytokin-Detektion mittels ELISA Die Cytokinsekretion wurde mit Hilfe des Enzymgekoppelten Immunadsorptionstests (engl. Enzyme Linked Immunosorbent Assay DUO Set ELISA Developpement System Kit von R&D Systems (Minneapolis, MN, USA) detektiert. Zunächst erfolgte das „Coating“, die Beschichtung der 96er Well-Platte (ELISA-Mikrotiterplatte), mit dem jeweiligen cytokinspezifischen Coating- Antikörper. Mittels einer Multikanalpipette wurden 100 μl Antikörperlösung zugegeben: Bei den Cytokinen TNF-α, IFN-γ, IL-6 und IL-17 jeweils [4 μg/ml] in PBS und bei dem Cytokin TGF-ß1 jeweils [2 μg/ml] in PBS. Die Inkubation erfolgte über Nacht bei +4˚C im Kühlschrank. Am folgenden Tag wurde die Antikörperlösung durch Invertieren der Platte verworfen. Im Anschluss folgte der Waschvorgang durch Pipettieren von je 200 μl Waschpuffer in jede Vertiefung der 96er Well-Platte mittels der Multikanalpipette. Durch anschließendes Verwerfen des Waschpuffers wurde die Flüssigkeit in den Vertiefungen entfernt. Das Waschen erfolgte bei jedem Waschvorgang jeweils dreimal. Die Zugabe von 200 μl Blockierungspuffer hatte zum Ziel spätere unspezifische falsch positive Cytokinbzw. Antikörperbindung an der Gefäßwand zu verhindern. Weitere drei Waschschritte folgten nach einer Stunde. Die Überstände des T-Zell Assays wurden zu jeweils 25-60 μl (abhängig von gemessenem Cytokin) in Vierfachbestimmung pipettiert, um eine Detektion der Cytokine TNF-α, IFN-γ, IL-6, IL-17 und TGF-β1 zu erzielen. Für die Detektion des Cytokins TGF-β1 musste zunächst eine Aktivierung der latenten Form dieses Cytokins erfolgen. Durch Zugabe von 6 μl 1M HCL zu 30 μl Überstand des T-Zell Assays und 10-minütiger Inkubation bei Raumtemperatur (RT) konnte die Dissoziation des Latenzassoziierten Peptids vom Homodimer TGF-β1 erzielt werden. Mittels 1,2 N NaOH und 0,5 M HEPES wurde die Lösung danach neutralisiert. Im Anschluss wurden die Überstände mit der nun immunreaktiven Form des TGF-β1 zu 35 μl pro Vertiefung in Vierfachbestimmung in die ELISA-Platte eingebracht. Um die Quantität der Cytokinkonzentration messen zu können, wurde das jeweilige Cytokin in definierter Konzentration als Standard in Doppelbestimmung auf die ELISA-Platte aufgebracht. Dazu erfolgte die Auftragung des Standards mit [1000 pg/ml] in 100 μl des jeweiligen Reagenzverdünnungsmittels (A, B, C, (siehe Tabelle)). Im Anschluss wurde eine Titration durchgeführt, indem 50 μl aus der [1000 pg/ml] Vertiefung entnommen, und mit 50 μl vorgelegten Reagenzverdünnungsmittel in der folgenden Vertiefung vermischt wurden. Mittels Titration konnte die spätere Software-gestützte Konzentrationskurve erstellt werden. Von einem Ausgangswert von [500 pg/ml] in der zweiten Vertiefung erfolgte anschließend eine zweifache serielle Verdünnung bis zu [15 pg/ml], die insgesamt sechsmal erfolgte. Nach 2-stündiger Raumtemperatur Inkubation folgten 3 der Überstände Waschschritte. und des Anschließend Standards wurden 100 bei μl Detektions-Antikörperlösung (mit Biotin konjugierte Antikörper) in jeweils jede Vertiefung pipettiert und in einem Zeitrahmen von 90 Minuten bei Raumtemperatur inkubiert. Weitere 3 Waschschritte folgten. Danach wurden pro Vertiefung 100 μl der Streptavidin-HRP-Lösung in 1/200 Verdünnung in Reagenzverdünnungsmittel pipettiert. Da die HRP eine Photosensitivität aufweist, musste die Inkubation in Dunkelheit und bei 20-minütiger Dauer erfolgen. Innerhalb dieses Zeitintervalls konnte die Bindung von Streptavidin an das an den Antikörper konjugierte Biotin erfolgen. 100 μl der Substratlösung wurden nach weiterem dreifachem Waschen in jede Vertiefung gegeben und bei Raumtemperatur für 20 min. in Dunkelheit inkubiert. Das Resultat war ein farbiges Reaktionsprodukt aus den zwei Substraten, das durch die katalytische Aktivität der HRP entstand. Die optische Dichte des Reaktionsproduktes konnte nach der Zugabe von 50 μl Stopplösung, bei einer Wellenlänge von 450 nm im ELISA-Lesegerät bestimmt werden. 2.2.4. FACS-Färbung und Analyse FACS steht für „Fluorescence activated cell sorting“ und wird im Deutschen allgemein als Durchflusszytometrie bezeichnet. Es dient u.a. der Untersuchung der Eigenschaften von verschiedenen Subpopulationen von Zellen, die durch monoklonale Antkörper gegen Zelloberflächenproteine identifiziert werden. Das Gemisch der markierten Zellen wird durch eine Düse gedrängt und es erfolgt eine Passage durch einen Laserstrahl, wobei es an den Zellen zu einer Lichtstreuung kommt. Sind Farbstoffmoleküle an eine Zelle gebunden entstehen Fluoreszenzimpulse. Durch Detektoren im FACS Gerät wird sowohl das gestreute als auch das emittierte Licht gemessen. Unterschiedliche Eigenschaften der Zellen können dadurch abgeleitet werden. Das gestreute Licht liefert Aussagen hinsichtlich der Größe und der Granularität der Zellen. Die Fluoreszenzsignale können Informationen über die Bindung der markierten monoklonalen Antikörper und somit über die Expression der Oberflächenproteine in jeder Zelle liefern. Nach einer Dauer von 3 Stunden wurden jeweils 350 μl Überstand von jedem Ansatz des T-Zell Assays abgenommen und verworfen. Das Zellpellet wurde im restlichen Medium resuspendiert und danach auf eine 96 Well-Platte transferiert. Anschließend wurde zentrifugiert um die Zellen zu waschen und das restliche Medium zu entfernen (Unter folgenden Bedingungen, die auch für die folgenden Zentrifugationsschritte gelten: 1700 U/min für 5 min bei Raumtemperatur). Der Überstand nach dem Zentrifugationsprozess wurde danach vorsichtig verworfen. Ein weiterer Waschschritt, bei dem das Zellpellet in 180 μl FACS-Puffer resuspendiert, wieder zentrifugiert und der Überstand vorsichtig dekantiert wurde, folgte. Um unspezifische Bindungen zu verhindern, wurden 50 μl FACS-Puffer zur Blockierung unspezifischer Bindungsstellen der Zellen hinzugegeben (10 min. bei Raumtemperatur). Die Färbung der zellulären Oberflächenantigene konnte durch Zugabe von 50 μl FACS-Puffer mit 2 μl Antikörpern mit anschließender Inkubation für 15 min. bei Raumtemperatur erfolgen. Zentrifugation und vorsichtiges Dekantieren des Überstandes folgten. Danach wurde das Zellpellet in 180 μl FACS-Puffer resuspendiert, zentrifugiert und der Überstand wieder vorsichtig dekantiert. Für die intrazelluläre Färbung Permeabilisierungslösung der zu Cytokine jedem wurden Ansatz 180 μl hinzugegeben Fixierungs-/ und bei Raumtemperatur für 20 min. inkubiert. Diese ermöglichte einerseits den Eintritt der Antikörper für die intrazelluläre Färbung und verhinderte andererseits den Eintritt bereits markierter Oberflächenmoleküle. Ein weiterer Zentrifugationsschritt mit vorsichtigem Dekantieren des Überstands und anschließender Resuspension der Zellen in 180 μl Waschpuffer mit Zentrifugation und erneuten vorsichtigem Dekantieren des Überstands folgten. Die Wiederholung dieses Perm/WashWaschvorgangs fand nach 10-minütiger Inkubation mit 180 μl des Blockierungspuffers (15 min bei Raumtemperatur) statt. Anschließend wurde zentrifugiert, der Überstand vorsichtig dekantiert und, die Zellen in 180 μl Waschpuffer resuspendiert, wieder zentrifugiert und der Überstand vorsichtig dekantiert. Durch Zugabe von 50 μl FACS-Puffer mit 2 μl Antikörpern und Inkubation für 15 min. bei Raumtemperatur konnte die intrazelluläre Färbung erfolgen. Nach dem letzten Perm/Wash-Waschvorgang wurde mit FACS-Puffer umgepuffert. Die Analyse wurde am FACS-Gerät durchgeführt. 2.2.5. Signifikanzanalyse Die Signifikanzanalyse der Ergebnisse aus ELISA und FACS erfolgte mit Hilfe des Student`s t-test und wurde mit der Software Graphpad Prism ermittelt. Signifikant waren Werte <0.05. Werte <0.01 waren hochsignifikant. Der Student`s t-test fand in dieser Arbeit Anwendung, da er für die statistische Analyse von kleinen Stichproben geeignet ist. Es handelt sich um eine Wahrscheinlichkeitsverteilung, die verwendet wird um Mittelwerte einer normal verteilten abzuschätzen, wenn die Anzahl der Messwerte relativ klein ist. Population 3. Ergebnisse 3.1. Vorarbeiten: Herstellung von P17-abgeleiteten proteaseresistenten Peptiden durch cleavage-site-directed amino-acid substitution (CSDAS) In vorangegangenen Publikationen wurde ein T-Zell Epitop des Proinsulins beschrieben, welches von T-Zellen von Typ1 Diabetes Patienten erkannt wurde. Die Peptidsequenz des T-Zell Epitops ist AGSLQPLALEGSLQKRK (Proinsulin 7490; P17). Diese genannte Peptidsequenz wurde herangezogen, um einen auf Proinsulin basierenden veränderten Peptidliganden, altered peptide ligand (APL) durch die cleavage-site directed amino-acid substitution (CS-DAS) zu generieren. Zunächst erfolgte eine Inkubation von P17 mit verschiedenen Cathepsinen, die normalerweise in den Prozess der Antigenprozessierung in antigenpräsentierenden Zellen (APCs) involviert sind. Die Fragmente wurden mittels LC-MS analysiert und die resultierenden Fragmente sind in einer Verdaukarte zusammengefasst (Abb.5). P17+CatG 74 90 A GS L Q P L A L EGS L Q K R G P17+CatV A GS L Q P L A L EGS L Q K R G P17+CatL A GS L Q P L A L EGS L Q K R G P17+CatS A GS L Q P L A L EGS L Q K R G P17+CatD/X/C/B/H A GS L Q P L A L EGS L Q K R G Abb. 5: Herstellung proteaseresistenter Peptide basierend auf der Proinsulinsequenz 74-90 Proinsulin Peptid 17 (P17) wurde mit ausgewählten Cathepsinen (Cat) in vitro inkubiert. Die daraus resultierenden Fragmente wurden durch Hochleistungsflüssigkeitschromatographie (HPLC) und Massenspektrometrie analysiert. Die Striche identifizieren Fragmente und die Pfeile kennzeichnen die Cathepsinschnittstellen. CatG verdaute P17 zwischen Leuzin 80 (L80) und Alanin 81 (A81). Die Spaltung von P17 durch CatS, L, und V erfolgte zwischen A81 und L82. Zusätzlich wurde P17 durch CatL und CatV zwischen Prolin 79 (P79) und L80 gespalten. P17 konnte von folgenden Cathepsinen nicht gespalten werden: CatD, X, C, B und H. Basierend auf den P17 Degradierungsdaten, wurden speziell diese Aminosäuren, die als potenzielle Spaltungsstellen für Cathepsine fungieren, durch natürliche sowie D-Aminosäuren ersetzt (Mu1 bis Mu5). Durch diese Methode wurde ein proteaseresistentes Peptid generiert und konnte in den folgenden Experimenten eingesetzt werden (Abb.6). BLC THP-1 P17 P17 A GS L Q P L A L EGS L Q K R G P17 Mu1 A GS L Q P I A L EGS L Q K R G A GS L Q P L A L EGS L Q K R G P17 Mu1 A GS L Q P I A L EGS L Q K R G P17 Mu2 P17 Mu2 A P GS L Q P I A L EGS L A K R G A P GS L Q P I A L EGS L A K R G P17 Mu3 P17 Mu3 A P GS L Q P E A L EGS L A K R G A P GS L Q P E A L EGS L A K R G P17 Mu4 P17 Mu4 A P S L Q P E A L E P S L A K DT G A P S L Q P E A L E P S L A K DT G P17 Mu5 P17 Mu5 A P S L Q P I P L E P S L A K DT G A P S L Q P I P L E P S L A K DT G Abb. 6: Herstellung proteaseresistenter Peptide basierend auf der Proinsulinsequenz 74-90 Herstellung proteaseresistenter Peptide durch Schnittstellen-spezifische Aminosären Substitution (CS-DAS). Mu-Peptide wurden für 6h bei 37°C mit lysosmalen Proteasen von B-lymphoiden Zellen (BLC) oder myelomonozytären Zellen (THP1) inkubiert. Die Verdauungsprodukte wurden über Hochleistungsflüssigkeitschromatographie (HPLC) und Massenpektrometrie analysiert. Experimente wurden durch PD Dr. T. Burster durchgeführt. Mu= Mu-Peptide 3.2. Cytokin-Detektion mittels ELISA Im ersten Ansatz wurden PBMCs von T1DM Patienten mit P17, in den weiteren Experimenten P17 mit verschiedenen Mu-Peptiden für 5 Tage kultiviert. Nach Abnahme der Überstände erfolgte die Detektion der Cytokinsekretion mit Hilfe von ELISA-Messungen. Ziel der vorliegenden Experimente war es, in Erfahrung zu bringen, ob Mu-Peptide in der Lage sind das Cytokinprofil von T-Zellen zu modifizieren. Insbesondere wurde darauf geachtet, ob Mu-Peptide die Sekretion proinflammatorischer Cytokine hemmen bzw. antiinflammatorische Cytokine induzieren. Diese Strategie stellt eine Möglichkeit dar, um autoreaktive T-Zellen zu manipulieren. 3.3. Ergebnisse TNF-α Peptide P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in pg/ml Donor 949A (DRB1*0101/0701) P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 1000 750 500 250 0 P17 TNF-a in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17+Mu2 P17+Mu1 P17 TNF-a in pg/ml P17+Mu5 Peptide 400 300 200 100 0 P17 TNF-a in pg/ml P17+Mu2 P17+Mu1 P17 TNF-a in pg/ml P17+Mu5 P17+Mu4 P17+Mu4 P17+Mu5 Peptide P17+Mu1 Peptide P17+Mu2 P17 TNF-a in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu4 P17+Mu3 P17+Mu2 600 450 300 150 0 Donor 945A (DRB1*0401/1302) P17+Mu5 P17+Mu4 P17+Mu1 P17 TNF-a in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 Donor 941A (DRB1*0401/0701) 1200 900 600 300 0 P17+Mu2 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17+Mu1 Donor 918A (DRB1*0401/0101) 280 210 140 70 0 ELISA ELISA Wdh. Peptide P17+Mu1 Peptide Donor 914A (DRB1*0401/0301) Donor 913A (DRB1*0401/0401) P17 400 300 200 100 0 Peptide 1200 900 600 300 0 P17 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 P17 TNF-a in pg/ml Donor 912A (DRB1*0401/1302) 60 45 30 15 0 Peptide TNF-a in pg/ml Peptide Donor 911A (DRB1*0404/0301) Donor 871A (DRB1*0401/0404) TNF-a in pg/ml 600 450 300 150 0 Peptide 100 75 50 25 0 ELISA ELISA Wdh. Donor 849A (DRB1*0401/0701) 120 90 60 30 0 Peptide ELISA Wdh. Peptide Donor 686A (DRB1*0401/0101) 100 75 50 25 0 ELISA 160 120 80 40 0 Peptide Donor 643A (DRB1*0401/0301) TNF-a in pg/ml P17+Mu2 Peptide P17+Mu1 P17 TNF-a in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in pg/ml a Donor 614A (DRB1*0401/0701) Donor 597A (DRB1*0401/1302) 800 600 400 200 0 P17+Mu1 Donor 211A (DRB1*0401/0301) 1200 900 600 300 0 Peptide P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in pg/ml Donor 952A (DRB1*0401/1303) 360 270 180 90 0 Peptide Abb. 7: Detektion von tumor necrosis factor α (TNF-α) in Zellkulturüberständen durch TNF-α spezifischen Enzym-gekoppelten Immunadsorptionstest (ELISA). Die ELISA Messungen erfolgten in Vierfachbestimmung. Die Stimulation der mononukleären Zellen des peripheren Blutes (PBMCs) von Typ 1 Diabetes mellitus (T1DM) Patienten mit Proinsulin Peptid 17 (P17) diente als Kontrolle. Mu= Mu-Peptide; Wdh.= Wiederholung Die TNF-α Produktion von PBMCs im T-Zell Assay konnte unter Verwendung der Mu-Peptide bei den Donoren 211A, 871A, 911A, 912A, und 918A nicht reduziert werden, sondern die TNF-α Produktion war erhöht (Abb. 7). Extremwerte mit einer Erhöhung der TNF-α Sekretion konnten mit folgenden Donoren gemessen werden: P17+Mu5 mit Donor 912A, P17+Mu4 mit Donor 914A, und P17+Mu5 mit Donor 941A. Lediglich drei Donoren zeigten mit einigen Mu-Peptiden eine verminderte TNF-α Sekretion: Donor 913A mit P17+Mu2, Donor 914A mit P17+Mu 1 und mit P17+Mu2 sowie Donor 941A mit P17+Mu1. Vier Donoren zeigten eine TNF-α Reduktion mit mehreren Peptiden: Donor 211A mit Mu1-Mu3, Donor 614A mit Mu2-Mu5, Donor 643A mit Mu1-Mu3 und Mu5, Donor 945A mit Mu1 und Mu35. Bei den drei Donoren 597A, 686A und 849A ist mit allen Mu-Peptiden außer Mu5 eine TNF-α Reduktion zu erkennen. Die TNF-α Reduktion wurde mit den MuPeptiden 1-4 gezeigt, während mit Mu5 bei diesen drei Donoren eine erhöhte TNF-α Produktion beobachtet wurde. Mit den beiden Donoren 949A und 952A konnte mit allen Mu-Peptiden eine deutliche Senkung der TNF-α Produktion detektiert werden. Donor 912A gab kein Signal wenn die PBMCs mit P17 stimuliert wurden. Ein nur schwaches Signal von P17 entstand mit Donor 871A, 911A, 918A und Donor 941A. Bei der Signifikanzanalyse konnte mit Mu5 mit den drei Donoren 914A, 945A und 949A eine signifikante Senkung gezeigt werden. Mit den vier Donoren 211A, 849A, 913A und 952A wurde kein signifikanter Unterschied gefunden und manche Donoren zeigten sogar einen erhöhten TNF-α Level (597A, 686A, 871A, 912A, 914A, 918A, 941A). IFN-γ P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IFN-g in pg/ml P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 p17+Mu3 P17+Mu2 P17+Mu1 40 30 20 10 0 P17 P17+Mu5 P17+Mu4 P17+Mu2 P17+Mu1 IFN-g in pg/ml Donor 952A (DRB1*0401/1303) 240 180 120 60 0 P17 60 45 30 15 0 Peptide Peptide Donor 945A(DRB1*0404/0301) IFN-g in pg/ml P17+Mu3 P17+Mu2 P17+Mu1 P17 IFN-g in pg/ml 160 120 80 40 0 Peptide Peptide Donor 918A (DRB1*0401/0101) Donor 871A (DRB1*0401/0404) P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IFN-g in pg/ml Donor 211A (DRB1*0401/0301) 120 90 60 30 0 Peptide Abb. 8: Detektion von Interferon-γ (IFN-γ) in Zellkulturüberständen durch IFN-γ spezifischen Enzymgekoppelten Immunadsorptionstest (ELISA). Die ELISA Messungen erfolgten in Vierfachbestimmung. Die Stimulation der mononukleären Zellen des peripheren Blutes (PBMCs) von Typ I Diabetes mellitus (T1DM) Patienten mit Proinsulin Peptid 17 (P17) dient als Kontrolle. Mu= Mu-Peptide; Wdh.= Wiederholung Die Analyse der IFN-γ Konzentration (Abb. 8) zeigte, dass bei zwei Donoren (211A, 945A) eine erhöhte IFN-γ Produktion zu beobachten war. Donor 871A wies eine erhöhte IFN-γ Produktion auf, außer bei P17+Mu3 und P17+Mu4, bei der sie unverändert blieb. Bei Donor 918A konnte eine verminderte IFN-γ Expression durch Einsatz von P17+Mu1 und P17+Mu4 verzeichnet werden. Bei den anderen Mu-Peptiden erhöhte sich die IFN-γ Expression. Eine Reduktion der IFN-γ Sekretion relativ zu P17 wies Donor 952A bei den P17+Mu2 bis Mu5 auf. Mu1 zeigte kein Signal. IL-6 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-6 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu3 P17+Mu2 P17 IL-6 in pg/ml P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-6 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu1 P17 IL-6 in pg/ml P17+Mu5 P17+Mu4 P17+Mu2 P17+Mu2 P17+Mu2 P17+Mu1 P17+Mu3 Peptide P17+Mu5 P17+Mu4 P17+Mu3 2600 1950 1300 650 0 P17 IL-6 in pg/ml P17+Mu5 P17+Mu4 1000 750 500 250 0 Donor 952A (DRB1*0401/1303) Donor 949A (DRB1*0101/0701) P17+Mu3 Donor 945A (DRB1*0401/1302) Peptide 200 150 100 50 0 P17+Mu2 Peptide 1200 900 600 300 0 Peptide P17+Mu1 P17+Mu3 P17+Mu2 P17+Mu1 Peptide P17 IL-6 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17+Mu1 P17 IL-6 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 280 210 140 70 0 Donor 941A (DRB1*0401/0701) 1200 900 600 300 0 P17 Donor 914A (DRB1*0401/0301) 1200 900 600 300 0 ELISA ELISA Wdh.1 ELISA Wdh.2 Peptide Peptide Peptide Donor 913A (DRB1*0401/0401) 1000 750 500 250 0 P17 Donor 875A (DRB1*0401/1102) 28 21 14 7 0 Peptide Peptide Donor 918A (DRB1*0401/0101) IL-6 in pg/ml Peptide 1200 900 600 300 0 Donor 912A (DRB1*0401/1302) IL-6 in pg/ml Peptide P17 IL-6 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-6 in pg/ml 400 300 200 100 0 ELISA ELISA Wdh. 1200 900 600 300 0 Donor 871A (DRB1*0401/0404) Donor 849A (DRB1*0401/0701) Il-6 in pg/ml P17+Mu2 ELISA ELISA Wdh.1 P17+Mu1 1000 750 500 250 0 P17 IL-6 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-6 in pg/ml Peptide ELISA ELISA Wdh. Donor 686A (DRB1*0401/0101) Donor 597A (DRB1*0401/1302) Donor 211A (DRB1*0401/0301) 1200 900 600 300 0 Peptide Abb. 9: Detektion von Interleukin-6 (IL-6) in Zellkulturüberständen durch IL-6 spezifischen Enzymgekoppelten Immunadsorptionstest (ELISA). Die ELISA Messungen erfolgten in Vierfachbestimmung. Die Stimulation der mononukleären Zellen des peripheren Blutes (PBMCs) von Typ 1 Diabetes mellitus (T1DM) Patienten mit Proinsulin Peptid 17 (P17) diente als Kontrolle. Mu= Mu-Peptide; Wdh.= Wiederholung. Eine Zunahme der IL-6 Produktion (Abb. 9) ließ sich bei den Donoren 597A, 912A und 914A verzeichnen. Im Vergleich hierzu zeigten sich die Donoren 849A, 871A und 918A unverändert oder wiesen eine minimal ansteigende Tendenz bei Einsatz der Mu-Peptide auf. Bei fünf Donoren wurde eine Erhöhung der IL-6 Produktion festgestellt mit Ausnahme jeweils eines Mu-Peptids (P17+Mu2), das bei Einsatz eine Senkung der IL-6 Produktion bewirkte: Donor 211A, 686A, 875A, 913A, und 941A. Zu bemerken ist, dass bei Donor 211A mit P17+Mu1 kein Signal zu beobachten war. Des Weiteren sollte festgehalten werden, dass bei einigen Donoren nur ein schwaches Signal für P17 erhalten wurde. Dies betrifft die Donoren 597A, 849A, 871A, 912A, 914A, und 918A. Ein Donor, Donor 945A, zeigte sowohl eine Zunahme als auch eine Abnahme der IL-6-Produktion: Dieser Donor wies bei Einsatz von P17+Mu1 und P17+Mu2 eine zunehmende und bei Einsatz von P17+Mu3-5 eine abnehmende IL-6 Produktion auf. Dagegen konnte bei den Donoren 949A und 952A eine deutliche Senkung der IL-6- Konzentration nach Zugabe der Mu-Peptide aufgezeigt werden. Bei der Signifikanzanalyse konnte bei 8 Donoren kein Unterschied zwischen P17 und P17+Mu5 erkannt werden. Im Gegensatz dazu konnte bei den anderen 8 Donoren eine höhere Konzentration IL-6 im Vergleich zur Kontrolle P17 beobachtet werden. IL-17 P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 80 60 40 20 0 P17 IL-17 in pg/ml Donor 952A (DRB1*0401/1303) P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu1 P17+Mu2 Peptide P17+Mu2 P17+Mu1 P17 P17 IL-17 in pg/ml P17+Mu5 Donor 941A (DRB1*0401/0701) Peptide 20 15 10 5 0 P17 IL-17 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17 IL-17 in pg/ml IL-17 in pg/ml P17+Mu5 P17+Mu4 p17+Mu4 P17+Mu3 Peptide 28 21 14 7 0 Donor 949A (DRB1*0101/0701) Donor 945A (DRB1*0401/1302) P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in pg/ml P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 p17+Mu3 P17+Mu3 ELISA ELISA Wdh. Peptide 40 30 20 10 0 p17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 200 150 100 50 0 Peptide P17+Mu2 600 450 300 150 0 Donor 918A (DRB1*0401/0101) Donor 914A (DRB1*0401/0301) P17+Mu1 P17+Mu2 P17+Mu1 Peptide 0,80 0,60 0,40 0,20 0,00 P17+Mu1 Donor 913A (DRB1*0401/0401) 80 60 40 20 0 P17 IL-17 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 Peptide Peptide Peptide P17 Donor 875A (DRB1* 0401/1102) 18 14 9 5 0 Donor 912A (DRB1*0401/1302) Donor 911A (DRB1*0404/0301) 120 90 60 30 0 P17 P17+Mu2 ELISA ELISA Wdh. P17+Mu1 400 300 200 100 0 P17 IL-17 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in pg/ml Peptide IL-17 in pg/ml Peptide Donor 849A (DRB1*0401/0701) 60 45 30 15 0 Peptide 60 45 30 15 0 Peptide Donor 686A (DRB1*0401/0101) IL-17 in pg/ml P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in pg/ml 240 180 120 60 0 Peptide IL-17 in pg/ml Donor 614A (DRB1*0401/0701) Donor 597A (DRB1*0401/0401) Donor 211A (DRB1*0401/0301) 80 60 40 20 0 Peptide Abb. 10: Detektion von Interleukin-17 (IL-17) in Zellkulturüberständen durch IL-17 spezifischen Enzymgekoppelten Immunadsorptionstest (ELISA). Die ELISA Messungen erfolgten in Vierfachbestimmung. Die Stimulation mononukleärer Zellen des peripheren Blutes (PBMCs) von Typ I Diabetes mellitus (T1DM) Patienten mit Proinsulin Peptid 17 (P17) diente als Kontrolle. Mu= Mu Peptide; Wdh.= Wiederholung Die Auswirkung auf die IL-17 Produktion (Abb. 10) lässt sich bei den Donoren 211A, 597A, 911A, 914A, 941A und 949A als relativ unverändert bezeichnen, wenn Mu-Peptide eingesetzt wurden. Bei zwei dieser genannten Donoren sind allerdings Extremwerte, die mit einer Erhöhung der IL-17 Sekretion korreliert, festzustellen; bei Donor 211A bei Einsatz von P17+Mu1 und bei Donor 597A bei Einsatz von P17+Mu5. Außerdem ist festzuhalten, dass bei den drei Donoren 875A, 912A, 918A die IL-17-Produktion eine aufsteigende Tendenz aufzuweisen war. Allerdings ist bei diesen Donoren auch zu vermerken, dass P17 nur ein sehr schwaches Signal zeigte. Bei fünf Donoren konnte eine reduzierte bzw. erhöhte IL-17-Produktion beobachtet werden, je nach Einsatz des jeweiligen Mu-Peptids. Bei Donor 614A konnte bei der jeweiligen Zugabe von Mu1, Mu4, und Mu5 zu P17 eine Senkung der IL-17 Produktion verzeichnet werden. Deutlich ist die verminderte IL-17 Sekretion nach dem jeweiligen Einsatz von Mu2 und Mu5 mit P17 auch bei Donor 686A zu sehen. P17+Mu1, P17+Mu3 und P17+Mu4 bewirken bei Donor 913A wiederholt eine verminderte IL-17-Produktion. Auch bei Donor 952A konnte bei Zugabe von jeweils drei Mu-Peptiden (Mu 1, 4, 5) zu P17 eine deutliche Senkung der IL-17 Konzentration gemessen werden. Auffallend ist, dass alle Mu-Peptide bei Donor 849A und 945A eine sehr deutliche Minderung der IL17-Produktion bei Zugabe zu P17 bewirkten. In der Signifikanzanalyse konnte eine deutliche Abnahme der IL-17 Konzentration von PBMCs, zu denen P17+Mu5 bzw. nur P17 hinzugegeben wurde, verzeichnet werden (Donor 614A, 849A, 945A, 952A). Dennoch handelte es sich bei zwei Donoren (Donor 911A, 913A) um keine signifikante Senkung der IL-17 Konzentration. TGF-β1 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in pg/ml P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu1 TGF-b1 in pg/ml P17 P17+Mu5 P17+Mu5 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 TGF-b1 in pg/ml P17+Mu5 P17+Mu4 P17+Mu2 P17+Mu3 P17+Mu3 P17+Mu4 P17+Mu2 P17+Mu3 P17+Mu3 ELISA ELISA Wdh. Peptide P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 200 150 100 50 0 P17 TGF-b1 in pg/ml P17+Mu5 P17+Mu4 800 600 400 200 0 Donor 952A (DRB1*0401/1303) Donor 949A (DRB1*0101/0701) P17+Mu3 Peptide Donor 945A (DRB1* 0401/1302) Peptide 600 450 300 150 0 P17+Mu2 P17+Mu2 P17+Mu1 Peptide P17+Mu1 ELISA ELISA Wdh. 400 300 200 100 0 P17 TGF-b1 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 Donor 914A (DRB1*0401/0301) 1200 900 600 300 0 Donor 941A (DRB1*0401/0701) 200 150 100 50 0 P17 Peptide 800 600 400 200 0 P17 Donor 918A (DRB1*0401/0101) P17+Mu1 ELISA ELISA Wdh.1 ELISA Wdh.2 Peptide Peptide Peptide Donor 913A (DRB1*0401/0401) P17 TGF-b in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 800 600 400 200 0 Peptide P17+Mu3 P17+Mu2 P17 TGF-b1 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in pg/ml ELISA ELISA Wdh. Donor 911A (DRB1*0404/0301) 100 75 50 25 0 Donor 912A (DRB1*0401/1302) 360 270 180 90 0 Peptide 800 600 400 200 0 Donor 875A (DRB1*0401/1102) 160 120 80 40 0 Peptide TGF-b1 in pg/ml P17+Mu2 P17+Mu1 P17 TGF-b1 in pg/ml Peptide Donor 871A (DRB1*0401/0404) TGF-b1 in pg/ml Donor 849A (DRB1*0401/0701) 600 450 300 150 0 Peptide TGF-b1 in pg/ml Peptide Donor 686A (DRB1*0401/0101) P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in pg/ml Donor 643A (DRB1*0401/0301) Donor 614A (DRB1*0401/0701) 1000 750 500 250 0 Peptide ELISA Wdh. 800 600 400 200 0 ELISA ELISA Wdh. P17+Mu2 ELISA P17+Mu1 P17 TGF-b1 in pg/ml P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in pg/ml Peptide 1000 750 500 250 0 TGF-b1 in pg/ml Donor 597A (DRB1*0401/1302) Donor 211A (DRB1*0401/0301) 280 210 140 70 0 Peptide Abb. 11: Detektion von transforming growth factor β1 (TGF-β1) in Zellkulturüberständen durch TGF-β1 spezifischen Enzym-gekoppelten Immunadsorptionstest (ELISA). Die ELISA Messungen erfolgten in Vierfachbestimmung. Die Stimulation mononukleärer Zellen des peripheren Blutes (PBMCs) von Typ I Diabetes mellitus (T1DM) Patienten mit Proinsulin Peptid 17 (P17) diente als Kontrolle. Mu= Mu-Peptide; Wdh.= Wiederholung Die Auswirkung auf die TGF-β1 Produktion (Abb.11) lässt sich bei den Donoren 211A, 597A, 643A, 686A, 849A, 871A, 875A, 918A, 941A, 949A, und 952A als relativ unverändert bezeichnen, wenn Mu-Peptide eingesetzt wurden. Bei einigen Donoren ist bei Zugabe von bestimmten Mu-Peptiden eine aufsteigende Tendenz zu beobachten: Bei Donor 614A nach Zugabe von Mu3 zu P17, bei Donor 913A nach Zugabe von jeweils Mu4 und Mu5 zu P17, bei Donor 914A nach Zugabe von Mu4 zu P17 und bei Donor 945A nach Zugabe von Mu4 zu P17. Eine deutliche Zunahme der TGF-β1 Produktion konnte bei Donor 911A und Donor 912A nur in der Wiederholung gezeigt werden. Festzuhalten ist, dass bei der Signifikanzanalyse des antiinflammatorischen Cytokins TGF-β1 bei acht Donoren kein Unterschied der TGF-β1 Produktion in PBMCs, die mit P17 und Mu5 versehen wurden im Vergleich zu PBMCs, die nur mit P17 kultiviert wurden, zu sehen war. Allerdings konnte bei sieben Donoren (Donor 614A, 849A, 911A, 912A, 914A, 941A, 952A) eine signifikante Erhöhung der Konzentration des antiinflammatorischen Cytokins TGF-β1 gemessen werden. 3.4. Intrazelluläre Cytokinbestimmung durch FACS-Analyse Zur weiteren Abklärung der mittels ELISA gewonnenen Daten über die Cytokinkonzentrationsänderungen von PBMCs nach Zugabe von Mu-Peptiden wurde die Cytokinproduktion der Donoren durch FACS-Analyse erhoben. PBMCs der Donoren wurden mit P17 und P17 mit den verschiedenen Mu-Peptiden für 5 Tage kultiviert. Anschließend wurde die Cytokinproduktion mittels FACS-Analyse ermittelt. Ziel war es zu untersuchen, ob die eingesetzten Mu-Peptide in der Lage sind das Cytokinprofil von T-Zellen zu modifizieren. Erreicht werden sollte eine Minderung der Produktion von proinflammatorischen Cytokinen und eine Erhöhung der Konzentration von antiinflammatorischen Cytokinen. Bei der FACSAnalyse wurde ausschließlich die veränderte Expression von Cytokinen in TGedächtniszellen, den CD4+, CD45RA--T-Zellen ermittelt. Die folgenden Diagramme spiegeln die mittels FACS-Analyse gemessenen Daten wieder. TNF-α TNF-a P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in arbiträren Einheiten P17+Mu5 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu1 P17 TNF-a in arbiträren Einheiten P17+Mu5 P17+Mu4 8 6 4 2 0 P17+Mu2 P17+Mu3 P17+Mu3 P17+Mu2 P17+Mu1 Peptide Peptide Donor 949A (DRB1*0101/0701) 8 6 4 2 0 P17 TNF-a in arbiträren Einheiten P17+Mu5 P17+Mu4 Peptide Donor 945A (DRB1*0401/1302) Donor 941A (DRB1*0401/0701) P17+Mu3 Donor 918A (DRB1*0401/0101) 24 18 12 6 0 Peptide 16 12 8 4 0 P17+Mu2 P17+Mu3 P17+Mu2 P17+Mu2 P17+Mu1 P17 TNF-a in arbiträren Einheiten 20 15 10 5 0 Peptide P17+Mu1 Peptide Donor 914A (DRB1*0401/0301) P17+Mu5 p17+Mu4 p17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in arbiträren Einheiten P17+Mu5 P17+Mu3 P17+Mu2 P17+Mu1 P17 Donor 912A (DRB1*0401/1302) 12 9 6 3 0 Peptide 20 15 10 5 0 P17 P17+Mu4 Donor 911A (DRB1*0404/0301) 4 3 2 1 0 P17 TNF-a in arbiträren Einheiten Peptide 16 12 8 4 0 Donor 913A (DRB1*0401/0401) TNF-a in arbiträren Einheiten 16 12 8 4 0 Peptide Donor 875A (DRB1*0401/1102) TNF-a in arbiträren Einheiten P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in arbiträren Einheiten P17+Mu5 p17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in arbiträren Einheiten 16 12 8 4 0 Peptide Peptide Donor 849A (DRB1*0401/0701) Donor 597A (DRB1*0401/1302) Donor 211A (DRB1*0401/0301) 16 12 8 4 0 Peptide P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TNF-a in arbiträren Einheiten Donor 952A (DRB1*0401/1303) 20 15 10 5 0 Peptide Abb. 12: + Detektion von intrazellulärem tumor necrosis factor α (TNF-α) in T-Gedächtniszellen (CD4 , CD45RA )־durch fluorescence activated cell sorting (FACS-Analyse). FACS-Messungen wurden in Doppelbestimmung durchgeführt. Mu= Mu- Peptide Die in Abbildung 12 detektierte TNF-α Produktion, der mit P17 stimulierten Zellen unterschied sich bei den Donoren 211A, 597A, 849A, 875A und 949A nicht wesentlich von den stimulierten Zellen, bei denen P17 zusammen mit den MuPeptiden eingesetzt wurde. Eine verminderte TNF-α Produktion konnte nicht verzeichnet werden. Bei Donor 211A konnte bei jeweiliger Zugabe der Mu-Peptide 1 und 2 sogar eine erhöhte TNF-α Konzentration gemessen werden. Die vier Donoren 911A, 913A, 914A und 941A zeigten sogar bei Zugabe aller Mu-Peptide zu P17 eine Zunahme der TNF-α Produktion. Anzumerken ist allerdings, dass bei einigen Donoren absteigende Tendenzen der TNF-α Sekretion beobachtet wurden: Donor 912A bei Einsatz von P17+Mu1 im Vergleich zu P17, Donor 918A bei Einsatz von jeweils P17+Mu1, Mu3 und Mu5 und Donor 945A bei Einsatz von P17+Mu1. Außerdem konnte eine deutliche Verringerung der TNF-α Konzentration bei Donor 952A durch Einsatz aller Mu-Peptide außer P17+Mu3 verzeichnet werden. Die Signifikanzanalyse wies eine signifikante Abnahme der TNF-α Produktion bei 8 Donoren (Donor 875A, 912A, 914A, 918A, 941A, 945A, 949A, 952A) auf, wenn P17 und Mu5 in den PBMC Zellen eingesetzt wurden, auch wenn im ELISA erhöhte TNF-α Sekretionen detektiert wurden, während bei 5 Donoren (Donor 211A, 597A, 849A, 911A und 913A) keine wesentliche Veränderung der TNF-α Produktion beobachtet wurde. INFIFN-g Donor 597A (DRB1*0401/1302) P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 8 6 4 2 0 P17 P17+Mu5 P17+Mu4 P17+Mu2 P17+Mu1 IFN-g in arbiträren Einheiten Donor 952A (DRB1*0401/1303) 24 18 12 6 0 P17 IFN-g in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu2 P17+Mu1 P17 IFN-g in arbiträren Einheiten P17+Mu5 Peptide Donor 945A (DRB1*0401/1302) Donor 941A (DRB1*0401/0701) P17+Mu2 P17+Mu1 P17 IFN-g in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu4 12 9 6 3 0 Peptide 12 9 6 3 0 P17+Mu2 P17+Mu1 P17 IFN-g in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu3 P17+Mu3 P17+Mu2 P17+Mu1 Peptide P17+Mu2 Donor 918A (DRB1*0401/0101) 16 12 8 4 0 P17 IFN-g in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 Peptide Donor 914A (DRB1*0401/0301) 16 12 8 4 0 P17+Mu1 8 6 4 2 0 Peptide Donor 912A (DRB1*0401/1302) P17 P17+Mu2 P17+Mu1 Peptide P17 Donor 871A (DRB1*0401/0404) 20 15 10 5 0 P17 IFN-g in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IFN-g in arbiträren Einheiten Peptide Donor 849A (DRB1*0401/0701) 8 6 4 2 0 IFN-g in arbiträren Einheiten 12 9 6 3 0 Peptide Donor 643A (DRB1*0401/0301) IFN-g in arbiträren Einheiten P17+Mu2 P17+Mu1 Peptide Peptide Donor 614A (DRB1*0401/0701) 28 21 14 7 0 P17 IFN-g in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IFN-g in arbiträren Einheiten Donor 918A (DRB1*0401/0101) 12 9 6 3 0 Peptide Peptide Abb. 13: + - Detektion von intrazellulären Interferon-γ (IFN-γ) in T-Gedächtniszellen (CD4 , CD45RA ) durch fluorescence activated cell sorting (FACS-Analyse). FACS-Messungen wurden in Doppelbestimmung durchgeführt. Mu= Mu-Peptide. Des Weiteren wurde die Produktion des proinflammatorischen Cytokins IFN-γ gemessen (Abb. 13). Eine gleichbleibende Konzentration des Cytokins IFN-γ konnte bei den Donoren 597A, 614A, 849A, 912A bei ELISA und Wiederholung und 952A nach Einsatz der Mu-Peptide im Vergleich zu P17 beobachtet werden. Eine Reduktion der Cytokinproduktion war bei den Donoren 918A bei P17+Mu5, bei Donor 643A bei Mu5, bei Donor 871A bei Mu1, bei Donor 914A bei Mu4 und 5, bei Donor 918A bei Mu5 und bei Donor 941A bei Mu5 festzustellen. Auffällig war, dass Donor 945A eine deutliche Abnahme des proinflammatorischen Cytokins IFN-γ nach Einsatz aller Mu-Peptide zeigte. Die Bestimmung der Signifikanz zeigte, dass bei sieben Donoren (643A, 912A, 914A, 918A, 941A, 945A und 952A) eine signifikante Reduktion der IFN-γ Konzentration detektiert wurde, im Gegensatz zu den sechs Donoren 211A, 597A, 614A, 849A, 871A, 912A, die vielmehr eine Erhöhung der IFN-γ Produktion aufwiesen. IL-6 Peptide Peptide P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-6 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu1 IL-6 in arbiträren Einheiten P17+Mu5 P17+Mu4 16 12 8 4 0 P17+Mu2 P17+Mu3 P17+Mu3 P17+Mu2 P17+Mu1 8 6 4 2 0 P17 IL-6 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 Donor 952A (DRB1*0401/1303) Donor 914A (DRB1*0401/0301) 12 9 6 3 0 P17+Mu1 Peptide Peptide Donor 912A (DRB1*0401/1302) P17 20 15 10 5 0 P17 Peptide P17+Mu2 P17+Mu1 12 9 6 3 0 P17 IL-6 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-6 in arbiträren Einheiten 16 12 8 4 0 IL-6 in arbiträren Einheiten Donor 871A (DRB1*0401/0404) Donor 643A (DRB1*0401/0301) Donor 614A (DRB1*0401/0701) Peptide Abb. 14: + - Detektion von intrazellulären Interleukin-6 (IL-6) in T-Gedächtniszellen (CD4 ,CD45RA ) durch fluorescence activated cell sorting (FACS-Analyse). FACS-Messungen wurden in Doppelbestimmung durchgeführt. Mu= Mu-Peptide In der folgenden IL-6-Konzentrationsbestimmung (Abb. 14) konnte bei Donor 643A und 871A nach Einsatz der Mu-Peptide kein Unterschied in der Konzentration gemessen werden. Lediglich eine leichte Erhöhung der Produktion von IL-6 wurde nach Zugabe von P17+Mu1 bei Donor 643A und nach Zugabe von P17+Mu5 bei Donor 871A verzeichnet. Eine Abnahme der IL-6 Konzentration konnte nicht beobachtet werden. Allerdings zeigten die Donoren 614A, 914A, und 952A eine deutliche Reduktion der IL-6-Produktion. Für Donor 614A galt dies für P17+Mu1, 2 und 3. Bei Donor 914A für P17+Mu2, 3, 4 und 5 sowie bei Donor 952A nur für P17+Mu1. Nach Einsatz von P17 und der Mu-Peptide, konnte bei Donor 912A eine im Trend geringere IL-6-Produktion beobachtet werden als bei jenen PBMCs, die nur mit P17 alleine stimuliert wurden. Es ist festzuhalten, dass keine signifikante Reduktion der IL-6 Konzentration bei den Donoren 643A, 871A, 912A, 914A und 952A gemessen wurde. IL-17 Donor 597A (DRB1*0401/1302) P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in arbiträren Einheiten P17+Mu5 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in arbiträren Einheiten P17+Mu5 P17+Mu5 p17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in arbiträren Einheiten P17+Mu5 P17+Mu4 Donor 952A (DRB1*0401/1303) P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 4 3 2 1 0 P17 IL-17 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu2 Peptide P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu4 P17+Mu3 P17+Mu3 P17+Mu2 P17+Mu1 P17+Mu1 12 9 6 3 0 P17 IL-17 in arbiträren Einheiten p17+Mu5 P17+Mu4 Peptide Donor 941A (DRB1*0401/0701) Donor 918A (DRB1*0401/0101) 8 6 4 2 0 P17+Mu3 8 6 4 2 0 Peptide Peptide P17+Mu2 Peptide Donor 914A (DRB1*0401/0301) 16 12 8 4 0 P17 IL-17 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 Peptide 4 3 2 1 0 Donor 913A (DRB1*0401/0401) Donor 912A (DRB1*0401/1302) 24 18 12 6 0 P17+Mu1 P17+Mu3 P17+Mu2 P17+Mu1 P17+Mu2 P17 IL-17 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 Donor 911A (DRB1*0404/0301) 4 3 2 1 0 Peptide P17 Peptide Donor 875A (DRB1*0401/1102) 12 9 6 3 0 P17 8 6 4 2 0 Peptide Donor 871A (DRB1*0401/0404) IL-17 in arbiträren Einheiten P17+Mu3 P17+Mu2 P17+Mu1 P17 P17 IL-17 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in arbiträren Einheiten 20 15 10 5 0 Donor 849A (DRB1*0401/0701) 12 9 6 3 0 Peptide IL-17 in arbiträren Einheiten Peptide Donor 686A (DRB1*0401/0101) Donor 643A (DRB1*0401/0301) IL-17 in arbiträren Einheiten 20 15 10 5 0 Peptide Peptide Peptide Donor 614A (DRB1*0401/0701) 16 12 8 4 0 Il-17 in arbiträren Einheiten P17+mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 IL-17 in arbiträren Einheiten Donor 211A (DRB1*0401/0301) 8 6 4 2 0 Peptide Abb.15: + - Detektion von intrazellulären Interleukin-17 (IL-17) in T-Gedächtniszellen (CD4 , CD45RA ) durch fluorescence activated cell sorting (FACS-Analyse). FACS-Messungen wurden in Doppelbestimmung durchgeführt. Mu= Mu-Peptide. Die Analyse der IL-17 Konzentration (Abb. 15) zeigte die Tendenz, dass die MuPeptide bei vier Donoren eine erhöhte IL-17-Produktion der PBMCs zur Folge hatten. Dies war bei den Donoren 614A, 643A, 912A und bei Donor 913A der Fall, allerdings nicht in der Wiederholung. Die Wiederholungen bei diesen genannten Donoren wiesen eine gleichbleibende Tendenz der IL-17-Konzentration auf. Ebenfalls wurde bei den Donoren 871A, 911A, 914A, und 952A kein Unterschied in der IL-17-Produktion gemessen. Des Weiteren ergab die Analyse der IL-17Produktion, dass bei vielen Donoren eine Abnahme der IL-17-Produktion verzeichnet werden konnte. Beobachtet wurde dies bei Donor 211A nach Einsatz von P17+Mu1 und P17+Mu3, bei Donor 597A nur bei Einsatz von P17+Mu5, bei Donor 686A bei Einsatz von P17+ Mu1,2 und 4, bei Donor 849A bei Einsatz von P17+Mu4 und Mu5 allerdings nur bei FACS und nicht bei der Wiederholung, bei Donor 918A nach Einsatz von P17+Mu2- 5 und zuletzt bei Donor 941A nach Einsatz von P17+Mu4 und 5. Die Bestimmung des proinflammatorischen Cytokins IL-17 ergab bei Donor 875A nach Zugabe von P17+Mu2, 3 und 5 zu den PBMCs eine Abnahme der IL-17 Produktion. In der Signifikanzanalyse konnte bei sieben Donoren (849A, 871A, 875A, 913A, 914A, 918A und 941A) eine signifikante Senkung der IL-17 Konzentration detektiert werden, wenn P17 mit Mu5 inkubiert wurde. Kein Unterschied wurde bei sechs Donoren verzeichnet und eine höhere IL-17-Produktion als bei der Referenz P17, wurde bei vier Donoren detektiert. TGF-β1 TGF-b1 P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 2,00 1,50 1,00 0,50 0,00 P17+Mu1 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 TGF-b1 in arbiträren Einheiten Donor 952A (DRB1*0401/1303) 8 6 4 2 0 P17 TGF-b1 in arbiträren Einheiten Donor 949A (DRB1*0101/0701) P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in arbiträren Einheiten TGF-b1 in arbiträren Einheiten Peptide P17 Donor 945A (DRB1*0401/1302) 8 6 4 2 0 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in arbiträren Einheiten P17+Mu5 P17+Mu5 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in arbiträren Einheiten 12 9 6 3 0 2,00 1,50 1,00 0,50 0,00 Peptide Peptide P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in arbiträren Einheiten p17+Mu5 P17+Mu4 P17+Mu4 P17+Mu4 P17+Mu3 P17+Mu2 Donor 941A (DRB1*0401/0701) Peptide P17+Mu2 Peptide Donor 918A (DRB1*0401/0101) P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 8 6 4 2 0 Peptide Donor 914A (DRB1*0401/0301) 4 3 2 1 0 P17+Mu1 P17+Mu3 P17+Mu2 P17+Mu1 P17+Mu1 P17 TGF-b1 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 Donor 913A (DRB1*0401/0401) 16 12 8 4 0 Peptide P17 Peptide Donor 912A(DRB1*0401/1302) 4 3 2 1 0 P17+Mu1 20 15 10 5 0 Peptide Donor 871A (DRB1*0401/0404) P17 Donor 849A (DRB1*0401/0701) 16 12 8 4 0 P17 TGF-b1 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in arbiträren Einheiten Peptide Donor 686A (DRB1*0401/0101) 12 9 6 3 0 Peptide TGF-b1 in arbiträren Einheiten 8 6 4 2 0 Peptide Donor 643A (DRB1*0401/0301) TGF-b1 in arbiträren Einheiten P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in arbiträren Einheiten 16 12 8 4 0 Peptide TGF-b1 in arbiträren Einheiten Donor 614A (DRB1*0401/0701) Donor 597A (DRB1*0401/1302) P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 TGF-b1 in arbiträren Einheiten Donor 211A (DRB1*0401/0301) 8 6 4 2 0 Peptide Peptide Abb. 16: Detektion von intrazellulären transforming growth factor β1 (TGF-β1) in T-Gedächtniszellen + (CD4 ,CD45RA ) durch fluorescence activated cell sorting (FACS-Analyse). FACS-Messungen wurden in Doppelbestimmung durchgeführt. Mu= Mu-Peptide Die T-Gedächtniszellen wurden weiter auf ihre Fähigkeit zur Produktion von antiinflammatorischen Cytokinen nach Zugabe von P17 und Mu-Peptiden untersucht (Abb.16). T-Zellen mit regulatorischem Phänotyp zeichnen sich dadurch aus, dass sie durch die Sekretion von antiinflammatorisch wirksamen Cytokinen wie TGF-β1 und IL-10 bzw. der Expression des Transkriptionsfaktors FoxP3, die Aktivität von autoaggressiven T-Zellen inhibieren können. In der Analyse der TGF-β1 Expression wurde bei zwei Donoren (Donor 941A und 949A) eine Reduktion der Cytokinproduktion festgestellt. Die Donoren 849A und 952A zeigten keinen Unterschied in der TGF-β1 Sekretion nach Zugabe der MuPeptide zu P17 der PBMCs. Die meisten Donoren wiesen eine im Trend höhere TGF-β1 Produktion auf als die PBMCs, die mit P17 alleine stimuliert wurden. So konnte bei Donor 211A, 597A, 614A, 643A, und 686A eine deutlich erhöhte Produktion des antiinflammatorischen Cytokins TGF-β1 nach Einsatz von P17 mit allen Mu-Peptiden gezeigt werden. Ferner war dies auch bei dem Donor 912A, aber nicht in der Wiederholung und mit Ausnahme von P17+Mu4 der Fall. Diese aufsteigende Tendenz der TGF-β1 Produktion wurde ebenfalls bei Donor 914A, bei Donor 918A und bei Donor 945A mit jeweiliger Ausnahme von P17+Mu1 gezeigt. Detektiert wurde bei Donor 871A eine Zunahme der TGF-β1 Sekretion bei Einsatz von P17 mit Mu1 und Mu2 und bei Donor 913A bei Einsatz von P17 mit Mu3 und Mu5. Eine signifikante Erhöhung des antiinflammatorischen Cytokins TGF-β1 konnte bei einer ausgesprochen hohen Zahl an Donoren (zwölf Donoren) detektiert werden. Dagegen konnten nur bei vier Donoren (912A, 941A, 949A und 952A) keine Unterschiede in der TGF-β1 Sekretion gezeigt werden, wenn P17 mit P17+Mu5 verglichen wurde. IL-10 P17+Mu5 P17+Mu4 P17+Mu2 P17+Mu1 P17 IL-10 in arbiträren Einheiten Donor 941A (DRB1*0401/0701) 4 3 2 1 0 Peptide Abb.17: + - Detektion von intrazellulären Interleukin-10 (IL-10) in T-Gedächtniszellen (CD4 , CD45RA ) durch fluorescence activated cell sorting (FACS-Analyse). Mu= Mu- Peptide. Donor 941A wies bei Einsatz von P17 mit Mu1 und 2 keine wesentliche Veränderung des IL-10 Niveaus auf (Abb. 17). Nach Zugabe von Mu4 und 5 zu P17 konnte eine minimale Verringerung der IL-10-Produktion verzeichnet werden, die aber nicht signifikant war. Die Detektion des 8 6 4 2 0 Peptide extrazellulären P17+Mu5 8 6 4 2 0 Peptide Oberflächenproteins cytotoxisches P17+Mu5 P17+Mu4 Peptide P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 Peptide P17+Mu3 Donor 941A (DRB1*0401/0701) P17+Mu1 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 Peptide P17+Mu2 Donor 913A (DRB1*0401/0401) P17+Mu1 P17 Donor 875A (DRB1*0401/1102) P17 Donor 686A (DRB1*0401/0101) P17+Mu1 8 6 4 2 0 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 Donor 597A (DRB1*0401/1302) P17 8 6 4 2 0 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 4 3 2 1 0 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 24 18 12 6 0 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 Peptide P17+Mu4 Peptide P17+Mu3 Peptide P17+Mu3 Donor 949A (DRB1*0101/0701) P17+Mu2 Peptide P17+Mu2 Peptide P17+Mu2 Donor 918A (DRB1*0401/0101) P17+Mu1 P17 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 CTLA-4 in arbiträren Einheiten Peptide P17+Mu2 Donor 912A (DRB1*0401/1302) P17+Mu1 P17 Donor 871A (DRB1*0401/0404) P17+Mu1 P17 Donor 643A (DRB1*0401/0301) P17+Mu1 8 6 4 2 0 P17 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 CTLA-4 in arbiträren Einheiten Donor 211A (DRB1*0401/0301) P17 8 6 4 2 0 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 CTLA-4 in arbiträren Einheiten 8 6 4 2 0 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 CTLA-4 in arbiträren Einheiten 8 6 4 2 0 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 CTLA-4 in arbiträren Einheiten 4 3 2 1 0 CTLA-4 in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 CTLA-4 in arbiträren Einheiten CTLA-4 4 3 2 1 0 Donor 614A (DRB1*0401/0701) Peptide 4 3 2 1 0 Donor 849A (DRB1*0401/0701) Peptide 8 6 4 2 0 Donor 911A (DRB1*0404/0301) Peptide 8 6 4 2 0 Donor 914A (DRB1*0401/0301) Peptide 8 6 4 2 0 Donor 945A (DRB1*0401/1302) Peptide 8 6 4 2 0 Donor 952A (DRB1*0401/1303) Peptide Abb. 18: Detektion von extrazellulärem Cytotoxischem T-Lymphozyten-Antigen 4 (CTLA-4) durch fluorescence activated cell sorting (FACS-Analyse). FACS-Messungen wurden in Doppelbestimmung durchgeführt. Mu= Mu-Peptide. T- Lymphozyten Antigen (CTLA-4), welches die T-Zellen Aktivierung reguliert, zeigte bei den meisten Donoren (211A, 614A, 643A, 686A, 849A, 875A, 912A, 913A, 945A, und 949A keinen Unterschied in der Expression, wenn P17 mit P17 und den Mu-Peptiden verglichen wurde. Einige Donoren wiesen sogar bei Zugabe bestimmter Mu-Peptide zu P17 eine verminderte CTLA-4 Expression auf. Z.B. Donor 614A nach Zugabe von P17+Mu2 und 5, Donor 849A nach Zugabe von P17+Mu2, Donor 912A nach Zugabe von P17+Mu1, 2 und 3, Donor 945A nach Zugabe von P17+Mu1 und 2 und Donor 949A nach Zugabe von P17+Mu2 und 4 zu P17. Allerdings ist festzustellen, dass bei einigen Donoren eine aufsteigende Tendenz der CTLA-4 Expression beobachtet werden konnte. Dies war bei Donor 597A nach Einsatz von Mu4 und 5 und bei Donor 952A nach Einsatz von P17+Mu4 der Fall. Das CTLA-4-Niveau war bei den Donoren 871A, 911A, 914A, 918A und 941A bei Einsatz aller P17+Mu-Peptide in Relation zu P17 erhöht. Die Signifikanzanalyse ließ bei vier Donoren keine signifikante Erhöhung der CTLA-4 Expression erkennen, während dagegen bei dreizehn Donoren (211A, 597A, 643A, 686A, 871A, 911A, 912A, 913A, 914A, 918A, 941A, 949A und 952A) eine signifikante Zunahme der CTLA-4 Expression verzeichnet werden konnte, wenn P17 mit P17 und Mu5 verglichen wurde. GITR P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 GITR in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 GITR in arbiträren Einheiten P17+Mu5 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 GITR in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 GITR in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 12 9 6 3 0 P17 P17+Mu5 GITR in arbiträren Einheiten Donor 952A (DRB1*0401/1303) 8 6 4 2 0 P17+Mu4 8 6 4 2 0 Peptide Peptide Donor 949A (DRB1*0101/0701) P17+Mu3 P17+Mu2 P17+Mu1 P17 GITR in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu3 P17+Mu3 P17+Mu2 P17+Mu1 P17+Mu1 P17 GITR in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 Donor 945A (DRB1*0401/1302) Donor 941A (DRB1*0401/0701) 8 6 4 2 0 Peptide P17+Mu3 Peptide P17 Donor 918A (DRB1*0401/0101) P17+Mu2 12 9 6 3 0 Peptide 8 6 4 2 0 P17+Mu2 P17+Mu3 P17+Mu2 P17 P17 GITR in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 Donor 914A (DRB1*0401/0301) 8 6 4 2 0 Peptide P17+Mu1 Peptide Donor 913A (DRB1*0401/0401) Donor 912A (DRB1*0401/1302) P17+Mu1 P17+Mu2 P17+Mu1 P17 GITR in arbiträren Einheiten GITR in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 12 9 6 3 0 Peptide 12 9 6 3 0 P17 Donor 911A (DRB1*0404/0301) 8 6 4 2 0 Peptide P17 Peptide Donor 875A (DRB1*0401/1102) 8 6 4 2 0 P17 8 6 4 2 0 Peptide Donor 871A (DRB1*0401/0404) GITR in arbiträren Einheiten Donor 849A (DRB1*0401/0701) 4 3 2 1 0 Peptide GITR in arbiträren Einheiten Peptide Donor 686A (DRB1*0401/0101) p17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 P17 GITR in arbiträren Einheiten Donor 643A (DRB1*0401/0301) GITR in arbiträren Einheiten 8 6 4 2 0 Peptide 8 6 4 2 0 Peptide P17+Mu2 P17+Mu1 P17 GITR in arbiträren Einheiten P17+Mu5 P17+Mu4 P17+Mu3 P17+Mu2 P17+Mu1 12 9 6 3 0 Peptide GITR in arbiträren Einheiten Donor 614A (DRB1*0401/0701) Donor 597A (DRB1*0401/1302) P17 GITR in arbiträren Einheiten Donor 211A (DRB1*0401/0301) 8 6 4 2 0 Peptide Abb. 19: Detektion von extrazellulärem glucocorticoid-induced TNFR-related protein (GITR) durch fluorescence activated cell sorting (FACS-Analyse). FACS-Messungen wurden in Doppelbestimmung durchgeführt. Mu= Mu- Peptide. Die Analyse des Zelloberfächenproteins glucocorticoid-induced TNFR-related protein (GITR) (Abb. 19) ergab keine Veränderung der GITR-Expression nach Zugabe der Mu-Peptide zu P17 bei Donor 211A, 597A, 849A, 871A, 875A, 912A, 913A und 952A. Es ist allerdings festzustellen, dass bei Untersuchung von Donor 643A mit P17+Mu5, von Donor 686A mit P17+Mu1, von Donor 911A mit P17+Mu1 und 3, bei Donor 914A mit P17+Mu1 und von Donor 949A mit P17+Mu5 eine erhöhte Expression von GITR zu sehen war. Im Gegensatz hierzu war bei Donor 945A nach Zugabe von P17+Mu1 und 3 und bei Donor 949A nach Zugabe von P17+Mu1, 2 und 4 eine Abnahme der GITR Expression festzustellen. Drei Donoren zeigten eine leicht aufsteigende Tendenz der Expression des antiinflammatorischen Zelloberflächenmoleküls GITR. Dies war der Fall bei Donor 614A, 918A und 941A. Die Detektion der GITR-Expression wies bei 16 Donoren keine signifikante Veränderung auf. 4. Diskussion Der T1DM ist eine chronische Autoimmunerkrankung, der sich in einer Zerstörung der Insulin-produzierenden ß-Zellen des Pankreas durch autoreaktive bzw. diabetogene T-Zellen äußert. Es gibt mehrere Autoantigene, die in der T1DM Pathogenese eine Rolle spielen. Hierzu zählen das Proinsulin, GAD65 und IA2. Das Hauptzielautoantigen beim T1DM ist das Proinsulin. Autoantigene wie das Proinsulin werden in endozytischen Kompartimenten von APCs in Peptide gespalten und anschließend auf MHC Klasse II Moleküle beladen. Der AntigenMHC Klasse II Komplex wird dann durch den T-Zell Rezeptor von CD4+-T-Zellen erkannt und aktiviert. Folglich führt deren Sekretion von proinflammatorischen Cytokinen zur Zerstörung der ß-Zellen des Pankreas. Im Rahmen dieser Arbeit wurde P17 mit verschiedenen proteaseresistenten veränderten Peptidliganden (prAPLs) mit PBMCs von T1DM Patienten kultiviert, um herauzufinden, ob diese prAPL in der Lage sind die Sekretion proinflammatorischer Cytokine bzw. eine Induktion von antiinflammatorischen Cytokinen zu bewirken. Das wesentliche Ziel wäre, die Zerstörung der pankreatischen ß-Zellen, die in einer Manifestation des T1DM resultiert, künftig verhindern zu können. Zunächst erfolgte in Vorarbeiten die Herstellung eines auf Proinsulin basierenden APL durch eine Schnittstellen-spezifische Aminosäuren Substitution (CS-DAS) durch Austausch bestimmter Aminosäuren, die spezifisch durch Cathepsine erkannt wurden und an dieser Stelle das Peptid hydrolisieren, gegen, andere natürliche, bzw. D-Aminosäuren. Dadurch konnte ein proteaseresistentes Peptid synthetisiert werden. Dieses fand in dieser Arbeit Einsatz und könnte eine immunmodulatorische Funktion zur Verhinderung eines T1DM einnehmen. Im ersten Schritt wurde die Funktionalität der prAPLs (Mu Peptide) bestimmt. Ein fünftägiger T-Zell Assay wurde durchgeführt, bei dem PBMCs von T1DM Patienten durch Einsatz von P17 und P17 mit je einem Mu-Peptid in gleicher Konzentration, kultiviert wurden. Als Referenz diente die Stimulation von PBMCs mit P17. Einsatz fanden PBMCs von 17 verschiedenen T1DM Patienten, deren MHC Genotyp HLA-DR4 heterozygot und lediglich in einem Fall HLA-DR4 homozygot war. Ferner ein weiterer T1DM Patient, der HLA-DR1 heterozygot war. Nach erfolgter Inkubation wurde mit den Zellkulturüberständen des T-Zell Assays die Sekretion proinflammatorischer Cytokine wie TNF-α, IFN-γ, IL-6 und IL-17, und des antiinflammatorischen Cytokins TGF-ß1 mittels ELISA gemessen. Zusammenfassend konnte im ELISA durch den Einsatz des P17+Mu5 Peptids eine Reduktion der proinflammatorischen Cytokinsekretion von TNF-α bei bis zu 20% der Donoren, von IFN-γ ebenfalls bei bis zu 20% der Donoren, und von IL-17 bei bis zu 65% der Donoren erreicht werden. Ein Anstieg des antiinflammatorischen Cytokins TGF-ß1 konnte bei 47% der Donoren beobachtet werden. Eine präzisere Beurteilung des Cytokinprofils, das mit ELISA gewonnen wurde, erfolgte mit der intrazellulären FACS-Analyse. In der FACS-Analyse konnte durch den Einsatz des P17+Mu5 Peptids eine Reduktion der proinflammatorischen Cytokinsekretion von TNF-α bei bis zu 60% der Donoren, von IFN-γ bei bis zu 55% der Donoren, und von IL-17 bei bis zu 40% der Donoren erreicht werden. Ein signifikanter Anstieg des antiinflammatorischen Cytokins TGF-ß1 konnte bei bis zu 75% der Donoren beobachtet werden. Zusätzlich konnte die Expression des Zelloberflächenmoleküls CTLA-4 bei bis zu 75% der Donoren erhöht werden. 4.1. Vorarbeiten: Herstellung von P17 proteaseresistenten Peptiden durch cleavage-site-directed amino-acid substitution (CS-DAS) Eine Strategie in der Prävention des T1DM könnten Peptide darstellen, die an MHC Klasse II Moleküle binden und eine Aktivierung von diabetogenen T-Zellen hemmen. In den 80er Jahren gelang es einen veränderten Peptidliganden (APL), der mit dem MHC-Peptid-TCR Komplex interferiert und eine T-Zell Aktivierung hemmt, zu entwickeln [13]. Ein wesentlicher Grund für die geringe Halbwertszeit der APLs ist sicherlich die fehlende Resistenz gegenüber proteolytischer Aktivität im Serum oder im endozytischen Kompartiment. Aufgrund dieser Tatsache wurde das kürzlich entdeckte T-Zell Epitop des Proinsulins mit der Peptidsequenz Proinsulin74-90 (P17) (AGSLQPLALEGSLQKRK), das von T-Zellen von T1DM Patienten erkannt wird, herangezogen um einen veränderten Peptidliganden herzustellen und mit der Schnittstellen-spezifischen Aminosäuren Substitution (CS-DAS) wurde dessen Proteaseresistenz erzielt. Um diese Proteaseresistenz zu erreichen, wurde P17 mit verschiedenen Cathepsinen, die am häufigsten in den Antigenprozessierungsweg involviert sind, inkubiert. Weder CatD, X, C, B noch H waren in der Lage P17 zu verdauen. Allerdings spaltete CatG, S, L, V P17. Die AS, die durch Cathepsine erkannt wurden, wurden durch natürliche oder D-AS ersetzt (Mu1-Mu5) und mit lyosomalen Cathepsinen der B-Zelllinie (BLC) und mit einer CatG exprimierenden myelomonozytären Zelllinie (THP-1) inkubiert. Mit der Inkubation von Mu2 und Mu3 mit lysosomalen Cathepsinen von THP-1 konnte keine wesentliche Proteaseresistenz erreicht werden. Erst der Austausch von DThreonin am C-terminalen Ende von Mu4 und Mu5 erbrachte die Generierung eines proteaseresistenten Peptids. 4.2. T-Zell Assay und Cytokin-Detektion mittels ELISA Um proteaseresistente APLs zu testen, wurde P17 mit Mu-Peptiden und PBMCs von T1DM Spendern oder Kontrollprobanden (HLA-DRB1*0401) in einem funktionellen T-Zell-Assay kokultivert. P17 alleine fungierte als Positivkontrolle, welches T-Zellen aktiviert. Ziel war es zu prüfen, ob das Cytokinprofil von T-Zellen verändert wird. Dabei konnte beobachtet werden, dass die Sekretion des proinflammatorischen Cytokins IL-17, das bei der Pathogenese von Autoimmunerkrankungen eine entscheidende Rolle spielt [10], bei PBMCs, die mit P17 und Mu5 inkubiert wurden reduziert war, im Vergleich zu PBMCs, die nur mit P17 alleine inkubiert wurden. Bei TNF-α konnte in einigen Fällen (n=3) eine Reduktion beobachtet werden, in anderen Fällen wurden keine signifikanten Unterschiede (n=4) gefunden. Einige Donoren wiesen sogar einen erhöhten TNFα Spiegel auf (n=7). Bei der IL-6 Sekretion wurde kein Unterscheid zwischen P17 kokultiviert mit Mu5 festgestellt (n=8). Bei 8 Donoren war die IL-6 Sekretion sogar höher als bei der Kontrolle. Neben der Sekretion von proinflammatorischen Cytokinen wurde das antiinflammatorische Cytokin TGF-ß1 untersucht. Hier wurden beim TGF-ß1-spezifischen ELISA keine signifikanten Unterschiede zwischen der TGF-ß1 Konzentration in PBMCs, die mit P17 und Mu5 bzw. denen, die nur mit P17 kultiviert wurden (n=8) detektiert. Auffallend war die signifikante Erhöhung des von vermutlich Treg-Zellen produzierten antiinflammatorischen Cytokins TGF-ß1 bei n=7 T1DM Donoren. Allerdings gibt es außer TGF-ß1 noch andere antiinflammatorische Cytokine wie z.B. IL-10. Dieses Cytokin lässt sich aber nur schwer im ELISA nachweisen, was in dieser Arbeit ebenfalls der Fall war. Der IL-10-spezifische ELISA ergab kein Signal. Jedoch kann durch den ELISA nicht detektiert werden, welche Zellen das Cytokin TGF-ß1 sekretieren. Daher wurde eine genauere Analyse mit Hilfe der Durchflusszytometrie durchgeführt (siehe 4.3.). 4.3. Intrazelluläre Cytokinbestimmung durch FACS-Analyse Eine umfassendere und quantitative Aussage zum Cytokinprofil, der im T-Zell Assay stimulierten PBMCs konnte durch die Durchflusszytometrie bestimmt werden. Hierbei kamen Oberflächenmarker spezifisch für die Gedächtnis-T-Zellen, die CD4+-und CD45RA--T-Zellen, sowie auch intrazelluläre Marker, zur CytokinDetektion von IL-17, TNF-α, IFN-γ, IL-6, TGF-ß1, zum Einsatz. Der Vorteil der FACS-Analyse ist hierbei, dass bestimmte CD4+-T-Zellen direkt auf ihre Cytokinsekretion untersucht werden können. Vermutetet wurde, dass die Detektion von Cytokinen nicht unbedingt von T-Zellen stammt. PBMCs enthalten neben den CD4+-T-Zellen weitere cytokinausschüttende Zellen. Somit ist die Cytokinsekretion nicht nur den CD4+-TZellen zuzuordnen. Deshalb wurde die Durchflusszytometrie zur Bestimmung der Cytokinexpression von Gedächtnis-CD4+-T-Zellen eingesetzt. Nach Quantifizierung der Ergebnisse mittels Flowjo Software konnten bis zu 40% verminderte IL-17 Konzentrationen gefunden werden, wenn P17+Mu5 im T-ZellAssay untersucht wurde. Die niedrigen Level von IL-17 bestätigten, dass die Sekretion von IL-17 durch CD4+-T-Zellen, vermutlich TH17-Zellen, durch Einsatz von Mu5 gehemmt wurde. Erklären lässt sich dies durch die Tatsache, dass die Mu-Peptide die Interaktion zwischen APCs und TH17-Zellen durch Bindung an MHC Klasse II Moleküle hemmen. Eine andere Erklärung hierfür könnte sein, dass TH17-Zellen indirekt durch Cytokine gehemmt werden, die von T-regulatorischen Zellen (Tregs) ausgeschüttet wurden. Bei TNF-α konnte eine Verminderung der Cytokinsekretion beobachtet werden, wenn P17 und Mu5 in der PBMC Kultur eingesetzt wurden (n=8), obwohl die TNF-α Sekretion beachtlich höher war als beim TNF-α spezifischen ELISA. Ein verminderter TNF-α Spiegel wurden bei bis zu 60% der Donoren (n=13) gemessen. Die Messung der IFN-γ Konzentration nach Stimulation wies verminderte IFN-γ Werte bei 7 Donoren und erhöhte IFN-γ Werte bei 6 Donoren auf. Insgesamt wurden verminderte IFN-γ Level bei bis zu 55% (n=13) der Donoren detektiert. Die Ausschüttung von IL-6 änderte sich nicht signifikant (n=5). IL-1 und IL-6 induziert in Kombination mit dem antiinflammatorischen Cytokin TGF-ß1 TH-17-Zellen, und TH17-Zellen hemmen die Funktion von Treg-Zellen [40]. Dies erklärt jedoch nicht die reduzierte IL-17 Detektion, die durch Mu5 gefunden wurde. Daher sind wohl weitere Mechanismen notwendig um TH17 zu induzieren. Auffallend war die TGF-ß1 Sekretion. Diese war zwar bei 4 Donoren unverändert, allerdings bei einer Vielzahl von Donoren (n=12) ausgesprochen erhöht nach Inkubation von P17 und Mu5. Insgesamt konnte ein erhöhter TGF-ß1 Level bei bis zu 75% der Donoren detektiert werden bei einer Gesamtzahl von 16 Donoren. Die Analyse des Zelloberflächenmoleküls CTLA-4 ergab keine signifikante Reduktion bei 4 Donoren und sogar eine erhöhte Expression bei 13 Donoren. Dies war bei einer Gesamtanzahl von 17 Donoren eine erhöhte Zelloberflächenexpression an CTLA-4 bei bis zu 75% der Fall. Ebenso war die Beobachtung bei GITR, bei der bei 16 Donoren nicht signifikante Ergebnisse erhalten wurden. Allgemein ist festzuhalten, dass die Sensitivität der Cytokin-Detektion mit der FACS-Analyse höher war, als mit ELISA. Dies lässt sich vermutlich durch die Fähigkeit des FACS Gerätes erklären, dass jede einzelne Zelle auf ihre intrazelluläre Cytokinproduktion analysiert wurde. 4.4. Regulatorische T-Zellen (Treg-Zellen) Die Treg-Zellen sind entscheidend für die periphere Toleranz [84]. Eine Einteilung der Treg-Zellen erfolgt in 2 Untergruppen, die CD4+-CD25+-FoxP3 exprimierenden Treg-Zellen (FoxP3 Treg-Zellen) und CD4+-IL-10 sezernierenden Treg-Zellen (Tr1) [49]. FoxP3 positive Treg-Zellen sind wesentlich für die Immunhomöostase sowie zum Schutz vor Autoimmunkrankheiten [82]. Naive CD4+-T-Zellen können sich bei Anwesenheit von IL-10 in der Peripherie durch APCs zu Tr1 (Treg-Zellen) differenzieren. Diese schütten dann antinflammatorische Cytokine TGF-ß1 und IL10 aus, die eine Hemmung autoaggressiver T-Zellen bewirken [9, 12, 25, 57]. Sie sind somit Hauptregulatoren der autoreaktiven T-Zellen [5] und können folglich autoimmune Prozesse verhindern. Bei unseren Experimenten war ein deutlicher Anstieg des antiinflammatorischen Cytokins TGF-ß1 in Gedächtnis T-Zellen zu verzeichnen, nachdem PBMCs mit P17 und Mu5 kokultiviert wurden (Abb.20). Ferner konnte eine erhöhte Expression des Zelloberflächenmoleküls CTLA-4 in TZellen, die mit P17 und Mu5 behandelt wurden, gefunden werden. Die Vermutung liegt nahe, dass Treg-Zellen durch Mu5 in unseren Experimenten aktiviert wurden. CTLA-4 spielt in der Entwicklung der Autoimmunität eine wichtige Rolle [94]. Eine erhöhte Expression von CTLA-4 auf Treg-Zellen ist von Relevanz für die Interaktion zwischen B7 Molekülen und dendritischen Zellen [88]. Diese B7 Moleküle spielen als kostimulatorische Signale der Treg-Zellen eine wichtige Rolle in der Kontrolle, Initierung, Progression und Pathogenese von Autoimmunkrankheiten wie dem T1DM [11]. Die Induktion von Treg-Zellen ist somit von großer Bedeutung in der Aufrechterhaltung der Autoimmunkrankheiten. Immunhomöostase Auch ein neuer der T-Zell kostimulatorischen Signale vielversprechenden Therapiestrategie und in der Therapieansatz Regulation resultieren, da Prävention im Bereich könnte in T-Zellen von der einer besser verschiedenen Stadien der Aktivierung bezielt werden könnten [11]. Abb. 20: Haupthistokompatibilitätskomplex (MHC) und T-Zellrezeptor (TCR) Bindungsstellen In blau Aminosäuren, die an Haupthistokompatibilitätskomplex (MHC) binden. In rot Aminosäuren, die an T-Zellrezeptor (TCR) binden. TGF-β1= transforming growth factor β1 in 4.5. Perspektive Die aktuelle Therapie des T1DM beinhaltet die Substitution des fehlenden Insulins, allerdings greift diese Therapie erst nach Zerstörung der ß-Zellen und ist somit nur eine symptomatische Therapie. Die Ursache der Erkrankung wird somit nicht behoben. Interessant sind deshalb Therapieansätze, die die Ursache der Entstehung des T1DM behandeln und nicht erst die Symptome. Hierzu zählen antigenspezifische Therapieansätze, die ein vielversprechendes Instrument in der gezielten Suppression der autoaggressiven bzw. diabetogenen T-Zellen darstellen und keine generelle Immunsuppression induzieren. Der Ansatz der antigenspezifischen Therapie, der in dieser Arbeit gezeigt wurde, beinhaltet den Einsatz von prAPLs (Mu-Peptide), die sich ähnlich verhalten wie TZell Epitope aber anstelle des antigenen Peptids an das MHC Klasse II Molekül binden und eine T-Zell-Antwort unterbinden bzw. eine TGF-ß1 Sekretion induzieren. Basierend auf diesen Daten könnten die Mu-Peptide als Immunmodulatoren angesehen werden, die autoaggressive T-Zellen hemmen bzw. CD4+-T-Zellen mit antiinflammatorischer Funktion stimulieren [15]. Somit sind diese Peptide, besonders Mu5, ein vielversprechendes Instrument in der Suppression einer autoaggressiven T-Zell-Antwort und können als Immunmodulatoren niedrigdosiert therapeutisch eingesetzt werden, da sie proteolytisch nicht abgebaut werden. Eine weitere Möglichkeit, um eine noch effektivere Reduktion der autoaggressiven T-Zellen zu erreichen, ist der Einsatz von verschiedenen und zellgängigen prAPLs. Hierdurch kann die intrazelluläre Kompetition um die Peptidbindungsstelle des MHC zugunsten der prAPLs verschoben werden und somit können weniger Autoantigenpeptide an MHC II binden. Nach erfolgreichem Einsatz der prAPLs in vitro wie in dieser Arbeit gezeigt werden konnte, könnten diese prAPLs in einem geeigneten Tiermodell getestet werden. 5. Zusammenfassung Proinsulin ist das wichtigste Autoantigen in der Pathogenese des Typ 1 Diabetes mellitus (T1DM). Die Proinsulin basierende Sequenz 74-90 (P17), ein T-Zell Epitop, welches autoaggressive bzw. diabetogene T-Zellen aktiviert, diente in dieser Arbeit als Basis zur Generierung proteaseresistenter veränderter Peptidliganden (prAPLs). Die Aktivierung von diabetogenen T-Zellen führt zur Sekretion von proinflammatorischen Cytokinen, die mitverantwortlich sind für die Zerstörung der insulinproduzierenden -Zellen des Pankreas. In diesem Zusammenhang wurde experimentell die Hypothese bearbeitet, ob prAPLs zum einen an das Humane-Leukozyten-Antigen-(HLA) DRB1*0401 binden und zum anderen eine Suppression proinflammatorischer bzw. eine Induktion antiinflammatorischer Cytokine bewirken könnten, welche direkt regulatorisch auf diabetogene T-Zellen wirken z.B. durch Aktivierung von regulatorischen T-Zellen (Treg-Zellen). Dabei wurden periphere mononukleären Zellen des Blutes (PBMCs) von T1DM Patienten mit P17 und je einem prAPL im T-Zell-Assay kokultiviert. Die Analyse des Cytokinprofils erfolgte mittels cytokinspezifischem ELISA und Durchflusszytometrie. Der Einsatz eines der prAPLs (P17+Mu5) zeigte eine Reduktion der proinflammatorischen Cytokinsekretion von TNF- bei bis zu 60%, IFN- bei bis zu 55% und IL-17 bei bis zu 40% der Donoren. Ein signifikanter Anstieg des antiinflammatorischen Cytokins TGF-ß1 konnte bei bis zu 75% der Donoren beobachtet werden, wenn P17 mit P17+Mu5 eingesetzt wurde. Zusätzlich konnte die Expression des cytotoxischen T-Lymphozyten-Antigens 4 (CTLA-4), welches regulatorisch auf T-Zellen wirkt, bei bis zu 75% der Donoren erhöht werden. Diese Ergebnisse demonstrieren, dass P17+Mu5 eingesetzt werden kann, um diabetogene T-Zellen zu supprimieren bzw. ein regulatorisches Cytokinmilieu zu induzieren. Daher besitzt P17+Mu5 ein großes Potenzial für den Einsatz in der immunmodulatorischen Therapie zur Protektion von -Zellen und eventuell zur Vermeidung eines T1DM. 6. 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. Literaturverzeichnis Adorini, L., S. Muller, F. Cardinaux, P.V. Lehmann, F. Falcioni, and Z.A. Nagy, In vivo competition between self peptides and foreign antigens in T-cell activation. Nature, 1988. 334: p. 623-5. Allen, J.S., K. Pang, A. Skowera, R. Ellis, C. Rackham, B. Lozanoska-Ochser, T. Tree, R.D. Leslie, J.M. Tremble, C.M. Dayan, and M. Peakman, Plasmacytoid dendritic cells are proportionally expanded at diagnosis of type 1 diabetes and enhance islet autoantigen presentation to T-cells through immune complex capture. Diabetes, 2009. 58: p. 138-45. Anderton, S.M. and D.C. Wraith, Selection and fine-tuning of the autoimmune T-cell repertoire. Nat Rev Immunol, 2002. 2: p. 487-98. Arif, S., T.I. Tree, T.P. Astill, J.M. Tremble, A.J. Bishop, C.M. Dayan, B.O. Roep, and M. Peakman, Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest, 2004. 113: p. 451-63. Bach, J.F., Autoimmune diseases as the loss of active "self-control". Ann N Y Acad Sci, 2003. 998: p. 161-77. Baekkeskov, S., H.J. Aanstoot, S. Christgau, A. Reetz, M. Solimena, M. Cascalho, F. Folli, H. Richter-Olesen, and P. De Camilli, Identification of the 64K autoantigen in insulindependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature, 1990. 347: p. 151-6. Banchereau, J. and R.M. Steinman, Dendritic cells and the control of immunity. Nature, 1998. 392: p. 245-52. Barrat, F.J., D.J. Cua, A. Boonstra, D.F. Richards, C. Crain, H.F. Savelkoul, R. de WaalMalefyt, R.L. Coffman, C.M. Hawrylowicz, and A. O'Garra, In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med, 2002. 195: p. 603-16. Bettelli, E., T. Korn, M. Oukka, and V.K. Kuchroo, Induction and effector functions of T(H)17 cells. Nature, 2008. 453: p. 1051-7. Bettelli, E., M. Oukka, and V.K. Kuchroo, T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol, 2007. 8: p. 345-50. Boehm, B.O. and J.A. Bluestone, Differential roles of costimulatory signaling pathways in type 1 diabetes mellitus. Rev Diabet Stud, 2004. 1: p. 156-64. Boehm, B.O., S. Rosinger, G. Sauer, B.J. Manfras, D. Palesch, S. Schiekofer, H. Kalbacher, and T. Burster, Protease-resistant human GAD-derived altered peptide ligands decrease TNF-alpha and IL-17 production in peripheral blood cells from patients with type 1 diabetes mellitus. Mol Immunol, 2009. 46: p. 2576-84. Brusko, T.M., A.L. Putnam, and J.A. Bluestone, Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev, 2008. 223: p. 371-90. Burster, T., A. Beck, E. Tolosa, V. Marin-Esteban, O. Rotzschke, K. Falk, A. Lautwein, M. Reich, J. Brandenburg, G. Schwarz, H. Wiendl, A. Melms, R. Lehmann, S. Stevanovic, H. Kalbacher, and C. Driessen, Cathepsin G, and not the asparagine-specific endoprotease, controls the processing of myelin basic protein in lysosomes from human B lymphocytes. J Immunol, 2004. 172: p. 5495-503. Burster, T. and B.O. Boehm, Processing and presentation of (pro)-insulin in the MHC class II pathway: the generation of antigen-based immunomodulators in the context of type 1 diabetes mellitus. Diabetes Metab Res Rev. 26: p. 227-38. Burster, T., V. Marin-Esteban, B.O. Boehm, S. Dunn, O. Rotzschke, K. Falk, E. Weber, S.H. Verhelst, H. Kalbacher, and C. Driessen, Design of protease-resistant myelin basic protein-derived peptides by cleavage site directed amino acid substitutions. Biochem Pharmacol, 2007. 74: p. 1514-23. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. Chapman, H.A., Endosomal proteases in antigen presentation. Curr Opin Immunol, 2006. 18: p. 78-84. Concannon, P., H.A. Erlich, C. Julier, G. Morahan, J. Nerup, F. Pociot, J.A. Todd, and S.S. Rich, Type 1 diabetes: evidence for susceptibility loci from four genome-wide linkage scans in 1,435 multiplex families. Diabetes, 2005. 54: p. 2995-3001. Congia, M., S. Patel, A.P. Cope, S. De Virgiliis, and G. Sonderstrup, T cell epitopes of insulin defined in HLA-DR4 transgenic mice are derived from preproinsulin and proinsulin. Proc Natl Acad Sci U S A, 1998. 95: p. 3833-8. Cucca, F., R. Lampis, M. Congia, E. Angius, S. Nutland, S.C. Bain, A.H. Barnett, and J.A. Todd, A correlation between the relative predisposition of MHC class II alleles to type 1 diabetes and the structure of their proteins. Hum Mol Genet, 2001. 10: p. 2025-37. D'Cruz, L.M. and L. Klein, Development and function of agonist-induced CD25+Foxp3+ regulatory T cells in the absence of interleukin 2 signaling. Nat Immunol, 2005. 6: p. 1152-9. Di Lorenzo, T.P., M. Peakman, and B.O. Roep, Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol, 2007. 148: p. 1-16. Dunne, D.W. and A. Cooke, A worm's eye view of the immune system: consequences for evolution of human autoimmune disease. Nat Rev Immunol, 2005. 5: p. 420-6. Durinovic-Bello, I., Autoimmune diabetes: the role of T cells, MHC molecules and autoantigens. Autoimmunity, 1998. 27: p. 159-77. Durinovic-Bello, I., S. Rosinger, J.A. Olson, M. Congia, R.C. Ahmad, M. Rickert, J. Hampl, H. Kalbacher, J.W. Drijfhout, E.D. Mellins, S. Al Dahouk, T. Kamradt, M.J. Maeurer, C. Nhan, B.O. Roep, B.O. Boehm, C. Polychronakos, G.T. Nepom, W. Karges, H.O. McDevitt, and G. Sonderstrup, DRB1*0401-restricted human T cell clone specific for the major proinsulin73-90 epitope expresses a down-regulatory T helper 2 phenotype. Proc Natl Acad Sci U S A, 2006. 103: p. 11683-8. Eizirik, D.L., M.L. Colli, and F. Ortis, The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol, 2009. 5: p. 219-26. Endl, J., S. Rosinger, B. Schwarz, S.O. Friedrich, G. Rothe, W. Karges, M. Schlosser, T. Eiermann, D.J. Schendel, and B.O. Boehm, Coexpression of CD25 and OX40 (CD134) receptors delineates autoreactive T-cells in type 1 diabetes. Diabetes, 2006. 55: p. 50-60. Fontenot, J.D., J.P. Rasmussen, M.A. Gavin, and A.Y. Rudensky, A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol, 2005. 6: p. 1142-51. French, M.B., J. Allison, D.S. Cram, H.E. Thomas, M. Dempsey-Collier, A. Silva, H.M. Georgiou, T.W. Kay, L.C. Harrison, and A.M. Lew, Transgenic expression of mouse proinsulin II prevents diabetes in nonobese diabetic mice. Diabetes, 1997. 46: p. 34-9. Geluk, A., K.E. van Meijgaarden, N.C. Schloot, J.W. Drijfhout, T.H. Ottenhoff, and B.O. Roep, HLA-DR binding analysis of peptides from islet antigens in IDDM. Diabetes, 1998. 47: p. 1594-601. Grinberg-Bleyer, Y., D. Saadoun, A. Baeyens, F. Billiard, J.D. Goldstein, S. Gregoire, G.H. Martin, R. Elhage, N. Derian, W. Carpentier, G. Marodon, D. Klatzmann, E. Piaggio, and B.L. Salomon, Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. J Clin Invest. 120: p. 4558-68. Groux, H., A. O'Garra, M. Bigler, M. Rouleau, S. Antonenko, J.E. de Vries, and M.G. Roncarolo, A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature, 1997. 389: p. 737-42. Herold G: Endokrinologie. In: Herold G (Hrsg) Innere Medizin, Herold Köln, p. 658-685 (2008). Herr, M., F. Dudbridge, P. Zavattari, F. Cucca, C. Guja, R. March, R.D. Campbell, A.H. Barnett, S.C. Bain, J.A. Todd, and B.P. Koeleman, Evaluation of fine mapping strategies for a multifactorial disease locus: systematic linkage and association analysis of IDDM1 in the HLA region on chromosome 6p21. Hum Mol Genet, 2000. 9: p. 1291-301. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. Hof H, Dörries R: Mechanismen der angeborenen u. der erworbenen Immunität. In: Bob A, Bob K (Hrsg) Duale Reihe Medizinische Mikrobiologie, 3. Aufl., Georg Thieme Verlag Stuttgart, p. 98-131 (2005). Honkanen, J., J.K. Nieminen, R. Gao, K. Luopajarvi, H.M. Salo, J. Ilonen, M. Knip, T. Otonkoski, and O. Vaarala, IL-17 immunity in human type 1 diabetes. J Immunol. 185: p. 1959-67. Howson, J.M., D.B. Dunger, S. Nutland, H. Stevens, L.S. Wicker, and J.A. Todd, A type 1 diabetes subgroup with a female bias is characterised by failure in tolerance to thyroid peroxidase at an early age and a strong association with the cytotoxic T-lymphocyteassociated antigen-4 gene. Diabetologia, 2007. 50: p. 741-6. Jasinski, J.M. and G.S. Eisenbarth, Insulin as a primary autoantigen for type 1A diabetes. Clin Dev Immunol, 2005. 12: p. 181-6. Jiang, H. and L. Chess, An integrated view of suppressor T cell subsets in immunoregulation. J Clin Invest, 2004. 114: p. 1198-208. Johnson, K.P., B.R. Brooks, J.A. Cohen, C.C. Ford, J. Goldstein, R.P. Lisak, L.W. Myers, H.S. Panitch, J.W. Rose, and R.B. Schiffer, Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology, 1995. 45: p. 1268-76. Jun, H.S., C.S. Yoon, L. Zbytnuik, N. van Rooijen, and J.W. Yoon, The role of macrophages in T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Exp Med, 1999. 189: p. 347-58. Kared, H., A. Masson, H. Adle-Biassette, J.F. Bach, L. Chatenoud, and F. Zavala, Treatment with granulocyte colony-stimulating factor prevents diabetes in NOD mice by recruiting plasmacytoid dendritic cells and functional CD4(+)CD25(+) regulatory T-cells. Diabetes, 2005. 54: p. 78-84. Kelly, M.A., M.L. Rayner, C.H. Mijovic, and A.H. Barnett, Molecular aspects of type 1 diabetes. Mol Pathol, 2003. 56: p. 1-10. Kent, S.C., Y. Chen, L. Bregoli, S.M. Clemmings, N.S. Kenyon, C. Ricordi, B.J. Hering, and D.A. Hafler, Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature, 2005. 435: p. 224-8. Knip, M. and H.K. Akerblom, Environmental factors in the pathogenesis of type 1 diabetes mellitus. Exp Clin Endocrinol Diabetes, 1999. 107 Suppl 3: p. S93-100. Kockum, I., C.B. Sanjeevi, S. Eastman, M. Landin-Olsson, G. Dahlquist, and A. Lernmark, Complex interaction between HLA DR and DQ in conferring risk for childhood type 1 diabetes. Eur J Immunogenet, 1999. 26: p. 361-72. Kyvik, K.O., A. Green, and H. Beck-Nielsen, Concordance rates of insulin dependent diabetes mellitus: a population based study of young Danish twins. BMJ, 1995. 311: p. 913-7. Lafaille, J.J., The role of helper T cell subsets in autoimmune diseases. Cytokine Growth Factor Rev, 1998. 9: p. 139-51. Lamont, A.G., M.F. Powell, S.M. Colon, C. Miles, H.M. Grey, and A. Sette, The use of peptide analogs with improved stability and MHC binding capacity to inhibit antigen presentation in vitro and in vivo. J Immunol, 1990. 144: p. 2493-8. Lande, R. and M. Gilliet, Plasmacytoid dendritic cells: key players in the initiation and regulation of immune responses. Ann N Y Acad Sci. 1183: p. 89-103. Lee, K.H., K.W. Wucherpfennig, and D.C. Wiley, Structure of a human insulin peptideHLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol, 2001. 2: p. 501-7. Lehuen, A., J. Diana, P. Zaccone, and A. Cooke, Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. 10: p. 501-13. Li, Q., B. Xu, S.A. Michie, K.H. Rubins, R.D. Schreriber, and H.O. McDevitt, Interferonalpha initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A, 2008. 105: p. 12439-44. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. 64. 65. 66. 67. 68. 69. 70. Ludewig, B., B. Odermatt, S. Landmann, H. Hengartner, and R.M. Zinkernagel, Dendritic cells induce autoimmune diabetes and maintain disease via de novo formation of local lymphoid tissue. J Exp Med, 1998. 188: p. 1493-501. Luo, X., K.V. Tarbell, H. Yang, K. Pothoven, S.L. Bailey, R. Ding, R.M. Steinman, and M. Suthanthiran, Dendritic cells with TGF-beta1 differentiate naive CD4+CD25- T cells into islet-protective Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A, 2007. 104: p. 2821-6. Malek, T.R. and A.L. Bayer, Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol, 2004. 4: p. 665-74. Max, H., T. Halder, M. Kalbus, V. Gnau, G. Jung, and H. Kalbacher, A 16mer peptide of the human autoantigen calreticulin is a most prominent HLA-DR4Dw4-associated selfpeptide. Hum Immunol, 1994. 41: p. 39-45. McKinney, P.A., R. Parslow, K.A. Gurney, G.R. Law, H.J. Bodansky, and R. Williams, Perinatal and neonatal determinants of childhood type 1 diabetes. A case-control study in Yorkshire, U.K. Diabetes Care, 1999. 22: p. 928-32. Mohan, J.F., M.G. Levisetti, B. Calderon, J.W. Herzog, S.J. Petzold, and E.R. Unanue, Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes. Nat Immunol. 11: p. 350-4. Mosmann, T.R., H. Cherwinski, M.W. Bond, M.A. Giedlin, and R.L. Coffman, Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol, 1986. 136: p. 2348-57. Nakayama, M., N. Abiru, H. Moriyama, N. Babaya, E. Liu, D. Miao, L. Yu, D.R. Wegmann, J.C. Hutton, J.F. Elliott, and G.S. Eisenbarth, Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature, 2005. 435: p. 220-3. Nejentsev, S., J.M. Howson, N.M. Walker, J. Szeszko, S.F. Field, H.E. Stevens, P. Reynolds, M. Hardy, E. King, J. Masters, J. Hulme, L.M. Maier, D. Smyth, R. Bailey, J.D. Cooper, G. Ribas, R.D. Campbell, D.G. Clayton, and J.A. Todd, Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature, 2007. 450: p. 887-92. Nejentsev, S., N. Walker, D. Riches, M. Egholm, and J.A. Todd, Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science, 2009. 324: p. 387-9. Olmos, P., R. A'Hern, D.A. Heaton, B.A. Millward, D. Risley, D.A. Pyke, and R.D. Leslie, The significance of the concordance rate for type 1 (insulin-dependent) diabetes in identical twins. Diabetologia, 1988. 31: p. 747-50. Ou, D., L.A. Mitchell, D.L. Metzger, S. Gillam, and A.J. Tingle, Cross-reactive rubella virus and glutamic acid decarboxylase (65 and 67) protein determinants recognised by T cells of patients with type I diabetes mellitus. Diabetologia, 2000. 43: p. 750-62. Oyer, P.E., S. Cho, J.D. Peterson, and D.F. Steiner, Studies on human proinsulin. Isolation and amino acid sequence of the human pancreatic C-peptide. J Biol Chem, 1971. 246: p. 1375-86. Pedotti, R., M. Sanna, M. Tsai, J. DeVoss, L. Steinman, H. McDevitt, and S.J. Galli, Severe anaphylactic reactions to glutamic acid decarboxylase (GAD) self peptides in NOD mice that spontaneously develop autoimmune type 1 diabetes mellitus. BMC Immunol, 2003. 4: p. 2. Peng, Y., Y. Laouar, M.O. Li, E.A. Green, and R.A. Flavell, TGF-beta regulates in vivo expansion of Foxp3-expressing CD4+CD25+ regulatory T cells responsible for protection against diabetes. Proc Natl Acad Sci U S A, 2004. 101: p. 4572-7. Pugliese, A., D. Brown, D. Garza, D. Murchison, M. Zeller, M.J. Redondo, J. Diez, G.S. Eisenbarth, D.D. Patel, and C. Ricordi, Self-antigen-presenting cells expressing diabetesassociated autoantigens exist in both thymus and peripheral lymphoid organs. J Clin Invest, 2001. 107: p. 555-64. Redondo, M.J., J. Jeffrey, P.R. Fain, G.S. Eisenbarth, and T. Orban, Concordance for islet autoimmunity among monozygotic twins. N Engl J Med, 2008. 359: p. 2849-50. 71. 72. 73. 74. 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 86. Redondo, M.J., L. Yu, M. Hawa, T. Mackenzie, D.A. Pyke, G.S. Eisenbarth, and R.D. Leslie, Heterogeneity of type I diabetes: analysis of monozygotic twins in Great Britain and the United States. Diabetologia, 2001. 44: p. 354-62. Reijonen, H., T.L. Daniels, A. Lernmark, and G.T. Nepom, GAD65-specific autoantibodies enhance the presentation of an immunodominant T-cell epitope from GAD65. Diabetes, 2000. 49: p. 1621-6. Risch, N., Assessing the role of HLA-linked and unlinked determinants of disease. Am J Hum Genet, 1987. 40: p. 1-14. Roncarolo, M.G., S. Gregori, M. Battaglia, R. Bacchetta, K. Fleischhauer, and M.K. Levings, Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev, 2006. 212: p. 28-50. Rudensky, A. and C. Beers, Lysosomal cysteine proteases and antigen presentation. Ernst Schering Res Found Workshop, 2006: p. 81-95. Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone, B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity, 2000. 12: p. 431-40. Sarvetnick, N., J. Shizuru, D. Liggitt, L. Martin, B. McIntyre, A. Gregory, T. Parslow, and T. Stewart, Loss of pancreatic islet tolerance induced by beta-cell expression of interferongamma. Nature, 1990. 346: p. 844-7. Schloot, N.C., B.O. Roep, D. Wegmann, L. Yu, H.P. Chase, T. Wang, and G.S. Eisenbarth, Altered immune response to insulin in newly diagnosed compared to insulin-treated diabetic patients and healthy control subjects. Diabetologia, 1997. 40: p. 564-72. Sercarz, E.E., P.V. Lehmann, A. Ametani, G. Benichou, A. Miller, and K. Moudgil, Dominance and crypticity of T cell antigenic determinants. Annu Rev Immunol, 1993. 11: p. 729-66. Sgouroudis, E. and C.A. Piccirillo, Control of type 1 diabetes by CD4+Foxp3+ regulatory T cells: lessons from mouse models and implications for human disease. Diabetes Metab Res Rev, 2009. 25: p. 208-18. Starr, T.K., S.C. Jameson, and K.A. Hogquist, Positive and negative selection of T cells. Annu Rev Immunol, 2003. 21: p. 139-76. Tang, Q. and J.A. Bluestone, The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol, 2008. 9: p. 239-44. Tran, D.Q., H. Ramsey, and E.M. Shevach, Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood, 2007. 110: p. 2983-90. Tselios, T., V. Apostolopoulos, I. Daliani, S. Deraos, S. Grdadolnik, T. Mavromoustakos, M. Melachrinou, S. Thymianou, L. Probert, A. Mouzaki, and J. Matsoukas, Antagonistic effects of human cyclic MBP(87-99) altered peptide ligands in experimental allergic encephalomyelitis and human T-cell proliferation. J Med Chem, 2002. 45: p. 275-83. Turner, R., I. Stratton, V. Horton, S. Manley, P. Zimmet, I.R. Mackay, M. Shattock, G.F. Bottazzo, and R. Holman, UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. UK Prospective Diabetes Study Group. Lancet, 1997. 350: p. 1288-93. Ueda, H., J.M. Howson, L. Esposito, J. Heward, H. Snook, G. Chamberlain, D.B. Rainbow, K.M. Hunter, A.N. Smith, G. Di Genova, M.H. Herr, I. Dahlman, F. Payne, D. Smyth, C. Lowe, R.C. Twells, S. Howlett, B. Healy, S. Nutland, H.E. Rance, V. Everett, L.J. Smink, A.C. Lam, H.J. Cordell, N.M. Walker, C. Bordin, J. Hulme, C. Motzo, F. Cucca, J.F. Hess, M.L. Metzker, J. Rogers, S. Gregory, A. Allahabadia, R. Nithiyananthan, E. TuomilehtoWolf, J. Tuomilehto, P. Bingley, K.M. Gillespie, D.E. Undlien, K.S. Ronningen, C. Guja, C. Ionescu-Tirgoviste, D.A. Savage, A.P. Maxwell, D.J. Carson, C.C. Patterson, J.A. Franklyn, D.G. Clayton, L.B. Peterson, L.S. Wicker, J.A. Todd, and S.C. Gough, Association of the T- 87. 88. 89. 90. 91. 92. 93. 94. 95. 96. 97. cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature, 2003. 423: p. 506-11. Ueno, H., E. Klechevsky, R. Morita, C. Aspord, T. Cao, T. Matsui, T. Di Pucchio, J. Connolly, J.W. Fay, V. Pascual, A.K. Palucka, and J. Banchereau, Dendritic cell subsets in health and disease. Immunol Rev, 2007. 219: p. 118-42. van Aalst, D., H. Kalbacher, D. Palesch, F. Zou, A. Spyrantis, S. Rosinger, B.O. Boehm, and T. Burster, A proinsulin 74-90-derived protease-resistant, altered peptide ligand increases TGF-beta 1 secretion in PBMC from patients with type 1 diabetes mellitus. J Leukoc Biol. 87: p. 943-8. van Endert, P. and J.A. Villadangos, Antigen processing and recognition. Curr Opin Immunol, 2007. 19: p. 63-5. Vinay Kumar, R.S.C., Stanley L. Robbins, Robbinss basic pathology. 2003. 7: p. 635-655. Wang, B., A. Gonzalez, C. Benoist, and D. Mathis, The role of CD8+ T cells in the initiation of insulin-dependent diabetes mellitus. Eur J Immunol, 1996. 26: p. 1762-9. Wasmuth, H.E., G. Hess, C. Viergutz, H.R. Henrichs, S. Martin, and H. Kolb, Non-specific viral infections as possible synchronising events of the manifestation of type 1 diabetes. Diabetes Metab Res Rev, 2000. 16: p. 177-8. Windhagen, A., C. Scholz, P. Hollsberg, H. Fukaura, A. Sette, and D.A. Hafler, Modulation of cytokine patterns of human autoreactive T cell clones by a single amino acid substitution of their peptide ligand. Immunity, 1995. 2: p. 373-80. Wing, K., Y. Onishi, P. Prieto-Martin, T. Yamaguchi, M. Miyara, Z. Fehervari, T. Nomura, and S. Sakaguchi, CTLA-4 control over Foxp3+ regulatory T cell function. Science, 2008. 322: p. 271-5. You, S. and L. Chatenoud, Proinsulin: a unique autoantigen triggering autoimmune diabetes. J Clin Invest, 2006. 116: p. 3108-10. Zavasnik-Bergant, T. and B. Turk, Cysteine cathepsins in the immune response. Tissue Antigens, 2006. 67: p. 349-55. Ziegler, R., C.A. Alper, Z.L. Awdeh, L. Castano, S.J. Brink, J.S. Soeldner, R.A. Jackson, and G.S. Eisenbarth, Specific association of HLA-DR4 with increased prevalence and level of insulin autoantibodies in first-degree relatives of patients with type I diabetes. Diabetes, 1991. 40: p. 709-14. 7. Abbildungsverzeichnis Abbildung 1: Lehuen A, Diana J, Zaccone P, Cooke A (2010): Immune cell crosstalk in type I diabetes. Nature Reviews Immunology, 10: S.501-513, S.503 Reprinted by permission from Macmillan Publishers Ltd.: [NATURE], copyright (2010), License number 3273300528609 Abbildung 2: Abbildung durch PD Dr. T. Burster Abbildung 3: Burster T., Boehm B.O. (2010): Processing and presentation of (pro) insulin in the MHC class II pathway: the generation of antigen-based immunmodulators in the context of type 1 diabetes mellitus. Diabetes Metabolism Research and Reviews. 2010; 26: 227-238 Reprinted by permission from John Wiley & Sons Ltd.: [WILEY INTERSCIENCE], copyright (2010), License number 3273310467761 Abbildung 4: Abbildung durch PD. Dr. T. Burster Abbildung 5: van Aalst D., Kalbacher H., Palesch D., Zou F., Spyrantis A., Rosinger S, Boehm B.O., Burster T. (2010): A proinsulin 74-90 derived protease-resistant altered peptide ligand increases TGF-ß1 secretion in PBMC from patients with type 1 diabetes mellitus. Journal of Leukocyte Biology Reprinted by permission from [JOURNAL OF LEUKOCYTE BIOLOGY], copyright (2010), permission granted 24.11.2013 Abbildung 6: van Aalst D., Kalbacher H., Palesch D., Zou F., Spyrantis A., Rosinger S, Boehm B.O., Burster T. (2010): A proinsulin 74-90 derived protease-resistent altered peptide ligand increases TGF-ß1 secretion in PBMC from patients with type 1 diabetes mellitus. Journal of Leukocyte Biology Reprinted by permission from [JOURNAL OF LEKOCYTE BIOLOGY], copyright (2010), permission granted 24.11.2013 Abbildung 7: ELISA TNF-α Abbildung 8: ELISA IFN-γ Abbildung 9: ELISA IL-6 Abbildung 10: ELISA IL-17 Abbildung 11: ELISA TGF-β1 Abbildung 12: FACS TNF-α Abbildung 13: FACS IFN-γ Abbildung 14: FACS: Il-6 Abbildung 15: FACS IL-17 Abbildung 16: FACS TGF-β1 Abbildung 17: FACS IL-10 Abbildung 18: FACS CTLA-4 Abbildung 19: FACS GITR Abbildung 20: Burster T., Boehm B.O. (2010): Processing and presentation of (pro) insulin in the MHC class II pathway: the generation of antigen-based immunmodulators in the context of type 1 diabetes mellitus. Diabetes Metabolism Research and Reviews. 2010; 26: 227-238 Reprinted by permission from John Wiley & Sons Ltd.: [WILEY INTERSCIENCE], 3273310467761 copyright (2010), License number 8. Tabellenverzeichnis Tabelle 1: Liste der beim ELISA verwendeten Lösungskonzentrationen Tabelle 2: Genotypen der untersuchten T1DM Patienten Tabelle 3: Verwendete Antikörper für die FACS-Analyse 9. Danksagung Die Danksagung wurde aus Gründen des Datenschutzes entfernt. 10. Curriculum Vitae Der Lebenslauf wurde aus Gründen des Datenschutzes entfernt.