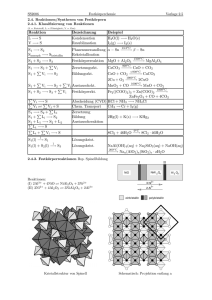
Lehrbücher:
Werbung

Literatur: • Hollemann-Wiberg, Lehrbuch der Anorganischen Chemie, 102. Auflage. Berlin, New York: de Gruyter, 2007. • W. Kaim, B. Schwederski: Bioanorganische Chemie. Stuttgart: Teubner, 1995. • L. Sigg, W. Stumm: Aquatische Chemie - eine Einführung in die Chemie wässriger Lösungen und natürlicher Gewässer. vdf Hochschulverlag an der ETH Zürich. Stuttgart: Teubner, 1996. Umweltchemie Die Zukunft soll man nicht voraussehen wollen, sondern möglich machen. Antoine de Saint-Exupery Die Forschungsziele der Umweltchemie begründen sich auf dem in den vergangenen Jahrzehnten erwachten Umweltbewusstsein der Gesellschaft und der damit verbundenen neuen Verantwortung des Chemikers. Umweltchemische Forschung ist eine Zukunftsdisziplin, die entscheidend dazu beitragen wird, die Tendenz zur Degradation der Ökosysteme und zum Schwund der Artenvielfalt unseres Planeten wirksam zu stoppen, und zugleich die Gesundheit und Lebensqualität der Menschen nachhaltig zu sichern. Viele gravierende Umweltprobleme, welche die Funktionsfähigkeit des Ökosystems Erde beeinträchtigen und die Menschheit in Zukunft zunehmend zu belasten drohen, harren derzeit noch einer Lösung. Diese Lösung kann nur auf Basis eines vertieften Verständnisses der vielfach vernetzten chemischen Vorgänge in der natürlichen Umwelt gefunden werden. Die Entwicklung neuartiger Technologien zur Lösung von Umweltproblemen gehört mit zu den Forschungszielen. Die beobachteten natürlichen Systeme sind äußerst komplex und nur ein interdisziplinärer Ansatz kann zum Erfolg führen. Umweltchemische Forschungen bauen daher auf Kooperationen mit anderen Forschungsdisziplinen auf wie z.B. Analytische Chemie, Geologie, Hydrologie, Meteorologie, Mikrobiologie, Ökologie. Transport und Verteilung von Metallen in Gewässern und Sedimenten Fällung und Auflösung von Carbonaten Hydrolyse und Bildung schwerlöslicher Oxide und Hydroxide Komplexbildung mit gelösten organischen Liganden Adsorption an Partikeloberflächen Redoxprozesse Biomethylierung CO2 • Kohlendioxid steht im Zentrum der geochemischen Kreisläufe • spielt eine zentrale Rolle in der Biosphäre: Photosynthese – Respiration • und in geochemischen Prozessen, welche Gesteine auflösen und Minerale bilden • In der Hydrosphäre wird Kohlenstoff vor allem als Ca(HCO3)2 transportiert • CaCO3 wird in Seen und im Ozean ausgefällt Kohlenstoff-Flüsse (1015 g/a) • • • • • • Netto-Primärproduktion der terrestrischen Biosphäre 57 – 58 Netto-Primärproduktion der marinen Biosphäre 54 – 59 Bildung von Kalkskeletten im Ozean (Coccolithophoriden, Foraminiferen, Kalkalgen, Korallen) 1 Marine physikalische Kohlenstoffpumpe 40 Marine biologische Kohlenstoffpumpe 11 – 16 C-Speicherung in Böden 1 • Diese natürlichen Senken werden durch natürliche Quellen (Atmung, Vulkanismus, Ausgasen aus dem Meer) kompensiert • In 5 Jahren wird das gesamte CO2 der Atmosphäre ausgetauscht • Anthropogene CO2 Freisetzung aus fossilen Brennstoffen 7 Monatsmittelwerte des global gemittelten atmosphärischen CO2 über der Ozeanoberfläche Burning peat bogs set alight by rainforest clearance in Indonesia are releasing up to a seventh of the world's total fossil fuel emissions in a single year Anthropogene CO2 Quellen (Gt C/a) 2008 • Verbrennen fossiler Energieträger: 7 • Brandrodung tropischer Wälder, Torfbrände, • Tauender Permafrostboden, Entwässerung von Mooren 0,0016% der Masse der Erde ist Kohlenstoff Das Carbonatsystem der Ozeane und Binnengewässer HCO3-, CO32- Enthält 50x soviel Kohlenstoff wie die Atmosphäre Ist das größte Kohlenstoffreservoir der Biosphäre Quelle: Ott Bei der Verwitterung der Silicate der Erdkruste wird CO2 verbraucht: z.B. Kalkfeldspat (Anorthit) CaAl2Si2O8 + 2 H2O + 2 CO2 = H2Al2Si2O8 + Ca(HCO3)2 (gelöst) Beim Ausfallen der Carbonate im Ozean wird die Hälfte des bei der Verwitterung der Silicate verbrauchten CO2 wieder frei: Ca2+ + 2 HCO3- = CaCO3 + H2O + CO2 Meerwasser: ________ Süßwasser: --------- Coccolithophoriden einzellige Algen, besitzen ein Calcit- oder Aragonit-Skelett Quelle: Ott Geochemischer Carbonat-Silicat Kreislauf Verwitterung von Kalk- oder Silicatgestein verbraucht CO2 Ca2SiO4+4 H2O+4 CO2→2 Ca(HCO3)2+Si(OH)4 (gelöst) Im Meer scheidet sich Kalk ab 2 Ca(HCO3)2→2 CaCO3↓+2 H2O+2 CO2↑ Es bilden sich Kalksedimente. In diesem Reservior verbleibt der Kohlenstoff Jahrmillionen. An den Kontinentalrändern schiebt sich der Meeresboden unter die Landmassen (Subduktion), unter hohen Drucken und Temperaturen reagiert dort das Calciumcarbonat mit Quarz 2 CaCO3+SiO2→Ca2SiO4+2 CO2↑ Verwitterung: Ursache der Wasserhärte und der Fruchtbarkeit von Boden und Gewässern • „Temporäre Härte“ (= Carbonathärte): Anteil an Calcium- und Magnesiumhydrogencarbonat • „Permanente Härte“ (=Sulfathärte): erfasst die gelösten Calcium- und Magnesiumsalze der Salzsäure, Schwefelsäure, Salpetersäure u.a. • Pflanzennährstoffe werden freigesetzt: Sulfat, Phosphat, Kieselsäure, K+, Mg2+, Cl-, Fe2+/3+,Mn2+, Zn2+, Cu2+ u. a. mol L-1 atm-1 Verwitterung von Calcit, offenes System (bei 101 325 Pa = Normaldruck) • 0.038% v/v CO2 in der Atmosphäre pH=8.3 [Ca2+] = 5x10-4 mol/L entspricht 2.8°dH • 3% v/v CO2 in der Bodenluft pH = 7.02 [Ca2+] = 2.8x10-3 mol/L entspricht 15.7°dH Effekt der Landpflanzen auf die Verwitterung • Durch den (mikrobiellen) Abbau von Pflanzenresten im Boden wird CO2 produziert und in der Bodenlösung angereichert • Die Anwesenheit der Vegetation beschleunigt die CO2-Verwitterung daher um das 100 – 150 fache • Dadurch entzieht ein Wald-Ökosystem in Österreich der Atmosphäre im Jahr ca. 20 g C/m2 also 200 kg C/ha • Dieser Kohlenstoff wird als Ca(HCO3)2 ins Meer transportiert und dort als CaCO3 ausgefällt. Die Hälfte des gebundenen CO2 wird dabei frei, die andere Hälfte in den Carbonatsedimenten dauerhaft gespeichert. Quelle: Raymond Die Flüsse sind “netto-heterotroph“, d.h. sie sind gegenüber der Atmosphäre CO2 übersättigt und können daher viel Ca(HCO3)2 aus der kontinentalen Kohlensäure-Verwitterung in Lösung halten und in die Ozeane transportieren. Carbonatsystem der Gewässer: Geschwindigkeit der Gleichgewichtseinstellung • Thermodynamisches Gleichgewicht stellt sich in der wässrigen Lösung im Allgemeinen rasch ein • Gewässer sind meist nicht im Gleichgewicht mit der Atmosphäre, weil biologische Prozesse im Wasser CO2 schneller produzieren oder konsumieren als der CO2 Transfer zwischen der Atmosphäre und dem Wasser erfolgt. • Bildung und Auflösung von CaCO3 können verzögert erfolgen • Metastabile Gleichgewichte: Aragonit (orthorhombisch) ist in einem natürlichen Wasser thermodynamisch weniger stabil als Calcit (trigonal). Unter bestimmten Bedingungen kann sich Aragonit gegenüber Calcit metastabil verhalten. Metallionen in Gewässern • Durch die zivilisatorischen Tätigkeiten sind die geochemischen Kreisläufe einer Anzahl metallischer Elemente beschleunigt. • Natürliche Flüsse: durch Verwitterung der Gesteine, vulkanische Emissionen, Verbreitung natürlicher Aerosole aus Böden und Meerwasser. • Die anthropogenen Flüsse übersteigen oft die natürlichen Flüsse. Die Gewässer sind dadurch besonders betroffen. • Anthropogene Quellen für Schwermetalle sind z.B. Erzgewinnung, metallverarbeitende Industrien, Verbrennung fossiler Brennstoffe, Zementproduktion. • Durch die Verbrennung fossiler Brennstoffe werden z.B. die Flüsse von As, Cd, Se, Hg, Zn in die Atmosphäre stark erhöht. Dadurch werden auch die Konzentrationen dieser Elemente im Wasser und in den Böden erhöht. Schwermetall-Entgiftungsstrategien der Organismen • • • Enzymatische Umwandlungen von toxischen zu weniger toxischen oder flüchtigen Spezies: Hg2+→Hg0 As(OH)3→(CH3)3As+-CH2COO- “Arsenobetain“ • Spezielle Membranen können den Durchtritt von Metallionen in besonders gefährdete Bereiche wie Gehirn, Fetus verhindern • Hochmolekulare Verbindungen wie z. B. die Metallthionein-Proteine (=kleine Proteine mit hohem Cysteinanteil von 20-30%) können toxische Metallionen binden und damit aus dem Verkehr ziehen Speziierung der Schwermetalle in der Umwelt • Gelöst oder an feste bzw. kolloidale Phasen gebunden • Komplexbildung mit verschiedenen Liganden in Lösung • Verschiedene Redoxzustände • Metallorganische Verbindungen • Das Schicksal von Schwermetallen in den Gewässern hängt von der Speziierung ab (z.B. Transport in die Sedimente, Mobilisierung aus Sedimenten, Infiltration ins Grundwasser, Anreicherung in Organismen) • Die Toxizität ist stark von der jeweiligen chemischen Spezies abhängig. • Dreiwertige lösliche Verbindungen des Arsens sind z.B. krebserregend. Dabei kommt es vermutlich nicht zu einer direkten Einwirkung auf die DNA, sondern einer Verdrängung des Zink-Ions aus seiner Bindung zu Metallothioneinen und damit Inaktivierung von Tumor-Repressor-Proteinen. Hg als umweltrelevantes Schwermetall 4x10-5 Massen% Hg in der Erdkruste Hg22+ ↔Hg0 + Hg2+ sehr schwerlösliche Minerale: Hg2Cl2 und HgS HSAB-Prinzip → Thiophilie des Quecksilbers: Starke Wechselwirkung mit schwefelhaltigen Liganden z.B. Thiole = „Mercaptane“ = Quecksilberfänger Quecksilberdampfdruck in Abhängigkeit von der Temperatur Metallisches Quecksilber ist gegenüber Luftsauerstoff bei Raumtemperatur stabil: keine Oxidhaut Bei 20°C im Gleichgewicht 13,6 mg/m3 Hg in der Luft MAK-Wert: 0,1 mg/m3 Chronische Quecksilbervergiftung z.B. durch Einatmen von Quecksilberdämpfen oder Stäuben : • Durchblutungsstörungen • Beeinträchtigung der Koordination z.B. beim Schreiben • Erregbarkeit “Mad Hatter Syndrom“ der Hg(NO3)2 verwendenden Hutmacher im 19. Jh (vgl. Lewis Carrols Alice im Wunderland) • Gedächtnisverlust • Bei extrem hoher Belastung: Lähmung, Taubheit, Blindheit, Tod Quecksilberverbindungen haben nur mit sehr harten Basen wie Fluorid und Nitrat den Charakter von Ionenkristallen. In anderen Fällen besitzen die Bindungen des Quecksilbers einen ungewöhnlich hohen kovalenten Anteil → geringe elektrische Leitfähigkeit der wässrigen Lösungen, gute Löslichkeit auch in organischen Lösungsmitteln, z.B. Löslichkeit von HgCl2: Quelle: Heiko Potgeter Quecksilber(II)verbindungen bilden mit vielen in Gewässern vorkommenden Anionen stabile Komplexe, dadurch kann die Wasserlöslichkeit stark erhöht werden. mit Halogenidionen: HgX3-, HgX42- (vor allem im Meerwasser) Ungewöhnlich stabile Komplexe mit Huminstoffen („gelöste“ kolloidale Phase in Fließgewässern, Seen, Grundwasser) Partikelgrößenverteilung von Huminstoff-Kolloiden Huminstoffe: ubiquitär in aquatischen Systemen, binden Schwermetalle Funktionen: 1. Entgiftung 2. Transport Bindungsformen: 1. Sorption 2. “non-exchangable binding“ Organische Quecksilberverbindungen Wichtig ist ausschließlich die Oxidationsstufe +II Monoorganyle RHgX, Diorganyle R2Hg linear gebaut Kovalente Bindung Hg-C relativ geringe Bindungsenergien 50 – 200 kJ/mol kann leicht homolytisch unter Bildung von Radikalen gespalten werden (thermische oder photolytische Zersetzung) → Lebensdauer in der Atmosphäre nur wenige Stunden Synthesechemie: die einfach durchzuführende homolytische Spaltung der Hg-C Bindung wird für die Bildung von Radikalen ausgenutzt. In der Umwelt ist die Hg-C Bindung gegenüber Wasser und Luft weitgehend inert (aus kinetischen Gründen) Charakteristische Abbaureaktionen in der Umwelt: Acidolyse: RHgX + HA → RH + AHgX Reduktion: RHgX + 2 H2O + 2 e- → RH + HX + 2 OH- + Hg0 Homolyse: R2Hg → 2 R· + Hg0 Methylcobalamin Entstehung von Dimethylquecksilber bei der Zersetzung von Monomethylquecksilber in Anwesenheit von H2S (H2S wird durch die mikrobielle Sulfatreduktion in reduzierenden Sedimenten gebildet): 2 CH3Hg+ + S2- → CH3Hg-S-HgCH3 → (CH3)2Hg + HgS Eigenschaft Hg0 Wasserlöslichkeit g/L bei 25°C 6·10-5 5 Dampfdruck Pa bei 25°C 0,25 1,76 CH3HgCl (CH3)2Hg HgCl2 Hg2Cl2 HgS 3 73 4·10-4 1,9·10-24 8300 0,016 1,3·10-8 nicht messbar Quelle: Heiko Potgeter Verwendung von Quecksilber: •Chloralkalielektrolyse –Amalgamverfahren (Hg-Kathoden). Wird zunehmend ersetzt (Diaphramaverfahren) •Dentaltechnik für Zahnplomben (Amalgam), wird zunehmend ersetzt. •Goldgewinnung durch Amalgamierung des Erzes, auch heute noch im Amazonasgebiet, sehr problematisch. •Batterien, Schaltelemente, Meßtechnik (Thermometer). •Organische Quecksilberverbindungen wurden als Saatbeizmittel und Fungizide in der Landwirtschaft eingesetzt (heute verboten). •Quecksilberorganische Verbindungen noch immer im medizinischen und kosmetischen Bereich verwendet. •Die Quecksilber-Fördermenge betrug zwischen 1900 und 1940 jährlich ca. 4 000 t und stieg 1973 bis auf 10 000 t an. Heute beträgt sie weltweit 4 000-6 000 Tonnen/Jahr. Zudem werden jährlich bis zu 3 000 Tonnen bei der Verbrennung fossiler Brennstoffe freigesetzt. Aufnahme mit der Nahrung: gelangt vor allem beim Verzehr von Pilzen, Fischen und Meerestieren über organische Quecksilberverbindungen in den Körper. Mit der Magensäure entsteht aus CH3Hg+ das wenig dissoziierte CH3HgCl Molekül, das wegen seiner Fettlöslichkeit gut resorbierbar ist Grenzwerte • Die Weltgesundheitsorganisation (WHO) hat eine „vorläufig duldbare wöchentliche Aufnahmemenge“ für Methylquecksilber von 1,6 μg/kg Körpergewicht festgelegt, • ein Erwachsener mit 70 kg Körpergewicht kann von Fisch mit einem Quecksilbergehalt am gesetzlich festgelegten Grenzwert von 1 mg/kg fettreichem Fisch nur 112 g pro Woche essen, um die wöchentlich tolerierbare Dosis von 1,6 μg/kg Körpergewicht nicht zu überschreiten. Toxische Wirkung Anorganische Hg-Verbindungen: Giftigkeit hängt von der Wasserlöslichkeit der jeweiligen Verbindung ab. Hg2+ ist bei pH 7 in Wasser leicht löslich und bildet mit den in Körperflüssigkeiten häufiger vorkommenden Anionen keine unlöslichen Verbindungen. Hg2+ tritt mit den Thiol- und Disulfideinheiten der Proteine in Wechselwirkung , blockiert aktive Zentren, verändert Strukturen von Enzymen. Körpereigene Entgiftung durch Binden an Metallthioneine: Proteine, Molekulargewicht ca. 6500 g/mol, 35% Cystein-Anteil. Organische Quecksilberverbindungen Größte toxische Wirkung wegen des ambivalent lipophilen/hydrophilen Charakters Resorptionsrate bei oraler Aufnahme bis 95% Am giftigsten ist Methylquecksilber(II) wegen der (kinetisch) stabilen Hg-C Bindung Weniger stabile quecksilberorganische Verbindungen werden im Körper zu anorganischem Quecksilber metabolisiert und wirken daher eher wie dieses Wegen seines lipophilen/hydrophilen Charakters ist Methylquecksilber(II)chlorid in der Lage, biologische Membranen zu durchdringen und sogar die Blut-HirnSchranke und Plazenta-Membran zu überwinden. Chronische Quecksilbervergiftung durch Methylquecksilber • Müdigkeit, Kopf- und Gliederschmerzen, Zahnfleischentzündungen, Zahnlockerung, vermehrter Speichelfluss, Durchfälle und Nierenentzündungen • Schädigung des Nervensystems wie Muskelzuckungen, Stimmungsschwankungen, Erregungs- und Angstzustände, • Hör-, Seh- , Gefühls-, Sprach- und Gangstörungen, bei extrem hohen Belastungen Tod. • Methylquecksilber kann sowohl die Blut-Hirnschranke als auch die Plazentaschranke überwinden. • Die Anreicherung im Gehirn kann auch schon bei relativ geringen Belastungen zu geistigen Störungen und Entwicklungshemmungen bei Kindern führen. Mutagene Wirkung durch die Bindung von CH3Hg+ an Nukleobasen, z.B. 8-Aza-modifiziertes Adenin: Quelle: Kaim/Schwederski Wirkungsweise von Methylquecksilber im menschlichen Körper Bioakkumulation und Biomagnifikation von Quecksilber Bioakkumulation = Anreicherung von Toxinen gegenüber dem Medium (Wasser). Biomagnifikation = Anreicherung von Toxinen mit steigendem trophischem Niveau, z.B. lipophile Organometallverbindungen, die sich der Exkretion über Metallthioneine entziehen.