7.9 Reaktionen mit Kohlenstoff-Nucleophilen
Werbung

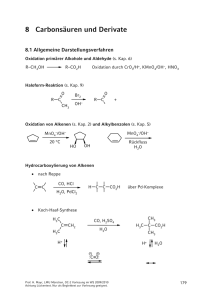
7.9 Reaktionen mit Kohlenstoff-Nucleophilen Die Addition von Cyanwasserstoff ist reversibel und wird durch Basen katalysiert. + HCN – CN– O R C + C N H Cyanhydrin Beim Behandeln der Cyanhydrine mit einer stöchiometrischen Menge Base – zerfallen die Cyanhydrine unter Rückbildung der Carbonylverbindung und von CN . Modifiziertes Kiliani-Fischer-Verfahren zur Verlängerung der Kohlenstoffkette in Zuckern. H CN H C O H C OH CH2OH D-Glycerinaldehyd C NH H C O H C OH H C OH H C OH H C OH H C OH H C OH CH2OH CH2OH CH2OH HCN CN H2, Pd/BaSO4 H+, H2O Druck HO C H H C NH HO C H H2O, – NH3 D-Erythrose H C O HO C H H C OH H C OH H C OH CH2OH CH2OH CH2OH D-Threose Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 161 Umpolung der Carbonyl-Reaktivität nach Hünig: O + R C LiN(iPr)2 (LDA) [CN–] Me3SiCN OSiMe3 R C: CN bzw. [ZnI2] H R'–Br F Et3N·2HF normale Reaktivität O Nu R C O umgepolte Reaktivität R C = H OSiMe3 R C: CN Streckersche Aminosäure-Synthese durch Umsetzung von Aldehyden mit HCN in Gegenwart von Ammoniak und nachfolgende Hydrolyse. O + NH3 H – H 2O R C + + H–CN + NH3 O R C C OH H H3O+ Benzoin-Kondensation O Ph C + :C N : Ph H OH C: CN Ph O C H ·· : :O Ph C N Ph OH H Benzoin Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 162 Cyanid-Ionen (CN–) katalysieren nur die Benzoin-Kondensation aromatischer Aldehyde. Bei aliphatischen Aldehyden nicht möglich, da die Ausbildung des Carbanions nicht gelingt. Statt CN– können hier Thiazolium-Ionen eingesetzt werden, die sich leicht deprotonieren lassen. + R + R N N + OH– H S S + H2O O : H C R Wirkprinzip von Vitamin B1 (Thiamin) Reaktionen mit Acetyliden R'' R C C: Na H3O+ C O + R'' R C C C OH R' R' Grignard-Verbindungen Bei Metallorganischen Verbindungen besitzt Kohlenstoff eine negative Partialladung, weil Metalle weniger elektronegativ als der Kohlenstoff sind. Elektronegativitätsunterschiede in C-Metall-Bindungen: C 2.5 Li 1.0 C 2.5 Mg 1.3 C 2.5 Zn 1.7 C 2.5 Sn 2.0 Die Reaktivität metallorganischer Verbindungen hängt von vielen Faktoren ab. Eine erste Orientierung über ihre Reaktivität liefert die Elektronegativitäts-Differenz. Organolithium- und Organomagnesium-Verbindungen sind besonders reaktiv. Organomagnesium-Verbindungen lassen sich durch Umsetzung Halogenalkanen, -alkenen und –arenen mit Magnesium herstellen. Reaktivität: R–I > Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. von R–Br > R–Cl 163 H 3C CH2 Br + Et2O oder THF Mg 20 °C Ethylmagnesiumbromid Magnesium geht hier von der Oxidationsstufe 0 in die Oxidationsstufe +II über, sodass man diese Einschiebung auch als oxidative Addition bezeichnet. Aryl- und Alkenyl-Grignard-Verbindungen werden auch durch Brom-MagnesiumAustausch hergestellt. iPrMgCl / LiCl X Br Turbo-Grignard (P. Knochel) + iPrBr X MgBr Organomagnesiumverbindungen (Grignard-Reagenzien) werden üblicherweise in Diethylether-Lösung oder Tetrahydrofuran-Lösung hergestellt, wobei die EtherSauerstoffe die freien Koordinationsstellen am Metall besetzen. R' : .O. R' : .O. R Mg Hal R Mg Hal .. : O R' .. O: R' Die in Lösung tatsächlich vorliegende Situation ist jedoch viel komplizierter, da neben dem nachstehend formulierten Schlenk-Gleichgewicht auch verbrückte Strukturen, wie A, eine wichtige Rolle spielen. R Mg R–Mg–R 2 R–Mg–Hal + Hal–Mg–Hal Schlenk-Gleichgewicht Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. Hal Hal Mg R A 164 Grignard-Verbindungen reagieren rasch mit Brønsted-Säuren. Häufig ist dies eine unerwünschte Störung, manchmal dient diese Reaktion auch zur gezielten Herstellung von Amid-Anionen bzw. Acetylid-Anionen. RCO2H H2O CH3CH2–MgBr ROH HNR2 R C C H Durch Umsetzung von Grignard-Reagenzien mit Carbonylverbindungen lassen sich primäre, sekundäre oder tertiäre Alkohole erzeugen. H RMgX C O H2O primärer Alkohol H Formaldehyd R' C O RMgX H2O RMgX H2O sekundärer Alkohol H Aldehyd R' C O tertiärer Alkohol R' Keton Die Grignard- Verbindung fungiert sowohl als Nucleophil als auch als Lewis-Säure zur Aktivierung der Carbonyl-Gruppe. R R δ+ δ– Mg O δ+ R' H + X Mg δ– O + δ– δ+ Mg X O + Mg Mg R X X R' H R R Mg X R' H R X An die Stelle der zweiten Grignard-Verbindung kann auch MgBr2 treten. Die Reaktion kann auch über einen Elektronentransfer-Mechanismus erfolgen, wobei Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 165 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. der aus dem Grignard-Reagens stammende Alkylrest in das Radikal-Kation R·+ übergeht, das isomerisieren kann. Nebenreaktionen bei Grignard-Additionen spielen insbesondere bei sterisch anspruchvollen Systemen eine wichtige Rolle: Enolisierung und Reduktion. Enolisierung: Das Grignard-Reagens fungiert als Brønsted-Base, wird protoniert, und die Carbonylverbindung wird nach saurer Aufarbeitung unverändert zurückgewonnen. a O H Hauptprodukt a CH3–MgBr + b b CH4 + Nebenprodukt H3O+ Reduktion: Der β-Wasserstoff der Organomagnesium-Verbindung hat hydridischen Charakter. X Mg O δ+ C R' R" + H CR2 CR2 δ– Organolithium-Verbindungen Deutlich reaktiver als Grignard-Verbindungen sind Organolithium-Verbindungen. Ihre Herstellung erfolgt i. A. auf einem der drei folgenden Wege: • Oxidative Addition Bu–Cl + 2 Li • Brom-Lithium-Austausch Br + BuLi OMe THF –80 °C Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 166 • Deprotonierung CH-acider Verbindungen (Corey-Seebach-Reaktion, s. oben) S S H Ph S + BuLi S Li Ph Zinkorganische Verbindungen • Reformatsky-Reaktion O O R + Zn Br H OEt + H2O β-Hydroxycarbonsäureester • Anders als Oranomagnesium-Verbindungen Verbindungen nicht mit Estern. • Moderne Entwicklungen der Zink-Organischen Chemie: P. Knochel reagieren Organozink- Elektronenreiche Arene als Nucleophile: Elektrophile aromatische Substitutionen mit Carbonylverbindungen Carbonylverbindungen sind zu wenig elektrophil, um an aromatischen Verbindungen angreifen zu können. Die elektrophile aromatische Substitution an elektronenreichen Arenen gelingt jedoch, wenn die Elektrophilie der Carbonylverbindung durch Brønsted oder Lewis-Säuren erhöht wird. NMe2 O R + C H H+ elektrophile aromatische Substitution oft nicht isolierbar, da rasche Folgereaktion mit weiteren Arenen Erinnerung an OC1: Die Bildung von Bakeliten (Phenol-Formaldehyd-Harze) erfolgt nach dem gleichen Mechanismus. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 167 7.10 Reaktionen mit H – Reduktionen mit komplexen Hydriden Li + H Na + H H Al H H B H O H EtOH R' C H R in Et2O oder THF EtOH analog mit 3 weiteren Carbonylgruppen H2O H Li + R' Al O C R 4 • Aldehyde und Ketone werden durch komplexe Hydride rasch zu primären bzw. sekundären Alkoholen reduziert. Während Natriumborhydrid in alkoholischer Lösung eingesetzt wird, darf LiAlH4 nicht mit protischen Lösungsmitteln zusammengebracht werden (heftige Reaktion unter Freisetzung von Wasserstoff, Brandgefahr). • Während LiAlH4 eine Vielzahl funktioneller Gruppen reduziert (wenig selektiv), reagiert NaBH4 bevorzugt mit Aldehyden und Ketonen (sowie Carbonsäurechloriden, s. Kap. 8). • Da Aldehyde reaktiver als Ketone sind, kann mit dem milden Reduktionsmittel NaBH4 eine Aldehyd-Gruppe in Gegenwart einer Ketogruppe reduziert werden. O H • O NaBH4 MeOH/CH2Cl2 –78 °C OH H O H Bei der Luche-Reduktion wird eine Ketofunktion chemoselektiv in Gegenwart einer Aldehyd-Gruppe reduziert, weil durch Zusatz eines Equivalents CeCl3 das Keton als stärkere Lewis-Base selektiv aktiviert wird. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 168 O O H CeCl3 NaBH4 OH O MeOH/CH2Cl2 –78 °C H H + O O CeCl3 über H aktiviert • L-Selektrid (Li+ –BH(sekBu)3) vermag wegen seines großen sterischen Anspruchs chemoselektiv zwischen Ketonen unterschiedlicher Größe zu differenzieren. O O Li+ –BH(sek-Bu)3 Diastereoselektive und enantioselektive Reduktionen von Carbonylgruppen sind Gegenstand der Vorlesung „Stereoselektive Reaktionen“. Die Meerwein-Ponndorf-Verley-Reduktion mit iPrOH in Gegenwart von Aluminium-isopropylat wurde bereits im Zusammenhang mit der OppenauerOxidation behandelt. Cannizzaro-Reaktion (Disproportionierung von Aldehyden ohne α-H-Atom) – ·· :O–H ·· Ph O C H C O H Ph Ph–CO2– + Ph–CH2OH Direkte Hydridübertragung nachgewiesen, da kein D-Einbau bei der Durchführung der Reaktion in D2O erfolgt. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 169 7.11 Reduktive Kupplungen Ph C O + K Dieses Radikalanion reagiert rasch mit Sauerstoff und Wasser Ether Ph Benzophenon-Kalium tiefblau Nur teilweise Dimerisierung dieses Radikal-Anions Pinakolbildung Me2C=O :Mg Me2C=O · ·· Me2C–O : ·· Mg2+ · ·· Me2C–O : ·· Me2C O Me2C Mg2+ H3O+ Me2C Me2C O OH OH Einschiebung: Pinakol-Pinakolon-Umlagerung OH OH H 3C C H 3C C CH3 CH3 + H+ – H 2O – H+ O CH3 H 3C C C CH3 CH3 McMurry-Reaktion: Bildung von Alkenen aus Aldehyden und Ketonen in einem Schritt ohne Isolierung des intermediären Diols. 2 C O TiCl3/K oder TiCl3/LiAlH4 Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. C C 170 Reaktion verläuft wie oben über Radikalanionen, wahrscheinlich auf der Oberfläche von fein verteiltem Titan, das durch Reduktion von TiCl3 entsteht: Titan-Metall O 2 Ti(0) C R R O Ti O O C· · C R R R R Ti C O O O C R R R R R R + C C R R 7.12 Oxidationen von Carbonylverbindungen O R C H CrVI H2O O R C OH Der über das Aldehyd-Hydrat verlaufende Mechanismus wurde bereits besprochen (s. Kap. 6). Baeyer-Villiger-Oxidation Keton R' Peroxysäure Ester O C R" + H O O O C R chiraler Rest R" wandert mit Konfigurationserhalt O R' C O HO R" + C R O Wanderungsneigung der Substituenten: H > tertiär > sekundär ≈ Phenyl > primär Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 171 O O C Ph CH3 C O OH O C CH3 O 7.13 α,β-ungesättigte Carbonylverbindungen O O ΔH° = –25 kJ mol–1 ähnlicher Wert für die Isomerisierung von 1,4-Pentadien zu 1,3-Pentadien α,β-ungesättigte Carbonylverbindungen gehen 1,2- und 1,4-Additionen ein. 1 2 3 4 3 2 4 1 · ·· ·O ·· ·· O: ··O·· ·· + + O OH Nu: O :Nu 1,2-Addition H Nu OH + NuH – Nu– Nu O Nu 1,4-Addition Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 172 Vgl. auch Kap. 4 „Nucleophile Additionen an CC-Mehrfachbindungen“ H2O [Ca(OH)2] MeOH [MeO–] RSH [Base] O R–NH2/H2O HCN 1,4- vs 1,2-Addition von metallorganischen Reagenzien • Grignard-Reagenzien können 1,2- und 1,4-Additionen eingehen Bei ungesättigten Aldehyden ist immer die 1,2-Addition bevorzugt, bei ungesättigten Ketonen ist die Voraussage schwierig. O + EtMgBr H Ph O + EtMgBr H3O+ H3O+ 37 % 1,2-Addukt 57 % 1,4-Addukt Bei großen Substituenten an der Carbonylgruppe ist die 1,4-Addition günstiger. Ph O + PhMgBr Ph Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. Ph H 3O + O Ph Ph 173 • Dasselbe Keton liefert mit PhLi nur 1,2-Addition Ph O + Ph–Li H3O+ Ph Es ist daher günstig, Organolithium-Verbindungen zu verwenden, wenn 1,2Addition angestrebt wird. • Dagegen wird mit Organokupfer-Verbindungen 1,4-Addition erreicht. 2 CH3Li + CuI CH3 n-Alkyl H – LiI (CH3)2CuLi = (CH3)2Cu– Li+ Gilman-Cuprat O (CH3)2CuLi H3O+ –78 °C, 4 h Neben den aus Organolithium-Verbindungen zugänglichen Gilman-Cupraten werden auch Normant-Cuprate (aus RMgBr und CuBr·SMe2) oder Knochel-Cuprate (aus RZnI und CuCN · 2 LiCl) eingesetzt. Details dazu in der Fortgeschrittenen-Vorlesung „Metallorganische Chemie“. Arbeitet man die bei der konjugaten Addition an α,β-ungesättigte Carbonylverbindungen zunächst erhaltenen Enolate nicht hydrolytisch auf, lassen sich durch Umsetzung mit einem elektrophilen Alkylierungsmittel in einem Eintopf-Verfahren auch zwei neue CC-Bindungen knüpfen. OLi O O Me2CuLi RX O R (±) Me Me Hauptprodukt Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. + R (±) Me Nebenprodukt 174 Michael-Additionen (d. h. 1,4-Additionen CH-acider Verbindungen: s. Kap. 9) Reduktionen α,β-ungesättigter Carbonyl-Verbindungen • Reduktion der Carbonylgruppe meist mit LiAlH4 möglich O LiAlH4 H3O+ OH Ether (NaBH4 ist demgegenüber weniger selektiv!) • Reduktion der CC-Doppelbindung (unter Erhalt der CO-Doppelbindung) mit H2/Pd bzw. Li/fl. NH3 O + Li NH3 – NH2– · O Li H in fl. NH3 stabil H3O+ H O H Isolierte CC-Doppelbindungen werden durch Li/NH3 nicht reduziert, während isolierte CO-Doppelbindungen ebenfalls reduziert werden. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 175 8 Carbonsäuren und Derivate 8.1 Allgemeine Darstellungsverfahren Oxidation primärer Alkohole und Aldehyde (s. Kap. 6) R–CH2OH R–CO2H Oxidation durch CrO3/H+, KMnO4/OH–, HNO3 Haloform-Reaktion (s. Kap. 9) O Br2 CH3 OH– R C O + R C Oxidation von Alkenen (s. Kap. 2) und Alkylbenzolen (s. Kap. 5) Hydrocarboxylierung von Alkenen • nach Reppe C • C CO, HCl H2O, PdCl2 H C C CO2H über Pd-Komplexe Koch-Haaf-Synthese H3C C CH2 CO, H2SO4 H2O H3C H+ CH3 H3C C CO2H CH3 H+ H2O + :C O: Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 176 Hydrolyse von Nitrilen R C N H2O H 2O O R C OH Nitril Amid Carbonsäure Die Hydrolyse erfordert langzeitiges Erhitzen mit starker Säure oder Base. Die Isolierung der intermediären Amide ist nicht erforderlich. Nitrile sind aus Alkylhalogeniden leicht zugänglich. Ph–CH2–Cl + :C N: S N2 40%ige H2SO4 100 °C / 3 h O PhCH2 C OH verwandt: Strecker-Synthese von Aminosäuren (s. Kap. 7) Carboxylierung metallorganischer Verbindungen O δ– R–MgX + C δ+ H 3O + O R C OH O δ– R–Li + CO2 H 3O + O R C OH R–Hal kann somit auf zwei verschiedene Weisen in R–CO2H überführt werden. Meist ist der Weg über R–MgX einfacher. Mechanistisch verwandt: Carboxylierung nach Kolbe (vgl. Kap. 5) Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 177 8.2 Reaktionsmöglichkeiten der Carbonsäuren ·· O: R C ·· H ·O · elektrophiles Zentrum ·· O: basische Zentren R C saures Proton E+ ·· H ·O · X: Die Acidität der Carbonsäuren ist durch die Resonanzstabilisierung der CarboxylatIonen bedingt. Elektronenziehende Substituenten erhöhen die Acidität. O R C + OH H3O+ + H2O ; Ka = pKa O H OH O OH O OH O OH 3.75 4.76 4.87 4.82 pKa = –lg Ka pKa O F OH OH O Br OH O I O O 2.59 pKa OH 4.82 Cl OH 1.25 Cl O O O Cl pKa OH 2.86 OH 2.89 OH 0.65 Cl3C Cl 2.90 3.18 Cl Cl O O OH O OH 4.06 4.52 F3C OH O NC OH 0.23 2.46 Ph–CO2H 4.22 Dicarbonsäuren sind acider als Monocarbonsäuren (induktive Effekte bzw. Stabilisierung der Anionen durch intramolekulare H-Brücken). Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 178 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. Carbonsäuren sind weniger basisch als Alkohole 8.3 Herstellung und Reaktionen von Carbonsäurederivaten mit Nucleophilen Allgemeines Reaktionsmuster: Vergleich von Carbonsäurederivaten mit Aldehyden und Ketonen OH + NuH – Nu– ·· O: R C Nu für X = Alkyl, Aryl, H X + :Nu– R C X Säure-Derivat bzw. Aldehyd, Keton – X– ·· O: R C Nu für X = elektronegative Gruppierungen Bei Reaktionen mit der Carbonsäure (X = OH) konkurriert die Deprotonierung der Carboxy-Gruppe mit dem Angriff des Nucleophils am Carboxy-Kohlenstoff. ·· – :O : R C ·· O H Nu ·· a Nu:– reversible nucleophile Addition (Weg a) ·· ·· a O : b O: :Nu– + H–Nu R C R C ·· – ·· H ·O ·O · ·· · reversible b Säure-BaseReaktion (Weg b) Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 179 Additions-Eliminierungs-Reaktionen können durch Säuren oder Basen katalysiert werden. :NuH + H+ ·· O: R C X :Nu– ·· O: – X– R – H+ C Nu Nu:– + HB+ NuH + B Reaktivitätsreihung gegenüber Nucleophilen O R C Cl > O R C O O C > R' O R C > O R C SR' > O R C OAr O > R > C OR' O R NR'2 Ketone C O Wegen der hohen Reaktivität von Säurechloriden können aus diesen alle genannten Carbonsäure-Derivate sowie Aldehyde und Ketone hergestellt werden. O R O O C C O R + HCl C SR' R R' + NaCl R O H2O C OR' R'OH O R O R'NH2 C R Cl OH R'–MgX H2/Pd, BaSO4 O C R H + LiAl(OR)3Cl + HCl C N R' H LiAl(OR)3H R + HCl C R'SH R'–CO2– Na+ HCl + O R' O R O C + MgXCl + HCl C H Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 180 Darstellung von Carbonsäure-halogeniden O SOCl2 + SO2 + HCl R C O C Cl Cl Cl O PCl5 + POCl3 + HCl R C O R C Cl R C 1/3 PCl3 + 1/3 H3PO3 O R + CO2 + CO + HCl C Cl O P(Ph)3 CCl4 Cl + CO2 + HCl Cl O O C C Cl Cl OH O R C O R + Ph3P=O + HCCl3 C Cl Mechanismus: Darstellung von Carbonsäure-bromiden: O 3 R C O 3 R + PBr3 OH C + H3PO3 Br Carbonsäureanhydride • Herstellung im Labor aus RCOCl: s. oben • Cyclische Carbonsäureanhydride werden auch aus Dicarbonsäure + Anhydrid oder durch Erhitzen der Dicarbonsäure hergestellt. • Technisch: Wacker-Verfahren O H2C=C=O + H3C C OH O H3C C O O C CH3 Hoechst-Knapsack-Verfahren: Oxidation von Acetaldehyd (CH3CHO) mit O2 (Luft) Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 181 Oxidation von Benzol bzw. Naphthalin O O2 O O2 O [V2O5] O [V2O5] O Maleinsäureanhydrid O Phthalsäureanhydrid • Carbonsäureanhydride sind etwas weniger reaktive Acylierungsmittel als Carbonsäurechloride • Umsetzungen mit Alkoholen, Aminen und Wasser möglich. Carbonsäureester Veresterung von Carbonsäuren unter H+-Katalyse. Basen-Katalyse nicht möglich, weil dabei RCO2– gebildet wird. • Mechanismus (bei primären und sekundären Alkoholen): Schritt 1: Protonierung der Carboxylgruppe O R C O + H+ R C H H H H O O O O R C H O R C H O H Dihydroxycarbenium-Ion Schritt 2: H + R C O + O H O R' – H+ H + H+ Schritt 3: H O H+ R C O H O –H2O R' – H+ Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 182 Alles Gleichgewichtsreaktionen: K= [Ester][Wasser] [Carbonsäure][Alkohol] • Umkehr: Säure-katalysierte Esterhydrolyse • Mechanismus im Einklang mit dem Befund, dass markierten Ester ergibt. • Bei tertiären Alkoholen verläuft die Reaktion über tert.-Alkyl-Kationen. R3C–OH H+ O-markiertes Methanol 18 O- + H 2O ·· O: R' 18 + R3 C ··O H ·· – H+ C+ O CR3 R' C O • Nach dem Prinzip der mikroskopischen Reversibilität verläuft die H+-katalysierte Hydrolyse von tert.-Alkylestern ebenfalls über eine O-AlkylSpaltung (tBoc-Schutzgruppe). • Die Veresterung sterisch anspruchsvoller Carbonsäuren verläuft über AcyliumIonen, die in einem langsamen Schritt gebildet werden und anschließend rasch mit Alkoholen weiter reagieren. O OH HO + H+ OH O [H+] OH2 O – H2O langsam • O – H+ Basische Ester-Hydrolyse (Verseifung) ·· O: R OR + ROH ·· + : O–H C ·· ··O R' ·· + –OR' R ·· O: C + HOR' ··O·· ·· Da equimolare Mengen an OH– verbraucht werden, handelt es sich hier um eine „basische“, nicht „basen-katalysierte“ Ester-Hydrolyse. Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 183 • Herstellung von Estern aus Alkoholen und Carbonsäurechloriden bzw. –anhydriden: s. oben Arbeitsweise nach Schotten-Baumann O Ph C + Ph-OH Cl H2O K2CO3 O Ph C OPh Der Alkohol bzw. Phenol wird durch das in H2O unlösliche PhCOCl aus der wässrigen Lösung extrahiert, sodass es zur Veresterung, nicht zur Hydrolyse kommt. Die Hydrolyse findet nur an der Phasengrenze statt. Arbeitsweise nach Einhorn: wasserfrei O Ph C + + R'-OH + N Cl Pyridin fungiert oft nicht nur als Base (um HCl zu binden), sondern auch als nucleophiler Katalysator. Als nucleophiler Katalysator besonders aktiv ist 4-Dimethylamino-pyridin (DMAP, Steglich-Base), sodass sich in gleicher Weise auch Carbonsäure-anhydride für die Estersynthese nutzen lassen (Zusatz einer kostengünstigen Hilfsbase). N(CH3)2 O R kat. C O + R'–OH N O R C NEt3 R C + + RCO2– HNEt3 O R' O :N N(CH3)2 + R'–OH – DMAP – H+ O R C O R C + H+ – RCO2H O Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 184 • Ester aus Carbonsäure-Salzen und Alkylhalogeniden (vgl. Kap. 2) O O R • Na C :O : – ·· + X–CH2–R' + S N2 R X = Br, I Stereochemie! C O CH2 R' Methylester aus Carbonsäuren und Diazomethan (vgl. Kap. 2) O R .–. + H2C N N: + O H O R O CH3 Wegen der Toxizität und Explosivität von CH2N2 nur eingesetzt, wenn wertvolle Carbonsäure verestert werden soll. • Mitsunobu-Reaktion (s. Kap. 1) zum Mechanismus: s. S. 41 O R1–OH 2 R + CO2Et C N N + Ph3P + OH EtO2C O 2 R C 1 N N + Ph3P=O + O R • CO2Et H EtO2C H Veresterung nach Mukaiyama Zunächst wird durch Umsetzung mit 2-Chlor-1-methylpyridinium-iodid ein Pyridiniumderivat erzeugt (Aktivester), das anschließend mit dem Alkohol zum Ester reagiert. Aktivester besitzen ein höheres Acylierungsvermögen als Phenylester. Weitere gebräuchliche Aktivester sind: O R F F O R C F O F C N O N N F Prof. H. Mayr, LMU München, OC-2 Vorlesung im WS 2010/2011 Achtung Lückentext. Nur als Begleittext zur Vorlesung geeignet. 185