UZL News Nr. 22, Juli 2010 - Universitären Zentrum für
Werbung

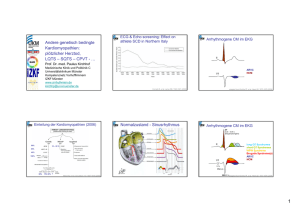
UniversitätsSpital Zürich UZL Universitäres Zentrum für Labormedizin und Pathologie NEWS Nr. 22 Verbund von Kerninstitutionen für Labormedizin und Pathologie der Universität und des UniversitätsSpitals Zürich mit Vertretung von assoziierten USZ-Speziallabors Hämatologie UniversitätsSpital Zürich Prof. Dr. M. Manz Klinische Chemie UniversitätsSpital Zürich Prof. Dr. A. v. Eckardstein Klinische Immunologie UniversitätsSpital Zürich Leiter a.i. Prof. Dr. Ch. Renner Medizinische Genetik Universität Zürich Prof. Dr. W. Berger, Frau Prof. Dr. A. Rauch Medizinische Mikrobiologie Universität Zürich Prof. Dr. E. C. Böttger Medizinische Virologie Universität Zürich Frau Prof. Dr. A. Trkola Pathologie UniversitätsSpital Zürich Prof. Dr. H. Moch Speziallabors Verschiedene Kliniken und Departemente UZL-Geschäftsstelle Christine Genné Institut für Klinische Chemie, UniversitätsSpital Zürich Kontaktpersonen: Dr. Katharina Spanaus, Tel. 044 255 22 93, E-mail: [email protected]; Andrea Wampfler, Tel. 044 255 96 43, E-mail: [email protected] «CRP sensitiv»: Methodenumstellung und Umbenennung in «CRP, KHK-Risiko» Das Institut für Klinische Chemie (IKC) hat Anfang Mai 2010 die Methode zur Bestimmung von «CRP sensitiv» umgestellt. Bisher wurden für die CRP-Bestimmung, abhängig von der Indikation, zwei unterschiedliche Methoden verwendet. Dabei wurde «CRP sensitiv» als kardiovaskulärer Risikofaktor mittels Chemilumineszenzimmunoassay und «CRP» als Entzündungsmarker mittels Immunturbidimetrie bestimmt. Im Rahmen der Testoptimierung durch den Hersteller konnte der Messbereich der Immunturbidimetrie erweitert werden, so dass das CRP mit dieser Methode nun auch im «sensitiven» Bereich zuverlässig gemessen werden kann (Nachweisgrenze neu 0,3 mg/l). Die dadurch möglichen Synergien möchte das IKC gerne nutzen, weshalb nun beide Parameter, «CRP» und «CRP sensitiv», mit der selben Methode (Immunturbidimetrie) bestimmt werden. Parallelmessungen zwischen dem Chemilumineszenzimmunoassay und der Immunturbidimetrie haben gezeigt, dass die beiden Methoden äusserst gut korrelieren und die CRP-Resultate direkt vergleichbar sind. In Bezug auf die Referenz- und Idealwerte ergeben sich keine Änderungen. Wegen der unterschiedlichen Indikationen werden die beiden Parameter weiterhin separat verordnet und berichtet. Aus patentrechtlichen Gründen wird das bisherige «CRP sensitiv» auf den Befunden künftig umbenannt in «CRP KHK-Risiko». Institut für Klinische Chemie, UniversitätsSpital Zürich Kontaktperson: Prof. Dr. Katharina Rentsch, Tel. 044 255 22 90 E-mail: [email protected] Tägliche Bestimmung von Antidepressiva und Neuroleptika Aufgrund einer technologischen Modernisierung in der Abteilung für Medikamentenanalysen und Toxikologie sind wir seit einigen Monaten in der Lage, einen Teil der Antidepressiva und Neuroleptika täglich zu bestimmen. Das heisst, dass wir von Montag bis Samstag alle bis 14 Uhr im Labor eintreffenden Proben für die folgenden Analysen über Nacht analysieren und Ihnen die Resultate am nächsten Tag übermitteln können. Falls Verdünnungen oder Wiederholungen notwendig sind, wird sich die Abgabe des Resultates um einen Tag verzögern. Antidepressiva: Citalopram, Escitalopram, Fluoxetin und Metabolit, Fluvoxamin, Mirtazapin, Paroxetin, Venlafaxin und Metabolit. Neuroleptika: Aripiprazol, Clozapin und Metabolit, Haloperidol, Olanzapin, Paliperidon, Quetiapin, Risperidon und Metabolit. Institut für Medizinische Mikrobiologie, Universität Zürich Kontaktpersonen: Prof. Dr. Brigitte Berger-Bächi, Tel. 044 634 26 50, E-mail: [email protected] Prof. Dr. Reinhard Zbinden, Tel. 044 634 26 08, E-mail: [email protected] Anpassung der Verpackung für den Postversand von diagnostischen Proben In der Ausgabe 18 der UZL News haben wir unseren Einsendern die neue Verpackung zum Postversand von infektiösem Material vorgestellt. Gleichzeitig haben wir allgemein über die Einteilung der Mikroorganismen in Risikogruppen wie auch über die entsprechenden Vorschriften für den Versand von Patientenproben und Kulturen gemäss unseren damaligen Recherchen informiert. Inzwischen sind die dort angegebenen Internetadressen wie auch die damalige Kennzeichnung mikrobiologisch-diagnostischer Patientenproben als «freigestellte medizinische Proben» überholt. Wir haben in den letzten zwei Jahren für den Versand von Patientenproben mit der abgebildeten normgerechten Einweg-Verpackung sehr gute Erfahrungen gemacht. Wir benutzten – wie abgebildet – als Sekundärverpackung einen wasserdichten Plastiksack mit saugfähigem Material (P650 zertifiziert), welche die Kennzeichnung UN 3373 trug. Diese Kennzeichnung UN 3373 wird in Zukunft auch auf der Kartonschachtel (entspricht der drittfesten Verpackung) erscheinen und ersetzt unsere frühere Aufschrift «freigestellte medizinische Probe». Wir machen diese Anpassung, um den aktuellen Interpretationen der Behörden gerecht zu werden, welche sich auf den Standpunkt stellen, dass ein Versand an ein mikrobiologisches Labor immer mit einem Verdacht auf einen Krankheitserreger verbunden sei. Bei dieser Gelegenheit möchten wir Sie auf folgende Internetadressen hinweisen, bei welchen die geltenden Transportvorschriften zu finden sind: www.biosicherheit.zh.ch www.efbs.admin.ch Disclaimer: Diese Information dient unseren Kunden und ersetzt nicht die verbindlichen Gesetze. Internetadressen wie auch gesetzliche Grundlagen werden laufend angepasst, so dass wir jegliche Haftung für Schäden ablehnen müssen, welche im Zusammenhang mit dem Gebrauch dieser wie auch früherer Informationen (UZL-News Nr. 18) stehen könnten. Institut für Medizinische Genetik, Universität Zürich Kontaktpersonen: PD Dr. Dagmar Keller Lang, UniversitätsSpital Zürich, Tel. 044 255 56 63, E-mail: [email protected] Prof. Dr. Wolfgang Berger, Tel. 044 655 70 31, E-mail: [email protected] Molekulare Diagnostik von hereditären Arrhythmien Das Long QT Syndrom und das Brugada Syndrom gehören zu den häufigsten und molekulargenetisch am besten untersuchten vererbbaren Arrhythmien. Eine hereditäre Arrhythmie ist definiert als eine primär elektrische kardiale Erkrankung ohne makroskopischen Nachweis einer strukturellen Veränderung. Klinisch können sich diese Syndrome leider mit dem plötzlichen Herztod (Sudden Cardiac Death, SCD) als fatale Erstmanifestation äussern. Deshalb ist die klinische und EKG-basierte Diagnose von grosser Wichtigkeit und kann durch die molekulare Untersuchung bestätigt werden. Beim Long QT Syndrom und beim Brugada Syndrom werden die Mutationen meistens dominant vererbt. Bei beiden Syndromen kodieren die Mutationen-tragenden Gene Proteine der kardialen Ionenkanäle. Es sind monogen komplexe Erkrankungen mit inkompletter Penetranz, wobei eine einzelne Mutation zum klinischen Phänotyp führen kann, welcher durch andere genetische Varianten (z.B. Polymorphismen) beeinflusst werden kann. Long QT Syndrom Das Long QT Syndrom (LQTS) ist charakterisiert durch eine Repolarisationsstörung mit verlängertem korrigiertem QT-Intervall (QTc) auf dem Oberflächen-EKG und klinisch durch polymorphe ventrikuläre Tachykardien, die über Kammerflimmern zum SCD führen können (1). Die Prävalenz liegt bei ca. 1:5000 –1:10000. Eine pathologische QTc-Zeit ist je nach Alter und Geschlecht unterschiedlich definiert. Bei Männern ist diese ab 450 ms, bei Frauen ab 470 ms verlängert. Ein kongenitales LQTS liegt dann vor, wenn eine medikamentös-induzierte QT-Verlängerung ausgeschlossen wurde. Die autosomal-dominante Form des LQTS wird als Romano-Ward-Syndrom bezeichnet, die weitaus seltenere autosomal-rezessive Form als Jervell-LangeNielsen-Syndrom. Bis heute sind 12 autosomal-dominante Formen bekannt, sowie 2 autosomal-rezessive, wobei die Typen 1– 3 für die grosse Mehrzahl aller Formen verantwortlich sind. Die Typen 1– 3 zeichnen sich durch spezifische T-Wellen-Morphologien zusätzlich zur QTc-Zeit-Verlängerung aus: Beim LQTS Typ 1 ist die T-Welle breit; eine doppel-gipflige T-Wellen-Morphologie mit niedriger T-Wellen-Amplitude findet sich beim LQTS Typ 2; eine sehr spät auftretende T-Welle mit variabler T-Wellen-Morphologie ist wegweisend für ein LQTS Typ 3 (2). Diese 3 Typen können routinemässig molekulargenetisch untersucht werden: Mutationen im Gen KCNQ1 führen zum LQTS Typ 1; das Gen kodiert die α-Untereinheit des kardialen Kalium-Kanals IKs. Das LQTS Typ 2 wird bedingt durch Mutationen im Gen KCNH2, welches den kardialen Kalium-Kanal IKr kodiert. Das Gen SCN5A kodiert den späten kardialen Natrium-Kanal Ina; SCN5A-Mutationen führen zum LQTS Typ 3. Da das genetische Screening beim LQTS bei max. 70% aller Patienten mit einem phänotypisch gesicherten LQTS zu einer spezifischen Diagnose führt, ist die Gendiagnostik nur dann indiziert, wenn der Phänotyp so genau wie möglich definiert ist und nie zur Ausschlussdiagnostik (3). Findet man keine Mutation, ist der genetische Hintergrund des LQTS nicht ausgeschlossen. Brugada Syndrom Das Brugada Syndrom (BrS) manifestiert sich durch eine spezifische ST-Streckenhebung in den rechtspräkordialen Ableitungen im Oberflächen-EKG als Zeichen einer gestörten Repolarisation. Klinisch kann das Syndrom über polymorphe ventrikuläre Tachykardien und Kammerflimmern zum SCD führen. Die Vererbung ist autosomal-dominant mit inkompletter Penetranz; die Prävalenz ist geschätzt auf 5:10000 (4). Für die Diagnosestellung braucht es das Vorliegen eines Typ 1 EKG (siehe unten) in Kombination mit Synkopen, Kammerflimmern, polymorphe ventrikuläre Tachykardien, Familienanamnese für den SCD oder Familienmitglieder mit einem BrS. Das typische BrS zeigt ein Typ 1 EKG in den Ableitungen V1– 2 mit einer zeltförmigen deszendierenden ST-Streckenhebung, präterminal negativer T-Welle und J-Punkt-Amplitude von ≥ 2 mm (5). Neben diesem klassischen EKG-Bild können weniger eindeutige EKG Typen 2 und 3 vorliegen, welche eine sattelförmige ST-Streckenhebung zeigen. Um die Diagnose eines BrS zu stellen, muss ein Typ 1 EKG vorliegen nebst entsprechender Klinik. Die EKG Typen 2 oder 3 EKG müssen mittels eines Natrium-Kanalblockers gemäss spezifischem Protokoll in ein Typ 1 EKG demaskiert werden. Neben der Gabe von Natrium-Kanalblockern kann Fieber ein BrS Typ 1 EKG demaskieren (6). Oft werden Patienten identifiziert, die während Fieber ein Typ 1 EKG zeigen, welches sich wieder (teil)normalisiert in afebrilem Zustand. Auch bei diesen Patienten liegt ein genetischer Hintergrund vor. Das BrS wird am häufigsten bedingt durch Mutationen im Gen SCN5A, welches ja auch zum LQTS Typ 3 führen kann (siehe oben), jedoch über einen anderen pathophysiologischen Mechanismus. Beim Screening von SCN5A findet sich jedoch nur bei 10 bis 30% der Patienten eine Mutation aufgrund der grossen genetischen Heterogenität. Molekulargenetische Diagnostik Am Lehrstuhl für Medizinische Molekulargenetik und Gendiagnostik, Institut für Medizinische Genetik der Universität Zürich, wird unter der Leitung von Prof. Wolfgang Berger in Kollaboration mit PD Dr. Dagmar Keller Lang mittels eines ’candidate gene approach’ vorgegangen, wobei das zu untersuchende Gen anhand der Klinik, des Ruhe-EKG und anderer kardiologischer Untersuchungen, wie einer Ergometrie oder einem Natrium-Kanalblocker-Test, bestimmt wird. Genmutationen können entdeckt werden nach Amplifizierung aller kodierenden Exone der Gene mittels ’Polymerase Chain Reaction’ (PCR), gefolgt von der DNA-Sequenzierung. Diese Methoden sind hoch spezifisch, wenn die PCR Primer das gesamte Exon und wichtige intronische Anteile umfassen. Die Gene KCNQ1 und KCNH2 umfassen jeweils 15 Exone, SCN5A ist mit 28 Exonen das grösste dieser drei Gene. Zum jetzigen Zeitpunkt sind im Gen KCNQ1 403 Mutationen, in KCNH2 606 Mutationen und in SCN5A 559 Mutationen bekannt (Quelle: Human Gene Mutation Database, HGMD Pro). Da jedoch immer wieder neue Sequenzvarianten entdeckt werden, wird jeweils das gesamte Gen auf genetische Varianten untersucht, nicht nur bekannte Mutationen. Wird eine Genmutation identifiziert, bestätigt dies die klinische Diagnose eines LQTS oder BrS. Bezüglich der Familienabklärung ist die Empfehlung, die Mutation auch bei den zumindest erstgradig verwandten Familienmitgliedern zu suchen. Die Untersuchungskosten für den Indexpatienten betragen ca. CHF 3400. Die Kosten für die nachfolgende selektive Mutationssuche bei Familienangehörigen beträgt zwischen CHF 400 und 600. Literatur: 1. Diagnostic criteria for the long QT syndrome. An update. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS. Circulation 1993;88:782-4 2. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Moss AJ, Zareba W, Benhorin J, et al. Circulation 1995;92:2929-34 3. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Tester DJ, Will ML, Haglund CM, Ackerman MJ. Heart Rhythm 2005;2:507-17 4. B rugada syndrome: report of the second consensus conference. Antzelevitch C, Brugada P, Borggrefe M, et al. Heart Rhythm 2005;2:429-440 5. P roposed diagnostic criteria for the Brugada syndrome: consensus report. Wilde AA, Antzelevitch C, Borggrefe M, et al. Circulation 2002;106:2514-9 6. B rugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Keller DI, Rougier JS, Kucera JP, et al. Cardiovasc Res 2005;67:510-9 Benötigtes Material Für die Gendiagnostik werden 5 –10 ml nicht geronnenes EDTA-Blut benötigt sowie das ausgefüllte Anmeldeformular und die Einverständniserklärung des Patienten. Die entsprechenden Formulare sind auf Anfrage erhältlich. UniversitätsSpital Zürich UZL-Geschäftsstelle UniversitätsSpital Zürich OPS D 23, Rämistrasse 100, 8091 Zürich Sekretariat: Tel. 044 255 87 31, Fax 044 255 45 90 [email protected]