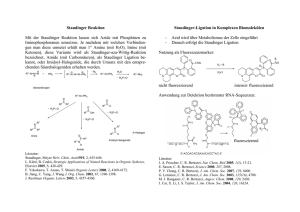
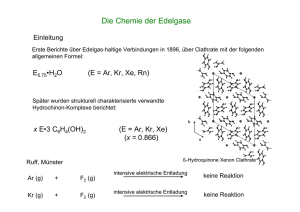
Tagungsband 2010 - Johannes Gutenberg
Werbung

Programm Sonntag, 21.02. 15:00 – 16:00 Ankunft und Registrierung im Foyer des Hörsaal C03 16:00 – 16:10 Begrüßung und Eröffnung (K. Heinze, E. Rentschler) Vorsitz: Anne Westphal (Christian-Albrechts-Universität zu Kiel) 16:10 – 16:30 V1 Ulf-Peter Apfel (Friedrich-Schiller-Universität Jena) 16:30 – 16:50 V2 Veronika Hoeke (Universität Bielefeld) 16:50 – 17:10 V3 Christine Männel-Croisé (Universität Zürich) 17:10 – 17:30 V4 Andrea Schindler (Universität Regensburg) 17:30 – 18:00 Modelle für das aktive Zentrum der [FeFe] Hydrogenase mit Bis-, Trisund Tetrakis(mercaptomethyl)silanen Einfluss der Umgebung auf die Eigenschaften von Einzelmolekülmagneten Cobalt(III)-basierte Indikatorsysteme Koordinationschemie mit Phosphaferrocenen Pause Vorsitz: Anne Dovalil (Universität Heidelberg) 18:00 – 18:20 V5 Christian Unkelbach (Technische Universität Dortmund) 18:20 – 18:40 V6 Markus Schmitz (Technische Universität Kaiserslautern) 18:40 – 19:00 V7 Philipp Stock (Technische Universität Berlin) 19:00 – 19:20 V8 Boris Burger (Georg-August-Universität Göttingen) ab 19:20 α-Silylated Secondary Alkyllithiums via Carbolithiation Deprotonation Reactions of Functionalized Silanes Koordinationsmoden eines Diazapyridinophanliganden unsymmetrisch and substituierten Eisen(II)-Spin-Crossover-Komplexe polypodaler Liganden für die Verankerung auf Oberflächen Neue zweikernige Eisenkomplexe bioinspirierte Oxidationschemie Geselliger Abend 1 als Modellsysteme für die Montag, 22.02. Vorsitz: Thomas Jozak (Technische Universität Kaiserslautern) 8:30 – 8:50 V9 Dominik Lieb (Universität Erlangen-Nürnberg) 8:50 – 9:10 V10 Malte Rolff (Christian-Albrechts-Universität zu Kiel) 9:10 – 9:30 V11 Carola Vogel (Universität Erlangen-Nürnberg) 9:30 – 9:50 V12 Burkhard Butschke (Technische Universität Berlin) 9:50 – 10:10 V13 Johannes Schnödt (Universität Stuttgart) 10:10 – 10:40 III Elementary reaction steps on Mn centers – towards understanding of enzymatic and non-enzymatic catalysis The First Catalytic Tyrosinase Model System Based Mononuclear Copper(I) Complex: Kinetics and Mechanism on a Molecular and Electronic Structure of tris-N-Heterocyclic Carbene Iron Complexes Oxygen versus Sulfur: On the Differences in the Ion/Molecule + Reactions of „Rollover“ Cyclometalated [Pt(bipy – H)] with (CH3)2X (X = O, S) – A Mechanistic Study Eigenschaften redoxaktiver Kupfer(I)-Komplexe mit biomimetischen N,S-Ligandensystemen Pause Vorsitz: David Schweinfurth (Universität Stuttgart) 10:40 – 11:00 V14 Thomas Jozak (Technische Universität Kaiserslautern) 11:00 – 11:20 V15 Gunnar Werncke (Humboldt-Universität zu Berlin) 11:20 – 11:40 V16 Alexander Döring (Georg-August Universität Göttingen) 11:40 – 12:00 V17 Natascha Kempf (Justus-Liebig-Universität Gießen) 12:00 – 12:20 V18 Sebastian Marks (Karlsruher Inst. f. Technologie (KIT)) 12:20 – 13:50 Mittagspause C2-Symmetric Dipyrazolyl Ligands Oxovanadiumthiobisphenolatkomplexe als Molekülmodelle für geträgertes Vanadiumpentoxid in der Oxidativen Dehydrogenierung von Alkanen Vergleichende Untersuchungen an Paaren von Molybdän- und Wolframverbindungen Untersuchung tripodaler Kupferkomplexe unter Verwendung guanidinhaltiger Liganden Bisphosphiniminomethanid als Ligand in der Koordinationschemie des Zinks 2 Vorsitz: Christian Unkelbach (Technische Universität Dortmund) 13:50 – 14:10 V19 Dirk Schuch (Friedrich-Schiller-Universität Jena) 14:10 – 14:30 V20 Anne Westphal (Christian-Albrechts-Universität zu Kiel) 14:30 – 14:50 V21 Philipp Mücke (Universität Regensburg) 14:50 – 15:10 V22 Nina Dovalil (Universität Heidelberg) 15:10 – 15:30 V23 Sandra Kisslinger (Justus-Liebig-Universität Gießen) 15:30 – 16:50 Synthese und magnetische Eigenschaften C3-symmetrischer triaminoguanidinbasierter Übergangsmetallkomplexe MCD Spectra of Mononuclear Manganese(III) Complexes Intramolekularer Elektronentransfer in [2.2]Paracyclophanen: Durch den Raum oder über die Brücke? Coordination chemistry of cyclic peptides Spin Crossover Properties of Iron(II) Complexes Posterpräsentation Vorsitz: Jeroen Volbeda (Technische Universität Braunschweig) 16:50 – 17:10 V24 Alexander Hoffmann (Technische Universität Dortmund) 17:10 – 17:30 V25 Thomas Schwarze (Universität Potsdam) 17:30 – 17:50 V26 Florian Blasberg (Goethe-Universität Frankfurt am Main) 17:50 – 18:10 V27 Thorsten Gehrmann (Universität Heidelberg) 18:10 – 18:30 V28 Subrata Kundu (Humboldt-Universität zu Berlin) ab 19:00 Geselliger Abend im „Eisgrubbräu“ Synthese und Charakterisierung Poly(pyrazolyl)methanliganden von Cobalt(II)-Komplexen mit Fluoroionophore für Palladiumdichlorid und Kupfer(II)-Ionen Quinone-substituted Bis(pyrazol-1-yl)methane Ligands and a Cu(II)Hydroquinone Coordination Polymer Zirkonium(IV)-Hydrazindiidokomplexe: N-N-Bindungsspaltung versus Cycloadditionsreaktionen Synthesis of Multinuclear Fe(II) Complexes 3 Dienstag, 23.02. Vorsitz: Natascha Kempf (Justus-Liebig-Universität Gießen) 8:30 – 8:50 V29 Johanna Niesel (Ruhr-Universität Bochum) 8:50 – 9:10 V30 David Schweinfurth (Universität Stuttgart) 9:10 – 9:30 V31 Christoph Topf (Johannes Kepler Universität Linz) 9:30 – 9:50 V32 Sebastian Lange (Universität Paderborn) 9:50 – 10:10 V33 Birte Haberlag (Technische Universität Braunschweig) 10:10 – 10:40 Untersuchungen zur Toxizität und intrazellulären Verteilung von + [Mn(CO)3(tpm)] Strukturelle, elektrochemische und spektroskopische Untersuchungen neuer triazolhaltiger Liganden und ihrer Übergangsmetallkomplexe Synthese und Charakterisierung von Ag(I)-, Au(I)- und Au(III)Komplexen funktionalisierter N-heterocyclischer Carbene Sauerstoffaktivität von Gruppe-9-Elementen in Komplexen mit NHCPhosphan-Liganden Alkyne Metathesis with Well-Defined Imidazoline-2-iminato Tungsten & Molybdenum Benzylidyne Complexes Pause Vorsitz: Ulf-Peter Apfel (Friedrich-Schiller-Universität Jena) 10:40 – 11:00 V34 Jabadurai Jayapaul (RWTH Aachen) 11:00 – 11:20 V35 Lam Oanh (Universität Erlangen-Nürnberg) 11:20 – 11:40 V36 Dr. Peter Kunz (Heinrich-Heine-Universität Düsseldorf) 11:40 – 12:00 V37 T. David Pilz (Universität Erlangen-Nürnberg) 12:00 – 12:20 Schlusswort und Verabschiedung (K. Heinze, E. Rentschler) Novel non-polymeric fluorescent bimodal contrast agent for magnetic resonance and optical imaging Activation of Carbon Dioxide and Related Small Molecules at Reactive U(III) Centers Wasserlösliche (Di)Phosphane - Neue Liganden für metallbasierte Wirkstoffe The first photo-redox-active N-heterocyclic carbene – a new class of bridging ligands suitable for photo- and redoxcatalysis 4 Posterpräsentation P1 Carlos Enrique Abad Andrade (Georg-August Universität Göttingen) The role of Sulfur and Selenium in the DMSO-reductase family of enzymes: getting answers from small models P2 Martin Bernard (Universität Paderborn) Kupferkomplexe mit Guanidin-Schwefel-Hybridliganden P3 Olga Bienemann (Technische Universität Dortmund) Neue hochaktive Kupfer-Guanidin-Komplexe in der Atomtransfer-RadikalpolymerisationsKatalyse (ATRP) P4 Aaron Breivogel (Johannes Gutenberg-Universität Mainz) Neue Rutheniumchromophore mit maßgeschneiderten Eigenschaften P5 Dr. Luca Carrella (Johannes Gutenberg-Universität Mainz) New class of heterometallic pivalate complexes -Study of their magnetic properties- P6 Victoria Colquhoun (Technische Universität Dortmund) Structural control in deprotonation and substitution reactions P7 Hari Sankar Das (Universität Stuttgart) Mono- & Dinuclear Ruthenium(II & III) Complexes With Noninnocent Quinonoid Ligands: Consequences For Structural, Redox, Electronic and Spectroscopic Properties P8 Maria Eckhardt (Universität Regensburg) Phosphaalkine als synthetische Bausteine zur Bildung von Oligomeren und Polymeren P9 Jan Falkenhagen (Humboldt-Universität zu Berlin) Die Synthese neuartiger Fe-O-Mo-Komplexe und deren Anwendung in der Oxygenierungskatalyse P10 Felix Friedel (Friedrich-Alexander-Universität Erlangen Nürnberg) Tuning of redox potentials of seven-coordinate iron and manganese complexes for superoxide dismutation and nitric oxide transformation P11 Felix Fuge (RWTH Aachen) Catalysis optimization of cyclopalladated nitrosamines for Heck- and Suzuki-coupling reactions P12 Simon-Andreas Gentschow (Technische Universität Berlin) Bindungsaktivierung in Eisen(II)-Komplexen polypodaler Phosphane P13 Sonja Hammer, Thomas Bernert (Universität Frankfurt) Strukturlösung und magnetische Eigenschaften von trans-[NiCl2(C5H5N)2] aus Pulverdaten P14 Dr. Christina Hauser (Friedrich-Alexander-Universität Erlangen-Nürnberg) Seeing is Believing – Spectroscopic Determination of Oxidation States in Non-Oxo Uranium (III) to (VI) complexes 5 P15 David Hornig (Friedrich-Schiller-Universität Jena) DFT-Studien zu den magnetischen Eigenschaften oligonuklearer Kupferkomplexe P16 Rajkumar Jana (Universität Stuttgart) Variable binding of heterodinuclear (Cu, Fe) organometallics to N and O donor functions of guanine, pterin, lumazine and alloxazine heterocycles P17 Dr. Marat Khusniyarov (Friedrich-Alexander-Universität Erlangen-Nürnberg) Can one oxidize a NacNac ligand? P18 Matthias Kruck (Universität Heidelberg) Bis(2-pyridylimino)isoindole als Steuerliganden in der asymmetrische Übergangsmetallkatalyse P19 Martin Maurer (Universität Heidelberg) Oxidation catalysis by high-valent iron bispidines P20 Yakiv Melomedov (Johannes Gutenberg-Universität Mainz) Neue Porphyrinaminosäuren mit einstellbaren elektronischen Eigenschaften für Untersuchung photoinduzierter Elektronentransfer und Energietransfer-Prozesse P21 Anja Molter (Bergische Universität Wuppertal) P22 Dr. Uwe Monkowius (Johannes Kepler Universität Linz) Synthesen und Strukturen von Phosphinogold(I)komplexen mit Seleno- und Thiocarbamatderivaten Synthese und Charakterisierung von Kupfer(I)-Komplexen mit chromophoren 1,2-Bis(arylimino)acenaphthen -Akzeptorliganden P23 Dr. Chandan Mukherjee (Universität Bielefeld) A Chiral Triplesalen Ligand: From Enantioselective Oxidation Catalysis to Chiral SingleMolecule Magnets P24 Kevin Muller (Technische Universität Kaiserslautern) 2-(3-Pyrazolyl)pyridines as Ligands in the Ruthenium Catalysed Hydrogenation of Carbon Dioxide P25 Dr. Alexander Petrov (Technische Universität Braunschweig) gem-Diamino-Dicarben-Komplexe des Rutheniums P26 Florian Pevny (Universität Regensburg) 3 Better Pt-based MLCT-Sensibilisators by Dye-Functionalisation P27 Anna-Maria Pütz (Johannes Gutenberg-Universität Mainz) New Model Complexes for Nickel and Copper Containing Enzymes P28 Sabine Reh (Technische Universität Kaiserslautern) Spincrossover in Cobalt(II)-Komplexen 6 P29 René Römer (Christian-Albrechts-Universität zu Kiel) P30 Dimitri Sakow (Philipps-Universität Marburg) Molybdän-Oxo-Komplexe mit einem tetradentaten Phosphan Liganden: Synthese und spektroskopische Untersuchung Koordinationschemie der 10-Thiacorrole P31 Anne Scheja (Philipps-Universität Marburg) Spincrossover bei BAI-Eisen(II)-Komplexen P32 Dr. Maria Schlangen (Technische Universität Berlin) On the formation and structure of 'bare' [M,C,H3O]+ complexes (M = Fe, Co, Ni) in the gas phase P33 Esther Schuh (Bergische Universität Wuppertal) Synthese und Charakterisierung von N-heterocyclischen Gold(I) Komplexen mit ThiolatLiganden P34 Elisabeth Seikel (Philipps-Universität Marburg) Synthetic Challenges Towards New Phthalocyanine Materials P35 Dr. Carsten Streb (Friedrich-Alexander-Universität Erlangen-Nürnberg) Supramolekulare Metalloxidkomplexe als Vorstufen für nanostrukturierte Komposite P36 Dominique Thielemann (Karlsruher Institut für Technologie) New Amino Acid Ligated Yttrium Hydroxy Clusters P37 Lars Valentin (Justus-Liebig-Universität Gießen) 2 Synthese und Charakterisierung des zweikernigen Ni(0)-Komplexes [Ni2(η -C4H6)2(O-BPY)] unter Verwendung von Ni(COD)2 und 1,2-Bis(2,2´-bipyridyl-6-yl)ethan (O-BPY) P38 Jeroen Volbeda (Technische Universität Braunschweig) Coordination Chemistry of Multidentate Imidazolin-2-imine Based Ligands P39 Jan Wimberg (Georg-August-Universität Göttingen) Übergangsmetallkomplexe mit neuen asymmetrischen NHC/Pyrazol-Liganden 7 V1 Modelle für das aktive Zentrum der [FeFe] Hydrogenase mit Bis-, Tris-, und Tetrakis(mercaptomethyl)silanen Ulf-Peter Apfel,‡ Dennis Troegel,§ Yvonne Halpin,† Stefanie Tschierlei,†† Ute Uhlemann,†† Helmar Görls,‡ Michael Schmitt,†† Jürgen Popp,†† Manfred Rudolph,‡ Johannes G. Vos,† Reinhold Tacke,§ Wolfgang Weigand‡ ‡ Institut für Anorganische und Analytische Chemie, Friedrich-Schiller-Universität Jena, August-Bebel-Straße 2, D-07743 Jena, Germany, § Institut für Anorganische Chemie, Universität Würzburg, Am Hubland, D-97074 Würzburg, Germany, † Solar Energy Conversion SRC, School of Chemical Sciences, Dublin City University, Dublin 9, Ireland, †† Institut für Photonische Technologien, Friedrich-Schiller-Universität Jena, Albert-EinsteinStraße 9, D-07745 Jena, Germany, Ausgehend von Chlormethylsilanen wurde eine Reihe von funktionalisierten Mercaptomethylsilanen des allgemeinen Typs R4-nSi(CH2SH)n (n = 2–4; R = organyl) synthetisiert. Die oxidative Addition an Fe3(CO)12 führte zu Komplexen, die Mercaptomethylsilane als verbrückende Liganden enthalten. Es soll getestet werden, welchen Einfluß die “Sila-Substitution” auf mögliche Hydrogenase Eigenschaften ausübt. Die chemische Zusammensetzung der Komplexe konnte durch Elementaranalyse, NMR Spektroskopie (1H, 13 C, 29 Si), und Röntgenbeugungsuntersuchungen bestätigt werden. Elektrochemische Untersuchungen mittels Zyklischer Voltammetrie deuten auf einen neuartigen Mechanismus für die Entstehung von molekularem Wasserstoff an [FeFe] Hydrogenasemodellen hin. Dabei ist der Reduktion des [2Fe2S(Si)] Clusters ein dynamisches Protonierungs- gleichgewicht an einem der verbrückenden Schwefelatome vorgelagert. Zudem wurden Reaktion eines [2Fe2S] Clusters mit HBF4 und Natriumamalgam IR- spektroskopisch untersucht sowie Digitalsimulationen der voltamogramme durchgeführt, welche den Zyklo- postulierten Mechanismus bestätigten. 8 V2 Einfluss der Umgebung auf die Eigenschaften von Einzelmolekülmagneten Hoeke, V.; Glaser, T.; Bielefeld/D Thorsten Glaser, Lehrstuhl für Anorganische Chemie I, Fakultät für Chemie, Universität Bielefeld, Universitätsstraße 25, 33615 Bielefeld Einzelmolekülmagnete sind eine Klasse von Koordinationsverbindungen, die einen hohen Spingrundzustand St in Kombination mit einer großen negativen axialen Anisotropie DSt aufweisen. Infolgedessen besitzen sie eine Energiebarriere für die Spinumkehr, die in einer magnetischen Hysterese rein molekularen Ursprungs resultiert. Wir haben das Ligandensystem Tripelsalen entwickelt, mit dem Einzelmolekülmagnete des Typs Mt6Mc (t: terminal; c: zentral) zugänglich sind. Unsere siebenkernigen Komplexe werden unter Ausnutzung der molekularen A O Abb. 1 A Mt O Erkennung aus drei Bausteinen B N aufgebaut (Abb. 1): zwei termi- N nale dreikernige Tripelsalen- N N Mt O Einheiten A und ein zentrales O O N M t N A O Hexacyanometallat B.[1-3] Eine Optimierung der magnetischen Eigenschaften der Komplexe ist durch eine gezielte Variation des Tripelsalenliganden und der Metallionen möglich. Die strukturellen und magnetischen Eigenschaften ausgewählter Mt6Mc-Komplexe werden vorgestellt. Anhand einer Reihe von MnIII6CrIII-Verbindungen wird der Einfluss der Umgebung auf die Eigenschaften von Einzelmolekülmagneten diskutiert. Ein Schwerpunkt liegt dabei auf der Auswertung der AC-Suszeptibilität nach Cole und Cole,[4] die eine exakte und umfassende Charakterisierung des Relaxationsverhaltens von Einzelmolekülmagneten erlaubt. [1] T. Glaser, M. Heidemeier, T. Weyhermüller, R.-D. Hoffmann, H. Rupp, P. Müller, Angew. Chem. Int. Ed. 2006, 45, 6033-6037. [2] T. Glaser, M. Heidemeier, E. Krickemeyer, H. Bögge, A. Stammler, R. Fröhlich, E. Bill, J. Schnack, Inorg. Chem. 2009, 48, 607-620. [3] E. Krickemeyer, V. Hoeke, A. Stammler, H. Bögge, J. Schnack, T. Glaser, Z. Naturforsch. B (Rolf W. Saalfrank Honorary Issue) 2010, 65b, zur Veröffentlichung angenommen. [4] K. S. Cole, R. H. Cole, J. Chem. Phys. 1941, 9, 341-351. 9 V3 Cobalt(III)-basierte Indikatorsysteme Männel-Croisé, C.; Zelder, F.; Zürich/ CH Christine Männel-Croisé, Institut für anorganische Chemie, Universität Zürich, Winterthurerstraße 190, 8057 Zürich Cyanid wirkt in physiologischen Systemen als hochwirksames Gift1, wird jedoch von etwa 2000 Pflanzen synthetisiert und in Form biologisch inaktiver cyanogener Glykoside gespeichert. Eine Zellbeschädigung, beispielsweise bei der Verwendung solcher Pflanzen als Nahrungsmittel, führt zur enzymatischen Freisetzung des Cyanids und stellt ein Gesundheitsrisiko dar.2 Ausgehend von Vitamin B12 setzten wir Corrinoide als hoch sensitive und selektive Chemosensoren zur kolorimetrischen Detektion von Cyanid in Wasser ein.3 Die Energiedifferenz der π-π* Übergänge des Corrin-chromophors wird durch die axialen Liganden des sechsfach koordinierten Co(III)-Ions stark beeinflusst. Die Substitution des Co(III) gebundenen Wassers durch Cyanid wird durch einen Farbumschlag von orange nach violett begleitet. Mechanistische Untersuchungen zeigten, dass die Sensitivität und Selektivität dieser metall-basierten Chemosensoren gegenüber Anionen durch Veränderung der Seitenketten gesteuert werden kann.4 Kinetische Experimente mit stopped-flow Technik gaben einen ersten Einblick in den Reaktionsmechanismus. Zum ersten Mal konnte mit einem optischen Chemosensor die enzymatische Freisetzung endogener 10 Cyanide mittels diffuse reflectance UV-vis Spektroskopie biologischen Oberflächen in Echtzeit verfolgt werden. [1] [2] [3] [4] [5] (DRUV-Vis) 5 Solomonson, L. P. Cyanide in Biology; Academic Press: London, 1981. Zelder, F. H.; Männel-Croisé, C. Chimia 2009, 63, 58-62. Zelder, F. H. Inorg. Chem. 2008, 47, 1264-1266. Männel-Croisé, C.; Zelder, F. H. Inorg. Chem. 2009, 48, 1272-1274. Männel-Croisé, C.; Probst, B.; Zelder, F. H. Anal. Chem. 2009, 81, 9493-9498. 11 an V4 Koordinationschemie mit Phosphaferrocenen Schindler, A.; Scheer, M. Prof. Dr. Manfred Scheer, Institut für Anorganische Chemie, Universität Regensburg, Universitätsstrasse 31, 93053 Regensburg Die Selbstanordnung molekularer Einheiten zu ein- oder mehrdimensionalen Koordinationspolymeren ist ein faszinierendes Gebiet moderner chemischer Forschung. Die Phosphaferrocene [Cp*Fe( 5-P3C2tBu2)], [Fe( 5-P3C2tBu2)2] und [Cp*Fe( 5-P5)] stellen in Kombination mit Cu(I)-Halogeniden sehr vielfältige Bausteine koordinationschemischer Untersuchungen dar. Neben der Bildung strukturell interessanter Koordinationspolymere ist unter optimalen Reaktionsbedingungen sogar der Aufbau sphärischer supramolekularer Aggregate zu beobachten. Durch den Zusatz geeigneter Template kann die Größe der Aggregate gesteuert werden (Abbildung 1).[1] Abbildung 1: Sphärische Koordinationsverbindung aus Pentaphosphaferrocen- und CuCl-Einheiten. Im Inneren des Moleküls befindet sich ortho-Carboran als Gast. [1] M. Scheer, A. Schindler, C. Gröger, A. V. Virovets, E. V. Peresypkina, Angew. Chem. 2009, 121, 4969-4980. 12 V5 α-Silylated Secondary Alkyllithiums via Carbolithiation and Deprotonation Reactions of Functionalized Silanes Unkelbach, C.; Strohmann, C.; Dortmund/D Carsten Strohmann, Anorganische Chemie, Technische Universität Dortmund, Otto-Hahn-Straße 6, 44227 Dortmund Silylated alkyllithium compounds apply as versatile nucleophilic synthons in organic and inorganic synthesis, for example in the Peterson olefination reaction or, after consecutive Tamao oxidation, as masked “α-hydroxycarbanions”.[1] Therefore, our objective is the access to stereochemically defined α-silylated alkyllithiums bearing a variety of side-chains. We investigated both carbolithiation and deprotonation reactions of respective silane substrates as possible routes to the desired class of compounds. The key concept is the inclusion of a stereochemically pure, coordinating side-arm in the substrate. This serves both to enhance the reactivity at low temperatures and also to dictate the configuration of the stereogenic metalated centre generated in the course of the reaction, resulting in diastereomeric ratios of up to 98:2 in the respective quenching products.[2] Figure 1: Left: General routes to α-silylated alkyllithiums via carbolithiation and deprotonation; Right: Crystal structure of an α-silylated alkyllithium compound bearing a lithiated ethyl group next to silicon. [1] a) T. H. Chan, P. Pellon, J. Am. Chem. Soc. 1989, 111, 8738; b) T. H. Chan, K. T. Nwe, J. Org. Chem. 1992, 57, 6107. [2] a) D. J. Ager, J. Chem. Soc. Perkin Trans. 1 1983, 1131; b) S. Murai, I. Ryu, J. Iriguchi, N. Sonoda, J. Am. Chem. Soc. 1984, 106, 2440; c) M. van der Leij, B. Zwanenburg, Tetrahedron Lett. 1978, 36, 3383. 13 V6 Koordinationsmoden eines unsymmetrisch substituierten Diazapyridinophanliganden Schmitz, M.; Krüger, H.-J.; Kaiserslautern/D Markus Schmitz, Fachbereich Chemie, TU Kaiserslautern, Erwin-Schrödinger-Str., 67663 Kaiserslautern Diazapyridinophanliganden, die neben den Pyridinstickstoffdonoratomen noch Aminstickstoffdonoratome enthalten, bilden in den meisten Fällen mit Metallionen cisoktaedrische Koordinationsumgebungen, wobei der Ligand über all seine vier Stickstoffdonoratome gebunden vorliegt[1]. In dem Liganden L-N4tBuTos ist eines der beiden Amine durch eine Tosylamidfunktion ersetzt worden, dessen σ-Donoreigenschaften als schwach einzuschätzen sind. In Komplexen mit diesem Liganden bestimmen die Ladungen der cis-ständigen Koliganden die Koordinationsweise des Makrozyklus. [Ni(L-N4tBuTos)Cl2] · MeCN [Ni(L-N4tBuTos)(pyc)](ClO4) · MeCN So koordiniert der makrozyklische Ligand mit zwei negativ geladenen oder einem dianionischen Koliganden nur über drei seiner vier potentiellen Stickstoffdonoratome an das Metallion, nämlich über die beiden Pyridinstickstoffatome und das Aminstickstoffatom. Dabei nimmt der Ligand eine syn-chair-boat-Konformation ein. Mit neutralen oder monoanionischen Koliganden hingegen wird nur die vierzähnige Bindungsweise des Makrozyklus beobachtet. [1] H.-J. Krüger, Chem. Ber., 1995, 128, 531 14 V7 Eisen(II)-Spin-Crossover-Komplexe polypodaler Liganden für die Verankerung auf Oberflächen Stock, P. a; Hörner, G. b; Gieb, K. c; Grohmann, A. a a Institut für Chemie, Technische Universität Berlin, Straße des 17. Juni 135, D-10623 Berlin b Fakultät für Chemie, Adam Mickiewicz Universität Poznań, Grunwaldzka 6, PL-60-780 Poznań c Physikalisches Institut, Universität Erlangen-Nürnberg, Erwin-Rommel-Straße 1, D-91058 Erlangen Viele Eisen(II)-Komplexe mit N6-Donorsatz zeigen Spin-Crossover. Darunter versteht man den reversiblen Übergang vom diamagnetischen low-spin zum paramagnetischen high-spin Zustand. Dieser Vorgang des Schaltens kann durch externe Stimuli, wie die Änderung des Drucks oder der Temperatur hervorgerufen werden.[1] Wir befassen uns mit der Aufbringung derartiger Komplexe in Form von self-assembled-monolayers (SAMs) auf Oberflächen. Zur Verankerung der Komplexe auf Au(111) verwenden wir Thiocyanatgruppen.[2] Es wird die Synthese zweier tripodaler Liganden, N[(CH2)2N=CHpy-O(CH2)4SCN]3 und (S)P[N(Me)N=CHpy-O(CH2)4SCN]3 vorgestellt. Diese „wickeln“ sich bei der Komplexierung mit Eisen(II)-Salzen schraubenartig um das Zentralion, was die Ausrichtung der Thiocyanatgruppen zur Folge hat. Die entstandenen Komplexe A und B wurden mittels SQUID, Hochtemperatur-NMR-Spektroskopie und LaserPhotolyse untersucht. [1] J. Pitarch López, H. Kämpf, M. Grunert, P. Gütlich, F. W. Heinemann, R. Prakash, A. Grohmann, Chem. Commun. 2006, 1718-1720 [2] J. W. Ciszek, M. P. Stewart, J. M. Tour, J. Am. Chem. Soc. 2004, 126, 13172-13173 15 V8 Neue zweikernige Eisenkomplexe als Modellsysteme für die bioinspirierte Oxidationschemie Burger, B.; Wöckel, S.; Dechert, S.; Meyer,F.; Göttingen/D Prof. Dr. Franc Meyer, Institut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstr. 4, 37077 Göttingen Zweikernige nichthäm-Eisen-Oxo-Proteine sind in der Natur sehr weit verbreitet und besitzen, trotz ihrer engen strukturellen Analogien, eine Vielzahl biorelevanter Funktionen. Die meisten der carboxylatverbrückten Dieisen(II)-Zentren solcher Proteine sind in der Lage, innerhalb ihrer funktionellen Prozesse kontrolliert mit molekularem Sauerstoff zu reagieren und diesen für weitere Umsetzungen zu aktivieren.[1, 2] Wir entwickeln und nutzen pyrazolbasierte Liganden, welche in 3- und 5-Position des heterozyklus mit chelatisierenden Seitenarmen substituiert sind, um ein rigdes, präorganisiertes Gerüst für entsprechende zweikernige Modellkomplexe solcher Proteine bereitzustellen.[3] N N Fe Fe X Allgemeine schematische Darstellung eines pyrazolat-basierten Dieisen-Komplexes Nach dieser Strategie konnten verschiedene biomimetische Diesen(II)-Komplexe dargestellt und strukturell charakterisiert werden. Einige Komplexe waren zur Aktivierung von Sauerstoff geeignet und konnten in exemplarischen Oxidationsreaktionen verschiedener Substrate eingesetzt werden. Wir berichten über die Darstellung der Komplexe sowie erste Ergebnisse zur Oxidationschemie. [1] D. M. Kurtz Jr., J. Biol. Inorg. Chem. 1997, 2, 159. [2] M. Merkx, D. A. Kopp, M. H. Sazinsky, J. L. Blazyk, J. Müller, S. J. Lippard, Angew. Chem. 2001, 113, 2860. [3] J. Klingele, S. Dechert, F. Meyer, Coord. Chem. Rev. 2009, 253, 2698. 16 V9 Elementary reaction steps on MnIII centers – towards understanding of enzymatic and non-enzymatic catalysis Dominik Lieb, Achim Zahl, Milos Filipovic, Ivana Ivanović – Burmazović Department of Chemistry and Pharmacy, University of Erlangen-Nürnberg, Egerlandstraße 1, 91058 Erlangen. The chemistry of manganese(III) is not yet fully explored and elementary reaction steps behind its activity in enzymatic and non-enzymatic redox catalysis need to be elucidated. For that reason we have investigated in detail the water exchange process on Mn(III) centres offering the better understanding of the reaction steps within the water splitting cycle of the Photosystem II oxygen evolving cluster (OEC)1 and superoxide dismutation by manganese centres.2 16.0 15.9 15.8 ln (1 / T2r) 15.7 15.6 15.5 15.4 15.3 15.2 0.0029 0.0030 0.0031 0.0032 0.0033 0.0034 0.0035 0.0036 1/T Chart 2. Time-resolved spectra of catalytic decomposition Chart 1. Water exchange on [MnIII(TMpyP)(H2O)OH] 4+ at of 500 μM ONOO- by SOD mimic at pH 10.0 (inset: for pH 10. comparison, spontaneous decomposition of 500 μM ONOO- at pH 10.0). In order to shed light on the real reaction pathways that are responsible for the beneficial effects of well established seven-coordinate manganese macrocyclic SOD mimetics3 we have studied in detail their cross-reactions towards OH, NO2 and NOO-. 1 Hiller, W.; Wydrzynski, T. Coord. Chem. Rev. 2008, 252, 306–317. 2 Salvemini, D.; Riley, D. P.; Cuzzocrea, S. Nat. Rev. Drug Discovery 2002, 1, 367–374. 3 Filipovic, M. R.; Duerr, K.; Mojovic, M.; Simovic, V.; Zimmerman, R.; Niketic, V.; Ivanovic-Burmazovic, I. Angew. Chem. 2008, 47 (45), 8735–8739. 17 V10 The First Catalytic Tyrosinase Model System Based on a Mononuclear Copper(I) Complex: Kinetics and Mechanism Rolff, M.; Tuczek, F.; Kiel/D Malte Rolff, Felix Tuczek, Institut für Anorganische Chemie, Christian-AlbrechtsUniversität zu Kiel, May-Eyth-Straße 2, 24118 Kiel Tyrosinase (Ty) is an ubiquitous copper enzyme mediating the o-hydroxylation of monophenols to catechols and the subsequent two-electron oxidation to quinones. These are the first steps of melanine synthesis. The reactivity of Ty towards phenolic or aromatic substrates has successfully been reproduced with small-molecule copper complexes. Nevertheless, the number of catalytic model systems of tyrosinase is scarce. We here present the first Ty model system based on a mononuclear copper(I) complex that is both catalytic and in a stoichiometric mode allows to individually address the two consecutive stages of the Ty reaction, phenol hydroxylation and product release as quinone.[1] Apart from the synthesis and characterisation of the ligand L1 and the corresponding copper(I) complex the electronic and structural factors that determine whether a model system is catalytic or not were investigated. The hydroxylation of the substrate in the stoichiometric reaction was shown to follow a first order kinetics in relation to copper. The resulting dinuclear Cu(II)2-µ-catecholato-µ-hydroxo species could be isolated and analysed demonstrating the dinuclear mechanism of this reaction. [1] M. Rolff, J. Schottenheim, G. Peters, F. Tuczek, submitted. 18 V11 Molecular and Electronic Structure of tris-N-Heterocyclic Carbene Iron Complexes Carola S. Vogel, Frank W. Heinemann, Jörg Sutter, and Karsten Meyer Department of Chemistry and Pharmacy, Friedrich-Alexander-University Erlangen-Nuremberg, Egerlandstraße 1, 91058 Erlangen Coordination compounds of iron in high oxidation states have been invoked as intermediates in biocatalysis, the iron nitride synthon has been implicated in nitrogenase at the site of biological nitrogen reduction.[1] In order to gain understanding of iron nitride reactivity and the possible role of such species in catalysis, insight into the molecular structure of complexes stabilizing the [FeN] moiety is highly desirable. We herein present the synthesis, spectroscopy, and the Xray structure determination of a discrete iron nitride complex stabilized by the sterically encumbering N-anchored tris-carbene ligand, tris[2-(3-aryl-imidazol-2ylidene)ethyl]amine (TIMENR, R = 2,6-xylyl (2,6xyl), mesityl (mes)).[2] Treatment of TIMENR with FeCl2 yields four-coordinated Fe(II) complexes [(TIMENR)Fe(Cl)]Cl. Reduction leads to the formation of the Fe(I) compounds [(TIMENR)Fe]BPh4. Addition of TMS-N3 to Fe(I) complexes results in formation of light yellow divalent [(TIMENR)Fe(N3)]BPh4. These azide compounds release dinitrogen to form deeply colored Fe(IV) nitride complexes [(TIMENR)Fe(N)]BPh4 when irradiated with UV light. Attempts to generate an Fe(V) species via oxidation yielded an Fe(II) insertion product [(TIMENmes)*Fe(MeCN)(NH)’](BPh4)2. The proposed mechanism is the oxidation of [(TIMENmes)Fe(N)]+, followed by nitride insertion into one of the iron carbene bonds accompanied by hydrogen abstraction. Aiming at iron complexes which show diverse reactivity we synthesized a series of new tripodal carbene ligands with sterically less demanding and electronically different aryl substituents, namely TIMENR (R = tolyl (tol), 3,5-xylyl (3,5xyl), 3,5(CF3)phenyl (3,5CF3)). Coordination of these new ligands to iron and the characterization of the resulting complexes were achieved. [1] O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard, D. C. Rees, Science 2002, 297, 1696 − 1700. [2] C. Vogel, F. W. Heinemann, J. Sutter, C. Anthon, K. Meyer, Angew. Chem. 2008, 120, 2721 – 2724. 19 V12 Oxygen versus Sulfur: On the Differences in the Ion/Molecule Reactions of „Rollover“ Cyclometalated [Pt(bipy – H)]+ with (CH3)2X (X = O, S) – A Mechanistic Study B. Butschke, H. Schwarz Institut für Organische Chemie, Technische Universität Berlin, Straße des 17. Juni 115, 10623 Berlin "Rollover" cyclometalated [Pt(bipy – H)]+ (1) [1] can be generated via electrospray ionization of methanolic solutions of [Pt(CH3)2(µ-(CH3)2S)]2 [2] and 2,2'-bipyridine. The ion/molecule reactions of 1 with different thioethers were investigated earlier and in these experiments dehydrosulfurization is observed as the main process in nearly all cases [1,3]. Particularly interesting is the reaction with (CH3)2S where dehydrogenative C–C-bond coupling of the methyl groups to produce neutral C2H4 is observed as the main channel (55 %). However, in the reactions with the oxygen analogues [4,5] no similar processes are observed. In the reaction of [Pt(bipy – H)]+ with (CH3)2O the formation of neutral CH2O is the main process (58 %) while the formation of CH2S in the case of (CH3)2S was only a minor process (5 %). The mechanistic details of the reactions of [Pt(bipy – H)]+ with (CH3)2X (X = O, S) were uncovered by means of DFT calculations as well as deuterium-labeling experiments. Common to both substrates is the initial Pt-mediated C–H-bond activation. [1] B. Butschke, M. Schlangen, D. Schröder, H. Schwarz, Chem. Eur. J. 2008, 14, 11050-11060. [2] G. S. Hill, M. J. Irwin, C. J. Levy, L. M. Rendina, R. J. Puddephatt, Inorg. Synth. 1998, 32, 149-153. [3] B. Butschke, M. Schlangen, D. Schröder, H. Schwarz, Int. J. Mass Spectrom. 2009, 283, 3-8. [4] B. Butschke, Shadan Ghassemi Tabrizi, H. Schwarz, Chem. Eur. J., accepted. [5] B. Butschke, Shadan Ghassemi Tabrizi, H. Schwarz, Int. J. Mass Spectrom., accepted. 20 V13 Eigenschaften redoxaktiver Kupfer(I)-Komplexe mit biomimetischen N,S-Ligandensystemen J. Schnödt,a W. Kaim,a M. Sieger,a J. Fiedler,b C.-Y. Su,c J. Manzur,d a Institut für Anorganische Chemie, Universität Stuttgart, Institut für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70550 Stuttgart, Germany, Email: [email protected] b J. Heyrovský Institute of Physical Chemistry, v.v.i., Academy of Sciences of the Czech Republic, Dolejškova 3, CZ-18223 Prague, Czech Republic c MOE Laboratory of Bioinorganic and Synthetic Chemistry, State Key Laboratory of Optoelectronic Materials and Technologies, School of Chemistry and Chemical Engineering, Sun Yat-Sen University, Guangzhou, 510275, China d Universidad de Chile, Facultad de Ciencias Físicas y Matemáticas, Santiago de Chile, Casilla 2777, Chile Biorelevante Stickstoff- (→Histidin) und Schwefel- (→Methionin) Ligandensysteme sind in der Lage, sowohl CuI als auch CuII zu stabilisieren, vergleichbar mit den Typ1Zentren von blauen Kupferproteinen sowie der Peptidylglycin--hydroxylierenden Monooxygenase (PHM)1. Verschiedene Kupfer(I)-Komplexe mit N,S-Ligandensystemen werden vorgestellt. Besonders diskutiert wird die Untersuchung spektroelektrochemischen Verhaltens des elektrochemischen hinsichtlich Elektronenübertragung. [1] S. T. Prigge, B. A. Eipper, R. E. Mains, L. M. Amzel, Science, 2004, 304, 864. 21 und reversibler V14 C2-Symmetric Dipyrazolyl Ligands Jozak, T.; Sun Y.; Thiel, W.; Kaiserslautern/D Thomas Jozak, Anorganische Chemie, TU Kaiserslautern, Erwin-Schrödinger Str., Geb. 54, D-67663 Kaiserslautern For quite some time we are interested in pyrazoles as the central motif in novel ligands and the applications of such systems in catalysis. Pyrazoles can be synthesized following simple procedures which allow further modification of the heterocycle in terms of steric and electronic features. We present here the synthesis of two chiral dipyrazolyl ligands (Figure 1), related to the well-known BINAP and DIOP systems. Figure1: Molecular structures of chiral dipyrazolyl ligands in the solid state; trimer of 2,2’-di(1,2-pyrazol-3-yl)-1,1’-binaphthyl formed by intermolecular hydrogen bonds (left), 3,3’-[(4S,5S)-2,2,-dimethyl-1,3-dioxolane-4,5-diyl]dipyrazolyl (right). Starting from the enantiomerically pure bidentate ligands, a series of coordination compounds could be obtained. The dipyrazolylbinaphthyl ligand coordinates efficiently to Cu(I), Ag(I) and Au(I) leading to hexanuclear, strongly fluorescent M6 structures similar to the structure of the free ligand (see above). The dipyrazolyldioxolane ligand undergoes coordination to palladium(II) sites. [1] T. Jozak, M. Fischer, J. Thiel, Y. Sun, H. Kelm, W. R. Thiel, Eur. J. Org. Chem. 2009, 1445-1452. 22 V15 Oxovanadiumthiobisphenolatkomplexe als Molekülmodelle für geträgertes Vanadiumpentoxid in der Oxidativen Dehydrogenierung von Alkanen Werncke, G.; Limberg, C.*; Berlin/D Institut für Chemie, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany; *[email protected] In einer Vielzahl von wichtigen industriellen Prozessen werden heterogene Katalysatoren eingesetzt. Ein weiteres Verfahren, das den Sprung zu einem kommerziellen Prozess noch nicht ganz bewältigt hat, ist die oxidative Dehydrogenierung (ODH) von Alkanen zu den entsprechenden Alkenen in Gegenwart von Sauerstoff. Dabei stellten sich Vanadiumoxide, geträgert auf z. B. SiO2, Al2O3 oder CeO2, als besonders geeignete Katalysatorsysteme dar, welche darüber hinaus auch in der Lage sind, die ODH von Methanol zu Formaldehyd zu vermitteln. Fragen hinsichtlich der aktiven Spezies, des Einflusses der verschiedenen Trägeroxide sowie der mechanistischen Abläufe werden dabei zum Teil noch kontrovers diskutiert.[1] Bei der Klärung dieser Punkte können molekulare Modellsysteme, die eindeutig definierte Strukturen aufweisen, entscheidende Hinweise geben. Anhand von Oxovanadium(thia)calixarenkomplexen als Modellverbindungen konnten bereits strukturelle und mechanistische Erkenntnisse bezügliche der ODH von Alkoholen gewonnen werden.[2] Darauf aufbauend berichten wir hier über die Synthese verschiedener Oxovanadiumthiobisphenolatkomplexe sowie deren Verhalten bei der katalytischen Oxidation geeigneter Alkohole. Weiterhin werden die Ergebnisse zu Reaktivitätsuntersuchungen dieser Verbindungen mit Hydroperoxiden vorgestellt, im Zuge derer eine reaktive Vanadiumperoxidverbindung isoliert und charakterisiert werden konnte. PPh 4+ tBu t Bu V V O O O O H2O2 tBu t Bu V O V O O V O S 9-Fluorenol VO O tBu t Bu tBu tBu [ SO2LVO(O2)]PPh4 O IV H O V O O O H S IVO V O O S O tBu tBu 2- 2 PPh 4+ S O O O tBu t Bu tBu tBu 2- 2 PPh4+ - O tBu tBu S tBu t Bu tBu t Bu [SLVO2] 2(PPh 4) 2 [ SLVO(OH)]2(PPh4)2 [1] a) A. Khodakov, J. Yang, S. Su, E. Iglesia, A. T. Bell, J. Catal. 1998, 177, 343-351; b) O. R. Evans, A. T. Bell, T. D. Tilley, J. Catal. 2004, 226, 292-300; c) O. L. J. Gijzeman, J. N. J. van Lingen, J. H. van Lenthe, S. J. Tinnemanns, D. E. Keller, B. M. Weckhuysen, Chem. Phys. Lett. 2004, 397, 277281; d) D. E. Keller, F. M. F. de Groot, D. C. Koningsberger, B. M. Weckhuysen, J. Phys. Chem. B 2005, 109, 10223-10233. [2] a) E. Hoppe, C. Limberg, B. Ziemer, Inorg. Chem. 2006, 45, 8308-8317; b) E. Hoppe, C. Limberg, Chem. Eur. J. 2007, 13, 7006-7016. 23 V16 Vergleichende Untersuchungen an Paaren von Molybdän- und Wolframverbindungen Alexander Döring, Carola Schulzke Institut für Anorganische Chemie, Universität Göttingen, Tammannstr. 4, 37077 Göttingen, Deutschland, email: [email protected] Zu den vielleicht interessantesten Bakteriengattungen gehören die extremophilen Bakterien, die in Lebensräumen gedeihen, die aus menschlicher Sicht als lebensfeindlich einzustufen sind. Diese Bakterienarten leben z.B. in sehr hohen oder sehr niedrigen Temperaturbereichen. Organismen mit einem Wachstumsoptimum im Temperaturbereich von 50-130°C werden als thermophile bzw. hyperthermophile Bakterien bezeichnet. Der Unterschied zwischen den thermophilen Bakterien zu den mesophilen Organismen (leben unter Normalbedingungen) ist, dass sie Wolfram in die aktiven Zentren ihrer Oxidoreduktasen einbauen statt Molybdän.[1] Die Oxidoreduktasen katalysieren Sauerstoff-Übertragungen auf Substrate oder die Abspaltungen von Sauerstoff unter Bildung von Wasser (R+ H2O RO + 2H+ + 2 e-). Dabei ist das Metallzentrum an den in Abb. 1 gezeigten Molybdopterin-Liganden koordiniert. Die Anbindung an das Metall findet über die Dithiolenfunktion statt[2]. Unser Ziel ist es nun herauszufinden, ob die Redoxpotentiale einen Einfluss auf die unterschiedliche Verwendung von Wolfram und Molybdän in den jeweiligen Habitaten haben. Dazu wurden Komplexe von Molybdän und Abb.1: Molybdopterin-Ligand Wolfram in der Oxidationsstufe IV-VI synthetisiert und die Redoxpotentiale temperaturabhängig gemessen. Hierbei konnte festgestellt werden, dass die Redoxpotentiale der Wolframverbindungen stärker von der Temperatur abhängig sind, als die der analogen Molybdänverbindungen. Diese stärkere Temperaturabhängigkeit konnte sowohl bei nicht Dithiolenkomplexen, als auch durch temperaturabhängige katalytische Untersuchungen bestätigt werden[3]. Zu verstehen, weshalb zwei verschiedene Metalle in den aktiven Zentren verwendet werden, ist von großem Interesse, um einen Einblick in den katalytischen Prozess dieser Enzyme und ihre Evolution zu bekommen und dieses Wissen dann für die Optimierung industrieller Katalysatoren einzusetzen. O S S N HN O O H2 N N N O O O O P P O O R [1] a) J. H. Enemark, C. G. Young, Adv. Inorg. Chem., 40, 1993, 1; b) R. Hille, Chem. Rev., 96, 1996, 2757; c) C. Kisker, H. Schindelin, D.C. Rees, Annu. Rev. Biochem., 66, 1997, 233; [2] C. Schulzke, Dalton Trans., 4, 2005, 713; [3] A. Döring, C. Schulzke, Dalton Trans., submitted 24 V17 Untersuchung tripodaler Kupferkomplexe unter Verwendung guanidinhaltiger Liganden Kempf, N.; Würtele, C.; Gießen/D Natascha Kempf, Dr. Christian Würtele, Justus-Liebig-Universität Gießen, Heinrich-Buff-Ring 58, 35392 Gießen Die Synthese und Charakterisierung neuer synthetischer „Kupfer-Sauerstoff-Adduktkomplexe“ ist nach wie vor von großem Interesse, da durch die Verwendung solcher niedermolekularer Modellverbindungen natürlich ablaufende Prozesse simuliert und untersucht werden können. Kupferkomplexe tripodaler Aminliganden gelten seit langem als ideale Modellverbindungen, da sie analog der natürlichen Vorbilder Sauerstoff reversibel aufnehmen und wieder abgeben können. In Abb.1 ist ein solcher reversibler Prozess schematisch dargestellt.[1] LCuI + O2 + LCuI O LCu O LCu O O CuL Abb. 1: Ausbildung von „Kupfer-Sauerstoff-Adduktkomplexe“ Kupfer(I)-Komplexe des Liganden tmpa bilden mit Sauerstoff zunächst einen Superoxokomplex, welcher als reaktives Intermediat durch Anlagerung eines weiteren Kupfer(I)-Komplexes einen Peroxokomplex ausbildet. Dieses System konnte in der Vergangenheit als Modellverbindung für das Protein Hämocyanin verwendet werden.[2] Die Verwendung des sterisch anspruchsvollen Liganden TMG3tren verhindert das Anbinden eines zweiten Kupfer(I)-Komplexes an das reaktive Intermediat, wodurch es zu einer Stabilisierung des Superoxokomplexes kommt. Unter Verwendung dieses Liganden konnte der erste kristallographische Beweis für einen synthetisch hergestellten „end-on“-koordinierten Kupfer-Superoxokomplex erbracht werden und erstmals eine Ligand-Hydroxiliering nachgewiesen werden, wie sie auch im Katalysezyklus der PHM vorkommt.[1,3] Betrachtet man den rein aliphatischen Liganden TMG3tren und den aromatischen Liganden tmpa als Grenzfälle, kann man eine Reihe von systematisch gemischten Liganden aufstellen. In Abb. 2 ist diese Ligandenreihe dargestellt, welche in Kooperation mit der Arbeitsgruppe von Dr. Herres-Pawlis (Uni Dortmund) synthetisiert wurde. N N N N N N N N TMG3tren N N N N N N N N N N N N N N N N N TMGuns-penp TMG2apme N N N tmpa Abb. 2: Variation der Ligandenreihe Ziel ist es, den Einfluss der Guanidin-Gruppen auf die Ausbildung der „KupferSauerstoff-Adduktkomplexe“ systematisch zu untersuchen und mit den bereits bekannten Systemen TMG3tren und tmpa zu vergleichen. [1] C. Würtele, E. Gaoutchenova, K. Harms, M. C. Holthausen, J. Sundermeyer, S. Schindler, Angew. Chem. 2006, 118, 3951., [2] R. R. Jacobson, Z. Tyeklar, A. Farooq, K. D. Karlin, S. Liu, J. Zubieta, J. Am. Chem. Soc. 1988, 110, 3690., [3] D. Maiti, D.-H. Lee, K. Gaoutchenova, C. Würtele, M. C. Holthausen, A. A. Narducci Sarjeant, J. Sundermeyer, S. Schindler, K. D. Karlin, Angew. Chem. 2007, 120, 88. 25 V18 Bisphosphiniminomethanid als Ligand in der Koordinationschemie des Zinks Marks, S.; Roesky, P. W.; Karlsruhe/D Peter W. Roesky, Institut für Anorganische Chemie, Karlsruher Institut für Technologie (KIT), Engesserstraße 15, 76131 Karlsruhe Der monoanionische Bisphosphiniminomethanid-Ligand {CH(PPh2NSiMe3)2}- ist in der Organometallchemie der Hauptgruppen- und Übergangsmetalle sowie der Lanthanoide weit verbreitet.[1] Die heteroleptischen Komplexe [{CH(PPh2NSiMe3)2}ZnR] (R = Me, Ph, N(SiMe3)2) sind aus der Reaktion des Neutralliganden mit den homoleptischen Zinkorganylen/ -amiden zugänglich.[2],[3] Ph P Ph SiMe3 Ph SiMe 3 Ph N P N P N Z nR 2 P Zn N Ph Ph SiMe3 Ph Ph R SiMe 3 R = Me , Ph, N(SiMe 3) 2 a Ph P .T HF Zn(O i Pr) 2 1.) BH 3 2.) KBIPM H B P Ph Ph Zn SiMe 3 Ph N H H Ph SiMe 3 Ph R R = BH 4, Me a N P N BH 3.THF Zn N SiMe 3 P Ph Ph R SiMe 3 R = Me , N(SiMe3)2 a Bisphosphiniminomethanid-Zink-Komplexe zeichnen sich durch eine ungewöhnliche Reaktivität des Ligandenrückgrates gegenüber Elektrophilen aus. So erhält man aus der Reaktion von in situ erzeugtem [Zn(BH4)2(THF)2] mit dem Kaliumsalz des Liganden einen heteroleptischen Boranat-Komplex, in welchem ein BH3-Molekül zwischen dem Metallzentrum und dem Brückenkohlenstoffatom des Liganden überkappend koordiniert. Die Bindungsverhältnisse in den Bisphosphin- iminomethanid-Zink-Komplexen wurden zudem anhand von quantenchemischen Rechnungen auf DFT-Niveau untersucht.[3] [1] T. K. Panda, P. W. Roesky Chem. Soc. Rev. 2009, 38, 2782; [2] A. Kasani, R. McDonald, R. G. Cavell Organometallics, 1999, 18, 3775; [3] S. Marks, T. K. Panda, R. Köppe, P. W. Roesky in Vorbereitung. 26 V19 Synthese und magnetische Eigenschaften C3-symmetrischer triaminoguanidinbasierter Übergangsmetallkomplexe Schuch, D.; Plaul, D.; Spielberg, E. T.; Plass, W; Jena/D Dirk Schuch, Institut für Anorganische und Analytische Chemie, Friedrich-SchillerUniversität, Carl-Zeiss-Promenade 10, 07745 Jena Auf dem Gebiet der Magnetochemie ist die gezielte Synthese von mehrkernigen Übergangsmetallkomplexen mit starker magnetischer Wechselwirkung von zentraler Bedeutung. Durch die Verwendung von tritopen triaminoguanidinbasierten SchiffBase-Liganden können gezielt C3-symmetrische dreikernige Übergangs- metallkomplexe mit sehr geringem Metall-Metall-Abstand synthetisiert werden.[1][2] Über die zentrale Wechselwirkungen Diazin-Einheit vermittelt. Unter werden starke Verwendung antiferromagnetische von Metallzentren mit ungeradzahligem Gesamtspin wird auf diese Weise ein S = 1/2 Grundzustand generiert. Derartige Systeme sind besonders für die Untersuchung von Spinfrustrationsphänomenen und als potentielle Bausteine für Quantencomputer interessant. Bei der Charakterisierung der Komplexe im Festkörper werden intermolekulare Wechselwirkungen beobachtet. Ziel der Untersuchungen ist es, durch die Einführung von Sulfonat- und tert-Butylgruppen am Ligandgrundgerüst, den Abstand zwischen den Dreikerneinheiten zu vergrößern und damit die intermolekularen Wechselwirkungen zu verringern. R O M R N O N N N M M N N O R [1] W. Plass, Coord. Chem. Reviews 2009, 253, 2286-2295. [2] A. E. Ion, E. T. Spielberg, H. Görls, W. Plass, Inorg. Chem. Acta 2007, 360, 3925-3931. 27 V20 MCD Spectra of Mononuclear Manganese(III) Complexes Westphal, A., Berends, H.- M., Kurz, P.*, Tuczek, F.*, Kiel Institut für Anorganische Chemie, Christian-Albrechts-Universität zu Kiel, Max-Eyth-Straße 2, 24118 Kiel In magnetic circular dichroism (MCD) spectroscopy the differential absorption of left (lcp) and right (rcp) circular polarized light induced by a longitudinal magnetic field is measured. It is a very powerful technique to gain insight into the electronic structures of paramagnetic transition metal complexes. The detailed understanding of the MCD spectra of mono- and dinuclear manganese complexes is still lacking and only few examples for MCD spectra of mononuclear manganese complexes can be found in the literature[1]. To develop fundamental methods for analyzing and interpreting manganese MCD data we first turned our attention to mononuclear manganese(III) complexes. We measured MCD spectra of five-coordinate [Mn(acen)X] complexes (acen = N,N’-ethylenebis(acetylacetone imine)). The effect of a distinct ligand field strength within the same coordination geometry (square pyramidal) on the MCD spectra can be explored by the variation of the axial co-ligand X (X = Cl-, Br-, I-, SCN-, etc.). The spectra show various prominent features (Fig. 1), which might provide a wealth of information about the electronic structure of the complexes. It is our task now to develop the theoretical tools to extract this information from the – for now – nonunderstood spectra using both conventional ligand-field, DFT and ab initio methods. a b Figure 1. MCD-spectra of a) [Mn(acen)Br] and b) [Mn(acen)I] in polystyrene films measured at T = 2 K. The magnetic fields were varied from 3 T to -3 T. The spectra for B = 1 T are shown as bold lines. [1] J. W. Whittaker, M. M. Whittaker, J. Am. Chem. Soc. 1991, 113, 15, 5528-5540. 28 V21 Intramolekularer Elektronentransfer in [2.2]Paracyclophanen: Durch den Raum oder über die Brücke? Mücke, P.; Winter, R.; Regensburg/D Mücke Philipp, Prof. Dr. R. Winter, Institut für Anorganische Chemie, Universität Regensburg, Universitätsstraße 31, 93053 Regensburg Wir berichten über die abgebildeten ein- und zweikernigen Rutheniumvinyl-Komplexe mit [2.2]Paracyclophanliganden. Die Komplexe wurden durch Insertion des jeweiligen Alkins in die Ru-H-Bindung von [RuClH(CO)(PiPr3)2] synthetisiert. Elektrochemischen Untersuchungen zu Folge lassen sich die Komplexe jeweils reversibel zu Radikalkationen und Dikationen (Komplexe 1 und 3) oxidieren. Die jeweiligen oxidierten Formen wurden in einer optisch transparenten Dünnschichtzelle in situ generiert und mittels der IR-, der UV/Vis/NIR- und der ESR-Spektroskopie untersucht. Im Zentrum stand dabei die Frage nach der elektronischen Kommunikation zwischen den Styryl-Rutheniumeinheiten in den Komplexen 1 und 3. Komplex 2 wurde als Referenz für eine einkernige Verbindung ohne intramolekularen Elektronentransfer herangezogen. Zur quantitativen Analyse wurden die Carbonylliganden an den Rutheniumatomen als ladungssensitives IR-Label sowie die auf der Stufe der Radikalkationen beobachteten IVCT-Banden genutzt. Komplex 3 als „geöffneter“ Cyclophankomplex erlaubt es schließlich, den Beitrag der elektronischen Kopplung durch die gesättigten Ethylenbrücken von dem der über stacking vermittelten Kopplung zu separieren. 1 2 3 29 V22 Coordination chemistry of cyclic peptides Comba, P;1 Dovalil, N.,1; Hanson, G. R.;2 Seibold, B.1 1 Anorganisch-Chemisches Institut, Universität Heidelberg, INF 270, D-69120 Heidelberg, Germany, 2 Centre for Magnetic Resonance The University of Queensland, Brisbane, Queensland 4072, Australia Patellamides (PatH4) are cyclic peptides present in the ascidians of the Indian and Pacific Oceans. Although the coordination chemistry of Ascidiacyclamide (AscH4) and the Pat(AF)H4 macrocycles (Scheme 1) as well as their pharmaceutical activities have been investigated, little is known about their biological function. Scheme 1: H4L4, Ascidiacyclamide and Patellamides A-F. Computed structures of H4L4 and its mono- and dinuclear CuII complexes. The pseudooctapeptide H4L4 was synthesized as a simplified model for the natural cyclic peptides. It was found, that H4L4 is highly preorganized for the formation of dinuclear CuII complexes. These complexes where found to bind and hydrolyze CO2 from air under neutral conditions (6.5 < pH < 7.5). The resulting bridging carbonate is only weakly bound and can easily be substituted by water to reform the hydroxo complex. The structure of the various CuII complexes and the mechanism of CO2 fixation have been established with ESI-MS, NMR, EPR, CD, UV-vis, IR spectroscopy in conjunction with 13CO2 and H218O labeling experiments and computational methods. Preliminary experiments with closer structural analogs of the Patellamides suggest a similar behavior. These results give insights into the possible Patellamides’ biological function in the ascidians. 30 V23 Spin Crossover Properties of Iron(II) Complexes Kisslinger, S.; Schindler, S. Sandra Kisslinger and Siegfried Schindler, Institut für anorganische und analytische Chemie, Justus-Liebig-Universität, Heinrich-Buff-Ring 58, 35392 Gießen Iron(II) complexes with tripodal tetradentate ligands such as tmpa are well known to show interesting spin crossover behaviour.1-3 With this background we have started to investigate systematically the influence of chelate ring size and donor atom modification in this type of iron complexes. The ligands applied are shown in the following figure. (CH2)n N R (CH2)n N N N R N n(H2C) N n = 1 (tmpa) n= 1,2 N N R NH2 N R = NH2, N(CH3)2, N NH2 uns-penp (DPEA) apme The iron(II) complexes with thiocyanate as a co-ligand have been synthesized and structurally characterized. In the complexes with the tripodal ligands the thiocyanate ions are cis coordinated. In contrast the ligand O-BPY enforces the trans position.4 The molecular structure of the [Fe(O-BPY)(SCN)2] is presented in the following figure. The current results of our investigations will be presented. [1] Toftlund, H.; McGarvey, J. J. Top. Curr. Chem. 2004, 233, 151. [2] Gütlich, P.; Hauser, A.; Spiering, H. Angew. Chem. 1994, 106, 2109. [3] Matouzenko, G. S.; Bousseksou, A.; Lecoq, S.; Koningsbruggen, P. J. v.; Perrin, M.; Kahn, O.; Collet, A. Inorg. Chem. 1997, 36, 2975. [4] Garber, T., van Wallendael, S., Rillema, D. P., Kirk, M., Hatfield, W. E., Welch, J. H., Singh, P.; Inorg. Chem. 1990, 29, 2863. 31 V24 Synthese und Charakterisierung von Cobalt(II)-Komplexen mit Poly(pyrazolyl)methanliganden Hoffmann, A., Dortmund/D; Flörke, F., Paderborn/D; Schürmann, M., Dortmund/D; Herres-Pawlis, S., Dortmund/D Alexander Hoffmann, Fakultät Chemie, Technische Universität Dortmund, OttoHahn-Straße 6, 44227 Dortmund Die Synthese der Bis(pyrazolyl)methane[1] erfolgt in einer Eintopf-Synthese. Zwei substituierte Pyrazole werden mittels Natriumhydrid und Thionylchlorid zu der Sulfonylverbindung umgesetzt. Diese reagiert im nachfolgenden Schritt mit der jeweiligen Carbonylverbindung bei Anwesenheit katalytischer Mengen Cobalt(II)chlorid zu dem Bis(pyrazolyl)methan in guten Ausbeuten (Abb. 1). Mit dieser Synthesestrategie können substituierte Bis(pyrazolyl)methane hergestellt werden, ohne auf den Einsatz von Phosgen zurückzugreifen. Die Tris(pyrazolyl)methane werden nach den bekannten Verfahren synthetisiert.[2] Abb. 1: Synthese von Bis(pyrazolyl)methanen Die Poly(pyrazolyl)methane sind mit Cobalt(II)-Salzen zu neuen Poly(pyrazolyl)methan-Cobaltkomplexen umgesetzt worden. Aus den Röntgenstrukturen geht hervor, dass diese Komplexe ungewöhnliche Koordinationsmotive und Reaktionspfade zeigen. Für die Bis(pyrazolyl)methankomplexe wird zudem in Lösung eine Cis-Trans-Isomerisierung beobachtet. [1] A. Hoffmann, U. Flörke, S. Herres-Pawlis, Manuskript in Vorbereitung. [2] C. Pettinari, R. Pettinari, Coord. Chem. Rev. 2005, 249, 525. 32 V25 Fluoroionophore für Palladiumdichlorid und Kupfer(II)-Ionen Schwarze, T.; Potsdam / D Thomas Schwarze, Anorganische Chemie, Institut für Chemie, Universität Potsdam, Karl Liebknecht Str. 24-25, Haus 26, 14476 Golm-Potsdam Schwer-und Übergangsmetallionen sind typische Fluoreszenzlöscher. Für den selektiven fluoreszenzspektroskopischen Nachweis dieser Ionen durch Fluoreszenzsteigerung sind Fluoroionophore erforderlich, die eine Metallionenselektive Rezeptoreinheit besitzen und bei denen es in der komplexierten Form zu keiner oder nur geringer Fluoreszenzlöschung durch das Metallion kommt. Ein zunehmendes Fluoreszenzsignal kann auch beobachtet werden, wenn eine Verbindung metallionenselektiv in ein Fluorophor umgewandelt wird.[1] Die Fluoroionophore 1 und 2 enthalten als selektive Rezeptoreinheit für Palladiumdichlorid die Dithiomaleonitrileinheit. fluoreszenzspektroskopischen Nachweis von 1 und 2 lassen Palladiumdichlorid den durch Fluoreszenzsteigerung zu (Fluoreszenzsteigerungsfaktor (FSF) für 1 : 3,2; FSF für 2 > 250).[2,3,4] 2-Pyridylmethylen- und Aryl- oder Heteroarylmethylen funktionalisierte Diaminomaleonitrile bilden nur mit Kupfer(II)-Ionen Imin-Amin-Komplexe. Kupfer(II)Ionen können die Luftoxidation von Liganden, wie z. B. 3, katalysieren. Der Einsatz von 3 für den Nachweis von Kupfer(II)-Ionen führt zu einem FSF von 1,6. [1] K. Rurack, Spectrochimica Acta Part A 2001, 57, 2161-2195. [2] T. Schwarze, H. Müller, C. Dosche, T. Klamroth, W. Mickler, A. Kelling, H.-G. Löhmannsröben, P. Saalfrank, H.-J. Holdt, Angew. Chem. 2007, 119, 1701-1704; Angew. Chem. Int. Ed. 2007, 46, 16711674. [3] T. Schwarze, W. Mickler, C. Dosche, R. Flehr, T. Klamroth, H.-G. Löhmannsröben, P. Saalfrank, H.-J. Holdt, Chem. Eur. J. 2010, 16, 1819-1825. [4] T. Schwarze, C. Dosche, R. Flehr, T. Klamroth, H.-G. Löhmannsröben, P. Saalfrank, E. Cleve, H.J. Buschmann, H.-J. Holdt, Chem. Commun. 2010, in press. 33 V26 Quinone-substituted Bis(pyrazol-1-yl)methane Ligands and a Cu(II)-Hydroquinone Coordination Polymer F. Blasberg, M. Wagner*, Frankfurt am Main, Germany Group of Prof. Dr. Matthias Wagner, Institut für Anorganische und Analytische Chemie, Goethe-Universität, Max-von-Laue-Straße 7, 60438 Frankfurt am Main The ability of transition metals to adopt different oxidation states plays a vital role in homogeneous catalysis and in materials sciences. This redox capacity can be extended further by coordinating the metal ion to a redox non-innocent ligand. Based on the para-hydro-/para-semi-/para-benzoquinone redox system, we have developed ligands bearing bis(pyrazol-1-yl)methane[1] or alkylimine[2, 3] anchor groups. The presentation addresses two aspects of the coordination chemistry of these ligand classes: (1) In the context of ligand development studies we observed the unexpected formation of a methoxy-ortho-benzoquinone 2 from the sterically encumbered para-hydroquinone-based bis(pyrazol-1-yl)methane 1 upon oxidation with [Ce(NH4)2(NO3)6] (CAN). Mechanistic details of this reaction will be discussed. (2) The Cu(II) coordination polymer 3[2, 3] shows a dramatic change in its magnetic behavior upon successive removal of DMF ligands. The underlying structure-property relationships will be described. [1] S. Scheuermann, T. Kretz, H. Vitze, J. W. Bats, M. Bolte, H.-W. Lerner, M. Wagner, Chem. Eur. J. 2008, 14, 2590-2601. [2] T. Kretz, J. W. Bats, S. Losi, B. Wolf, H.-W. Lerner, M. Lang, P. Zanello, M. Wagner, Dalton Trans. 2006, 4914-4921. [3] Y. Tsui, A. Brühl, K. Removic-Langer, V. Pashchenko, B. Wolf, G. Donath, A. Pikul, T. Kretz, H.-W. Lerner, M. Wagner, A. Salguero, T. Saha-Dasgupta, B. Rahaman, R. Valenti, M. Lang, J. Magn. Magn. Mater. 2007, 310, Part 2, 1319-1321. 34 V27 Zirkonium(IV)-Hydrazindiidokomplexe: N-N-Bindungsspaltung versus Cycloadditionsreaktionen T. Gehrmann, J. Lloret Fillol, H. Wadepohl, L. H. Gade; Heidelberg / D Thorsten Gehrmann, Anorganisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg Übergangsmetall-Hydrazidokomplexe wurden bisher hauptsächlich aufgrund ihres Auftretens bei der katalytischen und stöchiometrischen Reduktion von Distickstoff zu Ammoniak untersucht.[1] Neben den schwereren Gruppe VI Metallen wurden hierzu in jüngerer Zeit auch vermehrt Metalle der vierten Nebengruppe eingesetzt deren Komplexe verschiedene neuartige Reaktionsmuster zeigten.[2] Im Mittelpunkt des Interesses steht vor allem die katalytische Hydrohydrazinierung von Alkinen und verwandten Reaktionen.[3] Andererseits können Hydrazindiidokomplexe der Gruppe IV Metalle unter N-N-Bindungsspaltung als Metallnitrenäquivalente reagieren.[4] N Ph Ph N Zr N NPh2 TBDMS N py R N3 TBDMS R = TMS, Adamantyl 1 R N3 N Zr TBDMS N HN TBDMS Ph N Zr TBDMS N N TBDMS R Ph N N N Ph NPh2 N R = Mesityl N Zr TBDMS N N TBDMS Mes N N N NPh2 N Wir untersuchen Reaktionen des Zirkonium-Hydrazindiidokomplexes 1 gegenüber ungesättigten Substraten. Das Interesse richtet sich dabei sowohl auf neue Reaktionsprodukte und –pfade, als auch auf experimentelle und theoretische mechanistische Untersuchungen.[5] [1] a) R. R. Schrock, Acc. Chem. Res. 2005, 38, 955, und dort zitierte; b) A. MacKay, M. D. Fryzuk, Chem. Rev 2004, 104, 385. [2] Y. Ohki, M. D. Fryzuk, Angew. Chem. 2007, 119, 3242. [3] a) A. L. Odom, Dalton Trans. 2005, 225; b) K. Weitershaus, H. Wadepohl, L. H. Gade, Organometallics 2009, 28, 3381. [4] a) P. J. Walsh, M. J. Carney, R. G. Bergman, J. Am. Chem. Soc. 1991, 113, 6343; b) H. Herrmann, J. L. Fillol, H. Wadepohl, L. H. Gade; Angew. Chem. 2007, 119, 8578. [5] a) T. Gehrmann, J. L. Fillol, H. Wadepohl, L. H. Gade, Angew. Chem. 2009, 121 2186. b) T. Gehrmann, J. L. Fillol, H. Wadepohl, L. H. Gade, Organometallics 2010, 29, 28. 35 V28 SYNTHESIS of MULTINUCLEAR Fe(II) COMPLEXES Kundu, S.; Ray, K. * Institut für Chemie, Humboldt-Universitӓt zu Berlin, Brook-Taylor-Strasse 2, 12489 Berlin, Germany In photosynthesis, water is oxidized at a protein-bound Mn4Ca complex. Artificial water-oxidation catalysts that are similarly efficient and based on inexpensive and abundant materials are of great interest. The reactivity of the metal-oxo (Mn-Oxo in photosynthesis) is central to the formation of O-O bonds, which is the essential step for oxygen generation. Two basic strategies for the formation of O-O bonds at metal-oxo active sites are proposed. The acid–base (AB) strategy (reaction 1) involves the attack of a nucleophilic oxygen species (e.g., hydroxide) on an electrophilic metal-oxo. For the radical coupling (RC) strategy (reaction 2), two high-valent metal-oxos containing sufficient spin densities on the oxygen atoms are needed to drive O-O coupling. Within the last few years, a number of synthetic mononuclear nonheme iron(IV) oxo complexes bearing tetra- and pentadentate N-donor ligands have been synthesized as models for the high-valent iron(IV) oxo intermediates found in many mononuclear nonheme iron enzymes that activate dioxygen. Considerable spin densities (> 80 %) have been predicted on the oxygen atoms of the FeO units of many of these model complexes from DFT calculations. Thus, the Fe(IV)=O unit is calculated to possess a lot of Fe(III)-O• character. Whereas radical character at the oxygen of the Fe-O center is critical to advancing the RC strategy for O-O bond coupling, this criterion alone is insufficient. A RC strategy demands that two oxos be sufficiently close to each other for their effective coupling. In this project we report the synthesis and characterization of a number of multinuclear Fe(II) complexes supported on stannoxane cores, which not only ensure good solubility of the complexes in common organic solvents, but also help to bring the individual Fe centres sufficiently close to each other for undergoing electronic interactions. Efforts are being made to generate the corresponding multinuclear Fe(IV)oxo complexes by reaction of the Fe(II) complexes with iodosobenzene, and the possibility of undergoing the radical coupled O-O bond coupling within the system are also explored. Reaction 1: 36 Reaction 2: Hexameric Fe(II) Complex 37 V29 Untersuchungen zur Toxizität und intrazellulären Verteilung von [Mn(CO)3(tpm)]+ Johanna Niesela, Konrad Meisterb, Martina Havenith-Newenb and Ulrich Schatzschneidera a Lehrstuhl für Anorganische Chemie I und bLehrstuhl für Physikalische Chemie II, Ruhr Universität Bochum, Universitätsstraße 150, 44801 Bochum [email protected] Kohlenstoffmonoxid ist ein wichtiger biologischer Botenstoff. Um eine kontrollierte Gabe dieses Gases zu ermöglichen werden seit einigen Jahren Übergangsmetallcarbonylkomplexe als feste Speicherformen von CO untersucht. Es konnte gezeigt werden, dass diese sogenannten CORMs (CO releasing molecules) die gleiche gefäßerweiternde, entzündungshemmende und anti-apoptotische Wirkung haben wie CO selbst. Neben diesen positiven Effekten können CORMs auch dazu dienen die Toxizität von CO gezielt zu nutzen. [Mn(CO)3(tpm)]+ (tpm = tris(1-pyrazolyl)methane) ist im Dunkeln stabil, setzt aber bei Belichtung CO frei. Zudem wird es effektiv in Zellen aufgenommen und während es im Dunkeln keinen Einfluß auf die Zellviabilität von HT29 Darmkrebszellen hat so reduziert es diese bei Belichtung effektiv. Um einen Einblick in die intrazelluläre Verteilung von [Mn(CO)3(tpm)]+ zu erhalten wurde diese mittels Ramanmikroskopie untersucht. Zudem wurden weitere Mangantricarbonyle synthetisiert, um zu untersuchen wie das Ligandensystem die CO Freisetzung beeinflusst. [1] S.W. Ryter, J. Alam, A.M.K. Choi, Physiol. Rev. 2006, 583-650. [2] T.R. Johnson, B.E. Mann, J.E. Clark, R. Foresti, C.J. Green, R. Motterlini, Angew. Chem. Int. Ed. 2003, 42, 3722-3729. [3] J. Niesel, A. Pinto, H. W. Peindy N'Dongo, K. Merz, I. Ott, R. Gust, U. Schatzschneider, Chem. Commun. 2008, 1798-1800 38 V30 Strukturelle, elektrochemische und spektroskopische Untersuchungen neuer triazolhaltiger Liganden und ihrer Übergangsmetallkomplexe Schweinfurth D., Stuttgart David Schweinfurth, Fritz Weißer, Biprajit Sarkar, Institut für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart. Die durch Kupfer(I) katalysierte Cycloaddition von Aziden und Alkinen (CuAAC), auch als Click-Reaktion bekannt, führt zur Bildung von 1,4-substituierten 1,2,3Triazolen und ist zum Aufbau neuer Ligandensysteme geeignet, die eine Vielzahl von Koordinationsmöglichkeiten zeigen [1,2]. So wurden 2-pyridylsubstituierte Triazole für den Fluoreszenznachweis von Metallkationen eingesetzt. Die PdCl2- und PtCl2Komplexe dieser Liganden wurden strukturell und spektroskopisch untersucht. Mittels Cyclovoltammetrie konnte gezeigt werden, dass sich das Reduktionspotential dieser Liganden nach Komplexierung stark anodisch verschiebt [3]. Ebenso wurden die NiCl2-Komplexe dieser Liganden synthetisiert und als Präkatalysatoren in der Ethylenoligomerisierung getestet. [1] V. V. Rostovtsev, L. G. Green, V.V. Fokin und K. B. Sharpless, Angew. Chem. 2002,114, 2708-2711. [2] H. Struthers, Th. L. Mindt und R. Schibli, Dalton Trans. 2010, 675-696. [3] D. Schweinfurth, R. Pattacini, S. Strobel und B. Sarkar, Dalton Trans. 2009, 9291-9297. 39 V31 Synthese und Charakterisierung von Ag(I)-, Au(I)- und Au(III)-Komplexen funktionalisierter N-heterocyclischer Carbene Topf C.; Knör G.; Monkowius, U.; Linz/A Institut für Anorganische Chemie, Johannes Kepler Universität Linz, Altenbergerstr. 69, 4020 Linz, Austria N-heterocyclische Carbene (NHC) sind etablierte Liganden für Komplexe der Münzmetalle (Cu, Ag, Au). Die Koordinationschemie von funktionalisierten NHCs insbesondere für Au(III) Zentren ist jedoch weniger gut untersucht. Komplexe der Form [(NHC)Au(III)X3] können aus den entsprechenden Au(I)-Verbindungen durch Oxidation mit X2 (X = Cl, Br, I) hergestellt werden, die wiederum über die Silbersalzmethode ausgehend von [(NHC)Ag(I)X] und (R2S)AuCl (R2S = Me2S or tetrahydrothiophen) zugänglich sind [1]. In diesem Beitrag wird eine Reihe strukturell verwandter [M(DN ∩NHC)]-Komplexe vorgestellt (M = Ag(I), Au(I), Au(III)). Die NHC-Gruppe ist (un)symmetrisch mit unterschiedlichen N-Donorfunktionen DN substituiert (DN = 2-Picolyl bzw. R2NCH2CH2–, R = Me, i-Pr). Durch Reaktion der synthetisierten Imidazoliumsalze mit Silber(I)oxid können die entsprechenden Silbersalze erhalten werden. Die weitere Umsetzung von (DN ∩NHC)AgX mit (R2S)AuX und folgender Oxidation mit Br2 liefert die u.a. durch Röntgenstrukturanalyse charakterisierten Komplexe [AuBr2Cl(Me2NCH2CH2ImMe)], [AuBr2Cl(i-Pr2NCH2CH2ImMe)] und [AuBr3(2-Picolyl-ImMe)] (Im = Imidazol-2-yliden). Bei der Einwirkung von Säure auf den Komplex [AgCl(Me2NCH2CH2ImMe)] bildet sich die einzigartige Clusterstruktur [Ag10Cl10(Me2NCH2CH2ImMe)4], in welcher zwei Silberatome durch das Carbenkohlenstoffatom verbrückt sind. Asymmetrische Einheit der Clusterverbindung [Ag10Cl10(Me2NCH2CH2ImMe)4] [1] I. J. B. Lin, C. S. Vasam, Can. J. Chem. 83 (2005) 812-825. 40 V32 Sauerstoffaktivität von Gruppe-9-Elementen in Komplexen mit NHC-Phosphan-Liganden Sebastian Lange, Aziza Ahmida, Hans Egold, Ulrich Flörke, Gerald Henkel* Department Chemie, Universität Paderborn, Warburger Str. 100, D-33098 Paderborn *[email protected] Die Aktivierung kleiner Moleküle wie z. B. Sauerstoff ist eine herausragende Eigenschaft einiger Übergangsmetall-Komplexe.[1] Bei der reversiblen Bindung des Sauerstoffs wird dieser in einen aktivierten Zustand überführt.[2] Während Rhodium-Komplexe, welche Sauerstoff irreversibel binden, in der Literatur häufig beschrieben werden, sind entsprechende Komplexe mit reversibler Sauerstoffbindung selten. Unsere Forschung auf dem Gebiet neuartiger Chelatkomplexe von Gruppe-9-Elementen mit NHC-Phosphan-Hybridliganden führte zu der Synthese einkerniger Komplexionen des Typs cis-[M(EtImCH2CH2PPh2)2]+ (mit M = Ir, Rh)[3], wobei die Rhodiumvariante den Sauerstoff reversibel binden kann. N Cl N N Rh N P Ph P Ph Ph N + O2 - O2 N Rh N O P O P Ph Cl N Ph Ph Ph Ph Reversible Sauerstoffbindung an cis-[Rh(EtImCH2CH2PPh2)2]Cl Geringste Spuren von Sauerstoff führen zu einer oxidativen Addition an das quadratisch-planare System zur Peroxo-Verbindung [Rh(2-O2)(EtImCH2CH2PPh2)2]Cl. Über 31 P-NMR-Spektroskopie kann verfolgt werden, dass sich der ursprüngliche cis- Komplex sowohl bei thermischer Belastung als auch bei reduziertem SauerstoffPartialdruck zurückbildet. In diesem Vortrag berichten wir über die Synthesen von cis-[M(EtImCH2CH2PPh2)2]+Komplexionen und deren Reaktivität in Bezug auf Sauerstoff. [1] E. I. Solomon, U. M. Sundaram, T. E. Machonkin, Chem. Rev., 1996, 96, 2563. [2] Für Beispiele siehe: B. R. James, Y. Xiao-Yan, B. O. Patrick, Organometallics, 2006, 25, 4870. [3] A. Ahmida, Dissertation, Universität Paderborn, 2009. 41 V33 Alkyne Metathesis with Well-Defined Imidazoline-2-iminato Tungsten & Molybdenum Benzylidyne Complexes Birte Haberlag, Stephan Beer, Sergej Lysenko, Xian Wu, Matthias Tamm*; Braunschweig/D Matthias Tamm, Institut für Anorganische und Analytische Chemie, Technische Universität Carolo-Wilhelmina, Hagenring 30, 38106 Braunschweig; *[email protected] In recent years, alkene metathesis has established itself as one of the primary tools in both organic synthesis and polymer chemistry.[1] This is exemplified by the award of the 2005 Nobel prize in chemistry to Chauvin, Grubbs and Schrock.[2] In contrast, the closely related metathesis of alkynes is a far less developed synthetic method, although it has been known since 1974.[3] Only a limited number of active catalyst systems are known to date that fulfill the expectations for an alkyne metathesis catalyst with regard to its activity, substrate compatibility, and required reaction temperature.[4] Recently, we presented a novel highly active tungsten alkylidyne complex which contains a subtly balanced ligand set with a highly π-basic imidazoline-2-iminato ligand and electron-withdrawing fluorinated alkoxides.[5] This catalyst shows very high activities towards various alkyne metathesis reactions such as ACM (alkyne cross metathesis), RCAM (ring-closing alkyne metathesis) and ROMP (ring-opening alkyne metathesis polymerization) already at room-temperature.[5],[6] Ph CH3 F3C F3C tBu W O N N O + R F3C F3C N CH3 R R R tBu Furthermore, as the most active catalysts in alkene metathesis contain molybdenum as active metal center, e.g. Schrock-Type molybdenum alkylidene complexes[1] the syntheses of novel molybdenum benzylidyne complexes and their application in alkyne metathesis in comparison to corresponding well-defined imidazoline-2-iminato tungsten alkylidyne complexes will be presented. [1] R. R. Schrock, A. H. Hoveyda, Angew. Chem. Int. Ed. 2003, 42, 4592. [2] Nobel lectures: (a) Chauvin, Y. Angew. Chem. Int. Ed. 2006, 45, 3741-3747. (b) Schrock, R. R.; Angew. Chem. Int. Ed. 2006, 45, 3748-3759. (c) Grubbs, R. H. Angew. Chem. Int. Ed. 2006, 45, 3760-3765. [3] A. Mortreux, M. Blanchard, J. Chem. Soc. Chem. Commun. 1974, 786. [4] A. Fürstner, P. W. Davies, Chem. Commun. 2005, 2307; R. R. Schrock, C. Czekelius, Adv. Synth. Catal. 2007, 349, 93; W. Zhang, J. S. Moore, Adv. Synth. Catal. 2007, 349, 93. [5] S. Beer, C. G. Hrib, P. G. Jones, K. Brandhorst, J. Grunenberg, M. Tamm, Angew. Chem. Int. Ed. 2007, 46, 8890; S. Beer, K. Brandhorst, C. G. Hrib, X. Wu, B. Haberlag, J. Grunenberg, P. G. Jones, M. Tamm, Organometallics 2009, 28, 1534; M. Tamm, X. Wu, Chemistry Today (Chimica Oggi) 2009, in press. [6] S. Beer, K. Brandhorst, J. Grunenberg, C. G. Hrib, P. G. Jones, M. Tamm, Org. Lett. 2008, 10, 981; S. Lysenko, B. Haberlag, X. Wu, M. Tamm, Macromol. Symp. 2009, in press. 42 V34 Novel non-polymeric fluorescent bimodal contrast agent for magnetic resonance and optical imaging Jayapaul, J.; Hodenius, M.; Buhl, A.; Comba, P.; Kiessling, F.; Gaetjens, J.; Aachen/Germany Jabadurai Jayapaul, Fabian Kiessling, Jessica Gaetjens, Department of Experimental Molecular Imaging, Medical Faculty, RWTH Aachen University, Pauwelsstrasse 30, 52074 Aachen Michael Hodenius, Applied Medical Engineering, Medical Faculty, Helmholtz Institute, RWTH Aachen University, Pauwelsstrasse 20, 52074 Aachen, Alexandra Buhl, Department of Diagnostic Radiology, Medical Faculty, RWTH Aachen University, Pauwelsstrasse 30, 52074 Aachen Jabadurai Jayapaul, Peter Comba, Institute of Inorganic Chemistry, University of Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg We report here the first successful nonpolymeric fluorescence coating of iron oxide nanoparticles without any chemical surface modification. In this project, we have developed novel ultrasmall, fluorescent and superparamagnetic iron oxide particles (FLUSPIOs) as dual-mode contrast agents for magnetic resonance and optical imaging. We have focused on flavin mononucleotide (FMN) as fluorescent, and guanosine monophosphate (GMP) as nonpolymeric coating molecules, which bind strongly to the iron oxide surface due to their inherent phosphate groups. The USPIOs were synthesized by the chemical co-precipitation method. Nonpolymeric mixed coating was achieved by bath sonication of nanoparticles with FMN and GMP for 1h each and purified by high gradient magnetophoresis. The average size and fluorescence of the FLUSPIOs were evaluated by Transmission Electron Microscopy (TEM), fluorescence spectrometry and fluorescence microscopy, respectively. The relaxivities r2 and r1 were determined at room temperature using a clinical 3T MR scanner. The Vaskovsky phosphate estimation method was carried out for determining the amount of phosphonic acid coupled to the iron oxide surface. The cytotoxicity of the FLUSPIOs was evaluated by trypan blue staining in which PC-3 cells were incubated with our particles (c(Fe) = 3.0, 0.3 and 0.03 µmol/mL) for 3, 12 and 24 h. In vitro cellular uptake of FLUSPIOs was determined by using MRI and TEM techniques. TEM measurements showed an average particle size of 5 ± 1 nm. The FLUSPIOs intense green luminescence was evaluated by fluorescence microscopy. Fluorescence spectrometry of FMN on FLUSPIOs displayed emission at 530 nm in buffer solution, independent of pH. The drop of emission intensity is negligible after 3 days. The Vaskovsky phosphate determination method showed that 4 µmol/mL of FMN and GMP was bound to 40 µmol/mL of the iron core. MR measurements showed the expected relaxivity values r2 and r1 for FLUSPIOs as 148 s-1mM-1 and 2.138 s-1mM-1. No toxic effects of FLUSPIOs on PC-3 cells could be detected by trypan blue staining. Both MRI and TEM in vitro results showed very high cell labelling efficiency and cellular uptake of FLUSPIOs. FMN and GMP coated USPIOs displayed excellent stability at physiological pH and provide the possibility of further functionalization with specific antibodies. This novel fluorescent nonpolymeric coating allows us to tailor the properties of magnetofluorescent nanoparticles in a simple way without any chemical surface modification and it offers an excellent opportunity for various biomedical applications. Acknowledgment: DFG USPIO KI 1072/1-3 for the financial support. 43 V35 Activation of Carbon Dioxide and Related Small Molecules at Reactive U(III) Centers Lam, O.P.; Bart, S.C.; Meyer, K. Department of Chemistry and Pharmacy, Inorganic Chemistry, University ErlangenNürnberg, Egerlandstraße 1, D-91058 Erlangen, Germany We have demonstrated that the U(III) complexes, [((RArO)3tacn)U], supported by hexadentate triazacyclononane derivatized ligands with coordinating aryloxide arms (1,4,7-tris(3-R-5-tert-butyl-2-hydroxybenzyl)-1,4,7-triazacyclononane, R = t-Bu, Ad; Figure 1, A), can reduce CO2 to form the end-on bound η1-OCO•─ complex [((AdArO)3tacn)U(OCO•─)],[1] or doubly reduce CO2 to form the bridging oxo complex [{((tBu ArO)3tacn)U}2(μ-O)] and CO.[2] Here, the new low valent U(III) complexes [((t- Bu ArO)3mes)U] and [((AdArO)3N)U] react with CO2 to form the bridging carbonate [{((t-BuArO)3mes)U}2(μ-κ2:κ2-CO3)] complexes and [{((AdArO)3N)U}2(μ-η1:κ2-CO3)]. Uranium(IV) bridging oxo complexes have been determined to be the intermediate in these transformations.[3] activation forming Ad 2 the Trivalent [((AdArO)3N)U] also undergoes carbon disulfide dinuclear U(IV)/U(IV) bridging thiooxalate complex, 2 [{(( ArO)3N)U}2(μ-κ :κ -C2S4)], from reductive coupling of CS2. [1]. Castro-Rodríguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757-1759. [2]. I. Castro-Rodríguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242-11243. [3]. O. P. Lam, S.C. Bart, H. Kameo, F.W. Heinemann, and K. Meyer, Chem. Commun., 2010, in press. 44 V36 Wasserlösliche (Di)Phosphane - Neue Liganden für metallbasierte Wirkstoffe Kunz, P. C.; Wetzel, C. Düsseldorf/D Peter C. Kunz, Corinna Wetzel, Institut für Anorganische Chemie und Strukturchemie I, Heinrich-Heine-Universität, Universitätsstr. 1, 40225 Düsseldorf Cisplatin und analoge Verbindungen stellen heutzutage einen zentralen Bestandteil in der Kombinationstherapie verschiedener Krebserkrankungen dar. Neben den häufig sehr starken Nebenwirkungen ist die Ausbildung von Cisplatin-Resistenzen der schwerwiegendste Nachteil dieser Therapieform und die Überwindung ebendieser Resistenzen ein zentraler Aspekt in der Entwicklung neuer metallbasierter Chemotherapeutika.[1] Die Untersuchung von Goldverbindungen, ursprünglich für die Therapie der rheumatoiden Arthritis entwickelt, an Krebszelllinien in vitro und in vivo zeigte ein großes Potential insbesondere von Gold-Phosphanen als Tumortherapeutika.[2, 3] Die Balance zwischen Löslichkeit, Hydrophilie und Hydrophobie dieser Verbindungen beeinflusst maßgeblich ihre Pharmakokinetik und -dynamik. Hier präsentieren wir die Synthese neuer (Di)Phosphanliganden[4], Untersuchungen zu ihrem Verhalten in wässrigen Lösungen sowie einige Gold(I)- und Silber(I)-Komplexe. [1] B. A. Teicher, Clin. Cancer Res. 2008,14, 1610-1617. [2] I. Ott, Coord. Chem. Rev. 2009, 253, 1670-1681. [3] P. C. Kunz, M. U. Kassack, A. Hamacher, B. Spingler, Dalton Trans. 2009, 7741-7747. [4] P. C. Kunz, C. Wetzel, M. Bongartz, A.-L. Noffke, J. Organomet. Chem., submitted. 45 V37 The first photo-redox-active N-heterocyclic carbene – a new class of bridging ligands suitable for photo- and redoxcatalysis Pilz, T. D.; Rau. S.*; Erlangen/D T. David Pilz, Sven Rau, Department Chemie und Pharmazie, Friedrich-AlexanderUniversität Erlangen-Nürnberg, Egerlandstr. 1, 91052 Erlangen Light induced catalysis with molecular devices requires the synthesis and understanding of photoredox active metal complexes containing bridging ligands capable of binding potentially catalytically active metals.[1-3] Towards this aim it is of paramount importance to create supramolecular systems with independent subunits capable of long lived 3MLCT activation and other properties like redox activity. 2+ hν N 3 N N N + N MLCT e- N N N N Ru N N Ag Cl N Ru(bbip)Ag Ru(bbip) Starting from 1,10-phenanthroline, in a four step synthesis the bidentate bridging ligand bbip (1,3-bisbenzoyl-1H-imidazo[4,5-f][1,10]phenanthrolinium) is prepared, containing the phenanthroline coordination sphere and an imidazolium salt, easily forming N-heterocyclic carbenes (NHC). This ligand and its related precursors compounds are used to prepare and compare the series of Ru-complexes Ru(ip), Ru(bip), Ru(bbip) carrying the [Ru(tbbpy)2]2+-fragment in the diazadiene site. Furthermore we present detailed investigations of the formation of NHC complexes and we will discuss electrochemical and photophysical properties and the bridging abilities of this interesting new class of photo redox active metal complexes. [1] S. Rau et al. Angew. Chem. (Int. ed.) 2006, 45, 6215-6218 [2] M. Schwalbe et al. Dalton Trans. 2009, 4012-4022. [3] T. D. Pilz et al. J. Coord. Chem. submitted 46 P1 The role of Sulfur and Selenium in the DMSO-reductase family of enzymes: getting answers from small models Carlos E. Abad Andrade1, Carola Schulzke2 1 Institute for Inorganic Chemistry, Georg-August University of Goettigen, Tammanstr. 4, 37077, Goettingen, Germany 2 School of Chemistry, Trinity College Dublin, College Green, Dublin 2, Ireland [email protected] / [email protected] Some enzymes are interesting since they catalyze reactions that chemists find challenging. These reactions include the partial oxidation of a hydrocarbon by methane monooxygenase (MMO), the oxidation and production of hydrogen by hydrogenase and with the exception of the molybdenum-containing nitrogenase, molybdenum and tungsten cofactors (MoCo) relating in the oxygen atom transfer (OAT). As these kinds of enzymes own the ability to transfer an oxygen atom as shown in the overall reaction [1], they are important not only in biology but also for industrial and commercial applications. X + H2O XO + 2H+ + 2e- [1] The DMSO-reductase family is a specific group of enzymes at which the coordination to the active site from the peptide chain is achieved through specific aminoacids, serine (O), cysteine(S) or selenocysteine(Se). The reason of this natural selection is not well known and some studies were made to have an approach [2,3]. In order to compare the role of the aminoacid in the MoCo, small complexes were synthesized with S or Se containing ligands.1,4substituted- thiosemicarbazides and 1,4-substituted-selenosemicarbazides Schiff bases were chose as our systems (figure 1). H N N N O Mo S/Se S O O Figure 1. Some of the analogues Mo-Sulfur and Mo-Selenium complexes used to compare the dependence in the coordinated atom and the x-ray crystal structure of the sulfur one. Redox behavior were estimated as well as oxotransfer abilities of these complexes were screened in the reaction DMSO + PPh3 DMS + OPPh3 References: [1] P.P. Samuel, C. Schulzke, Molybdenum and tungsten cofactor model chemistry: past, present and future. In Handbook of Inorganic Chemistry, Nova Sci. Publ. NY 2009 (in press) [2] X. Ma, C. Schulzke, H.-G. Schmidt and M. Noltemeyer. Dalton Trans., 2007, 1773–1780. [3] C.E. Abad Andrade, X. Ma, W. Meyer-Klaucke, C. Schulzke. Polyhedron, 2010, 29, 664-668. 47 P2 Kupferkomplexe mit Guanidin-Schwefel-Hybridliganden Martin Bernard, Adam Neuba, Hans Egold, Ulrich Flörke, Gerald Henkel* Department Chemie, Universität Paderborn, Warburger Str. 100, D-33098 Paderborn *[email protected] Mit dem Fokus auf Modellkomplexe für das biologische Cu A-Zentrum (z. B. in den Cytochrom-c-Oxidasen) haben wir u. a. die Guanidin-Thioether-Disulfid-Hybride L1 und L2 entwickelt, um daraus Liganden mit den passenden N,S,S-Donorsätzen zu gewinnen. Versuche zur reduktiven Spaltung der Disulfid-Gruppe durch Cu(I)Halogenide führten allerdings nicht zu den erwarteten Thiolatkomplexen des Kupfers, sondern stattdessen zu zweikernigen Verbindungen, wenn die Disulfid-Komponente L1 eingesetzt wird. Als ein Beispiel ist die Chlorvariante K1 abgebildet.[1,2] N N N S N N S L1 S S N 2 2 L2 K1 Die insgesamt sechs funktionellen Gruppen des Liganden bilden zwei identische N,S,S-Donorsätze, die chelatförmig an unterschiedliche Kupferatome binden. Die Koordinationssphären dieser Kupferatome werden von zwei Chloridliganden zu verzerrten Tetraedern ergänzt. Die unerwartet ausgebliebene Redoxreaktion lässt sich dadurch erklären, dass die Edukte im Verhältnis 2:1 eingesetzt wurden und deshalb bei reduktiver Aktivierung der Disulfidbindung reine Kupfer(II)-Komplexe entstehen müssten, die vermutlich weniger stabil sind als die hier beobachteten Reaktionsprodukte. Die Transformation zu gemischtvalenten Thiolatkomplexen mit zentralen Cu2S2-Rauten sollte deshalb durch Umsetzung mit zusätzlichen Kupfer(I)Halogeniden erreicht werden können. Entsprechende Untersuchungen sind bereits im Gange. [1] S. Herres-Pawlis, A. Neuba, O. Seewald, T. Seshadri, H. Egold, U. Flörke, G. Henkel, Eur. J. Org. Chem. 2005, 4879 [2] A. Neuba, Dissertation, Paderborn, 2009 48 P3 Neue hochaktive Kupfer-Guanidin-Komplexe in der AtomtransferRadikalpolymerisations-Katalyse (ATRP) Bienemann, O., Dortmund/D; Haase, R., Paderborn/D; Flörke, F., Paderborn/D; Schürmann, M., Dortmund/D; Döring, A., Paderborn/D; Kuckling, D., Paderborn/D; Herres-Pawlis, S., Dortmund/D Olga Bienemann, Fakultät Chemie, Technische Universität Dortmund, Otto-HahnStraße 6, 44227 Dortmund Seit der Entwicklung der “lebenden” / kontrollierten Radikalpolymerisationsmethode ATRP im Jahr 1995 wurde eine Vielzahl neuer Katalysatoren für diese Reaktion intensiv untersucht. Kupfer-Komplexe mit polyfunktionellen N-Donor-Liganden waren die effizientesten Katalysatoren.[1,2] Hier berichten wir von neuen Kupfer-Komplexen mit Guanidinliganden, die auf ihre ATRP-Aktivität getestet wurden. Der Komplex [(TMGqu)2Cu]Cl mit dem aromatischen Hybridguanidinligand 1,1,3,3-Tetramethyl-2-(quinolin-8-yl)guanidin (TMGqu) hat sich als effizientester Katalysator erwiesen; dieser produziert Polystyrol mit einer sehr engen Molmassenverteilung (PD = 1,1) in hohen Ausbeuten. Kinetische Studien zeigen, dass die Atomtransfer-Radikalpolymerisation „lebenden” Charakter hat. Die strukturelle Charakterisierung mittels Einkristallröntgenstrukturanalyse führte uns zu einem mechanistischen Strukturvorschlag für den Aktivator- und den DesaktivatorKomplex (Abb. 1).[3] Abb. 1: Chloratomtransfer-Gleichwicht zwischen [(TMGqu)2CuI]+ und [(TMGqu)2CuIICl]+ [1] J.-S. Wang, K. Matyjaszewski, J. Am. Chem. Soc. 1995, 117, 5614. [2] K. Matyjaszewski, J. Xia, Chem. Rev. 2001, 101, 2921. [3] O. Bienemann, R. Haase, U. Flörke, M. Schürmann, A. Döring, D. Kuckling, S. Herres-Pawlis, Manuskript in Vorbereitung. 49 P4 Neue Rutheniumchromophore mit maßgeschneiderten Eigenschaften Breivogel, A.; Förster, C.; Heinze K.; Mainz/D Aaron Breivogel, Institut für Anorganische Chemie und Analytische Chemie, Johannes Gutenberg-Universität, Duesbergweg 10-14, 55099 Mainz Farbstoffsensibilisierte Solarzellen (DSSCs) sind eine vielversprechende Alternative zur teuren siliciumbasierten Photovoltaic-Technik.[1] Als Farbstoffkomponenten in DSSCs haben sich Ruthenium-Polypyridinkomplexe als die bislang effektivsten herausgestellt.[2] Die Farbstoffe sollten eine Funktion zur Verankerung auf Halbleiteroberflächen (z. B. TiO2) und intensive Absorptionen im sichtbaren Bereich besitzen und aus den elektronisch angeregten Zuständen effektiv Elektronen in das Leitungsband des Halbleiters injizieren können. Die 3 MLCT-Zustände sollten möglichst langlebig sein und nicht strahlungslos über thermisch erreichbare 3MCZustände zur Abregung führen. Strategien zur Verbesserung der photophysikalischen Eigenschaften sind Donor/Akzeptor-Substitution und optimierte Ligand-Biss-Winkel. Wir stellen die Synthese eines neuen Liganden (dppd) und eines heteroleptischen Rutheniumkomplexes sowie die strukturellen, spektroskopischen, optischen und elektrochemischen Eigenschaften des Komplexes vor.[3] [1] a) M. Grätzel, Nature 2001, 414, 338-344, b) N. Robertson, Angew. Chem. 2006, 118, 23982405. [2] M. K. Nazeeruddin, F. D. Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru, M. Grätzel, J. Am. Chem. Soc. 2005, 16835-16847. [3] A. Breivogel, C. Förster, K. Heinze, 2010, in Vorbereitung. 50 P5 New class of heterometallic pivalate complexes Study of their magnetic properties L. M. Carrella, E. Rentschler; Mainz/D Institut für Anorganische Chemie und Analytische Chemie, Johannes Gutenberg Universität Mainz, 55128 Mainz One of the most fascinating properties of transition metal complexes to us are the magnetic ones. Since the discovery of slow relaxation of the magnetization in single molecules several applications like high density memory devices and quantum computing are proposed for so called single molecule magnets, SMMs. In this context particularly carboxylate complexes are attracting more and more interest due to their large structural variety and flexibility. One of the basic requirements for SMMs is a spin ground state S 2 0. However, it is still challenging to achieve and predict a high spin ground state in complexes with simple ligand systems. We therefore have synthesised a new class of heterometallic complexes to study the influence of several metal ions on the magnetic properties. In a rational approach we are able to predict and manipulate the spin ground state in dependency of the choice of metal ions. 51 P6 Structural control in deprotonation and substitution reactions Colquhoun, V. P.; Strohmann, C.; Dortmund/D Carsten Strohmann, Anorganische Chemie, Technische Universität Dortmund, Otto-Hahn-Str. 6, 44227 Dortmund Metal silylamides and metal silanolates are of great scientific interest due to their potential as alkylation reagents in preparative synthesis or precursors in materials chemistry for silicon-based ceramic materials.[1] Fig. 1. General scheme of ligand Our first aim was to clarify the deprotonation ability of a metallic reagent, specifically lithium, magnesium and zinc alkyls. The well known kinetic hindrance of the deprotonation of many silazane compounds can be bypassed via the precoordination of the metal alkyl. Therefore a ligand of the general form A was designed. Based on the polarity of the organometallic reagent, the deprotonation reaction proceeds with differing activation energy and reaction rates. Consequently, we have been able to isolate various compounds including the precoordinated metal alkyl and various deprotonation products. The structures of these are strongly dependent on the sterical demand of the ligand. To quantify the specific Fig. 2. Crystal structure of a reactive intermediate (adduct of ligand and diethyl zinc before the deprotonation has occurred) reactivity of the metal alkyl and to understand the structure of the deprotonation products activation energies for the deprotonations and reaction enthalpies were calculated using quantum mechanical methods. Secondly we observed a substitution reaction of a siloxane with trimethyl aluminium. This unexpected reaction has been clarified by structural investigations and quantum mechanical calculations. [1] (a) B. Conway, E. Hevia, A. R. Kennedy, R. Mulvey, S. Weatherstone, Dalton Trans. 2005, 8, 1532; (b) E. Kroke, Y.-L. Li, C. Konetschny, E. Lecomte, C. Fasel, R. Riedel, Mater. Sci. Eng. 2000, 26, 97. 52 P7 Mono- & Dinuclear Ruthenium(II & III) Complexes With Noninnocent Quinonoid Ligands: Consequences For Structural, Redox, Electronic and Spectroscopic Properties Hari Sankar Das., Stuttgart/D, Biprajit Sarkar., Stuttgart/D, Ralph Hübner., Stuttgart/D, Hari Sankar Das, Institut für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70550 Stuttgart, Germany Abstract: Mono and dinuclear Ru(II) & Ru(III) complexes of the noninnocent bis-chilating ligand N,N´-diisopropyl-2-amino-5-alcoholate1,4-benzoquinonemonoiminium and dinuclear Ru(II) & Ru(III) complexes of 2,5bis(isopropylamino)-1,4-benzoquinone were synthesized with bipyridine and acetylacetonate as ancillary ligands. Structural, electrochemical, UV/Vis/NIR, EPR spectroelectrochemical and SQUID magnetometry investigations were carried out on these complexes in order to determine localization vs. delocalization of the π system in the quinonoid ligand, the site of electron transfer in these complexes, antiferromagnetic coupling between the two Ru(III) center and the general utility of such ligands in mixed-valence chemistry. O O O O RuL2 L2 Ru L2 Ru N N N NH N O RuL2 L2 Ru O O , L= N N O _ N Reference:- 1) H.S. Das, A.K. Das, R. Paattacini, R. Hübner, B. Sarkar*, P. Braunstein* Chem. Commun., 2009, 4387. 2) H.S. Das, F. Weisser, D. Schweinfurth, C.Y. Su, L. Bogani, J. Fiedler, B. Sarkar* Chem. Eur. J. DOI: 10.1002/chem.200903409 in press. 53 P8 Phosphaalkine als synthetische Bausteine zur Bildung von Oligomeren und Polymeren Eckhardt, M.; Vogel, U.; Scheer, M. Prof. Dr. M. Scheer, Institut für Anorganische Chemie, Universität Regensburg, Universitätsstraße 31, 93040 Regensburg Phosphaalkine sind metastabile Verbindungen, die thermisch bzw. photochemisch zu oligomeren oder polymeren Verbindungen reagieren. Die Oligomerisierung ist auch in der Koordinationssphäre von Übergangsmetallen möglich. Es konnte gezeigt werden, dass die Reaktion von CH3C≡P und tBuC≡P mit [(Cp′′′Co)2(µ,η4:4-C7H8)] zu Kopf-Schwanz-Dimerisierungen des Phosphaalkins unter Ausbildung von 1,3Diphospheten führt, welche durch ein Cp'''Co-Fragment stabilisiert werden. Die weitere Umsetzung mit Kupfer(I)-Halogeniden führt zu verschiedenen Koordinationsverbindungen. Die Oligomerisierung von tBuC≡P kann auch von CuI induziert werden und führt zur Bildung von Käfigverbindungen, die in einer CuI-Matrix eingebettet werden[1]. [1] U. Vogel, J. F. Nixon and M. Scheer, Chem. Commun. 2007, 5055-5057. 54 P9 Die Synthese neuartiger Fe-O-Mo-Komplexe und deren Anwendung in der Oxygenierungskatalyse Falkenhagen, J., Limberg, C.* Humboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Str. 2, 12489 Berlin, Germany, *[email protected] Formaldehyd ist eine der wichtigsten organischen Grundchemikalien in der Chemischen Industrie und dient als vielseitiger C1-Baustein für die Synthese höherwertiger Produkte. Heute existieren mehrere großtechnisch eingesetzte heterogenkatalytische Verfahren zur Erzeugung von Formaldehyd aus Methanol, die entweder auf der Verwendung von Silber- oder Eisen-Molybdat-Katalysatoren beruhen. Der größte Teil der Weltjahresproduktion von derzeit ca. 20 Mt/a wird dabei im Perstorp-Formox-Prozess erzeugt.[1] Ein Methanol-Luft-Gemisch wird hier bei Atmosphärendruck und 270400 °C an Mischphasen-Fe-Mo-O-katalysatoren partiell oxidiert.[2] Vor diesem Hintergrund sollen lösliche Fe-Mo-O-Komplexe als Modellverbindungen für Strukturmotive der Katalysatoroberfläche synthetisiert und die Aktivität dieser potentiellen Katalysatoren für die Oxygenierung repräsentativer Substrate soll untersucht werden.[3, 4] O2, 80°C MeCN O Wir berichten hier über die Synthese heteronuklearer Komplexe in denen Fe- und Mo-Zentren bei variabler Stöchiometrie über Sauerstoff-Brücken miteinander verknüpft sind. Erste Ergebnisse von Untersuchungen zur Reaktivität gegenüber Disauerstoff sowie zu ihrem Potenzial als Präkatalysatoren für die Epoxidierung von Cycloocten zu Cyclooctenoxid werden gezeigt. [1] House, M. P.; Carley, A. F.; Bowker, M. J. Catal. 2007, 252, 88–96. [2] Soares, A. P. V.; Portela, M. F.; Kiennemann, A. Catal. Rev. 2005, 47, 125–174. [3] Schröder, K.; Enthaler, S.; Bitterlich, B.; Schulz, T.; Spannenberg, A.; Tse, M. K.; Junge, K.; Beller, M. Chem. Eur. J. 2009, 15, 5471–5481. [4] Schröder, K.; Tong, X.; Bitterlich, B.; Tse, M. K.; Gelalcha, F. G.; Brückner, A.; Beller, M. Tetrahedron Lett. 2007, 48, 6339–6342. 55 P10 Tuning of redox potentials of seven-coordinate iron and manganese complexes for superoxide dismutation and nitric oxide transformation. Felix Friedel, Milos Filipovic, Prof. Dr. Ivana Ivanović-Burmazović, Department of chemistry and pharmacy, University of Erlangen-Nürnberg, Egerlandstr. 1, 91058 Erlangen, Germany. The synthesis and characterisation of functionalised seven-coordinate iron and manganese complexes with the DAPSOX ligand (see below) was achieved. We’ve shown that the electron-withdrawing and electron-donating functional groups in 4-position of the pyridine core of the ligand system influence the redox potential of the coordinated iron center in linear dependence of the Hammett constant of the functional group. +/2+ R H2O N N N N N M O O O O OH2 NH2 NH2 M = Mn(II), Fe(III) R = H, CN, NO2, OMe We investigate the effect of the shift of the redox potential on the dismutation of superoxide radical anion and transformation of nitric oxide as actors in so called ] oxidative stress.[1,2 Results will be discussed in terms of possible mechanisms and reaction selectivity towards one of these radical species. [1] D. P. Riley, Chem. Rev., 1999, 99, 2573-2587. [2] I. Fridovich, Biol. Chem., 1989, 264 (14), 7761 - 7764. 56 P11 Catalysis optimization of cyclopalladated nitrosamines for Heck- and Suzuki-coupling reactions Fuge, Felix a; Mohr, Fabian b Felix Fuge, Present address: Department of Experimental Molecular Imaging, Medical Faculty, RWTH Aachen University, Pauwelsstr. 20, 52074 Aachen Fabian Mohr, Department of Inorganic Chemistry, Bergische Unversität Wuppertal, Gausstr. 20, 42119 Wuppertal In this project, we successfully used nitrosamine based palladacycles for Suzuki- and Heck-coupling, and carried out detailed screening studies to determine optimal reaction conditions. While oxime based palladacycles have been subject of intense investigation as catalysts, the structurally very similar nitrosamine based derivatives have never been used in Suzuki- and Heck-coupling reactions.[1,2] In our study, the acetate bridged cyclopalladated nitrosamine ([Pd(µ-O2CMe){κ2C,N4-MeC6H3N(Me)NO}]2) and various derivatives thereof were investigated catalysts for Suzuki- and Heck-reactions using some simple model compounds.[3] Aromatic compounds with different degree of activation were reacted with either methylacrylate or phenylboronic acid. Statistical methods allowed for the determination of the ideal reaction conditions by evaluation of the results. Turn-over number (TON) values of up to 330000 with catalyst loadings of 0.0001 mol% were achieved. The yields and TON reactions exceeded those reported for the oxime palladacycles in several cases and we were able to reach values reported for some of the conventionally used Pd(0) catalysts.[1,2,4] The main advantages of the nitrosamine systems are: i) the compounds are neither air nor moisture or light sensitive; ii) they are easily prepared in high yields, iii) different solvent systems including water can be employed in catalysis. [1] [2] [3] [4] D. A. Alonso, C. Najera, M. C. Pacheco, Organic Letters 2000, 2, 1823. D. A. Alonso, C. Najera, M. C. Pacheco, Journal of Organic Chemistry 2002, 67, 5588. F. Fuge, C. Lehmann, F. Mohr, Journal of Organometallic Chemistry 2009, 694, 2395 I. P. Beletskaya, A. V. Cheprakov, Journal of Organometallic Chemistry 2004, 689, 4055. 57 P12 Bindungsaktivierung in Eisen(II)-Komplexen polypodaler Phosphane Gentschow, S.-A.a; Heinemann, F. W.b, und Grohmann, A.a a Technische Universität Berlin, Institut für Chemie, Strasse des 17. Juni 135, D-10623 Berlin b Universität Erlangen-Nürnberg, Department Chemie und Pharmazie, Anorganische Chemie, Egerlandstrasse 1, D-91058 Erlangen Der Tetraphosphanligand C5H3N[CMe(CH2PMe2)2]2 (1) neigt zu bemerkenswert spezifischer C–P-Bindungsaktivierung, woraus ein Bindungsbruch und die Bildung des Triphosphanliganden C5H3N[CMe(CH2PMe2)2][CMe2(CH2PMe2)] (2) resultieren. Unter geeigneten Bedingungen kann dieser C–P-Bindungsbruch unter Neubildung von 1 rückgängig gemacht werden.[1] Um den Mechanismus dieser Rückbildung zu untersuchen, wurde 2 direkt synthetisiert und mit Fe(BF4)2 · 6 H2O und Methyldiethylphosphinit als zusätzlichem monodentaten Liganden umgesetzt. Dies lieferte den gewünschten Komplex 3 und das Nebenprodukt 4. Beide weisen zusätzlich zum koordinierten Liganden 2 eine Methylgruppe in engem Kontakt mit dem Eisenzentrum auf und unterscheiden sich nur in dem monodentaten P-Liganden (Et2POMe (3) vs. Et2PH (4)). Während sich die erhoffte Aufklärung des fraglichen Mechanismus mit Hilfe von 3 als unmöglich herausstellte, wurde bei der Reaktion von 4 mit CO die Bildung zweier Produkte beobachtet, welche durch Röntgendiffraktometrie charakterisiert wurden. Das eine ist der Eisen-Dicarbonylkomplex (5, linke Struktur) von 2, das andere der Eisen-Carbonylkomplex (6, rechte Struktur) des Liganden 1, was einen intermolekularen Mechanismus der fraglichen Rückbildungsreaktion nahelegt. 5 6 [1] S. W. Kohl, F. W. Heinemann, M. Hummert, H. Weißhoff, A. Grohmann, Eur. J. Inorg. Chem. 2006, 3901-3910. 58 P13 Strukturlösung und magnetische Eigenschaften von trans-[NiCl2(C5H5N)2] aus Pulverdaten Alig, E.; Bernert, T.; Fink, L.; Hammer, S. M.; Külcü, N.; Wolf, B. Lothar Fink, Institut für Anorganische und Analytische Chemie, Goethe-Universität, Max-von-Laue-Str. 7, 60438 Frankfurt am Main Die thermische Zersetzung von [NiCl2(C5H5N)4] führt zu neuen Verbindungen des Typs [NiCl2(C5H5N)x], mit x = 2, 1 und 2/3 [1]. [NiCl2(C5H5N)2] wurde über DTA/TG, Röntgenpulverdiffraktometrie und magnetische Messungen charakterisiert [2]. Die Kristallstruktur wurde mit DASH [3] durch Simulated Annealing gelöst. Eine anschließende Rietveldverfeinerung mit TOPAS [4] führt zu folgender Struktur: Fig. 1: Kristallstruktur von [NiCl2(C5H5N)2] Fig. 2: Inverse molare Suszeptibilität In der Raumgruppe P2/c besetzt Nickel zwei symmetrisch unabhängige Positionen (Wyckoff Positionen 2e und 2f). Nickel ist durch 4 Chloridionen und 2 Stickstoffatome der Pyridinringe oktaedrisch koordiniert. Innerhalb der Kette sind die Wechselwirkungen der Nickelionen ferromagnetisch. Unterhalb von ca. 8 K überwiegen antiferromagnetische Wechselwirkungen zwischen den Ketten [5]. [1] G. Liptay, T. Wadsten, A. Borbély-Kuszmann, Journal of Thermal Analysis 1981, 31 845-852. [2] E. Alig, T. Bernert, L. Fink, N. Külcü, T. Ye ilkaynak, Acta Cryst. 2010, E66, m239. [3] W. I. F. David, K. Shankland, J. van de Streek, E. Pidcock, S. Motherwell, DASH (2004), Version 3.0. Cambridge. Crystallographic Data Centre, 12 Union Road, England [4] A. A. Coelho, TOPAS Academic User Manual (2007), Coelho Software, Brisbane, Australia [5] H. T. Witteveen, W. L. C. Rutten, J. Reedijk, J. Inorg. Nucl. Chem. 1974, 37, 913-919. 59 P14 Seeing is Believing – Spectroscopic Determination of Oxidation States in NonOxo Uranium (III) to (VI) complexes Christina T. Hauser†, Ingrid Castro-Rodriguez‡, Steven D. Conradson¥, Frank W. Heinemann†, and Karsten Meyer†* ‡ Department of Chemistry & Biochemistry, University of California, San Diego ¥ Los Alamos National Laboratory, Los Alamos,NM 87545, USA, †* Institute of Inorganic Chemistry, Department of Chemistry and Pharmacy, Friedrich-Alexander-University Erlangen-Nürnberg The correct interpretation of spectroscopic data, influenced by complex geometry and ligand nature, is often limited by a lack of suitable calibration data. This is especially true for molecular complexes of actinides, many of which have ambiguous assignments of charge distribution because the degree of covalency of their metalligand bonds is unknown.1 The availability of a series of structurally comparable actinide coordination compounds with varying oxidation states therefore provides a unique opportunity for the detailed investigation of electronic properties. U(III), n=0 U(IV), n=0 U(V), n=0 U(VI), n=1 U(?), n=0 We here present a comprehensive XANES L3 absorption edge study of a complete series of non-oxo U(III) to U(VI) complexes of the general type [((AdArO)3tacn)U(L)]n+ (L = N-donor ligand or CO2). Combining XANES and XRD studies allows for unambiguous determination of the uranium oxidation states including the previously difficult to assign U(IV) oxidation state of the activated CO2-complex [((AdArO)3tacn)U(CO2)]. [1] Denning, R., Electronic Structure and Bonding in Actinyl Ions. Complexes, Clusters and Crystal Chemistry, Springer: Berlin/Heidelberg, 1992; Vol. 79, pp 215-276. [2] Castro-Rodríguez, I.; Nakai, H.; Meyer, K., Angew. Chem., Int. Ed. 2006, 45, 2389-2392. [3] Castro-Rodríguez, I.; Nakai, H.; Zakharov, L. N.; Rheingold, A. L.; Meyer, K., Science 2004, 305, 60 P15 DFT-Studien zu den magnetischen Eigenschaften oligonuklearer Kupferkomplexe David Hornig, Winfried Plass Institut für Anorganische und Analytische Chemie, Friedrich-Schiller-Universität Jena, Carl-Zeiss-Promenade 10, 07745 Jena Für oligonukleare Metallkomoplexe ist die Bestimmung der Kopplungskonstanten aus experimentellen Suszeptibilitätsmessungen bei entsprechend niedriger Symmetrie auf Grund der hohen Anzahl notwendiger Parameter in der Regel nicht eindeutig durchführbar. Zur Untersuchung der magnetischen Eigenschaften mehrkerniger Komplexe eignen sich insbesondere DFT-Methoden, da diese im Gegensatz zu ab initio-Methoden die Berechnung deutlich größerer Systeme, bei dennoch hinreichender Genauigkeit, erlauben. Dies wird anhand ausgewählter Beispiele gezeigt. Chirale tetranukleare Kupferkomplexe mit zuckerbasierten Liganden bilden Cu4O4Heterocubane, die lediglich C1-Symmetrie aufweisen. Diese Komplexe zeigen in Abhängigkeit des modifizierten Ligandsystems ein unerwartetes magnetisches Verhalten. Für eine Serie dieser Komplexe werden die Ergebnisse vorgestellt und mit den experimentellen Ergebnissen verglichen. Für einen sechskernigen Kupferkomplex mit N-tert-Butyl-diethanolamin als Ligand wird ein C3-symmetrischer Metallazyklus bestehend aus 6 Kupferionen beobachtet (siehe Abbildung), die antiferromagnetisch gekoppelt sind. Zusätzlich zu den direkten 1,2Wechselwirkungen zwischen Kupferionen im Ring werden 1,3- und 1,4-Kopplungen über die wasserstoffverbrückten H3O+-Ionen beobachtet. Die entsprechenden Kopplungskonstanten werden in diesem Falle aus DFT-Berechnungen ermittelt, die über einen diamagnetischen Ersatz der Kupfer(II)- durch Zink(II)-Ionen geeignet modifiziert wurden. 61 P16 Variable binding of heterodinuclear (Cu, Fe) organometallics to N and O donor functions of guanine, pterin, lumazine and alloxazine heterocycles Rajkumar Janaa, Biprajit Sarkara, Jan Fiedlerb, and Wolfgang Kaim*a a Institut für Anorganische Chemie, Universität Stuttgart; bJ. Heyrovsky Institute of Physical Chemistry. Czech Republic. Abstract Co-ordination compounds of the first row transition elements with small biorelevant heterocycles such as coenzymes, vitamins, and also nucleobases are frequently labile, precluding the isolation and structural identification of species present in solution equilibria. In contrast, complexes of the heavier d block elements are often inert as exemplified by the platinum and platinum metal coordination chemistry of nucleobases1 and coenzymes2. Herein we wish to report a number of first row transition metal compounds which owe their stability to the use of a particular kind of organometallic complex fragments, viz., [(dopf)Cu]+, dopf = 1,1`-bis(diorganylphosphino)ferrocene. Fe Fe R2 P R2 P O H PR 2 O H N N ButOC N H + Cu N N + Cu Fe PR 2 O Me N O N R2 P PR 2 N But OC N H + Cu Fe N Me N N R 2P + Cu O Me PR 2 N Me N Me N N Me N O N Me The dopf ligands are being extensively used as scaffolding chelate ligands in catalysis3, 4, sometimes using their propensity for reversible one-electron oxidation at the ferrocene iron site. Our experience especially with the dppf ligand (dppf = 1,1`- bis(diphenylphosphino)ferrocene) in heterobimetallic [(dppf)Cu]+ to stabilize complexes with unreduced strong acceptors such as α-azoimines5 or o-quinones6 has prompted us to explore the coordination of [(dppf)Cu]+ and [(dippf)Cu]+, dippf = 1,1`- bis(diisopropylphosphino)ferrocene, with other small biorelevant unsaturated molecules containing the lumazine, isoalloxazine, pterin and guanine heterocyclic structures. Synthesis and structural characterization will be reported as will be electrochemical studies on oxidation (ferrocene) and reduction (heterocycles). 62 References: 1. Montagner, D; Zangrando, E; Longato, B. Inorg. Chem. 2008, 47, 2688; Lippert, B., Ed.; Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug; WileyVCH, Weinheim, 1999. 2. Jaouen, G., Ed.; Bioorganometallics: Biomolecules, Labelling, Medicine; Wiley-VCH, Weinheim, 2006. 3. Alcazar-Roman, L. M.; Hartwig, J.F.; Rheingold, A. L; Liable-Sands, L. M.; and Guzei, I. A. J. Am. Chem. Soc. 2000, 122, 4618. 4. Driver, M. S.; Hartwig, J. F. J. Am. Chem. Soc. 1997, 119, 8232. 5. Roy, S.; Sieger, M.; Sarkar, B.; Schwederski, B.; Lissner, F.; Schleid, T.; Fiedler, J.; Kaim, W. Angew. Chem. Int. Ed. 2008, 47, 6192. 6. Roy, S.; Sarkar, B.; Bubrin, D.; Niemeyer, M.; Zalis, S.; Lahiri, G.K.; Kaim, W. J.Am.Chem.Soc. 2008, 130, 15230. 63 P17 Can one oxidize a NacNac ligand? Khusniyarov, M. M.a,b; Weyhermüller, T.a; Bill, E.a; Wieghardt, Ka; Mülheim an der Ruhr/D a b MPI for Bioinorganic Chemistry, Stiftstrasse 34-36, 45470, Mülheim an der Ruhr Friedrich-Alexander-University Erlangen-Nürnberg, Egerlandstr. 1, 91058, Erlangen We provide experimental and theoretical evidence that -diketoiminato(1–) (NacNac1–) ligands are redox-active (non-innocent) ligands. The neutral [Ni(MeNacNac)2] featuring electron-donating substituents can be electrochemically oxidized to the [Ni(MeNacNac)2]1+ cation at low potentials. According to EPR spectroscopy, the cation possesses a doublet ground state with a spin density predominantly located at the metal center. Due to the twisted tetrahedral geometry of [Ni(MeNacNac)2]0/1+ complexes and the EPR data the electronic structure of the cation should be described as [NiII(MeNacNac1–)(MeNacNac)]1+ containing a high-spin Ni(II) ion and one neutral NacNac ligand -radical. The doublet ground state (St = 1/2) is obtained via a strong antiferromagnetic coupling between the metal ion (SNi = 1) and the ligand radical (SR = 1/2). Broken symmetry density functional theory calculations confirm this assignment. More importantly, due to the odd-number of atoms in the NCCCN -system, the intra-ligand bond distances of a NacNac ligand are virtually not sensitive to its oxidation level. As a consequence, X-ray crystallography cannot be utilized to determine the oxidation level in a NacNac ligand class and possibly in other odd-numbered atom systems. This is in stark contrast to a large family of redoxactive -diimine, o-phenylenediimine,[1] and other related O- and S-donor ligands with even-numbered atom -systems, all of which show significant changes in intra-ligand bond distances upon ligand oxidation or reduction.[2] [1] M. M. Khusniyarov, T. Weyhermüller, E. Bill, K. Wieghardt, J. Am. Chem. Soc. 2009, 131, 1208. [2] P. Chaudhuri, C. N. Verani, E. Bill, E. Bothe, T. Weyhermüller, K. Wieghardt, J. Am. Chem. Soc. 2001, 123, 2213. 64 P18 Bis(2-pyridylimino)isoindole als Steuerliganden in der asymmetrische Übergangsmetallkatalyse Matthias Kruck, Désirée Sauer, Björn K. Langlotz und Lutz H. Gade Anorganisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg. In den letzten Jahren spielten tridentate Stickstoffdonorliganden wie Trispyrazolylborate oder Triazacyclanone eine bedeutende Rolle als Liganden für die Übergangsmetallkatalyse. 1,3-Bis(pyridylimino)isoindole (BPI)[1] gehören einer weiteren Klasse tridentater Stickstoffdonorliganden an, die bisher sowohl in Hinblick auf ihre katalytischen als auch auf ihre biomimetischen Eigenschaften kaum untersucht wurden. Diese monoanionischen, meridonalen Pincerliganden wurden zuerst von Linstead et al. dargestellt.[2] Im Gegensatz zu den meisten Phosphanliganden sind BPIs inert gegenüber einer Oxidation durch Sauerstoff oder Hydrolyse durch Wasser und bieten einen leichten synthetischen Zugang durch Kondensation von 1,2Dicyanobenzol mit 2-Aminopyridinderivaten.[3] Der modulare Charakter der Synthese ermöglicht die unproblematische Darstellung vieler verschiedener BPI-Derivate. Neben achiralen Liganden sind auch chirale Vertreter bekannt, die stereoselektive Katalysen ermöglichen.[4] Wir zeigen die Synthese von achiralen und chiralen BPI-Liganden sowie deren Koordinationschemie mit verschiedenen Übergangsmetallen der ersten Reihe. Die Anwendungsmöglichkeiten dieser Metallkomplexe als Katalysatoren für asymmetrische Transformationen werden anhand von Cyclopropanierung und Hydrosilylierung präsentiert. [1] a) M.B. Meder, L.H. Gade, Eur. J. Inorg. Chem. 2004, 2716. b) B.K. Langlotz, J. Lloret Fillol, J.H. Gross, H. Wadepohl, L.H. Gade, Chem. Eur. J. 2008, 14, 10267. [2] J.A. Elvidge, R.P. Linstead, J. Chem. Soc. 1952, 5000. [3] R.R. Gagné, W.A. Marritt, D.N. Marks, W.O. Siegl, Inorg. Chem. 1981, 20, 3260. [4] B.K. Langlotz, H. Wadepohl, L.H. Gade, Angew. Chem. 2008, 120, 4748; Angew. Chem. Int. Ed. 2008, 47, 4760. 65 P19 Oxidation catalysis by high-valent iron bispidines Comba, P.; Maurer, M. Institute of Inorganic Chemistry, University of Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg High-valent iron model complexes have raised attention due to their biomimetic behavior and their efficiency in the catalytic oxidation of alkanes and alkenes.[1] The iron bispidine complexes are highly efficient and selective catalysts for the oxidation of cyclohexane.[2,3] The iron-bispidine-catalyzed oxidation of cyclohexane with H2O2, where either a tetradentate or a pentadentate bispidine ligand is coordinated to the iron center, yields up to 35% cyclohexanol and cyclohexanone. The alcohol/ketone ratio of up to 4 and relatively high kinetic isotope effects (KIE) suggest that the hydrogen abstraction by a ferryl complex is the rate determining step. The 3°/2° ratio in experiments with adamantane as substrate supports the presence of high-valent iron oxo species. However studies with hydroxyl radical traps show that OH radicals are also involved as oxidant. Product distributions and 18O isotope labeling experiments reveal significant differences in the reaction pathways between the tetradentate and pentadentate ligand catalysts. Not only in the oxidation of alkanes and alkenes the iron bispidines have proven their efficiency, but also in the oxidation of sulfides to sulfoxides.[4] The tetradentate as well as the pentadentate iron(II)bispidine complexes yield up to 75% of sulfoxides product under ambient conditions, using PhI(OAc)2 oxidant. All experiments are fully supported by a DFT-based computational analysis. [1] Meunier, B., Biomimetic Oxidations Catalyzed by Transition Metal Complexes. Imperial College Press: 2000. [2] Comba, P.; Maurer, M.; Vadivelu, P., J. Phys. Chem. (A) 2008, 112, 13028. [3] Comba, P.; Maurer, M.; Vadivelu, P., Inorg. Chem. 2009, 48, 10389–10396. [4] Comba, P.; Maurer, M.; Madhavan, J., unpulished results. 66 P20 Neue Porphyrinaminosäuren mit einstellbaren elektronischen Eigenschaften für Untersuchung photoinduzierter Elektronentransferund Energietransfer-Prozesse Yakiv Melomedov, Dr. Wei Liu, A. Reinhart, Daniel Siebler, Prof. Dr. Katja Heinze Institut für Anorganische und Analytische Chemie Johannes Gutenberg-Universität Mainz Die Natur verwendet äußerst komplexe Anordnungen der Chromophore um die Sonnenenergie einzufangen und diese in ein chemisches Potential umzuwandeln, welches die chemischen Prozesse in den grünen Pflanzen, Algen und zur Photosynthese befähigten Bakterien antreibt. Die Chromophore sind in einem Proteingerüst eingebettet, das die nicht-kovalent gebundenen Pigmente für einen schnellen Energie- und gerichteten Ladungstransport im optimalen Abstand hält. Um die Prozesse in den photosynthetischen Reaktionszentren nachzuahmen, wurde eine große Anzahl biomimetischer Verbindungen entwickelt, die es ermöglichen den Primärschritt der Photosynthese zu erforschen. Mithilfe der hier vorgestellten [1-3] trans-AB2C-Porphyrin-Aminosäureester mit einstellbaren elektronischen Eigenschaften sollen Porphyrin-Peptide aufgebaut werden, wobei die Sequenz der Porphyrin-Aminosäuren derart eingestellt wird, dass ein Elektronentransfer entlang eines Redoxgradienten gewährleistet werden kann. Aufgrund des Donor-Akzeptor-Substitutionsmusters der Porphyrin-Aminosäuren ist die Vorzugsrichtung des Ladungstransfers bereits vorgegeben. An den funktionellen Gruppen der Porphyrin-Aminosäureester lassen sich sowohl Ferrocen[3] Bausteine als auch mit elektronenziehenden bzw. -liefernden Substituenten versehene Porphyrin- Aminosäureester anknüpfen. Die Eigenschaften dieser Dyaden werden auf dem Poster diskutiert. 1 K. Heinze, A. Reinhart, Dalton Trans. 2008, 469-480; A. Reinhart, Dissertation, Universität Heidelberg, 2008. 2 S. L. Gould, G. Kodis, R. E. Palacios, D. Gust, T. A. Moore, A. L. Moore, J. Phys. Chem. B, 2004, 108, 10566-10580. 3 K. Heinze, M. Schlenker, Eur. J. Inorg. Chem. 2004, 2974-2988. 67 P21 Synthesen und Strukturen von Phosphinogold(I)komplexen mit Seleno- und Thiocarbamatderivaten Molter, A.; Mohr, F.; Wuppertal/D Anja Molter, Fachbereich C - Anorganische Chemie, Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal Goldverbindungen mit schwefelhaltigen Liganden gehören zur ältesten und am besten untersuchten Klasse bekannter Goldkomplexe. Das Interesse an dieser Art Komplexe resultiert in erster Linie aus medizinischen Anwendungen und ihren Lumineszenzeigenschaften. Einige Goldthiolatkomplexe sind bereits seit über 30 Jahren im klinischen Einsatz, deshalb überrascht es, dass die analogen Selenogoldkomplexe[1] bis heute nur wenig Beachtung fanden. Aus diesem Grund wurden Gold(I)komplexe mit einer Vielzahl von Selenliganden synthetisiert sowie ihre Strukturen und Eigenschaften untersucht. Begonnen wurde mit Phosphinogold(I)komplexen, methylselenocarbamat gebunden an die das wurde.[2] Es deprotonierte folgten weitere N-phenyl-OSeleno- und Thiocarbamatderivate mit der gemeinsamen Einheit NH-C(E)- (E = S oder Se).[3] Sowohl mono- als auch dinukleare Gold(I)komplexe wurden aus den entsprechenden Phophinogold(I)chlorokomplexen mit Base erhalten (Schema). Alle Produkte wurden mit spektroskopischen Methoden umfassend charakterisiert, für einige konnten auch Kristallstrukturen erhalten werden. [1] A. Molter und F. Mohr, Coord. Chem. Rev. 2010, 254, 19-45. [2] D. Gallenkamp, E.R.T. Tiekink und F. Mohr, Phosphorus, Sulfur, and Silicon 2008,183, 10501056. [3] D. Gallenkamp,T. Porsch, A. Molter, E.R.T. Tiekink und F. Mohr, J. Organomet. Chem. 2009, 6944, 2380-2385. 68 P22 Synthese und Charakterisierung von Kupfer(I)-Komplexen mit chromophoren 1,2-Bis(arylimino)acenaphthen -Akzeptorliganden Granner, G.a; Kern, K.a; Monkowius, U.a; Zabel, M.b; Knör, Ga. a Johannes Kepler Universität Linz, Institut für Anorganische Chemie, Altenbergerstraße 69, 4040 Linz b Universität Regensburg, Zentrale Analytik, Röngtenstrukturanalyse, Universitätsstraße 31, 93053 Regensburg Kupferkomplexe spielen in der Natur eine wichtige Rolle als Bestandteile der aktiven Zentren von Elektronentransferproteinen und Oxidoreduktase-Enzymen [1]. Dabei ist das Redoxpaar von Cu(I)/Cu(II) von entscheidender Bedeutung. In synthetischen Modellverbindungen können Bis(arylimino)acenaphthen Speichermedium redoxaktive (Ar-BIAN) für Mehrelektronenübertragungen bei Elektronen begünstigen. π-Akzeptorliganden der dienen Oxidation [2], von und wie Cu(I) 1,2als dadurch Aufgrund dieser Fähigkeit haben Komplexe mit diesem Liganden ein großes Anwendungspotential in der Photo- und Elektrokatalyse. Beispiele für derartige Verbindungen sind die zweikernigen Komplexe der Form [Cu2(Pr2-Ph-BIAN)2]X2 (X = Cl, Br, I) sowie homoleptische Komplexe vom Typ [(BIAN)2Cu]PF6. Die Synthese, spektroskopische und strukturelle Charakterisierung dieser Komplexe, sowie erste elektrochemische Untersuchungen werden vorgestellt. [1] Y. Lee, K. Karlin in H.-B. Kraatz, N. Metzler-Nolte (Eds.): Concepts and Models in Bioinorganic Chemistry, Wiley-VCH, 2006. [2] I. Feduschkin, A. A. Sakatova, V. K. Cherkasov, V. A. Chudakova, S. Dechert, M. Hummert, H. Schuhmann, Chem. Eur.J. 9 (2003) 5778-5783. 69 P23 A Chiral Triplesalen Ligand: From Enantioselective Oxidation Catalysis to Chiral Single-Molecule Magnets. Mukherjee, C.; Stammler, A.; Bögge, H.; Glaser, T.; Bielefeld/D Thorsten Glaser, Lehrstuhl für Anorganische Chemie I, Fakultät für Chemie, Universität Bielefeld, Universitätsstr. 25, D-33615 Bielefeld, Germany Asymmetric oxidation of small organic molecules, like, olefins to epoxides, sulfides to sulfoxides, etc, is an important pathway for the construction of complex chiral molecules. The use of chiral metal salen complexes as enantioselective catalysts for the oxidation processes during the last two decades made a strong impact in this field. In this respect, we have synthesized the trinucleating chiral version of the salen ligand, H6chandRR.[1] This can coordinate three metal ions and hence, provides chiral trinuclear MnIII3 and FeIII3 triplesalen complexes. The trinuclear MnIII3 triplesalen complex acts as good catalyst for asymmetric epoxidation of unfunctionalized olefins, while the trinuclear FeIII3 triplesalen complex catalyzes asymmetric sulfoxidation. OH N OH N N N HO OH OH N HO N H6chandRR Due to our on going interest for a rational synthesis of cyanide-bridged singlemolecule magnets of M3M´M3[2] topology we used the chiral trinuclear MnIII3 and FeIII3 triplesalen complexes as building blocks to synthesize heptanuclear chiral complexes. Asymmetric oxidations by trinuclear chiral MnIII3 and FeIII3 triplesalen complexes and chiral SMMs will be presented. [1] C. Mukherjee, A. Stammler, H. Bögge and T. Glaser, Inorg. Chem. 2009, 48, 9476-9484. [2] T. Glaser, M. Heidemeier, T. Weyhermüller, R.-D. Hoffmann, H. Rupp and P. Müller, Angew. Chem. Int. Ed. 2006, 45, 6033-6037. 70 P24 2-(3-Pyrazolyl)pyridines as Ligands in the Ruthenium Catalysed Hydrogenation of Carbon Dioxide K. Muller; K. Roth; W. R. Thiel; Kaiserslautern/D Keven Muller, Katrin Roth, Anorganische Chemie, TU Kaiserslautern, ErwinSchrödinger-Str., Geb. 54, D-67663 Kaiserslautern We present here, ruthenium based catalysts [1] with pyrazolylpyridine ligands for the catalytic hydrogenation of CO2 to formic acid derivatives.[2] These ligands have already shown promising results in other catalytic transformations [3] that have been investigated in our group. A series of different ruthenium(II)phosphine complexes such as RuCl2(dppe)2, RuCl2(PPh3)4 had been used as catalyst precursors in the past.[4] We synthesized air stable RuCl2(PPh3)2(pyrazolylpyridine) (Figure 1) by simple ligand exchange. In a recent step we enhanced the activity of the system by substitution of the triphenylphosphine with less sterical demanding trimethylphosphine. Furthermore, the development of a simplified catalyst screening process, as well as a proposition of the hydrogenation mechanism will be presented. Figure 1: RuCl2(PPh3)2(2-(5-methylpyrazol-3-yl)pyridine) and RuCl2(PPh3)2(bis-(5-butyl-3-pyrazolyl)) 1 2 L. Schmid, A. Canonica, A. Baiker, Appl. Cat. A: General 2003, 255, 23–33. P. G. Jessop, F. Joó, C. Tai, Coord. Chem. Rev. 2004, 248, 2425–2442. 3 W. R. Thiel, J. Eppinger, Chem. Eur. J. 1997, 4 3(5), 696–705. L. Schmid, M. S. Schneider, D. Engel, A. Baiker, Catal. Lett. 2003, 88(3–4), 105–113. 71 P25 gem-Diamino-Dicarben-Komplexe des Rutheniums Petrov, A. R.; Tamm, M.; Braunschweig/D Dr. Alexander R. Petrov, Prof. Dr. Matthias Tamm, Institut für Anorganische und Analytische Chemie, Technische Universität Carolo-Wilhelmina, Hagenring 30, 38106 Braunschweig Design von freien Carbenen spielt in der Entwicklung von Carbenkomplexen und ihrer Anwendungen eine entscheidende Rolle. Eine direkte Verknüpfung von zwei Amin-stabilisierten Carbenen ergibt Diaminoacetylene. Die Koordination von solchen elektronenreichen Acetylenen an Metallzentren ist in der Literatur nur von wenigen Beispielen bekannt (M = Fe,[1] Cr, Mo und W [2]). M NR2 NR2 R2N R2N M NR2 M NR2 Metall induzierte Isomerisierung von linearen Acetylenen in geminale Dicarbene und die chemischen Eigenschaften der entstandenen Komplexe sind am Beispiel des Rutheniums untersucht worden. [1] A. C. Filippou, T. Rosenauer, Angew. Chem. Int. Ed. 2002, 41, 2393–2396. [2] a) A. C. Filippou, W. Grünleitner, C. Völkl, P. Kiprof, Angew. Chem. 1991, 103, 1188–1191; b) J. Heck, K.-A. Kriebisch, W. Massa, S. Wocadlo, J. Organomet. Chem. 1994, 482, 81–84. 72 P26 Better Pt-based 3MLCT-Sensibilisators by Dye-Functionalisation Pevny, F.; Winter, R.; Regensburg/D Florian Pevny, Institut für Anorganische Chemie, Universität Regensburg, Universitätsstraße 31, 93040 Regensburg Fig. 1: Complexes 1 (X = S), 2 (X=O) and 3 The new square-planar platinum complexes 1, 2 and 3 with coumarine-dyetypeligands were synthesized. To obtain more information about the nature of the frontier orbitals these chromophors were studied by (spectro-) electrochemistry. UV/visabsorption spectroscopy and luminescence measurements could demonstrate that the emitting 3MLCT-state is populated by irradiation into both the (LM)=>LCT band of the complex and the - *-absorption band of the attached dye. This implies a better utilization of the sunlight spectrum to generate the charge separated state. Fig.2: Generating the charge-separated state by using the - *-Transition of the Dye-Ligand. 400 nm 480 nm Excitation Fig. 3: Absorption spectrum of 1 Fig.4: Emission spectrum of 1 ( and 480 nm) 73 exc = 400 nm P27 New Model Complexes for Nickel and Copper Containing Enzymes Pütz, A.; Jung, M.; Rentschler, E.; Mainz/D Institute of Inorganic Chemistry, University of Mainz, Duesbergweg 10-14, 55099 Mainz, Germany, [email protected] The presented work focuses on the long range magnetic interaction between metal dimers (M = Ni, Cu) and nitroxide-nitronyl (NIT) radicals[1,2], as good model systems for some metalloproteins like laccase or tyrosinase where radicals play an important role. The polydentate ligand NPr-L (N, N’, N’- tetrakis (2‘-(1-n-propylbenz-imidazolylmethyl))-1,4-diethyleneaminoglycolether) is chosen, because it provides two dipodal N-N’-N moieties with a central amino nitrogen and two imidazole nitrogen atoms mimicking histidine rich environments. A flexible aliphatic chain between the nitrogen atoms including two ether groups allows different conformations of the metal dimers. The copper dimers [Cu2(NPr-L)(NO2C6H4COO)2](PF6)2 and [Cu2(NPr-L)(NIT-C6H4 COO)2](PF6)2 display an ‘open’ form with no additional bridging group between the two metal centers[3] resulting in weakly ferromagnetic intermolecular metal-metal coupling. In the nickel dimers [Ni2(NPr-L)(CH3COO)(Cl)](ClO4)2 and [Ni2(NPr-L)(NITC6H4COO)(Cl)](PF6)2 the carboxylate ligands bridge the two metal ions in a ‘closed’ form with an additional µ2-Chloridion[3] leading to pronounced intramolecular ferromagnetic metal-metal exchange interactions. In [Ni2(NPr-L)(CH3C6H4PO3)(H2O)2] (ClO4)2 and [Ni2(NPr-L)(NIT-C6H4PO3)(H2O)2](ClO4)2 the unique feature of a single µ2Phosphonate bridge is observed resulting again weakly antiferromagnetic interactions. In all cases the intramolecular exchange interaction with the NITradicals is weakly antiferromagnetic. [1] M. Jung, E. Rentschler et al, Inorganic Chemistry 2009, 48, 7244-7250 M. Jung, E. Rentschler et al, European Journal of Inorganic Chemistry 2009, 11, 1495-1502 [3] M. Jung, E. Rentschler et al, to be published [2] 74 P28 Spincrossover in Cobalt(II)-Komplexen Reh, S.; Graf, M.; Krüger, H.-J.; Kaiserslautern Sabine Reh, TU Kaiserslautern, Erwin-Schrödinger-Straße, 67663 Kaiserslautern Spincrossover wird hauptsächlich bei oktaedrisch koordinierten Eisen(II)-Komplexen beobachtet. Es gibt jedoch nur wenige Beispiele für Spincrossover-Komplexe mit Cobalt(II)-Ionen, da wegen des geringeren Beitrags des Cobalt(II)-Ions zur Ligandenfeldaufspaltung Liganden mit sehr starkem Ligandenfeld (z.B. bipy, phen, terpy) benötigt werden. Aufgrund der strukturellen Besonderheiten des Tetraazamakrozyklus̕ L-N4tBu2 ist es uns gelungen, auch mit schwächeren Liganden Cobalt(II)-Spincrossover-Komplexe zu synthetisieren. So weisen die beiden Verbindungen N R N N R N R = Me (L-N4Me2) R = tBu (L-N4tBu2) [Co(L-N4tBu2)(NCS)2] und [Co(L-N4tBu2)(NCSe)2] einen graduellen Spinübergang bei T1/2 = 120 bzw. 225 K auf. Der Makrozyklus L-N4Me2 besitzt aufgrund der geringeren sterischen Wechselwirkung zwischen den Methylsubstituenten und den Coliganden ein stärkeres Ligandenfeld als L-N4tBu2. Trotzdem wird bei [Co(L-N4Me2)(NCS)2] nur der reine high-spin Zustand beobachtet. Dagegen weist der Eisen(II)-Komplex [Fe(L-N4Me2)(NCS)2] einen Spinübergang (T1/2 = 233 K) auf. [Fe(L-N4tBu2)(NCS)2] hingegen bleibt im untersuchten Temperaturbereich im high-spin Zustand. Dieser ungewöhnliche Trend bei Cobalt lässt sich auf die tetragonale Verzerrung des Ligandenfeldes zurückführen, die der koordinierte makrozyklische Ligand auf das Cobalt(II)-Ion ausübt. Diese Verzerrung ist beim tert.-butylsubstituierten Liganden L-N4tBu2 deutlich stärker ausgeprägt als beim methylsubstituierten Derivat L-N4Me2. Weiterhin gelang es uns, mit [Co(L-N4tBu2)(dbsq)](B(p-C6H4Cl)4) [1] den ersten Cobalt(II)-Semichinonat-Komplex zu synthetisieren, der Spincrossover-Eigenschaft anstelle der bei solchen Komplexen üblicherweise beobachteten Valenztautomerie aufweist. [1] M. Graf, G. Wolmershäuser, H. Kelm, S. Demeschko, F. Meyer, H.-J. Krüger, Angew. Chem. 2010, 122, 962-965. 75 P29 Molybdän-Oxo-Komplexe mit einem tetradentaten Phosphan Liganden: Synthese und spektroskopische Untersuchung Römer, R.; Tuczek, F.; Kiel/D René Römer, Institut für Anorganische Chemie, Christian-Albrechts-Universität, Max-Eyth-Straße 2, 24118 Kiel Vor kurzem konnte durch die Umsetzung von [Mo(O)I2(PMe3)] mit dem vierzähnigen Phosphan-Liganden prP4 (1,1,4,8,11,11-Hexaphenyl-1,4,8,11-tetraphoshaundecan) der Komplex trans-[Mo(O)I(meso-prP4)]+ (1) erhalten werden[1]. Dieser konnte durch Reaktion in Methanol unter Zusatz von Natriumhydroxid in den Methoxo-Komplex trans-[Mo(O)(OCH3)(meso-prP4)]+ (2) überführt werden. Die anschließende Umsetzung mit HBF4-Etherat führte zu einer Abspaltung des Methoxo-Ligand und lieferte den Komplex trans-[Mo(O)F(meso-prP4)]+ (3). Anhand der vorliegenden Molybdän(IV)-Oxo-Komplexe mit dem vierzähnigen Phosphanliganden prP4 in der äquatorialen Koordinationssphäre kann sehr gut der Einfluss des trans-oxo-ständigen, anionischen Liganden auf die spektroskopischen und geometrischen Eigenschaften beobachtet werden. [1] R. Römer, G. Stephan, C. Habeck, C. Hoberg, G. Peters, C. Näther, F. Tuczek, Eur. J. Inorg. Chem. 2008, 3258-3263. 76 P30 Koordinationschemie der 10-Thiacorrole Sakow, D.; Bröring, M.; Marburg/D Dimitri Sakow, Martin Bröring, Philipps-Universität Marburg, Hans-Meerwein-Straße, 35032 Marburg. Das Gebiet methinsubstituierter 10-Heterocorrole ist bisher nur wenig erforscht. Dabei können aufgrund der Ähnlichkeit zum Porphyrin und zum Corrol ungewöhnliche Eigenschaften der entsprechenden Koordinationsverbindungen erwartet werden. Gegenwärtig sind die Halogenidoeisenkomplexe der Octaethyl-10-thiacorrole (OESCorFeX) synthetisch realisiert und mittels SQUID-Magnetometrie sowie Resonanzspektroskopien (1H-NMR, EPR) untersucht worden. Zudem gelang die Synthese von Eisenkomplexen mit zum Teil ungewöhnlichen Axialliganden: [OESCorFe·DMSO]+[BArF](Ausschnitt) OESCorFeN3 OESCorFeI3 Aktuelle Bemühungen konzentrieren sich auf die vollständige Untersuchung der kürzlich synthetisierten Mangan- und Kobaltkomplexe. Zusätzlich werden versuchte Wege zur Darstellung von meso-Aryl-substituierten 10-Thiacorrolen gezeigt. 77 P31 Spincrossover bei BAI-Eisen(II)-Komplexen Scheja, A.; Pietzonka, C.; Bröring, M.; Marburg/D Anne Scheja, Martin Bröring, Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Straße, 35032 Marburg Spincrossover-Phänomene an oktaedrisch koordinierten Eisen(II)-Komplexen werden seit längerer Zeit mit großem Interesse untersucht. Hierbei ist besonders der Einfluss von Kooperativität auf den Spincrossover noch nicht vollständig geklärt. Die Klasse der Bis(arylimino)isoindoline (BAI) bietet die Möglichkeit, eine Vielzahl von elektronisch und sterisch unterschiedlichen Liganden zu erzeugen, und zudem Verknüpfungen zwischen den Liganden sowie funktionelle Gruppen einzuführen. Auf diese Weise soll gezielt Einfluss auf die elektronische Dynamik von Eisen(II)Spincrossovern genommen werden. SQUID N N 4,5 N 4,0 N N N N 3,5 N N Fe N Cl Cl H H -1 N 2,5 3 χ T / cm mol K 3,0 2,0 N S 1,5 N 1,0 0,5 N 0,0 N 0 50 100 150 200 250 300 N N N Fe N N N N N S N Fe N N N N N N S 350 S T/K Durch Variation der Baugruppen der BAI-Liganden konnten erste Eisen(II)Verbindungen mit unterschiedlichen magnetischen Eigenschaften synthetisiert und mittels SQUID-Magnetometrie und Röntgenstrukturanalyse untersucht werden. Ein mit Dichlormethan cokristallisierter Komplex des unsubstituierten BPI-Liganden zeigt hierbei ein ungewöhnliches Verhalten, dessen Ursache noch nicht geklärt ist. (s. Abb.) Das Poster fasst die Ergebnisse der bisherigen Untersuchungen zusammen. 78 P32 On the formation and structure of 'bare' [M,C,H3O]+ complexes (M = Fe, Co, Ni) in the gas phase Schlangen, M.; Schwarz, H.; Berlin/D Institut für Chemie, TU Berlin, Straße des 17. Juni 135, 10623 Berlin Structural aspects of the 'bare' [M(C,H3,O)]+ complexes (M = Fe, Co, Ni), generated by electrospray ionization of aqueous methanolic solutions of MX2 (X = Br, I), have been probed in the gas phase by a combination of labeling studies and collisioninduced dissociation (CID) experiments. For M = Fe, the only species initially formed corresponds to [Fe(OCH3)]+; for M = Co, in addition to [Co(OCH3)]+ its isomer [Co(CH2OH)]+ is generated, and for M = Ni only the complex [Ni(CH2OH)]+ is accessible. Thus, in the course of ion formation both O−H and C−H bond activation of the ligand CH3OH takes place. Under CID conditions, while partial isomerization of the initially formed complexes precedes the dissociation the spectra nevertheless reveal structure-indicative decomposition pattern. Fig. 1 Partial ESI source spectra of the Fe, Co, and Ni halides NiX2 (X = Br, I) dissolved in a) CH3OH/H2O, b) CD3OH/H2O, and c) CH3OD/D2O. 79 P33 Synthese und Charakterisierung von N-heterocyclischen Gold(I) Komplexen mit Thiolat-Liganden Schuh, E.; Mohr, F.; Wuppertal/D Esther Schuh, Fachbereich C - Anorganische Chemie, Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal N-Heterocyclische Carbene sind für Gold relativ neue Liganden, die ersten Verbindungen wurden vor etwa 15 Jahren berichtet[1]. Seit dem wurden eine Vielzahl von NHC-Goldkomplexen publiziert, wobei diese Komplexe meist als Halogenderivate vom Typ [AuX(NHC)] erhalten wurden. Es gibt nur eine kleine Anzahl von Komplexen, in denen das Halogenid gegen andere Liganden substituiert wurde[2]. Da in unserer Gruppe schon lange Interesse an lumineszierenden und biologisch aktiven Phosphangold(I) Komplexen mit Thiolatliganden besteht, haben wir begonnen, analoge NHC Komplexe zu untersuchen. Eine Serie von NHC-Au-Halogen Komplexen wurde über Carben Transfer Reaktionen aus den entsprechenden Silberkomplexen dargestellt. In der sich anschließenden Reaktion mit einem Thiol in Gegenwart von Base konnte das Halogenid gegen das entsprechende Thiolat ausgetauscht werden (Schema 1). Die neuen Gold-Thiolat Komplexe [Au(SR)(NHC)] konnten mit spektroskopischen und strukturellen Methoden vollständig charakterisiert werden. Studien zur biologischen Aktivität der Verbindungen werden zurzeit durchgeführt. Schema 1 [1] a) I.J.B. Lin, C.S. Vasam, Can. J. Chem. 2005, 83, 812-825. b) N.M. Scott, S.P. Nolan, Eur. J. Inorg. Chem. 2005, 1815-1828. c) R.H. Crabtree, J. Organomet. Chem. 2005, 690, 5451-5457. [2] a) H.W.J. Wang, C.Y.L. Chen, I.J.B. Lin, Organometallics 1999, 18, 1216-1223. b) M.V. Baker, P.J. Barnard, S.J. Berners-Price, S.K. Brayshaw, J.L. Hickey, B.W. Skelton, A.H. White, J Organomet. Chem, 2005, 690, 5625-5635. c) P. de Fremont, E.D. Stevens, M.D. Eelman, D.E. Fogg, S.P. Nolan, Organometallics, 2006, 25, 5824-5828. 80 P34 Synthetic Challenges Towards New Phthalocyanine Materials Elisabeth Seikel, Wael Darwish, Michaela Grau, Ralf Käsmarker, Marat Khusniyarov, Jörg Sundermeyer; Marburg/D Elisabeth Seikel, Fachbereich Chemie, Philipps Universität Marburg, HansMeerwein-Straße, D-35037 Marburg E-Mail: [email protected] Phthalocyanines (Pcs) have been subject of ongoing research because of their potential applications such as in optical limiting devices or photoconducting materials. Their properties depend essentially on the intermolecular arrangement of the macrocycles. Different substituents at the axial and equatorial positions affect the lattice packing of the molecules. We are interested in the development of synthetic routes to novel substituted phthalocyanines, preparation of analytically pure crystalline substances and their characterization. The effects of different substituents are being investigated. O Ar N O ArNCO -CO2 Ti Ar Ar S S ArNCO [2+2 Add.] 2 H2S, air Ti DipNCO -CO2 N N Ti Ti 2 H2S, inert gas Ar = Ph, pTol, Mes, pClPh NH2 S NH2 Ti i Pr iPr N NH HN Ti Scheme 1: Left: Synthesis of axially substituted phthalocyanines. The ellipse represents the macrocyclic ligand. Right: Crystal structures of imido and ureato titanium phthalocyanines. Ti Two routes have proved to be successful, the first being the variation or exchange of an existing axial ligand (Scheme1). The second route involves the insertion of metal precursors bearing the desired axial ligand into the macrocycle cavity. We have investigated the photophysical properties (UV/vis, photoluminescence) of the new compounds as well as their incorporation into mesoporous silica materials. W. Darwish, S. Schlecht, A. Schaper, M. Fröba, K. Harms, W. Massa, J. Sundermeyer, Z. Anorg. Allg. Chem. 2009, 635, 1215-1224 W. Darwish, K. Harms, J. Sundermeyer, Acta Cryst., Sect. E 2005, E61(7), m1280-m1282. E. Seikel, Diplomarbeit, Philipps Universität Marburg, 2008. 81 P35 Supramolekulare Metalloxidkomplexe als Vorstufen für nanostrukturierte Komposite Carsten Streb, Erlangen/D Carsten Streb, Institut für Anorganische Chemie II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstr. 1, 91058 Erlangen Die kontrollierte, auf molekularen Vorstufen basierte bottom-up-Synthese nanostrukturierter Materialien ist eine der größten Herausforderungen der modernen Koordinationschemie.[1] Dieser Beitrag präsentiert eine neue Reaktivtemplatroute, bei der supramolekulare Metalloxidcluster kontrolliert in Nanokomposite überführt werden. In den letzten Jahren konnten wir durch Reaktion von Silber(I)-Kationen mit einer Reihe von Polyoxometalaten supramolekulare Architekturen mit 1-, 2- und 3dimensionalen Architekturen erzeugen.[2] Im hier präsentierten Beispiel führt die Reaktion von Silber(I) mit Dekavanadatclustern, [H3V10O28]3-, zur Bildung von 1DZickzack-Ketten und 2D-, planaren Netzwerken ( siehe Abb. 1).[3] Abb. 1: Bildungsmechanismus der Silber-Vanadiumoxid-Nanodrähte Die kontrollierte Zersetzung dieser kristallinen Materialien unter reduzierenden Bedingungen resultiert in der Bildung eng verwobener Vanadiumoxid-Nanodrähte mit einem Durchmesser von 20-30 nm und Längen im Bereich von mehreren Mikrometern. Eingebettet in diese Drähte sind metallische Silbernanopartikel von 525 nm, die in situ gebildet werden. Das halbleitende Komposit kann im Elektronenstrahl direkt zu metallischen V/Ag-Nanodrähten reduziert werden. Das Reaktivtemplat-Komzept wurde anschließend erweitert und erlaubt nun den Einbau verschiedener Metallnanopartikel wie z.B. Co, Ni und Cu in die VOx-Drähte. [1] [2] [3] D.-L. Long, E. M. Burkholder, L. Cronin, Chem. Soc. Rev. 2007, 36, 105. C. Streb, C. Ritchie, P. Kögerler, D.L. Long, L. Cronin, Angew. Chem. Int. Ed. 2007, 46, 7579. C. Streb, R. Tsunashima, D. A. MacLaren, T. McGlone, T. Akutagawa, T. Nakamura, A. Scandurra, B. Pignataro, N. Gadegaard, L. Cronin, Angew. Chem. Int. Ed. 2009, 48, 6490. 82 P36 New Amino Acid Ligated Yttrium Hydroxy Clusters Thielemann, D. T.; Fernández, I.; Roesky, P. W.; Karlsruhe/D Peter W. Roesky, Institut für Anorganische Chemie, Karlsruher Institut für Technologie, Engesserstrasse 15, 76131 Karlsruhe Targeting a partial functionalization of the previously published lanthanide clusters [Ln5(OH)5(Ph2acac)10] (Ln = Y,[1] Eu,[2] Dy,[3] Er; Ph2acac = dibenzoylmethanide), we introduced biologically active ligands in terms of α-amino acids into this chemistry. For this purpose, the amino acids D-phenyl glycine (D-PhGlyH), L-proline (L-ProH), L-valine (L-ValH) and L-tryphophan (L-TrpH) were reacted with yttrium trichloride hexahydrate [YCl3·(H2O)6] and dibenzoylmethane (Ph2acacH) under basic conditions to give the mixed pentanuclear yttrium hydroxy clusters [Y5(OH)5(α-AA)4(Ph2acac)6] (α-AA = D-PhGly (1); L-Pro (2); L-Val (3); L-Trp (4)).[4] The solid state structures of compounds 1-4 revealed the metal atoms to build up a square pyramidal scaffold which is quintuply capped by µ3-O and µ4-O moieties. All six Ph2acac ligands act as terminal chelates, whereas the amino acid ligands serve as bridging chelates (Fig. 1). Fig. 1 Compounds 3 (left) and 4 (right). Hydrogen atoms are omitted for clarity. Besides standard-spectroscopic characterization, the hydrodynamic radius of compound 3 was determined via pulse gradient spin-echo (PGSE) diffusion experiments. In addition, a novel 1H,89Y-gHMQC spectrum of compound 3 was recorded proving its permanent existence in solution.[4] [1] P. W. Roesky, G. Canseco-Melchor, A. Zulys, Chem. Commun. 2004, 738-739; [2] R.-G. Xiong, J.L. Zuo, Z. Yu, X.-Z You, W. Chen, Inorg. Chem Comm. 1999, 2, 490-494; [3] M. T. Gamer, Y. Lan, P. W. Roesky, A. K. Powell, R. Clérac, Inorg. Chem. 2008, 47, 6581-6583; [4] D. T. Thielemann, I. Fernández, P. W. Roesky, Dalton Trans. 2010, submitted. 83 P37 Synthese und Charakterisierung des zweikernigen Ni(0)-Komplexes [Ni2(η2-C4H6)2(O-BPY)] unter Verwendung von Ni(COD)2 und 1,2-Bis(2,2´-bipyridyl-6-yl)ethan (O-BPY) Valentin, L. ; Leibold, M.; Walter, O.; Schindler, S.; Gießen/D Lars Valentin, Siegfried Schindler, Justus-Liebig-Universität Gießen, Heinrich-BuffRing 58, 35392 Gießen Die Verbindung 1,2-Bis(2,2´-bipyridyl-6-yl)ethan (O-BPY, Abb. 1.1) ist ein interessanter Ligand für die Darstellung unterschiedlicher Metallkomplexe. O-BPY wird dabei ausgehend von 2,2´-Bipyridin synthetisiert. [1] Bei der Umsetzung von O-BPY mit [Ni(COD)2] wurde beobachtet, dass hier nicht der erwartete zweikernige [Ni2(COD)2(O-BPY)] gebildet wurde, sondern eine Spaltung von COD zu zwei Molekülen 1,3-Butadien erfolgte. Der Produktkomplex konnte kristallographisch charakterisiert werden und gezeigt werden, dass es sich dabei um [Ni2(η2-C4H6)2(O-BPY)] (Abb. 1.2) handelt. Im Gegensatz dazu bildeten Nickel(II)Salze mit dem Liganden O-BPY nur einkernige Komplexe. Untersuchungen sind im Gange, den Reaktionsmechanismus dieser interessanten Reaktion zu analysieren. Hierzu wird unter anderem versucht, die Länge der Alkylbrücke zwischen den beiden Bipyridin-Einheiten von -(CH2)- bis -(CH2)4- zu modifizieren. Erste Ergebnisse dieser Arbeiten werden präsentiert. Ni N N N N N N Ni N N Abb.1.1: 1,2-Bis(2,2´-bipyridyl-6yl) ethan (O-BPY) Abb.1.2: Struktur von [Ni2(η2-C4H6)2(O-BPY)] [1] Garber, T.; Van Wallendael, S.; Rillema, D. P.; Kirk, M., Inorg. Chem. 1990, 29, (15), 2863-8 84 P38 Coordination Chemistry of Multidentate Imidazolin-2-imine Based Ligands J. Volbeda, S. Filimon, T. Glöge, C. G. Hrib, C. Daniliuc, M. Tamm M. Tamm, Institut für Anorganische und Analytische Chemie, Technische Universität CaroloWilhelmina zu Braunschweig, Hagenring 30, 38106, Braunschweig, Germany Superbasic imines represent an interesting class of ligands, being more electron-donating than normal imines yet still being neutral. A well known class of superbasic imines are those based on guanidine moieties. Imidazoline-2-imines which are closely related to guanidines were first developed by Kuhn et al. [1] and can effectively stabilize a negative charge on the imine nitrogen leading to an enhanced electron donating capacity [2]. Two ethylene bridged bidentate [3] (BLMe and BLiPr) ligands and a pyridine bridged tridentate [4] (TLtBu) ligand based on imidazoline-2-imines have been developed in our group. Here we report on some of their applications in coordination chemistry. R R N N N BLMe: R= Me BL iPr N N N N R N N N N N N R TLtBu i : R= Pr Ruthenium complexes bearing the bidentate ligands (e.g. [(η6-C6H6)RuBLMe]Cl2) have proven to be effective catalysts in transfer hydrogenation of acetophenone with 2-propanol as hydrogen source [5]. We are currently developing imidazoline-2-imine based systems for the asymmetric hydrogenation of ketones. As imidazole-2-imines are good electron donors they should be able to stabilize electron deficient metal centers. In this context we decided to implement the tridentate ligand (TLtBu) in stabilizing metals in high oxidation states. [1] N. Kuhn, R. Fawzi, M. Steimann, J.Wiethoff, D. Bläser and R. Boese, Z. Naturforsch., B: Chem. Sci. 1995, 50, 1779-1784 [2] M. Tamm, D. Petrovic, S. Randoll, S. Beer, T. Bannenberg, P. G. Jones, J. Grunenberg, Org. Biomol. Chem. 2007, 5, 523-530. [3] D. Petrovic, T. Glöge, T. Bannenberg, C.G. Hrib, S. Randoll, P. J. Jones, M. Tamm, Eur. J. Inorg. Chem. 2007, 3472-3475. [4] D. Petrovic, T. Bannenberg, S. Randoll, P. G. Jones, M. Tamm, Dalton Trans. 2007, 2812-2822. [5] T. Glöge, D. Petrovic, C. G. Hrib, P. G. Jones, M. Tamm, Eur. J. Inorg. Chem. 2009, 4538-4546. 85 P39 Übergangsmetallkomplexe mit neuen asymmetrischen NHC/Pyrazol-Liganden Wimberg, J.; Dechert, S.; Meyer, F.; Göttingen/D Prof. Dr. Franc Meyer, Institut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstr. 4, 37077 Göttingen Die Vorteile heterobimetallischer Katalyse sind aus verschiedenen Enzymen bekannt.[1] Eine gezielte Synthese und Nutzung von heterobimetallischen Komplexen für katalytische metallorganische Transformationen ist daher von besonderem Interesse.[2] Zur Darstellung solcher Komplexe wurde die neue Ligandenklasse 1 entwickelt, die verschiedener zwei unterschiedliche aufweist.[3] Metallionen Kompartimente Hierbei für werden die Koordination einerseits ein N- heterozyklisches Carben (NHC) für ein metallorganisches Kompartiment und andererseits ein Pyrazol für ein N-Donor Kompartiment verwendet. Als Brücke ist das Pyridazin durch zwei Stickstoffatome in der Lage, die Metallionen in räumlicher Nähe zu binden und somit kooperative Effekte in heterobimetallischen Komplexen (2) zu ermöglichen. R2 R2 N 1 R N N N N N N 3 1 R R N N N N M1 M Ln 1 2 N R3 Ln 2 R1 = Pyridyl, Methyl R2 = Phenyl, Methyl, H R3 = Mesityl, t-Butyl, 2,6-Diisopropyl Wir berichten hier über die Herstellung der Liganden und erste Ergebnisse zur Koodinationschemie. [1] J.C. Mealli, T.B. Rauchfuss, Angew. Chem., Int. Ed. 2007, 46, 8942-8944. [2] S. Handa, V. Gnanadesikan, S. Matsunaga, M. Shibasaki, J. Am. Chem. Soc. 2007, 129, 49004901. [3] J. Wimberg, S. Dechert, F. Meyer, in Vorbereitung. 86 Teilnehmerliste Carlos Enrique Abad Andrade Georg-August Universität Göttingen Ulf-Peter Apfel Friedrich-Schiller-Universität Jena Martin Bernard Universität Paderborn Thomas Bernert Universität Frankfurt Olga Bienemann Technische Universität Dortmund Florian Blasberg Goethe-Universität Frankfurt am Main Aaron Breivogel Johannes Gutenberg-Universität Mainz Prof. Dr. Martin Bröring Philipps-Universität Marburg Boris Burger Georg-August-Universität Göttingen Burkhard Butschke Technische Universität Berlin Dr. Luca Carrella Johannes Gutenberg-Universität Mainz Victoria Colquhoun Technische Universität Dortmund Prof. Dr. Peter Comba Universität Heidelberg Hari Sankar Das Universität Stuttgart Alexander Döring Georg-August Universität Göttingen Nina Dovalil Universität Heidelberg Maria Eckhardt Universität Regensburg Jan Falkenhagen Humboldt-Universität zu Berlin Dr. Christoph Förster Johannes Gutenberg-Universität Mainz Felix Friedel Friedrich-Alexander-Universität Erlangen Nürnberg Felix Fuge RWTH Aachen Dr. Jessica Gätjens RWTH Aachen Thorsten Gehrmann Universität Heidelberg Simon-Andreas Gentschow Technische Universität Berlin Prof. Dr. Thorsten Glaser Universität Bielefeld Prof. Dr. Andreas Grohmann Technische Universität Berlin Birte Haberlag Technische Universität Braunschweig Sonja Hammer Universität Frankfurt Dr. Christina Hauser Friedrich-Alexander-Universität Erlangen-Nürnberg Prof. Dr. Katja Heinze Johannes Gutenberg-Universität Mainz Prof. Dr. Gerald Henkel Universität Paderborn Dr. Sonja Herres-Pawlis Technische Universität Dortmund 87 Veronika Hoeke Universität Bielefeld Alexander Hoffmann Technische Universität Dortmund Prof. Dr. Hans-Jürgen Holdt Universität Potsdam David Hornig Friedrich-Schiller-Universität Jena Rajkumar Jana Universität Stuttgart Jabadurai Jayapaul RWTH Aachen Thomas Jozak Technische Universität Kaiserslautern Prof. Dr. Wolfgang Kaim Universität Stuttgart Natascha Kempf Justus-Liebig-Universität Gießen Dr. Marat Khusniyarov Friedrich-Alexander-Universität Erlangen-Nürnberg Sandra Kisslinger Justus-Liebig-Universität Gießen Prof. Dr. Günther Knör Johannes Kepler Universität Linz Matthias Kruck Universität Heidelberg Prof. Dr. Hans-Jörg Krüger Technische Universität Kaiserslautern Subrata Kundu Humboldt-Universität zu Berlin Dr. Peter Kunz Heinrich-Heine-Universität Düsseldorf Prof. Dr. Doris Kunz Eberhard Karls Universität Tübingen Oanh Lam Friedrich-Alexander-Universität Erlangen-Nürnberg Sebastian Lange Universität Paderborn Dr. Michael Leibold Universität Kassel Dominik Lieb Friedrich-Alexander-Universität Erlangen-Nürnberg Prof. Dr. Christian Limberg Humboldt-Universität zu Berlin Christine Männel-Croisé Universität Zürich Sebastian Marks Karlsruher Institut für Technologie (KIT) Martin Maurer Universität Heidelberg Yakiv Melomedov Johannes Gutenberg-Universität Mainz Prof. Dr. Karsten Meyer Friedrich-Alexander-Universität Erlangen-Nürnberg Prof. Dr. Franc Meyer Georg August-Universität Göttingen Prof. Dr. Fabian Mohr Bergische Universität Wuppertal Dr. Andriy Mokhir Universität Heidelberg Anja Molter Bergische Universität Wuppertal Dr. Uwe Monkowius Johannes Kepler Universität Linz Philipp Mücke Universität Regensburg Dr. Chandan Mukherjee Universität Bielefeld 88 Kevin Muller Technische Universität Kaiserslautern Johanna Niesel Ruhr-Universität Bochum Dr. Alexander Petrov Technische Universität Braunschweig Florian Pevny Universität Regensburg T. David Pilz Friedrich-Alexander-Universität Erlangen-Nürnberg Prof. Dr. Winfried Plass Friedrich-Schiller-Universität Jena Anna-Maria Pütz Johannes Gutenberg-Universität Mainz Prof. Dr. Sven Rau Friedrich-Alexander-Universität Erlangen-Nürnberg Dr. Kallol Ray Humboldt Universität zu Berlin Prof. Dr. Eva Rentschler Johannes Gutenberg-Universität Mainz Sabine Reh Technische Universität Kaiserslautern Malte Rolff Christian-Albrechts-Universität zu Kiel René Römer Christian-Albrechts-Universität zu Kiel Dimitri Sakow Philipps-Universität Marburg Dr. Biprajit Sarkar Universität Stuttgart Dr. Ulrich Schatzschneider Ruhr-Universität Bochum Anne Scheja Philipps-Universität Marburg Prof. Dr. Siegfried Schindler Justus-Liebig-Universität Gießen Andrea Schindler Universität Regensburg Dr. Maria Schlangen Technische Universität Berlin Markus Schmitz Technische Universität Kaiserslautern Johannes Schnödt Universität Stuttgart Wolf Schorn Philipps-Universität Marburg Dirk Schuch Friedrich-Schiller-Universität Jena Esther Schuh Bergische Universität Wuppertal Thomas Schwarze Universität Potsdam David Schweinfurth Universität Stuttgart Elisabeth Seikel Philipps-Universität Marburg Dr. Monika Sieger Universität Stuttgart Philipp Stock Technische Universität Berlin Dr. Carsten Streb Friedrich-Alexander-Universität Erlangen-Nürnberg Prof. Dr. Matthias Tamm Technische Universität Braunschweig Prof. Dr. Werner Thiel Technische Universität Kaiserslautern Dominique Thielemann Karlsruher Institut für Technologie (KIT) 89 Christoph Topf Johannes Kepler Universität Linz Prof. Dr. Felix Tuczek Christian-Albrechts-Universität zu Kiel Dr. Sabrina Turba Wiley-VCH Verlag Christian Unkelbach Technische Universität Dortmund Lars Valentin Justus-Liebig-Universität Gießen Carola Vogel Friedrich-Alexander-Universität Erlangen-Nürnberg Jeroen Volbeda Technische Universität Braunschweig Prof. Dr. Matthias Wagner Goethe-Universität Frankfurt am Main Gunnar Werncke Humboldt-Universität zu Berlin Anne Westphal Christian-Albrechts-Universität zu Kiel Jan Wimberg Georg-August-Universität Göttingen Prof. Dr. Rainer Winter Universität Regensburg Dr. Christian Würtele Justus-Liebig-Universität Gießen Dr. Felix Zelder Universität Zürich 90