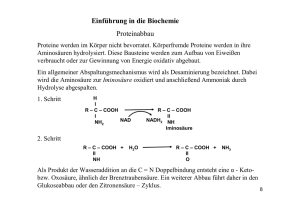
Aufgabe 3: Glutarazidurie Typ I bei den Amish
Werbung

90 Übungsaufgaben Molekulare Grundlagen: Proteinbiosynthese und Genwirkketten Angewandte Genetik: Gensonde als Werkzeug der Gentechnik Aspekte der Cytogenetik: Stammbaumanalyse Aufgabe 3: Glutarazidurie Typ I bei den Amish Einleitung Die Glutarazidurie Typ I (GA I) ist eine sehr seltene Stoffwechselerkrankung, die weltweit mit einer Häufigkeit von 1 auf 100 000 Neugeborenen vorkommt. Dagegen leidet eins von 400 Kindern bei den Amish an dieser Erbkrankheit. Die Amish gehören einer Glaubensgemeinschaft an, die heute vornehmlich in den USA und Kanada lebt. Sie sind bekannt dafür, dass sie viele Formen des technischen Fortschritts ablehnen und Neuerungen nur nach sorgfältiger Überlegung akzeptieren. Sie legen großen Wert auf Familie, Gemeinschaft und Abgeschiedenheit von der Außenwelt. Überwiegend stammen sie von Südwestdeutschen oder Deutschschweizern ab und sprechen untereinander meist Pennsylvaniadeutsch. Aufgabenstellung 1.Ermitteln Sie mithilfe der Hardy-Weinberg-Formel p2 + 2 pq + q2 = 1 den Anteil homozygoter und heterozygoter Merkmalsträger für das Gen für Glutarazidurie Typ 1 (a) weltweit und (b) bei den Amish. (c) Erklären Sie die abweichenden Ergebnisse bei den Amish. 2.Beschreiben Sie die Erbkrankheit Glutarazidurie Typ I durch Auswertung der Materialien M1 bis M5 und begründen Sie Ihre Schlussfolgerungen. Benennen Sie auch den genetischen Defekt. 3.Ermitteln Sie mithilfe der Materialien M5 und M7 die Unterschiede im Bau der Glutaryl-CoA-Dehydrogenase von Gesunden und GA I-Patienten. 4.Untersuchen Sie den Erbgang der Glutarazidurie Typ I aufgrund von M6, benennen und erläutern Sie ihn. 5.Erläutern Sie, wie mithilfe der Amniozentese und einer Gensonde festgestellt werden kann, ob ein menschlicher Embryo die Krankheit Glutarazidurie Typ I in den ersten Lebensjahren ausbilden oder nicht ausbilden könnte. 6.Begründen Sie mithilfe von M1, wie bei einem Neugeborenen die Krankheit Glutarazidurie Typ I biochemisch festgestellt werden könnte. 7.Beurteilen Sie die Aufnahme eines Glutarazidurie-Tests in die Testverfahren, die jedes Neugeborene durchlaufen muss (Neugeborenenscreening). 8.Stellen Sie eine Hypothese auf für eine Glutarazidurie-Typ I-Therapie. Genetik Material M1 Ausschnitt aus dem Aminosäurestoffwechsel Tryptophan Hydroxylysin Lysin Ketoadipinsäure 3-Hydroxyglutarsäure KetoglutaratDehydrogenase Glutaryl-CoA Glutaconyl-CoA Glutaryl-CoADehydrogenase Glutaconsäure Crotonyl-CoA Glutarsäure + Carnitin Acetyl-CoA Glutarylcarnitin M1 Abbauwege der Aminosäuren Tryptophan, Lysin und Hydroxylysin. Das Abbauprodukt Acetyl-CoA kann z. B. im Citratzyklus weiterverarbeitet werden. Bei Patienten mit GA I kommt es zur Anreicherung von Glutaryl-CoA. Im Urin dieser Kranken findet man vor allem das harmlose Glutarylcarnitin, aber auch die giftigeren Stoffe Glutarsäure, Glutaconsäure und die besonders gefährliche 3-Hydroxyglutarsäure. Hinweis: Enzyme in blauer Schrift M2 Symptome von GA I-Patienten Die meisten Patienten mit GA I erleiden in den ersten Lebensjahren eine einzige enzephalopathische Krise. In wenigen Minuten werden dabei bestimmte Neurone im Bewegungszentrum des Gehirns zerstört. Die Folge ist eine äußerst schwere Bewegungsstörung. Die Intelligenz der Kinder dagegen ist weitgehend unbeeinträchtigt. Bleibt die Erkrankung unbehandelt, entwickelt sich in späteren Jahren oft zusätzlich eine geistige Retardierung. Ungefähr 25 Prozent der Patienten erleiden keine enzephalopathischen Krisen, sondern entwickeln schleichend eine Bewegungsstörung unterschiedlichen Ausmaßes. M3 Strukturelle Ähnlichkeit des Neurotransmitters Glutamat COOH COOH CH2 HCNH2 HCOH CH2 CH2 CH2 COOH 3-Hydroxyglutarsäure COOH Glutaminsäure M3 Das Salz der Glutarsäure Glutamat ist ein wichtiger erregender Neurotransmitter im Gehirn. Die Glutarsäure ist der 3-Hydroxyglutarsäure strukturell sehr ähnlich. 19p13.2 M4 Lokalisation des Gens für das Enzym Glutaryl-CoA-Dehydrogenase M4 Chromosoms 19 (schematisch): Das Gen für das Enzym Glutaryl-CoA-Dehydrogenase GCGH befindet sich auf dem kurzen Arm (p) des Chromosoms 19 an Position 13.2. 91