carnot
Werbung

Inhalt: 6. Entropie und Zweiter Hauptsatz der Thermodynamik 2 6.1. Reversible – Irreversible Prozesse ............................................................................................. 4 6.1.1. Reversible Prozesse: ........................................................................................................... 4 6.1.2. Irreversible Prozesse ........................................................................................................... 4 6.1.3. Modellbeispiel: Joule´sche Wirbelmaschine ....................................................................... 5 6.1.4. Modellbeispiel: Wärmefluss ............................................................................................... 5 6.2. Wärmekraftmaschinen ⇒ 2. Hauptsatz ...................................................................................... 6 6.2.1. Historischer Exkurs: Wärmekraftmaschinen:...................................................................... 6 6.3. Idealisierte Wärmekraftmaschine – Carnot´scher Kreisprozess ................................................. 8 6.3.1. Effizienz einer Wärmekraftmaschine: ................................................................................. 8 6.3.2. Carnot´sche Behauptung: .................................................................................................. 12 6.3.3. Konkrete idealisierte "Realisation" einer Carnot´schen Maschine: Carnot-Zyklus ............. 9 6.4. Entropie, thermodynamische Definition................................................................................... 13 6.5. Entropieänderung reversibler Prozesse .................................................................................... 14 6.5.1. Reversibel adiabatisch (2. Carnot-Schritt): ....................................................................... 14 6.5.2. Reversible isotherme Expansion (1. Carnot-Schritt) ......................................................... 14 6.5.3. Reversible Zustandsänderung eines idealen Gases............................................................ 14 6.5.4. Reversibler Phasenübergang bei konstantem p ................................................................. 14 6.6. Entropieänderung irreversibler Prozesse .................................................................................. 17 6.6.1. Irreversible Zustandsänderung eines idealen Gases .......................................................... 17 6.6.2. Irreversibler und reversibler Wärmeaustausch (bei konstantem p).................................... 18 6.7. Entropie von System und Umgebung– Clausius´sche Ungleichung ......................................... 21 6.7.1. Reversibler Prozess:.......................................................................................................... 21 6.7.2. Verallgemeinerter irreversibler Modell-Zyklus: ............................................................... 22 6.7.3. Irreversibler Modell-Zyklus – spezielles Beispiel: ............................................................ 23 6.8. Messung der Entropie – Absoluter Bezugspunkt bei T→0 – 3. Hauptsatz ............................... 25 6.8.1. Beiträge zu qrev: ................................................................................................................ 25 6.8.2. Absoluter Bezugspunkt der Entropie – Beispiel Schwefel ................................................ 26 6.8.3. 3. Hauptsatz ...................................................................................................................... 28 6.9. Mikroskopische (statistische) Interpretation der Entropie ........................................................ 29 6.9.1. Beispiel: N Moleküle HCl bilden einen perfekten Kristall bei T=0 (starke WW) ............. 30 6.9.2. Beispiel: Abweichungen von S(T→0)=0 bei nicht perfekten Kristallen ........................... 30 6.9.3. Beispiel: 1mol = NA Moleküle CO bei T = 0K ................................................................. 30 6.9.4. Beispiel: Pokern ................................................................................................................ 30 6.10. Praktische Anwendung: Adiabatische Entmagnetisierung .................................................... 31 6.11. Entropiefluss in Wärmekraftmaschinen und Wärmepumpen ................................................ 33 6.11.1. Aus der Praxis: Moderne Dampfmaschinen .................................................................. 34 6.11.2. Kältemaschinen ............................................................................................................. 37 6.12. Die Clausius´sche Ungleichung............................................................................................ 38 6.13. Maximal mögliche Arbeit→Freie Energie ........................................................................... 39 6.14. Maximal mögliche Nicht-Volumenarbeit ⇒ Freie Enthalpie............................................... 40 6.15. Gibbs´sche Gleichungen oder Fundamentalgleichungen der Thermodynamik ..................... 41 6.15.1. Weitere Fundamentalgleichungen ⇒ die Gibbs´schen Gleichungen ............................. 41 6.15.2. Alle Gibbs´schen Gleichungen mit den dazugehörenden partiellen Ableitungen: ......... 42 6.15.3. Anwendungsbeispiel: Druckabhängigkeit der Freien Enthalpie .................................... 43 6.16. DieGibbs-Helmholtz Gleichung ........................................................................................... 44 6.17. Die Maxwellgleichungen...................................................................................................... 45 6.17.1. Anwendungsbeispiel: Innere Energie bei isothermer Expansion ................................... 46 ⎛ ∂H ⎞
6.17.2. Zusammenhang zwischen ⎜
⎟ und dem linearen Ausdehnungskoeffizienten α ..... 47 ⎝ ∂p ⎠ T
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 1
© Dr. Ogrodnik
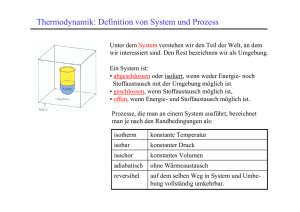
Überblick und Zusammenfassung:
6.
Entropie und Zweiter Hauptsatz der Thermodynamik
Suche: Wodurch wird die "Richtung" eines Prozesses bestimmt?
6.1.Wir vergleichen zuerst reversible Prozesse (im Gleichgewicht, der Prozess hat keine
Richtung) mit irreversiblen Prozessen, die spontan "vorwärts" aber nicht in
"Rückwärtsrichtung" laufen (Beispiele). Schlussfolgerung: die Energie wird möglichst
gleichmäßig auf alle "Freiheitsgrade" verteilt (=maximale Unordnung)!
6.2. Im engen Zusammenhang steht die Frage, wie sich die beiden Energieformen des 1.
Hauptsatzes Wärme q und mechanische Arbeit w ineinander überführen lassen ⇒ aus
der Effizienz von Wärmekraftmaschinen gewinnen wir die empirische Erkenntnis ⇒
2. HS: Es ist nicht möglich (in einem zyklischen Prozess) Wärme vollständig in Arbeit
umzuwandeln
6.3. Zur weiteren Untersuchung und Quantifizierung dient ein idealisiertes Modell eines
reversiblen Kreisprozesses ⇒ Carnotzyklus:
♦ Isotherme Expansion
♦ Adiabatische Expansion
♦ Isotherme Kompression
♦ Adiabatische Kompression
2 wichtige Schlussfolgerungen:
♦ Carnot´sche Behauptung: um maximale Arbeit
zu leisten muss der Prozess reversibel sein (⇒maximaler Wirkungsgrad!).
♦
Für den Quotienten
dqrev
dqrev
=0
gilt: v∫
T
T
reversibler
Zyklus
♦
und:
dq
<0
v∫
irreversibler T
dqirrev
⎛
⎞
⎜ Achtung: T ist nicht die Entropie!⎟
⎝
⎠
Zyklus
6.4.
6.5.
6.6.
6.7.
dqrev
T
Wir berechnen die Entropieänderung eines Systems für reversible Prozesse
(z.B. Phasenübergang ⇒ Trouton´sche Regel) und
und für irreversible Prozesse (da ist die Entropie erst mal nicht definiert, deshalb
Suche nach "äquivalentem" reversiblen Weg, da S=Zustandsfunktion, d.h.
wegunabhängig!)
Definition: Umgebung eines Systems: "unendliches" Wärmereservoir welches
stets im Gleichgewicht ist (=reversibel).
Erfahrungssatz: für das "Weltall" (=System + Umgebung) gilt:
Dies führt zur Definition der Zustandsfunktion Entropie S: mit dS ≡
⎧ = 0 für reversible Prozesse
ΔSWeltall ⎨
(Clausius´sche Ungleichung)
⎩> 0 für irreversible Prozesse
6.8.
d.h. Die Entropie im Universum nimmt aufgrund irreversibler Prozesse zu!
Messung der Entropieänderung anhand der reversibel umgesetzten Wärme ⇒
d.h. Integration über die spezifische Wärme. Aus dem Vergleich verschiedener
Substanzen folgt:
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 2
© Dr. Ogrodnik
♦
♦
6.9.
6.10.
6.11.
6.12.
6.13.
für T→0 geht Cp→0 ⇒ ΔS→0
Alle (vollkommen geordnete) Substanzen haben für T→0 die gleiche Entropie ⇒
wir definieren einen Nullpunkt: S(T→0)=0
Weitere empirische Beobachtung: T=0 kann nur in unendlich vielen Schritten
erreicht werden (3. HS). Anwendungsbeispiele:
Kühlung durch adiabatische Entmagnetisierung
Quantitativer Entropiefluss in Wärmekraftmaschinen und Wärmepumpen
Wir finden eine neue Formulierung der Clausius´schen Ungleichung, die nur
Systemvariable enthält, und die Umgebung nicht mehr explizit sondern implizit
berücksichtigt.
Schließlich suchen wir eine Zustandsfunktion die sowohl den 1. und 2. HS erfüllt
und uns für konst. T die maximal mögliche (Volumen-)Arbeit wmax liefert und
finden:
A =
U
− TN
⋅S mit wmax =−ΔA =−ΔU + T ⋅ΔS
N
N
freie Energie
(gesamte)
gebundene
innere Energie
Energie
6.14. Analog: die maximal mögliche Nicht-Volumenarbeit we,max
G
H − TN
⋅S mit w =−ΔG =− H + T ⋅ΔS
N = N
(gesamte) gebundene
freie
Enthalpie Enthalpie Enthalpie
e ,max
6.15. Die Gibbs´schen Gleichungen fassen den 1.und 2.HS (in Form der Clausius´schen
Ungleichung) in je eine Fundamentalgleichung für U, H, A und G zusammen:
6.16. Die Gibbs-Helmholtzgleichung zeigt, dass die Temperaturabhängigkeit der freien
Enthalpie G nur von der Enthalpie H abhängt:
⎛G⎞
∂⎜ ⎟
⎝ T ⎠ =− H
∂T
T2
p
6.17. Aus den Gibbs´schen Gleichungen lassen sich mit der Schwarz´schen Gleichung die
für vollständige Differentiale gilt die Maxwellgleichungen ableiten, anhand deren sich
beispielsweise der Binnendruck und der Joule-Thomson Koeffizient berechnen lassen.
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 3
© Dr. Ogrodnik
6 Reverrsible – Irrreversiblee Prozessee
6.1.
66.1.1.
Reeversible Prrozesse:
Z
Zustandsän
nderung, diie in infinitesimalen Schritten
S
ab
bläuft, und dabei langgsam genug
g abläuft, dass
d
s zu jedem
sie
m Zeitpunk
kt im Gleich
hgewicht mit
m der Umg
gebung ist. Wird die Ä
Änderung einer
e
Z
Zustandsva
ariablen wieeder rückggängig gemacht dann laufen auch
h alle andeeren Änderungen exak
kt
r
rückwärts
a (idealisierter Gren
ab
nzfall!).
B
Beachte:
Physikalisch
P
hen Grund
dgesetze (M
Mechanik, Quantenme
Q
echanik, Eleektrodynam
mik) sind
Z
Zeitumkehr
rinvariant, das heißt, sie ändern
n sich nicht wenn man
n die Zeitricchtung umk
kehrt ⇒
m
mikroskopi
ische Sichtw
weise!
a
aber:
Fundamenttale Erfahrrung: Unserr Leben un
F
nd die meistten Ereigniisse lassen sich zeitlich
h nicht
u
umkehren:
Die Zeit haat eine eind
deutige Ricchtung. Diee Zeit "verrrinnt", es ggibt (meist) kein zurücck!
⇒
⇒makrosko
opische Sicchtweise!
6
6.1.2.
Irrreversible Prozesse
P
Suche: konk
S
krete, einfaache (durch
hschaubaree) Beispielee
1 Modellbeeispiel: Hüp
1.
pfender Baall (=betrachhtetes Systeem) trifft auuf den Bodenn (=Umgeb
bung).
Beobaachtung:
♦ Stahlkugel (o
oder Hartguummiball) aauf festem Untergrund
U
(Stahllplatte) sprin
ngt fast auff gleiche Hööhe zurück (fast
(
reversibbel)
♦ Ball
B auf weicchem Unterrgrund sprinngt nach jed
dem Aufpralll
wenigger hoch (irrreversibel)
Analyyse:
Bei jedem Aufp
prall verliertt der Ball
einenn Teil seinerr kinetischeen Energie an
den Boden
B
(Eneergieerhaltunng gilt!!!)
⇒ Die Moleeküle im Booden könnenn aber die Bewegung
B
nicht
n
geradliinig fortsetzzen ⇒Stösse mit anderrn
M
Molekülen
⇒ elastischee Streuung nach allen Richtungeen ⇒ Umwaandlung in u
ungeordnette =
thermische Bewegung der Bodenaatome.
Richtung dieeses Prozesses führt sp
R
pontan (d.h.. ohne weiteere Einwirkkung von auussen) mehr oder wenigger
s
schnell
zur Ruhelage
R
dees Balls
D
Dabei
wird Ekin
E
der geerichteten Bewegung
B
v
vollständig
in kinetischhe Energie eeiner ungerrichteten
thermischen
n Bewegunng umgesetzzt.
Umgekehrtter Prozess wurde nochh nie beobachtet:
U
d ein ruheender Ball fängt
d.h.
f
nicht spontan
s
an zu
z springen, in dem er einem warm
men
U
Untergrund
Wärme entzieht und inn gerichtete kinetische Energie verrwandelt ⇒ irrversibeel!
S
SS2008
Entrropie, freie Enthalpie + Gibbs´schhe Gl. 4
© Dr. Ogrod
dnik
Grund: Waahrscheinlichhkeit, dass die
G
d ungeorddnete Beweg
gung der Attome gerichhtet wird, d.h. für eine
k
kurze
Zeit ettwa 1020 Attome paralleel auf den Ball
B zufliegeen ist sehr, sehr,
s
sehr, ssehr, sehr kllein.
6
6.1.3.
Moodellbeispiel: Joule´scche Wirbellmaschine
Arbeit volllständig in
n Wärme um
mgewandelt
Umkehrun
ng nicht möglich!
6
6.1.4.
Moodellbeispiel: Expansiion ins Vak
kuum
Eiin Gas im Vakuum
V
zieh
ht sich nich
ht spontan aauf ein klein
neres Volum
men
zuusammen, soondern es dehnt
d
sich auus ⇒ Unordnung nim
mmt zu!.
⇒ gleichmäß
ßige Verteiilung der Moleküle
M
übeers gesamtee Volumen
hrbar!
Niicht umkeh
6
6.1.5.
Moodellbeispiel: Wärmeefluss
Wärme fließt so lannge, bis Sysstem und
Umgebuung auf gleiccher Tempeeratur sind
Die heiß
ßen (schnelllen) Molekü
üle der
Umgebuung übertraggen mehr En
nergie ins
System hinein
h
als ddie kalten (langsamen))
Molekülle aus dem System herraus.
Gleichgeewicht: ⇒ gleiche
Geschwiindigkeitsveerteilung inn
nen und außßen
(Impulseerhaltung inn beiden Ricchtungen)
Umkehrrung: Es istt nicht mög
glich, selektiive
ddie starken Molekülstöß
M
ße (große kiinetische Ennergie ) durrch Impulsüübertrag aus dem System
m heraus unnd
d schwachhen Stöße deer langsameen Molekülee ins System
die
m hinein zu übertragen so dass die Temperatuur
n
niedriger
wiird!
S
SS2008
Entrropie, freie Enthalpie + Gibbs´schhe Gl. 5
© Dr. Ogrod
dnik
Kriterium für
K
f einen sp
pontanen Prozess
P
E Prozess läuft in die Richtung spontan
Ein
s
(freiwillig) ab, in der die Gesamtener
G
gie des abgeschlosseneen
S
Systems
mööglichst "gleichmäßig"
" verteilt isst (d.h. ungeeordnet).
6 Wärmekraftmasschinen ⇒ 2. Haupptsatz
6.2.
Frage: Wiee lassen sichh die beidenn Energieforrmen des 1. Hauptsatzees Wärme q (mikrosko
F
opisch,
s
statistisch
veerteilt, nichtt orientiert) und mechaanische Arb
beit w (makkrosopisch, in gleiche Richtung
R
o
orientiert)
inneinander übberführen?
Wir wollen dies
W
d am Beiispiel von Wärmekraf
W
ftmaschineen exemplarrisch untersuuchen. Wie führen die q in
w über?
6
6.2.1.
Hiistorischer Exkurs: Wärmekraft
W
tmaschinen
n:
Die ersten Dampfmasc
D
D
chinen:
A
Atmosphäris
sche Dampffmaschine von
v Newcom
men:
1 Schritt:: Ventil A wird
1.
w geöffneet, Dampf wird
w vom Kolben
K
angessaugt (der B
Balancier-Balken ist linnks
etwas scchwerer als rechts) bis er am obereen Totpunk
kt angekomm
men ist. Beaachte: es wird hier nichht
mit Übeerdruck gearrbeitet (=athhmosphärissche Dampfm
fmaschine)!
2 Schritt:: Das Ventiil B wird geschlossen, A wird geöfffnet (das mussten
2.
m
frühher Kinder gemachen)
g
u
und
es wird kaltes Wasser in den Zylinder
Z
einngesprüht biis Dampf koondensiert ((leider mussste dabei auch
der gesaamte Zylindder mitgeküühlt werden, was zu ern
normen Wärrmeverlusteen führte)⇒ bildet Vakuuum
(Dampffdruck von Wasser
W
bei Zimmertem
mperatur = 0.03bar)
0
⇒ Ventil A w
wird geschlossen, der
Kolben wird nach unten
u
gesauugt und ziehht an der Waasserpumpee am linken Balancier-B
Balken). ⇒
Schritt 1
W
Weiterer
Naachteil: Geggossener Zyllinder⇒rauuhe Innenflääche, sehr scchlechte Dicchtung
S
SS2008
Entrropie, freie Enthalpie + Gibbs´schhe Gl. 6
© Dr. Ogrod
dnik
W
Wirkungsgr
rad (ohne Verluste):
V
Z
Zugeführte
Wärme: C p ⋅ Δ
T +ΔHVerdampfung = 75.4
N
≈90 K
Vm
G
Geleistete
A
Arbeit:
J
kJ
kJ
⋅90 K + 40.66
= 47.4
K ⋅moll
mol
mol
wm =− ∫ p⋅dV = p⋅Vm = R⋅398 K =3.3
0
ε=
wgeleistet
qzugeführt
=
kJ
mol
3.3
=7% Realistisch
her Wirkun
ngsgrad: ε=
=0.5%
47.4
A
Animation:
http://www
w.keveney.ccom/newcom
mmen.html
W
Watt´sche
D
Dampfmasc
chine: 65 Jahre später::
Zweiterr externer Zylinder
Z
zum
m
Konden
nsieren des Dampfes
D
(bbei
konstan
nter niedererr Temperatuur)
= Kond
densator
Der Arb
beitszylindeer bleibt
dadurch
h bei ≈konsttant hoher
Temperratur
⇒ gerin
ngere Verluuste
Wirkungsgrrad: 1.5% Verdreifaacht!
W
http://ww
A
Animation:
ww.geocities.com/Ath
hens/Acrop
polis/6914/w
wvae.htm
http://en
n.wikipediaa.org/wiki/S
Steam_engine
W
Weiterer
Foortschritt: Arbeitszylinder gebohrrt (neue Tecchnik aus deem Kanoneenbau) ⇒ viiel größere
Vm
D
Dichtigkeit⇒
⇒man kannn mit Überdrruck arbeiteen⇒ wm =− ∫ p ⋅ dV steeigt enorm!
0
Zweiter Haauptsatz derr Thermod
Z
dynamik
M kann viiele derartigge Systeme untersuchenn und findeet als zusam
Man
mmenfasssenden
(
(empirisch
hern) Erffahrungsssatz:
2 HS: E
2.
Es ist nichtt möglich
h (in einem
m zyklisch
hen Prozeess) Wärm
me vollstä
ändig in
A
Arbeit
umzuwandelln!
(N
Nichtexsisstenz einees „perpettuum mob
bile 2. Arrt“)
Beachte: Deer 2. HS bezzieht sich auuf einen volllständigen geschlossen
B
nen Zykluss. In einem einzelnen
issothermen Schritt giltt ΔU=0 ⇒ q=-w
q
⇒ voollständige Umwandlu
ung von Wäärme in Arrbeit!
S
SS2008
Entrropie, freie Enthalpie + Gibbs´schhe Gl. 7
© Dr. Ogrod
dnik
6.3. Idealisierte Wärmekraftmaschine – Carnot´scher Kreisprozess
Frage: Wie kann man in Wärmekraftmaschinen die geleistete Arbeit maximieren?
Zu Beginn der 19. Jahrh. dachten viele, dass man bei maximalem Druck maximale Arbeit erhält (vgl.
Hochdruck-Dampfmaschine)! Das war aber eine gefährliche
Strategie ⇒ Explosionen⇒viele Tote! Stimmt das?
Sadi Carnot 1786-1832 (an der Cholera gestorben ⇒ alle
Aufzeichnungen verbrannt)
Historische Notiz: Als Carnot seinen berühmten Kreisprozess
1824 formuliert, war der Energieerhaltungssatz noch in den
Kinderschuhen und wurde kontrovers diskutiert. Der Erste
Hauptsatz wurde ein Jahrzehnt später formuliert. 2 Jahrzehnte
spater entdeckte und bestimmte Joule das mechanische
Wärmeäquivalent. Die kalorische Theorie war vorherrschend,
wonach die Wärme eine gewichtlose, unsichtbare Flüssigkeit ist,
zu fließen beginnt, wenn sie nicht im Gleichgewicht ist.
die
Carnot dachte sich folgende Idealisierung einer
Wärmekraftmaschine:
♦
♦
♦
♦
Das Arbeitsmedium ist vor dem Start kalt, Kontakt mit einem kalten (unendlich
großen) Wärmereservoir mit Temperatur TK (in der Praxis ist es die
Umgebungstemperatur )
Dem Arbeitsmedium der Maschine (=System) aus einem (unendlich großen)
heißen Wärmereservoir bei der Temperatur TH die Wärme qH zugeführt
(isotherm) (in der Praxis wird die Temperatur im heißen Reservoir durch
Verbrennung eines Energieträgers aufrechterhalten).
das Arbeitsmedium expandiert ⇒ die Maschine leistet die Arbeit w
Um auf die Ausgangstemperatur TK (geschlossener Zyklus) zurückzugelangen
muss das Arbeitmedium abgekühlt werden. Dies erfolgt über das kalte
Wärmereservoir der Umgebung
6.3.1.
ε=
System
Effizienz einer Wärmekraftmaschine:
wgeleistet
qzugeführt
=
− wsystem
qH
Wegen des 2.H.S. muss wgeleistet < qzugeführt sein! ⇒ ε<1!
Geschlossener Zyklus ⇒ ΔUZyklus=0 ⇒ − wSystem = qgesamt = qH + qK eingesetzt:
ε=
qH + qK
q
=1+ K (1)
qH
qH
Beachte: qW positiv, qK negativ, Wärme fließt von heißen Reservoir zur Maschine und von der
Maschine zum kalten Reservoir! Da wegen de 2. H.S. gilt ε<1 muss folglich qK>0. d.h. jede
Maschine muss Abwärme produzieren!
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 8
© Dr. Ogrodnik
6.3.2.
Konkrete "Realisation" einer idealen, d.h. reversiblen Carnot´schen Maschine: CarnotZyklus
p
2. Schritt:
isotherme Expansion
1
1. Schritt:
adiabatische
Kompression
wzyklus = Nutzarbeit
Kompressionsarbeit
< Expansionsarbeit
qH=-wH
0
4
2
4. Schritt:
isotherme Kompression
TH
3. Schritt:
adiabtische Expansion
qK=-wK
3
TK
V
0. Schritt (Ausgangssituation): Das Arbeitsmedium ist mit dem kalten Reservoir im Gleichgewicht ⇒
T1=TK, V1, p1
1. Schritt reversible adiabatische Kompression
das Arbeitsmedium wird in
diesem Schritt vom kalten
Reservoir thermisch
entkoppelt
Das Volumen wird von VK auf VH komprimiert ⇒
Das Medium läuft auf der Adiabaten von p1 nach p2
dabei wird das Gas wird von TK auf TH erwärmt
(p2V2 > p1V1)
Dabei muss dem Medium mechanische Arbeit
zugeführt werden (Drücken des Kolbens)
2. Schritt reversible isotherme Expansion ⇒
Medium wird an das heisse
Reservoir gekoppelt
T=TH ⇒ ΔU=0
ΔT=0
Durch die Expansionsarbeit wird Energie
des Mediums verbraucht. Da aber ΔU=0,
wird der mechanische Energieverlust durch
die aus dem Reservoir zugeflossene Wärme
kompensiert: q2=-w2
3. Schritt reversible adiabatische Expansion
thermische Entkoppelung
Man lässt das Medium weiter expandiern. Dabei wird
vom Medium immer noch Arbeit geleistet, und zwar
wegen der thermischen Entkoppelung auf Kosten der
inneren Energie. Deshalb fällt jetzt die Temperatur.
Wenn die Ausgangstemperatur TK erreicht ist, wird die
Expansion gestoppt.
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 9
© Dr. Ogrodnik
4. Schritt reversible isotherme Kompression (Zyklus abgeschlossen!)
Medium hat die gleiche
Temperatur wie das kalte
Reservoir und kann
reversibel an dieses
gekoppelt werden
T=TK ⇒ ΔU=0
Es muss Volumenarbeit ins System
gesteckt werden, um wieder auf das
Ausgangsvolumen zu kommen
Da ΔU=0 ist, geht diese Arbeit in
Form von Wärme an das kalte
Reservoir verloren
Frage: Kann man die maximale Arbeit bzw. die Effizienz quantifizieren?
Dazu benötigen wir den Arbeits- und den Wärmeumsatz!
Beide Terme stecken im 1. HS:
dU
N = dq + dw
N
∂U
− p ⋅ dV
⋅ dT
∂
T
CV ⋅dT
n ⋅ R ⋅T
Als Arbeitsmedium betrachten wir ein ideales Gas: p =
eingesetzt:
V
CV ⋅ dT = dq − n ⋅ R ⋅T ⋅
dV
V
wir teilen durch T (Variablen sortieren) und integrieren über den
Gesamtzyklus:
dT
dV
− n⋅ R⋅v∫
(2)
V
innere Energie Wärme Arbeit
T
T
T
C ⋅
v
∫
T
V
=
dq
vN
∫T
Als nächstes betrachten wir die einzelnen Carnotschritte die zum ersten Integral beitragen
T
T
T
T
H
K
K
H
dT
dT
dT
dT
dT
(3)
C
C
C
C
⋅
=
⋅
+
⋅
+
⋅
+
v∫ V T ∫ V T ∫ V T ∫ V T ∫ CV ⋅ T = 0
TK
TK
TH
TH
1.Schritt
2.Schritt 3.Schritt
4.Schritt
adiabatisch isotherm
isotherm
adiabatisch
TH
=0
=0
dT
− ∫ CV ⋅
T
T
K
dV
∫V =
Das letzte Integral: v
V2
∫ d ln(V )
V1
+
V3
∫ d ln(V )
V2
+
V4
∫ d ln(V )
V3
+
V1
∫ d ln(V )
V4
= 0 (4)
Schritt
Schritt
Schritt
1.
2.
4.
Schritt
3.
ln(V2 )−ln(V1 ) ln(V3 )−ln(V2 ) ln(V4 )−ln(V3 ) ln(V1 )−ln(V4 )
Beachte: da V eine Zustandsgrösse ist, ist auch jede Funktion von V eine Zustandsgröße, und es gilt:
v∫ funktion(V )dV = 0
Aus (2), (3) und (4) folgt:
dq
=0 (5)
v
∫
Carnot T
Zyklus
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 10
© Dr. Ogrodnik
Ohne das Integral explizit ausrechnen zu müssen sehen wir, dass für jeden geschlossenen Carnotzyklus
dq
obiges Wegintegral Null ist. Folglich verhält sich der Term
wie ein vollständiges Differential, bzw.
T
wie eine Zustandsfunktion!
Betrachten nun doch explizit die einzelnen Schritte im letzten Integral:
dq
v∫
Carnot T
Zyklus
=
dq
dq
dq
dq
+ ∫
+ ∫
+ ∫
T 2.Schritt T 3.Schritt T 4.Schritt T (6) ⇒
1. Schritt
adiabatisch
isotherm
adiabatisch
isotherm
0
0
q2
q4
∫
T2
qH
TH
T4
qH
TH
dq qH qK
= + = 0 (7)
TH TK
v∫
Carnot T
Zyklus
⇒
qH
T
=− H
qK
TK
Daraus ergibt sich mit ε =1+
qK
(8)
qH
Effizienz des Carnot-Zyklus: ε rev =1−
TK
TH
(9)
(Carnot´scher Wirkungsgrad)
♦
♦
Je größer die Temperaturspreizung desto größer ε
Je kleiner TK desto größer ε (kryogene Wärmekraftmaschine, Wärmekopplung ans kalte
Weltall?)
εrev ist unabhängig vom Arbeitsmedium, d.h. gilt auch für nicht ideale Gase. Dies gilt auch für Gl. (5)
6.3.3.
Wärmepumpe:
Eine reversibel arbeitende Maschine muss per definitionem auch rückwärts laufen können, d.h. alle
Arbeits- und Wärmeflüsse kehren sich um, d.h. Arbeit wird in die reversible Maschine gesteckt und
Wärme wird vom kalten Reservoir aufgenommen und an das heiße Reservoir abgegeben.
Beachte: Ohne Arbeitszufuhr würde dieser Prozess natürlich nicht spontan ablaufen.
Eine so betriebene Maschine nennt man Wärmepumpe. Sie findet Einsatz in Kältemaschinen und z.T.
in Heizsystemen mit Heizwirkungsgraden >100%.
Man kann beispielsweise den Carnotprozess rückwärts laufen lassen:
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 11
© Dr. Ogrodnik
p
2. Schritt:
isotherme Kompression
3. Schritt:
adiabatische
Expansion
wzyklus
Kompressionsarbeit >
Expansionsarbeit
⇒
Arbeit wird verbraucht
qH=-wH
TH
1. Schritt:
adiabtische Kompression
4. Schritt: isotherme Expansion
TK
qK=-wK
V
6.3.4.
Carnot´sche Behauptung:
Carnot´sche Behauptung:
Nur eine reversibel arbeitende Maschine produziert die maximal
mögliche nutzbare Arbeit
Folglich ist die Effizienz nicht reversibler Maschinen kleiner als der Carnot´sche
Beweis:
Dazu nehmen wir an, dass es eine Supermaschine
gibt, die besser als eine reversible Maschine ist:
-w super
-w
und ε rev = rev
ε super > ε rev wobei: ε super =
q H,super
q H,rev
Kopplung: Wir können nun die Supermaschine mit
einer reversibel arbeitenden Wärmepumpe zu einer
Gesamtmaschine koppeln. Die Wärmepumpe soll so
ausgelegt sein, dass sie die gesamte Abwärme der
Supermaschine vom kalten Niveau ins heiße
zurückpumpt, d.h.: q K,super = q K,rev . In der Bilanz wird
also keine Wärme ans kalte Reservoir abgegeben.
(Dies lässt sich durch Isolation der kalten Seite vom
kalten Reservoir erreichen. Durch richtige Wahl von
wrev muss die Wärmepumpleistung so eingestellt
werden, dass dabei die Temperatur TK konstant
bleibt.)
Die für den Betrieb der Wärmepumpe nötige Arbeit
-w rev =ε rev ⋅q H,rev soll von der Supermaschine
abgezweigt werden, die insgesamt die Arbeit
-w super =ε super ⋅q H,super zur Verfügung stellen kann.
TK
Isolator
Der Überschuss w ges =w super +w rev soll an die
Umgebung abgeführt werden (Beachte: wsuper
negativ, wrev positiv)..
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 12
© Dr. Ogrodnik
Da die innerre Energie im
D
m geschlossenen Zykluus sich nich
ht ändern daarf, d.h. ΔUZZyklus=0 erhaalten wir übber
d 1.HS folgende Beziehung zwisschen Arbeiit und Energ
den
gie ⇒ − wSyystem = q gesamt = qH + qK
Damit lässt sich jeweilss qH durch qK ausdrückken: qH =− w−
D
w qK und foolgende Eneergiebilanz aufstellen:
E
Energiebila
anz für die Gesamtmaaschine:
q H,ges =q H,supper +q H,rev = -w
- super -q K,supper -w rev -q K,,rev = -w super -w
- rev -q K,supeer -q K,rev = -w
w ges
-wges
0
D bedeuteet aber, dass die gesam
Dies
mte Wärmeeenergie in Arbeit
A
umgew
wandelt wirrd!
♦
Dies ist
i aber im Wiederspru
W
uch zum 2.. HS
♦
Folglicch gibt es keine
k
Superrmaschine mit höhereem Wirkun
ngsgrad als die reversiible! q.e.d.
6 Entroppie, therm
6.4.
modynamissche Defin
nition
Suche: Könnnen wir einne neue therm
S
modynamissche Zustandsfunktion mit Aussaggekraft über die "Richtuung"
e
eines
Prozessses finden?? ⇒ Entropiie S
g
griechisch:
trepein = drehen,
d
ändeern, trope=Verwandlu
ung (von Wäärme in Arb
beit )
♦
reversib
ble Prozessse: stets im Gleichgewiicht, läuft vorwärts geenauso wahrrscheinlich wie rückw
wärts
⇒ kein
ne Richtungg⇒ hier solll ΔS=0
♦
irreverssible Prozeesse: hier sooll sich S än
ndern ⇒ ΔS≠0
Δ
Auf der Suche nach einner derartigeen Funktionn kehren wirr zur Beziehhung (5) zuurück. Sie giilt allgemeinn
A
fü jeden revversiblen Krreisprozess,, da dieser stets
für
s
aus ein
ner Reihe voon Carnot-Z
Zyklen zusam
mmengesettzt
w
werden
kannn (vgl. Skizzze):
Zerlegung eiines
Z
beliebigen
geschlosseneen
K
Kreisweges
iin kleinste
addiabatische und
issotherme Scchritte
v∫
belliebiger
revversibler
Zykklus
dqrevv
=0
T
(10)
1 Wichtige Schlussfollgerung:
1.
dqrev
fü einen revversiblen Pffad ist T ein vollstäändiges Diff
für
fferential
o
oder:
S =∫
dqrev
T
ist eine
e Zustanndsfunktion
Clausius´sche (thermoodynamisch
C
he) Definitiion der Enttropie:
(uursprünglicch genannt: „Verwandluungsinhalt““)
q keine Zustandsfun
A
Achtung:
Z
nktion, aberr der Quotieent!!!
(mathem
matisch nennnt man 1/T den
d integrieerenden Fakktor)
S
SS2008
Entrropie, freie Enthalpie + Gibbs´schhe Gl. 13
dq
dSS ≡ rev
T
© Dr. Ogrod
dnik
(111)
z.B.: isochorer Prozess: dS =
dqrev
dT
=CV ⋅ =CV ⋅d ln (T ) . Da T eine Zustandsgröße, ist ln(T) auch eine
T
T
Zustandsfunktion!
6.5. Entropieänderung reversibler Prozesse
6.5.1.
Reversibel adiabatisch (2. Carnot-Schritt):
dqrev=0 ⇒ ΔS=0
6.5.2.
Reversible isotherme Expansion (1. Carnot-Schritt)
isotherm⇒T=const
V2
V
dq
q
1 2
ΔS = ∫ rev
=
⋅
dqrev = rev
N
∫
T
T V
T
V
da
1
1
T=const!
⇒
ΔS =
q rev
T
Berechnung von qrev für ideales Gas:
ΔU = qrev + wrev =
1HS:
qrev =− wrev =−
0N
da isotherm
"ideales Gas"⇒
⇒
6.5.3.
p=
nRT
V
⇒
VGas
∫
− pdV
⎛V ⎞
qrev = n⋅R⋅T ⋅ln ⎜ 2 ⎟
⎝ V1 ⎠
V flüssig
⎛V ⎞
ΔS = n⋅ R⋅ln ⎜ 2 ⎟
⎝ V1 ⎠
Reversible Zustandsänderung eines idealen Gases
dU
rev
N = dqrev + dw
N
∂U
−
p
⋅
dV
⋅dT
∂T
N
CV
nRT
ideales Gas:
p=
V
1. HS:
T2
dqrev = CV ⋅dT +
dS =
n⋅ R ⋅T
⋅dV
V
dqrev
dT
dV
= CV ⋅ + n ⋅ R ⋅
T
T
V
V
2
dT
dV
ΔS = ∫ CV (T )⋅ + n⋅ R ⋅ ∫
T
V
T
V
1
1
für kleine ΔT ist CV≈const
6.5.4.
ΔS ≈ CV ⋅
⎛T ⎞
⎛V ⎞
+ n ⋅ R ⋅ ln ⎜ 2 ⎟
ln ⎜ 2 ⎟
T1 ⎠
V1 ⎠
⎝
⎝
=0
=0
wenn isotherm
wenn isochor
(12)
Reversibler Phasenübergang bei konstantem p
Phasenübergang ⇒ T=const! (vgl. oben)
z.B. Schmelzen, Verdampfen...
Phase 2
ΔS Phasenübergang =
dq
∫ Trev
Phase1
ΔH Phasenübergang
q
1
⋅ ∫ dqrev = rev =
T Phase1
T
T
Phase 2
=
N
da
T=const!
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 14
© Dr. Ogrodnik
ΔHVerdampfung
ΔH Schmelz
ΔSVerdampfung =
T
T
und
ΔHschmelz=6.006 kJ/mol
Beispiel: Eis→Wasser:
TSchmelz=273K
ΔS Schmelz =
kJ
6.0006
ΔH Schmelz
mol = 20 J
ΔS Schmelz =
=
T
mol ⋅ K
273.15 K
Vergleich der Verdampfungsenthalpien und –Entropien verschiedener
Substanzen flüssig ↔ gas
Substanz
Tsiede [K]
He
4.206
H2
20.38
N2
77.33
HCl
188.1
NH3
239.72
SO2
263.13
CH3OH
337.85
CCl4
349.85
C2H5OH 351.65
C6H6
353.25
H2O
373.15
CH3COOH 391.45
Hg
629.72
Cs
963.15
Zn
1180.15
NaCl
1738.15
Pb
2023.15
Ag
2466.15
ΔH V [J/mol]
0.084
0.904
5.58
16.16
23.37
24.93
35.30
30.02
38.60
30.79
40.69
24.37
58.16
68.33
114.85
170.83
180.04
254.23
ΔS V =
ΔH V
[J/K ⋅ mol]
Tsiede
19.68
44.38
72.17
85.92
97.48
94.76
104.47
85.81
109.78
87.15
109.03
62.25
92.35
70.95
97.32
98.28
88.99
103.09
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 15
© Dr. Ogrodnik
1. Beobachtung: ΔHV ist für verschiedene Substanzen sehr unterschiedlich
Grund: ΔHV spiegelt den Unterschied in den Wechselwirkungen der Moleküle untereinander bei Gas und
Flüssigkeit wieder
Im Gas ist die WW vernachlässigbar. In der Flüssigkeit sind die Anziehungsenergien der Moleküle
verscheiden, da:
unterschiedliches Volumen ⇒ andere Gleichgewichtsabstand
unterschiedliche starke Anziehungskräfte
Potential von Molekülen in Flüssigkeit:
2. Beobachtung:
Trouton´sche Regel: Die Verdampfungsentropien der verschiedenen Substanzen sind fast gleich!
ΔSV ≈88J / K ⋅mol
unabhängig von der Substanz.
d.h. bei der Bildung von Gas aus der Flüssigkeit wird ähnlich viel Unordnung erzeugt!
Grund: Bei der Verdampfung erfolgt eine enorme isotherme Ausdehnung von der Flüssigkeit zum Gas.
Dies ist der wesentliche Beitrag zur Entropie.
⎛ V
ΔSVerdampfung ≈ n⋅ R ⋅ln ⎜ Gas
⎜ V flüssig
⎝
⎞ ← für alle Gase ≈ gleich (ideales Gas)
⎟⎟
⎠ ← für alle Flüssigkeiten ≈ gleich und sehr klein (dichteste Kugelpackung)
Bei der Verdampfung erfahren die meisten Substanzen eine ähnliche Volumenänderung und damit eine
ähnliche Entropieänderung
Ausnahmen:
♦ H2O, Alkohole: in der flüssigen Phase tragen Wasserstoffbrücken zu einer höheren Ordnung und
damit zu einer geringeren Entropie bei ⇒ grössere Entropieänderung beim Verdampfen
♦ He, H2:
sehr kleine WW
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 16
© Dr. Ogrodnik
Entropiebillanz:
E
reev
Z
Zum
Verdam
mpfen musss die Wärmeemenge qSy
r Umgebungg dem Systeem zugeführrt werden:
ystem aus der
ΔS Syystem =
rev
qSystem
T
r
rev
E
Entsprechen
nd wird − qSystem
der Um
mgebung enntzogen:
Sy
ΔSUmgebunng =
rev
−qSystem
T
=−ΔS System
Also ist die Gesamtenttropieänderrung ΔSGessamt =ΔS Systemm +ΔSUmgebunng = 0 , wie ees ja für eineen reversibllen
A
P
Prozess
gefoordert ist.
6 Entroppieänderu
6.6.
ung irreveersibler Prrozesse
6
6.6.1.
Irrreversible Zustandsän
Z
nderung ein
nes idealen
n Gases
p1, V1,T1 ⇒ p2, V2,T2
Es gibt imm
E
mer einen revversiblen Weg
W von p1, V1,T1 nach
h p2, V2,T2 (vgl.
(
Kap. 66.7.2)
D S Zustanndsfunktion ist hängt ΔS nur von AnfangsDa
A
un
nd Endzusttand, nichtt aber vom Weg ab.
ΔSirrev=ΔSrev
Wir können also die Enntropieänderrung auf einnem irreverrsiblen Pfadd, für den jaa qrev nicht definiert isst,
W
d
durch
die Enntropieändeerung auf einnem reverssiblen Pfad zwischen den gleichen
n Anfangs- und
E
Endpunkten
n beschreibben.
F
Folglich:
⎛T ⎞
⎛V ⎞
lnn ⎜ 2 ⎟
+ n⋅ R⋅ ln ⎜ 2 ⎟
T1 ⎠
V1 ⎠
⎝
⎝
=0
=0
wenn isochor
wennn isotherm
i
B
Beachte:
Daabei muss die
d auf dem
m irreversib
blen Pfad umgesetzte
u
Wärme qirrrev nicht glleich der au
uf
d
dem
reversiiblen Pfad umgesetzteen Wärme qrev sein!
m (12)⇒
mit
ΔSirrev ≈ CV ⋅
B
Beispiel:
Frreie Expanssion ins Vakuum (Jou
ule´sche Exp
periment): V2=2.V1
⇒ V2>V1
⎛ ∂T ⎞
T=coonst, da für ideales Gass μ JT =⎜
⎟ =0 , d.h T=konstantt
⎝ ∂V ⎠ H
W können
Wir
n als äquivaalenten revversiblen Weg
W eine iso
otherme Exxpansion wählen (12)::
ΔS m = R⋅ln ( 2 ) =5.76
5
S
SS2008
Entrropie, freie Enthalpie + Gibbs´schhe Gl. 17
J
mol ⋅ K
© Dr. Ogrod
dnik
ΔS
isotherm
rev
isotherm
adiab
⎛ VE ⎞
qrev
adiab qirrev
=
= n⋅R⋅ln ⎜ ⎟ =ΔSirrev ≠
T
T
⎝ VA ⎠
denn: dU=0 und wirrev=0 ⇒ qirrev=0
Da der Prozess aber irreversibel ist ⇒ ΔSirrev>0 (obwohl qirrev=0!!)
6.6.2.
Irreversibler und reversibler Wärmeaustausch zwischen heißem und kaltem Reservoir
(bei konstantem p)
Beispiel: Wasser 25oC ⇒ 50oC
T2
ΔS = ∫
C p (T )
T1
T
⋅dT unabhängig ob reversibel oder irreversibel!
Unabhängig ob ganz langsam oder ganz schnell mit Bunsenbrenner erwärmt oder mit Joule´scher
Wirbelmaschine
Reversible Erwärmung müsste differentiell erfolgen!
"Unendlich langsame Wärmebrücke"
heiss // Wärmebrücke // kalt
System T1 ⇔ Wärmebrücke T1+dT
Wärmebrücke T1+dT ⇔ Wärmebrücke T1+2.dT
Wärmebrücke T1+2dT ⇔ Wärmebrücke T1+3.dT ........ bis
Wärmebad T2
(Wie eine Folge von unendlich vielen Wärmebädern)
Aber:
Auch für die kleinsten dT ist die Fließrichtung der
Wärme eindeutig von warm nach kalt, nicht
umkehrbar!
⇒ irreversibel!!!
Beachte: es wird keine Arbeit geleistet!
“Unendlich kleiner Wärmelöffel“
Kann Wärme nur von warm nach kalt transportieren (löffeln),
nicht umgekehrt, auch wenn der „Löffel“ noch so klein ist.
„Reversible Wärmespritze“
reversibel:
Es soll in einem zyklischen Prozess die
Wärmemenge q von Reservoir T1 zu einem
Reservoir mit T2 übertragen werden.
Um diese Wärmemenge zu transportieren benutzen
wir ein Gasvolumen in einem Zylinder mit Kolben
1. Schritt:
Zylinder wird in Reservoir 1 eingetaucht ⇒
isotherme reversible Expansion ⇒ nimmt
Wärmemenge q auf
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 18
© Dr. Ogrodnik
ΔS Res =−
2. Schritt:
qrev
Gas qrev
⇒ ΔS1 =
S nimmt zu, da das Gas expandiert!
T1
T1 gas
Zylinder wird aus dem Reservoir entnommen und isoliert eingepackt ⇒ durch
adiabatische reversible Expansion wird das Gas auf die Temperatur T2 abgekühlt
qrev=0 ⇒ ΔS2Gas =0
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 19
© Dr. Ogrodnik
3. Schritt:
Zylinder wird in Reservoir 2 eingetaucht ⇒ isotherme reversible Kompression ⇒ gibt
Wärmemenge q ab
ΔS Res =
4. Schritt:
qrev
qrev
Gas
⇒ ΔS3 =−
T2
T2
Damit der Zylinder die nächste Wärmeportion abholen kann wird er wieder dem Reservoir
entnommen und isoliert eingepackt ⇒ das Gas wird solange adiabatisch reversibel
komprimiert bis es die Ausgangstemperatur T1 erreicht hat
ΔS4Gas =0
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 20
© Dr. Ogrodnik
Bilanz:
qrev qrev
+
>0
T1 T2
q
q
Gas
ΔS gesamt
= rev − rev < 0
T1 T2
Res
ΔS gesamt
=−
ΔS Univ =ΔS Syst +ΔS Umgeb = 0
⇒ reversibel, wie erwartet
⇒ Schritt 1-4 ergeben aber einen Carnot-Zyklus!
Im Verlauf des Gesamtzyklus wird Arbeit abgegeben ⇒
Carnotzyklus⇔maximale Arbeit⇔reversibel
6.7. Entropie von System und Umgebung– Clausius´sche Ungleichung
♦
Bisher nur ΔSsyst betrachtet
♦
ΔS =ΔS +ΔS
Jetzt:
Das „Universum“ sind alle Bereiche dieser Welt die mit dem System wechselwirken können!
ODER: die Umgebung ist per definitionem mit sich und dem System stets im Gleichgewicht,
folglich sind Prozesse innerhalb der Umgebung und zwischen System und Umgebung stets
reversibel!! (obwohl sie innerhalb des Systems irreversibel sein können!!!)
Univ
♦
♦
Syst
Umgeb
Umgeb
d.h. wir können anhand der mit der Umgebung stets reversibel ausgetauschten Wärme dqrev ,
die Entropieänderung der Umgebung berechnen, da:
die von der Umgebung aufgenommene Wärme = die vom System abgegebene Wärme
dqUmgeb
=− dq System
rev
⇒
6.7.1.
dS
Univ
rev
dSUmgeb =
− dq Syst
dqUmgeb
rev
=
T
T
Reversibler Prozess:
= dS
Syst
+ dS
Umgeb
Syst
Syst
dqrev
dqrev
Univ
=
−
= 0 Integration⇒ ΔS rev = 0
T
T
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 21
© Dr. Ogrodnik
6.7.2.
Verallgemeinerter irreversibler Modell-Zyklus:
Für jeden irreversiblen Pfad: 1⇒2 können wir immer einen reversiblen Pfad finden, der 1 und 2
miteinander verbindet!, (gibt zusammen einen geschlossenen Zyklus, vgl. speziellen Model-Zyklus 1 in
Paragraph 4):
2→3: Arbeit adiabatisch ⇒ Temperaturerhöhung TBad → ΔS2→3=0 da adiabtisch
3→4: Kontakt mit Wärmebad bei TBad ⇒ isotherme Wärmeumsatz q3rev
→4 bis die Entropie S1 erreicht ist
rev
⇒ ΔS
rev
3→4
q3→4
irrev
q3rev
irrev
Δ
=−
S
= →4 =ΔS 2irrev
=−Δ
S
1
→
2
⇒
→1
1→2
TBad
TBad
4 →1> isenthrop ⇒ dS = 0=
dq rev
rev
⇒ dq3rev
→4 = 0 ⇒ q3→4 = 0 ⇒adiabatisch
T
Für den geschlossenen Zyklus:
v∫
dU = 0=
1→2→3→4→1
v∫
( dq + dw)= q3→4 + w
1→2→3→4→1
w=−q3→4
Annahme: q3→4 > 0 ⇒ wUmgeb= − q3→4 ⇒ Die ganze aufgenommene Wärme wird als Arbeit an die
Umgebung abgegeben ⇒ Widerspruch zu 2. HS
q 3rev→4
⇒ Annahme falsch⇒ q3→4 ≤ 0 ⇒ ΔS1irrev
=−
≥0
→2
TBad
Dies gilt für jeden irreversiblen Prozess
⇒
ΔSirrev
ges ≥ 0
reversibel ⇒
irreversibel ⇒
= ⇒ gesamt Entropie bleibt konstant!
≥ ⇒ gesamte Entropie nimmt zu!
Clausius´sche Ungleichung
gilt allgemein (Erfahrungssatz): Die Entropie im Universum nimmt zu!
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 22
© Dr. Ogrodnik
Irreversibler Modell-Zyklus – spezielles Beispiel:
6.7.3.
Wir ersetzen im Carnot-Zyklus, die reversiblen adiabatischen Schritte durch irreversible isochore
Wärmeleitungsprozesse, d.h.
0. Schritt (Ausgangssituation): Das Arbeitsmedium ist mit dem kalten Reservoir im Gleichgewicht ⇒
T1=TK, V1, p1
1. Schritt
irreversible isochore Wärmeleitungsprozesse
Das Volumen wird festgehalten
das Arbeitsmedium wird an das heisse
Reservoir thermisch gekoppelt ⇒ Wärme
fließt irreversibel vom heissen Reservoir
zum kälteren Arbeitsmedium, bis die
Temperatur TH erreicht wird.
Wärmefluss nicht rückgängig
⇒ irreversibel
irreversibel übertragene Wärme:
q1=n.CV.(TH-TK)
Zum Vergleich die Entropieänderung
für den reversiblen Prozess:
TH
ΔS1rev =
2. Schritt
reversible isotherme Expansion ⇒
T
⎛T ⎞
dqrev H CV ⋅dT
∫ T = ∫ T =CV ⋅ln ⎜⎝ THK ⎟⎠
TK
TK
ΔT=0
Man lässt das Volumen isotherm expandieren: T=TH = konst.⇒ ΔU=0
⎛V ⎞
q2 =− w2 =+ n⋅R⋅TH ⋅ln ⎜ 2 ⎟ >0
V1 ⎠
⎝N
>1
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 23
© Dr. Ogrodnik
irreversible isochore Wärmeleitungsprozesse
3. Schritt
das Arbeitsmedium wird an das kalte Reservoir thermisch gekoppelt ⇒
Wärme fließt irreversibel zum kalten Reservoir, bis die Temperatur TK
erreicht wird
q3=n.CV.(TK - TH)
Zum Vergleich die Entropieänderung
für den reversiblen Prozess::
T
dq rev K CV ⋅dT
∫ T =∫ T
TH
TH
ΔS3rev =
TK
⎛T ⎞
=−CV ⋅ln ⎜ H ⎟ =−ΔS1rev
⎝ TK ⎠
4. Schritt
reversible isotherme Kompression (irreversibler Zyklus abgeschlossen!)
Das Medium bleibt am kalten Reservoir gekoppelt ⇒
isotherm, T=TK ⇒ ΔU=0
Übertragene Wärme:
⎛V ⎞
q 4 =− w = + n⋅ R ⋅TK ⋅ln ⎜ 1 ⎟ < 0
V2 ⎠
⎝N
<1
Es muss Volumenarbeit ins System
gesteckt werden, um wieder auf das
Ausgangsvolumen zu kommen
Für die reversiblen Schritte 2 und 4 ergibt sich:
ΔS 4 =
⎛V ⎞ q
q4
=− n⋅R⋅ln ⎜ 2 ⎟ = - 2 =−ΔS 2
TK
⎝ V1 ⎠ TH
Reversibler Kreisprozess
Es heben sich jeweils die Entropieänderungen im Schritt 1 und 3 bzw. 2 und 4 auf ⇒
v∫
dS = 0 wie
reversibler
Zyklus
erwartet!
Irreversibler Kreisprozess
-q2
n⋅CV ⋅(TK −TH ) T
n⋅CV ⋅(TH −TK )
P
P
PH
q
q3
q1
q
q
+ 2+
+ 4
∑ Ti =
TH
TK
TH
TK
Zyklus i
⎛ 1 1 ⎞
qi n⋅CV ⋅(TH −TK ) n ⋅CV ⋅(TK −TH )
=
+
= n⋅CV ⋅(TH −TK ) ⋅⎜
− ⎟
TH
TH TK ⎠
TK
Zyklus Ti
⎝
<0!
gilt unabhängig von der Umlaufrichtung!
∑
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 24
© Dr. Ogrodnik
2. Schlussfolgerung:
q
<0
für irreversible Prozesse:
T
irreversibler
v∫
Zyklus
Je grösser die Irreversibilität (d.h. je grösser TH-TK) desto negativer ist das Integral
Beachte: für irreversible Prozesse ist die Entropieänderung nicht! durch das Integral
q !!!
∫ T ≠ ΔS gegeben.
irreversibler
Weg
6.8. Messung der Entropie – Absoluter Bezugspunkt bei T→0 – 3. Hauptsatz
6.8.1.
Beiträge zu qrev:
⎛ dq ⎞
dqrev =⎜
⎟ ⋅T =cV ⋅dT (bei konst V)
⎝ dT ⎠V
♦ Schmelzwärme: ΔHSchmelz
Verdampfungswärme: ΔHVerdampfung
♦
♦ sonstige Phasenübergangsenthalpien
TSchmelz
T
C p ( s)
ΔHVerdampf T C p ( g )
ΔH Schmelz Siede C p ( fl )
S (T ) =
SN
(0)
+ ∫
⋅dT +
+ ∫
⋅dT +
+ ∫
⋅dT (13)
T
T
T
T
T
Schmelz
Verdampf
T
T
0
willkürlich
♦
Schmelz
Siede
gewählte
Bezugstemperatur
T =0 K
Problem: Bei sehr tiefen Temperaturen (<30K) ist es schwer die Cp zu bestimmen ⇒ Lösung: man
extrapoliert anhand einer theoretischen Näherung ⇒ Debye´sches T3 Gesetz: C p (T ) =const ⋅T 3 (hier
werden alle Schwingungen des gesamten Festkörpers mitberücksichtigt, die man sich als stehende Wellen
vorstellen kann, deren Anzahl quadratisch mit der Schwingungsfrequenz zunimmt⇒ siehe z.B. Kittel,
Festkörperphysik)
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 25
© Dr. Ogrodnik
Beispiel: Stickstoff
B
A
Aufgetragen
n ist die spezzifische Wäärme Cp getteilt durch T,
T so dass man
m durch Inntegration die
d Entropie
e
erhält:
Die Entrropie gemäß
ß Gl. (13):
Cpα
T3
extrapoliertt!
6
6.8.2.
Ab
bsoluter Beezugspunktt der Entropie – Beisp
piel Schwefe
fel
P
Problem
: Wir
W kennen die
d Entropieen bei T=0 nicht,
n
d.h. wir
w haben keeinen absoluuten Wert
W
Wichtiges
B
Beispiel:
Scchwefel: 2 Modifikation
M
nen: rhomb
bisch / monooklin
monoklinen Kristallsystem
m
K
m. Sie bildet nadelförmige
e Kristalle
rh
rhombisch.
M erkennt Doppelyrami
Man
D
iden, denen häufig noch die Spitzen fehlen
f
K
Kühlt
man mo
onokline Krisstalle ab, so zerfallen
z
die Nadeln (wen
nn auch lang
gsam) unter B
Bildung von rhombischen
n
D
Dipyramiden.
.
rhombisch-ddipyramidall
S
SS2008
Entrropie, freie Enthalpie + Gibbs´schhe Gl. 26
© Dr. Ogrod
dnik
Umwandluungstemperaatur TUmwanddlung=368.5K
K
s
gen
nug
Wenn mann die monokkline Form schnell
unter TUmwwandlung kühltt (Abschrecken), bleibtt sie
in der monnoklinen Phaase „gefang
gen“ und kann
bei beliebig tiefen Tem
mperaturen vermessen
werden.
rhoombisch
monoklin
n
SS2008 Entrropie, freie Enthalpie + Gibbs´schhe Gl. 27
S
© Dr. Ogrod
dnik
J
ΔHUmwanndlung 401 m
mol =1.09 J
=
U
Umwandlun
ngsentropie ΔSUmwandlunng =
TUmwanddlung
K ⋅mol
368..5K
Man kann diie spezifischhe Wärme beider
M
b
Form
men bis ≈13K
K vermesseen und anschhließend miit T3 bis T→
→0
e
extrapolieren
n (siehe Graaphik).
W lösen (113) nach S(oo) auf und erhalten
Wir
e
anhhand der exp
perimentelleen Werte:
ΔH Um
=ΔS Um =
TUm
TUm
Cp (s,monooklin)
⋅dT
∫
T
0 J
37.82
m
K ⋅mol
J
1.09
K ⋅mol
m
Smonooklin
Srhom
mb
ΔSUm
TUm
∫
Cp (s,rhomb
b)
T
⋅dT
0 3
36.86
J
ol
K ⋅mo
S rhomb (0)
( − S monoklinn (0)
Wir erhaltenn somit für T→0
W
T
folgennden
J
E
Entropieun
nterschied für
f beide Modifikationeen:
= 0.13
K ⋅ mol
S
Schlussfolge
erung: für T→0 sind die
d Entropieen der beideen Schwefellmodifikatioonen praktiisch gleich!
6
6.8.3.
3. Hauptsatz
H
Allgemeine Beobachtu
A
ung: Auch für
f andere Substanzen
S
extrapolierrt S auf
d gleichenn Wert bei T→0
den
T
D
Deshalb
deffiniert man
n:
P
Planck
[19111]: Thermodynamik, 3rdd Ed. Leipzigg: Veit & Co.,,
S →0)=0
S(T
für reine, perfekt
p
geoordnete Sub
bstanze)
Walther Nernst
N
[1864-19
941]
S
SS2008
Entrropie, freie Enthalpie + Gibbs´schhe Gl. 28
© Dr. Ogrod
dnik
Es gibt noch eine ganze Reihe anderer Formulierungen des 3.HS´s
Nernstsches Prinzip:
Die Entropieänderung eines reversiblen Prozesses geht gegen Null,
wenn die Temperatur gegen Null geht
Nernstsches Wärmetheorem:
Die maximale Arbeit die ein Prozess leisten kann, errechnet sich aus der Wärme, die dieser Prozess bei
T→0 freisetzt (vgl. Carnot´scher Wirkungsgrad:
ε rev =
− wsystem
qH
=1−
TK
→1 )
TH
Beobachtung:
Bei dem Versuch, immer tiefere Temperaturen zu erreichen, stellte man fast, dass es mit abnehmender
Temperatur immer schwieriger wurde, die Temperatur noch weiter abzusenken. ⇒
Nernstsche Behauptung:
Es ist unmöglich T=0 in endlich vielen Schritten zu erreichen!
6.9. Mikroskopische (statistische) Interpretation der Entropie
Erinnerung: Kriterium für einen spontanen Prozess
Ein Prozess läuft spontan so ab, dass die Gesamtenergie des abgeschlossenen Systems möglichst
"gleichmäßig" (d.h. ungeordnet) verteilt ist ⇒ Gleichgewicht
Das System strebt dem Zustand maximaler Wahrscheinlichkeit zu
Frage: Wie können wir das quantifizieren?
Die Wahrscheinlichkeit ist proportional zur Anzahl der verschiedenen Möglichkeiten Ω, die
Gesamtenergie auf die unterschiedlichen Zustände des Systems zu verteilen (vgl. Zahl der
Kombinationsmöglichkeiten eine bestimmte die Gesamtenergie zu erhalten bei der Ableitung des
Boltzmannfaktors!)
Entropie als Mass der Unordnung muss folglich eine Funktion von Ω sein:
S=(f(Ω)
ges
= qrev ,1 + qrev,2 .
Hat man zwei Systeme 1 und 2, dann ist die Gesamtwärmemenge qrev
Beziehen wir uns auf das gleiche Wärmereservoir (isotherm), dann ist die Gesamtentropie ebenfalls die
Summe:
ges
ges
qrev ,1 + qrev ,2 qrev ,1 qrev ,2
qrev
qrev
= S1 + S 2 (14)
=
=
+
= S1 + S 2 ⇒ S ges =
S ges =
T
T
T
T
T
N
N
S1
S2
Gesamtwahrscheinlichkeit:
Die Gesamtwahrscheinlichkeit für zwei Ereignisse ist das Produkt der Einzelwahrscheinlichkeiten
Ω ges =Ω1 ⋅Ω 2 (15)
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 29
© Dr. Ogrodnik
Beispiel:
41.1% der Bevölkerung in Deutschland ist weiblich
36 % der Bevölkerung in Deutschland ist blond
Ω(blond und weiblich)=Ω (blond)⋅Ω (weiblich)=41.1%⋅36%=18.6%
Suche: wir suchen eine Funktion f(Ω), die sowohl das Kriterium für die Gesamtentropie (14) als auch das
für die Gesamtwahrscheinlichkeit (15) erfüllt:
f (Ω ges ) = f ( Ω1 ⋅Ω2 ) = f ( Ω1 ) + f ( Ω2 )
Die einzige Funktion ist der Logarithmus: ln ( Ω1 ⋅Ω2 ) =ln ( Ω1 ) + ln ( Ω2 )
( )
Statistische Definition der Entropie: S = k B ⋅ln Ω (0.16)
Es stellt sich heraus, dass die Proportionalitätskonstante kB die Boltzmannkonstante ist
Statistische Sicht des 3.HS:
Bei T→0 liegt ein perfekter Kristall ohne thermisch induzierte Fehlstellen vor. In diesem Fall gibt es nur
1 einzige Möglichkeit einen perfekten Kristall anzuordnen ⇒ Ω=1 ⇒ S=0
6.9.1.
Beispiel: N Moleküle HCl bilden einen perfekten Kristall bei T=0 (starke WW)
Es gibt nur eine einzige Anordnung der Moleküle ⇒ Ω=1
S = k ⋅ln (1) =0
Stimmt mit der thermodynamischen Definition von S(T→0)=0 überein!
Beispiel: Abweichungen von S(T→0)=0 bei nicht perfekten Kristallen
6.9.2.
Es gibt Substanzen die bei Abkühlung T→0 sich nicht perfekt ordnen, weil während des Abkühlprozesses
das thermische Gleichgewicht nicht vollständig aufrecht erhalten werden kann (Unordnung wird
Eingefroren, bevor der niederste energetische Zustand eingenommen werden kann). Folglich ist die
Entropie nicht Null!
Beispiel: 1mol = NA Moleküle CO bei T = 0K
6.9.3.
Es gibt grundsätzlich für ein Paar jeweils zwei Möglichkeiten der relativen Orientierung im Kristall, d.h.
ΩPaar=2:
parallel: C ≡ O C ≡ O bzw. antiparallel: C ≡ O O ≡ C
Aufgrund des sehr kleinen Dipolmomentes ist die Wechselwirkung klein und somit der
Energieunterschied zwischen beiden Einstellungen sehr klein. Beim Kristallisieren ist die Temperatur und
damit die thermische Energie noch so hoch, dass die Moleküle in statistischer Unordnung eingebaut
werden. Kühlt man weiter ab, reicht aber die thermische Energie nicht mehr aus um ein Molekül im
Kristallverband umzudrehen, da man dafür sehr viel Energie aufwenden müsste. Die Entropie kann in
diesem Fall deshalb nicht weiter mit der Temperatur absinken (thermische Falle).
( )
J
N
Ω ges =Ω Paar ⋅Ω Paar ⋅Ω Paar ......= 2 N ⇒ S = k ⋅ln 2 A = k ⋅ N A ⋅ln ( 2 ) =5.76
K ⋅mol
N Moleküle
6.9.4.
Beispiel: Pokern
Ein Pokerspiel hat 4x13=52 Karten
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 30
© Dr. Ogrodnik
Es gibt 52!=8.066 x 1067 Möglichkeiten die Karten anzuordnen.
Im Prinzip kann man, wenn man lang genug mischt, ein völlig geordnetes Spiel "mischen", die
Wahrscheinlichkeit ist aber eher nicht ermutigend.
52!
= 2598960 verschiedene Blätter, davon sind die meisten bekanntermaßen wertlos.
Es gibt
5!⋅( 52−5 ) !
In nachfolgender Tabelle sind die (nicht normierten) Wahrscheinlichkeiten (Ω=Zahl der Möglichkeiten)
ein bestimmtes Blatt zu erhalten eingetragen, zusammen mit dessen Logarithmus.
Die Entropie eines wertlosen Blattes (kein Paar) ist folglich ca. 10 mal größer als die eines Royal flash.
Blatt
Ω
ln Ω
Royal flash (AKQJ10 gleicher Farbe)
4
1.39
Straight flash (5 Karten gleicher Farbe in Folge) 36
3.58
4 gleiche Karten
624
6.44
Full house (3 gleiche Karten + 1Paar)
3,744
8.23
Flush (5 Karten gleicher Farbe)
5,108
8.54
Straight (5 Karten beliebiger Farbe in Folge)
10,200
9.23
3 gleiche Karten
54,912
10.91
2 Paare
123,552
11.72
1 Paar
1,098,240 13.91
Kein Paar
1,302,540 14.08
Total 2,598,960
6.10. Praktische Anwendung: Adiabatische Entmagnetisierung
Eine magnetische Substanz besteht aus vielen "Elementarmagneten" (=Spins von Ionen).
Paramagnetismus: Diese Ionenspins können mit einem extern angelegten Magnetfeld wechselwirken, so
dass sie sich mehr oder weniger parallel dazu stellen. Bei höheren Temperaturen nimmt die
Wahrscheinlichkeit zu, dass ein Spin in den energetisch ungünstigeren antiparallelen Zustand geht, und
zwar liegt dieser antiparallele Zustand energetisch umso höher, je stärker das äußere Feld ist.
D.h. das Spinsystem (neuer Freiheitsgrad!) kann umso leichter thermische Energie aufnehmen, je kleiner
das äußere Feld ist (in anderen Worten: die spezifische Wärme des Spinsystems nimmt mit steigendem
äußeren Feld ab und mit zunehmender Temperatur zu). Der gesamte Wärmeinhalt eines Paramagneten ist
auf die Gitterfreiheitsgrade und die Spinfreiheitsgrade verteilt, wobei bei hinreichend tiefen Temperaturen
und hohen externen Feldern die Spinfreiheitsgrade weitgehend gesperrt werden können. Schaltet man das
Feld aus, so kann ein Teil der thermischen Energie aus dem Gitter ins Spinsystem fließen, die Zahl der
verfügbaren Freiheitsgarde nimmt zu, die mittlere thermische Energie pro Freiheitsgrad nimmt ab.
Folglich nimmt die Temperatur ab!!!
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 31
© Dr. Ogrodnik
Ausgangszustand:
Ungeordneter Magnet
große Entropie
1. Schritt:
Das System wird an ein
Reservoir gekoppelt ⇒
isotherm
Durch Anlegen eines
äußeren Magnetfeldes
werden die Magneten
orientiert ⇒ geordnet ⇒
Entropie qrev/T nimmt ab
Folglich muß das
"Spinsystem" Wärme qrev
ans Reservoir abgeben
2. Schritt:
Das System wird thermisch
entgekoppelt ⇒ adiabatisch
Das äußere Magnetfeld wird
abgeschaltet ⇒ die SpinOrdnung verschwindet ⇒ SpinEntropie qrev/T nimmt zu
Folglich muß das "Spinsystem"
Wärme qrev aufnehmen
Die einzige Wärmequelle die
zur Verfügung steht, sind die
Schwingungen des Gitters
⇒ dessen Temperatur und
Entropie nimmt ab
Die Gesamtentropie von Spin
und Gitter jedoch bleibt
konstant (oder nimmt bei einem
Wärmeleck zu)!
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 32
© Dr. Ogrodnik
6.11. Entropiefluss in Wärmekraftmaschinen und Wärmepumpen
Voraussetzung: Prozess zur Erzeugung von Arbeit muss freiwillig ablaufen.
⇒ Erhöhung der Entropie!
Hypothetische Maschine: Aus einem Wärmereservoir (bei der Temperatur Tw) wird die Wärmemenge qw
vollständig in Arbeit umgewandelt.
Wärmeabgabe⇒ Negative Entropie ⇒ Prozess nicht freiwillig ⇒ Maschine arbeitet nicht
⇒
Wärme kann nicht vollständig in Arbeit umgewandelt werden 2.HS!
⇒ Zweites Wärmereservoir notwendig (bei der Temperatur Tw), welches einen Teil der Wärme +qk
aufnehmen kann ⇒ ergibt positiven Entropiebeitrag ΔS =+
qw
Tw
ΔS ges =−
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 33
qw qk
+
Tw Tk
© Dr. Ogrodnik
Maschine läuft, falls ΔSges positiv ⇒
qk ≥
Tk
⋅q w
Tw
Wärmemenge qk abgegeben
Energieerhaltung⇒Differenz wird zu Arbeit
Wärmemenge qw aufgenommen
Maximale Arbeit:
⎛ T ⎞
wmax = q − qk ,min =⎜ 1− k ⎟⋅qw
⎝ Tw ⎠
Maximaler Wirkungsgrad:
ε max =
wmax
T
ΔT
=1− k oder =
qw
Tw
Tw
(0.17)
Für Tk→0 oder Tw→∞ wird ε→1
6.11.1.
Aus der Praxis: Moderne Dampfmaschinen
Exkurs
kaltes Reservoir ⇒ Flüsse, Seen oder Luft ⇒ Tw≈280-290K
Dampfmaschine:
Tw(Wasser)<Tkrit=647K (374oC)
ε max =57%
1985: Durchschnittlicher Wirkungsgrad aller Kraftwerke ε=38 %
Maximaler Wirkungsgrad ε=43 %.
1997: ein dänisches Kraftwerk: Welt-Bestwert von 47 %.
Hochdruck Dampf-Maschinen
Um die Effizient gegenüber er Watt´schen Dampfmaschine zu steigern wird das Wasser in geschlossenen
Druckkesseln erhitz (die früher oft explodierten!). Der Hochdruck-Dampf wird über ein Ventil (8) dem
Zylinder von einer Seite (hier links) zugeführt, so dass der Kolben bis zum rechten Totpunkt gedrückt
wird. Dabei kann in der rechten Kammer der Restdampf über das geöffnete Ventil entweichen. Dann wird
das Ventil umgestellt, der Dampf wird rechts zugeführt und der Restdampf kann links entweichen..
Animation: http://commons.wikimedia.org/wiki/Image:Steam_engine_in_action.gif
http://www.avero.de/?links/dampfmaschine/index.html
Mehrstufige Dampfmaschinen:
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 34
© Dr. Ogrodnik
Um den hohhen Druck möglichst
U
m
voollkommen auszunutzeen (es sollte ja ein grossses Endvolu
umen und
d
dadurch
ein einen mögllichst niederren Enddrucck erreicht werden)
w
kannn der Dam
mpf schließliich in
h
hintereinand
dergeschalteeten mehrstuufige Expannsionszylind
dern auf Athhmosphärenndruck (odeer mittels einnes
K
Kondensator
rs sogar darrunter) entsppannt werdeen⇒ Effizieenzoptimierrung.
E Kraftweerk mit Wärrmerückgew
Ein
winnung im Gegenstrom
mprinzip errreicht etwa 30% Effizieenz.
h
http://www.
answers.com
m/main/Reccord2?a=NR
R&url=http
p%3A%2F%
%2Fcommonns.wikimed
dia.org%2Fw
wiki
%
%2FImage%
%3ATriple+
+expansion+
+engine+animation.giff
D
Dampfturb
ine
Idealisierte Turbine
T
mitt thermodynnamischen Kreisprozes
K
ss (Brayton--Zyklus):
1→2 Adiaabatische Koompression (wenn irrevversibel: iseenthropisch,, s.später)
T3
2
2→3
Isobaare Erwärm
mung q2 = ∫ C Gas
T ⇒ Expanssion
p ( T ) dT
TT2
⎛ ⎛ V ⎞γ −1 ⎞
w=CV ⋅TA ⎜ ⎜ A ⎟ −1⎟
⎜ ⎝ VE ⎠
⎟
⎝
⎠
V3 T3
(ideal)
=
V2 T2
3
3→4
Adiaabatische (issenthropischhe) Expansiion
4
4→1
Isobaare Abkühluung (im Freeien)
S
SS2008
Entrropie, freie Enthalpie + Gibbs´schhe Gl. 35
© Dr. Ogrod
dnik
Entwicklungspotential: Kombinierte Gas- und Dampfturbinen (GuD) zielen auf Wirkungsgrade um
55 %.
Wasserkraftwerk mit 85 %
Solarzelle ⇒ elektronisch angeregter Zustand z.B. 1eV⇒96kJ/mol⇒T=109K
Aber Photonenspektrum mit Maximum bei 2eV ⇒ Rest geht als Wärme verloren! ⇒ Solar: 15%
Potential: 30%
Kraft-Wärme-Kopplung (KWK) :
durch die gleichzeitige Nutzung der Niedertemperatur-Wärme als Prozesswärme (z.B.BASF) ⇒ sehr viel
höherer Gesamt-Wirkungsgrad (bis zu 90 Prozent, d.h. nur 10% der Wärme geht ungenutzt in die
Umgebung verloren). Achtung: hier bezieht sich der Wirkungsgrad nicht auf die Arbeit sondern auf die
gesamte genutzte mechanische und thermische Energie.
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 36
© Dr. Ogrodnik
6.11.2. Kältemaschinen
Wärmefluss kalt ⇒ heiss
ist nicht spontan!!
q
Pk
q q
ΔS Ges = w − k <0
Tw Tk
Die Entropieabnahme bestätigt das.
Ausweg:
Arbeit zuführen um qw grösser zu machen ⇒ qW=qk+w
⇒ inverse Wärmekraftmaschine
ΔS ges =−
qk qk + w
+
≥0!
Tk
Tw
Die Entropiezunahme bestätigt, dass die zugeführte Energie w zu einem "spontanen" Wärmeentzug aus
dem kalten Reservoir führt
Aus
ΔS ges =−
qk qk + w
+
≥ 0!
Tk
Tw
folgt ⇒
⎛T
⎞
qk + w qk
≥ ⇒ w≥ qk ⋅⎜ w −1⎟
Tw Tk
⎝ Tk ⎠
Wir definieren den
Leistungskoeffizienten:
c≡
qk
w
qk
Tk
Tk
c
≡
=
=
max
⇒
wmin Tw −Tk ΔT
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 37
© Dr. Ogrodnik
6.12. Die Clausius´sche Ungleichung
Problem: Es ist umständlich immer System und Umgebung zu berücksichtigen um die Gesamtentropie
auszurechnen. Wir wollen deshalb nur das System allein betrachten:
der Umgebung zugeführte Wärme = die vom System abgegebene Wärme
qUmgebung =− q Syst
Syst
Mit ΔS
Umgebung
qUmgebung ⇒ ΔS ges =ΔS Syst − q
=
TUmgebung
TUmgebung
Im thermischen Gleichgewicht ⇒ TUmgebung=TSyst
Wir können die Clausius´sche Ungleichung neu formulieren:
ΔS
⇒
ges
=ΔS
Syst
q Syst
−
≥0
TSyst
(hier kommt nur noch das System vor)
oder differentiell:
dS −
Clausius´sche Ungleichung
isochorer Fall: V=konst. ⇒
dS −
dU
≥0
T
⇒
(18)
w=− p⋅dV =0 ⇒ dU = dqV eingesetzt:
T⋅dS ≥ dU
Wenn U=konst.
dq
≥0
T
Nur noch von Zustandsfunktionen abhängig
Wenn S=konst.
T⋅dS ≥ 0
dU ≤ 0
isobarer Fall: p=konst.
⎛ dH ⎞
dq p =C p ⋅dT =⎜
⎟ ⋅dT = dH
⎝ dT ⎠ p
in (18) ⇒
dS −
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 38
dH
≥0
T
⇒
T ⋅dS ≥ dH
© Dr. Ogrodnik
6.13. Maximal mögliche Arbeit→Freie Energie
Wir suchen eine Zustandsfunktion die uns die Maximal mögliche Arbeit bei konstanter Temperatur
angibt.
1 HS:
dU =δ q +δ w
(19) (abgegebene = geleistete Arbeit= -δw)
2. HS: die maximale Arbeit -δwmax wird dann geleistet, wenn in der
Clausius´schen Ungleichung (18) das Gleichheitszeichen gilt:
dS −
dq
=0
T
⇒
δ q =TdS
in (19) ⇒
dU =T ⋅dS +δ wmax
oder:
δ wmax = dU −T ⋅dS
(20)
da isotherm: d (T ⋅S ) =T ⋅dS + S ⋅dT
N (21) ⇒
0
δ wmax = dU − d (T ⋅S ) = d (U −T ⋅S )
≡A
U und S sind Zustandsfunktionen T Zustandsgröße ⇒
A ist eine Zustandsfunktion
Definition: Freie Energie
A≡U −T ⋅S
(22)
mit (21):
ΔA≡ΔU −TN
⋅ΔS
ist der Teil der inneren
Energie der maximal
in Arbeit umgesetzt
werden kann ⇒ frei!
Änderung der Inneren
Energie=
im System zur
Verfügung stehende
Energie die bei V=konst
in Form von Wärme
abgegeben werden kann
(23)
ΔS<0: gebundene
Energie=Anteil der nicht in
Arbeit umgesetzt werden kann
ΔS>0: maximale
Wärmemenge die aus der
Umgebung zufliessen kann
Erläuterung:
ΔS negativ: ⇒ ΔA<ΔU
Wir sehen, was wir bisher schon wiederholt festgestellt haben, dass nicht die gesamte innere Energie in
Arbeit umgesetzt werden kann.
Grund: ein Teil der inneren Energie muss in Form von Wärme abgegeben werden, um in der Umgebung
die Entropie zu vergrößern und somit die Entropieabnahme im System zu kompensieren.
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 39
© Dr. Ogrodnik
ΔS positiv: ⇒ ΔA>ΔU
In diesem Fall kann die geleistete Arbeit größer sein als die Änderung der inneren Energie. Das System
nimmt aus der Umgebung die dazu notwendige Energie in Form von Wärme auf (nicht mehr als TΔS!).
Dies wird dadurch ermöglicht, dass im System die Entropie zunimmt, und somit eine Entropieabnahme in
der Umgebung kompensiert wird.
6.14. Maximal mögliche Nicht-Volumenarbeit ⇒ Freie Enthalpie
Neben der Volumenarbeit –pdV kann die innere Energie dazu genutzt werden, auch NichtVolumenarbeit we zu leisten, z.B. elektrische Arbeit die benötigt wird um Elektronen durch einen
Stromkreis zu drücken ⇒ Batterie, Brennstoffzelle)
δ w= −
p⋅dV +
Volumenarbeit
δN
we
(24)
Nicht −Volumenarbeit
Um die maximale Nicht-Volumenarbeit zu berechnen schauen wir uns die Enthalpieänderung an:
dH =δ q +δ w+ d ( pV )
1 HS:
(25)
2. HS: die maximale Arbeit δwmax wird dann geleistet, wenn in der Clausius´schen Ungleichung (18) das
Gleichheitszeichen gilt:
dS −
dq
=0
T
Wir ersetzen δq durch
δ q =TdS
und δw durch (24) ⇒
dH =TN
⋅dS − p⋅dV +δ we,max + p⋅dV +V ⋅dp
δ q ⋅ )
d ( pV
δ wmax
Annahme: isotherm und isobar
isotherm:
d (T ⋅S ) =T ⋅dS + S ⋅dT
N
0
isobar:
dp=0
beides eingesetzt
dH = d (T ⋅S ) − p⋅dV +δ we ,max + p⋅dV ⇒
δ we,max = dH − d (T ⋅S ) = d ( H −T ⋅S )
H −T ⋅ S ist wieder eine neue Zustandsfunktion:
Analog zu Gl. (22) definieren wir:
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 40
© Dr. Ogrodnik
G ≡ H −T ⋅S
Definition: Freie Enthalpie:
(26)
ΔG ≡ΔH −T ⋅ΔS
und
-ΔG=we,max ist die maximal mögliche
Nicht-Volumenarbeit, die das System bei
konstantem Druck und konstanter
Temperatur leisten kann
Wir werden gleich sehen, dass ΔG eine enorm wichtige Größe im Zusammenhang mit dem
Gleichgewicht chemischer Reaktionen ist.
6.15. Gibbs´sche Gleichungen oder Fundamentalgleichungen der Thermodynamik
Wir haben aus dem 1. HS und 2. HS (Clausius´sche Ungleichung) die maximal mögliche Volumenarbeit
abgeleitet (18): δ wmax = dU − T ⋅ dS
Wir lösen nach dU auf und erhalten: dU = T ⋅ dS + δ wmax
Da U eine Zustandsfunktion ⇒ wegunabhängig ⇒ d. h. obige Gleichung muss auch für irreversible
Prozesse gelten.
dUirrev=dUrev
d.h. sie gilt auch auf Wegen, bei denen nicht die maximale Volumenarbeit δwmax geleistet wird.
Wir können also δwmax durch –p.dV für einen beliebigen Weg der Anfangs- und Endzustand verbindet
ersetzen und erhalten die sogenannte
dU = T ⋅ dS − p ⋅ dV
Fundamentalgleichung:
Sie enthält das "geballte" Wissen des 1. und 2. HS
(0.27) gilt auch für irreversible Prozesse!
Reversibler Fall: Der 1. Term der Fundamentalgleichung entspricht der reversibel umgesetzen Wärme:
T ⋅ dS = δ q rev , und der zweite Term der Arbeit, die gemäß der Carnot´schen Behauptung (→6.3.4) die
maximal mögliche ist δ w rev =−p⋅dV =δ w max . Setzen wir beide Terme in die FG ein, so erhalten wir den
1.HS: dU = δ q + δ w
(Erinnerung: Die Schreibweise δ q bzw. δ w bedeutet, das es sich nicht um ein vollständiges Differential
dq bzw. dw handelt).
rev
rev
Irreversibler Fall: In diesem Fall gilt die Clausius´sche Ungleichung und T ⋅ dS > δ q
Obwohl nun der erste Term größer ist als im reversiblen Fall, bleibt der 1.HS: nach wie vor erfüllt, da der
zweite Term entsprechend kleiner ist: − p ⋅ dV < δ wmax ist (hebt sich gegenseitig auf!).
Da dU ein vollständiges Differential sein muss (Zustandsfunktion), bedeutet Gl. (0.27), dass U eine
eindeutige Funktion der Variablen S und V ist (siehe 6.15.2 bis 6.16)
6.15.1. Weitere Fundamentalgleichungen der Thermodynamik ⇒ die Gibbs´schen Gleichungen
Ausgehend von Gl. (0.27) können wir weitere Fundamentalgleichungen ableiten, die anstatt der inneren
Energie U andere Zustandsfunktionen enthalten:
Enthalpie: H ≡U + p ⋅V ⇒ Differential: dH = dU + p⋅dV +V ⋅dp
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 41
© Dr. Ogrodnik
dU = T ⋅ dS − p ⋅ dV einsetzen: dH =T ⋅dS + p⋅dV +V ⋅dp − p⋅dV ⇒
dH =T ⋅dS +V ⋅dp
(0.28)
Freie Enthalpie: G ≡ H −T ⋅S ⇒ Differential: dG = dH −T ⋅dS − S ⋅dT
dH =T ⋅dS +V ⋅dp einsetzen: dG = T ⋅ dS − p ⋅ dV + p ⋅ dV +V ⋅ dp − T ⋅ dS − S ⋅ dT ⇒
dG =V ⋅dp − S ⋅dT (0.29)
Freie Energie: A≡U −T ⋅S ⇒ Differential: dA= dU −T ⋅dS − S ⋅dT ⇒ mit dU = T ⋅ dS − p ⋅ dV ⇒
dA=− S ⋅dT − p⋅dV (0.30)
Beachte die Vorgehensweise: Die "energetischen" Zustandsfunktionen H, A, und G werden
ausdifferenziert, die ursprüngliche Fundamentalgleichung (0.27) eingesetzt und alle sich aufhebenden
doppelten Terme gestrichen. Dies lässt sich leichter durchführen und merken, als gewisse kursierende
Merkregeln (Guggenheimschema), die zudem "inhaltsleer" sind.
Da dU, dH, dA und dG vollständige Differentiale sind (Zustandsfunktionen!!), können diese
vollständigen Differentiale anhand der zu den jeweiligen Variablen gehörenden partiellen Ableitungen
dargestellt werden:
⎛ ∂G ⎞
⎛ ∂G ⎞
z.B.:
dG =⎜
⎟ ⋅dp + ⎜
⎟ ⋅dT ).
⎝ ∂T ⎠ p
⎝ ∂p ⎠T
Diese können nun mit den Gibbs´schen Gleichungen verglichen werden, so dass wir daraus direkt diese
partiellen Ableitungen ablesen kann:
z.B.:
dG =V ⋅dp − S ⋅dT
⎛ ∂G ⎞
⎛ ∂G ⎞
und erhalten: ⎜
⎟ =− S (0.32)
⎟ =V (0.31) und ⎜
⎝ ∂T ⎠ p
⎝ ∂p ⎠T
Bevor wir uns einige Anwendungen dieser Gleichungen ansehen, können wir auch aus den anderen
Gibbs´schen Gleichungen nützliche partiellen Ableitungen ablesen:
6.15.2.
Übersicht über alle Gibbs´schen Gleichungen mit den dazugehörenden partiellen
Ableitungen:
(0.33)
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 42
© Dr. Ogrodnik
6.15.3.
Anwendungsbeispiel: Druckabhängigkeit der Freien Enthalpie
⎛ ∂G ⎞
Anhand von (0.31): ⎜
⎟ =V sehen wir, dass das Volumen die Druckabhängigkeit der Freien
⎝ ∂p ⎠T
Enthalpie bestimmt.
Fig. 6.1: Druckabhängigkeit
der freien Enthalpie
Ausgehend von dG =V ⋅dp − S ⋅dT können wir bei festgehaltener Temperatur dG auf integrieren:
p2
T =konst
p2
G ( p2 ) =G ( p1 ) + ∫ dG = G ( p1 ) + ∫ Vdp
p1
p1
Für Flüssigkeiten und Festkörper können wir V näherungsweise als konstant annehmen und erhalten:
G ( p2 ) =G ( p1 ) +V ⋅Δp
Für Gase nehmen wir das ideale Gasgesetz und integrieren:
p2
p2
⎛p ⎞
1
G ( p2 ) =G ( p1 ) + ∫ Vdp =G ( p1 ) + n⋅ R⋅T ∫ dp =G ( p1 ) + n⋅R⋅T ⋅ln ⎜ 2 ⎟
p
⎝ p1 ⎠
p1
p1
p2
Das Integral
( p)
anschaulich dargestellt. Da
∫ V ( p ) dp ist nebenan graphisch als Fläche im Vp-V-Diagramm
p1
der Druck p die Integrationsvariable ist, muss entsprechend die Fläche zwischen der p-Achse und der
Isothermen betrachtet werden (man könnte natürlich auch die Achsen vertauschen).
ΔG eines Gases zwischen zwei Drücken
entspricht der Fläche unter der zugehörigen
Isothermen - gilt auch für reale Gase!
Achtung: nicht mit der Volumenarbeit
V2
w=− ∫ pdV verwechseln!
V1
Wir sehen in Fig. 6.1 die schwache lineare
Abhängigkeit der freien Enthalpie vom Druck in
kondensierten Phase und die logarithmische
Abhängigkeit in Gasen.
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 43
der
© Dr. Ogrodnik
6.16. DieGibbs-Helmholtz Gleichung
⎛ ∂G ⎞
Analog sehen wir aus (0.32): ⎜
⎟ =− S , dass die Temperaturabhängigkeit der freien Enthalpie
⎝ ∂T ⎠ p
ausschließlich von der Entropie bestimmt ist (vgl.: G (T ) ≡ H (T ) −T ⋅S (T ) )
Da die Entropie (Unordnung!) eines Systems im festen Zustand kleiner ist als im flüssigen, und in der
Gasphase sehr viel grösser ist ergibt sich qualitativ folgende Temperaturabhängigkeit der Freien
Enthalpie (wobei der Einfachheit S temperaturunabhängig angenommen ist). Man sieht, dass G in der
Gasphase eine besonders starke Temperaturabhängigkeit aufweist:
Fig. 6.2 Temperaturabhängigkeit
der freien Enthalpie
Aus der Definition von G erhalten wir − S =
G−H
⎛ ∂G ⎞
und setzen in ⎜
⎟ =− S ein, und erhalten:
T
⎝ ∂T ⎠ p
∂G
G−H
∂G G
H
=−
− =−
⇒
∂T p
T
∂T p T
T
Wir versuchen die linke Seite der Gleichung als Funktion von
G
G
auszudrücken. Wir leiten deshalb
T
T
nach T ab und erhalten:
⎛G⎞
1
∂⎜ ⎟
∂
G
1
1 ⎛ ∂G G ⎞
∂
T
⎝ ⎠ = ⋅
+G⋅ T = ⋅⎜
− ⎟
T ∂T p
T ⎜⎝ ∂T p T ⎟⎠
∂T
∂T
p
Np
1
T2
Oben eingesetzt erhalten wir:
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 44
© Dr. Ogrodnik
⎛G⎞
∂⎜ ⎟
⎝ T ⎠ =− H
(0.34)
T2
∂T
Die Gibbs-Helmholtz Gleichung:
p
Folgerung: Temperaturabhängigkeit von G/T hängt nur von der Enthalpie H ab, und ist unabhängig
von der Entropie!
6.17. Die Maxwellgleichungen
⎛ ∂F ⎞
⎛ ∂F ⎞
Für jedes vollständige Differenzial (d.h. für jede Zustandsfunktion) dF =⎜
⎟⋅dy
⎟⋅dx +⎜
⎝ ∂x ⎠
⎝ ∂y ⎠
⎛ ⎛ ∂F ⎞ ⎞ ⎛ ⎛ ∂F ⎞ ⎞
⎜ ∂ ⎜ ∂x ⎟ ⎟ ⎜ ∂ ⎜ ∂y ⎟ ⎟
⎠⎟
⎠ ⎟ =⎜ ⎝
muss die Schwarze Gleichung gelten: ⎜ ⎝
⎜
∂x ⎟
⎜ ∂y ⎟
⎟
⎜
⎟ ⎜
⎝
⎠x ⎝
⎠y
Wir können nun aus den Gibb´schen Gleichungen die entsprechenden partiellen Differenziale einsetzen:
dU = T ⋅ dS − p ⋅ dV
⎛ ∂T ⎞
⎛ ∂p ⎞
Und erhalten die Maxwellgleichung: ⎜
⎟ =− ⎜ ⎟ (0.35)
⎝ ∂V ⎠ S
⎝ ∂S ⎠V
Entsprechend erhalten wir:
Aus dH =T ⋅dS +V ⋅dp ⇒
⎛ ∂T ⎞ ⎛ ∂V ⎞
⎜
⎟ =⎜
⎟ (0.36)
⎝ ∂p ⎠ S ⎝ ∂S ⎠ p
Aus dA=− S ⋅dT − p⋅dV ⇒
⎛ ∂S ⎞
⎛ ∂p ⎞
⎜
⎟ =− ⎜
⎟ (0.37)
⎝ ∂V ⎠T
⎝ ∂T ⎠V
Aus dG =V ⋅dp − S ⋅dT ⇒
⎛ ∂S ⎞
⎛ ∂V ⎞
⎜
⎟ =− ⎜ ⎟ (0.38)
⎝ ∂T ⎠ p
⎝ ∂p ⎠T
1 ⎛ ∂V ⎞
Im letzeren Fall können wir den Volumenausdehnungskoeffizienten α = ⋅⎜
⎟ einsetzen, und
V ⎝ ∂T ⎠ p
erhalten:
⎛ ∂S ⎞
⎜ ⎟ =V ⋅α (0.39)
⎝ ∂p ⎠T
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 45
© Dr. Ogrodnik
6.17.1.
Suche:
Anwendungsbeispiel: Innere Energie bei isothermer Expansion
∂U
=???
∂V T
Dazu beginnen wir mit der Gibbs´schen Gl. (0.27): dU = T ⋅ dS − p ⋅ dV
Wir halten T fest und leiten nach dV ab (formal durch dV teilen):
⎛ ∂U ⎞
⎛ ∂S ⎞
⎜
⎟ =T ⋅⎜
⎟ − p (0.40) (dies ist nur eine andere Schreibweise der Gibbs´schen Gl.)
⎝ ∂V ⎠T
⎝ ∂V ⎠T
⎛ ∂S ⎞
⎛ ∂p ⎞
Nun können wir die Maxwellgleichung (0.37) ⎜
⎟ =− ⎜
⎟ einsetzen und erhalten:
⎝ ∂V ⎠T
⎝ ∂T ⎠V
∂U
⎛ ∂p ⎞
=T ⋅⎜
⎟ − p (0.41)
∂V T
⎝ ∂T ⎠V
ideales Gas
⎛ ∂p ⎞ n⋅R
und setzen ein:
Wir nutzen das ideale Gasgesetz und erhalten: ⎜
⎟ =
⎝ ∂T ⎠V V
∂U
n⋅ R n⋅R⋅T
=T ⋅
−
=0
∂V T
V N
V
p
Wie erwartet erhalten wir, dass die innere Energie bei der isothermen Expansion eines idealen Gases
erhalten bleibt (hängt nur von T ab!!, da keine zwischenmolekulare Wechselwirkung vorhanden ist,
die sich bei der Expansion ändern könnte)!
reales Gas
⎛ ∂p ⎞
Wir berechnen ⎜
⎟ für ein reales Gas anhand der van der Waals Gleichung:
⎝ ∂T ⎠V
⎛
a ⎞
⎛ ∂p ⎞ ⎛ ∂
RT
a
− 2 ⇒⎜
⎜ p + 2 ⎟⋅(Vm −b ) = RT (0.42)⇒ p =
⎟ =⎜
⎝ ∂T ⎠V ⎜⎝ ∂T
(Vm −b ) Vm
⎝ Vm ⎠
⎛ RT
a ⎞⎞
R
− 2 ⎟⎟ =
⎜⎜
⎟
⎟
⎝ (Vm −b ) Vm ⎠ ⎠V (Vm −b )
(0.43)
wir setzen (0.42) und (0.43) in (0.41) ein:
⎡ R ⎤ ⎡ RT
a ⎤ a
⎛ ∂U ⎞
⎛ ∂p ⎞
T
p
T
=
⋅
−
=
⋅
−
−
= 2 = Binnendruck
⎢
⎥ ⎢
⎜
⎟
⎜
⎟
2⎥
(0.44)
⎝ ∂V ⎠T
⎝ ∂T ⎠V
⎢⎣ (Vm −b ) ⎥⎦ ⎢⎣ (Vm −b ) Vm ⎥⎦ Vm
⎛ ∂p ⎞
p
⎜
⎟
⎜ ∂T ⎟
⎝
⎠V
⎛ ∂U ⎞
⇒⎜
⎟ ist der Binnendruck! ⇒ Ist ein Maß für die Anziehungskräfte in einem realen Gas
⎝ ∂V ⎠T
Die Volumenänderung führt zu einer Änderung des molekularen Anziehungspotentials und damit zur
Änderung der inneren Energie.
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 46
© Dr. Ogrodnik
6.17.2.
⎛ ∂H
Zusammenhang zwischen ⎜
⎝ ∂p
⎞
⎟ und dem linearen Ausdehnungskoeffizienten α
⎠T
Exkurs:
Im Kapitel 1.HS hatten wir gezeigt, dass der Joule Thomson Koeffizient sich ausdrücken lässt durch:
1 ⎛ ∂H ⎞
⋅⎜
μ JT = −
⎟ (0.45)
C p ⎝ ∂p ⎠T
⎛ ∂H ⎞
Zur Berechnung von ⎜
⎟ gehen wir von der Gibbs´schen Gleichung (0.28) dH =T ⋅dS +V ⋅dp aus,
⎝ ∂p ⎠ T
und teilen sie formal durch dp ⇒
⎛ ∂H ⎞
⎛ ∂S ⎞
⎜
⎟ =T ⋅⎜
⎟ +V (0.46)
⎝ ∂p ⎠T
⎝ ∂p ⎠T
ein ⇒
⎛ ⎞
und setzen die Maxwellbeziehung (0.37) ⎜ ∂S ⎟ =− ⎛⎜ ∂V ⎞⎟
⎝ ∂p ⎠T ⎝ ∂T ⎠ p
⎛ ∂H ⎞
⎛ ∂V ⎞
⎜
⎟ =−T ⋅⎜
⎟ +V (0.47)
⎝ ∂T ⎠ p
⎝ ∂p ⎠T
⎧⎪ ⎛ ∂V ⎞
⎫⎪
⋅ ⎨T ⋅ ⎜
⎟ − V ⎬ (0.48)
⎪⎩ ⎝ ∂T ⎠ p
⎭⎪
1 ⎛ ∂V ⎞
α≡ ⎜
⎟
V ⎝ ∂T ⎠ p
Mit dem thermischen Ausdehnungskoeffizient (1. Kapitel):
ergibt sich:
V
μ JT =
{α ⋅ T − 1}
Cp
(0.49)
So dass wir mit (0.45) erhalten: μ JT =
1
Cp
SS2008 Entropie, freie Enthalpie + Gibbs´sche Gl. 47
© Dr. Ogrodnik