Kapitel28
Werbung

(404)
31 8. Nebengruppe
Als einzige Gruppe des PSE enthält die 8.NG Elemente verschiedener Elektronenzahl in der
zweitäußersten Schale. In ihr werden jeweils drei Elemente der drei Übergangsmetallreihen
aufgenommen, wodurch sich drei Triaden ergeben. Die erste Triade (Eisengruppe) unterscheidet sich
chemisch so erheblich von den restlichen sechs Elementen (Platinmetalle), dass eine getrennte
Behandlung sinnvoll erscheint.
Eisen Fe
Ruthenium Ru
Osmium Os
Cobalt Co
Rhodium Rh
Iridium Ir
Nickel Ni
Palladium Pd
Platin Pt
Eisengruppe, unedel
leichte Platinmetalle
schwere Platinmetalle
edel
31.1 Eisengruppe (Eisenmetalle: Fe, Co, Ni)
Ordnungszahl
Außenelektronen
Dichte bei 20oC/g · cm-3
Schmelzpunkt /oC
E°/V für M/M2+
für M2+/M3+
Ionenradien
(in pm)
Eisen
26
3d64s2
7,87
1535
-0,44
+0,77
M2+(KOZ = 6) 92(hs), 75(ls)
M3+(KOZ = 6) 79(hs), 69(ls)
Cobalt
27
3d74s2
8,70
1490
-0,28
+1,81
Nickel
28
3d84s2
8,90
1452
-0,25
89(hs), 79(ls)
75(hs), 69(ls)
83
ls: low spin
hs: high spin
Die Elemente der Eisengruppe (3d-Übergangsmetalle) besitzen ähnliche Dichten und Schmelzpunkte. Sie
sind alle ferromagnetisch.
Für das Verhalten von Übergangsmetallen ist die Beteiligung ihrer d-Elektronen bei der
Verbindungsbildung charakteristisch. Die Behandlung der übrigen Nebengruppen des PSE wird zeigen,
dass bis zur halben Füllung der d-Unterschalen (also bis d5) alle diese ungepaarten d-Elektronen
(zusammen mit den beiden äußersten s-Elektronen) sich mehr oder weniger leicht an der
Verbindungsbildung beteiligen (bis zur maximalen Oxidationsstufe +VII in der 7. NG.). Diese
vollständige Beteiligung der d-Elektronen gilt dann nicht mehr, wenn die d-Bahnen über die Hälfte
besetzt sind (vom Eisen ab in der ersten Übergangsreihe). Deshalb werden die formal denkbaren höchsten
Oxidationsstufen (Fe +VIII, Co +IX und Ni +X) nicht erreicht; die Elemente bilden vielmehr recht
niedrige Oxidationsstufen aus - von Fe und Co kennt man als wichtigste +II und +III, und von Ni +II.
Die Normalpotentiale der drei Elemente zeigen, dass alle drei zwar prinzipiell “unedel” sind, dass aber
nur Eisen schon an feuchter Luft - naturgemäß besonders von Säuren - leicht oxidiert wird, während
Cobalt und Nickel gegenüber H2O und nicht oxidierenden Säuren zunehmend beständiger werden. Von
oxidierenden Säuren werden dagegen alle drei Eisenmetalle erwartungsgemäß leicht angegriffen und
aufgelöst.
Die kleinen Ionenradien in den Oxidationsstufen +II und +III zeigen eine sehr hohe Ladungsdichte an.
Diese hohe Ladungsdichte einerseits und das Vorhandensein nur unvollständig gefüllter d-Orbitale
andererseits begünstigen in sehr starkem Maße die Bildung von zahlreichen Komplexverbindungen dieser
Elemente.
Das Redoxpotential Fe2+/Fe3+ zeigt, dass Fe2+ leicht zu Fe3+ zu oxidieren ist. Beim Cobalt ist in Salzen die
stabile Oxidationsstufe +II, Co(II)-Komplexe lassen sich aber leicht oxidieren, und in
Komplexverbindungen ist Co(III) beständiger.
(405)
Charakteristische Komplexverbindungen sind: oktaedrische diamagnetische Co(III)-Komplexe mit der
low-spin-Konfiguration t62g, tetraedrische Co(II)-Komplexe und planar-quadratische Pd(II)-Komplexe.
Die Salze und Komplexe sind meist farbig.
31.1.1 Eisen
Eisen(II)- und Eisen(III)-Verbindungen
Standardredoxpotentiale Eo (in V, pH = 0)
Fe
Fe2+ + 2e-
-0,41
Fe
Fe3+ + 3e-
-0,036
Fe2+
Fe3+ + e-
+0,77
Diese Standardredoxpotentiale verändern sich mit der Erhöhung des pH-Wertes und in Gegenwart
von Komplexbildnern.
Eisen(II)- und Eisen(III)-Salze hydrolysieren und bilden mit OH- schwerlösliche Hydroxide.
Fe2+ - und Fe3+ besitzen eine reiche Komplexchemie.
FeSO4 · 7H2O, Eisen(II)-Sulfat-Heptahydrat (blassgrün)
Hydrolyse (Reaktion von Fe2+ mit dem koordinierten H2O):
[Fe(H2O)6]2+ + H2O
blassgrün
[FeOH(H2O)5]+ + H3O+
K = 10-7
schwach saure Reaktion!
pH einer 0,1 molaren Lösung 4
Mit der Zeit wird die Lösung dunkler und trübe.
Die infolge Hydrolyse sauer reagierende Lösung oxidiert sich an der Luft unter teilweiser Abscheidung
von basischem Eisen(III)-Sulfat:
4Fe2+ + O2 + 4H3O+
4Fe3+ + 6H2O
Fe2+
Fe3+ + e-
+0,77 V
6H2O
O2 + 4H3O+ + 4e-
+1,23 V
FeSO4 · 7H2O “verwittert” an der Luft zu Fe(III) (Gelbbraunfärbung). Luftstabiler ist das Doppelsalz mit
Ammoniumsulfat (NH4)2 Fe(SO4)2 · 6 H2O “Mohrsches Salz”.
Fe2+ lässt sich leicht zu Fe3+ oxidieren.
(406)
Fe(NO3)3, Eisen(III)-nitrat (blassrosa)
Hydrolyse:
[Fe(H2O)6]3+ + H2O
[FeOH(H2O)5]2+ + H3O+
K = 10-3
gelbbraun
pH einer 0,1 molaren Lösung 2, vergleichsweise stark saure
Reaktion
-2H2O
4[FeOH(H2O)5]
2+
+2H2O
2[(H2O)5 Fe - O - Fe - (H2O)5]4+
gelbrauner, zweikerniger Eisenkomplex
6+
-H2O, -2H+
+H2O, +2H+
H2O
OH2
H2O Fe
H2O
H2O
O
OH2
OH2
Fe
H2O
H2O
O
OH2
OH2
Fe
H2O
H2O
O
OH2
OH2
Fe
H2O
OH2
OH2
rotbraun
Die Lösung färbt sich mit der Zeit immer mehr rotbraun und es kommt zur Niederschlagsbildung; aus den
mehrkernigen löslichen Eisenkomplexen (Isopolyoxykationen) haben sich schwerlösliche
Eisen(III)-oxid-Hydrate gebildet. Die Hydrolyse lässt sich mit Salpetersäure zurückdrängen (Bildung des
[Fe(H2O)6]3+ in stark sauer Lösung).
Fe(OH)3, Eisen(III)-hydroxid
Fe(OH)3 FeO(OH)x · xH2O Eisen(III)-oxid Hydrat
Versuch: Fe3+ + 3OH-
Lp {Fe(OH)3} = c (Fe3+) . c3(OH-) = 5 . 10-38 mol4/l4
Fe(OH)2, Eisen(II)-hydroxid
Versuch: Fe2+ + 2OH-
Fe(OH)2 weiß
(grünlich durch Anwesenheit von Fe3+, mit der Zeit
grün-schwarz-braun Fe(OH)3)
Lp {Fe(OH)2} = c (Fe2+) . c2(OH-) = 2 . 10-15 mol3/l3
Wie groß ist das Redoxpotential für das Redoxgleichgewicht Fe2+/Fe3+ bei pH = 14 {c(OH-) = 1 mol . l-1}?
Unter Berücksichtigung der Löslichkeitsprodukte für Fe(OH)3 und Fe(OH)2 kann mit der Nernstschen
Gleichung das Redoxpotential berechnet werden:
E(Fe2+/Fe3+) = E°(Fe2+/Fe3+) +
E°(Fe2+/Fe3+) = 0,77 V
0,059
1
lg
c(Fe3+)
c(Fe2+)
(407)
c(Fe3+) = 5 · 10-38 mol4 · l-4 / c3(OH-)
c(Fe2+) = 2 · 10-15 mol3 · l-3 / c2(OH-)
c(OH-) = 1 mol · l-1
2+
5 . 10-38
3+
E(Fe /Fe ) = 0,77 + 0,059 lg
2 . 10-15
= 0,77 + 0,059 lg (2,5 · 10-23) = 0,77 + 0,059 (0,398-23)
E(Fe2+/Fe3+) = -0,56 V
Aufgrund der erheblichen Unterschiede in den Löslichkeitsprodukten von Fe(OH)3 und Fe(OH)2 wird das
System Fe2+/Fe3+ im basischen Milieu zu einem starken Reduktionsmittel.
Versuche: Fe2+ als Reduktionsmittel
a) im basischen Milieu
+II
+V
8 Fe(OH)2 + NO3- + 6H2O
+III
-III
8Fe(OH)3 + NH3 + OH-
Fe(OH)2 kann Nitrat bis zum Ammoniak reduzieren. Entweichendes NH3 färbt feuchtes Lackmuspapier
blau.
b) im sauren Milieu
+II +V
3Fe2+ + NO3- + 4H+
+III +II
3 Fe3+ + NO + 2H2O
Die Bildung von NO wird durch die Bildung des braunen Pentaaquanitrosyleisen(II)-Komplexes
[Fe(H2O)5NO]2+ sichtbar.
In saurer Lösung ist Fe(II) ein deutlich schwächeres Reduktionsmittel. Es wird nur durch stärkere
Oxidationsmittel, wie HNO3 oder H2O2 in Fe(III) überführt.
Da Fe2+ im alkalischen Medium stärker reduzierend wirkt, muss umgekehrt Fe3+ in saurem Milieu stärker
oxidierend wirken.
Versuch:
H2O + HSO3Fe2+
+IV
2Fe3+ + HSO3- + H2O
SO42- + 3H+ + 2e-
Eo(V)
0,158
Fe3+ + e-
0,77
+VI
2Fe2+ + SO42- + 3H+
Die Bildung von Fe2+ kann durch die Zugabe von OH- sichtbar gemacht werden {Bildung von
“grünlichem” Fe(OH)2}.
(408)
Fe(II) und Fe(III)-Komplexverbindungen
+4CNVersuch: Fe2+ + 2CN-
K4[Fe(CN)6]
K3[Fe(CN)6]
Fe(CN)2
rotbraun
-4CNEisen(II)-cyanid
Kalium-hexacyanoferrat(II)
Kalium-hexacyanoferrat(III)
[Fe(CN)6]4gelbe Lösung
Hexacyanoferrat(II)-Komplex
gelbes Blutlaugensalz
rotes Blutlaugensalz
Blutlaugensalze - der Name rührt daher, dass sie früher u.a. durch Erhitzen von Blut (Fe-, C- und Nhaltig) mit K2CO3 und Auslaugen der dabei erhaltenen Schmelze mit Wasser gewonnen wurden.
Das Redoxpotential des Paares Fe2+/Fe3+ verändert sich, wenn Fe2+ und Fe3+ als Hexacyanokomplexe
vorliegen:
E(pH = 7)
2+
3+
Fe
Fe + e
~ -0,15 V
+II
[Fe(CN)6]4-
+III
[Fe(CN)6]3- + e-
+ 0,36 V
Durch die Komplexbildung mit CN- hat die Reduktionskraft von Fe2+ abgenommen und die
Oxidationskraft von Fe3+ zugenommen.
Die 18-Elektronen-Regel erklärt die relative Redoxstabilität von [Fe(CN)6]4- gegenüber [Fe(CN)6]3- .
[Fe(CN)6]4Fe2+(d6) 6 Elektronen
12 Elektronen
_____________________
18 Elektronen
Edelgaskonfiguration
(Krypton)
[Fe(CN)6]3-
- jeweils 6CN - Liganden
(koordinative Bindung
in Durchdringungskomplexen)
Fe3+(d5) 5 Elektronen
12 Elektronen
_______________________
17 Elektronen
Ein Elektron fehlt zur KryptonElektronenkonfiguration, deshalb ist
Hexacyanoferrat(III) ein mildes
Oxidationsmittel
Nachweise für Fe2+ und Fe3+
Fe3+ + gelbes Blutlaugensalz
tiefblaue Niederschläge von Berliner Blau
+II +III
Fe2+ + rotes Blutlaugensalz
Fe[FeFe(CN)6]3
Die Strukturen dieser Niederschläge leiten sich von einem einfachen Würfelgitter ab, in welchem die
Würfelecken von Fe-Ionen und die Würfelkanten zwischen den Fe-Ionen von längs dieser Kante
angeordneten CN-Ionen besetzt sind (Abb.). Das weichere Lewis-basische Kohlenstoffende von CN- ist
mit dem weicheren Lewis-sauren Fe2+ und das härtere Lewis-basische Stickstoffende mit dem härteren
Lewis-sauren Fe3+ verknüpft.
(409)
(NC)5Fe
low spin
C
N
-
Fe(NC)5
ambidentes high spin
Cyanidion
Ausschnitt aus der Kristallstruktur von Berliner Blau zur Illustration der Brückenbildung durch die
ambidenten Cyanidionen. Große Kreise: Fe(II), kleine Kreise: Fe(III), schwarzer Punkt: Sauerstoffatom
eines Wassermoleküls. Weitere, in den kubischen Hohlräumen eingelagerte Wassermoleküle wurden
nicht eingezeichnet, ebenso die meisten der Cyanidionen.
Ein sehr empfindlicher Fe3+-Nachweis ist die Reaktion mit Kaliumthiocyanat, KSCN.
Fe3+ + 3SCN-
Fe(SCN)3 + 3H2O
blutrot
Versuch:
Farbiges Zaubergemälde
Mit fünf Lösungen {Kaliumhexacyanoferrat(II), Kaliumthiocyanat, Gallusssäure, Salicylsäure und
Natriumcarbonat} wird ein Bild auf Filterpapier gemalt. Man lässt so lange trocknen, bis die Pinselstriche
fast unsichtbar geworden sind und besprüht dann das “latente” Gemälde mit FeCl 3-Lösung, worauf sich
ein Bild mit folgenden Farben entwickelt:
dunkelblau
blutrot
violett
schwarz
“Berliner Blau”
Thiocyanatoeisen(III)-Komplexe
Fe3+-Komplexe der Salicylsäure
Eisengallustinte - Fe3+-Komplexe der Gallusssäure
(410)
hellbraun
Eisen(III)-hydroxid (OH--Ionen werden durch die Hydrolyse von Na2CO3
gebildet).
Vorkommen
In der Erdhülle (Lithosphäre, Hydrosphäre, Biosphäre und Atomsphäre) ist Eisen das vierthäufigste
Element und zweithäufigste Metall (4,7 Gew.-% Fe, 8,13 % Al, 26,3 % Si, 48,9 % O).
Dagegen besteht der Erdkern mit einem Radius von 3500 km (mehr als die Hälfte des gesamten
Erdradius) aus etwa 86 % Fe (Eisen ist ein besonders beständiges Element nach der Kernbindungsenergie
pro Nukleon - siehe Punkt 1.8 (Massendefekt - S. 16).
Bezogen auf die ganze Erdkugel ist Fe mit 34,6 % das häufigste Element.
In der Erdkruste (Lithosphäre) liegt Eisen meist in Form von Oxiden, Sulfiden und Carbonaten vor. Das
aus Magma abgeschiedene Gestein enthält Eisen in der Regel in zweiwertiger Form, während die
Verwitterungsprodukte meist dreiwertiges Eisen aufweisen “Eisen(II) ist das Reduktionsmittel der
Erdoberfläche”. Die roten, braunen und gelben Farbtöne des Erdbodens rühren von Fe2O3 bzw.
Fe2O3 · xH2O her.
Wichtige Erze:
Magneteisenstein
Roteisenstein
Brauneisenstein
Spateisenstein
Eisenkies
Fe3O4 (= FeO · Fe2O3, Magnetit)
Fe2O3 (verschiedene Erscheinungsformen: Hämatit, Eisenglanz)
Fe2O3 · H2O (Limonit)
FeCO3 (Siderit)
FeS2 (Pyrit, Schwefelkies)
Der Name Eisen leitet sich von der gotischen Bezeichnung isarn für festes Metall ab (im Gegensatz zur
weichen Bronze), das Symbol Fe vom lateinischen Namen “ferrum” für Eisen.
Gewinnung
Die technische Darstellung von Eisen ist einfach und besteht in der Reduktion von oxidischen Eisenerzen
mit Koks im Hochofen. Das dabei entstehende Eisen enthält durchschnittlich 4 % Kohlenstoff und wird
„Roheisen“ genannt, wobei man ganz allgemein unter der Bezeichnung Roheisen Eisensorten mit einem
Kohlenstoffgehalt > 1,7 % versteht. Roheisen ist spröde, daher nicht schmiedbar und schmilzt beim
Erhitzen plötzlich. Durch Verringerung seines C-Gehaltes kann man es in den schmiedbaren und beim
Schmelzen allmählich erweichenden „Stahl“ (<1,7 % C) überführen.
Der Hochofen (h = 25-30 m) besteht aus zwei mit den breiten Enden zusammenstoßenden, abgestumpften
Kegeln. Der breiteste Teil des Ofens hat einen Durchmesser von rund 10 m. Eine gerade Zylinderform ist
für den Hochofen nicht möglich, weil die Beschickung während des Niedergehens (Zunahme der
Temperatur) anschwillt und ein “Hängen” des Hochofens verursachen würde, falls man nicht durch
Verbreiterung des Durchmessers nach unten dieser Volumenvergrößerung Rechnung trüge. Im unteren
Teil des Hochofens ist wiederum eine Verkleinerung des Durchmessers möglich, da hier wegen der noch
höheren Temperatur die Beschickung unter Volumenverminderung zum Schmelzen kommt.
Der Hochofen wird vom oberen Ende („Gicht“) her automatisch schichtweise mit einem entsprechend
dem Eisengehalt des Erzes zusammengesetzten Gemisch von Koks und Erz beschickt (Pyrit FeS 2 und
Eisenspat FeCO3 müssen vor ihrer Verarbeitung in das Oxid überführt werden „Rösten“). Dazu müssen
noch „Zuschläge“ gegeben werden, die zusammen mit der „Gangart“ (mineralische Beimengungen der
Erze) eine relativ niedrig schmelzende Schlacke bilden. Für basische Gangarten setzt man
kieselsäurereiche Gesteine, für saure Gangarten dagegen entsprechende Mengen an Kalk zu, so dass die
geschmolzene Schlacke ein Calciumaluminiumsilicat ist. Die Schlacke wird je nach ihrer
Zusammensetzung als Straßenbaumaterial oder zur Herstellung von Mörtel, Bausteinen bzw.
Eisenportlandzement oder Hochofenzement verwendet.
(411)
Aus der untersten Öffnung des Hochofens wird das flüssige Roheisen abgelassen. Darüber schwimmt die
leichtere, aber ebenfalls geschmolzene Schlacke. Sie wird daher aus einer höher gelegenen Öffnung
„abgestochen“. Unmittelbar über der geschmolzenen Schlacke wird heiße Gebläseluft zur Verbrennung
des eingebrachten Kokses eingepresst.
Während des Betriebes - ein Hochofen „läuft“ kontinuierlich mehrere Jahre lang und liefert dabei täglich
1000-1500 Tonnen Eisen und fast ebensoviel Schlacke (s. oben) - finden im Hochofen folgende
Vorgänge statt: Unterhalb der Gicht wird die Beschickung bei 200 °C getrocknet, aufgelockert und
vorgewärmt. Darunter folgt die „Reduktionszone“ mit einer Temperatur von 450-850 °C, in dieser
Schicht reduziert das im Rast aus Koks und heißer Luft gebildete Kohlenstoffmonoxid CO das Eisenoxid
zu sehr lockerem, oberflächenreichem metallischem Eisen nach:
Schematische Darstellung eines Hochofens:
Dieses so gebildete Eisen katalysiert noch die Einstellung des mit Temperaturerniedrigung ohnehin nach
rechts verschobenen Gleichgewichtes,
(412)
2CO
C + CO2
weshalb sich an ihm feinst verteilter Kohlenstoff abscheidet. Dieser löst sich zu einem erheblichen Anteil
im Eisen und erniedrigt dadurch dessen Schmelzpunkt so (von 1535 °C auf 1100 - 1200 °C), dass es in
der 1500 °C heißen Schmelzzone zusammenschmilzt. Die darauf schwimmende Schlacke schützt das
flüssige Roheisen vor der eingeblasenen Heißluft, die sonst das unedle Metall wieder verbrennen würde.
Das an der Gicht austretende „Gichtgas“ enthält 25 % CO; daher kann es noch zur Energiegewinnung
- bes. zum Erhitzen der notwendigen Gebläseluft im „Winderhitzer“ - herangezogen werden. Zur
Überführung von Roheisen in Stahl werden heute praktisch nur noch zwei Verfahren technisch
angewandt: das Konverterverfahren (Windfrischverfahren) und das Siemens-Martin-Verfahren.
Legierte Stähle: Für viele praktische Anwendungszwecke werden besondere Stahllegierungen hergestellt.
Sie können neben C, Si und Mn noch Cr, Ni, W, Mo, V und weitere Elemente enthalten. Der Stahl wird
mit den gewünschten Legierungsbestandteilen unter möglichst vollkommenem Ausschluss des
Luftsauerstoffs umgeschmolzen.
Ni-Zusatz macht den Stahl sehr zäh (mit 25 % Ni, kann er auf die doppelte Länge ausgezogen werden).
Cr-Zusätze machen Stahl besonders verschleißfest und korrosionsbeständig. So enthält beispielsweise
V2A-Stahl 0,2 % Kohlenstoff, 18 % Chrom und 8 % Nickel, V4A-Stahl dazu noch 2 % Molybdän.
Eigenschaften des Eisens
Reines Eisen ist silberweiß. Es tritt in drei verschiedenen enantiotropen Modifikationen auf. Bis 906 °C
als kubisch raumzentriertes -Eisen, von 906 °C bis 1401 °C als kubisch flächenzentriertes -Eisen oder
Austenit und von 1400 °C bis zum Schmelzpunkt bei 1535 °C als -Eisen:
906oC
1401oC
1535oC
- Eisen
-Eisen
-Eisen
krz
ferromagentisch,
kdp
paramagnetisch
krz
paramagnetisch
geschmolzenes Eisen
Wie auch seine beiden rechten Nachbarn im Periodensystem, Cobalt und Nickel, ist Eisen
ferromagnetisch. Für den an den kristallinen Zustand gebundenen Ferromagnetismus ist charakteristisch,
dass die magnetische Suszeptibilität ( Aufnahmefähigkeit) sehr hohe Werte annimmt.
Ferromagnetismus
- kooperatives bzw. kollektives magnetisches Phänomen, magnetische Spinmomente der
paramagnetischen Zentren (Fe, d6) richten sich unterhalb einer bestimmten Temperatur (ferromagnetische
Curie-Temperatur, Fe: 770°C, Co: 1130 °C, Ni: 358 °C) spontan parallel in kleinen Stoffbezirken
“Weiss’sche Bereiche” aus. Jedoch sind die Richtungen der Magnetisierung der einzelnen Weiss’schen
Bereiche statistisch im Raum verteilt, so dass sich die magnetischen Momente zu einem Gesamtmoment
von Null ergänzen. Eine Magnetisierung der Ferromagnetika (parallele Ausrichtung der Momente der
Weiss’schen Bereiche) erfolgt erst im äußeren Magnetfeld. Ferromagnetika besitzen eine magnetische
Aufnahmefähigkeit , die um 107 bis 1010 mal größer als die normaler Paramagnetika ist (sie werden
besonders stark von Magneten angezogen).
Ferrimagnetismus
Ferrimagnetische Stoffe (z. B. Fe3O4 = FeO . Fe2O3 enthalten zwei Sorten paramagnetischer Zentren (im
Falle Fe3O4*: Fe2+ und Fe3+). Unterhalb der ferrimagnetischen Curie-Temperatur (585oC für Fe3O4)
richten sich innerhalb Weiss’scher Bereiche die magnetischen Spinmomente gleichartiger Zentren
(413)
spontan parallel und ungleichartiger Zentren antiparallel zueinander aus. Sofern sich die antiparallel
orientierten magnetischen Momente wie etwa im Falle von Fe3O4 nicht kompensieren, resultieren
beachtliche magnetische Momente für die einzelnen Weiss’schen Bereiche, die aber wegen ihrer
statistischen Verteilung im Raum nach außen nicht in Erscheinung treten. Eine Magnetisierung der
ferrimagnetischen Stoffe (parallele Ausrichtung der einzelnen Momente der Weiss’schen Bereiche)
erfolgt ähnlich wie bei den ferromagnetischen Stoffen erst nach Einwirkung eines äußeren Magnetfeldes.
* Fe3O4 enthält pro Formeleinheit ein Fe2+-Ion (4 ungepaarte Elektronen, magnetisches Moment pro Ion
5.2 BM) und zwei Fe3+-Ionen (5 ungepaarte Elektronen; magnetisches Moment pro Ion 5.9 BM). Es sind
die Fe3+-Ionen auf Tetraederplätzen mit den Fe2+- und Fe3+-Ionen auf Oktaederplätzen des inversen
Spinells antiferromagnetisch gekoppelt, so dass das magnetische Moment der Fe2+- Ionen unkompensiert
bleibt. Der älteste bekannte magnetische Werkstoff ist “Magneteisenstein” (“Magnetit”) Fe 3O4. Er gab als
“lithos magnetis” (= Stein aus Magnesia) der Erscheinung Magnetismus ihren Namen. Ferro- und
ferrimagnetische Werkstoffe werden in der Stark- und Schwachstromtechnik, in der Nachrichtentechnik
und Elektronik und in der Tonaufzeichnungs- und Videotechnik verwendet. (Fe3O4 und -Fe2O3 als
Magnetpigmente in Audiocassetten, Ton- und Videobändern).
Chemische Eigenschaften
Eisen löst sich als unedles Metall in Säuren Eo(Fe/Fe2+) = -0,44 V.
Es bildet an trockener Luft und in luft- und kohlendioxidfreiem Wasser eine zusammenhängende OxidSchutzhaut (bildet sich auch in konz. H2SO4 und konz. HNO3, sowie beim Behandeln mit Mennige
Pb3O4-Rostschutzfarbe).
An feuchter, kohlendioxidhaltiger Luft oder in kohlendioxid- und lufthaltigem Wasser wird Eisen unter
Bildung von Eisen(III)-oxid-Hydrat FeO(OH) = “Fe2O3 · H2O” angegriffen (“Rosten”), indem sich
zunächst Eisencarbonate bilden, die dann der Hydrolyse unterliegen (besonders aggressiv verhält sich
elekrolythaltiges Meerwasser oder SO2-haltiges Wasser in Industriegebieten). Die auf diesem Weg
gebildete Oxidschicht stellt keine zusammenhängende festhaftende Haut dar, sondern springt in Schuppen
ab und legt dabei frische Metalloberflächen frei, so dass der Rostvorgang weiter in das Innere des Eisens
fortschreiten kann.
Für das Rosten sind Wasser, Sauerstoff und ein Elektrolyt vonnöten - fehlt nur eine dieser Komponenten,
ist kein merkliches Rosten feststellbar. Zum Schutz von Eisen und Stahl vor Rost geht man vielerlei
Wege
a) Aufbringung resistenter Schichten (Lackierung, Emaille) und b) Zulegieren von edleren Metallen
(Edelstähle) oder man macht sich die Elektrochemie zu Nutze.
Versuch (Korrosionsschutz):
In einer Schale mit einer K3[Fe(CN)6]-Lösung (rotem Blutlaugensalz) werden drei abgeschmirgelte FeNägel hineingelegt. Ein Fe-Nagel wird leitend mit einem Cu-Blech verbunden, ein anderer mit Zink.
Nach wenigen Minuten bildet sich in der Umgebung des mit Kupfer verbundenen Nagels eine
Blaufärbung, später (10-20 Min.) auch am „freien Nagel. An dem mit Zink verbundenen Nagel bleibt die
Blaufärbung aus.
Erklärung:
Werden zwei verschiedene Metalle leitend miteinander verbunden, bildet sich ein „Lokalelement“ aus.
Das unedlere Metall geht in Lösung (Anode) und überträgt die entstehenden Elektronen auf das edlere
Metall, das dann als Kathode wirkt und vor einem Angriff geschützt ist. Dieses Prinzip wird bei
Rohrleitungen, Brücken, Tanks und Schiffen ausgenutzt. An den zu schützenden stählernen Objekten
bringt man „Opferanoden“ an (meist aus Magnesium oder Zink).
(414)
Der umgekehrte Fall kann auftreten, wenn ein eisernes Bauteil mit einem Überzug eines edleren Metalls
(z.B. Zinn) versehen ist. So lange die Schutzschicht unversehrt ist, erfolgt keine Korrosion. Ist die
Oberfläche beschädigt, bildet sich ein Lokalelement aus, wobei das unedlere Eisen schneller angegriffen
wird.
Im Versuch wird die Auflösung des Eisens, d.h. die Freisetzung von Fe2+-Ionen, durch das
Hexacyanoferrat angezeigt: Es bildet sich „Berliner Blau“.
Bioanorganische Chemie des Eisens
Eisen übt unter allen Elementen besonders viele Funktionen in der lebenden Natur aus und ist in
Organismen in Form von Eisenproteinen wesentlich am Sauerstofftransport sowie an
Elektronenübertragungsreaktionen (“Elektronentransfer”) beteiligt. Man unterteilt die Eisenproteine in
Hämproteine, welche Eisen-Porphyrin-Komplexe enthalten, sowie in Nichthämproteine, welche
Eisen-Schwefel-Cluster oder reine Eisen-Protein-Komplexe aufweisen.
Eisenhaltige Wirkstoffe der Biosphäre:
Hämproteine
Nichthämproteine
Eisenporphinproteine Funktion
Eisenschwefelproteine
Funktion
Hämoglobin
Myoglobin
Cytochrome
Oxygenasen
Oxidasen
Peroxidasen
Catalasen
Rubridoxine
Ferredoxine
Nitrogenasen
Elektronentransfer (B)a)
Elektronentransfer (T, B, P)a)
N2-Redukt. zu NH3 (B, P)a)
Eisenproteine
Funktion
Transferrine
Ferritine
Tierischer Eisentransport
Eisenspeicherung (T, B, P)a)
Tierischer O2-Transport
Tierische O2-Speicherung
Elektronentransfer (T, B, P)a)
Oxygenierungen mit O2
O2-Redukt. zu O2-, O22-, O2Oxidation mit H2O2
H2O2-Dispr. zu H2O/O2
a) Tierische (T), bakterielle (B) und pflanzliche (P) Funktionen. -b) Ferredoxine sind in Kombination mit
anderen Enzymen u.a. an der „Stickstofffixierung“, der „Photosynthese“, der „Atmung“ in den
Zellmitochondrien, der „Kohlendioxidfixierung“ beteiligt. -c) Einführung von O-Atomen aus O2Molekülen in Biosubstrate {Monooxygenierungen: O2
O (inkorporiert) + H2O; Dioxygenierungen:
O2
2O (inkorporiert)}.
Der erwachsene Mensch enthält ca. 4,2 g chemisch gebundenes Eisen, wobei etwa 73 % des
Gesamtgehaltes auf das Hämoglobin entfallen. Bei der Atmung wird das Disauerstoffmolekül durch das
Hämoglobin (Hb) der roten Blutkörperchen (“Erythrocyten”), von der Lunge zum ortsfesten Myoglobin
(O2-Speicherung) in den Muskeln transportiert. Von dort gelangt das O2-Molekül bei Bedarf zur
Energiegewinnung zu den Cytochromen, einer Gruppe weiterer Hämproteine, die als Redoxkatalysatoren
die Endglieder der Atmungskette sind (Energiegewinnung durch metabolische Oxidation von Glucose).
(415)
Blut
Gewebe
Hb(O2)2-3 Mb
Lunge
O2
H2O
Cytochrome
Hb
Mb(O2) 2H+
Der Transport und die Speicherung von O2 mittels der Metalloproteine Hämoglobin und Myoglobin ist
eine ”stöchiometrische Funktion”.
Vereinfacht dargestellt ist Hämoglobin (2 x 141 und 2 x 136 Aminosäuren) ein tetrameres des
monomeren Hämoproteins Myoglobin, das aus einer Polypeptidkette von 153 Aminsäure besteht, die um
eine “Hämgruppe” herum gefaltet ist (s. Abb.) Die chemische Umgebung der Häm-Gruppe ist in
Myoglobin und Hämoglobin fast identisch.
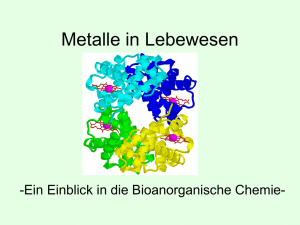
Schematische Strukturen des Myoglobins (links) und des tetrameren Proteins
Hämoglobin (rechts); jeweils mit Proteinfaltung und angedeuteter Häm-„Scheibe“:
(416)
Die Häm-Gruppe besteht aus einem Porphyrin-Eisen-Komplex (s. unten). Porphyrine sind substituierte
Porphine. In diesen Tetrapyrrol-Makrocyclen sind die Pyrrol-Ringe über Methin-Brücken (=CH-)
verknüpft. Es resultiert ein 18--Elektronensystem, wodurch diese Makrocyclen planar und relativ starr
werden. Deshalb ist die Größe des Hohlraumes des Porphyrins recht genau definiert (r = 60-70 pm) und
es können im Hohlraum nur Metallionen entsprechender Größe ungespannt koordiniert werden (durch
vier N-Atome quadratisch planar). Im Hämoglobin schirmt jeweils eine Polypeptidkette ein Eisenzentrum
weitgehend ab und fungiert mit einem Histidin-Imidazolring als axialer Ligand am Eisen, während die
zweite axiale Koordinationsstelle der Komplexeinheit im Desoxyhämoglobin nicht besetzt ist (s. Abb.).
In diesem Zustand erhält die Hämgruppe zweiwertiges Eisen in einem high-spin Zustand mit einem
Ionenradius von 92 pm, das nicht in den Hohlraum des Porphyrins hineinpasst. Folglich nimmt der
Komplex eine out-of-plane Struktur an, mit dem Fe-Atom 42 pm oberhalb der Ringebene.
eg
Fe2+ (d6)
Fe2+
high spin
t2g
r (Fe2+) = 92 pm
out-of-plane Struktur
Bei der end-on Koordination eines O2-Moleküls in der freien axialen Position ändert das Eisen seinen
Spinzustand und wird zu einem low-spin Zentrum mit einem Ionenradius von unter 75 pm. Durch das
“Schrumpfen” des Eisen-Kations erlaubt dieses eine in-plane Koordination (vergl. Abb. unten).
(417)
eg
Fe2+ (d6)
Fe2+
low spin
t2g
r(Fe2+) < 75 pm
in-plane Struktur
Das Fe(II)-Ion bewegt sich also infolge der O2-Koordination in Richtung der Porphyrinebene und “zieht”
dabei den Histidinrest des Proteins mit sich und löst dadurch eine teilweise Umordnung in der Peptidkette
aus. Diese Umordnung in einer Peptidkette löst eine Störung der Salzbrücken zwischen den vier
Häm-Protein-Ketten aus, wodurch die “Taschen” der drei verbleibenden Häm-Untereinheiten geöffnet
werden und die Affinität der betreffenden Häm-Gruppen für O2 zunimmt. (Kooperativität bei der
O2-Aufnahme durch Hämglobin; ist erst einmal ein O2 pro Hb aufgenommen, so wird die Bindung
weiterer O2-Moleküle erleichtert.).
Die außerordentliche Giftwirkung von CO beruht darauf, dass das Hämoglobin (weiches Fe2+-Zentrum)
CO noch stärker bindet als O2, wodurch ein O2-Transport unmöglich wird.
Die Giftigkeit von CN- geht wesentlich auf eine Blockierung von Cytochromoxidase (Fe3+-Zentrum)
zurück.
31.1.2 Cobalt
Cobalt verdankt seinen Namen dem bösen Erdgeist Kobalt. Ihn machten die Bergleute dafür
verantwortlich, dass die Co enthaltenden Erze, die ein schönes, vielversprechendes Aussehen besaßen,
beim Rösten einen üblen, knoblauchähnlichen Geruch entwickelten (Arsen-Gehalt!) und dass sich mit den
damaligen Verhüttungsmethoden kein wertvolles Metall gewinnen ließ. Mit Co-Verbindungen haben
bereits im Altertum die Ägypter, Griechen, Römer und Babylonier Gläser blau gefärbt.
Vorkommen
Co ist ein Spurenelement. Es ist in den meisten Böden anzutreffen und kommt in zahlreichen Mineralien,
fast immer in Begleitung von Nickel, vor.
Wichtigste Co-Erze:
Cobaltin (Kobaltglanz, CoAsS)
Skutterudit (Speiskobalt, Smaltin, CoAs3)
Erythrin (Kobaltblüte, Co3(AsO4)2 · H2O)
Verwendung
- korrosionsbeständige Legierungen
- gepulvertes Kalium-cobaltsilicat zur Blaufärbung, farbloser Glasflüsse („Cobaltblau“)
- Co2(CO)8 Katalysator Hydroformylierung (Alken + CO + H2 Aldehyd)
- Ein Sinterwerkstoff aus Wolframcarbid WC und 10 % Co dient als “Widia” (hart wie Diamant) zur
Herstellung
von
Schneidwerkzeugen
anstelle
von
Diamanten
für
Gesteinbohrer.
Co(II)- und Co(III)-Verbindungen
Die wichtigsten Oxidationsstufen von Co sind +II und +III. Bei den einfachen Co-Verbindungen ist die
zweiwertige Oxidationsstufe wesentlich beständiger als die dreiwertige, in Gegenwart von
Komplexbildner lässt sich Co(II) dagegen leicht zu Co(III) oxidieren.
(418)
2+
E°(V) bei pH = 0
-0,28
-
Co
Co
+ 2e
Co
Co3+ + 3e-
+0,41
Co2+
Co3+ +
+1,808
([Co(H2O)6]2+
rosa
Co2+(d7)
[Co(H2O)6]3+ + e-)
blau
Co3+(d6)
e-
- anders als beim Eisen ist in Wasser beim Cobalt die zweiwertige Oxidationsstufe wesentlich beständiger
als die dreiwertige, demzufolge gibt es eine große Zahl von Co(II)-Salz-Hydraten und nur wenige
Co(III)-Salze
Co(II)-Salze: meist rosa gefärbt durch das oktaedrische [Co(H2O)6]2+-Ion
Hydrate: CoSO4 · 7H2O, CoCl2· 6H2O, Co(NO3)2 · 6H2O
Co(III)-Salz: CoF3 braun, aus den Elementen erhältlich (Co(III)-Salze können nur H2O-frei existieren).
Wie im Falle des Redoxsystems Fe(III)/Fe(II) verschiebt sich das Potential auch für das Redoxsystem
Co(III)/Co(II) beim Übergang von pH = 0 zu pH = 14 auffallend zu weniger positiven Werten wegen der
geringen Löslichkeit der dreiwertigen Stufe im alkalischen Milieu, so dass frisch mit NaOH gefälltes
blaues Co(OH)2 an der Luft analog Fe(OH)2 zu braunem CoO(OH) oxidiert wird.
-
Eo(in V) pH = 14
+0,17
Co(OH)2
Co(OH)3 + e
4OH-
O2 + 2H2O + 4e-
+0,40
Versuch:
2+
2Co
-
+ 4OH
2Co(OH)2
1
2 O2
H2O
2CoO(OH) . H2O
blau
[Co(H2O)6]Cl2
H2O-Abgabe
2Co(OH)3
braun
[CoCl2(H2O)2]
H2O-Aufnahme
rosa
blau
Auf dem gleichen Vorgang beruht der Farbumschlag von blauem Kieselgel (“Blaugel”), der anzeigt, dass
das Trocknungsvermögen des Kieselgels erschöpft ist und das nunmehr rosafarbene Trockenmittel durch
Erhitzen (Wasserabgabe unter Rückkehr der blauen Farbe) wieder regeneriert werden muss.
(419)
Zwei- und dreiwertiges Cobalt weisen ähnlich wie zwei- und dreiwertiges Eisen eine hohe
Komplexbildungstendenz auf und treten auch in der lebenden Natur als Wirkstoffzentren auf.
Co(III)-Komplexe
Co3+ (isoelektronisch mit Fe2+ ) bildet in der Regel oktaedrische low-spin-Komplexe, da nur so hohe
Ligandenfeldstabilisierungsenergien (LFSE) erzielbar sind:
eg
t2g
low-spin
- 24Dq + 2P
LFSE:
high-spin
- 4Dq
In Komplexverbindungen ist Co(III) stabiler als Co(II). Die hohen Ligandenfeldstabilisierungsenergien
stabilisieren die Co(III)-Komplexe, und die meisten oktaedrischen Co(II)-Komplexe sind, wie die
Redoxpotentiale zeigen, instabil gegen Luftsauerstoff (in Anwesenheit von Komplexbildnern wird Co(II)
von Luftsauerstoff oxidiert).
[Co(CN)5]
3-
-
+ CN
[Co(CN)6]
3-
-
+ e
Eo(V)
-0,83
+ e-
+0,11
[Co(NH3)6]2+
[Co(NH3)6]3+
6H2O
O2 + 4H3O+ + 4e-
4[Co(H2O)6]2+ + 4NH4+ + 20NH3 + O2
rosa
4[Co(NH3)6]3+ + 26H2O
orangegelb
+1,23
Co(III) besitzt eine starke Affinität zu N-Liganden. Es sind etwa 2000 Komplexe mit NH3, Aminen und
Nitrogruppen bekannt. Paramagnetische high-spin Komplexe sind die Ausnahme (z. B. blaues [CoF6]3-).
Co(II)-Komplexe
Fast alle Co(II)-Komplexe sind oktaedrisch oder tetraedrisch gebaut und high-spin-Komplexe.
Co(II) bildet mehr tetraedrische Komplexe als die anderen Übergangsmetallionen. Für ein d 7-Ion ist die
Differenz zwischen oktaedrischer und tetraedrischer Ligandenfeldstabilisierung kleiner als für die meisten
d-Konfigurationen, die Benachteiligung der tetraedrischen Koordination also gering.
(420)
d7
oktaedrisch
(high-spin)
tetraedrisch
LFSE: -8DqO
LFSE: -12DqT
LFSE: -12DqT . 4
9
~ -5,3DqO
~
Tetraedrische Komplexe werden mit einzähnigen Liganden wie Cl-, Br-, SCN-, OH- gebildet. Der Wechsel
der Koordination führt auch zu einem Farbwechsel. Oktaedrische Co(II)-Komplexe sind im allgemeinen
rosa bis rot, tetraedrische Co(II)-Komplexe blau.
Versuch:
+4Cl- (konz. HCl)
[Co(H2O)6]
rosa
2+
[CoCl4]2tiefblau
-6H2O
Versuch:
Co2+-Nachweis neben Fe3+ (Maskierung durch F-)
+7SCN2+
Co + Fe
3+
+6F2-
[Co(SCN)4]2- + [FeF6]3-
[Co(SCN)4] + [Fe(SCN)3]
blau
blutrot
-3SCN-
blau
farblos
dunkel
Tetra(thiocyanato)cobaltat(II)-Komplex
Bioanorganische Chemie
Spuren von Cobalt (2,5 mg im erwachsenen Menschen) haben im lebenden Organismus u.a. in Form des
mit verschiedenen Apoenzymen gekoppelten Coenzyms B12 5-Desoxyadenosylcobalamin (s.Abb.), einem
dem Hämoglobin (FeII-Komplex, s.o.) verwandten Co(III)-Komplex mit hydriertem, leicht verändertem
Porphin-Liganden (Corrin-Ligand) eine Reihe von Funktionen bei der Erythrocytenbildung im
Knochenmark, ferner bei der Nervenleitung und beim Wachstum. Da das Cobalaminsystem von
Menschen und Tieren nicht selbst erzeugt werden kann, muss es u.a. in Form “von Vitamin B 12" von
außen zugeführt werden.
(421)
Das 5-Desoxyadenosylcobalamin (Coenzym B12) und das Methylcobalamin (s. Bioalkylierung von HgII)
besitzen in der axialen Metall-Koordinationsstelle eine primäre Alkylgruppe, wodurch diese Komplexe zu
den bislang einzigen gesicherten Beispielen für “natürliche” metallorganische Verbindungen in der
Biochemie werden.
Alkylcobalamine nehmen an Redoxreaktionen, Alkylierungen und Umlagerungen teil. An EinelektronenReduktionen und -Oxidationen können CoIII-, CoII- und CoI- Spezies teilnehmen. In der
Ausgangskonfiguration (Abb. unten) liegt dreiwertiges Cobalt (d6) in sechsfach koordinierter Form vor
(Corrin-Anion, carbanionischer Alkyl-Ligand und neutrales Dimethylbenzimidazol; negativ geladenes
Phosphat in der Seitenkette sorgt für den Ladungsausgleich). Hiervon ausgehend sind zwei
Einelektronen-Reduktionsschritte möglich, wobei für die Redoxpotentiale die Art und Anzahl der axialen
Liganden ausschlaggebend sind. Insbesondere tritt bei der Reduktion eine Tendenz zur Verringerung der
axialen Koordination bis hin zur völligen Abspaltung dieser Liganden ein.
X
CoIII
Y
-X-+e
+X-e-
CoII
Y
-Y-+e
+Y-e-
reduktive Eliminierung
oxidative Addition
CoI
(422)
31.1.3 Nickel
Vorkommen, Darstellung, Verwendung
NiS ist mit Magnetkies und Kupferkies vergesellschaftet, die arsenidischen Nickelerze meist mit Co, Cu
und Edelmetallen.
Über verschiedene Verfahren erhaltenes Rohnickel wird durch den Mond-Prozess (Carbonylverfahren)
gereinigt.
80oC
Ni + 4CO
Ni(CO)4
180-200oC
Tetracarbonylnickel, tetraedrische Struktur, Schmp. -19oC, Sdp. 43oC
stark giftig
Metallisches fein verteiltes Nickel wird in Türmen im Gegenstrom mit Kohlenmonoxid bei etwa 80 oC zu
Ni(CO)4 umgesetzt. Anschließend wird Ni(CO)4 dann bei höherer Temperatur thermisch zersetzt, wobei
sehr reines Nickel (99,99 %) erhalten wird.
Ni wird vorrangig für korrosionsbeständige Stähle und Legierungen (Monellmetall: 70 % Ni, 30 % Cu)
verwendet.
Nickel(II)-Verbindungen (d8)
Ni [Ar] 3d8 4s2
Ni2+ [Ar] 3d8
Die wichtigste Oxidationsstufe des Nickels ist +II.
In wässeriger Lösung ist Nickel nur in dieser Oxidationsstufe stabil. Die wasserhaltigen Nickel(II)-Salze
und ihre Lösungen sind grün.
[Ni(H2O)6]2+, Hexaaquanickel(II)-Komplex, blassgrün
z. B. in NiSO4 · 6H2O und Ni(NO3)2 · 6H2O
Nickel(II)-hydroxid, Ni(OH)2
Ni2+ + 2OH-
Ni(OH)2
Lp{Ni(OH).2} = 2 . 10-16 mol2/l2
voluminöses grünes Gel
Komplexverbindungen
Die einzige stabile Oxidationsstufe des Nickels ist +II. Nickel(II)–Komplexe sind daher redoxstabil.
Nickel(II)-Komplexe existieren mit unterschiedlichen Koordinationen. Typisch für Nicke(II) sind
oktaedrische, quadratisch-planare und tetraedrische Komplexe.
(423)
oktaedrisch
quadratisch-planar
H2O
NH3
Ethylendiamin
tetraedrisch
Als Liganden kommen entweder
starke Liganden wie das CN- in
Frage, oder solche, die die quadratisch-planare Konfiguration
erzwingen, wie z. B. Dimethylglyoxim
grün,
blau bis violett
gelb, rot
Cl-, Br-, I-
blau
dx2-y2
eg
t2
dxy
dz2
t2g
e
dxz,d yz
paramagnetisch
diamagnetisch
paramagnetisch
Versuche zum Chelateffekt:
[Ni(H2O)6]2+
+6NH3
[Ni(NH3)6]2+
-6H2O
hellgrün
+3en
[Ni(en)3]2+
-6NH3
blau
G = G = H - TS
- Ni stärker an N
als an O gebunden
H=S 0
violett
G = - in beiden Komplexen
6 N an Ni gebunden
H 0
- Zahl der Teilchen nimmt zu
S=+
Der Chelateffekt wird durch einen Entropiegewinn
verursacht.
(424)
Versuch zum Nachweis (qual. und quant.) von Ni2+:
H
O
H3C
2
C
+ Ni2+
C
H3C
O
H3C
N
N
O
H
Dimethylglyoxim
(Diacetyldioxim)
+2NH3
C
-2NH4+
C
H --
N
O
N
CH3
C
Ni
H3C
N
N
O -H
O
C
CH3
Bis(dimethylglyoximato)nickel(II)