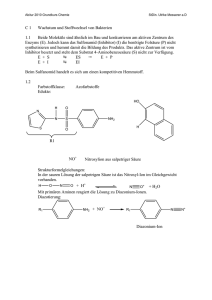
1 6. Stoffwechsel der Kohlenhydrate: 6.1. Glycolyse
Werbung

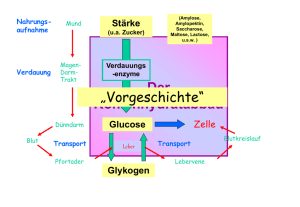
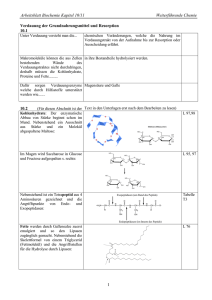
6. Stoffwechsel der Kohlenhydrate: 6.1. Glycolyse: Glucose ⇒ 2 * Pyruvat • • • • Glucose+ 2 NAD+ + 2 Pi + 2 ADP ⇒ 2* (Pyruvat + NADH + H+ +ATP +H2O ) Glycos: süß, lysis: Spaltung Ubiquitärer Stoffwechselweg: 10 enzymatische Schritte, Alles im Cytoplasma. Alle Enzyme sind löslich. Nur in einigen eukaryontischen Parasiten (z.B. Trypanosoma) sind die Glycolyseenzyme in spezialisierten Multienzymkomplexen kompartmentiert. 6.1.1. Aufnahme von Glucose in die Zelle A) Cotransport von Glucose und Na+: Indirekt aktiver Transport • Wichtig für die Resorption von Glucose. Wird im Abschnitt Verdauung/ Resorption besprochen. B) Glucosetransporter: Carrier • • • Transmembranproteine, enthalten 12 Transmembranhelices, transportieren Glucose entlang des GlucoseKonzentrationsgradienten, Sättigungskinetik, keine Diffusion!. Bilden eine Familie homologer Gene (GLUT x) mit gewebsspezifischer Expression. Isoformen (1...,12) unterscheiden sich in Affinität, Selektivität, Regulation! GLUT 4 wird u.a. in der Muskulatur und in Adipocyten exprimiert. Der Einbau in die Membran wird durch Insulin stimuliert. In gluconeogenetischen Organen werden GLUTs auch für den Export von Glucose genutzt! 6.1.2. Phasen der Glycolyse A) Stufe I (Investition: Energieaufwand) • • • Glucose wird phosphoryliert und in zwei Triosephosphate (Glycerinaldehyd-3-phosphat, GAP) gespalten. Zwei energiereiche Bindungen (ATP) werden investiert! 2 mol ATP/ mol Glucose werden hier verbraucht! Glucose + 2 ATP ⇒ 2 GAP + 2 ADP B) Stufe II (Ertrag: ATP-Bildung) • • • Umwandlung der Triosephosphate in Pyruvat 2 GAP + 4 ADP + 2 Pi => 2 Pyruvat + 4 ATP ATP-Synthese: 4 mol ATP/ mol Glucose werden in der Stufe II gebildet! 6.1.3. Glucose ⇒ 2* Pyruvat: 10 enzymkatalysierte Reaktionen 1.Glucose wird zu Glucose-6-phosphat (Glc-6-P) phosphoryliert Glucose + ATP ⇒ Glc-6-P + ADP; ∆G0r= - 16.7 kJ/mol; irreversibel! Enzym: Hexokinase. Gruppenübertragungsreaktion • • • • • ATP-Verbrauch (MgATP2-) Glucose induziert eine Konformationsänderung des Proteins! Das Enzym katalysiert auch die Phosphorylierung von D-Fructose und D-Mannose Leberzellen enthalten ein als Glucokinase bezeichnetes Isoenzym! Die Glucokinase unterscheidet von anderen Hexokinasen in der Organverteilung und regulatorischen Eigenschaften. Glc-6-P ist negativ geladen und kann (wie auch andere Zuckerphosphate) die Zelle nicht verlassen! 1 2. Glucose-6-phosphat wird zu Fructose-6-phosphat (Fru-6-P) isomerisiert. Glc-6-P == Fru-6-P ; ∆G0r= +1.7 kJ/mol, reversibel! Enzym: Phosphoglucose-Isomerase (PGI): eine Isomerase • • Glc-6-P (Aldose, Pyranosering) wird zu einer Ketose (Furanosering) isomerisiert. Die PGI ist ein moonlighting Enzyme (S D Copley: Enzymes with extra talents: moonlighting functions and catalytic promiscuity,Current Opinion in Chemical Biology 2003, 7:265–272). 3. Fructose-6-phosphat wird zu Fructose-1,6-bisphosphat (Fru-1,6-P2) Fru-6-P + ATP ⇒ Fru-1,6-P2 + ADP; irreversibel! Enzym: Phosphofructokinase (PFK, auch PFK-1), eine Kinase! • • ATP-Verbrauch (MgATP2-) Ansatzpunkt der Regulation der Glycolyse; ein allosterisches Enzym! 4. Fructose-1,6-bisphosphat wird zu Glycerinaldehyd-3-phosphat (GAP) und Dihydroxyacetonphosphat (DHAP) gespalten! Fru-1,6-P2 == GAP + DHAP; ∆G0r= +23.8 kJ/mol • GAP: Aldose. DHAP: Ketose; Durch die Aldolspaltung von Fru-1,6-P2 (C6) entstehen zwei C3Fragmente. Enzym: Aldolase (eine C-C-Lyase) • Stereospezifische Reaktion. Reversible basenkatalysierte Aldolspaltung! • Die Carbonylgruppe des Fru-1,6-P2 reagiert mit der ε-Aminogruppe eines Lysylrestes des Aldolaseenzyms unter Bildung einer Schiff-Base. • Isoenzyme: o Aldolase A ist für Fructose-1,6-bisphosphat spezifisch. o Aldolase B (Leber, Nieren) katalysiert auch die Spaltung von Fru-1-P (s. Fruktosestoffwechsel). 5. Dihydroxyacetonphosphat (DHAP) wird zu Glycerinaldehyd-3-phosphat (GAP) isomerisiert DHAP == GAP ; ∆G0r= +7.5 kJ/mol; Enzym: Triosephosphat Isomerase (TIM) • • reversibel! Nur Glycerinaldehyd-3-P (GAP) wird in der Glycolyse abgebaut werden! Ende der Investitionsphase, Beginn der Ertragsphase der Glycolyse! 6. Oxidation von Glycerinaldehyd-3-phosphat (GAP) zu 1,3-Bisphosphoglycerat (1,3-BPG) GAP + Pi + NAD+ == 1,3-BPG + NADH + H+ ; reversibel, endergon Enzym: Glyceraldehyd-3-phosphat-Dehydrogenase (GAPDH) • • • Oxidation der Aldehydgruppe des GAP und Kondensation der Carboxylgruppe mit Phosphat Energiekonservierung: 1,3-BPG- ein Acylphosphat- ist eine energiereiche Verbindung! Bildung von NADH! Dabei reagiert die Aldehydgruppe des GAP mit einem Cysteinylrest des Enzyms unter Ausbildung eines Thiohalbacetals. Die bei der Oxidation der Aldehydgruppe freiwerdende Energie wird in einem kovalent am Enzym gebundenen energiereichen Thioester konserviert, der dann unter Bildung von 1,3-BPG phosphorylytisch gespalten wird. Die endergone Entstehung des Acylphosphates wird mit der exergonen Aldehydoxidation durch den energiereichen Thioester am Enzym gekoppelt! 2 7. 1,3-Bisphosphoglycerat wird zu 3-Phosphoglycerat (3-PG) und ATP 1,3-BPG + ADP == 3-PG + ATP ; reversibel, ∆G0r= -18.5 kJ/mol • • Enzym: Phosphoglyceratkinase, Mg2+ Substratkettenphosphorylierung: ATP wird gebildet! Kopplung von GAPDH (endergon) und Phosphoglyceratkinase (exergon): GAP + ADP + Pi + NAD+ == 3-PG + ATP + NADH + H+ ; ∆G0r= -12 kJ/mol • • 1,3-BPG ist das gemeinsame Zwischenprodukt beider Reaktionen, dessen energiereiche Acylphosphatgruppe in der exergonischen PGK-Reaktion auf ADP übertragen wird! Entkopplung: Arsenat reagiert in der GAPDH-Reaktion unter Bildung von 1-Arseno-3-Phosphoglycerat, das spontan (ohne ATP-Bildung) zu Arsenat und 3-PG umgesetzt wird! 8. Mutase-Reaktion: 3-Phosphoglycerat wird zu 2-Phosphoglycerat (2-PG) isomerisiert 3-Phosphoglycerat == 2-Phosphoglycerat ; ∆G0r= + 4.4 kJ/mol • • • Enzym: Phosphoglycerat-Mutase Reversible intermolekulare Verschiebung der Phosphatgruppe! 2,3-Bisphosphat (2,3-BPG) wird als Cosubstrat benötigt! Bei der Reaktion entsteht intermediär ein phosphorylierter Histidinyl-Rest im aktiven Zentrum. Intermolekularer Phosphattransfer! 9. Dehydratisierung von 2-Phosphoglycerat zu Phosphoenolpyruvat (PEP) 2-PG == PEP + H2O ; reversibel, ∆G0r= + 7.5 kJ/mol Enzym: Enolase, Mg2+ -abhängig; Ein energiereiches Enolphosphat (PEP) entsteht! 10. Übertragung der Phosphatgruppe von Phosphoenolpyruvat (PEP) auf ATP. PEP + ADP ⇒ Pyruvat + ATP ; ∆G0r= - 31 kJ/mol; irreversibel! Enzym: Pyruvatkinase • • • Substratkettenphosphorylierung: ATP wird gebildet! Isoenzyme der Pyruvatkinase (L-Leber, M-Muskel und Gehirn) unterscheiden sich in der Regulation! Pyruvat tritt zunächst in der Enolform auf, die nichtenzymatisch in die Ketoform tautomerisiert! 6.1.4. Glycolyse: Bilanz Glucose+ 2 NAD+ + 2 Pi + 2ADP ⇒ 2 (Pyruvat + NADH + H+ + ATP + H2O ); ∆G0r= - 85 kJ/mol, irreversibel! A) Exergonischer Teilprozeß: Glucose+ 2 NAD+ => 2 (Pyruvat + NADH + H+ ); ∆G0r= - 140 kJ/mol B) Endergonischer Teilprozeß: 2 Pi + 2 ADP ⇒ 2 (ATP + H2O ); ∆G0r= +61 kJ/mol Ca. 95% der Energie (bezogen auf den physiologischen Brennwert der Glucose) verbleiben im Pyruvat ! 3 Die Glycolyse ist ein irreversibler Prozess: 3 irreversible enzymatische Reaktionen treten auf, katalysiert durch Hexokinase, Phosphofructokinase und Pyruvatkinase. 6.1.5. Metabolismus des Pyruvats: Regeneration von NAD+ Die Glycolyse benötigt: • NAD+ (GAPDH) und ADP (Phosphoglyceratkinase und Pyruvatkinase), ATP (Hexokinase und Phosphofructokinase) NAD+ muß reoxidiert werden! Je nach Stoffwechselsituation werden verschiedene Metabolite als Oxidationsmittel verwendet! 6.1.5.1 Lactatbildung: Lactatdehydrogenase (LDH) Pyruvat + NADH + H+ == Lactat + NAD+ ; reversibel, Oxidation von NADH, Bildung von Lactat! Lactat: • • Endprodukt des Glucoseabbaus in Erythrocyten; Retina,.. wird im Glucosestoffwechsel des Muskels bei O2 -Mangel gebildet! Die LDH-Reaktion ist reversibel • • Im Stoffwechsel des Erythrozyten und der Muskulatur wird Lactat aus Pyruvat gebildet! Der Herzmuskel und die Leber bilden Pyruvat aus Lactat! LDH-Isoenzyme Die LDH ist ein Tetramer, das aus zwei Typen von Untereinheiten aufgebaut ist ( H & M), die organspezifisch exprimiert werden. Das M4-Isoenzym arbeitet optimal unter anaeroben Bedingungen, während das H4-Isoenzym in aerobe Situationen exprimiert wird. Bei Organschäden können diese Isoenzyme im Serum nachgewiesen werden. Die organspezifische Verteilung und die unterschiedlichen Ladungseigenschaften der Isoformen erlaubt Hinweis auf die Zellschädigung. Die LDH-Diagnostik bei kardialen Schädigungen beruht auf der Bestimmung des Konzentrationsverhältnisses der Isoenzyme LDH-1 (H4) und LDH-2 (H3M) im Serum ( LDH1 > LDH2). Anaerober Abbau der Glucose zu Lactat: Bilanz Glucose + 2 Pi + 2 ADP ⇒ 2 (Lactat +ATP + H2O ) 6.1.5.2 Alkoholische Gärung: Bildung von Ethanol, CO2 (in Hefen) Kooperation von Pyruvatdecarboxylase (Thiaminpyrophosphat) und Alkoholdehydrogenase (NADHOxidation). In Hefen wird dadurch NADH reoxidiert und Ethanol gebildet, In der Leber wird Alkohol durch cytosolische Alkoholdehydrogenasen (ADH) und das MEOS (microsomal ethanol oxidizing system) abgebaut. Die Alkoholdehydrogenase ist ein dimeres Metalloenzym (mehrere Isoenzyme. (Km = 0.2...2 mmol/l)). Das MEOS ist ein mikrosomales Cytochrom P450-Enzym (eine Monooxygenase, Km =8...12 mmol/l). 4 6.1.5.3 Bildung von Acetyl-CoA durch den Pyruvatdehydrogenase-Komplex (PDH) Bildung von Acetyl-CoA aus Pyruvat durch den mitochondrialen Pyruvatdehydrogenase-Komplex (PDH)! Die Glycolyse findet im Cytoplasma, die Bildung von Acetyl-CoA und der Citratcyclus in den Mitochondrien statt. Ein Pyruvat-Carrier transportiert Pyruvat gegen OH (elektroneutral) in die Mitochondrien. Pyruvatdehydrogenase-Reaktion: Oxidative Decarboxylierung von Pyruvat: Pyruvat + CoA + NAD+ ⇒ Acetyl-CoA + CO2 + NADH + H+ ; Irreversibel! • • PDH: 3 Enzyme und 5 Coenzyme sind beteiligt! Coenzyme und prosthetische Gruppen: NADH, CoA, FAD, Thiamin-Pyrophosphat, Liponamid. Die Reaktion wurde durch Fritz Albert Lipmann (1899-1986) aufgeklärt, der dafür den Nobelpreis 1953 erhielt. Enzyme des PDK-Komplexes Enzym Abk. Coenzyme Pyruvat-Dehydrogenase (Decarboxylase) E1 Thiaminpyrophosphat (TPP), FAD Dihydrolipoyl-Transacetylase E2 Lipoamid Dihydrolipoyl-Dehydrogenase E3 FAD, NADH Reaktionen am PDH-Komplex E1: Decarboxylierung von Pyruvat und Bindung an Thiaminpyrophosphat (TPP) • Pyr + TPP ⇒ Hydroxyethyl-TPP + CO2 E2: Oxidation der Hydroxyethylgruppe, Transfer auf Liponamid • Hydroxyethyl-TPP + Liponamid ⇒ Acetyl-Liponamid + TPP E2: Transfer der Acetylgruppe auf CoA • Acetyl-Liponamid + CoA ⇒ Acetyl-CoA + Dihydroliponamid E3: Regeneration des Liponamids (FAD abhängige Teilreaktion an E3!!) • Dihydroliponamid + NAD+ ⇒ Liponamid + NADH + H+ Regulation der PDH-Aktivität • • • • Das Enzym wird durch Phosphorylierung von Serylresten des E1-Proteins inaktiviert.! Kinase und Phosphatase sind mit dem PDH-Komplex assoziiert. Die PDH wird durch die Produkte Acetyl-CoA und NADH und ATP gehemmt! Ligandenbindung induziert Konformationsänderungen, die die Aktivitäten von Proteinkinase und Proteinphosphatase beeinflussen: Die Proteinkinase wird durch ATP, Acetyl-CoA und NADH aktiviert und durch Pyruvat gehemmt. PDH: Pathobiochemie • Defekte in PDH-Genen bewirken Lactatcidose und neurologische Störungen(LEIGH-SYNDROM). 5 Anhang. Vitamin B1: Thiamin Bedarf: ca.1.5mg/Tag Der Bedarf an Vitamin B1 ist von der Menge der eingenommenen Kohlehydrate abhängig. Gute Quellen sind Fleisch, Getreide, Nüsse und Gemüse. Einige Lebensmittel ( z.B. Schwarzer Tee), enthalten Anti-Thiaminfaktoren. Aufnahme im Darm: Von den Darmepithelzellen wird Thiamin via einen Rezeptor aufgenommen oder passiv transportiert und ans Blut abgegeben. In den Geweben wird das Provitamin zu Thiamin-Pyrophosphat phosphoryliert. Funktionen: Thiaminpyrophosphat ist Coenzym bei Decarboxylierungen: Decarboxylierung von α-Ketosäuren zu Aldehyden (Pyruvat- und αKetoglutarat-Dehydrogenase-Komplex, Pyruvatdecarboxylase in Hefen), Coenzym der Transketolase (Pentosephosphatzyklus), Coenzym der Acetolactat-Synthase (Biosynthese von Valin und Isoleucin). Kovalente Katalyse: Bildung eines kovalentes Intermediates am TPP in der Pyruvatdehydrogenase-Reaktion. Klinische Bedeutung: Wernicke-Korsakoff-Syndrom, Beri Beri Syndrom. Vitamin B1-Mangel tritt hier am ehesten bei Säuglingen und bei Alkoholikern auf (Wernicke-Korsakoff-Syndrom). Alkohol vermindert die Resorption vonThiamin im Darm. In Gegenden, wo Reis als Hauptnahrungsmittel dient, kannVitamin B1-Mangel auftreten, wenn derReis geschält gegessen wird (Beri Beri-Syndrom). Thiaminmangel fördert auch zur Ausbildung einer metablischen Acidose (Lactatacidose). 6