Strukturmotive des CuAl2 Typs in Elementen und Verbindungen
Werbung

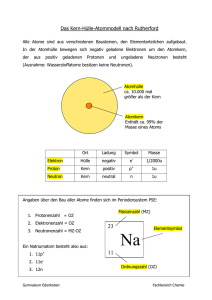
Tätigkeitsbericht 2003 Schwarz, Ulrich et al. | Strukturmotive des CuAl2 Typs in Elementen und ... 1 Strukturmotive des CuAl2 Typs in Elementen und Verbindungen Schwarz, Ulrich Max-Planck-Institut für chemische Physik fester Stoffe Forschungsbereich - Chemische Metallkunde Forschungsgebiet: Festkörperforschung/Materialwissenschaften Korrespondierender Autor: Schwarz, Ulrich E-Mail: [email protected] Zusammenfassung An einer Reihe von intermetallischen Phasen der Zusammensetzung AB 2, die im Strukturtyp von CuAl2 kristallisieren, wird die Relevanz des tetragonal-antiprismatischen Strukturmusters für die chemische Bindung in Vertretern unterschiedlicher Metalle A und B experimentell und theoretisch untersucht. Die quantenchemischen Berechnungen zeigen eindeutig, dass in Abhängigkeit vom Hauptgruppenelement B verschiedene Bindungsmuster ausgebildet werden. Dieses Ergebnis steht in Übereinstimmung mit Bestimmungen der Stärke ausgewählter Wechselwirkungen durch schwingungsspektroskopische Messungen, die eine Gruppierung der Verbindungen in Abhängigkeit von der chemischen Zusammensetzung erkennen läßt. Eine Variante des Strukturmusters wird auch vom Element Antimon bei hohen Drücken ausgebildet. Dabei verändert sich allerdings teilweise die Besetzung einiger Positionen der Atomsorte A. Dennoch belegt die Ähnlichkeit der atomaren Anordnung von Antimon zu einem AB 2 Strukturtyp, dass gleichartige Atome unterschiedliche Positionen von Strukturtypen besetzen können. Abstract In a series of intermetallic phases which crystallize in the CuAl2 type, the relevance of the tetragonal antiprismatic structure pattern is investigated. Chemical bonding in these compounds of composition AB 2 is characterized both experimentally and theoretically for different metals A and B. The quantum chemical calculations clearly show that dependent from the main group metal B different bonding patterns are formed. This result is in agreement with determinations of the magnitude of selected interactions by vibrational spectroscopic methods which evidence the formation of certain groups of compounds as a function of chemical composition. A variety of the structural pattern is also formed by elemental antimony at high pressures. Thereby, the occupation of some positions of the atom type A changes. However, the similarity of the atomic arrangement of antimony to that of an AB 2 structure type substantiates that one type of atom can be located on different positions of a structure type Einer der Schwerpunkte der Forschung an intermetallischen Verbindungen liegt in der Beschreibung der Bindungsverhältnisse mit experimentellen und theoretischen Methoden. Dazu wird oft eine große Anzahl intermetallischer Phasen und Verbindungen nach unterschiedlichen Merkmalen eingeteilt, um Stabilitätsfelder bestimmter Zusammensetzungen oder Kristallstrukturen einzugrenzen. Auch bei Untersuchungen der chemischen Bindung werden Verbindungen oft nach typischen atomaren Anordnungen untergliedert, da nach ähnlichen Strukturmotiven gesucht wird. Eine häufig auftretende Umgebung von Atomen in intermetallischen Verbindungen ist die tetragonal-antiprismatische, die am © 2003 Max-Planck-Gesellschaft 2 Schwarz, Ulrich et al. | Strukturmotive des CuAl2 Typs in Elementen und ... Tätigkeitsbericht 2003 einfachsten als ein verdrehter Würfel beschrieben werden kann und beispielsweise im CuAl2-Strukturtyp vorkommt. Zur Analyse der Bindungssituation in Verbindungen dieses Strukturtyps werden die Untersuchungen zunächst auf die AB2-Verbindungen CuAl2, MnSn2, FeSn2, CoSn2, TiSb2, und VSb2 konzentriert. Die Hauptgruppenelemente (B = Al, Sn oder Sb) bilden Antiprismen, die durch Übergangsmetallatome (A = Ti, V, Mn, Fe, Co oder Cu) zentriert sind. Kristallstrukturen vom CuAl2-Typ werden in der Literatur mit mindestens drei unterschiedlichen Modellen beschrieben. Bei Betonung der Wechselwirkungen A-B (entsprechend dem Abstand d5(CuAl) in Abb. 1a) heben sich Säulen gestapelter tetragonaler Antiprismen aus Atomen B besonders heraus. Diese Stapel sind über gemeinsame Kanten zu einem dreidimensionalen Netzwerk aneinander gefügt. Hervorhebung der kürzesten Abstände zwischen Atomen B (d1 und d2 in Abb. 1b) führt zu einer Beschreibung, bei der sich Graphit-artige Netze durchdringen. Durch Verbindung aller benachbarten Atome B (Abstände d1 - d4 in Abb. 1c) werden Tetraedersterne erzeugt, die ebenfalls über gemeinsame Kanten dreidimensional verbunden sind. In beiden Fällen befinden sich die Atome A in den Hohlräumen eines dreidimensionalen Verbunds. Abb. 1: (a-c) Drei unterschiedliche Modelle der CuAl2-Struktur. (d) Kraftkonstanten der Wechselwirkungen d1, d2 und d5 in Verbindungen der CuAl2-Familie, die aus orientierten und polarisierten Raman-Messungen an Einkristallen bestimmt werden. Zur Ermittlung einer Darstellung, die geeignet ist, die chemische Bindung in den Strukturen der CuAl2-Familie optimal zu veranschaulichen, werden zunächst quantenchemische Berechnungen durchgeführt. Zur Untersuchung der räumlichen Anordnung kovalenter Wechselwirkungen in den intermetallischen Verbindungen wird die Elektronen-Lokalisierungsfunktion (ELF) berechnet und analysiert. Die Ergebnisse zeigen, dass in Abhängigkeit vom Hauptgruppenmetall B drei verschiedene Bindungsmuster ausgebildet werden. In den Diantimoniden (Atome B = Sb) werden kovalente Wechselwirkungen entlang der Verbindungslinien d1 beobachtet, so dass sich hantelförmige Sb2-Baugruppen herausbilden (Abb. 2a). Zusätzliche Bindungen werden zwischen den A Atomen entlang der c Richtung der Elementarzelle gefunden (d(A-A) in Abb. 2a). Zwischen Antimon und dem © 2003 Max-Planck-Gesellschaft Tätigkeitsbericht 2003 Schwarz, Ulrich et al. | Strukturmotive des CuAl2 Typs in Elementen und ... 3 Übergangsmetall treten Dreizentren-Bindungen auf. In Distanniden (Atome B = Sn) treten zusätzliche Bindungen B-B entlang der Verbindungslinie d2 auf (Abb. 2b). Die Wechselwirkung A-A ist im Vergleich zu den Verbindungen des Antimons wesentlich schwächer ausgeprägt. Die Aluminiumverbindung CuAl2 zeigt ähnliche kovalente Wechselwirkungen. Im Gegensatz zu den AB2 Verbindungen des Zinns und des Antimons werden hier aber keine Wechselwirkungen zwischen den A Atomen beobachtet (Abb. 2c). Ein Vergleich der berechneten ELF-Darstellungen mit Modellen der Kristallstruktur von CuAl2 zeigt, dass die Abbildung mit durchdringenden Graphitnetzen bei Einbeziehung der Wechselwirkung d5 die Bindungsverhältnisse zutreffend widerspiegelt. Besonders die kovalenten Bindungen d1 und d2 werden durch die Berechnungen bestätigt. Abb. 2: Isoflächen der Elektronen-Lokalisierung-Funktion (ELF) für drei Repräsentanten des CuAl2-Strukturtyps: (a) TiSb2, (b) MnSn2 und (c) CuAl2. Die Farben der eingezeichneten Wechselwirkungen entlang d1, d2 oder d5 entsprechen denjenigen der Darstellungen in Abbildung 1. Isoflächen der ELF visualisieren die entsprechenden Bindungen. (Verwendete Isowerte: (a) TiSb2: ν(d1) = 0,57, ν(d5) = 0,57, ν(d[A-A]) = 0,32; (b) MnSn2: ν(d1) = 0,45, ν(d2) = 0,39, ν(d5) = 0,4, (d[A-A]) = 0,301; (c) CuAl2: ν(d1) = 0,57, ν(d2) = 0,54, ν(d5) = 0,51). Als experimentelle Methode zur Untersuchung der chemischen Bindung wird die optische Schwingungsspektroskopie gewählt. Dazu werden Raman-Messungen mit polarisierter Laserstrahlung an orientierten Einkristallen durchgeführt. Gemäß gruppentheoretischer Analyse zeigen die Verbindungen fünf Ramanaktive optische Moden (jeweils eine A1g, B1g und B2g sowie zwei Eg). Als erste Näherung kann angenommen werden, dass die Moden der Symmetrie A1g die Valenzschwingung der B-B Hanteln repräsentiert. Damit ist die entsprechende Frequenz ein Maß für die Stärke der Wechselwirkung zwischen zwei B Atomen, die den Abstand d1 aufweisen. Aus den Schwingungsfrequenzen werden die Kraftkonstanten der Wechselwirkung bestimmt. Durch diesen Schritt werden Beiträge eliminiert, die nur auf die unterschiedliche Masse der Atome zurückzuführen sind. Zum Vergleich der experimentellen Ergebnisse mit den quantenchemischen Resultaten werden die A1g Schwingungsfrequenzen mit Werten der Berechnungen korreliert. Dazu wird der Kehrwert gebildet, der aus dem Maximum der ELF ηmax in den einzelnen Bassins berechnet wird gemäß 1/χmax = (ηmax - 1) -1/ 2. Die Korrelation von 1/χ max und den Kraftkonstanten ist linear für die Verbindungen des Zinns und des Antimons, während der Wert von CuAl2 selbst deutlich abweicht. Auf diese Weise werden zum ersten Mal berechnete ELF-Werte auf experimentell beobachtete physikalischen Daten bezogen. Unsere bisherigen theoretischen Untersuchungen weisen darauf hin, dass ein Zusammenhang zwischen dem Wert 1/χmax und der Schwingungsfrequenz bzw. der Kraftkonstante für zweiatomige Moleküle von Elementen derselben Gruppe des Periodensystems besteht. © 2003 Max-Planck-Gesellschaft 4 Schwarz, Ulrich et al. | Strukturmotive des CuAl2 Typs in Elementen und ... Tätigkeitsbericht 2003 Das Strukturmotiv der tetragonalen Antiprismen tritt nicht nur in intermetallischen Phasen vom CuAl2-Typ auf, sondern bildet sich auch in einer Hochdruckphase des Elements Antimon. Während bei Normalbedingungen von Antimon eine leicht verzerrte, annähernd kubisch-primitive Atomanordnung realisiert wird, bildet sich bei Drücken oberhalb von 8,2 GPa die Modifikation Sb-II mit komplizierter Kristallstruktur aus. Diese Hochdruckphase ist seit geraumer Zeit Gegenstand experimenteller und theoretischer Untersuchungen. Zur Strukturlösung werden hochaufgelöste Röntgenbeugungsmessungen an pulverförmiger Proben mit Synchrotronstrahlung durchgeführt. Die Diagramme können nicht mit früher vorgeschlagenen Atomanordnungen in Einklang gebracht werden. Die Lagen der Reflexe weisen vielmehr darauf hin, dass eine tetragonale Kompositstruktur vorliegt, bei der die Atome zwei verschiedene Substrukturen ausbilden, die gemeinsame Gitterparameter a aufweisen, aber unterschiedliche Identitätsperioden in Richtung c zeigen: a = 805,53(4) pm, c1 = 389,91(2) pm und c2 = 297,33(4) pm. Neben den Hauptreflexen werden Satellitenreflexe mit geringer Intensität beobachtet, die auf zusätzliche, so genannte inkommensurable Modulationen der Kristallstruktur hinweisen. Das bedeutet, dass die Atomlagen der Kristallstruktur von den mittleren Positionen abweichen. Die Ergebnisse belegen diese Auslenkungen der Atompositionen, die nur mithilfe vierdimensionaler Symmetriegruppen in Abhängigkeit von einer beiden Untergittern gemeinsamen Koordinate zutreffend beschrieben werden können. Aus den Beugungsintensitäten werden die Positionen der Atome in beiden Teilstrukturen bestimmt, die mit drei unterschiedlichen vierdimensionalen Raumgruppen zu vereinbaren sind. Modulationswellen der Gastatome sind aber mit der Symmetrie von zwei dieser Gruppen nicht zu vereinbaren. Daher wird die Verfeinerung in der verbleibenden dritten Raumgruppe LI422-111:LI422-111 durchgeführt. Der verfeinerte Ortsparameter der Wirtsstruktur des Antimons zeigt eine überraschende Ähnlichkeit zu dem der B Atome im CuAl2-Typ. Das Vorliegen gleicher Teilstrukturen belegt die Relevanz intermetallischer Kristallstrukturen auch für Elementmodifikationen mit hoher Dichte. Allerdings besetzen beim CuAl2-Typ die Kupferatome alle Zentren tetragonaler Antiprismen, während die Gastatome des Antimons ein komplexes Besetzungsmuster der Lücken realisieren. Daher entspricht die Zusammensetzung auch nicht mehr exakt derjenigen des AB2-Typs, sondern der Summenformel (Sb1) 1-x (Sb2)2 mit χ ≈ 0,33. Die wellenförmige Modulation der Atomlagen ermöglicht eine kontinuierliche Anpassung der interatomaren Abstände als Funktion der gemeinsamen vierten Koordinate t = c1/c2 x z1 = c2/c1 x z2. Hier sind c1 und c2 die entsprechenden Gitterparameter und z1 sowie z2 die mittleren Lageparameter in der jeweiligen Elementarzelle. Bezüglich der Wechselwirkungen zwischen den beiden Teilgittern verursacht die modulierte Auslenkung der Atome eine zusätzliche Verkürzung im Bereich kleiner Abstände und eine Verlängerung bei großen Abständen (Abb. 3). Die Atome innerhalb der Antiprismen haben zwei Nachbarn innerhalb der Säule und - wegen des komplexen Besetzungsmusters - eine variierende Anzahl von Kontakten zu Wirtsgitteratomen. Zwei Extreme begrenzen den Bereich der Koordination. Die kleinste Zahl von insgesamt sechs Nachbarn haben Gastatome in den Zentren von Quadraten. Die größte Anzahl von zehn wird für Atome in den Zentren der tetragonaler Antiprismen beobachtet (t = 0). Die Wirtsatome sind von sechs anderen Gerüstatomen und zwei Gastatomen koordiniert. © 2003 Max-Planck-Gesellschaft Tätigkeitsbericht 2003 Schwarz, Ulrich et al. | Strukturmotive des CuAl2 Typs in Elementen und ... 5 Abb. 3: Interatomare Abstände von Sb-II beim Druck P = 12 GPa als Funktion der gemeinsamen vierten Koordinate t (siehe Text). Gestrichelte Linien repräsentieren Abstände, die für eine nicht-modulierte Kristallstruktur berechnet werden, durchgezogene Linien zeigen diejenigen der modulierten Anordnung. Die Farbcodierung der Abstände entspricht derjenigen der Abbildungen 1 und 2. Anhand der beschriebenen Lösung der Kristallstruktur kann ein Modell für quantenmechanische Berechnungen entwickelt werden, um die Stabilität dieser Atomanordnung vergleichend zu untersuchen. Folgende experimentell nachgewiesene Kristallstrukturen oder Varianten davon werden für Bestimmungen der Gesamtenergie verwendet: Die Normaldruckphase (A7), eine kubisch innenzentrierte Modifikation (BCC), eine in einer früheren Untersuchung vorgeschlagene tetragonal primitive Anordnung (TP) und schließlich ein tetragonaler Approximant des inkommensurablen Komposits (TA). Die Ergebnisse der Berechnungen der Gesamtenergie sind in Abbildung 4 zusammengefasst. Abb. 4: (a) Energiedifferenzen als Funktion des Volumens pro Atom für die untersuchten Kristallstrukturmodelle von Sb-Modifikationen. Die durchgezogenen Linien entsprechen kubischen Polynomen, die an die berechneten Daten angepasst werden. Die Geraden repräsentieren Tangenten, die an die berechneten Energie-Druck Kurven angelegt werden. Diese sogenannte Helmholtz- © 2003 Max-Planck-Gesellschaft 6 Schwarz, Ulrich et al. | Strukturmotive des CuAl2 Typs in Elementen und ... Tätigkeitsbericht 2003 Konstruktion dient der Ermittlung berechneter Übergangsdrücke. (b) Volumina pro Antimonatom bei Drücken bis 42 GPa. Symbole stellen experimentelle Daten dar. Die Kurven entsprechen BirchMurnaghan Gleichungen, die durch Anpassung an Ergebnisse quantenmechanischer Berechnungen erhalten werden. Zu Vergleichszwecken sind die Ergebnisse bezüglich Energie und Volumen pro Atom angegeben. Als Referenz-Nullpunkt wird das Minimum in E(V) des idealisierten Komposits TA gewählt. Aus Abbildung 4a ist ersichtlich, dass die anhand der Berechnungen bestimmte Abfolge der Phasenumwandlungen mit den experimentellen Ergebnissen übereinstimmt. Die experimentellen Daten zeigen, dass die Normaldruckstruktur A7 von Sb-I bei Drücken bis etwa 8.2 GPa stabil bleibt, der theoretisch berechnete Übergangsdruck beträgt 7.2 GPa. Die BCC Struktur von Sb-III besitzt die niedrigste Energie bei Volumina kleiner als etwa 75 % des theoretischen Gleichgewichtsvolumens. Im Bereich zwischen diesen Phasen stellt der Approximant TA der modulierte Struktur von Sb-II die stabilste Anordnung der Atome dar. Die früher vorgeschlagene Kristallstruktur TP entspricht einer viel höheren Energie und kann auf Basis der Berechnungen als mögliche Sb-II Modifikation definitiv ausgeschlossen werden. Der direkte Vergleich der experimentellen und berechneten Volumendaten in Abbildung 4b zeigt eine sehr gute Übereinstimmung für die Phase A7 von Sb-I und die Modifikation BCC von Sb-III. Nur für das angenäherte Modell des Approximanten TA treten etwas größere Abweichungen im Vergleich zur realen modulierten Kristallstruktur von Sb-II auf. Untersuchungen der chemischen Bindung durch Berechnungen der ELF werden zurzeit durchgeführt. Die gleichartige Organisation der Atome in den untersuchten intermetallischen Verbindungen und in SbII verdeutlicht, dass in Abhängigkeit von Zustandsparametern wie Druck oder Zusammensetzung die Atome einiger Elemente (kristall)chemisch unterschiedliche Positionen eines Strukturtyps besetzen können. Dieses Ergebnis steht in Einklang mit der Organisation der Atome in anderen Verbindungen vom CuAl2-Typ. Beispielsweise bildet beim GaZr2 - im Gegensatz zu CuAl2 selbst - das Übergangsmetall die Wirtsstruktur und das Hauptgruppenelement die Gaststruktur. © 2003 Max-Planck-Gesellschaft