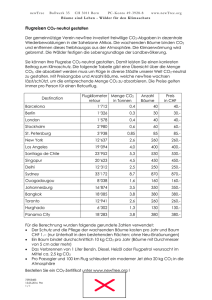
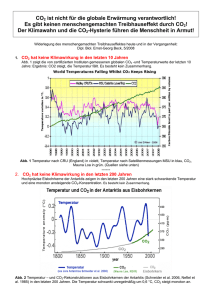
Lehrbücher
Werbung

Lehrbücher: Hollemann-Wiberg, Lehrbuch der Anorganischen Chemie, 102. Auflage. Berlin, New York: de Gruyter, 2007. W. Kaim, B. Schwederski: Bioanorganische Chemie. Zur Funktion chemischer Elemente in Lebensprozessen. Teubner, 4. Auflage 2005. L. Sigg, W. Stumm: Aquatische Chemie - eine Einführung in die Chemie wässriger Lösungen und natürlicher Gewässer. vdf Hochschulverlag an der ETH Zürich. Teubner, 1996. Quelle: Robert Kummert/Werner Stumm, Zürich 1987; Idee nach Tyler Miller, 1971 Quelle: Die Geschichte der Erdatmosphäre, von Manfred Schidlowski, 1987 Beweis dafür, dass es in der frühen Erdatmosphäre keinen Sauerstoff (O2) gab: In 2,5x109 Jahre alten Flussschottern (Konglomeraten) fand man Uraninit UO2. Die Körnchen wurden von Flüssen im Geröll weite Strecken transportiert und dabei charakteristisch abgerundet. Hätte die Atmosphäre Sauerstoff enthalten, so wäre das Uran(IV) rasch zu Uran(VI) oxidiert worden. Die Körnchen hätten sich aufgelöst, weil das entstehende [UO2]2+ (Uranyl- Ion) im Gegensatz zum Uraninit gut wasserlöslich ist. Auch Schottersteine aus Pyrit FeS2 in solchen Sedimenten beweisen die Abwesenheit von Sauerstoff. Wieso enthält die heutige Atmosphäre O2? Die heutige Zusammensetzung der Atmosphäre ist eine Folge des Lebens auf der Erde. Entscheidende Bedeutung der Ozeane für die Entstehung des Lebens Wasser ist der Träger aller chemischen Lebensvorgänge in den Organismen Wasser löste das atmosphärische CO2 und verhinderte zu großen Treibhauseffekt Wasser löste CO2 und alle anderen Nährstoffe Wasser wirkte aufgrund seiner hohen Wärmekapazität 4,1826 J/(K.g) stabilisierend auf das irdische Klima Wasser absorbierte das kurzwellige UV (200nm – 290nm) (die ersten Organismen lebten unter einer 10 m dicken Wasserschicht (UV Filter entspricht der heutigen Atmosphäre) Photosynthese CO2 + 2 H2S + Licht [CH2O] + H2O + 2 S 2 CO2 + 2 H2O + H2S + Licht 2 [CH2O] + H2SO4 CO2 + 2 H2 + Licht [CH2O] + H2O photoautotrophe schwefeloxidierende Bakterien (Grüne Schwefelbakterien und Schwefelpurpurbakterien, photoautotrophe Wasserstoffbakterien) CO2 + 2 H2O + Licht [CH2O] + H2O + O2 Cyanobakterien betreiben oxygene Photosynthese Viele Prozesse wie die N2- CO2- und S-Assimilation waren bereits von Anbeginn des Lebens mit Lichtreaktionen, einem Elektronenfluss und ATP verbunden. Aber erst die Kopplung zweier lichtgetriebener Reaktionen, d.h. die Kopplung zweier Photosysteme erzeugte die notwendige Energie für die Oxidation von Wasser und die Freisetzung von Sauerstoff. Die mikropaläontologisch-morphotypischen Funde in ältesten Gesteinen (3,5 Milliarden Jahre alt) weisen darauf hin, dass cyanobakterielle Ökosysteme sehr bald die frühe Erde beherrscht haben. (Manfred Schidlowski, 1998) Die biologische Kohlenstoff-Fixierung durch die archaischen Stromatolithen zeigt, dass prokaryotisches Mikrobenthos die Erde schon vor etwa 3,5 Milliarden Jahren beherrscht hat. Die Archaisch-Proterozoischen Funde bestehen hauptsächlich aus Cyanobakterien. Vergleichbare mikrobielle Gesellschaften gehören zu den produktivsten Ökosystemen der heutigen Biosphäre. Besonders die benthischen (= auf dem Meeresboden lebenden) Cyanobakterien können die erstaunliche Primärproduktivität von 10g C/m² am Tag erreichen. Die Photosynthese hat daher in quantitativer Hinsicht während der nachfolgenden Evolution des Leben nur wenig an Wichtigkeit dazu gewonnen. Quelle: Die Geschichte der Erdatmosphäre, von Manfred Schidlowski, 1987 Die Glykolyse ist der einzige metabolische Weg, den alle Organismen gemeinsam haben, was auf seine sehr frühe Entstehung hinweist; existierte bereits in den ersten Procaryoten vor 3,5-3,8 Milliarden Jahren. Die Sauerstoff-Atmung bedeutet gegenüber der Gärung eine 14x effektivere Ausnutzung der in den Kohlenhydraten gespeicherten Energie. Atmung ist schon bei 1% des heutigen O2 Partialdrucks möglich. Infolge dieses Fortschritts nahm die biologische Aktivität und damit auch die Sauerstoff-Produktion sprunghaft zu. Fe(III) ist im Meerwasser extrem schwer löslich Liu and Millero, 2002 Mangel an bioessentiellen Metallen im Ozean vor 2 Milliarden Jahren bis vor 1 Milliarde Jahren fehlte weitgehend eine globale Zirkulation des Weltozeans. Gleichzeitig begann vor 2 Milliarden Jahren die Sulfat-Zufuhr zum Ozean durch die einsetzende oxidative Verwitterung der Gesteine der Kontinente. Durch dissimilatorische Sulfatreduktion entstanden hohe H2S Gehalte in tieferen sauerstofffreien Wasserschichten, dies führte zur Bildung schwerlöslicher Sulfide von Mo, Fe, Cu, Zn. Durch den Mangel an Mikronährstoffen im Ozean verzögerte sich die Entwicklung der Eukaryoten, die erst vor 1,4 Milliarden Jahren die Bildfläche betraten. Transport und Verteilung von Metallen in Gewässern und Sedimenten •Fällung und Auflösung von Carbonaten •Hydrolyse und Bildung schwerlöslicher Oxide und Hydroxide • Komplexbildung mit gelösten organischen Liganden • Adsorption an Partikeloberflächen •Redoxprozesse •Biomethylierung CO2 Kohlendioxid steht im Zentrum der geochemischen Kreisläufe spielt eine zentrale Rolle in der Biosphäre: Photosynthese – Respiration und in geochemischen Prozessen, welche Gesteine auflösen und Minerale bilden In der Hydrosphäre wird Kohlenstoff vor allem als Ca(HCO3)2 transportiert CaCO3 wird in Seen und im Ozean ausgefällt Kohlenstoff-Flüsse (1015 g/a) Netto-Primärproduktion der terrestrischen Biosphäre 57 – 58 Netto-Primärproduktion der marinen Biosphäre 54 – 59 Bildung von Kalkskeletten im Ozean (Coccolithophoriden, Foraminiferen, Kalkalgen, Korallen) 1 Marine physikalische Kohlenstoffpumpe 40 Marine biologische Kohlenstoffpumpe 11 – 16 C-Speicherung in Böden 1 Diese natürlichen Senken werden durch natürliche Quellen (Atmung, Vulkanismus, Ausgasen aus dem Meer) kompensiert In 5 Jahren wird das gesamte CO2 der Atmosphäre ausgetauscht Anthropogene CO2 Freisetzung aus fossilen Brennstoffen 7 Es bedarf eines Überschusses der Photosyntheserate über die Atmungsrate: Der reduzierte Kohlenstoff wird nicht zur Gänze wieder zu CO2 oxidiert, sondern wandert zum Teil ins Sediment, wo er vor Oxidation geschützt ist. So kann der Sauerstoffverbrauch durch die Oxidation reduzierender Krustenbestandteile und reduzierender vulkanischer Gase ausgeglichen werden. Schematische Darstellung von Manfred Schidlowski 0,03% der Masse der Erdkruste ist Kohlenstoff Das Carbonatsystem der Ozeane und Binnengewässer HCO3-, CO32- Enthält 60x soviel Kohlenstoff wie die Atmosphäre Ist das größte Kohlenstoffreservoir der Biosphäre Quelle: Ott Bei der Verwitterung der Silicate der Erdkruste wird CO2 verbraucht: z.B. Kalkfeldspat (Anorthit) CaAl2Si2O8 + 2 H2O + 2 CO2 = H2Al2Si2O8 + Ca(HCO3)2 (gelöst) Beim Ausfallen der Carbonate im Ozean wird die Hälfte des bei der Verwitterung der Silicate verbrauchten CO2 wieder frei: Ca2+ + 2 HCO3- = CaCO3 + H2O + CO2 Meerwasser: ________ Süßwasser: --------- Geochemischer Carbonat-Silicat Kreislauf Verwitterung von Kalk- oder Silicatgestein verbraucht CO2 Ca2SiO4+4 H2O+4 CO2→2 Ca(HCO3)2+Si(OH)4 (gelöst) Im Meer scheidet sich Kalk ab 2 Ca(HCO3)2→2 CaCO3↓+2 H2O+2 CO2↑ Es bilden sich Kalksedimente. In diesem Reservior verbleibt der Kohlenstoff Jahrmillionen. An den Kontinentalrändern schiebt sich der Meeresboden unter die Landmassen (Subduktion), unter hohen Drucken und Temperaturen reagiert dort das Calciumcarbonat mit Quarz 2 CaCO3+SiO2→Ca2SiO4+2 CO2↑ The carbonate-silicate cycle, which plays a key role in stabilizing Earth's climate over long time scales Quelle: J. F. Kasting Verwitterung: Ursache der Wasserhärte und der Fruchtbarkeit von Boden und Gewässern „Temporäre Härte“ (= Carbonathärte): Anteil an Calcium- und Magnesiumhydrogencarbonat „Permanente Härte“ (=Sulfathärte): erfasst die gelösten Calcium- und Magnesiumsalze der Salzsäure, Schwefelsäure, Salpetersäure u.a. Pflanzennährstoffe werden freigesetzt: Sulfat, Phosphat, Kieselsäure, K+, Mg2+, Cl-, Fe2+/3+,Mn2+, Zn2+, Cu2+ u. a. mol L-1 atm-1 Verwitterung von Calcit, offenes System (bei 101 325 Pa = Normaldruck) 0.038% v/v CO2 in der Atmosphäre pH=8.3 [Ca2+] = 5x10-4 mol/L entspricht 2.8°dH 3% v/v CO2 in der Bodenluft pH = 7.02 [Ca2+] = 2.8x10-3 mol/L entspricht 15.7°dH Effekt der Landpflanzen auf die Verwitterung Durch den (mikrobiellen) Abbau von Pflanzenresten im Boden wird CO2 produziert und in der Bodenlösung angereichert Die Anwesenheit der Vegetation beschleunigt die CO2-Verwitterung daher um das 100 – 150 fache Dadurch entzieht ein Wald-Ökosystem in Österreich der Atmosphäre im Jahr ca. 20 g C/m2 also 200 kg C/ha Dieser Kohlenstoff wird als Ca(HCO3)2 ins Meer transportiert und dort als CaCO3 ausgefällt. Die Hälfte des gebundenen CO2 wird dabei frei, die andere Hälfte in den Carbonatsedimenten dauerhaft gespeichert. Quelle: Peter A. Raymond Nature 436, p. 469, 2005 Die Flüsse sind “netto-heterotroph“, d.h. sie sind gegenüber der Atmosphäre CO2 übersättigt und können daher viel Ca(HCO3)2 aus der kontinentalen Kohlensäure-Verwitterung in Lösung halten und in die Ozeane transportieren. Quelle: Biologische Station Lunz/See Carbonatsystem der Gewässer: Geschwindigkeit der Gleichgewichtseinstellung Thermodynamisches Gleichgewicht stellt sich in der wässrigen Lösung im Allgemeinen rasch ein Gewässer sind meist nicht im Gleichgewicht mit der Atmosphäre, weil biologische Prozesse im Wasser CO2 schneller produzieren oder konsumieren als der CO2 Transfer zwischen der Atmosphäre und dem Wasser erfolgt. Bildung und Auflösung von CaCO3 können verzögert erfolgen Metastabile Gleichgewichte: Aragonit (orthorhombisch) ist in einem natürlichen Wasser thermodynamisch weniger stabil als Calcit (trigonal). Unter bestimmten Bedingungen kann sich Aragonit gegenüber Calcit metastabil verhalten. Metallionen in Gewässern Durch die zivilisatorischen Tätigkeiten sind die geochemischen Kreisläufe einer Anzahl metallischer Elemente beschleunigt natürlichen Flüsse: durch Verwitterung der Gesteine, vulkanische Emissionen, Verbreitung natürlicher Aerosole aus Böden und Meerwasser Die anthropogenen Flüsse übersteigen oft die natürlichen Flüsse. Die Gewässer sind dadurch besonders betroffen Anthropogene Quellen für Schwermetalle sind z.B. Erzgewinnung, metallverarbeitende Industrien, Verbrennung fossiler Brennstoffe, Zementproduktion Durch die Verbrennung fossiler Brennstoffe werden z.B. die Flüsse von As, Cd, Se, Hg, Zn in die Atmosphäre stark erhöht. Dadurch werden auch die Konzentrationen dieser Elemente im Wasser und in den Böden erhöht. Schwermetall-Entgiftungsstrategien der Organismen Enzymatische Umwandlungen von toxischen zu weniger toxischen oder flüchtigen Spezies: Hg2+→Hg0 As(OH)3→(CH3)3As+-CH2COO- “Arsenobetain“ Spezielle Membranen können den Durchtritt von Metallionen in besonders gefährdete Bereiche wie Gehirn, Fetus verhindern Hochmolekulare Verbindungen wie z. B. die Metallthionein-Proteine (=kleine Proteine mit hohem Cysteinanteil von 20-30%) können toxische Metallionen binden und damit aus dem Verkehr ziehen Speziierung der Schwermetalle in der Umwelt Gelöst oder an feste bzw. kolloidale Phasen gebunden Komplexbildung mit verschiedenen Liganden in Lösung Verschiedene Redoxzustände Metallorganische Verbindungen Das Schicksal von Schwermetallen in den Gewässern hängt von der Speziierung ab (z.B. Transport in die Sedimente, Mobilisierung aus Sedimenten, Infiltration ins Grundwasser, Anreicherung in Organismen) Die Toxizität ist stark von der jeweiligen chemischen Spezies abhängig. Hg als umweltrelevantes Schwermetall 4x10-5 Massen% Hg in der Erdkruste Hg22+ ↔Hg0 + Hg2+ sehr schwerlösliche Minerale: Hg2Cl2 und HgS HSAB-Prinzip → Thiophilie des Quecksilbers: Starke Wechselwirkung mit schwefelhaltigen Liganden z.B. Thiole = „Mercaptane“ = Quecksilberfänger Quecksilberdampfdruck in Abhängigkeit von der Temperatur Metallisches Quecksilber ist gegenüber Luftsauerstoff bei Raumtemperatur stabil: keine Oxidhaut Bei 20°C im Gleichgewicht 13,6 mg/m3 Hg in der Luft MAK-Wert: 0,1 mg/m3 Chronische Quecksilbervergiftung z.B. durch Einatmen von Quecksilberdämpfen oder Stäuben : Durchblutungsstörungen Beeinträchtigung der Koordination z.B. beim Schreiben Erregbarkeit “Mad Hatter Syndrom“ der Hutmacher im 18. und 19. Jh (vgl. Lewis Carrols Alice im Wunderland), sie verwendeten Hg ( NO2 )2 2H 2O Gedächtnisverlust Bei extrem hoher Belastung: Lähmung, Taubheit, Blindheit, Tod Die außergewöhnlichen Eigenschaften des Quecksilbers gehen darauf zurück, dass die d- und s-Außenelektronen des Hg durch die f- Elektronen der drittinnersten Schale schlecht abgeschirmt werden Und dass durch relativistische Effekte die s-Außenelektronen eine zusätzliche Energieabsenkung erfahren Diese starke Bindung der s-Elektronen an den Kern ist u.a. der Grund dafür, dass im elementaren Quecksilber nur schwache Quecksilber-Quecksilber Wechselwirkungsenergien vorliegen Quecksilberverbindungen haben nur mit sehr harten Basen wie Fluorid und Nitrat den Charakter von Ionenkristallen. In anderen Fällen besitzen die Bindungen des Quecksilbers einen ungewöhnlich hohen kovalenten Anteil → geringe elektrische Leitfähigkeit der wässrigen Lösungen, gute Löslichkeit auch in organischen Lösungsmitteln Quecksilber(II)verbindungen bilden mit vielen in Gewässern vorkommenden Anionen stabile Komplexe, dadurch kann die Wasserlöslichkeit stark erhöht werden. mit Halogenidionen: HgX3-, HgX42- (vor allem im Meerwasser) Ungewöhnlich stabile Komplexe mit Huminstoffen („gelöste“ kolloidale Phase in Fließgewässern, Seen, Grundwasser) HS entstehen vor allem aus pflanzlichem Material wie Lignocellulose Huminstoffe: ubiquitär in aquatischen Systemen, binden Schwermetalle Funktionen: 1. Entgiftung 2. Transport Bindungsformen: 1. Sorption 2. “non-exchangable binding“ Organische Quecksilberverbindungen •Wichtig ist ausschließlich die Oxidationsstufe +II •Monoorganyle RHgX, Diorganyle R2Hg •linear gebaut •Kovalente Bindung Hg-C •relativ geringe Bindungsenergien 50 – 200 kJ/mol •kann leicht homolytisch unter Bildung von Radikalen gespalten werden (thermische oder photolytische Zersetzung) •Lebensdauer in der Atmosphäre nur wenige Stunden Synthesechemie: die einfach durchzuführende homolytische Spaltung der Hg-C Bindung wird für die Bildung von Radikalen ausgenutzt. In der Umwelt ist die Hg-C Bindung gegenüber Wasser und Luft weitgehend inert (aus kinetischen Gründen) Charakteristische Abbaureaktionen in der Umwelt: Acidolyse: RHgX + HA → RH + AHgX Reduktion: RHgX + 2 H2O + 2 e- → RH + HX + 2 OH- + Hg0 Homolyse: R2Hg → 2 R· + Hg0 Die Bildung des Methylquecksilbers in der Umwelt • kann auf biotischem als auch auf abiotischem Weg erfolgen, • wobei die biochemische Quecksilbermethylierung durch Mikroorganismen verursacht wird •Eine wichtige Bakteriengruppe für die Quecksilbermethylierung sind Sulfat-reduzierende Bakterien in Sedimenten Methylcobalamin Die Methylierung des Quecksilbers erfolgt mit Hilfe von Methylcobalamin. Wird ausschließlich von Mikroorganismen synthetisiert. Besitzt ein Corrin-Ringsystem. Im Fall des Methylcobalamins (MeCoB12) ist das Kobaltion mit einer Methylgruppe substituiert. Diese Methylgruppe kann als Carbanion (CH3-) auf Quecksilber(II) übertragen werden. Beim Übergang der Methylgruppe wird die Oxidationsstufe des Quecksilbers nicht verändert. Entstehung von Dimethylquecksilber bei der Zersetzung von Monomethylquecksilber in Anwesenheit von H2S (H2S wird durch die mikrobielle Sulfatreduktion in reduzierenden Sedimenten gebildet): 2 CH3Hg+ + S2- → CH3Hg-S-HgCH3 → (CH3)2Hg + HgS Eigenschaft Hg0 Wasserlöslichkeit g/L bei 25°C 6·10-5 5 Dampfdruck Pa bei 25°C 0,25 1,76 CH3HgCl (CH3)2Hg HgCl2 Hg2Cl2 HgS 3 73 4·10-4 1,9·10-24 8300 0,016 1,3·10-8 nicht messbar Aufnahme mit der Nahrung: gelangt vor allem beim Verzehr von Pilzen, Fischen und Meerestieren über organische Quecksilberverbindungen in den Körper. Mit der Magensäure entsteht aus CH3Hg+ das wenig dissoziierte CH3HgCl Molekül, das wegen seiner Fettlöslichkeit gut resorbierbar ist Grenzwerte Die Weltgesundheitsorganisation (WHO) hat eine „vorläufig duldbare wöchentliche Aufnahmemenge“ für Methylquecksilber von 1,6 μg/kg Körpergewicht festgelegt, ein Erwachsener mit 70 kg Körpergewicht kann von Fisch mit einem Quecksilbergehalt am gesetzlich festgelegten Grenzwert von 1 mg/kg fettreichem Fisch nur 112 g pro Woche essen, um die wöchentlich tolerierbare Dosis von 1,6 μg/kg Körpergewicht nicht zu überschreiten. Toxische Wirkung Anorganische Hg-Verbindungen: Giftigkeit hängt von der Wasserlöslichkeit der jeweiligen Verbindung ab. Hg2+ ist bei pH 7 in Wasser leicht löslich und bildet mit den in Körperflüssigkeiten häufiger vorkommenden Anionen keine unlöslichen Verbindungen. Hg2+ tritt mit den Thiol- und Disulfideinheiten der Proteine in Wechselwirkung , blockiert aktive Zentren, verändert Strukturen von Enzymen. Körpereigene Entgiftung durch Binden an Metallthioneine: Proteine, Molekulargewicht ca. 6500 g/mol, 35% Cystein-Anteil. Organische Quecksilberverbindungen Größte toxische Wirkung wegen des ambivalent lipophilen/hydrophilen Charakters Resorptionsrate bei oraler Aufnahme bis 95% Am giftigsten ist Methylquecksilber(II) wegen der (kinetisch) stabilen Hg-C Bindung Weniger stabile quecksilberorganische Verbindungen werden im Körper zu anorganischem Quecksilber metabolisiert und wirken daher eher wie dieses Wegen seines lipophilen/hydrophilen Charakters ist Methylquecksilber(II)chlorid in der Lage, biologische Membranen zu durchdringen und sogar die Blut-HirnSchranke und Plazenta-Membran zu überwinden. Wirkungsweise von Methylquecksilber im menschlichen Körper Bioakkumulation und Biomagnifikation von Quecksilber Bioakkumulation = Anreicherung von Toxinen gegenüber dem Medium (Wasser). Biomagnifikation = Anreicherung von Toxinen mit steigendem trophischem Niveau, z.B. lipophile Organometallverbindungen, die sich der Exkretion über Metallthioneine entziehen. Die häufigsten Elemente in der Erdkruste Element O Si Al Massen% 47,0 26,9 8,1 Vol% 88,2 0,32 0,55 Fe3+ 1,8 0,32 Fe2+ Ca Mg Na K 3,3 5,0 2,3 2,1 1,9 1,08 3,42 0,60 1,55 3,49 Tabelle Silicate Silicate sind die dominierenden gesteinsbildenden Minerale in der Erdkruste. Primäre Silicate: sind aus dem Magma durch Erstarrung hervorgegangen. Sekundäre Silicate: metamorphe Gesteine sowie die durch Verwitterung der primären Silicate entstandenen Tonminerale. Kugelmodelle: SiO4 Tetraeder und FeO6 Oktaeder nur die obere Darstellung ist maßstäblich, in den unteren Darstellungen sind die Sauerstoffionen verkleinert. Silicatstrukturen: Ketten- Band- und Schichtsilicate (Tetraedermodell) Quelle: Scheffer/Schachtschabel Quelle: Scheffer/Schachtschabel Glimmerstruktur Quelle: Scheffer/Schachtschabel Tetraedermodell eines Albits Natronfeldspat NaAlSi3O8 Quelle: Scheffer/Schachtschabel Primäre Silicate Erguss- oder Eruptivgesteine: rasch abgekühlt Tiefengesteine: langsam abgekühlt Für das Magma ist die Bindung großer Mengen leichtflüchtiger Bestandteile (Gase) charakteristisch, wie H2O, H2S, Cl2, Schwermetallchloride, CO2. Diese entweichen bei den Ergussgesteinen Quelle: Scheffer/Schachtschabel Bedeutung der Silicatverwitterung Natürliche Fruchtbarkeit und Elektrolytgehalt von Böden Überführung der Kieselsäure in Lösung Anreicherung von Alkali- und Erdalkalimetall-Ionen in Wässern Bildung von austauschaktiven Tonmineralen Gelöste Kieselsäure Flusswasser und Meerwasser enthalten gelöste Kieselsäure in sehr geringen Konzentrationen Daher keine chemische Ausfällung Im SiO2 Kreislauf des Ozeans ist Ausfällung von Kieselsäure nur durch Organismen möglich! Radiolarien, Diatomeen und Kieselschwämme bauen ihre Skelette aus Opal auf Es setzen sich schließlich Diatomeen- und Radiolarienschlämme ab (1010 Tonnen SiO2 jährlich) Im Süßwasser bildet sich poröse Diatomeenerde (Kieselgur) Diatomeen (=Kieselalgen) liefern 20 -25% der gesamten Primärproduktion der Erde! Tonminerale (Sekundärsilicate) Bilden sich inkongruent aus den Primärsilicaten im Kontakt mit der Verwitterungslösung Oder kongruent aus übersättigten Lösungen Ihre Zusammensetzung variiert über weite Bereiche, entsprechend dem Grad der Verwitterung