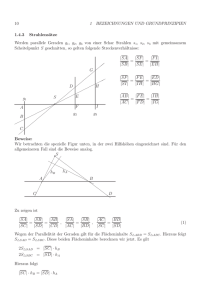
Theoretische Meteorologie I: Atmosphärische Thermodynamik
Werbung

Theoretische Meteorologie I: Atmosphärische Thermodynamik Skript Dieses Skript ist zu beziehen unter: http://www.staff.uni-mainz.de/curtius/J_Curtius_Vorlesungen.html Joachim Curtius [email protected] 06131-39 22862 IPA, Raum 505 Stand: 26. Oktober 2006 1 Inhalt 1 Einführung ...................................................................................................................... 4 2 Temperatur, Druck und ideales Gasgesetz................................................................... 7 2.1 2.2 2.3 2.4 2.5 2.6 Temperatur........................................................................................................................ 7 Druck ................................................................................................................................ 8 Ideales Gasgesetz............................................................................................................ 10 Barometrische Höhenformel........................................................................................... 12 Die Maxwell-Boltzmann Geschwindigkeitsverteilung................................................... 15 Zustandsgleichungen ...................................................................................................... 19 3 Erster Hauptsatz, innere Energie und Enthalpie ...................................................... 20 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 Arbeit und Wärme .......................................................................................................... 20 Innere Energie und Erster Hauptsatz .............................................................................. 23 Wärmekapazitäten .......................................................................................................... 26 Adiabaten ........................................................................................................................ 29 Isotherme Zustandsänderungen ...................................................................................... 31 Der trocken-adiabatische Temperaturgradient................................................................ 31 Auftrieb und atmosphärische Stabilität........................................................................... 32 Enthalpie ......................................................................................................................... 35 4 Entropie und Zweiter Hauptsatz................................................................................. 37 4.1 4.2 4.3 4.4 4.5 4.6 4.7 Zweiter Hauptsatz ........................................................................................................... 37 Carnot-Kreisprozess........................................................................................................ 38 Entropie........................................................................................................................... 41 Entropie des idealen Gases ............................................................................................. 48 Potentielle Temperatur: die Entropie der Meteorologen ................................................ 48 van der Waals-Gas .......................................................................................................... 52 Folgerungen aus den Hauptsätzen .................................................................................. 58 5 Thermodynamische Potentiale .................................................................................... 64 5.1 5.2 5.3 5.4 5.5 Natürliche Zustandsvariablen ......................................................................................... 64 Legendre-Transformationen ........................................................................................... 66 Die Gibbs-Duhem-Relation ............................................................................................ 71 Mischungsentropie.......................................................................................................... 72 Gleichgewichtsbedingungen........................................................................................... 74 5.5.1 Isolierte Systeme................................................................................................. 75 5.5.2 Geschlossenes System im Wärmebad ohne Arbeitsaustausch ........................... 76 5.5.3 Geschlossenes System im Wärmebad bei konstanten Kräften ........................... 77 2 6 Phasenübergänge .......................................................................................................... 79 6.1 6.2 6.3 6.4 6.5 6.6 6.7 6.8 6.9 6.10 Phasendiagramm des Wassers ........................................................................................ 81 Dampfdruckkurve (Clausius-Clapeyron)........................................................................ 82 Temperaturabhängigkeit der Verdampfungsenthalpie.................................................... 86 Sättigungsdampfdruck über Eis und über unterkühltem Wasser.................................... 89 Taupunkt-Temperatur ..................................................................................................... 90 Vertikalgradient des Siedepunkts ................................................................................... 90 Dampfdruckerniedrigung, Raoultsches Gesetz, ideale Lösungen .................................. 91 Henry-Gesetz .................................................................................................................. 94 Kelvin-Gleichung und Nukleation.................................................................................. 96 Sättigungsdampfdruck über Lösungströpfchen, Köhlerkurven .................................. 100 7 Wasserdampf und Wolken in der Atmosphäre ....................................................... 103 7.1 7.2 7.3 7.4 7.5 7.6 7.7 7.8 Definitionen und nützliche Gleichungen ...................................................................... 103 Vertikalgradient des Taupunkts: Wolkenbildung ......................................................... 104 Virtuelle Temperatur und Dichte der feuchten Luft ..................................................... 108 Die "wet-bulb"-Temperatur und die äquivalente Temperatur ...................................... 111 Vertikalgradient für isentropen Aufstieg eines gesättigten Luftpakets ........................ 114 Äquivalente potentielle Temperatur und potentielle Feuchtetemperatur ..................... 118 Thermodynamische Diagramme................................................................................... 122 7.7.1 skew T-log p Diagramm ................................................................................... 122 7.7.2 Tephigramm...................................................................................................... 128 7.7.3 Stüve-Diagramm............................................................................................... 129 Konvektion, Wolkenbildung, CAPE und CINE ........................................................... 130 8 Thermodynamische Potentiale in der physikalischen Chemie ............................... 137 8.1 8.2 8.3 8.4 8.5 Standardenthalpien........................................................................................................ 137 Der Dritte Hauptsatz der Wärmelehre .......................................................................... 141 Standardentropien ......................................................................................................... 143 Gibbsche Standardenthalpie ......................................................................................... 145 Chemische Gleichgewichte........................................................................................... 147 3 1 Einführung Inhalt des ersten Teils der Vorlesung ist die klassische Thermodynamik, als Teilgebiet der Physik. Der Inhalt dieses ersten Teils ist zwar noch relativ Physik-orientiert, die Beispiele werden aber schon im Hinblick auf atmosphärische Prozesse und Anwendungen ausgewählt: so werden wir uns mit den Phasenübergängen des Wassers und nicht mit den Phasenübergängen von idealen Paramagneten beschäftigen. Im zweiten Teil der Vorlesung wenden wir uns dann hauptsächlich konkreten Themen und Fragestellungen aus der Meteorologie und der Atmosphärenphysik zu, die mit Hilfe der Thermodynamik behandelt werden können. Die Thermodynamik ist in der Meteorologie schon wegen der Größen Temperatur und Druck von großer Bedeutung. Mit Hilfe der Thermodynamik können wesentliche Eigenschaften der Atmosphäre (z.B. Druck- und Temperaturgradienten) beschrieben und charakterisiert, Gesetzmäßigkeiten festgelegt, und damit letztendlich Prozesse verstanden werden, die dann Vorhersagen in Bezug auf Wetter und Klima ermöglichen. Unsere Vorlesung beschäftigt sich nur mit der Gleichgewichts-Thermodynamik, d.h. ihre Aussagen sind strenggenommen immer nur für Systeme im Gleichgewicht gültig. Ein thermodynamisches Gleichgewicht liegt vor, wenn alle thermodynamischen Variablen, wie Druck, Temperatur, innere Energie, Entropie, etc. sich mit der Zeit makroskopisch nicht mehr verändern. Es gibt innerhalb des Systems auch keine Gradienten dieser Variablen und „keine Abhängigkeit des Systems von seiner Geschichte“. Der Name Thermostatik wäre daher eigentlich passender. Obwohl die Situation des thermodynamischen Gleichgewichts in der Atmosphäre grundsätzlich - zumindest großräumig - nicht gegeben ist (Druck- und Temperaturgradienten, Wind, etc.), so können doch viele Aussagen der Thermodynamik auf die Atmosphäre angewandt werden, wenn man nicht die Atmosphäre als ganzes betrachtet, sondern den Begriff des lokalen Gleichgewichts einführt und einzelne Luftpakete untersucht. Wir werden diese beiden Begriffe noch genauer definieren. Zentrale Grundlage der Thermodynamik sind die sogenannten Hauptsätze. Alle weiteren Aussagen werden aus diesen Hauptsätzen hergeleitet. Der Nullte Hauptsatz postuliert die Existenz einer Temperatur. Der Erste Hauptsatz erklärt Wärme zu einer Energieform und fordert unter ihrer Einbeziehung die Gültigkeit des Energiesatzes. Der Zweite Hauptsatz handelt von der Unmöglichkeit, Wärme vollständig in andere Energieformen, wie z.B. mechanische Bewegungsenergie umzuwandeln. Der Dritte Hauptsatz betrifft die Unerreichbarkeit des absoluten Nullpunkts. Für uns wichtige Aussagen und Anwendungen ergeben sich aus dem Ersten und Zweiten Hauptsatz. Die statistische Mechanik ist die zur Thermodynamik komplementäre Theorie: Begriffe und Gesetze der Thermodynamik werden nicht als phänomenologische Tatsachen hingenommen, sondern mit statistischen Methoden aus der Quantenmechanik und der Klassischen Mechanik, aus der Mikrostruktur der dem System zu Grunde liegenden Elementareinheiten erklärt. Gelegentlich werden wir im Rahmen der Vorlesung auch auf die zu Grunde liegenden mikroskopischen Prozesse eingehen und so einige phänomenologische Erkenntnisse der Thermodynamik auch mikrophysikalisch erklären können. Zum Beispiel wird in Abschnitt 2.2 der Druck eines Gases aus dem Impulsübertrag der Gasmoleküle auf eine Wand abgeleitet. Weder in der Thermodynamik noch in der statistischen Mechanik ist es das Ziel irgendwelche Systemeigenschaften aus der Lösung der Bewegungsgleichungen der einzelnen Teilchen 4 abzuleiten. Dies würde bedeuten, dass man für einen Kubikzentimeter Gas bei Standardbedingungen ca. 2.5 × 1019 gekoppelte Bewegungsgleichungen lösen müsste! Dies ist nicht nur völlig unmöglich, die Vielzahl der Informationen wäre gar nicht zu überschauen und diese Informationen sind auch völlig unnötig. Die Mittelwerte, die aus der Vielzahl der mikroskopischen Daten folgen, reichen aus, um das makroskopische System vollständig zu charakterisieren. Es gibt zwar unablässig unzählige mikroskalige Bewegungen und Veränderungen der einzelnen Teilchen, diese Fluktuationen spielen aber für die makroskopische Messung keine Rolle. Um uns in der Begriffswelt der Thermodynamik bewegen zu können, beginnen wir mit einigen Definitionen. System Als thermodynamisches System bezeichnet man in der Physik jedes makroskopische System, das aus einer sehr großen Anzahl von Elementargebilden („Teilchen“ wie z.B. Atome, Moleküle, Elektronen, Photonen) aufgebaut ist. Hierbei interessieren uns in der Thermodynamik nicht die vielen Mikrozustände des Systems, sondern nur das Verhalten von makroskopisch beobachtbaren Mittelwerten. Das typische Beispiel für ein System aus der Meteorologie ist ein Luftpaket. Ein wichtiger Aspekt des Systems ist, dass es –zumindest in Gedanken – abgegrenzt werden kann gegen seine Umgebung. Alles außerhalb des Systems wird die Umgebung des Systems genannt. (Meistens interessiert jedoch nur die unmittelbare Umgebung, nicht der ganze Rest des Universums). Man unterscheidet: • Ein isoliertes (abgeschlossenes) System erlaubt keinerlei Teilchen- oder Energieaustausch mit der Umgebung. • Ein geschlossenes System hat keinen Teilchenaustausch mit der Umgebung. Ein solches System kann aber durchaus noch im Kontakt stehen mit der Umgebung. Besonders relevante Beispiele sind: 1. Wärmeaustauschkontakt (thermischer Kontakt). Dieser führt zum Temperaturausgleich zwischen System und Umgebung durch Austausch von Energie in Form von Wärme. Kann man die Umgebung als sehr großes System auffassen, dessen Temperatur sich bei Entnahme einer endlichen Wärmemenge praktisch nicht ändert, so sagt man, das System befinde sich in einem Wärmebad. Ein System ohne Wärmeaustauschkontakt heißt thermisch isoliert. 2. Arbeitsaustauschkontakt. Durch Arbeitsleistung vom System an der Umgebung oder umgekehrt werden Systemeigenschaften geändert. Es kann sich dabei um mechanische, elektrische, chemische oder andere Formen von Arbeit handeln. Beispiel für Arbeitsaustauschkontakt: die Bewegung eines Kolbens ändert das Volumen V eines Gases. • Ein offenes System unterliegt keinen Einschränkungen. Es steht im Energie- und Teilchenaustausch mit der Umgebung. Ein gedachtes, vollkommen isoliertes System ist somit ein „Universum für sich“, in der Realität existieren natürlich –außer dem gesamten Universum selbst– nur Annäherungen an ein solches System, da es z.B. aufgrund von Wärmestrahlung und Dampfdruck der Systemwände keine vollständige Isolierung geben kann. Zustandsgrößen, Zustand Zur Beschreibung eines thermodynamischen Systems werden die Resultate von Messungen an charakteristischen Observablen, den sogenannten Zustandsgrößen, benutzt. Welche Größen in Betracht kommen, ist nicht eindeutig vorgeschrieben, sondern richtet sich nach 5 Interesse und Zweckmäßigkeit (einfache Messung, unabhängige Observable, ausreichende (vollständige) Beschreibung). Von einem vollständigen Satz unabhängiger Variablen spricht man genau dann, wenn sich alle anderen thermodynamischen Größen des Systems als Funktionen dieser Variablen darstellen lassen. Beispiele für die Zustandsgrößen eines Gases sind der Druck p, die Temperatur T, die Teilchenzahl N, die Entropie S, die innere Energie U usw.. Nicht alle Zustandsgrößen sind unabhängig, es gibt Relationen zwischen ihnen. Man unterscheidet extensive und intensive Zustandsgrößen. Extensive Zustandsgrößen sind mengenproportional, d.h. sie verhalten sich additiv bei der Zusammensetzung von Systemen. Beispiele sind die das Volumen V, die Teilchenzahl N oder die Masse m. Intensive Zustandsgrößen, wie z.B. die Temperatur T, der Druck p oder die Dichte ρ sind mengenunabhängig. Der Zustandsraum ist der Raum, der von einem vollständigen Satz unabhängiger Zustandsgrößen aufgespannt wird. Einen Zustand nennt man das Ensemble von Werten eines vollständigen Satzes von Zustandsgrößen, also einen Punkt im Zustandsraum. Das thermodynamische Gleichgewicht ist der Zustand, in dem sich die Werte der (Basis-)Zustandsgrößen zeitlich nicht mehr ändern. Erfahrungsgemäß strebt jedes isolierte System von alleine in den Gleichgewichtszustand. Die Zeit, die benötigt wird, um diesen Zustand zu erreichen wird Relaxationszeit genannt. Die Relaxationszeit kann von System zu System um Größenordnungen variieren. Mit dem Begriff Zustandsänderung oder Prozess bezeichnen wir eine Folge von Zuständen, die ein System durchläuft. War der Ausgangszustand ein Gleichgewichtszustand, so kann eine Zustandsänderung nur durch Änderung der äußeren Bedingungen veranlasst werden. Die Zustandsänderung wird quasistatisch genannt, wenn sie so langsam gegenüber den Relaxationszeiten verläuft, dass sie praktisch aus einer Folge von Gleichgewichtszuständen besteht. Die Zustandsänderung wird reversibel genannt, wenn es sich um eine umkehrbare Folge von Gleichgewichtszuständen handelt, d.h. wenn einer zeitlichen Umkehr der Änderung der äußeren Bedingungen eine zeitliche Umkehr der vom System durchlaufenen Zustände entspricht. Eine irreversible Zustandsänderung ist demzufolge nicht umkehrbar. Paradebeispiel ist die Durchmischung zweier Gase (s. Abbildung 1.1). Der Umkehrprozess (Hereinschieben der Trennwand) führt nicht wieder zum Ausgangszustand. Reale Prozesse sind in der Regel weder quasistatisch noch reversibel. Ein Kreisprozess ist ein Prozess bei dem Anfangs- und Endzustand des Systems (nicht der Umgebung!) gleich sind, d.h. bei dem alle Zustandsgrößen (unabhängige und abhängige) zu den Ausgangswerten zurückkehren. Abbildung 1.1. Irreversible Durchmischung zweier verschiedener Gase durch das Herausziehen der Trennwand [Nolting]. 6 Mehrere Teile dieses Skripts sind dem Buch Atmospheric Thermodynamics von C.F. Bohren und B.A. Albrecht entnommen. Diese Autoren machen jedoch den Versuch die Thermodynamik ohne Differentiale darzustellen. Stattdessen werden die Gleichungen als zeitliche Ableitungen dargestellt. Diese Schreibweise ist einerseits unkonventionell und birgt auch einige Probleme. Wir werden im Allgemeinen die in den Standard-ThermodynamikBüchern übliche und dem Leser vermutlich schon geläufige Differentialdarstellung befolgen, an wesentlichen Stellen aber auch die Schreibweise von Bohren und Albrecht angeben. Die physikalische Aussage der beiden Schreibweisen ist natürlich die selbe. Wir können so in den Herleitungen einiger Aussagen der atmosphärischen Thermodynamik auch auf die ansonsten guten Darstellungen von Bohren und Albrecht zurückgreifen. 2 Temperatur, Druck und ideales Gasgesetz Temperatur und Druck spielen sowohl in der Thermodynamik als auch in der Meteorologie herausragende Rollen. Kein Wetterbericht ohne Temperaturvorhersagen, keine Wetterlage ohne Hoch- und Tiefdruckgebiete. Diese beiden Größen sind entscheidend für Wetter und Klima auf der Erde und sie sind gleichzeitig zentrale Größen der Thermodynamik. Grund genug, sich mit beiden Größen eingehend zu befassen. 2.1 Temperatur Die Temperatur erscheint uns vielleicht als die am unmittelbarsten erfahrbare physikalische Größe überhaupt, schließlich macht schon jedes Kind Erfahrungen mit „heiß“ und „kalt“. Daher erscheinen die sechs Axiome des sogenannten Nullten Hauptsatzes der Thermodynamik, die die Temperatur als physikalische Messgröße im Rahmen der Thermodynamik etablieren, fast selbstverständlich: 1.) Jedes makroskopische System besitzt eine Temperatur T. Die Temperatur ist eine intensive Zustandsgröße, die in einem sich selbst überlassenen, isolierten System überall den selben Wert annimmt, d.h. einem homogenen Gleichgewicht zustrebt. 2.) T ist durch eine Zahl gekennzeichnet, ist also eine skalare Größe. 3.) Von zwei sich im Gleichgewicht befindlichen Systemen kann stets gesagt werden: TA > TB oder TA < TB oder TA = TB (Anordnungsaxiom). 4.) A, B, C seien thermodynamische Systeme, dann folgt aus TA > TB und TB > TC stets TA > TC (Transitivität). 5.) Systeme A und B seien in thermischem Kontakt, das Gesamtsystem A ∪ B sei isoliert, dann gilt im Gleichgewicht: TA = TB = TA∪B. 6.) Sei für zwei zunächst getrennte Systeme TA(a) < TB(a) , dann gilt nach Herstellung des thermischen Kontakts im Gleichgewicht : 7 TA(a) < TA∪B < TB(a). Zur Messung der Temperatur eignet sich jede physikalische Eigenschaft, die sich monoton und eindeutig mit T ändert, z. B. das Volumen beim Quecksilberthermometer, der Druck in einem Gasthermometer oder der elektrische Widerstand in einem Widerstandsthermometer. Für jede Temperaturmessung wird die Eigenschaft des thermischen Gleichgewichts benutzt. Jedes Thermometer misst eigentlich seine eigene Temperatur , die erst im thermischen Gleichgewicht mit der des zu untersuchenden Systems übereinstimmt. Bei unterschiedlichen Ausgangstemperaturen tritt wegen 5.) eine gewisse Verfälschung der Systemtemperatur auf. In der Praxis ist für die Messung zu beachten, dass für die Angleichung von Systemtemperatur und Thermometertemperatur immer eine gewisse Einstellzeit erforderlich ist. 2.2 Druck Drei idealisierende Modellvorstellungen definieren das ideale Gas, das aus N Molekülen gebildet wird: 1) 2) 3) Die Moleküle der Masse m befinden sich in ständiger, regelloser Bewegung, die Moleküle haben keine Eigenvolumina, (Punktmassen) die Teilchen haben keine Wechselwirkungen untereinander, außer wenn sie miteinander elastisch zusammenstoßen. Eine Menge Gas aus N identischen Molekülen sei in einem würfelförmigen Behälter mit Volumen V eingeschlossen. Entlang der Kanten des Würfels wird ein kartesisches Koordinatensystem ausgerichtet. Obwohl die Moleküle des idealen Gases nicht untereinander wechselwirken, so kollidieren sie doch mit den Wänden des Behälters. Mit n bezeichnen wir die Dichte der Gasmoleküle im Behälter: n = N/V. Betrachten wir nun den Molekülfluss, der pro Zeiteinheit die Wand der Fläche A in der yz-Ebene trifft. Die Moleküle bewegen sich in alle Raumrichtungen mit gleicher Wahrscheinlichkeit, d.h. nur eine Hälfte der Moleküle bewegt sich auf die Wand zu, hat also eine Geschwindigkeitskomponente vx in Richtung der Wand. Damit ist der Fluss der Moleküle, die auf die Wand treffen, nvx/2 (Abbildung). Vor der Wandkollision haben die Moleküle in x-Richtung den Impuls px = mvx. Von der Wand werden die Moleküle in den Behälter zurückreflektiert Wir nehmen eine Spiegelreflexion mit „Einfallswinkel gleich Ausfallswinkel“ an und nehmen an, dass es sich um eine elastische Kollision handelt, bei der die kinetische Energie des Gasmoleküls unverändert bleibt. Nach der Kollision haben die Moleküle dann also den Impuls –mvx. Die Wechselwirkung mit der Wand führt zu einer Impulsumkehr, die Impulsänderung beträgt pro Molekül Δpx = 2mvx. Die y- und z-Komponenten des Impulses werden nicht verändert. Nach dem zweiten Newtonschen Gesetz gilt: (2.1) r dp r =F dt Die Impulsänderung des Teilchens wird durch eine Kraft hervorgerufen. Da die Kraft auf das Teilchen gleich einer entgegengesetzten Reaktionskraft Fx sein muss, wirkt auch auf die Wand eine Kraft. (2.2) Δp x = 2mv x = ∫ Fx dt . 8 Abbildung 2.1. Der Druck eines Gases hergeleitet aus dem Impulsübertrag der Moleküle auf eine Wand [Atkins]. Während der Zeit t erreichen nvxAt/2 Moleküle die Wand. so dass die gesamte zeit-integrierte Kraft auf die Wand t (2.3) 1 nvx 2 mvx At = ∫ Fx dt 2 0 ist, wobei Fx die instantane Kraft auf A ist. Die mittlere Kraft auf A innerhalb der Zeit t ist t (2.4) Fx = ∫ Fx dt 0 t . Die Kraft pro Fläche auf der die Kraft wirkt, ist der Druck p: (2.5) p = nmvx2 Hierbei wurde angenommen, dass alle Moleküle die gleiche Geschwindigkeit haben. Tatsächlich haben die Moleküle unterschiedliche Geschwindigkeiten. Daher müssen wir noch vx2 in der Gleichung (2.5) durch den Mittelwert ersetzen: (2.6) p = nm vx2 Auf diesen Mittelwert werden wir noch im weiteren Verlauf des Kapitels genauer eingehen. Wir können festhalten, dass der Druck eines Gases auf eine Wand im Impulsübertrag der Gasmoleküle auf die Wand begründet liegt. Da die Moleküle sich in den drei Raumrichtungen zufällig bewegen, muss für die Geschwindigkeitskomponenten 9 (2.7) vx2 = v y2 = vz2 gelten. Weiterhin gilt dann: (2.8) v 2 = vx2 + v y2 + vz2 = vx2 + v y2 + vz2 . Damit können wir Gleichung (2.6) umformen: (2.9) p= 1N mv 2 3V Der Druck eines Gases beträgt also 2/3 seiner kinetischen Energiedichte. An dieser Stelle können wir eine absolute Temperatur T definieren: 1 mv 2 = kT 3 (2.10) wobei k, die Boltzmann-Konstante, den Wert 1.38 × 10-23 J/K hat. Die Definition identifiziert die Temperatur direkt mit der kinetischen Energie der Moleküle; Temperatur ist also nichts anderes als ein Maß für die mittlere kinetische Energie der Moleküle, die ungeordnete Bewegung der Gasmoleküle bedingt die Temperatur des Gases: (2.11) Ekin = 1 2 3 mv = kT . 2 2 2.3 Ideales Gasgesetz Mit den Gleichungen (2.9) und (2.10) des vorhergehenden Abschnitts können wir jetzt sofort das ideale Gasgesetz formulieren: pV = NkT (2.12) Eine andere gebräuchliche Form des idealen Gasgesetzes lautet: p=m (2.13) N RT . V Hierbei wurde die Gaskonstante R = k/m eingeführt. Achtung: wir folgen hier der Notation, wie sie im Buch von Bohren und Albrecht verwendet wird: Es handelt sich bei R jeweils um eine gasspezifische Konstante, so beträgt Rd = 287 J kg-1 K-1, für trockene Luft und Rv = 461.5 J kg-1 K-1 für reinen Wasserdampf. Die den Physikern besser geläufige universelle Gaskonstante wird mit einem Stern versehen: R* = 8.3141 J mol −1 K −1 Mit ihr lautet das ideale Gasgesetz auch: 10 (2.14) pV = υ R* T . Hier ist ν die Anzahl der Mole. Beachten wir weiterhin, dass die Massendichte ρ = mN/V ist, so erhalten wir noch eine weitere Form des idealen Gasgesetzes: (2.15) p = ρ RT . Abbildung 2.2. p-V-T-Zustandsoberfläche für das ideale Gas [Atkins]. Abbildung 2.3. T-V Diagramm zur Veranschaulichung des Gay-Lussac Gesetzes. [Atkins] Weiterhin sei noch die Beziehung R* = kNA genannt, wobei NA = 6.02252 × 1023 mol-1 die Avogadro-Konstante bezeichnet. 11 Historisch gesehen wurde das ideale Gasgesetz aus zwei experimentell abgeleiteten Teilgesetzen zusammengesetzt: dem Boyle-Mariotte-Gesetz, das von Robert Boyle bereits 1660 veröffentlicht wurde, und dem Gay-Lussac Gesetz, das 1802 von Joseph-Louis GayLussac publiziert wurde. Das Boyle-Mariotte-Gesetz besagt: pV = const. (2.16) für T , N = const. und das Gay-Lussac-Gesetz, das die thermische Ausdehnung eines Gases bei konstantem Druck beschreibt, lautet: V = V0 (1 + α 0ϑ ) . (2.17) für p = const. V0 ist das Volumen des Gases bei 0°C, ϑ ist die Temperatur in °C (ϑ = T - 273.15), und α0 ist der Volumenkoeffizient der thermischen Ausdehnung: α= (2.18) 1 ∂V 1 = V ∂T T für ϑ = 0°C. Gay-Lussac bestimmte α0 = 1/273,15 K-1. Damit kann bereits eine sehr gute Abschätzung für die Temperatur des absoluten Nullpunkts extrapoliert werden, d.h. der Temperatur, bei der das Volumen eines idealen Gases verschwindet: ϑabs = -273,15°C. 2.4 Barometrische Höhenformel Der atmosphärische Luftdruck nimmt mit der Höhe ab. Wie sieht der Druckgradient mit der Höhe quantitativ aus? Dazu wollen wir eine einfache Näherungslösung herleiten. Man betrachte eine ebene, ruhende Luftschicht in der Höhe z über dem Erdboden. Die Schicht habe die Fläche A und die Dicke Δz. Das Luftpaket ist im (Kräfte-)Gleichgewicht mit seiner Umgebung. Das Luftpaket selbst drückt mit seiner Gewichtskraft Mg nach unten, wobei seine Masse M mit A Δz ρ(z) ausgedrückt werden kann. Dem entgegen steht die Kraft p(z) A, die der Luftdruck der darunterliegenden Luftsäule in der Höhe z ausübt; hinzu kommt noch die Kraft von der Luftsäule, die über der Luftschicht steht: p(z+Δz) A. Das Kräftegleichgewicht lautet also: (2.19) p( z ) A − p( z + Δz ) A − ρA Δz g = 0 Teilen wir beide Seiten durch A Δz und bilden den Limes für Δz → 0: (2.20) lim Δz → 0 p( z + Δz ) − p( z ) dp = = − ρg Δz dz Diese Gleichung wird auch hydrostatische Grundgleichung genannt. Kombinieren wir diese Gleichung mit dem idealen Gasgesetz (2.15) so erhalten wir eine Differentialgleichung (2.21) 1 dp g =− p dz RT mit der Lösung 12 (2.22) ⎛ z g ⎞ p dz ⎟ . = exp ⎜ − ∫ p0 ⎝ 0 RT ⎠ Hier ist p0 der Druck an der Stelle z = 0. Das einzige Problem bei der Lösung dieser Differentialgleichung ist, dass die Temperatur höhenabhängig ist: T = T(z). Näherungsweise können wir diese Abhängigkeit vernachlässigen und durch eine einzige mittlere Temperatur Tav ersetzen. Somit kann die Gleichung umgeformt werden: (2.23) ⎛ gz ⎞ ⎟⎟ p ≈ p0 exp⎜⎜ − ⎝ RTav ⎠ Der Faktor RTav/g muss die Dimension einer Länge haben, so dass wir auch schreiben können: (2.24) ⎛ z⎞ p ≈ p0 exp⎜ − ⎟ ⎝ H⎠ Die Größe H ist eine charakteristische Länge, die sogenannte Skalenhöhe. Setzt man Tav = 273 K ein, so erhält man eine Skalenhöhe von ~8 km. Wie gut die gemachte Näherung die Wirklichkeit beschreibt, zeigt der Vergleich unseres Exponential-Fits mit der US Standard Atmosphere, der „typischen Durchschnittsatmosphäre“. Abbildung 2.4. Höhenabhängigkeit des Luftdrucks im Vergleich von barometrischer Höhenformel mit der US Standard Atmosphere. Skalenhöhe hier 7.3 km [Bohren]. Der Luftdruck nimmt etwa innerhalb von 8 km auf den Wert 1/e von p0 ab. Ein Bergsteiger, der einen Achttausender ohne Sauerstoffgerät erklimmen will, muss also über längere Zeit mit einem Luftdruck von weniger als 370 mb auskommen. Analog zur Formel für den Druck ergibt sich mit der gleichen Näherung auch für die Dichte ρ(z) eine entsprechende exponentielle Abnahme mit der Höhe 13 (2.25) ⎛ z⎞ ⎟. ⎝ H⎠ ρ ≈ ρ0 exp⎜ − Mit der Definition der Gaskonstanten R = k/m können wir Gleichung (2.23) auch noch anders interpretieren: (2.26) ⎛ mgz ⎞ ⎛ gz ⎞ ⎟⎟ ⎟⎟ = p0 exp⎜⎜ − p ≈ p0 exp⎜⎜ − RT kT av ⎠ av ⎠ ⎝ ⎝ Das Verhältnis von Zähler und Nenner im Exponenten stellt das Verhältnis von potentieller Gravitationsenergie und kinetischer Energie der Moleküle dar. Der Luftdruckgradient, der sich mit der Höhe einstellt, ergibt sich gewissermaßen aus dem Wettstreit zwischen der Gravitation, die Moleküle zur Erde zieht, und kinetischer Energie, die dafür sorgt, dass die Moleküle in alle Raumrichtungen umherschießen. 14 2.5 Die Maxwell-Boltzmann Geschwindigkeitsverteilung In einem Gas haben die Moleküle nicht alle die gleiche Geschwindigkeit, sondern die Häufigkeit der Molekülgeschwindigkeiten folgt der Maxwell-Boltzmann-Verteilung. ⎛ mv 2 ⎞ ⎟⎟ f (v) = A4πv exp⎜⎜ − ⎝ 2kT ⎠ 2 (2.27) 32 ⎛ m ⎞ mit A = ⎜ ⎟ . ⎝ 2πkT ⎠ Wir wollen hier diese Verteilungsfunktion nicht herleiten (eine Herleitung findet sich z.B. in Physical Chemistry von Atkins). Stattdessen werden wir die Form der Verteilung analysieren. Die Verteilung enthält einen zu 4πv2 proportionalen Teil. Dieser Faktor stammt daher, dass die Wahrscheinlichkeit f(v)dv, dass die Moleküle eine Geschwindigkeit in einem Intervall [v,v+dv] haben, unabhängig von der Richtung des Geschwindigkeitsvektors ist. Die Summe der Wahrscheinlichkeiten bildet daher eine Kugelschale im Geschwindigkeitsraum (s. Abbildung 2.6) und das Volumen dieser Schale ist gerade 4πv2dv. Der quadratische Term dominiert das Verhalten der Funktion f(v) für kleine Geschwindigkeiten v (s. Abbildung 2.5). Der Exponential-Faktor in (2.27) erinnert an den Ausdruck, den wir bei der barometrischen Höhenformel kennengelernt haben. Diesmal steht im Zähler die kinetische Energie des einzelnen Moleküls (1/2mv2), während im Nenner kT steht, was die mittlere kinetische Energie aller Moleküle beschreibt (2.11). Der Exponential-Faktor stellt also die kinetische Enegie des einzelnen Moleküls ins Verhältnis zur mittleren kinetischen Energie. Für große Geschwindigkeiten v ist dieser Exponentialfaktor dominierend für das Verhalten von f(v) (s. Abbildung 2.5). Abbildung 2.5. [Atkins],[Bohren]. Die Maxwell-Boltzmann-Geschwindigkeitsverteilung 15 Abbildung 2.6. Die Vektoren gleicher Länge |v| bilden eine Kugelschale im Geschwindigkeitsraum [Bohren]. Der Faktor A ist so gewählt, dass f(v) immer die Normierungsbedingung ∞ ∫ f (v ) (2.28) dv = 1 0 erfüllt. Der Teil der Moleküle, die eine Geschwindigkeit zwischen v1 und v2 besitzen, ist v2 ∫ f (v ) (2.29) dv . v1 Die Geschwindigkeiten v1 und v2 sind beliebig, im allgemeinen muss dieses Integral jedoch numerisch gelöst werden. Nach dem Mittelwertsatz der Integralrechnung findet sich immer eine Geschwindigkeit v , für die gilt: v +Δv (2.30) ∫ v ⎛ mv 2 ⎞ f (v) dv = f (v )Δv = A4π v Δv exp ⎜ − ⎟ ⎝ 2kT ⎠ 2 Je kleiner Δv, desto besser können wir v durch v approximieren. Wie in Abbildung 2.5 zu sehen ist, erhält man für niedrige Temperaturen, bzw. für große Molekülmassen eine schmale Verteilung, für hohe Temperaturen oder kleine Molekülmassen hingegen ist die Verteilung relativ breit. Wir unterscheiden die folgenden maßgebliche Geschwindigkeiten: Die wahrscheinlichste Geschwindigkeit vmp (der Index mp steht für "most probable") ist die Geschwindigkeit bei der die Verteilung f(v) ihr Maximum annimmt. Wir finden den Wert von vmp indem wir die Ableitung df/dv bilden und gleich null setzten. (2.31) vmp = 2kT . m Um zu sehen, wie vmp von der Temperatur abhängt, bilden wir noch die Ableitung dvmp/dT: 16 dvmp (2.32) dT = 1 vmp 2 T Nähern wir die Ableitung durch das Verhältnis der Differenzen Δvmp/ΔT, so erhalten wir Δvmp (2.33) vmp = 1 ΔT 2 T An dieser Gleichung können wir zum Beispiel direkt ablesen, dass die wahrscheinlichste Geschwindigkeit um ~7 % sinkt, wenn die Temperatur in der Atmosphäre von +20°C auf -20°C fällt. Das heißt, im Mittel bewegen sich Luftmoleküle im Sommer nur ein paar Prozent schneller als im Winter. Weiterhin bestimmen wir die mittlere Geschwindigkeit ⟨ v⟩ aus: ∞ (2.34) ⟨ v⟩ = ∫ v f (v) dv = 0 2 π vmp Zur Bestimmung dieses Integrals haben wir Gleichung (2.31) und die aus Integral-Tafeln bekannte Beziehung ∞ (2.35) 3 2 ∫ x exp(ax ) dx = 0 1 2a 2 benutzt. Die mittlere Geschwindigkeit ist nicht identisch mit der wahrscheinlichsten Geschwindigkeit, da es sich bei f(v) nicht um eine symmetrische Verteilung handelt. Setzt man Molekülgewichte ein und bestimmt die mittlere Geschwindigkeit für T = 273 K, so erhält man für Stickstoff und Wasserstoff mittlere Geschwindigkeiten ⟨ v⟩ von 450 m/s und 1700 m/s. Weiterhin gilt für die Wurzel der quadratgemittelten Geschwindigkeit (root mean square): (2.36) vrms = v 2 = 3 vmp 2 wobei hierfür das Integral ∞ (2.37) ⟨ v ⟩ = ∫ v 2 f (v ) dv 2 0 zu lösen war. Das Mittel des Geschwindigkeitsquadrats kennen wir ja schon aus der Herleitung des Gasdrucks in Abschnitt 2.2. Die Abbildung 2.5 zeigt die Lage der drei verschiedenen gemittelten Geschwindigkeiten. Als Letztes geben wir noch die mittlere Relativgeschwindigkeit an, die Geschwindigkeit, mit der sich zwei Moleküle im Mittel aufeinander zubewegen: (2.38) vrel = 2 v 17 Abbildung 2.7. Bei zufälliger. regelloser Bewegung ist die Relativbewegung der Moleküle im Mittel senkrecht zueinander. Dies erklärt den Faktor 2 in der Gleichung für die Relativgeschwindigkeiten [Atkins]. Anhand der Abbildung 2.7 können wir den Faktor 2 plausibel machen. Aus der Moleküldichte n = N/V erhält man sofort das Volumen, das jedem Molekül zur Verfügung steht. Daraus ergibt sich auch der mittlere Abstand D zwischen zwei Molekülen im Gas: 1 ⎛ kT ⎞ 3 D=n =⎜ (2.39) ⎟ ⎝ p ⎠ In atmosphärischer Luft bei 1000 mb und 273 K besteht demnach ein Abstand von ~3 nm zum nächsten Nachbarmolekül. −1 3 Abbildung 2.8. Veranschaulichung der "Kollisionsröhre" zur Bestimmung der Kollisionsfrequenz. Stoßfrequenz Im folgenden bestimmen wir die Häufigkeit mit der die Moleküle eines Gases miteinander stoßen und die durchschnittliche Länge des Weges zwischen zwei Molekülstößen. Wir zählen einen Stoß immer dann, wenn die Zentren zweier Moleküle sich bis auf den Stoßdurchmesser d einander annähern. Der Stoßdurchmesser hat etwa die Größe des geometrischen Durchmessers, wenn man Moleküle als harte Kugeln ansieht (s. Abbildung 2.8). Zunächst bestimmen wir die Kollisionsfrequenz z. Hierfür stellen wir uns zunächst vor, alle Moleküle 18 bis auf eines „gefrieren“ in ihrer Bewegung. Wir beobachten was passiert während das eine mobile Molekül sich mit der mittleren (relativen) Geschwindigkeit vrel für eine Zeit Δt durch die anderen Moleküle hindurchbewegt (s. Abbildung 2.8). Es durchstößt eine "Kollisionsröhre" mit Querschnittsfläche σ = πd2 und Länge vrel Δt. Das Volumen des Zylinders ist daher σ vrel Δt. Die Anzahl der festgefrorenen Moleküle, deren Zentren innerhalb der Röhre liegen ist dann nσ vrel Δt , wobei n = N/V die Moleküldichte beschreibt. Dies ist die Anzahl der Stöße eines Moleküls innerhalb der Zeit Δt. Die Kollisionsfrequenz, also die Anzahl der Kollisionen pro Zeit, ist damit nσ vrel . Ersetzen wir noch n durch das ideale Gasgesetz (2.12), dann erhalten wir: (2.40) z = σ vrel n = σ vrel p kT Der Stoßquerschnitt σ eines Stickstoffmoleküls beträgt 0.43 nm2, damit ergibt sich die Stoßfrequenz eines N2-Moleküls bei 1000 mb und 273 K zu 7×109 s-1. Mittlere freie Weglänge Mit der Stoßfrequenz können wir auch sofort die mittlere freie Weglänge λ bestimmen, die ein Molekül im Mittel zurücklegt, bevor es mit einem anderen Molekül zusammenstößt. Die Zeitdauer zwischen zwei Stößen des Moleküls ist nämlich 1/z und die in dieser Zeit zurückgelegte Distanz ist dann: (2.41) λ= v kT = z 2σ p Setzen wir die Werte für N2 bei 273 K und 1000 mb ein, so erhalten wir ein λ von ~60 nm. Man beachte den Unterschied zwischen D und λ. Zusammenfassend können wir festhalten, dass Luft bei 1013 mb und 273 K aus Molekülen besteht, die sich mit Geschwindigkeiten von typischerweise ~400 m/s bewegen. Jedes Molekül kollidiert mit einem anderen innerhalb von ~1 ns und legt dabei eine Strecke von 100 bis 1000 Moleküldurchmessern zurück. Bei ~1×1019 Molekülen cm-3 resultieren insgesamt ~1×1028 Stöße s-1 cm-3. 2.6 Zustandsgleichungen Das ideale Gasgesetz (2.12) - (2.15) ist ein Beispiel einer sogenannten Zustandsgleichung. Unter Zustandsgleichungen verstehen wir Relationen zwischen gewissen extensiven und intensiven Zustandsvariablen Zi des Systems: (2.42) f ( Z1 , Z 2 ,..., Z n ) = 0 Sie müssen eindeutig umkehrbar, d.h. nach allen Variablen Zi auflösbar sein. Man kann mit ihrer Hilfe abhängige Zustandsvariablen in unabhängige umwandeln und umgekehrt. In der klassischen Thermodynamik folgen die Zustandsgleichungen in der Regel aus Modellvorstellungen über das zugrundeliegende physikalische System. 19 3 Erster Hauptsatz, innere Energie und Enthalpie 3.1 Arbeit und Wärme Zustandsänderungen eines thermodynamischen Systems sind im allgemeinen mit Energieänderungen verknüpft. In der Thermodynamik unterscheidet man nun typischerweise Energieänderungen, die durch am (oder vom) System geleistete Arbeit (ΔW) hervorgerufen werden, und solche, bei denen Wärme (ΔQ) auf das (oder vom) System übertragen wird. Hierbei legen wir folgende Vorzeichenkonvention fest: ΔW > 0 ΔW < 0 ΔQ > 0 ΔQ < 0 wenn am System Arbeit geleistet wird. wenn vom System Arbeit geleistet wird. wenn Wärme in das System hineingepumpt wird. wenn Wärme aus dem System herausgepumpt wird. Der Begriff der Arbeit wird der Klassischen Mechanik bzw. der Elektrodynamik entnommen: q1, ..., qm : F1, ..., Fm : generalisierte Koordinaten (zugeordnete) generalisierte Kraftkomponenten. Die qi müssen nicht notwendig die Dimension einer Länge haben, und die Kräfte müssen nicht unbedingt Kräfte im eigentlichen Sinne sein. Das Produkt qF hat aber in jedem Fall die Dimension einer Energie. (differentielle, quasistatische) Arbeit m δW = ∑ Fi dqi (3.1) i =1 Beispiele: Fi -p σ B0 E µ Druck Oberflächenspannung Magnetfeld elektrisches Feld chemisches Potential dqi dV dS dm dP dN δW -pdV σdS B0dm EdP µdN Wir wählen für die differentielle Arbeitsleistung bewusst den Buchstaben "δ" statt des üblichen "d", um anzudeuten, dass es sich bei δW nicht um ein totales Differential handelt. Das bedeutet, dass die Linienintegrale ∫ δW im allgemeinen wegabhängig sind. C 20 ----------------------------------------------------------------------------------------------------------------Zwischenbemerkung: Eine Differentialform m (3.2) δA = ∑ a j ( x1 , x 2 ,..., x m )dx j j =1 ist genau dann ein totales Differential (integrabel), wenn die folgenden Integrabilitätsbedingungen erfüllt sind (vgl. hierzu z.B. Lehrbücher der theoretischen klassischen Mechanik): (3.3) ⎛ ∂ai ⎛ ∂a j ⎞ = ⎜⎜ ⎜ ⎟ ∂ x ⎝ i ⎠ xm ,m≠i ⎝ ∂x j ⎞ ⎟⎟ ⎠ xm ,m≠ j ∀ i,j. r r r Beispiel: Für die Komponenten Fi einer konservativen Kraft (definiert durch F (r ) = −∇Φ (r ) ) gilt wegen der Vertauschbarkeit der zweiten Ableitungen: (3.4) ∂ 2Φ ∂ 2Φ = ∂x∂y ∂y∂x und es folgt (3.3): (3.5) ∂Fx ∂Fy = . ∂y ∂x Es gilt weiterhin für jeden geschlossenen Weg: (Herleitung über rot F = 0 und Stokes'schem Satz): (3.6) ∫ δA = ∫ dA =0 Sei m=2, wie häufig in der Thermodynamik, und (3.7) δA = a1 dx1 + a 2 dx 2 kein totales Differential, dann gibt es immer einen integrierenden Faktor µ(x1,x2), so dass: (3.8) df = μ δA = ( μ a1 ) dx1 + ( μ a 2 ) dx2 ein totales Differential wird. Dazu muss µ so festgelegt werden, dass (3.9) ⎛ ∂ ( μ a1 ) ⎞ ⎛ ∂ ( μ a2 ) ⎞ ⎜⎜ ⎟⎟ = ⎜⎜ ⎟⎟ ⎝ ∂ x2 ⎠ x1 ⎝ ∂ x1 ⎠ x2 21 gilt. Die Wahl von µ = µ(x1,x2) ist nicht eindeutig. Die Zustandsgrößen der Thermodynamik müssen eindeutig sein. Durchläuft das System im Zustandsraum einen geschlossenen Weg, so müssen am Ende alle abhängigen wie unabhängigen Zustandsgrößen wieder ihre Ausgangswerte angenommen haben. Von einer Zustandsgröße Z fordern wir also, dass ∫ dZ = 0 (3.10) für alle geschlossenen Wege im Zustandsraum gilt. Dies bedeutet aber, dass dZ ein totales Differential darstellt. In diesem Sinne ist Arbeit keine Zustandsgröße. Die generalisierten Kräfte Fi hängen im allgemeinen auch von der Zustandsgröße Temperatur ab (z.B. Druck p = p(T)). Das gilt dann auch für δW, so dass wir statt (3.2) eigentlich schreiben sollten: m (3.11) δW = ∑ Fi (q1 ,..., qm , T ) dqi + 0 dT i =1 Die Integrabilitätsbedingungen (3.3) lassen sich dann nicht erfüllen: (3.12) ⎛ ∂0 ⎞ ⎛ ∂ Fi ⎞ ⎟⎟ = 0 . ⎜⎜ ⎟⎟ ≠ ⎜⎜ ∂ ∂ T q ⎝ ⎠... ⎝ i ⎠... ----------------------------------------------------------------------------------------------------------------Die für uns wichtigste Form der Arbeit in der atmosphärischen Thermodynamik wollen wir nun noch etwas genauer untersuchen und die gemachten Aussagen anhand des Beispiels verdeutlichen. Volumenarbeit In einem zylindrischen Gefäß mit Querschnittsfläche A befinde sich ein Gas. Nach oben sei das Gefäß durch einen reibungslos laufenden Kolben begrenzt, auf dem sich ein Gewicht der Masse M befindet. Gleichgewicht liegt dann vor, wenn der Gasdruck p das Gewicht in der Schwebe hält: (3.13) pA = Mg Durch eine infinitesimale Verschiebung des Gewichts nach oben erhöht sich dessen potentielle Energie um (3.14) dE pot = Mg dx = pA dx = pdV 22 Die vom Gas am Gewicht geleistete, also nach außen abgegebene Arbeit ist demnach: δ W = − p dV (3.15) Für eine endliche Volumenänderung V1 → V2 mit V2 > V1 gilt dann: V2 (3.16) ΔW12 = − ∫ p(V ) dV V1 p Die geleistete Arbeit ΔW12 entspricht also der Fläche unter der Kurve im p(V)-Diagramm. Wie müssen wir uns so einen Prozess vorstellen? Damit das Gas überhaupt expandieren kann, muss der durch die Masse M hervorgerufene Kolbendruck etwas kleiner sein als der Gasdruck. Ist der Differenzdruck groß, so erfolgt eine zu rasche Expansion des Gases, das Gas gewinnt dadurch Strömungsenergie, die letztendlich in die - noch zu besprechende - Wärmeenergie umgewandelt wird (man kann sich z. B. den Extremfall vorstellen, die Masse M schlagartig ganz wegzunehmen, dann expandiert das Gas, ohne irgendwelche Arbeit zu leisten). Die tatsächliche Volumenarbeitsleistung ist dann kleiner als das Integral in (3.16). Nur bei einem ausreichend langsamen Verlauf der Volumenänderung, wenn man den Prozess also quasistatisch durchführt ("Gramm für Gramm die große Masse M verkleinern"), kann der Differenzdruck so klein gemacht werden, dass die Prozesskurve mit der p(V)-Kurve zusammenfällt. Dies entspricht dann der maximal möglichen Arbeitsleistung. V1 V2 V Wärmeaustauschkontakt In Erinnerung und Ergänzung zur Definition des geschlossenen Systems in Kapitel 1 wollen wir den Begriff Wärmeübertrag (Wärmeaustauschkontakt) noch einmal präzisieren: bei einem Wärmeübertrag δQ handelt es sich um einen Energieübertrag bei dem das System seine Temperatur ändert, ohne dass an ihm oder von ihm Arbeit geleistet wird. Wie bei der Arbeit handelt es sich bei δQ um kein vollständiges Differential. 3.2 Innere Energie und Erster Hauptsatz Weiterhin definieren wir die innere Energie U, die den gesamten Energieinhalt des Systems darstellt. Die innere Energie ist eine Zustandsgröße, sie kann also als eindeutige Funktion von unabhängigen Zustandsvariablen, z.B. T und V, dargestellt werden. Am Ende eines Kreisprozesses muss die innere Energie wieder ihren Ausgangswert annehmen, denn sonst könnte man innerhalb eines geschlossenen Systems Energie schaffen (oder vernichten). Dies entspräche dann einem perpetuum mobile 1. Art. Daher gilt für jeden Kreisprozess: (3.17) ∫ dU =0 dU ist also ein totales Differential. 23 Mit diesen Definitionen können wir nun den Ersten Hauptsatz der Thermodynamik formulieren. Es handelt sich dabei um den bekannten Energiesatz, unter Einbeziehung des Wärmeaustauschs: 1. Isoliertes System dU = 0 (3.18) 2. Geschlossenes System dU = δ Q + δ W (3.19) 3. Offenes System (3.20) dU = δ Q + δ W + δ EC Dabei gilt: (3.21) α δ EC = ∑ μi dN i i =1 Hierbei ist δEC der Teilchenaustauschkontakt und Ni die Zahl der Teilchen der Sorte i (i = 1, ... , α). Das chemische Potential µi ist die Energie, die bei δW = δQ = 0 benötigt wird, um dem System ein Teilchen der Sorte i hinzuzufügen. Wir werden uns mit dem Begriff des chemischen Potentials noch eingehender befassen. Das chemische Potential beinhaltet unter anderem z.B. die chemische Bindungsenergie von eingebrachten Molekülen. Der erste Hauptsatz besagt also, dass die Änderung der inneren Energie eines Systems gleich der ausgetauschten (zu- oder abgeführten) Wärme plus der ausgetauschten Arbeit und der ausgetauschten chemischen Energie ist. Wir können die Zustandsgröße U als unabhängige Variable auffassen oder aber als Zustandsfunktion anderer unabhängiger Variablen, z.B.: U = U(T, p, N), U = U(V, p, N) etc. Die Relation U = U(T, V, N) wird auch kalorische Zustandsgleichung genannt, die analoge Relation p = p(T, V, N) heißt thermische Zustandsgleichung. Für das ideale Gas im geschlossenen System gilt U = U(T), d.h. U ist unabhängig von V. Nach Gay-Lussac (2.17) ist ja das Volumen des idealen Gases direkt abhängig von der Temperatur, daher ist die innere Energie in diesem Fall nur von einer Variablen abhängig. 24 Abbildung 3.1. schematische Darstellung eines zwei-atomigen (homonuklearen) Moleküls. [Bohren] Es gilt für hinreichend ideale Gase bei atmosphärischen Temperaturen: (3.22) U= s NkT 2 Der Wert s beschreibt die Anzahl der Freiheitsgrade des Moleküls. Hierbei nimmt s den Wert 3 für ein-atomig Gase, den Wert 5 für zwei-atomige Gasmoleküle und den Wert 6 für nicht-lineare mehr-atomige Moleküle an. Um dies zu verstehen, müssen wir auf die Molekülstruktur eingehen. Ein ein-atomiges Gas kann (bei atmosphärischen Temperaturen) Energie nur in Form von kinetischer Translationsenergie aufnehmen oder abgeben (drei Translationsfreiheitsgrade für die drei möglichen Bewegungsrichtungen). Andere Formen der Energieaufnahme, wie z.B. elektronische Anregungen, d.h. das Anheben eines Elektrons in eine unbesetzte höhere Schale sind bei atmosphärischen Temperaturen nicht möglich). Ein zwei-atomiges Gasmolekül kann man sich vorstellen, wie in der Abbildung 3.1 gezeigt: die beiden Atomkerne werden durch die Bindungselektronen zusammengehalten, sie könnten aber um Ihre Ruhelage herumschwingen (als wäre die Verbindung nicht starr, sondern wie eine Feder). Ähnlich wie bei den Elektronenschalen mit ihren diskreten Energieniveaus sind auch für die Vibrationsschwingungen quantenmechanisch nur diskrete Niveaus "erlaubt" (s. Abbildung 3.2). Das erste angeregte Niveau ist bei atmosphärischen Temperaturen für die Moleküle ebenfalls nicht erreichbar, daher scheidet auch diese Form der Energieaufnahme aus. Weiterhin ist es möglich die Moleküle durch Stöße in Drehungen zu versetzen. Hierbei sind nur solche Drehungen um den Molekülschwerpunkt erlaubt, die nicht entlang der Molekülachse verlaufen, daher gibt es hier nur zwei unabhängige Rotationsfreiheitsgrade. Der Grund hierfür ist, dass die Massen der Atomkerne bei Drehung um die Molekülachse ja gerade auf der Drehachse liegen, es gibt dann keinen "Hebelarm" für eine Rotation. Auch Drehungen unterliegen quantenmechanischen Einschränkungen und es sind wieder nur diskrete Energieniveaus zugänglich, die Energieniveaus liegen aber so dicht beisammen, dass sie bei atmosphärischen Temperaturen besetzt werden können. Wir erhalten also s = 5 aus drei Translations- und zwei Rotationsfreiheitsgraden. Bei nicht-linearen mehr-atomigen Molekülen sind dann alle drei Freiheitsgrade der Rotation zugänglich und daher s = 6. Nach dem sogenannten Äquipartitionsprinzip wird die übertragene Energie im Mittel auf jeden der zugänglichen Freiheitsgrade gleich verteilt, d.h. in jedem Freiheitsgrad ist der gleiche Anteil der inneren Energie untergebracht, nämlich ½kT. 25 Abbildung 3.2. Schematische Darstellung der elektronischen, der Rotations-, und der Vibrations-Energieniveaus eines zweiatomigen Gasmoleküls (z.B. N2 oder O2). Zum Vergleich ist jeweils die Größe kT für T ≈ 300 K angegeben. Die Größe kT entspricht der Größenordnung des Energieübertrags bei den Molekülstößen. [Bohren] 3.3 Wärmekapazitäten Wärmekapazitäten geben an, mit welcher Temperaturänderung dT ein System auf eine differentielle Wärmezufuhr δQ reagiert. Da es neben der Temperatur T noch andere Zustandsvariablen gibt, müssen wir zusätzlich angeben, wie sich diese bei der Zustandsänderung verhalten. Definition der Wärmekapazität: (3.23) ⎛δQ ⎞ Cx = ⎜ ⎟ ⎝ dT ⎠ x Hierbei steht x für eine oder mehrere Zustandsgrößen, die bei der Wärmezufuhr δQ konstant gehalten werden. Weiterhin definieren wir die spezifische Wärmekapazität: (3.24) ⎛ δQ ⎞ cx = ⎜ ⎟ ⎝ M dT ⎠ x Hierbei ist M die Masse des Systems. Die molare Wärmekapazität (auch Molwärme genannt) ist mit der Anzahl der Mole des Systems ν analog definiert: (3.25) ⎛ δQ ⎞ cxmol = ⎜ ⎟ ⎝ ν dT ⎠ x 26 Man kann sich vorstellen, dass Wärmekapazität so etwas ist wie Trinkfestigkeit: ein System mit großer Wärmekapazität kann große Wärmemengen δQ aufnehmen, ohne dass die Temperatur des Systems stark ansteigt; eine Person mit großer Trinkfestigkeit kann große Alkoholmengen zu sich nehmen, ohne betrunken zu werden. Wir setzten ein geschlossenes System (Ni = const.) voraus, dessen innere Energie im allgemeinen von der Temperatur T und von den generalisierten Koordinaten qi abhängen wird: U = U (T , q1 , q2 ,..., qm ) (3.26) Das heißt, es gilt für dU: m ⎛ ∂U ⎞ ⎛ ∂U ⎞ + dU = ⎜ dT dqi ⎜ ⎟ ∑ ⎟ ⎝ ∂T ⎠qi i =1 ⎝ ∂qi ⎠T , q j , j ≠i (3.27) Wir lösen den Ersten Hauptsatz (3.19) nach δQ auf, und setzen (3.1) und (3.27) für δW und dU ein: m δ Q = dU − ∑ Fi dqi i =1 (3.28) ⎤ m ⎡ ⎛ ∂U ⎞ ⎛ ∂U ⎞ ⎢ ⎥ dT F =⎜ + − ⎜ ⎟ ∑ i dqi ⎟ ⎥ ⎝ ∂T ⎠{qi } i =1 ⎢⎝ ∂qi ⎠T , q j , j ≠i ⎣ ⎦ Hieraus lesen wir die folgenden Spezialfälle ab: 1.) {qi} = const. Es sind dann alle dqi gleich null, so dass bleibt: ⎛δQ ⎞ ⎛ ∂U ⎞ C{qi } = ⎜ ⎟ =⎜ ⎟ . ⎝ dT ⎠{qi } ⎝ ∂T ⎠{qi } (3.29) 2.) {Fi} = const. Hier müssen zunächst die Zustandsgleichungen Fj = Fj (q1 , q2 ,..., qm , T ) (3.30) j = 1,...,m nach qi aufgelöst werden: qi = qi ( F1 ,..., Fm , T ) (3.31) m ⎛ ∂q ⇒ dqi = ∑ ⎜ i ⎜ i =1 ⎝ ∂F j ⎞ ⎛ ∂q ⎞ dFj + ⎜ i ⎟ dT ⎟⎟ ⎝ ∂T ⎠{ Fi } ⎠T , Fk , k ≠ j 27 Dies ergibt für die Wärmekapazität: (3.32) ⎤ ∂q m ⎡ ⎛ ∂U ⎞ ⎛ i⎞ ⎛δQ ⎞ ⎛ ∂U ⎞ ⎢ ⎥ C{ Fi } = ⎜ F = + − ⎜ ⎟ ∑ i ⎜ ⎟ ⎜ ⎟ ⎟ ⎥ ⎝ ∂T ⎠{ Fi } ⎝ dT ⎠{ Fi } ⎝ ∂T ⎠{qi } i =1 ⎢⎝ ∂qi ⎠T , q j , j≠i ⎣ ⎦ Nach diesen allgemeinen Vorarbeiten können wir die für uns interessanten Beispiele sofort ablesen. Für ein Gas gilt: q=V und F = -p. Nach (3.29) gilt dann: (3.33) ⎛ δ Q ⎞ ⎛ ∂U ⎞ CV = ⎜ ⎟ =⎜ ⎟ ⎝ dT ⎠V ⎝ ∂T ⎠V und (3.32) liefert: (3.34) ⎤ ⎛ ∂V ⎞ ⎛ δ Q ⎞ ⎛ ∂U ⎞ ⎡⎛ ∂U ⎞ Cp = ⎜ ⎟ =⎜ ⎟ + ⎢⎜ ⎟ + p⎥ ⎜ ⎟ ⎝ dT ⎠ p ⎝ ∂T ⎠V ⎣⎝ ∂V ⎠T ⎦ ⎝ ∂T ⎠ p Dies ergibt: (3.35) ⎡⎛ ∂U ⎞ ⎤ ⎛ ∂V ⎞ C p − CV = ⎢⎜ ⎟ + p⎥ ⎜ ⎟ ⎣⎝ ∂V ⎠T ⎦ ⎝ ∂T ⎠ p Für den Spezialfall des idealen Gases gilt dann weiterhin nach (3.22) und (2.14): (3.36) (3.37) ⎛ ∂U ⎞ ⎜ ⎟ =0 ⎝ ∂V ⎠T und * ⎛ ∂V ⎞ ν R ⎜ ⎟ = p ⎝ ∂T ⎠ p ⇒ C p − CV = ν R* = Nk Mit den Definitionen der Gaskonstanten (S. 8), der spezifischen und der molaren Wärmekapazität ((3.24), (3.25)) folgt noch: (3.38) c p − cV = R − cVmol = R* c mol p Die Gleichung (∂U/∂V)T = 0 wird auch Joulesches Gesetz genannt und kann als Definition des idealen Gases benutzt werden. Aus (3.37) folgt, dass immer Cp > CV gelten muss. Diesen Sachverhalt können wir uns auch physikalisch plausibel machen. Man stelle sich einen Gasbehälter mit dem üblichen reibungslosen aber luftdichten Kolben vor (Abbildung 3.3). Zunächst sei der Kolben in einer Position fest fixiert. Nun heize man das Gas mit einem Wärmeübertrag δQ. Die Temperatur des Gases steigt in diesem isochoren Prozess. Der Versuch wird wiederholt, diesmal entfernen 28 wir aber die Fixierung des Kolbens (isobarer Prozess). Wir heizen das Gas wiederum mit δQ. Die Temperatur des Gases steigt, gleichzeitig dehnt sich das Gas aus und drückt den Kolben nach außen. Es wird Volumenarbeit gegen den externen Druck geleistet und die innere Energie nimmt nicht so stark zu, wie im ersten Prozess (1. HS). Daher nimmt auch die Temperatur des Gases nicht so stark zu, wie beim ersten Versuch (3.22). Dies bedeutet dann nach Definition der Wärmekapazitäten auch, dass Cp größer sein muss als Cv. Abbildung 3.3. Zum Vergleich von CV und Cp. Im 2. Versuch muss vom Gas Volumenarbeit geleistet werden, daher ist Cp > CV. 3.4 Adiabaten Wir wollen spezielle Arten von Zustandsänderungen mit Hilfe des Ersten Hauptsatzes diskutieren. Diese sind dadurch charakterisiert, dass bei ihrer Durchführung bestimmte abhängige oder unabhängige Zustandsgrößen konstant gehalten werden. Adiabatische Zustandsänderungen sind definiert durch δQ = 0 (3.39) Wir kennzeichnen adiabatische Zustandsänderungen durch den Index "ad". Die Zustandsgröße, die bei diesen Prozessen konstant bleibt, ist die – im folgenden Kapitel ausführlich behandelte – Entropie. Man kann daher einen reversiblen, adiabatischen Prozess auch isentrop nennen. Ausgangspunkt der Überlegungen ist der Erste Hauptsatz in der Form (3.28): (3.40) ⎤ m ⎡ ⎛ ∂U ⎞ ⎛ ∂U ⎞ ⎢ ⎥ = − ( ) (dqi ) ad dT F ⎜ ⎟ ∑ ad i ⎜ ⎟ ∂qi ⎠T , q i ⎥ ⎝ ∂T ⎠{qi } i =1 ⎢ ⎝ j , j ≠i ⎦ ⎣ Für ein Gas gilt dann: q=V und F = -p. 29 (3.41) ⇒ ⎡ ⎛ ∂U ⎞ ⎛ ∂U ⎞ ⎤ ⎜ ⎟ (dT ) ad = − ⎢ p + ⎜ ⎟ ⎥ (dV ) ad ⎝ ∂T ⎠V ⎝ ∂V ⎠T ⎦ ⎣ Mit (3.33) gilt dann: (3.42) ⎛ dT ⎞ ⎜ ⎟ ⎝ dV ⎠ ad ⎛ ∂U ⎞ p+⎜ ⎟ ⎝ ∂V ⎠T =− CV und für das ideale Gas gilt weiter wegen des Jouleschen Gesetzes (3.36): (3.43) p ν R*T ⎛ dT ⎞ = ⎜ ⎟ =− CV CV V ⎝ dV ⎠ ad Mit (3.37) folgt: (3.44) C p − CV ⎛ dV ⎞ ⎛ dT ⎞ ⎜ ⎟ ⎜ ⎟ =− CV ⎝ V ⎠ ad ⎝ T ⎠ad Wir definieren: γ= (3.45) Cp CV und erhalten: (3.46) (d ln T ) ad = −(γ − 1)(d ln V ) ad ⇒ d (ln TV γ −1 ) ad = 0 Dies bedeutet: (3.47) TV γ −1 = const1 Setzt man das ideale Gasgesetz ein, so erhält man zwei weitere Gleichungen: (3.48) pV γ = const2 1−γ (3.49) p γ T = const3 Diese drei Gleichungen nennt man Adiabatengleichungen oder auch Poisson-Gleichungen. 30 3.5 Isotherme Zustandsänderungen Isotherme Zustandsänderungen sind definiert durch dT = 0 (3.50) Der Erste Hauptsatz liefert dann: ⎤ m ⎡ ⎛ ∂U ⎞ (δ Q)T = ∑ ⎢⎜ − Fi ⎥ (dqi )T ⎟ ⎥ i =1 ⎢⎝ ∂qi ⎠T , q j , j ≠i ⎣ ⎦ (3.51) Für das Gas mit q = V und F = -p folgt: ⎛ δ Q ⎞ ⎛ ∂U ⎞ ⎜ ⎟ =⎜ ⎟ +p ⎝ dV ⎠T ⎝ ∂V ⎠T (3.52) und für das ideale Gas folgt: (3.53) ⎛ ∂U ⎞ ⎜ ⎟ =0 ⎝ ∂V ⎠T ⇒ (δ Q )T = ( pdV )T . 3.6 Der trocken-adiabatische Temperaturgradient Zur Bestimmung des trockenadiabatischen Temperaturgradienten gehen wir von folgendem Modell aus: Luft wird am Boden erwärmt, ein Luftpaket steigt auf, ohne durch Strahlung oder Wärmeleitung Energie aufzunehmen oder abzugeben. Wegen der Abnahme des Luftdrucks mit zunehmender Höhe expandiert das Luftpaket und leistet dabei Arbeit gegen den aktuellen Luftdruck. Der Luftdruck p des Pakets gleicht sich immer sofort dem Umgebungsluftdruck ps an (pp = ps). Da der Vorgang adiabatisch ist, muß die Energie für diese Arbeit der inneren Energie des Gases entzogen werden: das Luftpaket kühlt ab. Umgekehrt wird auch beim adiabatischen Absinken eines Luftpakets potentielle Energie in innere Energie umgewandelt: das Gas wird komprimiert und die Temperatur nimmt zu. Der im folgenden hergeleitete Temperaturgradient heißt trocken-adiabatisch, weil wir annehmen, dass keine Kondensation von Wasserdampf beim Abkühlen auftritt und damit auch keine latente Wärme (Verdampfungsenthalpie) freigesetzt wird. Die Überlegungen betreffen zwar ein einzelnes Luftpaket, da sich der Prozess im Laufe der Zeit aber typischerweise wiederholt, also immer neue Luftpakete am Boden aufgeheizt werden und dann aufsteigen, entsteht eine fortlaufende Konvektion und die ganze Atmosphäre nimmt nach und nach den Temperaturverlauf an, der dem trocken-adiabatischen Temperaturgradienten entspricht. Gehen wir der Einfachheit halber von einem Luftpaket aus, das ein Mol Luft enthält und behandeln wir die Luft als ideales Gas. Mit (3.19) und (3.33) können wir dann den Ersten Hauptsatz aufstellen: (3.54) δ Q = p dV + Cv dT 31 Die Zustandsgleichung des idealen Gases pV = R*T für ein Mol Gas in der differentiellen Form heißt: (3.55) pdV + Vdp = R * dT Damit können wir die Energiegleichung unter Verwendung von (3.37) und dem Gasgesetz für ein Mol umformen in (3.56) δ Q = −Vdp + ( R * +CV )dT = −Vdp + C p dT = − R *T dp + C p dT p Da wir uns mit einem adiabatischen Prozess beschäftigen gilt δQ = 0. Weiterhin setzen wir die barometrische Höhenformel (2.20) R *T dp / p = − gM dz ein und erhalten: (3.57) C p dT = − Mgdz An dieser Form der Energiegleichung sehen wir sofort, dass bei einem adiabatischen Prozess in der Atmosphäre die Summe aus thermischer Energie und potentieller Energie eine Erhaltungsgröße ist. Umgeformt erhalten wir: (3.58) dT Mg g =− = − = −Γ d dz Cp cp Hiermit haben wir den trocken-adiabatischen Temperaturgradienten Γd eingeführt. Mit den Zahlenwerten für Luft: Cp = 29 J K-1 mol-1 und M = 29 g mol-1 und g = 9.81 m s-2 ergibt sich: (3.59) Γ d = 9.8 K km −1 Somit ist die Faustregel von 1 °C Temperaturabnahme pro 100 m Höhe erklärt, sie gilt aber nur unter den gegebenen Voraussetzungen, vor allem darf kein Wasserdampf kondensieren. 3.7 Auftrieb und atmosphärische Stabilität In diesem Abschnitt wollen wir uns mit der Stabilität der atmosphärischen Luftschichten befassen. Jeder weiß, dass "warme Luft aufsteigt", oder dass es "Inversionswetterlagen" gibt, bei denen eine wärmere Luftschicht über einer kälteren liegt, und daher der konvektive Austausch und die Durchmischung der Luftschichten verhindert werden. Wir wollen nun herausfinden, welche vertikalen Temperaturprofile in der Atmosphäre zu einer stabilen oder einer instabilen Schichtung führen. Den Begriff der Stabilität entnehmen wir wieder der klassischen Mechanik. Ein System ist dann im stabilen Gleichgewicht, wenn das System bei einer kleinen Auslenkung aus der Ruhelage wieder in seine Gleichgewichtsposition zurückkehrt (s. Abbildung 3.4). Im instabilen Gleichgewicht führt eine Auslenkung hingegen zu einer Verstärkung des Effekts, das System entfernt sich von seiner ursprünglichen Lage. Ein neutrales Gleichgewicht nennt man den Fall, wenn das System bei einer Störung sich weder in den ursprünglichen Zustand zurückentwickelt, noch sich weiter davon entfernt. 32 Abbildung 3.4. Verdeutlichung der Definition von Stabilität in der klassischen Mechanik. Im stabilen Gleichgewicht kehrt das System nach einer kleinen Auslenkung aus der Gleichgewichtslage in den ursprünglichen Zustand zurück. [Bohren] Als nächstes müssen wir uns mit dem Begriff des Auftriebs näher auseinandersetzen. Man betrachte ein bestimmtes Volumen V eines Gases (oder einer Flüssigkeit) im Schwerefeld der Erde (Abbildung 3.5). Durch den umgebenden Luftdruck p wird eine Kraft von der Umgebung auf das Volumen V ausgeübt. Diese Kraft ist gegeben durch das Oberflächenintegral: r − ∫ pn dA (3.60) A r wobei n der Einheitsnormalenvektor, senkrecht zur Oberfläche A, und p der Außendruck ist. r Das Minuszeichen ist notwendig, da n nach außen zeigt. Mit Hilfe des Gaußschen Satzes können wir das Volumenintegral umwandeln: (3.61) r − ∫ pn dA = − ∫ ∇p dV A V Hydrostatisches Gleichgewicht bedeutet die Annahme, dass diese Kraft durch das Gewicht von V ausgeglichen wird: (3.62) ur − ∫ ∇p dV + ∫ ρ g dV = 0 V V Da das Volumen V beliebig gewählt ist, kann (3.62) nur erfüllt sein, wenn gilt: (3.63) ur ∇p = ρ g Dies ist eine generellere Formulierung der barometrischen Höhenformel, bzw. des hydrostatischen Gleichgewichts, als die in Abschnitt 2.4 gegebene Herleitung. 33 Abbildung 3.5. Auf ein Gas (in Ruhe) der Dichte ρ im Volumen V im Schwerefeld der Erde wirken zwei gleiche, entgegengesetzte Kräfte: seine Gewichtskraft (ρgV) und der integrale Druck über die gesamte Oberfläche A. Wird das Gas durch ein Gas der Dichte ρ' ersetzt, ohne die äußeren Druckkräfte zu verändern, so resultiert eine Nettokraft auf das Volumen: die Auftriebskraft. [Bohren] Nun nehmen wir an, dass das Gas im Volumen V durch ein Gas mit Dichte ρ' ≠ ρ ersetzt werde, während das Gas in der Umgebung unverändert bleibt. Da der äußere Druck nur durch die Dichte in der Umgebung und das Gravitationsfeld bestimmt wird, ändert sich der äußere Druck nicht. Das Gewicht des Volumen V ändert sich aber und daher resultiert jetzt eine Nettokraft, die Auftriebskraft. (3.64) ur r ur F = − ∫ pn dA + ∫ ρ ' g dV A V Mit (3.61) und (3.63) können wir umformen: (3.65) ur ur F = ∫ ( ρ '− ρ ) g dV V Für ρ' < ρ erhalten wir einen nach oben gerichteten positiven Auftrieb, bei ρ' > ρ einen nach unten gerichteten negativen "Auftrieb". Der in der Atmosphäre gemessene Temperaturgradient entspricht natürlich nicht generell dem trocken-adiabatischen Temperaturgradienten. Nah am Boden können z.B. sehr große Abweichungen auftreten, innerhalb der ersten zwei Meter über dem Boden kann der Gradient leicht 5 °C/m statt Γ = 1 °C/100 m betragen. Es können auch die schon erwähnten Inversionswetterlagen auftreten, bei denen die Temperatur mit zunehmender Höhe nicht absondern zunimmt. Wir wollen jetzt sehen, was wir über die Stabilität einer atmosphärischen Schichtung sagen können. Gehen wir zunächst von einer atmosphärischen Situation aus, bei der die Temperatur mit der Höhe schneller abnimmt als der trocken-adiabatische Temperaturgradient. Stellen wir uns nun ein Luftpaket im Gleichgewicht mit seiner direkten Umgebung vor, das wir ein Stück nach oben auslenken. Wie bisher gleicht sich der Druck des Pakets sofort an den Umgebungsdruck an. Wir nehmen weiterhin an, dass es keinen Energieaustausch mit der Umgebung gibt (adiabatischer Prozess) und , dass es keine Phasenübergänge von Wasserdampf im Luftpaket gibt (trocken). Unter diesen Annahmen folgt die Temperatur des ausgelenkten Pakets dem trocken-adiabatischen Gradienten und die Temperatur des Pakets ist immer größer als die Umgebungstemperatur. Daher ist auch die Dichte des Pakets kleiner als die Dichte der Umgebung und damit hat das Paket einen positiven Auftrieb. 34 Abbildung 3.6. Der trockenadiabatische Temperaturgradient als Grenze zwischen stabiler und instabiler Schichtung. [Bohren] Entsprechendes kann man sich für eine Auslenkung des Pakets nach unten überlegen. In diesem Fall wird das Paket einen negativen Auftrieb erfahren. Eine Region der Atmosphäre, die ein solches Temperaturprofil aufweist wäre also statisch instabil. Nun können wir den Fall betrachten, dass ein atmosphärisches Temperaturprofil vorliegt, bei dem die Temperatur mit der Höhe weniger abnimmt, als der trocken-adiabatische Gradient. Überlegungen, analog zu den oben gemachten, zeigen, dass in diesem Fall eine statisch stabile Schichtung vorliegt. Ein leicht ausgelenktes Luftpaket kehrt in seine Ausgangshöhe zurück. Der trocken-adiabatische Temperaturgradient selbst markiert genau die Grenze zwischen statisch stabiler und instabiler Schichtung (siehe Abbildung 3.6). Wir sehen hieran auch, dass für eine stabile Schichtung nicht unbedingt eine echte Inversionswetterlage vorliegen muss. Es gibt auch stabile Schichtungen, obwohl die Temperatur mit der Höhe abnimmt. Sie muss nur langsamer abnehmen als der trockenadiabatische Temperaturgradient. 3.8 Enthalpie An dieser Stelle wollen wir noch eine weitere thermodynamische Variable einführen, die sich im weiteren Verlauf der Vorlesung als nützlich und wichtig herausstellen wird. Wir definieren für ein Gas im geschlossenen System die Enthalpie H: (3.66) H = U + pV Für ein geschlossenes System (N = const.) können wir H als Funktion von den zwei thermodynamischen Variablen T und p auffassen: (3.67) H ( p, T ) = U (V ( p, T ), T ) + p V ( p, T ) mit dem totalen Differential: 35 dH = (3.68) ∂H ∂H dT + dp . ∂T ∂p Mit dem Ersten Hauptsatz und Gleichung (3.66) können wir schreiben δ Q = dH − Vdp (3.69) Setzen wir Gleichung (3.68) für dH ein, so erhalten wir: δQ = (3.70) ⎛ ∂H ⎞ ∂H dT + ⎜ − V ⎟ dp ∂T ⎝ ∂p ⎠ Für einen Prozess, der bei konstantem Druck abläuft (dp = 0), sehen wir dann mit (3.32): ⎛ ∂H ⎞ ⎛ δ Q ⎞ ⎜ ⎟ =⎜ ⎟ = Cp ⎝ ∂T ⎠ p ⎝ dT ⎠ p (3.71) analog zur Gleichung (3.33): (∂U/∂T)V = CV. Für ein ideales Gas können wir weiterhin mit Gleichung (3.22) feststellen: CV = s Nk = s ν R* und damit U = CV T . Setzen wir noch das ideale Gasgesetz pV = νR*T 2 2 ein, so erhalten wir: (3.72) H = U + pV = ( CV + ν R* ) T = C pT Für die Atmosphäre und den Bereich der in der Atmosphäre vorkommenden Temperaturen, können wir im allgemeinen in guter Näherung vom idealen Gas ausgehen, insbesondere dass Cp und CV nicht temperaturabhängig sind. Zweckmäßigerweise gehen wir noch auf spezifische Größen über. Es gilt dann näherungsweise in der Atmosphäre: (3.73) h ≈ c pT und u ≈ cV T . Mit diesem Ergebnis können wir noch einmal Temperaturgradienten (3.58) betrachten und schreiben: (3.74) den trocken-adiabatischen dh = −g dz Dies können wir umformen zu: (3.75) d ( h + gz ) = 0 dz Wir sehen, dass die Größe h + gz , die auf englisch dry static energy genannt wird, eine Erhaltungsgröße ist für trocken-adiabatischen Auf- oder Abstieg von Luftpaketen. 36 4 Entropie und Zweiter Hauptsatz 4.1 Zweiter Hauptsatz Der Erste Hauptsatz reicht offensichtlich zur Beschreibung thermodynamischer Systeme noch nicht aus. Wir können uns leicht Prozesse vorstellen, die zwar nach dem Energiesatz allein erlaubt sind, die wir aber nie beobachten können. Z. B. wird nie beobachtet, dass ein am Boden liegender Ball unter Abkühlung nach oben springt. Es zeigt sich, dass es Energieumwandlungen gibt, bei denen Wärme mit im Spiel ist, die nicht umkehrbar sind. Arbeit kann zwar vollständig in Wärme umgewandelt werden, z.B. durch Reibung, der umgekehrte Prozess, dass Wärme vollständig in Arbeit umgewandelt wird, d.h., dass ein ruhender Körper sich durch Abkühlung wieder in Bewegung setzt, ist zwar nach dem Ersten Hauptsatz denkbar, findet aber in der Natur nicht statt. Gäbe es diesen Prozess, so hätten wir ein perpetuum mobile zweiter Art. Das ist eine thermodynamische Maschine, die periodisch (zyklisch) arbeitet, die nichts anderes bewirkt, als dass bei einem Umlauf Arbeit verrichtet wird, wobei nur einem einzigen Wärmereservoir eine Wärmemenge ΔQ entnommen wird. Die Aussage des Zweiten Hauptsatzes ist somit: Ein perpetuum mobile zweiter Art gibt es nicht! Diese Formulierung des Zweiten Hauptsatzes nennt man die Kelvinsche Aussage. Sie besagt, dass es keine Zustandsänderung geben kann, deren einzige Wirkung darin besteht, eine Wärmemenge einem Reservoir zu entziehen und vollständig in Arbeit zu verwandeln. Es bestehen noch weitere äquivalente Formulierungen des Zweiten Hauptsatzes: Es gibt keine periodisch arbeitende Maschine, die lediglich einem kälteren Wärmebad Wärme entzieht und diese einem heißeren Wärmebad zuführt. Diese Formulierung nennt man die Clausiussche Aussage. Das Wort lediglich ist hier äußerst wichtig, denn es bedeutet, dass sonst, also auch in der Umgebung des Systems, nichts passiert. Bevor wir die Aussagen des Zweiten Hauptsatzes weiter betrachten führen wir den Begriff der Wärmekraftmaschine ein. Eine Wärmekraftmaschine ist ein thermodynamisches System, das einen Kreisprozess zwischen zwei Wärmebädern WB(T1) und WB(T2) mit T1 > T2 durchläuft, wobei genau das folgende passiert: 1) ΔQ1 > 0 2) ΔW < 0 durch Kontakt mit WB(T1), (das wärmere WB überträgt Wärme auf das System), (die Maschine verrichtet Arbeit), 3) ΔQ2 < 0 durch Kontakt mit WB(T2), (das kältere WB nimmt Wärme vom System auf). 37 Eine solche Maschine verletzt nicht den Zweiten Hauptsatz, da sie in Kontakt mit zwei Wärmebädern steht, wobei die dem ersten WB entzogene Wärme nicht vollständig in Arbeit umgewandelt wird. Es ist |ΔQ2| < |ΔQ1|, da sonst der Erste Hauptsatz verletzt wäre. Man definiert für eine solche Maschine den Wirkungsgrad: η= (4.1) vom System geleistete Arbeit −ΔW . = zugeführte Wärmemenge ΔQ1 Der Beweis der Äquivalenz der Clausiusschen und der Kelvinschen Aussage: 1. Behauptung: Wenn die Clausius-Aussage falsch ist, dann ist auch die Kelvin-Aussage falsch. a) Mit einer periodisch arbeitenden Maschine entnehmen wir ΔQ1 >0 aus dem Wärmebad WB(T2) und führen es dem Wärmebad WB(T1) zu, wobei T1 > T2 ist. Das geht, da die Clausius-Aussage falsch sein soll. b) Wir betreiben eine Wärmekraftmaschine so, dass ΔQ1WB(T1) entnommen und ΔQ2 < 0 (|ΔQ2| < ΔQ1) bei Arbeitsleistung ΔW < 0 an WB(T2) zurückgegeben wird. Insgesamt wurde also ΔQ = ΔQ1 + ΔQ2 > 0 aus WB(T2) vollständig in Arbeit umgewandelt. Sonst ist nichts passiert, da sowohl a) als auch b) Kreisprozesse sind. Damit ist auch die Kelvin-Aussage falsch. 2. Behauptung: Wenn die Kelvin-Aussage falsch ist, dann ist auch die Clausius-Aussage falsch. a) Wir entnehmen ΔQ > 0 dem Wärmebad WB(T2) und verwandeln es mit einer periodisch arbeitenden Maschine in Arbeit. Das geht, weil die Kelvin-Aussage falsch sein soll. b) Wir verwandeln die Arbeit aus a) vollständig in Wärme. Das geht immer, nur die umgekehrte Richtung nicht. Die so gewonnene Wärme übertragen wir auf WB(T1) mit T1 > T2. Insgesamt wurde lediglich ΔQ > 0 von WB(T2) auf WB(T1) trotz T1 > T2 übertragen. Damit ist die Clausius-Aussage falsch. Die beiden Aussagen ergeben kombiniert die Äquivalenz der Clausiusschen und der Kelvinschen Formulierung. 4.2 Carnot-Kreisprozess Bei einem Kreisprozess durchläuft das thermodynamische System verschiedene (Wärme-, Arbeits- und Teilchen-)Austauschkontakte und kehrt schließlich in seinen Ausgangszustand zurück. Es ist wichtig zu beachten, dass nur das thermodynamische System in seinen 38 Ausgangszustand zurückkehrt. Die Umgebung kann sich durchaus verändert haben, da z.B. Energie in Form von Arbeit oder Wärme zwischen verschiedenen Reservoiren ausgetauscht worden sein kann. Es gilt zwar nach dem Ersten Hauptsatz (4.2) 0= ∫ dU = ∫ δ Q + ∫ δ W die beiden einzelnen Terme auf der rechten Seite können aber von null verschieden sein. Wir wollen nun einen speziellen Kreisprozess und eine spezielle Wärmekraftmaschine diskutieren. Abbildung 4.1. Carnot-Prozess. [Nolting] Der Carnot-Kreisprozess ist ein reversibler Kreisprozess, der entlang zweier Adiabaten und zweier Isothermen zwischen zwei Wärmebädern WB(T1) und WB(T2) verläuft mit T1 > T2. Er besteht aus folgenden Teilstücken: a→b: Adiabatische Kompression mit ΔT = T1 – T2 > 0. b→c: Isotherme Expansion mit Wärmeaufnahme ΔQ1 > 0 aus WB(T1). c→d: Adiabatische Expansion mit ΔT = T2 – T1 < 0. d→a: Isotherme Kompression mit Wärmeabgabe ΔQ2 < 0 an WB(T2). Die bei einem Umlauf verrichtete Arbeit entspricht gerade der im p-V-Diagramm vom Weg a→b→c→d→a umschlossenen Fläche. Wir symbolisieren den Carnot-Prozess durch ein Diagramm, wie es in Abbildung 4.2 gezeigt wird. Abbildung 4.2. Symbolisches Carnot-Diagramm. [Nolting] Nach dem Ersten Hauptsatz gilt: 39 0= (4.3) ∫ dU = ΔQ + ΔQ 1 2 + ΔW Damit lautet der Wirkungsgrad dieser Wärmekraftmaschine: η= (4.4) ΔQ2 −ΔW ΔQ1 + ΔQ2 = = 1+ ΔQ1 ΔQ1 ΔQ1 Wegen ΔQ2 / ΔQ1 < 0 ist immer η < 1. Da der Carnot-Prozess reversibel sein soll, lässt sich sein Umlaufsinn umkehren (s. Abbildung 4.3): ΔQ2 > 0; ΔQ1 < 0; ΔW > 0; |ΔQ1| > ΔQ2. Abbildung 4.3. Umgekehrter Carnot-Prozess: Wärmepumpe. [Nolting] Eine solche Maschine arbeitet als Wärmepumpe. Die Arbeitssubstanz der Carnot-Maschine sei das ideale Gas. Damit wollen wir den Wirkungsgrad explizit ausrechnen. a→b (Adiabate): Adiabate ⇔ ΔQ = 0 ⇔ ΔW = ΔU ⇒ ΔWab = CV (ΔT ) = CV (T1 − T2 ) = −ΔWcd b→c (Isotherme) c Vc V dV = −νR *T1 ln c V Vb Vb ΔWbc = − ∫ p (V ) dV = −νR T1 ∫ * b c→d (Adiabate) s. Prozess a→b d→a (Isotherme) ΔWda = −νR *T2 ln Va Vd Auf den Adiabaten gilt nach (3.47): 40 T2Vaγ −1 = T1Vbγ −1 (4.5) T2Vdγ −1 = T1Vcγ −1 ⇒ Va Vb = Vd Vc damit ergibt sich für die gesamte Arbeitsleistung: (4.6) ΔW = ΔWab + ΔWbc + ΔWcd + ΔWda = −ν R* (T1 − T2 ) ln Vd <0 Va Auf der Isothermen b→c ist ΔU = 0 und damit: (4.7) ΔQ1 = −ΔWbc = ν R*T1 ln Vc V = ν R*T1 ln d > 0 Vb Va Damit ergibt sich nach (4.1) der Wirkungsgrad ηC der Carnot-Maschine: (4.8) η = 1+ ΔQ2 T = 1− 2 . T1 ΔQ1 Als Folge des Zweiten Hauptsatzes gilt auch noch: 1) Der Carnot-Prozess hat den höchsten Wirkungsgrad von allen periodisch zwischen zwei Wärmebädern arbeitenden Maschinen. 2) ηC wird von allen reversibel arbeitenden Maschinen erreicht. Der Wirkungsgrad ηC ist also universell für alle reversiblen Kreisprozesse. Den Beweis der Aussagen 1) und 2) kann man z.B. [Nolting] entnehmen. 4.3 Entropie ``In der Natur nimmt die Entropie die Rolle des Direktors ein, die Energie aber nur die eines Buchhalters.'' Sommerfeld Wir können nun die für die Thermodynamik sehr wichtige Zustandsgröße Entropie einführen. Wir hatten für den Wirkungsgrad der Carnot-Maschine gefunden: (4.9) η = 1+ ΔQ2 T = 1− 2 T1 ΔQ1 Das bedeutet: 41 ΔQ1 ΔQ2 + =0 T1 T2 (4.10) Dieses Ergebnis wollen wir weiter verallgemeinern. Abbildung 4.4. Kreisprozess. [Nolting] Ein thermodynamisches System durchlaufe quasistatisch einen (nicht notwendig reversiblen) Kreisprozess K. Zur Beschreibung der Temperaturänderung zerlegen wir den Zyklus in n Schritte, während derer die Temperatur des Systems durch dessen Kontakt mit einem Wärmebad WB(Ti) (i = 1, 2, ..., n) konstant ist. Dabei findet ein Wärmeaustausch δQi statt, der positiv wie negativ sein kann. Nach dem Ersten Hauptsatz gilt dann für die gesamte Arbeitsleistung: n ΔWK = −∑ δ Qi (4.11) i =1 Wir koppeln nun an jedes WB(Ti) eine Carnot-Maschine Ci , die zwischen diesem WB(Ti) und einem Wärmebad WB(T0) arbeitet, wobei T0 > Ti für alle i gelten soll. Jedes Ci kann sowohl als Wärmekraftmaschine als auch als Wärmepumpe arbeiten. Wir dimensionieren die Ci so, dass sie gerade die Wärmemenge von WB(Ti) aufnehmen, die von dem System an WB(Ti) abgegeben wurde (bzw. umgekehrt). (4.12) δ QC = −δ Qi i ∀i Für jede Carnot-Maschine gilt mit (4.10): (4.13) δ QC(0) = − i T0 T δ QCi = 0 δ Qi Ti Ti Abbildung 4.5. Kreisprozess mit n Carnot-Maschinen angekoppelt, die alle mit dem Wärmebad T0 arbeiten. [Nolting] 42 Das System der Carnot-Maschinen leistet dann insgesamt die Arbeit: n n ΔWC = ∑ δ Wi = −∑ηCi δ QC(0)i = (4.14) i =1 i =1 n ⎛ T ⎞T ⎛ T ⎞ = −∑ ⎜1 − i ⎟ 0 δ Qi = ∑ ⎜ 1 − 0 ⎟ δ Qi T0 ⎠ Ti Ti ⎠ i =1 ⎝ i =1 ⎝ n Bei dem gesamten Zyklus (4.15) K + {C1 + C2 + ... + Cn } wird die Wärmemenge (4.16) n n δ Qi i =1 i =1 Ti ΔQ (0) = ∑ δ QC(0)i = T0 ∑ mit WB(T0) ausgetauscht und dabei die Arbeit (4.17) n δ Qi i =1 Ti ΔW = ΔWK + ΔWC = −T0 ∑ geleistet. Sonst ist nichts passiert. Der Erste Hauptsatz ist offensichtlich erfüllt: (4.18) ΔW = −ΔQ (0) . Der Zweite Hauptsatz fordert nun aber, dass ΔW ≥ 0 (4.19) ist. Im umgekehrten Fall wäre nämlich nichts anderes passiert, als dass das thermodynamische Gesamtsystem Wärme ΔQ(0) aus WB(T0) aufgenommen und vollständig in Arbeit ΔW ≤ 0 verwandelt hätte. Das ist aber unmöglich. Damit folgt aus (4.17) und (4.19) das wichtige Ergebnis (4.20) n δ Qi i =1 Ti ∑ ≤0, das nur Daten des ursprünglichen Kreisprozesses K enthält, also unabhängig von den Daten der Carnot-Maschinen und insbesondere unabhängig von T0 ist. Ist der Kreisprozess K sogar reversibel, dann lässt sich der Umlaufsinn von K umkehren. An den obigen Überlegungen ändert das überhaupt nichts. Die Größen δQi in (4.20) haben jedoch ihr Vorzeichen geändert. Da (4.20) aber für beide Durchlaufrichtungen gleichermaßen richtig ist, führt nur das Gleichheitszeichen nicht zum Widerspruch: 43 (4.21) n δ Qi i =1 Ti ∑ =0 ⇔ K reversibel Durch den Übergang auf n → ∞ Teilschritte ergibt sich aus (4.20) und (4.21) die fundamentale Clausiussche Ungleichung: (4.22) δQ ∫T ≤0 Für reversible Prozesse gilt: (4.23) ∫ δ Qrev T =0 Diese letzte Beziehung definiert eine neue Zustandsgröße. Sei A0 ein fester Punkt des Zustandsraums, dann ist das Integral A (4.24) ∫ δ Qrev A0 T unabhängig vom Weg, auf dem wir im Zustandsraum vom Zustand A0 zum Zustand A gelangen, und bei festem A0 eine eindeutige Funktion des Zustands A. Die sogenannte Entropie S, A (4.25) S ( A) = ∫ A δ Qrev T 0 ist also eine bis auf eine additive Konstante festgelegte Zustandsgröße mit dem totalen Differential (4.26) dS = δ Qrev T 1/T ist somit der integrierende Faktor (3.8), der aus der nicht-integrablen Differentialform δQ ein totales Differential macht. Man beachte, dass die Entropie stets über einen reversiblen Weg von A0 nach A zu berechnen ist. Dabei ist es unerheblich, wie das System den Zustand tatsächlich erreicht hat, ob irreversibel oder reversibel. Man benötigt zur Bestimmung von S(A) also stets einen reversiblen Ersatzprozess. Für eine beliebige Zustandsänderung Z gilt: 44 A2 ∫ S ( A2 ) − S ( A1 ) ≥ (4.27) δQ A1 T (Z ) Beweis: Sei R ein reversibler Ersatzprozess für Z. Auf diesem gilt A2 S ( A2 ) − S ( A1 ) = (4.28) ∫ A 1 (R) δQ T Da der Weg R reversibel ist, lässt er sich umkehren und mit Z zu einem Kreisprozess kombinieren, für den dann nach der Clausiusschen Ungleichung (4.22) gelten muss: A2 ∫ A δQ 1 (Z ) (4.29) T A1 ⇔ − ∫ A2 (− R) A1 + ∫ A δQ 2 (− R) δQ T T A2 ≥ ∫ A1 ≤0 δQ T (Z ) A2 ⇔ S ( A2 ) − S ( A1 ) ≥ ∫ A 1 (Z ) δQ T q.e.d . Der Begriff der Entropie wurde 1850 von Rudolf Clausius eingeführt. Er ist abgeleitet aus dem griechischen Wort für 'Umwandlung'. Der Begriff ist bewusst so gewählt, dass er dem Begriff der Energie sehr ähnlich klingt, um die Verwandtschaft der physikalischen Größen und ihre ähnliche Wichtigkeit hervorzuheben. Oft wird die Entropie erklärt als "ein Maß für die Unordnung eines Systems". Mit dieser Erklärung wird zwar das Wesen der Entropie recht anschaulich wiedergegeben, es wird aber davor gewarnt, die Begriffe Entropie und Unordnung gleichzusetzen. Entropie ist ein genau definierter, und vor allem quantifizierter Begriff, Entropie ist eine messbare Größe, während Unordnung ein allgemeiner, nicht-quantitativer Begriff ist. Zur Ableitung der Ergebnisse (4.22) und (4.27) haben wir nur die Gültigkeit des Zweiten Hauptsatzes voraussetzen müssen. Wir erhalten aus diesen Resultaten deshalb umgekehrt eine mathematische Formulierung des Zweiten Hauptsatzes: (4.30) dS ≥ δQ T Wobei das Gleichheitszeichen nur für reversible Prozesse gilt. 45 Kombinieren wir nun den Ersten und den Zweiten Hauptsatz, so ergibt sich die Grundrelation der Thermodynamik, die auch Gibbsche Fundamentalgleichung genannt wird.: TdS ≥ dU − δ W − δ EC (4.31) Mit dieser Grundrelation und der Einführung der Entropie als Zustandsgröße sind nun die zentralen Begriffe der Thermodynamik aufgestellt. Alle folgenden Überlegungen stellen deshalb mehr oder weniger Folgerungen aus diesem Grundkonzept dar. Betrachten wir zum Abschluss dieses Abschnitts noch den Spezialfall des isolierten Systems. Da das isolierte System per Definition keine Wärme mit der Umgebung austauschen kann gilt δQ = 0 und daher: isoliertes System : dS ≥ 0 (4.32) Solange in einem solchen System noch (irreversible) Prozesse ablaufen können, kann die Entropie nur zunehmen. Sie ist deshalb maximal im Gleichgewichtszustand. Der Übergang ins Gleichgewicht ist irreversibel. Entropie-Zuwachs ohne Austausch kennzeichnet irreversible Prozesse. Da das Universum ein isoliertes System darstellt, kann seine Entropie nur ständig zunehmen. Dies definiert zum einen eine Zeitachse, bzw. eine Richtung der Zeit im bestehenden Universum. Bleibt diese Richtung der Zeit für immer erhalten und nimmt die Entropie tatsächlich immer weiter zu, so würde dies bedeuten, dass das Universum letztendlich selbst einem Zustand größter Unordnung, Durchmischung und gleichmäßiger Temperatur zustrebt. Man spricht dann vom "Wärmetod des Universums" als End- und Gleichgewichtszustand. Wir wollen die physikalische Bedeutung der Entropie an einem Beispiel – ähnlich dem Beispiel aus Abschnitt 3.1 – illustrieren, der isothermen Expansion des idealen Gases: 1) Reversibel Vakuum Das Gas verschiebe einen reibungslosen Kolben, der durch ein Gewicht belastet ist. Die Arbeit, die das Gas beim Verschieben des Kolbens leistet, ist in der potentiellen Energie des Gewichts gespeichert und kann im Prinzip dazu dienen, die Verschiebung wieder rückgängig zu machen. Die Expansion des Gases ist damit reversibel. – Das Gas befinde sich in einem Wärmebad WB(T), sämtliche Zustandsänderungen verlaufen damit isotherm: M Ideales Gas WB(T) U = U(T) ⇒ ΔU = 0 Nach dem Ersten Hauptsatz gilt dann: (4.33) ΔQ = −ΔW = V2 V2 ∫ p dV = ν R T ln V * V1 1 46 Bei dieser reversiblen Zustandsänderung ändert sich gemäß (4.30) die Entropie: (ΔS )Gas = (4.34) V ΔQ = ν R* ln 2 T V1 Die zur Arbeitsleistung nötige Wärmemenge ΔQ wurde dem Wärmebad entnommen und kann durch Kompression des Gases aus der potentiellen Energie des Gewichts an dieses wieder zurückgegeben werden. Auch die Vorgänge im Wärmebad sind deshalb reversibel. (ΔS )WB = (4.35) −ΔQ = −(ΔS )Gas T Die Entropie des Gesamtsystems hat sich also nicht geändert. Dies kann auch als Definition des reversiblen Prozesses benutz werden: bei einem reversiblen Prozess ändert sich die Entropie des Universums nicht! 2) Irreversibel Der analoge irreversible Prozess ist die freie Expansion des idealen Gases: Vakuum WB(T) WB(T) Bei der freien Expansion leistet das Gas keine Arbeit. Es wird deshalb dem Wärmespeicher keine Wärme entzogen. Den Zeitablauf dieses irreversiblen Prozesses können wir nicht beschreiben. Anfangs- und Endzustand sind jedoch Gleichgewichtszustände. Sie entsprechen denen des Vorgangs 1). Prozess 1) ist deshalb der reversible Ersatzprozess für 2). Die Entropieänderung des Gases ist deshalb dieselbe wie unter 1): (ΔS )Gas = ν R* ln V2 V1 Wegen ΔQ = 0 ist jedoch (ΔS )WB = 0 Die Entropie des Gesamtsystems hat sich demnach erhöht. T(ΔS)tot ist gerade der Energiebetrag, der im reversiblen Fall 1) in verwertbare Arbeit (-ΔW) umgewandelt wurde. Das bedeutet: Irreversibilität verschenkt verwertbare Energie. 47 4.4 Entropie des idealen Gases Für ein Gas (F = -p; q = V) als geschlossenes System (dN = 0) folgt aus der Grundrelation der Thermodynamik im reversiblen Fall (Gleichheitszeichen): dS = (4.36) dU pdV + T T Mit Gleichung (3.33) gilt für das ideale Gas (U = U(T)): dU = CV dT (CV unabhängig von T). Mit dem idealen Gasgesetz können wir dann schreiben: (4.37) dS = CV dT dV + ν R* T V Diese Gleichung lässt sich integrieren: (4.38) S = S0 + CV ln T V + ν R* ln T0 V0 wobei S0 = S(T0, V0) die Integrationskonstante ist. Wegen νR* = Cp-CV (3.37) und γ = Cp/CV (3.45) können wir dies weiter umformen in: (4.39) ⎛ T V γ −1 ⎞ S = S0 + CV ln ⎜ γ −1 ⎟ ⎝ T0 V0 ⎠ Wir können somit die Entropie des idealen Gases bis auf eine Konstante bestimmen. 4.5 Potentielle Temperatur: die Entropie der Meteorologen Wir betrachten eine Situation in der Atmosphäre bei welcher der Druck und die Temperatur der Luft in einer bestimmten Höhe mit p und T gegeben sind. Wir definieren nun die potentielle Temperatur Θ der Luft in dieser Höhe als die Temperatur, die ein Luftpaket hätte, wenn es adiabatisch und reversibel aus dieser Höhe auf einen Referenzdruck p0 (üblicherweise wählen wir p0 = 1000 mb) gebracht würde. Aus den Poisson-Relationen (3.49) folgt sofort: (4.40) ⎛p ⎞ Θ=⎜ 0 ⎟ ⎝ p⎠ γ −1 γ T Verwenden wir ein weiteres Mal das ideale Gasgesetz um die mit Gleichung (4.39) gegebene Entropie S des idealen Gases als Funktion der Variablen T und p darzustellen, dann gilt: 48 γ −1 γ −1 (4.41) T ⎛V ⎞ ⎜ ⎟ T0 ⎝ V0 ⎠ ⎛ T ⎞ T ⎜ p ⎟ = ⎜ ⎟ T0 ⎜ T0 ⎟ ⎝ p0 ⎠ γ γ −1 ⎛T ⎞ ⎛ p ⎞ =⎜ ⎟ ⎜ 0 ⎟ ⎝ T0 ⎠ ⎝ p ⎠ und damit: (4.42) γ −1 ⎡ ⎤ γ ⎛ ⎞ ⎛Θ⎞ ⎛ ⎞ p T 0 ⎢ S = S0 + CV γ ln ⎜ ⎟ ⎜ ⎟ ⎥ = S0 + C p ln ⎜ ⎟ ⎢⎝ T0 ⎠ ⎝ p ⎠ ⎥ ⎝ T0 ⎠ ⎣⎢ ⎦⎥ Schreiben wir die Gleichung noch auf die üblichen intensiven Variablen um, da dies für unseren Anwendungsfall Atmosphäre nützlicher ist (spezifische Entropie und spezifische Wärmekapazität): (4.43) ⎛Θ⎞ s − s0 = c p ln ⎜ ⎟ ⎝ T0 ⎠ Bis auf einen konstanten Faktor ist also die potentielle Temperatur, genaugenommen der Logarithmus der potentiellen Temperatur, das gleiche wie die Entropie! Jeder isentrope Prozess in der Atmosphäre ist dann auch ein Prozess, bei dem die potentielle Temperatur konstant bleibt. Den Gradient der Entropie mit der Höhe erhalten wir aus der Ableitung von (4.43) nach z: (4.44) ds c p d Θ = dz Θ dz Der vertikale Gradient von Θ aus der Ableitung von Gleichung (4.40), bzw. aus der Ableitung der Adiabatengleichung nach der Höhe (dT/dz) = ((γ-1)/γ)*(T/p)*(dp/dz), ist: (4.45) d Θ Θ ⎛ dT γ − 1 T dp ⎞ . = ⎜ − dz T ⎝ dz γ p dz ⎟⎠ Hier können wir den trocken-adiabatischen Temperaturgradienten (3.58) einsetzen: (4.46) d Θ Θ ⎛ dT ⎞ = ⎜ − Γd ⎟ dz T ⎝ dz ⎠ Wenn das tatsächliche Temperaturprofil gerade gleich dem trocken-adiabatischen Temperaturgradienten ist, dann ist die potentielle Temperatur mit der Höhe konstant. Wir können unser in Abschnitt 3.7 hergeleitetes Kriterium für statische Stabilität jetzt auch für den Gradienten der potentiellen Temperatur formulieren: (4.47) dΘ >0 dz stabil 49 (4.48) dΘ =0 dz neutral (4.49) dΘ <0 dz instabil Dieses Kriterium ist einfacher zu merken und man kann an einem Diagramm in dem die potentielle Temperatur gegen die Höhe aufgetragen ist, sofort ablesen, ob die Schichtung stabil ist oder nicht. Für einen reversiblen Prozess gilt dS = δQ/T und für einen adiabatischen Prozess gilt δQ = 0; d.h. ein für einen reversiblen, adiabatischen Prozess gilt dS = 0, ein solcher Prozess ist also isentrop. Aus der Äquivalenz von S und Θ folgt dann auch, dass für einen solchen Prozess dann auch die potentielle Temperatur konserviert ist. Der Begriff der potentiellen Temperatur ist nur in der Meteorologie gebräuchlich. In Physiklehrbüchern taucht dieser Begriff nicht auf. In der Meteorologie hingegen ist der Begriff der potentiellen Temperatur wesentlich häufiger gebraucht als der Begriff Entropie. Die potentielle Temperatur in Form von Gleichung (4.40) wurde von Herman von Helmholtz 1888 eingeführt. L.A. Bauer zeigte dann 1908 die Äquivalenz von potentieller Temperatur und Entropie. Oszillationen von Luftpaketen Befindet sich ein mechanisches System in einem stabilen Gleichgewicht, dann sind meist Oszillationen mit kleiner Amplitude um den Gleichgewichtszustand möglich. Man betrachte ein Luftpaket mit genügend kleinem Volumen V, so dass das Paket als homogen angesehen werden kann (ρ, p, etc. sind innerhalb des Pakets konstant). Die Bewegungsgleichung für vertikale Oszillationen, die der Schwerpunkt des Luftpakets ausführen kann, ist dann: (4.50) F = − g ( ρ p − ρ s )V = M d 2z d 2z = ρ V p dt 2 dt 2 wobei die Indexe p und s jeweils für das Paket und seine Umgebung stehen. Es sei z0 die Gleichgewichtsposition aus der das Paket leicht nach oben oder unten verschoben wird. Die Bewegungsgleichung kann man dann umformen: (4.51) g ( ρs − ρ p ) ρp = d2 ( z − z0 ) dt 2 50 Abbildung 4.6: Ein Luftpaket mit Volumen V ist in stabilem Gleichgewicht mit seiner Umgebung in der Höhe z0 Wird das Paket anfangs leicht vertikal verschoben, so oszilliert es anschließend mit der charakteristischen Brunt-VäisäläFrequenz, die vom Gradienten der potentiellen Temperatur abhängt. [Bohren] Der Druck des Pakets passt sich immer an seine Umgebung an: ps = pp = p: (4.52) p = ρ s RTs = ρ p RTp damit folgt: (4.53) ⎛ Tp − Ts ⎞ d2 z − z = g ( ) ⎜ ⎟ 0 dt 2 ⎝ Ts ⎠ da das Paket immer den gleichen Druck annimmt wie seine aktuelle Umgebung, ist das Verhältnis der Temperaturen gleich dem Verhältnis der potentiellen Temperaturen: (4.54) ⎛ Θ p − Θs ⎞ d2 ( z − z0 ) = g ⎜ ⎟ 2 dt ⎝ Θs ⎠ Wie wir im letzten Abschnitt gesehen haben, ist die potentielle Temperatur bei einem reversiblen, adiabatischen Prozess eine Erhaltungsgröße und daher ist die potentielle Temperatur des Pakets konstant. Bei z = z0 ist Θp = Θs, während der Oszillation ändert sich also nur die potentielle Temperatur in der Paketumgebung. Wir entwickeln die inverse potentielle Temperatur als Taylor-Reihe um z0: (4.55) 1 1 ⎛ 1 ∂Θ s ⎞ = −⎜ ⎟ ( z − z0 ) + ... Θ s Θ p ⎝ Θ 2s ∂z ⎠0 und vernachlässigen die Terme höherer Ordnung (Näherung für kleine Amplituden). Damit erhalten wir als genäherte Bewegungsgleichung: (4.56) ⎛ 1 ∂Θ s ⎞ d2 ( z − z0 ) = − g ⎜ ⎟ ( z − z0 ) 2 dt ⎝ Θ s ∂z ⎠ 51 Wie wir in Gleichung (4.47) gezeigt haben, ist die Bedingung für statische Stabilität ein positiver Gradient ∂Θ/∂z. Damit ist (4.56) die Bewegungsgleichung eines einfachen harmonischen Oszillators mit der charakteristischen Schwingungsfrequenz: (4.57) ⎛ 1 ∂Θ s ⎞ ⎟ ⎝ Θ s ∂z ⎠0 ω g2 = g ⎜ Diese Frequenz wird Brunt-Väisälä-Frequenz genannt. Sie stellt eine fundamentale Größe in der Atmosphäre dar, als Produkt aus der Erdbeschleunigung g und der relativen vertikalen Änderung der potentiellen Temperatur. Je größer der Vertikalgradient, desto stabiler die atmosphärische Schichtung, desto größer die Rückstellkraft, die auf das Paket wirkt und desto höher die Oszillationsfrequenz. Für einen typischen troposphärischen Gradienten der potentiellen Temperatur von 3°C/km beträgt die Schwingungsperiode ~10 Minuten. Die Brunt-Väisälä-Frequenz ist eng verwandt mit der Frequenz von atmosphärischen Schwerewellen ("internal gravity waves"; Schwerewellen sollten keinesfalls mit Gravitationswellen der allgemeinen Relativitätstheorie verwechselt werden!). Wie in unserem einfachen eindimensionalen Fall, ist auch bei Schwerewellen die Auftriebskraft die rücktreibende Kraft. Diese Wellen sind aber nicht auf rein vertikale Schwingungen begrenzt, sondern diese Transversalwellen können sich vertikal und horizontal in der Atmosphäre ausbreiten. Schwerewellen sind beispielsweise verknüpft mit den sogenannten Leewellen, die sich im Lee, also auf der windabgewandten Seite von Gebirgszügen bilden. Luftmassen, die vom Wind gegen ein Gebirgsbarriere geschoben werden und konvektive Luftmassen, die auf stabile Luftschichten auftreffen sind zwei Mechanismen, die Schwerewellen in der Atmosphäre generieren. Parallele Wolkenstreifen, die auch parallel zu den Bergkämmen auf der windabgewandten Seite verlaufen, bilden sich dann oft in den Bereichen, in denen sich die Luft in diesen Schwerewellen aufwärts bewegt. Der Abstand zwischen den Wolkenstreifen entspricht dann der Wellenlänge und die zugehörige Frequenz ist die Brunt-Väisälä-Frequenz. Schwerewellen können auch für die im Flugzeug gelegentlich wahrgenommenen Turbulenzen ("clear air turbulences", umgangssprachlich "Luftlöcher") verantwortlich sein. 4.6 van der Waals-Gas Die Zustandsgleichung des idealen Gases ist an die in Abschnitt 2.2 gemachten Voraussetzungen geknüpft und daher ist das ideale Gasgesetz nur im Grenzfall sehr kleiner Teilchendichte für reale Gase verwendbar. Insbesondere kann die Zustandsgleichung für ideale Gase nicht den Phasenübergang "Gas-Flüssigkeit" beschreiben. Wir wollen deshalb das ideale Gasgesetz so verallgemeinern, dass die Bedingungen für das ideale Gas (keine Eigenvolumina und keine Wechselwirkungen der Teilchen untereinander) wegfallen, im Grenzfall großer Verdünnung soll aber wieder das ideale Gasgesetz resultieren. Wir gehen daher von folgendem Ansatz aus: (4.58) peff Veff = ν R*T Eigenvolumina: Für p → ∞ gilt bei T = const. in der idealen Gasgleichung V → 0, d.h. die Gasmoleküle haben kein Eigenvolumen. Für ein reales Gas müssen wir das minimale Eigenvolumen der Moleküle berücksichtigen: 52 (4.59) (4.60) Vmin ≈ N Teilchenvolumen = N b NA Veff = V − Vmin = V −ν b Wechselwirkung der Teilchen: Die Teilchen des realen Gases wechselwirken miteinander, auch wenn sie nicht gerade miteinander stoßen: die Teilchen ziehen sich gegenseitig schwach an. Wegen der homogenen Verteilung der Teilchen heben sich die Wechselwirkungskräfte im Gefäßinneren im Mittel auf. Für ein Teilchen am Rand bleibt allerdings eine resultierende Kraftkomponente nach innen! Das vermindert den Druck des Gases auf die Gefäßwand, wo der Druck andererseits gemessen wird. (4.61) peff > "Wanddruck " p Die anziehenden Kräfte der Teilchen untereinander sind proportional zur molaren Teilchenkonzentration (ν/V). Die anziehende Kraft der Teilchen untereinander führt sowohl zu einer verminderten Frequenz, mit der die Teilchen auf die Gefäßwand stoßen, als auch zu einem geringeren Impulsübertrag pro Kollision mit der Wand. Daher ist die Druckdifferenz zwischen effektivem Druck und Wanddruck proportional zu (ν/V)2. Wir schreiben daher für den effektiven Druck: (4.62) ⎛ν ⎞ peff = p + a ⎜ ⎟ ⎝V ⎠ 2 Der Term a(ν/V)2 wird Binnendruck genannt. (4.62) und (4.60) in (4.58) eingesetzt, ergibt die van der Waals-Zustandsgleichung: (4.63) ⎛ ν2 ⎞ + p a V − ν b ) = ν R*T ⎜ 2 ⎟( V ⎠ ⎝ Die Proportionalitätskonstanten a und b sind experimentelle Materialkonstanten, wobei a stark, b weniger stark von Substanz zu Substanz variiert (s. Tabelle). Die hier gegebenen Argumente über die Auswirkungen der anziehenden Kräfte der Gasmoleküle auf den Binnendruck und das Eigenvolumen der Moleküle sind äußerst vage und "handwaving". Es soll nur das Zustandekommen der Gleichung plausibel gemacht werden. Die Konstanten a und b sind experimentell bestimmte Koeffizienten und gehen nicht exakt aus den molekularen Eigenschaften der unterschiedlichen Gase hervor. Wie wir sehen werden, ist die van der Waals-Zustandsgleichung auch nur eine (recht brauchbare) von vielen Näherungen, die das tatsächliche Verhalten von realen Gasen beschreiben. 53 Tabelle 4.1. Van der Waals-Koeffizienten. [Atkins] Abbildung 4.7. p-V-Diagramm für van der Waals-Gas. [Nolting] Abbildung 4.8. Experimentell bestimmtes pV-Diagramm für CO2. [Atkins] 54 Kritischer Punkt Man kann (4.63) umformen: (4.64) ⎛ ν R*T ⎞ aν 2 ν3 − ab = 0 V − V ⎜ν b + ⎟ +V p ⎠ p p ⎝ 3 2 Dies ist eine Gleichung dritten Grades für das Volumen V, die bei gegebenem p und T für p < pc, T < Tc drei reelle Lösungen, sonst eine reelle und zwei komplexe Lösungen hat. Es gibt einen kritischen Punkt (pc, Tc, Vc), bei dem die drei Lösungen zusammenfallen. In diesem speziellen Punkt muss gelten: (4.65) 0 = (V − Vc )3 = V 3 − 3V 2Vc + 3VVc2 − Vc3 Aus dem Koeffizientenvergleich mit (4.64) können die Parameter des kritischen Punktes sämtlich aus den Parametern a und b bestimmt werden: (4.66) Vc = 3bν pc = a 27b 2 R*Tc = 8a 27b Man kann aus diesen Gleichungen auch a und b eliminieren und erhält dann den kritischen Kompressionsfaktor Z (4.67) Zc = pcVc 3 = * ν R Tc 8 Experimentell findet man für praktisch alle realen Gase, dass Zc etwas kleiner als 3/8 ist, meist Zc,exp ≈ 0,3. Während für das ideale Gas Zc = 1 gilt, ist die Beschreibung des realen Gases als van der Waals-Gas also schon eine deutliche Verbesserung. Tabelle 4.2. Zustandsgleichungen für Gase. [Atkins] 55 Tabelle 4.3. kritische Konstanten einiger Gase. [Atkins] korrespondierende Zustände: Führt man die reduzierten Größen Vr = V Vc pr = p pc Tr = T Tc ein, so lässt sich die van der Waals-Gleichung in eine Form bringen, die keine Materialeigenschaften mehr enthält: (4.68) ⎛ 3 ⎞ ⎜ pr + 2 ⎟ ( 3Vr − 1) = 8Tr Vr ⎠ ⎝ Man sagt dann, dass zwei Substanzen mit den selben (pr,Vr,Tr)-Werten sich in korrespondierenden Zuständen befinden. Maxwell-Konstruktion Die pV-Isothermen zeigen für T < Tc ein unphysikalisches Verhalten, denn es gibt einen Bereich, in dem (4.69) ⎛ ∂p ⎞ ⎜ ⎟ >0 ⎝ ∂V ⎠T ist. Dieses kann nicht realistisch sein, da eine Volumenabnahme dV < 0 dann auch eine Druckabnahme dp < 0 zur Folge hätte Das System würde kollabieren. Die Ursache diese unphysikalischen Besonderheit liegt darin, dass wir implizit bei der Ableitung der van der Waals-Gleichung davon ausgegangen sind, dass das System aus genau einer homogenen Phase besteht. Wir bezeichnen eine Phase als homogen, wenn in ihr die intensiven Zustandsgrößen (z.B. ρ, T, p, ...) überall denselben Wert haben. Diese Annahme ist für T < Tc falsch. In dem gestrichelten Bereich der Abbildung 4.7 liegt vielmehr ein Zwei-PhasenBereich vor. Flüssigkeit und Gas stehen miteinander im Gleichgewicht. man hat hier die van der Waals-Isotherme durch eine Parallele zur V-Achse zu ersetzen, und zwar so, dass die in der Skizze angedeuteten Flächen A und B gleich sind. Man nennt dies die MaxwellKonstruktion. Bei der Temperatur T1, dem Druck p1 und dem Volumen V1 besteht das System aus einer homogenen Phase Flüssigkeit. Wird bei konstanter Temperatur das Volumen vergrößert, so bleibt der Druck konstant. Ein Teil der Flüssigkeit verdampft zu Gas. Bei V2 ist das gesamte System gasförmig, weitere Volumenvergrößerung führt dann zu einer Druckabnahme. 56 Virialentwicklung Phasenübergänge sind offensichtlich unstetiger Natur, wie wir am Übergang Flüssigkeit ↔ Gas gerade gesehen haben. Man kann deshalb nicht erwarten, dass exakte Zustandsgleichungen realer Gase einfache analytische Ausdrücke darstellen. Für eine angenäherte Beschreibung mussten wir ja bereits die Maxwell-Konstruktion zur van der Waals-Gleichung hinzunehmen. Man benutzt deshalb bisweilen die Reihenentwicklung nach der Teilchendichte: (4.70) p= NkT V 2 ⎪⎧ ⎪⎫ ⎛N⎞ ⎛N⎞ 1 + + B B ⎨ 1⎜ 2⎜ ⎟ ⎟ + ...⎬ ⎝V ⎠ ⎝V ⎠ ⎪⎩ ⎭⎪ Die sogenannten Virialkoeffizienten Bi drücken die Abweichung vom Verhalten des idealen Gases aus. Für das van der Waals-Gas findet man: (4.71) b a B1 = − 2 , N A N A kT ⎛ b ⎞ Bi = ⎜ ⎟ ⎝ NA ⎠ i für i ≥ 2 . 57 4.7 Folgerungen aus den Hauptsätzen Wir betrachten zunächst nur reversible Prozesse im geschlossenen System. Die Grundrelation der Thermodynamik lautet dann: TdS = dU − δ W (4.72) Einige wichtige Schlussfolgerungen ergeben sich bereits aus der Tatsache, dass dU und dS totale Differentiale sind. Das betrachtete System sei ein Gas. Als unabhängige Zustandsvariablen wählen wir T und V. S = S (T , V ) (4.73) U = U (T , V ) mit: (4.74) 1 p ⎛ ∂S ⎞ ⎛ ∂S ⎞ dS = ⎜ ⎟ dT + ⎜ ⎟ dV = dU + dV T T ⎝ ∂T ⎠V ⎝ ∂V ⎠T (4.75) ⎛ ∂U ⎞ ⎛ ∂U ⎞ dU = ⎜ ⎟ dT + ⎜ ⎟ dV ⎝ ∂T ⎠V ⎝ ∂V ⎠T Einsetzen von (4.75) in (4.74) ergibt: dS = (4.76) ⎤ 1 ⎛ ∂U ⎞ 1 ⎡⎛ ∂U ⎞ ⎜ ⎟ dT + ⎢⎜ ⎟ + p ⎥ dV T ⎝ ∂T ⎠V T ⎣⎝ ∂V ⎠T ⎦ da dS ein totales Differential ist, sind die Integrabilitätsbedingungen (3.3) erfüllt: ⎤ 1 ⎧⎪ ⎡ ∂ ⎛ ∂U ⎞ ⎤ ⎛ ∂p ⎞ ⎫⎪ 1 ⎡ ∂ ⎛ ∂U ⎞ ⎤ 1 ⎡⎛ ∂U ⎞ ⎜ ⎟ ⎥ = − 2 ⎢⎜ ⎟ + p ⎥ + ⎨⎢ ⎜ ⎟ ⎥ +⎜ ⎟ ⎬ ⎢ T ⎣ ∂V ⎝ ∂T ⎠V ⎦T T ⎣⎝ ∂V ⎠T ⎦ T ⎩⎪ ⎣ ∂T ⎝ ∂V ⎠T ⎦V ⎝ ∂T ⎠V ⎭⎪ Da auch dU ein totales Differential ist, d.h. die zweiten Ableitungen vertauschbar sind, vereinfacht sich der Ausdruck zu: (4.77) ⎛ ∂U ⎞ ⎛ ∂p ⎞ ⎜ ⎟ =T ⎜ ⎟ −p ⎝ ∂V ⎠T ⎝ ∂T ⎠V Die rechte Seite ist allein durch die Zustandsgleichung bestimmt. Das bedeutet, dass bei bekannter Wärmekapazität CV = ∂U/∂T (vgl. (3.33)) sich mit Hilfe von (4.77) die innere Energie U(T,V) allein aus der Zustandsgleichung herleiten lässt. Wir diskutieren kurz die Beispiele ideales Gas und van der Waals-Gas. Ideales Gas: (4.78) ⎛ ν R* ⎞ ⎛ ∂U ⎞ ⎛ ∂p ⎞ T p T = − = ⎜ ⎟− p = 0 ⎜ ⎟ ⎜ ⎟ ⎝ ∂V ⎠T ⎝ ∂T ⎠V ⎝ V ⎠ 58 Damit gilt in (4.75): (4.79) ⎛ ∂U ⎞ dU = ⎜ ⎟ dT = CV dT ⎝ ∂T ⎠V Hierbei haben wir das Ergebnis (3.33) verwendet. Die Gleichung (4.79) lässt sich sofort integrieren zu: (4.80) U = U (T ) = CV T + const. Wir haben also noch einmal bestätigt, dass die innere Energie des idealen Gases nicht vom Volumen abhängt, sondern nur von der Temperatur. Van der Waals-Gas Führt man die partielle Ableitung der Zustandsgleichung des van der Waals-Gases (4.63) durch, so erhält man aus (4.77): ν ⎛ ∂U ⎞ ⎜ ⎟ =a 2 V ⎝ ∂V ⎠T 2 (4.81) In diesem Fall ist die innere Energie nun volumenabhängig. Unter der Voraussetzung CV = const. ergibt die Integration von (4.75) nun: (4.82) U = U (T ,V ) = CV T − a ν2 V + const. Bevor wir weitere Folgerungen aus den Hauptsätzen formulieren, wollen wir noch zwei nützliche Theoreme herleiten. Zwischenbemerkung: Es gibt zwei einfache Theoreme, die im folgenden häufig verwendet werden, um partielle Ableitungen zueinander in Beziehung zu setzen. Man nehme an, dass eine Beziehung zwischen drei thermodynamischen Variablen x, y, z existiert mit der Form: f ( x, y , z ) = 0 (4.83) Wir können (4.83) z.B. entweder in der Form x = x(y,z) oder y = y(x,z) lösen. Die Differentiale dieser Funktionen sind: (4.84) (4.85) ⎛ ∂x ⎞ ⎛ ∂x ⎞ dx = ⎜ ⎟ dy + ⎜ ⎟ dz ⎝ ∂z ⎠ y ⎝ ∂y ⎠ z ⎛ ∂y ⎞ ⎛ ∂y ⎞ dy = ⎜ ⎟ dx + ⎜ ⎟ dz ⎝ ∂x ⎠ z ⎝ ∂z ⎠ x Einsetzen von (4.85) in (4.84) ergibt: 59 ⎤ ⎛ ∂x ⎞ ⎛ ∂x ⎞ ⎡⎛ ∂y ⎞ ⎛ ∂y ⎞ dx = ⎜ ⎟ ⎢⎜ ⎟ dx + ⎜ ⎟ dz ⎥ + ⎜ ⎟ dz ⎝ ∂z ⎠ x ⎦ ⎝ ∂z ⎠ y ⎝ ∂y ⎠ z ⎣⎝ ∂x ⎠ z (4.86) dies kann umgeschrieben werden zu: (4.87) ⎡⎛ ∂x ⎞ ⎛ ∂y ⎞ ⎛ ∂x ⎞ ⎤ ⎛ ∂x ⎞ ⎛ ∂y ⎞ dx = ⎜ ⎟ ⎜ ⎟ dx + ⎢⎜ ⎟ ⎜ ⎟ + ⎜ ⎟ ⎥ dz ⎝ ∂y ⎠ z ⎝ ∂x ⎠ z ⎣⎢⎝ ∂y ⎠ z ⎝ ∂z ⎠ x ⎝ ∂z ⎠ y ⎦⎥ Nur zwei von den drei Variablen sind unabhängig, wir wählen x und z. dann muss (4.87) wahr sein für alle dx und dz. Wenn dx ≠ 0 und dz = 0 ist, dann gilt: ⎛ ∂x ⎞ ⎛ ∂y ⎞ ⎜ ⎟ ⎜ ⎟ =1 ⎝ ∂y ⎠ z ⎝ ∂x ⎠ z (4.88) ⇒ ⎛ ∂x ⎞ 1 ⎜ ⎟ = ∂y ⎝ ∂y ⎠ z ⎛ ⎞ ⎜ ⎟ ⎝ ∂x ⎠ z Für den Fall dx = 0 und dz ≠ 0 folgt: ⎛ ∂x ⎞ ⎛ ∂y ⎞ ⎛ ∂x ⎞ ⎜ ⎟ ⎜ ⎟ +⎜ ⎟ = 0 ⎝ ∂y ⎠ z ⎝ ∂z ⎠ x ⎝ ∂z ⎠ y (4.89) Mit Hilfe von (4.88) können wir dies umschreiben in: ⎛ ∂x ⎞ ⎛ ∂y ⎞ ⎛ ∂z ⎞ ⎜ ⎟ ⎜ ⎟ ⎜ ⎟ = −1 ⎝ ∂y ⎠ z ⎝ ∂z ⎠ x ⎝ ∂x ⎠ y (4.90) Angewendet auf das ideale Gas bedeutet dies mit x = V, y = p und z = T: (4.91) ⎛ ∂V ⎞ V ⎜ ⎟ =− , p ⎝ ∂p ⎠T (4.92) T ⎛ ∂T ⎞ ⎜ ⎟ = , ⎝ ∂V ⎠ p V p ⎛ ∂p ⎞ ⎜ ⎟ = ⎝ ∂T ⎠V T ⎛ ∂V ⎞ ⎛ ∂T ⎞ ⎛ ∂p ⎞ ⎜ ⎟ ⎜ ⎟ ⎜ ⎟ = −1 ⎝ ∂p ⎠T ⎝ ∂V ⎠ p ⎝ ∂T ⎠V Als Folge des Ersten Hauptsatzes hatten wir in (3.35) gefunden: ⎡⎛ ∂U ⎞ ⎤ ⎛ ∂V ⎞ C p − CV = ⎢⎜ ⎟ + p⎥ ⎜ ⎟ ⎣⎝ ∂V ⎠T ⎦ ⎝ ∂T ⎠ p Daraus wird mit (4.77): (4.93) ⎛ ∂p ⎞ ⎛ ∂V ⎞ C p − CV = T ⎜ ⎟ ⎜ ⎟ ⎝ ∂T ⎠V ⎝ ∂T ⎠ p Die rechte Seite lässt sich durch die relativ leicht messbare Response-Funktion (vgl. Abschnitt 5.1) ausdrücken. 60 Wir erinnern an die Definition (2.18) des isobaren, thermischen Ausdehnungskoeffizienten: α= (4.94) 1 ⎛ ∂V ⎞ ⎜ ⎟ V ⎝ ∂T ⎠ p und definieren die isotherme (adiabatische) Kompressibilität: 1 ⎛ ∂V ⎞ κT (S ) = − ⎜ ⎟ V ⎝ ∂p ⎠T ( S ) (4.95) Mit (4.88) und (4.94) gilt: 1 1 ⎛ ∂T ⎞ = ⎜ ⎟ = ∂V Vα ⎞ ⎝ ∂V ⎠ p ⎛ ⎜ ⎟ ⎝ ∂T ⎠ p (4.96) aus (4.92) und (4.95) folgt dann: ⎛ ∂V ⎞ ⎛ ∂T ⎞ ⎛ ∂p ⎞ 1 ⎛ ∂p ⎞ = −1 ⎜ ⎟ ⎜ ⎟ ⎜ ⎟ = −κ T ⎜ α ⎝ ∂T ⎟⎠V ⎝ ∂p ⎠T ⎝ ∂V ⎠ p ⎝ ∂T ⎠V (4.97) ⇔ α ⎛ ∂p ⎞ ⎜ ⎟ = ⎝ ∂T ⎠V κ T Eingesetzt in (4.93) ergibt dies: (4.98) C p − CV = TV α 2 κT Die mechanische Stabilität des Systems erfordert κT ≥ 0, und daher können wir auch an dieser Gleichung Ablesen, dass Cp > CV gelten muss, was wir ja schon in Abschnitt 3.3 diskutiert hatten. TdS-Gleichungen Wir haben bisher T und V als unabhängige Zustandsvariablen vorausgesetzt. Experimentelle Bedingungen können jedoch T und p bzw. V und p bequemer messbar erscheinen lassen. Man hat dann die Zustandsfunktionen in dem betreffenden Variablensatz zu formulieren. Das wollen wir am Beispiel der Entropie demonstrieren. Wir leiten die sogenannten TdSGleichungen ab. 1) S = S (T , V ) Das ist der bereits diskutierte Fall. Setzt man (4.77) in (4.76) ein und nutzt (4.97), so erhält man: 61 TdS = CV dT + T (4.99) α dV κT Wie bei der inneren Energie, erfordert auch die Berechnung der Entropie nur die Kenntnis der thermischen Zustandsgleichung (→ α, κT) und die Kenntnis von CV. 2) S = S (T , p ) V = V (T , p ) ⇒ ⎛ ∂V ⎞ ⎛ ∂V ⎞ dV = ⎜ ⎟ dp ⎟ dT + ⎜ ⎝ ∂T ⎠ p ⎝ ∂p ⎠T Dies setzen wir in (4.76) ein: ⎡⎛ ∂U ⎞ ⎤ ⎛ ∂V ⎞ ⎛ ∂U ⎞ TdS = ⎜ ⎟ dT + ⎢⎜ ⎟ + p⎥ ⎜ ⎟ dT + ⎝ ∂T ⎠V ⎣⎝ ∂V ⎠T ⎦ ⎝ ∂T ⎠ p ⎡⎛ ∂U ⎞ ⎤ ⎛ ∂V ⎞ + ⎢⎜ ⎟ dp ⎟ + p⎥ ⎜ ⎣⎝ ∂V ⎠T ⎦ ⎝ ∂p ⎠T mit (3.34), (4.97) und (4.96) folgt dann: ⎛ ∂p ⎞ ⎛ ∂V ⎞ TdS = C p dT + T ⎜ ⎟ dp = ⎟ ⎜ ⎝ ∂T ⎠V ⎝ ∂p ⎠T ⎛α ⎞ = C p dT + T ⎜ ⎟ ( −V κ T ) dp ⎝ κT ⎠ Damit lautet die TdS-Gleichung in den Variablen T und p: TdS = C p dT − TV α dp (4.100) 3) S = S (V , p ) T = T (V , p ) ⇒ ⎛ ∂T ⎞ ⎛ ∂T ⎞ dT = ⎜ ⎟ dp ⎟ dV + ⎜ ⎝ ∂V ⎠ p ⎝ ∂p ⎠V Einsetzen in (4.99): TdS = CV dT + T α ⎛ ∂p ⎞ dV = CV dT + T ⎜ ⎟ dV κT ⎝ ∂T ⎠V ergibt: (4.101) ⎡ ⎛ ∂T ⎞ ⎛ ∂T ⎞ ⎛ ∂p ⎞ ⎤ TdS = CV ⎜ ⎟ dp + ⎢CV ⎜ ⎟ +T ⎜ ⎟ ⎥ dV ⎝ ∂T ⎠V ⎥⎦ ⎢⎣ ⎝ ∂V ⎠ p ⎝ ∂p ⎠V 62 Mit (4.97) folgt: ⎛ ∂T ⎞ κT , CV ⎜ ⎟ = CV α ⎝ ∂p ⎠V (4.102) ⎡ ⎛ ∂T ⎞ ⎛ ∂p ⎞ ⎤ ⎛ ∂T ⎞ ⎡ ⎛ ∂p ⎞ ⎛ ∂V ⎞ ⎤ ⎢CV ⎜ ⎟ +T ⎜ ⎟ ⎥=⎜ ⎟ ⎢CV + T ⎜ ⎟ ⎜ ⎟ ⎥= ⎝ ∂T ⎠V ⎦⎥ ⎝ ∂V ⎠ p ⎢⎣ ⎝ ∂T ⎠V ⎝ ∂T ⎠ p ⎦⎥ ⎣⎢ ⎝ ∂V ⎠ p Cp ⎛ ∂T ⎞ = Cp ⎜ ⎟ = ⎝ ∂V ⎠ p V α wobei wir in den letzten beiden Schritten (4.93) und (4.94) benutzt haben. Damit ergibt sich die letzte TdS-Gleichung: TdS = CV (4.103) κT 1 dp + C p dV α Vα Wertet man diese TdS-Gleichungen speziell für adiabatisch-reversible Prozesse (dS = 0) aus, so ergeben sich einige weitere nützliche Relationen. Aus (4.99) folgt: CV κ T ⎛ ∂V ⎞ ⎜ ⎟ =− Tα ⎝ ∂T ⎠ S (4.104) und aus (4.100): Cp ⎛ ∂p ⎞ ⎜ ⎟ = ⎝ ∂T ⎠ S TV α (4.105) Kombiniert ergibt dies: 1 ⎛ ∂p ⎞ ⎛ ∂T ⎞ ⎛ ∂p ⎞ = −⎜ ⎟ ⎜ ⎟ = −⎜ ⎟ = CV V κ T ⎝ ∂T ⎠ S ⎝ ∂V ⎠ S ⎝ ∂V ⎠ S V κ S Cp Damit folgt: (4.106) γ= Cp CV = κT κS Mit (4.93) können diese Gleichungen auch noch nach Cp und CV alleine aufgelöst werden. 63 5 Thermodynamische Potentiale 5.1 Natürliche Zustandsvariablen Für reversible Zustandsänderungen, die wie wir nun wissen, faktisch quasistatisch als Prozesse zwischen Gleichgewichtszuständen ablaufen müssen (denn der Übergang ins Gleichgewicht ist irreversibel), lautet die Grundrelation der Thermodynamik in der allgemeinsten Form (s. (4.31)): m α i =1 j =1 dU = TdS + ∑ Fi dqi + ∑ μ j dN j (5.1) Hier ist also r r U = U S , q, N ( (5.2) ) r r Speziell für Gase mit F , q → {− p,V } : { } α dU = TdS − pdV + ∑ μ j dN j (5.3) j =1 r U = U S ,V , N ( (5.4) ) Da dU ein totales Differential ist, kann man die innere Energie U in gleicher Form auch als die Erzeugende der abhängigen Variablen auffassen. An (5.3) liest man beispielsweise direkt ab: (5.5) ⎛ ∂U ⎞ T =⎜ ⎟ ⎝ ∂S ⎠V , Nr ; ⎛ ∂U ⎞ − p=⎜ ⎟ ⎝ ∂V ⎠T , Nr ⎛ ∂U ⎜ ∂N ⎝ j μj = ⎜ ; ⎞ ⎟⎟ ⎠ S ,V , Ni ,i≠ j Die experimentell wichtigen Response-Funktionen ergeben sich aus den zweiten Ableitungen: −1 (5.6) ⎡⎛ ∂S ⎞ ⎤ rev T ⎛ ∂ 2U ⎞ ⎛ ∂T ⎞ = = ⎢ ⎜ 2⎟ r ⎜ ⎟ ⎜ ⎟ ⎥ = CV ⎝ ∂S ⎠V , N ⎝ ∂S ⎠V , Nr ⎣⎝ ∂T ⎠V , Nr ⎦ (5.7) ⎡⎛ ∂ 2U ⎞ ⎤ ⇒ CV = T ⎢⎜ 2 ⎟ ⎥ ⎢⎣⎝ ∂S ⎠V , Nr ⎥⎦ −1 Die zweite Ableitung der inneren Energie nach dem Volumen führt auf die adiabatische Kompressibilität: 64 ⎛ ∂ 2U ⎞ 1 ⎛ ∂p ⎞ ⎜ 2 ⎟ r = −⎜ ⎟ r = ⎝ ∂V ⎠ S , N V κ S ⎝ ∂V ⎠ S , N (5.8) 1 ⇒ κS = V (5.9) ⎡⎛ ∂ 2U ⎞ ⎤ ⎢⎜ 2 ⎟ ⎥ ⎢⎣⎝ ∂V ⎠ S , Nr ⎥⎦ −1 Weitere nützliche Relationen ergeben sich noch aus der Tatsache, dass dU ein totales Differential ist, d.h. aus den entsprechenden Integrabilitätsbedingungen: ⎛ ∂T ⎞ ⎛ ∂μi ⎞ ⎛ ∂T ⎞ ⎛ ∂p ⎞ ⎟ r =⎜ ⎜ ⎟ r = −⎜ ⎟ r ; ⎜ ⎟ ⎝ ∂V ⎠ S , N ⎝ ∂S ⎠V , N ⎝ ∂Ni ⎠V , S , N j , j≠i ⎝ ∂S ⎠V , Nr (5.10) Diese Beziehungen werden Maxwell-Relationen genannt. Die Gleichungen (5.5) bis (5.7) machen klar, dass das gesamte Gleichgewichtsverhalten des Systems eindeutig festgelegt ist, z.B. auch die Zustandsgleichungen, sobald r r U = U S , q, N ( (5.11) ) bekannt ist. Eine Größe, die so etwas leistet nennt man ein thermodynamisches Potential Dessen unabhängige Zustandsvariablen heißen natürliche Variablen. { } r Die natürlichen Variablen der inneren Energie sind also S , q, N , und speziell für das Gas r S ,V , N . Die Bezeichnung Potential rührt von einer formalen Analogie mit dem Potential der Klassischen Mechanik her. Dort erhält man die Komponenten der Kräfte direkt als erste Ableitungen des Potentials nach den Koordinaten (vgl. (3.4),(3.5)). Von natürlichen Variablen eines thermodynamischen Potentials spricht man genau dann, wenn sich die entsprechenden abhängigen Variablen direkt durch Ableiten der Potentiale ergeben. Das ist nach (5.5) bei der inneren Energie U genau dann der Fall, wenn wir sie für ein Gas als Funktion von S, V und N darstellen. Das sind die Variablen, in denen die differentiellen Eigenschaften von U besonders einfach und vollständig sind. Es ist daher die kalorische Zustandsgleichung (vgl. Abschnitt 3.2): { (5.12) } r U = U T ,V , N ( ) kein geeignetes thermodynamisches Potential. Wegen (5.13) ⎛ ∂U ⎞ ⎛ ∂U ⎞ ⎛ ∂p ⎞ ⎜ ⎟ = CV ; ⎜ ⎟ =T⎜ ⎟ −p ⎝ ∂T ⎠V ⎝ ∂V ⎠T ⎝ ∂T ⎠V 65 folgen die abhängigen Zustandsvariablen S und p nicht unmittelbar aus den ersten Ableitungen von U. Es gibt weitere Gesichtspunkte, die die natürlichen Variablen auszeichnen. So werden wir später Gleichgewichtsbedingungen für thermodynamische Systeme formulieren, und zwar in dem Sinne, dass in Systemen, in denen die natürlichen Variablen konstant gehalten werden, alle irreversiblen Prozesse so ablaufen, dass das thermodynamische Potential im Gleichgewicht extremal wird. Die Einführung anderer thermodynamischer Potentiale, wie wir sie im nächsten Abschnitt durchführen, erfüllt dann lediglich den Zweck, andere Energiefunktionen (Enthalpie, freie Energie, etc.) zu finden, die in anderen Variablensätzen ähnlich einfach sind wie U als r r Funktion von S , q , N . { } Löst man die Grundrelation (5.1) nach dS auf, (5.14) dS = ( 1 1 m 1 α dU − ∑ Fi dqi − ∑ μ j dN j T T i =1 T j =1 ) r r so erkennt man, dass auch S = S U , q , N ein thermodynamisches Potential darstellt. 5.2 Legendre-Transformationen Ein Nachteil beim Gebrauch der inneren Energie U als thermodynamisches Potential ist offensichtlich. Die natürlichen Variablen sind sehr unbequem, da z.B. die Entropie S nicht leicht zu kontrollieren ist. Man führt deshalb, je nach den experimentellen Randbedingungen, andere thermodynamische Potentiale ein, die als natürliche Variablen gerade solche Größen verwenden, die dem Experiment direkter zugänglich sind. Der Übergang von einem Variablensatz zum anderen erfolgt mit Hilfe der aus der Klassischen Mechanik bekannten Legendre-Transformation. Diese wenden wir auf die innere Energie U an, wobei wir parallel stets das Gas als spezielle Anwendung diskutieren wollen. (1) Freie Energie: (5.15) r r F = F T , q, N r Gas : F = F T , V , N ( ( ) ) Damit ist der Buchstabe F unglücklicherweise doppelt belegt. F bedeutet die freie Energie, bzw. dF das totale Differential der freien Energie, die schon bekannten Fi sind nach wie vor r die Komponenten der generalisierten Kräfte, bzw. F ist die Gesamtheit der generalisierten r Kräfte: F = { F1 , F2 ,..., Fm } . Die ursprünglich, d.h. in Bezug auf U, unabhängige Variable S soll nun durch die Temperatur T ersetzt werden: (5.16) ⎛ ∂U ⎞ F =U − S ⎜ ⎟ = U − TS ⎝ ∂S ⎠ qr , Nr 66 Das totale Differential dF ergibt sich mit (5.1) zu: (5.17) dF = dU − d (TS ) = dU − S dT − T dS (5.18) dF = − S dT + ∑ Fi dqi + ∑ μ j dN j m α i =1 j =1 Dies bedeutet speziell für das Gas: α (5.19) dF = − S dT − p dV + ∑ μ j dN j j =1 Die natürlichen Variablen der freien Energie sind demnach (5.20) r r Gas : T ,V , N {T , qr, N } ; { } Die abhängigen Zustandsgrößen ergeben sich unmittelbar aus den ersten partiellen Ableitungen: (5.21) ⎛ ∂F ⎞ −S = ⎜ ⎟ ⎝ ∂T ⎠ qr , Nr ⎛ ∂F Fj = ⎜ ⎜ ∂q ⎝ j ; ⎞ ⎟⎟ ⎠T , Nr ,qi ,i≠ j Dies bedeutet speziell für das Gas: (5.22) ⎛ ∂F ⎞ −S = ⎜ ⎟ ⎝ ∂T ⎠V , Nr ⎛ ∂F ⎞ − p=⎜ ⎟ ⎝ ∂V ⎠T , Nr ; Ferner gilt z.B. die Maxwell-Relation: ⎛ ∂S ⎞ ⎛ ∂p ⎞ ⎜ ⎟ r =⎜ ⎟ ⎝ ∂V ⎠T , N ⎝ ∂T ⎠V , Nr (5.23) (2) Enthalpie r r H = H S, F, N r Gas : H = H S , p, N ( ( (5.24) ) ) r Ausgehend von U sollen nun die generalisierten Koordinaten q mit den generalisierten r Kräften F vertauscht werden: (5.25) m m ⎛ ∂U ⎞ H = U − ∑ qi ⎜ U qi Fi = − ⎟ r ∑ q ∂ i =1 i = 1 ⎝ i ⎠ S , N , q j ≠i Dies bedeutet speziell für das Gas: 67 H = U + pV (5.26) Zur Berechnung des totalen Differentials dH benutzen wir wieder (5.1): m dH = dU − ∑ ( dqi Fi + qi dFi ) i =1 (5.27) ⇒ m α i =1 j =1 dH = T dS − ∑ qi dFi + ∑ μ j dN j Speziell für das Gas: α dH = T dS + V dp + ∑ μ j dN j (5.28) j =1 Die natürlichen Variablen der Enthalpie sind also: r r r Gas : S , p, N {S , F , N } ; (5.29) { } Da auch H ein thermodynamisches Potential darstellt, ergeben sich die abhängigen Zustandsvariablen direkt aus den ersten partiellen Ableitungen: (5.30) ⎛ ∂H ⎞ T =⎜ ⎟ ⎝ ∂S ⎠ Fr , Nr ; ⎛ ∂H ⎞ qi = − ⎜ ⎟ ⎝ ∂Fi ⎠ S , Nr , F j , j≠i Die zweite Gleichung lautet für das Gas: (5.31) ⎛ ∂H ⎞ V =⎜ ⎟ ⎝ ∂p ⎠ S , Nr Es folgt für das Gas die Maxwell-Relation: (5.32) ⎛ ∂T ⎞ ⎛ ∂V ⎞ ⎜ ⎟ r =⎜ ⎟ ⎝ ∂p ⎠ S , N ⎝ ∂S ⎠ p , Nr (3) Gibbsche (freie) Enthalpie: (5.33) r r G = G T, F, N r Gas : G = G T , p, N ( ( ) ) r r Ausgehend von U sollen nun S und q gegen T und F vertauscht werden: 68 m ⎛ ∂U ⎞ ⎛ ∂U ⎞ G =U − S ⎜ qi ⎜ − ⎟ ∑ ⎟r r ⎝ ∂S ⎠ q , N i =1 ⎝ ∂qi ⎠ S , Nr ,q j≠i (5.34) m = U − TS − ∑ qi Fi i =1 Dies bedeutet speziell für das Gas: G = U − TS + pV (5.35) Zur Berechnung des totalen Differentials dG benutzen wir wieder (5.1): m dG = dU − TdS − SdT − ∑ ( dqi Fi + qi dFi ) i =1 (5.36) ⇒ m α i =1 j =1 dG = − S dT − ∑ qi dFi + ∑ μ j dN j Speziell für das Gas: (5.37) α dG = − S dT + V dp + ∑ μ j dN j j =1 Die natürlichen Variablen der Gibbschen Enthalpie sind: (5.38) r r r Gas : T , p, N {T , F , N } ; { } Da auch G ein thermodynamisches Potential darstellt, ergeben sich die abhängigen Zustandsvariablen direkt aus den ersten partiellen Ableitungen: (5.39) ⎛ ∂G ⎞ S = −⎜ ⎟ ⎝ ∂T ⎠ Fr , Nr ; ⎛ ∂G ⎞ qi = − ⎜ ⎟ ⎝ ∂Fi ⎠ S , Nr , F j , j≠i Die zweite Gleichung lautet für das Gas: (5.40) ⎛ ∂G ⎞ V =⎜ ⎟ ⎝ ∂p ⎠T , Nr Es folgt für das Gas aus (5.37) noch die Maxwell-Relation: (5.41) ⎛ ∂S ⎞ ⎛ ∂V ⎞ −⎜ ⎟ = ⎜ ⎟ r ⎝ ∂p ⎠T , N ⎝ ∂T ⎠ p , Nr U, F, G und H sind die wichtigsten thermodynamischen Potentiale. Eine Fülle von wichtigen Beziehungen resultieren allein aus der Tatsache, dass dU, dF, dG und dH totale Differentiale sind. Zum Schluss seien die soeben abgeleiteten Beziehungen für das aus einer Komponente (α = 1) bestehende Gas noch in einer Tabelle zusammengefasst: 69 Potential natürliche Variablen totales Differential innere Energie U ( S ,V , N ) dU = TdS − pdV + μdN freie Energie F (T , V , N ) dF = − SdT − pdV + μ dN Enthalpie H ( S , p, N ) Gibbsche freie Enthalpie G ( T , p, N ) konjugierte Variablen (T , − p, μ ) ( − S , − p, μ ) dH = TdS + Vdp + μ dN dG = − SdT + Vdp + μ dN MaxwellRelationen ⎛ ∂T ⎞ ⎛ ∂p ⎞ ⎜ ⎟ = −⎜ ⎟ ⎝ ∂V ⎠ S , N ⎝ ∂S ⎠V , N ⎛ ∂S ⎞ ⎛ ∂p ⎞ ⎜ ⎟ =⎜ ⎟ ⎝ ∂V ⎠T , N ⎝ ∂T ⎠V , N (T , V , μ ) ⎛ ∂T ⎞ ⎛ ∂V ⎞ ⎜ ⎟ =⎜ ⎟ ⎝ ∂p ⎠ S , N ⎝ ∂S ⎠ p , N ( − S ,V , μ ) ⎛ ∂S ⎞ ⎛ ∂V ⎞ −⎜ ⎟ = ⎜ ⎟ ⎝ ∂p ⎠T , N ⎝ ∂T ⎠ p , N Tabelle 5.1. Thermodynamische Potentiale Ohne weiteren Beweis wollen wir noch festhalten, dass alle thermodynamischen Potentiale extensive Zustandsgrößen sind. Für Gase gelten daher die folgenden Homogenitätsrelationen: (5.42) r r U λ S , λV , λ N = λU S ,V , N r r S λU , λV , λ N = λ S U , V , N r r F T , λV , λ N = λ F T ,V , N r r H λ S , p, λ N = λ H S , p, N r r G T , p, λ N = λ G T , p, N ( ( ( ( ( ) ) ( ( ) ) ) ( ( ( ) ) ) ) ) Wie bereits festgestellt, sind V, S und N extensive Größen, während T, p und µ intensive Größen sind. 70 5.3 Die Gibbs-Duhem-Relation Nach der Homogenitätsrelation (5.42) gilt: r r r r λ G (T , F , N ) = G (T , F , λ N ) (5.43) Wir differenzieren beide Seiten nach λ und setzen λ=1: r r r r d G T, F, N = G T, F,λN λ =1 dλ ⎛ α ⎛ ∂ ( λ N j ) ⎟⎞ ∂G ⎞ ⎜ ⎟ = ∑⎜ ∂λ ⎟⎟ ⎜⎜ j =1 ⎝⎜ ∂ ( λ N j ) ⎠⎟T , Fr , N i≠ j ⎝ ⎠ λ =1 ( (5.44) ) ( ) ⎛ α ⎛ ⎞ ∂G ⎞ ⎜ ⎜ ⎟ Nj⎟ = ∑ ⎜⎜ j =1 ⎜ ∂ ( λ N ) ⎟ r ⎟⎟ j ⎠ T , F , Ni≠ j ⎝ ⎝ ⎠λ =1 und es ergibt sich: α G = ∑μjNj (5.45) j =1 Diese Gleichung wird Gibbs-Duhem-Relation genannt. Für ein ein-komponentiges System (j = 1) gilt: μ= (5.46) G , N d.h. das chemische Potential ist die Gibbsche freie Enthalpie pro Molekül! Mit (5.47) folgt für das totale Differential: (5.47) α α j =1 j =1 dG = ∑ μ j dN j + ∑ N j d μ j Kombiniert mit (5.37) ergibt sich: α (5.48) − S dT + V dp = ∑ N j d μ j j =1 Diese Gleichung besagt, dass wenn sich die Temperatur und der Druck eines Systems ändern, damit auch eine entsprechende Änderung der chemischen Potentiale der verschiedenen Gaskomponenten einhergeht. Auch Gleichung (5.48) wird von verschiedenen Autoren GibbsDuhem-Relation genannt. Die Gibbs-Duhem-Relation gilt allgemein. 71 5.4 Mischungsentropie Wir wollen als nächstes die Entropieänderung berechnen, die auftritt, wenn man zwei ideale Gase, die aus unterschiedlichen Molekülsorten bestehen, miteinander mischt. (a) Die beiden Gase seien durch eine Wand getrennt. In jeder Kammer herrsche gleicher Druck p und gleiche Temperatur T. Es gelten die Zustandsgleichungen: pV1 = N1 kT pV2 = N 2 kT Thermodynamische Potentiale sind extensiv, deshalb gilt für die innere Energie U: U1 (T , N1 ) + U 2 (T , N 2 ) = U (T ) = CV ( N1 + N 2 ) T Hier haben wir mit CV die Wärmekapazität pro Teilchen eingeführt CV = CV N und wir haben angenommen, dass beide Teilchensorten das selbe CV haben. (b) Wir nehmen nun die Trennungswand heraus. Es setzt eine irreversible Durchmischung der beiden Gase bis zur homogenen Zusammensetzung des Gesamtsystems ein. Sonst passiert nichts! Es findet keine Arbeitsleistung und kein Wärmeaustausch statt. Nach dem Ersten Hauptsatz ist dann U = const. = U (T ) = CV ( N1 + N 2 ) T Da Temperatur und Druck konstant bleibt, gilt für die Zustandsgleichung p (V1 + V2 ) = ( N1 + N 2 )kT p, T, und U des Gesamtsystems ändern sich also nicht, möglicherweise aber die Entropie S. Über diese können wir nach (4.27) folgende Aussage machen: ΔS = S ( b ) − S ( a ) ≥ (5.49) b δQ ∫a T =0 Explizit können wir ΔS nur mit Hilfe eines reversiblen Ersatzprozesses ausrechnen: ( b1 ) ( b ) Das Gasgemisch befinde sich in zwei ineinandergeschobenen Behältern, die zu jeweils einer Seite durch eine semipermeable Wand abgeschlossen sind. Die linke Seite ist für die Teilchensorte 2 undurchlässig, während die Teilchen der Sorte 1 ungehindert hindurch diffundieren können. An der rechten Seite ist es umgekehrt. 72 ( b2 ) Wir ziehen nun die beiden Behälter quasistatisch auseinander: Dadurch werden die Gase reversibel entmischt, wobei jede Gassorte stets das konstante Volumen V beibehält. Die semipermeablen Wände bewegen sich widerstandslos durch das Gas. Die Entmischung bedarf also keiner Arbeitsleistung: ΔW = 0. Die Temperatur T ändert sich nicht, d.h. auch ΔU = 0, so dass auch ΔQ = 0 gilt. Der Prozess verläuft reversibel, daher ist: ΔSb1 →b2 = 0 Die Drucke haben sich geändert: (5.50) ( b3 ) ( a ) (a) ⎫ p1V = N1kT = pV1 ⎪ Vi ⇒ = ; p p ⎬ i (a) V p2V = N 2 kT = pV2 ⎪⎭ Dalton −Gesetz i = 1, 2 durch isotherme, reversible Kompressionen der beiden Teilsysteme, wie in Gleichung (4.33) beschrieben, führen wir das System in den Zustand (a) zurück. Dazu bringen wir das Gesamtsystem in Kontakt mit dem Wärmebad WB(T). Da der Prozess isotherm verläuft, ist ΔU = 0. Es muss jedoch Arbeit an den beiden Teilsystemen geleistet werden: V1 V2 ΔW = − ∫ p1 (V ') dV ' − ∫ p2 (V ') dV ' = V V V1 = − N1kT ∫ V V2 dV ' V' − N 2 kT ∫ dV ' V' = V V V ⎞! ⎛ = −kT ⎜ N1 ln 1 + N 2 ln 2 ⎟ =− ΔQrev V V ⎠ ⎝ Dies entspricht einer Entropieänderung: ΔSb2 →b3 = 1 ΔQrev T Die gesamte Entropieänderung von (b3) = (a) nach (b1) = (b) beträgt also: (5.51) ⎛ V V ⎞ ΔS = S ( b ) − S ( a ) = k ⎜ N1 ln + N 2 ln ⎟ . V1 V2 ⎠ ⎝ die Entropie hat also zugenommen! Aus der Zunahme der Entropie bei der Mischung von Gasen kann man vielleicht am besten die vereinfachende aber populäre Aussage "Entropie entspricht Unordnung" verstehen, denn, dass die "Ordnung" der beiden Gase durch die Durchmischung abgenommen hat, ist unmittelbar verständlich. 73 Die Verallgemeinerung der Aussage von zwei auf α verschiedene Gase lautet: Mischungsentropie: α ΔS = k ∑ N j ln (5.52) j =1 V Vj Man beachte, dass jedes Gas von jedem anderen unterscheidbar seien muss. Die Entropien vor und nach der Mischung unterscheiden sich nur durch die Volumina, die den Gassorten zur Verfügung stehen. Vor der Vermischung sind es die Teilvolumina Vj, hinterher ist es für alle Gassorten das Gesamtvolumen V. Für gleichartige Gase scheint die Gleichung (5.52) zu einem Widerspruch zu führen. Man betrachte z.B. zwei gleiche Gase mit N1 = N 2 = N ; 2 V1 = V2 = V . 2 dann ergibt (5.52) für die Durchmischung der beiden gleichen Gase ΔS = Nk ln 2 (5.53) obwohl die Entropie sich natürlich nicht geändert haben kann. Dies nennt man das Gibbsche Paradoxon. Der Widerspruch existiert in Wirklichkeit aber nicht, da (5.52) für gleiche Gase nicht gilt. Für solche Gase muss man wie folgt argumentieren: (5.54) S nachher ⎫ ⎛ V N⎞ Svorher = 2S ⎜ T , , ⎟ ⎪ ⎝ 2 2⎠ ⎪ ⎬ ⇒ ΔS = 0 ⎛ V N ⎞⎪ = S (T , V , N ) = 2 S ⎜ T , , ⎟ ⎝ 2 2 ⎠ ⎪⎭ Man muss also auch nach der Durchmischung von einer einzigen Gassorte ausgehen. Für gleiche Gassorten gibt es keine semipermeablen Wände, so dass der skizzierte Ersatzprozess zur Entmischung nicht durchführbar ist. 5.5 Gleichgewichtsbedingungen Die thermodynamischen Potentiale sind als Funktion ihrer natürlichen Variablen insbesondere dadurch ausgezeichnet, dass man durch Konstanthalten gewisser Variabler und Verändern der anderen sehr leicht erkennen kann, auf welche Art Energieaustausch mit der Umgebung erfolgt. Nehmen wir als Beispiel die innere Energie eines Gases: dU = TdS − pdV ⇒ 1) S = const.: dU = − pdV Arbeit 2) V = const.: dU = TdS Wärme Die Grundrelation der Thermodynamik 74 m α i =1 j =1 TdS ≥ δ Q = dU − ∑ Fi dqi −∑ μ j dN j lässt sich mit Hilfe der thermodynamischen Potentiale für die verschiedenen Kontakte von System und Umgebung in besonders einfache Formen bringen. Die Potentiale geben uns die Möglichkeit, die Entwicklung eines thermodynamischen Systems zum Gleichgewicht hin und das Gleichgewicht selbst zu beschreiben. Die verschiedenen Potentiale sind dabei verschiedenen experimentellen Situationen angepasst. Wir betrachten insbesondere die für die Meteorologie wichtigeren Spezialfälle. 5.5.1 Isolierte Systeme Diese Situation haben wir bereits im Zusammenhang mit dem Zweiten Hauptsatz diskutiert. Isolierte Systeme sind definiert durch: dU = 0 (δ Q = 0); (5.55) dqi = 0; dN j = 0 Dies bedeutet dS ≥ 0 dS = 0 (5.56) im Gleichgewicht Solange in einem isolierten System noch reale, irreversible Prozesse ablaufen, geschehen diese stets so, dass die Entropie dabei zunimmt. Die Entropie ist maximal im stationären Gleichgewicht. r r Solange wir nur den Gleichgewichtswert der Entropie S = S U , q , N ( ) zugrunde legen, können wir über (5.56) hinaus keine weiteren Schlußfolgerungen ziehen, da ja nach r r Voraussetzung U , q und N konstant sind. Wir erzeugen uns nun in einem Gedankenexperiment eine einfache Nicht-Gleichgewichtssituation, aus der sich weitere Informationen ableiten lassen. Das nach außen isolierte System (U = const., V = const., Nj = const.) werde durch eine Wand in zwei Teile zerlegt. Der Einfachheit halber nehmen wir an, dass das Gas in den Kammern aus nur einer Teilchensorte besteht (α = 1). Die Verallgemeinerung auf mehrere Komponenten ist nicht weiter schwierig. Die Wand sei beweglich und für Energie und Teilchen durchlässig! V1, V2 sowie U1, U2 und N1, N2 sind also noch variabel , allerdings unter den Randbedingungen: U = U1 + U 2 = const.; V = V1 + V2 = const.; N = N1 + N 2 = const.; Die Gesamtentropie ist additiv: S = S1 (U1 , V1 , N1 ) + S2 (U 2 , V2 , N 2 ) = S1 + S 2 Es gilt somit, da U, V, N konstant sind: 75 dU1 = −dU 2 ; dV1 = − dV2 ; dN1 = − dN 2 Die beiden Teilsysteme werden so lange reagieren, bis die Gleichgewichtsbedingung (5.56) erfüllt ist: 0 = dS = dS1 + dS2 = ⎛ ∂S ⎞ ⎪⎧⎛ ∂S ⎞ ⎪⎫ = ⎨⎜ 1 ⎟ −⎜ 2 ⎟ ⎬ dU1 + ∂ U ∂ U ⎩⎪⎝ 1 ⎠V1 , N1 ⎝ 2 ⎠V2 , N 2 ⎭⎪ ⎛ ∂S ⎞ ⎪⎧⎛ ∂S ⎞ ⎪⎫ + ⎨⎜ 1 ⎟ −⎜ 2 ⎟ ⎬ dV1 + ⎪⎩⎝ ∂V1 ⎠U1 , N1 ⎝ ∂V2 ⎠U2 , N 2 ⎪⎭ (5.57) ⎛ ∂S ⎞ ⎪⎧⎛ ∂S ⎞ ⎪⎫ + ⎨⎜ 1 ⎟ −⎜ 2 ⎟ ⎬ dN1 = N N ∂ ∂ 1 2 ⎝ ⎠ ⎝ ⎠ ⎪ U1 ,V1 U 2 ,V2 ⎭ ⎩⎪ ⎛ μ μ ⎞ ⎛1 1⎞ ⎛p p ⎞ = ⎜ − ⎟ dU1 + ⎜ 1 − 2 ⎟ dV1 + ⎜ − 1 + 2 ⎟ dN1 ⎝ T1 T2 ⎠ ⎝ T1 T2 ⎠ ⎝ T1 T2 ⎠ U1, V1 und N1 sind unabhängige Zustandsvariablen. Daher müssen die Klammern jede für sich verschwinden. Das Gleichgewicht ist also durch T1 = T2 = T ; (5.58) p1 = p2 = p ; μ1 = μ 2 = μ gekennzeichnet. – Wir können in dem Gedankenexperiment die Unterteilung weiter fortsetzen, um schließlich asymptotisch zu der Aussage zu kommen, dass in einem isolierten System im Gleichgewicht an allen Orten gleiche Temperatur, gleicher Druck und gleiches chemisches Potential vorliegen. 5.5.2 Geschlossenes System im Wärmebad ohne Arbeitsaustausch Damit ist im einzelnen gemeint: geschlossen ⇒ N j = const. ⇔ dN j = 0 im Wärmebad ⇒ T = const. ⇔ dT = 0 ohne Arbeitsaustausch ⇒ qi = const. ⇔ dqi = 0 Die Grundrelation lautet dann: TdS ≥ dU T = const. ⇒ TdS = d (TS ) ⇒ d (U − TS ) ≤ 0 Das bedeutet: (5.59) dF ≤ 0 dF = 0 im Gleichgewicht 76 Bei allen irreversiblen Prozessen, die unter den angegebenen Randbedingungen r r T = const.; q = const.; N = const. (5.60) noch ablaufen können, nimmt die freie Energie stets ab. F ist minimal im Gleichgewicht. 5.5.3 Geschlossenes System im Wärmebad bei konstanten Kräften Damit sind folgende Voraussetzungen gemeint: (5.61) dT = 0; dN j = 0; dFi = 0 ( einkompon. Gas : T = const.; N = const.; p = const.) Die Grundrelation lautet jetzt: (5.62) m ⎛ m ⎞ TdS = d (TS ) ≥ dU − ∑ Fi dqi = dU − d ⎜ ∑ Fi qi ⎟ i =1 ⎝ i =1 ⎠ m ⎛ ⎞ ⇒ d ⎜ U − ∑ Fi qi − TS ⎟ ≤ 0 i =1 ⎝ ⎠ Dies bedeutet: dG ≤ 0 (5.63) dG = 0 im Gleichgewicht Die Gibbsche freie Enthalpie G nimmt bei irreversiblen Prozessen, die unter obigen Bedingungen ablaufen, stets ab. Im Gleichgewicht ist G minimal! Ein ähnliches Gedankenexperiment wie eben wollen wir noch einmal durchführen. Diesmal befinde sich das System im Wärmebad, die Trennwand sei frei verschiebbar und durchlässig für Teilchen. Außerdem gilt die Zusatzbedingung p = const. in jeder Kammer. Das Experiment soll nun mit mehreren Teilchensorten durchgeführt werden. Die Nebenbedingungen (2) (2) N (1) = N j = const. ∀j ⇔ dN (1) j + Nj j = − dN j führen mit r r G = G T , p, N (1) + G T , p, N (2) = G1 + G2 ( ) ( ) auf den folgenden Ausdruck: 77 (5.64) ⎤ α ⎡⎛ ⎛ ∂G2 ⎞ ∂G1 ⎞ ⎥dN (1) − ⎜ (2) dG = ∑ ⎢⎜ (1) ⎟ ⎟ j = ⎜ ⎟ ⎜ ⎟ ⎢ ∂ ∂ N N r (2) ⎥ j =1 ⎝ j ⎠T , Fr , N (1) j ⎝ ⎠T , F , Ni ,i≠ j ⎦ i ,i ≠ j ⎣ α (2) ⎤ (1) = ∑ ⎡⎣ μ (1) j − μ j ⎦dN j = 0 j =1 Dies bedeutet, dass das chemische Potential (2) μ (1) = μj j = μj (5.65) im Gleichgewicht im ganzen System denselben Wert annimmt. Als Beispiel betrachte man die "Reaktion" A ↔ B bei konstanter Temperatur T und konstantem Druck p. Wir nehmen an, dass zunächst Na Moleküle von A und NB Moleküle B vorhanden sind. Die Gibbsche Freie Energie des Systems ist dann: G = µA N A + µB N B (5.66) Wenn man von einem geschlossenen System ausgeht, gilt: (5.67) N ges = N A + N B = const und wir können schreiben: (5.68) G = N ges ( x A µA + (1 − x A ) µB ) wobei (5.69) xA = NA N ν = A = A N A + N B N ges ν ges der Molenbruch von A ist (molare Fraktion von A am Gesamtsystem). Man nehme an, dass die Gibbsche Freie Energie des Gesamtsystems sich verhält, wie in der Abbildung 5.1 gezeigt. Wenn sich das System im Zustand K befindet, so besagt Gleichung (5.63), dass sich das System nach rechts, zum Zustand L entwickeln wird. G strebt dem Minimum zu, xA wird zunehmen und es gibt eine Netto-Reaktion von B nach A. Ebenso wird sich das System, wenn man von Punkt M ausgeht, nach links, ebenfalls zum Gleichgewichtspunkt L entwickeln und es gibt eine Netto-Reaktion von A nach B. Im Gleichgewicht findet dann keine NettoReaktion mehr statt und der Anteil der beiden Komponenten bleibt zeitlich konstant. 78 Abbildung 5.1. Gibbsche Freie Energie für ein geschlossenes System in dem die Reaktion A ↔ B abläuft als Funktion des Molenbruchs xA (bei konstanter Temperatur und konstantem Druck). [Seinfeld] 6 Phasenübergänge In der Atmosphäre sind die verschiedenen Aggregatsformen des Wassers sehr wichtig für den Energietransport, das Wettergeschehen und das Klima. Ständig verdunsten riesige Wassermassen aus den Ozeanen, kondensieren wieder in Wolken, es bilden sich Eisteilchen, Wolken verdunsten wieder usw. Um diese Prozesse zu verstehen, müssen wir uns eingehend mit der Thermodynamik der Phasenübergänge des Wassers beschäftigen. Die Ergebnisse des vorhergehenden Abschnitts enthalten bereits wichtige Aussagen, die wir im folgenden auf die Phasenübergänge erweitern und übertragen wollen. Wir hatten im vorhergehenden Abschnitt die Gleichgewichtsbedingungen für thermodynamische Systeme bestimmt. Im Rahmen von Gedankenexperimenten hatten wir hierfür das System in zwei fiktive Teilsysteme zerlegt und damit eine einfache Nichtgleichgewichtssituation geschaffen. Auf diese reagiert das System in gesetzmäßiger Weise und liefert damit Informationen über das Verhalten bestimmter Zustandsgrößen im Gleichgewicht. Eine solche Aufteilung des Systems realisieren wir im Folgenden ohne die bisherigen Trennwände durch verschiedene, nebeneinander existierende Phasen. Als Phasen bezeichnet man die möglichen, unterschiedlichen Zustandsformen einer makroskopischen Substanz, z.B. die verschiedenen Aggregatszustände: fest, flüssig, gasförmig. In den einzelnen Phasen können gewisse makroskopische Observable, wie z.B. die Teilchendichte, ganz unterschiedliche Werte annehmen. Als Meteorologen sind wir hauptsächlich an den drei Aggregatszuständen des Wassers interessiert und wir werden die Diskussion im Folgenden auf dieses System beschränken. Das Konzept der Phasenübergänge ist aber in der Physik auf viele andere thermodynamische Systeme übertragbar, wie z.B. Ferromagneten oder Supraleiter. Auch die Übergänge zwischen verschiedenen Kristallmodifikationen einer Substanz im festen Zustand können als Phasenübergänge beschrieben werden. 79 Wir beginnen unsere Diskussion mit der Betrachtung von nur zwei Phasen des Wassers (Indexe 1 und 2). Zunächst betrachten wir wieder das isolierte System: A) isoliertes System Genau wie in den Ausführungen von Abschnitt 5.5.1 gelten die selben Voraussetzungen: U = U1 + U 2 = const.; V = V1 + V2 = const.; N = N1 + N 2 = const. und die Gesamtentropie ist additiv: S = S (U1 , V1 , N1 ) + S (U 2 , V2 , N 2 ) = S1 + S 2 Es ergibt sich daher auch dieselbe Schlussfolgerung, dass im isolierten System im Gleichgewicht beide Phasen dieselbe Temperatur T, denselben Druck p und dasselbe chemische Potential µ haben. Diese Schlussfolgerungen lassen sich ohne weiteres – mathematisch mit Hilfe der Lagrangschen Multiplikatoren (s. z.B. Nolting) – auf π verschiedene Phasen (m = 1,2,...π) ausweiten (in der Meteorologie ist meist π = 3): In einem isolierten System haben im Gleichgewicht alle Phasen: 1) dieselbe Temperatur T, 2) denselben Druck p, 3) dasselbe chemische Potential µ. Wir wollen noch die für unsere Anwendungen wichtige Situation von Abschnitt 5.5.3 übertragen: B) Geschlossenes System mit p = const. und T = const. Wegen (5.63) gilt für ein System aus zwei Phasen: (6.1) Gleichgewicht ⇔ dG = 0 ; G ist minimal mit dem Ergebnis (vgl. (5.65)): μ1 = μ 2 = μ Wiederum kann das Ergebnis mit Hilfe der Lagrangschen Multiplikatoren auf π Phasen erweitert werden: In einem geschlossenen System mit p = const. und T = const. hat im Gleichgewicht in allen Phasen das chemische Potential denselben Wert. Auch eine Erweiterung auf mehrere Teilchensorten j (j = 1,2,...α) ist problemlos. Dann ist das chemische Potential jeder einzelnen Sorte j in den jeweiligen Phasen m (m = 1,2...π) gleich: (6.2) μ jm = μ j ∀m . 80 Abbildung 6.1. Phasendiagramm des Wassers. [Curry] 6.1 Phasendiagramm des Wassers Abbildung 6.1 zeigt das sogenannte Phasendiagramm des Wassers. Die Kurve, die den flüssigen vom gasförmigen Bereich trennt heißt Verdampfungskurve, die Trennung zwischen Gas und Festkörper heißt Sublimationskurve, die Trennung zwischen fest und flüssig heißt Schmelzkurve. Die drei Kurven treffen sich im Tripelpunkt. Dieser liegt für Wasser bei T0 = 0.0075°C und p0 = 6.11 hPa. Die Verdampfungskurve endet am kritischen Punkt. Dieser liegt für Wasser bei 374.2°C. Oberhalb dieser Temperatur ist keine Koexistenz der beiden Phasen Wasserdampf und Flüssigkeit mehr möglich. Der kritische Punkt entspricht dem kritischen Punkt aus dem p-V-Diagramm von Abschnitt 4.6. Es gilt die Gibbsche Phasenregel: (6.3) f = 2 +α −π wobei α die Zahl der Komponenten, π die Zahl der Phasen und f die Zahl der Freiheitsgrade des Systems ist (nicht zu verwechseln mit der Zahl der Freiheitsgrade s des Moleküls aus Abschnitt 3.2). Die Zahl der Freiheitsgrade f gibt an, wie viele unabhängig wählbare Variablen (z. B. Druck p und/oder Temperatur T) es gibt. So ist in den Einphasengebieten (fest, flüssig, gasförmig) des Phasendiagramms f = 2, weil dort eine Komponente 81 (Wasser) vorliegt, also α = 1 und nur eine Phase vorhanden ist, also π = 1. f = 2 bedeutet, dass beide Variablen Druck und Temperatur unabhängig gewählt werden können. Entlang der Koexistenzkurven ist f = 1, dort gibt es zwei koexistierende Phasen, es ist dann aber nur eine Variable frei wählbar, die andere ist dann festgelegt, z.B. p = p(T). Im Tripelpunkt schließlich ist π = 3, dort ist dann f = 0, es ist keine Variable frei wählbar, sondern es gibt genau ein (T0, p0), an dem alle drei Phasen im Gleichgewicht miteinander existieren können. Die Schmelzkurve verläuft fast parallel zur p-Achse, sie ist nur ganz leicht nach links geneigt: bei 1013 hPa ist die Schmelztemperatur genau 0°C, bei 20 MPa beträgt die Schmelztemperatur –1.52°C. Sehr häufig trifft man in der Atmosphäre auch den Fall der unterkühlten Flüssigkeit an, wenn sich mangels geeigneter Oberflächen und Eiskeimen keine Eisphase bilden kann (dieses Phänomen werden wir noch eingehender besprechen). Dann kann die Dampfdruckkurve über der Flüssigkeit auch für Temperaturen kleiner als T0 fortgesetzt werden (gestrichelte Linie in der Abbildung 6.1. 6.2 Dampfdruckkurve (Clausius-Clapeyron) Wir wollen nun das Gleichgewicht zwischen Flüssigkeit (f) und Dampf (g) und die Form der Verdampfungskurve des einkomponentigen Systems (also z.B. Wasser) genauer diskutieren. Abbildung 6.2. chemisches Potential der Flüssigphase und der Gasphase einer Substanz als Funktion der Temperatur. [Nolting] Wählt man p und T als Variable, dann gilt nach (6.2) im Gleichgewicht: (6.4) μ f (T , p ) = μ g (T , p ) 82 Aus dieser Beziehung muss sich im Prinzip eine Relation p = p(T) für die Zustände herleiten lassen, in denen Flüssigkeit und Dampf im Gleichgewicht stehen. Gilt (6.4) hingegen nicht, so folgt aus der Gibbs-Duhem-Relation (5.46), G ( T , p, N ) = N μ ( T , p ) , (6.5) dass sich das Gleichgewicht vollständig zu der Phase mit dem kleineren µ verlagert. Stabil ist jeweils die Phase mit minimaler Gibbscher Freier Enthalpie. Bei fester Teilchenzahl gilt nach (5.37): dG = − S dT + V dp = N d μ (T , p ) Wir betrachten eine Verschiebung (dp, dT) längs der Koexistenzlinie (Dampfdruckkurve). Dort ist wegen (6.4) d μ f (T , p ) = d μ g (T , p ) (6.6) und damit (6.7) − ( S f N f ) dT + (V f N f )dp = − ( S g N g )dT + (Vg N g )dp Damit erhalten wir die Steigung dp/dT der Dampfdruckkurve: dp ( S g N g ) − ( S f N f ) = dT (Vg N g ) − (V f N f ) (6.8) Üblicherweise bezieht man sich auf ein Mol statt durch die Anzahl der Moleküle zu teilen (Achtung Schreibweise spez. Größen: pro Mol oder pro kg!): dp sg − s f = dT vg − v f vg,f : sg,f : Molvolumina für Gas bzw. Flüssigkeit Entropien pro Mol. und definiert noch die molare Verdampfungswärme, auch molare Verdampfungsenthalpie, oder auch latente Wärme genannt: (6.9) lv = T ( sg − s f ) Diese Energiemenge wird beim Phasenübergang zur Überwindung der Kohäsionskräfte zwischen den Teilchen benötigt. Aus (6.8) wird damit die Clausius-Clapeyron-Gleichung (6.10) lv dp = dT T ( vg − v f ) 83 Bei der Ableitung von (6.8) und (6.10) mussten wir implizit Sg ≠ Sf und Vg ≠ Vf voraussetzen. Dies bedeutet wie man an Abbildung 6.2 sieht: μ g (T , p ) = μ f (T , p ) (6.11) ⎛ ∂μ f ⎞ ⎛ ∂μ g ⎞ ⎜ ⎟ ≠⎜ ⎟ ⎝ ∂T ⎠ p ⎝ ∂T ⎠ p ⎛ ∂μ f ⎞ ⎛ ∂μ g ⎞ ⎜ ⎟ ≠⎜ ⎟ ⎝ ∂p ⎠T ⎝ ∂p ⎠T ; Einen solchen Übergang Gas ↔ Flüssigkeit nennt man einen Phasenübergang erster Ordnung. Nur für einen solchen Übergang gilt die Clausius-Clapeyron-Gleichung. Um die Clausius-Clapeyron-Gleichung zu vereinfachen, können wir für unsere Zwecke das spezifische Volumen der Flüssigkeit vf gegen das spezifische Dampfvolumen vg vernachlässigen: (6.12) vg vf ⇒ l dp = v dT Tvg Mit dieser Vereinfachung können wir nun das ideale Gasgesetz anwenden: (6.13) pvg = RT Setzt man diesen Ausdruck in die Clausius-Clapeyron-Gleichung ein, dann erhält man: (6.14) l 1 dp = v2 p dT RT Strenggenommen ist die molare Verdampfungsenthalpie lv temperaturabhängig, lv = lv(T), doch in erster Näherung kann man, speziell für kleine Temperaturbereiche, lv als temperaturunabhängig annehmen. Dann kann Gleichung (6.14) integriert werden: (6.15) ⎛ l ⎞ ⎛ l ⎞ p = p0 exp ⎜ v ⎟ exp ⎜ − v ⎟ ⎝ RT ⎠ ⎝ RT0 ⎠ Wir erhalten also wieder einen typischen Exponentialausdruck, der die charakteristische Energie lv mit der kinetischen Energie der Moleküle (~ RT) in Beziehung setzt. Mit (6.15) haben wir somit in erster Näherung einen einfachen Ausdruck erhalten, der die Verdampfungskurve im Phasendiagramm beschreibt. Die Verdampfungsenthalpie Lv kann mit der Wärmemenge identifiziert werden, die benötigt wird, um bei konstantem Druck eine bestimmte Menge Wasser in Wasserdampf zu überführen. Sie stellt die Differenz der Enthalpien der beiden Phasen dar: (6.16) Lv = H g − H f Diese Beziehung lässt sich herleiten, wenn wir berücksichtigen, dass mit (6.9) und (4.28) gilt: g (6.17) Lv = T ( S g − S f ) = ∫ δ Qrev f 84 Da es sich um einen Prozess bei konstantem Druck handelt, gilt wegen Gleichung (3.69): δ Q = dH (6.18) und es folgt: g (6.19) Lv = ∫ dH = H g − H f f Der Phasenübergang von flüssig nach gasförmig bei konstanter Temperatur ist ein reversibler Prozess, denn beim Übergang erhöht sich zwar die Entropie der verdampften Wassermenge, gleichzeitig muss sich die verdampfende Wassermenge in einem Wärmebad befinden um die konstante Temperatur sicherzustellen. Diesem Wärmebad wird genau die Wärmemenge δQ entzogen, wodurch sich die Entropie des Wärmebads um genau dieselbe Entropiemenge erniedrigt. Die Gesamtentropie bleibt konstant, es handelt sich also um einen reversiblen Prozess. Natürlich müssen die üblichen Idealisierungen berücksichtigt werden: der Prozess muss als Folge von Gleichgewichtszuständen erfolgen. Analog zur Verdampfungsenthalpie Lv können wir auch noch die Schmelzenthalpie (Enthalpy of fusion) Lf und die Sublimationsenthalpie (Enthalpy of sublimation) Ls definieren, als diejenigen Wärmemengen, die benötigt werden, um eine Menge Substanz (z.B. Wasser) jeweils vom einen Aggregatszustand in den anderen zu überführen. Die drei Übergangsenthalpien stehen miteinander in Beziehung: (6.20) L f + Lv = Ls Es gelten die zu (6.9) analogen Gleichungen: (6.21) Ls = T ( S g − Si ) = H g − H i L f = T ( S f − Si ) = H f − H i . Sind Gasphase und die Flüssigkeit miteinander im Gleichgewicht, so wird der Dampfdruck, der sich einstellt, auch Sättigungsdampfdruck oder Gleichgewichtsdampfdruck genannt. In der Meteorologie wird speziell der Wasser-Dampfdruck mit dem Buchstaben e bezeichnet und der Wasser-Sättigungsdampfdruck mit es bezeichnet. Wir wollen uns dieser Nomenklatur im weiteren Verlauf der Vorlesung anschließen. Das Verhältnis (6.22) r= e es ist die relative Feuchte. Sie wird meist als Prozentwert angegeben (r × 100%). Es lässt sich zeigen, dass gilt: (6.23) C e = E es 85 Hierbei bezeichnet C die Rate mit der Moleküle aus der Gasphase kondensieren und E ist die Rate, mit der Moleküle aus der flüssigen Phase verdampfen (Evaporation). Diese Definition setzt voraus, dass bereits beide Phasen vorhanden sind (keine Nukleationsprozesse). Treten pro Zeiteinheit netto mehr Moleküle aus der Flüssigkeit aus, als Moleküle in die Flüssigkeit aufgenommen werden, so ist die Gasphase untersättigt (r < 1). Kondensieren mehr Moleküle aus der Gasphase als Moleküle, die aus der Flüssigkeit verdampfen, so ist (r > 1). 6.3 Temperaturabhängigkeit der Verdampfungsenthalpie Um die Clausius-Clapeyron-Gleichung integrieren zu können, hatten wir angenommen, dass Lv nicht von der Temperatur abhängt, was strenggenommen nicht richtig ist. Differenzieren wir Lv nach der Temperatur, so stellen wir fest, dass mit (3.71) gilt: (6.24) ∂Lv ∂H g ∂H f = − = C pg − C pf ∂T ∂T ∂T Da Lv nur von der Temperatur abhängt, können wir das partielle Differential auch als vollständiges Differential schreiben: ∂Lv/∂T = dLv/dT. Die spezifische Wärmekapazität von flüssigem Wasser beträgt 4209 J kg-1 K-1 bei 273 K und nimmt monoton ab auf 4180 J kg-1 K1 bei 323 K, eine Änderung um weniger als ein Prozent. Ähnlich gering ist die Änderung der spezifische Wärmekapazität von Wasserdampf. Daher ist der Fehler klein (innerhalb der Temperaturbereiche von Interesse in der Meteorologie), wenn wir die Wärmekapazitäten als von der Temperatur unabhängig annehmen, so dass wir die Gleichung (6.24) integrieren können: (6.25) Lv = Lv 0 + ( C pg − C pf ) (T − T ) 0 Hierbei ist T0 eine Referenztemperatur und Lv0 ist die zugehörige Verdampfungsenthalpie. Dividiert man beide Seiten der Gleichung durch die Masse (bzw. durch die Anzahl der Mole) so erhält man die spezifischen (bzw. die molaren) Größen: (6.26) lv = lv 0 + ( c pg − c pf ) (T − T ) 0 Berechnet man die Verdampfungsenthalpie des Wassers über einen Temperaturbereich von -20 bis +30°C mit 0°C als Referenztemperatur, bei der die Verdampfungsenthalpie 2.5×106 J/kg, cpf = 4200 J kg-1 K-1 und cpg = 1850 J kg-1 K-1 so stimmen die Ergebnisse mit gemessenen Verdampfungsenthalpien bis auf den Bruchteil eines Prozents überein. 86 Abbildung 6.3. Dampfdruckkurve des Wassers berechnet nach Gleichung (6.29) (durchgezogene Linie) und nach Gleichung (6.30) (gestrichelt). [Bohren] Wenn wir diese lineare Abhängigkeit von Lv von der Temperatur nun in die ClausiusClapeyron-Gleichung übernehmen, so kann auch diese Gleichung integriert werden. Wir erhalten aus der Kombination von (6.14) (in spezifischen Größen) und (6.26): 1 des lv 0 + ( c pf − c pg ) T0 c pf − c pg = − es dT RH 2OT 2 RH 2OT (6.27) Die Lösung dieser Gleichung ist: (6.28) ln es lv 0 + ( c pf − c pg ) T0 ⎛ 1 1 ⎞ c pf − c pg ⎛ T ⎞ = ln ⎜ ⎟ ⎜ − ⎟− es 0 RH 2O RH 2O ⎝ T0 T ⎠ ⎝ T0 ⎠ Der Einfachheit halber wählen wir die gleichen Referenztemperaturen (Integrationskonstanten) für die Verdampfungsenthalpie und für den Sättigungsdampfdruck. Wenn wir die oben angegebenen Parameter in (6.28) einsetzen, so erhalten wir: (6.29) ln ⎛ 1 1⎞ es T = 6808 ⎜ − ⎟ − 5.09 ln es 0 T0 ⎝ T0 T ⎠ Gleichung (6.15) mit der Verdampfungsenthalpie bei 0°C ergibt: (6.30) ln es 5417 = 19.83 − es 0 T Der Wasser-Sättigungsdampfdruck bei 0°C beträgt 6.11 hPa. Abbildung 6.3 zeigt die Gleichungen und (6.29) und (6.30) für den Temperaturbereich von –30 bis +30°C. Die Gleichung (6.29) ergibt fast vollständige Übereinstimmung mit den im Experiment ermittelten 87 Werten. Auch die einfache Näherung der Gleichung (6.30) liefert noch gute Übereinstimmung mit dem Experiment. Zur Veranschaulichung sind die Unterschiede zwischen gemessenen und den nach (6.29) und (6.30) berechneten Werten in der Abbildung 6.4 verdeutlicht. Abbildung 6.4. Prozentuale Differenz zwischen den gemessenen Dampfdrücken (Null-Linie), den nach (6.29) (gestrichelt) und nach (6.30) (durchgezogene Linie) berechneten Dampfdruckwerten. [Bohren] Um den Sättigungsdampfdruck über Eis bei Temperaturen unter 0°C zu erhalten, müssen wir nur die Notation der Größen von Gleichung (6.28) anpassen (Index i für "ice"): (6.31) ln esi ls 0 + ( c pi − c pg ) T0 ⎛ 1 1 ⎞ c pi − c pg ⎛ T ⎞ = ln ⎜ ⎟ ⎜ − ⎟− es 0 RH 2O RH 2O ⎝ T0 T ⎠ ⎝ T0 ⎠ wobei ls0 die Sublimationsenthalpie bei der Referenztemperatur T0 und cpi die spezifische Wärmekapazität von Eis bei konstantem Druck ist. Bei 0°C beträgt die Sublimationsenthalpie von Eis 2.834×106 J kg-1 und cpi = 2106 J kg-1 K-1. Damit ist (6.31): (6.32) ln ⎛ 1 1⎞ ⎛T ⎞ esi = 6293 ⎜ − ⎟ − 0.555ln ⎜ ⎟ es 0 ⎝ T0 T ⎠ ⎝ T0 ⎠ und die Näherungsgleichung Sublimationsenthalpie lautet: (6.33) für ln eine konstante, temperaturunabhängige esi 6142 = 22.49 − es 0 T In den Gleichungen (6.32) (6.33) ist es0 = 6.11 hPa der Sättigungsdampfdruck bei 0°C. 88 Abbildung 6.5. Sättigungsdampfdruck über Eis und unterkühltem Wasser. [Bohren] 6.4 Sättigungsdampfdruck über Eis und über unterkühltem Wasser Es gibt keinen Grund, Gleichung (6.29) nicht auch bei Temperaturen unterhalb von 0°C anzuwenden. In der Atmosphäre ist das Auftreten von unterkühlten Flüssigkeitströpfchen ein häufiges Phänomen, denn Wasser gefriert nicht automatisch, sobald die Temperatur unter 0°C fällt. Wie wir noch sehen werden, müssen, um zu gefrieren, Nukleationskeime vorhanden sein. Wenn diese fehlen, können die unterkühlten Tröpfchen als metastabile Phase bestehen bleiben. Die Abbildung 6.5 vergleicht den Sättigungsdampfdruck über Eis mit dem Dampfdruck über unterkühltem Wasser bis zu Temperaturen von –30°C. Der Dampfdruck der unterkühlten Flüssigkeit ist immer größer als der Dampfdruck über Eis. Wir erwarten, dass Wassermoleküle in der Flüssigkeit weniger fest mit ihren Nachbarn verbunden sind, und daher leichter aus der Flüssigkeit austreten können als Moleküle aus der festen Eiskristallstruktur. Obwohl der Unterschied zwischen den beiden Dampfdruckkurven relativ gering ist, führt der Unterschied zu deutlichen Konsequenzen in der Atmosphäre: Wenn in einer Wolke, die vorwiegend aus unterkühlten Flüssigkeitströpfchen besteht einige Tröpfchen gefrieren, da sie zufällig Eisnukleationskeime enthalten, so kann folgendes passieren: die Luftmasse kann untersättigt bezüglich der unterkühlten Flüssigkeit sein und übersättigt bezüglich Eis. Dann verdampfen die flüssigen Tröpfchen, während die Eiskristalle durch Deposition anwachsen. Eine umfassende Diskussion dieser Prozesse wird in der experimentellen Wolkenphysik-Vorlesung durchgeführt werden. Wasser kann, wenn keine Eisnukleationskeime vorhanden sind, ohne weiteres bei Temperaturen unterhalb von 0°C bestehen. Erst bei Temperaturen von etwa –40°C tritt sogenannte homogene Nukleation auf, bei der dann auch reines Wasser gefriert. 89 6.5 Taupunkt-Temperatur Als Taupunkttemperatur ist die Temperatur definiert, bei der sich Tau bildet. Dies ist natürlich eine Tautologie. Tau bildet sich dann an einer Oberfläche, wenn die Kondensation größer ist als die Evaporation. Die Netto-Kondensation kann dann als Tau sichtbar werden. Die Taupunkt-Temperatur Td (Index d für „dew“) kann bestimmt werden für eine aktuelle Temperatur T und einen aktuellen Wasserdampfpartialdruck e als: (6.34) es (Td ) = e Das heißt also, der Taupunkt Td ist die Temperatur, auf die Luft gekühlt werden muss, bei konstantem Wasserdampfpartialdruck, so dass der Dampfdruck gleich dem Sättigungsdampfdruck wird. Mit Hilfe der Clausius-Clapeyron-Gleichung (z.B. (6.29)) können wir also leicht den Taupunkt bestimmen, wenn die Temperatur T, der H2OPartialdruck e (oder die relative Feuchte r) bekannt sind. Analog zum Taupunkt definieren wir weiterhin den Frostpunkt Tf als die Temperatur, bei der Deposition von Wasserdampf als Eis auftritt. Der Frostpunkt entspricht somit dem Taupunkt bei Temperaturen unterhalb von 0°C. Tau- und Frostpunkt sind aus verschiedenen Gründen wichtig in der Meteorologie. Es wird manchmal vereinfachend gesagt, dass atmosphärische Temperaturen nicht unter ihre Taupunkt- bzw. Frostpunkt-Temperatur fallen können. Dies hat zwei Gründe. Einerseits wird der Wasserdampf auskondensieren (an Kondensationskeimen oder anderen Oberflächen) sobald der Taupunkt erreicht ist. Durch diese Kondensation verringert sich der Dampfdruck e und damit sinkt der Taupunkt. Weiterhin wird bei der Kondensation die Verdampfungsenthalpie freigesetzt. Diese Energiemenge erwärmt die Luft, was weiterhin verhindert, dass die Lufttemperatur unter die Taupunkttemperatur fällt. Aber es gibt auch in der Atmosphäre Situationen, in denen diese Aussage nicht richtig ist. Beispielsweise Luft, die frei von allen Kondensationskeimen ist, kann unter ihren Taupunkt abkühlen, oder Luftmassen, die so schnell konvektiv aufsteigen und dabei adiabatisch abkühlen, dass die Erwärmung durch die frei werdende Kondensationswärme nicht die adiabatische Abkühlung aufwiegen kann. Da die Kondensation Zeit braucht, kann das aufsteigende Luftpaket unter die Taupunktstemperatur abkühlen. 6.6 Vertikalgradient des Siedepunkts Der Siedepunkt Tb (Index b für boiling point) ist definiert als die Temperatur, bei welcher der Sättigungsdampfdruck gleich dem Umgebungsdruck p wird: (6.35) es (Tb ) = p Um den Vertikalgradienten des Siedepunkts zu ermitteln, differenzieren wir die Gleichung nach z: (6.36) des (Tb ) dTb dp = dTb dz dz Hieraus folgt, dass: 90 (6.37) 1 des (Tb ) dTb 1 dp = es (Tb ) dTb dz p dz Wir nähern den Druck mit der barometrischen Höhenformel (2.24) mit Skalenhöhe H: (6.38) ( p = p0 exp − z H ) Kombinieren wir (6.14) mit (6.37) und (6.38), so erhalten wir: (6.39) dTb RH 2OTb2 =− dz lv H Nehmen wir weiterhin die Verdampfungsenthalpie lv als temperatur-unabhängig an, so können wir die Gleichung integrieren zu: (6.40) Tb ⎛ z ⎞ ⎟ = ⎜⎜1 + Tb 0 ⎝ H b ⎟⎠ −1 wobei Tb0 die Referenz-Siedetemperatur am Boden ist, und die „Siedepunkt-Skalenhöhe“ Hb ist mit (6.41) Hb = H lv Tb 0 RH 2O gegeben. Nehmen wir Tb0 als 373 K und lv (Tb0) = 2.26×106 J kg-1 K-1 und H = 8 km, dann ist die Skalenhöhe des Siedepunkts etwa 107 km. Für die Troposphäre können wir daher die Gleichung (6.40) annähern: (6.42) ⎛ z ⎞ ⎟⎟ Tb ≈ Tb 0 ⎜⎜1 − H b ⎠ ⎝ z << H b und die Abnahmerate des Siedepunkts von Wasser wird damit (6.43) dTb T ≈ − b 0 ≈ −3.5°C / km dz Hb 6.7 Dampfdruckerniedrigung, Raoultsches Gesetz, ideale Lösungen Löst man irgendeine Substanz in Wasser, so erniedrigt sich in der Regel der Sättigungsdampfdruck des Wassers (bei konstanter Temperatur). Im Allgemeinen sind die physikalischen Eigenschaften einer Lösung von zwei Substanzen nicht einfach die gewichteten Mittel/Summen der beiden einzelnen Substanzen. Löst man eine Substanz 1 in einer Substanz 2, z.B. Salz in Wasser, und wird dieser Prozess adiabatisch (kein 91 Wärmeaustausch mit der Umgebung, δQ = 0, isentrop) und isobar durchgeführt, so ist die Gesamtenthalpie konserviert (vgl. (5.28)): H1 (Ti ) + H 2 (Ti ) = H12 (T f ) (6.44) wobei H1 und H2 die Enthalpien der beiden reinen Ausgangssubstanzen sind und H12 die Enthalpie der Lösung nach der Mischung. Ti ist die Anfangstemperatur (gleich für beide Komponenten) und Tf ist die Endtemperatur nach der Mischung. Führt man die gleiche Mischung der beiden Substanzen nun im Wärmebad bei der Temperatur Ti isobar durch, so gilt (vgl. (6.18) und (6.19)): ∫ δ Q = H ( T ) − H (T ) i (6.45) 12 f 12 i f Mit (6.45) können wir dies schreiben als: (6.46) ∫ δ Q = H (T ) − ⎡⎣ H (T ) + H (T )⎤⎦ = ΔH i 12 f 1 i 2 i i sol ΔHsol nennt man die Lösungsenthalpie. Sie stellt die Differenz der Enthalpien zwischen der Lösung und den einzelnen Komponenten bei gleichem Druck und gleicher Temperatur dar. Es stellt die Energiemenge dar, die benötigt wird/frei wird, um zwei Substanzen zu mischen. ΔHsol kann positiv oder negativ sein. Wenn man die Wärmekapazität bei konstantem Druck Cp12 der Mischung als temperatur-unabhängig annehmen kann, so können wir mit (3.71) schreiben: ΔH sol = −C p12 (T f − Ti ) . (6.47) Auch das Volumen einer Lösung ist nicht unbedingt die Summe der beiden Einzelvolumina. Die Masse des Lösung ist zwar konserviert, nicht aber das Volumen. Eine Lösung in der die Lösungsenthalpie gleich null ist und das Gesamtvolumen bei der Mischung strikt erhalten bleibt, nennt man ideal. Betrachten wir nun eine ideale Lösung aus zwei Komponenten A und B. Der Gleichgewichtsdampfdruck pA von A kann nur von der Temperatur und von den beiden Molfraktionen xA und xB abhängen: (6.48) xA = NA N A + NB xB = NB N A + NB Da xA+xB = 1 gilt, sind xA und xB nicht unabhängig. Wir können also die Dampfdrücke als Funktionen von nur zwei Variablen beschreiben, z.B.: (6.49) p A = p A ( T , xB ) pB = pB ( T , x A ) Wir betrachten den Spezialfall einer idealen Lösung, für die auch der Gesamtdruck konstant ist: 92 p = p A + pB (6.50) Dann gilt: (6.51) ∂p ∂p A ∂pB ∂p A ∂pB dx A = + = + =0 ∂xB ∂xB ∂xB ∂xB ∂x A dxB damit folgt: ∂p A ∂pB = ∂xB ∂x A (6.52) Die Randbedingungen lauten: (6.53) p A (T , 0 ) = p A0 = p, p A (T ,1) = 0 pB (T , 0 ) = pB0 = p, pB (T ,1) = 0 wobei pA0, und pB0 die Dampfdrück über den reinen Komponenten A und B sind. Die Funktionen (6.54) p A = p A0 (1 − xB ) , pB = pB0 (1 − xA ) erfüllen die Randbedingungen und (6.52). Umformen liefert das sogenannt Raoultsche Gesetz: (6.55) p A = p A0 x A pB = pB0 xB Dieses besagt, dass für ideale Lösungen der Dampfdruck über der Lösung jeweils proportional zum Molenbruch ist. Löst man beispielsweise NaCl in Wasser auf und bestimmt den Wasser-Gleichgewichtsdampfdruck über der Lösung so erhält man für kleine Beimengungen von NaCl eine gute Übereinstimmung mit dem Raoultschen Gesetz (vgl. Abbildung 6.6). Wir sehen hieran, dass der Dampfdruck sich tatsächlich vermindert bei steigendem Anteil der gelösten Substanz. Es gilt noch zu beachten, dass das Salz, wenn es aufgelöst wird, in Na+ und Cl⎯-Ionen dissoziiert, Daher müssen wir in der Formel für die Molenbrüche 2NB als Molekülanzahl einfügen. 93 Abbildung 6.6. Relative Feuchte über wässrigen Lösungen im Dampfdruckgleichgewicht bei 20°C. Durchgezogene Linien sind Messungen für Natriumchlorid und Ammoniumsulfat. Die gestrichelte Linie entspricht dem Raoultschen Gesetz. Die Signifikanz des Raoultschen Gesetzes für die Atmosphäre wird uns in den späteren Teilen dieses Kapitels klar werden. 6.8 Henry-Gesetz Wenn eine Wasseroberfläche der Luft ausgesetzt wird, so treffen ständig massenhaft Luftmoleküle auf die Oberfläche auf. Der Fluss der Moleküle jeder Komponente der Luft kann mit (6.56) 1 n v 4 angegeben werden. Hierbei ist n die Anzahldichte der Moleküle einer Sorte a und <v> ist die mittlere Geschwindigkeit (2.34). Der Faktor ¼ ergibt sich aus der Tatsache, dass sich im Mittel nur die Hälfte der Moleküle überhaupt auf die Fläche hinbewegen, und auch diese bewegen sich nicht alle genau in Richtung der Oberfläche, sondern haben nur eine Vektorkomponente senkrecht zur Oberfläche. Die exakte Rechnung ergibt einen weiteren Faktor ½, das Verhältnis der Projektion auf eine Ebene zur Oberfläche einer Halbkugel: πr2/2πr2. Luftmoleküle, die auf die reine Wasseroberfläche auftreffen, werden dort kurzzeitig adsorbiert, bevor sie wieder in die Luft zurückkehren. Einige der Moleküle diffundieren auch in das Wasser hinein. Dies passiert so lange, bis das Wasser überall eine gleichmäßige Konzentration ca bzw. eine gleichmäßige Molfraktion xa von Luftmolekülen enthält. Im Gleichgewicht gilt dann: (6.57) dca =C−E =0 dt 94 wobei C und E wieder die Kondensations- und die Evaporationsrate darstellen. Die Kondensationsrate ist: (6.58) C =α 1 n v 4 Dies ist der Fluss auf die Wasserfläche mal dem sogenannten Massenakkomodationskoeffizienten α, der das Verhältnis von Molekülen der Komponente a angibt, die an der Wasseroberfläche adsorbiert werden ("haften bleiben"), zur Gesamtanzahl von Luftmolekülen, die auf die Oberfläche treffen. Mit Hilfe des idealen Gasgesetzes und (2.34) können wir schreiben: (6.59) pα 2π mkT C= wobei p der Partialdruck der Luftkomponente ist und m die Masse der Komponente. Dies bedeutet, dass für eine konstante Temperatur und konstanten Partialdruck auch C konstant ist. Um in diesem Fall Gleichung (6.57) zu erfüllen, muss dann E entsprechend ca zunehmen. Die einfachste Funktion, die dies erfüllt ist: E = β ca (6.60) wobei der Koeffizient β von der Konzentration unabhängig ist, aber durchaus temperaturabhängig sein kann. Im Gleichgewicht ist dann: (6.61) ca = α β p 2π mkT Die Koeffizienten kann man kompakt zusammenfassen: (6.62) ca = p K (T ) mit (6.63) K (T ) = β 2π mkT α Gleichung (6.62) wird Henry-Gesetz genannt. Die Materialkonstanten K(T) werden HenryKonstanten genannt. Wegen der Proportionalität von Konzentration und Molenbruch bei kleiner Konzentration (xa << xw) können wir schreiben: (6.64) p ∝ K (T ) xa In dieser Form ist die Ähnlichkeit des Henry-Gesetzes zum Raoultschen Gesetz offensichtlich. Im allgemeinen gilt das Raoultsche Gesetz für das Lösungsmittel (große Konzentration bzw. große Molfraktion in der Lösung) während das Henry-Gesetz für die in niedriger Konzentration gelöste Komponente gilt. Die Abbildung 6.7 macht dies deutlich. 95 Abbildung 6.7. Experimentelle Dampfdrücke über Mischungen von Chloroform mit Aceton. Für das Lösungsmittel gilt jeweils das Raoultsche Gesetz, für die gelöste Komponente gilt das Henry-Gesetz. [Atkins] 6.9 Kelvin-Gleichung und Nukleation In der bisherigen Diskussion sind wir immer von ebenen Oberflächen an der Phasengrenze ausgegangen. In der Atmosphäre haben wir es aber meist mit kleinen Tröpfchen und Partikeln zu tun, die eine stark gekrümmte Oberfläche besitzen. In diesem Abschnitt wollen wir nun den Effekt der Krümmung der Oberfläche auf den Dampfdruck einer Substanz A untersuchen. Wir werden diesen Dampfdruck mit dem schon bekannten Dampfdruck über einer ebenen Oberfläche vergleichen. Wir gehen aus von der Änderung der freien Gibbschen Enthalpie die mit der Bildung eines einzelnen Tröpfchens mit Radius Rp (welches aus n Molekülen der reinen Substanz A besteht) einhergeht: (6.65) ΔG = GTröpfchen − GDampf Die Gesamtanzahl der Moleküle im Dampf betrage ursprünglich Nt. Nachdem das Tröpfchen gebildet wurde, ist die Anzahl der übrigen Dampfmoleküle N1 = Nt - n. Nennen wir gv und gl die Gibbschen freien Enthalpien eines einzelnen Moleküls in der Dampf-, bzw. in der flüssigen Phase: (6.66) ΔG = N1 g v + ngl + 4π R p2σ − N t g v Der Faktor 4πRp2σ entspricht der Gibbschen freien Enthalpie einer Oberfläche mit Radius der Krümmung Rp und Oberflächenspannung σ. Um eine Oberfläche aufzubauen, muss nämlich Arbeit geleistet werden (analog z.B. zur Volumenarbeit des Gases). Die Gleichung können wir umschreiben: 96 (6.67) ΔG = n ( g l − g v ) + 4π R p2σ Man beachte, dass die Anzahl der Moleküle n in einem Tröpfchen mit dem Radius Rp des Tröpfchens verknüpft ist: 4 π Rp n= . 3 vl 3 (6.68) Hier ist vl das Volumen eines Moleküls in der Flüssigphase. Damit ergibt sich für ΔG: (6.69) ΔG = 4π R 3p 3vl ( gl − gv ) + 4π Rp2σ Nun berechnen wir (gl – gv). Mit (5.37) und T = const., sowie ni = const., bleibt dg = v dp, bzw. hier: dg = (vl – vv) dp. da vv >> vl bei atmosphärischen Bedingungen können wir schreiben : dg = vvdp. Nehmen wir den Dampf als ideales Gas an (vv=kT/p), so können wir schreiben: ( gl − gv ) = −kT ∫p pA (6.70) 0 A dp p wobei pA0 der Dampfdruck der Substanz A über der ebenen Oberfläche ist, und pA ist der aktuelle Dampfdruck über der Flüssigkeit. Die Integration ergibt: (6.71) ( gl − gv ) = −kT ln pA p A0 definieren wir noch das Verhältnis pA/pA0 als Sättigungsverhältnis S. Durch Einsetzen in (6.69) folgt: (6.72) 4 kT ΔG = − π R 3p ln S + 4π R p2 σ 3 vl Die Abbildung 6.8 zeigt den Verlauf von ΔG als Funktion des Partikelradius. Für S < 1 steigen beide Terme in der Gleichung monoton an, es ist also bei Untersättigung die Bildung eines Tröpfchens aus der Gasphase energetisch nicht erlaubt. Bei Übersättigung S > 1 muss aber auch zunächst ein "Berg" überwunden werden: die sogenannte Nukleationsbarriere. Das kommt daher, dass der Oberflächenterm (~R2) zunächst größer ist, als der Volumenterm (~R3). Erst wenn ein kritischer Radius R* überwunden ist, wächst das Tröpfchen durch Kondensation spontan weiter, denn dann ist die flüssige Phase so lange energetisch bevorzugt, bis sich die Dampfphase abgebaut hat und S→1 geht. Entsprechende Nukleationsbedingungen gibt es für alle Phasenübergänge, wenn die zweite Phase nicht von Anfang an bereits vorhanden ist. Somit ist auch das zustande kommen der unterkühlten Flüssigkeitströpfchen vom Abschnitt 6.4 sofort verständlich. Die Nukleationsbarriere verhindert das Gefrieren der unterkühlten Tröpfchen, die Phase ist metastabil. Den kritischen Radius erhält man durch Nullsetzen der Ableitung nach Rp von (6.72): 97 R*p = (6.73) 2σ vl kT ln S Löst man Gleichung (6.72) nach pA auf, so erhält man: ⎛ 2σ vl ⎞ p A = p A0 exp ⎜ ⎜ kT R ⎟⎟ p ⎠ ⎝ (6.74) und in molaren Größen: (6.75) ⎛ 2σ M ⎞ p A = p A0 exp ⎜ * ⎜ R T ρ R ⎟⎟ l p ⎠ ⎝ wobei M das Molekulargewicht von A darstellt. Die Gleichung (6.75) heißt KelvinGleichung. Die Kelvin-Gleichung besagt, dass der Dampfdruck über einer gekrümmten Oberfläche immer größer ist, als der Dampfdruck über der entsprechenden ebenen Oberfläche. Als physikalische Veranschaulichung kann man sich Abbildung 6.9 ansehen. An einer gekrümmten Oberfläche werden die Moleküle, die aus der Oberfläche abdampfen von weniger bindenden Nachbarmolekülen umgeben, als bei einer ebenen Fläche. Sie können daher leichter der Flüssigkeit entkommen, der Dampfdruck steigt. Der Kelvin-Effekt wird bei Wassertröpfchen unterhalb von 50 nm Größe sehr relevant (Abbildung 6.10). Setzt man Werte ein für σ für reines Wasser bei 20°C (σ = 0.0073 N/m), dann gilt mit Rp in µm: ⎛ 0.00108 ⎞ (6.76) es = es0 exp ⎜ ⎟⎟ ⎜ R p ⎝ ⎠ Abbildung 6.8. Änderung der Gibbschen freien Enthalpie bei der Bildung eines Tröpfchens mit Radius Rp bei verschiedenen Sättigungs-Verhältnissen S. Das Maximum der Nukleationsbarriere ist eingezeichnet. [Seinfeld] 98 Abbildung 6.9. Veranschaulichung des Kelvin-Effekts. [Seinfeld] Abbildung 6.10. Sättigungsdampfdruck als Funktion der Tröpfchengröße für eine organische Flüssigkeit DOP (dioctyl phthalat) und Wasser bei 20°C. [Seinfeld] Ein gravierendes Problem der Kelvin-Gleichung ist, dass sie auf makroskopische Größen zurückgreift, wie z.B. die Oberflächenspannung oder die Dichte (steckt implizit in vl). Da es bei den Nukleations-Clustern meist um eine Zusammenballung von nur einigen Molekülen geht, sind diese makroskopischen Größen sicherlich schlecht geeignet. Verschiedenste Ansätze haben daher den klassischen Ansatz der Kelvin-Gleichung erweitert (z.B. molekulare Theorien und funktionale Dichtetheorien). Außerdem gibt es umfassende Erweiterungen der Kelvin-Gleichung für mehrkomponentige Systeme, die in der Atmosphäre besonders relevant sind. Eine weitere Besonderheit ist die sogenannte ionen-induzierte Nukleation. Sind Ionen in der Luft vorhanden, so können diese mit den übersättigten Molekülen wechselwirken (Wilsonsche Nebelkammer). Insbesondere können polare Moleküle, die sich an das Ion anlagern, nicht so leicht wieder abdampfen. Die Nukleationsbarriere wird dadurch abgesenkt und es gilt die Thomson-Gleichung: 2 q ⎛ 1 ⎞⎛ 1 1 ⎞ Δ G = − N k T ln ( S ) + 4 π r σ + ⎜ 1 − ⎟ ⎜⎜ − ⎟⎟ 2 ⎝ ε ⎠ ⎝ rp r0 ⎠ 2 p Den Unterschied zwischen neutraler und ionen-induzierter Nukleation veranschaulicht die Abbildung 6.11. 99 neutral nucleation ΔG ΔG*neutr. ΔG*ion ion-induced nucleation 0 r*ion r*neutr. requilib rp Abbildung 6.11. Vergleich zwischen neutraler und ionen-induzierter Nukleation nach Kelvin- und Thomson-Gleichung. 6.10 Sättigungsdampfdruck über Lösungströpfchen, Köhlerkurven Im letzten Abschnitt haben wir gezeigt, dass der Dampfdruck über gekrümmten Flächen ansteigt, ein Tröpfchen mit stark gekrümmter Oberfläche also auch bei 100% Sättigung verdunstet. Wenn aber das Tröpfchen eine Lösung ist, dann erniedrigt die gelöste Substanz den Dampfdruck nach dem Raoultschen Gesetz. Diese beiden gegenläufigen Effekte von Kelvin-Effekt und Raoultschem Gesetz wollen wir nun für die Lösung von NaCl und Wasser untersuchen. Wir gehen aus von einem zunächst ungelösten Salzkörnchen der Masse ms, Volumen Vs, Durchmesser ds und Dichte ρs = ms/Vs =2.17 g/ccm. Wassermoleküle in der Umgebung des Körnchen treffen auf die Oberfläche und beginnen das Salz aufzulösen. Es entsteht ein Partikel mit einem festen Salzkern und einem gesättigten Lösungsfilm an der Oberfläche. Die Löslichkeit von Salz in kaltem Wasser beträgt etwa 36 g Salz pro 100 ccm Wasser. Das Wasser-Volumen Vw, das benötigt wird, um das Körnchen Salz ganz aufzulösen erhält man aus c = ms/Vw, wobei c die (gesättigte) Konzentration der Lösung (in Einheiten Masse pro Einheit Lösungsmittel) darstellt. Erst wenn genügend Wasser kondensiert ist, so dass die ganze Salzmenge gelöst wird, ist das Partikel flüssig. Um die Abhängigkeit des Gleichgewicht-Dampfdrucks von der im Tröpfchen gelösten Salzmenge zu bestimmen benötigen wir noch die Molfraktion des Wassers : (6.77) xw = mw / M w mw / M w + 2ms / M s 100 mit den Molekulargewichten Mw und Ms. mw ist die Masse des Wassers. Diese ist variabel, während die Masse des NaCl fest ist. Der Faktor zwei berücksichtigt, dass das Salz in die beiden Ionen Na+ und Cl¯ dissoziiert. Wenn man mit V das Gesamt-Volumen des Tröpfchens bezeichnet, dann gilt: (6.78) xw = V − Vs ⎛ 2ρ M ⎞ V + Vs ⎜ s w − 1⎟ ⎝ ρw M s ⎠ und dieses kann noch auf die Durchmesser umgerechnet werden: (6.79) xw = 1 − ( ds / d ) 3 1 + 0.334 ( d s / d ) 3 Für stark verdünnte Lösungen (für die dann auch das Raoultsche Gesetz gilt) gilt schließlich d >> ds. Dann können wir die Näherung aufstellen: (6.80) ⎛d ⎞ xw ≈ 1 − ⎜ s ⎟ ⎝d ⎠ 3 Wir kombinieren nun das Raoultsche Gesetz (6.55) mit der Kelvingleichung für Wasser (6.76) und erhalten die Köhler-Gleichung für den -Wasser-Sättigungsdampfdruck über Tröpfchen einer Lösung von NaCl in Wasser bei 20°C: (6.81) 3 ⎞ es ⎛ 0.00216 ⎞ ⎛ 1 − ( d s / d ) = exp ⎜ ⎜ ⎟ ⎟⎜ 3 es∞ d ⎝ ⎠ ⎝ 1 + 0.334 ( d s / d ) ⎟⎠ Die Gleichung (6.81) ist das Produkt zweier Faktoren, wobei der eine monoton abnimmt, der andere monoton zunimmt. Das Verhältnis es/es∞ wird auch Sättigungsverhältnis genannt. Die Abbildung 6.12 zeigt Köhler-Kurven für NaCl-Wasser-Gemische für verschiedene ursprüngliche Durchmesser ds des NaCl Kristalls. Alle Kurven steigen über den Wert 1, also den Sättigungsdampfdruck für reines Wasser bei ebener Oberfläche. Das bedeutet, dass kleine Aerosolpartikel, wenn sie zu Wolkentröpfchen (>10 µm) anwachsen, nicht nur eine Übersättigung S > 1 benötigen, sondern eine Übersättigung, die höher ist, als das jeweilige Maximum der Köhlerkurve. Diese Tatsache wollen wir anhand der Abbildung 6.13 verdeutlichen. Wird ein Partikel, das sich am Punkt A bei einem Sättigungsverhältnis von 0.95 im Gleichgewicht mit seiner Umgebung befindet, plötzlich in eine übersättigte Umgebung gebracht (S = 1.01, gestrichelte Linie in der Abb.), so kondensiert Wasserdampf auf dem Partikel, bis es zu einer Größe angewachsen ist, die dem Punkt B entspricht. Weiter wird es nicht anwachsen! Ein anderes Partikel, zunächst im Gleichgewicht mit der Umgebung am Punkt C, wird, wenn die Sättigung sich plötzlich auf S = 1.01 ändert, unendlich anwachsen, jedenfalls bis durch die Kondensation der Umgebungsdampfdruck abgebaut wird. Als Dunst ("haze") bezeichnet man ein Aerosol, das links des Maximums der Köhler-Kurve liegt, Partikel, die rechts des Maximums liegen, werden als Wolkentröpfchen bezeichnet. Für allgemeinere Formen der Köhler-Gleichung siehe z.B. [Seinfeld]. 101 Abbildung 6.12. Köhlerkurven für trockene NaCl-Kristalle von unterschiedlichem ursprünglichem Durchmesser. [Bohren] Abbildung 6.13. Aktivierung von Wolkentröpfchen. [Bohren] 102 7 Wasserdampf und Wolken in der Atmosphäre Im vorhergehenden Kapitel haben wir uns hauptsächlich mit der Rolle des Wassers in der Atmosphäre aus mikrophysikalischer Perspektive beschäftigt. Im nun folgenden Kapitel wollen wir hauptsächlich die makroskopische Sicht hervorheben. 7.1 Definitionen und nützliche Gleichungen Bevor wir richtig loslegen, wollen wir einige Begriffe und Schreibweisen festlegen. Im Folgenden soll der Index d immer für trockene Luft ("dry") stehen, nur in Td steht er für ("dew"). Der Index v steht immer für den puren Wasserdampf ("vapor"), (Ausnahme: Tv steht für die virtuelle Temperatur), der Index w für das flüssige Wasser (Ausnahme: Tw und Θw wird für die "wet-bulb" Temperatur und die potentielle wet-bulb Temperatur stehen), der Index i für Eis ("ice"). Die Größen, die die Mischung von Wasserdampf und trockener Luft beschreiben werden ohne Index geführt. Es gilt somit für feuchte Luft (ohne auskondensiertes flüssiges oder eisförmiges Wasser): Für die Masse m der feuchten Luft: m = mv + md = ν v M v + ν d M d (7.1) Mit m sind die Gesamt-Massen bezeichnet, M bezeichnet die Molgewichte (Md = 29 g/mol, Mv = 18 g/mol). ν bezeichnet die jeweiligen Stoffmengen in Mol. Nach dem Dalton-Gesetz addieren sich die Partialdrücke: p = ∑ pj (7.2) j Das heißt in unserem Fall: p = e + pd (7.3) Der Wasserdampfpartialdruck wird ja mit e bezeichnet. Für die mittlere Dichte der feuchten Luft gilt: ρ= (7.4) m ν v M v +ν d M d = = ρv + ρd V V Das spezifische Volumen v ist definiert durch: v =V /m (7.5) Die spezifischen Gaskonstanten sind definiert als: (7.6) R* R= M R* J Rd = = 287, 0 Md kg K R* J Rv = = 461,5 Mv kg K Es ergibt sich das mittlere Molekulargewicht der feuchten Luft zu: 103 (7.7) M= M = ν v M v +ν d M d = xv M v + xd M d ν v +ν d wobei xv und xd die Molfraktionen des Wasserdampfs und der trockenen Luft bezeichnen. Die mittlere spezifische Gaskonstante der feuchten Luft ist dann: (7.8) R= R = mv Rv + md Rd . m In spezifischen Größen schreibt sich das ideale Gasgesetz als: pv = RT (7.9) Für den Wasserdampf und die trockene Luft gilt: (7.10) e = ρv RvT pd = ρ d Rd T Wir definieren ε als Verhältnis der spezifischen Gaskonstanten: (7.11) ε= Rd M v 18 = = = 0.62 Rv M d 29 In Ergänzung zur Definition der relativen Feuchte r = e/es definieren wir noch die spezifische Feuchte q und das Wasserdampfmischungsverhältnis w: (7.12) ρv ρ w= w w +1 w= q= ρv ρd Mit ρ = ρd + ρv folgt: (7.13) q= q 1− q Ersetzen wir die Dichten in (7.12) mit den allgemeinen Gasgleichungen (7.10), so bleibt: (7.14) w=ε e e e =ε ≈ε pd p−e p Die letzte Näherung ist brauchbar, da in der Atmosphäre p >> e recht gut erfüllt ist. 7.2 Vertikalgradient des Taupunkts: Wolkenbildung Wolken bilden sich in der Atmosphäre unter anderem dadurch, dass feuchte Luft von einer gegebenen Höhe aus aufsteigt. Wir werden nicht die dynamischen Details des Aufstiegs der Luftmassen untersuchen, da sie für uns nicht weiter von Belang sind, aber wir definieren als 104 "lifting condensation level" (LCL) die Höhe, auf die ein Luftpaket trocken-adiabatisch aufsteigen müsste, um eine Wasserdampf-Sättigung von 100% zu erreichen. Die Höhe der LCL ist für ein Luftpaket eine eindeutige Funktion seiner Temperatur und relativen Feuchte. Um die LCL-Höhe zu berechnen, gehen wir davon aus, dass das Luftpaket von seiner ursprüngliche Höhe z0 (oder auch p0) zur Höhe zLCL (entsprechend pLCL) angehoben wird, bei der seine Temperatur der Taupunktstemperatur entspricht. Während das Paket aufsteigt, sinkt seine Temperatur ungefähr mit dem trocken-adiabatischen Temperaturgradienten (solange das Wasser nicht auskondensiert, hat die Anwesenheit der kleinen Menge Wasserdampf fast keinen Einfluss auf Γ!), so dass bei der Höhe z gilt: (7.15) T = T0 − Γ d ( z − z0 ) mit T0 =T(z0) und Γd=g/cp. Der Taupunkt Td des Pakets variiert mit der Höhe gemäß: (7.16) Td = Td 0 − Γ dew ( z − z0 ) mit der Annahme, dass der Vertikalgradient des Taupunkts Γdew konstant ist. Um den zLCL zu erhalten, setzen wir die Temperatur T gleich der Taupunkts-Temperatur Td in (7.15) und (7.16): (7.17) z LCL = z0 + T0 − Td 0 Γ d − Γ dew Um den Gradienten des Taupunkts zu bestimmen, differenzieren wir zunächst die TaupunktsGleichung (6.34) nach der Höhe z: (7.18) des dTd de = dTd dz dz Der Dampfdruck e ist nach (7.14) direkt proportional zum Gesamtdruck: (7.19) e= w p w+ε Wir nehmen an, dass bis zum Erreichen von zLCL das Mischungsverhältnis w konstant bleibt. Dann gilt: (7.20) de w dp = dz w + ε dz Wenn wir die letzten drei Gleichungen kombinieren erhalten wir: (7.21) 1 des dTd 1 dp = es dTd dz p dz Mit der Clausius-Clapeyron-Gleichung ist: 105 (7.22) l 1 des = v2 es dTd RvTd und für einen adiabatischen Prozess gilt mit (2.21) und (3.58): (7.23) g 1 dp c p dT = =− p dz RT dz RT Die Gleichungen (7.21)-(7.23) ergeben kombiniert: (7.24) Γ dew = − dTd g Td2 = dz ε lv T Die Größe g/εlv ist ~6.3×10-6 m-1 und fast temperaturunabhängig. Obwohl der Faktor Td2/T von zwei (absoluten) Temperaturen abhängt, ist der Wertebereich, den diese Größe in der Troposphäre annimmt, relativ klein. Typische troposphärische Werte für Γdew liegen dann zwischen 1.7 und 1.9 °C/km. Damit ergibt der mittlere Wert von 1.8°C/km eine gute Abschätzung für den Vertikalgradienten des Taupunkts des Luftpakets. Berücksichtigen wir noch, dass Γ = 9.8°C/km ist, so erhalten wir aus (7.17): (7.25) z LCL − z0 ≈ T0 − Td 0 8 Ohne allzu große Näherungen haben wir somit eine hilfreiche Daumenregel abgeleitet: Wolken bilden sich, wenn man eine Luftmasse bei z0 um eine Höhe (in km) anhebt, die einem Achtel ihrer Taupunktdifferenz entspricht. An (7.25) sieht man noch etwas: die Tatsache, dass sich Wasserwolken in der Atmosphäre immer nur im Aufstieg bilden liegt daran, dass die Abnahme der Temperatur des Pakets mit der Höhe (Γd) größer ist als die Abnahme des Taupunkts (Γdew). Dies ergibt sich wiederum z.B. aus dem Wert von lv. Es sind andere Substanzen als Wasser denkbar, bei denen man eine Wolkenbildung im Absteigen der Luftmassen erwarten würde. Oft ist zLCL berechnet aus Temperatur und Taupunkt von Bodenluft eine gute Abschätzung für die Höhe der Basis von Kumuluswolken, die ihren Ursprung in der planetaren Grenzschicht besitzen. Wenn die Grenzschicht gut durchmischt ist, dann weicht zLCL der Oberflächenluft wenig von solchen zLCL ab, die irgendwo sonst in der planetaren Grenzschicht bestimmt wurden. Die potentielle Temperatur und das Mischungsverhältnis des Luftpakets sind Erhaltungsgrößen. Wenn die Luft in der Grenzschicht gut durchmischt ist, dann sind diese beiden Größen mit der Höhe konstant und daher haben alle Luftpakete, die aus irgendeiner Höhe in der Grenzschicht aufsteigen, dieselbe zLCL. Diesen Zusammenhang verdeutlicht die Abbildung 7.1. Zur Abschätzung der Temperatur TLCL in der Höhe zLCL brauchen wir nur die Gleichungen (7.15) und (7.16) etwas umzuformen. Bei zLCL gilt zLCL = z und TLCL = T = Td. Damit ist: (7.26) T0 − TLCL Γ = d Td 0 − TLCL Γ dew Aufgelöst nach TLCL: (7.27) TLCL = Td 0 Γ d − T0 Γ dew Γ d − Γ dew 106 Abbildung 7.1. Vergleich der Höhe der gemessenen Basis von Kumuluswolken (durchgezogene Linie) mit der berechneten zLCL (gepunktete Linie) für am Boden gemessene Lufttemperaturen und Taupunkte. [Bohren] und mit Γdew = 1.8 °C/km: TLCL = (7.28) 9.8Td 0 − 1.8T0 8 Es existieren wesentlich exaktere Formeln für die Berechnung von zLCL und TLCL. Eine Form lautet: 1 − ATd 0 (7.29) TLCL = T 1 + B ln 0 − A Td 0 Td 0 mit : c ⎞ ⎛c −c A = − ⎜ pv pw − p ⎟ , lr ε lr ⎠ ⎝ B= cp ε lr Die Herleitung dieser Gleichung findet sich in [Bohren und Albrecht]. 107 7.3 Virtuelle Temperatur und Dichte der feuchten Luft Wir untersuchen im folgenden die Temperaturgradienten für feuchte Luft, die aber noch untersättigt ist, d.h. es gilt zwar nicht der echte trocken-adiabatische Temperaturgradient, es kondensiert aber auch noch kein Wasser aus, d.h. wir müssen uns auch nicht um freiwerdende Verdampfungsenthalpien etc. kümmern. Hierbei wird uns die Einführung der virtuellen Temperatur helfen. In den Abschnitten 3.6 und 3.7 hatten wir anhand der Temperaturgradienten von trockener Luft Kriterien für die statische Stabilität von Luftmassen abgeleitet. Dies war möglich, da bei konstantem Druck, die Temperatur T äquivalent ist zur - eigentlich entscheidenden - Dichte ρ. Dieser Ansatz ist akzeptabel, solange das Wasserdampfmischungsverhältnis konstant ist. Da aber in der Atmosphäre das Wasserdampfmischungsverhältnis stark variiert, müssen wir nun bei den Stabilitätsbedingungen auf die Dichte selbst zurückgreifen. Die Bedingung für ein Luftpaket unter analogen Voraussetzungen wie in Abschnitt 3.7 (keine Mischung des Luftpakets mit der Umgebung; ps = pp; Paket bewegt sich adiabatisch reversibel, d.h. isentrop), lautet: (7.30) dρp dz > d ρs dz wobei ρp die Dichte des Pakets ist, welches adiabatisch angehoben wird bzw. adiabatisch absinkt. Wenn die Dichte des Pakets weniger stark abnimmt, wenn es angehoben wird, als die der umgebenden Luft, so hat es einen negativen Auftrieb und die Schichtung ist stabil. Aus der Poisson-Relation für Temperatur und Volumen (3.47) folgt: (7.31) Tp1/(1−γ ) ρp = const Dies abgeleitet nach der Höhe ist: (7.32) 1 d ρp 1 1 dTp =− ρ p dz γ − 1 Tp dz Den Temperaturgradienten dTp/dz können wir mit Hilfe des "trocken"-adiabatischen Temperaturgradienten ersetzen, wobei wir jetzt R statt Rd benutzen, also die spezifische Gaskonstante der feuchten Luft! Die Größe γ kann für feuchte Luft als dieselbe wie für trockene Luft angesehen werden: schon cp und cV ändern sich für feuchte Luft nur wenig (cpv weicht zwar deutlich von cpd ab, aber die Luft enthält ja nur einige Massenpromille Wasserdampf; das Verhältnis cp/cV ist noch weniger beeinflusst). (7.33) 1 dρp g 1 =− ρ p dz γ R Tp Das ideale Gasgesetz für die umgebende Luft lautet: 108 p = ρ s RTs = ρ s (7.34) R Ts Rd Rd Wir definieren nun als virtuelle Temperatur Tv: Tv = (7.35) M R T= dT Rd M wobei M das mittlere Molekulargewicht der feuchten Luft ist, Md das der trockenen Luft. Die virtuelle Temperatur ist die Temperatur, die Luft (mit gegebener fester Dichte und festem Druck) hätte, wenn sie ganz frei von Wasserdampf wäre. Man ersetzt sozusagen alle Wassermoleküle durch eine Anzahl von Luftmolekülen, so dass Druck und Dichte wie bei der feuchten Luft sind, und misst dann die Temperatur. Da das mittlere Molekulargewicht der feuchten Luft immer geringer ist als das Molekulargewicht der trockenen Luft, ist die virtuelle Temperatur Tv niemals kleiner als die Temperatur T. Wir wollen die Gleichung (7.35) noch etwas umformen. Das mittlere Molekulargewicht ist (vgl. (7.7)): (7.36) M = xd M d + xv M v mit : xd + xv = 1 Dann folgt, dass: (7.37) Md 1 = M 1 − xv (1 − ε ) mit ε = Mv/Md. Die spezifische Feuchte q = ρv/ρ kann auch geschrieben werden als: (7.38) q= xv M v xv M v + xd M d nach xv aufgelöst, ergibt sich: (7.39) xv = q ε + q (1 − ε ) Die Kombination von (7.37) und (7.39) liefert: (7.40) Md 1− ε = 1+ q = 1 + 0.608q ε M Damit folgt für Tv: (7.41) Tv = T (1 + 0.608q ) Nach Gleichung (7.34) und (7.35) können wir schreiben: (7.42) p = ρ s Rd Tv 109 Wir bilden die Ableitung nach z: 1 dp 1 dTv 1 d ρ s = + p dz Tv dz ρ s dz (7.43) Mit Hilfe der hydrostatischen Gleichung (2.21), können wir dies umformen: g 1 d ρs 1 dTv =− − RTs Tv dz ρ s dz (7.44) Nun ziehen wir die Gleichung (7.44) von der Gleichung (7.33) ab: g g 1 d ρ p 1 d ρs 1 dTv − =− + + ρ p dz ρ s dz γ RTp RTs Tv dz (7.45) Der Gradient der virtuellen Temperatur ist der, der Umgebung, nicht des Paketes. Atmosphärische Stabilität ist nach (7.30) abhängig vom Vorzeichen von: d ρp (7.46) dz − d ρs dz Wir bewerten diese Ableitungen nur in der Ausgangslage, in welcher zunächst Temperatur und Dichte des Pakets gleich der Umgebungstemperatur und Dichte sind (Ts = Tp und ρs = ρp)! Dann ist die statische Stabilität gegeben wenn: g g 1 dTv + + >0 γ RTp RTs Tv dz − (7.47) Dieses können wir umschreiben zu: dTv Tv ⎛ g γ − 1 ⎞ g γ −1 g > ⎜− =− = −Γ d ⎟=− dz Ts ⎝ R γ ⎠ Rd γ c pd (7.48) Hierbei wurde die Definition der virtuellen Temperatur Tv = RT/Rd und die Definition des trocken-adiabatischen Temperaturgradienten Γd nach (3.58) benutzt. Somit haben wir als Ergebnis analog zum Abschnitt 3.7: (7.49) dTv > −Γ d dz ( stabil ), dTv = −Γ d dz (neutral ), dTv < −Γ d dz (instabil ) Es handelt sich jetzt eben nur um den Gradienten der virtuellen Temperatur, Γd ist aber der echte trocken-adiabatische Gradient für die trockene Luft. Virtuelle Temperatur ist somit eine praktische Größe, um Stabilität von feuchten Luftschichten zu bestimmen, nicht jedoch eine messbare Temperatur. Wenn wir die Definitionsgleichung (7.35) nach der Höhe ableiten: 110 1 dTv 1 dT 1 dR = + Tv dz T dz R dz (7.50) so sieht man, dass der Gradient der virtuellen Temperatur sich zusammensetzt aus dem Gradienten der Temperatur und dem Gradienten der Feuchtigkeit, ausgedrückt durch die spezifische Gaskonstante der feuchten Luft. Nimmt man 0.01 als typischen wert für die spezifische Feuchte der Luft, dann ist die virtuelle Temperatur etwa ein halbes Prozent größer als die Temperatur, da es sich um absolute Temperaturen handelt, macht das typischerweise ein paar Grad Celsius aus. Wir definieren zum Abschluss noch die virtuelle potentielle Temperatur Θv: ⎛p ⎞ Θv = ⎜⎜ 0 ⎟⎟ ⎝ p⎠ (7.51) (γ −1) / γ Tv Die Stabilitätskriterien aus Abschnitt 3.7 gelten analog. 7.4 Die "wet-bulb"-Temperatur und die äquivalente Temperatur Die Feuchtetemperatur oder englisch wet-bulb Temperatur ist die niedrigste Temperatur, auf die Luft durch Verdunstungskühlung adiabatisch und isobar abgekühlt werden kann. Was bedeutet diese Definition? Wir stellen uns dazu folgenden Versuchsaufbau vor: Umgebungsluft wird in einen isolierten Zylinder eingeschlossen, der Kolben ist frei beweglich, so dass ein konstanter Druck gewährleistet ist. Am Boden des Zylinders füllen wir eine Schicht Wasser ein (bei gleicher Temperatur wie die Luft). Ist nun die in den Zylinder gesperrte Umgebungsluft nicht mit Wasserdampf gesättigt, so wird Wasser aus der Bodenschicht verdampfen, bis die Sättigung der Gasphase erreicht ist. Die Energie, die benötigt wird, um das Wasser zu verdampfen, wird der Flüssigkeit entzogen und führt dazu, dass das Wasser und damit dann auch die Luft im Zylinder sich abkühlen. Wie viel die Gleichgewichtstemperatur abnimmt, hängt davon ab, wie viel Wasser ich einfülle. Je mehr Wasser, desto höher die Gleichgewichtstemperatur. Wir können diesen Versuch mehrfach wiederholen, wobei wir die Flüssigwassermenge bei jedem Versuch weiter reduzieren. Die niedrigste Temperatur, die wir so erhalten können, ist dann die wet-bulb Temperatur. Wir wollen diese Temperatur im Zylinder nun quantitativ ausrechnen. In einem adiabatisch-isobaren Prozess ist die Enthalpie erhalten: H di + H vi + H wi = H df + H vf + H wf (7.52) in spezifischen Größen schreibt sich dies: (7.53) mdi hdi + mvi hvi + mwi hwi = mdf hdf + mvf hvf + mwf hwf Wir wissen aus (6.19): (7.54) lvi = hvi − hwi lvf = hvf − hwf und die Masse des Wassers (Dampf + Flüssigkeit) bleibt im geschlossenen System erhalten: 111 mvf + mwf = mvi + mwi = mtw (7.55) wie auch die Masse der trockenen Luft: md = mdi = mdf . (7.56) Somit folgt aus (7.53) md ( hdi − hdf ) = mtw ( hwf − hwi ) + mvf lvf − mvi lvi (7.57) und mit (3.73) folgt: (7.58) hdi − hdf = c pd (Ti − T f ) , hwi − hwf = c pw (Ti − T f ) wobei wir die Temperaturabhängigkeit der Wärmekapazitäten vernachlässigt haben. Wir vernachlässigen weiterhin die geringe Temperaturabhängigkeit von lv: lvi ≈ lvf = lv (7.59) Aus der Kombination der Gleichung (7.57) mit (7.58) und (7.59) folgt: (7.60) (m c d pd + mtwc pw )(Ti − T f ) = ( mvf − mvi ) lv Teilen wir beide Seiten durch md und verwenden die Definition des Mischungsverhältnisses: w = ρv/ρd = mv/md: (7.61) ⎛ ⎞ mtw c pw ⎟ ⎜ c pd + md ⎝ ⎠ T −T = w − w ( i f) f i lv Abbildung 7.2. Veranschaulichung der dimensionslosen Funktionen f(Tf) und g(Tf) zur Bestimmung der wet-bulb Temperatur. [Bohren] 112 Ursprünglich gegeben sind die Temperatur Ti und das Mischungsverhältnis wi für die Luft, die wir in den Zylinder sperren. Um die gesuchte End-Temperatur Tf zu erhalten, müsste die Gleichung (7.61) nach Tf aufgelöst werden. Das Wasserdampfmischungsverhältnis wf ist der Sättigungsdampfdruck des Wassers bei der End-Temperatur Tf : wf = ws(Tf). Wie eingangs vermutet, hängt Tf von der Gesamtmasse des Wassers mtw ab, bzw vom Verhältnis der Massen mtw/md. Da es sich um isobare Kühlung durch Verdampfen handelt muss gelten Tf ≤ Ti und wf ≥ wi. Wir lösen die Gleichung (7.61) indem wir den Schnittpunkt der beiden dimensionslosen Funktionen f(Tf) und g(Tf) bestimmen (s. Abbildung): (7.62) ⎛ ⎞ mtw c pw ⎟ ⎜ c pd + md ⎠ T −T f (T f ) = ⎝ (i f lv ) g (T f ) = ws (T f ) − wi (7.63) Beide Funktionen sind positiv für Tf ≤ Ti. f(Tf) = 0 für Tf = Ti und nimmt ab für Tf < Ti. g(Tf) = 0 für Tf = Td und nimmt zu mit höheren Temperaturen. Die Temperatur Tf am Schnittpunkt der beiden Funktionen nimmt zu, je größer das Verhältnis mtw/md ist. Der niedrigste Wert von Tf wird dann erreicht, wenn dieses Verhältnis gegen null geht. Da mtw nicht gleich null sein kann (irgendeine Menge flüssigen Wassers muss ja verdampfen, um die Kühlung zu bewirken), muss md gegen unendlich gehen. Diesen Minimalwert von Tf nennen wir die wet-bulb Temperatur oder Feuchtetemperatur Tw, die wir aus der Lösung der Gleichung (7.64) c pd lv (Ti − Tw ) = ws (Tw ) − wi bestimmen können. Nach (7.14) gilt, da e << p: (7.65) w= εe p−e ≈ εe p Mit dieser Näherung folgt die psychrometrische Gleichung: (7.66) e = es (Tw ) − pc pd ε lv (T − Tw ) Den Index i haben wir jetzt weggelassen, T ist einfach die ursprüngliche Lufttemperatur. Der Faktor (7.67) pc pd ε lv wird psychrometrische Konstante genannt. Der Wert dieser Konstanten liegt bei ~0.65 hPa/K bei Meereshöhe. 113 Wenn wir die gewöhnliche Temperatur T und die zugehörige wet-bulb Temperatur Tw gleichzeitig messen, so können wir nun den Wasserdampfdruck e und damit auch die relative Feuchte ausrechnen. Wir brauchen lediglich noch es(Tw) auszurechnen, beispielsweise mit Gleichung (6.29) oder (6.30). Die Messung der wet-bulb-Temperatur Tw mit einem Psychrometer (wet-bulb thermometer oder sling psychrometer) war in der Meteorologie ein weit verbreitetes Verfahren zur Bestimmung der relativen Feuchte. Hierbei werden zwei Thermometer benutzt, eines misst normal die Temperatur, während das andere mit einem in destilliertem Wasser getränkten Stoff überzogen ist. Die Thermometer werden gut ventiliert, nach einer Weile wird die Temperatur an beiden Thermometern abgelesen. Man kann zeigen (s. z. B. [Bohren]), dass diese Messung tatsächlich die wet-bulb Temperatur nach (7.64) liefert. Heute werden zur exakten Messung der relativen Feuchte meist präzisere und schnellere Verfahren eingesetzt (z.B. Taupunktspiegel oder Lyman-α-Hygrometer). Zum Schluss dieses Abschnitts definieren wir noch eine weitere Temperatur. Die äquivalente Temperatur ist die Temperatur auf die sich Luft erwärmen würde, wenn man allen in der Luft enthaltenen Wasserdampf in einem adiabatisch-isobaren Prozess auskondensieren würde. Die äquivalente Temperatur folgt sofort aus (7.61), wenn man dort wf= 0 setzt und Tf = Te sowie Ti = T setzt: (7.68) Te = T + lv w c pd + wc pw Diese Temperatur ist rein fiktiv, denn ein solcher Prozess ist nach dem Zweiten Hauptsatz verboten: die Entropie würde in diesem Prozess abnehmen. Für w = 0.001 (sehr trocken) beträgt Te - T ≈ 2°C. 7.5 Vertikalgradient für isentropen Aufstieg eines gesättigten Luftpakets Im weiteren Verlauf des Kapitels wollen wir uns solchen Fällen zuwenden, in denen ein Luftpaket mit Wasserdampf gesättigt ist und Wasser auskondensieren kann. Bei unserer bisherigen Herleitung des trocken-adiabatischen Temperaturgradienten für feuchte Luft unterhalb der Sättigung hatten wir auf die Gültigkeit der Poisson-Relation (7.31) zurückgegriffen. Diese gilt für das gemischte System aus Flüssigkeit, Dampf und trockener Luft nicht mehr. Deshalb müssen wir uns zunächst mit dem Verhalten der Entropie eines solchen Luftpaketes beschäftigen, um im feucht-adiabatischen Fall den vertikalen Temperaturgradienten usw. bestimmen zu können. Für die Gesamtentropie eines solchen Luftpakets aus trockener Luft, Wasserdampf und Flüssigwasser gilt: (7.69) S = mv sv + md sd + mw sw Differenzieren wir diese Gleichung nach der Höhe z und berücksichtigen, dass nach (6.9) lv = T(sv-sw) gilt, sowie die Tatsache, dass (7.70) dmv dm =− w dz dz Außerdem ist dmd/dz = 0. Damit folgt: 114 (7.71) ds ds ds dS dmv lv = + mv v + mw w + md d dz dz T dz dz dz Untersuchen wir nun die einzelnen Terme der Gleichung (7.71). Um den Term dsd/dz zu bestimmen, formen wir die Gleichung (4.38) mit Hilfe des idealen Gasgesetzes um: (7.72) ⎛T ⎞ ⎛V ⎞ S − S0 = Cv ln ⎜ ⎟ + Cv (γ − 1) ln ⎜ ⎟ ⎝ T0 ⎠ ⎝ V0 ⎠ ⎛T ⎞ ⎛ C p ⎞ ⎛ Tp0 ⎞ = Cv ln ⎜ ⎟ + Cv ⎜ − 1⎟ ln ⎜ ⎟ ⎝ T0 ⎠ ⎝ Cv ⎠ ⎝ T0 p ⎠ ⎛T ⎞ ⎛T ⎞ ⎛T ⎞ ⎛p ⎞ ⎛p ⎞ = Cv ln ⎜ ⎟ + C p ln ⎜ ⎟ − Cv ln ⎜ ⎟ + C p ln ⎜ 0 ⎟ − Cv ln ⎜ 0 ⎟ ⎝ p⎠ ⎝ p⎠ ⎝ T0 ⎠ ⎝ T0 ⎠ ⎝ T0 ⎠ ⎛T ⎞ ⎛ p⎞ = C p ln ⎜ ⎟ − ( C p − Cv ) ln ⎜ ⎟ ⎝ T0 ⎠ ⎝ p0 ⎠ Teilt man nun durch die Masse md um auf spezifische Größen zu kommen und benutzt (3.38: cpd - cvd = Rd), so erhält man für die spezifische Entropie der trockenen Luft: (7.73) ⎛T ⎞ ⎛p ⎞ sd − sd 0 = c pd ln ⎜ ⎟ − Rd ln ⎜ d ⎟ ⎝ T0 ⎠ ⎝ p0 ⎠ Um den Vertikalgradienten zu bestimmen, differenzieren wir diese Gleichung nach z: (7.74) dsd c pd dT Rd dpd = − dz T dz pd dz Ganz analog bestimmen wir den Gradienten dsv/dz: (7.75) dsv c pv dT Rv de c pv dT Rv des dT = − = − dz T dz e dz T dz es dT dz Nach Voraussetzung soll das Luftpaket ja immer gesättigt sein, d.h.: e = es , weiterhin können wir dann die Ableitung nach der Temperatur einschieben, da es und T festgelegte, eindeutige Funktionen der Höhe sind. Um die Entropieänderung des flüssigen Wassers zu bestimmen, gehen wir von Gleichung (4.36) in spezifischen Größen aus: (7.76) dsw = cvw p dT + dvw T T Nehmen wir das Wasser in guter Näherung als inkompressible Flüssigkeit an, so ändert sich das Volumen des flüssigen Wassers nicht bei Druckänderungen: dvw ≈ 0 und damit gilt dann für flüssiges Wasser (vgl. (3.35): 115 cw = c pw ≈ cvw (7.77) Es bleibt damit für den Gradienten von sw mit der Höhe: dsw cw dT = dz T dz (7.78) Setzt man die Ausdrücke (7.74), (7.75) und (7.78) in (7.73) ein, so erhält man (7.79) 1 dT mv Rv des dT dS lv dmv = + ( mwcw + mv c pw + md c pd ) − dz T dz T dz es dT dz − md Rd dpd pd dz Weiterhin nutzen wir mtw = mw + mv = const. , (7.80) die Beziehung (6.24): dlv = c pv − cw dT (7.81) und die Clausius-Clapeyron-Gleichung (6.14): l 1 des = v 2 es dT RvT (7.82) Damit folgt aus (7.79): (7.83) dS 1 dT d ⎛ lv mv ⎞ md Rd dpd = ( mtwcw + md c pd ) + − dz T dz dz ⎜⎝ T ⎟⎠ pd dz Da wir am adiabatischen Temperaturgradienten interessiert sind, setzen wir weiterhin dS = 0. Dann muss man "nur noch" die Gleichung (7.83) nach dem Temperaturgradienten dT/dz auflösen. Wir überspringen die diversen mathematischen Schritte und präsentieren direkt die Lösung: (7.84) ⎛ ⎜ 1 + lv ws dT g ⎜ RT Γs = − = ε lv2 ws dz c p ⎜ ⎜⎜ 1 + 2 ⎝ c p Rd T ⎞ ⎟ ⎟ ⎟ ⎟⎟ ⎠ Hierbei ist cp noch definiert als: 116 (7.85) cp = mtwcw + md c pd md Der Faktor vor der Klammer entspricht daher dem bekannten trocken-adiabatischen Temperatur-Gradienten Γd = g/cpd, nur cp ist nicht exakt gleich cpd. Die ausführliche Ableitung der Gleichung wird beispielsweise in [Bohren] S.288-291, oder in [Curry] S. 176-177 gegeben. Wir haben somit einen Ausdruck für den feucht-adiabatischen Temperaturgradienten. Wenn ein ungesättigtes Luftpaket isentrop aufsteigt, so sinkt seine (virtuelle) Temperatur mit dem trocken-adiabatischen Temperaturgradienten. Dabei nähern sich Taupunkt und Temperatur an. Ist schließlich am lifting condensation level LCL, Sättigung erreicht, so beginnt das Wasser auszukondensieren. Es bilden sich Wolken. An diesem Punkt ändert sich der adiabatische Temperaturgradient diskontinuierlich zum feucht-adiabatischen Gradienten Γs. Setzt man typische Zahlenwerte am LCL in Gleichung (7.84) ein, so erhält man Werte für Γs um 5°C/km. Der feucht-adiabatische T-Gradient ist hier also etwa einen Faktor 2 kleiner als der trocken-adiabatische. Eine Annahme, von der wir bisher immer ausgingen, ist, dass die Gesamtmasse des Wassers mtw sich beim Aufstieg des Luftpakets nicht ändert (es regnen also keine Tröpfchen aus der Wolke aus). Auch diese Einschränkung werden wir im weiteren Verlauf der Überlegungen wegfallen lassen. Als Beispiel für die Konsequenzen, die aus dem Unterschied zwischen trocken- und feuchtadiabatischem Temperaturgradienten erwachsen, sei qualitativ das Entstehen des Föhns in den Alpen erwähnt. Der Föhn ist als trockener und warmer Fallwind im Lee alpiner Gebirgsketten bekannt. Die Abbildung 7.3 zeigt ein vereinfachtes Schema, wie der Föhn entsteht: man sieht, wie die Temperatur zunächst mit ~10°C/km sinkt, sich dann am LCL Wolken bilden und die Temperatur nur noch mit ~5°C/km sinkt. Dann regnen die Wolken aus (bisher nicht berücksichtigt in unser Ableitung des feucht-adiabatischen Temperaturgradienten). Im Lee des Gebirgszuges sinken die Luftmassen dann wieder ab. Da die Luft nun nicht mehr mit Wasserdampf gesättigt ist, steigt die Temperatur nun entsprechend des trocken-adiabatischen Temperaturgradienten an und führt zur warmen und trockenen Luft auf der Gebirgs-Leeseite. Abbildung 7.3. Vereinfachtes Modell des alpinen Föhns. [Roedel] 117 7.6 Äquivalente temperatur potentielle Temperatur und potentielle Feuchte- Im vorhergehenden Abschnitt haben wir den feucht-adiabatischen Temperatur-Gradienten für eine gegebene Höhe eingeführt. Konvektionsereignisse können sich über einen größeren Höhenbereich erstrecken. Um das Temperaturprofil zu erhalten müsste man dann den Ausdruck für Γs über den ganzen Höhenbereich integrieren. Wir werden diesen Weg nicht verfolgen. Stattdessen suchen wir einen Temperatur-Parameter, der für den feuchtadiabatischen Anstieg über die ganze Höhe hinweg konstant bleibt. So wie die potentielle Temperatur im trocken-adiabatischen Fall zur (erhaltenen) Entropie äquivalent ist, und daher auch erhalten bleibt, suchen wir nun eine ähnliche Erhaltungsgröße für den feuchtadiabatischen Fall. Mit adiabatisch ist hier wie immer der reversibel-adiabatische, also isentrope, Fall gemeint. Gleichung (7.83) können wir umformen in: (7.86) 1 1 ⎛l w ⎞ R dS = ( wtwcw + c pd ) dT + d ⎜ v s ⎟ − d dpd md T ⎝ T ⎠ pd Hierbei sind wir wieder auf die Schreibweise als totales Differential übergegangen, haben die Gleichung durch md geteilt und haben benutzt, dass wtw = mtw/md = const, und ws = mv/md ist. Für isentrope Prozesse gilt wieder dS = 0. Gleichung (7.86) kann im folgenden integriert werden, um T als Funktion von pd zu erhalten für ein gegebenes konstantes wtw. Für ein Luftpaket, in dem der Wasserdampfdruck gesättigt ist, gilt p = pd + es(T), und daher legen pd und T den Druck p eindeutig fest. Wir definieren die Größe: ⎛p ⎞ Θd = T ⎜ 0 ⎟ ⎝ pd ⎠ (7.87) Rd / c p Hier ist p0 wieder ein Referenzdruck (üblicherweise 1000 hPa) und nach (7.85) wieder gilt: cp = cpd + wtwcw. Die Größe Θd entspricht der potentiellen Temperatur für feuchte ungesättigte Luft, nur mit pd statt p, Rd statt R und cp statt (cpd + cpv). Wir schreiben (7.86) um: (7.88) lw ⎞ 1 ⎛l w ⎞ ⎛ dS = c p d ( ln Θ d ) + d ⎜ v s ⎟ = d ⎜ c p ln Θ d + v s ⎟ md T ⎠ ⎝ T ⎠ ⎝ Setzen wir nun dS = 0 und integrieren die Gleichung von einem Anfangszustand bei T und pd(p) zu einem End-Zustand, gekennzeichnet durch den Index f: (7.89) c p ln ⎛ lv (T f ) wsf l w ⎞ = −⎜ − v s⎟ ⎜ Θd Tf T ⎟ ⎝ ⎠ Θdf wobei Θd und ws Funktionen von T und p sind. Bei einer genügend großen Höhe (niedriger Druck, z.B. nahe der Stratosphäre) nehmen wir an, dass aller Wasserdampf auskondensiert ist (wsf = 0). Θdf definiert dann die äquivalente potentielle Temperatur: (7.90) c p ln Θe lv ws = Θd T 118 beziehungsweise: (7.91) ⎛l w ⎞ Θe = Θ d exp ⎜ v s ⎟ ⎜ c pT ⎟ ⎝ ⎠ Äquivalente potentielle Temperatur ist also näherungsweise die potentielle Temperatur, die ein Luftpaket hätte, wenn man es zu einer so großen Höhe anhebt, dass aller Wasserdampf auskondensiert. Dies gilt nur näherungsweise, da die Definition des Exponenten von Θ nicht dem Exponenten von Θe entspricht, auch nicht für ws = 0. Für einen gesättigten Prozess (e = es) hat die Größe Θe die gleiche Beziehung zur Entropie, wie die potentielle Temperatur Θ im trockenen Fall: (7.92) 1 dS = c p d ln Θe md Während wir im trockenen Fall (vgl. (4.44)) gefunden hatten: (7.93) 1 dS = c pd d ln Θ md Diese Analogie erklärt den Namen "äquivalente" potentielle Temperatur. Im feuchtadiabatischen Fall ist nun also Θe die Erhaltungsgröße -ausgedrückt als eine Temperatur- für isentrope Prozesse. Die Tatsache, dass Θe erhalten bleibt, ist sehr praktisch, wenn Prozesse, in denen Kondensation und Evaporation auftreten, behandelt werden sollen. Obwohl wir Θe gerade für gesättigte Luft definiert hatten, kann man diese Größe auch für untersättigte Luft (mit Temperatur T, Druck p und Wasserdampfmischungsverhältnis w) definieren, indem man annimmt, dass die Luft zum LCL (dort wird e = es, und w = ws) angehoben wird. Dort wird Θd zu: (7.94) Θ d , LCL = TLCL ⎛ ⎞ p0 ⎜⎜ ⎟⎟ ⎝ pLCL − es (TLCL ) ⎠ Rd / c p und daher: (7.95) ⎛l w⎞ Θe = Θ d , LCL exp ⎜ v , LCL ⎟ ⎜ c pTLCL ⎟ ⎝ ⎠ da TLCL und pLCL für ein ungesättigtes Luftpaket eindeutig festgelegt sind durch den thermodynamischen Zustand des Pakets (T,p,w), und während eines trocken-adiabatischen Auf- oder Abstiegs des Luftpakets erhalten bleiben, ist Θe auch für ein ungesättigtes Luftpaket eine Erhaltungsgröße. Die äquivalente potentielle Sättigungs-Temperatur eines ungesättigten Luftpakets ist die äquivalente potentielle Temperatur, die das Paket hätte, wenn es gesättigt wäre. Das heißt: 119 ⎛ l (T ) ws (T ) ⎞ Θes = Θ d exp ⎜ v ⎟⎟ ⎜ c pT ⎝ ⎠ (7.96) Die Größe Θes bleibt jedoch beim isentropen Auf- oder Abstieg des Luftpaketes nicht erhalten. Für gesättigte Luft ist natürlich Θe = Θes. Die Größe ws spielt in den Gleichungen (7.91) und (7.96) eine unterschiedliche Rolle. Im ersten Fall ist es das tatsächliche Dampfdruckmischungsverhältnis des Pakets, in (7.96) ist es hingegen das zur aktuellen Temperatur T zugehörige Sättigungs-Dampfdruckmischungsverhältnis des ungesättigten Pakets. Werte von Θe und Θes für ungesättigte Luft auszurechnen, ist etwas mühsam, innerhalb von ~0.5°C Fehlergrenze gelten aber die folgenden Näherungen: (7.97) ⎛ w ⎞ Θe = Θ exp ⎜ 2.675 ⎟, TLCL ⎠ ⎝ w ⎞ ⎛ Θes = Θ exp ⎜ 2.675 s ⎟ . T ⎠ ⎝ Hier sind die Mischungsverhältnisse in g/kg einzusetzen und Θd ist durch Θ angenähert. Bisher sind wir immer von einem geschlossenen System ausgegangen, bei dem auch alles flüssige Wasser während des adiabatischen Aufstiegs im Luftpaket bleibt und mit aufsteigt. Tatsächlich kann es natürlich passieren, dass die flüssigen Tröpfchen als Niederschlag ausfallen und so aus dem Luftpaket entfernt werden. Wenn Niederschlag ausfällt, müssen wir das Luftpaket als offenes System behandeln. In einem pseudo-adiabatischen Prozess nehmen wir an, dass alles kondensierte Wasser sofort aus dem Luftpaket entfernt wird. So ein Prozess ist zwar noch adiabatisch in dem Sinne, dass δQ = 0 nach wie vor gilt, aber die Masse des Wassers mtw ist nicht mehr konstant. Die Gleichung (7.86) kann für einen solchen Prozess modifiziert werden, indem wir wtw durch ws ersetzen: (7.98) (w c s w + c pd ) 1 ⎛l w ⎞ R dT + d ⎜ v s ⎟ − d dpd = 0 T ⎝ T ⎠ pd Gleichung (7.86) und (7.98) haben beide die Form (7.99) 1 1 ⎡R ⎛ l w ⎞⎤ dT = ⎢ d dpd − d ⎜ v s ⎟ ⎥ T c p ⎣ pd ⎝ T ⎠⎦ wobei einmal cp = cpd + wtwcw (adiabatisch) und im zweiten Fall cp = cpd + wscw (pseudoadiabatisch) ist. Die Abnahme der Temperatur ist stärker im Falle des pseudo-adiabatischen Aufstiegs der Luft, als im adiabatischen Fall, da cp in diesem Falle kleiner ist. Die Abbildung 7.4 vergleicht die Temperatur als Funktion des Drucks für adiabatischen und pseudoadiabatischen Aufstieg ausgehend von einer ursprünglichen Temperatur von 25°C bei einem Druck von 900 hPa. Die Kurven wurden durch numerische Integration der Gleichung (7.99) bestimmt. 120 Abbildung 7.4. Vergleich von adiabatischem und pseudo-adiabatischem Aufstieg eines Luftpakets von 25°C und 900 hPa aus. [Bohren] Die Realität liegt irgendwo zwischen dem adiabatischen und dem pseudo-adiabatischen Fall. Für einige Arten von Wolkenbildung kann man davon ausgehen, dass sich kein Niederschlag bildet, z.B. flache Schönwetterwolken, so dass man vom adiabatischen Fall ausgehen kann; in hochreichender Konvektion von Kumulo-Nimbuswolken kann durchaus ein großer Teil des flüssigen Wassers durch Niederschlag aus der Luftmasse entfernt werden. Doch selbst in solch einem Fall erwartet man nicht, dass alles flüssige Wasser aus der Wolke entfernt wird. Man kann den Flüssigwassergehalt von Wolken abschätzen, indem man die Differenz wb - ws(T,p) bildet. Dabei ist ws das Sättigungsmischungsverhältnis für den adiabatischen Aufstieg und wb ist das Mischungsverhältnis an der Wolkenbasis (beide in g/kg). Diese Größe heißt adiabatischer Flüssigwassergehalt und stellt eine Obergrenze für den Flüssigwassergehalt einer Wolke dar. Zuletzt wollen wir noch einen weiteren Prozess betrachten: das feucht-adiabatische Absinken eines gesättigten Luftpakets zu einem Druck p0 (gewöhnlich 1000 hPa). Es kann sein, dass man zu einem solchen Luftpaket Wasser hinzufügen muss, um die Sättigung im Abstieg aufrecht zu erhalten. Die Temperatur des Pakets nach gesättigtem adiabatischem Abstieg auf p0 heißt dann potentielle Feuchtetemperatur oder wet-bulb potential temperature Θw. um einen genäherten Ausdruck für Θw zu erhalten gehen wir von Gleichung (7.89) aus, wobei cp als konstant angenommen wird Als Anfangszustand des Pakets nimmt man den Zustand des Pakets am LCL. Es gilt Θw = Tf = Θdf. (7.100) c p ln Θ w lv , LCL ws lv ( Θ w ) ws ( Θ w ) = − Θd Θw TLCL Diese Gleichung kann nur numerisch gelöst werden. Eine Näherungslösung lautet: 121 Θ w ≈ Θe − (7.101) lv ws ( Θ w ) cp Die potentielle wet-bulb Temperatur ist die potentielle Temperatur, die ein Paket hätte, wenn es während eines gesättigten Absinkens der Luftmasse auf p0 durch Verdunstung abgekühlt würde. Es lässt sich zeigen, dass auch Θw eine Erhaltungsgröße ist. Zum Überblick über die verschiedenen diskutierten Temperaturen fassen wir die wichtigsten Ergebnisse und in der folgenden Tabelle zusammen: T Temperatur Td Taupunkt-Temp. Td ≤ T Tv virtuelle Temp. Tv ≥ T Tw wet-bulb Temp. (Feuchtetemperatur) Td ≤ Tw ≤ T Te äquivalente Temp. Te ≥ T Tse gesätt. äquiv. Temp. Θ potentielle Temp. Θv Θw Θe pot. virt. Temp. pot. wet-bulb Temp. pot. äquivalente Temp. (auch pseudo-pot. Temp.) Θse pot. gesätt. äquiv. Temp. aktuelle Lufttemperatur Temperatur, bei der aktuelle Feuchte = 100% Feuchte wäre Temperatur, die ungesättigte Luft hätte, wenn sie frei von H2O-Dampf wäre, bei Druck und Dichte gleich minimale Temperatur, auf die man Luft abkühlen kann durch Evaporationskühlung Temperatur, die Luft hätte, wenn aller Wasserdampf adiabatisch, isobar auskondensiert würde äquival. Temperatur , die ungesättigte Luft hätte, wenn sie gesättigt wäre. Temperatur, die trockene Luft hätte, wenn adiabatisch zum Boden gebracht = Trocken-Adiabate virtuelle Temp. adiabatisch zum Boden gebracht. Temp der Feucht-Adiabate pot. Temp der Trocken-Adiabate, die gleich der Feucht-Adiabate für p → 0 pot. äquival. Temperatur , die ungesättigte Luft hätte, wenn sie gesättigt wäre. 7.7 Thermodynamische Diagramme Thermodynamische Prozesse in der Atmosphäre können sehr zweckmäßig mit Hilfe sogenannter thermodynamischer Diagramme visualisiert werden. Insbesondere können an diesen Diagrammen bestimmte Größen, wie z.B. einige von den Temperaturen, die in den vorhergehenden Abschnitten eingeführt wurden, direkt abgelesen werden. Thermodynamische Strukturen in der Atmosphäre, wie z.B. stabile Schichtungen oder instabile Konvektionssituationen können an den thermodynamischen Diagrammen dargestellt werden. Ausführlich diskutieren werden wir das am häufigsten gebräuchliche Diagramm: das skew Tlog p-Diagramm. 7.7.1 skew T-log p Diagramm Die Basiskoordinaten des skew T-log p Diagramm (schiefes Emagramm) sind die Temperatur T und der Druck p. Dabei wird die Temperatur T in einem Winkel von 45° relativ zur pAchse aufgetragen (schiefwinkliges Koordinatensystem). Dies hat vor allem den Vorteil, dass durch diese Auftragung der Winkel zwischen Adiabaten und Isothermen größer wird, wodurch die Analyse der Stabilität von Luftschichten erleichtert wird. Der Druck p wird als negativer Logarithmus (-ln p) aufgetragen, somit entspricht die Druckauftragung in etwa der Höhe. Die Abbildung zeigt die Basiskoordinaten des skew T-log p Diagramms. 122 Abbildung 7.5. Basiskoordinaten des skew T-log p Diagramms. [Bohren] Trocken-Adiabaten sind definiert als Kurven, für die gilt: (7.102) ⎛ p⎞ T = Θ⎜ ⎟ ⎝ p0 ⎠ Rd / c pd wobei Θ konstant ist. In erster Näherung können wir diese Gleichung auch schreiben als: (7.103) ⎛ p⎞ c − ln ⎜ ⎟ ≈ pd ⎝ p0 ⎠ Rd ⎛ T⎞ ⎜1 − ⎟ ⎝ Θ⎠ In dieser Näherung sieht man, dass -ln p als Funktion von T genähert einer Geraden mit negativer Steigung entspricht. Durch die Drehung der T-Achse um 45° werden die TrockenAdiabaten in etwa rechtwinklig zu den Isothermen. Wir können mit dem skew T-Diagramm also, wenn wir Temperatur und Druck einer Einzel-Messung oder einer ganzen Reihe von Messdaten, beispielsweise aus einem Ballonaufstieg, eintragen, direkt die zugehörigen potentiellen Temperaturen ablesen (s. Abbildung). Wir müssen zwei Dinge unterscheiden beim benutzen von thermodynamischen Diagrammen: Strukturen und Prozesse. Beides kann mit Hilfe von thermodynamischen Diagrammen gezeigt werden, man muss sich nur im klaren darüber sein, was gerade gezeigt wird: stellt z.B. die Linie AB in der Abbildung die Temperaturmesswerte eines Ballonaufstiegs (Vertikalprofil) dar, dann können wir sehen, dass es sich bei dieser Struktur um eine stabile Schichtung handelt, da der Temperatur-Gradient weniger mit der Höhe abnimmt, als trocken-adiabatisch. Wollen wir hingegen einen trocken-adiabatischen Prozess visualisieren, z.B. den trockenadiabatischen Aufstieg eines Luftpakets vom Punkt C aus, so müssen wir nur den Punkt C entlang der Trocken-Adiabate verschieben. Wir können dann direkt am Diagramm ablesen, wie sich Druck und Temperatur bei einem solchen trocken-adiabatischen Aufstieg ändern würden. 123 Abbildung 7.6. Skew T -ln p Diagramm mit Trocken-Adiabaten. [Bohren] Feucht-Adiabaten (Adiabaten für Prozesse, bei denen die Luftmasse mit Wasserdampf gesättigt ist) ergeben sich aus der numerischen Integration der Gleichung (7.99)für einen pseudo-adiabatischen Prozess mit cp = cpd + wscw. Feucht-Adiabaten sind also genaugenommen Pseudo-Feucht-Adiabaten! Die Feucht-Adiabaten sind meist mit Θw-Werten beschriftet, denn Θw ist Erhaltungsgröße für feucht-adiabatische Prozesse und Θw ist die Temperatur bei 1000 hPa. In Abbildung 7.7 sind die Feucht-Adiabaten, wie sie im skew TDiagramm erscheinen eingetragen. Für niedrige Drücke und Temperaturen (wenn fast aller Wasserdampf auskondensiert ist) werden die Feucht-Adiabaten parallel zu den TrockenAdiabaten. Man kann daher auch jede Feucht-Adiabate mit einem Θe-Wert belegen, der dem Wert der Trocken-Adiabaten entspricht, gegen den die Feucht-Adiabate konvergiert. Aus der Definition des Mischungsverhältnisses bei Sättigung folgt: (7.104) p = es (T ) + ε es (T ) ws Für konstantes ws definiert diese Gleichung eine weitere Kurve im skew T-Diagramm. Isolinien für konstante ws-Werte sind in Abbildung 7.8 gezeigt. Der Taupunkt von ungesättigter Luft legt beim Druck p das Mischungsverhältnis w fest: w = ws(Td,p). Im ungesättigten Aufstieg oder Abstieg eines Luftpakets ist das zugehörige w eine Erhaltungsgröße. Daher verschiebt sich die Taupunktstemperatur mit abnehmendem Druck entlang der ws-Isolinie. Wird, wie in der Abbildung 7.8 gezeigt, ein TaupunktsTemperaturprofil zwischen A und B gemessen und sind wir dann daran interessiert, wie die Taupunktstemperatur abnimmt, wenn man ein Luftpaket adiabatisch von p(C) nach p(D) verschiebt, dann muss man die Taupunktstemperatur von C → D entlang der ws-Isolinie überführen. Wie wir im folgenden sehen werden, kann man so leicht die zugehörige LCLHöhe finden, wenn T(p) und Td(p) bekannt sind, indem man für T der zugehörigen TrockenAdiabate folgt, und für Td der zugehörigen ws-Isolinie, am Schnittpunkt, wo dann Td = T ist, fängt das Wasser an auszukondensieren, dort ist das LCL. 124 Abbildung 7.7. Skew T -ln p Diagramm mit Feucht-Adiabaten. Die Isolinien sind mit den potentiellen wet-bulb Temperaturen bezeichnet. [Bohren] Nach Gleichung (7.14) ist w ≈ εe/p, mit ε = 0.622. Daher gilt: (7.105) w ≈ 622 e p Abbildung 7.8. Skew T -ln p Diagramm mit Iso-ws-Linien. [Bohren] 125 wenn man w in g/kg angibt. Das heißt aber auch, dass für p = 622 hPa, das Mischungsverhältnis gerade dem Wasserdampfdruck in hPa entspricht. Die 622 hPa-Linie ist in der Abbildung ebenfalls eingetragen und die ws-Linien sind dort mit ihren Werten bezeichnet. Man kann somit vom skew T-Diagramm unmittelbar den Sättigungsdampfdruck des Wassers ablesen: man folge z.B. der 0°C Temperaturlinie zum Schnittpunkt mit der 622 hPa Drucklinie. Der Wert von ws an dieser Stelle entspricht auch es, nämlich 6 g/kg bzw. 6 hPa, was dem uns nach Clausius-Clapeyron bekannten Wert entspricht. Ein skew T-log p Diagramm mit allen nun eingeführten Isolinien ist in Abbildung 7.9 gezeigt, inklusive Temperatur- und Taupunktprofil. Man kann sowohl die LCL-Höhe feststellen und auch stabile Inversionsschichten erkennen z.B. bei 850 hPa. Abbildung 7.9. Vollständiges skew T-ln p Diagramm mit gemessenem Vertikalprofil für Temperatur und zugehörigen Taupunkttemperaturen (gemessen über dem äquatorialen Pazifik). [Bohren] Abbildung 7.10. Thermodynamische Größen im skew T -ln p Diagramm. [Bohren] 126 Wir diskutieren die in den vorherigen Abschnitten eingeführten Temperaturen und anderen thermodynamischen Größen anhand der Abbildung 7.10 es handelt sich um ein skew T Diagramm, aus dem alle Isolinien, die nicht für unsere Analyse relevant sind, entfernt wurden. Man betrachte ein ungesättigtes Luftpaket mit Druck p, Temperatur T und Taupunkt Td, dann sind Θ und w schon eindeutig festgelegt. Man verschiebe nun das Paket adiabatisch nach oben. Da noch keine Sättigung erreicht ist, folgt die Temperatur der zunächst der TrockenAdiabate, während die Taupunktstemperatur der Linie konstanten Mischungsverhältnisses w folgt. An dem Punkt, an dem sich Td und T treffen, liegt das lifting condensation level LCL für unser Luftpaket. Setzt man den adiabatischen Aufstieg über das LCL hinaus fort, so folgen nun Temperatur und Taupunkt der Feucht-Adiabate. Die äquivalente Temperatur erhält man nun, indem man der Feucht-Adiabate zu niedrigen Drücken und niedrigen Temperaturen folgt, wo fast alles Wasser auskondensiert ist, und daher Trocken- und Feucht-Adiabate konvergieren. Die Temperatur dieser Trocken-Adiabate bei 1000 hPa entspricht der äquivalenten potentiellen Temperatur Θe für unser Luftpaket; der Punkt an dem diese Trocken-Adiabate das ursprüngliche p-Level schneidet ist die zugehörige äquivalente Temperatur Te. Auch eine Abschätzung für die adiabatische wet-bulb-Temperatur Tw können wir aus dem skew T-Diagramm ablesen, wenn wir von pLCL aus der Feucht-Adiabate zurück zum Druck p folgen (pseudo)-adiabatisch. Die adiabatische potentielle wet-bulb-Temperatur Θw liest man am Schnittpunkt der Feucht-Adiabaten mit der 1000 hPa-Linie ab. Leider entsprechen die hier eingeführten adiabatischen wet-bulb Temperaturen nicht exakt den isobar-adiabatischen wet-bulb Temperaturen, die wir in Abschnitt 7.4 eingeführt hatten! Die Arbeit, die in einem Kreisprozess verrichtet wird, wobei der Kreisprozess durch eine geschlossene Kurve im skew T-Diagramm repräsentiert wird, entspricht der umschlossenen Fläche. Wir können uns diese Tatsache plausibel machen, indem wir schreiben: (7.106) ∫ p dv = ∫ d ( pv ) − ∫ v dp = − ∫ v dp da für das Integral (7.107) ∫ d ( pv ) = ∫ d ( RT ) = 0 T gilt. Ebenfalls mit dem idealen Gasgesetz kann man weiter umformen: (7.108) − ∫ v dp = − ∫ RT dp = R ∫ T ( − d ln p ) p So, wie im V-p-Diagramm die Arbeit proportional zur umschlossenen Fläche ist, so ist auch im T -ln p -Diagramm die Arbeit proportional zur umschlossenen Fläche. Die schiefwinklige Anordnung der Koordinaten ändert daran nichts. Das einfache Emagramm entspricht dem skew T-log p Diagramm, nur wird die Temperatur im rechten Winkel zur Druckkoordinate aufgetragen. 127 Abbildung 7.11. Tephigramm, gepunktete Linien sind Linien konstanten ws in g/kg. Strich-Punkt-Linien sind Feucht-Adiabaten. Gestrichelte Linien sind Isobaren. [Bohren] 7.7.2 Tephigramm Im Tephigramm wird die Temperatur T rechtwinklig gegen den Logarithmus der potentiellen Temperatur aufgetragen. Der Logarithmus von Θ ist ja proportional zur Entropie s, daher heißt das Tephigramm auch T-s-Diagramm, bzw. die Größe s=cp ln Θ wird auch mit Φ bezeichnet, daher der Name Te-phi-gramm. Die Isobaren erhält man aus der Poisson-Relation (7.109) p (1−γ ) / γ T = const. Die Isobaren verlaufen ungefähr gradlinig im 45°-Winkel zu den T-Koordinaten. Daher ähnelt das Tephigramm stark dem skew T-Diagramm, wenn man dieses um 45° dreht. Auch im Tephigramm entspricht bei einem Kreisprozess die umschlossene Fläche der verrichteten Arbeit. Ein vollständiges Tephigramm mit Feucht-Adiabaten und ws-Isolinien ist in Abbildung 7.11 gezeigt. 128 Abbildung 7.12. Stüve-Diagramm. [Bohren] 7.7.3 Stüve-Diagramm Als letztes Diagramm wollen wir noch das Stüve-Diagramm, welches vom Deutschen Wetterdienst als thermodynamisches Diagramm verwendet wird, diskutieren. Hier ist die R/c Ordinate der inverse Druck, der mit p p skaliert. Als Abszisse wird die Temperatur verwendet. D.h. alle Trocken-Adiabaten sind dann gerade Linien, die im Punkt p = 0 zusammenlaufen (s. Abbildung). Dadurch werden die trocken-adiabatischen Stabilitätskriterien besonders einfach. Andererseits ist die Energie in diesem Bild nicht flächenproportional (es gibt aber Korrekturfaktoren). Auch die Linien konstanten ws sind gerade Linien. 129 7.8 Konvektion, Wolkenbildung, CAPE und CINE In Kapitel 3.7 hatten wir bereits die Stabilitätskriterien für trockene Luftmassen diskutiert und im Abschnitt 7.3 mit Hilfe der virtuellen Temperatur auf feuchte aber noch untersättigte Luftpakete ausgedehnt. Entscheidend für den Auftrieb eines Luftpakets war immer der Vergleich mit seiner Umgebung. Ergibt sich ein fortwährender Auftrieb auf Grund der unterschiedlichen Dichten, so steigt das Luftpaket konvektiv auf. Achtung: Um exakt zu beurteilen, ob sich ein Auftrieb ergibt, müsste die Adiabate der virtuellen Temperatur des Pakets mit dem virtuellen Temperaturprofil der umgebenden Luftmasse verglichen werden (vgl. Abschnitt 7.3). Man müsste also zunächst aus den Messungen der Temperatur und der Feuchten (bzw. Wasserdampfmischungsverhältnissen) die virtuellen Temperaturen sowohl des adiabatisch aufsteigenden Luftpakets als auch der Umgebung berechnen und anschließend vergleichen. Tatsächlich werden auf thermodynamischen Diagrammen meist nur die tatsächlichen Temperaturen der Umgebung mit den gewöhnlichen Adiabaten des interessierenden Luftpakets verglichen! Auf den Diagrammen sind auch nur die (Pseudo)Feucht- und Trockenadiabaten der Temperatur und nicht die der virtuellen Temperatur eingetragen. Weiterhin muss im gesättigten Fall noch die Definition der virtuellen Temperatur (vgl. (7.41)) erweitert werden um auch das Flüssigwasser mit einzuschließen. Die sogenannten liquid water virtual temperature lautet dann: Tvl = T (1 + 0.608wv − ww ) (7.110) und es ergibt sich für die allgemeinen Stabilitätskriterien: dTvl dz dT − vl dz dTvl Γs < − dz dT − vl dz dT − vl dz − (7.111) < Γs : absolut stabil = Γs : gesättigt neutral < Γd : bedingt stabil = Γd : trocken neutral > Γd : absolut instabil Die in Abbildung 7.13 gezeigten Fälle A und C sind schon hinreichend bekannt. Neu ist der Fall B: diese Situation nennt man bedingt stabil, denn wenn der Gradient von Tvl zwischen Γd und Γs liegt, dann hängt es davon ab, ob die Luftmasse mit Wasserdampf gesättigt ist oder nicht, ob die Luftschichtung stabil ist. Für den pseudo-adiabatischen Fall, den wir im folgenden annehmen wollen, ist ww = 0 und man kann bei der alten Definition der virtuellen Temperatur Tv bleiben, statt mit Tvl zu arbeiten. Um Konvektion zu beschreiben, und um festzustellen, wie hoch ein Luftpaket aufsteigt, müssen wir das ganze Vertikalprofil von Temperatur und Feuchte oberhalb des Luftpakets betrachten und nicht nur die Dichte/Temperatur von Paket und Umgebung in einer bestimmten Höhe vergleichen. Um die Konvektion eines Luftpakets zu beschreiben, beginnen 130 Abbildung 7.13. Veranschaulichung der Stabilitätskriterien für drei unterschiedliche Gradienten der virtuellen Temperatur; im Fall B entscheidet die Wasserdampfsättigung ob eine Schichtung stabil ist oder nicht. [Curry] wir wie in Gleichung (4.50) mit dem Kräfteansatz und betrachten die Beschleunigung des Luftpakets: (7.112) F = − g ( ρ p − ρ s )V = M d 2z d 2z = ρ V p dt 2 dt 2 ⎛ ρs − ρ p ⎞ d 2z = g⎜ 2 ⎜ ρ p ⎟⎟ dt ⎝ ⎠ (7.113) Diese Gleichung können wir nun verwenden um die Arbeit zu berechnen, die von den Auftriebskräften geleistet wird, um das Luftpaket von einer Ausgangshöhe zi zu einer Endhöhe zf anzuheben. Hierfür formen wir die Gleichung zunächst um, indem wir die Dichten durch spezifische Volumina ersetzen. und müssen dann die Gleichung von zi bis zf über die Höhe integrieren: zf ∫ (7.114) zi d 2z dz = dt 2 ⎛ v p − vs ⎞ g ∫z ⎜⎝ vs ⎟⎠dz i zf Mit Hilfe der hydrostatischen Grundgleichung dp/dz=-g/vs (vgl. (2.20)) können wir diese Gleichung umschreiben in: zf ∫ (7.115) zi pf d 2z dz = − ∫ ( v p − vs )dp dt 2 pi Mit Hilfe des idealen Gasgesetzes und unter Vernachlässigung des Beitrags des flüssigen Wassers zum spezifischen Volumen (pseudo-adiabatisch) gilt dann: zf (7.116) ∫ zi d 2z dz = − Rd dt 2 pf ∫ (T vp − Tvs )d ln p pi 131 wobei Tvp die virtuelle Temperatur des Pakets und Tvs die der Umgebung ist. An dieser Gleichung sehen wir, dass die durch den Auftrieb frei werdende Arbeit pro Einheitsmasse dem Integral der Temperaturdifferenzen zwischen Tvp und Tvs entspricht, wenn man über den Logarithmus des Drucks integriert. Das heißt, im skew T-ln p Diagramm gleicht die Fläche, die zwischen Tvp und Tvs eingeschlossen wird, der Arbeit pro Einheitsmasse, die frei/benötigt wird, um ein Luftpaket vertikal um die Distanz Δz zu verschieben. Abbildung 7.14. Veranschaulichung von Auftrieb, CAPE und CINE. Eigenen Auftrieb hat das vom Boden aus hochgehobene Luftpaket, wenn sein Temperaturgradient langsamer sinkt, als der Umgebungstemperaturgradient. Die dunkle Fläche entspricht der maximalen konvektiven potentiellen Energie CAPE des Pakets, die untere hellgraue Fläche entspricht CINE. [Curry] Zur Illustration dieses Sachverhalts betrachte man die Abbildung 7.14. Die gestrichelte Linie stellt das tatsächliche atmosphärische Temperaturprofil dar. Nun betrachten wir ein Luftpaket, das vom Boden bei 1000 hPa angehoben wird (durchgezogene Linie). Zunächst folgt es dem trockenadiabatischen Temperaturgradienten bis zum LCL, ab dort ist die Wasserdampfsättigung erreicht, es bilden sich Wolken und das Luftpaket folgt bei weiterer adiabatischer Hebung nun dem feuchtadiabatischen Temperaturgradienten. Es erreicht den Punkt LFC, das Level of Free Convection, ab hier ist die Abnahme der Temperatur mit der Höhe für das Paket geringer als für die Umgebung. Es erfährt also einen Auftrieb und würde von alleine in freier Konvektion aufwärts streben. Vom Boden bis zum LFC müsste das Paket jedoch dynamisch "aktiv" angehoben werden (in der Atmosphäre beispielsweise dadurch, dass das Luftpaket einen Berg hochgeschoben wird, oder dadurch, dass an einer Front sich kalte Luft unter das wärmere Luftpaket schiebt). Ab dem LFC steigt das Luftpaket nun in freier Konvektion ähnlich einem Heissluftballon auf. Es folgt weiterhin der Feuchteadiabaten und die durch Kondensation freiwerdende latente Wärme heizt unser Luftpaket und führt zu zusätzlichem Aufstieg. Spätenstens an der Tropopause, wenn die Umgebungstemperatur nicht mehr weiter abnimmt, kreuzen sich die Feuchteadiabate des Pakets und das Umgebungstemperaturprofil wieder. Hier befindet sich das Level of Neutral Buoyancy (LNB), an diesem Punkt erfährt das Paket weder positive noch negative Auftriebskräfte. Aufgrund seiner Vertikalgeschwindigkeit (s.u.) und seiner eigenen Trägheit steigt das Paket aber noch ein Stück über den LNB hinaus auf ("überschießen"). Die Wolkenoberkante wird also erst oberhalb des LNB zu finden sein. Die positive Fläche die von der Umgebungstemperatur und dem Luftpaket zwischen dem LFC und dem LNB eingeschlossen wird, definiert die maximale Arbeit, die von den Auftriebskräften verrichtet werden kann. 132 Diese Fläche ist ein Maß für die maximale Stärke der Konvektion, die ein Luftpaket haben kann. Sie ist damit gleichzeitig ein wichtiges Maß für die maximale Stärke, die ein Gewitter haben kann, das aus hochreichender Konvektion entsteht. Wie wir gleich sehen, kann daraus auch die maximale Geschwindigkeit des Aufwinds berechnet werden. Das Integral auf der linken Seite der Gleichung (7.116) repräsentiert auch die Änderung der kinetischen Energie pro Einheitsmasse, die das Paket zwischen z1 und z2 erfährt. Dies sieht man, wenn man die Integrationskonstante nach dt transformiert und die Vertikalgeschwindigkeit dz/dt=vz einsetzt: zf (7.117) ∫ zi tf d 2z dv 1 1 dz = ∫ z v z dt = v 2zf − v zi2 2 2 2 dt dt ti Wählen wir die Höhe z1=LFC als untere Integrationsgrenze und z2=LNB als obere Grenze, dann beinhaltet die Integration die maximale Arbeit, die durch konvektiven Auftrieb verrichtet werden kann. Diese Größe nennt man CAPE für Convection Available Potential Energy: (7.118) CAPE = − Rd pLNB ∫ (T vp − Tvs ) d ln p pLFC Die maximale potentielle Energie, die in dem Luftpaket steckt, bevor es angehoben wird, kann so also leicht bestimmt werden. Wir können die Situation mit einem Gewicht vergleichen, das auf einem Tisch liegt. Es enthält zunächst potentielle Energie, die dadurch, dass ich es über die Tischkante hinausschiebe, freigesetzt wird. Zunächst muss ich Energie hineinstecken, um das Gewicht über die Kante zu schieben, danach wird potentielle Energie in kinetische umgewandelt, wenn das Gewicht auf den Boden herunterfällt. Ebenso muss ich in unserer Situation zunächst Energie hineinstecken um das Luftpaket bis zum LFC anzuheben, ist das LFC erreicht, dann steigt das Paket von selbst weiter auf und die in ihm steckende potentielle Energie wird in kinetische Energie (Aufwind) umgewandelt. Starten wir bei vzi=0, dann ist die maximale Vertikalgeschwindigkeit, die das Luftpaket annehmen kann gegeben durch: (7.119) v zf = 2 CAPE Als große Werte für CAPE bezeichnet man Werte zwischen 1500 und 2500 J/kg, Werte darüber werden als extrem bezeichnet. Für einen Wert von 1800 J/kg ergibt sich bereits eine Maximalgeschwindigkeit des Aufwinds von 60 m/s! Damit diese potentielle Energie überhaupt freigesetzt werden kann, muss das Paket aber erst einmal bis zum LFC angehoben werden. Hierfür muss am Paket Arbeit geleistet werden, die der hellgrauen Fläche in der Abbildung 7.14 entspricht. Diese Größe nennt man CINE für Convection Inhibition Energy: (7.120) CINE = Rd pLFC ∫ (T vp − Tvs ) d ln p pi So kann beispielsweise eine kleine Inversionsschicht bedeuten, dass keine Konvektion entsteht, obwohl große positive CAPE-Wert vorhanden sind. Für pi wird meist der Luftdruck am Boden eingesetzt. Typischerweise werden auf thermodynamischen Diagrammen, auf denen durch Ballonaufstiege ermittelte Temperaturen und Taupunktstemperaturen dargestellt 133 Abbildung 7.15. Ballonsondierung Fort Worth, Texas, früher Morgen. Dicke Linie: Temperatur, dünne Linie: Taupunkttemperatur. [Bohren] Abbildung 7.16. Ballonsondierung, Fort Worth, Texas, nachmittags. Für ein Paket, das aus Bodennähe startet, besteht praktisch keine CINE-Schwelle für Konvektion mehr, und es besitzt einen großen CAPE-Wert. Obwohl es eine Inversionsschicht bei 730 hPa gibt, ist diese nicht stark genug ausgeprägt um eine hochreichende Konvektion zu verhindern. Ein Paket hingegen, das von 700 hPa aus adiabatisch angehoben würde (B), hätte sein zugehöriges LCL bei 600 hPa und es würde keinerlei Auftrieb erfahren, da die Adiabate des Pakets immer links von der Umgebungstemperaturkurve liegt. [Bohren] sind, auch Werte für CAPE und CINE angegeben. Werte für CINE haben natürlich nur Sinn, wenn auch positives CAPE vorhanden ist. Zur Verdeutlichung sehen wir uns Temperaturprofile aus zwei Ballonaufstiegen an: Abbildung 7.15 und Abbildung 7.16. Hier handelt es sich um zwei Profile, die in Fort Worth, Texas, gemessen wurden an einem Tag, an 134 dem nachmittags ein schweres Gewitter aus hochreichender Konvektion entstand. Das erste Profil wurde am frühen Morgen aufgenommen. Auch am Morgen besteht schon eine große CAPE-Fläche, aber es werden noch keine hochreichenden Kumuluswolken beobachtet, denn die stabile Schichtung unterhalb des LFC verhindert die Konvektion. Erst nachmittags, wenn die Sonne den Boden und die darüber liegende bodennahe Grenzschicht erwärmt hat, ist die CINE-Schwelle beseitigt, und es bildet sich hochreichende Konvektion, die zu großen Gewitterwolkentürmen führt. In der Wettervorhersage können geübte Meteorologen bereits aus den Temperaturprofilen am Morgen vorhersagen, ob und in welchem Umfang Konvektion im Verlaufe des Tages entsteht. Die in diesem Abschnitt vorgenommenen Überlegungen zur Konvektion geben die Wirklichkeit natürlich nur angenähert wieder. Insbesondere wurde die Einmischung von Umgebungsluft in die aufsteigenden Luftpakete bisher völlig vernachlässigt. Abbildung 7.17. Schemabild zur konvektiven Temperatur und zum Convective Condensation Layer CCL. [Lawrence] Zum Abschluss dieses Abschnitts wollen wir noch ein weiteres nützliches Konzept kurz erwähnen, das in der Meteorologie im Zusammenhang mit Konvektion häufig verwendet wird. Bisher sind wir immer davon ausgegangen, dass ein Luftpaket zunächst (erzwungenermaßen) angehoben wird, bevor es dann ab dem LFC von selbst weitersteigt. Kondensation tritt in diesem Konzept dann am LCL ein. Betrachten wir nun die Abbildung 7.17. Die Situation ist ganz ähnlich, wie im Fall der Abbildung 7.15 am frühen Morgen. Die dicke Linie symbolisiert das vorliegende Temperaturprofil. Es besteht wiederum eine Inversionsschicht über der Bodengrenzschicht, die sich durch Strahlungskühlung über Nacht gebildet hat. Diese Inversion verhindert zunächst wieder die Konvektion von Luft aus der Grenzschicht. Wir fragen uns nun, um wie viel die bodennahe Luft erwärmt werden muss (der Boden, der von der Sonne erwärmt wird, erwärmt die darüber liegende Luft), damit sie frei aufsteigt. Die sich erwärmende Luft mischt sich turbulent mit den darüber liegenden 135 Luftschichten (keine Pakete mehr!) und es entsteht eine Mischungsschicht mit trockenadiabatischem Temperaturprofil (parallele Linien für konstantes Γd). Die gestrichelte Linie gibt das gesättigte Mischungsverhältnis vor, das dem Gradienten des Taupunkts entspricht, wenn man annimmt, dass das Wasserdampfmischungsverhältnis vom Boden bis zum CCL konstant wäre. In diesem Fall entspricht die konstante wvs-Linie unserem Taupunktgradienten. In Wirklichkeit mischt sich meist trockenere Luft aus den höheren Luftschichten mit der feuchteren Bodenluft. Gleichzeitig verdampft bei den steigenden Temperaturen aber auch Wasser aus dem Boden in die Grenzschicht und erhöht damit wv in der Mischungsschicht. Der Schnittpunkt der Linie des gesättigten Mischungsverhältnisses und ursprünglichem Temperaturprofil bestimmt das CCL für Convective Condensation Level (deutsch: Kumulus-Kondensationsniveau). Das CCL liegt höher als das ursprüngliche LCL. Die Trockenadiabate, die durch diesen Schnittpunkt verläuft, bestimmt die konvektive Temperatur Tc, in der Abbildung mit TB gekennzeichnet. Sie ist die Temperatur, auf die die Bodenluft durch Sonneneinstrahlung erwärmt werden muss, damit die Luftmasse ohne weitere Hebungsmechanismen von selbst ansteigt. Die Wolken bilden sich erst am CCL, nicht schon am LCL, denn erst hier liegt der Schnittpunkt von Temperatur der erwärmten Luft und der Linie des gesättigten Mischungsverhältnisses. 136 8 Thermodynamische Potentiale in der physikalischen Chemie Die heutige Atmosphärenforschung beschäftigt sich in vielen Teilen mit chemischen Reaktionen. Als Beispiele seien nur die chemischen Reaktionszyklen, die zum Ozonabbau in der polaren Stratosphäre, oder die Reaktionen, die den sogenannten Sommersmog auslösen genannt. Auch die Spurenstoffkreisläufe in der Atmosphäre werden wesentlich durch chemische Reaktionen bestimmt. (Beispiel Schwefelkreislauf: Emission als SO2, dann Oxidation von Schwefeldioxid entweder in der Gasphase durch Reaktion mit dem OHRadikal, oder durch Oxidation in der Flüssigphase in Regentröpfchen, dann Ausregnen). Die Erforschung dieser chemischen Reaktionen ist ein weites Feld der aktuellen Atmosphärenforschung. Wesentliche Frage ist dabei meist, wie schnell bzw. wie effektiv läuft eine Reaktion ab. Diese Frage ist die Grundlage der sogenannten Reaktionskinetik, bzw. der Thermochemie von Reaktionen. Wir wollen im folgenden einige wichtige Grundlagen der Reaktionskinetik und der Thermodynamik von chemischen Reaktionen einführen und so das bisher erarbeitete Bild von den Thermodynamischen Potentialen abrunden. Wir beginnen mit einigen Definitionen. 8.1 Standardenthalpien Der Standardzustand einer Substanz bei einer bestimmten Temperatur ist ihr Zustand in reiner Form bei einem Druck von 1 bar. Der Standardzustand von flüssigem Ethanol bei 298 K ist also reines flüssiges Ethanol bei 298 K und 1 bar. Es darf also keine Mischung der Substanz mit einer anderen Substanz vorliegen (Mischungsenthalpie, man denke an die Mischung von Schwefelsäure und Wasser), weiterhin bezieht sich die Definition von Standardgrößen immer auf 1 bar Druck. Die Temperatur ist bei Standardzuständen nicht automatisch festgelegt, in der Regel wird aber immer ein Standardzustand bei 298,15 K, also 25,0 C, angegeben; es ist aber auch denkbar eine einen Standardzustand bei anderen Temperaturen anzugeben. Entsprechend definieren wir als Standardenthalpieänderung ΔH\ bei einem Prozess die Änderung der Enthalpie einer Substanz zwischen ihrem Standard-Anfangszustand und ihrem Standard-Endzustand. Bei einer chemischen Reaktion ist ΔH\ dann die Differenz der Enthalpien der Reaktanden in ihren Standardzuständen und der Produkte in ihren Standardzuständen, alle bei der selben Temperatur. Ein Beispiel für eine Standardenthalpieänderung kennen wir schon von den Phasenübergängen: die Standard-Verdampfungsenthalpie ΔvapH\ ist die Enthalpieänderung pro Mol, wenn eine Flüssigkeit bei 1 bar zu einem Gas bei 1 bar verdampft wird: H2O (l) → H2O (g) ΔvapH\ (373 K) = 40.66 kJ/mol H2O (l) → H2O (g) ΔvapH\ (298,15 K) = 44.02 kJ/mol bzw. bei 25 C: und analog die Standard-Schmelzenthalpie von Wasser: H2O (s) → H2O (l) ΔfusH\ (273 K) = 6.01 kJ/mol 137 Aus Gründen der Einheitlichkeit mit anderen Thermochemischen Größen der physikalischen Chemie wollen wir in diesem Kapitel die Verdampfungsenthalpien etc. nicht mit dem Buchstaben Lv sondern als ΔvapH bezeichnen. Da H eine Zustandsvariable ist muss auch immer ΔH\ (A→B) = -ΔH\ (B→A) sein. Weiterhin führen wir für chemische Reaktionen die Standard-Reaktionsenthalpie ΔrH\ ein. Dies ist die Änderung der Enthalpie von Reaktanden in ihren Standardzuständen zu Produkten in Standardzuständen. Beispiel: CH4 (g) + 2O2 → CO2 (g) + 2H2O (l) ΔrH\ (298 K) = -890 kJ/mol Eine Standard-Reaktionsenthalpie ist zu einem Prozess zugehörig, bei dem gilt: reine, ungemischte Reaktanden im Standardzustand → separierte, reine Produkte in ihren Standardzuständen. Für eine beliebige Reaktion: 2A + B → 3C + D gilt die Verallgemeinerung: Δr H Θ = (8.1) ∑ ν H mΘ − Produkte ∑ ν H mΘ Reaktanden wobei ν die jeweiligen stöchiometrischen Koeffizienten bezeichnet. Gehen wir weiter und verwenden die Konvention die stöchiometrischen Koeffizienten der Reaktanden als negativ zu verwenden, also in unserem Beispiel: νA = -2 νB = -1 νC = +3 νD = +1, dann können wir schreiben: (8.2) Δ r H Θ = ∑ν J H mΘ ( J ) J wobei J die jeweilige reine chemische Komponente darstellt. Bekannte Standard-Reaktionsenthalpien können kombiniert werden, um daraus neue Reaktionsenthalpien für andere Reaktionen zu bilden. Dies ist der Inhalt des Hess'schen Gesetzes: ΔrH\ /kJ mol-1 C3H6 (g) + H2 (g) → C3H8 (g) -124 -2220 C3H8 (g) + 5O2 (g) → 3CO2 (g) + 4H2O (l) +286 H2O (l) → H2 (g) + ½O2 (g) -2058 C3H6 (g) + 9/2 O2 (g) → 3CO2 (g) + 3 H2O (l) Somit kann aus bekannten Reaktionen auf das Verhalten von neuen Reaktionen geschlossen werden. Die sogenannte Standard-Bildungsenthalpie ΔfH\ einer Substanz ist die StandardReaktionsenthalpie, die nötig ist, um die Substanz aus ihren Elementen in deren 138 Referenzzuständen zu bilden. Der Referenzzustand eines Elements ist dabei der stabilste Zustand des reinen Elements bei der spezifizierten Temperatur und bei 1 bar. Beispielsweise ist der Referenzzustand von Stickstoff bei 298 K molekularer gasförmiger Stickstoff N2; der Referenzzustand von Kohlenstoff bei 298 K ist kristallines Graphit. Somit kann für jede beliebige chemische Verbindung mit Hilfe der Referenzzustände eine StandardBildungsenthalpie bestimmt werden. Die Standard-Bildungsenthalpien der Referenzzustände selbst sind natürlich null, denn man müsste sie aus Null-Reaktionen wie N2 (g) → N2(g) aufbauen. Mit Hilfe der Standard-Bildungsenthalpien können wir jede Reaktion schreiben als: (8.3) Δr H Θ = ∑ ∑ νΔ f H Θ − Produkte νΔ f H Θ Reaktanden bzw.: (8.4) Δ r H Θ = ∑ν J Δ f H Θ ( J ) J wenn man die stöchiometrischen Koeffizienten der Reaktanden negativ einsetzt. Als Beispiel nehmen wir die Reaktion: 2HN3 (l) + 2NO (g) → H2O2 (l) + 4N2 (g) dann ergibt sich aus den Standard-Bildungsenthalpien: ΔrH\ = {ΔfH\ (H2O2, l) + 4 ΔfH\ (N2, g)} -{2ΔfH\ (HN3, l) + 2ΔfH\ (NO, g)} = -187.78 + 0 – (2 × 264.0 + 2 × 90.25) kJ/mol = -892.3 kJ/mol Wir hatten in Abschnitt 3.8 bereits den Zusammenhang zwischen Enthalpie und Wärmekapazität kennen gelernt. Um die Temperaturabhängigkeit der Reaktionsenthalpien von der Temperatur zu bestimmen, stellen wir fest, dass mit (3.71) gilt: T2 (8.5) H ( T2 ) = H ( T1 ) + ∫ Cp dT T1 Hierbei ist allerdings angenommen, dass keine Phasenübergänge im interessierenden Temperaturbereich zwischen T1 und T2 stattfinden. Da diese Gleichung für jede chemische Komponente, die als Produkt oder Reaktand an einer Reaktion teilnimmt, gilt, folgt für die Standard-Reaktionsenthalpie: T2 (8.6) Δ r H Θ ( T2 ) = Δ r H Θ ( T1 ) + ∫ Δ r CΘp dT T1 wobei mit (8.7) Δ r CΘp = ∑ ν J CΘp,m ( J ) J 139 die Differenzen der Wärmekapazitäten bei Standardbedingungen gemeint sind. Die Gleichung (8.6) heißt Kirchhoffsches Gesetz. Der Zusammenhang wird in der Abbildung 8.1 verdeutlicht. Die Tabelle 8.1 listet die verschiedenen in der physikalischen Chemie üblichen Enthalpieänderungen. Abbildung 8.1. Temperaturabhängigkeit der Standard-Reaktionsenthalpien. [Atkins] Tabelle 8.1. Übergangsenthalpien. [Atkins] 140 Die Tabelle 8.2 listet einige Standardbildungsenthalpien bei 298 K. Tabelle 8.2. Standard-Bildungsenthalpien bei 298 K. [Atkins] 8.2 Der Dritte Hauptsatz der Wärmelehre Bevor wir uns mit der Rolle der Entropie in der Reaktionskinetik befassen, werden wir noch den Dritten Hauptsatz der Wärmelehre einführen. Als wir in Kapitel 4 die Entropie eingeführt haben, konnten wir diese nur bis auf eine additive Konstante definieren. Eindeutig sind deshalb nur Entropiedifferenzen zwischen zwei Punkten des Zustandsraums, wenn sich diese beiden Zustände durch eine reversible Zustandsänderung miteinender verbinden lassen. Der Dritte Hauptsatz, auch Nernstscher Wärmesatz genannt, macht eine Aussage über das Verhalten der Entropie für T → 0 und hebt damit teilweise diese Unbestimmtheit der Entropie auf. Auch dieser Hauptsatz beruht in der Thermodynamik auf unwiderlegten Erfahrungstatsachen, erst in der statistischen Mechanik wird er theoretisch begründet. Er lautet: Jede Entropieänderung ΔS eines Systems, die bei einer physikalischen oder chemischen Veränderung des Systems auftritt, geht gegen null, wenn die Temperatur gegen null strebt. Das gilt unabhängig von den Werten der anderen Zustandsvariablen: (8.8) lim ΔS = 0 T →0 Es folgt aus dem Nernstschen Satz, dass, wenn wir die Entropien der reinen chemischen Elemente in ihrer perfekten kristallinen Form mit null gleichsetzten, dann haben auch die reinen kristallinen Formen aller chemischen Verbindungen den Wert null am absoluten Nullpunkt. Dies gilt, denn nach dem Nernstschen Satz ist die Entropieänderung bei der Bildung der Verbindung am absoluten Nullpunkt ja ebenfalls gleich null. Also ist dann die Entropie aller reinen, perfekten Kristalle - egal ob der Kristall aus Elementen oder aus Verbindungen aufgebaut ist - am absoluten Nullpunkt gleich null. Damit kann der Dritte Hauptsatz auch folgendermaßen formuliert werden: Wenn die Entropie eines jeden chemischen Elements in seiner stabilsten Form bei T = 0 zu null gewählt wird, dann hat jede Substanz immer eine Entropie größergleich null, die für T = 0 auch null werden kann, und die für alle perfekten kristallinen Substanzen (inklusive perfekte Kristalle der chemischen Verbindungen) bei T = 0 tatsächlich gleich null wird. 141 Der Satz besagt nicht, dass die Entropien bei T = 0 auch null sind; es haben nur alle Entropien, egal welchen chemischen Elements oder welcher Verbindung in ihrer reinen perfekten Kristallform am Nullpunkt denselben universellen Wert, den man zweckmäßig zu null wählt. Die etwas umständliche Formulierung ist nötig um auch das korrekte Entropieverhalten von Substanzen wie z.B. superfluidem Helium und fehlerhaften Kristallen richtig wiederzugeben. Die Aussage des Dritten Hauptsatzes bedeutet auch die Unerreichbarkeit des absoluten Nullpunkts. Dies wollen wir näher erläutern. Tiefe Temperaturen erhält man durch Hintereinanderschalten von adiabatischen und isothermen Prozessen mit einer geeigneten Arbeitssubstanz, z.B. einem Gas (Linde-Verfahren) oder einem Paramagneten (adiabatische Entmagnetisierung). Wir erläutern kurz das Prinzip: Die Entropie muss außer von der Temperatur noch von einem anderen Parameter x abhängig sein, z.B. vom Druck p für ein Gas (p2 > p1) oder vom Feld H für einen Paramagneten (H2 > H1). Man führt dann folgenden Prozess durch: A → B: Entropieverminderung durch isotherme Änderung des Parameters x von x1 nach x2. Dabei muss eine bestimmte Wärmemenge abgeführt werden, die im reversiblen Fall gleich TaΔS ist. B → C: Das System wird thermisch isoliert und der Parameter x längs einer Isentrope auf den ursprünglichen Wert x1 zurückgebracht. dabei sinkt die Temperatur von Ta auf Te. Würden für T → 0 die Entropiekurven, wie skizziert, für verschiedene Werte des Parameters x gegen verschiedene Grenzwerte streben, so ließe sich der absolute Nullpunkt ohne Schwierigkeiten erreichen. ein solches ΔS-Verhalten widerspräche allerdings dem Dritten Hauptsatz, demzufolge die Entropieänderung für T → 0 immer kleiner werden muss. Man kann sich an der Abbildung unmittelbar klar machen, dass der Punkt T = 0 nur mit unendlich vielen Teilschritten asymptotisch erreicht werden kann. Man kann ihm beliebig nahe kommen, ihn aber nie erreichen. Zur Zeit liegt der "Kälterekord" bei ca. 0.5 nK (Ketterle et al., Science, 2003). 142 Eine weitere Folgerung aus dem Dritten Hauptsatz ist auch, dass für alle Wärmekapazitäten Cx → 0 für T → 0 gilt. Auch wenn Temperaturen T → 0 in der Atmosphärenforschung eigentlich keine Rolle spielen, so haben sie doch eine Auswirkung auf die Definition der Entropie. 8.3 Standardentropien Entsprechend dem Dritten Hauptsatz wollen wir die Wahl S(0) = 0 für perfekte Kristalle von nun an als Standard definieren. Dann ist S\ (T) die zugehörige Standardentropie bei einer Temperatur T. Einige Standardentropien für T = 298 K sind in Tabelle 8.3 aufgeführt. Analog zur Standard-Reaktionsenthalpie können wir nun auch für jede chemische Reaktion die Standard-Reaktionsentropie ΔrS\(T) definieren: (8.9) Δ r Sm Θ = ∑ Produkte ν SmΘ − ∑ ν S mΘ Reaktanden Die Reaktionsentropie entspricht der Differenz der molaren Entropien der reinen, separierten Reaktanden und Produkte. Tabelle 8.3. Standardentropien einiger Verbindungen bei 298 K. [Atkins] Änderung der Entropie bei Änderungen der Temperatur Für ein geschlossenes System können wir mit Hilfe der Gleichung (4.28) schreiben: 143 (8.10) S (T f ) = S (Ti ) + Tf ∫ Ti δ Qrev T Wir betrachten ein System bei konstantem Druck, dann gilt weiterhin nach (3.23): (8.11) ⎛δQ ⎞ Cp = ⎜ ⎟ ⎝ dT ⎠ p Somit gilt für die Entropie bei einer beliebigen Temperatur Tf: (8.12) S (T f ) = S (Ti ) + Tf ∫ Ti C p dT T Gehen wir von einer reinen kristallinen Substanz bei Ti = 0 aus (→ Si = 0), so können wir die Entropie bei jeder beliebigen Temperatur bestimmen, wenn wir nur die Wärmekapazitäten der Substanz zwischen T = 0 und Tf kennen. Treten Phasenübergänge auf, so müssen wir auch noch die Schmelz- und Verdampfungsentropien (vgl. (6.19) und (6.21), ΔStrs=ΔHtrs/Ttrs) berücksichtigen. Die Abbildung 8.2 macht den Zusammenhang deutlich. Die Messung von Wärmekapazitäten reiner Substanzen bei 1 bar über den gesamten Temperaturbereich T < Tf erlaubt also die Bestimmung der Standardentropien. Abbildung 8.2. Bestimmung der Entropie einer Substanz aus den Wärmekapazitäten. (a) Die Größe Cp/T für eine Substanz, (b) die zugehörige Entropie, die dem Integral der oberen Kurve entspricht, plus den Übergangsentropien bei Phasenübergängen. [Atkins] 144 8.4 Gibbsche Standardenthalpie Gibbsche Standard-Reaktionsenthalpie Mit Hilfe der Beziehung G = H - TS für die Gibbsche freie Enthalpie (vgl. (5.26) und (5.35)) können wir zum Schluss noch die Gibbsche Standard-Reaktionsenthalpie ΔrG\ einführen: ΔrGΘ = Δr H Θ − T Δr S Θ (8.13) Wie bei den Standard-Reaktionsenthalpien ist es weiterhin sinnvoll die Gibbsche StandardBildungsenthalpie ΔfG\ einzuführen: Die Gibbsche Standard-Bildungsenthalpie ΔfG\ ist die Gibbsche StandardReaktionsenthalpie ΔrG\ für die Bildung einer chemischen Verbindung aus ihren Elementen in deren Referenzzuständen. Somit lässt sich wiederum jede Gibbsche Standard-Reaktionsenthalpie darstellen als Summe der Gibbschen Standard-Bildungsenthalpien der einzelnen Reaktionspartner: (8.14) ΔrGΘ = ∑ νΔ f G Θ − Produkte ∑ νΔ f G Θ . Reaktanden Tabelle 8.4. Gibbsche Standard-Bildungsenthalpien bei 298 K. [Atkins] Die Gibbs-Helmholtz-Gleichung Für das geschlossene System bei konstantem Druck gilt nach (5.39) (8.15) ⎛ ∂G ⎞ S = −⎜ ⎟ ⎝ ∂T ⎠ p und mit G = H - TS folgt dann: 145 (8.16) G−H ⎛ ∂G ⎞ ⎜ ⎟ = T ⎝ ∂T ⎠ p Diese Gleichung können wir umformen zu: (8.17) H ⎛ ∂G ⎞ G ⎜ ⎟ − =− T ⎝ ∂T ⎠ p T und wegen: 1 ⎛ ∂G ⎞ d 1 ⎛ ∂ ⎛ G ⎞⎞ ⎜ ∂T ⎜ T ⎟ ⎟ = T ⎜ ∂T ⎟ + G dT T ⎝ ⎠⎠p ⎝ ⎠p ⎝ (8.18) = 1 ⎛ ∂G ⎞ 1 ⎜ ⎟ −G 2 T ⎝ ∂T ⎠ p T = 1 ⎡⎛ ∂G ⎞ G ⎤ ⎢⎜ ⎟ − ⎥ T ⎣⎢⎝ ∂T ⎠ p T ⎦⎥ folgt: (8.19) ⎛ ∂ ⎛ G ⎞⎞ H ⎜ ∂T ⎜ T ⎟ ⎟ = − T 2 ⎝ ⎠⎠p ⎝ Dieser Ausdruck heißt Gibbs-Helmholtz-Gleichung. Die Bedeutung dieser Gleichung wird im weiteren Verlauf noch deutlich werden. Druckabhängigkeit der Gibbschen freien Enthalpie Um die Druckabhängigkeit der Gibbschen Enthalpie in Abhängigkeit vom Wert von G beim Referenzdruck p\ zu bestimmen, gehen wir zur Gleichung (5.37) für konstante Temperatur (dT = 0) und konstante Teilchenzahl (dn = 0) zurück: (8.20) dG = Vdp Integriert man diese Gleichung von p\ bis zum gesuchten Druck p, so erhält man: (8.21) G ( p ) − G ( pΘ ) = p ∫ Vdp pΘ Für ein ideales Gas können wir weiter feststellen: (8.22) G ( p) − G ( p p Θ dp ) = ∫ nRT p pΘ und damit: 146 (8.23) ⎛ p ⎞ G ( p ) = G ( p Θ ) + nRT ln ⎜ Θ ⎟ . ⎝p ⎠ 8.5 Chemische Gleichgewichte Chemische Reaktionen laufen in Richtung des thermodynamischen Gleichgewichts ab. Diesen Sachverhalt hatten wir bereits in Abschnitt 5.5.3 aufgeführt. Im chemischen Gleichgewicht sind sowohl Reaktanden als auch Produkte vorhanden, die Konzentrationen sind aber zeitlich unveränderlich. Bei vielen Reaktionen liegt dieses Reaktionsgleichgewicht ganz auf der Seite der Produkte, d.h. nach einer Reaktion liegt das Gleichgewicht so weit auf der Produktseite und die Konzentration der Reaktanden ist so viel kleiner als die Konzentration der Produkte, dass man sagt, die Reaktion ist vollständig ("complete"). In vielen anderen Fällen ist das Reaktionsgleichgewicht nicht so eindeutig auf einer Seite, in solchen Fällen gibt es signifikante Mengen sowohl von Reaktanden als auch von Produkten. Wir werden nun untersuchen, wie wir mit Hilfe der Thermodynamischen Potentiale diese Reaktionsgleichgewicht bestimmen können. Zunächst eine Bemerkung zur Schreibweise: in diesem Kapitel sei µ das chemische Potential pro Mol (nicht wie bisher eingeführt chemisches Potential pro Molekül), weiterhin bezeichnen wir die Molmengen mit dem Buchstaben n statt des bisher üblichen ν. Mit dieser Schreibweise wird die Gibbs-Duhem-Relation (5.46) zu µ = G/n = Gm, d.h. das chemische Potential ist gleich der Gibbschen freien Enthalpie pro Mol. Damit wird (8.23) zu: (8.24) ⎛ p ⎞ Θ ⎟ ⎝p ⎠ μ ( p ) = μ ( p Θ ) + RT ln ⎜ Wenden wir uns zunächst wieder der einfachst möglichen chemischen Reaktion bei konstanter Temperatur und konstantem Druck zu: A ↔ B (vgl. Abschnitt 5.5.3). Dies könnte z.B. eine Isomerisierung sein zwischen Butan und Isobutan oder zwischen Pentan und 2-Methylbutan. Wir führen das Reaktionsmaß ξ ("extent of reaction") ein, das in der Einheit Mol angegeben wird. Eine Änderung dξ entspricht dann gerade: dξ = -dnA = dnB Entsprechend der Abbildung 8.3 bewegt sich die Reaktion auf das Minimum der Gibbschen freien Enthalpie zu. Die Gibbsche Reaktionsenthalpie ΔrG ist definiert als die Steigung der Gibbschen freien Enthalpie als Funktion des Reaktionsmaßes ξ: (8.25) ⎛ ∂G ⎞ ΔrG = ⎜ ⎟ ⎝ ∂ξ ⎠ p ,T Der Buchstabe Δ bezeichnet hier eine Ableitung, nicht wie sonst üblich eine Differenz, wie wir aber bereits wissen (vgl. (5.66)) ist ΔrG auch eine Differenz, nämlich die der chemischen Potentiale von A und B: (8.26) dG = μ A dnA + μ B dnB = − μ A dξ + μ B dξ = ( μ B − μ A ) dξ also: 147 (8.27) ⎛ ∂G ⎞ ΔrG = ⎜ ⎟ = μB − μ A ⎝ ∂ξ ⎠ p ,T Wie schon in Abschnitt 5.5.3 festgestellt, verläuft die Reaktion spontan in der Richtung A → B, wenn ΔrG < 0 und damit µA > µB gilt. Umgekehrt läuft die Reaktion in der Richtung B → A, wenn ΔrG > 0 und damit µB > µA ist. Das Gleichgewicht ist erreicht, wenn ΔrG = 0 und µA = µB. Abbildung 8.3. Gibbsche Energie als Funktion des Reaktionsmaßes. [Atkins] Chemische Gleichgewichte für ideale Gase Wenn die Substanzen A und B ideale Gase darstellen, dann können wir Gleichung (8.24) einsetzen, um zu schreiben: ΔrG = μB − μ A (8.28) ⎛ p ⎞ ⎛ p ⎞ = ⎜ μ BΘ + RT ln ΘB ⎟ − ⎜ μ AΘ + RT ln AΘ ⎟ pB ⎠ ⎝ pA ⎠ ⎝ ⎛p ⎞ = Δ r G Θ + RT ln ⎜ B ⎟ ⎝ pA ⎠ Hierbei ist die Gibbsche Standard-Reaktionsenthalpie ΔrG\ wiederum die Differenz der molaren Gibbschen Standardenthalpien von A und B: (8.29) Δ r G Θ = GBΘ, m − GAΘ,m = Δ f G Θ ( B ) − Δ f G Θ ( A ) Bezeichnen wir nun das Verhältnis der Partialdrücke mit dem Reaktionsquotienten Q: (8.30) Δ r G = Δ r G Θ + RT ln Q Nun hatten wir festgestellt, dass im Gleichgewicht gerade ΔrG = 0 ist. Im Gleichgewicht bezeichnen wir den Reaktionsquotienten Q mit der Gleichgewichtskonstanten K. Dann gilt also: 148 (8.31) Δ r G Θ = − RT ln K ⎛p ⎞ K =⎜ B ⎟ ⎝ p A ⎠equil . Hiermit haben wir die wichtigste Gleichung der chemischen Thermodynamik für den Spezialfall der idealen Gase hergeleitet! Sie stellt den Zusammenhang zwischen den Thermodynamischen Potentialen und der Gleichgewichtskonstanten her. Aus der Messung der Partialdrücke im Gleichgewicht kann man somit auf die Gibbsche StandardReaktionsenthalpie schließen. Wie wir sehen werden folgen daraus dann auch die anderen chemischen Standardpotentiale. Die Erweiterung auf reale Gase ersetzt dann nur noch die Partialdrücke durch die sogenannten Fugazitäten und die Erweiterung auf Flüssigkeiten für A und B wird mit Hilfe der Aktivitäten statt der Partialdrücke vorgenommen. Diese Erweiterungen, für die wir erst einmal Fugazität und Aktivität fundiert einführen müssten, wollen wir im Rahmen der Vorlesung nicht vornehmen, sie ist aber nicht weiter kompliziert. Auch die Erweiterung auf kompliziertere Reaktionen der Form νaA + νbB → νcC + νdD können problemlos vorgenommen werden. Umgekehrt können wir mit Gleichung (8.31) bei bekannten Gibbschen Standard-Bildungsenthalpien genau voraussagen, wo das Reaktionsgleichgewicht liegt, bzw. welche Partialdrücke die Reaktionspartner im Gleichgewicht haben müssen. Wenn man in der Atmosphäre davon ausgehen kann, dass ein Reaktionsgleichgewicht vorliegt, dann kann man z.B. mit Hilfe der Gibbschen StandardBildungsenthalpien vorhersagen, in welchem Maße eine emittierte chemische Substanz mit einer anderen Substanz weiterreagiert. van't Hoff-Diagramme Die Gleichung (8.31) können wir schreiben als: (8.32) ΔrGΘ ln K = − RT Die Ableitung nach der Temperatur ist dann: (8.33) Θ 1 d ( ΔrG / T ) d ln K =− dT R dT Mit Hilfe der Gibbs-Helmholtz-Gleichung (8.19) können wir ersetzen: (8.34) d (G / T ) H =− 2 dT T und damit: (8.35) d ln K Δ r H Θ = dT RT 2 und wegen: 149 d (1/ T ) 1 =− 2 dT T (8.36) gilt noch: (8.37) d ln K Δ HΘ =− r d (1/ T ) R Dies ist die van't Hoff-Gleichung. Sie besagt, dass wenn man die Gleichgewichtskonstante K als Funktion der Temperatur bestimmt und dann die Werte von -ln K gegen 1/T aufträgt, so erhält man ΔrH\. Dies ist ein gängiges Verfahren zur Bestimmung der StandardReaktionsenthalpie. Ein Beispiel zeigt die Abbildung 8.4. Abbildung 8.4. Van't Hoff-Diagramm: die Gradensteigung ergibt den Wert der Standard-Reaktionsenthalpie. [Atkins] 150 INDEX absoluter Nullpunkt.............................................. 12 Unerreichbarkeit ........................................... 142 Adiabate Feucht- .......................................................... 124 Pseudo-Feucht- ............................................. 124 Trocken- ........................................................ 123 Adiabatengleichungen.......................................... 30 adiabatische Entmagnetisierung........................ 142 Äquipartitionsprinzip ........................................... 25 Arbeit ................................................................... 20 Arbeitsaustauschkontakt ........................................ 5 Auftrieb ................................................................ 33 Auftriebskraft ....................................................... 34 Ausdehnungskoeffizient thermischer................................................ 12, 61 Avogadro-Konstante ............................................ 11 Barometrische Höhenformel ................................ 12 Boltzmann-Konstante ........................................... 10 Boyle-Mariotte-Gesetz ......................................... 12 Brunt-Väisälä-Frequenz....................................... 52 Carnot-Kreisprozess ............................................ 39 Chemisches Gleichgewicht................................. 147 ideales Gas .................................................... 148 Clausius-Clapeyron-Gleichung............................ 83 Clausiussche Ungleichung ................................... 44 Convection Available Potential Energy (CAPE) 133 Dalton-Gesetz .................................................... 103 Dampfdruckkurve................................................. 82 Diagramme skew T-log p .................................................. 122 Stüve-............................................................. 129 Tephigramm .................................................. 128 Thermodynamische........................................ 122 Dritter Hauptsatz ............................................... 141 Druck ..................................................................... 9 Dunst.................................................................. 101 Eisnukleationskeime............................................. 89 Energie innere .............................................................. 23 mittlere kinetische ........................................... 10 Enthalpie ........................................................ 35, 67 Entropie.................................................. 41, 44, 143 Erster Hauptsatz .................................................. 24 Feucht-Adiabaten............................................... 124 Feuchte relative............................................................. 85 spezifische ..................................................... 104 Föhn ................................................................... 117 Freie Energie ....................................................... 66 Freiheitsgrade des Moleküls ................................ 25 Frostpunkt ............................................................ 90 Gas ideales ............................................................... 8 van der Waals- ................................................ 52 Gaskonstante........................................................ 10 mittlere spezifische ........................................ 104 spezifische ....................................................... 10 universelle ....................................................... 10 Gay-Lussac-Gesetz .............................................. 12 Geschwindigkeit mittlere ............................................................ 17 wahrscheinlichste............................................ 16 Wurzel der quadratgemittelten........................ 17 Gibbsche freie Enthalpie...............................68, 146 Gibbsche Fundamentalgleichung ........................ 46 Gibbsche Paradoxon............................................ 74 Gibbsche Phasenregel ......................................... 81 Gibbsche Reaktionsenthalpie............................. 147 Gibbsche Standard-Bildungsenthalpie............... 145 Gibbsche Standard-Reaktionsenthalpie ......145, 148 Gibbs-Duhem-Relation ........................................ 71 Gibbs-Helmholtz-Gleichung .............................. 146 Gleichgewichtsbedingungen isotherm, bei konstanten Kräften .................... 77 isotherm, ohne Arbeitsaustausch..................... 76 Gleichgewichtsbedingungen ................................ 74 isoliertes System .............................................. 75 Gleichgewichtsdampfdruck.................................. 85 Gleichgewichtskonstante.................................... 148 Grundrelation der Thermodynamik ..................... 46 Henry-Gesetz ....................................................... 95 Henry-Konstante .................................................. 95 Hess'sches Gesetz............................................... 138 Homogenitätsrelationen....................................... 70 hydrostatische Grundgleichung ........................... 12 ideales Gas chemisches Gleichgewicht............................. 148 ideales Gas............................................................. 8 Entropie........................................................... 48 freie Expansion ............................................... 47 isotherme Expansion ....................................... 46 ideales Gasgesetz ................................................. 10 Integrabilitätsbedingungen .................................. 21 integrierender Faktor........................................... 21 ionen-induzierte Nukleation................................. 99 irreversibel............................................................. 6 isentrop ................................................................ 50 Joulesches Gesetz ................................................ 28 Kelvin-Gleichung ................................................. 98 Kirchhoffsches Gesetz........................................ 140 Köhler-Gleichung .............................................. 101 Köhler-Kurve ..................................................... 101 Kollisionsfrequenz ............................................... 18 Kompressibilität................................................... 61 korrespondierende Zustände................................ 56 Kraft konservative .................................................... 21 Kreisprozess........................................................... 6 kritischer Punkt.................................................... 81 kritischer Radius .................................................. 97 latente Wärme ...................................................... 83 Leewellen ............................................................. 52 Legendre-Transformation .................................... 66 Level of Free Convection (LFC) ........................ 132 Level of Neutral Buoyancy (LNB) ...................... 132 lifting condensation level (LCL) ........................ 105 Lösungsenthalpie ................................................. 92 Massenakkomodationskoeffizient ......................... 95 Massendichte........................................................ 11 Maxwell-Boltzmann-Verteilung ........................... 15 Maxwell-Konstruktion.......................................... 56 Maxwell-Relationen ............................................. 65 Mischungsentropie ......................................... 72, 74 Mittlere freie Weglänge ....................................... 19 Molekulargewicht mittleres......................................................... 103 Molenbruch .......................................................... 78 natürliche Variablen ............................................ 65 Nernstscher Wärmesatz...................................... 141 Nukleation ............................................................ 96 Nukleationsbarriere ............................................. 97 Nullter Hauptsatz ................................................... 7 perpetuum mobile 1. Art................................................................ 23 2. Art................................................................ 37 Phasen.................................................................. 79 homoge ............................................................ 56 Phasendiagramm ................................................. 81 Phasenübergänge................................................. 79 Poisson-Gleichungen ........................................... 30 Prozess ................................................................... 6 pseudo-adiabatisch........................................ 120 reversibel-adiabatisch ..................................... 50 pseudo-adiabatisch ............................................ 120 Pseudo-Feucht-Adiabate.................................... 124 psychrometrische Gleichung.............................. 113 psychrometrische Konstante .............................. 113 quasistatisch........................................................... 6 Raoultsches Gesetz......................................... 91, 93 Reaktionsmaß..................................................... 147 Reaktionsquotient............................................... 148 Referenzzustand ................................................. 139 relative Feuchte.................................................... 85 Relaxationszeit ....................................................... 6 Response-Funktion............................................... 64 reversibel................................................................ 6 reversibler Ersatzprozess ..................................... 44 Sättigungsdampfdruck.......................................... 85 Sättigungsdampfdruck über Eis ........................... 89 Sättigungsdampfdruck über unterkühltem Wasser89 Schichtung instabil............................................................. 35 stabile .............................................................. 35 Schmelzenthalpie.................................................. 85 Schmelzkurve........................................................ 81 Schwerewellen...................................................... 52 Siedepunkt ............................................................ 90 Vertikalgradient .............................................. 90 Skalenhöhe ........................................................... 13 spezifische Feuchte ............................................ 104 Stabilität............................................................... 32 Stabilitätskriterien.............................................. 130 Standard-Bildungsenthalpie............................... 138 Standardenthalpieänderung............................... 137 Standardentropie................................................ 143 Standard-Reaktionsenthalpie ............................. 138 Standard-Reaktionsentropie............................... 143 Standard-Verdampfungsenthalpie...................... 137 Standardzustand................................................. 137 statistische Mechanik ............................................. 4 stöchiometrischer Koeffizient............................. 138 Stoßquerschnitt .................................................... 19 Stüve-Diagramm ................................................ 129 Sublimationsenthalpie.......................................... 85 Sublimationskurve................................................ 81 System .................................................................... 5 geschlossenes .................................................... 5 isoliertes ............................................................ 5 offenes ............................................................... 5 Taupunkt-Temperatur .......................................... 90 TdS-Gleichungen ................................................. 61 Temperatur............................................................. 7 absolute ........................................................... 10 äquivalente .................................................... 114 äquivalente potentielle .................................. 118 äquivalente potentielle Sättigungs- ............... 119 Feuchte- ........................................................ 111 potentielle........................................................ 48 potentielle Feuchte-....................................... 121 potentielle wet-bulb....................................... 121 Taupunkt- ........................................................ 90 virtuelle ......................................................... 109 virtuelle potentielle........................................ 111 wet-bulb......................................................... 111 Temperaturgradient feucht-adiabatischer...................................... 117 trocken-adiabatischer ..............................32, 110 Tephigramm ....................................................... 128 Thermodynamische Diagramme ........................ 122 Thermodynamische Potentiale....................... 64, 70 thermodynamisches Gleichgewicht........................ 4 thermodynamisches System.................................... 5 Thomson-Gleichung............................................. 99 totales Differential ............................................... 21 Tripelpunkt........................................................... 81 Trocken-Adiabate............................................... 123 Übergangsenthalpien......................................... 140 van der Waals-Gas............................................... 52 kritischer Punkt ............................................... 55 van't Hoff-Diagramm ......................................... 150 van't Hoff-Gleichung.......................................... 150 Verdampfungsenthalpie ............................83, 84, 86 Verdampfungskurve ............................................. 81 Verdampfungswärme ........................................... 83 Virialentwicklung................................................. 57 Volumenarbeit...................................................... 22 Wärmeaustauschkontakt .................................. 5, 23 Wärmekapazität ................................................... 26 Wärmekraftmaschine ........................................... 37 Wärmepumpe ....................................................... 40 Wärmetod des Universums .................................. 46 Wasserdampfmischungsverhältnis ..................... 104 Wirkungsgrad....................................................... 38 Carnot-Maschine............................................. 41 Wolkentröpfchen ................................................ 101 Zustand .................................................................. 6 Zustandsänderung.................................................. 6 adiabatische .................................................... 29 isotherme......................................................... 31 Zustandsgleichung ............................................... 19 ideales Gas ...................................................... 10 kalorische ........................................................ 24 thermische ....................................................... 24 van der Waals.................................................. 53 Zustandsgröße........................................................ 5 Zustandsraum......................................................... 6 Zustandsvariablen................................................ 19 Zweiter Hauptsatz ................................................ 37 isoliertes System .............................................. 46 mathematische Formulierung ......................... 45 Literatur: C. F. Bohren and B. A. Albrecht, Atmospheric Thermodynamics, Oxford University Press, 1998 J. A. Curry and P. J. Webster, Thermodynamics of Atmospheres and Oceans, Academic Press, 1999 W. Nolting, Grundkurs Theoretische Physik Bd. 4: Thermodynamik, Springer, 2001 P. Atkins, Physical Chemistry, Freeman, 1999 J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics, Wiley, 1998 W. Zdunkowski and A. Bott, Thermodynamics of the Atmosphere, Skript, Uni Mainz