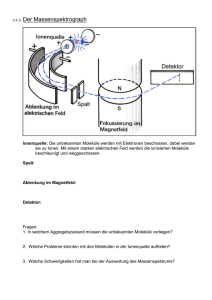
Lernskript Instrumentelle Analytik
Werbung

Instrumentelle Analytik Stephanie Negele Eine Zusammenfassung der Vorlesungen von Prof. Dr. W. Fink aus dem 4. Semester Naturwissenschaftliche Forensik an der Hochschule Bonn-Rhein-Sieg Im Frühjahr 2014 Inhaltsverzeichnis 1 Einführung 3 2 Chromatographie 2.1 Allgemeines . . . . . . . . . . . . . . . . . . . . . 2.1.1 Nernstscher Verteilungssatz . . . . . . . . 2.1.2 Bandenverbreiterung . . . . . . . . . . . . 2.1.3 Säulenchromatographie . . . . . . . . . . 2.2 Das Chromatogramm . . . . . . . . . . . . . . . 2.2.1 Qualitative Auswertung . . . . . . . . . . 2.2.2 Quantitative Auswertung . . . . . . . . . 2.2.3 Retentionszeit tR . . . . . . . . . . . . . . 2.2.4 Kapazität k 0 . . . . . . . . . . . . . . . . 2.2.5 Trennfaktor α . . . . . . . . . . . . . . . . 2.2.6 Phasenverhältnis β . . . . . . . . . . . . . 2.2.7 Peakbreiten σ . . . . . . . . . . . . . . . . 2.2.8 Auflösung Rs . . . . . . . . . . . . . . . . 2.3 Theoretische Bodenzahl . . . . . . . . . . . . . . 2.3.1 Van Deemter Kurve . . . . . . . . . . . . 2.4 Dünnschichtchromatographie . . . . . . . . . . . 2.4.1 Stationäre und mobile Phasen . . . . . . . 2.4.2 Funktionsprinzip . . . . . . . . . . . . . . 2.4.3 Hochleistungsflüssigkeitschromatographie 2.5 Gaschromatographie . . . . . . . . . . . . . . . . 2.5.1 Die Säule . . . . . . . . . . . . . . . . . . 2.5.2 Detektionsmethoden . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4 5 5 6 6 7 7 7 8 8 8 9 9 10 11 12 13 14 15 17 21 21 25 3 Spektrometrie 3.1 Strahlung . . . . . . . . . . . . . 3.2 Die Mikrowelle . . . . . . . . . . 3.3 Infrarotspektrometrie . . . . . . . 3.3.1 Theoretische Grundlagen 3.3.2 Fourier Transformation . 3.3.3 Das IR-Spektrum . . . . . 3.4 UV/vis-Spektrometrie . . . . . . 3.4.1 Chromophore . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27 28 29 31 32 36 37 38 39 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3.5 3.6 3.4.2 Das Lambert-Beer’sche Gesetz . . 3.4.3 Diodenarray-Detektor . . . . . . . 3.4.4 Fluoreszenz . . . . . . . . . . . . . Kernspinresonanzspektrometrie . . . . . . 3.5.1 Theoretische Grundlagen . . . . . 3.5.2 Chemische Verschiebung . . . . . . 3.5.3 Spin-Spin-Kopplung . . . . . . . . 3.5.4 Das NMR-Spektrum . . . . . . . . Massenspektrometrie . . . . . . . . . . . . 3.6.1 Theoretische Grundlagen . . . . . 3.6.2 Die Ionenerzeugung . . . . . . . . 3.6.3 Vorläufiges Interpretationsschema . 3.6.4 Kopplungsmöglichkeiten . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40 40 41 42 42 44 45 47 47 48 48 50 50 Abbildungsverzeichnis 52 Literaturverzeichnis 53 2 Kapitel 1 Einführung D ieses Skript ist eine von mir selbst verfasste Zusammenschrift der Vorlesungen des Prof. Dr. W. Fink im Fach der Instrumentellen Analytik an der Hochschule Bonn-Rhein-Sieg. Ich gebe keinerlei Garantie auf die Richtigkeit des hier Geschriebenen. Als Grundlage dient das von Prof. Dr. W. Fink veröffentlichte Skript der Vorlesung sowie die angegebene Literatur. Zitat- und Bildquellen sind stets angegeben und können im Literaturverzeichnis nachgesehen werden. Internetseiten wurden mit dem Datum des Aufrufs ebenfalls aufgeführt. 3 Kapitel 2 Chromatographie D ie Chromatographie gehört zu den chemisch-physikalischen Methoden, um Stoffe voneinander zu trennen. Sie wird sowohl in der analytischen als auch der präparativen Chemie eingesetzt. Ihr Haupteinsatzgebiet ist die Identifizierung und quantitative Bestimmung organischer und anorganischer Verbindungen. In der Analytik ist es häufig notwendig, einzelne Bestandteile in einer Lösung oder einem Gemisch von Stoffen zu isolieren und zu identifizieren. Hier bietet sich die Chromatographie als zuverlässiges und sehr präzises Mittel an. Selbst Stoffe in sehr geringen Konzentration können bestimmt werden. Meist ist die Analyse einzelner isolierter Stoffe mit relativ einfachen Methoden zu realisieren. Die Vorbereitung, die eigentliche Trennung der Stoffe voneinander ist vor allem durch die Chromatographie ermöglicht worden. Das Prinzip funktioniert in bei jeder Form der Chromatographie gleich. Eine Stoffgemischlösung strömt über ein festes, unlösliches (an)organisches Material, welches eine größtmögliche Oberfläche aufweist, in eine festgelegte Richtung. Es findet eine Wechselwirkung zwischen dem flüssigen Stoffgemisch und dem festen Material statt. Die einzelnen Komponenten der Lösung, welche als flüssige Phase bezeichnet wird, werden in unterschiedlichem Ausmaß von dem festen Material, der stationären Phase zurückgehalten. Der Trenneffekt beruht darauf, dass sich die Verbindungen mit unterschiedlichen, substanzspezifischen Verzögerungen über die chromatographische Trennstrecke bewegen1 . Jede chromatographische Trennung funktioniert nach diesem Prinzip des Unterschieds in den Wechselwirkungen des Stoffes mit den Phasen. Dabei ist die Wanderungsgeschwindigkeit aufgrund der konstanten Strömungsgeschwindigkeit der mobilen Phase stets gleich. Lediglich die unterscheidlichen Adsorptionszeiten des Stoffes an der stationären Phase bewirken die Auftrennung des Stoffgemischs. Es finden ständig Adsorptions- und Desorptionsvorgänge statt. Die Bindung des Stoffes an die stationäre Phase ist temporär und nur von kurzer Dauer. Das Gleichgewicht zwischen gelöstem und gebundenen Stoff stellt sich ständig neu ein. Man unterscheidet die beiden Extremfälle der totalen Lösung in der mobilen Phase, bei der keinerlei Bindung an die stationäre Phase stattfindet und die totale Adsorption, bei der der Stoff sich vollständig an die stationäre Phase bindet und keine Lösung in der mobilen Phase stattfindet. In beiden Fällen ist keine Trennung des Stoffgemischs möglich. Der Idealfall der chromatographischen Trennung liegt zwischen diesen beiden Extrema. Man kann die chromatographischen Methoden in drei Kategorien 1 J. Böcker, Chromatographie, S. 32 4 unterteilen: • nach dem Aggregatszuständen der mobilen oder stationären Phase (z.B. Gaschromatographie, Flüssigkeitschromatographie); • nach der Art des Trennvorgangs (allgemein die Adsorptionschromatographie, bei der die Wechselwirkung zwischen fester und flüssiger Phase stattfindet. Es gibt gasförmig-fest, flüssig-fest, gasförmig-flüssig und flüssig-flüssig Chromatographie); • nach der angewandten Technik (Säulen- und Papierchromatographie). 2.1 Allgemeines Gegen die fortschreitende Auftrennung des Stoffgemischs arbeitet die Diffusion2 , welche einen Konzentrationsausgleich anstrebt und dadurch die Umkehr der Auftrennung. Somit sollten chromatographische Vorgänge in möglichst kurzer Zeit ablaufen, um die Rückvermischung so gering wie möglich zu halten. Ist die Fließgeschwindigkeit der mobilen Phase jedoch zu schnell, kann keine vollständige Auftrennung stattfinden, da die leichten Bindungen zur stationären Phase gleich zerstört werden. Für jedes chromatographische System gibt es daher eine optimale Strömungsgeschwindigkeit, welche empirisch ermittelt werden muss. 2.1.1 Nernstscher Verteilungssatz Für die Verteilung zwischen mobiler und stationärer Phase gilt bei kleinen Konzentrationen der Nernstsche Verteilungssatz: Wenn ein Stoff in zwei Phasen löslich ist, so ist bei verdünnten Lösungen das Verhältnis der Konzentrationen in beiden Phasen konstant. Es gilt: cS Kc = (2.1) cM mit Kc als Verteilungskoeffizient oder -konstante cS als Konzentration des Stoffes in der festen (solid) Phase cM als Konzentration des Stoffes in der mobilen Phase. Die Effizienz, mit der eine stationäre Phase arbeiten kann, nennt man theoretische Böden. Die Distanz, die das Gleichgewicht braucht, um sich einzustellen, entspricht einer theoretischen Trennstufe. Je höher die Anzahl dieser Trennstufen ist, desto besser wird die Trennung der Stoffe. Allerdings werden bei erhöhender Trennstufenzahl (Länge der Säule) die Banden, also die Signale, breiter, weshalb die Trennstufen nicht ins unendliche wachsen sollten. 2 Physikalisch bedingte Tendenz zur gleichmäßigen Verteilung verschiedener Moleküle in größtmöglichem Abstand zueinander. 5 2.1.2 Bandenverbreiterung Banden können sich durch verschiedene Ursachen verbreitern. Kennt man diese, so kann man die angewendete Methode optimieren, sodass man eine möglichst hohe Trennstufenzahl bei geringster Bandenverbreiterung erreicht. • Eddy-Diffusion (Streudiffusion): Es kann passieren, dass manche Moleküle eines Stoffes vor allen anderen die stationäre Phase passieren, sodass sie verfrühte Banden erzeugen. Andere Moleküle benötigen vielleicht etwas länger als der Durchschnitt, sodass insgesamt die Banden unscharf werden und keine klaren Ränder haben. Die Eddy-Diffusion lässt sich mit folgender Gleichung beschreiben: 2 σE = L · dp (2.2) Mit der Säulenlänge L und dem Teilchendurchmesser dp . • Strömungsverteilung: Je weiter die mobile Phase von den Körnern der stationären Phase entfernt ist, desto höher ist die Fließgeschwindigkeit. In direkter Nähe zu den Körnern ist die Fließgeschwindigkeit erheblich niedriger. Auch hier werden die Banden unscharf und verbreitern sich. • Diffusion der Probenmoleküle in der mobilen Phase Durch das Konzentrationsgefälle, welches durch die Wechselwirkungen der Probenmoleküle mit der stationären Phase erzeugt wird, findet stets auch eine Diffusion statt. Je länger die Probenmoleküle in der mobilen Phase verweilen, desto höher ist die Diffusion. Dies ist vor allem in der Gaschromatographie von Bedeutung. Bei der Flüssigkeitschromatographie bilden die Körner der stationären Phase Hohlräume, in denen Diffusion vermehrt stattfinden kann. Dieses von den Körnern eingeschlossene Volumen bezeichnet man als Totvolumen. Dieses sollte möglichst gering sein. • Diffusion in der stationären Phase Die Körner der stationären Phase sind porös und können mobile Phase einschließen, wodurch enthaltene Probenmoleküle nicht mehr weitertransportiert werden können. Das Probenmolekül kann sich dann nur noch durch Diffusion aus dem Korn befreien. Deshalb sollte das Füllmaterial der stationären Phase eine möglichst enge Korngrößenverteilung haben und aus sehr kleinen Körnern bestehen. 2.1.3 Säulenchromatographie Bei diesem Trennverfahren befindet sich die stationäre Phase in einer Säule. Diese Technik wird häufig in der Gas- und Flüssigkeitschromatographie angewendet. Für die Aufnahme eines Chromatograms benötigt man eine besondere Apperatur, welche wie folgt aufgebaut ist. In der Gaschromatographie wird die mobile Phase als Trägergas, in der Flüssigkeitschromatographie als Eluent bezeichnet. Von einer Pumpe wird die mobile Phase kontinuierlich durch das System gepumpt. Die Probe wird vor der Säule appliziert und läuft, getragen von der mobilen Phase, durch die Säule, welche mit der stationären Phase gefüllt ist. Gleich hinter der Säule ist ein Detektor angeschlossen, der zeitabhängig das Ankommen der Probenbestandteile aufzeichnet und an einen Computer sendet, welcher das Chromatogramm erstellt. 6 Abbildung 2.1 – Prinzip eines Säulenchromatographen. [1, S. 38, Bild 2.5] Die einzelnen Substanzen erzeugen im Detektor ein elektrisches Signal, welches einen Schreiber ausschlagen lässt. Auf diesem Wege erhält man das klassische Chromatogramm mit verschiedenen Peaks. Dabei ist die Fläche des Peaks proportional zum Probenvolumen, das den Detektor erreicht. Zwischen den einzelnen Peaks erreicht die mobile Phase den Detektor und erzeugt im Chromatogramm die Basislinie. 2.2 Das Chromatogramm Ein Peak stellt die Probenkonzentration in Abhängigkeit von der Zeit dar und wird in seiner Gesamtheit mit allen anderen Peaks der Analyse im Chromatogramm dargestellt. Peaks haben in der Theorie die Form einer Verteilungsfunktion. In der Praxis sind sie leider häufig asymmetrisch. Ein Peak liefert sowohl qualitative als auch quantitative Informationen über die isolierte Substanz. 2.2.1 Qualitative Auswertung Die Zeit, die eine Substanz benötigt, um durch die Säule zum Detektor zu fließen, wird Retentionszeit genannt. Sie ist unter gleichen chromatographischen Bedingungen3 stoffspezifisch und kann daher dazu verwendet werden, Substanzen zu identifizieren. Für die Identifikation eines Stoffes ist es notwendig, zuvor einen Standard zu chromatographieren, um die stoffspezifische Retentionszeit zu ermitteln. In manchen Fällen kann es vorkommen, dass zwei Substanzen die gleichen Retentionszeiten haben. In diesem Falle lässt man die Standards in einem veränderten System laufen, um sie dort zu identifizieren. Ebenso wird mit den Proben verfahren. Zudem kann man einen speziellen Detektor verwenden, der weitere Informationen liefert und auf diesem Wege eine Identifizierung ermöglicht. Dies nennt man Kopplungstechnik. 2.2.2 Quantitative Auswertung Nachdem die Identifizierung der Stoffe stattgefunden hat, kann man die Peaks bezüglich der Quantität analysieren. Um dies zu ermöglichen, müssen die Peaks so weit voneinander getrennt sein, dass sie im Idealfall auf der Basislinie beginnen und enden. Je nach vorheriger Festlegung kann die Peakfläche 3 Dazu zählen die Art der Trennsäule, die Zusammensetzung und Fließgeschwindigkeit der mobilen Phase und die Temperatur. 7 oder die Peakhöhe4 proportional zur Stoffmenge sein. Man analysiert eine Standardlösung mit bekannten Stoffmengen und zeichnet mit den erhaltenen Peakflächen oder -höhen eine Kalibrationskurve, anhand derer man die Stoffmengen der Probenpeaks ermitteln kann. Beide Methoden sind anwendbar, unterscheiden sich jedoch manchmal in ihrer Reproduzierbarkeit. Eine schlechte Reproduzierbarkeit führt man allgemein auf eine fehlerhafte Injektion, Detektion oder Integration zurück. 2.2.3 Retentionszeit tR Bei der bereits angesprochenen Retentionszeit unterscheidet man zwei verschiedene Zeiten, die sich durch ihre Totzeit unterscheiden. • t0 : Totzeit der Trennsäule. Die Zeit, die die mobile Phase benötigt, um von der Probeninjektion bis zum Detektor zu gelangen. Eine Substanz, welche keine Wechselwirkung mit der stationären Phase zeigt, erreicht nach t0 den Detektor. • tR : Brutto-Retentionszeit. Die Zeit, die eine mit der stationären Phase wechselwirkende Substanz von der Probeninjektion bis zum Detektor benötigt. • t0R : Netto-Retentionszeit. Differenz aus tR und t0 . Die Totzeit t0 ist für alle eluierten Stoffe5 gleich und entspricht der minimalen Aufenthaltszeit aller Stoffe in der mobilen Phase. Die getrennten Stoffe unterscheiden sich in ihrer Netto-Retentionszeit t0R , die der Aufenthaltszeit in der stationären Phase entspricht. 2.2.4 Kapazität k 0 Die Retentionszeit ist abhängig von der Fließgeschwindigkeit der mobilen Phase und der Länge der Säule. Damit ist die Brutto-Retentionszeit tR für die Charakterisierung einer Substanz nicht geeignet. Günstiger ist dafür der Kapazitätsfaktor k 0 , welche wie folgt definiert ist: k0 = nstat t0 tR − t0 = R = nmob t0 t0 (2.3) Dieser Faktor ist von der Säulenlänge und Fließgeschwindigkeit unabhängig. Für gute Trennungen benötigt man einen Wert zwischen 1 und 5. Ist er <1, so eluieren die Verbindungen so schnell, dass eine Bestimmung der Retentionszeit schwierig wird. 2.2.5 Trennfaktor α Dieser gibt die relative Retention zweier benachbarter Peaks an. Es gilt α ≥ 1. α= t0 k2 tR2 − t0 = 0R2 = tR1 − t0 tR1 k1 (2.4) 4 In der Praxis wird in der Regel die Peakfläche genutzt, da sich die Peakhöhe durch Temperaturschwankung oder Änderung der Fließgeschwindigkeit verändern kann und nicht mehr proportional zur Stoffmenge ist. 5 von der mobilen Phase getragenen Stoffe. 8 Bei α = 1 erreichen beide Analyten ungetrennt den Säulenausgang. 2.2.6 Phasenverhältnis β Damit wird das Volumenverhältnis von mobiler zu stationärer Phase beschrieben. Je kleiner β, desto größer ist der Anteil der stationären Phase am Volumen der Säule. Die Retention der Analyten wird dadurch erhöht, da im Umkehrschluss weniger mobile Phase vorhanden ist. β= VM VS (2.5) Man kann nun die Kenngrößen von Kapazitätsfaktor k 0 Gl. 2.3 und Phasenverhältnis β über den Verteilungskoeffizienten Kc Gl. 2.1 miteinander verknüpfen: k= 2.2.7 Kc VS = Kc · β VM (2.6) Peakbreiten σ Die Form eines Peaks entspricht im Idealfall einer Gaußschen Glockenkurve, welche der mathematischen Formel −x2 y = y0 · e 2σ2 (2.7) y = Peakhöhe an jeder beliebigen Stelle des Peaks y0 = Höhe am Peakmaximum x = Abstand von der Ordinate σ = Standardabweichung der Verteilung σ 2 = Varianz der Verteilung Es lassen sich an drei Stellen die Peakbreiten ablesen. Zunächst die Stelle, in der Nähe der Ordinate, an denen die Wendetangenten liegen. An dieser Stelle wird die Basisbreite wb gemessen. Diese lässt sich durch die Standardabweichung der Verteilung ebenfalls berechnen, da man folgenden Zusammenhang festgestellt hat: wb = 4 · σ (2.8) Als zweites kann man die Peakbreite auf halber Peakhöhe bestimmen. Diese wird mit dem Formelzeichen wh abgekürzt: q wh = 2 · σ · 2 · ln(2) = 2, 354 · σ (2.9) Da man die Höhe eines Peaks leicht messen kann und dadurch die Bestimmung der Peakbreite auf halber Peakhöhe auch ohne weiteres zu ermitteln ist, wird wh in der Regel zur Berechnung der Standardabweichung der Verteilung genommen. Zuletzt gibt es noch einen weiteren Peakbreiten-Parameter, den man auf einer Höhe von 60,7% misst. Dieser entspricht dem Doppelten der Standardabweichung: wi = 2 · σ (2.10) Diese Berechnungen sind nur dann gültig, wenn die Peakform nicht signifikant von der Form einer Gaußschen Glockenkurve abweicht. 9 In der Praxis ist dies meist nicht anwendbar, da die Asymmetrie der Kurven recht offensichtlich ist. Dabei unterscheidet man zweierlei Fälle von Asymmetrie. Ist der Anstieg des Signals flacher als der Abstieg auf die Basislinie, so spricht man vom Fronting. Häufiger ist jedoch der Fall, dass der Anstieg des Signals steiler ist, als der Abfall, was man als Tailing bezeichnet. Der chromatographische Prozess beginnt mit der Probenaufgabe. Diese muss möglichst präzise geschehen, da sonst bereits vor der Säule eine Substanzzone entsteht, wodurch keine schmalen Banden mehr erzeugt werden können. Dies kann später nicht mehr rückgängig gemacht werden. Aus diesem Grund finden die Probenapplikation in der Regel automatisch statt, um eine hohe Reproduzierbarkeit zu erhalten. Durch die verschiedenen Wanderungsgeschwindigkeiten der Moleküle in der Säule erhält man die Gaußsche Verteilungsfunktion. Je länger sich eine Substanz in der Säule aufhält, desto geringer ist ihre Wanderungsgeschwindigkeit u und desto breiter erscheint die Bande im Chromatogramm. Der Zusammenhang zwischen der Wanderungsgeschwindigkeit und der Bodenhöhe H wurde von van Deemter erforscht (s. 2.3.1). 2.2.8 Auflösung Rs Durch den Parameter α (2.4) kann man die Auflösung zweier Peaks quantitativ bestimmen. Der erhaltene Wert gibt jedoch keine Information, ob die beiden Peaks auch wirklich gut voneinander getrennt sind, da die Peakbreite in der Formel 2.4 nicht miteinbezogen ist. Dafür wird zur Bewertung die Auflösung Rs herangezogen, in die sowohl die Retentionszeiten der Peaks als auch deren Basisbreiten miteinfließen. Rs = tR2 − tR1 wb1 +wb2 2 =2· (tR2 − tR1 ) (wb1 + wb2 ) (2.11) Betrachtet man zwei Peaks, die schmal und eng beieinander liegen, so kann man die folgende Näherung verwenden: Rs = Abbildung 2.2 – Verschiedene Auflösungen der gleichen chromatographischen Trennung.[2, S. 6-13, Abb. 6.7] tR2 − tR1 wb2 (2.12) In der Praxis werden zwei Peaks als getrennt gewertet, wenn die Auflösung Rs = 1 beträgt. Wirklich getrennt sind die Peaks aber erst ab einer Auflösung von Rs = 1, 5. Setzt man Gleichung 2.19 für wb 2 nun ein, so erhält man √ tR2 − tR1 N Rs = · (2.13) tR2 4 Nützlich ist dieser Ausdruck ebenfalls, wenn man die Retentionszeiten durch die entsprechenden Retentionsfaktoren (Kapazitätsfaktoren) k ausdrückt. Mithilfe von Gleichung 2.3 erhalten wir dann: √ k2 − k1 N Rs = · (2.14) 1 + k2 4 Mit Gleichung 2.4 erhalten wir √ α−1 k2 N Rs = ( )·( )· α 1 + k2 4 10 (2.15) Unter der Bedingung, dass zwei nahe beieinanderliegende Peaks betrachtet werden, kann man sehen, dass die Auflösung vom Trennfaktor α, vom Kapazitätsfaktor k2 des zweiten Peaks und von der Bodenzahl N der Säule abhängig ist. In Abb. 2.26 ist die Auflösung Rs < 1. Nun kann man an den Parametern Trennfaktor α, Kapazitätsfaktor k2 und Bodenzahl N der Säule etwas verändern, um die Auflösung zu erhöhen. In Abb. 2.2 wurde k2 vergrößert. Dies bewirkt eine beinahe Basislinientrennung, erhöht allerdings auch die Retentionszeit, wodurch die Peakbreite zunimmt. In Abb. 2.2 wurde die Bödenanzahl erhöht, wodurch die Trennung der Peaks besser wird. Die Veränderung der Bodenanzahl kann durch die Länge der Säule oder der Fließgeschwindigkeit der mobilen Phase beeinflusst werden. Eine Verlängerung der Säule bedeutet eine Erhöhung der Analysendauer, weshalb man dies stets zu vermeiden versucht. Die Erhöhung der Fließgeschwindigkeit ist kritisch, daher bietet sich eine Variation von Faktoren7 an, welche den Trennfaktor α verbessern, was zum gewünschten Ergebnis führen kann (s. Abb. 2.2). Eine Variation von einzelnen Faktoren ist nahezu unmöglich, da sie sich meist gegenseitig beeinflussen, sodass eine Optimierung meist schwierig und komplex wird. 2.3 Theoretische Bodenzahl Man stelle sich vor, dass eine stationäre Säule eine Distanz von 10 Körnern des Materials benötigt, um zwei Substanzen in einem Gemisch voneinander zu trennen. Diese Distanz nennen wir Trennstufe. Die Säule hat nun eine Länge x, welche nun durch die Länge der Trennstufe mit den 10 Körnern die Anzahl der Trennstufen ergibt. Diese werden auf die Länge von einem Meter Säule hochgerechnet und als Theoretische Bodenanzahl pro Meter angegeben. Diese Größe ist ein Maß für die Effizienz einer Trennsäule. Vergleicht man zwei Säulen mit identischen Phasen unter optimalen Bedingungen, so unterscheiden sie sich durch ihre Bodenzahl voneinander. Die mit der höheren Bodenzahl wird eine bessere Auflösung, eine bessere Auftrennung der Substanzen liefern. Die schlechtere wird möglicherweise keine Basislinientrennung liefern. Durch Säulen mit hoher Trennleistung erreicht man niedrigere Nachweisgrenzen und erzeugt geringere Bandenbreiten, wodurch schmalere, intensivere Peaks entstehen, welche ein besseres Signal-Rauschverhältnis haben. Die Anzahl der theoretischen Trennstufen Nth ergibt sich aus der Verbreiterung des Substanzpeaks und dessen Retentionszeit: tR Nth = 5, 54 · ( )2 (2.16) wh wobei tR der Retentionszeit der Testkomponente und wh der Peakbreite auf halber Peakhöhe entspricht. Die Kenngröße des Höhenequivalents eines theoretischen Bodens h wird auch als HEPT8 bezeichnet und wird mit dem Verhältnis von Säulenlänge zur Anzahl der theoretischen Böden angegeben: h = HEP T = l Nth (2.17) Die Angabe ist eine rein theoretische Größe. Je kleiner sie ist, desto höher die chromatographische Auflösung. 6 K. Cammann, Instrumentelle Analytische Chemie, S. 6-13, Abb. 6.7 Zusammensetzung der mobilen Phase, Säulentemperatur, Qualität der stationären Phase 8 Height equivalent to a theoretical plate 7 11 Nach Gleichung 2.9 lässt sich die Standardabweichung σ durch die Peakbreite auf halber Peakhöhe berechnen. Quadriert man σ, so erhält man die Varianz σ 2 , welche mit der Retentionszeit tR eines Analytpeaks im folgenden Zusammenhang zur Bodenzahl N der Säule steht: N= t2R σ2 (2.18) Substituiert man σ nun durch den Zusammenhang von Gl. 2.8 oder Gl. 2.9, so erhält man tR t2R 2 · 4 = 16 · ( )2 2 w wb b (2.19) tR t2R · 2, 3542 = 5, 54 · ( )2 wh wh2 (2.20) N= N= 2.3.1 Van Deemter Kurve Die theoretische Bodenzahl h ist abhängig von der Strömungsgeschwindigkeit u der mobilen Phase. Die Abhängigkeit wurde von Van Deemter erstmals beschrieben, der sie für die Gaschromatographie allgemein entwickelte: B HEP T = A + + C · u (2.21) u Die Van Deemter Kurve beschreibt die Abhängigkeit der Bodenhöhe von der Strömungsgeschwindigkeit. Die in schwarz dargestellte Kurve zeigt den Zusammenhang zwischen der linearen Strömungsgeschwindigkeit u der mobilen Phase und der Säule. Die Größen A, B und C beschreiben Einflüsse, welche Bandenverbreiterungen bewirken. Die Kurve hat die Form einer Hyperbel. A beschreibt sogenannte Dispersionseffekte9 . Es handelt sich hierbei um eine konstante Größe, welche nicht von der Strömungsgeschwindigkeit abhängig ist. Verringert man Abbildung 2.3 – Van Deemter Kurve.[2, S. die Trennstufenhöhe h, so erreicht man eine verbesserte 6-9, Abb. 6.5] Trennleistung, indem man den Teilchendurchmesser erniedrigt und die Homogenität der Packung der stationären Phase erhöht. In der Gaschromatographie kann A aufgrund seines geringen Einflusses vernachlässigt werden. Der isolierte Einfluss dieses Faktors wird durch die grüne Kurve beschrieben. B beschreibt die molekulare Diffusion entlang der Säulenachse. Die Größe der Körner der stationären Phase ist dabei nicht relevant, die Struktur der Körner jedoch umso mehr. Sind sie wenig verzweigt, so wird dieser Parameter besonders niedrig. In der Flüssigkeitschromatographie kann dieser Faktor vernachlässigt werden. Die rote Kurve repräsentiert diesen Einfluss. C enthält die Geschwindigkeit der Gleichgewichtseinstellung, welche dem Stofftransport in der mobilen und stationären Phase entspricht. Kleinere Teilchendurchmesser begünstigen den Massenaustausch. Bei größeren Durchmessern nimmt die Trennleistung erheblich ab. Man nennt dies auch Porendiffusion, da die Analytteilchen sich in den Poren der stationären Phase verfangen können und deshalb 9 Streu- und Eddy-Diffusion 12 unterschiedlich schnell durch die Säule wandern. Dieser Faktor ist sowohl in der GC als auch in der LC von entscheidender Bedeutung. Für diesen Einfluss steht die blaue Kurve im Diagramm ??. Der Faktor C setzt sich additiv aus den Massentransferkoeffizienten der stationären und mobilen Phase zusammen: C · u = Cs · u + Cm · u (2.22) Die Verzögerung des Massentransfers Cs durch die stationäre Phase kann für feste Phasen vernachlässigt werden, da die Übergange zwischen den Phasen in diesem Falle schnell verlaufen. Bei flüssigen stationären Phasen hängt der Massentransfer von der Diffusionsgeschwindigkeit Ds und der Filmdicke ab. Die Stoffaustauschvorgänge in der mobilen Phase werden durch den Koeffizienten Cm beschrieben. Dieser ist antiproportional zum Diffusionskoeffizienten DM . Man möchte einen möglichst kleinen Massentransferkoeffizienten haben, daher wählt man in der GC gern Trägergase mit hohem DM , wie Wasserstoff oder Helium. In der LC wird die antiproportionale Abhängigkeit jedoch noch stärker deutlich, da DM temperaturabhängig ist, erhöht man hier gern die Temperatur, um eine Optimierung der Lineargeschwindigkeit u zu erreichen. Zudem spielt die Porengröße der Körner der stationären Phase eine entscheidende Rolle. Die Analytteilchen bleiben in diesen Poren hängen und kommen stark verzögert am Ende der Säule an. Deshalb wählt man meist Körner mit möglichst glatten Oberflächen. Mithilfe der Van Deemter Gleichung 2.3.1 lassen sich chromatographische Systeme gezielt optimieren. Die optimale Strömungsgeschwindigkeit und damit die geringste Bandenverbreiterung erhält man am Minimum der schwarzen Kurve. Die Konstante C bestimmt die Steigung der Kurve bei hohen Geschwindigkeiten, also im rechten Teil. Bei zu hoher Fließgeschwindigkeit kann keine ausreichende Adsorption stattfinden. Zusammengefasst kann man sagen, dass die Eddy-Diffusion (s. Abschnitt 2.1.2), die Molekulardiffusion und die Stoffaustauschverzögerung die Hauptursachen für die Peakverbreiterung sind. 2.4 Dünnschichtchromatographie „In der Dünnschichtchromatographie (DC oder auch TLC10 ) werden Substanzgemische durch unterschiedliche Verteilung der Komponenten zwischen einer mobilen flüssigen und einer stationären Phase getrennt, wobei der Trennvorgang im Gegensatz zur bisher besprochenen Säulenchromatographie in einem offenen System, der planaren Trennschicht abläuft. Die etwa 200µm dicke stationäre Phase befindet sich auf einer meist quadratischen ebenen Platte, meist aus Glas. Am unteren Plattenrand wird eine Lösung der zu trennenden Probe mit einer Mikropipette aufgebracht. Nach dem Abdampfen des Lösungsmittels wird die planare Trennschicht in eine Trennkammer gestellt, an derem Boden sich die mobile Phase befindet, die hierbei als Fließ- oder Laufmittel bezeichnet wird. Durch Kapillarkräfte steigt die mobile Phase von der Eintauchzone über den Startfleck nach oben auf und nimmt dabei die Substanzen mit, die durch unterschiedliche Wechselwirkungen mit der stationären Phase retardiert werden. Zur Förderung der mobilen Phase wird keine Pumpe benötigt, die mobile Phase wandert durch einfach Kapillarwirkung über die trockene stationäre Phase. Nachdem die Front des Laufmittels eine ausreichende Strecke zurückgelegt hat, wird die Platte aus der Trennkammer genommen und getrocknet. Die augetrennten Substanzen verlassen nicht die Trennstrecke, sondern werden auf dem chromatographischen Bett nachgewiesen. Es handelt sich um ein sogenanntes inneres Chromatogramm11 .“ 10 11 Thin Layer Chromatography J. Becker, Chromatographie, S. 259 13 2.4.1 Stationäre und mobile Phasen In der Flüssigchromatographie unterscheidet man im wesentlichen zwischen zwei Arten von stationären Phasen. Die Normalphase (NP) und die Umkehrphase (RP12 ) sind in der DC und HPLC die wohl am weitesten verbreiteten. In den heutigen Anwendungen werden in der Regel nur die RPs eingesetzt, da man mit ihnen ein breites Spektrum an organischen Analyten mit unterschiedlicher Polarität trennen kann. Bei den Normalphasen trifft man im Allgemeinen auf poröse anorganische Adsorbenzien, wie Siliciumgel (Kieselgel, SiO2 ) oder Aluminiumoxid (Al2 O3 ), welche an ihrer Oberfläche polare Hydroxylgruppen angelagert haben. Siliciumdioxid tritt häufiger auf, da es kostengünstiger und leichter zu beschaffen ist. Die Probenkapazität ist zudem höher, als bei Aluminiumoxid, das zudem noch häufig mit den Analyten reagiert. Für ein optimales Trennergebnis ist es wichtig, dass die Körner möglichst gleichmäßig und vom Durchmesser nahezu identisch sind. In der Flüssigchromatographie (LC) wird die mobile Phase Eluent genannt. Dieser ist in der Normalphase stets unpolar (z.B. Pentan, Hexan, Heptan, Octan). Wird ein stark adsorbierter Analyt nicht mehr ausreichend von der stationären Phase gelöst, so kann dem Eluenten ein stärker polares Lösungsmittel13 hinzugegeben werden, um die Adsorbtion aufzuheben. Noch polarere Lösungsmittel sind Methanol oder Acetonitril. Die Polarität der mobilen Phase spielt eine entscheidende Rolle, da sie mit dem Verteilungskoeffizienten (vgl. Gl. 2.1) korreliert. Sie ist ein quantitatives Maß dafür, wie sehr das Gleichgewicht der Analytverteilung auf Seiten der mobilen Phase liegt. Ist ihre Elutionskraft zu stark, der Verteilungskoeffizient zu niedrig, so findet kaum noch eine Wechselwirkung mit der stationären Phase statt und der Analyt erreicht nahezu ungetrennt das Ende der Säule oder des Trennbettes. Die Elutionskraft hängt jedoch stets von der Polarität der stationären Phase ab. Es gibt sowohl für die RPs als auch die NPs sogenannte Elutrope Reihen, in denen man die Elutionskraft nachlesen kann. Für die NP sieht die Elutrope Reihe beispielsweise wie folgt aus: Pentan/Hexan < Cyclohexan < Toluen < Trichlormethan < Dichlormethan < Tetrahydrofuran < Dioxan < Acetonitril < Ethanol < Methanol Wasser Dies bedeutet, dass mit Pentan oder Hexan als Eluent die längsten Retentionszeiten erreicht würden, da die Analyten den Aufenthalt in der stationären Phase bevorzugen würden. Wasser hingegen würde die Analyten ohne nennenswerte Wechselwirkung ungetrennt durch die Säule transportieren. Die meisten stationären Phasen basieren auf Kieselgel (Siliciumdioxid, SiO2 ). Bei den sogenannten Umkehrphasen liegt ebenfalls eine Schicht von Kieselgel vor, welche jedoch durch lange Alkylketten chemisch modifiziert wurden, sodass nicht mehr die polaren Hydroxylgruppen die Oberfläche der festen Phase bedecken. Dadurch wird diese Phase unpolar. Entsprechend werden polare mobile Phasen benötigt. Die Länge der Alkylketten lässt sich beliebig variieren, wodurch eine hohe Präzision im Bezug auf das Analyseergebnis erzielt werden kann, weil man die Eigenschaften der stationären Phase sehr genau einstellen kann. Die gebräuchlichsten mobilen Phasen in der RP-Chromatographie sind Wasser, Methanol, Acetonitril und Tetrahydrofurat. Sie können in Reinform oder gemischt verwendet werden. Die oben gezeigte Elutrope Reihe hat in der RP-Chromatographe den umgekehrten Verlauf. Die Methode wird in der Regel zur Trennung von neutralen wasserlöslichen Verbindungen genutzt. Polare stationäre Phasen werden mit unpolaren Fließmittelsystemen (z.B. Chloroform) kombiniert. Unpolare stationäre Phasen werden mit polaren Fließmitteln verwendet. 12 13 Reversed Phase Tetrahydrofuran, Dichlormethan 14 Vor der Verwendung von Kieselgel als polare stationäre Phase (NP) wird dieses durch eine Erhitzung auf 150 ◦ C aktiviert. Dadurch wird das adsorbierte Wasser verdampft. Neben den durch Alkylketten (Silanen) modifizierten Kieselgele gibt es noch die CN -, Diol- und N H2 Phasen, die weniger polar als Kieselgel, jedoch polarer als die klassischen RP-Phasen sind. Bei diesen Phasen legt das Fließmittel fest, ob es sich um ein NP oder RP-System handelt. Cellulose wird in speziellen Fällen wie der Verteilungschromatographie hydrophiler Substanzen eingesetzt. 2.4.2 Funktionsprinzip In der DC wird die stationäre Phase in Form einer dünnen Schicht auf eine Trägerplatte aus Glas, Kunststoff oder Aluminium aufgebracht. In manchen Fällen ist eine Fixierung mit Bindemitteln notwendig. Bei der Verwendung von Kieselgel oder Cellulose ist dies nicht nötig. Nach erfolgter Trennung detektiert man die Analytsubstanzen meist mit UV-Indikatoren. Diese bestehen aus durch Mangan aktiviertes Zinksilikat, das durch UV-Bestrahlung grün-gelb fluoresziert. Der Bereich, in dem sich eine Substanz befindet, erscheint als dunkler Fleck, da die Analytsubstanz in diesem Bereich das UV-Licht absorbiert. Die mobile Phase wird durch Kapillarkräfte über das chromatographische Bett, die stationäre Phase gezogen. Durch das entstehen eines sogenannten inneren Chromatogramms auf dem chromatographischen Bett erhält man eine räumliche Auftrennung, anders als bei der GC oder HPLC. Deshalb gibt es in der DC spezielle Retentionsparameter. In der DC setzt man die Wanderstrecke der Substanz in ein Verhältnis zur Wanderstrecke des Fließmittels (Laufmittelfront). Dieser Parameter wird Verzögerungsfaktor oder RF -Wert14 genannt. RF = b a (2.23) wobei b die Wanderstrecke der Substanz ist und a die des Laufmittels ist. RF ist dimensionslos und stets < 1, da sonst die Substanz vor der Laufmittelfront wäre. Der Parameter ist abhängig 15 Abbildung 2.4 – RF -Wert in einem von der Schichtdicke der stationären Phase , der TempeDünnschicht-Chromatogramm.[6, Kapitel 1, ratur, bei der die Trennung durchgeführt wird, die SätS. 2] tigung des Kammervolumens mit den Lösemitteldämpfen der mobilen Phase und der aufgegebenen Probenmenge16 . Analog dazu kann man das Verhältnis einer Substanz zu einem Standard aufstellen. Hier wird der Standard als Laufmittelfront eingesetzt. Die Erhöhung der theoretischen Bodenzahl N kann aufgrund der fehlenden Variationsmöglichkeiten der Fließgeschwindigkeit der mobilen Phase nur durch die Teilchengröße der stationären Phase verändert werden. Je kleiner die Partikel und je enge ihre Korngrößenverteilung, desto besser die Trennlei14 relate to front In der DC liegt die Schichtdicke zwischen 0,20 und 0,25mm. 16 Kammersättigung: Sättigung der Kammer mit Lösungsmitteldämpfen. Ist die Kammeratmosphäre während der chromatographischen Trennung nicht gesättigt, nimmt die Konzentration durch das Verdampfen des Lösungsmittels zu und beeinflusst die Trennung. Für eine bessere Sättigung wird die Kammer mit einem mit Laufmittel getränkten Filterpapier ausgekleidet. 15 15 stung. Dadurch wird die Oberfläche erhöht, wodurch eine intensivere Wechselwirkung zwischen Probe und stationärer Phase resultiert. Die Adsorptionskraft nimmt zu und mit ihr die Retention. Die Probenaufgabe ist einer der Schritte, bei denen die schwerwiegensten Fehler geschehen können. Zunächst besteht die Gefahr des Aufbringens zu großer Probenflecken, sowie der Überladung der Platte durch eine zu große Probenmenge. Die Auftragung erfolgt in der Regel über eine Glaskapillare oder über einen Autosampler. Die Proben müssen mit etwas Abstand zur Kante des chromatographischen Betts aufgetragen werden, damit sie nicht in der Kammer in das Laufmittel eintauchen. Der Durchmesser des Probenflecks muss möglichst gering sein, damit der entstehende Substanzfleck bei der Entwicklung nicht zu diffus wird. Aus diesem Grund verwendet man in der Analytik heute auch in der DC Autosampler-Systeme. Als Entwicklung versteht man in der DC den Prozess der Auftrennung der Substanzen durch die Wanderung der Laufmittelfront entlang der stationären Phase. Man unterscheidet dabei die lineare und radiale Entwicklung, wobei erstere am weitesten verbreitet ist. Bei der linearen Entwicklung werden die Probensubstanzen als kleine Flecken oder schmale Streifen appliziert. Anschließend wird die Platte mit den applizierten Proben getrocknet und in eine Entwicklungskammer gegeben. Die Platten können dabei liegen (Linearkammer) oder aufgestellt sein (Flachboden-, Doppeltrog- oder Sandwichkammern). Gelingt eine Trennung nicht oder nur mit geringem Erfolg, so kann die Entwicklung mit dem selben oder mit anderen Fließmitteln wiederholt werden. Ebenso kann man zunächst ein starkes Elutionsmittel werden, die mobile Phase jedoch nur bis zur Hälfte der Platte wandern lassen und anschließend ein schwächeres Elutionsmittel einsetzen. Dadurch werden stark retardierende Substanzen gut separiert, die schwächeren werden erst durch den zweiten Schritt getrennt. Ähnlich funktioniert das Prinzip der Gradientenelution in der HPLC. Heute sind die Mehrfachentwicklungen durch sogenannte AMD17 -Geräte automatisiert. Die Elutionskraft der mobilen Phase nimmt stetig ab, sodass ein Stufengradient entsteht. Nach der Entwicklung folgt die Detektion. Meist weisen die Analyten keine besondere Eigenfärbung auf. Pflanzenpigmente, Lebensmittelfarben und Farbstoffe bilden hier die große Ausnahme. Manche Stoffe besitzen auch die Eigenschaft, unter bestimmten Wellenlängen zu fluoreszieren. Hierzu gehören die polycyclischen aromatischen Kohlenwasserstoffe (PAKs), Aflatoxine18 oder Riboflavin19 . Viele kommerziell erhältliche DC-Platten sind mit einem UV-Indikator beschichtet, sodass Verbindungen mit geeigneten Absorptionsmaxima bei 254 oder 366nm als dunkle Spots erscheinen. Manche Substanzen weisen diese Absorptionsmaxima jedoch nicht auf. In diesem Falle müssen sie durch chemische Behandlung sichtbar gemacht werden. Dies kann durch eine Sprühlösung oder ein Tauchbad geschehen. Bei organischen Verbindungen wendet man eine Sprühlösung mit Schwefel-, Essig- oder Salpetersäure bzw. Natriumdichromat an, sodass die Substanzen als braun-schwarze verkohlte Flecken erscheinen. Diese Anwendung bietet sich vor allem bei Proben unbekannter Zusammensetzung an. Außerdem gibt es noch Derivatisierungsreagenzien, welche spezifisch auf bestimmte funktionielle Gruppen reagieren. Die Kenntnis der chemischen Hintergründe ist dabei besonders wichtig, da es häufig zu Querempfindlichkeiten kommen kann. Die Anwendung von Derivatisierungschemikalien ist eine destruktive Methode und deshalb ungeeignet, wenn die Substanzen noch weitere Analyseschritte durchlaufen sollen, da ihre chemische Struktur durch die Derivatisierung verändert wird. Vor allem in der präparativen DC stellt dies ein Problem da, welches zum Teil durch die Anwendung von Iod vermieden werden kann. Das Iod löst sich in den Analysespots und färbt diese braun. Fast alle organischen Verbindungen bis 17 automated multiple development Pilzgifte 19 Häufig als Vitamin B2 bezeichnet. 18 16 auf einige gesättigte Alkane können so detektiert werden. Durch Abdampfen kann man die Färbung wieder vollständig rückgängig machen. Mithilfe der DC lassen sich besonders gut verschmutzte und schlecht detektierbare Proben untersuchen. Zudem kann man mehrere Proben simultan trennen. Die Probenvorbereitung ist weniger aufwändig als bei den anderen Analyseverfahren und wird daher besonders häufig in der Pharmazie zur Chargenüberprüfung und Reinheitskontrolle angewendet. In der Umweltanalytik wird in Europa die Bestimmung von Pflanzenschutzmitteln in Trinkwasser sogar nach DIN-Norm mittels DC nachgewiesen. Ebenso lassen sich anabole Steriode, welcher meist illegal als Wachstumsförderer in der Rinderzucht anwendet werden, durch die DC nachweisen. 2.4.3 Hochleistungsflüssigkeitschromatographie Die HPLC stellt eine ideale Ergänzung zur GC dar. Für die GC müssen die Analyten vollständig und unzersetzt verdampfen. Dies ist für die HPLC nicht notwendig, sodass die Probenaufgabe schonender gestaltet werden kann. Manche Analyte sind für die GC gänzlich ungeeignet. Dies ist besonders in der Bioanalytik häufig der Fall, da hier häufig thermisch labile Stoffe untersucht werden, die durch das Verdampfen beschädigt oder zerstört werden. Ein Lösungsmittelgemisch, die mobile Phase, wird nach der Entgasung durch eine oder mehrere Pumpen in das System befördert. Hinter der Pumpe wird die gelöste Probe injiziert. Das Lösungsmittel der Probe ist mit der mobilen Phase mischbar. Die chromatographische Trennung erfolgt auf der chromatographischen Säule, welche ggf. durch einen Säulenofen temperiert werden kann. Nach erfolgter Trennung können die Probenbestandteile durch einen entsprechenden Detektor nachgewiesen werden. Die mobile Phase Die Entgasung der mobilen Phase ist notwendig, da durch die Kompression durch die Pumpe gelöste Gase wie Stickstoff und Sauerstoff ausgasen können. Dies kann Auswirkungen auf die Reproduzierbarkeit der Analyseergebnisse haben. Ebenso können die gelösten Gase erst im Detektor ausgasen, sodass eine präzise Detektion nicht mehr möglich ist. Typisch dafür sind unruhige Grundlinien oder spontane, nicht reproduzierbare Peaks (Spikes). Im Falle der Fluoreszenzdetektion kann es durch den ausgegasten Sauerstoff zur Auslöschung der Fluoreszenz führen. Zum Entgasen eignen sich Ultraschallbäder. Teilweise sind diese on-line, also in das System bereits integriert, welche in der Regel mit hochreinem Helium betrieben werden. Bei der Pumpe hat man in der HPLC hohe Ansprüche, da es von entscheidender Bedeutung ist, dass die Flussrate konstant gehalten wird. Gerade bei Gradientenprogrammen muss es jedoch auch möglich sein, die Flussraten je nach Zusammensetzung der mobilen Phase variieren zu können. Die Bauteile der Pumpe müssen besonders korrosionsbeständig sein. In der Bioanalytik sind diese Voraussetzungen noch verschärft, die Bauteile sind entsprechend teurer. Die Salzbildung in den Bauteilen kann durch eine Kolbenhinterspülung bzw. durch Spülung mit destilliertem Wasser verringert oder sogar vermieden werden. Häufig wird in der HPLC die Gradientenelution eingesetzt. Die Elutionskraft der mobilen Phase wird dabei kontinuierlich in eine Richtung verändert. Dadurch erhält man schärfere Peaks bei kürzeren Laufzeiten, auch bei komplexen Gemischen mit stark unterschiedlich polaren Substanzen. Zunächst wird eine schwach eluierende Ausgangslösung eingesetzt, der nun eine steigende Konzentration von stärkeren Eluentien zugemischt wird. Dadurch werden zuerst die Substanzen detektiert, welche sich 17 wenig an die stationäre Phase binden. Steigt die Elutionskraft, so lösen sich im weiteren Verlauf auch zunehmend die Substanzen, welche eine stärkere Bindung zur stationären Phase aufgebaut haben. Sie werden durch die zunehmende Elutionskraft schneller eluiert und benötigen weniger Zeit, um den Detektor zu erreichen. Bei der Mischung der Lösungsmittelkomponenten unterscheidet man im Wesentlichen zwei Systeme. Im Hochdruckgradientensystem werden die Komponenten jeweils von einer separaten Pumpe angesaugt und in einer Mischkammer hinter dem Pumpensystem erst gemischt. Im Niederdruckgradientensystem findet die Mischung vor der Pumpe statt, wodurch man bis zu vier verschiedene Lösungsmittel miteinander mischen kann. Das Niederdrucksystem ist im Falle von ternären oder quarternären Eluentengemischen von Vorteil, da es deutlich preisgünstiger ist und mit nur einer Pumpe auskommt. Zudem bietet es den Einsatz einer Mischkammer mit sehr kleinem Volumen und dadurch geringer Verzögerung bis zur Einleitung in die Säule. Allerdings muss die mobile Phase in jedem Fall entgast werden, da das System empfindlicher ist. Bei sekundären Eluentengemischen genügt ein Hochdrucksystem. Die klassische Probenaufgabe in der HPLC geschieht mit einem Sechswege-Ventil. Dabei wird ein zunächst zu großes Volumen in die Probenschleife gespritzt. Diese Probenschleife hat ein bestimmtes Volumen (meist etwa 10µL), die überschüssige Probenflüssigkeit gelangt am Ende der Probenschleife in ein Abfallgefäß. Die mobile Phase strömt währenddessen kontinuierlich durch die Säule. Ist die Probenschleife vollständig gefüllt, stellt man das Ventil auf Injizieren. Nun wird die Probenschleife mit der Trennsäule verbunden und die bestimmte Menge Probenvolumen gelangt in das System. Die mobile Phase strömt von hinten durch die ProbenAbbildung 2.5 – Probenaufgabe mit einem schleife hinterher. Diese Ventile gibt es sowohl maSechswege-Ventil. [6, Kapitel 3, S. 2] nuell als auch im Autosampler automatisiert. Dennoch ist das Nennvolumen der Probenschleife recht ungenau, sodass man gerade bei Kalibrationsstandards auf ein exaktes Probenvolumina achten sollte. Die stationäre Phase Das Prinzip der stationären Säule ist in der HPLC ähnlich dem in der DC (vgl. 2.3.1). Auch hier wird als gängigsten Material Siliziumdioxid (SiO2 ) verwendet, welches in Form, Partikelgröße und Porositäten sehr variabel ist. Zudem kann es durch chemische Reaktionen modifiziert werden, wodurch man aus dem eigentlich polaren Siliziumdioxid eine Umkehrphase erzeugen kann, welche dann unpolar oder zumindest weniger polar ist. Die Eigenschaften der Umkehrphase lassen sich dann durch weitere chemische Veränderungen an die Analyse anpassen. Durch sogenanntes End-Capping können nicht modifizierte OH-Gruppen des Siliziumdioxids verringert werden, sodass die stationäre Phase noch unpolarer wird. Siliziumdioxid ist recht oberflächenaktiv und kann nur in einem bestimmten pH-Bereich eingesetzt werden. Im alkalischen Bereich (ab pH >8) müssen daher andere Materialien verwendet werden, da 18 sich sonst die stationäre Phase zersetzt. Durch den Einsatz von voluminösen Seitengruppen kann die ungewollte Empfindlichkeit gegen chemische Reaktionen verringert werden, da die mobile Phase mit den Substanzen nicht mehr an das Silica-Gerüst gelangt. Ein Beispiel dafür sind Ethylenbrücken, welche sich stabilisierend auswirken und somit den Anwendungsbereich auf einen pH-Bereich zwischen 1 und 12 erweitern. Neben Siliziumdioxid werden auch Aluminiumoxid (Al2 O3 ) oder Zirkonia (ZrO2 ) verwendet. Aluminiumoxid besitzt im Vergleich zum Kieselgel jedoch eine größere Oberflächenaktivität und wird daher nur selten als stationäre Phase eingesetzt. Zirkonia ist temperaturstabiler und erträgt einen größeren pH-Bereich. Die chemische Modifikation zur Umkehrphase ist ebenfalls möglich. In manchen Bereichen werden zudem Polymere eingesetzt. Sie sind zwar stabiler im Bezug auf den pH-Bereich, schwächer jedoch in der mechanischen Belastbarkeit bei hohen Drücken. Außerdem besteht die Gefahr der Reaktion mit der mobilen Phase, wodurch ein Schrumpfen oder Aufquellen der stationären Phase auftreten kann. Bei guter Pflege kann eine HPLC-Säule mehrere tausend Trennungen überstehen. Unabdingbar ist deshalb die ständige Kontrolle anhand von Standards im Bezug auf Peakform, Retentionszeit und Auflösung. In manchen Systemen werden zudem zur vorherigen Reinigung Vorsäulen mit identischer stationärer Phase eingesetzt, welche solche Partikel binden, die sich sonst an der Hauptsäule anlagern würden. Detektion In der HPLC kann man die Detektoren nach ihren Eigenschaften in zwei Gruppen unterteilen. Zum einen jene, die nur das Gelöste detektieren und zum anderen jene, die sogenannte Bulk-Eigenschaften, also die Summe der Eigenschaften von Gelöstem und Lösungsmittel nachweisen. Die Eignung des Detektors ist abhängig von der geplanten Anwendung und kann nicht universell festgelegt werden. Allgemein beurteilt man die Eignung eines Detektors nach den folgenden Kriterien: • Nachweisgrenze Die Nachweisgrenze eines der Leistungskriterien eines Detektors. Nicht immer ist die niedrigste Nachweisgrenze von Vorteil, da bei hohen Konzentrationen dann meist mehrere Verdünnungsschritte notwendig sind. • Linearer Messbereich Je nach Anwendung ist die Größe des linearen Messbereichs, in dem die Kalibrierkurve gilt, entscheidend. In der Umweltanalytik sind Schwankungen in Dekadengröße häufig, daher muss der Messbereich besonders groß gewählt sein. In der Medizin sind die Schwankungen deutlich geringer, sodass hier ein kleiner Messbereich bereits ausreichend ist. • Kompatibilität mit der mobilen Phase Diese ist vor allem beim Einsatz eines Konzentrationsgradienten bei der mobilen Phase wichtig. Verändert sich die Konzentration der mobilen Phase, darf dies keine Auswirkungen auf das Chromatogramm haben. Gerade bei Bulk-Detektoren treten hier Probleme auf, da diese auch die Veränderungen der mobilen Phase detektieren. Ähnliche Probleme treten bei der UV-vis-Spektrometrie auf, bei der spezielle UV-transparente Eluenten eingesetzt werden müssen. • Selektivität Je komplexer eine Probe zusammengesetzt ist, desto selektiver muss der Detektor sein, um eine Auftrennung noch erkennen zu können. Es gibt Detektoren, die selektiv auf bestimmte Moleküle, Atome oder funktionelle Gruppen reagieren. Ebenso der Nachweis von Chemilumineszenz, welche sehr selten ist, zählt zu den selektivsten Methoden. Massenspektrometrische Detektoren können atomselektiv detektieren und stellen die Königsklasse der Selektivität 19 von Detektoren dar. Bulk-Detektoren, welche den Brechungsindex oder das Verdampfungslicht messen, sind am wenigsten selektiv. • Flexibilität Meistens wird ein HPLC-System nicht nur für eine einzige Art von Analyse eingesetzt. Daher muss der Detektor flexibel auf die jeweilige Methode einstellbar sein. Bei UV-visDetektoren kann man so die gewünschte Wellenlänge einstellen, weshalb sie zu den Standarddetektoren in der HPLC gehören. • Robustheit und Preis Das Gerät muss einfach zu bedienen sein, um in der alltäglichen Laborroutine einsetzbar zu sein. Der Schulungsaufwand so wie die Wartungsarbeiten müssen möglichst gering gehalten werden. Ähnlich gilt dies für den Preis eines Detektors. Das kleinste vom Detektor erfassbare Signal kann nicht kleiner sein, als die doppelte Höhe des größten Rauschpeaks, da man sonst Peak und Rauschen20 nicht mehr voneinander unterscheiden kann. Die Detektorempfindlichkeit wird wie folgt berechnet: S= Ei ci (2.24) Wobei Ei die Peakhöhe und ci die Konzentration der Substanz in der mobilen Phase am Detektor darstellt. Beim Einschalten des Systems kann man das Driften der Nulllinie beobachten, die eine annähernd linieare Steigung hat. Dieser Anstieg der Nulllinie ist zu Beginn normal, da sich das System erst aufwärmen und equilibrieren muss. Der Arbeitsbereich, in dem der Detektor ein Signal in Abhängigkeit von der Stoffmenge erzeugt, entspricht theoretisch einer Geraden, die unten von der Nachweisgrenze begrenzt wird. Im oberen Bereich gibt es theoretisch keine Grenze, praktisch tritt diese jedoch auf, sodass man von einem Linearen Bereich spricht, in dem man detektieren kann. Der Dynamische Bereich beschreibt den kalibrierbaren Bereich des Detektors über den linearen Bereich hinaus. Auch, wenn dort keine Linearität mehr vorliegt, kann man dort bis zu einer gewissen Stoffmenge eine reproduzierbare Signaländerung beobachten. Es gibt eine ganze Reihe von Detektionsmöglichkeiten, auf die hier jedoch nur der Vollständigkeit halber eingegangen wird. Bei der Verdampfungs-Lichtsteudetektion (ELSD21 ) wird die Lichtstreuung an Partikeln gemessen, welche nach Verdampfung der mobilen Phase noch übrig bleiben. Es sind nur solche Analyten detektierbar, die einen signifikant höheren Siedepunkt als die mobile Phase haben. Bei schwerflüchtigen Analyten stößt diese Methode an ihre Grenzen. Die Brechungsindexdetektion ist eine klassische Universalmethode. In einem Durchfluss-Refraktometer22 wird der Brechungsindex des Eluats aus der chromatographischen Säule gemessen. Der erhaltene Brechungsindex setzt sich dann zum größten Teil aus dem Index der mobilen Phase und nur zu einem Teil aus dem Index des Analyten zusammen. Aus diesem Grund werden Differentialrefraktometer eingesetzt, welche das Gesamtsignal der mobilen Phase subtrahieren können. Diese Detektionsmethode ist dementsprechend nicht für die Gradientenelution einsetzbar. Diese Detektionsmethode ist daher in den letzten Jahren durch den ELSD größtenteils ersetzt worden. 20 Noise: Ein Detektor zeigt stets ein gewisses Rauschen, welches physikalisch begründet ist. Meist werden damit Verunreinigungen der mobilen Phase angezeigt. Es können aber auch Netzstörungen oder falsche Erdungen vorliegen. 21 Evaporative Light Scattering Detector 22 Häufig wird ein solches Gerät zur Bestimmung des Zuckergehalts in Wein, zur Wassergehaltsmessung bei Honig oder zur Bestimmung der Stammwürze bei Bier eingesetzt. 20 2.5 Gaschromatographie Die Gaschromatographie umfasst alle Varianten der Chromatographie, in denen die mobile Phase gasförmig vorliegt. Sie ist äußert leistungsstark und wird aufgrund der guten Automatisierbarkeit standardmäßig in industriellen Kontrolllabors eingesetzt. Durch die Kopplung mit entsprechenden Detektoren lassen sich Analyten nicht nur voneinander trennen, sondern auch identifizieren. In der Flüssigchromatographie tritt der Analyt in Wechselwirkung mit der stationären und der mobilen Phase. In der Gaschromatographie dient die mobile Phase lediglich dem Transport der Analyten vom Injektor durch die Säule bis zum Detektor. In der Regel werden deshalb inerte Trägergase eingesetzt, mit denen keine Wechselwirkung stattfindet. Es lassen sich im Prinzip alle Substanzen analysieren, welche in einem bestimmten Temperaturbereich einen entsprechenden Dampfdruck aufweisen. Ist dies nicht der Fall, so kann die Substanz durch Derivatisierung23 leichter flüchtig gemacht werden. Auf diese Weise können Metallionen gaschromatographisch untersucht werden, in dem man sie zu Komplexen umwandelt. Durch Pyrolyse24 bei einer definierten Temperatur können leichter flüchtige Bestandteile einer Substanz isoliert und gemessen werden, sodass Polymere und Kohle untersucht werden können. Die Nachweisgrenzen in der GC sind sehr niedrig. Durch Kapillartrennsäulen können hochauflösende Chromatogramme erzeugt werden, sodass die GC weitaus effektiver als die DC/HPLC ist. Wichtig bei der GC ist nicht, dass die zu analysierende Substanz gasförmig vorliegt. Sie wird meist ohnehin in flüssiger Phase injiziert. Entscheidend ist ihr Dampfdruck. Dieser muss im Temperaturbereich der Säule liegen. Dieser Temperaturbereich kann weit unterhalb der Siedetemperatur des Analyten liegen. 2.5.1 Die Säule In der Gas-Flüssig-Chromatographie (GLC) ist ein dünner Film einer polymeren Flüssigkeit auf einem inerten Trägermaterial oder direkt auf der Säule als stationäre Phase aufgetragen. In der Gas-FestChromatographie (GSC) liegt eine gepackte Säule vor, wie sie auch in der HPLC anzutreffen ist. Die Säule selbst ist ein Rohr, in dem stets die Gleichgewichtseinstellung zwischen mobiler und stationärer Phase stattfindet. Es gibt sowohl gepackte Säulen wie auch in der HPLC angewendet, aber auch Film (WCOT25 )- und Schichtkapillaren (SCOT26 /PLOT27 ). Bei der gepackten Säule befindet sich die stationäre Phase als Film auf den Partikeln des Trägermaterials. Als Trägermaterial wird häufig die natürlich vorkommende Kieselgur (auf Siliziumdioxidbasis) verwendet, welche industriell aufgereinigt wird. Es werden unterschiedliche Korngrößen verwendet, um eine möglichst dichte Packung zu erreichen. Fertig gepackte Säulen können dann mit beliebigen stationären Phasen beschichtet werden, in dem man die flüssige stationäre Phase durch die Säule laufen lässt. Da die Trennleistung im Vergleich zu den Kapillarsäulen jedoch gering ist, verlieren die gepackten Säulen in der GC inzwischen zunehmend an Bedeutung. 23 Derivatisierung beschreibt eine chemische Veränderung einer Verbindung, die Auswirkung auf ihre chemischen und physikalischen Eigenschaften hat. So kann zum Beispiel eine funktionielle Gruppe ersetzt/entfernt/hinzugefügt werden. Die Stammverbindung bleibt dabei jedoch unverändert. 24 Thermische Zersetzung 25 Wall coated open tubular columns, stationäre Phase liegt als dünne Filmschicht direkt auf der Glaskapillare. Die Streudiffusion (A-Term in der van-Deemter Gleichung) geht gegen 0, da keine Kornzwischenräume vorliegen. 26 Support coated open tubular columns, in der Verteilungs-GC, stationäre Phase liegt als Filmschicht auf dem feinkörnigen Trägermaterial vor. 27 Porous layer open tubular columns, in der Adsorptions-GC angewendet, mit durchgehend porösen Adsorbentien beschichtet. 21 Die Durchmesser der Kapillarsäulen sind ein Bruchteil der gepackten Säulen. Sie bestehen aus einem dünnen Rohr und haben eine wesentlich größere Länge, da durch den geringen Durchmesser kaum Druckverlust des Trägergases zu beobachten ist. Solche Kapillaren sind etwa 10-60m lang, was zu einer enormen Trennleistung führt. Die Säulen bestehen in der Regel aus hochflexiblen Quarzglas, welches aufgerollt im Säulenofen platziert werden kann. Um die Säule vor thermischen Einflüssen zu schützen, ist sie von einer Polyimidschicht ummantelt. Diese Schicht gibt der Kapillarsäule ihre charakteristische gelb-braune Färbung. Werden höhere Temperaturen in der GC gefordert, so verwendet man aluminiumbeschichtete Säulen. Für die Kapillarsäulen gilt die van-Deemter Gleichung in abgewandelter Form als sogenannte GolayGleichung. B HEP T = A + + C · u (2.25) u B + CM · u + CS · u (2.26) HEP T = u HEP T = CM CS rc k df DS DM d2f 1 r2 1 + 6k + 11k 2 2k 2DM + · c ·[ · u] + · ·u u 24 DM (1 + k)2 3(1 + k)2 DS (2.27) Massenaustauschterm der mobilen Phase (Gasphase) Massenaustauschterm der stationären Phase (Rohrwand und Wirkphase) Innenradius der Säule Kapazitäts- oder Retentionsfaktor vgl. Gl. 2.3 Filmdicke der stationären Phase Diffusionskoeffizient des Analyten in der stationären Phase Diffusionskoeffizient des Analyten in der mobilen Phase Bei nicht zu hohen Filmdicken ist der Massenaustauschterm der stationären Phase viel kleiner als der der mobilen Phase. Daher kann CS in der Kapillar-GC in den meisten Fällen vernachlässigt werden. Je dicker die Filmschicht, desto tiefer kann der Analyt in die stationäre Phase eindringen und mit ihr wechselwirken. Die Folge ist ein größerer Massenaustausch und eine Peakverbreiterung, da der Analyt erst verzögert die Säule passiert. Den gleichen Effekt hat ein großer Kapillarendurchmesser. HEP T = 2DM 1 r2 1 + 6k + 11k 2 + · c ·[ · u] u 24 DM (1 + k)2 (2.28) Die Dimension einer Kapillare ist stets ein Kompromiss bezüglich der maximalen Trenneffizienz (Hohe Bodenzahl N), möglichst geringer Analysendauer, ausreichender Belastbarkeit (durch das Phasenverhältnis β Gl. 2.5) und nicht zu hohem Druckabfall bei optimaler Strömung. 22 ID28 (mm) 0,10 0,18 0,32 Länge (m) 10 Filmdicke (µm) 0,10 Phasenverhältnis β 250 25 25 0,20 0,50 225 160 Eigenschaften der Säule Großes Trennvermögen, kurze Analysenzeiten, sehr kleine Kapazität, nicht für große Konzentrationsunterschiede geeignet Kapazität ist etwas höher Universalsäule, mittleres Trennvermögen, kurze Analysezeiten, gute Probenkapazität für große Konzentrationsunterschiede Je geringer die Filmdicke und der Innendurchmesser, desto größer ist das Trennvermögen. In der GC hängt die Bodenhöhe H vom Trägergas ab. Wasserstoff ist durch seine hohe Fließgeschwindigkeit wesentlich effizienter als Stickstoff, allerdings durch seine Explosionsgefahr nachteilig. Stickstoff ist am kostengünstigsten, hat jedoch die höchste Viskosität, weshalb die Diffusion dort am langsamsten geschieht. Man erhält eine höhere Trennleistung bei besserer Auflösung. Dies sieht man am niedrigsten Minimum der van-Deemter-Kurve des Stickstoffs. Arbeitet man ohne Temperaturgradienten, so bietet sich die Nutzung von Stickstoff an. Bei Helium und Wasserstoff sinkt die Trennleistung bei steigendem Trägerfluss jedoch deutlich geringer, weshalb man bei höheren Trägergasflussgeschwindigkeiten bevorzugt Helium oder Wasserstoff nutzt. Abbildung 2.6 – van-Deemter-Kurven für die drei Trägergase Stickstoff, Helium und Wasserstoff.[6, Kapitel 9, S. 5] Als stationärer Phasen sind vor allem die Polysiloxane am gebräuchlichsten. Sie sind gut verarbeitbar und können durch Polymerisation an die Innenwand der Quarzkapillare gebunden werden. Durch Bindung von polaren Seitengruppen kann eine Umkehrphase erzeugt werden. Das Einfügen von Phenylresten bewirkt eine Erhöhung der Temperaturstabilität. Wie auch sonst in der Chromatographie gilt: Unpolare Analyten werden auf unpolaren Säulen getrennt. Polare Analyten auf polaren Säulen. 23 Das fundamentale Prinzip der GC-Trennung sind Aktivität und Dampfdruck. Ein niedriger Dampfdruck bedeutet einen hohe Siedepunkt. Dies sorgt für große Retentionszeiten, da der Analyt ein geringes Betreben hat, in der mobilen Phase in Lösung zu gehen. Der Aktivitätskoeffizient γ sagt aus, wie ein Analyt mit der stationären Phase wechselwirkt. Große γ bedeuten große Abstoßung, kleine γ große Anziehung bzw. geringe Abstoßung. α= k t0R p γ k2 t0 γ1 · p 1 = R2 = 0 k1 tR1 γ2 · p 2 (2.29) Retentions-/Kapazitätsfaktor der zu trennenden Analyten Netto-Retentionszeiten der Analyten, also die Verweildauer der Analyten in der stationären Phase Dampfdruck der Analyten Aktivitätskoeffizienten der Analyten Man kann an der Trennformel nach Herington sehen, dass die gaschromatographische Trennung aufgrund verschiedener Dampfdrücke p oder bei ähnlichen Siedepunkten aufgrund der verschiedenen Wechselwirkungen (γ) der Analyten mit der stationären Phase getrennt werden können. Bei homologen Reihen29 unterscheiden sich die Moleküle nicht mehr in ihren Aktivitätskoeffizienten, da ihre funktionellen Gruppen immer identisch sind. Im Chromatogramm könnte man sie daran nicht unterscheiden. Sie können nur durch ihre verschiedenen Dampfdrücke/Siedepunkte getrennt werden. Sind die Dampfdrücke und Siedepunkte jedoch zu ähnlich, trennt man aufgrund der verschiedenen Aktivitätskoeffizi- Abbildung 2.7 – Trennung anhand verenten, hervorgerufen durch die unterschiedlichen Wechsel- schiedener Dampfdrücke. [6, Kapitel 9, S. wirkungen der funktionellen Gruppen. Im nebenstehenden 9] Chromatogramm sieht man die Trennung von verschiedenen Xylol-Isomeren. Sie unterscheiden sich nur durch die Anordnung der CH3 -Gruppen. Ihre Siedepunkte sind nahezu identisch. Abbildung 2.8 – Trennung anhand der Dipol-Dipol Wirkung. [6, Kapitel 9, S. 9] Durch die verschiedenen Anordnungen der unpolaren Alkangruppen wirkt sich die Dipolwirkung des Gesamtmoleküls auf die Wechselwirkungen mit der stationären Phase unterschiedlich aus. Daraus resultieren verschiedene Aktivitätskoeffizienten. Verbindungen mit niedrigem Dipolmoment werden als erstes eluiert. Hohe Dipolmomente benötigen am längsten, da ihre Wechselwirkungen mit der Dipolphase am stärksten sind. Ähnlich funktioniert die Trennung durch die unterschiedlichen Einflüsse der Wasserstoffbrückenbindungen bei Phenolen. Auch hier sind die Siedepunkte nahezu identisch. Die Moleküle unterscheiden sich nur durch die verschiedenen Aktivitätskoeffizienten. Je elektronegativer der Bindungspartner, desto stärker sind die Brückenbindungen. 29 Stoffe mit gleicher allgemeiner Summenformel. Durch Hinzufügen eines weiteren Kettengliedes entsteht das nachfolgende Molekül. Bsp. Alkane. 24 Beim Phenol (2) ist nur das H-Atom der Hydroxygruppe zur Brückenbindung befähigt, daher kommt der Analyt als erstes. Die Wechselwirkungen mit der stationären Phase sind am geringsten. Bei 2,4-Dimethylphenol (8) und 2,5-Dimethylphenol (7) liegen noch zwei weiter Methylsubstituenten vor, welche Brückenbindungen aufbauen können. Sie benötigen deutlich länger, um die Säule zu passieren. 2,6-Dimethylphenol benötigt aufgrund seiner sterischen Anordnung weniger Zeit, da die Brückenbindungen weniger stabil sind. Abbildung 2.9 – Trennung anhand von verschiedenen Einflüssen der Wasserstoffbrücken. [6, Kapitel 9, S. 9] Die Säulentemperatur hat einen nicht zu unterschätzenden Einfluss auf die Auflösung und Retentionszeit. Allgemein kann man sagen, dass eine Erhöhung der Temperatur um 25◦ C eine Halbierung der Retentionszeit bewirkt. Die Temperaturerhöhung ist endlich, da ab einer gewissen Temperatur die Analyten und Materialien zersetzt werden. Die Messung mit einem Temperaturgradienten zur Erhöhung der Effizienz ist daher nur in den Fällen ratsam, in denen die Probenmaterialien eine solche Temperierung ohne chemische Veränderung überstehen. 2.5.2 Detektionsmethoden Wie auch in der bereits besprochenen HPLC ist der Detektor zuständig für das Erfassen der getrennten Analyten. Das Detektorsignal kann hier auch sowohl quantitativ als auch qualitativ genutzt werden. Anders als in der Flüssigkeitschromatographie sind die GC-Detektoren jedoch teilweise in der Lage, die in einer Verbindung vorhandenen Elemente zu identifizieren. Dadurch erhöht sich der Nutzen der GC um ein Vielfaches. In der GC unterscheidet man zwischen destruktiven und nicht-destruktiven Detektionen. Wärmeleitfähigkeits- und Infrarotdetektoren gehören zur ersten Gruppe und werden häufig in der präparativen GC eingesetzt, wenn die Proben nach der Trennung noch weiterverarbeitet werden müssen und daher unverändert vorliegen müssen. Flammenionisations-, massensensitive und flammenphotometrische Detektoren zerstören die Probe jedoch durch die Detektion, da diese durch eine destruktive chemische Reaktion erfolgt. Der Flammenionisationsdetektor (FID) wird aufgrund seiner universellen Einsetzbarkeit und einfachen Handhabung häufig in der GC eingesetzt. Er weißt eine hohe Zuverlässigkeit, einen großen linearen Messbereich und gute Nachweisgrenzen auf. Die aus der Säule austretenden Analyten werden in einer Wasserstoff-Luft-Flamme verbrannt. Nahezu alle Kohlenstoffatome werden zu Methan reduziert, welches schnell mit dem Sauerstoffanteil der Luft über verschiedene Radikale verbrannt wird. Die angeregten Sauerstoffverbindungen reagieren dann mit nicht verbrannten Kohlenstoffen, sodass Elektronen freiwerden. Diese werden gemessen. Vor allem für leichtflüchtige Kohlenstoffverbindungen ist diese Detektionsmethode ideal. Substanzen, die vor der Detektion bereits thermisch zersetzt werden, oder Verbindungen, die keine Reaktion mit den angeregten Sauerstoffverbindungen eingehen, sind nicht detektierbar. Der Stickstoff-Phosphor-Detektor (NPD) funktioniert ähnlich wie der FID, ist allerdings selektiv für 25 Stickstoff und Phosphor Verbindungen. Durch eine Keramikperle werden Alkali Ionen erzeugt, welche selektiv mit Stickstoff und Phosphorverbindungen reagieren. Es werden Elektronen frei, welche dann gemessen werden können. Der Elektroneneinfangdetektor (ECD) ist der dritte Detektor. Er wird häufig in der Schadstoffanalyse eingesetzt, da er bei manchen Analyten extrem niedrige Nachweisgrenzen aufweist. Eine radioaktive Nickelfolie erzeugt Primärelelektronen, welche durch Kollision mit dem Trägergas Stickstoff ein elektrisches Feld aufbauen, in dem die entstandenen Sekundärelektrone zur Anode wandern. Dies nennt man den Ionisierungsgrundstrom. Gelangt nun eine Analytsubstanz in dieses elektrische Feld, so kann sie durch ihre hohe Elektronenaffinität das elektrische Feld verändern. Diese Veränderung entspricht dem Detektorsignal. Die detektierbaren Stoffe müssen eine hohe Elektronenaffinität besitzen, da sonst keine Reaktion stattfinden würde. Zu diesen Stoffen gehören alle halogenierten und nitrierten Verbindungen, sowie solche mit elektronenziehenden funktionellen Gruppen. Diese klassische Betriebsart hat einen nur sehr kleinen linearen Bereich. Deshalb verwenden heutige ECD-Geräte eine Pulstechnik, mit der man eine variable Frequenz einstellen kann. Eine festgelegte Spannung wird über einen winzig kleinen Zeitraum über die Elektrode gelegt. In dieser kurzen Zeit erreichen nur die thermisch entstandenen Elektronen die Anode, die Analytionen, welche wesentlich schwerer sind, bleiben zurück. Die nun erhaltene Stromstärke wird gemessen. Wird ein Elektronen einfangender Stoff aus der Trennsäule in den Detektorraum eluiert, so nimmt er die freien Elektronen vermehrt auf, sodass sie nicht bis zur Anode gelangen. Im Detektor sinkt der Elektronenstrom. Als Folge steigt die Pulsfrequenz soweit, bis der Strom wieder auf dem voreingestellten Level ist. Die Differenz in der Pulsfrequenz ist die signalgebende Größe. Das Detektorsignal gibt aufgrund der molekülspezifischen Response keinen Aufschluss über strukturelle Eigenschaften des Analyten. 26 Kapitel 3 Spektrometrie S pektroskopie ist die Wissenschaft über die Wechselwirkung zwischen Licht und Materie.1 Diese Wechselwirkungen zwischen Licht und Materie lassen sich mit der physikalischen Natur des Lichts erklären. Licht wird allgemein als elektromagnetische Wellenbewegung angesehen. Je nach seiner Frequenz erhält man verschiedene Eigenschaften des Lichts, wie das Sichtbare, die Röntgenstrahlung, die Ultraviolette oder Infrarote Strahlung. All dies sind elektromagnetische Wellen, die sich nur in ihrer Frequenz unterscheiden. Ihre Ausbreitungsgeschwindigkeit ist stets gleich, denn sie breiten sich mit Lichtgeschwindigkeit im ganzen Raum aus. In der Chromatographie haben wir bereits eindimensionale Analyseverfahren kennengelernt, die immer einen Messwert liefern, wie z.B. die Retentionszeit. Die Spektroskopie ist ein mindestens zweidimensionales Analyseverfahren, da für jedes auflösbare Wellenlängenintervall eines Spektrums mindestens ein Intensitätswert erhalten wird. Definitionsgemäß dient die Spektroskopie der Charakterisierung von Atomen, Ionen, Radikalen und Molekülen durch Aufnahme und Auswertung der erhaltenen Spektren. Der Grundaufbau eines Spektrometers ist immer die gleiche: Strahlungsquelle, spektrale Zerlegung und Detektor. Die heutige Spektroskopie lässt sich in die beiden großen Bereiche der Atom- und Molekülspektroskopie unterteilen. Bei der ersten geht es um die qualitative und quantitative Bestimmung von Elementen. Hierzu zählen die Verfahren der Atomabsorption, Atomemission und Röntgenfluoreszenz. Die Molekülspektroskopie ist der Überbegriff für Verfahren wie UV/vis, IR, Raman und NMR- Spektroskopie. Die dort erhaltenen Messwerte können zum qualitativen und quantitativen Nachweis einzelner Komponenten genutzt werden und geben Auskunft über Bindungen und die Struktur der Moleküle. Die Bereiche der Massenspektroskopie gelten als Sonderfall, da es sich hierbei um keine elektromagnetisch beeinflusste Wechselwirkung handelt, sondern um ein Trennverfahren. Ein weiterer Vorteil der Spektroskopie ist die Tatsache, dass die Messverfahren in der Regel nichtdestruktiv sind und daher für die präparative Analytik verwendet werden können. Viele spektroskopische Verfahren lassen sich vor Ort und in Echtzeit realisieren. Dadurch erhält die Spektroskopie eine große Bedeutung in der Routineanalytik, da Regelprozesse überwacht und direkt beeinflusst werden können. Als Spektrum bezeichnet man eine Darstellung der Intensität (Licht oder Teilchen) in 1 Böcker, Spektroskopie, S. 27 27 Abhängigkeit von der Wellenlänge, Schwingungsfrequenz, Energie, Masse oder einer anderen charakteristischen Größe. 3.1 Strahlung Abbildung 3.1 – Die Ausbreitung elektromagnetischer Felder durch elektromagnetische Strahlung.[6, Kapitel 10, S. 1] Licht ist physikalisch gesehen eine transversale2 elektromagnetische Wellenbewegung. Die Änderung der elektrischen und magnetischen Felder erfolgt periodisch. Unabhängig von ihrer Frequenz besitzen alle elektromagnetischen Wellen im Vakuum die gleiche Geschwindigkeit, die als Lichtgeschwindigkeit c bezeichnet wird. In Materie, wie z.B. Luft, verringert sich die Ausbreitungsgeschwindigkeit. Die Wellen unterscheiden sich in ihrer Länge λ bzw. ihrer Frequenz ν. Dies entspricht der Anzahl der Schwingungen pro Zeiteinheit, angegeben in s−1 bzw. [Hz]. Für Frequenz, Wellenlänge und Lichtgeschwindigkeit gilt der folgende Zusammenhang: c=λ·ν (3.1) Eine weitere Charakterisierungsmöglichkeit stellt die Wellenzahl ν̃ dar, welche der reziproke Wert der Wellenlänge ist: 1 ν̃ = [cm−1 ] (3.2) λ Die Wellenzahl gibt die Anzahl der Wellenlängen an, die auf einen cm entfallen. Sie darf nicht mit der Frequenz ν verwechselt werden. Max Planck entdeckte seiner Zeit, dass die elektromagnetische Welle nicht nur eine Wellennatur sondern im gewissen Rahmen eine Teilchennatur besitzt. Diese Lichtteilchen nannte er Photonen bzw. Quanten. Sie haben eine bestimmte Energie, welche durch die folgende Gleichung beschrieben wird: E =h·ν (3.3) h stellt hier einen Proportionalitätsfaktor dar, der als Plancksche Konstante eine allgemeine Naturkonstante darstellt. Mit Hilfe der Gleichung 3.1 erhalten wir E= h·c λ (3.4) Die Energie der Lichtquanten ist somit frequenzabhängig. Die größer die Frequenz, desto höher die Energie der Strahlung. Je kürzer die Wellenlänge, desto energiereicher ist das Licht.3 Diese Tatsachen macht man sich in der Spektroskopie zu Nutzen. Dafür teilt man die Wellen anhand ihrer Wellenlänge in Spektralbereiche. In der Regel werden die entsprechenden Wellenlängen λ zur Charakterisierung angeben. Nur im Infrarotbereich verwendet man die Wellenzahlen ν̃, bei Radiowellen hingegen die 2 3 Schwingungsrichtung ist senkrecht zur Ausbreitungsrichtung. vgl. Rücker/Neugebauer/Willems, Instrumentelle pharmazeutische Analytik, S. 36f 28 Frequenz ν. Der für den Menschen sichtbare Bereich des Lichts wird mit vis abgekürzt und umfasst einen sehr kleinen Wellenlängenbereich zwischen 400 und 800nm. Die Strahlung mit den kürzesten Wellen erscheint violett, die mit den längsten rot. Der Übergang zu den kürzesten Wellenlängen wird daher als Blauverschiebung, die zu den längsten Wellenlängen Rotverschiebung genannt. Die jeweils angrenzenden Bereiche nennt man den Ultravioletten (UV) und Infraroten (IR) Bereich. Mikrowellen und Radiowellen sind noch langwelliger als Infrarote Wellen. Es ist technisch möglich, Wellen einer bestimmten Länge zu erzeugen und für die Analyt nutzbar zu machen. Licht, welches aus nur einer einzigen Wellenlänge besteht, wird als monochromatisches Licht bezeichnet. Die unterschiedlichen Wellenlängen und damit die unterschiedlichen Energien der Wellen können unterschiedliche Wirkungen auf Moleküle und Atome haben. Daraus ergeben sich die vielfältigen Einsatzmöglichkeiten, die die Spektroskopie ausmachen. 3.2 Die Mikrowelle Im Frequenzbereich von 8 bis 40 GHz der elektromagnetischen Strahlung werden Moleküle zur Rotation angeregt. Die diskreten Übergänge zwischen den Energiezuständen der rotierenden Moleküle lassen sich messen, weshalb die Mikrowellenspektroskopie auch gern als Rotationsspektroskopie bezeichnet wird. Die Wellenlängen der Mikrowellen können im kleinen cm-Bereich bis in den µm-Bereich gehen. Zur analytischen Messung werden jedoch nur die Mikrowellen aus dem kurzwelligen Bereich genommen. Man kann hier sehr monochrome Strahlung erzeugen, die nur innerhalb weniger Frequenzen variieren, wodurch eine hohe Präzision erhalten wird. Es können bei diesem Verfahren nur verdünnte Gase oder verdampfbare Substanzen untersucht werden. Aus dem Rotationsspektrum kann man dann Rückschlüsse auf Bindungswinkel und Atomabstände, sowie auf Dipolmomente von Molekülen ziehen. Die Analytmoleküle dürfen zudem nicht zu groß sein, da die Auswertung der Spektren sonst zu komplex wird. Durch das schmale Anwendungsspektrum hat die Rotationsspektroskopie in Abbildung 3.2 – Modell eines rotierenden zweiatomigen, der Analytik keine breite Anwendung gefunheteronuklearen Moleküls. [6, Kapitel 11, S. 1] den. Die Rotationsspektroskopie wird als Grundlage für die IR-Spektroskopie genutzt, jedoch nicht mehr kommerziell angewendet. Zur Bestimmung von Bindungslängen ist dieses Verfahren jedoch noch immer das genaueste. Ein Molekül kann auf verschiedene Weisen Bewegungsenergie aufnehmen. Ein jedes Molekül hat 3N Freiheitsgrade der Bewegung, wobei N die Anzahl der im Molekül vorhandenen Atome ist. Drei Grade entfallen auf die Translation in die drei Raumkoordinaten. Die anderen 3N −3 Freiheitsgrade beschreiben die Rotation und Schwingung. Ein rotierendes Molekül kann aus einem elektromagnetischen Feld nur dann Energie aufnehmen, wenn es im Grundzustand ein permanentes elektrisches Dipolmoment besitzt, das sich beim Übergang zeitlich ändert. Aus diesem Grund haben Moleküle ohne permanenten Dipol (H2 , N2 , CH4 , CCl4 ) kein 29 Rotationsspektrum. Als Ausnahme gilt hier lediglich O2 . Lineare Moleküle haben zwei Rotationsfreiheitsgrade. Nicht lineare Moleküle haben drei Rotationsfreiheitsgrade. Lineare und symmetrische Kreiselmoleküle erzeugen ein Spektrum aus Linien mit nahezu konstanten Frequenzabständen. Asymmetrische Kreiselmoleküle erzeugen hingegen Spektren ohne erkennbare einfache Gesetzmäßigkeiten. Das Spektrum zeigt die Transmission [%]4 in Abhängigkeit von der Wellenzahl. Ein solch starrer linearer Rotator, wie er in Abb. 3.2 dargestellt ist, kann verschiedene Energiezustände annehmen. Die möglichen Energieniveaus ergeben sich aus der Schrödinger Gleichung: Erot = h I J h2 · J(J + 1) 8π 2 · I (3.5) Plancksche Konstante Trägheitsmoment ganzzahlige Rotationsquantenzahl Die Absorption der zugefügten Strahlung geschieht durch den Wechsel des Moleküls in ein höheres Energieniveau. Dafür benötigt es Energie, welches es sich aus der anglegten Strahlung nimmt. Ein Molekül kann dabei immer nur in das nächst höhere oder niedrigere Level wechseln. Dies ist in der Quantenmechanik begründet. ∆J = ±1 (3.6) Bei Raumtemperatur liegen Moleküle in verschiedenen Anregungsstufen vor, da die Differenzen zwischen den Energieniveaus durch die thermische Energie der Umgebung bereits überwunden werden kann. Die Differenzen zwischen den Energieniveaus sind stets die gleichen. Daher sind die Abstände zwischen den Linien im Spektrum auch stets konstant. Die Intensität der Linien spiegeln die Häufigkeit der Übergänge dar. Sie ist temperaturabhängig. Diese Häufigkeit lässt sich durch die BoltzmannVerteilung berechnen: −∆E NJ = (2J + 1) · e kT (3.7) NI NJ Zahl der Kerne im energiereicheren Zustand NI Zahl der Kerne im energieärmeren Zustand ∆E Energiedifferenz der Zustände bzw. Energiedifferenz zum Grundzustand Grundsätzlich lassen k Boltzmann-Konstante T Temperatur [K] sich alle Moleküle zur Rotation anregen. Nachweisbar und deshalb für die MW-Spektroskopie relevant sind jedoch nur Moleküle mit einem Dipolmoment. Die Intensität der Linien im Spektrum intensivieren sich durch niedrige Temperaturen. Daher wird in der MW in der Regel bei Raumtemperatur und mit gekühlten Proben gearbeitet und setzt die Proben unter Druck, um einen Wechsel in die Gasphase zu erreichen. Die erhaltenen Spektren der Proben werden mit Referenzspektren verglichen. In der Praxis erhält man keine klaren Signale sondern peakähnliche Signale mit einem gewissen SignalRausch-Verhältnis. Es ist technisch nicht möglich, eine Welle von genau einer bestimmten Frequenz zu senden. Meist werden die benachbarten Frequenzen mitgesendet. Außerdem kann man die natürliche Linienbreite beobachten, welche durch verschiedene Faktoren zustande kommt. In Flüssigkeiten wird 4 Durchlässigkeit eines Materials für (elektromagnetische) Wellen. 30 dies durch die Wechselwirkungen zwischen den vorhandenen Molekülen bedingt, welche sich gegenseitig anstoßen und deformieren. In Gasen verursacht der Dopplereffekt dieses Phänomen. Je nachdem, ob Teilchen sich auf die Strahlungsquelle hin- oder wegbewegen, absorbieren sie höhere oder niedriger frequente Strahlung. Zuletzt spielt auch die Heisenbergsche Unschärferelation eine gewisse Rolle, da der Energiezustand eines Teilchen nie vollkommen sicher bestimmt werden kann. 3.3 Infrarotspektrometrie In der Infrarotspektroskopie werden Moleküle durch infrarote Strahlung in mechanische Schwingung versetzt. Die Schwingung kann entlang von Bindungsachsen als Valenzschwingung oder Streckschwingung erfolgen, oder sich unter Deformation des Bindungswinkels als Deformations- oder Biegeschwingung äußern. Die Molekülschwingung bestimmter Atomgruppen oder funktioneller Gruppen ist dabei besonders charakteristisch und wird zu deren Identifizierung genutzt. Manche Moleküle geben sogar so charakteristische Spektren, dass sie als sogenannter fingerprint einzigartig sind und zur Identifizierung ganzer Moleküler herangezogen werden können. Die IR-Spektroskopie wird daher in der Regel als qualitative Methode und weniger als quantitative Methode eingesetzt. Es werden Wellenlängen zwisch 2,5 und 50µm verwendet, welches dem mittleren Infrarotbereich entspricht. Die aufgenommenen Spektren geben Auskunft über den Aufbau des Moleküls. Man vergleicht ein erhaltenes Spektrum mit denen aus einer Spektrenbibliothek und kann dadurch Rückschlüsse auf den vorliegenden Stoff ziehen. Diese Suche läuft heute in der Regel computergesteuert ab. Innerhalb eines Moleküls sind die Atome durch ihre Bindungen in ständiger minimaler Bewegung. Diese Schwingung geschieht in definierter Frequenz, den sogenannten Resonanzfrequenzen, die bedingt sind durch die Atommasse und die Stärke der chemischen Bindung. Die Schwingungsarten hängen dabei von der Anzahl der Atome im Molekül ab und stehen im direkten Zusammenhang mit den Rotationsfreiheitsgraden, die wir bereits in der Mikrowellenspektroskopie kennengelernt haben. Da sich die Schwingungen der Infrarotwellen und der Molekülbewegung in der gleichen Größenordnung befinden, kann ein Energieaustausch stattfinden, wenn beide Schwingungen dieselbe Frequenz haben. Absorbiert ein Molekül eine solche Schwingung, so verstärkt sich die Amplitude, also die Schwingungsstärke, nicht aber die Frequenz. Dies macht man sich in der Analytik zu Nutze, in dem man ein breites Spektrum infraroter Strahlung durch eine Probe schickt und die austretenden Frequenzen misst. Die absorbierten Frequenzen entsprechen den Resonanzfrequenzen der Schwingungsformen im Molekül. Ebenso kann man monochromatische Strahlung durch eine Probe schicken. Sie wird nur dann absorbiert, wenn die Frequenz der Schwingung mit der des Moleküls übereinstimmt. Der Zustand der Erregung eines Moleküls ist nur von sehr kurzer Dauer, da das Molekül die überschüssige Energie schnell an die umliegenden Moleküle weitergibt, was so eine Erwärmung der Probe verursacht. Alle Moleküle, bei denen sich durch Schwingung temporär das Dipolmoment ändert, sind IR-aktiv. Aus diesem Grund können Edelgase, Salze ohne kovalente Bindungen, Metalle und zweiatomige Moleküle aus gleichen Atomen (N2 , O2 , Cl2 ) mittels IR nicht nachgewiesen werden. Vor allem in der Anaylse von organischen Verbindungen findet die IR ihre größte Anwendung. Jedoch können auch alle anorganischen Verbindungen mit kovalenten Bindungen vermessen werden. Ein permanentes Dipol, wie es in der MW gefordert wird, ist nicht vonnöten. IR-inaktive Substanzen, wie Sauerstoff, Stickstoff oder die Edelgase dienen zum Spülen des Spektrometers, da Wasser und Kohlenstoffdioxid IR-aktiv sind. 31 3.3.1 Theoretische Grundlagen Es gibt verschiedene Arten von Schwingungen. Die einen betreffen das gesamte Molekül, die anderen auf Einzelbindungen oder funktionielle Gruppen (lokalisierte Schwingungen). Zu diesen lokalisierten Schwingungen zählen die Valenzschwingungen (asymmetrisch und symmetrisch), aber auch die Deformationsschwingungen wie Spreiz- (bending), Pendel- (rocking), Torsions- (twist) und Kippschwingung (wagging). Abbildung 3.3 – Schwingungsformen von Molekülen in der IR-Spektroskopie.[6, nach Kapitel 12] A B C D E F Symmetrische Streckschwingung, blaue Atome bewegen sich gleichzeitig vom gelben Atom weg. Asymmetrische Streckschwingung, blaue Atome bewegen sich abwechselnd vom gelben Atom weg. Schaukelschwingung, blaue Atome bewegen sich gleichmäßig nach links und rechts. Scherschwingung, blaue Atome bewegen sich voneinander weg und aufeinander zu. Drehschwingung, blaue Atome bewegen sich abwechselnd von hinten nach vorne. Wippschwingung, blaue Atome bewegen sich gleichzeitig nach vorne und hinten. Die Valenzschwingungen von Einfachbindungen zu Wasserstoffatomen absorbieren hochfrequente Schwingungen in Folge der geringen Masse des Wasserstoffatoms. In der Regel absorbieren Dreifachbindungen bei den höchsten Frequenzen, Einfachbindungen bei den niedrigen Frequenzen. Je größer also die Bindungsstärke, desto höher die benötigte Frequenz zur Anregung. Der bereits angesprochene Fingerprint 1 5 auf. tritt in der Regel bei Schwingungen unterhalb von 1500 cm Die IR wird vor allem zur Strukturanalyse von organischen Verbindungen genutzt, da diese Moleküle aus zahlreichen Bausteinen aufgebaut sind, die funktionellen Gruppen. Die Banden, die diese funktionellen Gruppen erzeugen liegen aufgrund der geringen Wechselwirkung zum restlichen Molekül meist an der gleichen Stelle im Spektrum, wodurch die Identifikation dieser Gruppen erleichtert wird. Dadurch kann man durch Vergleichen mit bekannten Spektren die Bausteine einer Verbindung identifizieren. Die Gerüstschwingung eines Gesamtmoleküls sind bei niederfrequenten Schwingungen unterhalb von 1 1500 cm zu beobachten. Diese Schwingungen sind charakteristisch für ein Molekül und erschweren die Identifikation von lokalisierten Schwingungen unterhalb dieses Bereichs, da sie meist von der größeren Gerüstschwingung überlagert wird. Ein IR-Spektrum besteht damit aus zwei Bereichen, die zur 1 Analyse verwendet werden können. Oberhalb von 1500 cm liegen die Absorptionsbanden, die man den funktionellen Gruppen zuordnet. Unterhalb liegt der fingerprint-Bereich, mit dem man das Molekül als Ganzes charakterisieren kann. Die Schwingungsspektren in der IR basieren auf den Übergängen zwischen den diskreten Energiezuständen schwingender und dabei strahlender Moleküle.6 Ein solches Spektrum kann bereits für kleine Moleküle sehr unübersichtlich und komplex sein, weshalb vereinfachende Modellbildungen und Bandenzuordnungen notwendig werden. 5 6 Wellenzahl, Umkehrterm der Wellenlänge λ. J. Böcker, Spektroskopie, S. 137 32 Abbildung 3.4 – Asymmetrische Valenzschwingung am Beispiel eines Kohlenstoffdioxid-Moleküls.[6, Kapitel 12, S. 1] Die Bindungen zwischen dem Kohlenstoffatom und den beiden Sauerstoffatomen hat einen bestimmten Gleichgewichtsabstand r0 . Durch äußere Einflüsse kann dieser Gleichgewichtsabstand verändert werden. Durch infrarote Strahlung können diese Bindungen gedehnt oder gestaucht werden. Eine andere Kraft versucht, das alte Gleichgewicht wieder herzustellen. Dadurch entsteht eine Schwingung der Atome um die Gleichgewichtslage. Dies kann man im oberen Teil der Abbildung 3.4 anhand der Position des Kohlenstoffatoms sehen. Durch die Schwingung verändern sich die lokalen Ladungsverteilungen im Molekül, was eine temporäre Veränderung des Dipolmomentes mit sich bringt (mittlerer Teil der Abbildung). Die temporäre Änderung des Dipolmoments lässt sich als Veränderung über die Zeit auftragen, sodass man das zuunterst stehende Diagramm erhält. Es handelt sich hierbei um eine harmonische, sinusförmige Schwingung. In der Physik vereinfacht man sich diese Vorstellung, in dem man sich auf nur einen schwingenden Massepunkt beschränkt und der andere als statisch festgelegt wird. Dies nennt man einen Oszillator. Für diese Veränderung der Bindungslänge, welche in der Physik als Auslenkung x beschrieben ist, gilt folgender Zusammenhang im Hookesches Gesetz: K = −k · x K −k x (3.8) Kraft, die das Gleichgewicht stört Kraft oder Federkonstante Auslenkung, Veränderung zum Gleichgewichtszustand Diese Beziehung kann man mithilfe der Bewegungsgleichung nach Newton zeitabhängig darstellen. Ein Körper verharrt im Zustand der Ruhe oder der gleichförmigen Translation, sofern er nicht durch einwirkende Kräfte zur Änderung seines Zustands gezwungen wird.7 7 I. Newton, Philosophiae Naturalis Principia Mathematica, Bd. 1, S.13 33 Demnach entspricht die Beschleunigung a der zweiten Ableitung des Ortes x. K =m·a=m· d2 x = −k · x dt2 (3.9) Aus der Physik kennen wir die Lösung des Differentailterms. Daher wissen wir, dass der Massenpunkt mit der Masse m mit der Frequenz ν schwingt: 1 ν= · 2π s k m (3.10) Da in der Realität jedoch im Molekül immer mehrere Atome gegeneinander schwingen, ersetzt man den Masseausdruck durch den gemittelten Massewert der beteiligten Atome. 1 ν= · 2π s k M (3.11) m2 ·m2 mit M = m11 ·m22 . Je größer die Bindungsenergie und kleiner die Atome, desto höher die Schwingungsfrequenz. Beim Schwingungsvorgang ändert sich die potentielle Energie des Oszillators wie folgt: Epot = 0, 5 · k · x2 (3.12) An dem Punkt des Gleichgewichts ist die potentielle Energie gleich 0. Erfährt der Massepunkt keinerlei Widerstand durch Reibung usw., so setzt er die ihm zugefügte Energie vollständig in Schwingung um. Löst man Gleichung 3.11 für k auf und setzt diesen in Gleichung 3.12 ein, so erhält man: Epot = 2π 2 · M · ν 2 · x2 Abbildung 3.5 – Potentialkruve eines harmonischen Oszillators.[6, Kapitel 12, S. 2] (3.13) Man erhält eine quadratische Funktion im Bezug auf die Auslenkung aus der Gleichgewichtslage. Daraus erhält man eine Parabel, deren Scheitel in der Gleichgewichtslage r0 liegt. Diese Gleichung gilt allgemein für Schwingungen. Führt man dem System dann Energie zu, so wird die Schwingungsamplitude größer. Dies geschieht kontinuierlich, atomare Systeme können jedoch nur diskrete Energiebeträge aufnehmen. Daher muss die Gleichung 3.13 entsprechend angeglichen werden. Ein Molekül kann nicht jede beliebige Energie durch Strahlungsabsorption aufnehmen, sondern nur Energiebeträge, die die Größe hν oder ein Vielfaches davon sind8 . Deshalb lautet die Gleichung für die Schwingungsenergie für Atome ESchw = hν · (υ + 0, 5) (3.14) Hierbei steht υ für die Schwingungsquantenzahl und darf nicht mit der Schwingungsfrequenz ν verwechselt werden! 8 Der Energiebedarf einer harmonischen Molekülschwingung wurde von Max Planck berechnet. 34 Setzt man υ = 0, so erhält man die Nullpunktenergie des Oszillators, die 0, 5hν entspricht. Für υ = 1 erhält man die Grundschwingung und für υ = 2 die erste Oberschwingung. Die Schwingungsquantenzahl darf nur ganzzahlige Werte über 0 annehmen. Eine negative Schwingungsquantenzahl ist nicht definiert. Atome können immer nur in den nächstliegenden Zustand wechseln. Ein direkter Wechsel über zwei Stufen ist nicht möglich. Außerdem kann eine Schwingung nur dann angeregt werden, wenn das schwingende Molekül dadurch sein Dipolmoment ändert. Je größer die Änderung des Dipolmoments durch die Anregung, desto wahrscheinlicher ist sie und desto größer ist der Extinktionskoeffizient der entsprechenden Schwingungsbande. Neben dem harmonischen Oszillator, der in Form einer Sinuskurve schwingt, gibt es aber noch den anharmonischen Oszillator. Dieser gilt jedoch nur bei einer groben Beschreibung von Molekülschwingungen. Die Annahme bei der sinusförmigen Schwingung erlaubt, dass der Abstand zwischen zwei Atomen null oder negativ wird und dass ein Atom beliebig stark angeregt werden kann, ohne dass dies Auswirkungen auf die Bindung hat. Ein Molekül ist somit also kein harmonischer Oszillator. Zu starke Anregung führt irgendwann zum Bindungsbruch, also zur Dissoziation. Einer Bindungsstauchung setzt ein Molekül eine größere Widerstandskraft entgegen als einer Dehnung. Die Annahme eines harmonischen Oszillators gilt also nur für kleine Anregungen, nicht für höhere oder gar grenzwertige. Bei der gegenseitigen Annäherung zweier Atome steigt die Potentialkurve demnach wegen der sehr erheblichen Abstoßung stärker an, als in Abb. 3.5 gezeigt. Zur Korrektur wurden von P. M. Morse die Berechnung der Schwingungsenergie wie folgt modifiziert: h2 · ν 2 ESchw = hν · (υ + 0, 5) − · (υ + 0, 5)2 (3.15) 4D Die Abstände zwischen den Energieniveaus sind damit nicht mehr gleich, sondern konvergieren gegen die Dissoziationsenergie D, die spektroskopisch bestimmt werden kann. Es dürfen zudem auch Sprünge über mehrere Niveaus stattfinden, jedoch werden die mit zunehmender Stufe seltener. Die benötigte Energie für Atomschwingungen ist weitaus höher als die für Rotationsbewegungen. Daher liegen bei Raumtemperatur kaum thermisch angeregte Atombindungen vor. Die Absorptionen erfolgen von Übergängen aus dem untersten Energieniveau. Die intensivsten Absorptionen entsprechen den Grundschwingungen. Die Oberschwingungen der nächsthöheren Energieniveaus sind weitaus weniger intensiv und liegen bei kürzerwelligen Frequenzen aufgrund der höheren Energie, die für die Anregung notwendig ist. Die notwendigen Energien sind in der nebenstehenden Abbildung zu erkennen. Die Atomschwingung ist ständig von der Rotationsbewegung der Moleküle begleitet, da diese schon durch die Abbildung 3.6 – Potentialkruve eines anharthermische Energie bei Raumtemperatur angeregt wird. monischen Oszillators. [6, Kapitel 12, S. 2] Eine Änderung des Schwingungsniveaus ist somit nicht ohne gleichzeitige Änderung des Rotationsniveaus möglich. Dies sieht man an der fehlenden 0-0-Linie im Spektrum. Im rechten Teil des Spektrums sieht man die kürzeren Wellenlängen (dargestellt mit R0 bis R5 ). In diesem Teil addiert sich die Energie des Rotationsübergangs zur Anregungsenergie der Schwingung hinzu. 35 Auf der linken Seite sieht man die längeren Wellenlängen. In diesem Bereich finden die Übergänge von höheren auf niedrigere Rotationsniveaus statt, sodass die Schwingungsenergie um die Energie der Rotationsanregung verringert ist. Die Intensitäten der Banden erklärt sich durch die unterschiedliche Verteilung der Moleküle auf die Rotationsausgangszustände im Schwingungsgrundzustand. Die reine Rotationsstruktur lässt sich nur in der Gasphase beobachten. In diesem Falle kann man eine feine Aufspaltung der Absorptionsbanden erkennen. In kondensierten Phasen9 , wie sie in der Regel vorkommen, kann man nur Absorptionsbanden erkennen, da die Feinstruktur der Rotationsbanden überlagert werden. Die funktionellen Gruppen eines Moleküls können sich in ihren Schwingungen auch gegenseitig beeinflussen. Man nennt sie deshalb auch gekoppelte Oszillatoren. Durch diese Kopplung können Verschiebungen des charakteristischen Absorptionsmaximums im Spektrum auslösen. Dadurch wird die Wellenzahl ν̃ für eine Atomgruppe umso charakteristischer, Abbildung 3.7 – Rotationsschwingspektrum eines je weniger deren Schwingung mit den Schwingun- zweiatomigen Moleküls.[6, Kapitel 12, S. 3] gen anderer Atomgruppen koppelt. Umgekehrt erlauben verschobene Absorptionsmaxima auch Rückschlüsse auf solche Kopplungseinflüsse. IR-aktiv sind nur jene Stoffe, die durch Anregung eine Veränderung ihres Dipolmomentes erfahren. Ist ein Molekül symmetrisch (z.B. Kohlenstoffdioxid), so erfährt es keine Dipolveränderung durch die Anregung und ist IR-inaktiv. Solche Moleküle sind jedoch Raman-aktiv, worauf hier jedoch nicht näher eingegangen wird. Die Kopplungseinflüsse der funktionellen Gruppen macht es kompliziert, eine solche Gruppe isoliert zu betrachten. Damit ist eine exakte Berechnung des Absorptionsmaximums nicht möglich. Stattdessen wurden diese Maxima empirisch ermittelt und tabelliert. Somit ist eine Zuordnung der funktionellen Gruppen auf diesem Wege durch Vergleichen mit bekannten Spektren möglich. Je nach Aufnahmetechnik kann der Substanzbedarf für eine IR-Analyse zwischen 1 und 15mg liegen. Eine Substanz kann in der Gasphase gemessen werden, in flüssiger bzw. geschmolzener Phase als dünner Film zwischen zwei Kochsalzscheiben, in Lösung oder im festen Zustand als Suspension in flüssigem Paraffin bzw. im Gemisch mit KBr oder KCl als Pressling. Die beiden Trägesubstanzen sind IR-inaktiv, daher erhält man nur das Spektrum des Analyten. 3.3.2 Fourier Transformation Bei der Fourier Transformations-IR wird das gesamte Spekrum des infraroten Lichts durch die Probe geschickt. Alle für die Probe charakteristischen Wellenzahlen werden absorbiert und erreichen nicht 9 bei flüssigen und festen Proben. 36 mehr den Detektor. Die aus der Probe austretende Lichtintensität wird in Abhängigkeit von der Zeit gemessen. Dadurch erhält man ein sogenanntes Interferogramm. Durch eine Rechenoperation (Fourier Transformation) wird daraus dann das Spektrum berechnet. Man erreicht mit diesem Verfahren eine wesentlich höhere Empfindlichkeit und somit einer niedrigeren Nachweisgrenze, da die gesamte Leistung der Lichtquelle genutzt wird und nicht bloß eine Wellenzahl. Dadurch dass das gesamte Spektrum gemessen wird, benötigt die Analyse weniger Zeit. Durch interne Kalibrierungsmöglichkeiten wird die Präzision erhöht. Die FTIR wird heute großflächig in der Gasanalyse, bei geringen, stark oder schwach absorbierenden Proben angewendet, Häufig wird sie mit einem GC oder HPLC System gekoppelt. Eine schonendere Präparation für Festkörper stellt die Präparation mit der Nujol10 -Technik dar. Bei diesem Verfahren werden etwa 2mg des Probenpulvers mit einigen Tropfen Nujol im Mörser verrieben. Der Öl vermindert die Strahlungsreflexion an der Oberfläche der so entstehenden Mikrokristalle. Dieses Öl sollte daher den gleichen Brechungsindex wie die Probe haben. Diese Methode empfiehlt sich immer dann, wenn außer Wasser kein passendes Lösungsmittel für diese Probe vorhanden ist oder diese Probe mit KBr reagiert. Zudem ist diese Methode apparativ weniger aufwendig als die Herstellung eines Presslings. 3.3.3 Das IR-Spektrum Im IR-Spektrum ist die Transmission T (Durchlässigkeit von Schwingungen) in aufsteigender Richtung gegen die absteigende Wellenzahl ν̃ (bzw. die aufsteigende Wellenlänge λ) aufgetragen. Die Bereiche großer Absorption erscheinen als große, nach oben geöffnete Banden. Zur Anregung von Valenzschwingungen (Schwingungen in Richtung der Bindungsachse) sind größere 1 Energiebeträge notwendig als zur Anregung von Deformationsschwingungen. Oberhalb von 1500 cm liegen deshalb vornehmlich Valenzschwingungen vor, mit Ausnahme der N H-Deformationsschwingung, welche ebenfalls in diesem Bereich liegt. Man unterscheidet grundlegend die folgenden Bereiche: • 4000-2800 1 cm : Valenzschwingungen, an denen Wasserstoffatome beteiligt sind11 . 1 • 2800-2100 cm : Valenzschwingungen von dreifach gebundenen Atomgruppen und kumulierten12 Doppelbindungen. • 2100-1500 1 cm : Valenzschwingungen von Doppelbindungen. • 1600-1000 1 cm : Gerüstschwingungen des gesamten Moleküls, Fingerprint-Region. Das Absorptionsmaximum kann sich aufgrund von Einflüssen umliegender Atome und deren Bindungen verschieben (s.o.). Je stärker beispielsweise die Elektronegativität eines Halogensubstituenten ist, desto höher liegt die Frequenz. Zusätzlich zu berücksichtigen ist der Einfluss des Aggregatszustandes auf die zwischenmolekularen Kräfte. Dies wirkt sich in der Feinstruktur der Rotationsbanden aus, welche im IR-Spektrum in der Regel von den Schwingungsbanden überlagert werden. Aufgrund der Kopplungseinflüsse ist ein Vergleichsspektrum zur Analyse und Identifikation des Stoffes notwendig. 10 Gemisch langkettiger flüssiger Paraffine. Dies liegt an der vergleichweise geringen Masse des Wasserstoffs. Starke chemische Bindungen und Atome kleiner Massen verursachen im IR-Spektrum Absorptionsmaxima bei großen Wellenzahlen. Diese Bindungen anzuregen benötigt viel Energie, also höhere Frequenzen bzw. kürzere Wellenlängen. 12 benachbarte, am selben Kohlenstoff vorliegende Doppelbindungen. −C = C = C− 11 37 3.4 UV/vis-Spektrometrie In der UV/vis-Spektrometrie wird Strahlung der Wellenlängen zwischen (100nm: Vakuum UV, 200nm: UV) 400 und 800nm eingesetzt. In Form eines Photons wird ein Molekül aus seinem elektrischen Grundzustand in einen angeregten Zustand überführt. Dieser Übergang wird durch kleinere Energiedifferenzen der Schwingungs- und Rotationsübergänge überlagert. Dadurch erhält man bei flüssigen Proben sehr breite Absorptionsbanden, wohingegen man in der Gasphase noch die schmaleren Banden erkennen kann. Dies kommt dadurch zustande, dass in der flüssigen Phase mehr Molekülkollisionen stattfinden, sodass die Auflösung zu gering wird, um die Banden dieser Stoßvorgänge separiert darstellen zu können. Im Regelfall gibt ein Molekül die ihm zugeführte Energie in Form von Wärme (Zusammenstöße mit anderen Molekülen) wieder ab. Eine Abgabe der Strahlung als Emission findet nicht statt. Die Absorption von Licht im sichtbaren Bereich wird durch sogenannte Chromophore13 hervorgerufen (s. 3.4.1). Meist wird bei der Anregung eines Moleküls nur seine chromophore Gruppe durch das Photon angeregt. Im Orbitalmodell gibt es verschiedene Energieniveaus für Elektronen, die σ, π und n14 -Orbitale, welche energetisch angeregt werden können. Diese werden dann im angeregten Zustand mit einem Stern (*) markiert. Durch die Anregung werden bindende Orbitale zu antibindenden Orbitalen. Die nichtbindenden Orbitale werden zu antibindenden π*-Orbitalen. Die zur Farbe beitragenden Elektronen befinden sich im chromophoren Teil des Moleküls. Die Elektronenübergänge von einem Niveau zum anderen sind kompliziert und daher im folgenden Schema dargestellt. Die Elektronen liegen in der Regel im Grundzustand S0 vor, in dem sie mit antiparaleller Spinrichtung sind (Singulett-Zustand). Von dort erreichen sie durch Anregung die energiereicheren Singulett-Zustände S1 oder S2 , usw. Die Rückkehr in den Grundzustand erfolgt durch Abgabe von Energie in Form von Wärme oder in selteneren Fällen durch Emission von Licht (natürliche Fluoreszenz). Ebenso können die Elektronen in den angeregten Singulett-Zuständen S1 oder S2 durch strahlungslose Spin-Umkehr in die energieärmeren Triplett-Zustände wechseln. In diesem Zustand sind die Spins der Elektronen parallel ausgerichtet. Von hier aus fallen die Elektronen unter Spinumkehr und Lichtemission in ihren Grundzustand zurück (Phosphoreszenz). Die Zustände S0 , S1 , S2 , T1 , T2 sind in ihren Energieniveaus nicht einheitlich, da sie verschiedene Schwingungsquantenzahlen ν beinhalten. Diese Schwingungsniveaus werden noch weiter in Rotationsniveaus I unterteilt. Der Wechsel von einem Zustand Abbildung 3.8 – Vereinfachtes Jablonski in den anderen benötigt je nach Schwingungsniveau des Schema.[4, S.99, Abb. 11.1] Moleküls unterschiedliche Energiebeträge. Dabei sind die Übergänge, die ohne Änderung des Abstands der Elektronen zum Atomkern geschehen am wahr13 Moleküle, die als Teil eines Farbstoffes für das Vorhandensein von Farbe verantwortlich sind. Dieser Teil sorgt durch seine charakteristische Reflexion und Streuung des einstrahlenden Lichts und der selektiven Absorption, welche ihn in einen angeregten Zustand versetzt, sodass Farbe erscheint. 14 n-Orbitale sind die nichtbindenden Orbitale. 38 scheinlichsten15 . Für die UV/vis-Spektroskopie relevant sind jedoch nur die Übergänge n → π* und π → π*, da diese Übergänge durch UV-Wellen zwischen 200 und 700nm angeregt werden können. Dies bedeutet somit, dass Moleküle ohne freie Elektronenpaare oder Doppelbindungen durch die UV/vis-Spektroskopie nicht nachgewiesen werden können. Die σ-Elektronen sind zu dicht am Atomkern gelegen, sodass zu ihrer Anregung ein hoher Energiebetrag benötigt wird. Derartige Moleküle, wie gesättigte Kohlenwasserstoffe absorbieren erst unterhalb von 150nm. Sie können nur mit speziellen Vakuumspektrometern nachgewiesen werden. Anders ist dies bei konjugierten π-Elektronensystemen, wie sie in aromatischen Ringen vorliegen. Je stärker sie konjugiert sind, desto weiter wird das Absorptionsmaximum zu den größeren Wellenlängen verschoben. Dies nennt man bathochrome Verschiebung. Die Verweildauer der Elektronen in den höheren Zuständen ist weitaus kürzer als in den niedrigeren. Die Schwingungsniveaus kann man in der Gasphase durch sehr schmale Banden erkennen. Im flüssigen Aggregatzustand bilden sie mit der Gesamtabsorption lediglich eine breitere Bande. Die Absorptionsbanden in der UV/vis-Spektroskopie sind charakteristisch durch ihre Lage (λmax), ihre Höhe (max) und ihre Form bzw. Feinstruktur. Die Lage des Absorptionsmaximum hängt von der Anregungsenergie ab, die notwendig ist, um das Molekül von einem Grundzustand in einen Anregungszustand zu versetzen. Die Intensität einer Absorptionsbande ist proportional zur Übergangswahrscheinlichkeit in einen anderen Zustand. Bei zunehmender Intensität steigt die Übergangswahrscheinlichkeit und das Molekül kann eher mit der Strahlung in Wechselwirkung treten. Je mehr sich die Ladungsverteilung im Molekül durch einen solchen Übergang ändert, desto größer ist die Wechselwirkung mit dem elektromagnetischen Feld des Lichtes und desto stärker wird es angeregt. Die Bandenbreite nimmt bei abnehmender Anregungsdauer zu, ebenso wie bei steigender Wechselwirkung mit dem Lösungsmittel, da dadurch die Energieabgabe erleichtert wird. Bei polaren Lösungsmitteln verbreitern sich die Banden noch zusätzlich. 3.4.1 Chromophore Als Chromophor (Farbträger) bezeichnet man den Teil eines Moleküls, der für eine bestimmte Absorption verantwortlich ist. Chemischer Aufbau des β-Carotins. Bathochrome Verschiebung am Beispiel von Benzol, Benzoesäure und Phenylproensäure (Zimtsäure).[6, Kapitel 13, S. 3] Chromophore Systeme aus π-Elektronen kommen bei Alkenen, Alkinen und aromatischen Verbin15 Frank-Condon-Prinzip 39 dungen vor. Mit zunehmender Konjugation bzw. Anzahl der Doppelbindungen verschieben sich die Absorptionsmaxima dieser Verbindungen zu den längeren Wellenlängen. Es findet die bereits angesprochene bathochrome Verschiebung statt. Bei genügender Länge des Systems aus konjugierten Doppelbindung kann eine bathochrome Verschiebung bis in den sichtbaren Bereich stattfinden. Dies geschieht z.B. beim β-Carotin. Dieses Molekül absorbiert Licht der Wellenlänge 451nm, welches dem blauen Licht entspricht, wodurch es in seiner Komplementärfarbe orange erscheint. Buten (H2 C=CHCH2 CH3 ) hat ein Absorptionsmaximum bei etwa 190nm und ist damit zu niedrig für eine konventionelle UV/vis-Messung. Butadien (H2 C=C=CHCH3 ) hat eine weitere Doppelbindung und ist damit höher konjugiert als Buten. Es hat sein Absorptionsmaximum bei 217nm und ist damit mittels UV/vis messbar. Hier wird das höchstbesetzte Orbital energetisch erhöht, während das niedrigste unbesetzte antibindende Orbital erniedrigt wird. Bei Dreifachbindungen tritt dieser Effekt noch stärker auf, da die Differenzen zwischen den Energieniveaus hier noch geringer sind und weniger Energie notwendig ist, um diese Differenzen zu überwinden. 3.4.2 Das Lambert-Beer’sche Gesetz dI ∼ Idx (3.16) Das Bouguer-Lambert-Gesetz besagt, dass die Schwächung dI eines Lichtstrahls in einer Küvette mit absorbierendem Inhalt proportional zur Intensität I und der optischen Weglänge x in der Küvette (Schichtdicke) ist. Johann Heinrich Lambert stellte zudem fest, dass die Schwächung des Lichtstrahls auch proportional zur Konzentration c ist. dI = α(λ) · c · Idx (3.17) Hierbei sei α(λ) der Absorptionskoeffizient, der als Proportionalitätsfaktor gilt. Die Veränderung der Intensität des Lichtstrahls beim Durchgang durch die Küvette erhält man durch Integration der Gleichung 3.17 über die Schichtdicke x: I = I0 · e−α(λ)·x·c (3.18) Man kann einen exponentiellen Zusammenhang zwischen Konzentration und Schichtdicke zur Veränderung der Intensität erkennen. I0 ist die unveränderte Intensität des einfallenden Lichtstrahls. Durch Umformung und Überführung in den dekadischen Logarithmus erhält man die folgende Gleichung: log I0 = α(λ) · x · c · log(e) I (3.19) Es sei nun der molare Extinktionskoeffizient (λ) = α(λ) · 0, 4343. Damit erhält man das Lambert-Beer’sche Gesetz in seiner häufigsten verwendeten Variante: log 3.4.3 I0 = E = (λ) · c · x I (3.20) Diodenarray-Detektor Die UV/vis-Detektion funktioniert über die Lichtabsorption in einem festgelegten Spektralbereich. Es gibt Detektoren, die mit Festwellendetektion durch eine spezielle Lampe genau ein Spektrum erzeugen können. Dies ist das einfachste und preiswerteste Verfahren. Werden nur Substanzen mit einem Absorptionsmaximum in diesem Wellenlängenbereich untersucht, so reicht diese Art der Detektion aus. 40 Die mangelnde Selektivität bei komplexen Probenmatrices macht diese Methode jedoch nur bedingt ideal. Die heutigen UV/vis-Detektoren sind in der Regel mit einer Deuterium- oder Wolframlampe ausgestattet, welche ein kontinuierliches Spektrum im ultravioletten und sichtbaren Bereich erzeugen können. Durch einen Monochromator kann die Wellenlänge eingestellt werden, bei der die zu analysierende Substanz ihr Absorptionsmaximum hat. Noch leistungsfähiger sind nur noch die DiodenarrayDetektoren. Sie verwenden die gleichen Lichtquellen wie die gängigen UV/vis-Detektoren, sind jedoch durch einen Polychromator in der Lage ist, das austretende Licht in seine Spektralfarben aufzuteilen. Durch den Einsatz von 512 Dioden werden die einzelnen Wellenlängen simultan gemessen, wodurch man eine dreidimensionale Abbildung in Abhängigkeit von Zeit und Wellenlänge erhält, da der Detektor nicht kontinuierlich, sondern in einstellbaren Zeitintervallen misst. So nimmt der Detektor für jede Wellenlänge des Spektrums ein Chromatogramm auf. Diese Abbildung 3.9 – Aufbau eines DiodenarrayMethode ermöglicht genauere Messwerte und eine bessere Detektors. [6, Kapitel 13, S. 4] Identifikation von Substanzen. Außerdem können Chromatogramme durch Verbesserung des Signal-Rausch-Verhältnisses durch Substraktion von Referenzwellen optimiert werden. Dadurch kann die Nachweisgrenze signifikant erhöht werden. Mit Hilfe einer Spektrenbibliothek kann das erhaltene Spektrum verglichen werden und so die Identität oder auch nicht-Identität eines Analyten bestimmt werden. Die eingesetzte Küvette oder auch Messzelle soll vernachlässigbar wenig zur Bandenverbreiterung beitragen, als einen möglichst geringen eigenen Brechungsindex haben. Daher wird in der Regel hochreines Quarzglas verwendet. Die Peak Purity kann ebenfalls mit dem DAD gemacht werden. Hierfür werden mehrfach Spektren der gleichen Sustanz gemessen und die Peakformen und deren Reproduzierbarkeit bestimmt. 3.4.4 Fluoreszenz Die Fluoreszenz wurde bereits in der Einleitung erwähnt. Sie ist eine ganz ähnlich der Wirkungsweise der Chromophore. Durch die Absorption von Photonen werden atomare Bindungen in höhere Energieniveaus gehoben. Fluoreszenz entsteht durch den Übergang vom niedrigsten Schwingungsniveau des ersten angeregten Zustandes S1 zu einem Schwingungsniveau des elektronischen Grundzustandes S0 . Fluorophore sind Moleküle mit einem ausgeprägten π-Elektronensystem, die im UV/vis-Bereich Licht absorbieren. Meist sind sie cyclisch, wenn nicht sogar aromatisch und organisch. In der Regel wird die durch das Zurückfallen in den Grundzustand freiwerdende Energie in Form von Wärme an die umliegenden Moleküle abgegeben. Die Emission von Licht ist ein seltenes Phänomen, das nur bei manchen Molekülen auftritt. Die Wechselwirkung mit umliegenden Molekülen wie dem Lösungsmittel wird externe Umwandlung genannt. Die Fluoreszenz-Messung wird meist zur quantitativen Analyse eingesetzt, da die Fluoreszenzemission proportional zur Analytkonzentration ist. Sie ist im Vergleich zur UV-Absorptionsmessung wesentlich empfindlicher. Durch die Energiedifferenz zwischen den Schwingungsniveaus resultiert eine Rotverschiebung der Fluoreszenzemission. Das einfallende Licht ist somit kurzwelliger als das ausfallende 41 Licht. Dies nennt man die Stokes-Verschiebung. 3.5 Kernspinresonanzspektrometrie Bei der Kernspinresonanzspektrometrie (NMR16 ) registriert man unter bestimmten Bedingungen eintretende Wechselwirkungen zwischen Atomkernen und elektromagnetischer Strahlung im Radiowellenbereich im starken homogenen Magnetfeld. Die NMR wird heute zur Strukturaufklärung von gelösten Substanzen genutzt. Es werden hochauflösende Spektren benötigt, welche durch den Einsatz von hohen Magnetfeldern erzeugt werden. Diese Magnetfelder werden durch supraleitende17 Spulen erzeugt. Die NMR ist ein relativ unempfindliches Verfahren, da die Energiedifferenzen zwischen den Kernspineinstellungen in einem Magnetfeld sehr gering sind. Deshalb benötigt man für die Anregung auch nur energiearme Wellen im Radiowellenbereich. 3.5.1 Theoretische Grundlagen Ein Atomkern besteht aus Nukleonen. Diese wiederum setzen sich aus positiv geladenen Protonen und ungeladenen Neutronen zusammen. Die Energie eines Atomkerns kann in einem äußeren Magnetfeld verschiedene Werte annehmen. Die Nukleonen haben genauso wie Elektronen bestimmte Elementareigenschaften wie Bahndrehimpuls und Eigendrehimpuls, die zusammen als Gesamtdrehimpuls I bezeichnet werden. Zwischen diesem und dem magnetischen Moment µ gilt das folgende Verhältnis: µ=γ·I (3.21) wobei γ das gyromagnetische Verhältnis beschreibt, das eine Stoffkonstante darstellt. Dadurch ist das magnetische Moment γ eines Atomkerns isotopenspezifisch, es ändert sich je nach Anzahl der Neutronen. Der resultierende Spin der Kerne setzt sich aus den Spins der Protonen und Neutronen zusammen und kann ganz- oder halbzahlige Werte zwischen 0 und 7 annehmen. Alle Kerne mit gerader Anzahl an Protonen und Neutronen nennt man g,g-Kerne. Sie haben einen Kernspin von I = 0 und zeigen keine kernmagnetische Resonanz. Auf etwa 60% der stabilen Atomkerne trifft dies zu. Nur Kerne, bei denen die Anzahl der Protonen und/oder Neutronen ungerade sind, haben magnetische Momente. Kerne, die eine ungerade Protonen- und Neutronenzahl haben, heißen u,u-Kerne und haben ganzzahlige Kernspins. Sie haben gerade Massenzahlen. Bei Atomen mit ungerader Massenzahl, den g,u- oder u,g-Kernen liegen halbzahlige Spins vor. Diese Kerne sind zur kernmagnetischen Resonanzabsorption fähig. Peter Zeeman entdeckte 1896, dass Atome in einem Magnetfeld so beeinflusst werden, dass sie aufgespaltene Linien emittieren. Normalerweise sind Elektronenzustände mit gleicher Haupt- und Nebenquantenzahl energetisch völlig identisch. Man nennt diese Elektronenzustände entartet. Die einzige Unterscheidungsmöglichkeit ist die räumliche Orientierung ihrer Orbitale. Durch ein äußeres Magnetfeld wird diese Entartung aufgehoben, da sich nun die Orientierung der einzelnen Orbitale im Bezug auf dieses Magnetfeld energetisch unterscheidet. Man kann sich die Atomkerne auch gut als Stabmagneten vorstellen, die man ohne das äußere Magnetfeld nicht voneinander unterscheiden könnte. Innerhalb des Magnetfeldes gibt es zwei Zustände, zwischen denen Übergänge möglich sind. 16 Nuclear magnetic resonance Supraleiter sind Materialien, deren elektrischer Widerstand beim Unterschreiten der Sprungtemperatur auf null abfällt. 17 42 Beispiel: Bei 1 H ist der Kernspin I = 21 . Der Atomkern besteht aus einem Proton und hat keine Neutronen. Legt man nun ein äußeres Magnetfeld an, richtet sich der Kern nach diesem Feld aus. Er kann deshalb eine Magnetquantenzahl von m = 12 oder m = − 12 annehmen, da sie sich entweder parallel oder antiparallel zum magnetischen Feld ausrichten. Um einen Kern auf ein höheres Energieniveau zu bringen muss die Differenzmenge an Energie, die die beiden Energieniveaus unterscheidet, überwunden werden. Dies geschieht durch die Energiezufuhr ∆E, welche durch Absorption eines Energiequants hν der elektromagnetischen Strahlung geschieht. ∆E = h · ν = γ · h · B0 2π (3.22) wobei B0 die Magnetflussdichte oder Induktion darstellt. Die Differenz der Energieniveaus ∆E und die Resonanzfrequenz ν sind proportional zur Induktion B0 . Entspricht die Energiedifferenz zwischen den Niveaus der Frequenz der eingestrahlten Radiowelle, so tritt eine magnetische Resonanz auf. Das magnetische Moment klappt bei der richtigen Frequenz in die energiereichere Form um. Diese ist isotopenspezifisch im Bezug auf das angelegte elektromagnetische Feld. Beim Rückfall in die energieärmere Form wird keine Emission von Radiowellen beobachtet, sondern die Freiwerdung thermischer Energie. Dies wird Relaxion genannt. Im Resonanzfall nimmt das Atom Energie aus dem magnetischen Feld auf, der für das Feld notwendige Strom verändert sich und dient als Messgröße. γ · B0 ν= (3.23) 2π Die Resonanzfrequenz berechnet sich aus dem gyromagnetischen Verhältnis γ und der Stärke des angelegten Magnetfeldes B0 . Kerne, deren Kernspin I = 0 ist, ergeben kein Signal. Zu diesen Kernen zählen die Isoptope 16 O und 12 C. Die Isotope 1 H und 13 C sind die wichtigsten Isotope und werden am häufigsten untersucht, da sie in vielen Verbindungen vorkommen. 13 C ist im Vergleich zum leichteren 12 C-Isotop selten, da es nur zu 1,1% auftritt. Dennoch sieht man es häufig in den NMR-Spektren. Die eingesetzten Magnete müssen sehr leistungsstark sein. Je stärker das angelegte Magnetfeld ist, desto geringer ist das Besetzungsverhältnis, welches die Verteilung der Atomkerne im angeregtem und normalem Zustand beschreibt. Es wird von der Boltzmann-Verteilung bestimmt: −∆E Ni = e k·T Nj Ni Nj ∆E k T (3.24) Grundzustand eines Kerns angeregter Zustand eines Kerns Differenz der Energieniveaus, entspricht der Energie eines Energiequants (h · ν), der für die Anregung sorgt. Boltzmann-Konstante Temperatur Durch Einsetzen der entsprechenden Werte erhält man die Verteilung der sich im Grundzustand befindlichen und angeregten Kerne. Über 99% der Kerne liegen im Grundzustand vor. Je größer dieser Besetzungsunterschied ist, desto empfindlicher wird die NMR. Dies geschieht zum Beispiel durch ein starkes äußeres Magnetfeld. In organischen Molekülen besitzen die Protonen ja nach ihrer Lage im Molekül verschiedene Resonanzfrequenzen. Dies wird die chemische Verschiebung genannt. 43 3.5.2 Chemische Verschiebung Die Elektronen einer Bindung in einem Molekül bewirken durch ihre elektromagnetische Abschirmung die chemische Verschiebung. Durch das angelegte Magnetfeld B0 zirkulieren die Elektronen in der Elektronenwolke der Bindung und erzeugen dadurch ein magnetisches Moment µ, welches dem angelegten Magnetfeld entgegengerichtet ist. Die Lenzsche Regel beschreibt den Zusammenhang zwischen dem lokalen Magnetfeld Blokal , der Abschirmkonstante σ 18 und dem äußeren Magnetfeld B0 . Blokal = B0 · (1 − σ) (3.25) Das magnetische Feld B0 hat eine bestimmte Richtung und wird von einem sogenannten Erregerfeld19 Blokal umgeben. Ändert man nun den magnetischen Fluss des Feldes B0 , so ändert sich auch die Flussrichtung des Erregerfeldes. Nach außen hin wirkt die chemische Verschiebung, als würde das Erregerfeld das von außen angelegte magnetische Feld überlagern. Je größer die Elektronendichte einer Bindung ist, desto stärker ist die Abschirmung. Es müssen deshalb stärkere Magnetfelder eingesetzt werden, um diese Abschirmung zu überwinden, was sich in einer Erhöhung der Resonanzfrequenz äußert. Eine Absolutmessung der chemischen Verschiebung ist nicht möglich, weshalb man auf Vergleichsmessungen zu Standardsubstanzen (z.B. Te- Abbildung 3.10 – Abschirtramethylsilan (CH3 )4 Si) zurückgreift. Diese Verbindung hat 12 chemisch mung von einem Magnetfeld durch die Elektronen in eiäquivalente20 Protonen und daher eine scharfe Linie im NMR-Spektrum. nem Benzolring. [6, Kapitel Man nimmt diese Linie als Nulllinie im NMR-Spektrum. 14, S. 3] Die NMR-Spektren sind von der Stärke der angelegten Magnetfelder abhängig. Um z.B. 60 und 100 MHz-Geräte miteinander vergleichen zu können, muss die Skala der Spektren von dem angelegten Magnetfeld unabhängig sein. Deshalb legte man den Parameter δ für die chemische Verschiebung fest. δ= Resonanzfrequenz der Substanz in Hz Betriebsfrequenz des Gerätes in MHz (3.26) Die Angabe von δ erfolgt in ppm21 . Kerne mit gleicher magnetischer Abschirmung und somit gleicher Signallage werden dann als chemisch äquivalent bezeichnet. Die chemische Verschiebung hängt somit direkt mit der magnetischen Abschirmung zusammen. Diese wiederum hängt von der Elektronenkonfiguration, also der chemischen Umgebung zusammen. Anhand des Spektrums und der Lage der Signale lässt sich dadurch eine Aussage über die Struktur des untersuchten Stoffes machen. Zu diesem Zweck gibt es empirisch ermittelte Verschiebungstabellen, ähnlich den Spektrenbibliotheken aus der IR. Das Sekundärfeld von π-Elektronen bei Alkenen, Alkinen und Aromaten kann das angelegte Magnetfeld am Ort des Protons sowohl schwächen als auch verstärken. Dies wird Anisotropie-Effekt22 genannt. 18 Die Abschirmkonstante ist von der Art des Protons abhängig. In Abb. ?? wird dieses Feld Sekundärfeld genannt. 20 Die Protonen liegen alle in der gleichen chemischen Umgebung. 21 parts per million 22 Anisotropie bedeutet wörtlich übersetzt, die Richtungsabhängigkeit einer Eigenschaft oder eines Vorgangs. Eine Bindung schirmt nicht in alle Raumrichtungen gleichmäßig ab, daher entstehen Richtungen, bei denen das Magnetfeld stärker oder schwächer wirkt. 19 44 Neben aromatischen Ringen und Mehrfachbindungen haben aber auch intermolekulare Wechselwirkungen wie die Van-der-Waals-Kräfte und Wasserstoffbrückenbindungen Einflüsse auf die Elektronendichte einer Bindung und somit auf die Stärke des induzierten Sekundärfelds. Im Grunde setzt sich die Abschirmung eines Kerns aus drei Einflüssen zusammen: • Diagmagnetische Verschiebungen sind solche zu niedrigeren δ-Werten welche aus einer verstärkten Abschirmung resultieren. Diese Verschiebung kommt durch Elektronenströme um den Kern zustande, die bei wachsender Elektronendichte zunehmen. Dies tritt vor allem bei kugelsymmetrischen Orbitalen auf. Das äußere Feld wird geschwächt. • Paramagnetische Verschiebung kommt durch die Behinderung der Elekronenbeweglichkeit zustande und sorgt für größere δ-Werte. Durch elektronegative Substituenten nimmt die Elektronendichte der Bindungen ab, wodurch der paramagnetische Abschirmungsbetrag am Kern größer wird. Dies tritt vor allem bei nicht kugelsymmetrisch verteilten Ladungen auf. Das äußere Feld wird verstärkt. • Die Anisotropie hat einen weiteren Einfluss auf die chemische Verschiebung. Je nach der Anordnung der Atome im Molekül bewirkt sie eine Verstärkung oder Schwächung des äußeren Magnetfeldes. Meist ist dieser Effekt bei Aromaten und Mehrfachbindungen zu beobachten. 3.5.3 Spin-Spin-Kopplung Bei ausreichend hoher Auflösung eines NMR-Spektrum sieht man, dass die einzelnen Signale in mehrere Einzelsignale aufgespalten werden. Dies nennt man häufig die Feinstruktur eines Spektrums. Grund dafür sind die 2I + 1 verschiedenen Orientierungsmöglichkeiten, die die magnetischen Momente der Kerne zum äußeren Magnetfeld einnehmen können. Hierdurch entstehen entsprechend viele Zusatzfelder, die das äußere Magnetfeld überlagern. Die magnetische Wechselwirkung zwischen den Kernen erfolgt über die Elektronen der Bindung. Dies nennt man die (elektronengekoppelte) SpinSpin-Wechselwirkung, die dann zu einer Aufspaltung der Signale führt. Chemisch äquivalente Protonen zeigen keinerlei Wechselwirkung miteinander. Nimmt man wieder an, dass es sich bei den Kernen um kleine Stabmagnete handelt, so wird deutlich, dass sie in Wechselwirkung miteinander treten, wenn ihr Abstand zu gering wird. Nehmen wir einmal das Beispiel mit zwei Atomkernen. Es ergeben sich insgesamt vier Möglichkeiten für deren Ausrichtung zum angelegten Magnetfeld. Beide in die gleiche Richtung und die beiden Abbildung 3.11 – Schematiantiparallelen Stellungen. Die antiparallelen Stellungen sind durch sche Darstellung der Spin-Spindie verschiedenen Resonanzfrequenzen unterscheidbar. Üblicherwei- Kopplung zweier benachbarter Kerse zeichnet man für diese Spin-Stellungen ein Energieschema, in ne. das alle Kernspineinstellungen und deren Energieniveaus eingetragen werden. Anhand dieser Darstellung kann man gut sehen, dass die Zustände b und c energetisch verschieden voneinander sind. Die Übergänge von einem zum anderen Energieniveau sind durch Absorption von Energie möglich. Der direkte Übergang von a nach d ist nicht erlaubt. Durch die verschiedenen Zustände, die die Kerne annehmen können, ergeben sich unterschiedliche Aufspaltungen der Signale. Je nach Anzahl der möglichen Kopplungen spalten sich die Resonanzsignale in Dubletts, Tripletts, Quartetts usw. Die Entstehung dieser Feinstruktur folgt den folgenden beiden Regeln: 45 • Besitzt ein Proton mehrere Arten von chemisch äquivalenten Protonen na , nb , ... in seiner direkten Nachbarschaft, so ist die Multiplizität seiner Resonanz (na + 1) · (nb + 1)·... • Die Intensitäten der Teilsignale eines Multiplett kann man anhand der Terme von (x + 1)n bestimmen, wobei n für die Anzahl der chemisch äquivalenten Protonen der benachbarten Gruppe stehen. Die Größe und Struktur bestimmter Molekülgruppen ist charakteristisch und können als weitere Strukturinformationen Aussagen über die Anzahl der koppelnden Nachbarkerne machen. Ethanol (CH3 CH2 OH) hat drei Gruppen mit Protonen. Die drei Protonen der CH3 -Gruppe sind chemisch äquivalent, ebenso wie die beiden Protonen der CH2 -Gruppe. Die Protonen der CH3 Gruppe befinden sich im äußeren, von dem äußeren Magnetfeld gestörten Bereich und werden durch die benachbarte CH2 -Gruppe noch zusätzlich gestört. Die Spins der Protonen der beiden Gruppen koppeln miteinander. Dadurch ergibt Abbildung 3.12 – NMR-Spektrum von Ethanol.[7] die CH3 -Gruppe ein Triplett. Die CH3 -Gruppe wirkt elektronenschiebend, weshalb die Elektronendichte der Bindung zur CH2 -Gruppe zunimmt. Somit findet eine diagmagnetische Verschiebung statt. Das Signal ist bei kleinen δ-Werten zu erwarten. Im Spektrum kommt diese Gruppe tatsächlich auch als erstes bei 1,22ppm. Wenden wir nun die beiden Regeln an, um das Ergebnis zu verifizieren: Ausgehend von der Methylgruppe (CH3 ) liegen in unmittelbarer Nachbarschaft die chemisch äquivalenten Protonen der Methylengruppe (CH2 ). Somit ist na = 2, da zwei chemisch äquivalente Protonen vorliegen. Für die Multiplizität der Resonanz der Nachbargruppe gilt (na + 1), da keine weiteren chemisch äquivalenten Protonen innerhalb von drei Bindungen vorliegen. Es ergibt sich eine Multiplizität von 2 + 1 = 3, weshalb im Spektrum ein Triplett zu erwarten ist. Die Intensitäten der drei Linien können durch die zweite Formel berechnet werden. Zwei benachbarte Protonen ergeben mit na = 2 eine quadratische Gleichung (x + 1)2 = 1x2 + 2x + 1, was anhand der Koeffizienten einem Intensitätsverhältnis von 1:2:1 entspricht. Dies kann man im Spektrum im Triplett der Methylgruppe gut erkennen. Nun schauen wir uns die nächste Gruppe an, die Methylengruppe, CH2 . Sie ist umgeben von der Methylgruppe und der Hydroxygruppe. Die Methylgruppe hat einen elektronenschiebenden Effekt, wohingegen die Hydroxygruppe einen elektronenziehenden Effekt hat, welcher einen größeren Einfluss hat und deshalb die Resonanz in Richtung zunehmendes δ verschiebt. Auch hier wenden wir nun die beiden Regeln an. Es liegen drei Protonen der Methylgruppe vor, somit ist na = 3. Für die Formel zur Berechnung der Multipletts dürfen nur Protone an direkt benachbarten Kohlenstoffen verwendet werden. Deshalb lautet die Formel zur Berechnung des Multipletts der Methylengruppe 3 + 1 = 4, womit ein Quartett vorliegen wird. Die Intensität der Teilsignale liegt im Verhältnis (x+1)3 = x3 +3x2 +3x2 +x weshalb das Signalverhältnis entsprechend der Koeffizienten 1:3:3:1 ist. Aufgrund des Sauerstoffs findet keinerlei Kopplung mit dem Proton der Hydroxygruppe statt. Daher erscheint diese Gruppe als Singulett im Spektrum an der Stelle 2,58ppm. 46 3.5.4 Das NMR-Spektrum Die am häufigsten betrachteten NMR-Spektren sind die 1 H-NMR-Spektren von organischen Verbindungen. Die Verbindungen liegen in Lösung vor und müssen protonenfrei oder in perdeuterierten Lösungsmitteln aufgenommen werden, da man sonst nur die Protonen des Lösungsmittels messen würde. Die Protonen des Analyten würden überlagert werden. Die Analyten müssen in Lösung vorliegen, da in der Gasphase die Teilchendichte für eine Anregung zu gering ist. Die Signalintensität ist direkt proportional zur Ge- Abbildung 3.13 – Strukturformel des psamtzahl der Teilchen, weshalb man die Signale in- Ethylisopropylbenzols. tegrieren kann, um eine quantitative Aussage bezüglich der Protonenanzahl machen zu können. In der Regel verfügt der elektronische Integrator über die Funktion Stufenkurven in das Spektrum einzufügen, um die Integration zu erleichtern. Kennt man die Summenformel des Analyten, so lassen sich über die Integration die Verhältnisse der Protonen in den funktionellen Gruppen bestimmen. Entscheidend bei der Intergration ist das Konstanthalten der Einflussgrößen wie Temperatur, magnetische Induktion und Frequenz. Beispiel einer Spektrenauswertung: p-Ethylisopropylbenzol. In der obenstehenden Graphik sieht man p-Ethylisopropylbenzol. Es wurde bereits in nach den Kohlenstoffen in Gruppen eingeteilt und mit griechischen Buchstaben versehen. Die Methingruppe (orange) wird im Folgenden mit α abgekürzt, die Methylgruppen (violett) mit β, der Benzolring (grün) mit γ, die Methylengruppe (blau) mit δ und die dritte Methylgruppe (rot) mit ε. Die Farben markieren die chemisch äquivalenten Protonen. Im 1 H-NMR Spektrum werden aufgrund der 5 Kohlenstoffgruppen ebenso viele Signale erwartet. Diese Signale werden die folgende Feinstruktur aufweisen: • 1α H ist von 6 chemisch äquivalenten β H umgeben. Damit ist na = 6 und (na + 1) ergibt 7. Das Signal von 1α wird im Heptett vorliegen. • 1α spaltet 6β zu einem Duplett. • 4γ liegt aufgrund der vielen Bindungen als Singulett vor. Es können keine Kopplungen stattfinden. • 3ε spalten 2δ zum Quartett. • 2δ spalten 3ε zum Triplett. 3.6 Massenspektrometrie Die Massenspektrometrie (MS) ist ein physikalisches Verfahren, bei dem im Vakuum stabile Ionen nach ihrem Verhältnis von Masse zu Ladung (m/z) getrennt werden. Es können sowohl Elemente als auch ganze Moleküle mit diesem Verfahren gemessen und identifiziert werden. Im Massenspektrum sieht man dann auf der Abszisse das Masse-Ladungs-Verhältnis gegen die Intensität auf der Ordinate aufgetragen. Die Messwerte sind konzentrationsabhängig und können so auch zur quantitativen Analyse verwendet werden. 47 Die zentrale Aufgabe der MS ist die Strukturaufklärung von organischen und metallorganischen Verbindungen, insbesondere von großen Molekülen. Die Probe wird durch die Messung fragmentiert bzw. atomatisiert und ionisiert. Somit handelt es sich um eine destruktive Messmethode. Je nach Aggregatzustand der Probe wird ein geeignetes Ionisationsverfahren gewählt, bei dem die Probe zunächst fragmentiert wird. Bei dieser Fragmentierung entstehen Kationen und Anionen, aber auch Radikalionen, die mit dem nachgeschalteten Massenspektrometer detektiert werden. Die erhaltenen Spektren werden dann mit Vergleichsspektren einer Bibliothek verglichen. Massenspektrometer werden in der Regel nicht alleinstehend verwendet, sondern in Kopplung mit einer Kapillarelektrophorese, Flüssigoder Gaschromatographiesystemen. 3.6.1 Theoretische Grundlagen Die Lorentz-Kraft beschreibt die Ablenkung eines geladenen Teilchens in seiner Flugbahn durch ein äußeres Magnetfeld. Auf diesem Grundprinzip basiert die Massenspektrometrie. Deshalb müssen die zu untersuchenden Teilchen als Ionen vorliegen. Die Elektronenstoßionisation ist die klassische Methode zur Ionisierung. Die Ionen werden dann in einem starken magnetischen Feld auf Kreisbahnen abgelenkt. Die Ablenkungsradien sind proportional zur Quadratwurzel der Ionenmassen. Je größer die Masse eines Ions, desto stärker wird es abgelenkt. Ein Massenspektrum wird in der Regel von kleineren zu größeren Massen aufgenommen. Auf der x-Achse befindet sich das Masse zu Ladungsverhältnis, auf der y-Achse die relative Intensität. Den intensivsten Peak nennt man Basispeak. Dessen Intensität wird als 100% gesetzt und alle anderen Peaks in Relation gesetzt. Bei besonders stabilen Verbindungen entspricht der Basispeak auch dem Molekülpeak, in der Regel ist dieser Peak jedoch jener mit dem höchsten Masse-Ladungsverhältnis. Die chemischen Elemente bestehen aus einem Isotopengemisch, welches sich durch die Massenspektrometrie nachweisen lässt. Neben den Peaks der Fragmente eines Moleküls finden sich daher auch stets die Isotopenpeaks. Die Intensitätsverhältnisse dieser Peaks spiegeln die natürliche Häufigkeit der jeweiligen Isotope wieder. Anhand dieser Isotopenzusammensetzung lassen sich Summenformeln der analysierten Substanzen ermitteln. Durch die Ionisierung kann es aber vorkommen, dass sich die Fragmente eines Moleküls derart verändern, dass sie dem Ausgangsmolekül kaum noch zuordbar sind. Ein jedes Fragmention besitzt ein bestimmes Verhältnis von Masse zu Ladung. Daher werden im allgemeinen die m/z-Werte mit den relativen Atommassen gleichgesetzt. Molekülionen sind positive Radikalionen. AB•+ → A+ + B• oder A • +B + Bei der Bildung von Fragmenten werden stets die stabileren Radikalionen bevorzugt. Bei der Fragmentbildung kann es jedoch auch passieren, dass sich neue Bindung zwischen Atomen bilden. Dies nennt man dann Umlagerung. Meist werden Wasserstoffe zur Stabilisierung neu angebunden, aber auch Methyl- und Phenylreste neigen gern zur Umlagerung. 3.6.2 Die Ionenerzeugung Es gibt verschiedene Methoden, ein Molekül zu fragmentieren, sodass ionisierte Teilgruppen entstehen. Die gängigste Methode ist die Elektronenstoßionisation (EI). Hier werden ein paar Milligramm Dampf einer Probe im Hochvakuum mit einem Elektronenstrahl von ca. 70eV beschossen. Dadurch werden Elektronen aus den Bindungen des Moleküls geschossen, wodurch ein positiv geladenes Molekülion entsteht. M + e− → M + + 2e− (3.27) 48 Dabei werden π-Elektronen und freie Elektronenpaare leichter zum Austritt angeregt, als σ-Elektronen. Organische Moleküle haben in der Regel eine gerade Anzahl von Elektronen, weshalb bei dem Beschuss solcher Verbindung häufig Radikalionen entstehen. Diese werden mit einem • markiert, da hier ein Elektron ungepaart vorliegt. Die Fragmentierung erfolgt nach bestimmten Abbaureaktionen und kann wegen des Auftretens charakteristischer Bruchstücke auf die Anwesenheit spezieller funktioneller Gruppen hinweisen. Aufgrund der hohen Elektronenenergie entsteht ein sehr fragmentreiches Spektrum, bei dem es vorkommen kann, dass kein Molekülionensignal mehr vorhanden ist. Ist dies der Fall, lässt sich keine Aussage über das eigentliche Molekülion machen und auch das Molekulargewicht der Verbindung kann nicht mehr ermittelt werden. Daher gibt es ein weiteres, schonenderes Verfahren zur Erzeugung von Molekülionen. Zu diesem Zwecke wurde das Verfahren der Chemischen Ionisierung (CI) entwickelt. Als ReaktandGas wird häufig Methan verwendet, welches zunächst konventionell ionisiert wird. Die zu untersuchenden Moleküle werden dann durch Protonenübertragung von Reaktandionen ionisiert. Die Fragmentierung ist bei diesem Verfahren sehr gering und deshalb besonders schonend. Im Falle von Methan als Reaktand-Gas laufen die beiden folgenden Reaktionen ab: + + CH4 + H → CH5 + + CH5 + M → MH + CH4 Durch die Protonierung der Substanzmoleküle erhält man das um 1 vergrößerte Molekulargewicht der Ausgangssubstanz. Diese Methode wird häufig gekoppelt mit einem Gaschromatographie-System verwendet, da durch die chromatographische Trennung das erhaltene Massenspektrum entzerrt und übersichtlicher wird. Man erreicht so eine hohe Selektivität bei komplexen Gasgemischen. Die Fragmentierung bei dieser Methode hängt vom Reaktand-Gas ab. Man kann je nach Basizität des Gases verschiedene Verbindungen ionisieren. In der Vorlesung betrachteten wir die Fragmentierung des 2-Butanon mit einer Molekülmasse von 72. Dieses Ausgangsmolekül wird nun mit Elektronen beschossen und verliert dadurch ein Elektron, sodass es positiv geladen ist. In dieser Form bildet es den Molekülion-Peak an der Stelle m/z= 72. Für die Fragmentierung gibt es nun zweierlei Möglichkeiten. Das Sauerstoffteilchen in der Verbindung ist das elektronegativste Teilchen und hat einen star- Abbildung 3.14 – Die Fragmentierung von ken elektronenziehenden Einfluss. Eine positive Ladung 2-Butanon.[6, Kapitel 15, S. 1] ist da sehr ungünstig. Daher zieht es aus der Bindung zur n nächstgelegenen Methylengruppe ein Elektron in seine Bindung zum Kohlenstoff. Die Methylengruppe sondert sich mit einem einzelnen freien Elektron als Radikalion ab und das Sauerstoffteilchen baut mit dem Kohlenstoffteilchen eine Dreifachbindung. Ebenso könnte der Sauerstoff ein Elektron der Bindung zwischen Kohlenstoff und Methylgruppe ziehen, sodass ein Ethan-Radikal abgespalten wird. Die letztere Abspaltung ist aufgrund der höheren Stabilität des Fragments die wahrscheinlichere. Die positive Ladung am Sauerstoff ist in beiden Fällen nicht stabil und kann daher auch am abgespaltenen Fragment vorliegen, weshalb im Massenspektrum auch die beiden eigentlich ungeladenen Fragmente 49 zu sehen sind. In der MS ist die Wahrscheinlichkeit, dass ein Molekül überhaupt vom Elektronenstrahl getroffen wird, sehr gering. Sie liegt bei 0,001%. Der Abstand zwischen den Atomen ist aufgrund des gasförmigen Aggregatzustands so gering, dass keinerlei Wechselwirkungen zwischen den Molekülen stattfinden können. In der NMR begegneten uns bereits die verschiedenen Isotope von Molekülen. Etwa 1,1% des Kohlenstoffs liegt als schweres 13 C-Isotop vor. Mit dieser Tatsache lassen sich Peaks in unmittelbarer Umgebung von größeren Peaks erklären. Peaks, die jenseits des Molekülion-Peaks liegen werden bei nicht signifikanter Größe als Verschmutzung gewertet und nicht für die Analyse verwendet. Bei kleineren Molekülen kann man aufgrund der Fragmentierung die Peaks im Massenspektrum zuordnen. Bei größeren Molekülen wird dies schnell kompliziert, weshalb man hier ein Interpretationsschema benötigt. 3.6.3 Abbildung 3.15 – Massenspektrum des 2-Butanon. [6, Kapitel 15, S. 1] Vorläufiges Interpretationsschema • Die höchste im Massenspektrum auftetende Massenzahl entspricht meist der relativen Molekülmasse des unfragmentieren Molekülions, wenn es sich um ein Spektrum einer reinen Substanz handelt. Außerdem muss es sich um das Ion handeln, aus dem alle Fragmente durch Verlust neutraler Teilchen23 • Wenn eine Substanz eine ungerade Anzahl von Stickstoff-Atomen besitzt, so hat sie ein ungeradzahliges Molekülion. Ebenso gilt der Umkehrschluss: Besitzt die Substanz ein ungeradzahlige Molekülmasse, so wird eine ungerade Zahl von Stickstoffatomen angenommen. • Bei brom- und chlorhaltigen Verbindungen treten charakteristische Isotopenpeaks auf, die durch leichtere und schwerere Isotope der Elemente verursacht werden. Ebenso gilt dies für das schwerere Kohlenstoffisotop 13 C. Die Anwendung der Isotopenmuster ist auf die Betrachtung des Molekülionpeaks beschränkt und darf nicht für andere Peaks verwendet werden. Die Intensität des 13 C-Peaks gibt Aufschluss über die Anzahl der vorhandenen Kohlenstoffatome. Teilt man den Anteil des 13 C-Peaks durch die relative Häufigkeit des Isotops bezogen auf 12 C (1,1%), so erhält man die in der Verbindung vorhandenen Kohlenstoffatome. 3.6.4 Kopplungsmöglichkeiten In der Regel werden niedrigauflösende MS-Geräte angewendet. Es gibt jedoch auch hochauflösende Geräte, die Massen mit Dezimalstellen angeben können. Dies sind die Hochauflösenden MS. Mit dieser Messmethode kann man Isotope direkt messen. Außerdem häufig angewendet sind Kopplungen wie die GC/MS. Der Analyt wird chromatographisch getrennt und anschließend massenspektrometrisch vermessen. Vergleichbar ist dies mit dem HPLC/DAD, bei dem man ein UV-Spektrum erhält. 23 Bei dem Verlust neutraler Teilchen sind nur folgende Differenzen zum Molekülion M + erlaubt: 1-4, 15-20 und >24. Diese Differenzen kommen dadurch zustande, dass es keine Moleküle aus reinen Kohlenstoffketten gibt. 50 Dies ist weniger aussagekräftig als ein MS-Spektrum, aber vergleichbar. Als Detektor wird ein MSSpektrometer eingesetzt. Die HPLC/MS ist wesentlich teuer und anspruchsvoller, da die Analyten erst vom Lösungsmittel getrennt werden müssen, in die Gasphase gebracht werden müssen, um ionisiert werden zu können. Bei diesen Kopplungen erhält man dreidimensionale Spektren, die im Grunde zeitabhängige Massenspektren sind. Sie geben Informationen über die Zeit der Detektion, die Auftrennung nach den Massen der Fragmente und deren Intensität. Zu jedem Zeitpunkt lässt sich so ein Chromatogramm mit dem jeweiligen Massenspektrum des ausgewählten Analyten erstellen. Eine Koelution24 lässt sich so entdecken, da das Massenspektrum die Elution aller Stoffe detektiert. So können Stoffe, die sich durch Chromatographie nicht trennen lassen durch den Detektor, ein Massenspektrometer trotzdem noch trennen. Dies geschieht im Totalionenchromatogramm (TIC), welches der Computer auch als Selected Ion Monitoring (SIM) ausgeben kann, sodass die Chromatogramme der beiden zeitgleich eluierten Stoffe erhalten wird. Da der Detektor die Anzahl der Ionen aufnimmt, lässt sich ein solches Spektrum sowohl qualitativ als auch quantitativ auswerten. 24 Peak-Überlagerung: Gleichzeitige Elution zweier Stoffe aus dem chromatographischen System. 51 Abbildungsverzeichnis 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.9 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 3.9 3.10 3.11 3.12 3.13 3.14 3.15 Prinzip eines Säulenchromatographen. [1, S. 38, Bild 2.5] . . . . . . . . . . . . . . . . . Verschiedene Auflösungen der gleichen chromatographischen Trennung.[2, S. 6-13, Abb. 6.7] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Van Deemter Kurve.[2, S. 6-9, Abb. 6.5] . . . . . . . . . . . . . . . . . . . . . . . . . . RF -Wert in einem Dünnschicht-Chromatogramm.[6, Kapitel 1, S. 2] . . . . . . . . . . Probenaufgabe mit einem Sechswege-Ventil. [6, Kapitel 3, S. 2] . . . . . . . . . . . . . van-Deemter-Kurven für die drei Trägergase Stickstoff, Helium und Wasserstoff.[6, Kapitel 9, S. 5] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Trennung anhand verschiedener Dampfdrücke. [6, Kapitel 9, S. 9] . . . . . . . . . . . . Trennung anhand von verschiedenen Einflüssen der Wasserstoffbrücken. [6, Kapitel 9, S. 9] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Die Ausbreitung elektromagnetischer Felder durch elektromagnetische Strahlung.[6, Kapitel 10, S. 1] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Modell eines rotierenden zweiatomigen, heteronuklearen Moleküls. [6, Kapitel 11, S. 1] Schwingungsformen von Molekülen in der IR-Spektroskopie.[6, nach Kapitel 12] . . . . Asymmetrische Valenzschwingung am Beispiel eines Kohlenstoffdioxid-Moleküls.[6, Kapitel 12, S. 1] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Potentialkruve eines harmonischen Oszillators.[6, Kapitel 12, S. 2] . . . . . . . . . . . Potentialkruve eines anharmonischen Oszillators. [6, Kapitel 12, S. 2] . . . . . . . . . . Rotationsschwingspektrum eines zweiatomigen Moleküls.[6, Kapitel 12, S. 3] . . . . . . Vereinfachtes Jablonski Schema.[4, S.99, Abb. 11.1] . . . . . . . . . . . . . . . . . . . . Aufbau eines Diodenarray-Detektors. [6, Kapitel 13, S. 4] . . . . . . . . . . . . . . . . Abschirmung von einem Magnetfeld durch die Elektronen in einem Benzolring. [6, Kapitel 14, S. 3] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Schematische Darstellung der Spin-Spin-Kopplung zweier benachbarter Kerne. . . . . NMR-Spektrum von Ethanol.[7] . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Strukturformel des p-Ethylisopropylbenzols. . . . . . . . . . . . . . . . . . . . . . . . . Die Fragmentierung von 2-Butanon.[6, Kapitel 15, S. 1] . . . . . . . . . . . . . . . . . Massenspektrum des 2-Butanon. [6, Kapitel 15, S. 1] . . . . . . . . . . . . . . . . . . . 52 7 10 12 15 18 23 24 25 28 29 32 33 34 35 36 38 41 44 45 46 47 49 50 Literaturverzeichnis [1] Jürgen Becker, Chromatographie (LaborPraxis), 1. Auflage, 1997, Vogel Buchverlag, Würzburg. [2] Karl Cammann, Instrumentelle Analytische Chemie, 1. Auflage, 2001, Spektrum Akademischer Verlag, Heidelberg. [3] Dr.-Ing. Dipl.-Chem. Jürgen Becker, Spektroskopie, 1. Auflage, 1997, Vogel Buchverlag, Würzburg. [4] G. Rücker/M. Neugebauer/G.G. Willems, Instrumentelle pharmazeutische Analytik, 3. Auflage, 2001, Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart. [5] Isaac Newton/Volkmar Schüller, Philosophiae Naturalis Principia Mathematica, Bd. 1: Tomus Primus, 1726, London. [6] Prof. Dr. Wolfgang Fink, Skript zur Vorlesung der Instrumentellen Analytik an der Hochschule Bonn-Rhein-Sieg, 2014, Rheinbach. [7] http://de.wikipedia.org/wiki/Ethanol. Download am 17.6.2014. 53