Erste Gentherapie am Auge - klinische Phase in Sicht
Werbung

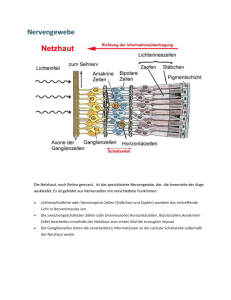
Powered by Seiten-Adresse: https://www.gesundheitsindustriebw.de/de/fachbeitrag/aktuell/erste-gentherapie-am-augeklinische-phase-in-sicht/ Erste Gentherapie am Auge - klinische Phase in Sicht Erblich bedingte Erkrankungen der Netzhaut des Auges gibt es sehr viele verschiedene, da unterschiedliche Schritte des Sehvorganges betroffen sein können. Gemeinsam ist jedoch allen, dass momentan die einzige Chance auf dauerhafte Heilung in einer Korrektur des zugrundeliegenden fehlerhaften Gens besteht. Mit solchen Gentherapien für neurodegenerative Augenerkrankungen beschäftigt sich Prof. Dr. Mathias Seeliger mit seiner Arbeitsgruppe am Forschungsinstitut für Augenheilkunde des Universitätsklinikums in Tübingen. Die dort zusammen mit der LMU München entwickelten Techniken waren bei Mäusen bereits lebenslang erfolgreich. Bei einem ersten Krankheitsbild steht dieser Ansatz nun kurz vor der Anwendung beim Menschen in einer ersten klinischen Phase. Prof. Dr. Mathias Seeliger, Leiter der Arbeitsgruppe Neurodegeneration des Auges am Forschungsinstitut für Augenheilkunde der Universität Tübingen. © privat Seit über 15 Jahren sind erbliche Netzhauterkrankungen ein bevorzugtes Forschungsthema 1 von Prof. Dr. Mathias Seeliger. Der Ingenieur und Doktor der Medizin leitet die Arbeitsgruppe Neurodegeneration des Auges am Forschungsinstitut für Augenheilkunde der Universität Tübingen. Er beschäftigt sich unter anderem mit Netzhauterkrankungen, deren Ursache genetisch bedingt ist. Bei den Patienten findet man Veränderungen in solchen Genen, die für das intakte Funktionieren der Netzhautzellen verantwortlich sind. Dabei handelt es sich meist um Punktmutationen, die je nach Position zum Komplettabbruch der Proteinsynthese führen oder dafür verantwortlich sind, dass nur ein kleiner Teil der eigentlich benötigten Proteinmenge in der entsprechenden Zelle verfügbar ist oder ein falsches, „störendes“ Protein erzeugt wird. Dies hat Auswirkungen auf das Sehvermögen der Netzhaut und resultiert in verschieden schweren Krankheitsbildern. Ursachen liegen häufig in Störungen des Sehvorgangs Beim gesunden Sehvorgang werden Lichtreize von den Sinneszellen der Netzhaut – den Stäbchen und den Zapfen – registriert: Photonen werden in den Fotorezeptoren vom Sehfarbstoff aufgenommen und die Aktivität zunächst in einer biochemische Kaskade verstärkt und dann in ein elektrisches Signal umgewandelt. Dieses wird an die nächste Ebene, die Bipolarzellen, weitergereicht. Am Ende der retinalen Verarbeitung stehen Amakrin- und Ganglienzellen, die die Signale in die universelle Sprache umsetzen, die die meisten Nervenzellen verstehen. „Die Sehinformation liegt nun in einer Erhöhung oder Erniedrigung einer Anzahl von Aktionspotenzialen in einem bestimmten Zeitraum“, erklärt Seeliger. „An jeder Stelle dieser Reaktionskette sind Fehler möglich, die das Sehvermögen des Patienten bis hin zur Erblindung beeinträchtigen können. Aber auch wenn generelle Körperfunktionen gestört sind, kann das Sehen in Mitleidenschaft gezogen werden, beispielsweise bei einem Vitamin-A-Mangel, Diabetes mellitus oder einer Gefäßerkrankung.“ Spezialsprechstunde für erbliche Netzhauterkrankungen in Tübingen Beim gesunden Sehvorgang werden Lichtreize von den Sinneszellen der Netzhaut registriert und als elektrische Signale an die Nervenzellen weitergeleitet. An jeder Stelle dieser komplexen Reaktionskette sind Fehler möglich, die das Sehvermögen des Patienten bis hin zur Erblindung beeinträchtigen können. © Seeliger An der Tübinger Augenklinik gibt es eine Spezialsprechstunde für erbliche Netzhauterkrankungen – eine von sehr wenigen in Deutschland. Dort wird bei den Patienten zunächst festgestellt, um was für eine Erkrankung es sich überhaupt handelt, also welcher Schritt des komplexen Sehvorgangs beeinträchtigt ist. Schwerpunkte der Diagnostik sind neben der ophthalmologischen Untersuchung die Elektrophysiologie und die Bildgebung. „Heute helfen uns auch genetische Tests; vor noch zehn Jahren war es gar nicht so einfach herauszufinden, mit welcher Form einer Krankheit wir es zu tun haben“, sagt der Professor. Insbesondere wenn die Patienten schon fortgeschrittene Stadien zeigen, ist die rein klinische Diagnose oft schwierig. Heute kann man, abhängig von der Erkrankung, bei einem großen 2 Prozentsatz der Betroffenen die ursächliche genetische Veränderung feststellen. Mausmodelle helfen Gendefekte der Patienten zu verstehen und zu behandeln Für viele dieser Erkrankungen existieren Mausmodelle: Die Tiere tragen genau den jeweiligen genetischen Defekt, den auch die Patienten in der Sprechstunde haben. Sie dienen dazu, die neurodegenerativen Augenerkrankungen auf molekularer Ebene zu erforschen und Therapien zu entwickeln. Seit etwa zehn Jahren beschäftigen sich die Tübinger Wissenschaftler speziell mit Ansätzen, um die Fehler im erkrankten Gen auszutauschen – der Gentherapie – mit Erfolg. Dabei kommt Seeliger und seinen Mitarbeitern auch ein weiteres Tübinger Spezialgebiet zugute, die Diagnostikentwicklung. So haben sie die meisten diagnostischen Geräte, die bei menschlichen Patienten eingesetzt werden, auch für Mäuse verfügbar gemacht. Damit können Befunde besser mit Humandaten verglichen und erzielte Ergebnisse leichter auf den Menschen übertragen werden. Für die Gentherapie bei Augenerkrankungen haben sich Adeno-assoziierte Viren (AAV) am erfolgreichsten erwiesen, weil sie an die Rezeptoren dort gut binden. „In der Virenhülle steckt nur das drin, was man auch haben will“, erklärt Seeliger. „Die Partikel binden an die Zellen und entleeren den gewünschten Inhalt – ein Ersatzgen – zur Ergänzung der fehlenden Information“. Ein spezifischer Promotor legt fest, dass die gewünschte Information wirklich nur in den Netzhautzellen abgelesen wird, für die sie auch bestimmt ist. War die Therapie erfolgreich, kommt es nach wenigen Wochen zu einer Verbesserung der Funktion, die dann dauerhaft anhalten kann. Mit Hilfe von spezifischen histologischen Markern weisen die Forscher später das korrekt synthetisierte Protein nach und bestätigen damit den Erfolg der Therapie. Bei Mäusen funktioniert dieses Verfahren bei einigen Erkrankungsformen mittlerweile gut. Nun steht die Übertragung auf den Menschen an. Therapie der Achromatopsie bei Mäusen lebenslang erfolgreich 3 Verschiedene elektronenmikroskopische Aufnahmen des Auges © Seeliger Für die Übertragung des Gentherapie-Verfahrens auf den Menschen wurde eine Erkrankung gewählt, die die Arbeitsgruppe Seeligers schon seit 15 Jahren zusammen mit einer Münchner Arbeitsgruppe erforscht. Bei der Achromatopsie fehlen die Kanäle in der Netzhaut, die für die Umwandlung der biochemischen in elektrische Signale verantwortlich sind. Somit kann kein elektrisches Signal generiert werden, es kann kein Sehen mit Zapfen stattfinden. „Dieses Leiden ist für die Betroffenen extrem belastend. Da im Sehzentrum, der Makula, ganz in der Mitte nur Zapfen vorkommen und auch in der Umgebung eine hohe Zapfendichte besteht, ist die Sehschärfe auf etwa 1/10 reduziert. Weiterhin stark belastend sind ein Augenzittern (Nystagmus) und eine extreme Blendempfindlichkeit, weiterhin fehlt jegliches Farbensehen. Dabei funktionieren die Zellen ja eigentlich, nur die Ionenkanäle zur Umsetzung der biochemischen in die elektrische Aktivität fehlen“, erklärt Seeliger. Bei der Gentherapie kommt es allerdings dann zu Problemen, wenn die behandelte Zelle selbst die Proteinmenge nicht regulieren kann. Denn zu viel des fehlenden Proteins kann im schlimmsten Fall zu einem Absterben der Zellen führen. Bei der Achromatopsie bestehen die Kanäle aus den zwei Untereinheiten α und β, die von unterschiedlichen Genen codiert werden. Da der genetische Defekt nur eine der beiden Untereinheiten betrifft, wird die Menge an produzierten Kanälen durch die andere, nicht betroffene Untereinheit reguliert. Eine Überdosierung ist also ausgeschlossen. Dies ist sicher einer der Gründe dafür, warum die Gentherapie der Achromatopsie im Mausmodell lebenslang erfolgreich war. Erste Gentherapie am Auge in Deutschland in den Startblöcken Der nächste logische Schritt wäre nun die Übertragung auf den Menschen. Für eine Gentherapie gelten aber dieselben Regeln wie für die Zulassung eines Medikamentes gegen Kopfschmerzen oder Herzprobleme. Umfangreiche Unterlagen sind zu erstellen. Zum Beispiel muss die toxikologische Unbedenklichkeit nachgewiesen werden und der Wirkstoff muss in ganz bestimmten zertifizierten Anlagen unter GMP-Bedingungen hergestellt worden sein. Für diese Dinge ist mit Kosten von mehreren Millionen Euro zu rechnen – eine Hürde, die solche Projekte oft an dieser Stelle stoppt. Es sei denn, es findet sich ein Investor, der finanzielles Interesse an der Therapie hat. Da es sich aber bei der Achromatopsie und vielen anderen erblichen Netzhauterkrankungen um seltene Erkrankungen handelt, ist das Interesse seitens der Medikamentenhersteller eher gering. „Im Falle einer Therapie spezifischer genetischer Defekte ist das besonders problematisch, denn bei Erfolg hat man kein Medikament an der Hand, das für mehrere Krankheiten gleichzeitig verordnet werden kann. Die Therapie ist ja nur bei einem einzelnen Gendefekt wirksam. Das heißt, die gleiche Summe muss dann gegebenenfalls nochmals für das nächste Gen aufgebracht werden und so weiter“, erklärt Seeliger. Im Fall des Tübinger Instituts ließ sich glücklicherweise ein privater Sponsor finden: Eine Stiftung unterstützt das Projekt finanziell für fünf Jahre, von denen gerade die zweite Hälfte angebrochen ist. Die ursprünglich für das Mausmodell konstruierten Vektoren wurden nun durch solche mit dem menschlichen Gen ersetzt. Da der humane Promotor auch noch bei den Mäusen funktioniert, haben die Wissenschaftler die Möglichkeit einer Qualitätskontrolle für die im französischen Nantes produzierten Vektorchargen. „Bisher hat bei den Vorserien alles sehr 4 gut funktioniert, wir erwarten die erste klinische Charge in diesen Tagen“, so der Tübinger Professor. „Diese testen wir dann am Mausmodell und gehen davon aus, dass sie für den Einsatz am Patienten freigegeben werden kann.“ Momentan werden unter einer großen Anzahl an infrage kommenden Patienten aus dem In- und Ausland diejenigen ausgesucht, bei denen das Studienziel am besten erreichbar erscheint. Dies wäre in Deutschland das erste Mal, dass eine Gentherapie am Auge stattfindet. Bleibt zu hoffen, dass sich auch für die weiteren Phasen Sponsoren finden. Fachbeitrag 20.10.2014 pbe BioRegio STERN © BIOPRO Baden-Württemberg GmbH Weitere Informationen Prof. Dr. Mathias Seeliger Schleichstr. 4/3 72076 Tübingen Tel.: 07071-29 80718 E-Mail: see(at)uni-tuebingen.de AG Neurodegeneration des Auges Der Fachbeitrag ist Teil folgender Dossiers Zell- und Gentherapien: Aus der Forschung in die Klinik 5