8.5. Pankreastumore Entsprechend der funktionellen
Werbung

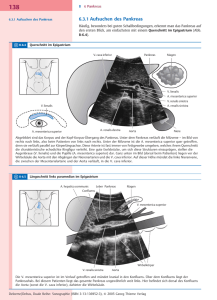
Ringel, Mayerle, Lerch Pankreastumoren 8.5. Pankreastumore Entsprechend der funktionellen Differenzierung des Pankreas unterscheidet man exokrine und endokrine Tumoren. Das duktale Adenokarzinom ist mit ca. 85% der häufigste Pankreastumor. Zu den selteneren epithelialen Neoplasien des exokrinen Teils zählen Azinuszell-Karzinome, serös-zystische und muzinös-zystische Tumore, intraduktale papillär-muzinöse Neoplasien (IPMN) und ampulläre Karzinome. Etwa 1 bis 2 % der Tumore sind endokrine Tumore. 8.5.1 Adenokarzinom des Pankreas Das Pankreaskarzinom gehört zu den Neoplasien mit besonders schlechter Prognose und relativ hoher Inzidenz. In Deutschland versterben daran jedes Jahr mehr als 11.000 Menschen [1]. In den USA steht es an 4. Stelle in der Statistik der Krebstodesursachen. Das mediane Erkrankungsalter liegt in der 6. – 8. Lebensdekade. Die 5 Jahresüberlebensrate beträgt nach optimistischen Studien 10% bei einer Resektionsrate zwischen 14-20 %. Insgesamt versterben 65% aller Patienten während der ersten 6 Monate nach Diagnosestellung [1]. Vom histologischen Typ handelt es sich bei etwa 70-80% der Tumoren des exokrinen Pankreas um duktale Adenokarzinome. Etwa 70% der Karzinome treten im Kopfbereich des Pankreas und im Processus uncinatus auf. Im Korpusbereich findet sich etwa ein Fünftel der Tumore. Tumoren im Schwanzbereich sind seltener aber wegen der erst spät einsetzenden Symptome mit einer besonders schlechten Prognose vergesellschaftet. Die Prognose für Patienten mit Pankreaskarzinomen wird vorrangig durch das Tumorwachstum, die lokale Infiltration und das Metastasierungsverhalten bestimmt. 8.5.1.1 Risikofaktoren und Pathogenese Mehrere Risikofaktoren gelten als ursächlich beteiligt an der Entstehung des Pankreaskarzinoms. Einerseits sind dies Umweltfaktoren und Lebensgewohnheiten, andererseits Vor- und Begleiterkrankungen. Rauchen gilt als der am besten belegte und in Studien reproduzierbare Risikofaktor [1]. Daneben gelten Pestizide und eine fettreiche Ernährung als Risikofaktoren. Wie bei anderen malignen Neoplasien wie dem kolorektalen Karzinome und dem Mammakarzinom geht eine Adipositas mit Ringel, Mayerle, Lerch Pankreastumoren einem erhöhten Pankreaskarzinomrisiko einher. Studien haben nachgewiesen, dass Diabetiker gegenüber Nicht-Diabetikern ein etwa doppelt so hohes Risiko für ein Pankreasadenokarzinom hatten. Eine langjährige chronische Pankreatitis ist ebenfalls mit einem erhöhten Risiko assoziiert [2,3]. Hier steigert sich das Risiko nach 20 Jahren auf 4%. Wahrscheinlich die größten Risikofaktoren sind definierte Erberkrankungen und Syndrome (Abb 1). Die Gründe für das aggressive Wachstum und die frühe Metastasierung der Pankreasadenokarzinome werden bis heute nur teilweise verstanden. Die derzeitigen Vorstellungen zur Ätiologie/Pathogenese des duktalen Adenokarzinoms basieren auf einem sequenziellen Progressionsmodell ähnlich der von Vogelstein für das kolorektale Karzinom postulierten Adenom-Karzinom-Sequenz. Über die Zeit akkumulieren Mutationen in Protoonkogenen und Tumorsuppressorgenen, so dass sich aus morphologisch definierten präneoplastischen Läsionen über das Stadium der Hyper- und Dysplasie ein Pankreaskarzinom entwickeln kann. Noch nicht geklärt ist, ob es sich dabei wirklich um eine lineare Sequenz handelt und wie die zeitliche Progression der einzelnen Grade verläuft. Als Ausgangspunkt für das duktale Adenokarzinom wurden hyperplastisch-papilläre Läsionen des Gangepithels identifiziert. Davon ausgehend können pankreatische intraepitheliale Neoplasien (PanINs) der Grade 1 bis 3 unterschieden werden, die durch eine bestimmte Morphologie sowie assoziierte molekulare Veränderungen charakterisiert sind. Verschiedende molekularbiologische Studien und Untersuchungen zeigen das gehäufte Auftreten von Mutationen des Ki-Ras Onkogens und der Tumorsuppressorgene p53, p21, p16, DPC4 und BRCA2. Neben den genetischen Veränderungen zeigen Pankreasadenokarzinome eine Deregulation von Wachstumsfaktoren und ihrer Rezeptoren. Intensiv wurde die Expression und der Einfluß des Transforming Growth Factor (TGF-), der TGF- Familie und des Epidermal Growth Factor (EGF) und deren spezifischen Rezeptoren untersucht. 8.5.1.2 Klinik und Diagnostik Die klinischen Symptome sind unspezifisch und äußern sich meistens in abdominellen (epigastrischen) Schmerzen und/oder Rückenschmerzen, Inappetenz, einem Gewichtsverlust sowie einem Ikterus. Im manchen Fällen manifestiert sich im Vorfeld ein Diabetes mellitus. Aufgrund der unspezifischen Symptomatik gestaltet Ringel, Mayerle, Lerch Pankreastumoren sich die Diagnostik insbesondere die die Differenzialdiagnostik zwischen chronischer Pankreatitis und Pankreaskarzinom schwierig. Bisher ist kein Laborverfahren geeignet eine sichere Differenzierung zwischen chronischer Pankreatitis und Pankreaskarzinom zu ermöglichen. Somit eignen sich derzeit Tumormarker im Serum auch nicht zur Diagnostik oder zur Differenzierung. Auch die einzelnen bildgebenden Verfahren lassen oftmals keine genaue Differenzierung zu und deshalb werden verschiedene Bildgebungsverfahren im Einzelfall kombiniert. Die Abdomensonographie stellt als einfaches, leicht zugängliches und preiswertes Verfahren die bildgebende Basisuntersuchung dar. Hierbei sind aber die bekannten Einschränkungen wie Erfahrung des Untersuchers, Patientenkonstitution und Luftüberlagerung limitierend. Eine deutlich bessere räumliche Auflösung lässt sich mit dem endoskopischen Ultraschall (EUS) erzielen (Abb. 2). Die Sensitivität des endoskopischen Ultraschalls beim lokalen Staging von Pankreaskarzinomen, liegt für T1-Tumore bei 88%, T2 bei 100%, und T3 bei etwa 93%. Die Anwendung einer Kontrastmittel-verstärkten Endosonographie kann die Differenzialdiagnose solider Pankreastumoren verbessern. Zusätzlich besteht bei dem Einsatz von linearen Sonden die Möglichkeit der Punktion. Entsprechend der Literatur sind die Multislice-Computertomographie (CT) und die Magnetresonanztomographie (MRT) inklusive Magnetresonanzcholangio- pankreatikographie sehr sensitive Verfahren zur Diagnostik und zum Staging von Pankreastumoren [4]. Mittels der Bildgebung kann dann die TNM-Klassifikation (Abb. 3) und somit eine Stadieneinteilung erfolgen. Insbesondere die Multislice-Computertomographie stellt ein Standardverfahren in vielen Zentren dar, da hiermit relativ kostengünstig Pankreastumore mit einer Sensitivität von etwas über 90% detektiert werden können. Im Gegensatz zur Endosonographie können mittels CT Fernmetastasen zuverlässig nachgewiesen werden. Dagegen scheint die Sensitivität der Multislice- Computertomographie bei T1 Tumoren mit 80% etwas geringer als die der Endosonographie. MRT und CT sind im T-Staging und N-Staging etwa gleichwertig. Durch die diffusionsgewichtete Analyse ist das MRT bei der Detektierung auch kleinster Lebermetastasen dem CT überlegen. Derzeit kann noch keine eindeutige Bewertung hinsichtlich der Überlegenheit der einzelnen Verfahren abgegeben Ringel, Mayerle, Lerch Pankreastumoren werden. Deshalb müssen die Methoden auch ggf. komplimentär eingesetzt werden [1,4]. 8.5.1.3 Therapie Die Therapie und Prognose des Pankreasadenokarzinoms hängt vom Stadium zum Zeitpunkt der Diagnose ab. Da die R0 Resektion die einzige potentiell kurative Therapie darstellt, muß zunächst anhand der Bildgebung die Operabilität festgelegt werden [1]. Wie oben beschrieben ist hier in Abhängigkeit von den lokalen Möglichkeiten und Expertisen die Kombination von verschiedenen bildgebenden Methoden von Vorteil. Entsprechend der aktuellen Leitlinien gelten im Allgemeinen Patienten mit Fernmetastasen oder mit einer Infiltration der arteriellen Gefäße als inoperabel [1,5]. In einzelnen Zentren gibt es dabei insbesondere im Rahmen von Studien abweichende Vorgehensweisen. Chirurgische Standardverfahren von Pankreaskopfkarzinomen sind die partielle Duodenopankreatektomie (klassische Kausch-Whipple OP) und die pyloruserhaltende partielle Duodenopankreatektomie (PP-Whipple), wobei als Therapieprinzip eine radikale Tumorentfernung mit einer Lymphknotendissektion peripankreatisch, im Bereich des Ligamentum hepatoduodenale sowie entlang des Truncus coeliacus und der Arteria und Vena mesenterica superior erfolgt. Ziel ist eine R0 Resektion. Die Mortalität dieser Operationen liegt heute in erfahrenen Zentren unter 5%. Bei Pankreaskorpus- und kaudatumoren wird eine Pankreaslinksresektion erforderlich. Die Komplikationsrate liegt hier zwischen 15-20% [1,5]. Vielfältig diskutiert wird das präoperative Vorgehen bei Ikterus. Liegt zum Diagnosezeitpunkt ein cholestatischer Ikterus vor, sollte entsprechend den Empfehlungen der Leitlinie nur bei Zeichen einer Cholangitis oder einer zeitlichen Verzögerung der Operation eine Galleableitung mittels Stent- oder Drainageeinlage erfolgen [1]. Aufgrund verschiedener Studien (CONKO-1, ESPAC-1 und 3v2) gilt eine adjuvante Chemotherapie nach R0 Resektion heute als Therapiestandard [1,6]. Auf Grundlage der CONKO-1 und der ESPAC-3v2 Studie bildete die 6-monatige Gemcitabinegabe die Standardchemotherapie [6]. Eine 5-Fluorouracil-basierte adjuvante Chemotherapie ist ähnlich effektive (Steigerung der 5-Jahres Überlebensrate von ca 10% auf über 20%) hat aber etwas ausgeprägtere Nebenwirkungen. Trotz verschiedener kleiner Untersuchungen empfiehlt die aktuelle Leitlinie keine Ringel, Mayerle, Lerch Pankreastumoren Radiochemotherapie sowohl in der adjuvanten als auch in der palliativen Situation weil die ESPAC-1 Studie einen Überlebens-Nachteil für Patienten nach Bestrahlung gezeigt hat. Obwohl in einer kleinen Studie vielversprechende Ergebnisse für eine intensivierte neoadjuvante Chemotherapie demonstriert wurden, kann diese derzeit nur unter Studienbedingungen empfohlen werden. Beim inoperablen Patienten (lokal fortgeschritten oder metastasiert) ist eine histologische Sicherung Tumorerkrankungen des steht Karzinoms eine obligat. Wie patientenzentrierte und bei allen malignen symptomorientierte Behandlung mit optimaler Schmerz- und Ernährungstherapie dabei im Mittelpunkt [1,7]. Daneben sollte bei entsprechendem körperlichen Allgemeinzustand eine palliative Chemotherapie erfolgen. Diese besteht standardmäßig aus einer Gemicabine Monotherapie (1000mg/m2 i.v., 1x wöchentlich) bis zum Progress des Tumorleidens [1,7]. Studien haben für diese Therapie eine Lebensverlängerung, eine verbesserte Lebensqualität, einen verringerten Schmerzmittelbedarf und einen geringeren Gewichtsverlust im Vergleich zur best-supportive-care erbracht. Die Studie von Moore et al. erbrachte einen signifikanten Vorteil für die Kombination von Gemcitabine mit dem Tyrosinkinaseinhibitor Erotinib bei Patienten mit metastasiertem Pankreaskarzinom [1]. Dabei korreliert das Ansprechen eng mit Ausbildungsgrad eines Exanthems (Rush) als typische Nebenwirkung. So verlängerte sich die Einjahresüberlebensrate von 17% bei Genitabine Monotherapie auf 23% bei der Kombination mit Erlotinib. Auf der anderen Seite kann die Erlotinibgabe nach etwa 6 Wochen beendet werden, wenn sich kein Exanthem (Rush) entwickelt hat, weil sie dann als wirkungslos gelten muss. Alle anderen bisherigen Chemotherapien/kombinationen erbrachten insbesondere unter Berücksichtigung der Toxizitäten keinen relevanten Vorteil. Als Zweitlinientherapien können 5-Flurouracil oder Oxaliplatin-basierte Therapien bei Patienten mit gutem körperlichem Zustand angewendet werden. Die CONKO-003 Studien zeigte eine Verdoppelung der Überlebenszeit unter dem OFF-Regime (Oxaliplatin, Folinsäure, 5-Flurouracil) als Zweitlinientherapie. Desweiteren kommen komplikationsorientierte Maßnahmen wie endoskpische Drainage bei Verschlußikterus oder Parazentesen bzw. lokale Chemotherapie bei maligem Aszites in Betracht [1,7]. Die Gastroenterostomie bei Infiltration bzw. Stenosierung des Duodenum und die biliodigestive Anastomose bei sonst nicht drainierbarer Cholestase stellen palliativchirurgische Maßnahmen dar. Ringel, Mayerle, Lerch Pankreastumoren Zusammenfassend ist zu sagen, dass nur eine kleine Gruppe von Patienten mit früh diagnostiziertem lokal-begrenzten Tumor Aussicht auf eine Heilung durch R0Resektion hat. Der Mehrzahl der Erkrankten kann aufgrund der späten Diagnose nur eine palliative Therapie angeboten werden mit dem Ziel der Verbesserung der Lebensqualität und einer Lebensverlängerung von einigen Monaten. Ziel derzeitiger Studien ist die Etablierung einer hochwirksamen neoadjuvanten Chemotherapie bei einem selektioniertem Patientengut. 8.5.2 Zystische Tumore des Pankreas Zystische Pankreasläsionen stellen eine sehr heterogene Gruppe von Neoplasien und oft eine diagnostische Herausforderung dar. 85 bis 90% aller Pankreaszysten sind Pseudozysten. Kongenitale Pankreaszysten z.B. im Rahmen von polyzystischen Syndromen oder der von-Hippel-Lindau Erkrankung machen etwa 2-3% der zystischen Läsionen aus. Die restlichen etwa 10% gilt es zu differenzieren in seröse oder muzinöse Zystadenome, IPMNs und Zystadenokarzinome sowie solidpseudopapilläre Tumore des Pankreas [8,9]. Ausgangspunkt bei der Diagnostik von zystischen Pankreasläsionen ist meist die Computertomographie. Aufgrund der hohen lokalen Auflösung hat die Endosonographie die höchste Sensitivität (93-100%) und Spezifität (92-98%) [4]. Die diagnostische Punktion einer zystischen Läsion mittels EUS ermöglicht durch die Bestimmung von Viskosität, Amylase/Lipase, CEA und einer zytologischen Diagnostik zwischen Pseudozysten und zystischen Neoplasien. So weisen z.B. Pseudozysten im Gegensatz zur serösen oder mucinösen Zystadenomen sehr hohe Konzentrationen an Amylase oder Lipase auf. Im Gegensatz dazu ist die CEA Konzentration in mucinösen Zystadenomen/-karzinomen signifikant höher als bei Pseudozysten oder serösen Zystadenomen (Abb. 4). Zur Differenzialdiagnose zystischer Pankreasläsionen insbesondere zum Nachweis eines Ganganschlusses ist die MRT kombiniert mit einer MRCT geeignet [4]. Auf eine invasive endoskopische retrograde Pankreatikographie (ERP) kann deshalb in vielen Fällen verzichtet werden. Daneben stellen Alter, Geschlecht, Anamnese (z.B. vorbekannte Pankreatitis oder Trauma, Gewichtsabnahme) wichtige differenzialdiagnostische Kriterien dar. So sind Ringel, Mayerle, Lerch Pankreastumoren zystische Läsionen vor dem Hintergrund einer bildgebend akuten oder chronischen Pankreatitis meist Pseudozysten [8,9]. Die Abbildung 4 fast schematisch ein differenzialdiagnostischen Algorhythmus für zystische Pankreasläsionen zusammen. Klinisch handelt es sich meist um asymptomatische Zufallsbefunde. Daneben können aber auch unspezifische Symptome wie Abdomenschmerzen, Völlegefühl, Übelkeit und bei entsprechender Lokalisation selten ein Ikterus auftreten. Gewichtsverlust oder ein palpabler Tumor weisen eher auf ein malignes Geschehen hin. In der Abbildung 5 sind häufigsten zystischen Pankreasneoplasien mit ihren wichtigsten Charakteristika zusammengefasst. Im Folgenden soll nun auf die einzelnen zystischen Neoplasien eingegangen werden. 8.5.2.1 Seröses Zystadenom Das seröse Zystadenom gehört mit etwa 30% zu den häufigsten zystischen Tumoren und wird als benigne Neoplasie eingeschätzt [8,9]. Es existieren nur einzelne Fallberichte über seröse Zystadenokarzinome. Wie in der Tabelle dargestellt, sind häufiger Frauen ab dem 6. Lebensjahrzehnt betroffen. Diese mikrozystischen Läsionen mit oft mehr als fünf Zysten mit einer Größe von <2cm sind meist im Pankreaskopf lokalisiert (Abb. 6). Häuft zeigt sich auch eine typische zentrale, sternförmige teilweise kalzifizierte Narbe. Die seröse Zystenflüssigkeit ist die Konzentration von CEA, Mucin und Amylase gering bzw. negativ [8,9]. Aufgrund der grundsätzlich benignen Biologie der serösen Zystadenome ist ein konservatives Vorgehen mit Verlaufskontrollen sinnvoll. Bei bestehenden klinischen Symptomen und/oder Gang-oder Gefäßobstruktion sollte eine Operation erfolgen. 8.5.2.2 Muzinöses Zystadenom Das muzinöse Zystadenom (oder mucinöse zystische Neoplasie - MCN) oder ist definiert als Muzin-produzierende, septierte, zystenbildende epitheliale Neoplasie des Pankreas mit einem Ovar-typischen Stroma [8,9]. An diesem Tumor erkranken eher jüngere Patienten im Alter von vierzig bis sechzig Jahren. Frauen sind zehn Mal häufiger vom Zystadenom betroffen als Männer. Muzinöse Zystadenome sind in etwa 95% der Fälle im Pankreaskorpus und Kaudabereich lokalisiert. In der Bildgebung können sich solitäre als auch multilokuläre Läsionen mit einer durchschnittlichen Größe von 7-8cm darstellen (Abb. Ringel, Mayerle, Lerch Pankreastumoren 7 (Herr Ringel, Abb 7 sieht aus wie ein IPMN oder angeborene Zysten, bitte überprüfen was das wirklich war kein gutes Beispiel) und 8). Die Zysten sind von einer dicken fibrotischen Wand umgeben und haben im Gegensatz zu den intraduktalen papillär-muzinösen Neoplasien keinen Ganganschluss. Aufgrund des möglichen Überganges muzinöse Zystadenom in ein invasives muzinöse Zystadenokarzinom gilt es als prämaligne Neoplasie. So können benigne und maligne Areale eng nebeneinander lokalisiert sein. Als wichtiges differenzialdiagnostisches Mittel dient die Zystenflüssigkeitsanalyse. Die mucinösen Adenome haben im Gegensatz zu serösen Zysten und Pseudozysten sehr visköse Flüssigkeit, wodurch die Aspiration oftmals erschwert ist [8,9]. Mucinöse Adenome zeichnen sich durch einen hohen CEA Gehalt aus, wobei Studien als cut-off Grenze eine Konzentration von 192ng/ml zur Differenzierung zu nicht-mucinösen Neoplasien angeben. Weiterhin ist eine CEA Konzentration >400ng/ml als guter Prädiktor für Malignität mit einer Spezifität von 95-100% und einer Sensitivität von 45-50%. Die operative Resektion stellt das Mittel der Wahl bei dieser potentiell malignen Neoplasie dar. Dabei ist mit dem Nachweis eines invasiven Karzinoms ein onkologisches Vorgehen notwendig. Nach operativer Resektion eines mucinösen Zystadenoms ist Überlebenswahrscheinlichkeit 100% und entsprechend der Studienlage ist kein follow up notwendig. Bei der R0 Resektion eines mucinösen Zystadenokarzinoms ist die 5-Jahresüberlebensrate mit 20-60% deutlich besser als beim duktalen Adenokarzinom [8,9]. Hinsichtlich des Nutzens einer Chemotherapie bei Zystadenokarzinomen gibt es neben Einzelfallbeschreibungen keine gesicherten Daten. 8.5.2.3 Intraduktale papillär-muzinöse Neoplasie Während der letzten Jahre gewannen die intraduktalen papillär-muzinösen Neoplasien (IPMNs) eine zunehmende klinische Bedeutung. Dabei handelt es sich um eine potentiell maligne Neoplasie, die von einem Adenom über ein Carcinoma in situ bis zum invasiv wachsenden Karzinom reicht. IPMNs treten in beiden Geschlechtern mit annährend gleicher Häufigkeit auf und das mediane Alter zum Diagnosezeitpunkt liegt bei 65 Jahren [8,9,10]. Klinisch imponieren IPMNs sehr unspezifisch meist mit rezidivierenden Oberbauchschmerzen. In der Bildgebung zeigt sich oftmals ein zystischer Ringel, Mayerle, Lerch Pankreastumoren Pankreasprozess mit Pankreasgangerweiterungen (Abb. 9). Diese werden durch ausgeprägte Mucinproduktion hervorgerufen. Dadurch kann es zu rezidivierenden akute Pankreatitiden bis hin zum Vollbild einer chronischen Pankreatitis kommen. Mittels ERC und MRCP kann der Ganganschluss der Zysten dargestellt werden. Ein typisches Bild bei der ERCP ist das sogenannte „fish eye“ Zeichen, wobei es durch die enorme Produktion von stark viskösen Mucus zur Protrusion der Papilla Vateri kommt [8,9,10]. Auf Grundlage der Histologie unterscheidet man folgende Gruppen: 1. benigne IPMN, 2. borderline und 3. maligne IPMN [10]. Die malignen IPMN werden noch in zwei Subklassen das Carinoma in situ und das invasive Karzinom unterteilt. Imsgesamt machen die malignen IPMNs etwa 60% der intraduktalen papillär-muzinösen Neoplasien aus, wovon etwa zwei Drittel ein invasives Verhalten zeigen. Bei etwa 33 bis 51% dieser Gruppe lassen sich Lymphknotenmetastasen nachweisen. Aus klinischer Sicht werden drei IPMN Subtypen unterschieden: der Hauptgang-Typ („main duct“, prädominant vom Pankreashauptgang ausgehend) und Seitengang-Typ („branch duct“, prädominant in den Seitenästen) und ein gemischter Typ. Entsprechend einer Studie beträgt die 5 Jahres Überlebensrate beim Hauptgang-Typ 47% versus 90% beim Seitengang-Typ. Bei Hauptgang-IPMNs ohne Nachweis eines invasiven Tumors liegt die 5 Jahres Überlebensrate nach Operation bis zu 100% [10]. Präoperative Prädiktoren für Malignität sind wandständige Knoten mit einer Größe 10mm sowie eine Gangweite von 10mm, zystische Seitenastdilatation >30mm (muss das nicht 3mm heissen?), ein Diabetes mellitus sowie in der Zytologie höhergradigen Dysplasien (Abb. 9) [11]. Entsprechend der internationalen (Hauptgang Konsensusleitinien und Seitengang) sollen alle IPMNs, Hauptgang IPMNs, symptomatische und gemischte IPMNs mit Malignitätsprädiktoren chirurgisch entfernt werden [11]. Der Umfang der Resektion hängt dabei von der Lokalisation und der intraoperativen Schnellschnittanalyse ab. Aufgrund des multifokalen Auftretens sind auch totale Pankreatektomien beschrieben. Ein weiteres therapeutisches Problem stellt die postoperative Rezidivrate von ca. 20% dar. Deshalb werden jährliche Kontrolluntersuchungen mittels CT oder MRT empfohlen. Bei einem Karzinomnachweis sollte diese alle 6 Monate erfolgen [11,12]. Die Patienten können beim Rezidivnachweis von einer erneuten Resektion profitieren. Aufgrund der aktuellen Datenlage kann keine adjuvante Chemotherapie empfohlen werden. Ringel, Mayerle, Lerch Pankreastumoren Da Patienten mit IPMNs (30-35%) ein erhöhtes Risiko für extrapankreatische Neoplasien haben, sollte insbesondere auf kolorektale und Magenkarzinome geachtet werden. Die Leitlinien zur Therapie der IPMNs schlagen weiterhin vor, dass Tumoren < 3 cm, die von Seitenästen ausgehen und bekanntermassen selten maligne transformieren engmaschig beobachtet werden können (Abb. 10) [11,12]. 8.5.2.4 Solide pseudopapilläre Neoplasie Bei der soliden pseudopapillären Neoplasie handelt es sich um einen Tumor mit niedrigem malignen Potential wobei aber von der WHO zwei Subtypen unterschieden werden: 1. soliden pseudopapillären Neoplasie mit borderline malignen Potential und 2. das solide pseudopapilläre Karzinom. Betroffen sind mit etwa 82% Frauen mit einem medianem Alter von etwa 30 Jahren. Solide pseudopapilläre Neoplasien mit einer Größe von 1 bis 30cm (muss das nicht 3cm heissen?) können in jedem Pankreasabschnitt auftreten. In der Bildgebung und bei der Punktion zeigt sich oft ein hämorrhagisch-zystischer Tumor umgeben von einer Pseudokapsel [8,9,12]. Die operative Resektion mit einem guten Langzeitüberleben stellte die Therapie der Wahl dar. Insgesamt muss festgehalten werden, dass mit keiner bildgebenden Methode auch unter Einbeziehung von Anamnese und Analyse des Zystenpunktats immer eine adäquate Differenzierung von zystischen Pankreasläsionen erzielt werden kann. Deshalb sollte im Zweifel eine operative Resektion angestrebt werden. 8.5.3 Neuroendokrine Tumore des Pankreas Nur 1-2% der Pankreastumore sind neuroendokrine Tumore (NET), wobei aber aufgrund der verbesserten bildgebenden Diagnostik die Inzidenz funktionell inaktiver Tumore leicht steigt. Neuroendokrine Tumore des Pankreas können in zwei Gruppen eingeteilt werden: 1. funktionell aktive NET 2. inaktive NET Bei etwa 50% NET des Pankreas kommt es zu einer Überproduktion von funktionell aktiven Hormonen, welche eine charakteristische klinische Symptomatik auslösen Ringel, Mayerle, Lerch Pankreastumoren [13,14]. An erster Stelle steht dabei das Insulinom mit einem Anteil von etwa 70%, wobei Frauen häufiger betroffen sind. An zweiter Stelle findet sich mit einem Anteil von etwa 20% das Gastrinom, welches bei Männern etwas häufiger auftritt. Die NET gelten im Vergleich zum Pankreasadenokarzinom als vergleichsweise wenig aggressiv verlaufende Neoplasien mit guter Gesamtprognose. Die 5-JahresÜberlebensraten liegen je nach Studie zwischen 45 und 77%. Im metastasiertem Stadium liegt das mediane Überleben jedoch für Patienten mit gut differenzierten Karzinomen bei 33 Monaten bzw. bei 5 Monaten mit schlecht differenzierten Tumoren [13,14]. NET des Pankreas treten in etwa 10% im Rahmen familiären Tumorerkrankungen wie der multiplen endokrinen Neoplasie (MEN)Typ 1, der Von-Hippel-LindauErkrankung oder einer Neurofibromatose Typ 1 [13,14]. 8.5.3.1. Klinik und Diagnostik Bei inaktiven neuroendokrinen Tumoren kann die Symptomatik unspezifisch mit Abdomenschmerzen oder bei entsprechender Lokalisation mit Ausbildung eines Ikterus einhergehen[13,14]. Oft handelt es sich aber um Zufallsbefunde im Rahmen einer bildgebenden Diagnostik. Funktionell aktive NETs sind durch typische klinische Symptomkomplexe charakterisiert, welche durch die vermehrte Sekretion der entsprechenden Hormone hervorgerufen werden [13,14]. Ein Überblick wird in der Abbildung 11 gegeben. Für das Insulinom ist die Whipple`sche Trias diagnostisch wegweisend: - Hypoglykämiesyndrom - Blutzucker ≤50 mg/dl - Prompte Rückbildung der Symptomatik unter Glukosegabe Zum Nachweis der inadäquaten Insulinausschüttung dient der 72h Hungertest unter stationärer Beobachtung. Zur Diagnosesicherung sind folgende sechs Kriterien definiert [15]: - dokumentierter Blutzucker unter ≤2,2mmol/l - gleichzeitiger Insulinspiegel ≥36pmol/l - C-Peptidspiegel ≥200pmol/l - Proinsulin-Spiegel ≥5pmol/l - Β-Hydroxybutyrat-Spiegel ≤2,7mmol/l - Fehlender Nachweis von Sulfonylharnstoffen im Plasma/Urin Ringel, Mayerle, Lerch Pankreastumoren Beim Gastrinom ist nach Pausierung der Protoneninhibitorentherapie für mindestens eine Woche eine Hypergastrinämie von >1000pg/ml diagnostisch wegweisend. Bei gering erhöhten Gastrinspiegeln sollte ein Sekretintest durchgeführt werden. Neben den spezifischen Markerhormonen ist die Bestimmung von Chromogranin A insbesondere bei der Verlausbeurteilung von funktionell inaktiven NETs hilfreich. Im Rahmen der bildgebenden Diagnostik ist wie auch bei den anderen Tumoren des Pankreas bildet die Abdomensonographie die Basisdiagnostik. Als Methode der Wahl gilt aber auch hier die Endosonographie mit der Möglichkeit der Biopsiegewinnung. Daneben stellen die Computertomographie und auch das MRT Standardverfahren bei der Diagnostik und Verlaufskontrolle dar [14]. Aufgrund der speziellen Somatostatinrezeptorszintigraphie Biologie eine der zentrale NETs Rolle beim spielt die Nachweis des Primärtumors als auch bei der Ausbreitungsdiagnostik. Die Sensitivität liegt hier zwischen 64% und 100%. Die Lokalisationsdiagnostik kann durch die Verwendung von Bildfusionstechniken (SPECT/CT/MRT) verbessert werden. Die Rolle der Positronen-Emissions-Tomographie (PET) insbesondere mit speziellen Tracern wird derzeit nur an spezialisierten Zentren und wegen der hohen Kosten meist im Rahmen von Studien durchgeführt. Insbesondere aufgrund der oft kleinen (Primär-)Tumore ist bei der Diagnostik von NETs des Pankreas ein interdisziplinäres Vorgehen notwendig. 8.5.3.2. Therapie Aufgrund der biologischen Besonderheiten wurde in den letzten Jahren ein spezifisches TNM Klassifikationssystem für Pankreas NETs entwickelt. Dabei gibt es aber Unterschiede zwischen der 2010 veröffentlichten TNM Klassifikation der UICC (Union Internationale Contre le Cancer) und der in Europa besser etablierten ENETS (Europäischen Neuroendokrinen Tumorgesellschaft)-Klassifikation (Abb. 12) [17]. Zusammen mit dem für das Grading von neuroendokrinen Tumoren entscheidenende Proliferationsverhalten (Abb. 13) kann so eine Risikostratifizierung vorgenommen werden [17]. Die Operation mit kurativer Zielsetzung ist die Primärtherapie bei NETs des Pankreas. Hierfür gibt es verschiedenen Methoden wobei insbesondere beim Insulinom die Enukleation oder Pankreassegmentresektion angewandt wird [18]. Daneben Ringel, Mayerle, Lerch Pankreastumoren kommen aber neben der klassischen Kausch-Whipple Operation modifizierte Varianten wie die pyloruserhaltende partielle Duodenopankreatetomie zur Anwendung. Entsprechend der Lokalisation des Primärtumor kann auch eine Linksresektion oder bei dissiminiertem Auftreten eine totale Pankreatektomie durchgeführt werden. Leider liegt bei vielen NETs zum Diagnosezeitpunkt ein metastasiertes Stadium insbesondere mit Lebermetastasen vor (Abb. 14). Trotz vielfältiger Studien und Leitlinien sind derzeit keine optimalen evidenzbasierten Strategien zur Behandlung von Lebermetastasen etabliert. Es besteht Konsens darüber, dass eine R0 Resektion des Primärtumors inklusive regionaler Lymphknotenmetastasen und Lebermetastasen zu einer Prognoseverbesserung und Symptomlinderung führen. Nach Resektion von Lebermetastasen wird das 5-Jahresgesamtüberleben je nach Studie mit 46-86% beziffert. Als allgemein anerkannte Voraussetzungen für eine Lebermetastasenresektion werden folgende Kriterien angesehen [18]: - gut differenzierter NET (G1 oder G2) - akzeptabler Allgemeinzustand des Patienten inklusive Komorbititäten - Ausschluss nichtresektabler extrahepatischer Metastasen - Möglichkeit einer R0 Resektion mit einem funktionellem Leberrestparenchym von ≥30% - Akzeptable Morbidität und Mortalität der Operation Für nicht kurativ resektable Patienten stehen systemisch-medikamentöse Therapien und lokal-interventionelle Methoden zur Verfügung. Die medikamentöse Therapie umfasst Somatostatinanaloga (SSA) (und hier insbesondere Depotpräparate), Interferon –alpha, systemische Chemotherapien und in Rahmen von Studien zielgerichtete molekulare Therapien wie m-TOR Inhibitoren und Multityrosinkinaseinhibitoren. Durch die antiproliferative Wirkung der SSA konnte in placebokontrollierten Studien die Zeit zur Progression von 6 auf 14,3 Monate erhöht werden. Bei VIPomen und Glucagonomen lässt sich die hormonassoziierte Symptomatik mit SAA gut behandeln, wogegen Insulinome nur in etwa 50% der Fälle ansprechen. Als Alternative kann beim Insulinom eine Therapie mit Diazozid Diazoxid? (50-300mg/d) zur Insulinsekretionshemmung eingesetzt werden. Bei Gastrinomen erfolgt eine Ringel, Mayerle, Lerch Pankreastumoren hochdosierte dem Schweregrad der Erkrankung angepasste Therapie mit Protonenpumpeninhibitoren. Dagegen besteht keine Empfehlung für den Einsatz von SAA. Bei hochdifferenzierten funktionell nicht aktiven NETs kann bei stabiler Erkrankung ein follow-up bis zur Progression erfolgen. Die Standardchemotherapie bei progredienten neuroendokrinen Karzinomen des Pankreas besteht aus Streptozotocin (500mg/m2 i.v. Tag 1-5) und 5-Flurouracil (400mg/m2 i.v. Tag 1-5; beides Wiederholung alle 6 Wochen). Alternativ kann bei guter Herzfunktion Streptozotocin (500mg/m2 i.v. Tag 1-5) mit Doxorubizin (50 mg/m2 i.v, Tag 1 und 22; beides Wiederholung alle 6 Wochen) genutzt werden [19]. Erste kleine Studien haben gezeigt, dass Tomozolomid in der Monotherapie zu einer 30% Remission führen. Die Kombination mit Capecitabin wird in kleinen Studien untersucht. Oxaliplatinhaltige Chemotherapieregime werde in erster Linie als Reserveoption genutzt [19]. Entsprechend der ENETS radionulkeotidmarkierten Konsensusleitlinie Somatatostatinanaloga 90 0 stellt die (sogenannte 3 radionuclide therapy-PRRT) mit [ Y-DOTA , Tyr ] Octreotid oder [ Therapie peptide mit receptor 177 Lu-DOTA0, Tyr3] Octreotat eine vielversprechende Alternative bei inoperablen oder metastasierten neuroendokrinen Tumoren dar [20]. Analog zur Therapie von kleinzelligen Bronchialkarzinomen werden schlecht differenzierte neuroendokrinen Karzinomen des Pankreas (G3) mit einer 2 Kombinationstherapie mit Cisplatin (130mg/ m i.v. Tag 1-3) und Etoposid (45mg/ m2 i.v. Tag 2-3; beides Wiederholung alle 4 Wochen) mit relativ hohen partiellen Remissionsraten aber hohem Rezidivrisiko behandelt. Neben der systemischen medikamentösen Therapie bestehen verschiedene lokal interventinelle Verfahren insbesondere zur symptomatischen Therapie und Behandlung von Lebermetastasen. Dazu gehören die transarterielle Embolisation mit z.B. Lipiodol, transarterielle Chemoembolisation (TACE), wobei verschiedene Protokolle mit verschiedenen Chemotherapeutika verwendet werden können, oder die selektive interne Radiotherapie (SIRT) mit Yttrium-90 beladenen Mikrosphären. Aufgrund der häufigen hepatischen Metastasierung und bei geringer Proliferation können auch lokal ablative Verfahren wie Radiofrequenzablation und die laserinduzierte Thermoablation eingesetzt werden. Insgesamt gibt es drei Indikationsgruppen für diese Therapieansätze: Ringel, Mayerle, Lerch Pankreastumoren 1. Lokaltherapie mit kurativer Intention 2. zytoreduktive Lokaltherapie, wobei eine Reduktion von >90% der hepatischen Tumormasse als sinnvoll erscheint 3. palliative Indikation zur Erreichung der Symptomfreiheit Insgesamt gibt es zu diesen Verfahren keine randomisierten prospektiven Studien und die Datenlage beruht meist auf kleinen Kohortenstudien. 8.5.4 Seltene Tumore des Pankreas Zu den seltenen soliden Neoplasien des Pankreas gehört das Azinuszellkarzinom, welches meist im Alter von 50 bis 70 Jahren mit einer Häufung bei Männern auftritt. Es zeigt sich keine Prädominanz hinsichtlich der Lokalisation im Pankreas. In der Bildgebung aber insbesondere bei der histologischen Aufarbeitung zeigen die Tumoren oft Nekrosen. Azinuszellkarzinom Eosinophilie Klinisch Arthralgien, beschrieben. wurden subkutane Die im Zusammenhang Fettgewebsnekrosen Prognose entspricht etwa mit und den dem einen duktalen Adenokarzinom. Etwa 4% der exogenen Pankreastumoren weisen eine adenosquamöse Differenzierung auf. Das sehr seltene Plattenepithelkarzinom ist häufig im Pankreaskaputbereich lokalisiert. Etwas häufiger lässt sich meist im Korpus- und Kaudabereich das pleomorphe großzellige Karzinom nachweisen. Diese Neoplasie besteht aus mehrkernigen Riesenzellen und kann klarzellige Areale wie bei einem Nierenzellkarzinom enthalten und ist von Pankreasmetastasen des Nierenkarzinoms (s.u.) zu differenzieren. Eine hämatogene Metastasierung wurde beschrieben. Das pleomorphe großzellige Karzinom mit osteoklastenartigen Zellen (Osteoklastom) ist ein sehr seltener Tumor, welcher bei geringer Mitoserate kontinuierlich die Umgebung infiltriert. Das sowohl infiltrativ wachsende als auch in die Leber metastasierende Pankreatoblastom macht nur etwa 0,2% der Pankreaskarzinome aus, ist aber häufiger bei Kindern und stellt in dieser Altersgruppe praktisch den einzigen malignen Pankreastumor dar. In seltenen Fällen lassen sich auch Lymphome im Pankreas nachweisen. Pankreaslymphome representieren weniger als 1-2% aller malignen Pankreastumore und weniger als 1% aller extranodalen Non-Hodgkin Lymphome. Wenn differentialdiagnostisch der hochgradige Verdacht auf ein Lymphom besteht, sollte Ringel, Mayerle, Lerch Pankreastumoren auch bei einem operablem Stadium eine gezielte histologische Materialgewinnung erfolgen, da diese Tumore unter Radiochemotherapie eine sehr gute Prognose haben. Neben diesen seltenen Pankreasneoplasien lassen sich immer wieder Metastasen anderer Tumor insbesondere vom Nierenzellkarzinom im Pankreas nachweisen. Diese oft im Kaudabereich gelegenen Nierenzellkarzinommetastasen weisen eine starke Vaskularität auf und ihre Prognose ist nach erfolgreicher Resektion (R0) besser als die des genuinen Adenokarzinoms des Pankreas. Aber auch anderer Tumorentitäten wie z. B. Mammakarzinome, Melanome oder gastrointestinale Neoplasien metastasieren in seltenen Fällen in das Pankreas. Die Therapie entspricht der des Primarius. Referenzen 1. Adler G, Seufferlein T, Bischoff SC. S3-Leitlinie „Exokrines Pankreaskarzinom. Z Gastroenterol 2007; 45: 487-523 2. Brand RE, Lerch MM, Rubinstein WS, et al. Advances in counselling and surveillance of patients at risk for pancreatic cancer. Gut. 2007;56:1460-9 3. Howes N, Lerch MM, Greenhalf W et al. Clinical and genetic characteristics of heriditary pancreatitis in Europe. Clin Gastroenterol Hepatol 2004; 2: 252-261 4. Holzapfel K, Fingerle A, Rummeny E. Aktueller Stand der bildgebenden Diagnostik des Pankreaskarzinoms. Onkologe 2010; 16: 568-579 5. Keck T. Pankreaskarzinom – Operationsindikation und chirurgische Radikalität. Onkologe 2010; 16: 580-587 6. Neoptolemos JP, Stocken DD, Bassi C, et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: a randomized controlled trial. JAMA. 2010;304:1073-81. Ringel, Mayerle, Lerch Pankreastumoren 7. Hoffmeister A, Mössner J. Palliative Therapie des fortgeschrittenen Pankreaskarzinoms. Onkologe 2010; 16: 604-609 8. Kosmahl M, Pauser U, Peters K, et al. Cystic neoplasms of the pancreas and tumor-like lesions with cystic features: a review of 418 cases and a classification proposal. Virchows Arch. 2004;445:168-78. 9. Stamatakos M, Sargedi C, Angelousi A. Management of rare entity of primary pancreatic cystic neoplasms. J Gastroenterol Hepatol 2009; 24: 1203-1210 10. Longnecker DS, Adler G, Hruban RH et al. Intraductal papillary mucinous neoplasms of the pancreas. In: Hamilton SR, Aaltonen LA, eds Pathology and Genetics of Tumors of the Digestive System. WHO Classification of Tumors. Lyon IARC Press 200: 237-240 11. Tanaka M, Chari S, Adsay V et al. International consensus guidelines for management of intraductal papillary mucinous neoplasms and mucinous cystic neoplasms of the pancreas. Pancreatology 2006; 6: 17-32 12. Werner JB, Bartosch-Harlid A, Andersson R. Cystic pancreatic lesions: Current evidence for diagnosis and treatment. Scan J Gastroenterol 2011; 116 13. Perren A, Schmitt A, Komminoth P et al. Klassifikation und Pathologie gastroenteropankreatischer neuroendokriner Tumoren. Viszeralmedizin 2010; 26: 234-240 14. Klöppel G. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: towards a standardized approach to the diagnosis of the gastroentropancreaticic neuroendocrine tumors and theis prognostic stratification. Neuroendocrinology 2009; 90: 162-166 Ringel, Mayerle, Lerch Pankreastumoren 15. Plöckinger U. Neuroendokrine Tumoren des Gastrointestinaltraktes: Klinik und Diagnostik. Viszeralmedizin 2010; 26: 242-252 16. De Herder W. ENETS consensus guidelines. Well-differenciated pancreatic tumor/carcinoma: insulinoma. Neuroendocrinology 2006; 84: 183-188 17. Klöppel G, Rindi G, Perren A. Die TNM-Klassifikation der NET des Gastrointestinaltrakts und des Pankreas von ENETS und UICC. Der Pathologe 2010; 31: 353-354 18. Frilling A, Sotiropoulus G, Kornasiewicz O et al. Chirurgische Therapie neuroendokriner Lebermetastasen – Resektion, Transplantation. Viszeralmedizin 2010; 26: 253-261 19. Eriksson B, Annibale B, Bajetta E et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: Chemotherapy in patients with neuroendocrine tumors. Neuroendocrinology 2009; 90: 214-219 20. Kwekkeboom D, Krenning E, Lebtahi R et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: Peptide receptor radionuclide therapy with radiolabeld somatostatin analogs. Neuroendocrinology 2009; 90: 220-226 Ringel, Mayerle, Lerch Pankreastumoren Abbildung 1 Heriditäre Tumorsyndrom und ihr Pankreaskarzinomrisiko (nach) Syndrom/Erkrankung Hereditäres Brust-Ovarialkarzinom Syndrom (HBOC) Heriditäre Pankreatitis Peutz-Jeghers Syndrom Familial Atypical Multiple Mole Melanoma Syndrom (FAMMM) Familiäres Pankreaskarzinom Pankreaskarzinomrisiko 3 bis 4-fach 70 bis 100-fach Über 100-fach 13 bis 52-fach Etwa 57-fach Abbildung 2 Endosonographisches Bild eines Pankreaskopfkarzinoms Abbildung 3 TNM Klassifikation des Pankreaskarzinoms T – Primärtumor Tis T1 T2 T3 T4 Carinoma in situ Tumor ≤ 2cm, begrenzt auf das Pankreas Tumor >2cm, begrenzt auf das Pankreas Tumor überschreitet Pankreas, invadiert aber nicht Truncus coeliacus oder A. mesenterica Tumor überschreitet Pankreas, invadiert aber Truncus coeliacus oder A. mesenterica Ringel, Mayerle, Lerch Pankreastumoren N – regionale Lymphknotenmetastasen N0 N1 keine regionären LK Metastasen regionäre LK Metastasen M – Fernmetastasen M0 M1 keine Fernmetastasen Fernmetastasen Abbildung 4 Darstellung eines möglichen differenzialdiagnostischen Algorhythmus für zystische Pankreasläsionen. Detailierte Ausführung im Text. Darstellung siehe Anlage Abb 4 Abbildung 5 Charakterisierung zystischer Pankreasneoplasien hinsichtlich Geschlechtsverteilung, medianes Alter und hinsichtlich des Malignitätsgrades (nach 8) Seröses Zystadenom Mucinöse zystische Neoplasie IPMN Anteil Geschlechtsverteilung Altersdurchschnitt in % Jahre 30Frauen 60-70 40 Bis 45 Frauen 40-60 2033 <10 Frauen Männer 60-70 Frauen 30-40 Frauen Männer 50-60 Männer etwas bevorzugt 60-70 Solide pseudopalilläre Neoplasie Zystische <1 endokrine Tumore Duktales <1 Adenokarzinom mit zystischer Degeneration Malignes Potential Kurative OP, selten Karzinome Prämaligne Läsion Potentiell maligne Borderline/ selten Metastasen wie solide endokrine Tumore Wie Adenokarzinom Schlechte Prognose Ringel, Mayerle, Lerch Pankreastumoren Abbildung 6: MRT Darstellung eines polyzystischen Prozesses im Pankreaskopfbereich. Bildmorphologisch wäre differenzialdiagnostisch auch eine IPMN möglich. Die histologische Aufarbeitung erbrachte ein seröses Zystadenom. Abbildung 7: MRCP Darstellung multilokulär über das Pankreas verteilter zystischer Läsionen ohne Anschluss an das Pankreasgangsystem als Beispiel für ein mucinöses Zystadenom. Bei einem CEA Gehalt von >800ng/ml in der endosonographisch gewonnenen viskösen Zystenflüssigkeit kann auch schon ein mucinöses Zystadenokarzinom vorliegen. War das wirklich ein muzinöses Zystadeno-CA und welche der vielen Zysten war der Tumor? Abbildung 8: MRCP Darstellung einer großen solitären zystischen Läsion im Kaudabereich, die schwer von eine Pseudozyste zu differenzieren ist. In der histologischen Aufarbeitung nach Pankreaslinksresektion bei hohem CEA Spiegel zeigte sich ein muzinöses Adenom ohne Hinweis für Malignität. Das hier habe ich ergänzt. Bitte überprüfen und sicher stellen, dass dies wirklich ein muzinöses Adenom war. Abbildung 9: Endosonographische Darstellung einer IPMN mit stark erweitertem Pankreasgang mit nodulärer Struktur, welche in den Gang reichen und ein Malignitätsprädiktor darstellt (D1). Daneben zeigen sich zystischen Gangerweiterungen. Abbildung 10: Empfohlenes Vorgehen bei dem Vorliegen von Seitenast IPMNs. Größe der Läsion < 1cm > 1-3 cm ohne Malignitätskriterien* > 1-3 cm mit V.a. invasives Wachstum Empfohlenes Procedere MR/CT/EUS nach 1 Jahr Bildgebung alle 6 Monate Resektion *Die Malignitätskriterien umfassen –wie im Text beschrieben: wandständige Knoten mit einer Größe 10mm und eine Gangweite von 10mm, zystische Seitenastdilatation >30mm, höhergradige Dysplasien in der Zytologie Ringel, Mayerle, Lerch Pankreastumoren Abbildung 11 Die häufigsten funktionell aktiven NET des Pankreas mit entsprechenden Syndromen NET/Syndrom Leitsymptome Insulinom Zollinger-Ellison-Syndrom (Gastrinom) Hypoglykämie Ulcusleiden, Diarrhoen, Reflux Verner-Morrison-Syndrom (VIPom) Diarrhoe, Hypokaliämie Glucagonom Erythema Glucagon necrolyticans migrans, Diabetes, Gewichtsverlust Akromegalie GHRH GHRHom/Akromegalie CRHom/ACTHom/Cushing- CushingSyndrom Syndrom Verantwortliches Hormon (Pro-) Insulin Gastrin VIP (vasoaktives intestinales Polypeptid) CRH, ACTH Weitere Hormone Glucagon, PP Insulin, PP, Glucagon, ACTH, Chromogranin A PP, Glucagon, Somatostatin, Chromogranin A PP, Insulin, Somatostatin, Chromogranin A Malignität, % 5-10 50-80 75 80 Somatostatin, 100 Gastrin, Insulin, Chromogranin A Gastrin, PP, >90 Chromogranin A Abbildung 12 TNM Klassifikation neuroendokriner Tumore des Pankreas entsprechend der Europäischen Neuroendokrinen Tumorgesellschaft (ENETS) (nach 14) T – Primärtumor T1 T2 T3 T4 Tumor ≤ 2cm, begrenzt auf das Pankreas Tumor 2-4cm, begrenzt auf das Pankreas Tumor >4cm begrenzt auf das Pankreas, oder Invasion des Duodenums oder des Gallenganges peripankreatische Ausbreitung mit Invasion der großen Gefäße und der angrenzenden Organe N – regionale Lymphknotenmetastasen N0 N1 keine regionären LK Metastasen regionäre LK Metastasen M – Fernmetastasen Ringel, Mayerle, Lerch Pankreastumoren M0 M1 keine Fernmetastasen Fernmetastasen Abbildung 13 Grading neuroendokriner Tumore des Pankreas entsprechend ENETS (nach 14) Grad Mitose Zahl (10 HPF)a MIB-1-Index, %b G1 <2 ≤2 G2 2-20 3-20 G3 >20 >20 a b HPF = high power field, Zur Bestimmung wird empfohlen 3x100 Zellen im Bereich der stärksten Proliferation auszuzählen Abbildung 14 Computertomograpische Darstellung eines funktionell inaktiven hepatisch metastasierten neuroendokrinen Karzinoms des Pankreas hier sehe ich den Primärtumor nicht gut. Bitte in Photo markieren.